
The Serpin α1Proteinase Inhibitor Is a Critical Substrate for Gelatinase B/MMP-9 In Vivo
10
Cell, Vol. 102, 647–655, September 1, 2000, Copyright 2000 by Cell Press The Serpin a1-Proteinase Inhibitor Is a Critical Substrate for Gelatinase B/MMP-9 In Vivo intracellular protein BP230 (Sawamura et al., 1991; Ta- naka et al., 1991) and the 180 kDa transmembrane pro- tein BP180 (BPAG2, or type XVII collagen) (Diaz et al., Zhi Liu,*# Xiaoye Zhou,* Steven D. Shapiro, ‡ J. Michael Shipley, Sally S. Twining, § Luis A. Diaz,* Robert M. Senior, 1990; Giudice et al., 1992). In BP the separation of the and Zena Werb k epidermis from the dermis occurs within the lamina lu- * Department of Dermatology cida of the basement membrane and is accompanied University of North Carolina by destruction of hemidesmosomal and extracellular Chapel Hill, North Carolina 27599 matrix components and formation of blisters (Schaum- Department of Medicine burg-Lever et al., 1972; Dvorak et al., 1982). Proteinases ‡ Departments of Pediatrics and Cell Biology released from infiltrating inflammatory cells have been Washington University School of Medicine implicated in the tissue damage in these lesions (Gam- St. Louis, Missouri 63110 mon, 1989): BP blister fluids and lesional/perilesional § Department of Biochemistry sites contain both serine proteinases, such as neutrophil Medical College of Wisconsin elastase (NE) and plasmin, and MMPs, such as GB and Milwaukee, Wisconsin 53226 gelatinase A/MMP-2 (Oikarinen et al., 1983; Welgus et k Department of Anatomy al., 1986; Grando et al., 1989a, 1989b; Gissler et al., University of California, San Francisco 1992; Kramer and Reinartz, 1993; Stahle-Backdahl et San Francisco, California 94143 al., 1994). We used a mouse model that recapitulates the key features of human BP (Liu et al., 1993). Subepidermal blister formation triggered by anti-mouse BP180 IgG Summary (anti-mBP180) depends on activation of complement and polymorphonuclear leukocyte (PMN) infiltration (Liu We have identified the key protein substrate of gela- et al., 1995b, 1997). Mice with targeted null mutations tinase B/MMP-9 (GB) that is cleaved in vivo during in either GB (Liu et al., 1998) or NE (Liu et al., 2000) do dermal–epidermal separation triggered by antibodies not form blisters. These results demonstrate that GB to the hemidesmosomal protein BP180 (collagen XVII, and NE are required for blister formation. Although GB BPAG2). Mice deficient in either GB or neutrophil elas- and NE degrade numerous extracellular components of tase (NE) are resistant to blister formation in response the basement membrane zone (Liu et al., 1998, 2000; to these antibodies in a mouse model of the autoim- Vu and Werb, 1998), the critical substrates for these two mune disease bullous pemphigoid. Disease develops enzymes involved in the blister formation have not been upon complementation of GB 2/2 mice with NE 2/2 neu- determined. In this study we have determined functional trophils or NE 2/2 mice with GB 2/2 neutrophils. Only interactions between GB and NE in dermal–epidermal separation (blistering) and identified the key substrate NE degrades BP180 and produces dermal–epidermal for GB in experimental BP. separation in vivo and in culture. Instead, GB acts upstream to regulates NE activity by inactivating a1- proteinase inhibitor (a1-PI). Excess NE produces le- Results sions in GB 2/2 mice without cleaving a1-PI. Excess a1-PI phenocopies GB and NE deficiency in wild-type Both GB and NE Are Required for Subepidermal mice. Blister Formation We first assessed the relationship between GB and NE during the skin disease process by injecting GB 2/2 and Introduction NE 2/2 mice with pathogenic anti-mBP180 IgG, then 2 hr later reconstituting them with 5 3 10 5 PMN from Matrix metalloproteinases (MMPs) are critical regulators GB 1/1 , GB 2/2 , NE 1/1 , or NE 2/2 mice. When these mice of physiologic and pathologic events (Werb, 1997). One were then assessed for subepidermal blistering 12 hr of the outstanding questions in disease pathogenesis after IgG injection, GB 2/2 mice reconstituted with PMN mediated by MMPs is the identity of their critical sub- from either GB 1/1 or NE 2/2 mice developed subepider- strate targets in vivo. Gelatinase B/MMP-9 (GB) is in- mal blisters (Figure 1, Table 1). In contrast, GB 2/2 mice volved in a relatively confined set of such events in reconstituted with PMN from GB 2/2 mice remained vivo, including angiogenesis during endochondral bone resistant. NE 2/2 mice reconstituted with PMN from formation and autoimmune subepidermal blistering (Liu NE 1/1 mice or PMN from GB 2/2 mice also developed et al., 1998; Vu et al., 1998). In the present study, we blisters. In contrast, the anti-mBP180 IgG failed to in- set out to identify the in vivo substrates in an immune duce skin lesions in NE 2/2 mice reconstituted with PMN inflammatory skin disease, called bullous pemphigoid from NE 2/2 mice. The cross-complementation suggests (BP), that is initiated by deposition of autoantibodies and that NE and GB act in the same pathway. complement components at the basement membrane zone (Jordon et al., 1985). BP autoantibodies recognize two major hemidesmosomal components, the 230 kDa Degradation of BP180 Antigen Depends on Both NE and GB BP180 antigen was cleaved during subepidermal blis- # To whom correspondence should be addressed (e-mail: zhiliu@ med.unc.edu). tering, as determined by immunoblotting of extracts of
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of The Serpin α1Proteinase Inhibitor Is a Critical Substrate for Gelatinase B/MMP-9 In Vivo

Cell, Vol. 102, 647–655, September 1, 2000, Copyright 2000 by Cell Press
The Serpin a1-Proteinase Inhibitor Is a CriticalSubstrate for Gelatinase B/MMP-9 In Vivo
intracellular protein BP230 (Sawamura et al., 1991; Ta-naka et al., 1991) and the 180 kDa transmembrane pro-tein BP180 (BPAG2, or type XVII collagen) (Diaz et al.,
Zhi Liu,*# Xiaoye Zhou,* Steven D. Shapiro,†‡
J. Michael Shipley,† Sally S. Twining,§Luis A. Diaz,* Robert M. Senior,†
1990; Giudice et al., 1992). In BP the separation of theand Zena Werb‖epidermis from the dermis occurs within the lamina lu-*Department of Dermatologycida of the basement membrane and is accompaniedUniversity of North Carolinaby destruction of hemidesmosomal and extracellularChapel Hill, North Carolina 27599matrix components and formation of blisters (Schaum-†Department of Medicineburg-Lever et al., 1972; Dvorak et al., 1982). Proteinases‡Departments of Pediatrics and Cell Biologyreleased from infiltrating inflammatory cells have been
Washington University School of Medicineimplicated in the tissue damage in these lesions (Gam-
St. Louis, Missouri 63110 mon, 1989): BP blister fluids and lesional/perilesional§Department of Biochemistry sites contain both serine proteinases, such as neutrophilMedical College of Wisconsin elastase (NE) and plasmin, and MMPs, such as GB andMilwaukee, Wisconsin 53226 gelatinase A/MMP-2 (Oikarinen et al., 1983; Welgus et‖Department of Anatomy al., 1986; Grando et al., 1989a, 1989b; Gissler et al.,University of California, San Francisco 1992; Kramer and Reinartz, 1993; Stahle-Backdahl etSan Francisco, California 94143 al., 1994).
We used a mouse model that recapitulates the keyfeatures of human BP (Liu et al., 1993). Subepidermalblister formation triggered by anti-mouse BP180 IgGSummary(anti-mBP180) depends on activation of complementand polymorphonuclear leukocyte (PMN) infiltration (LiuWe have identified the key protein substrate of gela-et al., 1995b, 1997). Mice with targeted null mutations
tinase B/MMP-9 (GB) that is cleaved in vivo during in either GB (Liu et al., 1998) or NE (Liu et al., 2000) dodermal–epidermal separation triggered by antibodies not form blisters. These results demonstrate that GBto the hemidesmosomal protein BP180 (collagen XVII, and NE are required for blister formation. Although GBBPAG2). Mice deficient in either GB or neutrophil elas- and NE degrade numerous extracellular components oftase (NE) are resistant to blister formation in response the basement membrane zone (Liu et al., 1998, 2000;to these antibodies in a mouse model of the autoim- Vu and Werb, 1998), the critical substrates for these twomune disease bullous pemphigoid. Disease develops enzymes involved in the blister formation have not beenupon complementation of GB2/2 mice with NE2/2 neu- determined. In this study we have determined functionaltrophils or NE2/2 mice with GB2/2 neutrophils. Only interactions between GB and NE in dermal–epidermal
separation (blistering) and identified the key substrateNE degrades BP180 and produces dermal–epidermalfor GB in experimental BP.separation in vivo and in culture. Instead, GB acts
upstream to regulates NE activity by inactivating a1-proteinase inhibitor (a1-PI). Excess NE produces le- Resultssions in GB2/2 mice without cleaving a1-PI. Excessa1-PI phenocopies GB and NE deficiency in wild-type Both GB and NE Are Required for Subepidermalmice. Blister Formation
We first assessed the relationship between GB and NEduring the skin disease process by injecting GB2/2 andIntroductionNE2/2 mice with pathogenic anti-mBP180 IgG, then2 hr later reconstituting them with 5 3 105 PMN fromMatrix metalloproteinases (MMPs) are critical regulatorsGB1/1, GB2/2, NE1/1, or NE2/2 mice. When these miceof physiologic and pathologic events (Werb, 1997). Onewere then assessed for subepidermal blistering 12 hrof the outstanding questions in disease pathogenesisafter IgG injection, GB2/2 mice reconstituted with PMNmediated by MMPs is the identity of their critical sub-from either GB1/1 or NE2/2 mice developed subepider-strate targets in vivo. Gelatinase B/MMP-9 (GB) is in-mal blisters (Figure 1, Table 1). In contrast, GB2/2 micevolved in a relatively confined set of such events inreconstituted with PMN from GB2/2 mice remainedvivo, including angiogenesis during endochondral boneresistant. NE2/2 mice reconstituted with PMN fromformation and autoimmune subepidermal blistering (LiuNE1/1 mice or PMN from GB2/2 mice also developedet al., 1998; Vu et al., 1998). In the present study, weblisters. In contrast, the anti-mBP180 IgG failed to in-set out to identify the in vivo substrates in an immuneduce skin lesions in NE2/2 mice reconstituted with PMNinflammatory skin disease, called bullous pemphigoidfrom NE2/2 mice. The cross-complementation suggests(BP), that is initiated by deposition of autoantibodies andthat NE and GB act in the same pathway.complement components at the basement membrane
zone (Jordon et al., 1985). BP autoantibodies recognizetwo major hemidesmosomal components, the 230 kDa Degradation of BP180 Antigen Depends on Both
NE and GBBP180 antigen was cleaved during subepidermal blis-# To whom correspondence should be addressed (e-mail: zhiliu@
med.unc.edu). tering, as determined by immunoblotting of extracts of

Cell648
Figure 1. Histological Evaluation of NeonatalGB2/2 and NE2/2 Mice Injected with Patho-genic anti-mBP180 IgG and Reconstitutedwith Mouse PMN
(A) Pathogenic rabbit anti-murine BP180 IgG(i.d. injection, 2.5 mg/g body weight) pro-duced subepidermal blistering in neonatalGB2/2 mice reconstituted with 5 3 105 PMNfrom (a) GB1/1, (c) NE2/2, but not (b) GB2/2
mice.(B) Pathogenic rabbit anti-murine BP180 IgG(i.d. injection, 2.5 mg/g body weight) pro-duced clinical (data not shown) and histologi-cal subepidermal blistering in neonatal NE2/2
mice reconstituted with 5 3 105 PMN from(a) NE1/1, (c) GB2/2, but not (b) NE2/2 mice(3184). Insets, higher magnifications of H&Estaining sections, demonstrate infiltratingPMN in the dermis (3920). Basal keratinocyte(arrow), dermis (d), epidermis (e), blister vesi-cle (v), PMN (arrowhead).
the lesional and nonlesional skin of the injected animals NE2/2 mice reconstituted with PMN from NE1/1 mice,NE2/2 mice reconstituted with PMN from GB2/2 miceusing anti-mBP180 IgG (Figure 2A). This raises the ques-
tion of which enzyme degrades BP180 during blister and GB2/2 mice reconstituted with PMN from NE2/2
mice (Figure 2A). In contrast, the nonlesional skin ex-formation. Both intact (180 kDa) and degraded mBP180(100 kDa) were present in lesional skin from NE1/1 mice, tracts of NE2/2 mice, NE2/2 mice reconstituted with PMN
Table 1. Summary: The Relationship between GB and NE in Experimental BP
Genotype of Genotype of PMN Number of Mean DiseaseHost Micea Reconstituted Mice Activity Scoreb
GB1/1 — 12 3GB2/2 — 12 0
5 3 105 GB1/1 8 35 3 105 GB2/2 8 02.5 3 106 GB2/2 8 25 3 105 NE2/2 8 3
NE1/1 — 12 3NE2/2 — 12 0
5 3 105 NE1/1 8 35 3 105 NE2/2 8 02.5 3 106 NE2/2 8 05 3 105 GB2/2 8 3
a Neonatal GB-sufficient (GB1/1), GB-deficient (GB2/2), NE-sufficient (NE1/1), and NE-deficient (NE2/2) mice were injected i.d. with pathogenicanti-mBP180 IgG. Purified mouse PMN from GB1/1, GB2/2, NE1/1, or NE2/2 mice were given i.d. 2 hr after IgG injection.b Injected animals were examined clinically 12 hr after IgG injection and disease activity scored as described in Experimental Procedures.

MMP-9 Inactivates Serpins In Vivo649
test the proteinase dependency of subepidermal blis-tering directly, neonatal mouse skin sections were incu-bated with purified human NE or mouse GB and exam-ined histologically at various time points over a 24 hrperiod. The skin sections treated with NE, but not withGB or MEM alone, showed complete detachment of theepidermis from the dermis at 24 hr (Figure 2B). The NE-induced dermal–epidermal separation was completelyblocked by an NE inhibitor, a1-PI, but not by a metallo-proteinase inhibitor, EDTA. These results suggest thatNE, and not GB, is critical for mBP180 antigen degrada-tion and dermal–epidermal separation in experimentalBP. This raises the question of how GB functions in thisprocess.
GB and NE Facilitate PMN RecruitmentOne possibility is that GB is required for recruitmentof PMN into the lesions. Therefore, we determined therelative number of PMN in skin from GB1/1, GB2/2,NE1/1, and NE2/2 mice injected with pathogenic anti-mBP180 IgG, and examined clinically for blisters 12 hrlater. As expected, GB1/1 and NE1/1 mice developedsubepidermal blisters, while GB2/2 and NE2/2 mice wereresistant (Figure 3 and Table 1). When skin sampleswere analyzed for PMN infiltration by assay of MPO, wefound significantly more PMN were recruited into thelesional skin sites in the GB1/1 and NE1/1 mice than intothe nonlesional sites in GB2/2 and NE2/2 mice (Figure3A). Moreover, GB was readily detected in the skin sam-ples from GB1/1 and NE1/1, and weakly in NE2/2, but notGB2/2 mice by gelatin zymography (Figure 3B). Similarly,NE activity was significantly elevated in the skin ofGB1/1 and NE1/1, but not of GB2/2 and NE2/2 mice(Figure 3C). However, at 4 hr after IgG injection, GB2/2
and NE2/2 mice had a similar number of infiltrating PMNFigure 2. BP180 Cleavage and Epidermal–Dermal Separation in the skin as compared to wild-type control mice, as(A) To identify BP180 degradation in the lesional skin of experimental shown previously (Liu et al., 1998, 2000). These dataBP neonatal NE2/2 (lanes 1–4), NE1/1 (lane 5), and GB2/2 (lanes 6–7) indicate that both GB and NE contribute to the recruit-mice were injected i.d. with anti-mBP180 IgG. 2 hr later, the IgG- ment of PMN to form the blisters. In their absence, re-injected mice were reconstituted with 5 3 105 mouse PMN. Skin
cruitment of PMN and, therefore available NE and GB,samples were obtained at 12 hr after IgG injection and protein ex-are reduced.tracts (15 mg/lane) were analyzed by immunoblotting using the anti-
mBP180 IgG. Both the intact and degraded BP180 were seen inlesional skin samples of IgG-injected NE1/1 mice (lane 5), NE2/2
GB Acts Upstream of NEmice injected with IgG and reconstituted with PMN from NE1/1 (laneOur data show that NE alone is able to produce dermal–2) and GB2/2 (lane 4) mice, and GB2/2 mice injected with IgG andepidermal separation in vitro. If this is the case in vivo,reconstituted with PMN from NE2/2 (lane 6) mice. In contrast, nothen we should be able to get blistering induced by NEdegraded BP180 was found in skin samples of IgG-injected NE2/2
mice (lane 1), IgG-injected NE2/2 mice reconstituted with PMN from in the absence of GB. Although blisters do not form inNE2/2 mice (lane 3), or GB2/2 mice reconstituted with PMN from GB2/2 mice, recruitment of PMN is also reduced inGB2/2 mice (lane 7). GB2/2 mice, and 5 3 105 GB2/2 PMN are unable to rescue(B) To generate BP-like dermal–epidermal separation in mouse skin the GB2/2 phenotype. However, when we reconstitutedby NE neonatal mouse skin sections were incubated in (a) MEM anti-mBP180 treated GB2/2 mice with five times as manyalone with (b) 25 mg/ml NE or (c) 80 mg/ml GB at 378C for 24 hr and
(2.5 3 106) GB2/2 PMN, skin lesions appeared in thewere examined by H&E staining. Dermal–epidermal separation wasabsence of GB (compare Figure 4A with Figure 1A; Tableobserved in sections treated with NE, but not GB. Control sections1) and cleaved mBP180 antigen was detected in skinincubated in MEM without proteinases showed no dermal–lysates (Figure 4B). In contrast, 2.5 3 106 NE2/2 PMNepidermal separation (panel A). Site of basal keratinocytes (arrow),
epidermis (e), dermis (d), blister vesicle (V) (3368). did not reconstitute NE2/2 mice, and extracts from theirskin showed only intact mBP180 antigen. These resultsdemonstrate that GB2/2 mice can develop skin blistersfrom NE2/2 mice and GB2/2 mice reconstituted withwhen compensated with a sufficient number of GB2/2PMN from GB2/2 mice showed only intact mBP180 an-(but NE1/1) PMN as a source of NE, while same numbertigen.of NE2/2 (but GB1/1) PMN did not induce blisters inNE2/2 mice. Thus, GB acts upstream of NE, but playsNE, but not GB, Produces Dermal–Epidermala minimal role in the direct degradation of extracellularSeparationproteins that cause subepidermal blistering in experi-Both NE (Liu et al., 2000) and GB (Stahle-Backdahl et
al., 1994) degrade recombinant BP180 in solution. To mental BP in this inflammatory cascade.

Cell650
NE, in turn, could degrade extracellular components inthe basement membrane zone, leading to subepidermalblister formation. In GB2/2 mice, the high level of activea1-PI is due to lack of GB activity, while in NE2/2 miceit is due to the decreased levels of GB because of fewerPMN present in the skin at 12 hr (see Figure 3A) andthe lack of NE (which forms a complex with a1-PI).
Proteolytic Inactivation of a1-PI Is GB DependentTo determine if GB is involved in a1-PI inactivation dur-ing lesion formation, we analyzed the skin extracts ofGB2/2 mice injected with pathogenic IgG and reconstitu-ted with PMN for a1-PI by immunoblotting. Both nativeand degraded a1-PI were present in lesional skin ofGB1/1 mice and GB2/2 mice reconstituted with 5 3 105
PMN from GB1/1 or NE2/2 mice (Figure 4D). In contrast,the nonlesional skin extracts of GB2/2 mice and GB2/2
mice reconstituted with 5 3 105 PMN from GB2/2 miceshowed only native a1-PI. Significantly, GB2/2 mice,which were reconstituted with 2.5 3 106 PMN from GB2/2
mice and developed subepidermal blisters, but showedonly native a1-PI. These results reveal the GB-depen-dent proteolytic inactivation of a1-PI during lesion for-mation. The finding that excess NE is able to circumventthe requirement for GB further suggests that active NEis limiting in GB2/2 mice and that NE is the major enzymein basement membrane zone destruction in experimen-tal BP.
a1-PI Inhibits Development of Experimental BlistersIf a1-PI is the critical substrate of GB, then administra-tion of excess a1-PI to wild-type mice should pheno-copy the response of GB2/2 and NE2/2 mice. Injectionof a1-PI into mice injected with anti-mBP180 blockedsubepidermal blistering (Figure 5A; mean diseaseFigure 3. Comparison of Levels of PMN Infiltration in the Lesionalscore 5 3 for controls [n 5 8]; disease score 5 0 forSkin in Experimental BPa1-PI injected [n 5 8]). In contrast, administration ofGB1/1, GB2/2, NE1/1, and NE2/2 mice were injected with pathogenica1-antichymotrypsin (a1-AC), an inhibitor of the PMNanti-mBP180 IgG. Recruitment of PMN (as assayed by MPO, GBenzyme cathepsin G was without effect. At 4 hr afterand NE in the IgG injection sites) was analyzed at 12 hr after IgG
injection. injection, there were similar numbers of PMN in animals(A) MPO activity assay showed significantly higher levels of PMN with or without a1-PI pretreatment, as monitored byrecruitment in the lesional skin of GB1/1 (bar 1) and NE1/1 (bar 3) extractable MPO activity (Figure 5B). However, at 12 hr,mice as compared to GB2/2 (bar 2) and NE2/2 (bar 4) mice. the animals treated with a1-PI had significantly lower(B) Gelatin zymography revealed GB bands in lesional skin samples levels of MPO activity than control mice and no blisters.of GB1/1 (lane 1), NE1/1 (lane 3), and nonlesional skin samples of
These data indicate that initial PMN recruitment is inde-NE2/2 (lane 4), but not in nonlesional skin samples of GB2/2 (lanependent of a1-PI, but later, PMN recruitment is not sus-2). 12 mg protein/lane for lanes 1 and 2; 30 mg protein/lane for lanestained in the presence of excess a1-PI and blisters do not3 and 4.develop. This is exactly the phenotype seen in GB2/2(C) NE activity assay was significantly higher in the lesional skin of
GB1/1 (bar 1) and NE1/1 (bar 3) mice, as compared to the skin of and NE2/2 mice (Figure 3). Pretreatment of a1-PI withGB2/2 (bar 2) and NE2/2 (bar 4) mice. (n 5 8 for each group, *p , gelatinase B eliminated its inhibitory activity on blister0.001, for paired samples: bar 1 versus 2 and 3 versus 4). formation (data not shown),
Discussiona1-Proteinase Inhibitor (a1-PI) Is Elevated in Skinof GB2/2 and NE2/2 Mice Treated with Anti-mBP180 Our study definitively establishes a1-PI as a key sub-
strate for GB in vivo. GB was discovered as an MMPThe experiments described above indicate that NE isnecessary to produce blistering. Why, then, are 5 3 105 that can cleave type IV and V collagens and denatured
collagens (gelatin) in vitro (reviewed in Vu and WerbGB2/2 PMN insufficient as a source of NE in GB2/2 mice?One possibility is that endogenous inhibitors of NE are 1998). Subsequently, GB has been shown to cleave
other ECM substrates, such as BP180, and non-ECMpresent in excess. The serpin a1-PI, which is the princi-pal plasma inhibitor of NE, was present at significantly proteins, such as serpins, and to activate TGF-b in vitro
(Vu and Werb 1998; Yu and Stamenkovic, 2000). Thehigher levels in the nonlesional skin of GB2/2 and NE2/2
mice, when PMN proteinases are deficient, than in blis- pathogenic anti-mBP 180 IgG dictates the localized de-granulation of proteinases at high concentration fromters of GB1/1 and NE1/1 mice (Figure 4C). A possible
explanation is that the role of GB in blister formation is PMN adhering to the immune complexes formation atthe dermal–epidermal junction. The surprising outcometo inactivate a1-PI, allowing NE activity to function. Free
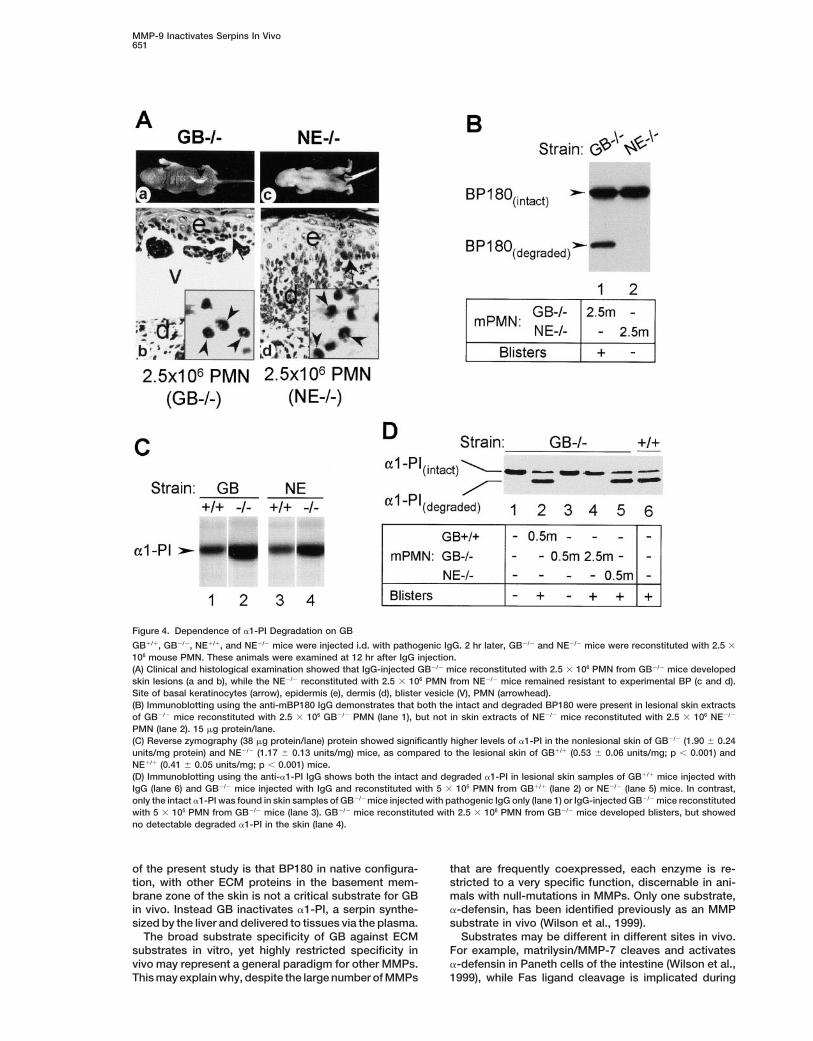
MMP-9 Inactivates Serpins In Vivo651
Figure 4. Dependence of a1-PI Degradation on GB
GB1/1, GB2/2, NE1/1, and NE2/2 mice were injected i.d. with pathogenic IgG. 2 hr later, GB2/2 and NE2/2 mice were reconstituted with 2.5 3
106 mouse PMN. These animals were examined at 12 hr after IgG injection.(A) Clinical and histological examination showed that IgG-injected GB2/2 mice reconstituted with 2.5 3 106 PMN from GB2/2 mice developedskin lesions (a and b), while the NE2/2 reconstituted with 2.5 3 106 PMN from NE2/2 mice remained resistant to experimental BP (c and d).Site of basal keratinocytes (arrow), epidermis (e), dermis (d), blister vesicle (V), PMN (arrowhead).(B) Immunoblotting using the anti-mBP180 IgG demonstrates that both the intact and degraded BP180 were present in lesional skin extractsof GB2/2 mice reconstituted with 2.5 3 106 GB2/2 PMN (lane 1), but not in skin extracts of NE2/2 mice reconstituted with 2.5 3 106 NE2/2
PMN (lane 2). 15 mg protein/lane.(C) Reverse zymography (38 mg protein/lane) protein showed significantly higher levels of a1-PI in the nonlesional skin of GB2/2 (1.90 6 0.24units/mg protein) and NE2/2 (1.17 6 0.13 units/mg) mice, as compared to the lesional skin of GB1/1 (0.53 6 0.06 units/mg; p , 0.001) andNE1/1 (0.41 6 0.05 units/mg; p , 0.001) mice.(D) Immunoblotting using the anti-a1-PI IgG shows both the intact and degraded a1-PI in lesional skin samples of GB1/1 mice injected withIgG (lane 6) and GB2/2 mice injected with IgG and reconstituted with 5 3 105 PMN from GB1/1 (lane 2) or NE2/2 (lane 5) mice. In contrast,only the intact a1-PI was found in skin samples of GB2/2 mice injected with pathogenic IgG only (lane 1) or IgG-injected GB2/2 mice reconstitutedwith 5 3 105 PMN from GB2/2 mice (lane 3). GB2/2 mice reconstituted with 2.5 3 106 PMN from GB2/2 mice developed blisters, but showedno detectable degraded a1-PI in the skin (lane 4).
of the present study is that BP180 in native configura- that are frequently coexpressed, each enzyme is re-stricted to a very specific function, discernable in ani-tion, with other ECM proteins in the basement mem-
brane zone of the skin is not a critical substrate for GB mals with null-mutations in MMPs. Only one substrate,a-defensin, has been identified previously as an MMPin vivo. Instead GB inactivates a1-PI, a serpin synthe-
sized by the liver and delivered to tissues via the plasma. substrate in vivo (Wilson et al., 1999).Substrates may be different in different sites in vivo.The broad substrate specificity of GB against ECM
substrates in vitro, yet highly restricted specificity in For example, matrilysin/MMP-7 cleaves and activatesa-defensin in Paneth cells of the intestine (Wilson et al.,vivo may represent a general paradigm for other MMPs.
This may explain why, despite the large number of MMPs 1999), while Fas ligand cleavage is implicated during

Cell652
the proteolytic inactivation of a1-PI by GB, underlies thepathology of an inflammatory skin disorder, BP. Ourstudy elucidates the mechanisms by which GB and NEpromote dermal–epidermal separation and blistering ina mouse model of BP and establishes an epistatic rela-tionship between the two enzymes: The key role of GBis the inactivation of a1-PI, whereas NE is the majortissue-damaging enzyme that produces BP180 degra-dation and dermal–epidermal junction separation. Al-though, in vitro, both GB and NE are able to cleavethe components of extracellular matrix and recombinantBP180 antigen, which is the main target of pathogenicantibodies (Stahle-Backdahl et al., 1994; Liu et al., 2000),GB does not cleave mBP180 in vivo or produce dermal–epidermal separation in mouse skin sections. Instead,GB contributes to tissue damage indirectly by inactivat-ing a1-PI, allowing unchecked activity of NE to cleavestructural proteins, including BP180 antigen, in the der-mal–epidermal junction. Reconstitution of wild-typemice with excess a1-PI develops the resistance to blis-ter formation seen in GB2/2 and NE2/2 mice.
Our data also point to a second function of GB inregulating experimental blister formation, that of sus-taining recruitment of inflammatory PMN. GB is not re-quired for acute recruitment of PMN in lungs, perito-neum, or skin (Betsuyaku et al., 1999). However,although initial recruitment of PMN at 4 hr is normalin both NE- and GB-deficient mice, recruitment is notsustained in the absence of either NE or GB. Thus, GBcollaborates with NE in generating and/or maintainingchemoattractant levels in the lesions. While the comple-ment fragments C5a and C3a, generated immediately
Figure 5. In Vivo Inhibition of Dermal–Epidermal Separation by after the administration of the toxic antibody are potenta1-PI chemoattractant, other factors probably take over as(A) Histology of mouse skin sections: (a) Blister in mice injected with the reaction develops. Fragments of cleaved proteinspathogenic IgG alone. (b) No blister in mice pretreated with a1-PI. may contribute to the later phase of PMN recruitment.(c) Blister in mice pretreated with the neutrophil cathepsin G inhibi-
Indeed, cleaved a1-PI is a potent chemoattractant fortor, a1-antichymotrypsin (a1-AC). Site of basal keratinocytesPMN (Banda et al., 1988).(arrow), epidermis (e), dermis (d), blister vesicle (V).
Several MMPs including collagenase, macrophage(B) MPO assay at 4 hr (hatched bars) after injection of wild-typeelastase, matrilysin, stromelysin-1, and stromelysin-3mice with pathogenic IgG, shows that the same number of PMN
were present in mice pretreated with a1-PI (bar 3), as those injected also can cleave a1-PI and destroy its NE inhibitory activ-with PBS alone (bar 1) and a1-AC (bar 5). However, by 12 hr (black ity (Banda et al., 1980, 1987; Ossanna et al., 1986; Po-bars) the mice pretreated with a1-PI (bar 4) had significantly fewer tempa et al., 1986; Knauper et al., 1990; Mast et al.,PMN than controls (bars 2 and 6) (*, p , 0.001). n 5 8 for each 1991; Okada et al., 1992; Pei et al., 1997). GB is notgroup. particularly efficient in a1-PI inactivation, compared to
other MMPs (Mast et al., 1991), yet in this specific siteit is most effective. Whether MMPs can contribute to
prostate involution following castration (Powell et al., experimental blister formation remains to be deter-1999). During healing of skin wounds, stromelysis-1/ mined. a1-PI can be inactivated not only by proteolysis,MMP-3 mediates wound closure by regulating the for- but also by the reactive oxidant HOCl (Morihara et al.,mation of an actin “purse-string” by dermal fibroblasts 1984; Banda et al., 1987; Weiss, 1989). Although methio-(Bullard et al., 1999), through an unknown substrate, nine, an HOCl scavenger, can reduce the NE-mediatedwhile in the mammary gland cell surface molecules, such subepidermal blistering and alter inactivation of a1-PIas E-cadherin on epithelial cells are the implicated tar- by z23% in vivo in experimental BP (data not shown),gets of stromelysin-1 (Sternlicht et al., 1999). proteolytic inactivation clearly is the major mechanism.
The present study shows a1-PI is the substrate for The mechanisms documented in this study are appli-GB in skin blisters. However, it is unlikely to be the cable to human BP, which is rich in eosinophils thatGB target resulting in VEGF bioavailability required for also express GB, and to PMN-mediated inflammatoryangiogenesis during endochondral ossification. (Vu et diseases in general. Indeed, inactivated a1-PI has beenal., 1998). What all of these studies suggest is that ECM detected in fluids recovered from inflammatory sites inmolecules are not the rate-limiting substrates for MMPs vivo, in patients with rheumatoid arthritis, adult respira-in many situations. tory distress syndrome and emphysema (Wong and
It has long been hypothesized that NE-mediated tis- Travis, 1980; Cochrane et al., 1983; Janoff, 1983; Wei-sue destruction in certain inflammatory diseases, such land et al., 1986; Zhang et al., 1990; Belaaouaj et al.,as emphysema, is caused by an imbalance in the ratio 1998). The involvement of both GB and NE in the patho-of NE to a1-PI (Weiss, 1989). Our findings provide the genesis of the blistering disease BP give us new insights
into the immunopathogenic mechanisms in autoimmunefirst in vivo evidence that this mechanism, mediated by

MMP-9 Inactivates Serpins In Vivo653
(OD460 nm/mg protein of the mouse skin injected with pathogenic anti-diseases and implies both enzymes are potential targetsmBP180 IgG minus OD460 nm/mg protein of the mouse skin injectedfor therapeutic intervention.with control IgG). Protein concentrations were determined by theOur results demonstrate that the important activitiesBio-Rad dye binding assay using BSA as a standard.of GB in physiology and pathology in vivo are dramati-
GB in protein extracts of PMN and skin sections from injectedcally different from those postulated from in vitro stud- animals was determined by gelatin gel zymography as describedies. Moreover the substrates can be synthesized in dis- previously (Liu et al., 1998). The gels were stained with 0.125%tant tissues, and thus be invisible to mRNA microarray Coomassie Brilliant blue. Areas of gelatinolytic activity appear asand related analyses. Elucidating the precise substrates clear zones against a dark blue background.
NE activity in skin and PMN extracts of mice was measured usingof GB and other MMPs in vivo has the promise of uncov-the NE-specific substrate methoxysuccinyl-Ala-Ala-Pro-Val-p-nitro-ering other novel extracellular regulatory and signalinganilide (Met-O-Suc-Ala-Ala-Pro-Val-pNA, Enzyme Systems Prod-pathways.ucts, Dublin, CA; Nakajima et al., 1979).
Experimental ProceduresAnalysis of BP180 CleavageIntact mBP180 and its degradation products in mouse skin were
Animalsidentified by SDS-PAGE (7%) followed by immunoblotting (Liu et
BALB/c mice were purchased from Jackson Laboratories (Bar Har-al., 1995a), using rabbit anti-mBP180 IgG and monospecific FITC-
bor, ME). GB2/2 and matched normal control (GB1/1), NE2/2 andconjugated goat anti-rabbit IgG (Kirkeggard & Perry Laboratories
matched normal control (NE1/1) mice were generated as describedInc., Gaithersburg, MD).
(Belaaouaj et al., 1998; Vu et al., 1998). Breeding pairs of thesemice were maintained at the Medical College of Wisconsin Animal
Analysis of Dermal–Epidermal Separation In VitroResource Center. Neonatal mice, 24–36 hr old, weighing 1.4–1.6 gMouse skin sections were obtained from neonatal BALB/c mice (36were used for passive transfer experiments.to 48 hr old) and cut into 2 3 2 mm strips with a razor blade. Theskin strips were then incubated in Minimum Essential Medium (MEM)
BP 180 Antibodies and Induction of Experimental BP Blisters either with or without 25 mg/ml human NE (Athens Research andFor preparation of pathogenic rabbit anti-murine BP180 IgG, recom- Technology Inc., Athens, GA) or 80 mg/ml mouse gelatinase B (Triplebinant murine BP180 and the immunization of rabbits were prepared Point Biologics, Forest Grove, OR) at 378C for various periods ofand the titers of rabbit anti-mBP180ABC antibodies in the rabbit time. At the end of the incubation, the skin strips were rinsed insera and in the purified IgG fractions were assayed by indirect IF fresh MEM, fixed in 10% formalin, and embedded in paraffin, sec-using mouse skin cryosections as substrate, as reported previously tioned and stained with H&E.(Liu et al., 1993). The pathogenicity of IgG preparations was testedby passive transfer experiments. IgG from several rabbits were used Analysis of a1-PIduring these experiments with similar results.
a1-PI in the skin was detected by SDS-PAGE (10%) followed byFor induction of experimental BP and clinical evaluation, neonates immunoblotting using a rabbit anti-human a1-PI antiserum (Athens
were given one intradermal (i.d.) injection (50 ml each, 2.5 mg/g body Research and Technology Inc., Athens, GA) that cross-reacts withweight) of a sterile solution of IgG in PBS, as described elsewhere murine a1-PI. To determine the levels of native (active) a1-PI in(Liu et al., 1993, 1995b). The skin of neonatal mice from the test and the skin, the skin extracts were analyzed by reverse casein gelcontrol groups was examined 4, 12, or 24 hr after the IgG injections. zymography (Twining et al., 1996). Briefly, protein extracts of skinThe activity of cutaneous disease was scored as follows: “0”, no sections from injected animals were subjected to SDS-PAGE ondetectable skin disease; “1”, mild erythematous reaction with no polyacrylamide gels containing casein (10% acrylamide and 1%evidence of epidermal detachment elicited by gentle friction of the casein) under nonreducing conditions. After electrophoresis gelsskin, which, when positive, produced fine, persistent wrinkling of were washed twice with 2.5% Triton X-100 for 30 min to remove SDS.the epidermis; “2”, intense erythema and epidermal detachment Gels were then rinsed briefly with water and incubated overnight atinvolving 10%–50% of the epidermis in the injection site; and “3”, 378C in chymotrypsin digestion solution (50 mg chymotrypsin/mlintense erythema with frank epidermal detachment involving more casein gel reaction buffer). The gels were stained with 0.125% Coo-than 50% of the epidermis in the injection site. massie Brilliant blue. a1-PI-protected areas appeared as blue bands
against a transparent background. The band density was quantifiedIsolation of PMN by densitometric scanning using the Molecular Dynamics Storm 860PMN were isolated from heparinized blood from GB2/2, GB1/1, scanner (Molecular Dynamics, Inc., Sunnyvale, CA). The level of a1-NE2/2, or NE1/1 mice by dextran sedimentation followed by separa- PI was expressed as relative a1-PI units/mg protein (OD units/mgtion on a density gradient (Coligan et al., 1994). PMN purity of the protein of the mouse skin injected with pathogenic anti-mBP180final cell preparation was consistently .96% as determined by cell- IgG minus OD units/mg protein of the mouse skin injected withcytospin and LeukoStat staining (Fisher Diagnostics, Orangeburg, control IgG).NY). The viability of the PMN was .96% as determined by trypanblue exclusion. For in vivo reconstitution of PMN mice were injected Injection of a1-PIi.d. with pathogenic anti-mBP180 IgG (2.5 mg/g body weight in 50 Wild-type mice were injected with human a1-PI or a1-AC (i.p. 100ml PBS). Two hours later, these mice were injected with 5 3 105 or mg/g body weight in PBS; Athens Research and Technology Inc.,2.5 3 106 PMN i.d. (in 100 ml of PBS/10 mM glucose) at the same Athens, GA) prior to injection of pathogenic IgG. Skin was examinedsite (Liu et al., 1998). The animals were analyzed 4 or 12 hr after the at 4 and 12 hr for clinical lesions. Injected areas were excised andIgG injections. extracted for analysis of PMN infiltration by MPO assay.
Statistical AnalysisMPO, GB, and NEFor statistical analysis, the data were expressed as mean 6 SEMFor quantification of PMN accumulation in the skin, tissue myeloper-and were analyzed using Student’s t test. A p value less than 0.05oxidase (MPO) activity in skin sites of the injected animals waswas considered significant.assayed as described (Bradley et al., 1982), using purified MPO
(Athens Research and Technology Inc., Athens, GA) as standard.The skin sites were excised and extracted by homogenization in an Acknowledgmentsextraction buffer containing 0.1 M Tris-Cl, pH 7.6, 0.15 M NaCl, 0.5%hexadecyl trimethyl ammonium bromide. MPO activity in superna- We thank Drs. George J. Giudice and Janet A. Fairley for helpful
discussion. This work was supported in part by U.S. Public Healthtants was measured by the change in optical density at (OD)460 nm resulting from decomposition of H2O2 in the presence of Service NIH grants AI 40768 (Z. L.), EY12731 (S. S. T.), AR32599
and AR32081 (L. A. D.), HL47328 (R. M. S.), HL54853 and HL56414o-dianisidine. MPO content was expressed as relative MPO activity

Cell654
(S. D. S.), and CA72006 (Z. W.), by a VA Merit Review Grant (L. A. D.), mechanisms in pemphigus and bullous pemphigoid. J. Invest. Der-matol. 85, 72s–78s.and by the Alan A. and Edith L. Wolff Charitable Trust (R. M. S.).
Knauper, V., Reinke, H., and Tschesche, H. (1990). Inactivation ofhuman plasma 1-proteinase inhibitor by human PMN leucocyte col-Received April 5, 2000; revised June 12, 2000.lagenase. FEBS Lett. 263, 355–357.
Kramer, M.D., and Reinartz, J. (1993). The autoimmune blisteringReferencesskin disease bullous pemphigoid. The presence of plasmin/2-anti-plasmin complexes in skin blister fluid indicates plasmin generationBanda, M.J., Clark, E.J., and Werb, Z. (1980). Limited proteolysis byin lesional skin. J. Clin. Invest. 92, 978–983.macrophage elastase inactivates human alpha 1-proteinase inhibi-
tor. J. Exp. Med. 152, 1563–1570. Liu, Z., Diaz, L.A., Troy, J.L., Taylor, A.F., Emery, D.J., Fairley, J.A.,and Giudice, G.J. (1993). A passive transfer model of the organ-Banda, M.J., Clark, E.J., Sinha, S., and Travis, J. (1987). Interactionspecific autoimmune disease, bullous pemphigoid, using antibodiesof mouse macrophage elastase with native and oxidized humangenerated against the hemidesmosomal antigen, BP180. J. Clin.1-proteinase inhibitor. J. Clin. Invest. 79, 1314–1317.Invest. 92, 2480–2488.
Banda, M.J., Rice, A.G., Griffin, G.L., and Senior, R.M. (1988). TheLiu, Z., Diaz, L.A., Swartz, S.J., Troy, J.L., Fairley, J.A., and Giudice,inhibitory complex of human alpha 1-proteinase inhibitor and humanG.J. (1995a). Molecular mapping of a pathogenically relevant BP180leukocyte elastase is a neutrophil chemoattractant. J. Exp. Med.epitope associated with experimentally induced murine bullous167, 1608–1615.pemphigoid. J. Immunol. 155, 5449–5454.
Belaaouaj, A., McCarthy, R., Baumann, M., Gao, Z., Ley, T.J., Abra-Liu, Z., Giudice, G.J., Swartz, S.J., Fairley, J.A., Till, G.O., Troy, J.L.,ham, S.N., and Shapiro, S.D. (1998). Mice lacking neutrophil elastaseand Diaz, L.A. (1995b). The role of complement in experimentalreveal impaired host defense against gram negative bacterial sepsis.bullous pemphigoid. J. Clin. Invest. 95, 1539–1544.Nat. Med. 4, 615–618.Liu, Z., Giudice, G.J., Zhou, X., Swartz, S.J., Troy, J.L., Fairley, J.A.,Betsuyaku, T., Shipley, J.M., Liu, Z., and Senior, R.M. (1999). Neutro-Till, G.O., and Diaz, L.A. (1997). A major role for neutrophils in experi-phil emigration in the lungs, peritoneum, and skin does not requiremental bullous pemphigoid. J. Clin. Invest. 100, 1256–1263.gelatinase B. Am. J. Respir. Cell. Mol. Biol. 20, 1303–1309.Liu, Z., Shipley, J.M., Vu, T.H., Zhou, X., Diaz, L.A., Werb, Z., andBradley, P.P., Priebat, D.A., Christensen, R.D., and Rothstein, G.Senior, R.M. (1998). Gelatinase B-deficient mice are resistant to(1982). Measurement of cutaneous inflammation: estimation of neu-experimental bullous pemphigoid. J. Exp. Med. 188, 475–482.trophil content with an enzyme marker. J. Invest. Dermatol. 78,
206–209. Liu, Z., Shapiro, S.D., Zhou, X., Twining, S.S., Senior, R.M., Giudice,G.J., Fairley, J.A., and Diaz, L.A. (2000). Neutrophil elastase playsBullard, K.M., Lund, L., Mudgett, J.S., Mellin, T.N., Hunt, T.K., Mur-a critical role in experimental bullous pemphigoid. J. Clin. Invest.phy, B., Ronan, J., Werb, Z., and Banda, M.J. (1999). Impaired wound105, 113–123.contraction in stromelysin-1 deficient mice. Ann. Surg. 230, 260–265.Mast, A.E., Enghild, J.J., Nagase, H., Suzuki, K., Pizzo, S.V., andCochrane, C.G., Spragg, R., and Revak, S.D. (1983). PathogenesisSalvesen, G. (1991). Kinetics and physiologic relevance of the inacti-of the adult respiratory distress syndrome. Evidence of oxidant ac-vation of alpha 1-proteinase inhibitor, alpha 1-antichymotrypsin, andtivity in bronchoalveolar lavage fluid. J. Clin. Invest. 71, 754–761.antithrombin III by matrix metalloproteinases-1 (tissue collagen-
Coligan, J.E., Kruisbeek, A.M., Margulies, D.H., Shevach, E.M., and ase), -2 (72-kDa gelatinase/type IV collagenase), and -3 (stromely-Strober, W. (1994). PMN isolation. Curr. Protocols Immunol. 3, sin). J. Biol. Chem. 266, 15810–15816.7.23.1.
Morihara, K., Tsuzuki, H., Harada, M., and Iwata, T. (1984). Purifica-Diaz, L.A., Ratrie, H.D., Saunders, W.S., Futamura, S., Squiquera, tion of human plasma alpha 1-proteinase inhibitor and its inactiva-H.L., Anhalt, G.J., and Giudice, G.J. (1990). Isolation of a human tion by Pseudomonas aeruginosa elastase. J. Biochem. (Tokyo) 95,epidermal cDNA corresponding to the 180-kD autoantigen recog- 795–804.nized by bullous pemphigoid and herpes gestationis sera. Immuno-
Nakajima, K., Powers, J.C., Ashe, B.M., and Zimmerman, M. (1979).localization of this protein to the hemidesmosome. J. Clin. Invest.Mapping the extended substrate binding site of cathepsin G and86, 1088–1094.human leukocyte elastase. Studies with peptide substrates related
Dvorak, A.M., Mihm, M.C., Jr., Osage, J.E., Kwan, T.H., Austen, K.F., to the 1-protease inhibitor reactive site. J. Biol. Chem. 254, 4027–and Wintroub, B.U. (1982). Bullous pemphigoid, an ultrastructural 4032.study of the inflammatory response: eosinophil, basophil and mast
Oikarinen, A.I., Zone, J.J., Ahmed, A.R., Kiistala, U., and Uitto, J.cell granule changes in multiple biopsies from one patient. J. Invest.(1983). Demonstration of collagenase and elastase activities in theDermatol. 78, 91–101.blister fluids from bullous skin diseases. Comparison between der-
Gammon, W.R. (1989). Immune complex and complement-mediated matitis herpetiformis and bullous pemphigoid. J. Invest. Dermatol.leukocyte recruitment in bullous pemphigoid. Immunol. Ser. 46, 81, 261–266.509–525.
Okada, Y., Gonoji, Y., Naka, K., Tomita, K., Nakanishi, I., Iwata, K.,Gissler, H.M., Simon, M.M., and Kramer, M.D. (1992). Enhanced Yamashita, K., and Hayakawa, T. (1992). Matrix metalloproteinaseassociation of plasminogen/plasmin with lesional epidermis of bul- 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibro-lous pemphigoid. Br. J. Dermatol. 127, 272–277. sarcoma cells. Purification and activation of the precursor and en-Giudice, G.J., Emery, D.J., and Diaz, L.A. (1992). Cloning and primary zymic properties. J. Biol. Chem. 267, 21712–21719.structural analysis of the bullous pemphigoid autoantigen BP180. Ossanna, P.J., Test, S.T., Matheson, N.R., Regiani, S., and Weiss,J. Invest. Dermatol. 99, 243–250. S.J. (1986). Oxidative regulation of neutrophil elastase-alpha-1-pro-Grando, S.A., Glukhenky, B.T., Drannik, G.N., Epshtein, E.V., Kos- teinase inhibitor interactions. J. Clin. Invest. 77, 1939–1951.tromin, A.P., and Korostash, T.A. (1989a). Mediators of inflammation Pei, D., Majmudar, G., and Weiss, S.J. (1994). Hydrolytic inactivationin blister fluids from patients with pemphigus vulgaris and bullous of a breast carcinoma cell-derived serpin by human stromelysin-3.pemphigoid. Arch. Dermatol. 125, 925–930. J. Biol. Chem. 269, 25849–25855.Grando, S.A., Glukhenky, B.T., Drannik, G.N., Kostromin, A.P., and Potempa, J., Watorek, W., and Travis, J. (1986). The inactivation ofChernyavsky, A.I. (1989b). Cytotoxic proteases in blister fluid of human plasma 1-proteinase inhibitor by proteinases from Staphylo-pemphigus and pemphigoid patients. Int. J. Tissue React. 11, coccus aureus. J. Biol. Chem. 261, 14330–14334.195–201.
Powell, W.C., Fingleton, B., Wilson, C.B., Boothby, M., and Matrisian,Janoff, A. (1983). Do neutrophils play a major role in elastin turnover L.M. (1999). The metalloproteinases matrilysin proteolytically gener-of normal tissues? Am. Rev. Respir. Dis. 127, 782–783. ates active soluble Fas ligand and potentiates epithelial cell apopto-
sis. Curr. Biol. 9, 1441–1447.Jordon, R.E., Kawana, S., and Fritz, K.A. (1985). Immunopathologic

MMP-9 Inactivates Serpins In Vivo655
Sawamura, D., Li, K., Chu, M.L., and Uitto, J. (1991). Human bullouspemphigoid antigen (BPAG1). Amino acid sequences deduced fromcloned cDNAs predict biologically important peptide segments andprotein domains. J. Biol. Chem. 266, 17784–17790.
Schaumburg-Lever, G., Orfanos, C.E., and Lever, W.P. (1972). Elec-tron microscopic study of bullous pemphigoid. Arch. Dermatol. 106,662–667.
Stahle-Backdahl, M., Inoue, M., Guidice, G.J., and Parks, W.C.(1994). 92-kD gelatinase is produced by eosinophils at the site ofblister formation in bullous pemphigoid and cleaves the extracellulardomain of recombinant 180-kD bullous pemphigoid autoantigen. J.Clin. Invest. 93, 2022–2030.
Sternlicht, M.D., Lochter, A., Sympson, C.J., Huey, B., Rougier, J.P.,Gray, J.W., Pinkel, D., Bissell, M.J., and Werb, Z. (1999). The stromalproteinase MMP3/stromelysin-1 promotes mammary carcinogene-sis. Cell. 98, 137–146.
Tanaka, T., Parry, D.A., Klaus-Kovtun, V., Steinert, P.M., and Stanley,J.R. (1991). Comparison of molecularly cloned bullous pemphigoidantigen to desmoplakin I confirms that they define a new family ofcell adhesion junction plaque proteins. J. Biol. Chem. 266, 12555–12559.
Twining, S.S., Zhou, X., Schulte, D.P., Wilson, P.M., Fish, B., andMoulder, J. (1996). Effect of vitamin A deficiency on the early re-sponse to experimental pseudomonas keratitis. Invest. Ophthalmol.Vis. Sci. 37, 511–522.
Vu, T.H., and Werb, Z. (1998). Gelatinase B: structure, regulationand function. In Matrix Metalloproteinases, W.C. Parks and R. P.Mecham, eds. (San Diego, CA: Academic Press), pp. 115–148.
Vu, T.H., Shipley, J.M., Bergers, G., Berger, J.E., Helms, J.A., Hana-han, D., Shapiro, S.D., Senior, R.M., and Werb, Z. (1998). MMP-9/gelatinase B is a key regulator of growth plate angiogenesis andapoptosis of hypertrophic chondrocytes. Cell 93, 411–422.
Weiland, J.E., Davis, W.B., Holter, J.F., Mohammed, J.R., Dorinsky,P.M., and Gadek, J.E. (1986). Lung neutrophils in the adult respira-tory distress syndrome. Clinical and pathophysiologic significance.Am. Rev. Respir. Dis. 133, 218–225.
Weiss, S.J. (1989). Tissue destruction by neutrophils. N. Engl. J.Med. 320, 365–376.
Welgus, H.G., Bauer, E.A., and Stricklin, G.P. (1986). Elevated levelsof human collagenase inhibitor in blister fluids of diverse etiology.J. Invest. Dermatol. 87, 592–596.
Werb, Z. (1997). ECM and cell surface proteolysis: regulating cellularecology. Cell 91, 439–442.
Wilson, C.L., Ouellete, A.J., Ayabe, T., Lopez-Boado, Y.S., Stratman,J.L., Hultgren, S.J., Matrisian, L.M., and Parks, W.C. (1999). Regula-tion of intestinal-defensin activation by the metalloproteinase matri-lysin in innate host defense. Science 286, 113–117.
Wong, P.S., and Travis, J. (1980). Isolation and properties of oxidized1-proteinase inhibitor from human rheumatoid synovial fluid. Bio-chem. Biophys. Res. Commun. 96, 1449–1454.
Yu, Q., and Stamenkovic, I. (2000). Cell surface-localized matrixmetalloproteinase-9 proteolytically activates TGF-beta and pro-motes tumor invasion and angiogenesis. Genes Dev. 15, 163–176.
Zhang, Z., Winyard, P.G., Chidwick, K., Farrell, A., Pemberton, P.,Carrell, R.W., and Blake, D.R. (1990). Increased proteolytic cleavageof 1-antitrypsin (1-proteinase inhibitor) in knee-joint synovial fluidfrom patients with rheumatoid arthritis. Biochem. Soc. Trans. 18,898–899.





















