
Transduction of Cell Lines and Primary Cells by FIV-Packaged HIV Vectors
10
Transduction of Cell Lines and Primary Cells by FIV-Packaged HIV Vectors Kevin V. Morris, 1 James Gilbert, 2 Flossie Wong-Staal, 3 Mehdi Gasmi, 4 and David J. Looney 1, * 1 Department of Medicine, University of California at San Diego, Stein Clinical Research Building, Room 302, La Jolla, CA 92093-0665, USA 2 Health Science, Gene Therapy Program, Louisiana State University, 533 Bolivar Street, Room 628, New Orleans, LA 70112, USA 3 Immusol Corporation, 9381 Judicial Drive, Suite 130, San Diego, CA 92121, USA 4 Ceregene, Inc., 11075 Roselle Street, San Diego, CA 92121, USA *To whom correspondence and reprint requests should be addressed. Fax: (858) 552-7416. E-mail: [email protected]. Available online 14 May 2004 Human immunodeficiency virus type 1 (HIV-1), simian immunodeficiency virus, and feline immunodeficiency virus (FIV) are capable of packaging viral RNA derived from heterologous as well as homologous lentiviruses, a phenomenon referred to as ‘‘cross packaging.’’ To remove the possibility of seroconversion to HIV proteins, and to avoid potential problems arising due to targeting of vector or packaging construct by antiviral genes, we investigated the feasibility of using an FIV-based packaging system to deliver human immunodeficiency virus type 2 (HIV-2)- based vectors bearing anti-HIV-1 RNA expression cassettes to target cells. In the absence of FIV rev, FIV was packaged by HIV-2 at only 3% the efficiency of FIV packaging by FIV, but this was increased to 39% of homologous controls by supplying FIV rev in trans. HIV-2 vectors were packaged by FIV at levels equal to or exceeding the homologous HIV-2 packaging system in the absence of HIV-1 tat and rev, and levels increased approximately four- to fivefold with the addition of tat and rev in trans. HIV-2 vectors bearing a polyribozyme cassette targeting multiple regions of HIV RNA were efficiently packaged by FIV and transferred to target cells. Upon challenge with cell-free HIV-1 (m.o.i. = 0.1) a significant reduction in replication was observed. These findings demonstrate that packaging HIV vectors with FIV is a viable alternative, which avoids use of HIV structural proteins. Key Words: lentivirus encapsidation, feline immunodeficiency virus, human immunodeficiency virus type 2, lentiviral vectors INTRODUCTION Cross packaging between human immunodeficiency vi- rus type 1 (HIV-1) and HIV-2 has been observed to be asymmetric, and qualitatively different, in that HIV-1 can package both HIV-2 genomic and spliced RNAs efficient- ly, whereas HIV-2 packages HIV-1 genomic RNA consid- erably less well [1]. Feline immunodeficiency virus (FIV) is genetically more distant from HIV-1, HIV-2, and simian immunodeficiency virus (SIV) than they are from one another, being more closely related to ungulate lentivi- ruses such as equine infectious anemia virus and visna virus [2,3]. Unlike HIV-1, HIV-2 and SIV sequences locat- ed between the major splice donor and the start codon of gag contribute little to encapsidation of the FIV genome. However, sequences upstream of the major splice donor are important for FIV encapsidation [4]. Packaging of all primate lentiviruses (HIV-1, HIV-2, and SIV) by FIV has been documented [5], suggesting that FIV encapsidation requirements may represent those of a more ‘‘primordial’’ lentiviral system, though the mechanism(s) by which FIV packages heterologous virus is not known. Of the nonprimate vector systems, feline immunodefi- ciency virus-based vectors have proven quite promising and have been used in both small- and large-animal gene ther- apies for several different purposes [6,7]; while other lenti- viral vector systems, including equine infectious anemia virus and bovine immunodeficiency virus, have also been developed, problems with stable transgene expression have been observed [8,9]. The ability to use an FIV packaging system with a primate lentiviral vector is technically desir- able, as the use of a primate lentiviral packaging construct entails the theoretical possibility of seroconversion to HIV/ SIV structural proteins [10], and furthermore the genetic divergence between FIV and HIV reduces the likelihood of recombination between vector and packaging constructs. MOLECULAR THERAPY Vol. 10, No. 1, July 2004 181 Copyright B The American Society of Gene Therapy 1525-0016/$30.00 doi:10.1016/j.ymthe.2004.03.019 METHOD
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Transduction of Cell Lines and Primary Cells by FIV-Packaged HIV Vectors

Transduction of Cell Lines and Primary Cells byFIV-Packaged HIV Vectors
Kevin V. Morris,1 James Gilbert,2 Flossie Wong-Staal,3
Mehdi Gasmi,4 and David J. Looney1,*1 Department of Medicine, University of California at San Diego, Stein Clinical Research Building, Room 302,
La Jolla, CA 92093-0665, USA2 Health Science, Gene Therapy Program, Louisiana State University, 533 Bolivar Street, Room 628, New Orleans, LA 70112, USA
3 Immusol Corporation, 9381 Judicial Drive, Suite 130, San Diego, CA 92121, USA4 Ceregene, Inc., 11075 Roselle Street, San Diego, CA 92121, USA
*To whom correspondence and reprint requests should be addressed. Fax: (858) 552-7416. E-mail: [email protected].
Available online 14 May 2004
Human immunodeficiency virus type 1 (HIV-1), simian immunodeficiency virus, and felineimmunodeficiency virus (FIV) are capable of packaging viral RNA derived from heterologous aswell as homologous lentiviruses, a phenomenon referred to as ‘‘cross packaging.’’ To remove thepossibility of seroconversion to HIV proteins, and to avoid potential problems arising due totargeting of vector or packaging construct by antiviral genes, we investigated the feasibility ofusing an FIV-based packaging system to deliver human immunodeficiency virus type 2 (HIV-2)-based vectors bearing anti-HIV-1 RNA expression cassettes to target cells. In the absence of FIV rev,FIV was packaged by HIV-2 at only 3% the efficiency of FIV packaging by FIV, but this was increasedto 39% of homologous controls by supplying FIV rev in trans. HIV-2 vectors were packaged by FIV atlevels equal to or exceeding the homologous HIV-2 packaging system in the absence of HIV-1 tatand rev, and levels increased approximately four- to fivefold with the addition of tat and rev in trans.HIV-2 vectors bearing a polyribozyme cassette targeting multiple regions of HIV RNA wereefficiently packaged by FIV and transferred to target cells. Upon challenge with cell-free HIV-1(m.o.i. = 0.1) a significant reduction in replication was observed. These findings demonstrate thatpackaging HIV vectors with FIV is a viable alternative, which avoids use of HIV structural proteins.
Key Words: lentivirus encapsidation, feline immunodeficiency virus, human immunodeficiencyvirus type 2, lentiviral vectors
INTRODUCTION
Cross packaging between human immunodeficiency vi-rus type 1 (HIV-1) and HIV-2 has been observed to beasymmetric, and qualitatively different, in that HIV-1 canpackage both HIV-2 genomic and spliced RNAs efficient-ly, whereas HIV-2 packages HIV-1 genomic RNA consid-erably less well [1]. Feline immunodeficiency virus (FIV)is genetically more distant from HIV-1, HIV-2, and simianimmunodeficiency virus (SIV) than they are from oneanother, being more closely related to ungulate lentivi-ruses such as equine infectious anemia virus and visnavirus [2,3]. Unlike HIV-1, HIV-2 and SIV sequences locat-ed between the major splice donor and the start codon ofgag contribute little to encapsidation of the FIV genome.However, sequences upstream of the major splice donorare important for FIV encapsidation [4]. Packaging of allprimate lentiviruses (HIV-1, HIV-2, and SIV) by FIV has
been documented [5], suggesting that FIV encapsidationrequirements may represent those of a more ‘‘primordial’’lentiviral system, though the mechanism(s) by which FIVpackages heterologous virus is not known.
Of the nonprimate vector systems, feline immunodefi-ciency virus-based vectors have proven quite promising andhave been used in both small- and large-animal gene ther-apies for several different purposes [6,7]; while other lenti-viral vector systems, including equine infectious anemiavirus and bovine immunodeficiency virus, have also beendeveloped, problems with stable transgene expression havebeen observed [8,9]. The ability to use an FIV packagingsystem with a primate lentiviral vector is technically desir-able, as the use of a primate lentiviral packaging constructentails the theoretical possibility of seroconversion to HIV/SIV structural proteins [10], and furthermore the geneticdivergence between FIV and HIV reduces the likelihood ofrecombination between vector and packaging constructs.
MOLECULAR THERAPY Vol. 10, No. 1, July 2004 181Copyright B The American Society of Gene Therapy
1525-0016/$30.00
doi:10.1016/j.ymthe.2004.03.019 METHOD

To define further the nature of cross packaging thatoccurs between HIV-1, HIV-2, and FIV, and to determinethe feasibility of using a cross-packaged primate lentiviralvector to transfer therapeutic genes to target cells, wecharacterized the cross packaging of FIV, HIV-1, and HIV-2 vectors by HIV-2 and the newly constructed FIV pack-aging system (pC34N).
RESULTS
Differences in Vector TiterFirst, we compared homologous and heterologous pack-aging of comparable HIV-2 (p4RzCGFP) and FIV (pVC-
GFPwP) vectors (Fig. 1) by an FIV packaging system(pC34N) to that of a previously documented [11] HIV-2packaging system (Fig. 1, bottom). The FIV packagingconstruct pC34N contains a CMV promoter preciselyfused to the FIV U5 LTR and FIV gag/pol, vif, and rev,with deletions in the core packaging region (C) andenvelope (see Experimental Procedures; Fig. 1, bottom),while the HIV-2 packaging construct pHIV-2D4 containsdeletions in vpr, vpx, vif, nef, env, and the core packagingregion. A major difference is that pC34N transcription isdriven by the CMV I/E promoter, while pHIV-2D4 tran-scription is driven by the native LTR. Both vectors con-tain EGFP expression cassettes driven by the CMV I/E
FIG. 1. Vectors used in this study. FIV-based vectors have a uniform nomenclature, in which the first letter designates vector status and the second the transgene
promoter (C or CMV for the cytomegalovirus immediate early promoter), followed by the transgene (GFP or EGFP, enhanced green fluorescent protein; LacZ, h-
galactosidase gene) abbreviation and enhancer element (wP or wPRE, woodchuck hepatitis virus posttranslational response element [33]) if present. HIV-2 vector
nomenclature includes presence or absence of a four-ribozyme cassette (4Rz/D4Rz), followed by a letter indicating transgene promoter and the abbreviated
transgene (as above). Other elements shown include the rev-response element (RRE, of homologous origin from FIV or HIV-2 in respective vectors), HIV-2 long
terminal repeat (5VLTR), splice donor (SD), primary packaging domain core region (C), gag coding region remnant (Gag(p17)), and HIV-1 cPPT/CTS (FLAP).
Packaging plasmids pHIV-2D4 (HIV-2) and pC34N (FIV) incorporate the complete gag/pol reading frames of the respective virus backbones, envelope and
primary packaging domain core region (C) deletions, both exons of rev (and tat for HIV-2), and heterologous polyadenylation (poly(A)) signal sequences from
bovine growth hormone (BGH, HIV-2) or simian virus 40 (SV40, FIV). The vesicular stomatitis virus G protein (VSV-G) was used to pseudotype all vectors. (Top)
FIV-based (pVC-GFPwP) and HIV-2-based (p4RzCGFP, pD4RzCGFP) GFP vectors and FIV-based (pVC-LacZwP) and HIV-2-based (p4RzLacZwP) LacZ vectors. The
HIV-1-based vector pHIV-7-GFP [27] is also shown. (Bottom) Schematic maps of FIV (pC34N) and HIV-2 (pHIV-2D4) packaging constructs used in this study, as
well as the VSV-G protein expression plasmid.
MOLECULAR THERAPY Vol. 10, No. 1, July 2004182Copyright B The American Society of Gene Therapy
METHOD doi:10.1016/j.ymthe.2004.03.019
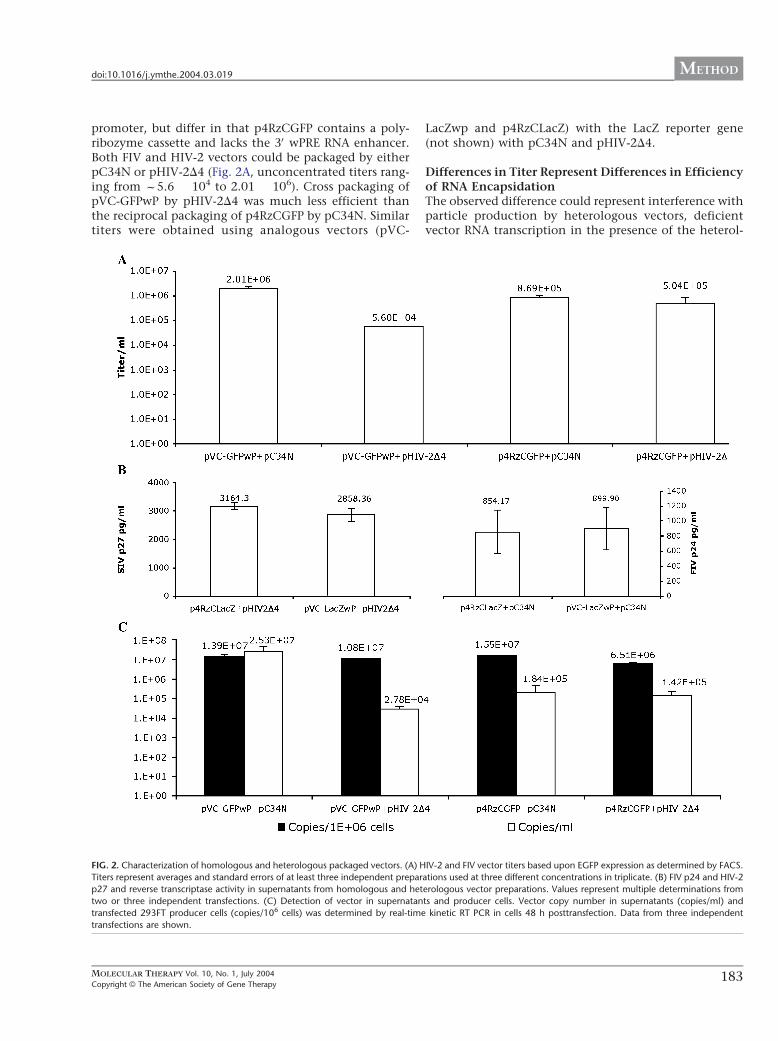
promoter, but differ in that p4RzCGFP contains a poly-ribozyme cassette and lacks the 3V wPRE RNA enhancer.Both FIV and HIV-2 vectors could be packaged by eitherpC34N or pHIV-2D4 (Fig. 2A, unconcentrated titers rang-ing from f5.6 � 104 to 2.01 � 106). Cross packaging ofpVC-GFPwP by pHIV-2D4 was much less efficient thanthe reciprocal packaging of p4RzCGFP by pC34N. Similartiters were obtained using analogous vectors (pVC-
LacZwp and p4RzCLacZ) with the LacZ reporter gene(not shown) with pC34N and pHIV-2D4.
Differences in Titer Represent Differences in Efficiencyof RNA EncapsidationThe observed difference could represent interference withparticle production by heterologous vectors, deficientvector RNA transcription in the presence of the heterol-
FIG. 2. Characterization of homologous and heterologous packaged vectors. (A) HIV-2 and FIV vector titers based upon EGFP expression as determined by FACS.
Titers represent averages and standard errors of at least three independent preparations used at three different concentrations in triplicate. (B) FIV p24 and HIV-2
p27 and reverse transcriptase activity in supernatants from homologous and heterologous vector preparations. Values represent multiple determinations from
two or three independent transfections. (C) Detection of vector in supernatants and producer cells. Vector copy number in supernatants (copies/ml) and
transfected 293FT producer cells (copies/106 cells) was determined by real-time kinetic RT PCR in cells 48 h posttransfection. Data from three independent
transfections are shown.
MOLECULAR THERAPY Vol. 10, No. 1, July 2004 183Copyright B The American Society of Gene Therapy
doi:10.1016/j.ymthe.2004.03.019 METHOD
ogous packaging construct, failure to encapsidate heter-ologous vector RNA, or defects in the infectivity of vectorpackaged by the heterologous viral proteins. To deter-mine which of these mechanism(s) were operative, wequantitated gag protein concentration, reverse transcrip-tase (RT) activity, and vector RNA in producing cells andsupernatant.
Similar concentrations of gag (Fig. 2B) and RT werepresent in supernatants when packaging constructs weretransfected with either heterologous or homologousexpressing vectors, indicating that heterologous vectordid not interfere with structural protein production. Notethat while absolute levels of RT activity (1.8–2.8 ng/Al forFIV-packaged vectors vs 0.27–0.34 ng/Al for HIV-2-pack-aged vectors, not shown) and core protein (329–626 vs2858–3164 pg/ml, Fig. 2B) produced by FIV and HIV-2packaging systems differ, the antigen detection assays arestandardized differently and buffers not adjusted foroptimal divalent cation preference for each virus, makingcomparison between rather than within packaging sys-tems difficult to interpret.
Production of vector copies in transfected producercells, as determined by real-time kinetic PCR for EGFP-containing message, did not differ significantly in ho-mologous and heterologous systems (Fig. 2C, closed bars,total copies ranging from 1.1 to 1.4 � 107 for pVC-GFPwPand 6.5 � 106 to 1.5 � 107 for p4RzCGFP), confirmingthat vector transcription proceeds normally in the pres-ence of the heterologous packaging construct.
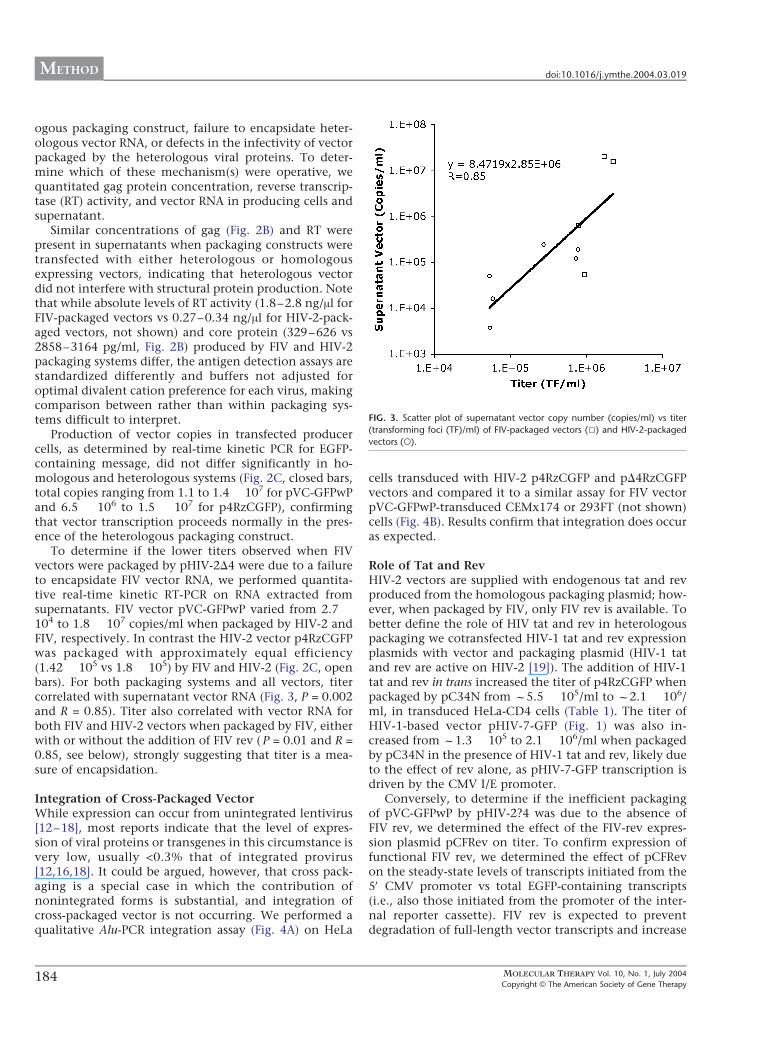
To determine if the lower titers observed when FIVvectors were packaged by pHIV-2D4 were due to a failureto encapsidate FIV vector RNA, we performed quantita-tive real-time kinetic RT-PCR on RNA extracted fromsupernatants. FIV vector pVC-GFPwP varied from 2.7 �104 to 1.8 � 107 copies/ml when packaged by HIV-2 andFIV, respectively. In contrast the HIV-2 vector p4RzCGFPwas packaged with approximately equal efficiency(1.42 � 105 vs 1.8 � 105) by FIV and HIV-2 (Fig. 2C, openbars). For both packaging systems and all vectors, titercorrelated with supernatant vector RNA (Fig. 3, P = 0.002and R = 0.85). Titer also correlated with vector RNA forboth FIV and HIV-2 vectors when packaged by FIV, eitherwith or without the addition of FIV rev ( P = 0.01 and R =0.85, see below), strongly suggesting that titer is a mea-sure of encapsidation.
Integration of Cross-Packaged VectorWhile expression can occur from unintegrated lentivirus[12–18], most reports indicate that the level of expres-sion of viral proteins or transgenes in this circumstance isvery low, usually <0.3% that of integrated provirus[12,16,18]. It could be argued, however, that cross pack-aging is a special case in which the contribution ofnonintegrated forms is substantial, and integration ofcross-packaged vector is not occurring. We performed aqualitative Alu-PCR integration assay (Fig. 4A) on HeLa
cells transduced with HIV-2 p4RzCGFP and pD4RzCGFPvectors and compared it to a similar assay for FIV vectorpVC-GFPwP-transduced CEMx174 or 293FT (not shown)cells (Fig. 4B). Results confirm that integration does occuras expected.
Role of Tat and RevHIV-2 vectors are supplied with endogenous tat and revproduced from the homologous packaging plasmid; how-ever, when packaged by FIV, only FIV rev is available. Tobetter define the role of HIV tat and rev in heterologouspackaging we cotransfected HIV-1 tat and rev expressionplasmids with vector and packaging plasmid (HIV-1 tatand rev are active on HIV-2 [19]). The addition of HIV-1tat and rev in trans increased the titer of p4RzCGFP whenpackaged by pC34N from f5.5 � 105/ml to f2.1 � 106/ml, in transduced HeLa-CD4 cells (Table 1). The titer ofHIV-1-based vector pHIV-7-GFP (Fig. 1) was also in-creased from f1.3 � 105 to 2.1 � 106/ml when packagedby pC34N in the presence of HIV-1 tat and rev, likely dueto the effect of rev alone, as pHIV-7-GFP transcription isdriven by the CMV I/E promoter.
Conversely, to determine if the inefficient packagingof pVC-GFPwP by pHIV-2?4 was due to the absence ofFIV rev, we determined the effect of the FIV-rev expres-sion plasmid pCFRev on titer. To confirm expression offunctional FIV rev, we determined the effect of pCFRevon the steady-state levels of transcripts initiated from the5V CMV promoter vs total EGFP-containing transcripts(i.e., also those initiated from the promoter of the inter-nal reporter cassette). FIV rev is expected to preventdegradation of full-length vector transcripts and increase
FIG. 3. Scatter plot of supernatant vector copy number (copies/ml) vs titer
(transforming foci (TF)/ml) of FIV-packaged vectors (5) and HIV-2-packaged
vectors (o).
MOLECULAR THERAPY Vol. 10, No. 1, July 2004184Copyright B The American Society of Gene Therapy
METHOD doi:10.1016/j.ymthe.2004.03.019

the fraction of transcripts containing FIV RRE. We deter-mined levels of total RNA (EGFP containing) and full-length RNA (RRE containing) transcripts using real-timekinetic PCR on RNA extracted from 293FT cells trans-fected with pVE-GFPwP and pCFRev or pUC19 control.Cotransfection with pCFRev increased the relative frac-tion of transcripts containing RRE f5-fold, confirmingactivity (Fig. 5), although levels of total EGFP transcriptswere not significantly altered (1.6 vs 1.5 � 106 copies/ngRNA). Next, we transfected 293FT cells with or withoutpCFRev and pHIV-2D4 and pVC-GFPwP. We collectedsupernatants from these cultures and used them totransduce target 293FT cells. We observed a 14-foldincrease in titer (5.4 � 104 to 7.8 � 105) and 51-foldincrease (3.7 � 103 F 788 to 1.9 � 105 F 3.6 � 104) invector supernatant copies in heterologous packagedpVC-GFPwP vectors when cotransfected with pCFRev in293FT producer cells relative to the non-pCFRev-trans-fected controls (Table 1), indicating that the lack ofexpression of full-length vector transcripts in the absenceof FIV rev in pHIV-2D4-packaged FIV vectors is responsi-ble for the observed discrepancy in heterologous pack-aged vector titers (Fig. 2A).
FIV-Packaged HIV-2 Vector-Transduced HeLa-CD4 andHuman PBMCs Are Resistant to HIV-1 InfectionTo demonstrate that HIV-2 vectors packaged by FIVremain functional in target cells (i.e., can efficientlytransfer resistance to viral infection mediated by a four-ribozyme cassette targeting HIV-1 RNA), we transducedHeLa-CD4 cells with FIV-packaged vectors (m.o.i. = 3),including pVC-GFPwP (FIV control), the HIV-2 vector
p4RzCGFP (containing a four-ribozyme cassette), orpD4RzCGFP (lacking the 4Rz cassette), and then chal-lenged them with HIV-1 HXB2 (m.o.i. = 0.1). Vectorp4RzCGFP-transduced HeLa-CD4 cultures displayed thegreatest inhibition of HIV-1 replication, 61.2%, comparedto the FIV vector control (Fig. 6A). Next, to determine ifthese heterologous packaged vectors were effective atinhibiting HIV-1 infection in the cells naturally infectedwith HIV-1, we transduced human PBMCs with FIV-packaged vectors (m.o.i. = 10) and 3 days later infectedthem with HIV-1 (HXB2, m.o.i. = 0.1). Expression of HIV-
TABLE 1: The effects of the addition of HIV-1 tat and rev orFIV rev in trans on heterologous packaged vector titers in
heterologous vs homologous packaged vectors
Vector PackagingAddition oftat or rev Titer/ml
p4RzCGFP(HIV-2)
pC34N (FIV) None 5.5 F 0.5 � 105
p4RzCGFP(HIV-2)
pC34N (FIV) HIV-1 tat/rev 2.1 F 0.02 � 106
pHIV-7-GFP(HIV-1)
pC34N (FIV) None 1.3 F 0.02 � 105
pHIV-7-GFP(HIV-1)
pC34N (FIV) HIV-1 tat/rev 2.1 F 0.07 � 106
pVC-GFPwP(FIV)
pHIV2-D4(HIV-2)
None 5.4 F 2.0 � 104
pVC-GFPwP(FIV)
pHIV2-D4(HIV-2)
FIV pCFRev 7.8 F 0.96 � 105
Titers represent averages from two or three separate vector preparations measured in
triplicate at various dilutions on CrFK cells.
FIG. 4. Detection of integrated HIV-2 and FIV
vectors. (A) Schematic of nested PCR integra-
tion assay. Alu-specific and either FIV- or HIV-
2-specific primers are used in PCR 1 followed
by PCR 2, using 5 Al of PCR 1 reaction mix and
either HIV-2- or FIV-specific LTR primers. (B)
HeLa-CD4 cells were transduced with HIV-2
vectors p4RzCGFP and pD4RzCGFP packaged
by FIV pC34N and genomic DNA was isolated
12 days later, and CEMx174 cells were
transduced with pVC-GFPwP packaged by
FIV pC34N, sorted by FACS, and genomic
DNA was isolated f45 days later. HIV-2
vector integration assay is shown in lanes
1–4 and FIV vector integration assay in lanes
5 and 6. (Lane 1) HIV-2 vector control (100
ng) p4RzCGFP plasmid control for PCR 2;
(lanes 2 and 3) p4RzCGFP and pD4RzCGFP
vector-transduced HeLa-CD4 cells, respec-
tively; (lane 4) untransduced HeLa-CD4 cells;
(lane 5) FIV vector control (100 ng) pVC-
GFPwP; and (lane 6) pVC-GFPwP-transduced
CEMx174 cells. The lower lanes show second-
stage PCR negative control on equivalent
amounts of genomic DNA (refer to Fig. 3).
MOLECULAR THERAPY Vol. 10, No. 1, July 2004 185Copyright B The American Society of Gene Therapy
doi:10.1016/j.ymthe.2004.03.019 METHOD
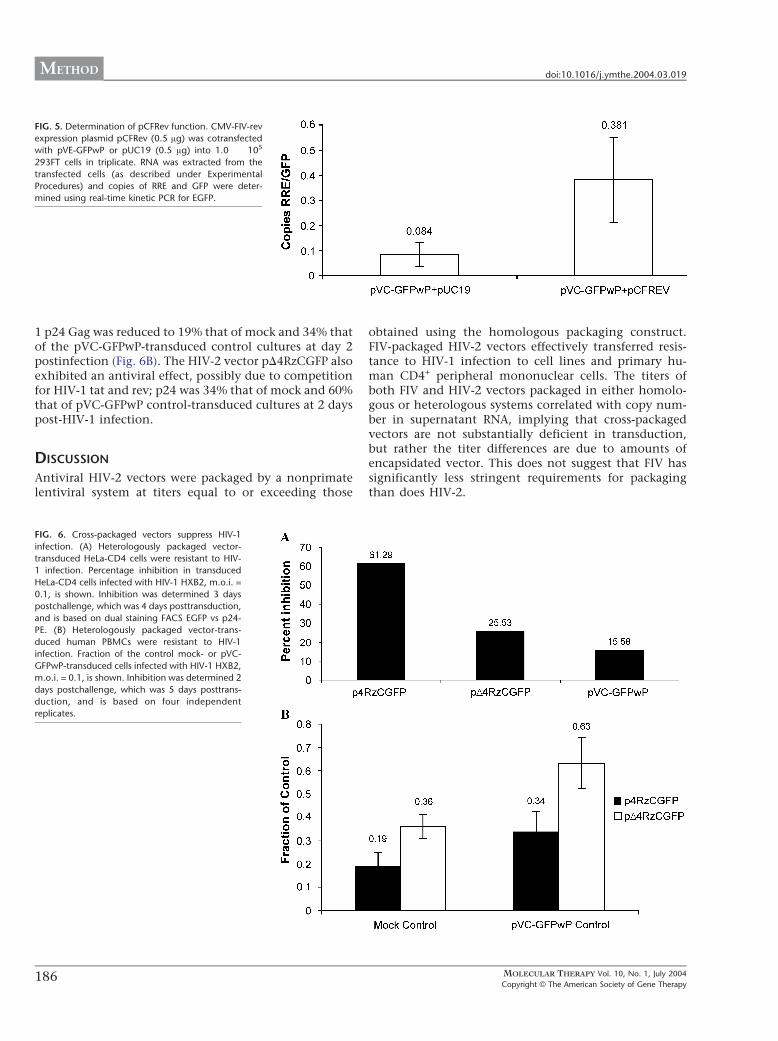
1 p24 Gag was reduced to 19% that of mock and 34% thatof the pVC-GFPwP-transduced control cultures at day 2postinfection (Fig. 6B). The HIV-2 vector pD4RzCGFP alsoexhibited an antiviral effect, possibly due to competitionfor HIV-1 tat and rev; p24 was 34% that of mock and 60%that of pVC-GFPwP control-transduced cultures at 2 dayspost-HIV-1 infection.
DISCUSSION
Antiviral HIV-2 vectors were packaged by a nonprimatelentiviral system at titers equal to or exceeding those
obtained using the homologous packaging construct.FIV-packaged HIV-2 vectors effectively transferred resis-tance to HIV-1 infection to cell lines and primary hu-man CD4+ peripheral mononuclear cells. The titers ofboth FIV and HIV-2 vectors packaged in either homolo-gous or heterologous systems correlated with copy num-ber in supernatant RNA, implying that cross-packagedvectors are not substantially deficient in transduction,but rather the titer differences are due to amounts ofencapsidated vector. This does not suggest that FIV hassignificantly less stringent requirements for packagingthan does HIV-2.
FIG. 5. Determination of pCFRev function. CMV-FIV-rev
expression plasmid pCFRev (0.5 Ag) was cotransfected
with pVE-GFPwP or pUC19 (0.5 Ag) into 1.0 � 105
293FT cells in triplicate. RNA was extracted from the
transfected cells (as described under Experimental
Procedures) and copies of RRE and GFP were deter-
mined using real-time kinetic PCR for EGFP.
FIG. 6. Cross-packaged vectors suppress HIV-1
infection. (A) Heterologously packaged vector-
transduced HeLa-CD4 cells were resistant to HIV-
1 infection. Percentage inhibition in transduced
HeLa-CD4 cells infected with HIV-1 HXB2, m.o.i. =
0.1, is shown. Inhibition was determined 3 days
postchallenge, which was 4 days posttransduction,
and is based on dual staining FACS EGFP vs p24-
PE. (B) Heterologously packaged vector-trans-
duced human PBMCs were resistant to HIV-1
infection. Fraction of the control mock- or pVC-
GFPwP-transduced cells infected with HIV-1 HXB2,
m.o.i. = 0.1, is shown. Inhibition was determined 2
days postchallenge, which was 5 days posttrans-
duction, and is based on four independent
replicates.
MOLECULAR THERAPY Vol. 10, No. 1, July 2004186Copyright B The American Society of Gene Therapy
METHOD doi:10.1016/j.ymthe.2004.03.019

While supplying tat and rev in trans was required toachieve optimal titers, vector preparations did not con-tain any HIV-1 structural proteins, eliminating any pos-sibility of exposure during therapeutic or laboratory use.Furthermore, we have been unable to detect mobilizationof FIV vectors after challenge of transduced cells withHIV-1 (Morris et al, manuscript in preparation), possiblydue to the inability of HIV-1 rev to complement FIVreplication or the relative transcriptional silence of theFIV LTR in human cells ([30] and reviewed in [27]). In anycase, we also cannot detect pC34N producer constructtranscripts in supernatants by RT-PCR (not shown), indi-cating that the use of a nonprimate lentiviral packagingsystem should markedly reduce the potential for recom-bination in vitro.
Limited data suggest that FIV may not be completelyinnocuous in primates, despite the absence of clinicaldisease in the face of extensive human exposure [20]. FIVhas been reported to infect primary human cells produc-tively in vitro [21] and to be capable of transient replica-tion in cynomolgus macaques (Macaca fasicularis), causingweight loss and CD4 decline [22]. We have not observedexpression of the FIV packaging construct to produce anytoxicity on a cellular basis, and even if FIV is capable oflimited infection in humans, this should not be a substan-tial drawback to the use of an FIV packaging construct.
The use of this or similar heterologous packagingsystems should also circumvent potential restrictions onantiviral gene targets due to homology with messages ofrequired homologous viral structural proteins, effectivelyexpanding the choice of antiviral transgenes that can beused. Conversely, the use of FIV-packaged HIV-basedvectors for specific purposes (e.g., transduction ofCD34+ hematopoietic stem cells) may avoid problemswith expression seen with FIV-based vectors [23].
The work presented here suggests that heterologouspackaging of primate lentiviral vectors by FIV may be auseful alternative for the delivery of HIV-1 and HIV-2vectors both in vitro and in vivo.
EXPERIMENTAL PROCEDURES
Vectors and Packaging ConstructsThe HIV-2 packaging construct (pHIV-2D4) was con-structed from pE41 [11,24,25] by restriction digest withBglII and HindIII to remove the vif, vpx, and part of vpraccessory genes. The BsrGI site within the nef codingregion was digested and filled in with Klenow and anXbaI linker (New England Biolabs), 5V-CTAGTCTAGAC-TAG-3V, was inserted into this site to generate a frameshiftand translational stop codons in all three nef readingframes (Fig. 1).
The HIV-2 vector (p4RzCLacZ) was constructed frompJG1. Plasmid pJG1 was generated by PCR amplificationof the HIV-2 3VLTR with 5V-ACGCGTTGGATGGGATGAT-TACA-3V and 5V-GCCGGCGGTGCTGCTGGTCTACTT-3V
and then ligated into SacI Klenow filled-in and NaeI-digested pUC19 to generate pUC-3VLTR. Next a fragmentof HIV-2 containing the 5VLTR, leader sequence, and p17of gag was amplified by PCR with 5VLTR, 5V-GTTAACTG-GATGATGTAGATGCAGATAATG-3V, and 3VLTR, 3V-CTTAACTTGCTTCTAACTGGCAGC-3V, and cloned intothe SphI-digested Klenow filled-in and the NaeI-digestedsites in pUC-3VLTR. Simultaneously, the HIV-2 RRE se-quence was ampl i f i ed us ing 5 VRRE, 5 V-TCTA-GAGTGTTTGTGCTAGGAGTC-3 V, and 3 VRRE, 5 V-GATATCTTCCAATCAGGTACTATG-3V, XbaI and EcoRVdigested, and ligated into pz-LacZ [26], and a fragmentconsisting of RRE-CMV-LacZ was then XbaI and BamHIdigested and ligated into pUC-3VLTR specifically betweenp17 and the 3VLTR to generate pJG1. Finally, a 4Rz hairpintRNA-expressed ribozyme cassette targeting HIV-1 pol (n =2 bp 2770, 2491), U5 (8905), and vif (bp 4713) was XbaIdigested from pT7.1 (gift from Mark Leavitt, Immusol,Inc.) and ligated into the similarly digested pJG1 to derivep4RzCLacZ. The GFP-containing vectors p4RzCGFP andpD4RzCGFP were constructed in a fashion similar to thatof p4RzCLacZ except the HIV-2 RRE was PCR amplifiedand cloned into the AseI site of peGFP-N1 (Clontech) andthen an XbaI and BamHI fragment spanning the RRE-pCMV-eGFP was cloned into the similarly digestedp4RzCLacZ to generate p4RzCGFP. To generatepD4RzCGFP the XbaI- and BamHI-digested RRE-pCMV-eGFP fragment was ligated into pJG1 prior to the insertionof the 4Rz cassette. The HIV-1 vector, pHIV-7-GFP (Fig. 1)[27], was provided by J. Rossi (Beckman Institute) [27].
To generate an FIV packaging construct, Petalumastrain clone p34TF10 [34] was first digested with Acc65Iand SpeI, and the 1893-bp env fragment cloned into pSK(�)(Stratagene) was digested with KpnI and SpeI to createplasmid pS-AccS. An 833-bp fragment that contained thesecond exon of rev was then amplified from pS-AccS usingprimers Env-Spe, 5V-GAGCATCAAGTACTAGTAATAGG-3V, and Rev3-Not, 5V-CCGCGGCCGCTAGTCCATAAG-CAT-3V. The product was digested with SpeI and NotI andcloned back into pS-AccS digested with AvrII and NotI tocreate pSDenv, deleting 1569 bp of env coding sequenceswhile preserving both exons of rev and associated splicingsignals. Next, a 4584-bp EcoRI–BglII fragment fromp34TF10 (containing pol and the 5V end of env) and a1145-bp BglII–NotI fragment from pSDenv were clonedinto pBSKS(�) (Stratagene) digested with EcoRI and NotI,creating pSPE. A 1.2-kb fragment of the 5VUTR and gag wasthen amplified from p34TF10 using 5V primer fp ak-Sal, 5V-GGAGTCGACGTCAACAAGGTAGGAATGGGGAATGG-3V, and 3V primer F gag3-RI, 5V-CCTTCCAGTTTCCC-GAATTCTTTCTATTTC-3V, preserving the 5V major splicedonor while deleting the putative core packaging signal.This PCR product was then digested with SalI and EcoRIand cloned into similarly digested pSPE to generate pSal-PakN. Finally, a 5756-bp SalI–NotI fragment from pSal-PakN (containing the 5V major splice donor, gag, pol, env
MOLECULAR THERAPY Vol. 10, No. 1, July 2004 187Copyright B The American Society of Gene Therapy
doi:10.1016/j.ymthe.2004.03.019 METHOD

deletion, and both exons of rev) was cloned intopCMVbGal (Clontech) digested with XhoI and NotI togenerate pC34N.
To produce an FIV vector with a CMV promoter andEGFP reporter each flanked by unique sites, the intronA-containing CMV promoter from pCTRZLb [25] wasamplified with primers 3346, 5V-TTTTTAGATCTTCAGA-GACTGACACGGACTC-3V, and 3936, 5V-TTTTTAAGCT-TACCGGTTTAAACGGCGCGCCGACGACGGTGACTG-CAGAAAAGA-3V, and digested with BglII and HindIII,and the 629-bp fragment was cloned into pRc/CMV2(Invitrogen) to create pRc/CMV-L (replacing the mini-mal CMV promoter). Next EGFP was amplifiedfrom pEGFP-C1 (Clontech) using primers 525,5V-TTTTTAAGCTTATGGTGAGCA-3V, and 554, 5V-TTTTTGCGGCCGCGTACGTTA-3V, digested with HindIIIand NotI, and cloned into similarly digested pRc/CMV-Lto create pRC-EGFP3. This plasmid was then cut withAccIII and BsiWI, and the 1.3-kb band containing CMV-EGFP was cloned into similarly digested pCTRZLb [2], tocreate vector construct pCTR-EGFP-PL, with the CMVpromoter flanked by a BglII site 5V and a HindIII site 3Vand EGFP flanked by AscI, PmeI, AgeI, and HindIII sites5V and PacI, BsiWI, and NotI sites 3V.
The FIV vector pVC-GFPwP was constructed by digest-ing pCTR-EGFP-PL with SpeI, blunt ending, and thendigesting with MfeI. The resulting fragment contained aCMV enhancer/promoter in lieu of the FIV 5V U3 andencompassed the packaging signal and 250 bp of FIV gag.This fragment was then inserted into pBluescript SK(�)(Stratagene), which was Acc65I digested, Klenow treated,and EcoRI digested to derive pSLDG. Next the RRE (f170bp) was amplified from p34TF10 using FRRE-5, 5V-GGATGCATTGAGGAGAAATGGTAGGC-3V, and FRRE-3,5V-GGCAGGCCTACAATACATACTTTATTAG-3V, digestedwith NsiI, and inserted into the PstI- and SmaI-digestedpSLDG to generate pSLDGR. The wPRE fragment wasamplified from the WHB genome (ATCC) with Pre-5, 5V-CGGAGATCTAACCTCTGGATTAC-3V, and Pre-3, 5V-TGTCTCCCGTGACGCCGAGGCGAAACAGG-3V, andused in a fusion PCR with the 3V PPT and LTR, whichwere amplified using LTR-5V, 5V-ACGTGGAGACAGCA-CAGTAGATTC-3V, and LTR-3V, 5V-AATGAGCTCGCTGC-GAAGTTCTCGG-3V, and p34TF10 as template. Thef1040-bp fusion PCR wPRE-3VLTR product was digestedwith BglII and cloned into pCMV-EGFP-C1 (Clontech),which was Acc65I digested, blunt-ended, and furtherdigested with BglIII, generating pC-GFPwPL. Finally, pC-GFPwPL was digested with AseI, Klenow treated, anddigested with Bsp1201 to generate an f2377-bp frag-ment, which was inserted into pSLDGR, which had beenBamHI digested, Klenow treated, and digested with NotI,to generate pVC-GFPwP.
To construct FIV vector pVE-GFPwP, the EF1a (EF1a)promoter was PCR amplified from pEF/His-C(Invitrogen) with primers EF1-5, 5 V-TTACTAG-
TCCCCGAGAAGTTGGGGG-3 V, and EF1-3 , 5 V-TTGGCGCGCCGGTCTCCCTATAGTGAG-3V, and clonedinto EcoRV-digested blunt-ended pSK(�) (Stratagene) togenerate pSEF1a. pVC-GFPwP was then digested withAgeI and BamHI and the subsequent product, contain-ing EGFP-wPRE-3VLTR, was inserted into pSEF1adigested with XmaI and BamHI to produce pSE-F1aGFPwPL. Next pSEF1aGFPwPL was digested with SalIand then blunt ended followed by BspEI digestion torelease the EF1a-EGFP fragment, which was insertedinto pVC-GFPwP digested with StuI and BspEI to gen-erate pVE-GFPwP.
The FIV vector pVC-LacZwP was generated by digest-ing pVC-GFPwP with AgeI and BglII to remove the GFP,while the LacZ gene was isolated from pCMV-BetaGal(Clontech) by XmaI and BamHI digestion. The LacZ genewas then inserted into the AgeI and BglII sites of thesimilarly digested pVC-GFPwP.
The HIV-1 rev expression plasmid, pCMV-rev, wasprovided by the AIDS Research and Reference ReagentProgram and the HIV-1 tat (pHIV-Tat) expression plas-mid was provided by J. Rappaport [28]. The FIV revexpression plasmid was constructed by cloning the PPR-Rev cDNA (NIH AIDS Research and Reference ReagentProgram) into expression plasmid pRcCMV2 (Invitro-gen). Briefly, pRCMV was digested with HindIII and NotIand the FIV rev insert was PCR amplified from the PPRcDNA template using 5V primer 741, which contains aHindIII site, 5V-CCCAAGCTTATGGCAGAAGGGTTTG-CAGCCAA-3V, and the 3V primer 740, which contains aNotI site, 5V-CATTTGTTTGGCGGCCGCCTTAGTCCA-TAA-3V. The amplified FIV rev was digested with HindIIIand NotI and then ligated into the similarly digestedpRCMV plasmid to make the FIV rev eukaryotic expres-sion vector pCFRev.
Vectors and PackagingTo prepare vectors, the FIV packaging construct pC34N (10Ag) or the HIV-2 packaging construct pHIV2D4 (10 Ag) anda VSV-G-pseudotyped envelope (5 Ag) (Fig. 1), togetherwith the relevant vector (p4RzCLacZ, pVC-LacZwP, pVC-GFPwP, pD4RzCGFP, p4RzCGFP, pHIV-7-GFP [27], orpLNXC2, 5 Ag) and, when described, pHIV-Tat (HIV-1tat, 2.5 Ag) and pCMV-rev (HIV-1 rev, 2.5 Ag) expressionplasmids, were transfected (calcium phosphate) in tripli-cate (4 � 106 293T cells/plate). These transfection condi-tions were not selected to produce optimal titers, butrather to allow comparison between titers ranging over>4 logs. The transfected cultures were incubated at 37jC inan atmosphere of 5% CO2. Forty-eight hours later trans-fected cell supernatants were collected and titered bytransducing various concentrations of vector-containingsterile-filtered (0.45 AM) supernatant of Crandell felinekidney (CrFK) cells (1.0 � 105/well in 12-well plates inthe presence of 8 Ag/well Polybrene). The titers weredefined by counting LacZ-positive cells following 2–5
MOLECULAR THERAPY Vol. 10, No. 1, July 2004188Copyright B The American Society of Gene Therapy
METHOD doi:10.1016/j.ymthe.2004.03.019

min incubation with fixing solution (2% v/v formalde-hyde and 0.2% v/v glutaraldehyde in PBS), followed by a1� PBS wash and incubation with fresh X-gal stainingsolution; blue cells were scored as transduced. GFP-expressing vectors were titered by FACS on CrFK,HT1080, and HeLa-CD4 cells (1.0 � 105/ml): 48 h post-transduction between 1.0 � 104 and 1.0 � 105 cells werewashed three times with 1� PBS + 1% FBS and thenresuspended in 1� PBS + 1% FBS and 1% EM gradeparaformaldehyde and assessed for GFP expression byFACS (CFAR Flow Cytometry core).
FIV p24 and SIV p27 ELISA and HIV-1 RT AssaysPackaged vector was collected from 750 Al of 293FT vector-containing supernatants pelleted by centrifugation at5000g for 10 min and sterile filtered through a 0.45-Am-pore-size membrane. The filtrate was then concentrated bycentrifugation at 20,000g for 1 h at 4jC. RT activity in viralpellets was determined (Boehringer Mannheim RT Kit).Viral core protein concentrations were determined usingEIA. FIV p24 was determined using the IDEXX PetChek FIVp24 assay modified to produce quantitative values basedupon an antigen standard [29]. HIV-2 p26 was quantifiedusing the cross-reactive Coulter SIV p27 EIA kit.
Real-time Kinetic PCR and RT PCRReal-time kinetic PCR was run on isolated vector RNA(Qiagen RNeasy) either from 1 ml of supernatant or from1.0 � 106 293FT cells used in the vector preparations.Cellular and vector RNA (f1 Ag) was DNase treated (f5–10 units/Ag of RNA, 2 h) and used as template for the RTreaction (20 Al). RT reaction mix contained 1 Al (2 pM) ofprimers specific for GFP (814, 5V-GGTGGTGCAGAT-GAACTTCAGGGTC-3V) and FIV RRE (5V-TTGATATGG-CAATTCCTGCATT-3V) along with AMV RT (5 U/Al), 2 Al ofdNTP’s (1.25 mM), 2 Al of RT buffer (100 mM Tris–HCl (pH9.0 at 25jC), 500 mM KCl, 15 mM MgCl2) 1% Triton X-100,4 Al of MgCl2 (25 mM), and 0.6 Al of rRNasin (20–40 U/Al).TheresultantcDNA(1h/42jCand10min/95jC,50ng)wasthen used for PCR. Real-time kinetic PCR was done on RT-amplified and a control equivalent amount of non-reverse-transcribed sample RNA, following previously establishedprotocols[30].PrimersusedforGFPwere625F,5V-AGCAAA-GACCCCAACGAGAA-3V; 684R, 5V-GGCGGCGGTCAC-GAA-3V; and 5V-CGCGATCACATGGTCCTGCTGG-3V 646T(Tet probe) and for FIV RRE primers 13, 5V-AGATACTTCAT-CATTCCTCCTCTTTTT-3 V; 14 , 5 V-TTGATATGG-CAATTCCTGCATT-3 V; and the RRE Tet probe ,5V-AGGAGAAATGGTAGGCAA-3V. In the case of RT-PCR,template cDNA for cellular RNA or pelleted (20,000g for 1hat4jC) vector supernatantwas amplified with5VGFP xba-I, 5V-GCGTCTAGAATGGTGAGCAAGGGCGAGGAG-3V,and 3V GFP 776, 5V-AACTTCAGGGTCAGCTTGCCG-TAGGTGGCATCGCC-3V, under the following conditions:1 cycle of 95jC (8 min); 35 cycles of 95jC (45 s), 55jC (45 s),and 72jC (45 s); followed by 1 cycle of 72jC (8 min).
Vector Integration AssaysQualitative vector integration assays were based on apreviously published protocol [31]. The assays were com-posed of a nested PCR using an Alu repeat-specific primeralong with an LTR-specific primer as a first stage, followedby a second-stage amplification using two LTR-specificprimers. Controls for this assay included untransfectedgenomic cell DNA, LTR plasmid controls, and equivalentamounts of transduced input genomic DNA not amplifiedin the Alu PCR. For HIV-2, first-stage primers included Aluprimer 638, 5V-TGCTGGGATTACAGGCGTGAG-3V [31],and HIV-2 LTR-specific primer 274, 5V-AAAACTC-GAGCTGGTGAGAGTCTAGCAGGGAAC-3V (Fig. 4A, PCR1). A second, nested PCR was then performed using HIV-2-specific primers 372, 5V-GGAACGCCCTCATACTTCTG-TATA-3V, and 274 (Fig. 4A, PCR 2). For FIV, the first-stageprimers included Alu primer 637, 5 V-TCCCAGC-TACTCGGGAGGCTGAGG-3V [32], and FIV LTR-specificprimer 670, 5V-GAGACTCCTCGAAGTTTCACA-3V. A sec-ond, nested PCR was then performed using FIV primers669, 5V-GGGACTGTTTACGAACAAATG-3V, and 670(above). Controls for amplification of nonintegrated viralDNA forms consisted of equivalent amounts of inputgenomic DNA (not amplified in PCR 1).
HIV-1 ChallengeHeLa-CD4 cells (1.0 � 105) were transduced with pC34N-packaged vectors (p4RzCGFP, pD4RzCGFP, and pVC-GFPwP, m.o.i. = 3.0 for 24 h). washed in 1� PBS, andthen infected with HIV-1 HXB2, m.o.i. = 0.1. Three daysfollowing transduction and 2 days following HIV-1 chal-lenge the cells were stained with Cytofix/Cytoperm(PharMingen) and exposed to PE-labeled mouse anti-p24 KC57-RDI (Coulter), and relative PE vs GFP expres-sion was determined by FACS. When PHA-stimulatedhuman PBMCs were used, 1.0 � 105 cells were transducedwith pC34N-packaged vectors (p4RzCGFP, pD4RzCGFP,and pVC-GFPwP, m.o.i. = 1.0 for 24 h), washed in 1� PBS,and then infected at day 3 posttransduction with HIV-1HXB2, m.o.i. = 0.1. The cultures were assayed for p24 onday 2 post-HIV-1 infection, which corresponds to day 5posttransduction.
ACKNOWLEDGMENTS
This project was supported by NIH AIDS Training Grant AI07384 (D. Richman),
program project NIH P01 A145992 (D. Looney), and assisted by the UCSD
Center for AIDS Research, P30 AI36214 (D. Richman). The authors thank
Christopher Chung, Werner Witke, Charlene Baroga, and Laura Innis for their
technical assistance and critical review of the manuscript.
RECEIVED FOR PUBLICATION JANUARY 2, 2004; ACCEPTED MARCH 24,
2004.
REFERENCES
1. Lever, A. M. (2000). HIV RNA packaging and lentivirus-based vectors. Adv. Pharmacol.
48: 1 – 28.
2. Poeschla, E., Wong-Staal, F., Looney, D. J. (1998). Efficient transduction of nondividing
human cells by feline immunodeficiency virus lentiviral vectors. Nat. Med. 4: 354 – 357.
MOLECULAR THERAPY Vol. 10, No. 1, July 2004 189Copyright B The American Society of Gene Therapy
doi:10.1016/j.ymthe.2004.03.019 METHOD

3. Schnolzer, M., et al. (1996). Comparative properties of feline immunodeficiency virus
(FIV) and human immunodeficiency virus type 1 (HIV-1) proteinases prepared by total
chemical synthesis. Virology 224: 268 – 275.
4. Kemler, I. R. B., Poeschla, E. M. (2002). Encapsidation signal of feline immunodefi-
ciency virus (FIV): mapping and significance for FIV vectors. Mol. Ther. 5: S174. [Con-
ference Abstract 528].
5. Browning, M. T., Schmidt, R. D., Lew, K. A., Rizvi, T. A. (2001). Primate and feline
lentivirus vector RNA packaging and propagation by heterologous lentivirus virions. J.
Virol. 75: 5129 – 5140.
6. Loewen, N., et al. (2002). Preservation of aqueous outflow facility after second-gen-
eration FIV vector-mediated expression of marker genes in anterior segments of human
eyes. Invest. Ophthalmol. Visual Sci. 43: 3686 – 3690.
7. Haskell, R. E., Hughes, S. M., Chiorini, J. A., Alisky, J. M., Davidson, B. L. (2003). Viral-
mediated delivery of the late-infantile neuronal ceroid lipofuscinosis gene, TPP-I to the
mouse central nervous system. Gene Ther. 10: 34 – 42.
8. O’Rourke, J. P., Newbound, G. C., Kohn, D. B., Olsen, J. C., Bunnell, B. A. (2002).
Comparison of gene transfer efficiencies and gene expression levels achieved with
equine infectious anemia virus- and human immunodeficiency virus type-1-derived
lentivirus vectors. J. Virol. 76: 1510 – 1515.
9. Berkowitz, R., et al. (2001). Construction and molecular analysis of gene transfer sys-
tems derived from bovine immunodeficiency virus. J. Virol. 75: 3371 – 3382.
10. White, S. M., et al. (1999). Lentivirus vectors using human and simian immunodefi-
ciency virus elements. J. Virol. 73: 2832 – 2840.
11. Cheng, L., et al. (2002). Human immunodeficiency virus type 2 (HIV-2) vector-medi-
ated in vivo gene transfer into adult rabbit retina. Curr. Eye Res. 3: 196 – 201.
12. Poon, B., Chen, I. S. (2003). Human immunodeficiency virus type 1 (HIV-1) Vpr en-
hances expression from unintegrated HIV-1 DNA. J. Virol. 77: 3962 – 3972.
13. Stevenson, M., Haggerty, S., Lamonica, C. A., Meier, C. M., Welch, S. K., Wasiak, A. J.
(1990). Integration is not necessary for expression of human immunodeficiency virus
type 1 protein products. J. Virol. 2421 – 2425.
14. Pauza, C. D., Galindo, J. (1989). Persistent human immunodeficiency virus type 1
infection of monoblastoid cells leads to accumulation of self-integrated viral DNA
and to production of defective virions. J. Virol. 63: 3700 – 3707.
15. Masuda, T., et al. (1995). Genetic analysis of human immunodeficiency virus type 1
integrase and the U3 att site: unusual phenotype of mutants in the zinc finger-like
domain. J. Virol. 69: 6687 – 6696.
16. Nakajima, N., Lu, R., Engelman, A. (2001). Human immunodeficiency virus type 1
replication in the absence of integrase-mediated DNA recombination: definition of
permissive and nonpermissive T-cell lines. J. Virol. 75: 7944 – 7955.
17. Wu, Y., and Marsh, J. W. (2001). Selective transcription and modulation of resting T cell
activity by preintegrated HIV DNA. Science 293: 1503 – 1506.
18. Cara, A., et al. (1995). Self-limiting, cell type-dependent replication of an integrase-
defective human immunodeficiency virus type 1 in human primary macrophages but
not T lymphocytes. Virology 208: 242 – 248.
19. Arya, S. K. (1988). Human and simian immunodeficiency retroviruses: activation and
differential transactivation of gene expression. AIDS Res. Hum. Retroviruses 4: 175 – 186.
20. Butera, S. T., et al. (2000). Survey of veterinary conference attendees for evidence of
zoonotic infection by feline retroviruses. J. Am. Vet. Med. Assoc. 217: 1475 – 1479.
21. Johnston, J., Power, C. (1999). Productive infection of human peripheral blood mono-
nuclear cells by feline immunodeficiency virus: implications for vector development. J.
Virol. 73: 2491 – 2498.
22. Johnston, J. B., Olson, M. E., Rud, E. W., Power, C. (2001). Xenoinfection of nonhuman
primates by feline immunodeficiency virus. Curr. Biol. 11: 1109 – 1113.
23. Price, M. A., et al. (2002). Expression from second-generation feline immunodeficiency
virus vectors is impaired in human hematopoietic cells. Mol. Ther. 6: 645 – 652.
24. Gilbert, J. R., Wong-Staal, F. (2001). HIV-2 and SIV. In Lentiviral Vector Systems for Gene
Transfers (G.L Buchschacher, Ed.), pp. 78 – 93.
25. Poeschla, E., Gilbert, J., Li, X., Huang, S., Ho, A., Wong-Staal, F. (1998). Identification of
a human immunodeficiency virus type 2 (HIV-2) encapsidation determinant and trans-
duction of nondividing human cells by HIV-2-based lentivirus vectors. J. Virol. 72:
6527 – 6536.
26. Ferrandez, A., Garcia, J. L., Diaze, E. (2000). Transcriptional regulation of the divergent
PAA catabolic operons for phenylacetic acid degradation in Escherichia coli. J. Biol.
Chem. 275: 12214 – 12222.
27. Banerjea, A., et al. (2003). Inhibition of HIV-1 by lentiviral vector-transduced siRNAs in T
lymphocytes differentiated in SCID-hu mice and CD34+ progenitor cell-derived macro-
phages. Mol. Ther. 8: 62 – 71.
28. Rappaport, J. S. J. L., Kihalili, K., Wong-Staal, F. (1989). The acidic amino-terminal
region of the HIV-1 Tat protein constitutes an essential activating domain. New Biol.
1: 101 – 110.
29. George, J. W., Pedersen, N. C., Higgins, J. (1993). The effect of age on the course of
experimental feline immunodeficiency virus infection in cats. AIDS Res. Hum. Retrovi-
ruses 9: 897 – 905.
30. Sheeter, D., Du, P., Rought, S., Richman, D., Corbeil, J. (2003). Surface CD4 expression
modulated by a cellular factor induced by HIV type 1 infection. AIDS Res. Hum. Retro-
viruses 19: 117 – 123.
31. Butler, S. L., Hansen, M. S. T., Bushman, F. D. (2001). A quantitative assay for HIV DNA
integration in vivo. Nat. Med. 7: 631 – 634.
32. Chun, T. W., et al. (1997). Presence of an inducible HIV-1 latent reservoir during highly
active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 94: 13193 – 13197.
33. Zufferey, R., et al. (1999). Woodchuck hepatitis virus posttranscriptional regulatory
element enhances expression of transgenes delivered by retroviral vectors. J. Virol.
73: 2886 – 2892.
34. Talbott (1989).
MOLECULAR THERAPY Vol. 10, No. 1, July 2004190Copyright B The American Society of Gene Therapy
METHOD doi:10.1016/j.ymthe.2004.03.019




















