
Thesis: Selective Association Between the Free-Living Nematode Acrobeloides maximus and Soil...
108
eScholarship provides open access, scholarly publishing services to the University of California and delivers a dynamic research platform to scholars worldwide. Electronic Theses and Dissertations UC Riverside Peer Reviewed Title: Selective Association Between the Free-Living Nematode Acrobeloides maximus and Soil Bacteria Author: Sedky, Sammy Farid Acceptance Date: 2013 Series: UC Riverside Electronic Theses and Dissertations Degree: M.S., Genetics, Genomics and Bioinformatics UC Riverside Advisor(s): De Ley, Paul Committee: Orwin, Paul , Crowley, David Permalink: https://escholarship.org/uc/item/14r816m2 Abstract: Copyright Information: All rights reserved unless otherwise indicated. Contact the author or original publisher for any necessary permissions. eScholarship is not the copyright owner for deposited works. Learn more at http://www.escholarship.org/help_copyright.html#reuse
Transcript of Thesis: Selective Association Between the Free-Living Nematode Acrobeloides maximus and Soil...

eScholarship provides open access, scholarly publishingservices to the University of California and delivers a dynamicresearch platform to scholars worldwide.
Electronic Theses and DissertationsUC Riverside
Peer Reviewed
Title:Selective Association Between the Free-Living Nematode Acrobeloides maximus and SoilBacteria
Author:Sedky, Sammy Farid
Acceptance Date:2013
Series:UC Riverside Electronic Theses and Dissertations
Degree:M.S., Genetics, Genomics and BioinformaticsUC Riverside
Advisor(s):De Ley, Paul
Committee:Orwin, Paul, Crowley, David
Permalink:https://escholarship.org/uc/item/14r816m2
Abstract:
Copyright Information:All rights reserved unless otherwise indicated. Contact the author or original publisher for anynecessary permissions. eScholarship is not the copyright owner for deposited works. Learn moreat http://www.escholarship.org/help_copyright.html#reuse

UNIVERSITY OF CALIFORNIA RIVERSIDE
Selective Association Between the Free-Living Nematode Acrobeloides maximus
and Soil Bacteria
A Thesis submitted in partial satisfaction of the requirements for the degree of
Master of Science
in
Genetics, Genomics and Bioinformatics
by
Sammy Farid Sedky
June 2013
Thesis Committee:
Dr. Paul De Ley, Chairperson Dr. Paul Orwin Dr. David Crowley

Copyright by Sammy Farid Sedky
2013

The Thesis of Sammy Farid Sedky is approved:
Committee Chairperson
University of California, Riverside

iv
ACKNOWLEDGMENTS
This master thesis would not have been possible without the help of
several individuals who contributed their time, assistance, and expertise to the
completion of this work:
• To my advisor, Dr. Paul De Ley: Thank you for giving me a lab I could call
home, where I was welcome and where I could grow as a researcher and
scholar. Your assistance in navigating the labyrinth of challenges involved in the
writing and completion of this document has been invaluable. Thank you for your
guidance and support.
• To Dr. Paul Orwin: I will always be grateful for the kind tutelage you provided
during your sabbatical stay at UCR. This study could not have been completed
without your vigilance.
• To Dr. David Crowley: Thank you for allowing us to use Science Lab I 308
to perform most of the wet lab work and for your assistance as a committee
member in the review and editing of this thesis.
• To my colleagues, Dr. JP Baquiran and Brian Thater: It was a blast working
with you both. Thanks for the laughs and good times.

v
UCR DEDICATIONS
• To my friend and protégé, Rosalynn Duong: I could not have chosen a
better undergraduate assistant. Thank you for all your help, the late hours you
put in taking care of the nematodes and bacterial cultures, your assistance with
molecular work, and for most of all sticking with me on this journey.
• To Dr. Irma Tandingan De Ley: Thank you for always being generous with
your time and for your help and technical expertise in the lab.
• To Dr. Cecilia Ritzinger: Thanks for showing me that sometimes we just
need to step back and take a coffee break! Obrigado!
• To the wonderful GGB graduate students: Amanda Hollowell, Yifan Lii,
Stephanie Coffman, Keira Lucas, Hagop Atamian, Patrick Schacht,
Meenakshi Kagda, Tusar Saha, Divya Sain, Ritu Chaudhary, Venkatesh
Sivanandam: I wish you all nothing but the best in your future endeavors! It’s
been a pleasure being your social committee chair these last two years.
• To Dr. Michael Fugate, Dr. John Oross, Dr. Laura Abbot, Dr. Brian
Brady, Dr. Gregory Filatov, Devon Duron Ehnes-Zepeda: Being a TA was one
of the highlights of my graduate career. It was a pleasure to work alongside you
while developing my pedagogical skills.
• To Tiago Pereira and Ran Sun: I always enjoyed your visits to the lab.
Thanks for taking time out of your busy schedules to chat with me. All the best!

vi
DEDICATIONS TO FAMILY AND FRIENDS
• To my loving parents, Soha and Mohamed: Thank you for your endless
encouragement, love, and support. This one’s for you!
• To my aunt and uncle, Siham and Fathy: Thanks for always believing in me
and for your love, encouragement, and support.
• To my sister, Sally: You are never far from my thoughts. Your strength and
courage has always been a source of inspiration.
• To the friends that cheered me on, encouraged me, and believed in me:
Jorge Flores, Ryan Lee, Johnny Tang, Arif Mosavi, Samuel Koo, Deep
Shah, Anand Panchal, Mrunal Patel, Jason Pico, Edgar Alvarado, David
Benitez, Max Flores, Jasmine Flores, Dynasty Carfangnia, Jose Hernandez,
Stephen Clawson: Thank you !!!

vii
TABLE OF CONTENTS
GENERAL INTRODUCTION IMPORTANCE OF NEMATODE- BACTERIA SYMBIOSIS RESEARCH
1
RECENT STUDIES OF NEMATODE-BACTERIAL ASSOCIATIONS IN SOIL MICROBIAL COMMUNITIES
4
Radopholus similis and Wolbachia 4
Entomopathogenic nematodes and γ-Proteobacteria 5
Steinernema carpocapsae and Xenorhabdus nematophila
6
Heterorhabditis bacteriophora and Photorhabdus luminescens
6
Bacillus thuringiensis Cry5B crystal proteins and free living nematodes
7
Caenorhabditis elegans 7
Pristionchus pacificus 8
SYNOPSIS OF THIS THESIS 8 REFERENCES 10 CHAPTER ONE: Survey of the microbiota associated with A. maximus in soil microcosm
ABSTRACT 15 INTRODUCTION 16 MATERIALS AND METHODS 18 Nematode cultivation and harvest 18
Rhizosphere soil collection and exposure 19
Long-term culture of A. maximus 20
Isolation of associated bacteria 20
DNA extraction and PCR analysis 21
16S rRNA gene library construction, sequencing and analysis 22
Preparation for Fluorescence In Situ Hybridization (FISH) 23
FISH Probes 24
FISH- Hybridization, Mounting, Microscopy 24
RESULTS 25 16S rDNA analysis of extracts 25
Co-cultures 26
Isolation and culture 26
Fluorescence In Situ Hybridization 27
DISCUSSION 27 REFERENCES 31 FIGURES AND TABLES 34

viii
CHAPTER TWO: Phylogenetic Analysis of Bacterial Genera Detected At Significant Levels Within The Microbiota Associated With A. maximus
ABSTRACT 41 INTRODUCTION 42 MATERIALS AND METHODS 43 Data Preparation 43
Multiple sequence alignments: T-Coffee, MAFFT, Kalign, and WebPRANK
44
Phylogenetic inference tools: Maximum Likelihood, PhyML, Mr. Bayes
45
LALIGN 45
RESULTS 46 Ochrobactrum sp. clones 46
Chitinophaga sp. clones 46
Pedobacter sp. clones 47
LALIGN plots 47
DISCUSSION 48 Comparison of multiple sequence alignment algorithms 48
Comparison of phylogenetic reconstruction methods 49
Ochrobactrum sp. clones 50
Chitinophaga sp. clones 51
Pedobacter sp. clones 53
LALIGN analysis 54
Requirements for describing a new bacterial species 55
Genotypic approaches 55
Phenotypic approaches 56
Chemotaxonomic approaches 56
FINAL THOUGHTS 57 REFERENCES 58 FIGURES AND TABLES 62 CONCLUDING COMMENTS BACTERIAL COMMUNITY IDENTIFICATION: 16S rRNA GENE MARKER SURVEY VERSUS SHOTGUN METAGENOMICS
88
16S rRNA Gene Marker Surveys 88
Shotgun Metagenomics 90
PCR-amplified 16S rRNA and Pyrosequencing 91
Sequencing Technologies and Future Considerations 92
REFERENCES 95

ix
LIST OF FIGURES
CHAPTER ONE
Figure 1. Soil exposed A. maximus populations were found to be associated with a consistent bacterial population
34
Figure 2A. Ochrobactrum is shown to be the predominate alpha-proteobacteria within A. maximus
35
Figure 2B. Pedobacter is shown to be the primary Spingobacteria within A. maximus, along with a consistent representation of Chitinophaga at a lower level
36
Figure 3A-C. Long term culture experiments appear to show stability at the phylum level (A) but reveal shifts in population as the worm population ages at the genus level (B,C)
37
Figure 4A-F. Fluorescence in situ hybridization was used to identify the presence of both Ochrobactrum sp. (green) and Pedobacter sp. (red) in the gut of formaldehyde fixed A. maximus after recovery from soil microcosm
38
CHAPTER TWO
Figure 1. Ochrobactrum: T-Coffee-PhyML Phylogram 67-68 Figure 2. Ochrobactrum: MAFFT-ML Phylogram 69-70 Figure 3. Ochrobactrum: Kalign-MrBayes Phylogram 71-72 Figure 4. Chitinophaga: T-Coffee-ML Phylogram 73-74 Figure 5. Chitinophaga: MAFFT-ML Phylogram 75-76 Figure 6. Chitinophaga: Kalign-ML Phylogram 77-78 Figure 7. Pedobacter : T-Coffee-PhyML Phylogram 79-80 Figure 8. Pedobacter : MAFFT-PhyML Phylogram 81-82 Figure 9. Pedobacter : Kalign-PhyML Phylogram 83-84 Figure 10A. LALIGN Plot: Ochrobactrum sp. clone Acro231
compared against the consensus sequence of Ochrobactrum clones
85
Figure 10B. LALIGN Plot: Chitinophaga sp. clone Acro210 compared against the consensus sequence of Chitinophaga clones
86
Figure 10C. LALIGN Plot: of Pedobacter sp. clone Acro279 compared against the consensus sequence of Pedobacter clones
87

x
LIST OF TABLES
CHAPTER ONE
Table 1. List Of Sequences Deposited Into GenBank 39
Table 2. Shannon-Wiener Population Statistics for Microcosm Samples
39
Table 3. Identified Isolates from association from A. maximus microcosm and long term culture
40
CHAPTER TWO
Table 1A. Ochrobactrum Sequences Used In Phylogenetic Analyses
62
Table 1B. Pedobacter Sequences Used In Phylogenetic Analyses 62
Table 1C. Chitinophaga Sequences Used In Phylogenetic Analyses 63
Table 2. Pedobacter Settings For Phylogenetic Trees 64
Table 3. Ochrobactrum Settings For Phylogenetic Trees 65
Table 4. Chitinophaga Settings For Phylogenetic Trees 66

1
GENERAL INTRODUCTION
“The soils of our yards, gardens, and fields swarm with thousands of kinds of minute animals and plants of which we know little or nothing. We depend on the soil for our very existence, and it may seem that this fact should have caused us long ago to make ourselves thoroughly acquainted with it and all its inhabitants; yet the truth is otherwise. Here beneath our very feet are microbes, protozoa, fungi, and many other kinds of small organisms, thousands of species, of which we know hardly the first thing beyond the mere fact of their existence. . . . Inhabiting the soil in myriads, hidden behind this veil of ignorance, there is a group of organisms known as nematodes.” (6) Nathan A. Cobb, 1914 IMPORTANCE OF NEMATODE-BACTERIA SYMBIOSIS RESEARCH
Nematodes are among the most abundant metazoans on Earth, inhabiting
terrestrial, marine, and freshwater ecosystems (7). They can be found in
mountains, grasslands, forests, rivers, lakes, oceans and even in inhospitable
environments such as arid deserts and icy tundras. Some parasitize animals and
plants, making their homes within these unsuspecting organisms (25, 28, 44).
Nematodes have evolved a myriad of fascinating survival strategies, allowing
them to adapt to virtually all environments to increase their chances of survival,
especially under adverse conditions. For example, nematodes temporarily enter
different quiescent states in response to environmental stresses, such as
anhydrobiosis in response to desiccation and cryobiosis in response to low
temperatures (27). Furthermore they also utilize several reproductive strategies
ranging from dioecous to protandric hermaphroditism to parthenogenesis (9).

2
Some species are easily culturable bacterial feeders and are therefore highly
suitable for eukaryotic-prokaryotic association studies.
Bacteria are the most abundant and ubiquitous of all organisms found in
nature. Together with nematodes they make up two of the most prominent groups
of organisms in the biosphere. Bacteria and nematodes have long interacted and
evolved alongside one another, resulting in the creation of many different types of
associations, along a continuum ranging from mutualistic and beneficial to
antagonistic and detrimental through for example competition, pathogenesis, and
parasitism (29). There are many free-living nematode species which obtain their
energy entirely or partially from the consumption of bacteria (54). These
nematodes can be non-selective in the bacteria they ingest, such as with
laboratory grown Caenorhabditis elegans, or they can be particular in the
bacteria they choose, allowing them to differentiate between bacteria with
different nutritional qualities and to select for specific genera or species of
bacteria to consume or cultivate (18, 19, 31, 48). This selectivity allows
nematodes to avoid dangerous pathogens while picking up bacteria that promote
development, fecundity, and survival (20, 35, 41). Investigating nematode-
bacteria symbioses can pave the way for comparative analyses with other
eukaryotic host-bacteria associations and will enhance our understanding in the
areas of developmental and evolutionary biology while also leading to
improvements in agriculture, ecology, and health. For example, it will allow for
new insights into the genes implicated in the evolution of symbiosis and those

3
involved in horizontal transfer between bacteria and eukaryotes (11, 12, 53).
Furthermore, it will aid in elucidating the metabolic pathways and chemical
interactions between symbiont and host and can thus lead to the creation of new
drug targets for invasive pathogens (32, 41).
Genetic and molecular tools are being applied to provide new insights into
nematode-bacterial interactions. Metagenomics, the direct sequencing of
environmental samples, has allowed for simultaneous sequencing of symbionts
and their hosts (24). Utilizing the power of the latest genome deep sequencing
platforms, these studies can now be performed on a genome-wide level as
opposed to more traditional gene-centric approaches focusing for example on
16S ribosomal RNA (16S rRNA) based molecular methods. Such high-
throughput molecular tools allow for comprehensive taxonomic and phylogenetic
classification of the diverse bacteria nematodes associate with in soil
communities (45, 46).
The remainder of this discussion will focus on some particularly interesting
recent studies of soil nematode-bacterial associations in which the participating
organisms engage in specific and persistent relationships. Understanding the
mechanisms involved in nematode-bacterial recognition may lead to functional
studies and practical applications through genetic manipulation of these
interactions (38).

4
RECENT STUDIES OF NEMATODE-BACTERIAL ASSOCIATIONS IN SOIL
MICROBIAL COMMUNITIES
Radopholus similis and Wolbachia
Wolbachia are a group of gram negative intracellular α-Proteobacteria in
the order Rickettsiales, the group from which mitochondria and similar organelles
are thought to have originated under the endosymbiotic theory. The latter
postulates symbioses between separate single-celled organisms leading to the
formation of organelles within eukaryotic organisms (50). In 2009, Haegeman et
al. confirmed the presence of a Wolbachia endosymbiont in Radopholus similis, a
burrowing plant-parasitic nematode which attacks the roots of banana plants
(21). This is the first and so far only reported occurrence of Wolbachia being
found in a non-zooparasitic soil-dwelling nematode.
The role Wolbachia plays in R. similis is not yet known. In filarial
nematodes however, Wolbachia is known to be an obligatory mutualist;
eradication of Wolbachia from the host via the antibiotic tetracycline has been
shown to cause adverse effects such as abnormal growth, eventually leading to
death of the nematodes (5, 14, 15, 22, 23). Conversely in arthropods, Wolbachia
takes on the role of a reproductive parasite, imparting a selective advantage to
infected females via feminization, parthenogenesis, male killing, or cytoplasmic
incompatibility, all leading to a decrease of the uninfected portion of the host
population while the endosymbionts are vertically transmitted to the offspring of

5
the infected portion (2, 47, 52). Due to the high infection rate of Wolbachia in R.
similis, it is believed the endosymbiont also provides the nematode with essential
metabolites and is necessary for survival (21), although no studies have as yet
been published documenting the effects on Radopholus of targeted Wolbachia
eradication.
Finding Wolbachia inside R. similis raises many unanswered questions. Do
other plant-parasitic nematodes carry Wolbachia endosymbionts? Can it be
found in nematode species or genera that are easy to culture and maintain in a
laboratory setting? What experiments can be performed to determine the role the
endosymbiont plays inside Radopholus similis?
Entomopathogenic nematodes and γ-Proteobacteria
Entomopathogenic nematodes are soil-dwelling insect parasites that work
in concert with insect-pathogenic bacteria to cause rapid insect death, usually
within 48 hours of infection (10). Model systems are being developed in which
entomopathogenic nematodes and their bacterial symbionts are used as
environmentally-friendly biological control agents against insect pests, providing
an alternative to the spraying of ecologically harmful chemical pesticides into the
environment (8, 4).

6
Steinernema carpocapsae and Xenorhabdus nematophila
Xenorhabdus are gram-negative γ-Proteobacteria in the order
Enterobacteriales (49). The bacterium Xenorhabdus nematophila is a mutualist of
the entomopathogenic nematode Steinernema carpocapsae and a pathogen of
many types of insects including weevils, caterpillars, moths, beetles, fleas, flies,
crickets, and ants (19).
S. carpocapsae ambush and infect highly mobile insects near the soil
surface (3). Once inside, the nematodes ingest the insect’s body liquid and
release X. nematophila into the hemocoel, where together they suppress the
insect’s immune system, leading to its eventual death (40). X. nematophila will
continue to grow and multiply in the insect cadaver, converting the cadaver into
substrates capable of supporting S. carpocapsae reproduction; the resulting new
nematode juveniles will ingest and store the endosymbiont in their digestive tract,
allowing the cycle to repeat (19).
Heterorhabditis bacteriophora and Photorhabdus luminescens
Like Xenorhabdus, Photorhabdus are also gram-negative γ-Proteobacteria
in the order Enterobacteriales (1, 16, 34). The bacterium Photorhabdus
luminescens is an endosymbiont of the entomopathogenic nematode
Heterorhabditis bacteriophora and a pathogen of many insect species (33).
H. bacteriophora typically infects insects that reside deep within the soil
(3). The cycle and mechanism of infection is similar to that utilized by S.

7
carpocapsae and X. nematophila; infected H. bacteriophora invade unsuspecting
insects and release P. luminescens within the insect, causing its death within a
few days (17).
Bacillus thuringiensis Cry5B crystal proteins and free living nematodes
Bacillus thuringiensis (Bt) is a gram positive soil-dwelling bacterium in the
order Bacillales known for producing crystal toxin proteins which are used as a
natural biological pesticide against insects in crop fields (39). Recent studies
have been undertaken to learn about the roles which these bacteria and their
crystal proteins play in the presence of nematodes and in soil microbial
communities (36, 37, 51). The Bt crystal protein, Cry5B has been shown to be
toxic towards Caenorhabditis elegans and Pristionchus pacificus (37). Cry5B
along with other nematicidal Bt crystal proteins have the potential to be utilized
as biological control agents against nematodes which parasitize plants and
animals (26, 51).
Caenorhabditis elegans
Caenorhabditis elegans, is a free-living rhabditid commonly used as a
model organism for genetics research because of its ease to cultivate and
maintain under laboratory conditions. Cry5B has been shown to have adverse
effects on C. elegans; when ingested the Bt toxin causes extensive damage to
the gut, a decrease in progeny production, and leads to death (26).

8
Pristionchus pacificus
Pristionchus pacificus, a free-living diplogastrid and a necromenic
associate of beetles is emerging alongside the popular model C. elegans to
become a commonly used tool in studying evolutionary biology, ecology, and
population genetics (42). Chemosensory studies have shown that P. pacificus
avoids Bt and other insect pathogenic bacteria (36, 37). P. pacificus is
susceptible to the Bt Cry5B crystal protein; when force fed E. coli expressing the
Bt toxin, the nematodes were unable to develop past the L1 larvae stage and
their intestinal tract exhibited signs of thinning, constriction, and degeneration
(51).
SYNOPSIS OF THIS THESIS
In the following chapters we will discuss our work investigating the soil
microbial community associated with the nematode Acrobeloides maximus, a
nonpathogenic bacterial-feeding cephalobid. Cephalobids occur in almost all
soils and are phylogenetically much closer to plant parasitic nematodes than the
model nematodes C. elegans and P. pacificus (7). Moreover, cephalobids have
developed ecological adaptations to desiccation and have slower generation
times at room temperature when compared with C. elegans and P. pacificus (30).
A sealed plate of A. maximus can last in culture well over six months whereas
faster growing species like C. elegans and P. pacificus require subculturing every
few weeks to avoid population collapse (13, 43). These differences make

9
cephalobids like A. maximus a much more suitable model for interaction studies
over C. elegans and P. pacificus. Chapter one discusses the results of our 16S
rRNA gene survey of soil microbiota associated with A. maximus while chapter
two takes a closer phylogenetic look at the bacterial genera detected at
significant levels within the nematode.

10
REFERENCES 1. Boemare NE, Akhurst RJ, Mourant RG. 1993. DNA relatedness between Xenorhabdus spp. (Enterobacteriaceae), symbiotic bacteria of Entomopathogenic Nematodes, and a proposal to transfer Xenorhabdus luminescens to a new genus, Photorhabdus gen. nov. International Journal of Bacteriology. 43:249-255. 2. Bourtzis K, O'Neill S. 1998. Wolbachia infections and arthropod reproduction. BioScience. 48(4): 287-293. 3. Campbell JF, Gaugler R. 1993. Nictation behavior and its ecological implications in the host search strategies of enomopathogenic nematodes. Behavior. 126:155-169. 4. Campos-Herrera R, Barbercheck M, Hoy CW, Stock SP. 2012. Entomopathogenic nematodes as a model system for advancing the frontiers of ecology. Journal of Nematology. 44(2):162-176. 5. Casiraghi M, McCall JW, Simoncini L, Kramer LH, Sacchi L, Genchi C, Werren JH, Bandi C. 2002. Tetracycline treatment and sex-ratio distortion: a role for Wolbachia in the moulting of filarial nematodes? International Journal for Parasitology. 32(12):1457-1468. 6. Cobb NA. 1914. Nematodes and their relationships. U.S. Department of Agriculture Yearbook. U.S. Government Printing Office, Washington D.C.. 457-490. 7. De Ley P. 2006. A quick tour of nematode diversity and the backbone of nematode phylogeny. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.41.1, http://www.wormbook.org. 8. Denno RF, Gruner DS, Kaplan I. 2008. Potential for Entomopathogenic Nematodes in Biological Control: A Meta-Analytical Synthesis and Insights from Trophic Cascade Theory. The Journal of Nematology. 40(2):61–72. 9. Denver DR, Clark KA, Raboin MJ. 2011. Reproductive mode evolution in nematodes: insights from molecular phylogenies and recently discovered species. Molecular Phylogenetics and Evolution. 61(2):584-592. 10. Dillman AR, Chaston JM, Adams BJ, Ciche TA, Goodrich-Blair H, Stock SP, Sternberg PW. 2012. An entomopathogenic nematode by any other name. PLOS Pathogens. 8(3):e1002527. 11. Dunning Hotopp JC, Clark ME, Oliveira DC, Foster JM, Fischer P, Muñoz Torres MC, Giebel JD, Kumar N, Ishmael N, Wang S, Ingram J, Nene RV, Shepard J, Tomkins J, Richards S, Spiro DJ, Ghedin E, Slatko BE, Tettelin H, Werren JH. 2007. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 317(5845):1753-1756. 12. Dunning Hotopp JC. 2011. Horizontal gene transfer between bacteria and animals. Trends in Genetics. 27(4):157–163.

11
13. Eyualem A, Blaxter M. 2003. Comparison of Biological, Molecular, and Morphological Methods of Species Identification in a Set of Cultured Panagrolaimus Isolates. Journal of Nematology. 35(1): 119–128. 14. Fenn K, Blaxter M. 2004. Are filarial nematode Wolbachia obligate mutualist symbionts? Trends in Ecology & Evolution. 19(4):163-166. 15. Fenn K, Blaxter M. 2006. Wolbachia genomes: revealing the biology of parasitism and mutualism. Trends in Parasitology. 22(2):60-65. 16. Fischer-Le Saux M, Viallard V, Brunel B, Normand P, Boemare NE. 1999. Polyphasic classification of the genus Photorhabdus and proposal of new taxa: P. luminescens subsp. luminescens subsp. nov., P. luminescens subsp. akhurstii subsp. nov., P. luminescens subsp. laumondii subsp. nov., P. temperata sp. nov., P. temperata subsp. temperata subsp. nov. and P. asymbiotica sp. nov. International Journal of Systematic Bacteriology. 49(4):1645-1656. 17. Forst S, Nealson K. 1996. Molecular biology of the symbiotic-pathogenic bacteria Xenorhabdus spp. and Photorhabdus spp. Microbiological Reviews. 60:21–43. 18. Freyth K, Janowitz T, Nunes F, Voss M, Heinick A, Bertaux J, Scheu S, Paul RJ. 2010. Reproductive fitness and dietary choice behavior of the genetic model organism Caenorhabditis elegans under semi-natural conditions. Molecules and Cells. 30(4):347-353. 19. Goodrich-Blair H. 2007. They’ve got a ticket to ride: Xenorhabdus nematophila-Steinernema carpocapsae symbiosis. Current Opinion in Microbiology. 10:225–230. 20. Goodrich-Blair H, Clarke DJ. 2007. Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Molecular Microbiology. 64(2):260-268. 21. Haegeman A, Vanholme B, Jacob J, Vandekerckhove TT, Claeys M, Borgonie G, Gheysen G. 2009. An endosymbiotic bacterium in a plant- parasitic nematode: member of a new Wolbachia supergroup. International Journal for Parasitology. 39(9):1045-1054. 22. Hoerauf A, Nissen-Pähle K, Schmetz C, Henkle-Dührsen K, Blaxter M, Büttner DW, Gallin MY, Al-Qaoud KM, Lucius R, Fleischer B. 1999. Tetracycline therapy targets intracellular bacteria in the filarial nematode Litomosoides sigmodontis and results in filarial infertility. The Journal of Clinical Investigation. 103(1):11–18. 23. Hoerauf A, Volkmann L, Nissen-Paehle K, Schmetz C, Autenrieth I, Büttner DW, Fleischer B. 2000. Targeting of Wolbachia endobacteria in Litomosoides sigmodontis: comparison of tetracyclines with chloramphenicol, macrolides and ciprofloxacin. Tropical Medicine & International Health. 5(4):275-279. 24. Kumar S, Blaxter ML. 2011. Simultaneous genome sequencing of symbionts and their hosts. Symbiosis. 55(3):119-126.

12
25. Lambert K, Bekal S. 2002. Introduction to plant-parasitic nematodes. The Plant Health Instructor. doi: 10.1094/PHI-I-2002-1218-01. 26. Marroquin LD, Elyassnia E, Griffitts JS, Feitelson JS, Aroian RV. 2000. Bacillus thuringiensis (Bt) Toxin Susceptibility and Isolation of Resistance Mutants in the Nematode Caenorhabditis elegans. Genetics. 155(4):1693–1699. 27. McSorley, R. 2003. Adaptations of nematodes to environmental extremes. Florida Entomologist. 86:138-142. 28. Mitreva M, Zarlenga DS, McCarter JP, Jasmer DP. 2007. Parasitic nematodes - from genomes to control. Veterinary Parasitology. 148(1):31-42. 29. Murfin KE, Dillman AR, Foster JM, Bulgheresi S, Slatko BE, Sternberg PW, Goodrich-Blair H. 2012. Nematode-bacterium symbioses--cooperation and conflict revealed in the "omics" age. The Biological Bulletin. 223:85–102. 30. Nicholas WL. 1962. A Study of a Species of Acrobeloides (Cephalobidae) in Laboratory Culture. Nematologica. 8(2):99-109. 31. Ott JA, Novak R, Schiemer F, Hentschel U, Nebelsick M, Polz MF. 1991. Tackling the sulfide gradient: a novel strategy involving marine nematodes and chemoautotrophic ectosymbionts. Marine Ecology. 12:261–279. 32. Pfarr KM, Hoerauf AM. 2006. Antibiotics which target the Wolbachia endosymbionts of filarial parasites: a new strategy for control of filariasis and amelioration of pathology. Mini-Reviews in Medicinal Chemistry. 6(2):203-210. 33. Poinar, GO Jr. 1975. Description and biology of a new insect parasitic rhabditoid Heterorhabditis bacteriophora new genus new species (Rhabditida; Heterorhabditidae new family). Nematologica. 21:463–470. 34. Poinar GO Jr, Thomas GM, Hess R. 1977. Characteristics of the specific bacterium associated with Heterorhabditis bacteriophora (Heterorhabditidae; Rhabditida). Nematologica. 23:97–102. 35. Poinar GO Jr, Hansen EL. 1986. Associations between nematodes and bacteria. Helminthological Abstracts, Series B (Plant Nematology). 55(3):61-81. 36. Rae R, Riebesell M, Dinkelacker I, Wang Q, Herrmann M, Weller AM, Dieterich C, Sommer RJ. 2008. Isolation of naturally associated bacteria of necromenic Pristionchus nematodes and fitness consequences. Journal of Experimental Biology. 211:1927–1936. 37. Rae R, Iatsenko I, Witte H, Sommer RJ. 2010. A subset of naturally isolated Bacillus strains show extreme virulence to the free-living nematodes Caenorhabditis elegans and Pristionchus pacificus. Environmental Microbiology. 12:3007–3021.

13
38. Rae R, Witte H, Rödelsperger C, Sommer RJ. 2012. The importance of being regular: Caenorhabditis elegans and Pristionchus pacificus defecation mutants are hypersusceptible to bacterial pathogens. International Journal for Parasitology. 42(8):747-753. 39. Roh JY, Choi JY, Li MS, Jin BR, Je YH. 2007. Bacillus thuringiensis as a specific, safe, and effective tool for insect pest control. Journal of Microbiology and Biotechnology. 17(4): 547–559. 40. Sicard M, Brugirard-Ricaud K, Pages S, Lanois A, Boemare NE, Brehelin M, Givaudan A. 2004. Stages of infection during the tripartite interaction between Xenorhabdus nematophila, its nematode vector, and insect hosts. Applied and Environmental Microbiology. 70:6473-6480. 41. Slatko BE, Taylor MJ, Foster JM. 2010. The Wolbachia endosymbiont as an anti-filarial nematode target. Symbiosis. 51(1):55–65. 42. Sommer RJ, McGaughran A. 2013. The nematode Pristionchus pacificus as a model system for integrative studies in evolutionary biology. Molecular Ecology. 22(9):2380-2393. 43. Stiernagle T. 2006. Maintenance of C. elegans. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.101.1 http://www.wormbook.org. 44. Stock SP. 2005. Insect-parasitic nematodes: from lab curiosities to model organisms. Journal of Invertebrate Pathology. 89(1):57-66. 45. Stock SP, Bird DM, Ghedin E, Godrich-Blair H. 2011. Abstracts of NEMASYM: The Second Nematode-Bacteria Symbioses Research Coordination Network Meeting. The Journal of Nematology. 43(1): 49–60. 46. Stock SP, Bird DM, Ghedin E, Godrich-Blair H. 2011. Abstracts of NEMASYM: The Third Nematode-Bacteria Symbioses Research Coordination Network Meeting. Symbiosis. 55(3): 97-109. 47. Stouthamer R, Breeuwer JA, Hurst GD. 1999. Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annual Review of Microbiology. 53:71-102. 48. Tan MW, Shapira M. 2011. Genetic and molecular analysis of nematode- microbe interactions. Cellular Microbiology. 13(4):497-507. 49. Thomas GM, Poinar GO Jr. 1979. Xenorhabdus gen. nov., a genus of entornopathogenic, nematophilic bacteria of the family Enterobacteriuceme. International Journal of Systematic Bacteriology. 29:352–360. 50. Thrash JC, Boyd A, Huggett MJ, Grote J, Carini P, Yoder RJ, Robbertse B, Spatafora JW, Rappé MS, Giovannoni SJ. 2011. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Scientific Reports. 1(13):1-9. 51. Wei JZ, Hale K, Carta L, Platzer E, Wong C, Fang SC, Aroian RV. 2003. Bacillus thuringiensis crystal proteins that target nematodes. Proceedings of the National Academy of Sciences U S A. 100:2760–2765.

14
52. Werren JH, Baldo L, Clark ME. 2008. Wolbachia: master manipulators of invertebrate biology. Nature Reviews Microbiology. 6(10):741-51. 53. Woolfit M, Iturbe-Ormaetxe I, McGraw EA, O'Neill SL. 2009. An Ancient Horizontal Gene Transfer between Mosquito and the Endosymbiotic Bacterium Wolbachia pipientis. Molecular Biology and Evolution. 26(2):367-374. 54. Venette RC, Ferris H. 1998. Influence of bacterial type and density on population growth of bacterial-feeding nematodes. Soil Biology and Biochemistry. 30(7):949–960.

15
CHAPTER ONE:
Survey of the microbiota associated with A. maximus in soil microcosm
ABSTRACT
Microbial populations observed in the microbiome of Acrobeloides
maximus consistently comprised a set of different microorganisms compared to
the microbiome of Caenorhabditis elegans or the metagenome of the soil sample
to which nematodes were exposed. Using 16S rRNA gene amplification, the
bacterial genera Ochrobactrum, Pedobacter, and Chitinophaga were identified at
significant levels within the A. maximus communities only and were found to be
consistently present during each of the three sampling periods over six months.
We were able to isolate and grow colonies of Ochrobactrum and Pedobacter
from the microbiome of the nematode on Miller’s Lysogeny Broth-Miller (LB) and
Yeast Extract (YE) agar plates. We were unable to isolate the associate from the
genus Chitinophaga. The results obtained suggest a possible specific and
persistent association between A. maximus and the two bacterial isolates we
were able to culture.
Using fluorescence in situ hybridization (FISH) with probes specific for
Ochrobactrum and Pedobacter, we were able to stain these bacterial cells and
show that they reside within the intestinal tract of A. maximus. It is possible that
these gut bacteria may be mutualistic by providing A. maximus with essential
metabolites, by aiding the nematode in the digestion of other bacteria, or by
providing it with protection against pathogens.

16
INTRODUCTION
Microbes are ubiquitous within metazoans. Understanding the relationship
between a multicellular organism and its microbiome will allow for a better
understanding of how these interactions have evolved over time and how
conserved these associations are across diverse populations. The biological
interactions that occur within these host-microbe symbioses are complex; the
abundance and diversity of microbes present and the totality of their interactions
between one another and as they relate to their host and environment are often
poorly understood. These host-bacterial associations can be neutral and
facultative, positive in the form of for example mutualistic cooperation or
commensalism, or negative involving for example competition, pathogenesis or
parasitism. Mutualistic microbes provide increased fitness benefits to their host,
whereas those that are pathogenic compromise the well-being of the host and
decrease its fitness. Commensal relationships occur when microbes use their
metazoan host for shelter or as a vehicle for transportation; some microbes even
linger around waiting for the death of their host to use the remains to their
advantage (3, 5, 32, 33).
Studying these networks of interactions will allow for a better
understanding of rhizosphere ecology in which bacteria, fungi, nematodes,
arthropods, and plants associate intimately with one another. Past studies often
lacked analytical focus on the distribution and abundance of organisms that are

17
part of these microbiomes. 16S rRNA based metagenomic studies are beginning
to elucidate the population composition of these microbiomes and the functions
they perform. This is not just important for our scientific understanding, but also
allows for practical applications such as the creation of microbial inoculants and
soil amendments to promote plant health & growth, and the utilization of
nematodes that promote soil nutrient cycling (14, 15, 23). It can also allow for
improvements in the prevention and treatment of microbial disease, for example
by enabling us to identify the mechanisms used by bacterial symbionts to protect
their eukaryotic host. This may in turn allow us to develop molecular tools and
inoculate soils, plants or animals with suppressive organisms to target those
microbes that might otherwise be harmful and pathogenic.
Nematodes are used as laboratory model systems to study microbial
associations. Several important and intricate microbial associations have been
studied among entomopathogenic and filarial nematodes. For example, the
insect-pathogenic bacteria Photorhabdus and Xenorhabdus are necessary
partners for the worm's life cycle in the entomopathogenic nematodes
Heterorhabditis and Steinernema nematodes, respectively (24). In a different
form of mutualism, treatment with the antibiotic tetracycline will eliminate
Wolbachia, an obligatory endosymbiont that provides essential nutrients and
metabolites for filarial nematodes such as Brugia malayi and Litomosoides
sigmodontis (11, 18).

18
In this study our efforts focus on the microbiome of Acrobeloides maximus,
a true soil inhabiting member of the nematode family Cephalobidae and a non-
pathogenic free-living bacterivore common in sandy soils that are subject to heat
and protracted desiccation. Using microcosms, we perform a 16S rRNA gene
survey of bacteria of our soil sample and compare that to the bacteria in the
microbiome of A. maximus extracted after exposure to a soil slurry for 24h. A
similar cultivation, exposure, and extraction was performed using monoxenically
grown cultures of Caenorhabditis elegans N2, a free-living bacterivorous
rhabditid specialized in rapid colonization of decomposing fruits and commonly
used as a model organism for molecular genetics research (10 ). For both
species, the nematodes extracted from the soil slurry were maintained on agar
plates supplemented with cholesterol and sampled over a period of two, four, and
six months. These samples were also analyzed using 16S rRNA amplification
and sequencing methods to monitor for changes in the number of species and
abundance of microbes present in the population over time.
MATERIALS AND METHODS
Nematode cultivation and harvest
15 mm and 90 mm petri dishes containing 1.5% nutrient-free agar
supplemented with cholesterol (1 µg/ml) were prepared and seeded with
Escherichia coli S17-1, which contains a constitutive red fluorescent protein
(RFP) expression plasmid for easy detection with fluorescence microscopy.

19
Exogenous cholesterol is necessary for nematode survival; most nematodes are
assumed to be incapable of de novo cholesterol biosynthesis and omission of
cholesterol has been shown to have adverse effects on growth, development,
molting, and brood size in C. elegans (9, 17, 25). A. maximus was grown in a
28°C incubator for a period of at least two weeks and C. elegans was cultivated
at room temperature for up to a week until the nematode populations reached
high density. Worm populations were counted directly and diluted to 104
worms/ml for subsequent soil exposure.
Rhizosphere soil collection and exposure
Rhizospheric soil samples were collected from wild sunflowers at the
California State University San Bernardino (CSUSB) Natural Preserve (San
Bernardino, CA) and placed in 50 ml sterile conical tubes, weighed, and stored at
4o C until processing. A worm slurry was created by rinsing with an adequate
amount of sterile water the agar plates on which nematodes were growing. For
every gram of soil, 1ml of the worm slurry was added to each of three conical
tubes after filling them with approximately 5g of soil each. In the same manner,
we also exposed C. elegans to the same soil in triplicate, and as control three
more tubes received 1 ml of sterile water without nematodes. Samples were
allowed to incubate for 24h at room temperature prior to worm extraction.
Employing a density gradient approach, we recovered the nematodes from
the soil samples: MgSO4 heptahydrate was added to the samples to a

20
concentration of 409 g/L followed by centrifugation at 1800g for 3 minutes (30).
The soil pelleted to the bottom and the above supernatant containing nematodes
was removed and then vacuum filtered using Millipore’s Stericup and Steritop
system through a 0.22 micron polyvinylidene difluoride (PVDF) membrane to
remove external bacterial contaminants. Nematodes were rinsed off the
membrane and collected in 3 ml of sterile water. The remaining soil pellets were
washed twice in sterile water and aliquots were frozen at -20 o C for later DNA
extraction and analysis for our long term culture investigation.
Long-term culture of A. maximus
Recovered nematodes were grown on 1.5% nutrient-free agar
supplemented with cholesterol (1 µg/ml) with no other food sources. The
nematodes were collected for analysis and subcultured at two, four, and six
months. Material from the plates was collected using sterile 10x phosphate buffer
saline (PBS) and washed as previously described to remove free-living bacteria.
Continuing cultures were seeded with 50µl of this washed worm suspension. The
remaining suspension was frozen at -20 o C for subsequent DNA analysis.
Isolation of associated bacteria
Individual nematodes from the initial culture of worms recovered from the
soil slurry were transferred onto petri plates containing 1.5% agar with 5g/L of
yeast extract (YE). The worms were allowed to wander about on the plates for up

21
to 24h, they were then removed and any released bacteria were allowed to grow.
All bacterial isolates were subsequently identified by PCR amplification and
sequencing of their 16s rRNA genes.
DNA extraction and PCR analysis
Using the FastDNA Spin Kit for Soil (Qbiogene), we extracted DNA from
the original rhizosphere soil as well as from soils exposed to A. maximus or C.
elegans and from the long term cultures. With the GenomiPhi V2 DNA
Amplification Kit (GE Healthcare Life Sciences) we individually amplified DNA
from all the samples, which we then used for PCR analysis. Utilizing the 27F (5'-
AGAGTTTGATCMTGGCTCAG-3') and 1492R (5'-GGTTACCTTGTTACGACTT-3') universal
bacteria primers, we next amplified near full length 16S rRNA gene fragments
from our samples (19, 29).
PCR reactions were performed in 50 µl reactions consisting of 25 µl of 1X
concentration of GoTaq Green PCR Master Mix (Promega), 0.5 µm of both 27F
and 1492R primers, and at least 20 ng of template DNA. Thermal cycling
conditions consisted of 5 minutes at 95˚C followed by 30 cycles of 30 sec at
94˚C, 30 sec at 55˚C, 1 min at 72˚C, followed by a final extension step for 10 min
at 72˚C. Gel electrophoresis was performed using a 1% agarose gel stained with
0.5µg/ml 1% ethidium bromide. PCR products were visualized using the BioRad
Gel Doc XR+ System.

22
16S rRNA gene library construction, sequencing and analysis
PCR products were purified using the Wizard SV Gel and PCR Clean-up
System (Promega) and subsequently ligated into the pCR 2.1-TOPO vector and
cloned with the TOPO TA Cloning Kit using One Shot TOP10 Electrocompetent
E. coli. Sets of 94 clones from each of the A. maximus triplicates, 94 clones from
the C. elegans triplicates, and 94 clones from the triplicate controls without
nematodes underwent rolling circle amplification in which the bacterial DNA
inserts within these recombinant plasmids were first amplified then sequenced
(Laragen, CA). In addition, 94 clones from the two, four, and six month long-term
culture studies were also sequenced in one direction, using the M13 forward
primer site in pCR2.1.
Sequences were compiled into FASTA formatted files and aligned against
the prokaryotic multiple sequence alignment (prokMSA) 16S rRNA gene
sequence database by applying the near alignment space termination (NAST)
algorithm (7). The Bellerophon program was used to detect and filter 16S rRNA
gene chimeras (13). The aligned set was then compared against the Ribosomal
Database Project (RDP) phylogeny database (rdp.cme.msu.edu) at 97%
similarity using the classifier on the Greengenes website (greengenes.lbl.gov) (4,
8). The 97% cutoff level is commonly used to differentiate bacterial organisms at
the species level and to distinguish between organisms that may represent
different strains or subgroups (16). To determine operational taxonomic units
(OTUs), SimRank, a general similarity measure was used to find the nearest

23
neighbors to our sequences and calculate divergence (8). Using online
calculators at Chang Bioscience (changbioscience.com), the Shannon-Wiener
diversity index and Shannon-Wiener evenness were calculated based on the
Greengenes assigned OTUs. The sample A. maximus 2 was later resequenced
in the reverse direction to verify error rates. All of our newly generated 16S rRNA
sequences have been deposited into Genbank (Table 1).
Refer to Chapter 2 for phylogenetic analysis and discussion regarding the
identities of the three most interesting bacterial genera found: Ochrobactrum,
Pedobacter, and Chitinophaga.
Preparation for Fluorescence In Situ Hybridization (FISH)
The previously described rhizosphere soil collection and exposure
methods were repeated for fluorescence in situ hybridization (FISH) probing of
exposed worms: worms were extracted after exposure to a soil slurry for 24h,
along with worms collected from agar plates without soil exposure. The FISH
protocol used was largely adopted from the work of Vandekerckhove et al. (31):
Preparation of samples was performed in triplicates. Samples were rinsed twice
in sterile water, fixed for 10 minutes in a 1:1 mixture of glacial acetic acid and
ethanol, rinsed twice for 5 minutes in pure ethanol, transferred to a mixture a 1:1
mixture of methanol and PBS w/Tween 20 (PBT; 50 mM NaCl, 10 mM Na3PO4,
0.1% (wt/vol) Tween 20 [pH 7.4]) for 10 minutes, and fixed for 30 minutes in a
PBT solution containing 1% (vol/vol) paraformaldehyde.

24
FISH Probes
The oligonucleotide probes used were based on probeBase’s 16S rRNA
data (microbial-ecology.net/probebase) and were designed to specifically anneal
to either Pedobacter or Ochrobactrum (21). The probe specific to the Pedobacter
sp., PED672 (5'-GCTACTTGTCACATTCCG-3') (12) was linked with sulfoindocyanine
chromophore Cy3, which fluoresces green and the probe specific to the
Ochrobactrum sp., Ochro3 (5'-AACTCCCCAACGGCTAACAT-3') (6) was linked to Cy5,
which fluoresces red. Each probe was dissolved in TE buffer (10 mM Tris-HCl, 1
mM EDTA [pH 7.4]) to a concentration of 10 μM where 10 μl was used for each
100 μl of the hybridization mixture.
FISH- Hybridization, Mounting, Microscopy
The hybridization mixture consisted of 20 mM Tris-HCl (pH 7.4), 0.02%
(wt/vol) sodium dodecyl sulfate (SDS), 0.9 M NaCl, 5 mM EDTA, 60% (vol/vol)
formamide, and a 1 μM concentration of the two fluorochrome-labeled probes.
Each of the fixed paraformaldehyde samples were added to this hybridization
buffer in sealed conical tubes at 42°C for 3 hours followed by two 30 minute
washes at 42°C in a hybridization buffer containing no formamide. Nematodes
were pipetted out of this wash onto microscope slides. DIC and fluorescence
images from the samples were collected using a Zeiss Axioplan2 equipped with a
CoolSnapFX cooled CCD camera and ImageProExpress 2.0 capture software.

25
RESULTS
16S rDNA analysis of extracts
Nematodes were recovered from soil samples after 24h using a density
gradient extraction, washed to remove free-living bacteria, and examined using
culture-independent 16S rRNA sequencing to taxonomically analyze the bacteria
comprising their microbiomes (Figure 1). Analysis revealed that the A. maximus
triplicates were dominated by α-Proteobacteria and Sphingobacteria, while C.
elegans associates showed a more broad distribution across taxa with no clear
dominant clade. The soil sample to which no nematodes were exposed had a
much higher percentage of bacilli compared to the other samples. Shannon-
Wiener diversity index calculations and analysis of population structures in the
samples showed marked differences among the samples (Table 2). The
α-Proteobacteria clade of the A. maximus triplicates was dominated by the genus
Ochrobactrum, while the Sphingobacteria clade was dominated by the genus
Pedobacter ; these two genera were not detected in the soil samples (data not
shown) and the samples from C. elegans gave no clear indication of any
dominant genera within these two clades (Figure 2). In subsequent experiments,
we were unable to recover transmitted E. coli S17-1 from the initial inoculants of
our samples, and were also unable to observe its red fluorescence via
fluorescence and confocal microscopy (data not shown).

26
Co-cultures
For a period of six months we maintained co-cultures with bacteria from
the soil, with bimonthly collecting and subculturing. Utilizing 16S rRNA methods
we examined the microbiome of a three week old co-culture which showed a
stable population similar in composition to the samples recovered from soil
exposure (Figure 3). Another co-culture was analyzed in the same way, which
again yielded a similar population makeup (data not shown). Over the six month
period we observed a shift away from the initial population structure however
(Fig 3A). Pedobacter and Chitinophaga persisted in the population for up to six
months, with Chitinophaga peaking at four months (Fig 3B) while Ochrobactrum
persisted in the population for up to four months only (Fig 3C). Throughout the
six months, the bacterial population displayed noticeable increases in the
abundance of Sphingobium and a decrease in Brevundimonas.
Isolation and culture
Bacterial isolates were obtained during various time points from the co-
cultures by inoculation onto different kinds of agar media or by isolating individual
nematodes on different media. Applying 16S rRNA gene sequencing we
identified these bacterial isolates (Table 3). Pedobacter was isolated by
transferring nematodes onto fresh agar plates lacking nutrients for bacterial
growth and isolating the microcolonies that formed, while Ochrobactrum isolates
were recovered by transferring individual nematodes onto YE agar. Both

27
Pedobacter and Ochrobactrum isolates were cultivated axenically at 37°C and
had aliquots frozen at -80°C. Attempts to isolate Chitinophaga isolates using
colloidal chitin media were unsuccessful.
Fluorescence In Situ Hybridization
After 24h of soil exposure, nematodes were removed from the soil, and
using FISH, were probed for the presence of Ochrobactrum and Pedobacter.
FISH signals corresponding to both genera were simultaneously identified in the
guts of the nematodes (Fig 4 A -D). Control experiments using cultured strains of
the two bacterial isolates showed no cross reactivity of the probes under fixation
and hybridization conditions and no background staining was observed in worms
fed only E. coli S17-1 (Fig 4E & F).
DISCUSSION
Bacterial isolates from Acrobeloides maximus recovered from soil
microcosms consistently yielded DNA sequences most closely matching with
known sequences of Ochrobactrum, Pedobacter, and Chitinophaga suggesting
that these are clade specific associations. The sequences belonging to these
genera were largely enriched in all A. maximus samples compared to similar
sampling from Caenorhabditis elegans exposed to soil and to direct sampling
from the soil, which showed differing association patterns. Furthermore, these
three bacterial genera persisted in long term co-culture with the nematodes on

28
agar plates where no external nutrients were provided to the nematodes or the
soil microbiota. These associations were adequately stable at the phylum and
subphylum level, but at the class level (alphaproteobacteria and
sphingobacteriia) the populations shifted substantially over time, most likely due
to nutrient consumption and dissipation. Bacterial isolates of Ochrobactrum and
Pedobacter were shown via FISH to colonize the gut of A. maximus.
Other studies describing the microbiota of other nematodes have provided
similar results to our own. Ochrobactrum and Pedobacter were both identified as
genera associated with wild C. elegans (28). In another study, these two genera
were also identified as associates of the soybean root cyst nematode Heterodera
glycines, and those investigators were able to isolate and culture Pedobacter
from soybean cysts (22). Pedobacter was also recently identified as a bacterial
associate of the plant parasitic nematode Bursaphelenchus mucronatus (27).
These investigations along with our own suggest a broad association of
Pedobacter with many nematodes, and that its occurrence is not just specific to
only A. maximus. The replicates we performed during our investigation show that
a quick selection process takes place in the A. maximus microbiome when the
nematodes are introduced to the soil, relative to the soil microbial community
structure. Conversely, it appears from our work and others that laboratory bred C.
elegans are usually unselective in its microbiota, consuming an array of
phylogenetically diverse bacteria which it isolates from the soil (1, 26). It is
important to note the E. coli strain S17-1, used to monoxenically farm both A.

29
maximus and C. elegans before soil inoculation, was quickly displaced by soil
bacteria as a result of nematode exposure to the soil.
The bacterial associations we observed in A. maximus were consistent and
reproducible; however, they were also prone to population shifts in our long term
culture analysis. In our culture based and culture independent tests we were able
to repeatedly recover Ochrobactrum and Pedobacter in association with A.
maximus, and using FISH were able to show that these two genera are localized
primarily in the gut of the nematode. We were unable to find an appropriate FISH
probe to test for the presence of Chitinophaga, and were also unable to culture it
from our enrichments.
Our observations suggest a symbiotic nematode-microbe interaction
between A. maximus and Ochrobactrum and Pedobacter, perhaps involving
mutualism or a phoretic association. These bacteria may for example act as
mutualists by aiding the nematode in the digestion of other microbes and toxins
through secretion of digestive enzymes or they may play a role in protecting the
nematode against attacks by pathogenic bacteria by producing antibiotics.
Research has shown various strains of Pedobacter to be associated with
antifungals and probiotics acting against pathogenic bacteria and fungi (20) and
there have been reports of Ochrobactrum strains protecting entomopathogenic
nematodes from harmful microbes (2). Thus, these two bacterial genera appear
to have potentially widespread associations with a number of taxonomically
different nematode species, suggesting a possibly important evolutionary

30
relationship that warrants further study to elucidate the occurrence and role of
symbiosis in the behavior and ecology of soil nematodes.

31
REFERENCES 1. Avery L, Shtonda B. 2003. Food transport in the C. elegans pharynx. Journal of Experimental Biology. 206:2441–2457. 2. Babic I, M Fischer-Le Saux, E Giraud, N Boemare. 2000. Occurrence of natural dixenic associations between the symbiont Photorhabdus luminescens and bacteria related to Ochrobactrum spp. in tropical entomopathogenic Heterorhabditis spp.(Nematoda, Rhabditida). Microbiology 146:709. 3. Campos-Herrera R, El-Borai FE, Duncan LW. 2012. Real-time PCR as an effective technique to assess the impact of phoresy by Paenibacillus sp. bacteria on Steinernema diaprepesi nematodes in nature. Molecular Ecology Resources. 12(5):885-893. 4. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed- Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research. 37:D141–D145. 5. Dedeine F, Ahrens M, Calcaterra L, Shoemaker DD. 2005. Social parasitism in fire ants (Solenopsis spp.): a potential mechanism for interspecies transfer of Wolbachia. Molecular Ecology. 14:1543–1548. 6. Demanèche S, H Sanguin, J Poté, E Navarro, D Bernillon, P Mavingui, W Wildi, TM Vogel, and P Simonet. 2008. Antibiotic-resistant soil bacteria in transgenic plant fields. Proceedings of the National Academy of Sciences 105:3957-3962. 7 DeSantis TZ Jr, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL. 2006. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Research. 34:W394-99. 8. DeSantis TZ Jr, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera- checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology. 72:5069–5072. 9. Entchev EV, Kurzchalia TV. 2005. Requirement of sterols in the life cycle of the nematode Caenorhabditis elegans. Seminars in Cell and Developmental Biology. 16:175-182. 10. Félix MA, Braendle C. 2010. The natural history of Caenorhabditis elegans. Current Biology. 20(22):R965-969. 11. Fenn K, Blaxter M. 2004. Are filarial nematode Wolbachia obligate mutualist symbionts? Trends in Ecology & Evolution. 19(4):163-166. 12. Friedrich U, H. Van Langenhove, K Altendorf, A Lipski. 2003. Microbial community and physicochemical analysis of an industrial waste gas biofilter and design of 16S rRNA-targeting oligonucleotide probes. Environmental Microbiology. 5:183-201.

32
13. Huber T, Faulkner G, Hugenholtz P. 2004. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 20(14):2317-2319. 14. Ingham RE, Trofymow JA, Ingham ER, Coleman DC. 1985. Interactions of bacteria, fungi, and their nematode grazers: Effects on nutrient cycling and plant growth. Ecological Monographs. 55:119–140. 15. Irshad U, Villenave C, Brauman A, Plassard C. 2011. Grazing by nematodes on rhizosphere bacteria enhances nitrate and phosphorus availability to Pinus pinaster seedlings. Soil Biology & Biochemistry. 43:2121–2126. 16. Janda JM, Abbott SL. 2007. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. Journal of Clinical Microbiology. 45(9): 2761–2764. 17. Kurzchalia TV, Ward S. 2003. Why do worms need cholesterol? Nature Cell Biology. 5: 684–688. 18. Landmann F, D Voronin, W Sullivan, MJ Taylor. 2011. Anti-filarial Activity of Antibiotic Therapy Is Due to Extensive Apoptosis after Wolbachia Depletion from Filarial Nematodes. PLOS Pathogens. 7(11): e1002351 19. Lane DJ. 1991. 16S/23S rRNA sequencing. Nucleic acid techniques in bacterial systematics. Stackebrandt, E., and Goodfellow, M., eds., John Wiley and Sons. New York, NY.115-175. 20. Lauer A, M Simon, J Banning, B Lam, R Harris. 2008. Diversity of cutaneous bacteria with antifungal activity isolated from female four-toed salamanders. ISME Journal. 2:145-157. 21. Loy A, F Maixner, M Wagner, M Horn. 2007. probeBase—an online resource for rRNA-targeted oligonucleotide probes: new features 2007. Nucleic Acids Research. 35:d800-d804. 22. Nour SM, JR Lawrence, H Zhu, GDW Swerhone, M Welsh, TW Welacky, E Topp. 2003. Bacteria associated with cysts of the soybean cyst nematode (Heterodera glycines). Applied and Environmental Microbiology. 69:607-615. 23. Postma-Blaauw MB, de Vries FT, de Goede RG, Bloem J, Faber JH, Brussaard L. 2005. Within-trophic group interactions of bacterivorous nematode species and their effects on the bacterial community and nitrogen mineralization. Oecologia 142:428-439. 24. Ruby EG. 2008. Symbiotic conversations are revealed under genetic interrogation. Nature Reviews Microbiology. 6:752-762. 25. Shim YH, Chun JH, Lee EY, Paik YK. 2002. Role of cholesterol in germ- line development of Caenorhabditis elegans. Molecular Reproduction and Development. 61:358-366. 26. Tan MW, Shapira M. 2011. Genetic and molecular analysis of nematode- microbe interactions. Cell Microbiology. 13(4):497–507.

33
27. Tian X, X Cheng, Z Mao, G Chen, J Yang, B Xie. 2011. Composition of Bacterial Communities Associated with a Plant Parasitic Nematode, Bursaphelenchus mucronatus. Current Microbiology. 6:117-125. 28. Troemel ER, MA Felix, NK Whiteman, A Barriere, FM Ausubel. 2008. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLOS Biology. 6:2736-2752. 29. Turner S, Pryer KM, Miao VPW, Palmer JD. 1999. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. Journal of Eukaryotic Microbiology. 46:327-338. 30. van Bezooijen J. 2006. Methods and Techniques for Nematology. Wageningen University. 27-51. 31. Vandekerckhove T, A Coomans, K Cornelis, P Baert, M Gillis. 2002. Use of the Verrucomicrobia-Specific Probe EUB338-III and Fluorescent In Situ Hybridization for Detection of “Candidatus Xiphinematobacter” Cells in Nematode Hosts. Applied and Environmental Microbiology. 68:3121- 3125. 32. Wade WN, Beuchat LR. 2003. Metabiosis of proteolytic moulds and Salmonella in raw, ripe tomatoes. Journal of Applied Microbiology. 95:437–450. 33. Waid J. 1999. Does soil biodiversity depend upon metabiotic activity and influences? Applied Soil Ecology. 13(2):151-158.

34
FIGURES AND TABLES
Figure 1. Soil exposed A. maximus populations were found to be associated with a consistent bacterial population. After 24h soil exposure, the A. maximus samples were dominated by α-Proteobacteria and Sphingobacteria, while C. elegans sample was shown to be associated with a broader bacterial distribution. A high percentage of Bacilli is shown to be associated with the bacteria identified from the soil sample.

35
Figure 2A. Within the dominant subgroups the A. maximus populations were consistent and limited compared to the C. elegans associated bacteria which were shown to be more diverse at the genus level. Ochrobactrum is shown to be the predominate alpha-proteobacteria within A. maximus.

36
Figure 2B. Within the dominant subgroups the A. maximus populations were consistent and limited compared to the C. elegans associated bacteria which were shown to be more diverse at the genus level. Pedobacter is shown to be the primary Spingobacteria within A. maximus, along with a consistent representation of Chitinophaga at a lower level.

37
A
B C
Figure 3. Long term culture experiments appear to show stability at the phylum level (A) but reveal shifts in population as the worm population ages at the genus level (B,C). At 6 months post exposure, the initially dominant clades have been supplanted in both the Sphingobacteria (B) and Alpha-Proteobacteria (C).

38
A B
C D
E F Figure 4. Fluorescence in situ hybridization was used to identify the presence of both Ochrobactrum sp. (green) and Pedobacter sp. (red) in the gut of formaldehyde fixed A. maximus after recovery from soil microcosm. Samples from soil microcosms were imaged by DIC (A,C,E) and epifluorescence (B,D,F). A. maximus recovered from soil microcosms (A-D) are compared with A. maximus grown on a monoxenic culture on E. coli DH5α (E,F) to show the specificity of the FISH probes. Scale bar = 10 microns.

39
Table 1. List Of Sequences Deposited Into GenBank Accession
Pedobacter KC110983, KC110977, KC110967, KC110963, KC110947, KC110933, KC110921, KC110919, KC110917, KC110913, KC110909, KC110905, KC110895, KC110984, KC110978, KC110968, KC110966, KC110958, KC110934, KC110904
Ochrobactrum KC110896, KC110901, KC110903, KC110910, KC110914, KC110916, KC110918, KC110923, KC110926, KC110929, KC110931, KC110932, KC110935, KC110936, KC110939, KC110940, KC110941, KC110944, KC110949, KC110951, KC110956, KC110957, KC110960, KC110961, KC110962, KC110970, KC110973, KC110975, KC110976, KC110980, KC110985
Chitinophaga KC110981, KC110971, KC110965, KC110955, KC110943, KC110915, KC110907, KC110897, KC110950, KC110908
Sphingobacteriales bacterium
KC110912, KC110922, KC110946
Niastella
KC110902, KC110979
Other Genera KC110924, KC110969, KC110945, KC110952, KC110959, KC110920, KC110953, KC110982, KC110954, KC110972, KC110898, KC110938, KC110974, KC110906, KC110948, KC110987, KC110899, KC110900, KC110925, KC110930, KC110937, KC110928, KC110911, KC110927, KC110942, KC110964, KC110986
Table 2. Shannon-Wiener Population Statistics for Microcosm Samples
A. maximus 1 A. maximus 2 A. maximus 3 C. elegans Soil
SW Diversity Index
1.78 2.26 1.96 3.41 1.31
SW-Evenness 0.63 0.71 0.64 0.9 0.53

40
Table 3. Identified Isolates from association from A. maximus microcosm and long term culture
Accession Isolate name OTUa Source(s) KC687078 Ochrobactrum sp.
W2P 1108 Nematode long term culture
and isolation on YE KC687079 Pseudomonas sp.
JPB-PO4 Massilia sp. PO12
18131396 Nematode long term culture
KC687080 Bacillus sp. JPB-PO2 758 Dead worm in long term culture
KC687081 Pedobacter sp. API 534 Nematode long term culture, oligotrophic medium
KC687082 Brevundimonas sp. JPunk3-2
1066 Acrobeloides stock plate
KC687083 Salmonella sp. JPunk1-2
1607 Nematode associated isolation on YE
KC687084 Achromobacter sp. JPB-PO7
1317 Nematode associated isolation on YE
KC687085 Pseudomonas sp. JPunk3-1
1813 Nematode associated isolation on YE
KC687086 Pseudomonas sp. PO7
1813 Nematode associated isolation on YE
KC687087 Bacillus sp. JPB-PO3 758 Dead worm long term culture Nematode associated isolation on YE
KC687088 Achromobacter sp. JPB-PO7
1317 Nematode associated isolation on YE
a OTU assigned using RDP phylogeny. Isolates assigned to the same OTU were verified to be distinct using direct sequence comparison (bl2seq)

41
CHAPTER TWO:
Phylogenetic Analysis of Bacterial Genera Detected At Significant Levels Within The Microbiota
Associated With A. maximus
ABSTRACT
Using 16S rRNA amplification and sequencing, three bacterial strains
belonging to the genera Ochrobactrum, Pedobacter, and Chitinophaga were
detected at significant levels within cultured Acrobeloides maximus populations
exposed to soil for 24 hours before extraction and re-inoculation on agar. All
three strains were found to persist in re-inoculated A. maximus cultures over a
period of six months. For each strain, sequences from our A. maximus
associated clones along with relevant 16S rRNA sequences from congeners or
related genera available in GenBank were subjected to comprehensive
phylogenetic analyses. Sequences were aligned using the multiple sequence
alignment algorithms implemented in T-Coffee, MAFFT, Kalign, and WebPRANK,
and phylogenetic trees were constructed using Bayesian and maximum-
likelihood methods. Similarities and differences among the different alignment
and tree-building methods suggest tree building methods with T-Coffee
performed more poorly while maximum-likelihood methods with MAFFT and
Kalign produced more mutually consistent topologies that correspond better to
recognized genus boundaries. Inferences are made about the known species

42
that are phylogenetically closest to each of our three strains. Our Ochrobactrum
strains appear to be most closely related to Ochrobactrum tritici, while our
Chitinophaga strains are most similar to a bacterium previously identified as
“Flexibacter cf. sancti.” Our Pedobacter strains seem to be a previously
undescribed sphingobacterium within the genus.
INTRODUCTION
During the last decades of the 20th century, Carl Woese pioneered the idea
of using small subunit ribosomal RNAs (SSU rRNA) as reliable molecular
markers to indicate relatedness among different organisms (45). SSU rRNAs are
thought to be reliable for discerning phylogenetic relatedness because they can
easily be amplified and sequenced directly and are one of the few genes which
can be found within all uni- or multicellular organisms. Furthermore, they encode
for highly conserved ribosomal structures yet usually exhibit sufficient nucleotide
sequence variation to allow for distinction between different species (36).
Sequencing of 16S rRNA has become an established method for identifying
bacteria and a standard tool for discerning their phylogenetic relationships (20,
34).
As discussed in Chapter 1, we used microcosms and performed a 16S
rRNA gene survey of bacteria of our soil sample, and then compared that to the

43
bacteria in the microbiome of Acrobeloides maximus extracted after 24h of
exposure to soil. Clones from our A. maximus sample were sequenced and
generated 16S rRNA sequences from dominant strains belonging among others
to the genera Ochrobactrum, Pedobacter, and Chitinophaga. In this study, a
phylogenetic analysis is undertaken to examine in greater detail how sequences
from our clones of these three bacterial genera relate to known species, and
particularly to clarify whether they most likely represent previously named
species versus as yet unidentified species.
MATERIALS AND METHODS
Data Preparation
Our 16S rRNA amplification via PCR and direct sequencing methods are
described in Chapter 1. Relevant 16S rRNA sequences for each bacterial genus
were gathered from GenBank using NCBI’s Reference Sequence (RefSeq)
database collection as well as the International Nucleotide Sequence Databases
(INSD) and compiled into a FASTA file along with sequences belonging to our
own clones (Table 1 A-C). For purposes of rooting trees and gauging quality of
formal identifications for previously published 16S sequences, outgroup
sequences were also included from genera closely related to each of our three
associates of A. maximus : sequences from the genus Brucella were used for the

44
outgroup for Ochrobactrum; sequences from Chitinophagaceae and
Sphingobacteriales along with sequences from the genera Segetibacter,
Parasegitibacter, and Niastella were used for the outgroup for Chitinophaga; and
sequences from the genera Sphingobacterium and Mucilaginibacter were used
for the outgroup for Pedobacter (Table 1 A-C).
Multiple sequence alignments: T-Coffee, MAFFT, Kalign, and WebPRANK
The following multiple sequence alignment (MSA) tools located on EMBL-
EBI (www.ebi.ac.uk/Tools/msa/) were used to align 16S rRNA sequences for
each genus: T-Coffee, MAFFT, Kalign, and WebPRANK. T-Coffee is an algorithm
that attempts to lessen the faults of other popular progressive alignment methods
by using a consistency based approach (32). By contrast, MAFFT utilizes fast
Fourier transforms to perform rapid alignments and when using more than 60
sequences is shown to be 100 times faster than T-Coffee without sacrificing
accuracy (19). Kalign uses the Wu-Manber string-matching algorithm to perform
quick and accurate sequence alignments (26). WebPRANK is a relatively new
tool which employs a phylogeny-aware progressive algorithm to perform its
sequence alignments (29). Each alignment algorithm ran at its respective default
settings on EMBL-EBI for all alignments.

45
Phylogenetic inference tools- Maximum Likelihood, PhyML, Mr. Bayes
Alignment files generated from the MSA tools were examined in MEGA 5, a
software program for molecular evolutionary genetic analysis (38). Maximum
likelihood (ML) analyses were performed in MEGA 5. The sequence alignment,
assembly and analysis software Geneious 6.0.5 (Biomatters Ltd.) incorporates a
MrBayes plugin which was used for Bayesian phylogenetic analyses (16). PhyML
analyses were carried out via the interface of the HIV database website
(hiv.lanl.gov) (13). The settings for substitution models, rate variation, gamma
categories, and branch support methods are presented in Tables 2-4.
LALIGN
LALIGN, a tool on the EMBL-EBI website (www.ebi.ac.uk/Tools/psa/lalign/)
for identifying regions of sequence similarity between two sequences (15) was
used to compare our clones which had noticeably longer branch lengths against
a consensus sequence of the other clones in their respective genus generated
using Consensus Maker v2.0.0 with Kalign on the HIV database website
(hiv.lanl.gov).

46
RESULTS
Ochrobactrum sp. clones
ML, PhyML, and MrBayes trees generated with T-Coffee and WebPRANK
alignments generally did not group the Ochrobactrum clones together;
furthermore, sequences from Ochrobactrum tritici strains did not form a
monophyletic group and were dispersed throughout the trees (Figure 1). The
trees produced using MAFFT and Kalign alignments yielded better results,
clustering the Ochrobactrum clones and the Ochrobactrum tritici sequences
together in their own respective clades, which jointly comprise a monophyletic
group (Figure 2, 3). The ML tree created with MAFFT shows a 99% bootstrap
support value for the monophylum of these two clades (Figure 2). In all trees,
Ochrobactrum sp. clone Acro231 (designated with a blue asterisk) is placed on a
noticeably longer branch than the other Ochrobactrum clones (Figures 1-3).
Chitinophaga sp. clones
Trees constructed with T-Coffee alignments were poorly resolved and
show low branch support values (Figure 4). Trees generated with the other three
sequence alignment tools produced better and similarly comparable results. The
Chitinophaga clones clustered together in their own monophyletic group along
with Flexibacter cf. sancti with 99-100% bootstrap support in the ML and PhyML
trees generated using MAFFT, Kalign, and WebPRANK alignments. ML trees
created with MAFFT and Kalign alignments show identical topology to one

47
another (Figures 5, 6). In a majority of the trees, the Chitinophaga sp. clone
Acro210 (designated with an orange asterisk) has a significantly longer branch
than the other clones (Figures 5, 6). Our Sphingobacteriales clones Acro262 and
Acro263 cluster together in the Segetibacter clade, while our Sphingobacteriales
clone Acro264 is placed in a Niastella clade along with known Niastella
sequences, as well as our Niastella clones Acro224 and 225 (Figures 5, 6).
Pedobacter sp. clones
In all trees, the Pedobacter clones grouped together and formed a sister
clade to Pedobacter africanus strains K1/K2 and NBRC 10065. PhyML trees
were the only ones to separate the Pedobacter strains and the Sphingobacterium
outgroup into distinct clades (Figures 7-9). The topology of the generated
Pedobacter trees is poorly resolved, disallowing clear distinctions or well
supported groupings at the species level. Pedobacter sp. clone Acro279
(designated with a green asterisk) represents a longer branch than the rest of the
Pedobacter clones in nearly all trees (Figures 7-9).
LALIGN plots
Analyses were performed for comparing Ochrobactrum sp. clone Acro231
(blue asterisk), Chitinophaga sp. clone Acro210 (orange asterisk), and
Pedobacter sp. clone Acro279 (green asterisk) against a generated consensus
sequences of their respective clones because they showed longer branch

48
lengths that their counterparts in the phylogenetic trees. A "dot-plot" like graph of
these sequence alignments is reported (Figure 10A-C).
DISCUSSION
Comparison of multiple sequence alignment algorithms
Many factors influence the performance of alignment algorithms, including
the number of sequences used, the length of the sequences, the occurrence and
lengths of insertions & deletions, and the degree of similarity among the
sequences (31). Benchmark criteria have been developed to compare the
performance of MSA algorithms against one another; such as for example
BAliBASE (Benchmark Alignment dataBASE) (bips.ustrasbg.fr/en/Products/
Databases/BAliBASE/) which examines problems commonly associated with
MSAs such as small numbers of sequences, unequal phylogenetic distributions,
internal insertions, repeats, inverted regions and so forth (39, 40, 41). An
alignment benchmark investigation performed by Thompson et al. in 2011
revealed key differences between some of the more popular alignment
algorithms: MAFFT and T-Coffee performed better with more divergent
sequences, whereas Kalign performed better with sequences containing multiple
conserved regions (41). Fletcher and Yang in 2010 performed their own
investigation and showed that WebPRANK consistently outperforms MAFFT in
their branch site test analysis (10).

49
Comparison of phylogenetic reconstruction methods
Computing time continues to be a major factor when comparing different
phylogenetic tree building tools. Standard maximum likelihood analyses with
bootstrapping suffer from the burden of lengthy computational time. Bayesian
inference methods, like MrBayes, tend to perform computationally faster than
standard maximum likelihood methods while still having comparable phylogenetic
accuracy (16, 25). PhyML utilizes approximate likelihood ratio test branch support
methods based on a Shimodaira-Hasegawa-like procedure (aLRT SH-like branch
support methods), making it extremely fast compared to standard maximum
likelihood bootstrapping methods: 1000 bootstrap iterations were used in each of
our standard ML analyses, whereas PhyML-aLRTs only required a single iteration
per analysis (2, 13).
Studies undertaken to compare Bayesian posterior probabilities against
maximum likelihood bootstrap support values show that both are overly
supportive in their measure of phylogenetic accuracy, with Bayesian support
values tending to be higher than values obtained from bootstrapping (6, 9, 27, 43,
44).

50
Ochrobactrum sp. clones
The genus Ochrobactrum is comprised of six species: O. anthropi, O.
intermedium, O. tritici, O. grignonense, O. gallinifaecis, and O. ciceri, which can
be found in plants and their rhizospheres, soil, wastewater, or animal feces (4).
The genus Brucella consists of highly pathogenic strains which infect humans
and livestock (e.g. cattle, sheep, goats, pigs) and is the closest relative to
Ochrobactrum among those available from published sequence databases; it
was therefore used as the outgroup in our phylogenetic analyses (Table 1).
ML, PhyML, and MrBayes trees generated with MAFFT and Kalign
converge on a consensus placing our Ochrobactrum clones and Ochrobactrum
tritici sequences together in their own monophylum (Figure 2 and 3), suggesting
our Ochrobactrum clones are closely related to, or are a new variant of
Ochrobactrum tritici. O. tritici was first found and isolated over a decade ago from
the rhizoplane of wheat (28) and isolates of O. tritici have also been recovered
from wastewater (11).
Some of the O. tritici sequences are very divergent from the others within
the monophylum; most divergent are O. tritici AS1 and the clade comprised of O.
tritici strains TA 93, W19-1, W19-2, A4, and W49-1 (Figures 2 and 3). From our
trees it is reasonable for us to identify our Ochrobactrum clones as being the
same species; it however draws into question whether all the sequences
gathered from GenBank as O. tritici are really one and the same organism.

51
In order to decide whether or not two bacterial strains are the same
species, microbiologists first use 16S rRNA sequence-based phylogeny to check
if bacteria are similar enough to be assigned the same name. Having greater
than 97% 16S rRNA sequence identity similarity is the recommended cut-off for
species delineation (8, 12, 21, 22). There are drawbacks to using 16S rRNA
sequencing to delineate species. The >97% threshold is not universally accepted
and by itself is not sufficient enough for accurate species determination,
moreover 16S rRNA sequencing has poor resolving power for discerning
differences at and below the species level (17). In addition, 16S rRNA genes can
be subject to horizontal gene transfer; this raises a slew of hotbed questions
regarding species concept and phylogenetic reconstruction, including questioning
the validity of 16S rRNA constructed phylogenetic trees (3, 5, 14).
Chitinophaga sp. clones
The genus Chitinophaga was first proposed in 1981 by Sankhobol and
Skerman, and contained only a single species, Chitinophaga pinensis (35). In
2006, Kämpfer et al. proposed reclassifying Cytophaga arvensicola, Flexibacter
filiformis, Flexibacter japonensis, and Flexibacter sancti into the genus
Chitinophaga (18). Thus, the genus now consists of six species: C. pinensis, C.
arvensicola, C. filiformis, C. japonensis, C. sancti, and C. skermanii. Members of
this genus are of special interest because of their ability to degrade the
polysaccharide chitin, a β-1,4-glycosidic linked homopolymer of N-acetyl-D-

52
glucosamine know to be the main component of arthropod exoskeletons and
fungal cell walls (7).
ML and PhyML trees generated using MAFFT, Kalign, and WebPRANK
cluster our Chitinophaga clones and an organism previously identified as
“Flexibacter cf. sancti” (Accession AF181568) together in their own monophylum
(Figure 5 and 6), suggesting our clones are novel strains of this species. From
our trees, it appears that "Flexibacter cf. sancti" is clearly a different organism
from the clade of virtually identical sancti sequences that are presumably
genuine and correctly identified (C. sancti strains NBRC 16033, IFO 15057,
NBRC 15057 and F. sancti strains IFO 16033, IFO 15057, ATCC 23092) (Figure
5 and 6). Most likely "Flexibacter cf. sancti" is an earlier discovery of the same as
yet unnamed Chitinophaga in our experiments to be an associate of A. maximus.
The soil bacterium “Flexibacter cf. sancti” (reclassified as Chitinophaga cf. sancti)
was first discovered as part of a consortium of bacteria comprising a biofilm that
degrades diclofop-methyl, an active ingredient found in many commercial
herbicides, although by itself "Flexibacter cf. sanct" is not known to be a diclofop-
methyl degrader (24).
The trees also group our Sphingobacteriales bacterium clones Acro262
and Acro263 in a Segetibacter specific clade, suggesting the sequences
represent this genus (Figure 5 and 6). The trees moreover group all the Niastella
sequences together into their own clade, which includes our Niastella sp. clones
Acro224 and Acro225. Furthermore, our Sphingobacteriales bacterium clone

53
Acro264 also falls into this Niastella clade, suggesting it too might be a member
of this genus. Parasegetibacter luojiensis RHYL-37 (Accession NR044576) is
placed into our Niastella clade as well, which is not surprising as it was first
classified as being a Niastella sp. in 2008 before being reclassified as
Parasegetibacter in 2009 (46) (Figure 5 and 6).
Pedobacter sp. clones.
The genus Pedobacter was proposed in 1998 and consisted initially of four
species: P. heparinus (formerly Sphingobacterium heparinurn), P. piscium
(formerly S. piscium), P. africanus, and P. saltans (37). As of date, there are no
fewer than 42 named species within the genus: P. africanus, P. agri, P.
alluvionis, P. aquatilis, P. arcticus, P. borealis, P. boryungensis, P. caeni, P.
composti, P. cryoconitis, P. daechungensis, P. daejeonensis, P. duraquae, P.
ginsengisoli, P. glucosidilyticus, P. hartonius, P. heparinus, P. himalayensis, P.
insulae, P. jejuensis, P. koreensis, P. kribbensis, P. kwangyangensis, P. lentus,
P. metabolipauper, P. nyackensis, P. oryzae, P. panaciterrae, P. piscium, P.
rhizospharae, P. roseus, P. saltans, P. sandarakinus, P. soli, P. steynii, P.
suwonensis, P. terrae, P. terricola, P. wanjuense, P. westerhofensis, P.
xinjiangensis, and P. yonginense. This large number of Pedobacter species
makes 16S rRNA phylogenetic analysis difficult and is probably one of the
reasons why the topologies of our Pedobacter trees provide little support for clear
distinctions and groupings at the species level (Figures 7-9). In all the trees our

54
Pedobacter clones cluster together and usually formed sister clades or
monophyla with Pedobacter africanus strains K1/K2 and NBRC 10065 (Figures
7-9). Pedobacter africanus is known to occur in soil and activated sludge in dry
regions, particularly within South Africa (37). There is evidence to suggest that
our Pedobacter clones may be a novel species, but further work will be required
to prove or disprove this assumption.
LALIGN analysis
LALIGN analysis of Ochrobactrum sp. clone Acro231 compared against
the consensus sequence of Ochrobactrum clones shows a 98.6% similarity
among the sequences (Figure 10A). This is above the 97% cutoff and suggests
the longer branch of Ochrobactrum sp. clone Acro231 seen in our trees (Figures
1-3) may be an artifact due to pitfalls intrinsic to the different tree-building
methods used (1).
LALIGN analysis of Chitinophaga sp. clone Acro210 compared against the
consensus sequence of Chitinophaga clones shows a 94.3% similarity (Figure
10B) and the analysis of Pedobacter sp. clone Acro279 compared against the
consensus sequence of Pedobacter clones shows a 94.0% similarity among the
sequences (Figure 10C). Both of these fall slightly below the 97% cuttoff
indicating they are dissimilar from the rest of their respective clones. Further
protocols need to be implemented (discussed below) to determine if they are
truly dissimilar.

55
Requirements for describing a new bacterial species
A combination of phenotypic, genotypic, and chemotaxonomic approaches
is required for a formal description of a new bacterial species. In addition, the
rules for nomenclature and guidelines for publication discussed in the
Bacteriological Code (1990 Revision) (23) must be followed. A multitude of
different diagnostic approaches is required in order to have sufficient descriptive
information to allow for characterization of a novel species.
Genotypic approaches
Use 16S rRNA gene sequencing information and phylogenetic analysis is
the first step to determine the phylogenetic placement of an organism down to at
least the family and genus level. This is commonly followed by a DNA-DNA
hybridization test in order to decide whether two isolates should be considered as
similar or different species. If their 16S rRNA sequences show > 97% identity and
if their genomes show >70% DNA-DNA hybridization the two strains are said to
belong to the same species (14). Multilocus sequence analysis can also be
carried out in which internal fragments of several housekeeping genes are
analyzed to characterize the species (30). Additionally, if the whole genomes of
reference strains are available, then whole genome sequencing of the isolate can
be undertaken. Examining the % G+C content of DNA might be helpful as well;
deviations from expected values may help in elucidating information about the
strain being studied.

56
Phenotypic approaches
Microscopy is the oldest method for obtaining information about the
morphological features of bacteria. Light microscopy may be sufficient for
visualizing and describing the cell shape, size, pigment color, and staining
characteristics. Utilizing different differential staining dyes and techniques such
as Gram staining, can aid in the characterization of the novel species. The
presence and/or absence of certain characteristics structures like flagella,
capsules, polyphosphate granules, and spores also can aid in the description
and identification of a new species. Notes regarding the motility and speed (or
lack thereof) of the bacterium should be recorded as it may be of significance.
Furthermore, it may be necessary to utilize scanning electron microscopy to
show morphology of whole cells or transmission electron microscopy to view
intracellular structures (42).
Physiological characteristics such as pH and temperature range for optimal
growth and its effect on phenotype should be recorded. Cells should be cultured
using various media in nutrient broths and on agar plates to determine carbon
and nitrogen source utilization.
Chemotaxonomic approaches
Chemotaxonomy typically deals with using chemical approaches to study
the composition of different cellular components, such as the cell membrane,
outer cellular layers, and the different organelles residing in the cytoplasm (42). It

57
is common to start with a commercially available cell-based phenotypic assay kits
such as those available from Biolog (Biolog, Inc.) or API (bioMérieux). Common
tests include the oxidase test (to detect cytochrome c oxidases), catalase test (to
determine presence of catalase enzymes using hydrogen peroxide), testing the
strains relationship to oxygen (is the strain aerobic and/or anaerobic?), testing
the strains relationship to carbon dioxide and hydrogen (via manometric
methods), and tests to determine the fatty acid and polar lipid profile of the strain
(33).
FINAL THOUGHTS
Our phylogenetic analysis suggests that two of the three most common
and persistent bacterial associates of A. maximus are unnamed species:
"Flexibacter cf. sancti" appears to be an earlier discovery of the same as yet
unnamed Chitinophaga from our experiments while our Pedobacter strains
appear to be a novel species within the genus. Our Ochrobactrum strains also
appear to be a new variant of Ochrobactrum tritici.
As discussed in the last chapter, the Pedobacter associate of A. maximus
is available in pure culture. This allows for the opportunity for the Pedobacter sp.
to be potentially named and characterized using the discussed phenotypic,
genotypic, and chemotaxonomic approaches.

58
REFERENCES 1. Altaba CR. 2009. Universal artifacts affect the branching of phylogenetic trees, not universal scaling laws. PLOS ONE. 4(2):e4611. 2. Anisimova M, Gascuel O.2006. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic Biology. 55(4):539-552. 3. Badger JH, Eisen JA, Ward NL. 2005. Genomic analysis of Hyphomonas neptunium contradicts 16S rRNA gene-based phylogenetic analysis: implications for the taxonomy of the orders 'Rhodobacterales' and Caulobacterales. International Journal of Systematic and Evolutionary Microbiology. 55(3):1021-1026. 4. Bathe S, Achouak W, Hartmann A, Heulin T, Schloter M, Lebuhn M. 2006. Genetic and phenotypic microdiversity of Ochrobactrum spp. FEMS Microbiology Ecology. 56(2):272-280. 5. Boto L. 2010. Horizontal gene transfer in evolution: facts and challenges. Proceedings of the Royal Society B: Biological Sciences. 277(1683):819- 827. 6. Buckley TR, Arensburger P, Simon C, Chambers GK. 2002. Combined data, Bayesian phylogenetics, and the origin of the New Zealand cicada genera. Systematic Biology. 51(1):4-18. 7. Del Rio TG, Abt B, Spring S, Lapidus A, Nolan M, Tice H, Copeland A, Cheng J-F, Chen F, Bruce D, Goodwin L, Pitluck S, Ivanova N, Mavromatis K, Mikhailova N, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang Y-J, Jeffries CD, Chain P, Saunders E, Detter JC, Brettin T, Rohde M, Göker M, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk H-P, Lucas S. 2010. Complete genome sequence of Chitinophaga pinensis type strain (UQM 2034T). Standards in Genomic Sciences. 2(1): 87–95. 8. Doolittle WF, Papke RT. 2006. Genomics and the bacterial species problem. Genome Biology. 7(9):116. 9. Douady CJ, Delsuc F, Boucher Y, Doolittle WF, Douzery EJ. 2003. Comparison of Bayesian and maximum likelihood bootstrap measures of phylogenetic reliability. Molecular Biology and Evolution. 20(2):248-254. 10. Fletcher W, Yang Z. 2010. The effect of insertions, deletions and alignment errors on the branch-site test of positive selection. Molecular Biology and Evolution. 27: 2257–2267. 11. Goris J, Boon N, Lebbe L, Verstraete W & De Vos P. 2003. Diversity of activated sludge bacteria receiving the 3-chloroaniline degradative plasmid pC1gfp. FEMS Microbiology Ecology. 46: 221–230. 12. Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. International Journal of Systematic and Evolutionary Microbiology. 57(1):81-91.

59
13. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology. 59(3):307-321. 14. Hollricher K. 2007. “Species Don‘t Really Mean Anything in the Bacterial World”. Lab Times. 5:22-25. 15. Huang X, Miller W. 1991. A Time-efficient, Linear-Space Local Similarity Algorithm. Advances in Applied Mathematics. 12: 373-381. 16. Huelsenbeck J, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17(8): 754-755. 17. Janda JM, Abbott SL. 2007. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. Journal of Clinical Microbiology. 45(9): 2761–2764. 18. Kämpfer P, Young CC, Sridhar KR, Arun AB, Lai WA, Shen FT, Rekha PD. 2006. Transfer of [Flexibacter] sancti, [Flexibacter] filiformis, [Flexibacter] japonensis and [Cytophaga] arvensicola to the genus Chitinophaga and description of Chitinophaga skermanii sp. nov. International Journal of Systematic and Evolutionary Microbiology. 56:2223–2228. 19. Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research. 30(14):3059–3066. 20. Kolbert CP, Persing DH. 1999. Ribosomal DNA sequencing as a tool for identification of bacterial pathogens. Current Opinion in Microbiology. 2:299–305. 21. Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proceedings of the National Academy of Sciences USA. 102(7): 2567–2572. 22. Konstantinidis KT, Ramette A, Tiedje JM. 2006. The bacterial species definition in the genomic era. Philosophical Transactions of the Royal Society B: Biological Sciences. 361(1475): 1929–1940. 23. Lapage SP, Sneath PHA, Lessel EF, et al., editors. 1992. International Code of Nomenclature of Bacteria (1990 Revision). Bacteriological Code. Washington, DC: American Society for Microbiology. 24. Laramée L, Lawrence JR, Greer CW. 2000. Molecular analysis and development of 16S rRNA oligonucleotide probes to characterize a diclofop-methyl-degrading biofilm consortium. Canadian Journal of Microbiology. 46(2):133-42. 25. Larget B, Simon DL. 1999. Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Molecular Biology and Evolution. 16: 750-759. 26. Lassmann T, Sonnhammer EL. 2005. Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 6:298.

60
27. Leaché AD, Reeder TW. 2002. Molecular systematics of the Eastern Fence Lizard (Sceloporus undulatus): a comparison of Parsimony, Likelihood, and Bayesian approaches. Systematic Biology . 51(1):44-68. 28. Lebuhn M, Achouak W, Schloter M, Berge O, Meier H, Barakat M, Hartmann A, Heulin T. 2000. Taxonomic characterization of Ochrobactrum sp. isolates from soil samples and wheat roots, and description of Ochrobactrum tritici sp. nov. and Ochrobactrum grignonense sp. nov. International Journal of Systematic and Evolutionary Microbiology. 50: 2207–2223. 29. Löytynoja A, Goldman N. 2010. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics. 11:579. 30. Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proceedings of the National Academy of Sciences USA. 95(6):3140-3145. 31. McClure MA, Vasi TK, Fitch WM. 1994. Comparative analysis of multiple protein-sequence alignment methods. Molecular Biology and Evolution. 11: 571–592. 32. Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology. 302(1):205-217. 33. Rainey FA, Oren A, editors. 2011. Taxonomy of Prokaryotes. Methods in Microbiology. Academic Press. 38:1-473. 34. Rosselló-Mora R, R Amann. 2001. The species concept for prokaryotes. FEMS Microbiology Letters. 25:39–67. 35. Sankhobol V, Skerman VBD. 1981. Chitinophaga, a new genus of chitinolytic myxobacteria. International Journal of Systematic Bacteriology. 31:285-293. 36. Smit S, Widmann J, Knight R. 2007. Evolutionary rates vary among rRNA structural elements. Nucleic Acids Research. 35(10):3339–3354. 37. Steyn PL, Segers P, Vancanneyt M, Sandra P, Kersters K, Joubert JJ. 1998. Classification of heparinolytic bacteria into a new genus, Pedobacter, comprising four species: Pedobacter heparinus comb. nov., Pedobacter piscium comb. nov., Pedobacter africanus sp. nov. and Pedobacter saltans sp. nov. proposal of the family Sphingobacteriaceae fam. nov. International Journal of Systematic Bacteriology. 48(1):165-177. 38. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, and Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular Biology and Evolution. 28: 2731-2739.

61
39. Thompson JD, Plewniak F, Poch O. 1999.BAliBASE: a benchmark alignment database for the evaluation of multiple alignment programs. Bioinformatics. 15(1):87-88. 40. Thompson JD, Koehl P, Ripp R, Poch O. 2005. BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins. 61(1):127-36. 41. Thompson JD, Linard B, Lecompte O, Poch O. 2011. A comprehensive benchmark study of multiple sequence alignment methods: current challenges and future perspectives. PLOS ONE. 6(3). 42. Tindall BJ, Rosselló-Móra R, Busse HJ, Ludwig W, Kämpfer P. 2010. Notes on the characterization of prokaryote strains for taxonomic purposes. International Journal of Systematic and Evolutionary Microbiology. 60(1):249-266. 43. Whittingham LA, Slikas B, Winkler DW, Sheldon FH. 2002. Phylogeny of the tree swallow genus, Tachycineta (Aves: Hirundinidae), by Bayesian analysis of mitochondrial DNA sequences. Molecular Phylogenetics and Evolution. 22(3):430-41. 44. Wilcox TP, Zwickl DJ, Heath TA, Hillis DM. 2002. Phylogenetic relationships of the dwarf boas and a comparison of Bayesian and bootstrap measures of phylogenetic support. Molecular Phylogenetics and Evolution. 25(2):361-71. 45. Woese CR. 1987. Bacterial evolution. Microbiological Reviews. 51(2):221- 271. 46. Zhang K, Tang Y, Zhang L, Dai J, Wang Y, Luo X, Liu M, Luo G, Fang C. 2009. Parasegetibacter luojiensis gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from a forest soil. International Journal of Systematic and Evolutionary Microbiology. 59(12):3058-3062.

62
FIGURES AND TABLES Table 1A. Ochrobactrum Sequences Used In Phylogenetic Analyses
Table 1B. Pedobacter Sequences Used In Phylogenetic Analyses
Clone Sequences Relevant Sequences from GenBank KC110896, KC110901, KC110903, KC110910, KC110914, KC110916, KC110918, KC110923, KC110926, KC110929, KC110931, KC110932, KC110935, KC110936, KC110939, KC110940, KC110941, KC110944, KC110949, KC110951, KC110956, KC110957, KC110960, KC110961, KC110962, KC110970, KC110973, KC110975, KC110976, KC110980, KC110985
AB091758, AB681865, AF508089, AF508090, AF508091, AF508092, AJ242579, AJ864999, AJ865000, AM114403, AM490634, AM490635, AM490636, AY429607, DQ647056, EU999218, GU171382, GU345782, JX010950, JX393006, JX393007, JX393011, NR028902, NR026039, NR042447, U88440 Outgroup sequences: L26166, L26168, L37584, NR042460, NR042462, NR042463, X13695
Clone Sequences Relevant Sequences from GenBank KC110895, KC110904, KC110905, KC110909, KC110913, KC110917, KC110919, KC110921, KC110933, KC110934, KC110947, KC110958, KC110963, KC110966, KC110967, KC110968, KC110977, KC110978, KC110983, KC110984, KC119214, KC157040
AB365796, AB680215, AB680732, AB680733, AB680841, AB681143, AB681144, AB681397, AB682400, AJ438170, AM237384, AM279214, AM279215, AM279217, AM279218, AM491368, EF660750, EF660752, EF693742, EU169155, EU734803, EU834277, FJ263019, GQ225687, GU391547, HM051286, HQ824869, JF915326, JQ236820, JQ342863, JX122142, JX122157, JX133186, JX401449, JX827217, JX869971, JX971539, JX971546, JX971550, JX971552, NR025534, NR025535, NR025536, NR028977, NR041371, NR041374, NR041506, NR041507, NR042133, NR042204, NR042323, NR042446, NR042601, NR042602, NR042603, NR042604, NR042605, NR043538, NR043543, NR043555, NR043665, NR044005, NR044083, NR044218, NR044219, NR044339, NR044380, NR044381, NR044382 Outgroup sequences: AJ459411, NR025537, NR042134, NR042135, NR044404

63
Table 1C. Chitinophaga Sequences Used In Phylogenetic Analyses
Clone Sequences Relevant Sequences from GenBank KC110897, KC110907, KC110908, KC110915, KC110943, KC110950, KC110955, KC110965, KC110971, KC110981
AB078049, AB078066, AB078067, AB078068, AB267723, AB267724, AB278570, AB680728, AB680761, AB680762, AB681008, AB681009, AB681041, AB681043, AB681047, AB681048, AB681049, AB681050, AB681051, AB681052, AB681053, AB682428, AB682429, AF181568, AM237311, AM237312, AM237313, AM237314, AM237315, JQ659521, M62795, NR028638, NR028720, NR040909, NR040917, NR041375, NR041486, NR042512, NR043516, NR044559, NR044560, Outgroup sequences: AB682366, GQ421847, KC110912, KC110922, NR041501, FN665659, FN665660, FN665661, KC110902, KC110979, NR044576, KC110946, AB682649, AB682408, AB682409, NR043672, NR043673

64
Table 2 Pedobacter Settings For Phylogenetic Trees
Multiple Sequence Alignment
Tool
Tree Building
Algorithm Substitution
Model Rates
Variation Gamma
Categories Branch Support
T-Coffee Maximum Likelihood K80
Gamma Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
T-Coffee PhyML K80 Uniform Rates 4 aLRT (SH-
like supports)
T-Coffee Mr. Bayes HKY85 Gamma
Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
MAFFT Maximum Likelihood K80
Gamma Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
MAFFT PhyML K80 Uniform Rates 4 aLRT (SH-
like supports)
MAFFT Mr. Bayes HKY85 Gamma
Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
Kalign Maximum Likelihood K80
Gamma Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
Kalign PhyML K80 Uniform Rates 4 aLRT (SH-
like supports)
Kalign Mr. Bayes HKY85 Gamma
Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
WebPRANK Maximum Likelihood K80
Gamma Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
WebPRANK PhyML K80 Uniform Rates 4 aLRT (SH-
like supports)
WebPRANK Mr. Bayes HKY85 Gamma
Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)

65
Table 3. Ochrobactrum Settings For Phylogenetic Trees
Multiple Sequence Alignment
Tool
Tree Building
Algorithm Substitution
Model Rates
Variation Gamma
Categories Branch Support
T-Coffee Maximum Likelihood GTR Uniform
Rates 5 Bootstrap
(1000 replicates)
T-Coffee PhyML GTR Uniform Rates 4
aLRT (SH-like
supports)
T-Coffee Mr. Bayes GTR Uniform Rates 5
Bootstrap (1000
replicates)
MAFFT Maximum Likelihood K80 Gamma
Distributed 5 Bootstrap
(1000 replicates)
MAFFT PhyML K80 Uniform Rates 4
aLRT (SH-like
supports)
MAFFT Mr. Bayes HKY85 Gamma Distributed 5
Bootstrap (1000
replicates)
Kalign Maximum Likelihood HKY85 Gamma
Distributed 5 Bootstrap
(1000 replicates)
Kalign PhyML HKY85 Uniform Rates 4
aLRT (SH-like
supports)
Kalign Mr. Bayes HKY85 Gamma Distributed 5
Bootstrap (1000
replicates)
WebPRANK Maximum Likelihood K80 Gamma
Distributed 5 Bootstrap
(1000 replicates)
WebPRANK PhyML K80 Uniform Rates 4
aLRT (SH-like
supports)
WebPRANK Mr. Bayes HKY85 Gamma Distributed 5
Bootstrap (1000
replicates)

66
Table 4. Chitinophaga Settings For Phylogenetic Trees
Multiple Sequence Alignment
Tool
Tree Building
Algorithm Substitution
Model Rates
Variation Gamma
Categories Branch Support
T-Coffee Maximum Likelihood GTR Uniform Rates 5
Bootstrap (1000
replicates)
T-Coffee PhyML GTR Uniform Rates 4 aLRT (SH-
like supports)
T-Coffee Mr. Bayes GTR Uniform Rates 5 Bootstrap
(1000 replicates)
MAFFT Maximum Likelihood K80
Gamma Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
MAFFT PhyML K80 Uniform Rates 4 aLRT (SH-
like supports)
MAFFT Mr. Bayes HKY85 Gamma
Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
Kalign Maximum Likelihood K80
Gamma Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
Kalign PhyML K80 Uniform Rates 4 aLRT (SH-
like supports)
Kalign Mr. Bayes HKY85 Gamma
Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
WebPRANK Maximum Likelihood K80
Gamma Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)
WebPRANK PhyML K80 Uniform Rates 4 aLRT (SH-
like supports)
WebPRANK Mr. Bayes HKY85 Gamma
Distributed with Invariant Sites
5 Bootstrap
(1000 replicates)

67

68
Figure 1. Ochrobactrum: T-Coffee-PhyML Phylogram

69

70
Figure 2. Ochrobactrum: MAFFT-ML Phylogram

71

72
Figure 3. Ochrobactrum: Kalign-MrBayes Phylogram

73

74
Figure 4. Chitinophaga: T-Coffee-ML Phylogram

75

76
Figure 5. Chitinophaga: MAFFT-ML Phylogram

77
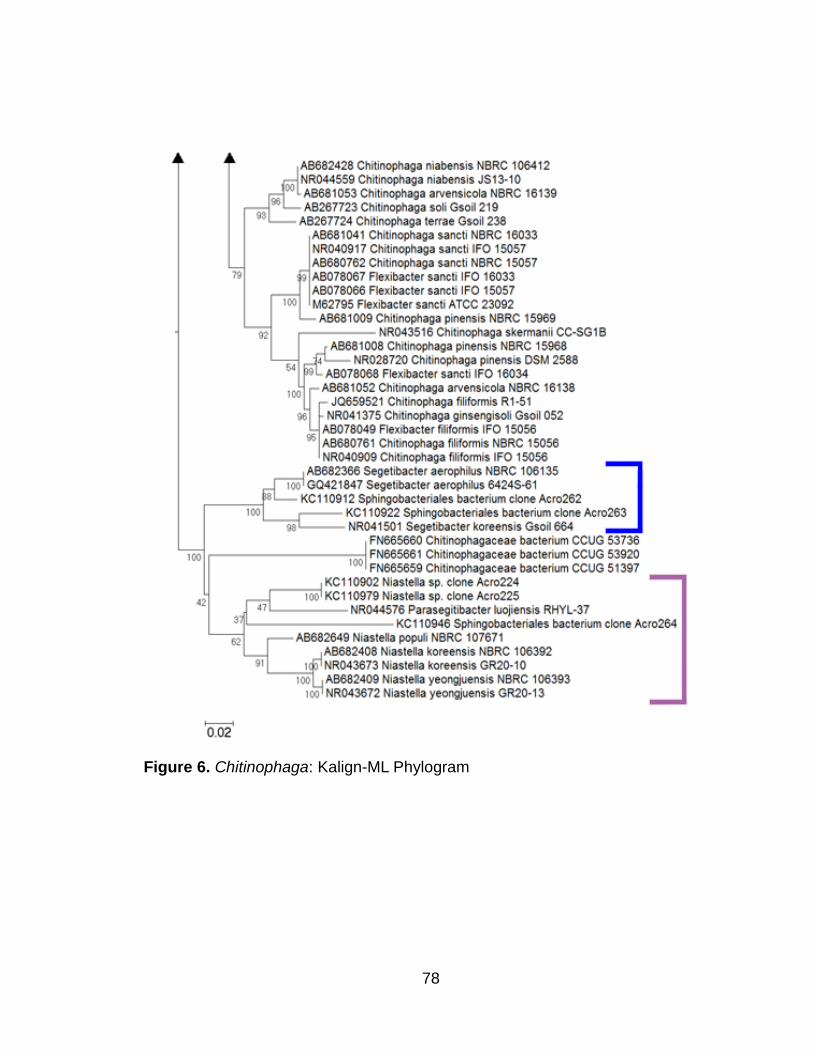
78
Figure 6. Chitinophaga: Kalign-ML Phylogram

79

80
Figure 7. Pedobacter : T-Coffee-PhyML Phylogram

81

82
Figure 8. Pedobacter : MAFFT-PhyML Phylogram

83

84
Figure 9. Pedobacter : Kalign-PhyML Phylogram
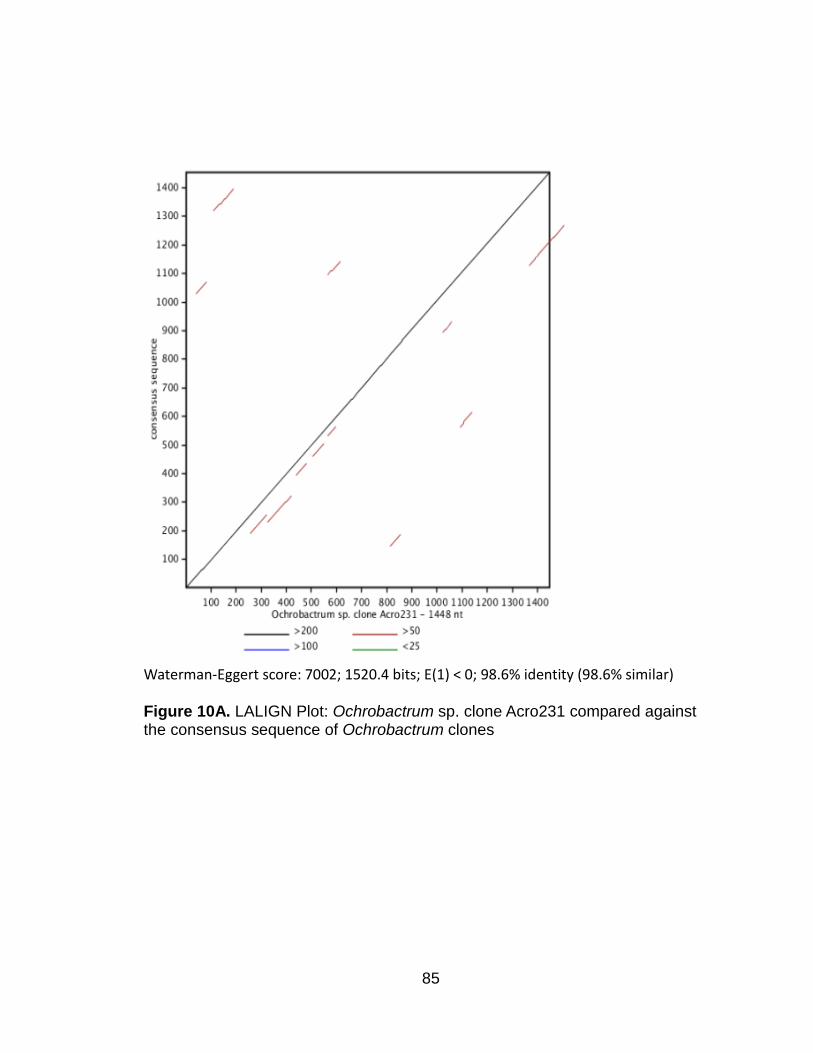
85
Waterman-Eggert score: 7002; 1520.4 bits; E(1) < 0; 98.6% identity (98.6% similar) Figure 10A. LALIGN Plot: Ochrobactrum sp. clone Acro231 compared against the consensus sequence of Ochrobactrum clones

86
Waterman-Eggert score: 6666; 1540.9 bits; E(1) < 0; 94.3% identity (94.3% similar) Figure 10B . LALIGN Plot: Chitinophaga sp. clone Acro210 compared against the consensus sequence of Chitinophaga clones

87
Waterman-Eggert score: 6532; 1447.5 bits; E(1) < 0; 94.0% identity (94.0% similar) Figure 10C. LALIGN Plot: of Pedobacter sp. clone Acro279 compared against the consensus sequence of Pedobacter clones

88
CONCLUDING COMMENTS
BACTERIAL COMMUNITY IDENTIFICATION: 16S rRNA GENE MARKER SURVEY VERSUS SHOTGUN METAGENOMICS
The following discussion will compare differences between 16S rRNA gene
marker surveys and shotgun metagenomics, including the advantages, pitfalls,
and considerations to be taken before deciding which method to utilize in a
bacterial community identification study. The primary difference between the two
methods is that the 16S rRNA gene survey focuses on a single gene to identify
members of a bacterial community, whereas metagenomics focuses on the
totality of all genomic information in a sample. The first step involved in any
bacterial community identification project is to strategically plan what questions
are to be answered and the goals to be accomplished, then to decide which of
these methods are most applicable given time considerations and financial
resources available.
16S rRNA Gene Marker Surveys
The 16S rRNA gene is about 1500 nucleotides long and has become the
marker of choice for profiling bacterial communities since the mid-1990s (18).
There are universal primers available that allow for near full-length amplification
of the 16S rRNA gene (14). The advantage of having these universal primers for
amplification is that it permits for simple gene library construction and

89
sequencing, allowing for rapid identification of unknown isolates. A query
sequence can be input into NCBI’s BLAST to yield fast approximate matching or
exact identification without the need for additional laboratory work (1).
Furthermore, separate forward and reverse sequencing reactions are possible
using each PCR primer as well as multiple robust forward or internal primers,
leading to more refined sequence data in terms of both accuracy and ability to
generate robust assemblies of the entire gene.
On the other hand, single gene approaches are hampered by various
limitations. A common problem associated with creating clone libraries of PCR-
amplified 16S rRNA genes is that it may not give an accurate representation of
the microbial community. High species diversity, taxon-specific amplification
problems, and undersampling can lead to some members of the microbial
community not being represented in the clone library (2, 7, 27). Another
drawback is that copy number of the 16S rRNA gene varies between bacterial
species. Species with low abundance of the gene may not be as readily amplified
during PCR as those with a high copy number, creating an amplification bias (9,
24). Despite these minor drawbacks, 16S rRNA gene surveys continue to be
performed to determine the microbial community composition in a variety of
samples and substrates (4, 11, 21).

90
Shotgun Metagenomics
Shotgun metagenomics involves using high throughput sequencing or
massively parallel pyrosequencing to sequence DNA directly from environmental
samples in order to study community structure and microbial biodiversity (5). As
with single-gene methods, shotgun metagenomics allows for the discovery of
bacterial species that cannot be cultured in the lab and as an additional
advantage it can aid in elucidating the genomes of unculturable species provided
those are represented with high abundance in the sample (23). Common
problems associated with shotgun metagenomics are that it applies a general
DNA extraction method to a sample containing many microbial organisms and
that a sample with low biomass may yield insufficient quantities of DNA for proper
analysis, letting rare species go undetected (13).
A limitation of shotgun metagenomics is that it is still very challenging and
cost-prohibitive to sequence microbial communities that are closely associated
with eukayotes, due to large eukaryote genome size and low microbial gene
coding density (13). During post-sequencing it is often difficult to separate
eukaryotic DNA from DNA belonging to the microbial community. This is one of
the reasons why we opted to perform a 16S rRNA survey of the bacterial
community associated with A. maximus rather than take the shotgun
metagenomics approach. In addition, while the wet lab chemistry steps can be
performed more efficiently and much faster relative to total species richness,
shotgun metagenomics presents many downstream bioinformatics challenges in

91
processing and robust assembling of short sequencing reads into longer contigs
(19). Chimeric sequences caused by PCR amplification, intracellular sequence
polymorphisms, pseudogenes, sequence contamination, and sequencing errors
continue to pose particular challenges to assembly algorithms and procedures for
assigning contigs to operational taxonomic units (20).
PCR-amplified 16S rRNA and Pyrosequencing
Recently pyrosequencing techniques have been applied to PCR-amplified
16S rRNA, bypassing the need to create traditional clone libraries (13). This low-
cost high-throughput technology is allowing for a rapid increase in the amounts of
16S rRNA being generated. Traditional PCR cloning methods of the past looked
at only a few dozen 16S rRNA sequences, whereas it is now possible to survey
thousands of 16S rRNA sequences at a time (26).
Pyrosequencing technology, such as the 454 GS FLX Titanium XL+
System developed by Roche, allows for generation of about 1,000,000 shotgun
reads per run, produces average read lengths up to 1,000 base pairs, and yields
about 700 Mbs per run. Past problems with pyrosequencing technologies are that
they produce short sequence read lengths ranging from 250 to 500 base pairs;
reads of less than 500 base pairs are insufficient for phylogenetic analysis as
they cannot be used to discern taxnonomic information down to the genus or
species level (24). Other drawbacks of pyrosequencing include the high cost of
reagents and high error rates associated with homopolymer repeats (15). Despite

92
these drawbacks, the long read lengths and deep sequencing capabilities of the
454 GS FLX Titanium XL+ have made it one of the most widely utilized
sequencing technologies (10). Other popular second generation technologies,
like the Illumina HiSeq 2500 are not ideal for 16S rRNA microbial community
analysis. While the Illumina HiSeq 2500 can generate more than ten times the
number of reads per run of the 454 GS FLX Titanium XL+, it suffers from short
read lengths of about 100 nucleotides (3).
Sequencing Technologies and Future Considerations
The field of metagenomics is still in its infancy, however sequencing
technologies and bioinformatics tools are developing at a rapid rate to
accommodate the challenges of obtaining and analyzing large volumes of data in
meaningful ways (8).
As third generation sequencing technologies develop and emerge, second
generation technologies continue to improve in throughput and reliability, while
becoming more cost-efficient. Second generation sequencing technologies
developed by Roche, Illumina, Applied Biosystems, and IonTorrent make use of
many individual cloned DNA fragments which are massively sequenced in
parallel using enzymatic replication, whereas third generation sequencing
technologies being developed by Helicos and Pacific Biosciences utilize single
molecule sequencing methods (16). The Pacific Biosciences single molecule real
time (SMRT) sequencing platform is the first sequencing technology to offer read

93
lengths averaging between 2000 to 3000 base pairs, however it suffers from error
rates much higher than those seen in the second generation technologies,
making it as of this writing not ideal for 16S rRNA microbial community surveys
(6).
If the third generation single molecule sequencing platforms overcome
their high error rate hurdles, they might be able to supersede the second
generation pyrosequencing technologies for microbial community studies. In
addition to the much longer read lengths and bypassing the biases associated
with an enzymatic amplification step, other advantages single molecule
sequencing has over massively parallel sequencing is the ability to sequence the
same DNA molecule multiple times for improved accuracy and the ability to easily
sequence molecules with high GC content, homopolymer regions, and complex
secondary structure (22). Detection of base modifications, methylation, and
single nucleotide polymorphisms can be achieved much more readily with third
generation single molecule sequencing platforms than with second generation
sequencing technologies; recently the methylomes of six different bacteria was
elucidated using SMRT sequencing (17). Ideally, single molecule sequencing
could develop to the point where complete microbial genomes can be
reconstructed from a single fragment library, greatly reducing the time and cost
associated with post-sequencing assembly (12).
Another equally likely possibility is that as second generation
pyrosequencing technologies continue to decrease in cost and increase in

94
throughput, they will make the development of these third generation sequencing
platforms practically moot. Researchers of tomorrow have many considerations
to take into account: the importance of sequencing longer reads, the potential for
sequencing whole DNA strands, and cost-benefit analysis of chasing third and
fourth generation sequencing technologies versus using new improvements
associated with second generation technologies.

95
REFERENCES
1. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. Journal of Molecular Biology. 215(3):403-410. 2. Baker GC, Smith JJ, Cowan DA. 2003. Review and re-analysis of domain- specific 16S primers. Journal of Microbiological Methods. 55(3):541-555. 3. Claesson MJ, Wang Q, O'Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O'Toole PW. 2010. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Research. 38(22):e200. 4. Davidson SK, Powell R, James S. 2013. A global survey of the bacteria within earthworm nephridia. Molecular Phylogenetics and Evolution. 67(1):188-200. 5. Eisen JA. 2007. Environmental shotgun sequencing: its potential and challenges for studying the hidden world of microbes. PLOS Biology. 5(3):e82. 6. English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, Gibbs RA. 2012. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLOS ONE. 7(11):e47768. 7. Forney LJ, Zhou X, Brown CJ. 2004. Molecular microbial ecology: land of the one-eyed king. Current Opinion in Microbiology. 7(3):210-220. 8. Glenn TC. 2011. Field guide to next-generation DNA sequencers. Molecular Ecology Resources. 11(5):759-769. 9. Gonzalez JM, Portillo MC, Belda-Ferre P, Mira A. 2012. Amplification by PCR artificially reduces the proportion of the rare biosphere in microbial communities. PLOS ONE. 7(1):e29973. 10. Hall N. 2007. Advanced sequencing technologies and their wider impact in microbiology. Journal of Experimental Biology. 210(9):1518-1525. 11. Han Y, Li L, Liu J. 2013. Characterization of the airborne bacteria community at different distances from the rotating brushes in a wastewater treatment plant by 16S rRNA gene clone libraries. Journal of Environmental Sciences. 25(1):5-15

96
12. Koren S, Harhay GP, Smith TPL, Bono JL, Harhay DM, Mcvey DS, Radune D, Bergman NH, Phillippy AM. 2013. Reducing assembly complexity of microbial genomes with single-molecule sequencing. eprint arXiv:1304.3752. 13. Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P. 2008. A bioinformatician's guide to metagenomics. Microbiology and Molecular Biology Reviews. 72(4):557-578. 14. Lane DJ. 1991. 16S/23S rRNA sequencing. Nucleic acid techniques in bacterial systematics. Stackebrandt, E., and Goodfellow, M., eds., John Wiley and Sons. New York, NY.115-175. 15. Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT. 2012. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLOS ONE. 7(2):e30087. 16. McGinn S, Gut IG. 2013. DNA sequencing - spanning the generations. New Biotechnology. 30(4):366-372. 17. Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. 2012. The methylomes of six bacteria. Nucleic Acids Research. 40(22):11450-11462. 18. Muyzer G, de Waal EC, Uitterlinden AG. 1993. Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S rRNA. Applied and Environmental Microbiology. 59(3): 695–700. 19. Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. 2009. Metagenomic pyrosequencing and microbial identification. Clinical Chemistry. 55(5):856-866. 20. Quince C, Lanzén A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT. 2009. Accurate determination of microbial diversity from 454 pyrosequencing data. Nature Methods. 6(9):639-641. 21. Ren T, Glatt DU, Nguyen TN, Allen EK, Early SV, Sale M, Winther B, Wu M. 2013. 16S rRNA survey revealed complex bacterial communities and evidence of bacterial interference on human adenoids. Enviornmental Microbiology. 15(2):535-547. 22. Schadt EE, Turner S, Kasarskis A. 2010. A window into third-generation sequencing. Human Molecular Genetics. 19(R2):R227-240. 23. Streit WR, Schmitz RA. 2004. Metagenomics – the key to the uncultured microbes. Current Opinion in Microbiology 7:492-498.

97
24. Suzuki MT, Giovannoni SJ. 1996. Bias Caused by Template Annealing in the Amplification of Mixtures of 16S rRNA Genes by PCR. Applied and Enivornmental Microbiology. 62(2):625-630. 25. Tamaki H, Wright CL, Li X, Lin Q, Hwang C, Wang S, Thimmapuram J, Kamagata Y, Liu WT. 2011. Analysis of 16S rRNA amplicon sequencing options on the Roche/454 next-generation titanium sequencing platform. PLOS ONE. 6(9):e25263. 26. Tringe SG, Hugenholtz P. 2008. A renaissance for the pioneering 16S rRNA gene. Current Opinion in Microbiology. 11(5):442-446. 27. Wintzingerode FV, Göbel UB, Stackebrandt E. 2006. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiology Reviews. 21(3):213-229.




















