
The tetratricopeptide repeats (TPR)-like superfamily of proteins in Leishmania spp., as revealed by...
12
The tetratricopeptide repeats (TPR)-like superfamily of proteins in Leishmania spp., as revealed by multi-relational data mining Michely C. Diniz, Ana Carolina L. Pacheco, Karen T. Girão, Fabiana F. Araujo, Cezar A. Walter, Diana M. Oliveira * Núcleo Tarcisio Pimenta de Pesquisa Genômica e Bioinformática, NUGEN, Faculdade de Veterinária, Universidade Estadual do Ceara – UECE, Av. Paranjana, 1700, Campus do Itaperi, Fortaleza, CE 60740-000, Brazil article info Article history: Available online 4 May 2010 Keywords: Multi-relational data mining Hidden Markov models Viterbi algorithm Tetratricopeptide repeat motif Leishmania proteins abstract Protein sequence analysis tasks are multi-relational problems suitable for multi-relational data mining (MRDM). Proteins containing tetratricopeptide (TPR), pentatricopeptide (PPR) and half-a-TPR (HAT) repeats comprise the TPR-like superfamily in which we have applied MRDM methods (relational associ- ation rule discovery and probabilistic relational models) with hidden Markov models (HMMs) and Viterbi algorithm (VA) in genome databases of pathogenic protozoa Leishmania. Such integrated MRDM/HMM/ VA approach seeks to capture as much model information as possible in the pattern matching heuristic, without resorting to more standard motif discovery methods (Pfam, SMART, SUPERFAMILY) and it has the advantage of incorporation of optimized profiles, score offsets and distribution to compute probability, as a more recently reported tool (TPRpred) in order to take in account the tendency of repeats to occur in tandem and to be widely distributed along the sequences. Here we compare such currently available resources with our approach (MRDM/HMM/VA) to highlight that the latter performs best into the TPR- like superfamily assignment and it might be applied to other sequence analysis problems in such a way that it contributes to tight-fit motif discoveries and a better probability that a given target sequence is, indeed, a target motif-containing protein. Ó 2010 Elsevier B.V. All rights reserved. 1. Introduction There are many data mining problems in genomics and post- genomics that involve representing and reasoning about biological sequences (DNA, RNA and protein). Among these problems are gene (Burge and Karlin, 1997) and protein motif discovery (Law- rence et al., 1993), and protein structure prediction (Schmidler et al., 2000). It has been argued that such sequence-analysis tasks are becoming multi-relational problems because other sources of evidence, besides the sequences themselves, are being leveraged in the learning and inference processes (Page and Craven, 2003) which are core parts of the bioinformatics scenario. In addition, many sequence elements of interest are composed from other se- quence elements. In this paper, we present and discuss results regarding motif discovery methods, focusing in particular on our recent work in analyzing the tetratricopeptide repeat (TPR)-like superfamily of proteins in Leishmania spp. (Girão et al., 2008). Early efforts in bioinformatics concentrated on finding the internal structure of individual genome-wide data sets and solving problems with the explosion of the ‘omics’ technologies. Since comprehensive coverage of the multiple aspects of cellular/organ- ellar physiology is also progressing rapidly, there is a need for inte- gration of vast amounts of biological data encompassing a systems-level approach that requires integrating all of the known properties of a given class of components (e.g., protein abundance, localization, physical interactions, etc.) with computational meth- ods able to combine large and heterogeneous sets of data (Ideker et al., 2007). One of these techniques for the generation of unified mechanistic models of cellular/organellar processes (a major chal- lenge for all who seek to discover functions of many yet unknown genes) is the multi-relational data mining (MRDM) approach, which looks for patterns that involve multiple input tables (rela- tions) from a relational database (DB) made of complex/structured objects whose normalized representation requires multiple tables (Getoor et al., 2001a). MRDM extends association rule mining to search for interesting patterns among data in multiple tables rather than in one input table (Dehaspe and De Raedt, 1997). Two major challenges, however, that sequence analysis applica- tions highlight for the area of MRDM are computational complexity and ease of use. Post-genomics databases are growing at an expo- nential rate and the task of determining whether a protein se- quence contains a given substructure (or a motif) is an optimization problem that falls into the NP-complete (Garey and 0167-8655/$ - see front matter Ó 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.patrec.2010.04.008 * Corresponding author. Tel./fax: +55 85 31019842. E-mail address: [email protected] (D.M. Oliveira). Pattern Recognition Letters 31 (2010) 2178–2189 Contents lists available at ScienceDirect Pattern Recognition Letters journal homepage: www.elsevier.com/locate/patrec
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of The tetratricopeptide repeats (TPR)-like superfamily of proteins in Leishmania spp., as revealed by...

Pattern Recognition Letters 31 (2010) 2178–2189
Contents lists available at ScienceDirect
Pattern Recognition Letters
journal homepage: www.elsevier .com/locate /patrec
The tetratricopeptide repeats (TPR)-like superfamily of proteinsin Leishmania spp., as revealed by multi-relational data mining
Michely C. Diniz, Ana Carolina L. Pacheco, Karen T. Girão, Fabiana F. Araujo, Cezar A. Walter,Diana M. Oliveira *
Núcleo Tarcisio Pimenta de Pesquisa Genômica e Bioinformática, NUGEN, Faculdade de Veterinária, Universidade Estadual do Ceara – UECE, Av. Paranjana, 1700, Campus do Itaperi,Fortaleza, CE 60740-000, Brazil
a r t i c l e i n f o a b s t r a c t
Article history:Available online 4 May 2010
Keywords:Multi-relational data miningHidden Markov modelsViterbi algorithmTetratricopeptide repeat motifLeishmania proteins
0167-8655/$ - see front matter � 2010 Elsevier B.V. Adoi:10.1016/j.patrec.2010.04.008
* Corresponding author. Tel./fax: +55 85 31019842E-mail address: [email protected] (D.M. O
Protein sequence analysis tasks are multi-relational problems suitable for multi-relational data mining(MRDM). Proteins containing tetratricopeptide (TPR), pentatricopeptide (PPR) and half-a-TPR (HAT)repeats comprise the TPR-like superfamily in which we have applied MRDM methods (relational associ-ation rule discovery and probabilistic relational models) with hidden Markov models (HMMs) and Viterbialgorithm (VA) in genome databases of pathogenic protozoa Leishmania. Such integrated MRDM/HMM/VA approach seeks to capture as much model information as possible in the pattern matching heuristic,without resorting to more standard motif discovery methods (Pfam, SMART, SUPERFAMILY) and it has theadvantage of incorporation of optimized profiles, score offsets and distribution to compute probability, asa more recently reported tool (TPRpred) in order to take in account the tendency of repeats to occur intandem and to be widely distributed along the sequences. Here we compare such currently availableresources with our approach (MRDM/HMM/VA) to highlight that the latter performs best into the TPR-like superfamily assignment and it might be applied to other sequence analysis problems in such away that it contributes to tight-fit motif discoveries and a better probability that a given target sequenceis, indeed, a target motif-containing protein.
� 2010 Elsevier B.V. All rights reserved.
1. Introduction
There are many data mining problems in genomics and post-genomics that involve representing and reasoning about biologicalsequences (DNA, RNA and protein). Among these problems aregene (Burge and Karlin, 1997) and protein motif discovery (Law-rence et al., 1993), and protein structure prediction (Schmidleret al., 2000). It has been argued that such sequence-analysis tasksare becoming multi-relational problems because other sources ofevidence, besides the sequences themselves, are being leveragedin the learning and inference processes (Page and Craven, 2003)which are core parts of the bioinformatics scenario. In addition,many sequence elements of interest are composed from other se-quence elements. In this paper, we present and discuss resultsregarding motif discovery methods, focusing in particular on ourrecent work in analyzing the tetratricopeptide repeat (TPR)-likesuperfamily of proteins in Leishmania spp. (Girão et al., 2008).
Early efforts in bioinformatics concentrated on finding theinternal structure of individual genome-wide data sets and solvingproblems with the explosion of the ‘omics’ technologies. Since
ll rights reserved.
.liveira).
comprehensive coverage of the multiple aspects of cellular/organ-ellar physiology is also progressing rapidly, there is a need for inte-gration of vast amounts of biological data encompassing asystems-level approach that requires integrating all of the knownproperties of a given class of components (e.g., protein abundance,localization, physical interactions, etc.) with computational meth-ods able to combine large and heterogeneous sets of data (Idekeret al., 2007). One of these techniques for the generation of unifiedmechanistic models of cellular/organellar processes (a major chal-lenge for all who seek to discover functions of many yet unknowngenes) is the multi-relational data mining (MRDM) approach,which looks for patterns that involve multiple input tables (rela-tions) from a relational database (DB) made of complex/structuredobjects whose normalized representation requires multiple tables(Getoor et al., 2001a). MRDM extends association rule mining tosearch for interesting patterns among data in multiple tablesrather than in one input table (Dehaspe and De Raedt, 1997).
Two major challenges, however, that sequence analysis applica-tions highlight for the area of MRDM are computational complexityand ease of use. Post-genomics databases are growing at an expo-nential rate and the task of determining whether a protein se-quence contains a given substructure (or a motif) is anoptimization problem that falls into the NP-complete (Garey and

Table 2Comparative results of half-a-TPR (HAT) motif finding in Leishmania genes obtainedwith three standard HMM tools (SUPERFAMILY, GeneDB and Pfam), with TPRpred andwith our method (MRDM/HMM/VA). Numbers are shown for HAT-containingproteins in three species (L. major, L. infantum and L. braziliensis).
HAT Motif GeneDB Pfam TPRpred MRDM
L. major 3 1 – 3L. infantum 2 1 – 3L. braziliensis 1 1 – 2
M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189 2179
Johnson, 1979) task of subgraph isomorphism (Page and Craven,2003). Any biologist can run a decision-tree learner or a linearregression routine on a given data with relatively little trainingand preprocessing, but running a MRDM system typically requiresa much larger knowledge engineering effort, what reinforces theneed for biologist-friendlier MRDM systems.
Keeping this in mind, we have applied MRDM methods (rela-tional association rule discovery – RARD and probabilistic rela-tional models – PRMs) combined with hidden Markov models(HMMs) (Eddy, 1998; Winters-Hilt, 2006; Madera, 2008) and theViterbi algorithm (VA) (Forney, 1973) to mine the tetratricopeptiderepeat (TPR) (Blatch and Lässle, 1999; D’Andrea and Regan, 2003;Das et al., 1998) and related motifs (pentatricopeptide repeat(PPR) (Small and Peeters, 2000; Kotera et al., 2005) and half-a-TPR (HAT) (Preker and Keller, 1998) in pathogenic protozoa Leish-mania spp. (Girão et al., 2008). We have, then, built a panel of theTPR-like superfamily of proteins, whose members can be furtherassigned functional roles for their containing motifs. TPR motifswere originally identified in yeast as protein-protein interactionmodules (Blatch and Lässle, 1999), but now they are known to oc-cur in a wide variety of proteins (over 12,000 as included in SMARTnrdb) present in prokaryotic and eukaryotic organisms (D’Andreaand Regan, 2003), being involved in protein–protein and protein–lipid interactions in cell cycle regulation, chaperone function andpost-translation modifications (Blatch and Lässle, 1999; D’Andreaand Regan, 2003; Das et al., 1998).
TPRs exhibit a large degree of sequence diversity and structuralconservation (two antiparallel alpha-helices separated by a turn)that might act as scaffolds for the assembly of different multipro-tein complexes (Scheufler et al., 2000), including the peroxisomalimport receptor and the NADPH oxidase (Koga et al., 1999). Similarto TPR, PPR and HAT motifs also have repetitive patterns character-ized by the presence of a tandem array of repeats, where the num-ber of motifs seems to influence the affinity and specificity of therepeat-containing protein for RNA (Preker and Keller, 1998; Lurinet al., 2004; Rivals et al., 2006). PPR-containing proteins (PPRPs)occur predominantly in eukaryotes (Small and Peeters, 2000) (par-ticularly abundant in plants), while it has been suggested that eachof the highly variable PPRPs is a gene-specific regulator of plantorganellar RNA metabolism. HAT repeats are less abundant andHAT-containing proteins (HATPs) appear to be components of mac-romolecular complexes that are required for RNA processing(Small and Peeters, 2000; Kotera et al., 2005; Preker and Keller,1998; Lurin et al., 2004; Rivals et al., 2006).
TPR-containing proteins (TPRPs) have recently attracted inter-est because of their versatility as scaffolds for the engineering ofprotein-protein interactions (Main et al., 2005; Karpenahalliet al., 2007). TPRPs are characterized by homologous, repeatingstructural units stacked together to form an open-ended superhe-lical structure. Such an arrangement is in contrast to the structureof most proteins, which fold into a compact shape (Groves and Bar-ford, 1999). The curvature created by the superhelical nature ofthese proteins predetermines the target proteins that can bind tothem (Kobe and Kajava 2000). TPRs, PPRs and HAT (all together re-ferred as TPR-like motifs) form a large superfamily or the clan TPR-like (Blatch and Lässle, 1999; D’Andrea and Regan, 2003; Das et al.,
Table 1Comparative results of TPR-like motif finding in Leishmania genes obtained with three smethod (MRDM/HMM/VA). Numbers are shown for TPR- and PPR-containing proteins in t
Motif Superfamily GeneDB
TPR PPR TPR PPR
L. major 95 – 62 12L. infantum 98 – 37 11L. braziliensis 99 – 53 8
1998; Scheufler et al., 2000; Preker and Keller, 1998; Lurin et al.,2004; Rivals et al., 2006). Homologous structural repeat units areoften highly divergent at the sequence level, a feature that makestheir prediction challenging. Currently, several web-based re-sources are available for the detection of TPRs, including Pfam(Finn et al., 2008) SMART (Schultz et al., 1998; Letunic et al.,2009), and SUPERFAMILY (Gough et al., 2001; Madera et al.,2004), that use HMM profiles, which are constructed from the re-peats trusted to belong to the family. However, the profiles usedare constructed from closely homologous repeats; therefore, diver-gent repeat units often get a negative score and are not consideredin computing the overall statistical significance, even though theyare individually significant (Karpenahalli et al., 2007; Madera,2008). For this reason, Pfam, SMART, and SUPERFAMILY performwith limited accuracy in detecting remote homologs of knownTPRPs and in delineating the individual repeats within a protein.A new profile-based method, TPRpred (Karpenahalli et al., 2007),uses a P-value-dependent score offset to include divergent repeatunits and to exploit the tendency of repeats to occur in tandem.
Although TPRpred indeed performs significantly better indetecting divergent repeats in TPRPs, and finds more individual re-peats than the afore mentioned methods, we have noticed that it(TPRpred) still fails to detect some members of the TPR-like super-family, such as 03 TPRPs, 02-05 PPRPs and 02-03 HATPs in a total of26 TPR-like proteins in the three examined species of Leishmania(Leishmania major, Leishmania infantum and Leishmania braziliensis)shown in Tables 1 and 2. The characterization of the proteins of agiven family often relies on the detection of regions of their se-quences shared by all family members, while computing the con-sensus of such regions provides a motif that is used to recognizenew members of the family. Our integrated approach of HMMs/VA with MRDM was suitable to detect 104 TPRPs, 36 PPRPs and08 HATPs in Leishmania spp. genomes, a greater number than cur-rently available resources (Pfam, SMART, SUPERFAMILY) are ableto yield and slightly higher than TPRpred. Our results are, there-fore, given as an illustration of how powerful MRDM algorithmsand techniques might be when conveniently applied to data-drivenknowledge discovery problems in bioinformatics.
2. Methods
2.1. Data sources and bioinformatics tools
We have used publicly available datasets of individual or clustersof gene/protein data on Leishmania spp., mainly L. major, L. brazilien-
tandard HMM tools (SUPERFAMILY, GeneDB and Pfam), with TPRpred and with ourhree species (L. major, L. infantum and L. braziliensis).
Pfam TPRpred MRDM
TPR PPR TPR PPR TPR PPR
55 12 101 31 104 3651 11 101 31 104 3442 8 100 31 103 33

2180 M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189
sis, L. infantum and related trypanosomatids (GeneDB (Hertz-Fowleret al., 2004), TryTrypDB (Aurrecoechea et al., 2007) and NCBI/Entrez– www.ncbi.nlm.nih.gov/sites/gquery). Variants of BLAST (Altschulet al., 1997), COBALT (Papadopoulos and Agarwala, 2007) and Glim-merHMM (Majoros et al., 2004) were widely used for sequence sim-ilarity searches, comparisons and gene predictions. External DBsearches were performed against numerous collections of proteinmotifs and families. Gene ontology (GO) (The Gene Ontology Con-sortium, 2000) terms were assigned, based on top matches to pro-teins with GO annotations from Swiss-Prot/trEMBL (www.expasy.org/sprot) and AMIGO after GeneDB (www.genedb.org/amigo/perl)access. Functional assignment of genes and gene products was in-ferred using the RPS-BLAST search against conserved domain DB(CDD) (Marchler-Bauer et al., 2007). For protein domain identifica-tion and analysis of protein domain architectures, Simple ModularArchitecture Research Tool (SMART) (Letunic et al., 2009), Pfam23.0 (Finn et al., 2008), SUPERFAMILY (Madera et al., 2004; Wilsonet al., 2009) and TPRpred (Karpenahalli et al., 2007) were used. Fordocumentation entries describing protein domains, families andfunctional sites, as well as associated patterns and profiles to iden-tify them we have used Prosite (Sigrist et al., 2010). For multiplealignments we used MUSCLE (Edgar, 2004) and a constraint basedalignment tool COBALT (Papadopoulos and Agarwala, 2007).
A detailed description of Leishmania protein datasets is givenelsewhere (TriTrypDB version 2.0 – http://TriTrypDB.org). Our ori-ginal dataset consisted of 25,756 unique sequences (Dataset 1 insupplementary material). The base of the negative samples in alltests consisted of randomly chosen 100 sequences of 300–500 res-idues returned as not-containing TPR motifs. Then, for each exper-iment, sequences matching the studied pattern (TPR-like motifs), ifany, were removed from the negative set. The TPR family cannot becovered by a single PROSITE pattern. Therefore, the positive datasetincluded two extended patterns: PDOC51375 – Pentatricopeptide(PPR) repeat profile (PS51375) and PDOC50005 – TPR repeat pro-files, the latter which include at least two profiles (PS50005; TPRrepeat profile and TPR_REGION, PS50293; TPR repeat region circu-lar profile), as provided by the Expasy Proteomics Server(www.expasy.org). The training set consisted of 5 different in-stances of the 31–35 residue long Prosite patterns (see Section 2.2,for details) and the 34-residue long pattern of SUPERFAMILY (Wil-son et al., 2009). We have added 94 Leishmania sequences from theUniProt matching PS50005 to the positive part of the testing set(Dataset 2 in supplementary material) (The UniProt Consortium,2010). The training set consisted of 11 representative instances(chosen on the basis of low sequence similarity) of these patternsand sequences found in the PROSITE false negative set. Therefore,286 complete sequences matching the patterns formed the posi-tive testing set. The TPR-like consensus pattern had 286 true posi-tive hits, while 34 sequences were missed and there were 76 falsepositives. According to our observations, some residues taking partin the TPR motif might not be covered by the 31–35 residue longpattern. Hence, we extended the length of the training sequencesup to 40 amino acids, according to multiple sequences alignment(see below). Eventually, the training set consisted of 68 represen-tative sequences of the pattern instance and its neighborhood(40 residues altogether).The positive test set consisted of 120non-redundant sequences which belong to the TPR-like superfam-ily (true positive and false negative PROSITE pattern PS50005matches from UniProt database) (The UniProt Consortium, 2010).The patterns that our meta-pattern is based on are given below.
2.2. Definitions of a TPR-like regular expression and of a TPR-likeprotein
As detailed previously (Girão et al., 2008), we have used theconsensus sequence for TPR-like motif as a regular expression (1)
to fully exploit the TPR motif finding problem in Leishmania spp.genomes. A regular expression for a protein sequence defines themost probable amino acid (aa) residue to appear at each positionwithin the protein sequence. Briefly, a TPR motif sequence is a min-imally conserved (degenerate and variable) region of 34-residuelong extension (with exceptions accepted to the range of 31 resi-dues (Koga et al., 1999). A PPR motif is a degenerate 35-residue se-quence, closely related to the 34-residue TPR motif. As reported inour earlier studies (Pacheco et al., 2007; Girão et al., 2008; Pachecoet al., 2009), we systematically solved inconsistencies in the motifannotation by manual expertise. Since motif occurrences are adja-cent in sequences, we could define the motif sequence of a proteinas the succession of motifs read from the N toward the C terminus.Therefore, we have defined a TPR-like protein as any protein se-quence containing a TPR-like motif that fits in our regular expres-sion (1), which also, by reference, confirms to a set of known bonafide domains contained in the TPR-like superfamily [a.118.8] ofSCOP (version 1.73) (Murzin et al., 1995; Karpenahalli et al.,2007), SMART v.6.0 (Letunic et al., 2009), TPRpred (Karpenahalliet al., 2007) and SUPERFAMILY v1.73 (Wilson et al., 2009). Classifi-cation criteria are supported by structural and sequence similarity,plus searches with remote homology prediction.
½WLF� � Xð2Þ � ½LIM� � ½GAS� � Xð2Þ � ½YLF� � Xð8Þ � ½ASE�� Xð3Þ � ½FYL� � Xð2Þ � ½ASL� � Xð4Þ � ½PKE� ð1Þ
As mentioned, lengths of those patterns vary from 30 to 40 aminoacids. Also numbers of true positive hits in different sequences inthe UniProtKB/Swiss-Prot database differ slightly between patterns(The UniProt Consortium, 2010). In order to produce a meta-pat-tern, a representative set of 11 instances of those patterns was cho-sen as a positive training set on the basis of 30% or lower similarityof the pattern. The positive tesst set of 68 sequences was pickedrandomly from the set of 94 sequences (Dataset 2). We must recallthat some sequences contain instances of more than one patternfrom the set. Although none of the PROSITE patterns involved inthe TPR-like meta-pattern was found in the negative dataset, afew sequences described in either PDB or GeneDB as containingTPR-like motifs had to be manually excluded from the set.
2.3. Profile generation for TPR-like superfamily assignment
Performance dependence on sequence profiles relies on eitherthe selectivity or sensitivity of its regime, respectively dependingon the number of close or remote homologues used (Karpenahalliet al., 2007). Here we have established a fixed threshold value toinclude a minimum number of remote homologues, thus avoidingtoo many false positives. Initial profiles were generated by iterativesearches against non-redundant DBs (nrdbs) at NCBI and GeneDB,filtered to a maximum pairwise sequence identity of 60% (nr-60)by CD-HIT (Li et al., 2001, 2002), slightly modified after (Karpenah-alli et al., 2007) in a sense that we have extracted sequences con-servatively with PSI-BLAST through multiple iterations using theTPR-like regular expression (1) as a query sequence. We, then, per-formed iterative searches to convergence on nr-60 minus TPRPs(detected by Pfam, SMART, SUPERFAMILY and TPRPred) with vari-ous threshold parameters to test the resulting profiles on a positive(TPR-like) or negative set (non TPR-like). Best profiles were se-lected based on its performance on a predicted family assignment.We must mention that a large protein can be split into smaller do-mains or motifs, which can occur by themselves or in combinationwith other domains. By definition, a superfamily will group to-gether domains of different families, which have a common evolu-tionary ancestor based on structural, functional and evolutionarydata. In other words, a superfamily contains all proteins for whichthere is structural evidence of a common evolutionary ancestor.

M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189 2181
To provide structural (and hence implied functional) assign-ments to TPR-like proteins at the superfamily level, structured se-quences from available Leishmania and trypanosomatid genomeswere randomly selected and parsed into unique 25,756 Leishmaniasequences (original dataset). Each sequence was a labeled input toa multi-class motif classifier. To pick the best method to representone or more of the three target motifs, we compared the results ofmotif classifiers when the sequence was presented as a (I) TPR-containing, (II) PPR-containing, (III) HAT-containing, (IV) combina-tion of any two or three motifs and (V) not-containing target mo-tifs. Performance was measured (as in Xu et al., 2006) byclassification precision, recall and F1 measure (a composite mea-sure of classification precision and recall).
2.4. Hidden Markov models (HMMs) and the Viterbi algorithm (VA)
An HMM is a probabilistic network of nodes, so called states.One state qi is connected to another state qj by a transition proba-bility ij. Non-silent states are able to emit an alphabet of symbols(Eddy, 1998; Winters-Hilt, 2006). A special topology of HMMs,termed pHMM, is frequently used in homology detection of proteinfamilies (Karplus et al., 1998). Transition and emission probabili-ties are estimated by a maximum likelihood approach combinedwith a standard dynamic programming algorithm for decodingHMMs, the Viterbi (VA) (Forney, 1973) to get site and path depen-dent probabilities for every hidden state in the posterior decoding.In a first validation step we used the feature of trained HMMs toemit domain-specific sequences according to their model parame-ters. Sequences were compared with generated state paths in thesame way as described earlier (Girão et al., 2008; Pacheco et al.,2009). The process of generation was repeated 10 times for everyTPR-like motif. To fully exploit the sequential ordering of motifsin a set, we used pHMMs to label motif types. We have trans-formed the motif categorization problem into an HMM sequencealignment problem. The HMM states correspond to the motif types.Labeling motifs in a sequence is equivalent to aligning the se-quences to HMM states. There are five states in our HMM model:(I) TPR-containing, (II) PPR-containing (II) HAT-containing, (IV)combination of any two or all motifs and (V) not-containing targetmotifs. Transition probabilities between these states were esti-mated from the training data by dividing the number of times eachtransition occurs in the training set by the sum of all the transi-tions. The state emission probabilities were calculated from thescore output reported by the multi-class classifiers. Given theHMM model (Karplus et al., 1998), state emission probabilitiesand state transition probabilities, VA was used to compute most-likely sequence of states that emit (any of the target) motifs in se-quences. Subsequently, the state associated with the motif was ex-tracted from the most-likely sequence of states (Friedrich et al.,2006).
2.5. Stochastic context free grammar (SCFG) Framework
Since an important drawback of HMM profiles is that they arenot human-readable, these descriptors cannot provide biologicalinsight by themselves. The expressive power of HMMs, in the fieldof protein sequence analysis and annotation, is similar to a sto-chastic regular grammar in which they have limitations regardingthe types of patterns they are able to encode (such as nested andcrossing relationships that are very common in proteins) (Dyrkaand Nebel, 2009). The use of SCFGs (formal definition and imple-mentation were elegantly provided by Dyrka and Nebel, 2009)can overcome such HMM limitations (Madera, 2008). Since eachSCFG deals with one amino acid property at a time, scores obtainedby several grammars need to be combined to obtain optimal re-sults. Since a protein can be fully defined by a string composed
of 20 different characters, protein grammar is expected to rely ona large set of terminals (20 aa or residues). Therefore, the spacecontaining the possible rules needed to describe the protein lan-guage is enormous and cannot be searched in a finite time by cur-rent induction techniques. To deal with the size of the proteinalphabet, we have used quantitative properties of amino acids intoa SCFG framework to reduce the number of symbols in a grammar.For each given property, we have defined all the terminal rules ofthe form A ? a, thus associating them with proper probabilities.Three non-terminal symbols (Low, Medium and High) were cre-ated to represent low, medium and high level of the property ofinterest, e.g., the presence of the residue valine in position 4 ofthe scanned motif. These non-terminals are also called ‘propertynon-terminals’ or pNT in a rule where each amino acid (ai) is asso-ciated with each of these non-terminals with a probability (Pr). Fora given property, probabilities were calculated using the knownquantitative values, pval, associated to the amino acids, ai, and,then, normalized so that the following Eq. (2) is true:X
i
PrLowðaiÞ ¼X
i
PrMediumðaiÞ ¼X
i
PrHighðaiÞ ¼ 1: ð2Þ
As proposed by Dyrka and Nebel (2009) in considering that all ter-minal rules are fixed with given probabilities, only the probabilitiesof the subset of rules in the form of A ? BC are subject to evolution.To avoid trivial solutions, non-terminals and Left-Hand Side (LHS)symbols, as well as ‘independent non-terminals’ or iNT were usedto further reduce the size of the possible rule space. Usually, prop-erties of each amino acid can be represented by a single feature inthe induced grammar, but here we have generated one grammarper relevant physiochemical property. Protein sequences wereparsed for each grammar and their parsing scores were combinedto achieve more robust results: the final score is an arithmetic aver-age of the scores obtained for each single property grammar.
The rates for grammars, based on different properties, theircombinations and their comparison with scores, were obtainedby PROSITE patterns (Sigrist et al., 2010). Overall, SCFGs were ableto detect relevant sequences missed by the PROSITE patterns forTPR and TPR-like motifs. The modified version of the Cocke–Kas-ami–Younger (CKY) algorithm, called Stochastic CKY (imple-mented by Dyrka and Nebel (2009)), was used to allow thatSCFGs can be parsed. The outcome of this procedure is the proba-bility that a given sequence was generated by a certain grammar G(i.e. the sequence belongs to the language L(G)) instead of a Bool-ean value as in the case of non-probabilistic CKY. This probabilityis defined as a product of probabilities of all grammar rules in-volved in the construction of the corresponding parse tree. A gen-eral approach to scaling within Viterbi-style CKY parsing scheme isto calculate normalized scores (Score’):
Score0 ¼ Score� A� ðRAÞi; ð3Þ
where A compensates the decrease in score caused by linking an-other terminal (A ? a), R compensates the decrease in score causedby invoking a rule (A ? BC) and i is the parsing level. As it is difficultto calculate the values of A and R, an empirical scaling factor, F = RA,was introduced. It is based on the change in average raw (notscaled) scores with increasing parse tree spans calculated over alarge dataset for a given grammar. If a grammar is correctly in-duced, the most likely parse tree of a binding site sequence reflectsits structural features (Sakakibara et al., 1994), whereas the basicversion of the parser returns a score which is the log of probabilityrather than the probability itself. Therefore, it is the average log ofprobabilities for all training samples which is optimized in thetraining step. A Genetic algorithm (GA) was used for grammarinduction in which a single individual represents a whole grammar.This strategy evolves probabilities of grammar rules, and the

2182 M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189
implementation of such grammar induction algorithm is based onMatthew Wall’s GAlib library, which provides a set of C++ geneticalgorithm objects (Dyrka, 2007). To normalize results regardingsequence length, the objective function is defined as an arithmeticaverage of logs of probability returned by the parsing algorithmfor each positive sample:
f ðXÞ ¼
PjSj
ilog PrðWijGÞ
jSj ; ð4Þ
where S is the positive sample, Wi is a sequence from S, G is a givengrammar and Pr(Wi|G) is the probability that Wi belongs to L(G), asposed by Dyrka and Nebel (2009). The algorithm stops when thereis no further significant improvement in the score.
2.6. Multi-relational data mining (MRDM) method fundamentals
Algorithms for relational association rule discovery (RARD) arewell suited for exploratory data mining due to the flexibility re-quired to experiment with examples more complex than featurevectors and patterns more complexes than item sets (Friedmanet al., 1999), such as the case with TPR-like motifs. An adequate ap-proach of machine learning (Getoor et al., 2001a) focuses on learn-ing a complex web of relationships among a collection of diverseobjects rather than supervised learning from independent andidentically distributed training examples (a classifier f that givenan object x would produce as output a classification labely = f(x)). Such formalism, developed as probabilistic relationalmodels (PRMs) (Getoor et al., 2001a,b), can represent these websof relationships and support learning and reasoning with them(Craven et al., 2000). PRMs are a multi-relational form of Bayesiannetworks that allow descriptions of a template for a probabilitydistribution. This, together with a set of motif objects, defines a dis-tribution over the attributes of the objects (Getoor, 2001). Such amodel can, then, be used for reasoning about an entity, using theentire rich structure of knowledge encoded by the relational repre-sentation (Getoor et al., 2001b; Segal et al., 2001; Getoor and Grant,2006). For each PRM, we were interested in constructing a modelwhose trades off fit to data with the TPR-like motif model com-plexity. This tradeoff allows us to avoid fitting the training datatoo closely, which would reduce our ability to predict unseen data.
2.7. Computational complexity
To deal with the size of the protein alphabet, we have treatedeach physiochemical property separately and introduced 3 non-terminal rules which express their quantitative values. Nonethe-less, the computational complexity of parsing a SCFG remains anissue, since it is cubic in time and square in space regarding thelength of the input sentence Dyrka and Nebel, 2009). For proteinsequence scanning (the average length of a sequence is around350 amino acids), the time complexity is a critical issue in partic-ular during the process of grammar induction. Typically a popula-tion made of hundreds of grammars needs to be parsed at eachstep of the iterative process. Moreover, since the structure of thefinal grammar is initially unknown, the initial population of gram-mars should have a set of rules large enough so that it has the po-tential to express the properties of the pattern of interest, in thepresent case the TPR motif. To better exploit capabilities and man-ageability, we have chosen to induce grammars using a constantpopulation of 30 sequences, because, for such a population size,the total number of rules of each sequence should not exceed 30to be able to generate a grammar within a reasonable amount oftime (a few hours). This constraint was made by supplying each se-quence with a complete set of rules associated with random prob-
abilities in order not to introduce any bias in the evolution ofgrammars. Since the Chomsky Normal Form for rules was used,the number of possible rules is bounded by the cube of numberof non-terminals. Therefore, in this scheme no more than 3 non-terminals can be introduced to keep the number of rules below30. For each grammar generation, we produced several grammars(usually 3) whose scanning results were combined using arithmet-ical averaging of scores. Using an advanced PC workstation (Intel�
Xeon� Quad-Core with 6,4 GT/s (Intel QuickPath), 2.8 GHz, 8 MBcache processor), the time needed for producing all grammarsassociated to a given TPR-like pattern was typically of 6–8 h.
3. Results and discussion
While data mining and annotation of the genomes of threeLeishmania species (at GeneDB, Sanger Center) has provided a com-plete inventory of predicted proteins and a fairly good estimativeof their containing motifs and associated roles and functions, re-sources for integrating this information into effective knowledgeare still greatly needed and currently underway. Given an originaldataset of 24,708 protein sequences (Dataset 1 in supplementarymaterial), there are a number of prediction tasks to be addressed.These include predicting additional genes, and detecting howgenes and proteins are organized into functional substructures(motifs or domains), in cases where this is not known. Note thatthese tasks are clearly multi-relational in that the representationincludes attributes of genes (e.g. sequence coordinates), relationsamong genes and proteins (e.g. containing the same motifs), andrelations between genes and other entities, such as cellular locali-zation and GO terms. Additionally, the records in the tables arelinked to these entities through their sequence coordinates. Re-cently we have considered various approaches to recognizing func-tional elements in flagellar proteins from Leishmania genomes. Inour recent works (Pacheco et al., 2007, 2009; Girão et al., 2008),we have employed probabilistic language models to simulta-neously predict TPR, PPR and HAT motifs, whereas now we extendthis research to a preliminary benchmarking with existing HMMmethods. Our model is, for the most part, an HMM, although acomponent of it is actually stochastic context free grammar (SCFG)(Dyrka and Nebel, 2009). The Viterbi path (Pacheco et al., 2007,2009) is the most likely derivation (parse) of the sequence, bythe given SCFG, to compute the total probability of all derivationsthat are consistent with a given sequence, based on some SCFGs.This is equivalent to the probability of the SCFG generating the se-quence, and is intuitively a measure of how consistent the se-quence is with the given grammar (Rivas and Eddy, 2001). Weargue that most of the relational structure in protein motif discov-ery within sequence analysis is sequential. Therefore, probabilisticlanguage models are especially suited to the task. Since the expres-sive power of an HMM is similar to a stochastic regular grammar(Durbin et al., 1998), they have limitations regarding the types ofpatterns they are able to encode. Context Free Grammars (CFGs)have the potential to overcome some of the limitations of HMMbased schemes because they have the next level of expressivenessin Chomsky’s classification and produce human-readable descrip-tors. CFGs comprise a reduced complexity that makes them morepractical and allows the possibility of learning grammar structurefrom examples (Dyrka and Nebel, 2009), such as we here applyto a group of TPR-like motifs/domains. Moreover, CFGs can be usedto model dependencies between different parts of a protein, suchas binding sites, motifs and domains, by using branching rules.Thus, the interest in development of grammars which have theabilities to model branched and nested relationships should permitto improve modeling of several motif-containing proteinsequences.

M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189 2183
3.1. TPR-like motif localization task
We have applied two MRDM methods (RARD and PRMs) afterHMMs/VA to mine TPR, PPR and HAT repeats in protein sequencesof Leishmania spp. Provided six variants of the data set for the TPR-likemotif localization task (considering four TPRs, one PPR and one HATin the TPR-like clan), the first version consisted of a single table with25,756 attributes and the second consisted of two tables with 26attributes in total. We used a normalized version of the data set withtwo tables. The names of the two original tables are motifs_relationand interactions_relation. The motifs_relation table contained 120different motifs but there could be more than one row in the tablefor each motif. The attribute motif_id identifies a motif uniquely.Since our current implementation of MRDM requires that the targettable must have a primary key, it was necessary to normalize themotifs_relation table before we could use it as the target table. Thisnormalization was achieved by creating the tables named motif,interaction, and composition as follows. Attributes in themotifs_relation table that did not have unique values for each motifwere placed in composition table and the rest of attributes wereplaced in motif table. The motif_id attribute is a primary key in themotif table and a foreign key in composition table. The interaction ta-ble is identical to the original interactions_relation table. This repre-sents one of several ways of normalizing the original table andrenormalization of the relational DB has an impact on the entity-rela-tion diagram for the renormalized version of Motif Localization db.Thus, for TPR-like motif localization task, the target table is motifand the target attribute is localization. From this point of view, thetraining set consists of 120 motifs and the test set 68. The experi-ments described here focused on building a classifier for predictingthe localization of motif-containing proteins by assigning the corre-sponding instance to one of six possible localizations. For this motiflocalization task, we have chosen to construct a classifier using alltraining data and test the resulting classifier on the test set providedby Leishmania sequences (original dataset). The design of an unbiasednegative sample is particularly difficult in protein sequence analysis,what prompted us to use an alternative approach based on stochasticgrammars (SCFGs) which, in principle, do not require a negative setfor their inference, as recently revisited by Dyrka and Nebel (2009)in their SCFG-based framework that is based on its fitting to the train-ing sample of sequences, i.e. the selected grammar is the one whichgenerates maximal log probabilities over the whole set. In order tonormalize results regarding sequence length, the objective functionwas defined as an arithmetic average of logs of probability returnedby the parsing algorithm for each positive sample.
This task presents significant challenges because many attributevalues in training instances corresponding to the 120 training mo-tifs are missing. Initial experiments using a special value to encodea missing value for an attribute resulted in classifiers whose accu-racy is around 40% on the test data. This prompted us to investigateincorporation of other approaches to handling missing values.Replacing missing values by the most common value of the attri-bute for the class during training resulted in an accuracy of around68%. This shows that providing reasonable guesses for missing val-ues can significantly enhance the performance of MRDM on ourdata sets. However, in practice, since class labels for test data areunknown, it is not possible to replace a missing attribute valueby the most frequent value for the class during testing. Hence,there is a need for better ways of handling missing values (e.g., pre-dicting missing values based on values of which attributes?).
3.2. Identification of the TPR-like superfamily in Leishmania spp.genomes
The percentage of repeat-containing proteins, such as TPR-like,grows with the complexity of the organism, with repeat proteins
being particularly abundant in multicellular organisms (Bjorklundet al., 2006). Genomes of unicellular eukaryotes, as Leishmania,usually possess a relatively high number of putative encodinggenes (around 8000 genes in L. major, e.g.) (Hertz-Fowler et al.,2004). Analyses of such a large number of coded proteins requirethat the characterization of a given family of proteins be dependenton detection of regions of their sequences shared by all familymembers. Computing the consensus of such regions provides amotif that is used to recognize new members of the family (Servantet al., 2002).
With the sequencing completion of 03 Leishmania and severaltrypanosomes genomes (Hertz-Fowler et al., 2004), we were ableto search for all TPR-like genes in Leishmania using the definingcharacteristic of a TPR-like protein. As depicted by Table 1, num-bers of members detected through different tools (GeneDB/SMART,Pfam, SUPERFAMILY and TPRpred) are shown in comparison to ourmethod (MRDM/HMM/VA), which is able to assign a significantlylarger number of TPR-like motif-containing proteins in Leishmania:104 TPRs and 36 PPRs at the most and 08 HATs in total (Table 2).These members are elements putatively involved in several keycellular processes, such as glycosome biogenesis (PEX5 andPEX14) and flagellar pathways (IFT subunits, cyclophilins, phos-phatases), besides binding partners of either motor or cargo pro-teins (kidins220/ARMS and other members of the KAP family ofP-loop NTPases) or those involved with assembly or disassemblyof protein complexes. The resulting descriptions of the familiesand its members, a good example of relevant patterns found alongwith reasonable assignment of family members with our approach,should provide a solid and unified platform on which future genet-ic and functional studies regarding Leishmania TPRPs can be based.
3.3. TPR-encoding genes in Leishmania spp. genomes
We first used the alignment of 275 sequences previously iden-tified as putative strict TPR-containing motifs (obtained fromSUPERFAMILY, SMART, Pfam and TPRpred) to obtain the consensusmodel. This TPR signature matrix was subsequently used to searchfor TPR motifs in the six reading frames of whole Leishmania gen-omes. Multiple alignment of Leishmania TPRP sequences revealedthat most of substitutions in the TPRs occur at nonconsensus posi-tions; consensus residues are selectively conserved between ortho-logues (particularly in Trypanosoma spp.). Because TPR motifs arehighly degenerate, a fairly large number of false positive hits wereexpected. However, because TPR motifs appear usually as tandemrepeats, we could remove most random uninteresting matches byomitting all orphan TPR motifs that were found farther than 200nucleotides from any other TPR motif. The 465 TPR motifs retainedformed 132 clusters, each of which comprised a putative TPR gene.Each TPR motif cluster was then investigated in detail by manuallyanalyzing the positions and reading frames of the TPR motifs com-pared with available Leishmania genomes (1) open reading frame(ORF) models and (2) predicted protein sequences within potentialcoding sequences. From this analysis, 104 putative TPR ORF modelswere constructed (i.e., 28 motif clusters were discarded or fusedwith other clusters). TPR genes are fairly evenly distributedthroughout the 36 mini-chromosomes of L. major, with little inthe way of obvious clusters. The densest grouping of TPR genes lieson chromosomes 30, 32 and 36, the latter which contains 13 genes,the maximum number found in any isolate chromosome of L. major(Table S1).
The number of proteins containing TPR motifs and the distribu-tion of individual repeats detected confidently by our MRDM/HMM/VA method are illustrated and tabulated in Figs. 1 and 2.We were able to detect more proteins as TPR-containing pro-teins in Leishmania genomes than all compared resources (Pfam,SUPERFAMILY, SMART and TPRpred) and over 2 fold more individual

2184 M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189
repeats than Pfam and SMART, but not TPRpred, which, as a profile-sequence comparison method, shows a marked improvement overexisting methods based on regular HMM (such as HMMER thatruns on Pfam and SMART web-servers and SAM that runs on Super-family web-server), particularly in the detection of non-canonical,divergent repeats. We have benefited from all TPRpred improve-ments (Karpenahalli et al., 2007), extending its scope to includeHAT motifs.
3.4. Functional features of TPR-like Motifs
The preliminary functional predictions of a range of familymembers performed here, together with the sparse data on theseproteins in Leishmania published so far, allows us to propose puta-tive models in which TPR-like proteins might play the role of se-quence-specific adaptors for a variety of other RNA-associatedproteins. Structural models of Leishmania TPRPs were predicted(Fig. 3) in an attempt to aid such studies, yet requiring further test-able hypothesis. Nonetheless, one can surround a testable predic-tion: that TPR-like proteins in Leishmania might be directly orindirectly associated with specific RNA sequences and with definedeffector proteins, as previously suggested in Arabidopsis (Lurinet al., 2004). The rational for utilizing SCFGs to produce motif find-ing descriptors is that not only they have the power to expressbranched and nested like dependencies, but also their rules canbe analyzed to acquire biological knowledge about recognizingmotifs of interest. In this study, we illustrate how analysis of se-
Fig. 1. Schematic diagram of TPR-containing genes in genomes of Leishmania major, L. iMarkov model/Viterbi algorithm approach (MRDM/HMM/VA). Shared orthologues amoidentifiers (GeneDB IDs) for putative TPRPs are given as lateral tables. (For interpretatioversion of this article.)
quence based SCFGs allows gaining additional insight into theidentification of TPR-like motifs in Leishmania protein sequences(mostly annotated as hypothetical conserved). We have analyzedprobabilistic grammars to extract biologically meaningful featuresfocusing on either parse trees or grammar rules. We have startedby providing an in-depth study of grammars produced to describetwo extended patterns: PDOC51375 – Pentatricopeptide (PPR) re-peat profile (PS51375) and PDOC50005 – TPR repeat profiles, thelatter which include at least two profiles (PS50005; TPR repeat pro-file and TPR_REGION, PS50293; TPR repeat region circular profile).These two profiles were developed for the module PDOC50005, thefirst one picks up TPR repeat units while the second profile is ‘cir-cular’ and will, thus, detects a region containing adjacent TPR re-peats (Expasy Proteomics Server – www.expasy.org). Throughthis analysis, we have used all the sequence information of Leish-mania genomes (coding sequences and predicted proteins), i.e.the original dataset (Supplementary Dataset 1), to visualize thegeneral structure of the motif and its description as provided bythe grammar parse trees. We have focused our attention on gram-mars based on residue features (small and large hydrophobic ami-no acids), propensity to turn-positions both between the twohelices of a single TPR and between the two adjacent TPRs, andpropensity to conservation of helix-breaking residues. These gram-mars are known as being the most informative to describe thePDOC50005 pattern.
Future work needs to be directed toward the identification ofthese factors to elucidate the precise functions of one of the largest
nfantum and L. braziliensis, detected after a multi-relational data mining and hiddenng the three species are illustrated as colored circles intersections and individualn of the references to colour in this figure legend, the reader is referred to the web
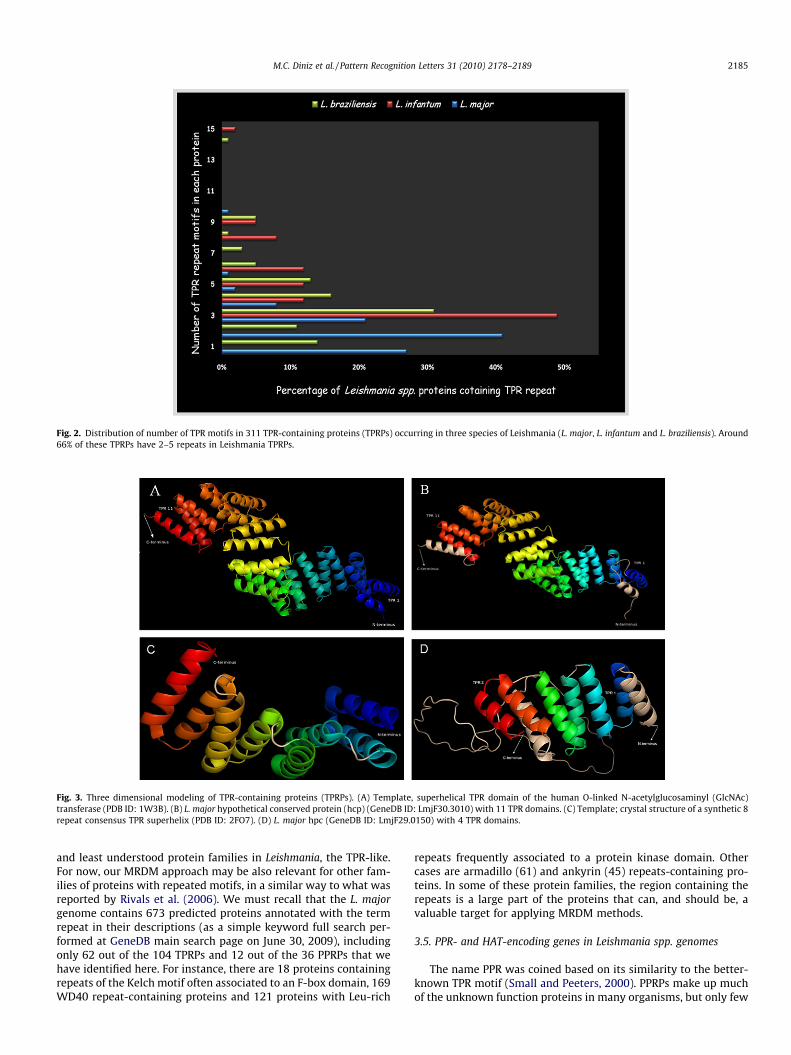
Fig. 2. Distribution of number of TPR motifs in 311 TPR-containing proteins (TPRPs) occurring in three species of Leishmania (L. major, L. infantum and L. braziliensis). Around66% of these TPRPs have 2–5 repeats in Leishmania TPRPs.
Fig. 3. Three dimensional modeling of TPR-containing proteins (TPRPs). (A) Template, superhelical TPR domain of the human O-linked N-acetylglucosaminyl (GlcNAc)transferase (PDB ID: 1W3B). (B) L. major hypothetical conserved protein (hcp) (GeneDB ID: LmjF30.3010) with 11 TPR domains. (C) Template; crystal structure of a synthetic 8repeat consensus TPR superhelix (PDB ID: 2FO7). (D) L. major hpc (GeneDB ID: LmjF29.0150) with 4 TPR domains.
M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189 2185
and least understood protein families in Leishmania, the TPR-like.For now, our MRDM approach may be also relevant for other fam-ilies of proteins with repeated motifs, in a similar way to what wasreported by Rivals et al. (2006). We must recall that the L. majorgenome contains 673 predicted proteins annotated with the termrepeat in their descriptions (as a simple keyword full search per-formed at GeneDB main search page on June 30, 2009), includingonly 62 out of the 104 TPRPs and 12 out of the 36 PPRPs that wehave identified here. For instance, there are 18 proteins containingrepeats of the Kelch motif often associated to an F-box domain, 169WD40 repeat-containing proteins and 121 proteins with Leu-rich
repeats frequently associated to a protein kinase domain. Othercases are armadillo (61) and ankyrin (45) repeats-containing pro-teins. In some of these protein families, the region containing therepeats is a large part of the proteins that can, and should be, avaluable target for applying MRDM methods.
3.5. PPR- and HAT-encoding genes in Leishmania spp. genomes
The name PPR was coined based on its similarity to the better-known TPR motif (Small and Peeters, 2000). PPRPs make up muchof the unknown function proteins in many organisms, but only few

2186 M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189
of these PPRPs have functional roles ascribed, although a putativeRNA-binding function is widely accepted (reviewed in Lurinet al., 2004) and one PPRP is involved in RNA editing (Koteraet al., 2005). The existence of a large family of PRPPs only becameapparent with the Arabidopsis Genome Initiative that revealed 446PPR coding genes – 6% of its entire genome (Lurin et al., 2004). ThePPRP family has been divided, according to their motif content andorganization, into two subfamilies: the PPRP-P and the exclusiveplant combinatorial and modular proteins (PCMPs). PPR motifshave been found in all eukaryotes analyzed to date, but with anextraordinary discrepancy in numbers between plant and nonplantorganisms (the human genome encodes only six putative PPRPs).Trypanosomatids and other flagellated organisms are expected tohave an intermediate number (around one hundred PPR genes), anumber still far from the 36 PPR-encoding genes we have foundhere (12 of them annotated at GeneDB as conserved hypotheticalproteins of L. major) (Fig. 4). Recent reports (Mingler et al., 2006;Pusnik et al., 2007) mention less than thirty (respectively 23 and28) PPRPs identified in Trypanosoma brucei (with at least 25 ortho-logues found in L. major) and with a predicted indication that mostof these proteins are targeted to mitochondria.
The HMMs used here were identical to those used for detectionof PPR motifs in plants (Lurin et al., 2004; O’Toole et al., 2008). Asmultiple models based on PPR variants were used, multiple over-lapping hits were usually obtained. In these cases, the highest scor-ing chain of nonoverlapping hits was retained and the alternativeoverlapping hits discarded. As an initial screen, genomic nucleotidesequence data in Leishmania were translated in all six frames. Thesearch was applied to the translated sequence data to identify clus-ters of all PPR motifs separated by fewer than 200 base pairs. Theseclusters correspond to putative PPR genes. As of Release 2.1 of Gen-eDB (www.genedb.org) with curated annotations of Leishmaniagenes, 13 of the GeneDB ORFs are annotated as conserved hypo-thetical proteins that contain PPR motifs based on matches with
Fig. 4. Schematic diagram of PPR-containing genes in genomes of Leishmania major, L. iMarkov model/Viterbi algorithm approach (MRDM/HMM/VA). Shared orthologues amoidentifiers (GeneDB IDs) for putative PPRPs are given as lateral tables. (For interpretatioversion of this article.)
the PFAM profile PF01535 or SMART profile IPR002885. None ofLeishmania GeneDB models are annotated as homologues of knownPPRPs. Of the two sets of ORF models (ours and GeneDB’s), 12 areidentical (i.e., our analysis agreed with the GeneDB model). The13th GeneDB model does not have an equivalent in our set becausewe did not consider it to be a PPRP by our criteria (lacking tandemmotifs matching our HMM profiles) 21 of our models have no Gen-eDB equivalent and correspond to genes apparently overlookedduring annotation or considered to be pseudogenes. In all, 22 ofour 36 models differ in at least some respects from the correspond-ing GeneDB model, but correspond quite well to the 28 PPRPs iden-tified in T. brucei (Pusnik et al., 2007), what reinforces how wellconserved PPR genes seem to be in trypanosomatids. It should benoted that in very few of these cases are molecular data availablethat can be used to decide between discordant models. Our choicehas been generally made by comparison with other genes in thefamily and a general expertise with these proteins. A noticeablecharacteristic of PPR genes is that they rarely contain introns with-in coding sequences even in higher eukaryotes (more than 80% ofknown PPR genes of plants unexpectedly do not contain introns),what is also true for Leishmania ORF models (an obvious extensionfor trypanosomatid genes that usually do not contain introns any-way). This feature might explain why PPR genes are relatively short(on average <2 kb) although PPRPs are comparatively large pro-teins (680 aa on average).
HAT repeats have three aromatic residues with a conservedspacing, being structurally and sequentially similar to TPRs/PPRs,although they lack the highly conserved alanine and glycine resi-dues found in TPRs. The number of HAT repeats found in differentproteins varies between 9 and 12. HATPs appear to be componentsof macromolecular complexes that are required for RNA processingand the HAT motif has striking structural similarities to HEAT re-peats (IPR000357), being of a similar length and consisting oftwo short helices connected by a loop domain, as in HEAT repeats
nfantum and L. braziliensis, detected after a multi-relational data mining and hiddenng the three species are illustrated as colored circles intersections and individualn of the references to colour in this figure legend, the reader is referred to the web

Fig. 5. Multiple sequence alignment of two HAT (half-a-TPR) motifs present in predicted proteins of Leishmania major (LmjF28.1020 and XP_001684383.1), Leishmaniainfantum (XP_001470154.1), Leishmania braziliensis (XP_001566140.1) and Trypanosoma cruzi strain CL Brener (XP_809989.1). HAT motif residues are shown with dark andlight gray shaded boxes for identical or similar residues, respectively. Well conserved aromatic residues are commonly observed at positions 11 and 25 of HAT motifs. Aschematic consensus for HAT (Champion et al., 2008) is aligned with TPR consensus to illustrate Leishmania predicted HAT motif-containing proteins (HATPs). Boxessurround residues in HATPs that are predicted to form alpha helices based on the TPR structure. Note that in TPRs, the conserved W is at position 4, and the conserved P is atposition 32, resulting in a repeat that encodes helix A followed by helix B. In HATs, helix B is followed by helix A and the alignment reflects the HAT consensus.
M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189 2187
(Preker and Keller, 1998; Small and Peeters, 2000; Kotera et al.,2005; Lurin et al., 2004; Rivals et al., 2006). Our survey identifieda total of 08 putative HATPs (Table 2) in the three species of Leish-mania analyzed, but the lack of general information on HATPs doesnot allow any further indication on their definite significance onthe protozoan genome.
As the first in silico identification of HAT-containing genes inLeishmania, we provide a multiple alignment in Fig. 5 to illustratetwo HAT repeats in full trypanosomatid HATP homologues. Thedetection of such a small, but significant, presence of HATPs inLeishmania (Table 2) is certainly an issue for future investigation.Several proteins have been found to interact with HATPs, but nospecific ligand for a HAT domain has been identified so far (Cham-pion et al., 2008).
4. Conclusions
We have performed bioinformatics analyses of Leishmania TPR,PPR and HAT genes/proteins (the TPR-like Superfamily) with anintegrated MRDM/HMM/VA approach that, in contrast to othercurrently available resources (more general HMMER resources asPFAM, SMART, SUPERFAMILY, or specific tools such as TPRpred),seeks to capture as much model information as possible in the pat-tern matching heuristic, without resorting to more standard motifdiscovery methods. TPR genes are ubiquitous, whereas PPRs andHATs are mostly found in eukaryotes, but, in common, they havethe fact of being largely unexplored in Leishmania parasites. Thediffusion of new developments and applications of MRDM algo-rithms and techniques to data-driven knowledge discovery prob-lems in bioinformatics and computational biology is a futuredirection towards better power of biological inference after se-quence and structural analyses. The advantages of our MRDMmethod in detecting protein motifs of TPR-like superfamily mem-bers could be explained by the fact that we have incorporated opti-mized profiles, score offsets and distribution to compute
probability, as deployed by TPRpred, altogether with a Viterbi algo-rithm improved HMM method which, in turn, has been used in or-der to take in account the tendency of repeats to occur in tandemand to be widely distributed along the sequences. This integratedMRDM/HMM/VA approach might be applied to other sequenceanalysis problems in such a way that it contributes to motif discov-eries with a better probability that a target sequence is, indeed, atarget motif-containing protein.
5. Authors’ contributions
MCD, ACLP and KTG were involved in the generation, analysisand interpretation of the data. FFA implemented the PERL scripts,programmed the HMM improvement with Viterbi algorithm andthe integration of MRDM/SCFGs. ACLP, CAW and DMO draftedthe manuscript. DMO planned and supervised the overall work.MCD, CAW and DMO critically revised the manuscript. All authorsread and approved the final manuscript.
Acknowledgements
Financial support for this study was provided by the Brazilianfunding agencies CNPq (National Council for Scientific and Techno-logical Development, Grant# 486266/2006-0) and FUNCAP (TheState of Ceará Research Foundation) through graduate fellowshipsto MCD and ACLP; as well as from Universidade Estadual do Ceará(UECE) internal funds. The funders had no role in study design,data collection and analysis, decision to publish, or preparationof the manuscript.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.patrec.2010.04.008.

2188 M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189
References
Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., Lipman, D.J.,1997. Gapped BLAST and PSI-BLAST: A new generation of protein databasesearch programs. Nucleic Acids Res. 25, 3389–3402.
Aurrecoechea, C., Heiges, M., Wang, H., Wang, Z., Fischer, S., Rhodes, P., Miller, J.,Kraemer, E., Stoeckert Jr., C.J., Roos, D.S., Kissinger, J.C., 2007. ApiDB: Integratedresources for the apicomplexan bioinformatics resource center. Nucleic AcidsRes. 35, D427–D430.
Blatch, G.L., Lässle, M., 1999. The tetratricopeptide repeat: A structural motifmediating protein–protein interactions. BioEssays 21, 932–939.
Bjorklund, A.K., Ekman, D., Elofsson, A., 2006. Expansion of protein domain repeats.PLoS Comput. Biol. 2, e114.
Burge, C., Karlin, S., 1997. Prediction of complete gene structures in human genomicDNA. J. Mol. Biol. 268, 78–94.
Champion, E.A., Lane, B.H., Jackrel, M.E., Regan, L., Baserga, S.J., 2008. A directinteraction between the Utp6 half-a-tetratricopeptide repeat domain and aspecific peptide in Utp21 is essential for efficient pre-rRNA processing. Mol.Cell. Biol. 28, 6547–6556.
Craven, M., Page, D., Shavlik, J., Bockhorst, J., Glasner, J., 2000. A probabilisticlearning approach to whole-genome operon prediction. In: Proc. EighthInternat. Conf. on Intelligent Systems for Molecular Biology, AAAI Press, LaJolla, CA, pp. 116–127.
D’Andrea, L.D., Regan, L., 2003. TPR proteins: The versatile helix. Trends Biochem.Sci. 28, 655–662.
Das, A.K., Cohen, P.W., Barford, D., 1998. The structure of the tetratricopeptiderepeats of protein phosphatase 5: Implications for TPR-mediated protein–protein interactions. EMBO J. 17, 1192–1199.
Dehaspe, L., De Raedt, L., 1997. Mining association rules in multiple relations. In:Proc. Sevneth Internat. Workshop on Inductive Logic Programming, vol. 1297,Springer-Verlag, LNAI, Heidelberg.
Durbin, R., Eddy, S.R., Krogh, A., Mitchison, G.J., 1998. Biological Sequence Analysis:Probabilistic Models of Proteins and Nucleic Acids, first ed. CambridgeUniversity Press, Cambridge, UK.
Dyrka, W., 2007. Probabilistic Context-Free Grammar for Pattern Detection inProtein Sequences. In: MSc Thesis. Kingston University, London.
Dyrka, W., Nebel, J.-C., 2009. A stochastic context free grammar based frameworkfor analysis of protein sequences. BMC Bioinf. 10, 323.
Eddy, S.R., 1998. Profile hidden Markov models. Bioinformatics 14, 755–763.Edgar, R.C., 2004. MUSCLE: Multiple sequence alignment with high accuracy and
high throughput. Nucleic Acids Res. 32, 1792–1797.Finn, R.D., Tate, J., Mistry, J., Coggill, P.C., Sammut, J.S., Hotz, H.R., Ceric, G., Forslund,
K., Eddy, S.R., Sonnhammer, E.L., Bateman, A., 2008. The Pfam protein familiesdatabase. Nucleic Acids Res. 36 (Database Issue), D281–D288.
Forney, G.D. Jr., 1973. The Viterbi algorithm. In: Proc. IEEE, vol. 61. p. 268.Friedman, N., Getoor, L., Koller, D., Pfeffer, A., 1999. Learning probabilistic relational
models. In: Proc. Internat. Joint Conf. on Artificial Intelligence, MorganKaufman, Stockholm, Sweden, pp. 1300–1307.
Friedrich, T., Pils, B., Dandekar, T., Schultz, J., Müller, T., 2006. Modelling interactionsites in protein domains with interaction profile hidden Markov models.Bioinformatics 22, 2851–2857.
Garey, M.R., Johnson, D.S., 1979. Computers and Intractability: A Guide to theTheory of NP-Completeness. W.H. Freeman, p. 202. ISBN:0-7167-1045-5. A1.4:GT 48.
Getoor, L., 2001. Multi-relational data mining using probabilistic relational models:Research summary. In: Knobbe, A.J., van der Wallen, D.M.G. (Eds.), Proc. FirstWorkshop in Multi-relational Data Mining, KDD, 2001.
Getoor, L., Friedman, N., Koller, D., Pfeffer, A., 2001a. Learning probabilisticrelational models. In: Dzeroski, S., Lavrac, N. (Eds.), Relational Data Mining.Kluwer, pp. 307–335.
Getoor, L., Grant, J., 2006. PRL: A probabilistic relational language. Mach. Learn. J. 62,7–31.
Getoor, L., Taskar, B., Koller, D., 2001. Using probabilistic models for selectivityestimation. In: Proc. ACM SIGMOD Internat. Conf. on Management of Data, ACMPress, 2001, pp. 461–472.
Girão, K.T., Oliveira, F.C.E., Farias, K.M., Maia, I.M.C., Silva, S.C., Gadelha, C.R.F.,Carneiro, L.D.G., Pacheco, A.C.L., Kamimura, M.T., Diniz, M.C., Silva, M.C.,Oliveira, D.M., 2008. Multi-relational Data Mining for TetratricopeptideRepeats (TPR)-Like Superfamily Members in Leishmania spp.: Acting-by-Connecting Proteins. Lecture Notes in Computer Science, vol. 5265, PatternRecognition in Bioinformatics, 2008, pp. 359–372. doi:10.1007/978-3-540-88436-1.
Gough, J., Karplus, K., Hughey, R., Chothia, C., 2001. Assignment of Homology toGenome Sequences using a Library of Hidden Markov Models that Represent allProteins of Known Structure. J. Mol. Biol. 313, 903–919.
Groves, M.R., Barford, D., 1999. Topological characteristics of helical repeat proteins.Curr. Opin. Struct. Biol. 9, 383–389.
Hertz-Fowler, C., Peacock, C.S., Wood, C., Aslett, M., Kerhornou, A., Mooney, P., Tivey,A., Berriman, M., Hall, N., Rutherford, K., Parkhill, J., Ivens, A.C., Rajandream,M.A., Barrel, B., 2004. GeneDB: A resource for prokaryotic and eukaryoticorganisms. Nucleic Acids Res. 32, D339–D343.
Ideker, T., Bafna, V., Lemberger, T., 2007. Integrating scientific cultures. Mol. Syst.Biol. 3, 105–112.
Karpenahalli, M.R., Lupas, A.N., Söding, J., 2007. TPRpred: A tool for prediction ofTPR-, PPR- and SEL1-like repeats from protein sequences. BMC Bioinf. 8, 2.
Karplus, K., Barrett, C., Hughey, R., 1998. Hidden Markov models for detectingremote protein homologies. Bioinformatics 14, 846–856.
Kobe, B., Kajava, A.V., 2000. When protein folding is simplified to protein coiling:The continuum of solenoid protein structures. Trends Biochem. Sci. 25, 509–515.
Koga, H., Terasawa, H., Nunoi, H., Takeshige, K., Inagaki, F., Sumimoto, H., 1999.Tetratricopeptide repeat (TPR) motifs of p67phox participate in interaction withthe small GTPase Rac and activation of the phagocyte NADPH oxidase. Biol.Chem. 274, 25051–25060.
Kotera, E., Tasaka, M., Shikanai, T., 2005. A pentatricopeptide repeat protein isessential for RNA editing in chloroplasts. Nature 433, 326–330.
Lawrence, C., Altschul, S., Boguski, M., Liu, J., Neuwald, A., Wootton, J., 1993.Detecting subtle sequence signals: A Gibbs sampling strategy for multiplealignment. Science 262, 208–214.
Letunic, I., Doerks, T., Bork, P., 2009. SMART 6: Recent updates and newdevelopments. Nucleic Acids Res. 37 (Database issue), D229–D232.
Li, W., Jaroszewski, L., Godzik, A., 2001. Clustering of highly homologous sequencesto reduce the size of large protein databases. Bioinformatics 17, 282–283.
Li, W., Jaroszewski, L., Godzik, A., 2002. Tolerating some redundancy significantlyspeeds up clustering of large protein databases. Bioinformatics 18, 77–82.
Lurin, C., Andrés, C., Aubourg, S., et al., 2004. Genome-wide analysis of arabidopsispentatricopeptide repeat proteins reveals their essential role in organellebiogenesis. The Plant Cell 16, 2089–2103.
Madera, M., 2008. Profile Comparer (PRC): A program for scoring and aligningprofile hidden Markov models. Bioinformatics 24, 2630–2631.
Madera, M., Vogel, C., Kummerfeld, S.K., Chothia, C., Gough, J., 2004. The superfamilydatabase in 2004: Additions and improvements. Nucleic Acids Res. 32, D235–D239.
Main, E.R.G., Lowe, A.R., Mochrie, S.G.J., Jackson, S.E., Regan, L., 2005. A recurringtheme in protein engineering: The design, stability and folding of repeatproteins. Curr. Opin. Struct. Biol. 15, 464–471.
Majoros, W.H., Pertea, M., Salzberg, S.L., 2004. TigrScan and GlimmerHMM: Twoopen source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879.
Marchler-Bauer, A., Anderson, J.B., Derbyshire, M.K., Derbyshire, M.K., DeWeese-Scott, C., Gonzales, N.R., Gwadz, M., et al., 2007. CDD: A conserved domaindatabase for interactive domain family analysis. Nucleic Acids Res. 35, D237–D240.
Mingler, M.K., Hingst, A.M., Clement, S.L., Yu, L.E., Reifur, L., Koslowsky, D.J., 2006.Identification of pentatricopeptide repeat proteins in Trypanosoma brucei. Mol.Biochem. Parasitol. 150, 37–45.
Murzin, A.G., Brenner, S.E., Hubbard, T., Chothia, C., 1995. SCOP: A structuralclassification of proteins database for the investigation of sequences andstructures. J. Mol. Biol. 247, 536–540.
O’Toole, N., Hattori, M., Andres, C., Iida, K., Lurin, C., Schmitz-Linneweber, C., Sugita,M., Small, I., 2008. On the expansion of the pentatricopeptide repeat gene familyin plants. Mol. Biol. Evol. 25, 1120–1128.
Pacheco, A.C.L., Araujo, F.F., Kamimura, M.T., Medeiros, S.R., Viana, D.A., Oliveira,F.C.E., Araújo-Filho, R., Costa, M.P., Oliveira, D.M., 2007. Following the ViterbiPath to deduce flagellar actin-interacting proteins of Leishmania spp.: Report oncofilins and twinfilins. In: Pham, T. (Ed.), Computer Models for Life Sciences,CMLS’07, AIP Proc., vol. 952. American Institute of Physics, Australia, 2007, pp.315–324.
Pacheco, A.C.L., Araujo, F.F., Kamimura, M.T., Silva, S.C., Diniz, M.C., Oliveira, F.C.E.,Araujo Filho, R., Costa, M.P., Oliveira, D.M., 2009. Hidden Markov models andthe Viterbi algorithm applied to integrated bioinformatics analyses of putativeflagellar actin-interacting proteins in Leishmania spp. Internat. J. Comput. AidedEng. Technol. (IJCAET) 1, 420–436.
Page, D., Craven, M., 2003. Biological applications of multi-relational data mining.SIGKDD Explorations 5, 69–79.
Papadopoulos, J.S., Agarwala, R., 2007. COBALT: Constraint-based alignment tool formultiple protein sequences. Bioinformatics 23, 1073–1079.
Preker, P.J., Keller, W., 1998. The HAT helix, a repetitive motif implicated in RNAprocessing. Trends Biochem. Sci. 23, 15–16.
Pusnik, M., Small, I., Read, L.K., Fabbro, T., Schneider, A., 2007. Pentatricopeptiderepeat proteins in Trypanosoma brucei function in mitochondrial ribosomes.Mol. Cell. Biol. 27, 6876–6888.
Rivas, E., Eddy, S.R., 2001. Noncoding RNA gene detection using comparativesequence analysis. BMC Bioinf. 2 (1), 8.
Rivals, E., Bruyère, C., Toffano-Nioche, C., Lecharny, A., 2006. Formation of thearabidopsis pentatricopeptide repeat family. Plant Physiol. 141, 825–839.
Sakakibara, Y., Brown, M., Hughey, R., Mian, I.S., Sjolander, K., Underwood, R.,Haussler, D., 1994. Stochastic context-free grammars for tRNA. Nucleic AcidsRes. 22, 5112–5120.
Scheufler, C., Brinker, A., Bourenkov, G., Pegoraro, S., Moroder, L., Bartunik, H., Hartl,F.U., Moarefi, I., 2000. Structure of TPR domain-peptide complexes: Criticalelements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell101, 199–210.
Schmidler, S., Liu, J., Brutlag, D., 2000. Bayesian segmentation of protein secondarystructure. J. Comput. Biol. 7, 233–248.
Schultz, J., Milpetz, F., Bork, P., Ponting, C.P., 1998. SMART, a simple modulararchitecture research tool: Identification of signaling domains. Proc. Natl. Acad.Sci. USA 95, 5857–5864.
Segal, E., Taskar, B., Gasch, A., Friedman, N., Koller, D., 2001. Rich probabilisticmodels for gene expression. Bioinformatics 1, 1–10.

M.C. Diniz et al. / Pattern Recognition Letters 31 (2010) 2178–2189 2189
Servant, F., Bru, C., Carrère, S., Courcelle, E., Gouzy, J., Peyruc, D., Kahn, D., 2002.ProDom: Automated clustering of homologous domains. Brief. Bioinf. 3, 246–251.
Sigrist, C.J., Cerutti, L., de Castro, E., Langendijk-Genevaux, P.S., Bulliard, V., Bairoch,A., Hulo, N., 2010. PROSITE, a protein domain database for functionalcharacterization and annotation. Nucleic Acids Res. 38 (Database issue),D161–D166.
Small, I.D., Peeters, N., 2000. The PPR motif – a TPR-related motif prevalent in plantorganellar proteins. Trends Biochem. Sci. 25, 46–47.
The Gene Ontology Consortium, 2000. Gene ontology: Tool for the unification ofbiology. Nature Genet. 25, 25–29.
The UniProt Consortium. The universal protein resource (UniProt) in 2010. NucleicAcids Res. (2010) D142–D148.
Xu, R., Supekar, K., Huang, Y., Das, A., Garber, A., 2006. Combining text classificationand Hidden Markov modeling techniques for structuring randomized clinicaltrial abstracts. In: AMIA Annu. Symp. Proc. 2006, pp. 824–828.
Wilson, D., Pethica, R., Zhou, Y., Talbot, C., Vogel, C., Madera, M., Chothia, C.,Gough, J., 2009. SUPERFAMILY – comparative genomics, datamining andsophisticated visualisation. Nucleic Acids Res. 37 (Database issue) D380–D386.
Winters-Hilt, S., 2006. Hidden Markov model variants and their application. BMCBioinf. 7, S14.
Web servers/resources
NCBI (National Center for Biotechnology Information/Entrez/Cn3D (All Databases)<http://www.ncbi.nlm.nih.gov/sites/gquery>.
The Pathogen Sequencing Unit – Wellcome Trust Sanger Institute – GeneDB –<www.genedb.org>.
TriTrypDB version 2.0 - <http://TriTrypDB.org>.The UniProt Consortium – <www.uniprot.org>.Swiss-Prot/trEMBL <www.expasy.org/sprot>.AMIGO after GeneDB access. <www.genedb.org/amigo/perl>.SMART <http://smart.embl.de>.SUPERFAMILY, <http://supfam.cs.bris.ac.uk>.TPRpred <http://toolkit.tuebingen.mpg.de/tprpred>.Arabidopsis Genome Initiative (AGI, 2000) <http://www.arabidopsis.org/portals>.Pfam <http://pfam.wustl.edu/hmmsearch.shtm>.Gene Ontology <www.geneontology.org>.




















