
Transport and interfacial properties of composite polymer electrolytes
10
Electrochimica Acta 45 (2000) 1481 – 1490 Transport and interfacial properties of composite polymer electrolytes G.B. Appetecchi, F. Croce, L. Persi, F. Ronci, B. Scrosati * Department of Chemistry, Electrochemical Section, Uni6ersity ‘La Sapienza’, P. le A. Moro 5, I -00185 Rome, Italy Received 2 November 1998; received in revised form 5 April 1999 Abstract Lithium polymer electrolytes formed by dissolving a lithium salt LiX in poly(ethylene oxide) PEO, may find useful application as separators in lithium rechargeable polymer batteries. The main problems, which are still to be solved for a complete successful operation of these materials, are the reactivity of their interface with the lithium metal electrode and the decay of their conductivity at temperatures below 70°C. In this paper we demonstrate that a successful approach for overcoming these problems, is the dispersion of selected ceramic powders in the polymer mass, with the aim of developing new types of composite PEO – LiX polymer electrolytes characterized by enhanced interfacial stability, as well as by improved ambient temperature transport properties. © 2000 Elsevier Science Ltd. All rights reserved. Keywords: Composite polymer electrolytes; Nanocomposite; Interfacial properties; Conductivity www.elsevier.nl/locate/electacta 1. Introduction The well known lithium polymer electrolytes formed by the dissolution of a lithium salt LiX in poly(ethylene oxide) PEO [1] are of practical importance, since they may find important application as separators in lithium rechargeable polymer batteries, LPBs. Indeed, large LPB research and development programs are presently in progress in North America and Europe [2–4]. How- ever, although successful materials, PEO – LiX polymer electrolytes are still affected by some problems, namely (i) a reactivity towards the lithium metal electrode; and (ii) a low conductivity at ambient temperature. Thus, work is needed to find the proper approaches for solving or at least minimizing these drawbacks. Indeed, the optimization and control of the electrode/polymer electrolyte interface is a key requisite for success in LPB development. Similarly, the enhancement of conductiv- ity to acceptable values at ambient temperature is of obvious importance for widening the application scopes of the polymer electrolytes. In this paper we demon- strate that the dispersion of selected ceramic powders in the polymer mass produces composite PEO – LiX poly- mer electrolytes showing consistent improvements in both interfacial and transport properties. 2. Experimental 2.1. Synthesis The procedures used for the synthesis of the com- posite polymer electrolytes were discussed in detail in previous works [5,6]. The basic goal was that of obtain- ing liquid-free, ceramic-added PEO-based composite electrolytes. This was achieved by a modification of the hot-pressing technique originally proposed by Vincent and co-workers [7]. Proper amounts of highly purified poly(ethylene oxide) PEO (either Aldrich Chemical Co. * Corresponding author. Tel./fax: +39-6-4462866. E-mail address: [email protected] (B. Scrosati) 0013-4686/00/$ - see front matter © 2000 Elsevier Science Ltd. All rights reserved. PII:S0013-4686(99)00363-1
Transcript of Transport and interfacial properties of composite polymer electrolytes

Electrochimica Acta 45 (2000) 1481–1490
Transport and interfacial properties of composite polymerelectrolytes
G.B. Appetecchi, F. Croce, L. Persi, F. Ronci, B. Scrosati *Department of Chemistry, Electrochemical Section, Uni6ersity ‘La Sapienza’, P. le A. Moro 5, I-00185 Rome, Italy
Received 2 November 1998; received in revised form 5 April 1999
Abstract
Lithium polymer electrolytes formed by dissolving a lithium salt LiX in poly(ethylene oxide) PEO, may find usefulapplication as separators in lithium rechargeable polymer batteries. The main problems, which are still to be solvedfor a complete successful operation of these materials, are the reactivity of their interface with the lithium metalelectrode and the decay of their conductivity at temperatures below 70°C. In this paper we demonstrate that asuccessful approach for overcoming these problems, is the dispersion of selected ceramic powders in the polymermass, with the aim of developing new types of composite PEO–LiX polymer electrolytes characterized by enhancedinterfacial stability, as well as by improved ambient temperature transport properties. © 2000 Elsevier Science Ltd.All rights reserved.
Keywords: Composite polymer electrolytes; Nanocomposite; Interfacial properties; Conductivity
www.elsevier.nl/locate/electacta
1. Introduction
The well known lithium polymer electrolytes formedby the dissolution of a lithium salt LiX in poly(ethyleneoxide) PEO [1] are of practical importance, since theymay find important application as separators in lithiumrechargeable polymer batteries, LPBs. Indeed, largeLPB research and development programs are presentlyin progress in North America and Europe [2–4]. How-ever, although successful materials, PEO–LiX polymerelectrolytes are still affected by some problems, namely(i) a reactivity towards the lithium metal electrode; and(ii) a low conductivity at ambient temperature. Thus,work is needed to find the proper approaches forsolving or at least minimizing these drawbacks. Indeed,the optimization and control of the electrode/polymerelectrolyte interface is a key requisite for success in LPBdevelopment. Similarly, the enhancement of conductiv-
ity to acceptable values at ambient temperature is ofobvious importance for widening the application scopesof the polymer electrolytes. In this paper we demon-strate that the dispersion of selected ceramic powders inthe polymer mass produces composite PEO–LiX poly-mer electrolytes showing consistent improvements inboth interfacial and transport properties.
2. Experimental
2.1. Synthesis
The procedures used for the synthesis of the com-posite polymer electrolytes were discussed in detail inprevious works [5,6]. The basic goal was that of obtain-ing liquid-free, ceramic-added PEO-based compositeelectrolytes. This was achieved by a modification of thehot-pressing technique originally proposed by Vincentand co-workers [7]. Proper amounts of highly purifiedpoly(ethylene oxide) PEO (either Aldrich Chemical Co.
* Corresponding author. Tel./fax: +39-6-4462866.E-mail address: [email protected] (B. Scrosati)
0013-4686/00/$ - see front matter © 2000 Elsevier Science Ltd. All rights reserved.
PII: S 0013 -4686 (99 )00363 -1

G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–14901482
or BDH Polyox WSR301, both 4 000 000 averagemolecular weight), LiCF3SO3 (Aldrich R.G Selectipur)or, alternatively, lithium tetrafluoborate, LiBF4 (MerckSelectipur, battery grade) and low particle size (about 4mm) gLiAlO2 (Cyprus Foote Mineral Co., HSA-10)were intimately mixed, heated and hot-pressed at110°C. After cooling in liquid nitrogen, the membraneswere dried at 55°C under vacuum for at least 48 h andstored in an argon atmosphere dry box. This methodproduced homogeneous, mechanically stable electrolytemembranes of 200 mm average thickness and with acomposition of either PEO20LiCF3SO3+20w/ogLiAlO2 or PEO20LiBF4+20w/o gLiAlO2 composition(w/o is the weight percent).
The nanocomposite polymer electrolytes were ob-tained by combining PEO with LiClO4 and using TiO2,Al2O3 or SiO2, respectively, as the ceramic filler. TheLiClO4/PEO concentration ratio was fixed to 1/8 con-centration and the amount of added ceramic to 10% ofthe total PEO8–LiClO4 weight. The preparation of thenanocomposite electrolyte samples involved first thedispersion of the selected ceramic powder and of theLiClO4 lithium salt in acetonitrile, followed by theaddition of the PEO polymer component and by athorough mixing of the resulting slurry. The homoge-neous slurry was then cast between two glass platesprovided by spacers to set thickness, to finally yieldmechanically stable membranes of average thickness ofabout 150 mm. The same casting procedure was alsoused to prepare ceramic-free PEO–LiClO4 samples forcomparison purpose.
2.2. Characterization
The various composite polymer electrolyte sampleswere characterized by conductivity, lithium ion trans-ference number and compatibility with the lithiummetal electrode. The conductivity was measured byplacing the given samples in a two stainless-steel elec-trode cell of 1 cm2 active area The measurements werecarried out both in heating and cooling scans using aSolartron Frequency Response Analyzer (Mod 1255)over a 0.1 Hz–100 kHz frequency range. The data wereprocessed by using an appropriate fitting program [8].
The lithium transference number, TLI+ , was evaluated
using two independent methods, i.e. the method pro-posed by Vincent and co-workers [9] and the refinedmethod proposed by Abraham and co-workers [10].According to these methods, the TLi
+ values were deter-mined by imposing ac and dc polarization pulses tocells of the Li/electrolyte sample/Li) type and by fol-lowing the time evolution of the resulting current flow.To improve the accuracy of the results, we have devel-oped a special software capable of acquiring a verylarge number of current data per second immediatelyafter the application of the voltage pulse.
The lithium electrode–polymer electrolyte interfacialstability was evaluated by monitoring the impedanceresponse of cells again formed by sandwiching the givenelectrolyte sample between two lithium electrodes.These cells were stored under open circuit conditionsand their impedance response was analyzed using thecited fitting program [8]. This procedure allows theseparation of contributions from the various phenom-ena which are observed, to determine the interfacialresistance. The interfacial stability and the associatedcyclability of the lithium electrode were further studiedand evaluated by imposing galvanostatic pulses on atwo-electrode cell using a finely polished nickel working(substrate) electrode, a lithium counter electrode, andthe selected composite membrane sample as the elec-trolyte. First, a known amount of charge (depositioncharge, QD) was passed through the cell in order topromote the ongoing of lithium deposition on thenickel substrate. Then, a fraction of this charge (cyclingcharge, QC) was alternately cycled across the cell topromote lithium deposition-stripping cycles and thelithium stripping overvoltage was monitored upon cy-cling. The test was assumed to be completed when asignificant increase in overvoltage was detected. Themean value of the lithium electrode cycling efficiency, h,was finally calculated by using the equation:
h=nQC
nQC+QD
(1)
where n represents the total number of cycles the cellhas undergone. The experiment was run and controlledusing a MACCOR 4000 battery cycler.
3. Results and discussion
Fig. 1 shows the Arrhenius plot of thePEO20LiCF3SO3+20w/o gLiAlO2 and of thePEO20LiBF4+20w/o gLiAlO2 polymer electrolyte sam-ples. It may be clearly seen that the conductivity curvesof both samples show a break around 60–70°C due tothe well-known crystalline-amorphous transition of thePEO component [1]. Useful conductivity (i.e. in the10−4 S cm−1 range) was generally obtained at temper-atures higher than ambient and typically around 80–90°C. Thus, this was assumed as the testingtemperature range for monitoring the stability of thelithium electrode/polymer electrolyte interface.
As well known, the reactivity of the lithium electrodecan affect this interface due to uncontrolled passivationphenomena which result in the formation of thick andnot-uniform surface layers [11]. These layers may inturn cause uneven lithium deposition in the course ofthe charge process, this leading to dendritic growth andeventually to cell short-circuiting. Therefore, the inves-tigation of the lithium interfacial characteristics is of
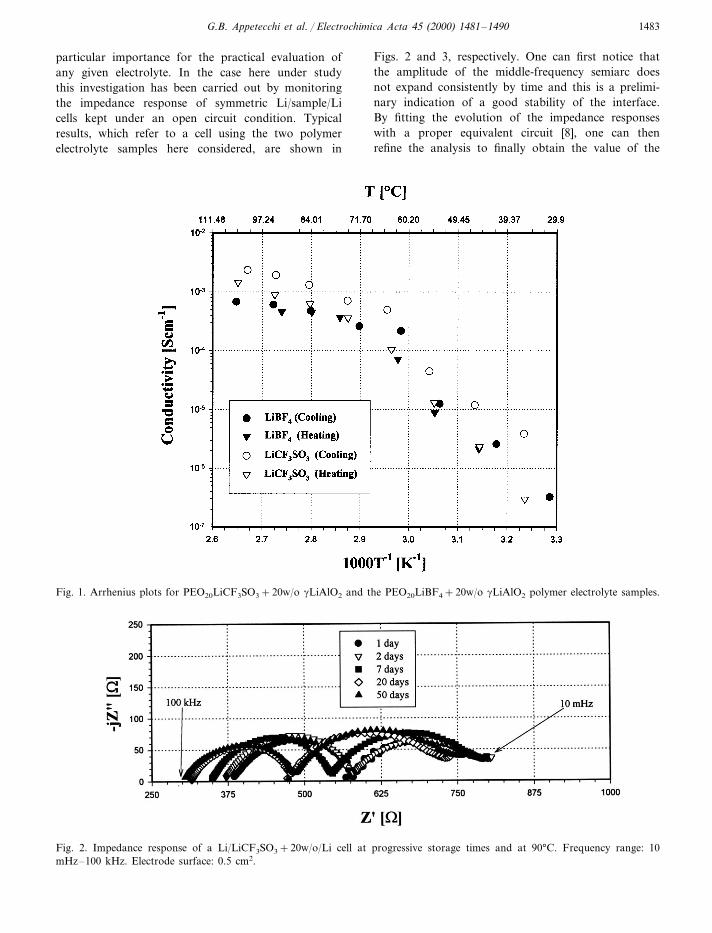
G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–1490 1483
particular importance for the practical evaluation ofany given electrolyte. In the case here under studythis investigation has been carried out by monitoringthe impedance response of symmetric Li/sample/Licells kept under an open circuit condition. Typicalresults, which refer to a cell using the two polymerelectrolyte samples here considered, are shown in
Figs. 2 and 3, respectively. One can first notice thatthe amplitude of the middle-frequency semiarc doesnot expand consistently by time and this is a prelimi-nary indication of a good stability of the interface.By fitting the evolution of the impedance responseswith a proper equivalent circuit [8], one can thenrefine the analysis to finally obtain the value of the
Fig. 1. Arrhenius plots for PEO20LiCF3SO3+20w/o gLiAlO2 and the PEO20LiBF4+20w/o gLiAlO2 polymer electrolyte samples.
Fig. 2. Impedance response of a Li/LiCF3SO3+20w/o/Li cell at progressive storage times and at 90°C. Frequency range: 10mHz–100 kHz. Electrode surface: 0.5 cm2.
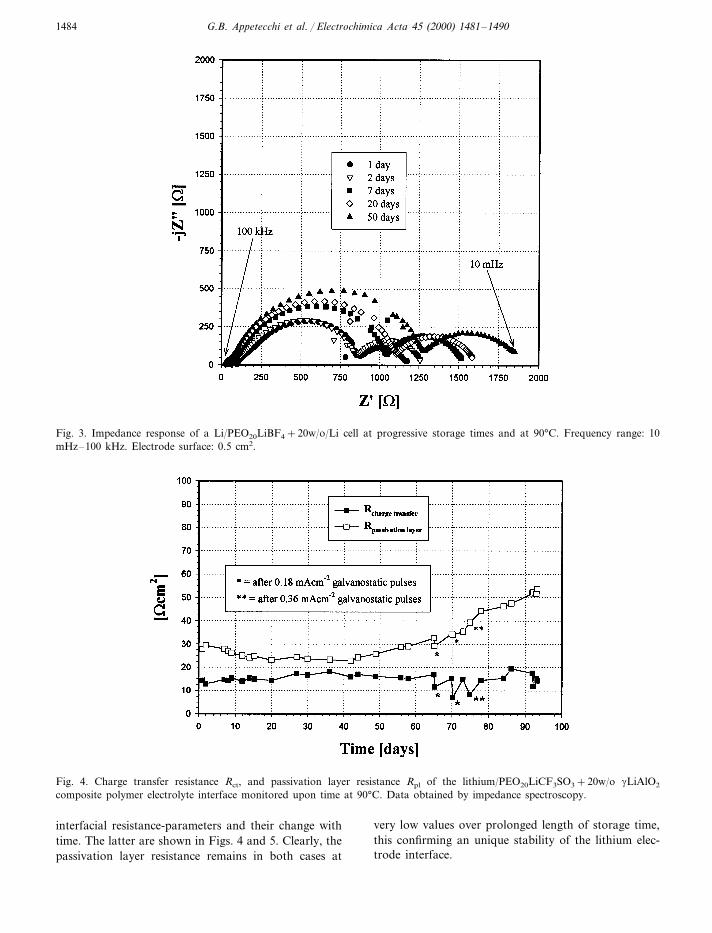
G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–14901484
Fig. 3. Impedance response of a Li/PEO20LiBF4+20w/o/Li cell at progressive storage times and at 90°C. Frequency range: 10mHz–100 kHz. Electrode surface: 0.5 cm2.
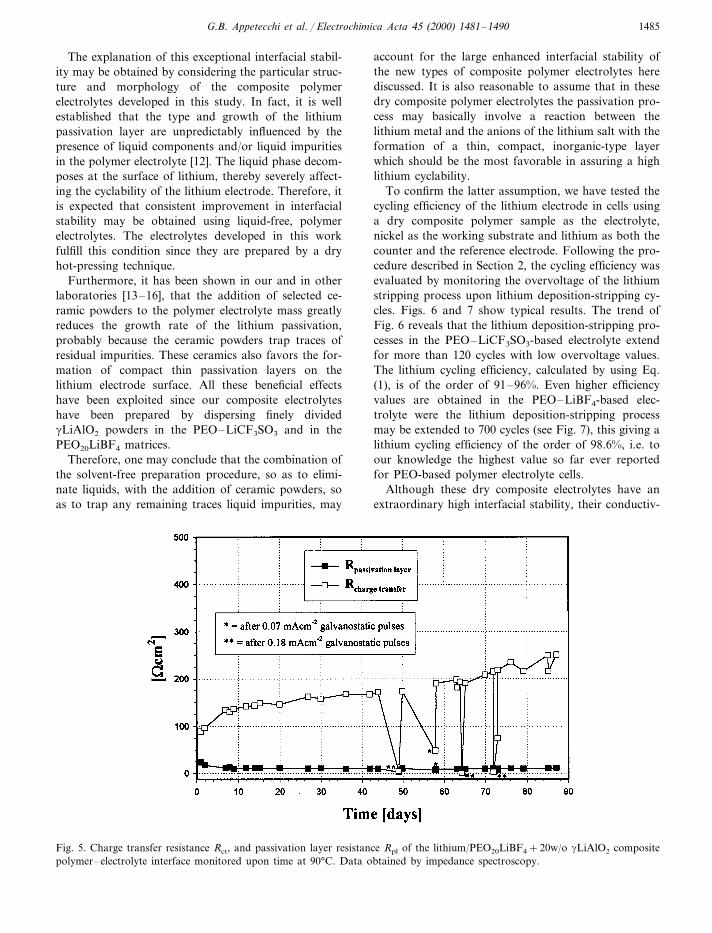
Fig. 4. Charge transfer resistance Rct, and passivation layer resistance Rpl of the lithium/PEO20LiCF3SO3+20w/o gLiAlO2
composite polymer electrolyte interface monitored upon time at 90°C. Data obtained by impedance spectroscopy.
interfacial resistance-parameters and their change withtime. The latter are shown in Figs. 4 and 5. Clearly, thepassivation layer resistance remains in both cases at
very low values over prolonged length of storage time,this confirming an unique stability of the lithium elec-trode interface.
G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–1490 1485
The explanation of this exceptional interfacial stabil-ity may be obtained by considering the particular struc-ture and morphology of the composite polymerelectrolytes developed in this study. In fact, it is wellestablished that the type and growth of the lithiumpassivation layer are unpredictably influenced by thepresence of liquid components and/or liquid impuritiesin the polymer electrolyte [12]. The liquid phase decom-poses at the surface of lithium, thereby severely affect-ing the cyclability of the lithium electrode. Therefore, itis expected that consistent improvement in interfacialstability may be obtained using liquid-free, polymerelectrolytes. The electrolytes developed in this workfulfill this condition since they are prepared by a dryhot-pressing technique.
Furthermore, it has been shown in our and in otherlaboratories [13–16], that the addition of selected ce-ramic powders to the polymer electrolyte mass greatlyreduces the growth rate of the lithium passivation,probably because the ceramic powders trap traces ofresidual impurities. These ceramics also favors the for-mation of compact thin passivation layers on thelithium electrode surface. All these beneficial effectshave been exploited since our composite electrolyteshave been prepared by dispersing finely dividedgLiAlO2 powders in the PEO–LiCF3SO3 and in thePEO20LiBF4 matrices.
Therefore, one may conclude that the combination ofthe solvent-free preparation procedure, so as to elimi-nate liquids, with the addition of ceramic powders, soas to trap any remaining traces liquid impurities, may
account for the large enhanced interfacial stability ofthe new types of composite polymer electrolytes herediscussed. It is also reasonable to assume that in thesedry composite polymer electrolytes the passivation pro-cess may basically involve a reaction between thelithium metal and the anions of the lithium salt with theformation of a thin, compact, inorganic-type layerwhich should be the most favorable in assuring a highlithium cyclability.
To confirm the latter assumption, we have tested thecycling efficiency of the lithium electrode in cells usinga dry composite polymer sample as the electrolyte,nickel as the working substrate and lithium as both thecounter and the reference electrode. Following the pro-cedure described in Section 2, the cycling efficiency wasevaluated by monitoring the overvoltage of the lithiumstripping process upon lithium deposition-stripping cy-cles. Figs. 6 and 7 show typical results. The trend ofFig. 6 reveals that the lithium deposition-stripping pro-cesses in the PEO–LiCF3SO3-based electrolyte extendfor more than 120 cycles with low overvoltage values.The lithium cycling efficiency, calculated by using Eq.(1), is of the order of 91–96%. Even higher efficiencyvalues are obtained in the PEO–LiBF4-based elec-trolyte were the lithium deposition-stripping processmay be extended to 700 cycles (see Fig. 7), this giving alithium cycling efficiency of the order of 98.6%, i.e. toour knowledge the highest value so far ever reportedfor PEO-based polymer electrolyte cells.
Although these dry composite electrolytes have anextraordinary high interfacial stability, their conductiv-
Fig. 5. Charge transfer resistance Rct, and passivation layer resistance Rpl of the lithium/PEO20LiBF4+20w/o gLiAlO2 compositepolymer–electrolyte interface monitored upon time at 90°C. Data obtained by impedance spectroscopy.

G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–14901486
Fig. 6. Lithium stripping overvoltage upon galvanostatic cycling of a lithium metal electrode in a PEO20LiCF3SO3+20w/o gLiAlO2
composite polymer electrolyte cell. Ni substrate. QD=1C; QC=0.1C. Temperature: 90°C. Current density: 0.1 mA cm−2.
Fig. 7. Lithium stripping overvoltage upon galvanostatic cycling of a lithium metal electrode in a PEO20LiBF4+20w/o gLiAlO2
composite polymer electrolyte cell. Ni substrate. QD=1C, QC=0.1C. Temperature: 90°C. Current density: 0.1 mA cm−2.
ity reaches useful values only at medium-high tempera-tures (see Figs. 1 and 2), and this may limit theirapplicability. Therefore, it would be quite useful tolower to the ambient region the temperature of opera-tion of the electrolytes. Large research efforts have beendevoted in the past to reach this goal for the generalclass of the PEO–LiX polymer electrolytes. The mostcommonly used approach has been the addition ofliquid plasticizers, e.g. low molecular weight
polyethylene glycols or aprotic organic solvents, to thePEO–LiX matrix. However, the addition of liquidsbeside resulting in a deterioration of the electrolyte’smechanical properties, greatly increases its reactivitytowards the lithium metal anode (see above). Therefore,the ideal achievement in electrolytes of the PEO–LiXtype would be the enhancement of low temperatureionic conductivity by modifications which avoid anyliquid contamination. This goal is not easily achievable
G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–1490 1487
since fast ion transport in PEO–LiX is a characteristicof the amorphous state which is intrinsically reachedabove 70°C or artificially induced at lower temperatureby the addition of liquid plasticizers.
We have considered to solve this problem by extend-ing our composite electrolyte approach to the use ofselected ceramic fillers, e.g. TiO2, Al2O3 or SiO2, at thenanoscale particle size. The idea is that these fillers mayact as solid plasticizers, capable of enhancing the com-posite polymer electrolyte’s transport properties with-out affecting its mechanical and interfacial stability [6].
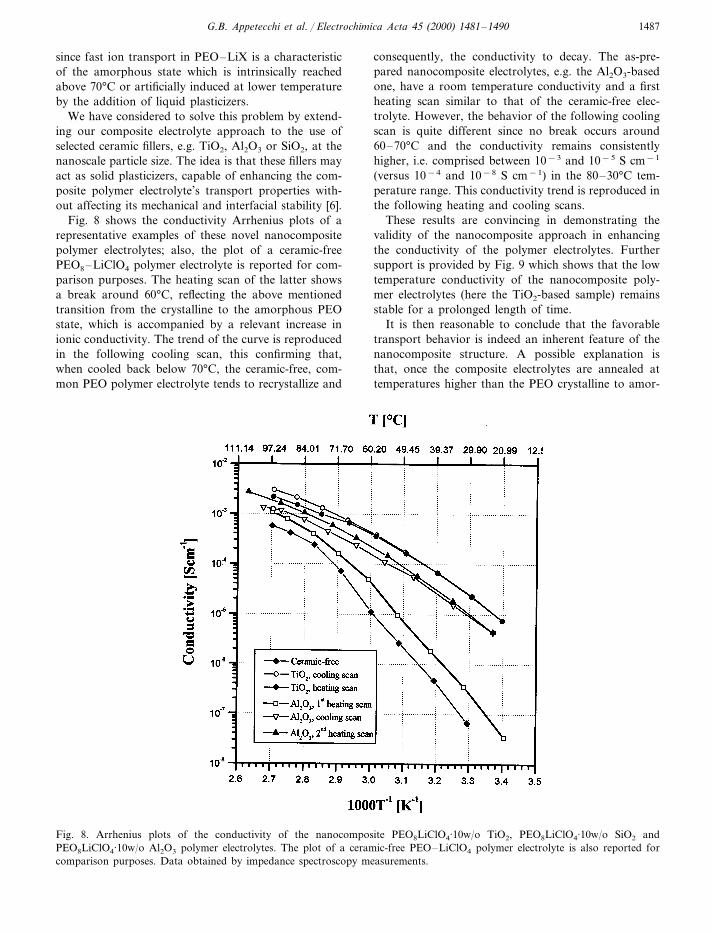
Fig. 8 shows the conductivity Arrhenius plots of arepresentative examples of these novel nanocompositepolymer electrolytes; also, the plot of a ceramic-freePEO8–LiClO4 polymer electrolyte is reported for com-parison purposes. The heating scan of the latter showsa break around 60°C, reflecting the above mentionedtransition from the crystalline to the amorphous PEOstate, which is accompanied by a relevant increase inionic conductivity. The trend of the curve is reproducedin the following cooling scan, this confirming that,when cooled back below 70°C, the ceramic-free, com-mon PEO polymer electrolyte tends to recrystallize and
consequently, the conductivity to decay. The as-pre-pared nanocomposite electrolytes, e.g. the Al2O3-basedone, have a room temperature conductivity and a firstheating scan similar to that of the ceramic-free elec-trolyte. However, the behavior of the following coolingscan is quite different since no break occurs around60–70°C and the conductivity remains consistentlyhigher, i.e. comprised between 10−3 and 10−5 S cm−1
(versus 10−4 and 10−8 S cm−1) in the 80–30°C tem-perature range. This conductivity trend is reproduced inthe following heating and cooling scans.
These results are convincing in demonstrating thevalidity of the nanocomposite approach in enhancingthe conductivity of the polymer electrolytes. Furthersupport is provided by Fig. 9 which shows that the lowtemperature conductivity of the nanocomposite poly-mer electrolytes (here the TiO2-based sample) remainsstable for a prolonged length of time.
It is then reasonable to conclude that the favorabletransport behavior is indeed an inherent feature of thenanocomposite structure. A possible explanation isthat, once the composite electrolytes are annealed attemperatures higher than the PEO crystalline to amor-
Fig. 8. Arrhenius plots of the conductivity of the nanocomposite PEO8LiClO4·10w/o TiO2, PEO8LiClO4·10w/o SiO2 andPEO8LiClO4·10w/o Al2O3 polymer electrolytes. The plot of a ceramic-free PEO–LiClO4 polymer electrolyte is also reported forcomparison purposes. Data obtained by impedance spectroscopy measurements.

G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–14901488
Fig. 9. Conductivity of the nanocomposne PEO8LiClO4·10w/o TiO2 polymer electrolyte monitored upon time at 31°C. Dataobtained by impedance spectroscopy measurements.
phous transition (i.e. above 70°C), the ceramic additive,due to its large surface area, prevents PEO chain reor-ganization with the result of freezing at ambient tem-perature a high degree of disorder which is likely to beaccompanied by a consistent enhancement of the ionicconductivity.
Accordingly, one may assume that the structuralmodifications are induced via Lewis acid–base reac-tions between the ceramic surface states and the PEOsegments, as in fact already proposed by Wieczorek etal. [17,18]. According to this model, the Lewis acidcharacter of the added ceramics would compete withthe Lewis acid character of the lithium cations for theformation of complexes with the PEO chains. Thus, theceramics would act as cross-linking centers for the PEOsegments, this lowering the polymer chain reorganiza-tion tendency and promoting an overall structure stiff-ness. Such a structure modification would provide Li+
ions conducting pathways at the ceramics surface, thisaccounting for the improvement in ionic transport.
We have undertaken a series of complementary teststo support this model. An important one was thedetermination of the lithium ion transference numberTLi
+ which, according to the model, should be consis-tently higher in the nanocomposite electrolytes than inceramic-free electrolytes. In fact, if the action of theceramic filler is that of promoting conducting pathways as a result of its Lewis acid type interactions with thePEO chains, the lithium ions are expected to movefreely along these ceramic surface pathways and thus, aconsistent enhancement of their transference number isexpected. Indeed, the results have been consistent indemonstrating an increase in TLi
+ when passing from theceramic-free to the nanocomposite polymer electrolytes[6]. In addition and to further confirm the model, theTLi
+ values consistently vary according to the Lewis acidcharacter of the added ceramic. For the most acidic
type, i.e. the TiO2 one, transference number values ofthe order of 0.5–0.6 in the 45–90°C temperature rangewere obtained. To our knowledge, such a high valuehas never been reported for common PEO-basedelectrolytes.
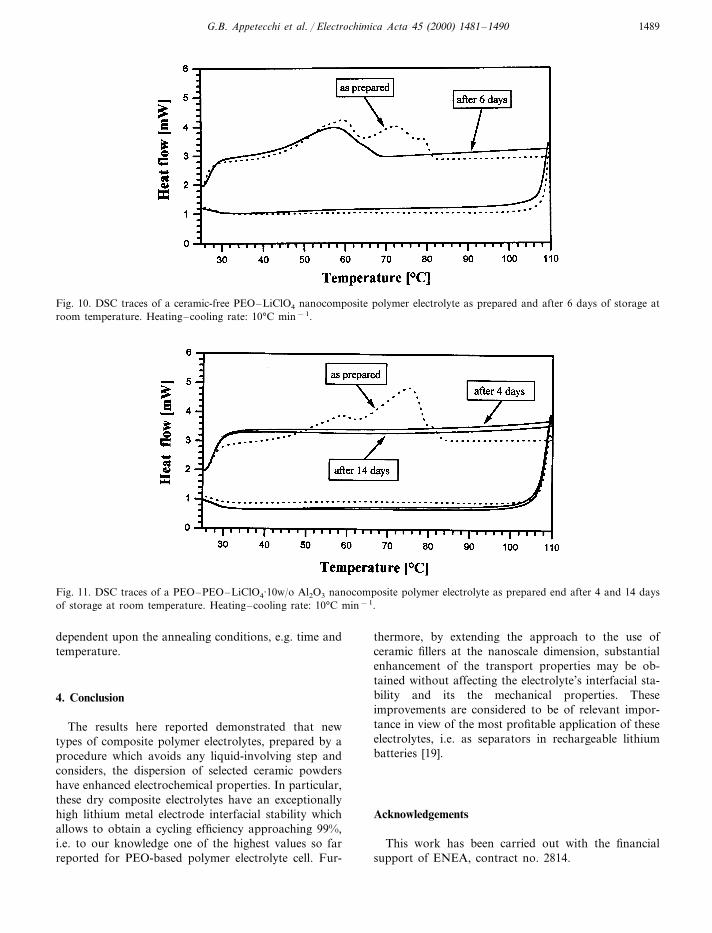
Another important test was the control of the crystal-lization kinetics of the nanocomposite samples. Figs. 10and 11 compare the differential scanning calorimetry,DSC, heating– –cooling traces of a ceramic-free poly-mer electrolyte with those of an Al2O3-based nanocom-posite electrolyte. The heating scan of an as-preparedceramic-free PEO8LiClO4 polymer electrolyte sample(Fig. 10), shows at 60–70°C, the peak series due to thecrystalline to amorphous transition. The following traceis the cooling scan from 100°C to room temperature; nopeak is observed since even ceramic-free electrolyteshave a relatively slow recrystallization kinetics [1].However, the electrolyte does recrystallize, as demon-strated by the peak shown in the trace obtained after 6days of storage at room temperature. There is quite adifferent DSC response for the nanocomposite elec-trolyte (Fig. 11). The heating scan of a as-preparedsample also shows a peak due to the crystalline toamorphous transition around 60–70°C. However, incontrast with the previous case, in all the followingcooling and heating traces no peaks are revealed evenafter prolonged storage times (i.e. exceeding 2 weeks).
These results support the conductivity results (seeFigs. 8 and 9) and, ultimately the transport model sincethey confirm that, after being annealed at temperaturesabove the PEO transition, the nanocomposite polymerelectrolytes retain their amorphous state even if kept atroom temperature for several days. A key question iswhether the annealed nanocomposite sample may even-tually recrystallize. Preliminary results suggest that afterprolonged storage some recrystallization may occur.however, with kinetics which appear to be critically
G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–1490 1489
Fig. 10. DSC traces of a ceramic-free PEO–LiClO4 nanocomposite polymer electrolyte as prepared and after 6 days of storage atroom temperature. Heating–cooling rate: 10°C min−1.
Fig. 11. DSC traces of a PEO–PEO–LiClO4·10w/o Al2O3 nanocomposite polymer electrolyte as prepared end after 4 and 14 daysof storage at room temperature. Heating–cooling rate: 10°C min−1.
dependent upon the annealing conditions, e.g. time andtemperature.
4. Conclusion
The results here reported demonstrated that newtypes of composite polymer electrolytes, prepared by aprocedure which avoids any liquid-involving step andconsiders, the dispersion of selected ceramic powdershave enhanced electrochemical properties. In particular,these dry composite electrolytes have an exceptionallyhigh lithium metal electrode interfacial stability whichallows to obtain a cycling efficiency approaching 99%,i.e. to our knowledge one of the highest values so farreported for PEO-based polymer electrolyte cell. Fur-
thermore, by extending the approach to the use ofceramic fillers at the nanoscale dimension, substantialenhancement of the transport properties may be ob-tained without affecting the electrolyte’s interfacial sta-bility and its the mechanical properties. Theseimprovements are considered to be of relevant impor-tance in view of the most profitable application of theseelectrolytes, i.e. as separators in rechargeable lithiumbatteries [19].
Acknowledgements
This work has been carried out with the financialsupport of ENEA, contract no. 2814.

G.B. Appetecchi et al. / Electrochimica Acta 45 (2000) 1481–14901490
References
[1] F.M. Gray, Polymer Electrolytes, Royal Society of Chem-istry Monographs, Cambridge, 1997.
[2] (a) G. Kobe, Automotive Industries, 1996. (b) C. Don-nelly, L. Christensen, D. Kuller, Electric and HybridVehicle Technology, 1996.
[3] P. Baudry, S. Lascaud, H. Majastre, D. Bloch, J. PowerSour. 68 (1997) 432.
[4] G.B. Appetecchi, F. Croce, M. Mastragostino, B.Scrosati, F. Soavi, F. Zanelli, J. Electrochem. Soc. 145(1998) 4133.
[5] G.B. Appetecchi, F. Croce, G. Dautzenberg, M. Mas-tragostino, F. Ronci, B. Scrosati, F. Soavi, F. Zanelli, F.Alessandrini, P.P. Prosini, J. Electrochem. Soc. 145 (1998)4126.
[6] F. Croce, G.B. Appetecchi, L. Persi, B. Scrosati, Nature394 (1998) 456.
[7] F.M. Gray, J.E. MacCallum, C.A. Vincent, Solid StateIon. 18–19 (1986) 282.
[8] B.A. Boukamp, Solid State Ion. 20 (1986) 31.[9] J. Evans, C.A. Vincent, P.G. Bruce, Polymer 28 (1987)
2325.[10] K.M Abraham, Z. Jiang, B. Carroll, Chem. Mater. 9
(1997) 1918.[11] F. Croce, B. Scrosati, J. Power Sour. 43–45 (1993) 9.[12] I.M. Ismail, U. Kadiroglu, N.D. Gray, J.R. Owen, Fall
Meeting of the Electrochemical Society, San Antonio,TX, 1996, abstract no. 63.
[13] M.C. Borghini, M. Mastragostino, S. Passerini, B.Scrosati, J. EIectrochem. Soc. 142 (1995) 2118.
[14] B. Scrosati, F. Croce, Poll. Adv. Technol. 4 (1993) 198.[15] F. Capuano, F. Croce, B. Scrosati, US Patent no.
5576115, 1996.[16] B. Kumar, L.G. Scanlon, J. Power Sour. 52 (1994) 261.[17] W. Wieczorek, Z. Florjancyk, J.R. Stevens, Electrochim.
Acta 40 (1995) 2251.[18] J. Przyluski, M. Siekierski, W. Wieczorek, Electrochim.
Acta 40 (1995) 2101.[19] B. Scrosati, Nature 373 (1995) 557.
.




















