
Tomaculous neuropathy in chromosome 1 Charcot-Marie-Tooth syndrome
7
Acta Neuropathol (1994) 87:91-97 Springer-Verlag 1994 Florian R Thomas Roger V. Lebo Gorazd Rosoklija Xin-Sheng Ding Robert E. Lovelace Norman Latov Arthur E Hays Tomaculous neuropathy in chromosome 1 Charcot-Marie-Tooth syndrome Received: 1 March 1993 / Revised, accepted: 17 September 1993 Abstract We performed morphological and immuno- histochemical studies on sural nerve biopsies from two members of a Charcot-Marie-Tooth type 1B famib; in which a mutation of the P0 gene on chromosome 1 had been found. Biopsies showed a tomaculous neuropathy with loss of myelinated fibers and frequent small onion bulbs. Immunofluorescence with antibodies to P0 showed this protein to be present in tomaculous and non-tomaculous areas of the myelin sheath. The sever- ity of the myelin abnormalities suggests that in this family Charcot-Marie-Tooth disease may result from a generalized disturbance of Schwann cells as a result of an abnormal P0 protein. Key words Charcot-Marie-Tooth disease HMSNIB CMT1B P0 " tomacula Hereditary motor and sensory neuropathies of the Charcot-Marie-Tooth or peroneal muscular atrophy type (CMT) have been classified according to mode of inheritance, natural history, clinical findings and elec- Supported by the Aaron Diamond Foundation and center grants from the MDA and from NINCDS to R.V. Lebo (NS25541) and N. Latov (NSl1761). ER Thomas is a fellow of the Medical Research Council of Canada ER Thomas G. Rosoklija - Xin-Sheng Ding - A.E Hays Department of Pathology (Division of Neuropathology), College of Physicians & Surgeons, Columbia University, New York, NY, USA EE Thomas - R.E. Lovelace N. Latov Neurological Institute, College of Physicians & Surgeons, Columbia University, New York, NY, USA EE Thomas ([~) Clinical Research Institute of Montreal, 110 Pine Avenue West, Montreal, Quebec, Canada H2W 1R7 R.V. Lebo Departments of Obstetrics, Gynecology and Pediatrics, University of California, San Francisco, CA, USA trophysiological and histopathological characteristics [17]. Genetic studies have distinguished clinically simi- lar slow nerve conduction CMT by linkage to chromo- some 1 (CMT1B) [5, 14, 26, 27, 29, 31-33, 48, 49] chromosome 17 (CMT1A) [55], chromosome X (CMTX) [2, 21-23, 28, 61], and another autosome (CMT1C) [11]. CMT associated with myotonic dystro- phy cosegregates with chromosome 19 polymorphisms [8]. The CMT1A locus has been mapped to distal band 17p11.2 [34], where a duplication including the periph- eral myelin protein-22 gene (PMP22) has been found in a majority of patients [37, 43, 52, 53], while a point mutation in the transmembrane domain of PMP22 was reported in one family [46]. Recently, independent mutations in the P0 gene, located in the q21.3--+q22 region on chromosome 1, have been identified in three families with CMT1B, indicating that an abnormal P0 may be responsible for this syndrome [27, 31, 49]. Immediately adjacent to P0, the three Fcy receptor type II genes (FcyRI]) are located, reported to be expressed in peripheral nerve myelin [57, 58]. In sural nerve biop- sies from two members of one of these families we found pathological changes of a neuropathy with sausage-shaped enlargements (tomacula) in all teased myelinated nerve fibers. Case reports We examined two patients, father and son, from the largest Duffy- linked CMTIB family [27, 32, 33, 48, 49]. Both ]had slowly pro- gressive weakness since childhood, affecting the lower extremities more than the upper extremities. They complained of numbness in the hands and feet and denied recurrent focal weakness or pain. There was no liability to pressure palsies. Eighteen living male and female relatives from four generations are affected. The physical examination was similar in father and son, except that the findings were more pronounced in the former. Both had pes cavus deform- ity. The father had enlarged, firm peripheral nerves. Muscle strength was reduced to 4/5 (British Medical Research Council Classification), distally more than proximally. Deep tendon reflexes were absent. Plantar responses were flexor. All sensory modalities were impaired. Laboratory and electrophysiological
Transcript of Tomaculous neuropathy in chromosome 1 Charcot-Marie-Tooth syndrome

Acta Neuropathol (1994) 87:91-97 �9 Springer-Verlag 1994
Florian R Thomas �9 Roger V. Lebo �9 Gorazd Rosoklija Xin-Sheng Ding �9 Rober t E. Lovelace �9 Norman Latov Ar thur E Hays
Tomaculous neuropathy in chromosome 1 Charcot-Marie-Tooth syndrome
Received: 1 March 1993 / Revised, accepted: 17 September 1993
Abstrac t We per formed morphological and immuno- histochemical studies on sural nerve biopsies from two members of a Charcot-Marie-Tooth type 1B famib; in which a mutat ion of the P0 gene on chromosome 1 had been found. Biopsies showed a tomaculous neuropathy with loss of myelinated fibers and frequent small onion bulbs. Immunofluorescence with antibodies to P0 showed this protein to be present in tomaculous and non-tomaculous areas of the myelin sheath. The sever- ity of the myelin abnormalities suggests that in this family Charcot-Marie-Tooth disease may result from a generalized disturbance of Schwann cells as a result of an abnormal P0 protein.
K e y words Charcot-Marie-Tooth disease �9 HMSNIB CMT1B �9 P0 " tomacula
Heredi tary motor and sensory neuropathies of the Charcot-Marie-Tooth or peroneal muscular atrophy type (CMT) have been classified according to mode of inheritance, natural history, clinical findings and elec-
Supported by the Aaron Diamond Foundation and center grants from the MDA and from NINCDS to R.V. Lebo (NS25541) and N. Latov (NSl1761). ER Thomas is a fellow of the Medical Research Council of Canada
ER Thomas �9 G. Rosoklija - Xin-Sheng Ding - A.E Hays Department of Pathology (Division of Neuropathology), College of Physicians & Surgeons, Columbia University, New York, NY, USA
EE Thomas - R.E. Lovelace �9 N. Latov Neurological Institute, College of Physicians & Surgeons, Columbia University, New York, NY, USA
EE Thomas ([~) Clinical Research Institute of Montreal, 110 Pine Avenue West, Montreal, Quebec, Canada H2W 1R7
R.V. Lebo Departments of Obstetrics, Gynecology and Pediatrics, University of California, San Francisco, CA, USA
trophysiological and histopathological characteristics [17]. Genet ic studies have distinguished clinically simi- lar slow nerve conduction CMT by linkage to chromo- some 1 (CMT1B) [5, 14, 26, 27, 29, 31-33, 48, 49] chromosome 17 (CMT1A) [55], chromosome X (CMTX) [2, 21-23, 28, 61], and another autosome (CMT1C) [11]. CMT associated with myotonic dystro- phy cosegregates with chromosome 19 polymorphisms [8]. The CMT1A locus has been mapped to distal band 17p11.2 [34], where a duplication including the periph- eral myelin protein-22 gene (PMP22) has been found in a majority of patients [37, 43, 52, 53], while a point mutat ion in the t ransmembrane domain of PMP22 was repor ted in one family [46]. Recently, independent mutations in the P0 gene, located in the q21.3--+q22 region on chromosome 1, have been identified in three families with CMT1B, indicating that an abnormal P0 may be responsible for this syndrome [27, 31, 49]. Immediately adjacent to P0, the three Fcy receptor type II genes (FcyRI]) are located, repor ted to be expressed in peripheral nerve myelin [57, 58]. In sural nerve biop- sies from two members of one of these families we found pathological changes of a neuropathy with sausage-shaped enlargements (tomacula) in all teased myelinated nerve fibers.
Case reports
We examined two patients, father and son, from the largest Duffy- linked CMTIB family [27, 32, 33, 48, 49]. Both ]had slowly pro- gressive weakness since childhood, affecting the lower extremities more than the upper extremities. They complained of numbness in the hands and feet and denied recurrent focal weakness or pain. There was no liability to pressure palsies. Eighteen living male and female relatives from four generations are affected. The physical examination was similar in father and son, except that the findings were more pronounced in the former. Both had pes cavus deform- ity. The father had enlarged, firm peripheral nerves. Muscle strength was reduced to 4/5 (British Medical Research Council Classification), distally more than proximally. Deep tendon reflexes were absent. Plantar responses were flexor. All sensory modalities were impaired. Laboratory and electrophysiological

92
Fig. 1 A, g Sural nerve in CMTiB. A Semithin section of the father's nerve shows a marked depletion of myelinated nerve fibers. Scattered onion bulbs consist of concentrically arranged Schwann cell processes (arrowheads), some without a central mye- linated fiber. Two myelinated fibers have a very thick myelin sheath and an irregular contour, suggesting that they are tomacula (open arrows). B In the son, the density of myelinated fibers is greater than in the father. Many of the large myelin structures are tomacula (open arrows). In many tomacula axons cannot be recognized, but axons could be identified in virtually all of them by electron microscopy (see Fig. 4). Other myelinated fibers have an abnormally thin sheath. Scattered small onion bulbs are indi- cated by arrowheads. A, B x660
studies were done in the father. B~, folate and lead levels, anti- body titers to MAG and GM1 (measured by high-performance thin layer chromatography and ELISA, respectively) and serum protein electrophoresis were all normal. On electrophysiological examination no sensory or motor responses were obtained with surface recordings. By needle examination of the left median nerve, the motor conduction velocity was slowed to 11 m/s (lower normal value 49 m/s), and the amplitude was reduced to 0.3 mV (lower normal value 5 mV). Sensory responses and F wave were absent. Electromyography revealed minimal spontaneous activity with high amplitude motor unit potentials. Sural nerve biopsies were performed in both patients.
Materials and methods
Paraffin-embedded nerve was stained with hematoxylin and eosin. A portion of the sural nerve was fixed in 2 % paraformaldehyde/ i % glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.3, poststained with 1% osmium tetroxide and embedded in Epon 812 for preparation of sections for light and electron microscopy. Myelinated fiber diameters and densities were obtained from digi- talized images of transverse semithin sections of nerve using a Leitz 100• oil immersion objective, a video camera and a Loats Association image analysis system. Diameters were derived from the transverse fiber areas under the assumption that they are per- fect circles. Radial myelin periodicity was measured in electron micrographs at an overall magnification of 367,500 using an ocular with a micrometer. The period distance of successive 10 to 20 mye- lin lamellae was measured and the periodicity expressed as the mean. One fiber was analyzed in the father, seven in the son, and five in each of three controls. For preparation of teased nerve fibers, 1 cm of nerve was dehydrated with increasing concentra- tions of ethanol and infiltrated with Spurt resin without the ac- celerator. Analysis was carried out according to Asbury and John- son [1]. Another portion was frozen in liquid nitrogen and cryo- sections were examined by immunofluorescence [51] using poly- clonal antibodies to P0 (gift of Dr. David Colman) and a monoclo- nal antibody to the FcyRII [57, 58] (gift of Dr. Christian Vedeler). Sural nerves from normal controls and from two patients with chronic neuropathies of unknown etiology were used in parallel.
Results
T h e b iopsy f indings were s imilar in fa ther and son. Semi th in cross-sect ions of ne rve showed a r educ t ion of m y e l i n a t e d f iber dens i ty which was m o d e r a t e in the son and severe in the fa ther [Fig. 1]. M a n y r e m a i n i n g f ibers had very th in mye l in sheaths. F r e q u e n t small o n i o n bu lbs and scat tered t o m a c u l a were found . T h e myel in - a ted f iber dens i ty was 250 /mm 2 in the fa ther and 3,147/ m m 2 in the son. H i s tome t r i c m e a s u r e m e n t s showed a
u n i m o d a l d i s t r ibu t ion of mye l ina t ed f ibers with a shift of the peak to d iamete r s b e t w e e n 1 and 4 ~m in the fa ther a nd a b i m o d a l d i s t r ibu t ion with one peak b e t w e e n 1 a nd 4 ~tm a nd a second peak at 6 ~tm in the son (Fig. 2). Near ly all of the fibers larger t han 5 ~tm in

diameter had features of tomacula . In teased-f iber prepara t ions and longitudinal semithin sections virtu- ally all fibers showed sausage-shaped expansions of myelin, located in bo th the paranodal and internodal regions (Fig. 3). Segmental remyelinat ion was found in all teased myel inated nerve fibers. Ultrathin sections conf i rmed the finding of tomacula (Fig. 4). These struc- tures consisted of closely apposed redundant loops of myelin sheath wound around or layered on one side of a thinly myel inated fiber. Radial periodicity of myelin sheaths including the tomacula was normal and varied f rom 13.5 to 14.7 nm in both patients. Three controls had a periodicity of 13.6 to 14.7 nm. The per iod dis- tance did not vary by more than 1.3 nm f rom the mean of each fiber. Myelin-like debris was demons t ra ted by electron microscopy within the cytoplasm of Schwann cells, chiefly in such cells that gave rise to a tomacu lum
93
% E
. .Q
LL
A
0 2 i
4 6 8 i i i I i I
10 12 14 16 18 20
%
. .Q
iT
840
720
600
48O
360
240
120
0 0
B
d i J i I
2 14 16 18 20 i i i i i
4 6 8 10 12
Fiber diameter (lam)
431.573.2
Fig. 2 A, B Histogram of diameters of myelinated nerve fibers. More large diameter than small diameter fibers are lost. In the son (B), nearly all fibers with a diameter of > 5 ~m represent toma- cula; A father; B son
Fig. 3 A, B Longitudinal view of tomacula. A A semithin section of the son's biopsy demonstrates a tomaculum in continuity with a myelinated fiber (arrow). The myelin sheath is inappropriately thin, probably as a result of segmental remyelination or hypomye- lination. B A teased myelinated nerve fiber contains tomacula consisting of globular expansions of myelin measuriLng 30 to 50 ~m in length. Some of the tomacula seem to be located at nodes of Ranvier (arrows), while others are located within internodes. Seg- ments of the myelin sheaths between tomacula vary in thickness and many of them are abnormally thin. All teased myelinated nerve fibers in both biopsies exhibit tomacula. A >'430; B • 120
and enclosed an identifiable axon. By indirect immu- nofluorescence, the ant ibody to P0 bound to myelin sheaths including the tomacula (Fig. 5). Immunos ta in- ing of the FcyRII had a similar pat tern. The staining characteristics did not differ f rom normal and disease controls.
Discussion
Sural nerve biopsies f rom two m e m b e r s of this family with C M T I B showed a tomaculous neuropa thy with normal spacing of the myelin lamellae; there was no
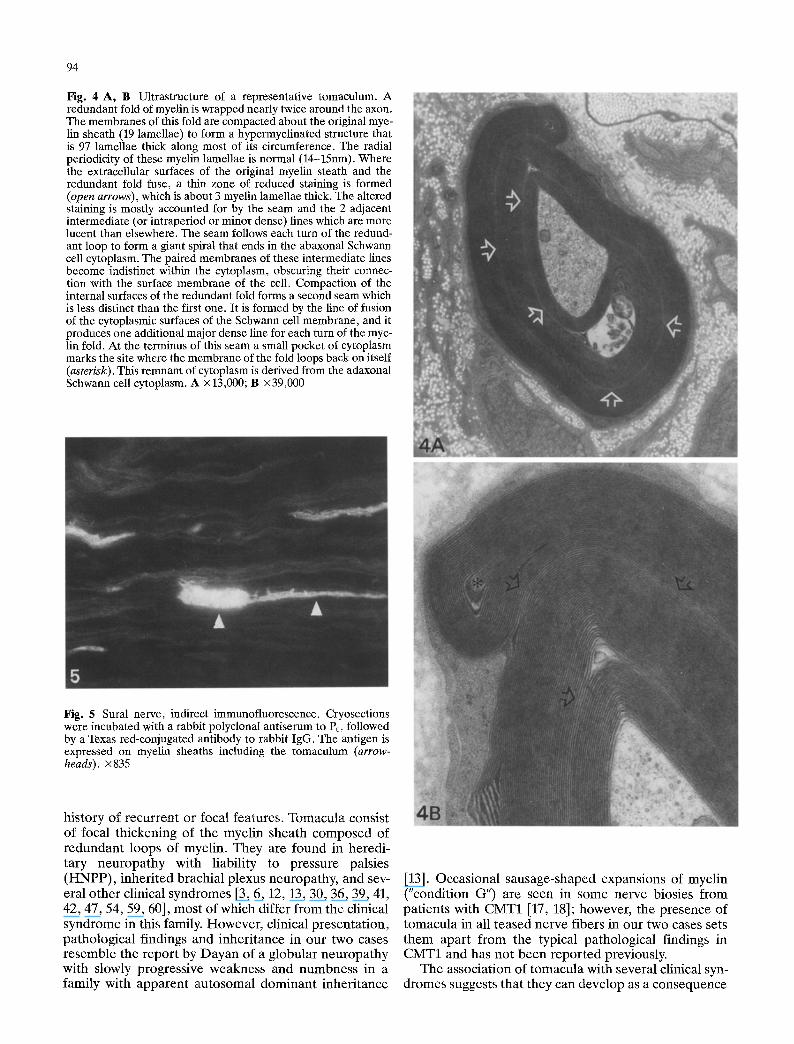
94
Fig. 4 A, B Ultrastructure of a representative tomaculnm. A redundant fold of myelin is wrapped nearly twice around the axon. The membranes of this fold are compacted about the original mye- lin sheath (19 lamellae) to form a hypermyelinated structure that is 97 lamellae thick along most of its circumference. The radial periodicity of these myelin lamellae is normal (14-15nm). Where the extracellular surfaces of the original myelin steath and the redundant fold fuse, a thin zone of reduced staining is formed (open arrows), which is about 3 myelin lamellae thick. The altered staining is mostly accounted for by the seam and the 2 adjacent intermediate (or intraperiod or minor dense) lines which are more lucent than elsewhere. The seam follows each turn of the redund- ant loop to form a giant spiral that ends in the abaxonal Schwann cell cytoplasm. The paired membranes of these intermediate lines become indistinct within the cytoplasm, obscuring their connec- tion with the surface membrane of the cell. Compaction of the internal surfaces of the redundant fold forms a second seam which is less distinct than the first one. It is formed by the line of fusion of the cytoplasmic surfaces of the Schwann cell membrane, and it produces one additional major dense line for each turn of the mye- lin fold. At the terminus of this seam a small pocket of cytoplasm marks the site where the membrane of the fold loops back on itself (asterisk). This remnam of cytoplasm is derived from the adaxonal Schwann cell cytoplasm. A x 13,000; B •
Fig. 5 Sural nerve, indirect immunofluorescence. Cryosections were incubated with a rabbit polyclonal antiserum to P0, followed by a Texas red-conjugated antibody to rabbit IgG. The antigen is expressed on myelin sheaths including the tomaculum (arrow- heads), x835
history of recurrent or focal features. Tomacula consist of focal thickening of the myelin sheath composed of redundant loops of myelin. They are found in heredi- tary neuropathy with liability to pressure palsies (HNPP), inherited brachial plexus neuropathy, and sev- eral other clinical syndromes [3, 6, 12, 13, 30, 36, 39, 41, 42, 47, 54, 59, 60], most of which differ from the clinical syndrome in this family. However, clinical presentation, pathological findings and inheritance in our two cases resemble the report by Dayan of a globular neuropathy with slowly progressive weakness and numbness in a family with apparent autosomal dominant inheritance
[13]. Occasional sausage-shaped expansions of myelin ("condition G") are seen in some nerve biosies from patients with CMT1 [17, 18]; however, the presence of tomacula in all teased nerve fibers in our two cases sets them apart from the typical pathological findings in CMT1 and has not been reported previously.
The association of tomacula with several clinical syn- dromes suggests that they can develop as a consequence

95
of several different pathogenic mechanisms. It has been suggested that tomacula are the result of an abnormal response to nerve injury or abnormal axon-myelin inter- actions [60]. In three families H N P P has been associ- ated with a deletion in chromosome 17p11.2 [12]. The same 1.5-Mb region contains the gene for PMP22, which when duplicated or carrying a point mutat ion, can also result in CMT1A [37, 43, 46, 52, 53]. These findings suggest that abnormal myelin membrane prote- ins can lead to the formation of tomacula. The resem- blance of tomacula to myelin folding and loops during early development may also reflect immature or abnor- mal axon-myelin interaction [15, 40]. It is of interest that myelin outfolding and hypermyelination, in addi- t ion to uncompacted myelin, occur in human and experimental neuropathies associated with antibodies to myelin proteins including epitopes shared by P0, that presumably interfere with the interaction between adhesion molecules [9, 10, 38, 44, 45, 56, 59].
In this family a substitution of a positively charged lysine for a negatively charged glutamate at codon 96 in the extracellular region of the myelin t ransmembrane P0 gene on chromosome 1 has been found [27, 49]. P0 is expressed only in Schwann cells and constitutes more than 50 % of all myelin protein [25]. It belongs to the immunoglobulin supergene family, whose members are involved in cell adhesion and recognition [35]. The extracellular domain of P0 appears to behave like a homophilic adhesion molecule [16, 19], and a similar funcion has been proposed for the intraceltular domain; thus P0 may have a role in myelin compaction at both the major and minor dense lines [20]. The cytoplasmic domain undergoes phosphorylat ion and dephospho- rylation on serine residues, consistant with a possible function in signal transduction [7, 50]. Transgenic mice homozygous for an inactivated P0 gene developed a hypomyelinat ion neuropathy with axonal degenerat ion and abnormally high expression of several myelin anti- gens, including N-Cam, MAG, L1, and the low-affinity nerve growth factor receptor [24]. Fur thermore , a severe demyelinating neuropathy with onion bulb formation was repor ted in a patient with anti-P0 anti- bodies [4]. It has, thus, been proposed that P0 may con- trol the myelination program [20]. The expression of a mutated P0 could lead to the tomaculous neuropathy found in the described family, while its absence, at least in the transgenic animals, causes hypomyelinat ion [24]. Interestingly, both a deletion of the PMP22 gene and a point mutat ion of the P0 gene are associated with toma- culum formation [12, 27, 49], suggesting that their gene products act either in parallel in a noncomplementary fashion or that they have different roles along one func- tional pathway.
Acknowledgement Sincere appreciation is extended to the CMT patients who participated in this study, for their continued cooperation.
References
1. Asbury AK, Johnson PC (1978) Pathology of peripheral nerve. Saunders, Philadelphia
2. Beckett J, Holden JJA, Simpson NE, White BN, MacLeod PM (1986) Localization of X-linked dominant Charcot-Marie- Tooth disease (CMT2) to Xq13.J Neurogenet 3:225-231
3. Behse E Buchthal F, Carlsen F, Knappels GG (1972) Heredi- tary neuropathy with liability to pressure palsies. Electrophys- iological and histopathological aspects. Brain 95:777-794
4. Ben Jelloun-Dellagi S, Dellagi K, Burger D, Ben Younes- Chennoufi A, Hentati FF, Steck A, Ben Hamida M (1992) Childhood peripheral neuropathy with autoantibodies to myelin glycoprotein P0- Ann Neurol 32:700-702
5. Bird TD, Ott J, Giblett ER (1982) Evidence for linkage of Charcot-Marie-Tooth neuropathy to the Duffy locus on chromosome 1. Am J Hum Genet 34:388-394
6. Bradley WG, Madrid R, Thrush DC, Campbell MJ (1975) Recurrent brachial plexus neuropathy. Brain 98:38!-398
7. Brunden KR, Poduslo JF (1987) A phorbol ester-sensitive kinase catalyzes the phosphorylation of P0 glycoprotein in myelin. J Neurochem 49:1863-1872
8. Brunner HG, Spaans F, Smeets HJM, Coerwinkel-Driessen M, Hulsebos T, Wieringa B, Ropers H-H (1991) Genetic link- age with chromosome 19 but not chromosome 17 in a family with myotonic dystrophy associated with hereditary motor and senory neuropathy. Neurology 41:80-84
9. Burger D, Perruisseau G, Simon M, Steck AJ (1992) Com- parison of the N-linked oligosaccharide structures of the two major human myelin glycoproteins MAG and P0: assessment and relative occurrence of oligosaccharide structures by serial lectin affinity chromatography of [14C]glycopeptides. J Neuro- chem 58:845-853
10. Burger D, Perruisseau G, Simon M, Steck AJ (1992) Com- parison of the N-linked oligosaccharide structures of the two major human myelin glycoproteius MAG and P0: assessment of the structures bearing the epitope for HNK-1 and human monoclonal immunoglobulin M found in demyelinating neu- ropathy. J Neurochem 58:854-861
11. Chance PE BirdTD, O'Connell P, Lipe H, Lalouel J-M, Lep- pert M (1990) Genetic linkage and heterogeneity in type I Charcot-Marie-Tooth disease (hereditary motor and sensory neuropathy type I). Am J Hum Genet 47:915-925
12. Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsu- nami N, Smith B, Swanson PD, Odelberg SJ, Disteche CM, Bird TD (1993) DNA deletion associated with hereditary neu- ropathy with liability to pressure palsies. Cell 72:143-151
13. Dayan AD, Graveson GS, Robinson PK, Woodhouse MA (1968) Globular neuropathy. A disorder of axons and Schwann cells. J Neurol Neurosurg Psychiatry 31:552-560
14. Defesche JC, Hoogendijk JE, de Visser M, Ongerboer de Vis- ser BW, Bolhuis PA (1990) Genetic linkage of hereditary motor and sensory neuropathy type I (Charcot-Marie-Tooth disease) to markers of chromosomes 1 and 17. Neurology 40: 1450-1453
15. Dunn JS (1970) Developing myelin in human peripheral nerve. Scott Med J 15:108-117
16. D'Urso D, Brophy PJ, Staugaitis SM, Gillespie CS, Frey AB, Stempak JG, Colman DR (1990) Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evi- dence for homotypic interaction. Neuron 2:449-460
17. Dyck PJ Chance R Lebo R, Carney JA (1993) Hereditary motor and sensory neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduseo JF (eds) Peripheral neuropathy, 3rd edn. Saunders, Philadelphia, pp 1094-1136
18. Dyck PJ, Giannini C, Lais A (1993) Pathologic alterations of nerves. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Po- duseo JF (eds) Peripheral neuropathy, 3rd edn. Saunders, Phi- ladelphia, pp 514-595

96
19. Filbin MT, Walsh FS, Trapp BD, Pizzey JA, Tennekon GI (1990) Role of myelin P0 protein as a homophilic adhesion molecule. Nature 344:871-872
20. Filbin MT, Tennekoon GI (1992) Myelin P0-protein, more than just a structural protein? BioEssays 14:541-547
21. Fischbeck KH, ar-Rushdi N, Pericak-Vance M, Rozear M, Roses AD, Fryns JP (1986) X-linked neuropathy: gene local- ization with DNA probes. Ann Neurol 20:527-532
22. Fryns JP, van den Berghe H (1980) Sex-linked recessive in- heritance in Charcot-Marie-Tooth disease with partial clinical manifestations in female carriers. Hum Genet 55:413-415
23. Gal A, Mucke J, Theile H, Wieacker PF, Ropers HH, Wienker TF (1985) X-linked dominant Charcot-Marie-Tooth disease: suggestion of linkage with a cloned DNA sequence from the proximal Xq. Hum Genet 70:38-42
24. Giese KP, Martini R, Lemke G, Soriano P, Schachner M' (1992) Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degenera- tion of myelin and axons. Cell 71:565-576
25. Greenfield S, Brostoff S, Eylar EH, Morrell P (1973) Protein composition of myelin of the peripheral nervous system. J Neurochem 20:1207-1216
26. Guiloff RJ, Thomas PK, Contreras M, Armitage S, Schwartz G, Sedgwick EM (1982) Linkage of autosomal dominant type I hereditary motor and sensory neuropathy to the Duffy locus on chromosome 1. Ann Hum Genet 46:25-27
27. Hayasaka K, Himoro M, Sato W, Takada G, Uyemura K, Shi- mizu N, Bird T, Conneally M, Chance PF (1993) CMT neu- ropathy 1B is associated with mutations of the myelin P0 gene. Nature Genet 5:31-34
28. Ionasescu VV, Burns TL, Searby C, Ionasescu R (1988) X-linked dominant Charcot-Marie-Tooth neuropathy with 15 cases in a family genetic linkage study. Muscle Nerve 11: 1154-1156
29. IonasescuVV, Trofatterr J, Haines JL, Ionasescu R, Searby C (1992) Charcot-Marie-Tooth disease related to chromosome 1. Am J reed Genet 42:728-732
30. Joy JL, Oh SJ (1989) Tomaculous neuropathy presenting as acute recurrent polyneuropathy. Ann Neurol 26:98-100
31. Kulkens T, Bolhuis PA, Wolterman RA, Kemp S, te Nijenhuis S, Valentijn LJ, Hensels GW, Jennekens FGI, de Visser M, Hoogendijk JE, Baas F (1993) Deletion of the serine 34 codon from the major peripheral myelin protein P0 gene in Charcot- Marie-Tooth disease type lB. Nature Genet 5:35-39
32. Lebo RV, Chance PF, Dyck PJ, Redila-Flores MT, Lynch ED, Golbus MS, Bird TD, King MC, Anderson LA, Hall J, Wie- gant J, Jiang Z, Dazin PF, Punnett HH, Schonberg SA, Moore K, Shull MM, Gendler S, Hurko O, Lovelace RE, Latov N, Trofatter J, Conneally PM (1991) Chromosome 1 Charcot-Marie-Tooth disease (CMT1B) locus in the Fcu receptor gene region. Hum Genet 88:1-12
33. Lebo RV, Lynch E, Wiegant J, Moore K, Trounstine M, van der Ploeg M (1991) Multicolor fluorescence in situ hybridiza- tion and pulsed-field electrophoresis dissect CMT1B gene region. Hum Genet 88:13-20
34. Lebo RV, Lynch ED, Bird TD, Golbus MS, Barker DF, O'Connell P, Chance PF (1992) Multicolor in situ hybridiza- tion and linkage analysis order Charcot-Marie-Tooth type 1 (CMTIA) gene-region markers. Am J Hum Genet 50:42-55
35. Lemke G, Lamar E, Patterson J (1988) Isolation and analysis of gene encoding peripheral myelin protein zero. Neuron 1: 73-83
36. Madrid R, BradleyWG (1975)The pathology ofneuropathies with focal thickening of the myelin sheath (tomaculous neu- ropathy). Studies on the formation of the abnormal myelin sheath. J Neurol Sci 25:415-448
37. Matsunami N, Smith B, Ballard L, Lensch MW, Robertson M, Albertsen H, Hanemann CO, Miiller HW, Bird TD, White R, Chance PF (1992) Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot-Marie-Tooth 1A. Nature Gene 1:176-179
38. Nardelli E, Pizzighella S, Tridente G, Rizzuto N (1981) Peripheral neuropathy associated with immunoglobulin disor- ders. An immunological and ultrastructural study. Acta Neu- ropathol (Berl) [suppl] VII: 258-261
39. Nordborg C, Conradi N, Sourander P, Hagberg B, Westerberg B (1984) Hereditary motor and sensory neuropathy of demye- linating and remyelinating type in children. Ultrastructural and morphometric studies on sural nerve biopsy specimens from 10 sporadic cases. Acta Neuropathol (Bed) 65:1-9
40. Ochoa J (1971) The sural nerve of the human foetus: electron microscope observations and counts of axons. J Anat 108: 231-245
41. Oh SJ, Joy JL, Sunwoo IN (1989) Tomaculous neuropathy presenting as demyelinating mononenropathy. Ann Neurol 26:168-169
42. Ohnishi A, Murai Y, Ikeda M, Fujita T, Furuya H, Kuroiwa Y (1989) Autosomal recessive motor and sensory neuropathy with excessive myelin outfolding. Muscle Nerve 12:568-575
43. Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask B J, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM, Lupski JR, Suter U (1992) The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth dis- ease type 1A. Nature Genet 1:159-165
44. Raine CS, Bornstein MB (1979) Experimental allergic neuri- tis. Ultrastructure of serum-induced myelin aberrations in peripheral nervous system cultures. Lab Invest 40:423-432
45. Raine CS, Wisniewski H, Prineas J (1969) An ultrastructural study of experimental demyelination and remyelination. II. Chronic experimental allergic encephalomyelitis in the peripheral nervous system. Lab Invest 21:316-327
46. Roa BH, Garcia CA, Suter U, Kulpa DA, Wise CA, Mueller J, Welcher AA, Snipes GJ, Shooter EM, Patel PI, Lupski JR (1993) Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 Gene. N Engl J Med 329:96-101
47. Schady W, Ochoa J (1984) Ehlers-Danlos in association with tomaculous neuropathy. Neurology 34:1270
48. Stebbins NB, Conneally PM (1982) Linkage of dominantly inherited Charcot-Marie-Tooth neuropathy to the Duffy locus in an Indiana family. Am J Hum Genet 34: 195A
49. Su Y, Brooks DG, Li L, Lepercq J, Trofatter JA, Ravetch JV, Lebo RV (1993) Myelin protein zero gene mutated in Charcot-Marie-Tooth disease type 1B patients. Proc Natl Acad Sci USA 90:10856-10860
50. Suzuki M, Sakamoto Y, Kitamura K, Fukunaga K, Yamamoto H, Miyamoto E, Uyemura K (1990) Phosphorylation of P0 glycoprotein in peripheral nerve myelin. J Neurochem 55: 1966-1971
51. Takatsu M, Hays AP, Latov N, Abrams GM, Nemni R, Sher- man WH, Nobile-Orazio E, Saito T, Freddo L (1985) Immu- nofluorescence study of patients with neuropathy and IgM M- proteins, Ann Neurol 18:173-181
52. Timmermann V, Nelis E, van Hul W, Nieuwenhuijsen BW, Chen KL, Wang S, Othman KB, Cullen B, Leach RJ, Hane- mann CO, de Jonghe P, Raeymaekers P, van Ommen G-JB, Martin J-J, Miiller HW, Vance JM, Fischbeck KH, van Broeckhoven C (1992) The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nature Genet 1:171-175
53. Valentijn LJ, Bolhius PA, Zorn I, Hoogendijk JE, van den Bosch N, Hensels GW, Stanton VP Jr, Housman DE, Fisch- beck KH, Ross DA, Nicholson GA, Meershoek EJ, Dau- werse HG, van Ommen G-JB, Baas F (1992) The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie- Tooth disease type 1A. Nature Genet 1:166-170
54. Vallat JM, Gil R, Leboutet MJ, Hugon J, Moulies D (1987) Congenital hypo- and hyperrayelination neuropathy. Acta Neuropathol (Berl) 74:197-201
55. Vance JM, Nicholson GA, Yamaoka LH, Stajich J, Stewart CS, Speer MC, Hung WY, Roses AD, Barker D, Pericak- Vance MA (1989) Linkage of Charcot-Marie-Tooth neuropa- thy type la to chromosome 17. Exp Neurol 104:186-189

97
56. van den Berg LH, Sadiq SA, Thomas FP, Latov N (1990) Characterization of HNK-1 bearing glycoproteins in human peripheral nerve myelin. J Neurosci Res 25:295-299
57. Vedeler CA (1987) Demonstration of Fcy receptors on human peripheral nerve fibers. J Neuroimmunol 15:207-216
58. Vedeler CA, Matre R, Kristoffersen EK (1989) Solubilization of human peripheral nerve Fcy receptors and purification of a functional 40-kDA receptor. Immunol Lett 22:281-286
59. Vital C, Pautrizel B, Lagueny A, Vital A, Bergouignan FX, David B, Loiseau P (1985) Hypermydlinisation dans un cas de neuropathie pdriph6rique avec gammapathie monoclonale b6nigne ~t IgM. Rev Neurol (Paris) 141:729-734
60. Windebank AJ (1993) Inherited recurrent focal neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF (eds) Peripheral neuropathy, 3rd edn. Saunders, Philadelphia, pp 1137-1148
61. Woratz G (1964) Neurale Muskelatrophie mit dominantem X-chromosomalem Erbgang. Abh Dtsche Akad Wiss Berl 2




















