
The promoting effect of noble metal addition on niobia-supported cobalt catalysts
11
The promoting effect of noble metal addition on niobia-supported cobalt catalysts F.B. Noronha a,b, A. Frydman a,c, D.A.G. Aranda a, C. Perez a, R.R. Soares a B. Morawek b, D. Castner c, C.T. Campbell c, R. Frety b, M. Schmal a,· • NUCAT I PEQ I COPPE-Escola Quimica, Univ. Federal Rio de Janeiro, Rio de Janeiro, Brazil b Inst. Recherches Caralyse and LACE I CNRS, Villeurbanne, France c Inst, Chemistry. University a/Washington, Washington, DC, USA The promoting effects of a noble metal (Pd, Pt, Rh) added to Co/Nb 2 0 s catalysts were studied by varying the Me/Co atomic ratios. Acid niobium was calcined to niobium pentoxide. The surface and bulk structures of the calcined materials were characterized by XPS and TPR techniques. The catalytic performance was obtained with CO hydrogenation. The addition of a noble metal promoted the reduction of C0 3 + and Co 2 + phases at the surface. XPS results revealed that Co 2 + species are well dispersed as a thin layer around the niobium support together with C0 3 0 4 crystallites islands. The C0304/C02~ ratio depends on the surface area of the support. XPS measurements also revealed that Pd~, Rh 2 0 3 and Pt0 2 are the main phases in the mono and bimetallic catalysts. The activity of the bimetallic catalysts increased and the stability was already attained. The selectivities towards C t and oxygenates increased with the addition of Rh up to an atomic ratio of 0.5 and decreased beyond that. This behavior is similar for both temperatures of reduction at 573 and 773 K. was that the product selectivity changed, de- pending on the reduction temperature and the metal loading. The niobia-supported catalysts showed a higher selectivity toward saturated hydrocarbons compared to the alumina sup- ported ones, after reduction at high temperatures [5-7]. The important feature is that Nb 2 Os in the presence of a metal presents a SMSI effect [8,9]. Silva et al. [5] showed that Co/Nb 2 0 s after reduction at 773 K presents a strong SMSI effect and that it is destroyed after oxidation in agreement with the literature [8,10]. In particu- Cobalt based catalysts have been frequently used in the Fischer- Tropsch synthesis. The na- ture of the support has a strong influence on the catalytic properties for the CO hydrogenation [1,2]. This reaction has also been studied on metal supported reducible oxides, like titanium and niobium oxides [3-7] and the main observation
Transcript of The promoting effect of noble metal addition on niobia-supported cobalt catalysts

The promoting effect of noble metal addition on niobia-supportedcobalt catalysts
F.B. Noronha a,b, A. Frydman a,c, D.A.G. Aranda a, C. Perez a, R.R. Soares a
B. Morawek b, D. Castner c, C.T. Campbell c, R. Frety b, M. Schmal a,·
• NUCAT IPEQ I COPPE-Escola Quimica, Univ. Federal Rio de Janeiro, Rio de Janeiro, Brazilb Inst. Recherches Caralyse and LACE ICNRS, Villeurbanne, France
c Inst, Chemistry. University a/Washington, Washington, DC, USA
The promoting effects of a noble metal (Pd, Pt, Rh) added to Co/Nb20s catalysts were studied by varying the Me/Coatomic ratios. Acid niobium was calcined to niobium pentoxide. The surface and bulk structures of the calcined materialswere characterized by XPS and TPR techniques. The catalytic performance was obtained with CO hydrogenation. Theaddition of a noble metal promoted the reduction of C03 + and Co2 + phases at the surface. XPS results revealed that Co2 +
species are well dispersed as a thin layer around the niobium support together with C0304 crystallites islands. TheC0304/C02~ ratio depends on the surface area of the support. XPS measurements also revealed that Pd~, Rh203 and Pt02are the main phases in the mono and bimetallic catalysts. The activity of the bimetallic catalysts increased and the stabilitywas already attained. The selectivities towards C t and oxygenates increased with the addition of Rh up to an atomic ratio of0.5 and decreased beyond that. This behavior is similar for both temperatures of reduction at 573 and 773 K.
was that the product selectivity changed, de-pending on the reduction temperature and themetal loading. The niobia-supported catalystsshowed a higher selectivity toward saturatedhydrocarbons compared to the alumina sup-ported ones, after reduction at high temperatures[5-7].
The important feature is that Nb2 Os in thepresence of a metal presents a SMSI effect [8,9].Silva et al. [5] showed that Co/Nb20s afterreduction at 773 K presents a strong SMSIeffect and that it is destroyed after oxidation inagreement with the literature [8,10]. In particu-
Cobalt based catalysts have been frequentlyused in the Fischer- Tropsch synthesis. The na-ture of the support has a strong influence on thecatalytic properties for the CO hydrogenation[1,2].
This reaction has also been studied on metalsupported reducible oxides, like titanium andniobium oxides [3-7] and the main observation

Pt wt.-% Co wt.-% Pt/Co ratio a Rhwt.-% Cowt.-% Pd/Rh ratio •
0.5 0.90.5 5.0 0.03 0.3 2.0 0.09
5.0 0.6 1.9 0.190.7 1.8 0.241.9 2.3 0.72
1.9
lar for the CO hydrogenation, Haller and Re-sasco [to], as well as Kunimori et al. [3], haveshown that the SMSI effect is destroyed duringthe reaction itself, due to the presence of water.However, Silva et al. [5] and Frydman et al. [6]found that the activity was not completely re-versed and that the selectivity was drasticallymodified, enhancing the longer carbon chainsafter high temperature of reduction. These re-sults demonstrate strong interaction of cobaltwith the support and its rearrangement, suggest-ing that new species promoted the sites whichincrease the hydrocarbons toward longer chains.
Now, the question is what happens when asecond metal is added to Co/Nb20s catalysts?Noronha et al. [11] showed that the addition ofcopper to Pd/Nb20s diminished the SMSI ef-fect. The addition of a noble metal can alsochange the cobalt properties [12-14]. Lapidus etal. [12] reported data of the CO hydrogenationwith Ru, Pd and Pt added to Co/ A1203 cata-lysts and found that they are also more selectivein C; hydrocarbons.
The main objective of this work is to deter-mine the effect of the addition of a second metalto the Co/Nb20s catalysts and to understandbetter the surface properties of the metallicphases due to the interaction with the support byevaluating their performance in the CO hydro-genation reaction. Besides the reaction itself, thecatalysts were characterized by XPS, TPR andDRS techniques.
Table IComposition of the bimetallic catalysts
Pd wt.-% Co wt.-% Pd/Co ratio a
1.3
2.11.6
0.530.17
Niobium pentoxide was obtained by calcina-tion of niobium acid (CBMM) at two differenttemperatures: 873 K for 4 h, resulting in acrystalline form, T or IT, of niobium pentoxide(25 m2/ g) and 823 K for 2 h (50 m2/ g).
The catalysts were prepared by impregnationof cobalt, palladium and rhodium nitrates orplatinum hexachloride on both supports, as de-scribed elsewhere [15-17]. The cobalt loadingwas 5% on Pd-Co and Pt- Co and differentMe/Co atomic ratios. On the Rh-Co systemthe cobalt content was 2%, keeping atomic ra-tios of the same order as above. The followingcatalysts and their compositions are summarizedin Table I
TPR experiments were performed in a con-ventional apparatus, as described elsewhere [11],using mixtures of 1.73% H2/ Ar and 2% 02/He,respectively. In addition, DRS measurementswere carried out in the oxide or passivated formin a Cary 5 UV-DRS system. XPS measure-ments were performed using monochromatic AlK a radiation. The Nb 3d = 206.4 eV of thesupport was used as reference.
The reaction was performed with Pt-Co andRh-Co supported on Nb20s catalysts, after be-ing treated at different reduction temperatures:573 K (LTR) and 773 K (HTR). The followingconditions were used in the microreactor, keep-ing conversions below 20%: reactant mixture of4% He/H2/CO (H2/CO = 2); total pressure0.1 MPa, temperature 533 K and time on streamof approximately 30 h. Data were taken everyhour in the first 8 h and after running 25-30 h.Selectivities were obtained at isoconversion af-ter attaining steady state condition.
The formation of a bimetallic system and thetransformations occurring during the reductionwere studied, using different temperature pro-grammed techniques, like TPR and TPO. Thestates of the intermediate oxide forms as iso-lated or bimetallic and alloy species were ana-lyzed by complementary techniques to achieve areal understanding of the effect of a secondmetal supported on niobium oxide, known as areducible oxide at elevated temperatures.
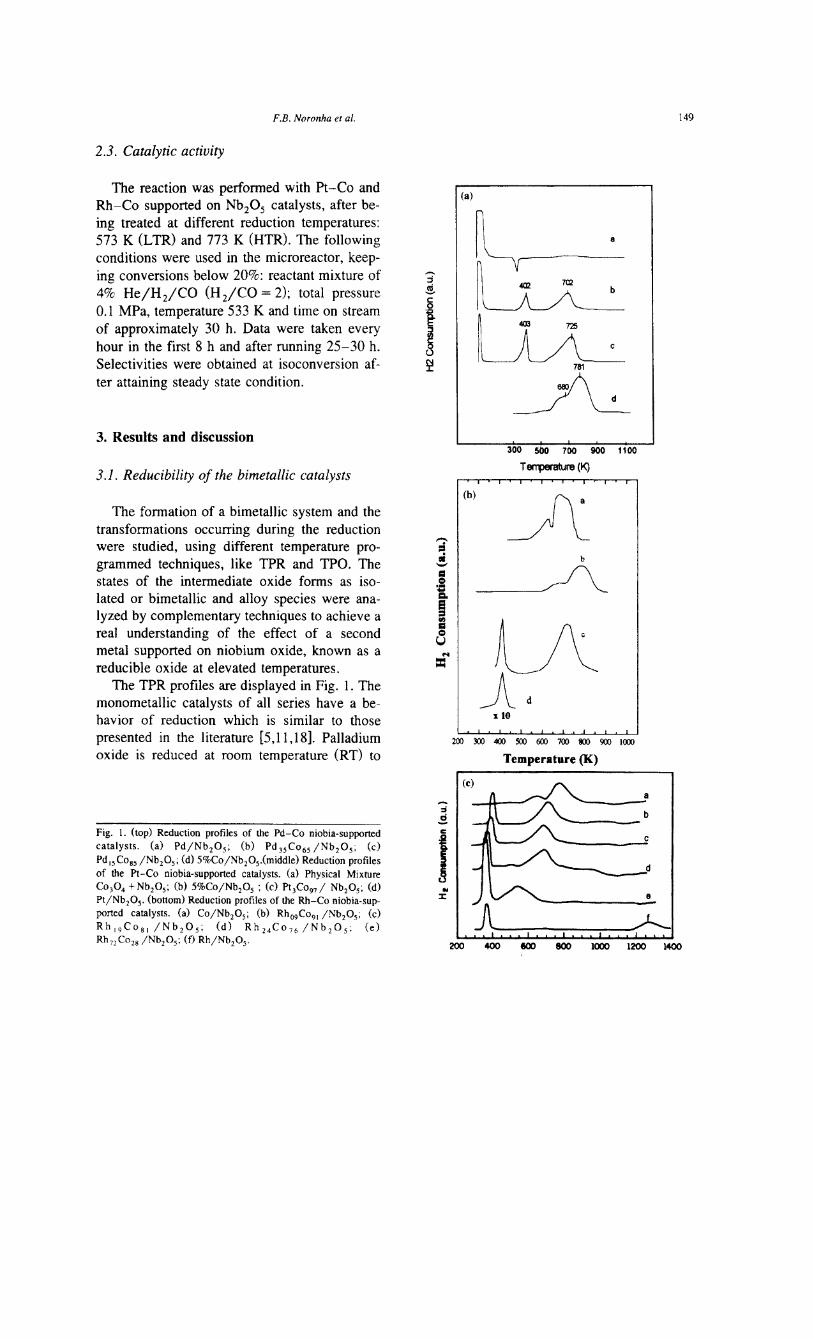
The TPR profiles are displayed in Fig. 1. Themonometallic catalysts of all series have a be-havior of reduction which is similar to thosepresented in the literature [5,11,18]. Palladiumoxide is reduced at room temperature (RT) to
Fig. 1. (top) Reduction profiles of the Pd-Co niobia-supportedcatalysts. (a) Pd/Nb20S; (b) Pd3SC06S /Nb20S: (c)Pd ISC08S /Nb20S; (d) 5%Co /Nb20s.(middle) Reduction profilesof the Pt-Co niobia-supported catalysts. (a) Physical MixtureC0304 + Nb20S; (b) 5%Co/Nb20S ; (c) Pt3C09d Nb20S: (d)Pt/Nb20S' (bottom) Reduction profiles of the Rh-Co niobia-sup-ported catalysts. (a) Co/Nb20S; (b) Rho9C091/Nb20S; (c)Rh 19C081 /Nb20S; (d) Rh 24C076 /Nb20S; (e)RhnC028 /Nb20S; (f) Rh/Nb20S'
h a
781
.~
300 5110 700 900 1100
TElf1llllr8luTe(K). ,
(b)
A'":'=• b~
~=~ig
WJ t_A=~U..•= --Ad
II0,
200 300 400 SOO 600 700 llX) 900 100>
Temperature (K)
(c)a
:;b~
c:
I c
d
••:I: eJ\. /--200 400 eoo 800 1000 1200 l400

metallic palladium and displays one desorptionpeak at 341 K, which is attributed to the desorp-tion of reversible hydrogen from the palladiumsurface and to the decomposition of palladiumhydride [19]. In addition, H2 consumption isalso observed at elevated temperatures (473 K),in general attributed to a partial reduction of thesupport [20].
Rhodium supported catalyst (Rh/Nb20s)profile shows a sharp peak at 367 K, suggestinga single reduction step of Rh3 + to Rho, which isin good agreement with van't Blick onRh/ Al203 [18]. It means that, after calcination,rhodium oxide is in the Rh203 form. ThePt/Nb20s profile is similar, showing a welldefined peak at 411 K, indicating that it hasonly one Pt structure in the calcined form. DRSresults show indeed the presence of Pt02 at thesurface [15].
Noteworthy is the reduction of Co/Nb20scatalyst. For the 5% Co/Nb20s catalyst, thereare two main peaks at 680 and 781 K (Fig. la).Recently, Frydman et al. [17] reported the pres-ence of both C0304 particles and Co2+ species,probably linked to the support. Then, the Hzconsumption at 680 K could be attributed to thereduction of C0304 particles and the peakaround 781 K due to the reduction of C02+species. The profile of a physical mixture ofC0304 + Nb20s and DRS spectra confirms thatC03 04 is the main phase in the cobalt catalyst[15].
The TPR profiles of the bimetallic catalystsdisplay quite different behaviors from themonometallics. The shapes and temperatures ofreduction are dependent on the second metaladded to cobalt. The profiles are not only asingle superposition of the monometallic pro-files, showing different temperatures of reduc-tion peaks. The Hz consumptions varied as afunction of the metal added to cobalt.
Profiles of the Pd-Co/NbzOs catalysts areshown in Fig. 1a for different Pd/Co ratios.They show typically three temperatures of re-duction, namely at RT, 400 and around 700-725
K. These peaks are shifted to lower tempera-tures when compared to the Co/Nb20s cata-lyst. It seems that the addition of palladiumpromotes the reduction of C0304•
The Pt-Co/Nb20s and Rh-Co/Nb20s pro-files are displayed in Fig. 1b and Fig. 1c, fordifferent Me/Co atomic ratios together with themonometallic catalysts. The reduction occurs attwo different temperature ranges. The first onearound 400 K, corresponding to the simultane-ous reduction of Pt02 or RhZ03 with C0304--+ CoO. The second at elevated temperatures,presented only one broad peak between 600 and800 K corresponding to C02+ --+ CoO reduc-tion, shifted down 70 K when compared to theCo/Nb20s catalyst. The band at 728 nm relatedto Co3+ disappeared from DRS spectra [15].Thus, PtO promotes the reduction of C0304after low temperature (510 K), as shown in Fig.1b. These profiles also reveal a bimetallic for-mation and a strong influence of metallic Pt andRh on the reduction of cobalt oxide.
For the Rh/NbzOs and PtjNb20s catalysts,the hydrogen consumptions correspond exactlyto the theoretical values and, thus, indicatingfull reduction. It is more complex for theCo/NbzOs catalyst. In the 2% cobalt catalyst,we observed an excess of Hz consumption ofapproximately 23% of Hz above 623 K, due tothe partial reduction of NbzOs in contact withthe metal.
Now, analyzing the Hz consumption onbimetallic catalysts we have the following ob-servations:
For the Pd-Co/NbzOs catalysts, the Hzconsumption at RT is higher than the theoreticalamount needed to reduce Pd~ to Pdo. It is alsohigher in the region between 298-473 K whereC0304 reduces to CoO. Therefore, it seems thatPdo promotes the reduction of C0304 particlesat lower temperatures. Magnetic measurementsconfirm that 3% of cobalt at RT and 12% afterreduction at 473 K are in the metallic form [16].However, the promoting effect of Pdo decreaseswith higher cobalt loadings. Similar considera-

tions should be extended to the Pt -Co andRh-Co catalyst for the temperature range be-tween 340-430 K.
For Pd-Co, Pt-Co and Rh-Co catalysts, theamount of H 2 consumed in the high temperaturerange is higher than that necessary to reduceCoO and C02+ to Coo. Magnetic measurementsshowed that, after reduction at 873 K, only Coois present. This was also verified by TPO forPt-Co and Co/Nb20s catalysts [15]. Therefore,this excess of hydrogen consumption can beascribed to a partial reduction of the support, asit was seen for Co/Nb20s catalyst. The behav-ior of the Rh-Co/Nb20s catalyst was some-what surprising, because the H 2 consumptionexceeded extremely the stoichiometric value forthe reduction of the oxides to the metallic phase.This excess decreases as rhodium content in-creases and disappears with rhodium alone.
Figs. 2 and 3 show the XPS lineshapes of Co2p spectra for Rh-Co/Nb20s, Pd-Co/Nb20sand Pt-Co/Nb20s calcined catalysts, respec-tively.
For the Pd/Nb20s and Pd3sC06s/Nb20scatalysts, the binding energies for the Pd 3ds/2peak were 337.0 and 336.9 eV, respectively
•••• :,It" .,,/ ···vo--''·"! :.'. Co,O•....--..",....-"J' '---
. ~\! .~ J,,-:' CoNb 0
f ./'I, ./J'" '.-":::I •J''\ , ..~ \ 'fI\ ••
_ ._~ ...•..••.~!~\~/Nben • .,......". '"",~. I '.;RII Co /Nb~ ~,..."..,....,.""!,'\ . .:"~' It. --""-'" ~ :.... .'-,r .•••.• \ .. ~i '. .\ Rtt"CoIl/Nb.. ,,': ' :.~
. .::.'"".'",,-"·".v :'.R1114Co7l/Nb.-',,-: ..~."~ ..'.." ,.' ..,.....".-. ..;"-..:f' •.•.;",... \ Rb Co /Nb.,.."••••~".,- • .........:;., M
120 ItO 800 7llO 780 770 1'80Bindino Eneroy l.v)
Fig. 2. XPS Co 2p spectra of the calcined Rh-CojNb205 cata-lysts.
Fig. 3. XPS Co 2p spectra of the calcined Pd-CojNb205 andPt-CojNb205 catalysts.
(Table 2). According to the literature, thesevalues correspond to the presence of PdO [21].On the other hand, on the Rh-Co and Pt-Cocatalysts, two peaks were observed for the Rh3ds/2 and Pt 4f7/2 suggesting a partial reduc-tion of both Rh203 and Pt02 by the X-raybeam.
In order to identify the cobalt species byXPS, the binding energy (BE) of Co 2P3/2 peakand spin-orbit splitting values of reference com-pounds are listed in Table 2. The BE valuesfound for the two different cobalt oxides (CoO
Table 2Binding energies of the main elements in the niobia-supportedcatalysts and reference compounds
PdPdJ5C065RhRhSC092Rh,6COS4
Rh,9CoslRh42C05S
PtPtJC097
2% Co5% CoC0304
CoOCoNb206
Binding Energy (e V)
Pd 3dl/2 Rh 3d5/2
337.0336.9
308.6 (310.2)308.7 (310.2)308.5 (310.3)308.6 (31 0.2)308.9 (31 0.1)
780.8 15.4780.3 15.3780.4 15.4780.8 15.6
71.6 780.4 15.4(75.1)
780.5 15.4781.6 15.8779.9 15.1780.4 16.0780.3 16.1

and CO,04) are very similar and it does notallow a distinction between them. On the otherhand, the secondary features (shake-up, spin-orbit splitting) of the spectra are different andcan be used to distinguish them. On the C0304,the satellite is practically absent while CoO andCoNb206 show a strong satellite peak. Further-more, the energy separation between the Co2P3/2 peak and the Co 2PI/2 is close to 15.2 eVfor C0304 but substantially larger for pure Co2+oxides like CoO (15.9 eV), as shown in Table 2.
The Co 2p lineshapes of the Co/Nb20s cata-lysts suggest the presence of different Co specieson the surface. The broadening of the Co 2P3/2line and the spin-orbit splitting values indicatesthat the main cobalt species on both Co/Nb20scatalysts was C0304•
Several cobalt species have been detected inthe precursors of supported cobalt catalysts[2,22,23]. The case of alumina-supported cobalthas been well described. At least, three differentcobalt species have been identified: C0304 par-ticles, Co2+ species and CoAl 204 , a surfacecompound with the spinel structure. In addition,the fraction of each phase can vary with cobaltloading. Thus, the amount of C0304 particlesincreases at higher cobalt loading [23). Ho et al.[22] used XPS to characterize the state of cobaltin a series of Co /Ti02 catalysts with differentcobalt contents. They showed that the cobaltphase was primarily present as a highly dis-persed surface cobalt species in the 0.5 and 1.0wt.-% Co/Ti02 samples. By increasing theamount of cobalt, discrete C0304 particles wereformed, in addition to C02+ species.
Therefore, our results are in good agreementwith the model proposed in the literature. Insuch a model, a Co/Nb20s catalyst with asupport having a low surface area would show ahigher amount of C0304 particles. This canexplain the similar fraction of C0304 particleson both CojNb20S catalysts with differentcobalt loadings.
The Co 2p XPS spectra of the bimetalliccatalysts also suggest the presence of differentcobalt species (Fig. 2). In the case of Rh-
Co/Nb20S catalysts, an experimental proce-dure, described elsewhere [17), was used toresolve the Co 2p lineshapes of the catalystsinto contributions from C0304 and C02+ fea-tures. The Co 2p spectrum of the catalysts wascompared to a Co 2p spectrum of pure C0304•
The difference between these two spectra wasthen compared to a Co 2p spectrum of pureCoNb206. It was clear that the difference spec-trum was essentially identical to the CoNb206reference spectrum meaning that in all catalysts,the Co 2p spectra were well fit by a simplesuperposition of C0304 and C02+ (speciallyCoNb206) features. Since, the lineshapes ofother Co2 + oxides like CoO are very similar tothat of CoNb206, we could not definitivelydetermine whether the C02
+ species detected onthe supported catalysts is Co niobate or anotherC02+ oxide.
From the experimental XPS intensity ratiosfor CojNb, Rh/Nb and Rh/Co, it was seenthat Rh is surface-enriched relative to Co, since,the Rh/Co surface atomic ratios are larger thanthe bulk ratios by up to a factor of 2 [17].Through XPS intensities, several model struc-tures were postulated to represent the surface ofthe calcined bimetallic catalysts and to rule outothers. The model that best described the exper-imental XPS intensities considered a Co2 + sur-face phase covering the surface of the Nb20sparticles in the spaces between bilayer islandsof Rh203 on top of C0304. The amount of Coin the C02+ phase was very small, so that78-90 wt.-% of the total Co was present in theCo 3°4 phase.
On the Pd-CojNb20s catalyst, the XPSanalysis also revealed the presence of C0304
particles and a C02+ surface phase (Fig. 3) [16].The Co 2p lineshape of the Pd3sCo6sjNb20sbimetallic catalyst showed a Co 2P3/2 peak at781.3 eV, a strong shake up satellite around 786eV and a spin-orbit coupling of 16.1 eV sug-gesting that the fraction of the Co2+ surfacephase was quite pronounced.
The TPR and XPS results allowed us topostulate a model which represents these

0 Co/NlILlRl
• Co/MIIHTRl
•• ~IRaI+ RII/Nl(LTRIx RI\IN)( HTRI* ItIINIlRa)
.,+:~41~-..x !Iii'
*cc"Al~ m.itJ~[] Cb c
•• •• ••••
I
bimetallic catalysts. From the TPR profiles ofthe Pd-Co/Nb20s catalysts, it was seen thatthe presence of palladium promoted not only thereduction of C0304 particles but also the cobaltsurface phase, represented by the second TPRpeak. These results can be explained by thepresence of the palladium oxide particles spreadover the Co2+ layer or interfaced with C0104•
The same model can be proposed for the Pt-Co
catalysts, since, similar TPR and XPS resultswere obtained.
Figs. 4 and 5 show the rate of CO conversionas a function of time for all Rh-CO and Pt-COcatalysts reduced at 573 and 773 k. The steadystate was obtained very quickly for the bimetal-
2.0 [] RIl.eo./Nb (lJ1l)
• •••• eo./Nb (HTR)
• AII.CD.I PI! 1lJ1l)
0OJ ~/Nb(HTR1~ ~"IlJ'R)":t~ + ~Nbtmll~Cl x AIIrfA.I Nb llJ'R )i * ffIrPJ.INb tmt I
.: 'f*oxlJ! YxXx II PlP.r/NbI HT'R)11.0 x~~ Il~
o PtPw/NbILTllI
v vv;j1Jf • Cag,fNbI511LTll)
8 •••• t9. • CouINIIl511 HTR)
l5 •• •Io.~II
1~ 20 2~RIacIlon lime lh I

lic catalysts already after 8 h. The activity ofRh/Nb20S was 3 fold higher than forCo/Nb20; after LTR. The activities decreasedwith the increase of the temperature of reduc-tion and increases with the addition of a secondmetal. No significant deactivation was observedon the bimetallic catalysts, only the initial activ-ity of Rh/Nb20S catalyst decays drastically,but stabilizes after 8 h.
The effect of the temperature of reductionwas significant for the monometallics. Compar-ing the activities after LTR and HTR the decaywas 7 fold for Co/Nb20S while for Rh/Nb20sit was only 1.5. In order to verify the SMSIeffect, we checked its reversibility by perform-ing the reaction after HTR and then after asequence of oxidation at 673 K and reduction atLTR. Results show, in accordance with formerdata [5,6], that the reversibility of the SMSIeffect on the Co/Nb20s is only partially re-stored, however, it was completely reverted onRh/Nb20;, in agreement with Kunimori's ob-servations [3].
The addition of Pt to Co/Nb20S' as dis-played in Fig. 5 increases the activity that de-cays drastically after HTR. This is expected
Table 3Product selectivity for the CO hydrogenation
Catalyst f"'duc,;on Selectivity (%)------------C1 C2-4 C;
LTR'HTRo
h09 -Co9, LTRHTR
1'9-C081 LTRHTR
14-C076 LTRHTR
! -C028 LTRHTRLTRHTR
C097 LTRHTR
CpH C; /Cz CO2
I J.4 c 2.317.1 c 2.30.97 0.691.03 0.860.76 0.231.26 0.341.47 0.192.J9 1.36I.S3 1.724.31 5.851.56 6.132.12 9.41
835
52.3 29.3 16.J14.1 34.8 48.8 -26.5 26.42 37.44 8.9417.5 14.08 62.26 5.6523.49 20.53 51.85 2.3715.09 12.43 67.1 4.0522.99 19.03 53.92 3.9614.89 11.74 68.64 3.3720.15 13.17 63.05 1.9 JI 1.86 9.34 71.28 1.6727.89 22.62 38.58 4.7826.46 24.01 35.38 4.7455 5 3220 7 38
low temperature reduction = 573 K.high temperature reduction"" 773 K.:2: ethenejethane ratio.
because Pt favors the spillover of H2
neighborhood of the metals [23], promotireduction of cobalt. Unfortunately, afterthe active bimetallic species are not prenant.
On the contrary, the effect of the redutemperature on the bimetallic catalyst Rhwas rather weak in the CO conversion.reaction rate is approximately of the same o~at LTR and HTR. We proposed from XPS c.a model system assuming that rhodium is c.persed over C0304 islands and on dispers.Co2+ species covering the support. TherefOlafter reduction the surface of the bimetall.system is not substantially modified, and thes,sites, even if some alteration occurs with respecito the initial state, are able to recover a perma-nent equilibrium state with time on stream.
The product seleCtivity obtained at isoconver-sion varied with the addition of a second metaland with the reduction temperature, as presentedin Table 3. In Table 3 are given the oxygenatedproducts, namely ethanol, isopropanol, propanoland butanol together with the selectivities ofC2--4' C; /C2 ratio and CO2,
Noteworthy are the behaviors of the CH 4 andC~ selectivities with the reduction temperatureand the rhodium content added to cobalt. Whileat LTR the selectivity of C J on Co /Nb20S ishigh and C; is low, this situation is reversedafter HTR. These results show strong interac-tion of cobalt with the support and its rearrange-ment, suggesting that new species promoted thesites which increase the hydrocarbons towardslonger chains [6,12].
Results reported in [5} have shown that theselectivity changes with time on stream. Theselectivity towards C; increases significantlyduring the first 8 h on stream before stabilizingfor more than 50 h. Therefore, the reactionmodifies the nature of the sites during the acti-vation with time on stream. Lee andBartholomew [24] proposed mechanisms involv-

ing the formation of an intermediate (CH xO) orformiate methoxy species, on different sites ofthe metal, in the presence of the support, due tothe spillover of Hz and CO over the support.This model is consistent for catalysts with lowcobalt loading which contains a large fraction ofaluminates or Coz+ species dispersed over thesupport and which are unable to dissociate COand Hz.
Nevertheless, the selectivity on Rh/NbzOs isdifferent, presenting large molecules of olefinsand alcohols. Iizuka et al. [25] reported theformation of such products on a Rh/ AlzO,catalyst.
For bimetallic catalysts, by increasing the Rhcontent, C I and C; selectivities attained mini-mum and maximum values, respectively, at aCo/(Rh + Co) ratio of about 0.5. Methane waslow while C; was enhanced by a factor 6-7,together with formation of oxygenates due tothe presence of rhodium. The C; products in-crease markedly with the increase of Rh, attain-ing a maximum selectivity of ca. 70% anddecrease with higher contents, as shown in Fig.6. The same behavior is seen for C; /Cz ratioand COz selectivities after HTR. These resultsare in good agreement with Iglesia et aI. [26] onRu-Co/ AI20, catalyst. They obtained, at simi-lar conditions, 85% of C; products, rather closeto our results. With respect to the formation ofalcohols, it attains a maximum with the additionof small amounts of Rh in the bimetallic cata-lyst but decreases to a minimum around theratio 0.5. This shows synergetic effects of Rh inthe presence of Co. In the present case, theselectivity changed drastically. On the contrary,van't Blik [19] did not observe marked changeson selectivity for Rh-Co supported on AlzO,and TiOz in the CO/Hz reaction at 523 K.
Lee et al. [27] suggested that rhodium in theionic state (Rhn+) favors the formation ofmethanol while Rho favors the formation ofethanol. However, they demonstrated that Co-Rh particles after reduction were enriched withCo and that rhodium initially present as RhO isoxidized due to CO adsorption. The Rh-Rh
Fig. 6. Product selectivities of c; and C I as a function of cobaltatomic ratio. (.~.) Rh-Co: C; after LTR; ("') Rh-Co: C; afterHTR; (.) Rh-Co: C 1 after LTR; (D) Rh-Co: C 1 after HTR;(~) Pt-Co C; after LTR; ('7) Pt-Co: C; after HTR; (e)PI-Co: C, after LTR; (0) Pt-Co: C, after HTR.
bond breaks, forming carbonyls (Rh(CO)z), inwhich Rh is in the ionic state Rh I+. Our resultsare consistent with the conclusion of Lee et aI.Therefore, with increasing Rho at the surface,during CO adsorption it provides Rh 1+, favor-ing the formation of oxygenated compounds.Nevertheless, the CO on platinum particles isbasically adsorbed in the linear form [13], whichexplains the lower selectivity towards the oxy-genated products in Pt-Co/NbzOs.
We try to explain this selectivity behavior onthe basis of our TPR and XPS results discussedabove. The surface model presented by Fryd-man et aI. [17] suggests schematically the pres-ence of Coz+ species covering practically thewhole niobium oxide surface as a thin and welldispersed layer, together with isolated islands ofCO,04 crystals covered with rhodium particles.As shown in Fig. 6, the selectivity is a functionof Rh added to Co and present similar shapesafter LTR and HTR. The TPR results suggest aneasy reduction of the supported phase, togetherwith a small fraction of the support. After LTR,an important amount is consumed at lower tem-

peratures on the bimetallic catalysts whereas onCo/Nb20s alone it is rather limited. Moreover,the selectivity towards C; increases signifi-cantly. Therefore, these results provided us aninsight to improve our model and to predict thatRho is well dispersed over the C02+ surfacelayer covering the support, being less dependentor the reduction temperature. Thus, the C02+species play a preferential role over niobiumsurfaces which corroborates with the proposedmodel of Lee et al. [27]. The presence of Rho(MeO) helps to reduce these C02+ species andC0304 particles at LTR and HTR. Therefore,during the reaction the reduced particles caneasily be transformed to Rhn+ and Con+ in thebimetallic catalysts, which are potential sites forthe methylene insertion, as well as the COinsertion to account for oxygenates and C;products. However, at higher content ofrhodium, larger particles may occur decreasingthe efficiency of formation of these ionic speciesdue to a better organization of both Rh and Coparticles in the bimetallic system.
(1) The addition of a noble metal to theCo/Nb20s catalyst promoted pronouncedchanges in the reduction of cobalt. It helps toreduce the C02+ and mainly the CoH speciesof C0304•
(2) XPS results made possible to confirm thatthe C02+ species are well dispersed on a thinlayer around the niobium oxide together withisolated C0304 crystallites. The C0304/C02+ratio depends on the surface area of the support.It has been shown that the noble metals are welldispersed over the C0304 and Co2+ species. Insummary, C0304' Co2+ as well as Pd~, Rh203and Pt02 are the main phases identified.
(3) The CO hydrogenation performed at twodifferent reduction temperatures showed goodstability for bimetallic catalysts, held constantup to 50 h with time on stream.
(4) The CO hydrogenation showed that the
addition of Rh to Co/Nb20s in differentamounts increased the C; hydrocarbons and theformation of oxygenated compounds after 573and 773 K. However, they attained maximumyields around an atomic ratio of 0.5.
(5) On the basis of XPS, TPR and CO hydro-genation data, we have a good proposal for amodel system. The C02+ species are well dis-persed as a thin layer at the surface and thenoble metal is also well dispersed over theC02+ species and CO,04 islands, promotingtheir reduction. Therefore, during the reaction,CO promotes the ionic state of Men+ and Con+
forming sites for the insertion of methylene andCO to account for the growth of carbon chainsand oxygenates. However, with increasing load-ing of noble metal it induces agglomeration ofthe particles, blocking the ionic sites.
We acknowledge CNPq, Pronac and Finepfor the financial support of this work.
[I] R.C Reuel and CH. Bartholomew, J. Calal., 85 (1984) 63.[2] D.G. Castner and D.S. Santilli. ACS Symp. Ser., 248 (1984)
39.[3] K. Kunimori, H. Abe, E. Yamaguchi, S. Matsui and T.
Uchijima, VIII Int. Congo Calal., Berlin, 1984, Vol. 5, VerlagChemie, Weinheim, 1984, p. 251.
[4] M.A. Vannice. J. Calal., 50 (1977) 228.[5] R.R.C.M. Silva, M. Schmal, R. Frety and J.A. Da1mon, J.
Chern. Soc., Faraday Trans., 89(2]) (1993) 3975.[6] A. Frydman, R.R. Soares and M. Schmal, Stud. Surf. Sci.
Calal., C (1993) 2797.[7] R.R. Soares, A. Frydman and M. Schmal, Catal. Today, 16
(1993) 361.[8] SJ. Tauster, S.C Fung and R.L. Garten, 1. Calal., 55 (1978)
29.[9] SJ. Tauster and S.c. Fung, J. Calal., 55 (1978) 29.
[10] G.L. Haller and D.E. Resasco, Adv. Calal., 36 (1989) 173.[11] F.B. Noronha, M. Schmal, M. Primet and R. Frely, Appl.
Calal., 78 (I99J) 125.[12] A.L. Lapidus, A.Y. Krylova, J. Rathousky, A. Zukal and M.
Jancalkova, Appl. Calal., 80 (1992) I.[13] E. Iglesia, S.c. Reys, S.L. Soled and RJ. Madon. Adv.
Calal. Related Subjects, 39 (I993) 221.

[14] A. Fukuoka, F.S. Xiao, M. Ichikawa, D.F. Shriver and W.Hendeson, Shokubai, 32 (1990) 368.
[15] R.R. Soares, D.A.G. Aranda, A,1. Almeida and M. Schmal,XIV Congo Ibero-Americano Cata!., I (1994) 189.
[161 F.B. Noronha, P. Delichere, G. Bergeret, B. Moraweck, R.Frety and M. Schmal, to be published.
[17] A. Frydman, D.G. Castner, M. Schmal and e.T. Campbell, J.Cata!., 152 (1995) 164.
[18J H,1.F. van't Blik, DSc.-Thesis, University of Technology,Eindhoven, 1984.
[19] T.e. Chang, J,1. Chen and e.T. Yeh, J. Catal., 96 (1985) 51.[20] Z. Hu, K. Kunimori and T. Uchijima, App!. Catal., 69 (1991)
253.
[21] L. Otto, L.P. Haack and J.E. de Vries, Appl. Cata!' B, 1(1991) I.
[22] SW. Ho, M. Houalla and D.M. Hercules, J. Phys. Chem., 94(1990) 63%.
[23] R.L. Chin and D.M. Hercules, J. Chem., 86 (1982) 360.[24] W.H. Lee and e.H. Bartholomew, Spring Meet. California
Catal. Soc., 1986.[25] T. Iizuka, Y. Tanaka and K,1. Tanabe, J. Mol. Catal., 17
(1982) 381.[26] E. Iglesia, S.L. Soled, A. Fiato and G.H. Via, J. Cata!., 143
(1993) 345.[27] G.V. Lee, B. Schuller, H. Post, T.L.F. Favre and V. Ponec. J.
Catal., 98 (1986) 522.




















