
Thermally induced self-assembly of cylindrical nanodomains in low molecular weight PS- b -PMMA thin...
7
This content has been downloaded from IOPscience. Please scroll down to see the full text. Download details: IP Address: 193.205.155.85 This content was downloaded on 27/05/2014 at 08:46 Please note that terms and conditions apply. Thermally induced self-assembly of cylindrical nanodomains in low molecular weight PS-b- PMMA thin films View the table of contents for this issue, or go to the journal homepage for more 2014 Nanotechnology 25 045301 (http://iopscience.iop.org/0957-4484/25/4/045301) Home Search Collections Journals About Contact us My IOPscience
Transcript of Thermally induced self-assembly of cylindrical nanodomains in low molecular weight PS- b -PMMA thin...

This content has been downloaded from IOPscience. Please scroll down to see the full text.
Download details:
IP Address: 193.205.155.85
This content was downloaded on 27/05/2014 at 08:46
Please note that terms and conditions apply.
Thermally induced self-assembly of cylindrical nanodomains in low molecular weight PS-b-
PMMA thin films
View the table of contents for this issue, or go to the journal homepage for more
2014 Nanotechnology 25 045301
(http://iopscience.iop.org/0957-4484/25/4/045301)
Home Search Collections Journals About Contact us My IOPscience

Nanotechnology
Nanotechnology 25 (2014) 045301 (6pp) doi:10.1088/0957-4484/25/4/045301
Thermally induced self-assembly ofcylindrical nanodomains in low molecularweight PS-b-PMMA thin films
Gabriele Seguini1, Tommaso J Giammaria1,2, Federico Ferrarese Lupi1,Katia Sparnacci2, Diego Antonioli2, Valentina Gianotti2, Francesco Vita3,Immacolata F Placentino3, Jan Hilhorst4, Claudio Ferrero4,Oriano Francescangeli3, Michele Laus2 and Michele Perego1
1 Laboratorio MDM, IMM-CNR Via C. Olivetti 2, I-20864 Agrate Brianza, Italy2 Dipartimento di Scienze e Innovazione Tecnologica (DISIT), Universita del Piemonte Orientale‘A. Avogadro’, INSTM, UdR Alessandria, Viale T. Michel 11, I-15121 Alessandria, Italy3 Dipartimento di Scienze e Ingegneria della Materia, dell’Ambiente ed Urbanistica and CNISM,Universita Politecnica delle Marche, Via Brecce Bianche, I-60131 Ancona, Italy4 European Synchrotron Radiation Facility, Boıte Postale 220, F-38043 Grenoble, France
E-mail: [email protected]
Received 9 September 2013, revised 30 October 2013Accepted for publication 11 November 2013Published 6 January 2014
AbstractThe phase behaviour in thin films of an asymmetric polystyrene-b-polymethylmethacrylate(PS-b-PMMA) block copolymer with a molecular weight of 39 kg mol−1 was assessed at awide range of temperatures and times. Cylindrical PMMA structures featuring a diameterclose to 10 nm and perpendicularly oriented with respect to the substrate were obtained at180 ◦C in relatively short annealing times (t ≤ 30 min) by means of a simple thermaltreatment performed in a standard rapid thermal processing machine.
Keywords: PS-b-PMMA, block copolymer, rapid thermal processing (RTP), self-assembling,nanolithography
(Some figures may appear in colour only in the online journal)
1. Introduction
Block copolymers (BCPs) consisting of two covalently end-joined homopolymers [1] can phase-segregate, giving rise toa variety of ordered self-assembled domains whose structureand inherent periodicity (L0) are controlled by the relativevolume fraction (f ) and chain length (N) of the two chemicallydifferent blocks [2]. This peculiar behaviour makes the BCPsa paradigmatic system for investigating the interplay betweenthe information encoded in the molecular characteristics of themacromolecule and the process parameters within the spatialand thermal boundaries dictated by the system dimensionality(bulk versus thin film) [3].
The self-assembling phase separation originates fromthe competition between the repulsive driving force and the
chain packing resistance. The critical dimension, for whichthe former is not enough to counter the latter, is dictated bythe strength of the repulsive interaction between the blocksand is characterized by the product χN, where χ is theFlory–Huggins parameter [4] and N is the polymer chainlength [5]. The minimum achievable L0 depends on N muchmore strongly than on χ . Thus, the most efficient way toreduce L0 is to decrease N while maintaining the product χNlarger than the χN value at the order–disorder transition [1].
The self-assembly of BCPs into a nanometric orderedstructure and the chemical similarity with the photoresistmaterials employed in standard photolithography havetriggered an in-depth investigation of the application ofBCPs as template masks [6]. In particular, BCPs formingcylindrical domains perpendicularly oriented with respect
10957-4484/14/045301+06$33.00 c© 2014 IOP Publishing Ltd Printed in the UK

Nanotechnology 25 (2014) 045301 G Seguini et al
to the substrate are particularly interesting because theselective etching of the cylinder-building component resultsin an array of hexagonally packed nanopores. This domainstructure provides a template for additive or subtractivenanofabrication strategies leading to dots and holes. Thecylindrical nanodomain morphology is also attractive forother applications such as electrical biomolecule detection [7].For all of these applications, the fabrication of smallerand smaller pores is required. The challenging need todrive the pore sizes close to or below 10 nm diametershould be addressed by finely tuning fundamental BCPmolecular properties such as low N or high χ togetherwith a good etch selectivity between the blocks [8]. Equallyimportant is the control of the orientation of BCP domainswith respect to the substrate, especially the propensity toform perpendicularly oriented nanodomains. Finally, thecommensurability between the film thickness and L0, as wellas the BCP interaction with the interfaces, substrate and air,poses additional constraints [9].
A proper selection of the polymer properties is crucial forthe macromolecular design in order to achieve an optimumtrade-off among the various and sometimes conflictingrequirements. Whilst hybrid organic–inorganic BCPs, suchas polystyrene-b-polydimethylsiloxane (PS-b-PDMS) [10],are appealing because of the large χ parameter (≈0.12 at150 ◦C) [9] and high etch selectivity between the blocks,it is difficult to obtain a perpendicular orientation of themicrodomains since there is a large difference betweenthe PS and PDMS surface energies. This mismatch givesrise to a preferential wetting of PDMS on both airand substrate interfaces. Another strategy for providing apronounced differential etch resistance and for achieving,at the same time, domain orientation control is theincorporation of an organometallic block, like polystyrene-b-polyferrocenyldimethylsilane (PS-b-PFS) [11]. On the otherhand, the use of organic BCPs with high χ (≈0.12 at 150 ◦C),such as polystyrene-b-poly(2-vinylpyridine) (PS-b-P2VP) [9,12], is disadvantageous owing to the low etch selectivity.
Furthermore, organic BCPs with good selectivity, suchas the widely studied polystyrene-b-polymethylmethacrylate(PS-b-PMMA), can form perpendicularly oriented nan-odomains since the weakly unbalanced surface interactionsare easily neutralized by grafting the appropriate P(S-r-MMA) random copolymer (RCP) to the surface. In thiscase, the lower bound to the domain sizes is determinedby the low χ (≈0.04 at 150 ◦C) [13]. Actually, in thissystem, it is quite difficult to decrease N in order to reduceL0, whilst maintaining χN high enough to achieve theself-assembling phase separation. It is worth noting thatthe minimum achievable diameter (d) for PS-b-PMMA waspredicted to be d ≈ 13 nm [9]. Experimentally, hexagonallypacked PMMA cylinders with d ≈ 14 nm have been obtainedonly by means of an electric-field-assisted thermal treatmentusing a PS-b-PMMA featuring a molecular weight (Mn)of 42 kg mol−1 [5]. Up to now, various strategies havebeen explored to overcome this size limit. The chemicalproperties of the constituent blocks have been modifiedfollowing different approaches [3, 6]. The sizes have
been changed, adjusting the RCP composition [14]. In thepostprocessing, the nanofeatures have been modified byoveretching [4], atomic layer deposition [15], or sequentialinfiltration synthesis [16, 17]. In comparison, the simpledecrease of N toward the lowest limit has received littleattention.
The present study is focused on the development of asimple and fast way to address the 10 nm diameter challengeusing low molecular weight PS-b-PMMA BCPs, that isby reducing N. In particular, the self-assembly and phasebehaviour characteristics of an asymmetric PS-b-PMMAwith Mn = 39 kg mol−1 were investigated using therapid thermal processing (RTP) technology. The successfuldeployment of RTP has been recently demonstrated todisclose unprecedented opportunities for manipulating andtuning the BCP self-assembling process owing to its precisetemperature control and extremely fast (up to 50 ◦C s−1)heating rate. Nanodomain orientation in thin films ofPS-b-PMMA (with Mn = 67 kg mol−1) was achieved withina few seconds without the assistance of external solventsor force fields, thus suggesting that RTP could become theleading technology for the exploitation of self-assemblingmaterials [18].
2. Experimental details
After cleaning of the SiO2-covered Si substrates with Piranhasolution (H2SO4:H2O2, 3:1, 80 ◦C, 40 min), a solution of18 mg P(S-r-MMA) RCP (fS = 62%, Mn = 13.5 kg mol−1,PDI = 1.26, Polymer Source) in 2 ml toluene was spun ontothe substrates (area ≈ 1 cm2) for 30 s at 3000 rpm. Allthe samples were then RTP-heated (heating rate 20 ◦C s−1)under N2 atmosphere, and maintained at the final temperature(T = 250 ◦C) for 10 min. This treatment allows achievingthe grafting of the OH end groups of the RCP chainsonto the activated SiO2, leading to the neutralization of thesubstrate surface. BCP films 30 nm thick were depositedon the neutralized substrates by spin coating a solution ofasymmetric PS-b-PMMA (fS = 68.7%, Mn = 39 kg mol−1,PDI = 1.07, Polymer Source) in toluene. In order topromote the self-organization of the PS-b-PMMA film intohexagonal arrangements of PMMA cylinders in the PSmatrix, the samples were RTP-heated (heating rate 20 ◦C s−1)under N2 atmosphere (N2 flux 1000 sccm) to the finalannealing temperature (TA), maintained at this temperaturefor well-defined time periods (tA) and then fast cooled down toroom temperature. Annealing temperatures TA ranging from160 to 200 ◦C, and annealing times tA lying between 1 and30 min were chosen.
The thickness of the polymeric films was measured via aM-200U spectroscopic ellipsometer (J A Wollam Co.) usinga Xe lamp at 70◦ incident angle. The thermal analysis wasperformed using a Mettler Toledo TGA/SDTA 851e apparatusin N2 atmosphere. The morphology of the BCP films wasdetermined by scanning electron microscopy (SEM; ZeissSupra 40 SEM). A selective removal of the cylindrical PMMAblocks was performed to enhance the contrast of the PS maskfor SEM and grazing incidence small angle x-ray scattering
2

Nanotechnology 25 (2014) 045301 G Seguini et al
Figure 1. TGA (left scale) and DTG (right scale) curves at20 ◦C min−1 heating rate for bulk PS-b-PMMA, Mn = 39 kg mol−1,under N2 atmosphere.
(GISAXS) characterizations. The GISAXS measurementswere carried out at the ID01 beamline of the EuropeanSynchrotron Radiation Facility (ESRF, Grenoble, France).The operating conditions were: incident wavelength λ =
0.155 nm, sample-to-detector distance D = 0.830 m, beamsize 0.1×0.1 mm2. Measurements were performed at incidentangles 0.12◦, i.e. below the measured critical angle αc =
0.136◦, and 0.15◦ in order to explore the morphology acrossthe entire film thickness. 2D patterns were recorded usingthe MAXIPIX fast-readout photon-counting pixel detector(516 × 516 pixels, pixel size = 55 µm) positioned at the endof a vacuum tube.
3. Results and discussion
In general, the lower temperature limit for the self-assemblyof the BCPs is the glass transition temperature (TG) andthe upper one is the degradation temperature (TD). TDwas measured by thermogravimetric analysis, as reported infigure 1, for the bulk BCP at a heating rate of 20 ◦C min−1
in N2 atmosphere. No detectable weight loss was observedup to about 300 ◦C, where a single loss, extending overa temperature range from 300 ◦C to about 450 ◦C, wasobserved. These results indicate that TD is actually above250 ◦C. According to the data supplied by the producer, TG forthe PS block is 101 ◦C while for the PMMA block is 122 ◦C.Therefore, the temperature range between 160 and 200 ◦C canbe considered to be well above TG and far below TD.
Accordingly, in the first set of experiments, TA was variedfrom 160 to 200 ◦C whereas tA was kept constant at tA =15 min. The morphology, developed at each specific TA, wasfrozen by rapidly cooling the system [18]. The SEM imagesof the film annealed at different temperatures are displayed infigure 2. At TA = 160 ◦C, the BCP morphology is the typicalone for a disordered state, thus indicating that this temperature
Figure 2. SEM images of the self-assembled PS-b-PMMA,Mn = 39 kg mol−1, as a function of TA, for tA = 15 min.
is too low to allow the block copolymer flow, leading toa rearrangement of the disordered structure frozen at roomtemperature during the spinning process. On the other hand, at
3
Nanotechnology 25 (2014) 045301 G Seguini et al
TA = 200 ◦C, the ordered phase vanishes, indicating that thistemperature is very close to the order-to-disorder transition(ODT) temperature. Within this temperature gap, when TAlies between 170 and 190 ◦C, the SEM images provideclear evidence of the perpendicularly oriented cylindricaldomains originating from the phase separation. Indeed,for TA = 170 ◦C the cylinder structure is very defective.Uniformly sized PMMA cylinders are clearly detectable buttheir in-plane order is only short ranged. Upon annealingat 180 and 190 ◦C, the PMMA cylinders self-organize on alarger length scale and form a two-dimensional hexagonallattice [19]. A careful investigation of the SEM images of thetwo samples annealed at 180 and 190 ◦C indicates that, apartfrom structural defects in the hexagonal arrangement, someof the pores in the lattice were not completely open. Thesedefective elements were already observed in samples preparedusing widely investigated BCPs (with Mn = 67 kg mol−1).A careful optimization of the PMMA block removal process,which is beyond the scope of the present work, is necessary tofully eliminate these defects [14].
In order to measure the characteristic size of theself-organized cylinders a twofold investigation on the sampleannealed at 180 ◦C for 15 min was performed. L0 and dfor the PMMA cylinders in the PS matrix were determinedby quantitative analysis of the SEM images. The d valuewas found to be 12.0 ± 2.0 nm, while the in-plane spacingL0 was evaluated to be 24.0 ± 1.0 nm. The latter wasdetermined by the GISAXS measurements as well. GISAXSmeasurements allow probing the morphology throughout theentire film thickness and extracting structural parameters withhigh accuracy. Furthermore, the x-ray beam in GISAXSexperiments covers large regions (square millimetres) of thesample and the measurements provide significantly betterstatistics than SEM. The above-mentioned peculiarities makethe GISAXS analysis particularly suited for the long-rangecharacterization of the self-assembled films.
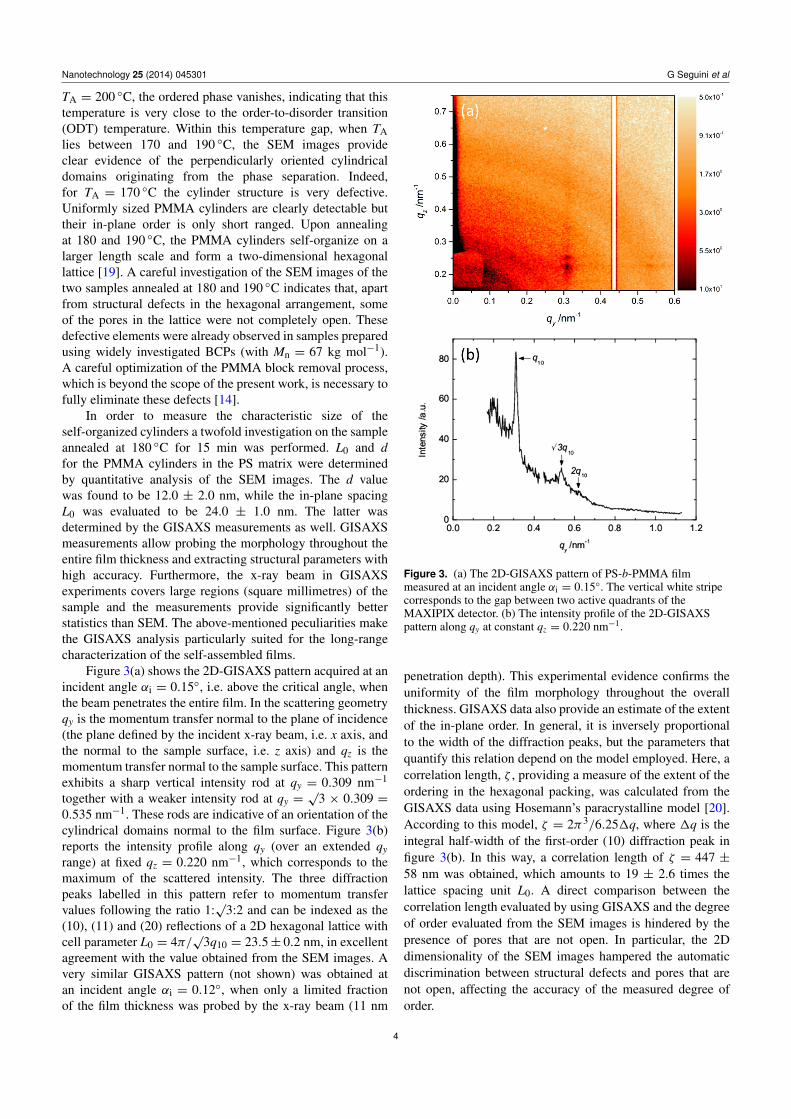
Figure 3(a) shows the 2D-GISAXS pattern acquired at anincident angle αi = 0.15◦, i.e. above the critical angle, whenthe beam penetrates the entire film. In the scattering geometryqy is the momentum transfer normal to the plane of incidence(the plane defined by the incident x-ray beam, i.e. x axis, andthe normal to the sample surface, i.e. z axis) and qz is themomentum transfer normal to the sample surface. This patternexhibits a sharp vertical intensity rod at qy = 0.309 nm−1
together with a weaker intensity rod at qy =√
3 × 0.309 =0.535 nm−1. These rods are indicative of an orientation of thecylindrical domains normal to the film surface. Figure 3(b)reports the intensity profile along qy (over an extended qyrange) at fixed qz = 0.220 nm−1, which corresponds to themaximum of the scattered intensity. The three diffractionpeaks labelled in this pattern refer to momentum transfervalues following the ratio 1:
√3:2 and can be indexed as the
(10), (11) and (20) reflections of a 2D hexagonal lattice withcell parameter L0 = 4π/
√3q10 = 23.5± 0.2 nm, in excellent
agreement with the value obtained from the SEM images. Avery similar GISAXS pattern (not shown) was obtained atan incident angle αi = 0.12◦, when only a limited fractionof the film thickness was probed by the x-ray beam (11 nm
Figure 3. (a) The 2D-GISAXS pattern of PS-b-PMMA filmmeasured at an incident angle αi = 0.15◦. The vertical white stripecorresponds to the gap between two active quadrants of theMAXIPIX detector. (b) The intensity profile of the 2D-GISAXSpattern along qy at constant qz = 0.220 nm−1.
penetration depth). This experimental evidence confirms theuniformity of the film morphology throughout the overallthickness. GISAXS data also provide an estimate of the extentof the in-plane order. In general, it is inversely proportionalto the width of the diffraction peaks, but the parameters thatquantify this relation depend on the model employed. Here, acorrelation length, ζ , providing a measure of the extent of theordering in the hexagonal packing, was calculated from theGISAXS data using Hosemann’s paracrystalline model [20].According to this model, ζ = 2π3/6.251q, where 1q is theintegral half-width of the first-order (10) diffraction peak infigure 3(b). In this way, a correlation length of ζ = 447 ±58 nm was obtained, which amounts to 19 ± 2.6 times thelattice spacing unit L0. A direct comparison between thecorrelation length evaluated by using GISAXS and the degreeof order evaluated from the SEM images is hindered by thepresence of pores that are not open. In particular, the 2Ddimensionality of the SEM images hampered the automaticdiscrimination between structural defects and pores that arenot open, affecting the accuracy of the measured degree oforder.
4

Nanotechnology 25 (2014) 045301 G Seguini et al
Figure 4. SEM images of the self-assembled PS-b-PMMA,Mn = 39 kg mol−1, as a function of tA, for TA = 180 ◦C.
In the second set of experiments, the annealingtemperature was kept constant, TA = 180 ◦C, whereas theordering evolution was monitored as a function of tA. SEMimages for tA varying from 1 to 30 min are shown in
figure 4. The phase separation was already detected for tA =1 min, although the cylindrical structure appeared to be quitedefective. At tA = 10 min the cylindrical morphology is stillpoorly organized and resembles the one observed after tA =15 min when TA is 170 ◦C (figure 2). This similarity couldbe understood by considering that in both cases there is adeficiency, either in terms of TA or in terms of tA, of theenergy supplied to the BCPs during the ordering process.This energy lack results in a small reservoir of enthalpyfor promoting the self-organization occurring upon phaseseparation. On the other hand, for tA = 30 min, the orderingis even better than the already ordered pattern obtained fortA = 15 min. Although the peculiar characteristics of the RTPheating process allow modulation of the heating and coolingrate to be performed, in the present study the heating andcooling times were determined by the heating ramp and theN2 flux respectively. These values were kept constant for allthe samples. This experimental procedure results in a constantand negligible contribution of the two ramps (around 10 s atTA = 180 ◦C) to the overall time spent by the system aboveTG.
4. Conclusions
The reported process conditions enable us to achieve ahexagonal packing of cylinders with L0 = 24 nm and d =12 nm using the full organic PS-b-PMMA with Mn of39 kg mol−1. This target was achieved using a simplethermal process performed in a standard RTP system [18].The self-assembling information encoded in the molecularcharacteristics of the BCP was properly transferred tothe resulting phase-separated cylindrical domains through afine-tuning of TA and tA within a relatively wide operationrange. The fabrication process was regulated at TA valuesbetween 170 and 190 ◦C and for tA periods between 1 and30 min demonstrating that, in this temperature range, it ispossible to drive the system toward the phase separation, whensufficiently long treatments are provided. It is worth notingthat previously reported experiments with low molecularweight (but still higher than that employed in the presentpaper) PS-b-PMMA BCPs required the introduction ofcomplex thermal processes assisted by electric field [5].This fact suggests that the experimental achievements hereinreported are strictly related to the peculiar process conditionsof RTP, which offer unprecedented opportunities for drivingthe self-assembling mechanism using a simple technologythat is fully compatible with the stringent industrialconstraints of lithographic applications. Further achievementsare expected from this specific thermal treatment in therace toward the goal of sub-10 nm pore diameter usingPS-b-PMMA BCP with even smaller molecular weight Mn.
Acknowledgments
This research was financially supported by the NANOBLOCKproject (‘NANO-device fabrication using block copolymerbased technology’). Patent protection related to this work ispending. The authors are grateful to Dr Tobias Schulli, ESRF,for his valuable support.
5

Nanotechnology 25 (2014) 045301 G Seguini et al
References
[1] Koo K, Ahn H, Kim S-W, Ryu D Y and Russell T P 2013 SoftMatter 9 9059
[2] Kim H-C, Park S-M and Hinsberg W D 2010 Chem. Rev.110 146–77
[3] Mastroianni S E and Epps T H 2013 Langmuir 29 3864–78[4] Rasappa S, Borah D, Senthamaraikannan R, Faulkner C C,
Shaw M T, Gleeson P, Holmes J D and Morris M A 2012Thin Solid Films 522 318–23
[5] Xu T, Kim H, Derouchey J, Seney C, Levesque C, Martin P,Stafford C M and Russell T P 2001 Polymer 42 9091–5
[6] Bang J, Jeong U, Ryu D Y, Russell T P and Hawker C J 2009Adv. Mater. 21 4769–92
[7] Jeong C K, Jin H M, Ahn J-H, Park T J, Yoo H G, Koo M,Choi Y-K, Kim S O and Lee K J 2013 Smalldoi:10.1002/smll.201301202
[8] Nunns A, Gwyther J and Manners I 2013 Polymer 54 1269–84[9] Bates C M, Seshimo T, Maher M J, Durand W J, Cushen J D,
Dean L M, Blachut G, Ellison C J and Willson C G 2012Science 338 775–9
[10] Son J G, Gotrik K W and Ross C A 2012 ACS Macro Lett.1 1279–84
[11] Ramanathan M, Nettleton E and Darling S B 2009 Thin SolidFilms 517 4474–8
[12] Shin D O, Lee D H, Moon H-S, Jeong S-J, Kim J Y, Mun J H,Cho H, Park S and Kim S O 2011 Adv. Funct. Mater.21 250–4
[13] Russell T P, Hjelm R P and Seeger P A 1990 Macromolecules23 890–3
[14] Andreozzi A, Poliani E, Seguini G and Perego M 2011Nanotechnology 22 185304
[15] Andreozzi A, Lamagna L, Seguini G, Fanciulli M,Schamm-Chardon S, Castro C and Perego M 2011Nanotechnology 22 335303
[16] Peng Q, Tseng Y-C, Darling S B and Elam J W 2010 Adv.Mater. 22 5129–33
[17] Tseng Y-C, Peng Q, Ocola L E, Elam J W and Darling S B2011 J. Phys. Chem. C 115 17725–9
[18] Ferrarese Lupi F, Giammaria T J, Ceresoli M, Seguini G,Sparnacci K, Antonioli D, Gianotti V, Laus M andPerego M 2013 Nanotechnology 24 315601
[19] Black C T and Guarini K W 2003 J. Polym. Sci. 42 1970–5[20] Vainshtein B K 1966 Diffraction of X-Rays by Chain
Molecules (Amsterdam: Elsevier)
6




















