
Theoretical calculations of the permeability of monensin–cation complexes in model bio-membranes
13
Theoretical calculations of the permeability of monensin^cation complexes in model bio-membranes Nir Ben-Tal a , Doree Sitko¡ b , Sharron Bransburg-Zabary a , Esther Nachliel a , Menachem Gutman a ; * a Department of Biochemistry, George S. Wise Faculty of Life Sciences, Tel Aviv University, Ramat Aviv 69978, Israel b Bristol-Myers Squibb, P.O. Box 4000, Princeton, NJ 08543-4000, USA Received 12 October 1999; received in revised form 6 January 2000; accepted 24 January 2000 Abstract Monensin is one of the best-characterized ionophores; it functions in the electroneutral exchange of cations between the extracellular and cytoplasmic sides of cell membranes. The X-ray crystal structures of monensin in free acid form and in complex with Na ,K and Ag are known and we have recently measured the diffusion rates of monensin in free acid form (Mo^H) and in complex with Na (Mo^Na) and with K (Mo^K) using laser pulse techniques. The results have shown that Mo^H diffuses across the membrane one order of magnitude faster than Mo^Na and two orders of magnitude faster than Mo^K. Here, we report calculations of the translocation free energy of these complexes across the membrane along the most favorable path, i.e. the lowest free energy path. The calculations show that the most favorable orientation of monensin is with its hydrophobic furanyl and pyranyl moieties in the hydrocarbon region of the membrane and the carboxyl group and the cation at the water^membrane interface. Further, the calculations show that Mo^H is likely to be inserted deeper than Mo^ Na into the bilayer, and that the free energy barrier for transfer of Mo^H across the membrane is V1 kcal/mol lower than for Mo^Na, in good agreement with our measurements. Our results show that the Mo^K complex is unlikely to diffuse across lipid bilayers in its X-ray crystal structure, in contrast to the Mo^H and Mo^Na complexes. Apparently, when diffusing across the membrane, the Mo^K complex assumes a different conformation and/or thinning defects in the bilayer lower significantly the free energy barrier for the process. The suitability of the model for treating the membrane association of small molecules is discussed in view of the successes and failures observed for the monensin system. ß 2000 Elsevier Science B.V. All rights reserved. Keywords : Transporter ; Continuum solvent model; Poisson^Boltzmann equation ; Di¡usion rate; Di¡usion across membrane ; Ionophore 1. Introduction Monensin, a disc-like molecule of about 50 heavy atoms, is a natural antibiotic produced by Strepto- myces sp. The antibiotic activity of monensin is at- tributed to its ability to exchange protons and cati- ons in an electroneutral process. As a result, the monensin dissipates the vpH term of the proton mo- 0005-2736 / 00 / $ ^ see front matter ß 2000 Elsevier Science B.V. All rights reserved. PII:S0005-2736(00)00156-5 Abbreviations: Mo^H, monensin in free acid form; Mo^Na, monensin in complex with Na ; Mo^K, monensin in complex with K ; PARSE, parameter for solvation energy; FDPB, ¢nite di¡erence Poisson^Boltzmann * Corresponding author. Fax : +972-3-6415053. Biochimica et Biophysica Acta 1466 (2000) 221^233 www.elsevier.com/locate/bba
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Theoretical calculations of the permeability of monensin–cation complexes in model bio-membranes

Theoretical calculations of the permeability of monensin^cationcomplexes in model bio-membranes
Nir Ben-Tal a, Doree Sitko¡ b, Sharron Bransburg-Zabary a, Esther Nachliel a,Menachem Gutman a;*
a Department of Biochemistry, George S. Wise Faculty of Life Sciences, Tel Aviv University, Ramat Aviv 69978, Israelb Bristol-Myers Squibb, P.O. Box 4000, Princeton, NJ 08543-4000, USA
Received 12 October 1999; received in revised form 6 January 2000; accepted 24 January 2000
Abstract
Monensin is one of the best-characterized ionophores; it functions in the electroneutral exchange of cations between theextracellular and cytoplasmic sides of cell membranes. The X-ray crystal structures of monensin in free acid form and incomplex with Na�, K� and Ag� are known and we have recently measured the diffusion rates of monensin in free acid form(Mo^H) and in complex with Na� (Mo^Na) and with K� (Mo^K) using laser pulse techniques. The results have shown thatMo^H diffuses across the membrane one order of magnitude faster than Mo^Na and two orders of magnitude faster thanMo^K. Here, we report calculations of the translocation free energy of these complexes across the membrane along the mostfavorable path, i.e. the lowest free energy path. The calculations show that the most favorable orientation of monensin is withits hydrophobic furanyl and pyranyl moieties in the hydrocarbon region of the membrane and the carboxyl group and thecation at the water^membrane interface. Further, the calculations show that Mo^H is likely to be inserted deeper than Mo^Na into the bilayer, and that the free energy barrier for transfer of Mo^H across the membrane is V1 kcal/mol lower thanfor Mo^Na, in good agreement with our measurements. Our results show that the Mo^K complex is unlikely to diffuseacross lipid bilayers in its X-ray crystal structure, in contrast to the Mo^H and Mo^Na complexes. Apparently, whendiffusing across the membrane, the Mo^K complex assumes a different conformation and/or thinning defects in the bilayerlower significantly the free energy barrier for the process. The suitability of the model for treating the membrane associationof small molecules is discussed in view of the successes and failures observed for the monensin system. ß 2000 ElsevierScience B.V. All rights reserved.
Keywords: Transporter; Continuum solvent model; Poisson^Boltzmann equation; Di¡usion rate; Di¡usion across membrane;Ionophore
1. Introduction
Monensin, a disc-like molecule of about 50 heavyatoms, is a natural antibiotic produced by Strepto-myces sp. The antibiotic activity of monensin is at-tributed to its ability to exchange protons and cati-ons in an electroneutral process. As a result, themonensin dissipates the vpH term of the proton mo-
0005-2736 / 00 / $ ^ see front matter ß 2000 Elsevier Science B.V. All rights reserved.PII: S 0 0 0 5 - 2 7 3 6 ( 0 0 ) 0 0 1 5 6 - 5
Abbreviations: Mo^H, monensin in free acid form; Mo^Na,monensin in complex with Na� ; Mo^K, monensin in complexwith K� ; PARSE, parameter for solvation energy; FDPB, ¢nitedi¡erence Poisson^Boltzmann
* Corresponding author. Fax: +972-3-6415053.
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
Biochimica et Biophysica Acta 1466 (2000) 221^233www.elsevier.com/locate/bba

tive force without a compensating increment of thecross-membranal electric ¢eld [1]. Monensin bindscations and the X-ray crystal structures of manymonensin^ion complexes have been determined [2^5]. One example, the monensin^sodium complex, isshown in Fig. 1. Monensin consists of three furanand two pyran rings. The rings are arranged in acircle with their polar oxygen atoms pointing to thecation that is buried at the center, partially screeningits electric charge. The hydrophobic regions of therings face outwards and mediate the interaction ofthe ion with the lipid bilayer.
Cation transport across cell membranes has beenstudied extensively, and the ability of monensin toaccelerate the process is attributed to its ability topartition into membranes [1]. Monensin, having anegative charge, can partition into the membraneonly in complex with cations. On each face of thebilayer, the monensin equilibrates with the ions ac-cording to their concentrations and their a¤nities forthe ionophore. The unequal concentrations of thevarious species drive a £ux of ions until the electro-chemical potentials on both sides of the membraneare equal. We recently studied the kinetics of mon-ensin-mediated cation transport across membranesusing laser-induced proton pulse techniques [6]. Ourmain conclusion was that the di¡usion rates of mon-ensin across the membrane are not the same for allthe complexes; protonated monensin (Mo^H) dif-fuses about 10 times faster than sodium^monensin(Mo^Na) and about 100 times faster than potassi-um^monensin (Mo^K). The variance in the di¡usionbehavior of molecules of comparable molecularweight and shape implies that their electrostatic in-teractions with the membrane are su¤ciently di¡er-ent to account for the observations.
In this work, we used continuum solvent modelsand calculated the free energy of the monensin^cat-ion complexes in di¡erent orientations with respectto lipid bilayers to ¢nd the minimal free energy pathof each complex across the bilayer. The calculationsare based on a simple model where the lipid bilayer isrepresented by a homogenous region of low dielectricconstant embedded in the high dielectric constant ofwater. The monensin^cation complex is the only spe-cies that is described in atomic detail in the model.We have recently used this model for calculating thefree energy of insertion of polyalanine K-helices into
lipid bilayers and the results were in good agreementwith experimental data [7,8]. Likewise, the presentcalculations are in good agreement with the kineticmeasurements for the Mo^H and Mo^Na complexes.However, the calculations are fundamentally incon-sistent with the available experimental data on theMo^K complex. The discrepancy may suggest thatthe Mo^K complex undergoes conformational rear-rangements when partitioning into bilayers.
2. Materials and methods
The total free energy di¡erence between a moleculein the membrane and in the aqueous phase (vGtot)can be decomposed into a sum of di¡erences of thefollowing energies: the electrostatic potential (vGelc),non-polar contributions (vGnp), molecule immobili-zation e¡ects (vGimm), lipid perturbation e¡ects(vGlip) and the free energy of the molecule's confor-mational changes (vGcon) [7,9^12]:
vGtot � vGelc � vGnp � vGimm � vGlip � vGcon �1�We evaluate the contributions of the last three termson the right hand side of Eq. 1 below, and in thefollowing we focus on the ¢rst two, which we de¢neas the solvation free energy, vGsolv
vGtotWvGelc � vGnprvGsolv �2�vGsolv is the free energy of transfer of monensin at agiven conformation from water to a bulk hydrocar-bon phase. It accounts for electrostatic contributionsresulting from changes in the solvent dielectric con-stant as well as for van der Waals and solvent struc-ture e¡ects, which are grouped in the non-polar termand together de¢ne the classical hydrophobic e¡ect.We calculate vGsolv using the continuum solventmodel. The method has been described in detail inour earlier study of the membrane association ofpolyalanine K-helices [7] and in the following sub-sections we present a brief outline, with emphasison the minor changes we made to adapt it to mon-ensin.
2.1. Electrostatic contributions
The calculations are based on a continuum modelin which electrostatic contributions are obtained
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233222

from ¢nite di¡erence solutions to the Poisson^Boltz-mann equation (the FDPB method) [13,14]. We haveused three monensin complexes: Mo^H, Mo^Na andMo^K, the three-dimensional (3D) structures of
which were retrieved from the Cambridge Catalogue.After retrieval, hydrogen atoms were added and themolecular structure was minimized as described be-low. The monensin complexes were represented in
Fig. 1. Space ¢lling model of the Mo^Na complex [4] in its most favorable orientation with respect to the lipid bilayer. Carbon atomsand methyl groups are black, oxygen atoms red, hydrogen atoms white and the Na� ion yellow. The white horizontal line representsthe boundary between the hydrocarbon region of the lipid bilayer, above the line, and the aqueous phase below it. Notice that eventhough the ion is above the line, it is partially shielded from the hydrocarbon environment. The chemical structure of monensin A ispresented in the frame at the bottom of the ¢gure. The atoms marked by asterisks are in contact with the metal ion.
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233 223

atomic detail, with atomic radii and partial chargesde¢ned at the coordinates of each nucleus. Thecharges and radii were taken from a parameter setfor solvation energy (PARSE), a parameter set thatwas derived to reproduce gas phase-to-water [15] andalkane-to-water [16] solvation free energies of smallorganic molecules. We recently used it to studyamide hydrogen bond formation [17], polyalanineK-helices insertion into lipid bilayers [7] and helix^helix interactions in lipid bilayers [18].
The parameters for the ether group are missing inPARSE, therefore we derived partial charges to re-produce its experimentally measured solvation freeenergy. In the absence of direct measurement of al-kane-to-water partitioning of simple ether-containingcompounds, we relied on vacuum-to-water data.Vacuum-to-water solvation free energies are reportedfor four ether-containing molecules: dimethylether,diethylether, 2-methoxypropane and 1,2-dimethoxy-ethane [19]. Comparisons of solvation data of smallorganic molecules have revealed that alkane-to-waterelectrostatic solvation free energies are smaller thanthe corresponding gas phase-to-water free energies oftransfer by a factor of 0.9 [16], and our parameter-ization scheme is based on this rule. Using the Pauliradii that are standard in PARSE, and by assigningpartial charges of 30.73 to the ether oxygen and+0.365 to each of the two ether carbons, we repro-duced the estimated experimental transfer free ener-gies of the above compounds with less than 10%error. Standard charges of +1 and radii of 1.13 Aî
for sodium and 1.53 Aî for potassium were assignedbased on the coordination state of the ions [20].
In the FDPB calculations reported here, theboundary between the monensin complexes and thesolvents (water or membrane) was set at the contactsurface between the van der Waals surface of thecomplex and a solvent probe (de¢ned here as havinga 1.4 Aî radius). The complexes and the lipid bilayerwere assigned a dielectric constant of 2, whereaswater had a dielectric constant of 80. The systemwas mapped onto a lattice of 1293 grid points, witha resolution of four points per Aî , and the Poisson^Boltzmann equation was numerically solved for theelectrostatic potential. The electrostatic free energywas calculated by integration over the potential timescharge distribution in space.
2.2. Non-polar contributions
The non-polar contribution to the solvation freeenergy, Gnp, was assumed to be proportional to thewater-accessible surface area of the monensin com-plex, A, through the expression
Gnp � Q A� b �3�We used the parameters Q= 0.0278 kcal/(mol Aî 2) andb =31.7 kcal/mol that have been derived from thepartitioning of alkanes between liquid alkane andwater [16], and have been successfully used in ourprevious study [7]. The total area of the monensincomplexes accessible to lipids in a particular con¢g-uration was calculated with a modi¢ed Shrake^Rup-ley [21] algorithm [22].
2.3. Models of monensin and the solvents
The 3D structures of the monensin^cation com-plexes available in the Cambridge Catalogue includeonly the heavy atoms, except for Mo^H [3], which isreported with all hydrogen atoms included. Weadded hydrogen atoms to Mo^Na [4] and Mo^K[5], and minimized the two structures using the In-sight-II set of molecular modeling tools (MSI, SanDiego, CA, USA). The minimization procedure, 100steps of steepest descend, hardly a¡ected the struc-tures.
In the following calculations, the complexes weredescribed in atomic detail, and were placed at di¡er-ent distances and orientations with respect to ourmodel of the lipid bilayer. The bilayer was repre-sented as a 30 Aî slab with a dielectric constant of2, known from a combination of thickness and ca-pacitance measurements [23,24]. This is a very sim-plistic model of the membrane that has many limi-tations as discussed in [7] and below. Nevertheless, itis a standard model for the dielectric properties ofthe bilayer and we use it since the experimental evi-dence suggests that the solvation free energy is thedominant contribution to the free energy of manysystems (e.g. [13,14,25^29]).
2.4. The lowest free energy path
The main objective of this work was to determine
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233224
the lowest free energy path for the transfer of themonensin^cation complexes across the bilayer. Thiscould, in principle, be done by exhaustively searchingin the con¢gurational space. To save time, it wasreasoned that the most stable orientations shouldbe those with maximum contact area between thehydrophobic groups and the lipid, and with minimalcontact area between the polar groups and the lipid.We choose the initial orientation of each monensin^cation complex accordingly, and sampled orienta-tions generated by small rotations and translationsaround the initial orientation relative to the mem-brane normal. We then used the orientation of themost negative solvation free energy from the probingcalculations (Fig. 1) throughout the study.
2.5. Estimate of vGimm, vGlip and vGcon
vGimm re£ects con¢nement of the external degreesof freedom of the monensin complex upon mem-brane association. Our calculations show that thetwo monensin^ion complexes that partition into themembrane, Mo^H and Mo^Na, are free to translateand rotate inside the lipid bilayer, suggesting that theimmobilization e¡ect should be negligibly small. Infact, one expects monensin to freely move inside thebilayer in view of its role as an ion exchanger.vGlip accounts for the perturbing e¡ect of the
monensin molecules on the lipid structure. It hasan entropic origin and is proportional to the lipid-accessible area of monensin (e.g. [8]). The latter issmall and thus the lipid perturbation e¡ect of mon-ensin should be rather small as well. We estimate itas zero.
The ring structure of monensin suggests rigiditybut monensin has many sp3^sp3 bonds, about whichthere is considerable rotational freedom. The consis-tency of the conformation of monensin in the threeNa� structures, hydrated and anhydrous forms [4]and the NaBr complex [30], indicates that the overallstructure of Mo^Na is hardly in£uenced by crystalpacking. It is, therefore, safe to assume that Mo^Nais unlikely to change its conformation upon mem-brane association, i.e. vGconV0. Unfortunately, itis di¤cult to estimate the stability of the Mo^H [3]and Mo^K [5] structures because each of them hasbeen crystallized only in one from. The results shownbelow suggest that vGcon should be approximately
zero for the Mo^H complex and very large forMo^K. We address this issue in Section 4 below.
3. Results
3.1. The insertion free energy
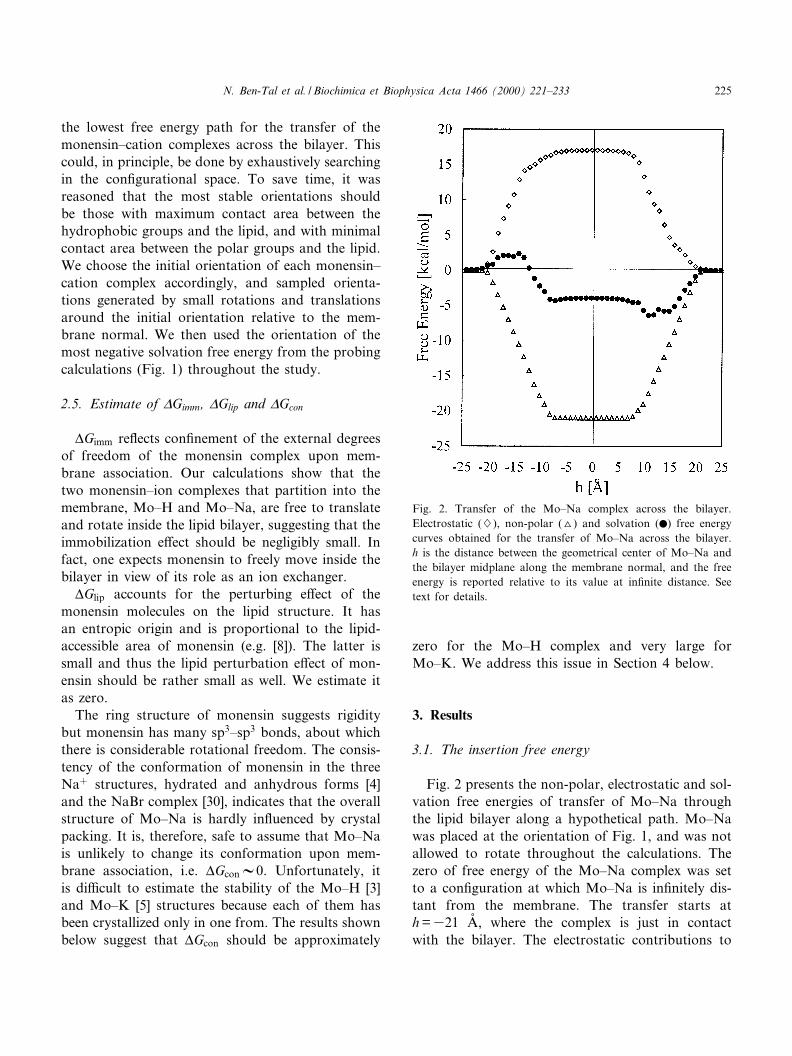
Fig. 2 presents the non-polar, electrostatic and sol-vation free energies of transfer of Mo^Na throughthe lipid bilayer along a hypothetical path. Mo^Nawas placed at the orientation of Fig. 1, and was notallowed to rotate throughout the calculations. Thezero of free energy of the Mo^Na complex was setto a con¢guration at which Mo^Na is in¢nitely dis-tant from the membrane. The transfer starts ath =321 Aî , where the complex is just in contactwith the bilayer. The electrostatic contributions to
Fig. 2. Transfer of the Mo^Na complex across the bilayer.Electrostatic (7), non-polar (O) and solvation (b) free energycurves obtained for the transfer of Mo^Na across the bilayer.h is the distance between the geometrical center of Mo^Na andthe bilayer midplane along the membrane normal, and the freeenergy is reported relative to its value at in¢nite distance. Seetext for details.
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233 225

the free energy become gradually more positive andthe non-polar contributions more negative during theprocess, until they reach their maximal absolute val-ues around h =37 Aî , where Mo^Na is entirelyburied in the bilayer. Mo^Na is fully buried in thebilayer at 37 Aî 6 h6 7 Aî , and the electrostatic andnon-polar contributions to the free energy are attheir saturation values in this region. As the trans-location process proceeds, the absolute values of theelectrostatic and non-polar contributions to the freeenergy of transfer gradually decrease from their max-imal values around h = 7 Aî to zero at h = 21 Aî , wherethe complex is just outside the bilayer and the pro-cess ends. The non-polar and electrostatic contribu-tions have opposite e¡ects on the transfer process,but their sum, the solvation free energy, is not zerobecause they do not fully balance each other.
Mo^Na is a fairly symmetric molecule as is evidentby the symmetry of the non-polar free energy curve(triangles, Fig. 2). However, the electrostatic freeenergy curve of monensin is directionally biased; itis steeper for insertion in the orientation wherethe polar region of the complex is facing the lipid(321 Aî 6 h637 Aî ) than for reversed polarity(7 Aî s hs 21 Aî ). The sum of the non-polar andelectrostatic contributions is the solvation free en-ergy, which gives a curve that is asymmetrical aswell. It has a free energy minimum around h = 12Aî , where the hydrophobic furanyl and pyranyl moi-eties of Mo^Na are buried in the hydrophobic coreof the membrane while the carboxyl group still pro-trudes into the aqueous phase (Fig. 1), and a max-imum at the reversed polarity of h =314 Aî . Thelatter orientation is obviously energetically unfavor-able and it is most likely that Mo^Na rotates in thehydrophobic region of the bilayer and protrudesfrom the other side of the membrane with its carbox-yl group ¢rst. Thus, the free energy curve for thetransfer of the complex across the bilayer is likelyto be fully symmetrical around the bilayer midplane.
3.2. The rotation of monensin^cation complexeswithin the membrane
The free energy minimum observed for the mon-ensin^sodium complex in the orientation of Fig. 1implies that the di¡usion of the complex across thelipid bilayer involves a 180³ rotation to reach the
symmetrical stable orientation at the other end ofthe bilayer. The experiments of Nachliel et al. [6]support this conclusion and even provide an estimateof the rotation rate. In that study, we noted that therate constants of the reactions between monensinand the ions are di¡usion-controlled, implying thatwhenever a cation encounters a monensin molecule,the ionophore is already in the right con¢guration tobind it, i.e. the carboxylate region is facing the aque-ous phase. Thus, the rotation rate of the monensin^cation complex inside the membrane is not a ratelimiting step in the overall process. Based on themeasured rate constants of ion binding (k = 1010
M31 s31) and the ion concentration used in the ex-periments (0.1 M), the rotation time should be short-er than the encounter rate in Nachliel's experiment,d= (0.1 k)31 = 1 ns. This rate is in accordance withthe rotational di¡usion of a sphere the size of mon-ensin in a matrix of the membrane viscosity (R6 2P)[31].
3.3. Comparison of the three complexes
We repeated the calculations of Fig. 2 using twoother monensin^cation complexes: Mo^H and Mo^K. Our calculations show that only one of them,Mo^H, is likely to di¡use across lipid bilayers. Incontrast, the crystal structure of Mo^K possesses alarge electrostatic dipole that prevents the complexfrom immersing inside lipid bilayers (vGsolV40 kcal/mol), in con£ict with experimental data. This issuewill be discussed further below.
The solvation free energy curves obtained for theMo^H and Mo^Na complexes are presented in Fig.3. It is evident from the ¢gure that the two com-plexes are likely to dissolve in the bilayer, but thatthey are unequal in their tendency to do so; Mo^H ismore likely to be inserted into the bilayer than Mo^Na. Besides the solubility di¡erence, the free energycurves of the two complexes have similar features.Each of them shows locations of maximal and min-imal stability, i.e. minima and maxima in the solva-tion free energy curves, respectively. The free energyminima correspond to the con¢gurations where thehydrophobic furanyl and pyranyl moieties of themonensin are buried inside the hydrocarbon regionof the bilayer while the charged or polar carboxylicgroup and the ion protrude into the aqueous phase
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233226

(Fig. 1). This orientation provides free energy bene-¢ts from the non-polar interactions and the leastpossible electrostatic free energy penalty. The reverseorientation, where the charged or polar carboxylicgroup is buried in the membrane and the furanyland pyranyl groups are in the aqueous phase, is ob-viously unfavorable and results in the observed freeenergy maximum.
From our point of view, the most interesting dif-ferences between the solvation free energy curves ofFig. 3 are the relative location and local depths ofthe free energy minima. The free energy minima ofMo^H and Mo^Na occur at h = 9 Aî and h = 12 Aî ,respectively. The depths of the corresponding localfree energy minima are 1.2 and 2.3 kcal/mol. Weshall relate these values to the measurements of Na-chliel et al. [6] in Section 4 below.
3.4. Convergence tests
We repeated the calculations of Fig. 2 using di¡er-ent grid sizes (973, 1133 and 1293) and scales (2, 3and 4 grids/Aî ) to test the convergence of our calcu-
lations. We also tested the e¡ect of changing theboundary conditions from `Coulombic' to `Dipolar'in DelPhi. Our results show that the calculations areconverged to less than 0.2 kcal/mol, which is morethan su¤cient for this study. Notice, however, thatthe high precision of our calculations is due to thesimpli¢ed model we used. The neglect of the polarhead groups region of the bilayer and the ¢xed con-formation of monensin in our model may result in anerror that is larger than 0.2 kcal/mol as discussedfurther below.
4. Discussion
Our calculations show that two of the three mon-ensin^cation complexes, Mo^H and Mo^Na, maypartition into the bilayer without deviating fromtheir X-ray crystal structure. For these two com-plexes, the calculations further show that the mini-mal free energy paths for the translocation across themembrane are qualitatively similar (Fig. 3) and con-form to the available experimental results. We focuson these two complexes in the beginning of this sec-tion and deal with the Mo^K complex below.
The calculations demonstrate that the most stableorientation of the Mo^H and Mo^Na complexes inthe membrane is with the hydrophobic furanyl andpyranyl rings buried in the hydrocarbon region ofthe lipid bilayer and the polar carboxyl group andion protruding into the aqueous phase (Fig. 1). Be-sides these common features, there are quantitativedi¡erences between the solvation free energy curvesof the Mo^H and Mo^Na complexes:
1. On setting the free energy of the complex in wateras a reference, the likelihood of the complexes topartition into the bilayer is not identical. Mo^H,with vGsol =39.4 kcal/mol, is more soluble in thebilayer than Mo^Na, with vGsol =36.4 kcal/mol.This pattern is in accord with experimental dataon the stability of the complexes in organic sol-vents [32,33] and on the relative membrane a¤n-ities of the cation complexes [1].
2. The most likely location of the complex in thebilayer depends on the cation. The Mo^H com-plex is almost fully buried inside the bilayer ath = 9 Aî , where only one of the etheric oxygen
Fig. 3. Translocation of the monensin^cation complexes acrossthe bilayer. Solvation free energy curves obtained for the trans-fer of Mo^H (O) and Mo^Na (8) across the bilayer. h is thedistance between the geometrical center of the complex and thebilayer midplane along the membrane normal. See text for de-tails.
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233 227

atoms extends into the aqueous phase. On theother hand, the Mo^Na complex has a preferredposition closer to the interface, where the ion is inthe low dielectric phase and the carboxylate pro-trudes into the high dielectric water phase.
3. The height of the free energy barrier for mem-
brane crossing varies in accordance with the ex-perimental results; the free energy barrier ob-served for the Mo^H complex is lower than thebarrier observed for Mo^Na. The local free energywell, out of which the Mo^H complex has toemerge in order to di¡use towards the midplane
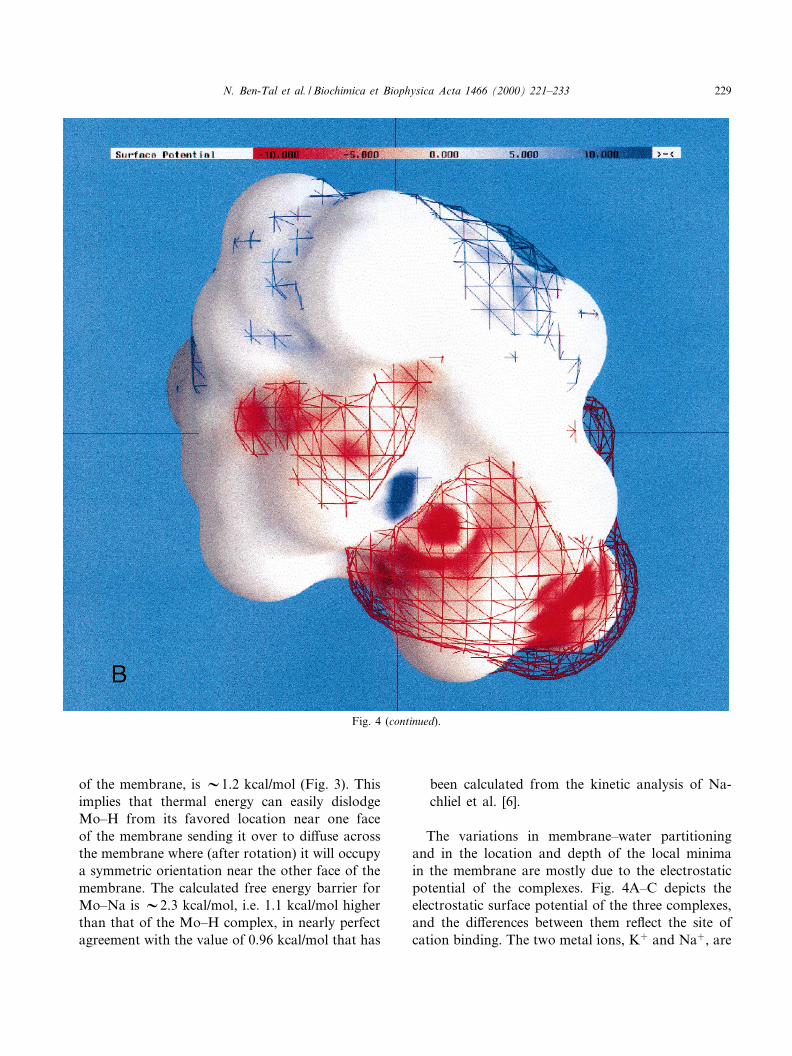
Fig. 4. Surface potential of the monensin complexes. (A) Mo^H, (B) Mo^Na and (C) Mo^K. Potentials more positive than +10 kT/eare deep-blue, potentials more negative than 310 kT/e are deep-red and neutral potentials (0 kT/e) are white. 3D contours of the po-tential at + and 31 kT/e are shown in blue and red mesh. The level of polarity of each complex is re£ected both in the ratio of col-ored vs. white region of the molecular surface and by the proximity of the contours to the molecular surface; polar complexes arecharacterized by large patches of red and blue on their molecular surface and by the fact that their contours are far from the molecu-lar surface. It is obvious from the ¢gure that Mo^K is signi¢cantly more polar than Mo^H and Mo^Na. The ¢gure was generated bythe GRASP program [38].
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233228
of the membrane, is V1.2 kcal/mol (Fig. 3). Thisimplies that thermal energy can easily dislodgeMo^H from its favored location near one faceof the membrane sending it over to di¡use acrossthe membrane where (after rotation) it will occupya symmetric orientation near the other face of themembrane. The calculated free energy barrier forMo^Na is V2.3 kcal/mol, i.e. 1.1 kcal/mol higherthan that of the Mo^H complex, in nearly perfectagreement with the value of 0.96 kcal/mol that has
been calculated from the kinetic analysis of Na-chliel et al. [6].
The variations in membrane^water partitioningand in the location and depth of the local minimain the membrane are mostly due to the electrostaticpotential of the complexes. Fig. 4A^C depicts theelectrostatic surface potential of the three complexes,and the di¡erences between them re£ect the site ofcation binding. The two metal ions, K� and Na�, are
Fig. 4 (continued).
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233 229

encapsulated by the furanyl and pyranyl rings ofmonensin and the carboxylate group is not in met-al-binding coordination. In contrast, the X-ray crys-tal structure of Mo^H shows that the proton is co-valently linked to the carboxylate, e¡ectivelyneutralizing its charge, and involved in a hydrogenbond in the hydrogen bonds network of Mo^H. Forthese reasons, the polarity of the protonated monen-sin is the smallest of all three, it has relatively smallpatches of positive and negative electrostatic poten-
tials, and its +1 kT/e and 31 kT/e iso-potential con-tours occur near its molecular surface (Fig. 4A). TheMo^Na complex (Fig. 4B) is slightly more polar thanthe Mo^H molecule, as evident by the larger patchesof positive and negative potential and from the factthat its iso-potential contours occur far from themolecular surface. The Mo^K complex (Fig. 4C) issigni¢cantly more polar than the other two com-plexes due to its larger ionic radius that distorts thestructure of monensin [5].
Fig. 4 (continued).
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233230

The present set of calculations is complementaryto our previous experimental study, in which wemeasured the rate constants of the elementary pro-cesses of the monensin-mediated ion exchange mech-anism. Evidently, the outcome of the free energypro¢les yields results that corroborate the experimen-tally derived estimates of the rate constants, and sug-gests a molecular interpretation of the elementaryprocesses involved in monensin-mediated cationtransport across biological membranes. However,the model has a number of limitations that we out-line in the following paragraphs.
The main limitation of our model is that the con-formation of the monensin^ion complex is taken as agiven, and thus we are practically bound to use onlythe crystal structures that were experimentally deter-mined by X-ray crystallography. Such conformationsare, presumably, the most stable under the speci¢cconditions selected for growing the crystals (e.g. thetype of solvent and the solute density), but may notnecessarily be the most stable in all phases. For ex-ample, our calculations show that the membranetranslocation of the Mo^K complex in its X-ray crys-tal structure involves a solvation free energy penaltyof about 40 kcal/mol. Since the available experimen-tal data indicate that Mo^K does di¡use across bi-layers [6], we conclude that the membrane-associatedconformation of the complex is di¡erent than that ofthe X-ray structure and/or thinning defects signi¢-cantly reduce the free energy barrier for the process[34].
A related shortcoming of our model is the assump-tion that the conformation of the monensin^ioncomplex remains unchanged throughout the transferfrom the aqueous phase to the bilayer. Monensin is asmall molecule and its membrane-associated confor-mation may be slightly di¡erent from its form inaqueous solutions. The smallness of monensin sug-gests that the energetic costs of such a conformation-al change may be small compared to its e¡ect on thesolvation free energy. The consistency of the monen-sin conformation in the three Mo^Na crystals indi-cates that the overall structure of Mo^Na is verylittle in£uenced by crystal packing. Thus, we feelsafe to assume that Mo^Na is unlikely to changeits conformation upon membrane association. Wecannot state with the same con¢dence that Mo^Hdoes not go through conformational changes in the
transition from the aqueous phase into the lipid bi-layer because only one crystal form of Mo^H hasbeen observed. Moreover, there is no direct evidencethat its conformation in any of these phases is similarto the X-ray structure. However, the agreement be-tween the calculations and the kinetic measurementsis an indirect support that Mo^H does not change itsconformation upon binding or that the conforma-tional changes have little e¡ect on the free energycurve of Fig. 3.
The description of the lipid bilayer as a slab of lowdielectric constant obscures all atomic detail aboutmonensin^bilayer interactions. However, this mini-malistic model of the dielectric properties of the hy-drocarbon region of the membrane successfully ac-counts for the e¡ects of the aqueous phase oncharged and polar groups that are buried in themembrane [7,35^36].
While the slab model is an acceptable approxima-tion for calculating the water^membrane partitioningof uncharged molecules, it might be inadequate forcalculating the water-to-membrane transfer free en-ergy of ion-containing complexes, such as Mo^Naand Mo^K. To explore this possibility, we repeatedthe calculations of Fig. 2 for the Mo^Na complexwith a di¡erent charge distribution between the cen-tral cation and the surrounding oxygen atoms. Bycomparing the results of these calculations with theoriginal free energy curve described above, we get anupper bound estimate of the contribution of the Na�
ion to solvation. To this end, we eliminated the +1charge of the Na� ion and in parallel changed thepartial charge of each of the four coordinated oxygenatoms from 30.73 to 30.48 for compensation. Thefree energy curve thus obtained was very similar tothe original curve; the minimum occurred at thesame con¢guration, i.e. h = 12 Aî , and its depthchanged by V1.5 kcal/mol only. We conclude that,due to the coordination of the oxygen atoms aroundthe cation, the monensin complex has practically noionic nature (Fig. 4B).
The greatest uncertainty in the slab model resultsfrom its complete neglect of the polar head groupsregion, which is presumably the most favorable siteof the monensin^cation complexes. Since the dielec-tric constant in this region is estimated to be between25 and 40 [37], the polar head groups region might
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233 231

most appropriately be regarded as part of the aque-ous phase de¢ned in this study.
The most crucial factor in the present calculationsis the charge distribution on the monensin^cationcomplexes, which is determined by the assignmentof the atomic partial charges from the PARSE pa-rameter set. However, because PARSE yields accu-rate transfer free energies between water and liquidalkane for small organic molecules including ethersand carboxylic acids (both charged and neutral), itseems reasonable to assume that it provides a goodapproximation to the water^membrane solvationproperties of monensin complexes that are con-structed from the same chemical groups.
Monensin is a small drug molecule that exerts itse¡ect by wrapping a hydrophobic surface aroundcharged ions to facilitate their di¡usion across thecell membrane. A large number of hydrophobic com-pounds, such as drugs, hormones and vitamins, usedto modulate biological activities have to propagatethrough membranes, to bind to their targets. Inthis respect, monensin can be viewed as a represen-tative of a large group of entities and our model mayenable us to screen their conformations with respectto the membrane association and permeation, e.g.the crystal structures of Mo^H and Mo^Na vs.that of Mo^K. Our experience from the studies onthe monensin^cation complexes is that relativelysmall conformational changes, involving minorchanges in the internal energy, lead to large changesin the solvation free energy. Thus, combining themodel with tools for sampling conformations, suchas molecular dynamics simulations or ab initio quan-tum mechanical calculations, may provide a valuablemethod for screening small molecule libraries to sug-gest candidates that are likely to partition into lipidbilayers and to estimate their rate of di¡usion acrossthe bilayer. The main strength of our model is that,while it takes into account the atomic details of thesmall drug molecule, it does not require a detaileddescription of the membrane. The neglect of detaileddescription of the lipid bilayer typically leads to lessaccurate calculations, but it also makes the modelgeneral and readily applicable for screening the mem-branal permeability of small molecules of potentialtherapeutic capabilities.
Acknowledgements
The work started while M.G. was a visiting Pro-fessor in the laboratory of Prof. Barry Honig. Wewish to thank Prof. Honig for his generous hospital-ity and for helpful discussions. This research wassupported by Grant number 683/97-1 from the IsraelScience Foundation and by fellowships from theWolfson and Alon Foundations to N.B.-T.
References
[1] B.C. Pressman, Ionophorous antibiotics as models for bio-logical transport, Fed. Proc. 27 (1968) 1283^1288.
[2] M. Pinkerton, L.K. Steinrauf, Molecular structure of mono-valent metal cation complexes of monensin, J. Mol. Biol. 49(1970) 533^546.
[3] W.K. Lutz, F.K. Winkler, J.D. Dunitz, Crystal structure ofthe antibiotic monensin. Similarities and di¡erences betweenfree acid and metal complex, Helv. Chim. Acta 54 (1971)1103^1108.
[4] W.L. Duax, G.D. Smith, P.D. Strong, Complexation of met-al ions by monensin Crystal and molecular structure of hy-drated and anhydrous crystals forms of Monensin^Na,J. Am. Chem. Soc. 102 (1980) 6725^6729.
[5] W. Pangborn, W.L. Duax, D. Langs, The hydrated Potassi-um complex of the ionophore Monensin A, J. Am. Chem.Soc. 109 (1987) 2163^2165.
[6] E. Nachliel, Y. Finkelstein, M. Gutman, The mechanism ofmonensin-mediated cation exchange based of real time mea-surements, Biochim. Biophys. Acta 285 (1996) 131^145.
[7] N. Ben-Tal, A. Ben-Shaul, A. Nicholls, B. Honig, Free-en-ergy determinants of K-helix insertion into lipid bilayers,Biophys. J. 70 (1996) 1803^1812.
[8] A. Ben-Shaul, N. Ben-Tal, B. Honig, Statistical thermody-namic analysis of peptides and protein insertion into lipidmembranes, Biophys. J. 71 (1996) 130^138.
[9] D.M. Engelman, T.A. Steitz, The spontaneous insertion ofproteins into and across membranes: the helical hairpin hy-pothesis, Cell 23 (1981) 411^422.
[10] F. Jahnig, Thermodynamics and kinetics of protein incorpo-ration into membranes, Proc. Natl. Acad. Sci. USA 80(1983) 3691^3695.
[11] R.E. Jacobs, S.H. White, The nature of the hydrophobicbinding of small peptides at the bilayer interface: implica-tions for the insertion of trans-bilayer helices, Biochemistry28 (1989) 3421^3437.
[12] M. Milik, J. Skolnick, Insertion of peptide chains into lipidmembranes: an o¡-lattice Monte Carlo dynamics model,Proteins 15 (1993) 10^25.
[13] B. Honig, K. Sharp, A.-S. Yang, Macroscopic models ofaqueous solutions: biological and chemical applications,J. Phys. Chem. 97 (1993) 1101^1109.
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233232

[14] B. Honig, A. Nicholls, Classical electrostatics in biology andchemistry, Science 268 (1995) 1144^1149.
[15] D. Sitko¡, K.A. Sharp, B. Honig, Accurate calculation ofhydration free energies using macroscopic solvent models,J. Phys. Chem. 98 (1994) 1978^1988.
[16] D. Sitko¡, N. Ben-Tal, B. Honig, Calculation of alkane towater solvation free energies using continuum solvent mod-els, J. Phys. Chem. 100 (1996) 2744^2752.
[17] N. Ben-Tal, D. Sitko¡, I.A. Topol, A.-S. Yang, S.K. Burt,B. Honig, The free energy of amide hydrogen bond forma-tion in vacuum, in water and in liquid alkane solutions,J. Phys. Chem. B 101 (1997) 450^457.
[18] N. Ben-Tal, B. Honig, Helix-helix interactions in lipid bi-layers, Biophys. J. 71 (1996) 3046^3050.
[19] C. Hansch and A. Leo, Substituent Constants for Correla-tion Analysis in Chemistry and Biology, Wiley, New York,1979.
[20] J.E. Huheey, E.A. Keiter and R.L. Keiter, Inorganic Chem-istry, Harper Collins, New York, 1993, p. 72.
[21] A. Shrake, J.A. Rupley, Environment and exposure to sol-vent of protein atoms. Lysozyme and insulin, J. Mol. Biol.79 (1973) 351^371.
[22] S. Sridharan, A. Nicholls, B. Honig, A new vertex algorithmto calculate solvent accessible surface area, Biophys. J. 61(1992) A174.
[23] R. Fettiplace, D.M. Andrews, D.A. Hatdon, The thicknesscomposition and structure of some lipid bilayers and nuturalmembranes, J. Membr. Biol. 5 (1971) 277^296.
[24] J.P. Dilger, R. Benz, Optical and electrical properties of thinmonoolein lipid bilayers, J. Membr. Biol. 85 (1985) 181^189.
[25] J. Kyte, R.F. Doolittle, A simple method for displaying thehydropathic character of a protein, J. Mol. Biol. 157 (1982)105^132.
[26] D. Eisenberg, A.D. McLachlan, Solvation energy in proteinfolding and binding, Nature 319 (1986) 199^203.
[27] A. Warshel, Computer Modeling of Chemical Reactions inEnzymes and Solutions, John-Wiley and Sons, New York,1991.
[28] K. Yue, A.K. Dill, Folding proteins with a simple energyfunction and extensive conformational searching, ProteinSci. 5 (1996) 254^261.
[29] J.A. McCammon, Theory of biomolecular recognition, Curr.Opin. Struct. Biol. 8 (1998) 245^249.
[30] D.L. Ward, K.-T. Wei, J.C. Hoogerheide, A.I. Popov, Thecrystal and molecular structure of the sodium bromide com-plex of monensin, C36H62O11.Na�Br3, Acta Cryst. B34(1987) 110^115.
[31] M.K. Jain and R.C. Wagner, Introduction to BiologicalMembranes, Wiley and Sons, New York, 1980.
[32] B.G. Cox, N.G. vanTruong, J. Rzeszotarska, H. Schneider,Rates and equilibria of alkali metals and silver ion complexformation with monensin in ethanol, J. Am. Chem. Soc. 106(1984) 5965^5969.
[33] P.J.F. Henderson, J.D. McGivan, J.B. Chappel, The actionof certain antibiotics on Mitochondrial, Erythrocyte and ar-ti¢cial phospholipids membranes, J. Biochem. 111 (1969)521^535.
[34] M.A. Wilson, A. Pohorille, Mechanism of unassisted iontransport across membrane bilayers, J. Am. Chem. Soc.118 (1996) 6580^6587.
[35] S. Berneche, M. Nina, B. Roux, Molecular dynamics simu-lation of melittin in a dimyristoylphosphatidylcholine bilayermembrane, Biophys. J. 75 (1998) 1603^1618.
[36] P. LaRocca, Y. Shai, M.S. Sansom, Peptide-bilayer interac-tions: simulations of dermaseptin B, an antimicrobial pep-tide, Biophys. Chem. 76 (1999) 145^159.
[37] R.G. Ashcroft, H.G. Coster, J.R. Smith, The molecular or-ganisation of bimolecular lipid membranes. The dielectricstructure of the hydrophilic/hydrophobic interface, Biochim.Biophys. Acta 643 (1981) 191^204.
[38] A. Nicholls, K.A. Sharp, B. Honig, Protein folding and as-sociation: insights from the interfacial and thermodynamicproperties of hydrocarbons, Proteins 11 (1991) 281^296.
BBAMEM 77825 17-5-00 Cyaan Magenta Geel Zwart
N. Ben-Tal et al. / Biochimica et Biophysica Acta 1466 (2000) 221^233 233




















