
TNF-α-mediated anxiety in a mouse model of multiple sclerosis
8
TNF-α-mediated anxiety in a mouse model of multiple sclerosis Nabila Haji a, b, 1 , Georgia Mandolesi a, 1 , Antonietta Gentile a , Lucia Sacchetti c , Diego Fresegna a , Silvia Rossi a, d , Alessandra Musella a , Helena Sepman a , Caterina Motta a, d , Valeria Studer a, d , Valentina De Chiara a, d , Giorgio Bernardi a, d , Piergiorgio Strata b , Diego Centonze a, d, ⁎ a Fondazione Santa Lucia/Centro Europeo per la Ricerca sul Cervello (CERC), Rome, Italy b National Institute of Neuroscience, University of Turin, Turin, Italy c Clinica Psichiatrica, Dipartimento di Neuroscienze, Università Tor Vergata, Rome, Italy d Clinica Neurologica, Dipartimento di Neuroscienze, Università Tor Vergata, Rome, Italy abstract article info Article history: Received 2 March 2012 Revised 6 July 2012 Accepted 18 July 2012 Available online 24 July 2012 Keywords: Anxiety-like behavior EPSC Experimental autoimmune encephalomyelitis Inflammation Striatum TNF-α Multiple sclerosis (MS) causes a variety of motor and sensory deficits and it is also associated with mood dis- turbances. It is unclear if anxiety and depression in MS entirely reflect a subjective reaction to a chronic disease causing motor disability or rather depend on specific effects of neuroinflammation in neuronal circuits. To an- swer this question, behavioral, electrophysiological, and immunofluorescence experiments were performed in mice with experimental autoimmune encephalomyelitis (EAE), which models MS in mice. First, we ob- served high anxiety indexes in EAE mice, preceding the appearance of motor defects. Then, we demonstrated that tumor necrosis factor α (TNF-α) has a crucial role in anxiety associated with neuroinflammation. In fact, intracerebroventricular (icv) administration of etanercept, an inhibitor of TNF-α signaling, resulted in anxiolytic-like effects in EAE-mice. Accordingly, icv injection of TNF-α induced per se overt anxious behavior in control mice. Moreover, we propose the striatum as one of the brain regions potentially involved in EAE anxious behavior. We observed that before disease onset EAE striatum presents elevated TNF-α levels and strong activated microglia, early signs of inflammation associated with alterations of striatal excitatory post- synaptic currents (EPSCs). Interestingly, etanercept corrected the synaptic defects of pre-symptomatic EAE mice while icv injection of TNF-α in non-EAE mice altered EPSCs, thus mimicking the synaptic effects of EAE. In conclusion, anxiety characterizes EAE course since the very early phases of the disease. TNF-α released from activated microglia mediates this effect likely through the modulation of striatal excitatory synaptic transmission. © 2012 Elsevier Inc. All rights reserved. Introduction Recent studies on clinical depression led to the development of a novel theory, which links proinflammatory cytokines and behavioral disorders. According to this hypothesis, during systemic inflammatory diseases, peripherally released cytokines circulate in the blood, reach the brain and cause anxiety, anhedonia, social withdrawal, fatigue, and sleep disturbances (Dantzer et al., 2008; Miller et al., 2009; Raison et al., 2006). Proinflammatory cytokines have been claimed to play a role also in stress-induced mood disturbances and in major depression, indicating a widespread role of classical mediators of inflammation in emotional control (Capuron et al., 2002; Maes et al., 1995). A large body of evidence reports TNF-α as one of the proinflammatory cyto- kines involved in emotionality control both in humans and rodents (Bercik et al., 2010; Fiore et al., 1996, 1998; Krishnan et al., 2007; Tyring et al., 2006; Yamada et al., 2000). Anxiety and depression are often diagnosed in patients with MS (Feinstein, 2007), a frequent and severe inflammatory disease of the central nervous system (CNS) caused by autoreactive T lymphocytes directed against myelin sheaths of a variety of central tracts. Whether mood disturbances in this disorder entirely reflect a subjective reac- tion to a chronic disease causing motor disability, or rather follow specific effects of neuroinflammation in neuronal circuits is still debated. MS, in fact, has an unpredictable course accompanied by weakness, visual loss, bowel and bladder incontinence, fatigue and sensory deficits, ultimately determining chronic disability and possi- bly explaining the high prevalence of mood alterations diagnosed in patients (Compston and Coles, 2008). On the other hand, the level of proinflammatory cytokines dramatically increases in the brains of MS patients and of EAE mice, and is in fact involved in oligodendro- cyte and neuronal death, as well as in astrogliosis and microglial acti- vation typical of the disease (Imitola et al., 2005). Recently, a strong interest has been placed on the role of inflam- matory and immune molecules in the modulation of central nervous Experimental Neurology 237 (2012) 296–303 ⁎ Corresponding author at: Clinica Neurologica, Dipartimento di Neuroscienze, Università Tor Vergata, Via Montpellier 1, 00133 Rome, Italy. Fax: +39 06 7259 6006. E-mail address: [email protected] (D. Centonze). 1 N.H. and G.M. are equally contributing first authors. 0014-4886/$ – see front matter © 2012 Elsevier Inc. All rights reserved. doi:10.1016/j.expneurol.2012.07.010 Contents lists available at SciVerse ScienceDirect Experimental Neurology journal homepage: www.elsevier.com/locate/yexnr
Transcript of TNF-α-mediated anxiety in a mouse model of multiple sclerosis

Experimental Neurology 237 (2012) 296–303
Contents lists available at SciVerse ScienceDirect
Experimental Neurology
j ourna l homepage: www.e lsev ie r .com/ locate /yexnr
TNF-α-mediated anxiety in a mouse model of multiple sclerosis
Nabila Haji a,b,1, Georgia Mandolesi a,1, Antonietta Gentile a, Lucia Sacchetti c, Diego Fresegna a,Silvia Rossi a,d, Alessandra Musella a, Helena Sepman a, Caterina Motta a,d, Valeria Studer a,d,Valentina De Chiara a,d, Giorgio Bernardi a,d, Piergiorgio Strata b, Diego Centonze a,d,⁎a Fondazione Santa Lucia/Centro Europeo per la Ricerca sul Cervello (CERC), Rome, Italyb National Institute of Neuroscience, University of Turin, Turin, Italyc Clinica Psichiatrica, Dipartimento di Neuroscienze, Università Tor Vergata, Rome, Italyd Clinica Neurologica, Dipartimento di Neuroscienze, Università Tor Vergata, Rome, Italy
⁎ Corresponding author at: Clinica Neurologica, DUniversità Tor Vergata, Via Montpellier 1, 00133 Rome,
E-mail address: [email protected] (D. Centonze1 N.H. and G.M. are equally contributing first authors
0014-4886/$ – see front matter © 2012 Elsevier Inc. Alldoi:10.1016/j.expneurol.2012.07.010
a b s t r a c t
a r t i c l e i n f oArticle history:Received 2 March 2012Revised 6 July 2012Accepted 18 July 2012Available online 24 July 2012
Keywords:Anxiety-like behaviorEPSCExperimental autoimmune encephalomyelitisInflammationStriatumTNF-α
Multiple sclerosis (MS) causes a variety of motor and sensory deficits and it is also associated with mood dis-turbances. It is unclear if anxiety and depression inMS entirely reflect a subjective reaction to a chronic diseasecausing motor disability or rather depend on specific effects of neuroinflammation in neuronal circuits. To an-swer this question, behavioral, electrophysiological, and immunofluorescence experiments were performedin mice with experimental autoimmune encephalomyelitis (EAE), which models MS in mice. First, we ob-served high anxiety indexes in EAE mice, preceding the appearance of motor defects. Then, we demonstratedthat tumor necrosis factor α (TNF-α) has a crucial role in anxiety associated with neuroinflammation. In fact,intracerebroventricular (icv) administration of etanercept, an inhibitor of TNF-α signaling, resulted inanxiolytic-like effects in EAE-mice. Accordingly, icv injection of TNF-α induced per se overt anxious behaviorin control mice. Moreover, we propose the striatum as one of the brain regions potentially involved in EAEanxious behavior. We observed that before disease onset EAE striatum presents elevated TNF-α levels andstrong activated microglia, early signs of inflammation associated with alterations of striatal excitatory post-synaptic currents (EPSCs). Interestingly, etanercept corrected the synaptic defects of pre-symptomatic EAEmice while icv injection of TNF-α in non-EAE mice altered EPSCs, thus mimicking the synaptic effects of EAE.In conclusion, anxiety characterizes EAE course since the very early phases of the disease. TNF-α releasedfrom activated microglia mediates this effect likely through the modulation of striatal excitatory synaptictransmission.
© 2012 Elsevier Inc. All rights reserved.
Introduction
Recent studies on clinical depression led to the development of anovel theory, which links proinflammatory cytokines and behavioraldisorders. According to this hypothesis, during systemic inflammatorydiseases, peripherally released cytokines circulate in the blood, reachthe brain and cause anxiety, anhedonia, social withdrawal, fatigue,and sleep disturbances (Dantzer et al., 2008; Miller et al., 2009; Raisonet al., 2006). Proinflammatory cytokines have been claimed to play arole also in stress-induced mood disturbances and in major depression,indicating a widespread role of classical mediators of inflammationin emotional control (Capuron et al., 2002; Maes et al., 1995). A largebody of evidence reports TNF-α as one of the proinflammatory cyto-kines involved in emotionality control both in humans and rodents
ipartimento di Neuroscienze,Italy. Fax: +39 06 7259 6006.)..
rights reserved.
(Bercik et al., 2010; Fiore et al., 1996, 1998; Krishnan et al., 2007;Tyring et al., 2006; Yamada et al., 2000).
Anxiety and depression are often diagnosed in patients with MS(Feinstein, 2007), a frequent and severe inflammatory disease of thecentral nervous system (CNS) caused by autoreactive T lymphocytesdirected against myelin sheaths of a variety of central tracts. Whethermood disturbances in this disorder entirely reflect a subjective reac-tion to a chronic disease causing motor disability, or rather followspecific effects of neuroinflammation in neuronal circuits is stilldebated. MS, in fact, has an unpredictable course accompanied byweakness, visual loss, bowel and bladder incontinence, fatigue andsensory deficits, ultimately determining chronic disability and possi-bly explaining the high prevalence of mood alterations diagnosed inpatients (Compston and Coles, 2008). On the other hand, the levelof proinflammatory cytokines dramatically increases in the brains ofMS patients and of EAE mice, and is in fact involved in oligodendro-cyte and neuronal death, as well as in astrogliosis and microglial acti-vation typical of the disease (Imitola et al., 2005).
Recently, a strong interest has been placed on the role of inflam-matory and immune molecules in the modulation of central nervous

297N. Haji et al. / Experimental Neurology 237 (2012) 296–303
system (CNS) function. TNF-α, through the action of its two receptors(TNFR1 and TNFR2) which are expressed on neurons and glial cells,has a broad range of actions on neurons which may be either neuro-protective or neurotoxic. Interestingly, it plays a facilitatory role inglutamate excitotoxicity, both directly and indirectly by inhibitingglial glutamate transporters on astrocytes. Additionally, TNF-α hasdirect effects on glutamate transmission, for example increasing ex-pression of AMPA receptors on synapses and also plays a role in syn-aptic plasticity, through the modulation of long-term potentiation(LTP) (Pickering et al., 2005). Interestingly, in EAE mice we observed,since the presymptomatic phase of the disease, an enhancement ofthe glutamate transmission likely dependent on TNF-α in the nucleusstriatum (Centonze et al., 2009), a brain area recently recognized toplay a substantial role in mood control (Favilla et al., 2008; Haroonet al., 2012; Kim et al., 2008; Nestler and Carlezon, 2006; Steineret al., 2010; Zhang et al., 2008).
Therefore, it can be hypothesized that in EAE cytokines may directlyalter neuronal activity and cause mood alterations independently oftheir destructive effects on myelinated axons and of the associateddisability. Previous works on EAE did not provide a conclusive an-swer to this important biological dilemma, because both sickness(Pollak et al., 2002, 2003) and anxiety were evaluated only duringthe symptomatic phase of the disease, in conjunction with motordisturbances (Peruga et al., 2011). On the other hand, Rodrigues etal. (2011) recently reported no sign of anxiety in EAE mice in boththe pre-symptomatic and symptomatic phases of the disease. There-fore, there is still a debate about the occurrence of anxiety-likebehavior in EAE mice.
Thus, to answer this critical issue, here we studied anxiety-relatedbehavior in EAE before the appearance of motor disturbances. We ob-served high anxiety indexes in EAEmice precedingmotor defects and,associated with increased TNF-α levels, microglial activation and al-tered spontaneous excitatory postsynaptic currents (sEPSCs) in thestriatum. By in vivo modulation of TNF-α we provided solid evidencethat it is one of the major proinflammatory cytokines responsible forthe emotional and synaptic alterations seen in early stage of EAE.
Materials and methods
Experimental animals
The subjects in this studywere 6 to 8 week old femalemice, C57BL/6,purchased from Charles-River (Italy). They were randomly assignedto standard cages, with four to five animals per cage, and kept atstandard housing conditions with a light/dark cycle of 12 h andfree access to food and water. All experiments were carried out in ac-cordance with the Guide for the Care and Use of Laboratory Animalsand the European Communities Council Directive of 24 November,1986 (86/609/EEC).
Induction and clinical assessment of EAE
After 1 week of acclimatization, mice were injected subcutaneous-ly at the flanks with 200 μg of myelin oligodendrocyte glycoproteinp35-55 (MOG35-55) emulsion to induce EAE by active immunization.The emulsion was prepared under sterile conditions using MOG35-55
(>85% purity, Espikem, Florence, Italy) in complete Freund's adjuvant(CFA, Difco), andMycobacterium tuberculosis H37Ra (8 mg/ml; strainH37Ra, Difco, Lawrence, KS, USA) emulsified with phosphate bufferedsaline (PBS). The control emulsion was prepared the same way with-out MOG35-55 for the control group (CFA group). All animals wereinjected with 500 ng pertussis toxin (Sigma, St. Louis, MO, USA) intra-venously on the day of immunization and 2 days later according tostandard protocols of EAE induction. Animals were scored daily forclinical symptoms of EAE, according to the following scale: 0, no clin-ical signs; 1, flaccid tail; 2, hind limb weakness; 3, hind limb paresis;
4, complete bilateral hind limb paralysis; 5, death due to EAE; inter-mediate clinical signs were scored adding 0.5 value (Aktas and Zipp,2003; Centonze et al., 2009; Rossi et al., 2010).
Stereotaxic surgery
Mice (n=12) were surgically implanted with a 22-gauge guidecannula (model C313G; Plastics One, Inc., Roanoke, VA) under ketamine(100 mg/kg) anesthesia. Cannulae were placed into the right lateralventricle using the following coordinates: anteroposterior: −0.4 mm;−1 mm lateral and 2 mm below the horizontal plane of bregma. Micewere given icv infusions of TNF-α 20 ng/μl (n=5) or vehicle (n=7)and tested 3 days later for their emotional behavior.
Minipump implantation
One week before immunization, a group of EAE (n=25) and CFAmice (n=20) were implanted with a minipump in order to allowcontinuous icv infusion of either vehicle or etanercept (10 μg/μl,Wyeth Lederle, Italy) starting the day of minipump implantation andover 4 weeks (two sets of experiments). Alzet osmotic minipumps(model 1004; Durect Corporation, Cupertino, CA) connected via cath-eter tube to intracranial cannula (Alzet Brain Infusion Kits 3) deliveredvehicle or etanercept into the right lateral ventricle at a continuousrate of 0.11 μl/h. The coordinates used for icv minipump implantationwere from bregma: anteroposterior=−0.4 mm and lateral=−1 mm;depth: 2.5 mm from the skull.
Behavior
Previous studies have already described the onset of motor deficitsin EAE mice starting from 10 to 11 days post immunization (dpi)(Centonze et al., 2009). Thus, anxiety was assessed in EAE and inthe respective CFA-treated control mice at 7 dpi and 8 dpi by meansof the open field test (OFT) and the elevated plus maze (EPM) respec-tively. At the OFT, intense anxiety causes mice showing thigmotaxis(the tendency to stay on the periphery of the open field). At 7 dayspost immunization (dpi), each mouse, 9 weeks old, was placed inthe center of the arena (40 cm×30 cm) and video-recorded duringexploration of the apparatus for 30 min. The apparatus was cleanedwith 10% ethanol between trials. The time spent in the center of thearena was measured by a dedicated software (Ethovision, Noldusand VideoTrack, ViewPoint).
EPM, which was conducted 1 day after the OFT (8 dpi), consistedof a center platform (5 cm×5 cm) 36.5 cm off the ground with fourbranching arms (30 cm×5 cm) where two of the arms are openand illuminated, while the other two arms are enclosed by plasticwalls of a standard height (16 cm). The test session began with themouse individually placed on the center square facing an open arm.Animals were allowed to freely explore the maze for 5 min for armentries and time spent in open arms (with all four paws) (expressedas a percentage of total time). The EPM was cleaned with 10% ethanolafter each trial to effectively remove the scent of the previously testedanimal. Behavioral parameters were recorded and subsequently ana-lyzed using automatic videotracking systems (Ethovision, Noldus Inc.,Leesburg, VA and VideoTrack, Viewpoint, France) for OFT and EPM.
We performed two different sets of experiments and both resultsfrom OFT and EPM were reproducible. Therefore, we pooled togeth-er the data except for the OFT due to different values between theexperiments.
ELISA assay
Ten dpi EAE (n=7) and CFA (n=7) mice from three different im-munizations were decapitated, brains were quickly removed and stri-ata were dissected on ice and frozen. Tissues were homogenized in

298 N. Haji et al. / Experimental Neurology 237 (2012) 296–303
ice-cold lysis buffer containing 20 mM Tris, 150 mMNaCl, 1% NonidetP-40, 0.5% sodium deoxycholate, 1 mM EDTA and 0.1% SDS (pH 7.5)with protease inhibitors cocktail (Sigma), and centrifuged for 20 minat 13,000 rpm at 4 °C. Supernatants were collected and the amountof proteins was quantified by Bradford colorimetric reaction (Bio-RadProtein Assay). The proteins were collected and used for quantifica-tion of TNF-α levels by commercially available ELISA kit (eBioscience,San Diego, CA, USA) according to manufacturer's instructions. Eachsample was loaded in duplicate and equal amount of proteins wereloaded per well (600 μg).
Immunohistochemistry and confocal microscopy
EAE and CFA mice were killed before the appearance of motor defi-cits at 7 dpi. They were deeply anesthetized with avertine and perfusedthrough the aorta with ice-cold 4% paraformaldehyde. Brains werepost-fixed for at least 4 h at 4 °C and equilibrated with 30% sucroseovernight. Thirty micrometer-thick coronal sections were perme-abilized in PBS with Triton-X 0.25% (TPBS). All following incubationswere performed in TPBS. Sections were pre-incubated with 10% normaldonkey serum solution for 1 h at room temperature and incubatedwiththe primary antibody rabbit anti-ionized calcium binding adaptormolecule 1 (Iba1; 1:500, WAKO) overnight at 4 °C. After being washedfor three times, 10 min each, sections were incubated with the second-ary antibody Cy3-conjugated donkey anti-mouse (1:200, Jackson) for2 h at RT, rinsed and DAPI counterstained. Sections were mountedwith Vecta-shield (Vector Labs, USA) on poly-L-lysine-coated slides,air-dried and coverslipped. Images were acquired using a LSM7 Zeissconfocal laser-scanner microscope (Zeiss, Göttingen, Germany). Byusing a 20× objective (zoom 1×, pixel resolution 1024×1024, 5 μmZ-step, pinhole of 1 airy units) immunostaining stacks of the striatumfrom coronal sections were z-projected and exported in TIFF file formatby NIH ImageJ software (http://rsb.info.nih.gov/ij/). Iba1-positive cellswere countedmanually on the z-projections and divided by the surfaceof striatal sections (microglial density). Microglial surface was mea-sured on the basis of anti-Iba1 staining, maintaining the same thresholdbetween the groups. Measurementswere repeated for 7–9 images from5 sections (n=3 mice per group).
Electrophysiology
Mice were killed by cervical dislocation under halothane anesthe-sia and corticostriatal coronal slices (200 μm) were prepared fromfresh tissue blocks of the brain by means of a vibratome (Centonzeet al., 2007, 2009). A single slice was then transferred to a recordingchamber and submerged in a continuously flowing artificial CSF(ACSF) (34 °C, 2–3 ml/min) gassed with 95% O2–5% CO2. The compo-sition of the control ACSF was (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2,1.2 NaH2PO4, 2.4 CaCl2, 11 Glucose, and 25 NaHCO3. Recording pipetteswere advanced towards individual striatal cells in the slice under posi-tive pressure and visual control (WinVision 2000, Delta Sistemi, Italy)and, on contact, tight GΩ seals were made by applying negative pres-sure. The membrane patch was then ruptured by suction and mem-brane current and potential monitored using an Axopatch 1D patchclamp amplifier (Molecular Devices, Foster City, CA, USA). Whole-cellaccess resistances measured in voltage clamp were in the range of5–20 MΩ. Whole-cell patch clamp recordings were made with borosil-icate glass pipettes (1.8 mm o.d.; 2–3 MΩ), in voltage-clamp mode, atthe holding potential (HP) of −80 mV. To study sEPSCs from mediumspiny neurons (MSNs), the recording pipettes were filled with internalsolution of the following composition (mM): K-gluconate (125), NaCl(10), CaCl2, (1.0), MgCl2 (2.0), 1,2-bis (2-aminophenoxy) ethane-N,N,N,N-tetraacetic acid (BAPTA; 0.5), N-(2-hydroxyethyl)-piperazine-N-s-ethanesulfonic acid (HEPES; 19), guanosine triphosphate (GTP;0.3), and Mg-adenosine triphosphate (Mg-ATP; 1.0), adjusted to pH7.3 with KOH. Bicuculline (10 μM)was added to the perfusing solution
to block GABAA-mediated transmission. Synaptic events were storedby using P-CLAMP 9 (Axon Instruments) and analyzed offline on a per-sonal computer with Mini Analysis 5.1 (Synaptosoft, Leonia, NJ, USA)software. The detection threshold of sEPSCs was set at twice the base-line noise. The fact that no false events would be identified was con-firmed by visual inspection for each experiment. Offline analysis wasperformed on spontaneous synaptic events recorded during fixedtime epochs (1–2 min), sampled every 2–3 min (5–12 samplings)(Centonze et al., 2009; Rossi et al., 2010). Only cells that exhibited stablefrequencies in control (less than 20% changes during the control sam-plings) were taken into account. For sEPSCs kinetic analysis, eventswith peak amplitude between 10 and 50 pA were grouped, aligned byhalf-rise time, and normalized by peak amplitude. In each cell, all eventsbetween 10 and 50 pAwere averaged to obtain rise times, decay times,and half widths (Centonze et al., 2009). The electrophysiological effectsof etanercept in EAE mice at 7–10 dpi were compared with those ofuntreated EAE and CFA at the same timepoints. One to nine cells per an-imalwere recorded. For each type of experiment and time point, at leastfour distinct animals were employed from each experimental group.Throughout the text “n” in electrophysiological experiments refers tothe number of cells. Drugs were applied by dissolving them to thedesired final concentration in the bathing ACSF. Drugs were (in μM)CNQX (10), MK-801 (30) (Tocris Cookson, Bristol, UK), and Bicuculline(10) (Sigma-RBI, St. Louis, USA).
Statistical analysis
Two-sample comparisons were carried out using the Student'st-test, while multiple comparisons were made using one-way ANOVAfollowed by followed by Holm–Sidak method.
Results
EAE causes anxiety before the appearance of motor deficits
To determine whether EAE mice develop an anxiety-like behaviorduring the pre-symptomatic phase of the disease, we performed twobehavioral tests, the OFT and the EPM on both CFA and EAE mice (n=14 mice for each group; two sets of experiments), at 7 and 8 dpi,respectively.
We observed that during the OFT (Fig. 1A), EAE mice spent sig-nificant less time in the center of the arena relative to CFA mice(CFA: 358.0±20.2 s; EAE: 172.6±21.2 s; pb0.001), suggesting an in-creased anxiety in these animals. The total distance traveled in thearena did not differentiate between EAE and CFA mice, thus excludingany changes in locomotor activity in the two groups (CFA: 3767.09±351.84; EAE: 3324.0±253.08; p>0.05). Accordingly, at the EPMtest (Fig. 1B) EAE mice exhibited a significant decreased preferencefor the open arm (EAE: 16.14±2.24%, CFA: 30.07±4.92%, pb0.05)over the close arm (EAE: 74.11±2.27%, CFA: 61.77±5.38%, pb0.05).
Altogether, these results demonstrate that the levels of anxiety-likebehavior were significantly different between EAE and CFA mice.
Effects of in vivo modulation of TNF-α signaling on anxiety-like behavior
A good candidate asmolecular player involved in the EAE-mediatedanxious behavior is the proinflammatory cytokine TNF-α. To addressthis issue we treated mice with etanercept, a TNF-α inhibitor func-tioning as a decoy receptor that binds to soluble TNF-α (Tyring et al.,2006). Etanercept or vehicle was delivered icv by osmotic minipumpfor 4 weeks in both CFA and EAE mice. One week after implantation,we induced EAE and behavioral tests were performed during thepre-symptomatic phase. We observed at the OFT that EAE mice treatedwith etanercept spent 2.5 fold greater time in the center of the arenathan EAE mice receiving icv vehicle (EAE-etanercept, n=8: 139.0±14.4 s, EAE vehicle, n=7: 52.4±14.9 s), and closely resembled both
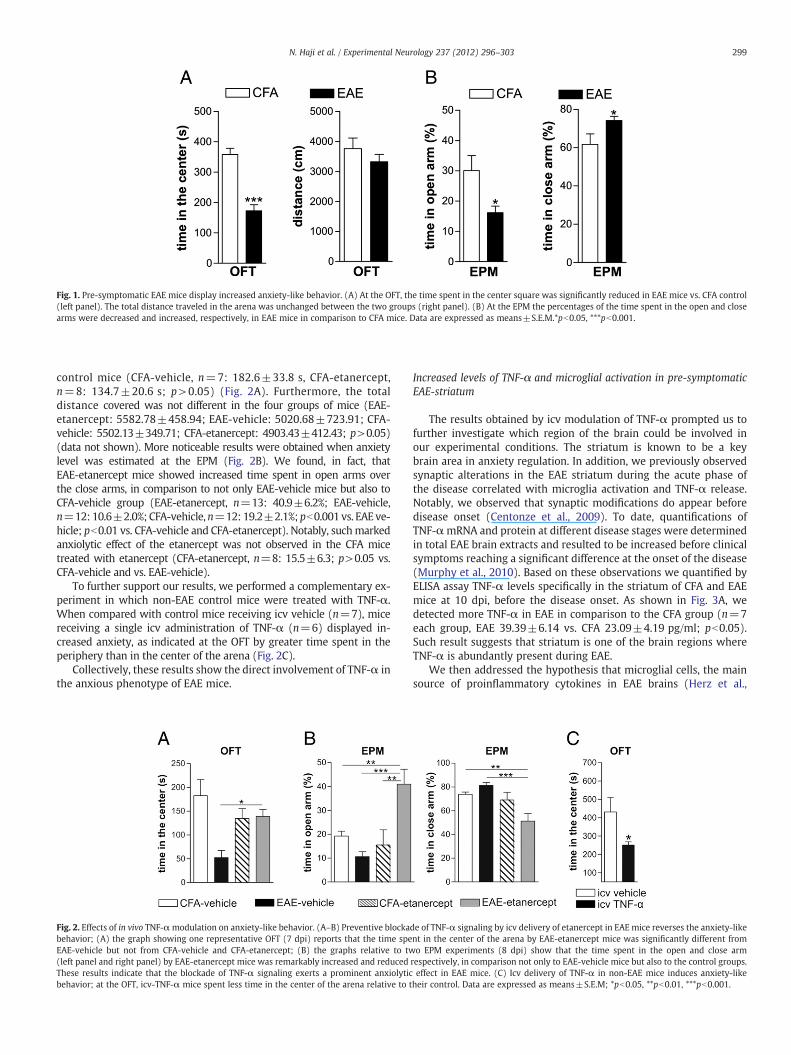
Fig. 1. Pre-symptomatic EAE mice display increased anxiety-like behavior. (A) At the OFT, the time spent in the center square was significantly reduced in EAE mice vs. CFA control(left panel). The total distance traveled in the arena was unchanged between the two groups (right panel). (B) At the EPM the percentages of the time spent in the open and closearms were decreased and increased, respectively, in EAE mice in comparison to CFA mice. Data are expressed as means±S.E.M.*pb0.05, ***pb0.001.
299N. Haji et al. / Experimental Neurology 237 (2012) 296–303
control mice (CFA-vehicle, n=7: 182.6±33.8 s, CFA-etanercept,n=8: 134.7±20.6 s; p>0.05) (Fig. 2A). Furthermore, the totaldistance covered was not different in the four groups of mice (EAE-etanercept: 5582.78±458.94; EAE-vehicle: 5020.68±723.91; CFA-vehicle: 5502.13±349.71; CFA-etanercept: 4903.43±412.43; p>0.05)(data not shown). More noticeable results were obtained when anxietylevel was estimated at the EPM (Fig. 2B). We found, in fact, thatEAE-etanercept mice showed increased time spent in open arms overthe close arms, in comparison to not only EAE-vehicle mice but also toCFA-vehicle group (EAE-etanercept, n=13: 40.9±6.2%; EAE-vehicle,n=12: 10.6±2.0%; CFA-vehicle,n=12: 19.2±2.1%; pb0.001 vs. EAE ve-hicle; pb0.01 vs. CFA-vehicle and CFA-etanercept). Notably, suchmarkedanxiolytic effect of the etanercept was not observed in the CFA micetreated with etanercept (CFA-etanercept, n=8: 15.5±6.3; p>0.05 vs.CFA-vehicle and vs. EAE-vehicle).
To further support our results, we performed a complementary ex-periment in which non-EAE control mice were treated with TNF-α.When compared with control mice receiving icv vehicle (n=7), micereceiving a single icv administration of TNF-α (n=6) displayed in-creased anxiety, as indicated at the OFT by greater time spent in theperiphery than in the center of the arena (Fig. 2C).
Collectively, these results show the direct involvement of TNF-α inthe anxious phenotype of EAE mice.
Fig. 2. Effects of in vivo TNF-αmodulation on anxiety-like behavior. (A–B) Preventive blockabehavior; (A) the graph showing one representative OFT (7 dpi) reports that the time speEAE-vehicle but not from CFA-vehicle and CFA-etanercept; (B) the graphs relative to tw(left panel and right panel) by EAE-etanercept mice was remarkably increased and reducedThese results indicate that the blockade of TNF-α signaling exerts a prominent anxiolyticbehavior; at the OFT, icv-TNF-α mice spent less time in the center of the arena relative to t
Increased levels of TNF-α and microglial activation in pre-symptomaticEAE-striatum
The results obtained by icv modulation of TNF-α prompted us tofurther investigate which region of the brain could be involved inour experimental conditions. The striatum is known to be a keybrain area in anxiety regulation. In addition, we previously observedsynaptic alterations in the EAE striatum during the acute phase ofthe disease correlated with microglia activation and TNF-α release.Notably, we observed that synaptic modifications do appear beforedisease onset (Centonze et al., 2009). To date, quantifications ofTNF-αmRNA and protein at different disease stages were determinedin total EAE brain extracts and resulted to be increased before clinicalsymptoms reaching a significant difference at the onset of the disease(Murphy et al., 2010). Based on these observations we quantified byELISA assay TNF-α levels specifically in the striatum of CFA and EAEmice at 10 dpi, before the disease onset. As shown in Fig. 3A, wedetected more TNF-α in ΕΑΕ in comparison to the CFA group (n=7each group, EAE 39.39±6.14 vs. CFA 23.09±4.19 pg/ml; pb0.05).Such result suggests that striatum is one of the brain regions whereTNF-α is abundantly present during EAE.
We then addressed the hypothesis that microglial cells, the mainsource of proinflammatory cytokines in EAE brains (Herz et al.,
de of TNF-α signaling by icv delivery of etanercept in EAE mice reverses the anxiety-likent in the center of the arena by EAE-etanercept mice was significantly different fromo EPM experiments (8 dpi) show that the time spent in the open and close arm
respectively, in comparison not only to EAE-vehicle mice but also to the control groups.effect in EAE mice. (C) Icv delivery of TNF-α in non-EAE mice induces anxiety-likeheir control. Data are expressed as means±S.E.M; *pb0.05, **pb0.01, ***pb0.001.

Fig. 3. Quantification of TNF-α protein and microglial activation in the pre-symptomatic EAE-striatum. (A) ELISA quantification of TNF-α shows a significant increase of TNF-α pro-tein in the EAE-striatum (10 dpi) versus CFA-striatum. (B–C) Microglia activation in EAE-striatumwas determined by Iba1-immunostaining and confocal analysis on coronal striatalsections from CFA and EAE mice (7 dpi). (B) Representative image of hypertrophic activated microglia expressing Iba1 in EAE-striatum in comparison to CFA. (C) A low magnifi-cation of striatal sections stained with anti-Iba1 antibody (red fluorescence) and counterstained with DAPI shows an increased number of microglial cells in EAE mice versus CFA.(D–E–F) Confocal quantitative analysis of Iba1+ activated microglia; the measurements of the density (D) and mean cell area (F) of Iba1+ cells and their total surface covered byIba1 staining (E) (a parameter indicative of both proliferation and morphological changes of microglia cells), indicate a strong microglial reaction in striatum pre-symptomatic EAErelative to CFA. Data are expressed as means±S.E.M ; *pb0.05, ***pb0.001. Scale bars: B, 10 μm; C, 50 μm.
300 N. Haji et al. / Experimental Neurology 237 (2012) 296–303
2010), were involved in the behavioral alterations of EAE mice. Fol-lowing activation, microglia cells and macrophages undergo prolifer-ation and morphological changes and overexpress Iba1 (Lynch, 2009;Mandolesi et al., 2012). Therefore, we evaluated microglial activationin the striatum of EAE mice at the same time points of the behavioralexperiments, by immunostaining with anti-Iba1 antibody (Figs. 3B–C).We then carried out a confocal quantitative analysis for the total sur-face covered by Iba1+ cells (a parameter indicative of both prolifer-ation and morphological changes of microglia cells), their densityand their mean cell area (parameters indicative respectively of pro-liferation and morphological change). We found that in EAE (7 dpi)striata the density of microglial cells, measured as number of Iba1+
cells per area, was 4 fold higher than that detected in CFA animals(n=3 mice each group, pb0.001) (Fig. 3D). In addition, we foundthat in EAE striatum microglia morphology becomes hypertrophic
being the total Iba1+ surface 6.4 fold higher in comparison to CFAgroup (pb0.001) (Fig. 3E) and the mean cell area double in EAEthan in CFA (pb0.05) (Fig. 3F).
These observations show that the early microglial activation inpre-symptomatic EAE-striatum is correlated with increased levels ofTNF-α.
In vivo modulation of TNF-α signaling alters glutamate transmission
In a previous work we suggested TNF-α as key mediator of theatypical spontaneous glutamatergic transmission observed in EAE(Centonze et al., 2009). In particular we observed that incubation ofcontrol brain slices with TNF-α mimicked the effects of EAE and ofactivated microglia on sEPSC kinetic properties, by increasing theirdecay time and duration. Moreover, blockade of TNF-α signaling
301N. Haji et al. / Experimental Neurology 237 (2012) 296–303
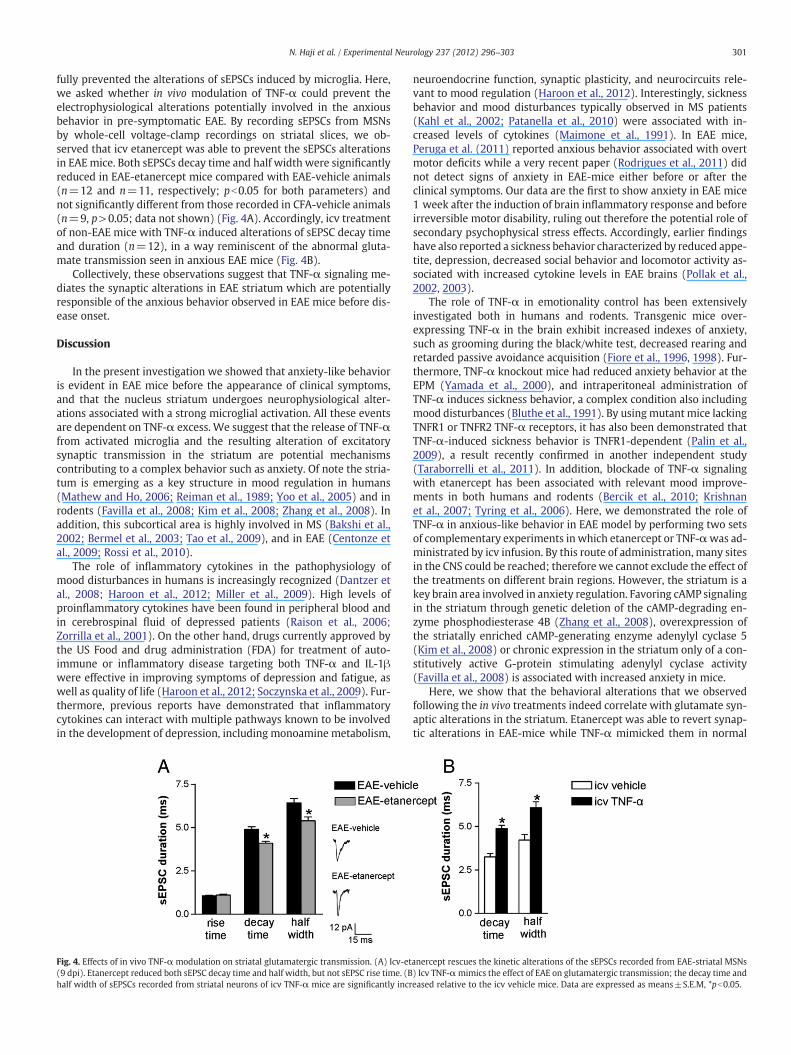
fully prevented the alterations of sEPSCs induced by microglia. Here,we asked whether in vivo modulation of TNF-α could prevent theelectrophysiological alterations potentially involved in the anxiousbehavior in pre-symptomatic EAE. By recording sEPSCs from MSNsby whole-cell voltage-clamp recordings on striatal slices, we ob-served that icv etanercept was able to prevent the sEPSCs alterationsin EAEmice. Both sEPSCs decay time and half width were significantlyreduced in EAE-etanercept mice compared with EAE-vehicle animals(n=12 and n=11, respectively; pb0.05 for both parameters) andnot significantly different from those recorded in CFA-vehicle animals(n=9, p>0.05; data not shown) (Fig. 4A). Accordingly, icv treatmentof non-EAE mice with TNF-α induced alterations of sEPSC decay timeand duration (n=12), in a way reminiscent of the abnormal gluta-mate transmission seen in anxious EAE mice (Fig. 4B).
Collectively, these observations suggest that TNF-α signaling me-diates the synaptic alterations in EAE striatum which are potentiallyresponsible of the anxious behavior observed in EAE mice before dis-ease onset.
Discussion
In the present investigation we showed that anxiety-like behavioris evident in EAE mice before the appearance of clinical symptoms,and that the nucleus striatum undergoes neurophysiological alter-ations associated with a strong microglial activation. All these eventsare dependent on TNF-α excess. We suggest that the release of TNF-αfrom activated microglia and the resulting alteration of excitatorysynaptic transmission in the striatum are potential mechanismscontributing to a complex behavior such as anxiety. Of note the stria-tum is emerging as a key structure in mood regulation in humans(Mathew and Ho, 2006; Reiman et al., 1989; Yoo et al., 2005) and inrodents (Favilla et al., 2008; Kim et al., 2008; Zhang et al., 2008). Inaddition, this subcortical area is highly involved in MS (Bakshi et al.,2002; Bermel et al., 2003; Tao et al., 2009), and in EAE (Centonze etal., 2009; Rossi et al., 2010).
The role of inflammatory cytokines in the pathophysiology ofmood disturbances in humans is increasingly recognized (Dantzer etal., 2008; Haroon et al., 2012; Miller et al., 2009). High levels ofproinflammatory cytokines have been found in peripheral blood andin cerebrospinal fluid of depressed patients (Raison et al., 2006;Zorrilla et al., 2001). On the other hand, drugs currently approved bythe US Food and drug administration (FDA) for treatment of auto-immune or inflammatory disease targeting both TNF-α and IL-1βwere effective in improving symptoms of depression and fatigue, aswell as quality of life (Haroon et al., 2012; Soczynska et al., 2009). Fur-thermore, previous reports have demonstrated that inflammatorycytokines can interact with multiple pathways known to be involvedin the development of depression, including monoamine metabolism,
Fig. 4. Effects of in vivo TNF-α modulation on striatal glutamatergic transmission. (A) Icv-et(9 dpi). Etanercept reduced both sEPSC decay time and half width, but not sEPSC rise time. (Bhalf width of sEPSCs recorded from striatal neurons of icv TNF-α mice are significantly incr
neuroendocrine function, synaptic plasticity, and neurocircuits rele-vant to mood regulation (Haroon et al., 2012). Interestingly, sicknessbehavior and mood disturbances typically observed in MS patients(Kahl et al., 2002; Patanella et al., 2010) were associated with in-creased levels of cytokines (Maimone et al., 1991). In EAE mice,Peruga et al. (2011) reported anxious behavior associated with overtmotor deficits while a very recent paper (Rodrigues et al., 2011) didnot detect signs of anxiety in EAE-mice either before or after theclinical symptoms. Our data are the first to show anxiety in EAE mice1 week after the induction of brain inflammatory response and beforeirreversible motor disability, ruling out therefore the potential role ofsecondary psychophysical stress effects. Accordingly, earlier findingshave also reported a sickness behavior characterized by reduced appe-tite, depression, decreased social behavior and locomotor activity as-sociated with increased cytokine levels in EAE brains (Pollak et al.,2002, 2003).
The role of TNF-α in emotionality control has been extensivelyinvestigated both in humans and rodents. Transgenic mice over-expressing TNF-α in the brain exhibit increased indexes of anxiety,such as grooming during the black/white test, decreased rearing andretarded passive avoidance acquisition (Fiore et al., 1996, 1998). Fur-thermore, TNF-α knockout mice had reduced anxiety behavior at theEPM (Yamada et al., 2000), and intraperitoneal administration ofTNF-α induces sickness behavior, a complex condition also includingmood disturbances (Bluthe et al., 1991). By using mutant mice lackingTNFR1 or TNFR2 TNF-α receptors, it has also been demonstrated thatTNF-α-induced sickness behavior is TNFR1-dependent (Palin et al.,2009), a result recently confirmed in another independent study(Taraborrelli et al., 2011). In addition, blockade of TNF-α signalingwith etanercept has been associated with relevant mood improve-ments in both humans and rodents (Bercik et al., 2010; Krishnanet al., 2007; Tyring et al., 2006). Here, we demonstrated the role ofTNF-α in anxious-like behavior in EAE model by performing two setsof complementary experiments in which etanercept or TNF-αwas ad-ministrated by icv infusion. By this route of administration, many sitesin the CNS could be reached; therefore we cannot exclude the effect ofthe treatments on different brain regions. However, the striatum is akey brain area involved in anxiety regulation. Favoring cAMP signalingin the striatum through genetic deletion of the cAMP-degrading en-zyme phosphodiesterase 4B (Zhang et al., 2008), overexpression ofthe striatally enriched cAMP-generating enzyme adenylyl cyclase 5(Kim et al., 2008) or chronic expression in the striatum only of a con-stitutively active G-protein stimulating adenylyl cyclase activity(Favilla et al., 2008) is associated with increased anxiety in mice.
Here, we show that the behavioral alterations that we observedfollowing the in vivo treatments indeed correlate with glutamate syn-aptic alterations in the striatum. Etanercept was able to revert synap-tic alterations in EAE-mice while TNF-α mimicked them in normal
anercept rescues the kinetic alterations of the sEPSCs recorded from EAE-striatal MSNs) Icv TNF-αmimics the effect of EAE on glutamatergic transmission; the decay time andeased relative to the icv vehicle mice. Data are expressed as means±S.E.M, *pb0.05.

302 N. Haji et al. / Experimental Neurology 237 (2012) 296–303
mice. Interestingly, we previously demonstrated that TNF-α-inducedpotentiation of striatal glutamate transmission is under the control ofcAMP signaling (Centonze et al., 2009). In particular, we showed thatthe changes in the shape of sEPSCs were associated with an upreg-ulation of both total GluA1 and GluA1-p-Ser845 expression in thestriatum of presymptomatic and symptomatic EAE mice. Interesting-ly, no difference was detectable in the hippocampus at both timepoints, suggesting a specific involvement of the striatum. Phosphory-lation at S845, is mediated by protein kinase A (PKA), whose activi-ty is known to be dependent on cellular levels of cyclic AMP (Luand Roche, 2011). Accordingly, TNF-α has been found to activatecAMP-dependent signaling in neurons (Balkowiec-Iskra et al., 2011;Pickering et al., 2005). In addition, it has been shown that AMPA re-ceptor exocytosis can be mediated by activation of TNFR1 through aphosphatidylinositol 3-kinase (PI3)-dependent pathway (Stellwagenet al., 2005).
Of note, recent studies demonstrated that the regulation of AMPAreceptor trafficking and synaptic plasticity by GluA1 phosphorylationhas important implications for animal cognition. In particular phos-phorylation at the S831 and S845 sites is implicated in both the reten-tion of newly acquired spatial memory and norepinephrine-mediatedenhancement of contextual learning (Hu et al., 2007; Lee et al., 2003).In addition, a recent study revealed an imperative role for GluA1 S845phosphorylation in fear memory erasure (Clem and Huganir, 2010),demonstrating how altered AMPA receptor trafficking induced byGluA1 phosphorylation modulates animal behavior.
It could be also hypothesized that TNF-α, which has a broad range ofactions on neurons, induces an anxious-like behavior as a result of aneurotoxic effect. Previous studies showed that TNF-α activates mi-croglia in an autocrine/paracrinemanner and exerts indirect excitotoxicneuronal damage through induction of glutamate release from activat-ed microglia (Shijie et al., 2009; Takeuchi et al., 2006). Thus, one possi-ble mechanism of anxiolytic effect by TNF-α blocking is that it may halta vicious spiral of microglial activation in EAE mice. However, such hy-pothesis is unlikely since the EAE-glutamate alterations here reportedreflect mainly a postsynaptic mechanism involving AMPA receptors.Moreover, we investigated the presymptomatic phase of the disease,which eventually triggers the neurodegenerative processes occurringlater on. We indeed observed a synaptopathy and a selective loss ofparvalbumin positive neurons both in striatum and cerebellum dur-ing the symptomatic phase of the disease (Centonze et al., 2009;Mandolesi et al., 2012; Rossi et al., 2010).
Finally, we observed a positive effect of icv etanercept treatmentalso onmotor deficits during the symptomatic phase of the disease, al-though the difference was not statistically significant (data notshown). Such partial beneficial effect could be due either to theroute of drug administration or to a dual role of anti TNF-α therapy ob-served by other groups (Kassiotis and Kollias, 2001; Taoufik et al.,2011). Accordingly, increasing body of evidence reported a dual roleof TNF-α in EAE model and in MS patients. Besides the proin-flammatory role and the detrimental effects that may exert TNF-αon myelin reactivity during disease progression, TNF-α seems to berequired for the spontaneous regression of myelin reactivity, andendowed of immunosuppressive properties at later stages of EAE(Kassiotis and Kollias, 2001). Anti-TNF-α therapy has also been asso-ciated with adverse events such as demyelination and exacerbationofMS course, whichwere reduced once the treatmentwas interrupted(Fromont et al., 2009).
Conclusions
In conclusion, despite the limitations of behavioral studies in ani-mal models and the consequent difficulty to extrapolate anxiousbehavior to MS patients, the present work provides evidencesupporting the involvement of TNF-α effects in anxiety-like behaviorassociated with neuroinflammatory diseases. Importantly, anxiety
and depression do not correlate with the degree of neurological dis-ability in MS patients (Rietberg et al., 2011; Suh et al., 2010),confirming the relevance of our findings for further understandingthe pathophysiology of mood alterations in this disorder.
In addition, a considerable literature connects cytokines, such asTNF-α, with cognitive impairments and mood disturbance not onlyin MS but also in neurodegenerative diseases such as Alzheimer andParkinson disease (Baune et al., 2010; Clark et al., 2010; Menza etal., 2010). Therefore, modulation of proinflammatory cytokine levelsand activity might open new avenues for better targeted therapiesagainst psychiatric disorders associated with inflammatory and neu-rodegenerative diseases of the CNS.
Potential conflict of interests
Silvia Rossi received honoraria for writing from Bayer Scheringand funding for traveling from Novartis, Teva, and Merck Serono.She is involved as sub-investigator in clinical trials for Novartis,Merck Serono, Teva, Bayer Schering, Sanofi-aventis, and Biogen Idec.
Valentina De Chiara received funding for traveling by Teva. She isinvolved as study coordinator in clinical trials for Novartis, MerckSerono, Teva, Bayer Schering, Sanofi-aventis, and Biogen Idec.
Giorgio Bernardi is the principal investigator in clinical trials forMerck Serono and Teva.
Diego Centonze is an Advisory Board member of Merck-Serono,Teva, Bayer Schering, and received funding for traveling and honorariafor speaking or consultation fees from Merck Serono, Teva, Novartis,Bayer Schering, Sanofi-aventis, and Biogen Idec. He is also an externalexpert consultant of the European Medicine Agency (EMA), and theprincipal investigator in clinical trials for Novartis, Merck Serono,Teva, Bayer Schering, Sanofi-aventis, and Biogen Idec.
The other authors have no conflict of interests to declare.
Acknowledgments
The authors thank Vladimiro Batocchi and Massimo Tolu for excel-lent technical assistance. Authors would also like to acknowledgeFlorent Varenne for assistance with behavioral videotracking. Thisinvestigation was supported by the Italian National Ministero dellaSalute and by Fondazione TERCAS to D.C.; by Fondazione ItalianaSclerosi Multipla (FISM) to S.R. and by a grant from the EuropeanCommunity (AXREGEN: Axonal regeneration, plasticity and stemcells—Grant agreement 21 4003) to P.S.
References
Aktas, O., Zipp, F., 2003. Regulation of self-reactive T cells by human immunoglobulins—implications for multiple sclerosis therapy. Curr. Pharm. Des. 9, 245–256.
Bakshi, V.P., Smith-Roe, S., Newman, S.M., Grigoriadis, D.E., Kalin, N.H., 2002. Reductionof stress-induced behavior by antagonism of corticotropin-releasing hormone 2(CRH2) receptors in lateral septum or CRH1 receptors in amygdala. J. Neurosci.22, 2926–2935.
Balkowiec-Iskra, E., Vermehren-Schmaedick, A., Balkowiec, A., 2011. Tumor necrosisfactor-alpha increases brain-derived neurotrophic factor expression in trigeminalganglion neurons in an activity-dependent manner. Neuroscience 180, 322–333.
Baune, B.T., Dannlowski, U., Domschke, K., Janssen, D.G., Jordan, M.A., Ohrmann, P.,Bauer, J., Biros, E., Arolt, V., Kugel, H., Baxter, A.G., Suslow, T., 2010. The interleukin1 beta (IL1B) gene is associated with failure to achieve remission and impairedemotion processing in major depression. Biol. Psychiatry 67, 543–549.
Bercik, P., Verdu, E.F., Foster, J.A., Macri, J., Potter, M., Huang, X., Malinowski, P., Jackson,W., Blennerhassett, P., Neufeld, K.A., Lu, J., Khan, W.I., Corthesy-Theulaz, I., Cherbut,C., Bergonzelli, G.E., Collins, S.M., 2010. Chronic gastrointestinal inflammation in-duces anxiety-like behavior and alters central nervous system biochemistry inmice. Gastroenterology 139, 2102–2112.
Bermel, R.A., Innus, M.D., Tjoa, C.W., Bakshi, R., 2003. Selective caudate atrophy in mul-tiple sclerosis: a 3D MRI parcellation study. Neuroreport 14, 335–339.
Bluthe, R.M., Dantzer, R., Kelley, K.W., 1991. Interleukin-1 mediates behavioural but notmetabolic effects of tumor necrosis factor alpha in mice. Eur. J. Pharmacol. 209,281–283.
Capuron, L., Hauser, P., Hinze-Selch, D., Miller, A.H., Neveu, P.J., 2002. Treatment ofcytokine-induced depression. Brain Behav. Immun. 16, 575–580.

303N. Haji et al. / Experimental Neurology 237 (2012) 296–303
Centonze, D., Rossi, S., Prosperetti, C., Gasperi, V., De Chiara, V., Bari, M., Tscherter, A.,Febbraro, F., Bernardi, G., Maccarrone, M., 2007. Endocannabinoids limit metab-otropic glutamate 5 receptor-mediated synaptic inhibition of striatal principalneurons. Mol. Cell. Neurosci. 35, 302–310.
Centonze, D., Muzio, L., Rossi, S., Cavasinni, F., De Chiara, V., Bergami, A., Musella, A.,D'Amelio, M., Cavallucci, V., Martorana, A., Bergamaschi, A., Cencioni, M.T.,Diamantini, A., Butti, E., Comi, G., Bernardi, G., Cecconi, F., Battistini, L., Furlan, R.,Martino, G., 2009. Inflammation triggers synaptic alteration and degeneration inexperimental autoimmune encephalomyelitis. J. Neurosci. 29, 3442–3452.
Clark, I.A., Alleva, L.M., Vissel, B., 2010. The roles of TNF in brain dysfunction anddisease. Pharmacol. Ther. 128, 519–548.
Clem, R.L., Huganir, R.L., 2010. Calcium-permeable AMPA receptor dynamics mediatefear memory erasure. Science 330, 1108–1112.
Compston, A., Coles, A., 2008. Multiple sclerosis. Lancet 372, 1502–1517.Dantzer, R., O'Connor, J.C., Freund, G.G., Johnson, R.W., Kelley, K.W., 2008. From inflam-
mation to sickness and depression: when the immune system subjugates thebrain. Nat. Rev. Neurosci. 9, 46–56.
Favilla, C., Abel, T., Kelly, M.P., 2008. Chronic Galphas signaling in the striatum increasesanxiety-related behaviors independent of developmental effects. J. Neurosci. 28,13952–13956.
Feinstein, A., 2007. Neuropsychiatric syndromes associated with multiple sclerosis.J. Neurol. 254, II73–II76.
Fiore, M., Probert, L., Kollias, G., Akassoglou, K., Alleva, E., Aloe, L., 1996. Neuro-behavioral alterations in developing transgenic mice expressing TNF-alpha in thebrain. Brain Behav. Immun. 10, 126–138.
Fiore, M., Alleva, E., Probert, L., Kollias, G., Angelucci, F., Aloe, L., 1998. Exploratory anddisplacement behavior in transgenic mice expressing high levels of brain TNF-alpha. Physiol. Behav. 63, 571–576.
Fromont, A., De Seze, J., Fleury, M.C., Maillefert, J.F., Moreau, T., 2009. Inflammatory demye-linating events following treatmentwith anti-tumornecrosis factor. Cytokine 45, 55–57.
Haroon, E., Raison, C.L., Miller, A.H., 2012. Psychoneuroimmunology meets neuro-psychopharmacology: translational implications of the impact of inflammationon behavior. Neuropsychopharmacology 37, 137–162.
Herz, J., Zipp, F., Siffrin, V., 2010. Neurodegeneration in autoimmune CNS inflammation.Exp. Neurol. 225, 9–17.
Hu, H., Real, E., Takamiya, K., Kang, M.G., Ledoux, J., Huganir, R.L., Malinow, R., 2007.Emotion enhances learning via norepinephrine regulation of AMPA-receptor traf-ficking. Cell 131, 160–173.
Imitola, J., Chitnis, T., Khoury, S.J., 2005. Cytokines in multiple sclerosis: from bench tobedside. Pharmacol. Ther. 106, 163–177.
Kahl, K.G., Kruse, N., Faller, H., Weiss, H., Rieckmann, P., 2002. Expression of tumor ne-crosis factor-alpha and interferon-gamma mRNA in blood cells correlates with de-pression scores during an acute attack in patients with multiple sclerosis.Psychoneuroendocrinology 27, 671–681.
Kassiotis, G., Kollias, G., 2001. Uncoupling the proinflammatory from the immunosup-pressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level:implications for pathogenesis and therapy of autoimmune demyelination. J. Exp.Med. 193, 427–434.
Kim, K.S., Lee, K.W., Baek, I.S., Lim, C.M., Krishnan, V., Lee, J.K., Nestler, E.J., Han, P.L.,2008. Adenylyl cyclase-5 activity in the nucleus accumbens regulates anxiety-related behavior. Neurochem. 107, 105–115.
Krishnan, R., Cella, D., Leonardi, C., Papp, K., Gottlieb, A.B., Dunn, M., Chiou, C.F., Patel,V., Jahreis, A., 2007. Effects of etanercept therapy on fatigue and symptoms of de-pression in subjects treated for moderate to severe plaque psoriasis for up to 96weeks. Br. J. Dermatol. 157, 1275–1277.
Lee, H.K., Takamiya, K., Han, J.S., Man, H., Kim, C.H., Rumbaugh, G., Yu, S., Ding, L., He, C.,Petralia, R.S., Wenthold, R.J., Gallagher, M., Huganir, R.L., 2003. Phosphorylation ofthe AMPA receptor GluR1 subunit is required for synaptic plasticity and retentionof spatial memory. Cell 112, 631–643.
Lu, W., Roche, K.W., 2011. Posttranslational regulation of AMPA receptor traffickingand function. Curr. Opin. Neurobiol. http://dx.doi.org/10.1016/j.conb.2011.09.008(2011 Oct 14).
Lynch, M.A., 2009. The multifaceted profile of activated microglia. Mol. Neurobiol. 40(2), 139–156.
Maes, M., Meltzer, H.Y., Bosmans, E., Bergmans, R., Vandoolaeghe, E., Ranjan, R.,Desnyder, R., 1995. Increased plasma concentrations of interleukin-6, solubleinterleukin-6, soluble interleukin-2 and transferrin receptor in major depression.J. Affect. Disord. 34, 301–309.
Maimone, D., Gregory, S., Arnason, B.G., Reder, A.T., 1991. Cytokine levels in the cerebrospi-nal fluid and serum of patients with multiple sclerosis. J. Neuroimmunol. 32, 67–74.
Mandolesi, G., Grasselli, G., Musella, A., Gentile, A., Musumeci, G., Sepman, H., Haji, N.,Fresegna, D., Bernardi, G., Centonze, D., 2012. GABAergic signaling and connectivityon Purkinje cells are impaired in experimental autoimmune encephalomyelitis.Neurobiol. Dis. 46, 414–424.
Mathew, S.J., Ho, S., 2006. Etiology and neurobiology of social anxiety disorder. J. Clin.Psychiatry 67, 9–13.
Menza, M., Dobkin, R.D., Marin, H., Mark, M.H., Gara, M., Bienfait, K., Dicke, A.,Kusnekov, A., 2010. The role of inflammatory cytokines in cognition and othernon-motor symptoms of Parkinson's disease. Psychosomatics 51, 474–479.
Miller, A.H., Maletic, V., Raison, C.L., 2009. Inflammation and its discontents: the roleof cytokines in the pathophysiology ofmajor depression. Biol. Psychiatry 65, 732–741.
Murphy, A.C., Lalor, S.J., Lynch, M.A., Mills, K.H., 2010. Infiltration of Th1 and Th17 cellsand activation of microglia in the CNS during the course of experimental autoim-mune encephalomyelitis. Brain Behav. Immun. 24, 641–651.
Nestler, E.J., Carlezon Jr., W.A., 2006. The mesolimbic dopamine reward circuit indepression. Biol. Psychiatry 59, 1151–1159.
Palin, K., Bluthe, R.M., McCusker, R.H., Levade, T., Moos, F., Dantzer, R., Kelley, K.W.,2009. The type 1 TNF receptor and its associated adapter protein, FAN, arerequired for TNFalpha-induced sickness behavior. Psychopharmacology 201,549–556.
Patanella, A.K., Zinno, M., Quaranta, D., Nociti, V., Frisullo, G., Gainotti, G., Tonali, P.A.,Batocchi, A.P., Marra, C., 2010. Correlations between peripheral blood mononuclearcell production of BDNF, TNF-alpha, IL-6, IL-10 and cognitive performances in mul-tiple sclerosis patients. J. Neurosci. Res. 88, 1106–1112.
Peruga, I., Hartwig, S., Thone, J., Hovemann, B., Gold, R., Juckel, G., Linker, R.A., 2011. In-flammation modulates anxiety in an animal model of multiple sclerosis. Behav.Brain Res. 220, 20–29.
Pickering, M., Cumiskey, D., O'Connor, J.J., 2005. Actions of TNF-alpha on glutamatergicsynaptic transmission in the central nervous system. Exp. Physiol. 90, 663–670.
Pollak, Y., Orion, E., Goshen, I., Ovadia, H., Yirmiya, R., 2002. Experimental autoimmuneencephalomyelitis-associated behavioral syndrome as a model of 'depression dueto multiple sclerosis'. Brain Behav. Immun. 16, 533–543.
Pollak, Y., Ovadia, H., Orion, E., Weidenfeld, J., Yirmiya, R., 2003. The EAE-associatedbehavioral syndrome: I. Temporal correlation with inflammatory mediators.J. Neuroimmunol. 137 (1–2), 94–99.
Raison, C.L., Capuron, L., Miller, A.H., 2006. Cytokines sing the blues: inflammation andthe pathogenesis of depression. Trends Immunol. 27, 24–31.
Reiman, E.M., Fusselman, M.J., Fox, P.T., Raichle, M.E., 1989. Neuroanatomical correlatesof anticipatory anxiety. Science 243, 1071–1074.
Rietberg, M.B., van Wegen, E.E., Uitdehaag, B.M., Kwakkel, G., 2011. The associationbetween perceived fatigue and actual level of physical activity in multiplesclerosis. Published online before print May 17, 2011. http://dx.doi.org/10.1177/1352458511407102.
Rodrigues, D.H., Vilela Mde, C., Lacerda-Queiroz, N., Miranda, A.S., Sousa, L.F., Reis, H.J.,Teixeira, A.L., 2011. Behavioral investigation of mice with experimental autoim-mune encephalomyelitis. Arq. Neuropsiquiatr. 69 (6), 938–942.
Rossi, S., Muzio, L., De Chiara, V., Grasselli, G., Musella, A., Musumeci, G., Mandolesi, G.,De Ceglia, R., Maida, S., Biffi, E., Pedrocchi, A., Menegon, A., Bernardi, G., Furlan, R.,Martino, G., Centonze, D., 2010. Impaired striatal GABA transmission in experimen-tal autoimmune encephalomyelitis. Brain Behav. Immun. 25, 947–956.
Shijie, J., Takeuchi, H., Yawata, I., Harada, Y., Sonobe, Y., Doi, Y., Liang, J., Hua, L.,Yasuoka, S., Zhou, Y., Noda, M., Kawanokuchi, J., Mizuno, T., Suzumura, A., 2009.Blockade of glutamate release from microglia attenuates experimental autoim-mune encephalomyelitis in mice. Tohoku J. Exp. Med. 217, 87–92.
Soczynska, J.K., Kennedy, S.H., Goldstein, B.I., Lachowski, A., Woldeyohannes, H.O.,McIntyre, R.S., 2009. The effect of tumor necrosis factor antagonists on mood andmental health-associated quality of life: novel hypothesis-driven treatments for bi-polar depression? Neurotoxicology 30, 497–521.
Steiner, H., Van Waes, V., Marinelli, M., 2010. Fluoxetine potentiates methylphenidate-induced gene regulation in addiction-related brain regions: concerns for use ofcognitive enhancers? Biol. Psychiatry 67, 592–594.
Stellwagen, D., Beattie, E.C., Seo, J.Y., Malenka, R.C., 2005. Differential regulation ofAMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha.J. Neurosci. 25 (12), 3219–3228.
Suh, Y., Motl, R.W., Mohr, D.C., 2010. Physical activity, disability, and mood in the earlystage of multiple sclerosis. Disabil. Health J. 3, 93–98.
Takeuchi, H., Jin, S., Wang, J., Zhang, G., Kawanokuchi, J., Kuno, R., Sonobe, Y., Mizuno,T., Suzumura, A., 2006. Tumor necrosis factor-alpha induces neurotoxicity via glu-tamate release from hemichannels of activated microglia in an autocrine manner.J. Biol. Chem. 281 (30), 21362–21368.
Tao, G., Datta, S., He, R., Nelson, F., Wolinsky, J.S., Narayana, P.A., 2009. Deep gray mat-ter atrophy in multiple sclerosis: a tensor based morphometry. J. Neurol. Sci. 282(1–2), 39–46.
Taraborrelli, C., Palchykova, S., Tobler, I., Gast, H., Birchler, T., Fontana, A., 2011. TNFR1is essential for CD40, but not for lipopolysaccharide-induced sickness behavior andclock gene dysregulation. Brain Behav. Immun. 25 (3), 434–442.
Taoufik, E., Tseveleki, V., Chu, S.Y., Tselios, T., Karin, M., Lassmann, H., Szymkowski, D.E.,Taraborrelli, C., Palchykova, S., Tobler, I., Gast, H., Birchler, T., Fontana, A., 2011.TNFR1 is essential for CD40, but not for lipopolysaccharide-induced sickness be-havior and clock gene dysregulation. Brain Behav. Immun. 25, 434–442.
Tyring, S., Gottlieb, A., Papp, K., Gordon, K., Leonardi, C., Wang, A., Lalla, D., Woolley, M.,Jahreis, A., Zitnik, R., Cella, D., Krishnan, R., 2006. Etanercept and clinical outcomes,fatigue, and depression in psoriasis: double-blind placebo-controlled randomisedphase III trial. Lancet 367, 29–35.
Yamada, K., Iida, R., Miyamoto, Y., Saito, K., Sekikawa, K., Seishima, M., Nabeshima, T.,2000. Neurobehavioral alterations in mice with a targeted deletion of the tumornecrosis factor-alpha gene: implications for emotional behavior. J. Neuroimmunol.111, 131–138.
Yoo, H.K., Kim, M.J., Kim, S.J., Sung, Y.H., Sim, M.E., Lee, Y.S., Song, S.Y., Kee, B.S., Lyoo,I.K., 2005. Putaminal gray matter volume decrease in panic disorder: an optimizedvoxel-based morphometry study. Eur. J. Neurosci. 22, 2089–2094.
Zhang, H.T., Huang, Y., Masood, A., Stolinski, L.R., Li, Y., Zhang, L., Dlaboga, D., Jin, S.L.,Conti, M., O'Donnell, J.M., 2008. Anxiogenic-like behavioral phenotype of micedeficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology 33,1611–1623.
Zorrilla, E.P., Luborsky, L., McKay, J.R., Rosenthal, R., Houldin, A., Tax, A., McCorkle, R.,Seligman, D.A., Schmidt, K., 2001. The relationship of depression and stressors toimmunological assays: a meta-analytic review. Brain Behav. Immun. 15, 199–226.




















