
Addition reactions of E-E and E-H bonds to triple ... - De Gruyter
Upload
khangminh22Category
view
2download
0
ADDITION TO CARBON : CARBON MULTIPLE BONDS • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • •
/ D•i / 1NTRODUCTION
. Carbon-carbon double bond consists of a strong sigma bond and a weaker pi-bond. The pi-bond IS represent~d by two rather diffuse Jobes of electron density, one above and one below the plane defined by six a~oms. The kinds of reactions that we would expect to occur with carbon-carbon double bond on the basis of thermodynamics are those in which the weak pi-bond is broken and is replaced by two new stronger sigma ~onds. A reaction of this type is called addition reaction. The reaction may be represented as follows :
....._C=C/ A -- / / - ,+ -B~.,,...T-T' A B
In the addition reaction, an unsaturated molecule and another reagent combine to give saturated compound. In addition reaction the sp2-hybrid ( or sp-hybrid) carbon atoms are rehybridised to sp3. Compound containing pi-bonds are of higher energy than comparable compound containing only sigma bonds, consequently an addition reaction is usually exothermic.
For example : CH2=CH2 + H2 CH3-CH3 Aff = - 32.7 kcal/mole
CH2=CH2 + Cl2 CH2Cl-CH2CI Aff = - 43.2 kcal/mole CH2=CH2 + HBr -~ CH3-CH2-Br Aff = - 19 keal/mole
CH2=CH2 + HOH CH3-CH2-OH Aff = - 10.9 kcal/mole Thus, addition reaction is the characteristic of reaction of a pi-bond. Ions and radicals may add
across a multiple bond; accordingly the additions are said to be : (i) ionic or polar addition and (ii) radical addition. . . . .
Ionic additions take place in polar solvents and the radical add1t1ons take place m n~n-polar solvents. In the case of an ionic addition reactions, an electrophile or ,a nucleophtl~ m~y initiate the process of addition. When an electr~phi!e. i~itiates the process._ th~ re~ct1on 1s termed as electrophilic addition; whereas a react10n m1t1ated by a nucleophtle 1s said to be a nucleophilic addition.
·•• I ELECTROPHILIC ADDITION TO CARBON-CARBON DOUBLE BOND
Th · b e and below a double bond is electron rich because of the pi-bond. Conse~uently . b e region a ov t Lewis bases Thus the pi-bond is particularly susceptible to r- onds have tendency to ac as · •

electrophiles. Electrophiles include positive electrophiles such as proton, neutral electrophiles such as • • EB ++
bromme and the Lewis a~ids, BF3 and AlX3. Metal ions that contain vacant d-orbitals-Ag, Hg and
Pt++ also act as electrophiles. To introduce the mechanism of electrophilic addition, let us consider the following general
reaction : I I
)C=C/ +E-Nu -C-C-Nu Substrate' Reagent I I
E Product (Adduct)
For E-Nu to react with substrate, there must be some attractive force that could cause the two species to come together.
EB •
The reagent, E-Nu can be thought of as being divisible into electrophile E and nucleophile e .
Nu. Double bonds have a tendency to act as Lewis bases. The 'c=C/ + :E® EB / ........ ••
electrophile E is attracted to cloud of pi-electrons and can be EB
embedded in it. Now the reacting species E has made contact and the reaction can commence. Since electrophile initiated this contact, the reaction is termed as ele~trophilic addition.
n-complex or
Fig. 9.1. Formation of n complex
The species that results from the interaction of pi-electron clouds of carbon-carbon double bond with an electrophile is known as pi-complex. During the interaction vacant orbital on the electrophile overlaps a pi-orbital of the double bond. In this complex electrophile is not located on a particular atom. the pi-complex can be represented as shown in Fig. 9.1.
Thus, the initial step of an electrophilic additio~ is the formation of a pi-complex. Such a complex does not actually involve a formal covalent bond and may be regarded as an association in which electrophile gets embeded in the pi-electron cloud of double bond. The pi-complex then may change to a carbocation through the formation of a sigma bond between the elecrophile and any on~ of the double bonded carbons as shown below :
I ® --+ -c-c-
1 I - E
n-complex Carbocation
If the electrophile has a lone pair of electrons, then a three . . in which the electrophile forms two bonds with tw b membered cychc cation results bears a positive charge. 0 car on atoms of the carbon-carbon double bond ao<l
The cyclic cation may either be an intermediate ( . I true cyclic product (e.g., addition products of carbe e_.g., m the case of polar addition of Brz) ~r a :
. cation has, to some extent, a carbocation charact n~ n_itrene, etc.). However, the cyclic intermediate 1 three resonating structures. er. at,ons such as these may be described in teflll
5;

T E E -C-c /\ Ell I I , - -C--C- -C-C- . If both (A) and (A) I I I I
contributor to the (C) correspond t (B) (C) If either (A) actual structure of th O un~table carbocation then structure (B) is a more important
or (C) c e cation more and the cation h orresponds to rela . . . .
The carbocaf as substantial carbo / 1vely stable carbocat1on, then these structures contnbute ion can then be att k ca 10n character without much cyclic ion character.
E + ac ed by a nuc~~ophile to yield the prcxluct.
e E +Nu ---. "---- / E~ I ~c, ---+ ~C-Nu
Nucleophilic attack on carbocation (:t) , ~r.,., , , _
.,,,,,..c~ ---+ c-c-~e - .,,,,,.., I
Nu Nu Thus, the overall . Nucleophilic attack on cyclic ion First step. mechanism can be represented as follows :
Second step.
\ I 1ctc\
E© 7t-complex
+ Nu8
"--c111111*c/ Slow
Fast
E* TS1
"--c-~/ Fast
/ I ~" E Nu0-
TS2
The free energy diagram can be represented by the Fig. 9.2.
I
" I C-C-Nu /I I E Product
M STRUCTURAL ORIENTATION AND REGIOSELECTIVITY
If an alkene is unsymmetrical, there is the possibility of two different products from the addition of unsymmetrical reagents.

TS1
Progress of the reaction --+ Fig. 9.2. Free energy diagram for electrophillc addition reaction
CH3-CH=CH2 + H-Nu CH3-CH2-CH2-Nu + CH3-CH-CH3 . . I
Nu
This problem of structural orientation does not arise in the cases of a?~ition of symmetr~cal reagents to both symmetrical and unsymmetrical alkenes and also the add1t1on of unsymmetrical reagents to the symmetrical alkenes. For example :
CH3-CH=CH-CH3 + HBr Symmetrical Unsymmetrical
CH3-CH-CH2-CH3 I Br
Only one possible product
CH3-CH=CH2 + Br2 CH3-CHBr-CH2Br Unsymmetrical Symmetrical Only one possible product
An electrophilic addition leads to the formation of two products, one product usually predominates over the other. This is known as structural orientation. Structural orientation in electrophilic addition reactions means as.:.:rtaining which one of the doubly bonded carbons ultimately forms bond with the electrophile and which one combines with the nucleophile. In most of the cases problems may be solved by emperical rule given by Markovnikov and this emperical rule is known as Markovnikov rule. This rule states that the positive part of the unsymmetrical reagent attaches itself to that doubly bonded carbon atom which has more number of hydrogen atoms. This means that negative part goes to that doubly bonded carbon which bears less hydrogen atoms.
For example : Cl I
CH3-CH=CH2 + HCI CH3-CH-CH3 . Markovnikov addition product
Reactions that illustrate Markovmkov rule are said to be Markovnikov additions.

I H I I THEORETICAL EX PLANATION OF MARKOVNIKOV RULE Markovnikov form .
explained h ulated his rule b . on t e basis the mech . ecause of expenmental observations, this rule can be If an unsymmetrical lk amsm of the reaction.
conceivabl I a ene undergoes actd· · · Y ead to two diffe t . itioo with an unsymmetrical reagent, then step-I could ren carbocations :
e CH 2 1 -<CH3-CH2-CH2
3-CH=CH2 + H-X 1°-car;acation
CH3-CH-CH3 . When propene reacts with HX h 2°-carbocation · It can add to the carbon-
2 t f: ' t e proton can add to carbon-I to form primary carbocation or
2-halopropane because it is i or~ a secondary carbocation. The chief product of the reaction is pi-complex collapes more rea~~; ty mo;e stable ~arbocati_on. This indicates that the first-formed
Y O orm 2 -carbocahon than 1t does to form 1 °-carbocation (Fig. 9.3)
w
r CH3-CHtCH2
H (±)
1t-complex
(±) CH3-CH-CH3
Progress of formation of carbocation ----Fig. 9.3. Decomposition of the 1l'"'Complex to protonated propene Formation of
2°-carbocation occurs more readily than does formation of 1 °-carbocation.
The major product obtained by addition of HBr to 2-methyl-2-butene is 2-bromo-2-methyl Jutane; only a small amount of 2-bromo-3-methyl butane is obtained. The major product obtained by lddition of HI to 1-methylcyclohexene is 1-iodo-1-methylcyclohexane. In both cases the more stable ertiary carbocation is formed more rapidly than the less _stable seco~dary carboc_ation, so the major ,roduct of each reaction is the one that results from formmg the tertiary carbocatton.
Br Br I I
CH3-C=CH-CHJ + HBr CH3-1-CH2-CH3 + CH3-,H-CH-CH3
I CH3 CH3 CH3 Major product Minor product

+
Major Minor product product
Th~s, add~tion of a reagent to an unsymmetrical alkene proceed~ by ":'ay of the m_ore st
able carbocation. This is the reason that Markovnikov rule is followed. With this understanding of the me~~anism f~r the ionic addition of unsymmetrical reagent to alkenes behin~ u~, we now in position to give following modern statement of Markovnikov rule. In the wmc additi?n of an unsymmetrical reagent to double bond, the positive portion of the addin? reagent ~ttaches ~tself to _a carbon atom of the double bond so as to yield the more stable carbocat1on as an intermediate. This statement can also be formulated as follows : Negative part of the reagent adds on to that doubly bonded carbon which forms more stable carbocation with electrophile.
Unsymmetrical alkenes give two different products with unsymmetrical reagents. These products are constitutional isomers.
A reaction (given above) in which two or more constitutional isomers could be obtained as product but one of them predominates is called a regioselective reaction. Thus, ionic addition of unsymmetrical reagents to unsymmetrical alkenes is regioselective reaction.
There are different degrees of regioselectivity. A reaction can be moderately regioselective, highly regioselective or completely regioselective.
In a completely regioselective reaction, one of the possible products is not formed at all. The addition of a HX to isobutene is more highly regioselective than the addition of a HX to 2-methyl-2-butene © ©
CH3-C=CH2 + HBr CH3-C-CH3 + CH3-CH-CH2 I I I
CH3 CH3 CH3 3°-carbocation 1 °-carbocation
There is great difference between the stability of 3° and 1 °-carbocation. HBr © e
CH3-T-CH2-CH3 + CH3-,H-CH-CH3 CH3-C=CH-CH3 I
CH3 CH3 CH3 3 ° -carbocation 2° -carbocation
These two carbocations are closer in stability. The addition of to 2-pentene is not regioselective. Both isomers are bt · d from the
dd. . f . h f h 2 b . o ame a 1t10n o proton e1t er o t sp -car on produces a secondary carbocation. Both carbocations
have the same stabihty, so both wil_I form equally easily, resulting in the formation of a roximately equal amounts of the two alkyl hahdes. PP .
EB H
CH3-CH=CH-CH2-CH3 EB
CH3-CH-CH2-CH2-CH3 + CH3-CH2-CH-CH2-CH3
I EB Both are 2°-carbocation I Br EB I
CH -C Br , 3 I H-CH2-CH2-CH3 CH3-CH2-CH-CH2-CH3 I I I
Br Br

ADDITION TO CARBON -CARBON MULTIPLE BONDS
Both alkyl bromides ar . Anti-Markovnikov . : form~? m comparable amounts.
a powe.rful electron w·thd•om~ addition : Anti-Markovnikov addition is observed if the alkene bears - 1 rawmg g . , , , roup attached directly to the doubly bonded carbon.
' .,. Br I e CH2-CH2-N(CH3)3
Br I
. CH2=CH-CF3 HBr CH2-CH2-CF3 These anfl-Markovnik dd .. the eJectron-withdra . ov a lt10ns do proceed through the most stable carbocations. However,
relative to the Jo wmdg gro~p (-I group) of the double bond destablise the higher order cation wer or er cation due to -I effect.
CH2=CH-CF3 + H CH2-CH2-CF3 + CH3-CH-CF3 1 ° -carbocation 2° -carbocation
(more stable due to (less stable due to
- I powcr r~~F3> - I poi~:fCF3)
Br I
Br I
CH2-CH2-CF3 CH3-CH--CF3 . . · Major product Minor product
Thus, while they violate the emperical Markonikov rule, they follow the modem Morkovnikov rule.
/@i/ REARRANGEMENTS IN IONIC ELECTROPHILIC ADDITION REACTIONS
Some e1ectrophi1ic addition reactions give product whose formation neither can be explained by Markonikov or modern Markonikov rule. For example, the addition of HBr to 3-methyl-l-butene results in the formation of two products : 2-bromo-3-methylbutane, the product expected from Markovnikov rule and 2-bromo-2-methyl-butane, an unexpected product. Furthermore, unexpected product is the major product of the reaction ..
CH3 CH3 CH3 I I I
CH3-CH-CH=CH2 + HBr CH3-CH-CH-CH3 + . I
CH3-C-CH2~CH3 I
Br Br · Minor product Major product
(normal product) (unexpected product) Similarly 3,3-dimethyl-1-butene also gives normal product and unexpected product with HBr.
CH CH3 Br Br I 3 I · I I -
CH3-C-CH-CH2 + HBr ---7 CH31-CH--::-CH3 + CH3-, y1H- CH3
/ CH3 CH3 CH3 CH3 Major product
(unexpected product) Minor producj (normal produt t)

F. c. Whitmore was the first to suggest that the "unexpected" product results from a rearrangement of the carbocation reaction intermediate. Carbocations rearrange only if they become more stable as a result of the rearrangement.
® CH3-CH-CH=CH2 + H ---.
I CH3
CH3-C-CH- CH3 I
CH3 A secondary carbocation
(less stable)
Br I
CH3-CH-CH- CH3 I . CH3
e a 1, 2 H shift ®
CH3-C-CH2-CH3 I
CH3 (a 3° carbocation)
(more stable)
Br le
Br I
CH -C- CH2-CH 3 I 3
CH3 Normal product Rearranged product (minor product) (major product)
Carbocation rearrangements can also occur by ring expansion. Which is another type of 1,2-shift.
(>+ CH3
ITCH3 @ 6GCH3 6 H CH l, 2-shift of CH=CH2 CH- 3 b CH3
® I carbon-car on 5 l 5 4 2 sigma bond 4
2°-carbocation 3°-carbocation
In the above example, the carboczrtion formed initially is a secondary carbocation. Ring expansion leads ·to a tertiary carbocation, which is more stable and, because of the five membered ring, has less angle strain.
It is important to remember that whenever a reaction leads to the formation of a carbocation you . must check its structure for the possibility of rearrangement.
/ n, / sTEREocHEMISTRv oF ADDITION REAcT,oNs
Stereochemistry of the addition reactions depends on two factors : (i) Whether both the electrophile and the nucleophile will join themselves to the two double
bonded carbons from the same side (syn addition) of the double bond or from the opposite I sides (anti-addition)
(ii) The geometrical orientation of the two parts of the a1ddendum E and Nu to each other and
the rest of the product molecule, i.e., the configuration of the addition product. [

ADDITION TO CARBON 'ARBON MULnPLE BONDS
(B 0 479 If E and Nu enter from th . ( · ) d e same side of th d cts an stereospecific· whe . e ouble bond (or triple bond) the addition will be syn . . , reas if they t f ' stereospec1f1c but anti (tr ) S . en er rom the opposite sides the addition will be
If h ans · omet1mes th dd · • ' t e electrophile forms . _e a tlon 1s found to be non-stereospecific. nucleophile has no other altem t cyclic cation as an intermediate in an electrophilic addition, the the addition is anti. · a ive but to attack the intermediate from the opposite side and naturally
(~u 'c ,,,,-- © ......___ \ ,,,,-- =c......___ + E --. c-c.,,,,,, ---+ /\E-)-......_
(±) On the other hand, if the intermediate is I . . . . . . .
stereochemistry of the dd • • a c ass1cal carbocat1on, 1t 1s difficult to predict the rotation about carbon- ab iti~n. When the carbocation has relatively long life, this may undergo Relativel short-I" d carbon st?ma bond and as a consequence the reaction will be non-stereospecific.
. Y • Ive . c_ar ocations undergo stereospecific reaction. For example, when the reagent is a dipole, after the addition of the electrophi'li'c t th I h·1 l' • ct· • • . . , . par e nuc eop 1 e may 1orm an mterme iate 1on-pa1r with the carbocation and m this case the addition will be syn.
_d e E Nu E Nu
"l ?, "I (±)/ ,,,,--c-c, ---... .,,,,,,c-c'-- ---. Ion-pair
To ascertain the nature of addition, syn or anti of a particular reagent (E-Nu), it is very often allowed to add to a substrate of the type abC = Cab (where E *a* b and Nu *a* b but E may or may not be identical to Nu.
Note: Reagents which form four membered cyclic intermediate (or TS) also give syn addition reaction. The main examples are addition of BH3 and addition ofH2 in the presence of a catalyst.
Since both the double bonded carbons are identical and since each of them has 50% probability of being attacked by E, the following results are obtained from cis- and trans-isomers :
Case-I : When E :t= Nu . and addition is syn. (i) Syn addition to cis-compound gives (d[) erythro-form. (Scheme-9.1)
Nu E
--+ a-0 + a~ ._ b b
(dl)-Erythro form
ea
a~h
b
Scheme 9_ 1 _ Syn-addition to a els-form
.. . . ompound gives (di) threo form (Scheme-9.2) (11) Syn-add1t10n to trans-c
.~· - .-0" + .:0 -- ~r· b ~~~form
Scheme 9.2. Syn-addition to a trans-form

Case-II : When E =t:- Nu and addition is anti. (i) Anti-addition to a cis-compound forms (dl) threo form (Scheme-9.3)
a
iAg aVNu b
a a
- kb + ~ukb _ a-{' Nu a-{' i: b b
(dl)-thero fonn Scheme 9.3. AntJ.additlon to a els-form
a j b
b
(ii) Anti-addition to a trans-compound forms (dl) erythro-form (Scheme-9.4). Note : When E = Nu, the (dl) erythro-form becomes a meso but the (d[) threo-form remains
the same. Thus, if the configuration of the substrate and that of the product are known, the mode of the
addition and the probable mechanism may be guessed. For example, addition of bromine to fumaric acid forms the meso product; it is therefore expected that the addition is anti which in turns indicates that the reaction may involve cyclic bromonium ion.
a a ~b b e Nu e~ b ::u (f~
a a
l ! a a -0:b -trb +
a a b b
(±) Erythro-form
Scheme 9.4. Anti-addition to a tran~form
/@I/ ADDITION OF BROMINE AND CHLORINE
Alkenes react with solutions of bromine and chlorine in an inert solvent to produce vic-dihalides. Addition reaction between an alkene and iodine is a reversible one and the equilibrium lies over the reactant side. ·
Example :
.........._C=C/ +X2 .........._C-C/ / ' /I 1'
X X vic-dihalide ·
X = Cl or Br
. Br2/CCI4 .. . CH3-CH=~H2 --- CH3-CHBr-CH2Br . ·
. ~he rate_ of add,1t10n ~f halogen mcreases with increasing alkylation at the double bond. Thus, react1v1ty of given alkenes m decreasilJi order is as follows :
R "C=CH2 > R-CH=CH2 > CH2-CH2 R/ I

•
Mechanism of Brom· . mat1on Step-I · Th h . ·t · · e alogen molecule i 1 · · s sigma bond, and the elect h T s po ansed by the electron rich carbon-carbon pi-bond along
1.e., _7t-complex. rop 1 ic halogen reacts with the pi-systerri to 'form a weak complex,
Step-II :
:Br-Br: . .. . .
o+ Br I Br &-
o+ &-Br-Br
1t-complex (I)
The 7t-complex breaks down to form a er-complex.
~+ Br I Br &-
, V,,,. e __. C--C +Br A'--
:~r: u\ sp- hybrid carbon
a-complex {II)
. .
Step-III : The vacant p-orbital on sp2 carbon overlaps with non-bonding electron pair on bromine to form a cyclic bromonium ion. Bromonium ion is a cation in which the bulk of the positive charge is on the bromine.
Cyclic bromomium ion (III)
_i I
Step-IV : The cyclic bromonium ion is attacked by a nucleophile at the carbon which are part (fl
of the three-membered ring. They are attacked because the Br makes them slightly positive. This (fl
particular step can be considered an SN2 reaction where the leaving group is Br.
(tr '--c c/ ___. / \ A'--By
(±)

Evidence for the mechanism E>
(i) Wh~n bromination is carried out in a solution containing a nucleophile other than Br (for
example, ~l), the product includes a mixed halogen product but does not include dichloride.
0 Br2/Cl CH2=CH2 --4 CH2Br-CH2Br + CH2Br-CH2Cl no CH2Cl-CH2Cl
Formation . of CH2Br--CH2Cl confirms that reaction intermediate is positively charged e
intermediate which is trapped by the nuceophile, Cl. This experimental result also ruled out a concerted addition of Br2 which can takes place as
follows : .......... / .......... / /C~C........_ Br ,........c-C........_Br
~Br (ii) The addition of bromine to cyclopentene provides evidence for bromonium ion intermediate
in bromine solution. When cyclopentene reacts with bromine in carbon tetrachloride, anti addition occurs and the products of the reaction are trans-1,2-dibromocyclopentane enantiomers. o ~~i.• U+Q
Br H H Br This anti-addition of bromine can be explained by a mechanism that involves the formation of
bromonium ion. · An open or free carbocation similar to (II) in step II is inconsistent with this stereochemistry.
If the intermediate had a vacant p-orbital on a carbocation, that vacant orbital would be subject to attack from either side. An attack on both sides of the p-orbital would mean that bromine addition to cyclopentene would have led to a mixture of cis- and trans-1,2-dibromocyclopentane.
+ Br a H b/0
,-----.1 H Br
~e Br
Open-carbocation
b
Br cis
~r
Br trans
This observat_ion confi~ms that pi-~om~lex does not convert into sigma complex. This 1t-complex directly converts_ mto cychc ~romomum 1~0 - A symmetric cyclic bromonium ion explains the formation of eqmmolar amount of the enanttomeric trans-1,2-dibromocyclopentane. Since attack at carbon I and 2 are equally probable.

Q . I
© r I 2
eJ te Br Br 2
Br H
H Br One of
the products of the addition of bromine to propene in the presence of chloride ion is
1-bromo-2-chloropropane. 2-Bromo-1-chloropropane is not formed in this reaction.
0 Cl Br Br/Cl / /
CH3-CH=CH2 ----4 CH3-CH-CH2Br but no CH3-CH-CH2-Cl To accou_nt for_ this we must modify the structure of cyclic bromonium ion. The above r~s~lt can
only be expla1?ed if we place more positive charge Rn carbon-2 than on carbon-I. This 1s not unreasonable, smce the methyl group should stabilise some positive charge at carbon-2. The net result is_ to produce an unsymmetrical bromonium ion.
6+ CH3-CH-CH2
I ¾ Br 6+
Unsymmetrical bridged bromonium ion
The incoming nucleophile, Cl, preferentially attacks the unsymmetrical bromonium ion at C-2 producing 1-bromo-2-chloropropane rather than 2-bromo-1-chloropropane.
~I CI 6+ I
CH3-CH-CH2 ----+ CH3-CH--CH2-Br I
CJ: On the basis of above results mechanism of the reaction can be as follows :
Step-I: ')C=C( +Br-Br ")C=tc~
Step-II :
Step-III :
'etc/_.. / .........
Br-Br 6+ 6-
Br-Br 1t-complex
'-- Ii+/ ./c\-p, +-+
Br Ii+
Br --..... I c-c-/1 I
Br

The following experimental results support the given mechanism : (~) T~e rate of the addition of bromine increases :
W~th increasing number of electron-donating groups on ooubly bonded carbons. (n) With the increasing polarity of the solvents. In carbon tetrachloride medium the reaction is
slow._ O~ the other hand, in the polar protic solv~nts like HOH., ROH and RCO~H. the , reaction 1s rapid and mixed products are obtained which consist of monobromo and dibromo derivatives.
Since the polarising effects increases the rate of the reaction, it proceeds through a polar mechanism. . .
(2) When cis,-2-butane is treated with bromine, cent percent racemic, 2,:}-dibromobutane (± threo form) is obtained; whereas bromination of trans-2-butene gives entirely meso-2,3-dibromobutane ( optically inactive, eryt~ro form)
CH3 CH3 I - I
H-C-Br Br-C-H I + 1-- ·
Br-C-H H-C-· Br I I CH3 CH3
(elf) mixture; ± threo fonn
CH3 I
CH3" /H H-C-Br
/C=C, +Br2 I
H . '-CH3 H-C-Br I CH3
, Meso-erythro form Sinc;e trans-add(tion of a symmetrical reagent to a cis-isomer forms (±) threo derivative and that
to a trans-isomer produces meso-erythro, the addition of bromine to a carbon-carbon double bond must be cent percent trans-addition. Cent percent trans-addition rules out the possibility of the formation of an intermediate carbocation because both the isomers, cis- and trans would then form one and the same carbocation which in turn would give rise to identical product distribution form both the isomers. Furthermore, to account for the cent percent stereospecificity and the product distribution, a bridged bromonium ion may be considered.
(B) Kinetics : The reaction follows the second order kinetics : Rate= K [Alkene] [Br2]
This confirms .that the transition state of the rate limiting step must involve both the substrate and the halogen. This is also consistent with the proposed mechanism.
In several situations bromonium ions have been observed. Under superacid conditions 1-bromo-2-fluoropropene gives a cation which in fact is a bromonium ion. Its formation is confirmed by NMR spectroscopy.
SbF5 0 CH3-CH-CH2-Br ---~ CH3--CH-CH2 + SbF6
I \/ F Br
EB

--, .. '-C OUNDS
Bromonium ion produced b . . . 1
bserved by NMR. Y species that should generate electrophilic bromme is a so 0
CH3, . CH "c-c / 3 o+ 0- Ell e CH3\ /CH3 e
CH / - "\ + Br-~N-SbF5 --) C-C, + CNSbFs 3 CH3 CH3/ \/ '-CH3
Br T.he highly hindered alk d . Ell • • h ·ch .
tallised as a trib 'd ene, a amantyhdene adamantane forms a bromomum 10n w 1 crys. h th c lll _romi e salt. An X-ray crystal structure determination of this compouod shows that 1t as e 101 owmg structure :
Fig. 9.4. · e
er®
e Br
In this case bromonium ion is not attacked by Br, the attack is completely prevented by the steric hindrance offered to the backside approach of the bromide ion by the extremely bulky cage like structure.
Whether the intermediate is halonium ion or an open carbocation, the mechanism is termed as AdE2. The kinetics of brominations are often complex and a third order reaction (AdE3) has been proposed namely the attack of a halide ion on the 1t-complex. The 1t-complex may collapse to an ion-pair which then gives the product (~cheme 9.5).
'-., / Br2 c-c ---'--"-+ / \/ '-..
Br® Br0 Anion pair
Scheme 9.5.
Effect of substituents on the mechanism . When an electron-donating group or electron-withdrawing groups are present on doubly bonded
carbon, the reaction is always anti-addition reaction. COOH I
- H H - Hi-Br "C=C/ +Br2
HOOC/ "COOH Br-C-H I
+ enantiomer
COOH
COOH I
H COOH H-C-Br . "-c=C/ + Br2 I / '--. H-C-Br HOOC H I
COOH
L

When the alkene has an aryl group attached to the double bond; the selectivity becomes less and both anti- and syn- adducts are formed. ·
C6Hs, / CH3 ' ~00 00 H/ C=C"-._ H + Br2 80% cis-product + 20% trans-pr uct
C6Hs Ii . "'--e=c/ + Br H/ "--.ctt3
2 AcOH 17% cis-product + 83% trans-proouct
C6Hs' / C6Hs AcOH /C=C" + Br2 90% cis-adduct + 10% trans-adduct H H
A common feature of the compounds that gi,;e extensive syn-addition is the presence of at least one phenyl substituent on the double bond. The presence of a phenyl group diminished the strength of bromonium ion bridging by stabilising the cation centre by delocalisation. A weakly bridged structure in equilibrium with an open benzylic carbocation can account for the loss in selectivity, i.e., streospecificity.
Br2 ..
. J
(ion pair) (II)
Rotation about carbon-carbon
single bond
(I) will give trans-adduct and (II) will give cis-adduct.
e Br
C6Hs, +o /o--I3 c-c H/ %;_ / '----H
Br +o
(Bridged structure)
e Br
C<iHs, ltt>. /CH3 c-c H/ ~"H
Br (ion pair)
(I)
Addition of chlorine : Aliphatic alkenes usually give trans-addition, syn-addition is often dominant for phenyl substituted alkenes.
CH3 \
H-C--Cl \ · + enantiomer
Cl-C-H \ CH3

C6Hs I
+ H-y-CI
H-C-CI I
C6H6 CH3 ami-product syn-product
(minor) (major)
The above results indicate that with unconjugated alkenes, there is strong bridging and high anti-stereospecificity. But phenyl substitution leads the formation of carbocation as reaction intermediate and there is more syn-addition due to the formation of ion pair. Chlorine is not as effective as bromine for the formation of halonium ion because of its smaller size and lesser polarisibility. Bromination therefore generally gives a higher degree of anti-addition than chlorination, all other factors being the same.
Proton loss and rearrangement reactions also take place in chlorination. These two reactions confirmed the formation of carbocation as reaction intermediate.
Cl CH3" Clz CH3" /
...... C=CH2 -- C-CH2Cl + CH2=C--CH2Cl CH3' AcOH CH3/ /
(1) CH3 . minor product (2)
major product
Formation of product (2) can be explained by formation of cyclic as well as open-chain cation.
AcOH
l l
On the other hand, formation of rearranged product can only be explained if the reaction intermediate is carbocation.
CH3 I AcOH
CH3-C-CH CH2 + Cl2 I
CH3
CH3 CH3 I I
CH -C-CHCI-CH2Cl + CH2=C-CH-CH2CI 3 I I
CH3 CH3

Reaction takes place as follows :
CH3 CH3 I I (t) c~3 shift CH3-C-CH=CH2 + Cl2 ---+ CH3-C-CH-CH2CI 7 I . I . .
CH3 CH3
Rearranged product
(t) CH3-c---CH-CH2Cl I I -
~HCH3
Addition of halogens to Alkynes · : The addition of bromine to alkynes is found to be stereoselective,, In this reaction trans-product is the prominent product. This result indicates that the addition takes' place via formation of bridged halonium ion in two steps :
R-C=C-R + Br2 ----+-
® ,,.-,Br
, Y \ ir R-C~
I DI I ADDITION OF HYDROGEN HALIDES
R._____ /Br C=C
Br/ "--R trans product
Hydrogen halides add to the pi-bond -of alkenes to yield alkyl halides. The addition of hydrogen halides to alkenes to prepare alkyl halides is often used as a synthetic reaction. Usually, the gaseous hydrogen halide is bubbled through a solution of the alkene. The reactivity of HX in this reaction is HI> HBr-0> HCl > HF. The strongest acid is the most reactive while the weakest acid is the least reactive. '"
""- / I /C=C.....,__ +HX -CH-C-X I I
Product Addition of HX takes place in two discrete steps :
EB Step-I : . Electrophile H attacks on pi-electron cloud of an alkene to form either a more stable
carbocation or a 1t-complex, i.e., protonium ion. Since there is no non-bonded electron pair on a proton, the formation of bridged protonium ion
is not possible. The electrophilic attack may or may not occur via a pi-complex formation as shown in Scheme 9.6.
The carbocation formation step is slow step-the rate determining step. E>
Step-II: The rapid attack of the carbocation by the nucleophile X to give the addition product. ........... EB / E> fast ........... / /CH-C, +X /CH-CX,
step
Since the first step, the rate determi:°in~ step, involves the substrate and the reagent, the reaction is expected to follow the second order kmet1cs. _
Rate= K [alkene] [HX]
Among the cases in which second order kinetics have been observed are the addition of HX tc styrene, stilbene, and isobutene .
•

-........ / /e: e, tt6+
I I x_&-
Polarisation of H - X by 7t-electrons
6+ H
.......... 6+/ e"""e / - '--T.S.
'ere/ / '--(:f) H
7t-complex Scheme 9.6. Electrophilic addition of HX
-
-In most of the cases the reaction also follows the third order kinetics.
Rate= K [Alkene] (HX]2
e H 0X '--' / ,,,c-o-----
Carbocation
(:f) H
'-..... ~/ e"""e / - '--Complex
protonium ion
Among the cases in which third order kinetics have been observed are the addition of halogen halides to 2-methyl-1-butene, 2-methyl-2-butene, 1-methylcyclopentene and 1-methylcyclohexene. The third order kinetics suggest that most of the alkenes react via a transition state that involves the alkene, hydrogen halide and a third species (which is also HX) which delivers the nucleophilic (Scheme 9.7).
-........ / /C=C-........ +HX
H-X -........= . / /C=C-........
X-H
Fast
Fast ,
Slow
-........= / e=c / (&+-' ~H OR
il-X
'e=c~ / -........
H
-........~....,....c/ / 1'
X Scheme 9.7.
Slow step
Stereochemistry : The stereochemistry of the addition of hydrogen halide to carbon-carbon double bond is not well established. The reaction is less stereoselective than the addition of halogens. There are cases where trans-addition occurs, at the same time predominant cis-products are also obtained in a number of cases. 1,2-Dimethylcyclohexene, cyclohexene, 1,2-dimethylcyclopentene, cyclopentene, cis- and trans-2- butene and cis- and trans-3-hexene give trans-addition with hydrogen bromide. On the other hand, cis- and trans-1-phenylpropene give predominentaly cis-addition with liBr and HCI.

For _the trans-addition, it is assumed that the nucleophile, X attacks the protonium ion complex from behmd at the more stable carbocation site (Scheme 9.8).
For example :
® H
H I 'c-C,.....-
/1 -........ (a)
X
(b)
H
X trans product
,..® ,H
C6Hs
Scheme 9.8.
0 Br
Br trans product
H
To account for the predominant cis-product in case of I-phenyl propene, it is supposed that the carbocation and the halide ion form an ion pair which then collapses to form C-X sigma bond from
e H X . H X
, / ® ,I ®/ ,I I/ . /c=c, + H --+ c-c --+ /c-c, / I Ro;on about cis product i C-C (major product)
H ,b_c/ / ®"
e X r
H
"6-c/ / I"
X trans product
Scheme 9.9. Formation of els product as main product
r

the same side of the double bond and thereby cis-prod.uct results. It is further assumed that when the
carbocation is stable, a portion of the carbocation rotates about C-C sigma bond then forms the trans-product. (Scheme 9.9)
Addition of hydrogen halide to carbon-carbGn double bonds can be controlled by altering the solvent _a~d the temperature. Addition in non-polar solvents is predominantly anti- while in polar ~olve~t It is ;yn. For ex~1?1ple, the addition of hydrogen chloride to 1,2-dimethylcyclohexane in ether is anfl at 25 C, syn-addition dominates in CH2c12 at -78°C. •
/II=•/ ACID-CATALYSED HYDRATION AND RELATED ADDITION REACTIONS
The_ acid-catalysed addition of water to the carbon-carbon double bond and triple bond is known ~s hydratwn of alkenes and_ alkynes. This reaction is very useful method for conversion of alkenes mto alco~ols. The acids most commonly used to catalyse the hydration of alkenes are dilute solutions of sulfunc and phosphoric acid. Hydration of alkenes are usually regioselective and it follows Markonikov rule.
OH I
R-CH=CH2 + HOH R-CH-CH3 Cone. H2SO4
The mechanism for the hydration of an alkene is simply the reverse of the mechanism for the dehydration of an alcohol. Since the reaction is a reversible one, the principle of microscopic reversibility may be applied to determine the mechanism of the hydration of alkenes. The principle tells us that for a reversible reaction, the mechanism for the forward reaction and the reverse reaction is one and the same provided the reaction conditions are identical. Naturally both the reactions must involve identical transit-ion states and reaction intermediates. The mechanism of the reaction is as follows :
First Step : The· first step is the protonation of the alkene to yield a carbocation. This may or may not occur via a pi-complex or protonium ion. This step is the rate determining step.
0
.. t H-O-H+H-O-S-O-H .. +
0 (±)
R-CH=CH2 + H-O-H I
H
Transition state of the first step is 6+ 6+
R-CH!!.!!.!!CH2 111111 H
® e H-O-H + O-SO3H
I H (±)
R-CH-CH3
1l

S · h cleophile, i.e., H20 to form a protonat d tep-11 : The carbocation of the reaction reacts wit n~ e alcohol.
. 8so3H is a weaker nuclephile than water, hence does not attack the carbocation. Furthermore,
water molecules envolve the 8so3H ion b:}' solvation and thus it fails to come in contact with the carbocation.
0000000 o e O 0 0 S03H ()4-Watermolecule 0000000
Step III : A transfer of a proton to a molecule of water leads to the formation of the product.
R-CH-CH3 I
H~~'-H
\__0-H I H
(t) R-CH-CH3 + H30
I OH
The proposed mechanism is supported by the following facts : e
(i) In the addition of water there is practically no OH -ions present in the solution at any moment because hydration does not occur in alkaline medium. This is strong evidence in favour of the first step which involves the electrophilic attack of a proton.
(ii) Water alone does not attack the carbon,..carbQJ1 double bond. W.ater is a weak electrolyte it does not dissociate appreciably. Thus, it cannot transfer a proton. Therefore, .the proton must come from the acid.
(iii) Cert1a~n dal~~~es bform .rear:ange~ products. ~o~ation of rearranged products can only be . I
exp ame .;, .car ocat1on 1ormat1on as reas;tton mtermediate. For example : CH3 CH3 OH I I H2S04 I I
CH3-C-CH=CH2 HOH CH3-C-CH-CH3 + CH3-C---CH-CH3 I I I I I
CH3 CH3 OH CH3 CH3 (minor product) (major product)
. normal product rearranged product (iv) With styrene, the rate of hydration is increased by electron-donating substitutents. A
substantial solvent isotope effect is also observed. Both of these observations are in accord with a rate determining protonation to give a carbocation intermediate.
OH HOH I
R-CH=CH2 -- R-CH-CH3 KH2o H2S04

R--CH=CH2 D20 D2S04
KH -=2-4.
Hd . Ko Y ration reaction is not a 1 .
0th 1 . . se ect1ve reaction er nuc eoph1hc solvents J"k . - . with alkenes in the presenc f I e RO_H, PhOH, RCOOH also give electrophilic addition reaction analogous to that for hydr ; 0 sstrong acid catalyst. The reaction and mechanisms in these cases are
a ion. ome examples are :
CH3-C=CH2 + CH3OH _H_B_F_44
I CH3
CH3-CH=CH2 + CH3COOH _H_B_F_44 CH3-CH-CH3 I OCOCH3
OCOCF3 CF3COOH I
CH3-C=CH2 + CF3COOH ----- CH3-C-CH3 I I
CH3 CH3
/II=•/ ELECTROPHILIC ADDITIONS INVOLVING METAL CATIONS
Certain metal cations which are electrophile in character are capable of elec.trophilic attack on alkenes. The cations are generally Hg++ and Pd++. ·
Hydration -,sing Hg++ (mercuric acetate) _ Mercuric acetate and water add to alkenes in a reaction called oxymercuration. Unlike the
hydration reaction, oxymercuration proceeds without rearrangement. The product of the oxymercuration is usually reduced with sodium borohydride in a subsequent . reaction called demercuration to yield an alcohol, the same alcohol that would be formed if water had been added across the double bond. Oxymercuration-demercuration reactions usually give better yields of alcohols than the addition of water/sulphuric acid. -
Hg(OAc)z!HOH NaBH4 R-CH=CH2 . J R-CH-CH2-Hg-O-COCH3 ---.-.4 R-CH-CH3 +Hg
(oxymercuration) / Demercurat10n I OH OH
The reductive replacement of mercury using sodiu~ borohydride is a free radical pr~.ess. R-TH-CH2-HgOCOCH3 == R1-HgOCOCH3
OH

Bu.~ R1-Hg-H R1-Hg-OCOCH3 + Na 1
-'4 R1-Hg-H R·1 + Hg(I)H R'1 + R l HgH R 1-H + Hg(II) + R·1
0 . . dd' · proceeds by electrophilic attack
e xymercurat1on IS two step process. The a ttion of Hg-O----COCH3 followed by nucleophilic attack of water. Because re~rrangements do not occur, the intermediate formed by electrophile attack cannot be a true carbocatt?n, on
the
0ther hand, since
Markovnikov rule is followed the intermediate must have some carbocation character. Both these facts are explained by postulating ; bridged ion or cyctiC ion, as the intermediate. The mechanism can be
represented as follows : @ e CH
3COO-Hg-OCOCH3 Hg-OCOCH3 + CH3COO
Electrophile @
First step : The electrophile in the first step of the reaction is HgOCOCH3. It is soft acid and strongly polarising. It polarises the pi-electrons of an alkene to the extent that a three-centre, two electron bond is formed between mercury and two carbons of the double bond.
9 R~CH=CH2 + Hg-O-COCH3- R-CH~CH2 __. R-CH-CH /.1, I 2 "1.1.
I'// ,. '2+ Hg 6+g
A three-center, two elect~on bond implies weaker bridging in the mercurinium ion than in the three center-four electron bondmg of the bromonium ion.
'-... / (t) /c=c, + Hg-OCOCH3
Three center, two Th · . . . electron bonding
e formation of mercunmum ion is usually fast and reversible st Second step : In the second step a water molec l k ep. charge. u e attac s the carbon bearing the partial positive ,
H ®I 0-H
I R-CH-CH2-HgOCOCH3
Step II~ : In this step an acid-b""e reaction tran to an acetate 10n). sfers a proton to another water molecule (or
This step produces the (hydroxyalkyl) me H rcury compound.
®IJr\~ O__LH H',......-••'-tt I
R-CH-CH2-HgOCOCH3
OH I
-... R-CH-CH - ©
Calculations indicate that mercury-brt'dg d 2
Hg-OCOCH3 + H30 · e carbo f · n retain much of the positive charge on the mere . ca tons such as those formed in this reactt
0
ury moiety Onl haro~ · Y a small portion of the positive c ii

h · h .is large enough the carbon w IC can form more stable carbocation. The charge on carbon "d carbon
resides ont for the observed Markonikov addition, but it is too small to allow the usual rapt ccoun • h k I · · s It to a arrangements t at ta e pace with more fully developed carbocatton · . This resu I ton re O . . . . ddition react10n. . ske e ochemistry : xymercuration 1s usually a stereospec1fic anti-a Stere ent with the involvement of bridged ion . . a<>reem OH
is 10
OH
CH3 Hg(OAc)z
HOH
HgOAc D

~,, •• I ADDITION OF BH3 HgOAc
Borane is neutral strong electrophile because it has empty p-orbital.
H,,,,, 0 11 B-H H"U
Borane
u
Free borane is unavailable as it spontaneously dimerises to diborane (BzJ¼). It is an unpleasantly flammable and toxic gas. When diborane dissolves in diethyl ether a complex of ether and borane is formed. This complex of borane and ether can be used as a source of BH3. Often the cyclic ether tetrahydrofuran is used.
B2H6+2Q -... 2Q.i:~H3 0 _, •• THF: BH3
Solution containing the THF : BH3 complex can be obtained commercially. When the borane-ether complex is allowed to react with an alkene, there is rapid addition across ·
the double bond. The initial product is an alkylborane and the reaction is called hydroboration. , /e®O II C=C + H B-0 __.. -C-C- + THF / '---
3 I I -H BH2
Borane undergoes rapid and quantitative reaction with alkenes to form organoboranes (R3B). The pverall reaction is the result of three separate reaction steps. In each step, one alkyl group is added to bornne unit until all the three hydrogens have been replaced by alkyl groups.
(i) R-CH=CH2 + BH3 ---? R-CH2-CH2-BH2 (ii) R-CH=CH2 + R-CH2-CH2-BH2 ---? (R-CH2-CH2½-BH
(iii) R-CH=CH2 + (R-CH2-CH2½-BH ---? (R-CH2-CH2½-B Trialkylborane
Borane is different from the other addition reagents we have mentioned because H is the electronegative portion of the molecule instead of the electropositive portion, as it is in HX or HOH.

496
Regioselectivity : When borane adds to a double bond, the hydrogen (which is electronegative in character) becomes bonded to the more substituted carbon. The result is what appears to be anti-Markonikov addition although addition reaction is Morkonikov reaction.
In other words. the boron atom becomes attached to the less substituted carbon atom of th
e double bond and hydrogen is transferred from the boron atom to the other carbon atom of the double bo
nd.
CH3
CH3 "- 6+ 5- I C=CH2 + BH3 ---7 CH3-C-CH2-BH2
CH3/ ;i Hon more
substituted carbon
Thus, hydroboration is regioselective reaction. Other examples that illustrate this tendency for the boron atom to become attached to the less
substituted carbon atom are : CH3-CH2-CH2-CH2-CH=CH2 i . i
6% 90%
CH3 I
CH3-C-CH-CH3 2% 98%
CH3-CH=CH-C(CH3)3 58% 42%
In the above examples the percentage designate where the boron atom becomes attached. This observed attachment of boron to the less substituted carbon atom of the double bond seems
to result in part from steric factors-. the bulky b~ron containing group can approach the less substituted
carbon atom more easily. Organoboranes are easily oxidised to alcohols by alkaline hydrogen peroxide. The final addition
of borane addition followed by H2O2 oxidation, appears as if water had been added to the double bond in an anti-Markonikov manner. Overall yields are often 95-100%.
BH3 3CH~:--CH2-CH=CH2 --? (CH3-CH2-· CH2-CH2hB l H202/0~
3CH3-CH2-CH2-CH20H + BO~-Beside, being oxidised to alcohols, organobcfranes can be converted to alkanes, alkyl halides or
other products.
CH3-CH2-CH2-H B is replaced by H ofCH3COOH
CH3-CH2-CH2-Br B is replaced by
Br ofBr2

/ - -·· UIUL.lltJLE BONDS Mechanism . Al ----..:.:...:::....:~~::__ _____ _______ ___ _ takes place th gh. 1 the avaifabl · . • d
1 . d
8 rou a four-me be e evidence suggests that hydroboration is a concerted process an
po ~n~e b -H bond in Which~ red cyclic transition state formed by addition to the double bond of _a v:::11
Yb 0nded to the less sub e _boron atom is the more positive. In this transition state boron atom 1s Ph 'ft . {h 0~?ed to the other doub~tituted carbon atom of the double bond and one hydrogen atom is
bs
01 d 10Th .e irection of the boro Y bonded carbon atom. As this transition state is approached, electrons n · IS makes th n atoms and f -f h d bl bears an el e more substitut away rom the more substituted carbon atom o t e ou _e
electronic =~~ron-~eleasing alkyl grou ed .;~rbon atom develop a partial positive charg~. and because 1t stenc factors account /• h is bet~er able to accommodate this positive charge. Thus, both o O or the antt-Markonikov orientation of the addition. ·
R-CH-CH O 0 U 02 R C 0"-0"2
0 . H-u::_: W':r-H
R-CHIII CH2 + BH Ill O 0 3 R-CH=tCH2 R-CH-CH2
H-~=3 {J = \J ........-H H-B · o~
111
. HrmuB-H 16-H
Four center transition state
/;
The above mechanism is supported by stereochemistry of the reaction (syn-addition of H and B) and by the directive effect of polar substituents.
(i) Stereochemistry : The transition state for hydroboration requires that the boron atom and the hydrogen atom add to the same face of the double q.ond. The addition is, therefore syn-addition. 1-Methylcyclohexene gives syn-addition with BH1.
, CH3
o-CH3 + BH3 --+
L{H J > Both are cis B 2

1-Methylcyclopentene also gives syn addition to BH3. D
(YD BH3'fHF+
\_J(_ _ BH2 CH3 H · h droxyl group ends up m the
When an organoborane is subsequently oxidised to an alcohol,_ the f~onfiguration at that carbon. . h . ith retention o same position as the boron atom that is replaced-t at is, w , CH3 e GCH3 n OHIH202 H
H BH2 tH
OH replaces BH2 wit_h retention of configuration
. duct Similarly (E)-2,3-dideutero-2-butene (Z)-2,3-Dideutero-2-butene gives racemic e'!th~0 pro . · These results also confirmed the
gives racemic threo-product in hydroboration ox1dat10n reactions. syn-j.ddition reaction.
OH
Z-2, 3-dideutero-2-butene
(i) BHlfHF e
(ii) H202/0H
(i) BH3/THF e
(ii) H202/0H
jcH3 + Enantiomer
D CH3 Racemic, erythro-2, 3-dideutero-2-butanol
OH .
t CH3 + Enantiomer
H3C D Racemic, thero-2, 3-dideutero-2-butanol
These results confirmed that product formation takes place by the formation of four-centered cyclic transition state in which addition is syn-addition.
(ii) In allyl derivatives and in nuclear substituted styrenes the proportion of product formed by addition of boron to the a-carbon atom increases with the electronegativity of the substituent.
CH3-o-@-cr~cH2
5%
Cl~ CH=CH ~t 2
25%

1 ;he above results can only be explained if product formation takes eplace by the formation °1
po ar our membered cyclic transition state in which carbon has carbocation character.
AD -- DITION OF HYDROGEN : CATALYTIC HYDROGENATION The catalytic add. • , · compou d Th" Ilion of hydrogen gas to an alkene or alkyne is a reduction of the pi-bonded
n · 1s reaction · kn The catal t is own as catalytic hydrogenation. . ( l-4 t ) ys s normally employed for the hydrogenation of alkenes to alkanes at low pressure
a m and moderate t · II d. h d
. R emperature (~100°C) contain noble metals such as platinum, pa a mm, or r O mm. aney nickel · f · · II . h , an active orm of nickel (obtained by reaction of a nickel-alumm1uw a oy wilt aqueous sodium hydroxide, which dissolves out the aluminium and leaves a nickel powder wi th a arge surface area) · • 1 . . . , is mam y used for medium to high pressure work. Reduct10n over platinum employ the fmely divided metal obtained by the reduction of platinum oxide. Palladium and rhodium catalysts are usually d · f • • . epos1ts o the metal on the surface of inert supports such as carbon. alumrmum or banum sulphate.
CH -CH Catalyst 2- 2 +H2 CH3-CH3 _All of these catalysts apparently · function by allowing the alkene and the hydrogen to be
chemically adsorbed (weak bonding) on the surface. This brings the two molecules into close contact. Reacti_on occu~s by simultaneous addition of a hydrogen atom to each carbon of the double bond; the alkane produced is then rapidly desorbed from the surface. :
The fact that catalytic hydrogenation is a reaction that takes place on a bulky metal surface has two other implications, each of which is well supported by experiment. The first is that the fewer the number and size of the substituents that are attached to the double bond, the more easily the compound will fit on to the catalytic surface, and the faster will be the rate of hydrogenation. Relative rates of hydrogenation of alkenes fall off in the order :
ethylene > monosubstiiuted > disubstituted > trisubstituted > tetrasubstituted double bonds. Thus selective hydrogenation of one double bond in the presence of another is often possible. For
example:
1 mole H2 catalyst
\ . 1. • · that both hydrogens become attached to the same side of the double
The second imp ication islt· from syn-addition. for example, cis-2,3-diphenyl-2-butene on b d t ;_\ give a product resu mg . . . on ° • d whereas trans-derivative gives racem1c mixture. hydrogenation gives meso-compoun
C6Hs I
CH3-C-H H2/Ni I \
\ CH3-C-H 1--C6lfs Meso

C6Hs I
CH3-C-H I + Enantiomer H-C-CH3
I C6Hs
cis-1,2-Dimeth y lcyclOhexene gives mesa-product.
~Q CH3 CH3
Syn prodt!Ct
· In most of the cases syn or cis-addition of hydrogen occurs from the less ster!c_ally_hindered side of the double bond. Of course, cent percent syn-addition does not occur. Syn-add1t1on ts about 80%. This observation supports that the two hydrogens do not add to the double bond at a time; aftt=r the addition of one hydrogen atom to a double-bonded carbon atom, the carbon with its group rotates about carbon-carbon single bond and then the other atom of hydrogen adds. and this gives rise to an anti- or trans-product. Since the addition of two hydrogens are rapid the addition is stereospecific and almost syn. :
Mechanism: , As mentioned earlier that the reaction is supposed to involve chemisorption-the process of exothermic adsorption of gases by solids in which a chemical link is fonned between the adsorbing and adsorbed materials. When a catalytic hydrogenation of an alkene involves gaseous reactant and product, it occurs through the following steps :
Experimental evidence supports the theory that first the hydrogen molecules are adsorbed on to the metallic surface, then the H2 sigma bonds are broken and metal hydrogen bonds are formed. The alkene is also absorbed on to the metallic surface, with its pi-bond interacting with the empty orbitals of the metal. · ·
H I
H R c~cR 2- - 2 I 22
Diffusion of the gases on I the metal surface +
·l
Rotation about
C-C single bond 1
R . 1'
R
trans~product cis-product Scheme 9.10 . . Hydrogenation of alkene

The alkene molecule moves around on the surface until it collides with a metal bonded hydrogen atom, undergoes reaction, and then leaves as the hydrogena.ted product (Scheme 9.10). !f ydrogenation reactions are exothermic, but they do not proceed spontaneously because the energies of activation are extremely high. Heating, cannot supply the energy needed to get t~e molecules to the transition state; however reaction proceeds smoothly when a catalyst is us~d: It _is the metal surface that speeds up the reaction. A metal atom at the surface has extra reactivity m comparison with an atom well within the metal, it lowers the energy of activation by activating ~he reactant making chemical link with the latter, the lowering of energy of activation makes the reactwn to occur even at room temperature (Fig. 9.5).
w
r
Ea: For the reaction without catalyst
-t----+-- Ea: For the reaction with catalyst
-------------------------- ---- .--------+-· CH2=CH2 + H2 ~H
----'-· CH3-CH3
Progress of the reaction ---Fig. 9.5. Energy diagram for a hydrogenation reaction
Heat of hydrogenation and stability of Alkenes The heat of hydrogenation of an alkene is the energy difference between the starting alkene and
the product alkane. It is calculated from the amount of heat released in a hydrogenation reaction. Table 9.1 lists the heats of hydrogenation of some alkenes.
Table 9.1. Heats of hydrogenation for some alkenes
Alkene -~H, kcal/mole
CH2=CH2 32.8 Propene 30.l I-Butene 30.3
cis-2-butene 28.6 trans-2-butene 27.6
2-Methyl-2-butene 26.9
'der the three alkenes that can be reduced to butane : Let us cons1 j CH3-CH2-CH=CH2
H2/catalyst cis-CH3-CH=CH-CH3 -----l. n-butane
trans-CH3-CH=CH-CH3

The product butane has the same energy regardless of the starting alkene. Any difference in ~H fo_r the three reactions reflect differences in energies of the starting alkene. The greater the numen~al_ value of the .1H of hydrogenation, the higher is the energy of the starting alkene and hence lower ts its stability (Fig. 9.6). Thus on the basis of heat of hydrogenation, the stability order of different substituted alkenes is as follows :
-30 Kcal/mole
___l ---------------~]-~------------~-I: ____________ ~Butene cis-2-Butene trans-2-butene
Fig. 9.6. Comparison of the heats of hydrogenation of the three butenes that give butane upon
Tetrasubstituted > Trisubstituted > Disubstituted > monosubstituted > ethylene. The relative heat of hydrogenation shows that trans-2-butene is the most stable of the -three
butene and that I-butene is the least stable (maximum energy).
I fltJ I HYDROXYLATION OF ALKENES
The conversion of alkenes . to vie diols by oxidation and subsequent hydrolysis is known as hydroxylation of alkenes. The most popular reagent used for this purpose is a cold alkaline aqueous solution of potassi~m permanganate, although this reagent usually gives low yield. The other reagent is osmium tetroxide. Although this reagent gives better yields of diols, use of this reagent is limited because it is both expensive and toxic. Beth the permanganate and the osmium tetroxide oxidation proceed by way of cyclic inorganic esters, which yield cis-diols if the product is capable of geometrical isomerism.
Mechanism : (A) Hydroxylation by Osmium tetroxide : The overall reaction takes place in two steps via
a cyclic intermediate, an ester of osmic acid and the alkene. Step-I : This consists of syn-addition of osmium(VIII) tetroxide to the alkene to form a
cis-cyclic osmonic(VI) ester. This step is rate determining step.
........... == / /c~c, o~ ,,-;po
Os o~ ~o
Slow step 'c111111111111,11c/
/~ ~' 0~ .,,<,6 ,.;os~ o?"' ~o
T.S. Cyclic intennediate (an osmate ester)

. . 8tep II : Cyclic ester on hydrolysis in the presence of aqueous ethanolic NaH~U3 g tvt;;s vi1..,
c1s-d1ol.
l OH, /OH '-- /
Os + C-C o~ ~o ,......., I' OH OH
Since both the OH groups form bond from the same side of the double bond, cis or syn addition products are obtained and hence the reaction is steroselective. It has also been found that reaction is stereospecific. The stereospecificity of the reaction is proved by the fact that (Z)-2-butene and (.E)-2-butene give optically inactive meso-2,3-butanediol and (dl) mixture of 2,3-butanediol, respectively.
o~ ~o OH HO=fH /os,
f Topside O:tH HOHIC2H50HI
attack NaHS03 = ¥ OH H
H
H)(H CH3 CH3
H CH3 (R,R)
H H CH3 CH3
Bottom side HOHIC2HsOHI =H~+~H attack He / NaS03H
0-0s=O II OH CH3 0 (S,S)
The formation of the cyclic intermediate, the osmic ester, has been proved by its isolation. When the addition of osmium tetroxide to an alkene i~ carried out !n the presence of pyridine, usually a coloured crystalline compound having the followmg structure 1s obtamed.
I -C-0" I /Os02(Py)i -C-0
I
4A4 St 4CU .-:rt

Osmium tetroxide is a highly specific reagent for syn-hydroxylation on the less hindered side of the alkene. For example :
. .
CH~(C1:~-CH3 H OH
(i) 0s04 (ii) NaS03H
(B) Hydroxylation by permanganate : Treatment of an alkene with a neutral aqueous or alkaline (pH 12-13) solution of potassium permanganate at room temperature forms a cis-diol. Wh~n , a neutral permanganate is used for the reaction, its purple colour disappears and a brown precipitate '\ of hydrated manganese dioxide forms. In alkaline solution, the colour changes from purple to green by this reaction. Since the reaction involves a visible change of colour, it is used to test the presence of a carbon-carbon pi-bond in an organic compound containing no other easily oxidisable groups by the reagent and the test is usually referred to as Baeyer's test for a carbon-carbon multiple bond.
Mechanism : The overall reaction takes place in two steps via a cyclic intermediate, an ester e
of Mn(V), H2MnO4 and the alkene. · Step-I : Syn-addition of permanganate ion [Mn(VII)] to the· carbon-carbon double bond to form
e a cyclic ester of H2MnO4 (Mn V). This is a slow step and rate determining step.
Mn(VII)
'c-c/ 111111 ..........
¾ 0~1 .,~o
Mn o~ 'o 0
Cyclic five membered transition state
Mn(V) Cyclic intermediate
Step-II : Co-ordination of the hydroxide ion from water with the Mn(V) of the cyclic ester and stepwise cleavage of O-Mn bonds give cis-1,2-diol. This step is fast step.
/OH HO-Mn +
II 'o o e Thus, the overall reaction involves the addition of two OH groups to an alkene from the same
side of the double bond, i.e., the addition is a stereoselective syn-addition. This is also a stereospecific reaction: (Z)-2-butene forms meso-2,3-butanediol and (E)-2-butene gives (dl)-2,3-butanediol.

\
Mn02 "" cf~o e OH Top side OH attack
H)(H H
KMn04 H COOH COOH
COOH (R,R)
H e H Bottom side
co~ OH
attack
c':H r H O-Mn02 OH e (S, S)-.
OH
Top side attack
COOH HOOC
COOH)(H H KMn04 Meso-tartaric acid
+ H Ill H
Bottom side attack
OH Mesa-tartaric acid
Similarly maleic acid gives meso tartaric acid and fumaric acid gives (dl)-tartaric acid. Cyclic ester of the reaction has not yet been isolated from the reaction mixture, but there is
strong evidence for the formation of this cyclic ester. When an alkene is treated with M~i~ instead 18
of ordinary ~o4, the isolated diol contains two -OH groups. This proves that two oxygen atoms
·~ 8 of the diol have come from the Mn04 and neither from the reagent (OH) nor from the solvent,
H20,
\
\
"' '-,.

I Elfil I ADDITION OF PER ACIDS : FORMATION OF OXIRANES
O~ida_tion of alkenes with an organic peroxy acid (sometimes called simply a per acid) is known as epox1dat1on reaction.
A_ nu~ber of peroxy-acids have been used in the past, including perbenzoic, performic. and per~cetic acid,_ but these have now been replaced by m-chloroperbenzoic acid. It is comm_ercially available and 1s an excellent reagent for lhe epoxidation of carbon-carbon double bond. It 1s more stable than other peroxy acids and has even been used at an elevated temperature in the presence of non-polar solvent (dichloromethane, 90°C).
Mec_hanism : Epoxidation reaction is believed to take place by electrophilic attack of the peroxy-ac1d on the double bond (Scheme 9.11).
'c''''''' o l~~o~'--C-R d,~Hdl ,., ,,,,,, 0
Alkene
T.S. Scheme 9.11. The peroxy acid transfers an oxygen atom to the carbon-carbon double bond in a cyclic. single step mechanism. The result is the syn-addition of the oxygen to the carbon-carbon
double bond, with the formation of an epoxide and a carboxylic acid In accordance with this mechanism the rate of the epoxidation is increased by
electron-withdrawing groups in the peroxy acid or electron-donating substitutents on the double bond. Thus, trifluoroacetic acid is more effective than per acid. Terminal mono-substituted alkenes
react only slowly with most peroxy acids. 1,2-Dimethyl-1, 4-cyclohexadiene reacts preferentially at the tetra substituted double bond. On the other hand, conjugation of the alkene double bond with other unsaturated groups reduces the rate of epoxidation because of the pi-electrons. These above results support the mechanism of the reaction.
Since the reaction involves the attack from one side of the double bond, it should give syn-addition product; in fact this is so. Thus, this reaction is highly stereoselective and highly stereospecific with those alkenes which show geometrical isomerism. For example, (Z)-2-butene yields only cis-2,3-dimethyl oxirane and (£)-2-butene yields only the racemic trans-2,3-dimethyloxiranes.
Z-2-butene
+RCOOOH
Z-2-butene
CH3
-----+ H3C 4 ~ . F-\ H
cis-2, 3-dimethyloxirane (a mesa compound)
Enantiomeric-trans-2, 3-dimethyloxiranes
With conformationally regid cyclic alkenes the reagent usually approaches from the less hindered side of the double bond (equation 1).

+ ... (I)
94% 6% Oxidation with peroxy acids is not the only way to convert an alkene into an epoxide. It has
recently been found that reaction of alkenes ~ith t-butylhydroperoxide (TBHP) in the presence of vanadium (V5+) or molybedenum (Mo6+) catalysts provides another excellent method ~or the prep~rati_on of ~poxides. It has been found tha\ the molybedenum catalysts are most effective for epox1dation of isolated double bonds and the van'~dium catalysts for ally! alcohols.
. The vandium catalyst shows remarkable reactivity towards the double bond of ally! al~ohols. This makes possible selective epoxidation which cannot be obtained with other reagents (Equation 2).
QC TBHP Qo VO(acach \
CH20H CH20H
... (2)
Another advantage of these metal catalysed reactions is that they are much more stereoselective than the reaction with peroxy acids. For example : \
TBHP VO(acac)i
The Sharpless Asymmetrici:poxidation
H -::-
CH3~§' OH \_\\\\\\
C4H9 0
Erythro (syn) epoxide 99% selectivity
K.B. Sharpless and co-workers reported a method that has since become one of the most valuable tools for chiral synthesis. The Sharpless asymmetric epoxidation is a m~thod for converting allyl alcohols to chiral expoxy alcohols with very high enatioselectivity, i.e., fith preference for one enatiomer rather than formation of a racemic. mixture. It involves treating ttie allylic alcohol with TBHP, titanium(IV) tetraisopropoxide (Ti(OPr'))4 and a specific stereoisomer 'Qf a tartrate ester. The esters most commonly used are(+) or(-) diethyl or dj-isopropyl tartrate (DET arld DIPT). The tartrate stereoisomer that is c~osen depends on the spe~ifi~ enantiomer o~ the eroxide d~ir~d.
The main attraction of the Sharpless epox1dat10n procedure 1s that 1t affords a\ smgle enantiomer of the epoxide in high enantiomeric excess, and in a predictable manner (Equation \ ,, 4).
~OH
j (-) DET , Ti(OPri)4
TBHP
~OH (R) Methylglycidol
(+) DET Ti(OPri)4
TBHP
,,,,Q ,,, =
~OH ... (3)
(S) Methylglycidol
\
... (4)
\

It can be applied to both primary and secondary allylic alcohols. • 1 1 . From a purely practical point of view it is found that the addition of 4 ft: m? ecu ar Sieves permits ·
the use of only catalytic amount of the titanium catalyst and the ~ate of o~tdatton c~ be accelerated four to five times by the addition of a drying agent such as cal~ium hydride and Siltca gel. .
Mechanism: The precise course of the reaction is not_enttrely cl~ar. but t~e rat~ of a~celeratt~n and the high stereoselectivity observed suggest the formation of an intermediate m which allyhc hydroxyl group is co-ordinated to the metal.
OH V B~OOH
Ti (OPri)4
(f) O-But
..,,,,,1. I tA .. 'Ti-O: ,, o\_j/
(I)
It seems to be generally agreed that transfer of oxygen to the double bond takes place in a titanium ester. possibly of the form (I) in a conformation which minimises steric interactions .among the various substituent groups.
~-But O-Bu1
,,,,,~ e I~ ,,,,,~I @ · ''Ti--0: __. ''Ti O __.
'I c·) 'IU~\ o o c~ - ~
,,,, 111 • 0 B 1 Ti-- - u 'I o\_Y
A number of different models have been proposed to account for the observed stereoselectivity of the reaction. One of which shows the ally lie alcohol being forced to adopt a conformation in which one face is preferentially exposed to the oxidant as shown in figure 9.7 (where E is an ester group).
R'
AR" E O • -:;;:P .
/Ti Ti E o I o.....,l "o · E Bu1
0~ /@
-~ . . , . Fig. 9.7. ~s shown m the ~gure given ~low~ if the allylic alcohol is drawn so that the hydroxymethyl
group 1s at the lo~er nght, oxygen 1s delivered at the bottom face in the .presence of ( +) DET nd from the top face m the presence of(-) DEf. a
D(-}DET 0
R,) R .Jr _R_;CoH 0
L(+) DET
-

For example :
CC4H19 OH
TBHP
(+) DET
(-) DET
0 ,,,,,,, C C4H 19 1111111 OH
82% 90% enantiomerie excess
82% A • . , 90% enantiomerie excess
. pphcation of Sharpless epoxidation : Numerous applications of the sharpless epoxidation r~action have been reported. The first example involves straight forward synthesis of gypsy moth P enomone (7R, 8S)-disparlur (Scheme 9.12).
(ii) BuLi CH3
(iii) 'cH-(CH-\4-CHO CHJ',.....- 21 .
H
H
lTBHP. Ti (OPr)4 (-) DET
(7R, 8S)-Disparlune Scheme 9.12.
A second example involves . straightforward syntheses of the 13-blocker (S)-propanolol (Scheme 9.13).
~OH TBHP (+) DET
~OH
OH _.........CH3 O NH-CH....____
ONa
©@
CH3 S-propanolol
Scheme 9.13. Synthesis of S-popanolol
OH
O~OH
lTsCl/Py
0~
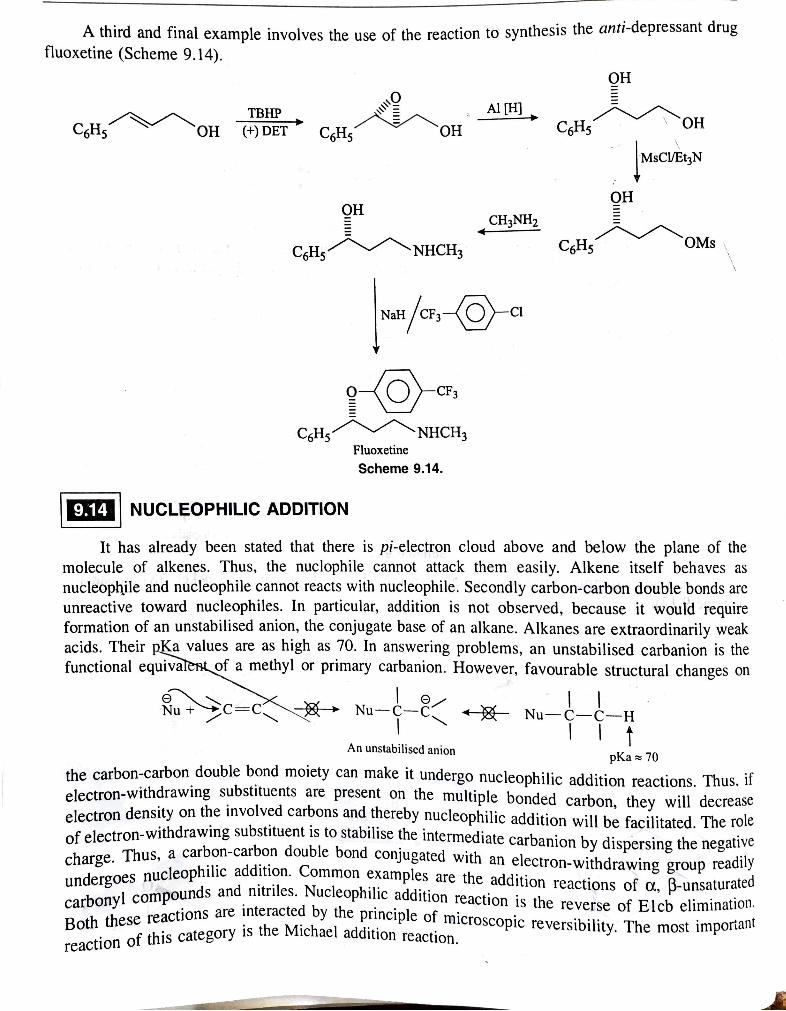
. . . . thesis the anti-depressant drug A third and fmal example involves the use of the reaction to syn fluoxetine (Scheme 9.14).
TBHP (+) DET
1--@-cF, C6Hs NHCH3
Fluoxetine Scheme 9.14.
I !Elt• I NUCLEOPHILIC ADDITION
Al [H]
OH
C6Hs ~OH
lMscJ,,N QH
C6Hs OMs \ \ \
It has already been stated that there is pi-electron cloud above and below the plane of the molecule of alkenes. Thus, the nuclophile cannot attack them easily. Alkene itself behaves as nucleophile and nucleophile cannot reacts with nucleophile·. Secondly carbon-carbon double bonds are unreactive toward nucleophiles. In particular, addition is not observed, because it would require formation of an unstabilised anion, the conjugate base of an alkane. Alkanes are extraordinarily weak acids. Their Ka values are as high as 70. In answering problems, an unstabilised carbanion is the functional equiva of a methyl or primary carbanion. However, favourable structural changes on
e~ ........__ Nu+~C=C
/ ' I e/
Nu-C-C )8( I --...._,_ An unstabiliscd anion
I I Nu-C-C-H
I I t pKa"" 70
the carbon-carbon double bond moiety can make it undergo nucleophilic addition reactions. Thus. if electron-with~rawing s~bstituents are present on the multiple bonded carbon, they will decrease electron density on the mvolved carbons and thereby nucleophilic addition will be facilitated. The role of electron-withdrawing substituent is to stabilise ~he intermediate carbanion by dispersing the negative charge. Thus, a car~~n-car~~n double bond conJugated with an electron-withdrawing group readily undergoes nucleophihc add_iti?n. Common ~~ampl~s. are the addition reactions of a, ~-unsaturated carbonyl compounds and_ mtnles. Nucleophihc addition reaction is the reverse of Elcb elimination. Both these reactions are ~nteracte? by the p_ri_nciple of microscopic reversibility. The most important
. f this category 1s the Michael addition reaction. react10n °

Michael Addition : Conjugate addition of enolate ion to a, P-unsaturated carbonyl compounds is called Mich I d · · b (08 H R O ec amines t ) ae a d1tion. This reaction takes place in the presence of a ase , - , s • e c. · Some chemists refer to all nucleophilic conjuoate additions as Michael addition.
The substrates of the reaction are the comp~unds containing activated carbon-carbon double bonds i e A ' • · A t d ·t ' · ·, a, p-unsaturated carbonyl compounds, esters, cyanides, qumones, a., p-unsatura e m ro compound R · ·d· h d s. eagents of the reaction are those compounds which have at least one ac1 IC Y rogen and conv · · · ert Into nucleophile in the presence of a base. Such compounds are compounds ha'\tmg active mlkethY1ene group, nitroalkanes, sulphones, indene, fluoenes, alcohols, thioalcohols and terminal a ynes.
Michael addition in which nucleophile is enolate ion Step I : Base removes a proton to form an anion (carbanion) of the compound having acidic hydrogen.
/COOR CH2 · "cooR
0 /COOR CH
"-.,COOR Nucleophile
Step II : Conjugate addition of the anion to the a, P-unsaturated carbonyl compound leads to a new enolate ion.
t 0
C2H5ooc, 8 II ,......-CH-CH2-CH-C-R
C2H500C
Step III : The enolate anion is protonated by an acid to give the product.
0
0 OH C2H500C" I C2H500C '\ I
/CH-CH2-CH=C-R /CH-CH2-CH=C-R C2H500C C2H500C (IA-Addition product)
Tautomerisation
C2H500C " II /CH-CH2--CH2-C-R
C2H500C 1,2-Addition product

Thus the net reaction can be writt~n as follows : 4 0
l 2 II Nu-H + CH2 CH-C-R
3
0 l 2 II
Base ~u-CH-CHi-C-R ·. i i
Additio'n of Addition of Nuon Hon
carbon-1 · , carbon-2
. Mic~ael additi_o~ is conjugate addition, i.e., 1 ,4-addition rea~tion but overall product of the reaction 1s . l ,2-add1t10n product which takes place on carbon-carbon double bond due to the tautomerisation. .
Michael addition of vinyl cyanide is also known as cyanoethylation reaction bec~use it_ introduces the -CH2-CH2-CN fliagment into a molecule. Cyanoethylation of the carbamons ts a useful method of lengthening a chitn by three carbon atoms (Sch~me 9 .15) shown on the next page.
Conjugate addition of a, ~-unsaturated carbonyl compounds with nitroalkane having atleast one alpha hydrogen is known as Nef reaction.
0 0 II iPr2NH/CHCl3 It ,
CH3-N02 + CH2-CH-C-CH3 -----+ 02N-CH2-CH2-CH2-C~H3
0 II .
R-C-CH2-CH2-CH2-CN H2N-CH2-CH2-CN
\
CH3-S-CH2-CH2CN CH3SH CH2=CH-C==N base, // 125°C · , R::,.,
C G ~J / ou ()'ry f
c~ / 4:J/J /j " &,.,
CH3-C........_ /CH-CH2-CH2-CN
CH3-C II 0
CH2OH I CHOH I CH2OH
base/50°c
TH2 - 0-CH2- CH2- CN
IH-O-CH2-CH2-CN
CH2 - 0-CH2- CH2- CN Scheme 9.15.

- - • _,,, IUV/V 11/IUL.llt'L.t: t:JU,VU:, »
[fi'4J FREE RADICAL ADDITION
d Radicals, like cations are electron-deficient species Thus radicals will attack the pi-syStem dof 3
ouble bond of an alkene because pi-electrons of an ·alkene can provide the electron neede . to complete the outer shell of a radical. The net result of this usually is the addition of some spe_cies across t_he d~uble bond. These reactions involve radical intermediate. Free radical addition reac~rons :cur either m the gas phase or in inert non-polar solvents in the presence of UV light or sunhght,
ea~ or catalytic amount of radical initiators such as organic peroxides, labile azo compounds like az~rsobutyro_nitrile (AIBN), etc. The mode of the addition reaction involves the general steps for the ra?rcal reactr~ns, i.e., initiation, chain propagation and termination. The most important reaction of th1s category 1s hydrobromination.
Hy~robromination : . The radical addition of HBr to a carbon-carbon double bond occurs in the pres~nce o~ UV light or a small amount of initiators such as dibenzoyl peroxide. HF, HCI and HI do not ,grve this addition.
When propene reacts with hydrogen bromide in the presence of dibenzoyl peroxide, the product is 1-bromopropane.
CH3-CH=CH2 + HBr dibenzoyl peroxide CH3-CH2-CH2-Br
In this addition the hydrogen that is being added ends up on the carbon bearing the smaller number of hydrogens. Therefore, this is an anti-Markonikov addition. In general. unsymmetrical alkenes add, HBr, in the presence of a radical initiator, in an anti-I¼arkonikov manner.
radical . R-CH=CH2 + HBr -- R-CH2-CH2-Br
source
Mechanism : The free radical mechanism starts with an initiation step that results in oxidation of HBr to bromine atom.
0 0 0 II II II •
C6Hs-C-O-O-C-C6Hs 2C~5-C-O benzoylperoxy radical
0 0 II • II •
C6H5-C-O + H-Br C6Hf---C-OH + Br
Propagation step I : The bromine radical lacks an octet of electrons in its valence shell, making it electron deficient and electrophile. It adds to a double bond, forming a new free radical with the odd electron on the carbon atom.
l • • R-CH=CH2 + Br R-CH-CH2-Br (addition step)
alkyl free radical
p pagation step II : The alkyl free radical of the propagation step I reacts with an HBr molecu:: to generate another bromine radical.
• R-CH-CHz-Br + H-Br ----? R-CH2-CH2-Br + Br (Chain transfer step)
...., . t· 0 • rrermination occurs by any ( or all) of the reactions which use up species .1 erm1na 10 • 1 '
nvolved in the propagation steps (I and II).

(i)
(ii)
• • Br+ Br Br2
• • ~HR R-CH-CH2Br + R-CH--CH2Br ---i> RjH -I -
CH2Br CH2Br
(iii) R-CH-CH2Br + Br R-CH-CHzBr . I
Since the first species that adds to the ~~kene is radical, the addition of HBr in the presence of peroxides is called a radical addition reaction. .
According to the mechanism, anti-Markonikov addition of HBr anses because :
(a) Br (and not H) is the first to react with the n-system, and (b) The most stable radical is formed most rapidly. Since the addition of HBr in the presence of peroxide involves the formation of a radical
intermediate rather than a carbocation intermediate, rearrangement of the intermediate does not occur. Radicals do not rearrange as readily as carbocations.
Peroxide CH3-CH-CH=CH2 + HBr --- CH3-CH-CH2-CH2-Br
I ·1 CH3 CH3
Stereochemistry : At room temperature the radical addition of HBr to an acyclic alkene is stereo- selective and not stereospecific because cis or trans alkenes give 80% of trans addition product and 20% cis addition product.
Q Q . R · H '--c=C/ + . R......_ / Br H Br H/ '-R . Br H,3-c,R-H R~o-c(R-H
Perhaps after the formation of bromoalk 1 ct· . . different conformation owing to the rotation ab~u/~~al t?ere is time for the radical to assume a
On the otherhand. when liqu'id H B . dd d C Sigma bond. - r is a e to an 1· . found to be almost stereospecific· in th' acyc ic alkene at -80°C the reaction 1s lk . ' is case stereosele t. I . . ' trans-a enes give 92% meso-product but . lk . c ive Y trans addition occurs whereby
f h d. l dd' . c1s a enes give aim t 100 . o t e r_a ic~ a. ttlon_ ca~ be explained in terms of a . os % (dl) pair. The stereospecific1ty bromonmm ton m the tome addition of HBr to alkenes. bndged structure in analogy to the bridged
. Backside ,-:=---:---.::::.:.... _ _J__ Backside
H3C attack at C-1 0 B
H~C .,,,,, r H.,,,,, --C111111 CH Br '°' H 3 CH311~c ,,,,,,CH3 H.-,, -C-H
(dl)pair ........_ H
H

Br _. Br
H3C1111 I/ '\ ,,,,H 11C-C'' H' 2 'cH3
H Backside Backside attack at C-1 attack at C-2
HC B 3111 / r 111 ,r H-C---C111111 H H/ 'cH3
meso form
d . T~ans-diaxial addition is the preferred stereochemical mode for addition to cyclohexene and its · envatives. This stereochemistry can be explained in terms of a bromine-bridged intermediate.
~I
HBr/hv -780
Br
H
H trans-addition product
Why is the peroxide effect observed for the addition of HBr but not for the addition of HCI or HI? This question can be answered by calculating the m 0 for the two propagation steps in the radical chain reaction (using the bond dissociation energies).
m 0 = -9 kcal/mole [
R----CH=CH2 + Br -----? R-CH-CH2Br
R----CH-CH2-Br + HBr -----? R-CH2-CH2-Br + Br m 0 = -7 kcal/mole
[ R----CH=CH2 + Cl - R-CH-CH2-CI
R-CH-CH2----Cl + HCl -----? R-CH2-CH2CI + Cl r R--CH=CH2 + i -----? R-CH-CH2-I
L R-CH-CH2-l + HI --4 R-CH2-CH2-I
.1H 0 = -22 kcal/mole
m 0 = +8 kcal/mole
.1H0 = +5 kcal/mole
m 0 = -24 kcal/mole
For the radical addition of HCl, the first propagation step is exothermic and the second one is endothermic. For the radical addition of HI. the first propagation step in endothermic and the second one is exothermic. Only for the radical addition of HBr, the both propagation steps are exothermic.
In a free radical reaction the steps that propagate the radical chain are always in competition with recombination reactions that terminate the radical chain. These recombination reactions (termination J and II) are so fast that only the exothermic propagation steps are fast enough to compete with them. In the HBr addition both propagation steps a~e exothermic and thus fast enough to prevent the termination of the radical chains. In addition of other hydrogen halides, one or the other propagation is endothermic _I? other words, there is an energy barri~r to th~se reactions that makes them relatively slow. In add1t10n o~ HCI or HI, theref?re, the f_r~e radical cham reaction is terminated
b eting recombination react10ns, and Markov11;1kov addition by the ionic pathway is observed Y comp · · k b t h b h · d Th free radical cham reactions wor es w en ot propagation steps are exothern1k:.
mstea . us 1 ·ct 'bl · An endothermic step corresponds to a s ow a11 revers1 e react10n that breaks the chain.

516 ADVANCED ORGANIC~ .
. Other compounds that have appropriate bond strengths can ad? to ~ouble bonds under f radical conditions. Example includes chlorine, bromine, hydro~en su.lphide, thiol_s and polyhaloalkanree
. Carbon tetrachloride and carbon tetrabromide react readily with alkenes 10 the presence of fes. rad 1 · · · ree 1ca m1t1ators (In) to give I : I adducts. •
Initiation : In+ CXi InX + CX3
Propagation I : • •
R-· CH=CHz + CX3 R-CH-CH2-CX3 X
Propagation II : . I •
R-CH-CH2-CX3 + C)(.i R-CH-CH2-CX3 + CX3
Termination :
• • (ii) CX3 + CX3 ---+ CX3-CX3
• • (iii) R-CH-CH2CX3 + CX3 ---+ R-CH-CH2-CX3 I
CX3 Other important radical addition reactions are given on the next page :
R'SH
R-CH =CH2 __ P_er_ox_id:..:.e_.~~(~fH~~~ 6 shSiH
CICOCI
0 II
R'-C-H
X I
R-CH-CH2--X
Br I
R-CH-CH2-Cct3
R-CH2-CH2-SR'
R-CH2-CH2-SH
R-CH2-CH2-Si(C6Hsh Cl I
R-CH-CH2COCl
0 II
H-C-NH 0 /Iii@/ ADDITION TO CVCLO . ' R-CH2-CH2-~-NH2 . . PROPANE RING
The bonds m cyclopropane a and 1t-bonds, so that cycJopropanare bebem (banana) b d ,, es h . on s and . b twee••
. . ave in some resp . are intermediate in character e fore, ects hke double bond compounds. There

ADDmON TO CARBON : CARBON MULT1PLE BONDS 517
cyclopropa I • · h . nes undergo addition reactions analogous to that of double bond compounds resu tmg m t e openmg of the three membered ring. For example :
Ni/H2 - CH3-CH2-CH3 80°C
Cl2/FeCl3 CICH2CH2CH2CI -(i) Cone. H2SO4 CH3CH2CH20H
(ii) H2O
HBr CH3CH2CH2Br
D_Bu F3CCOOH CH3CH2CHBu
I OCOCF3
In general, cyclopropane undergoes addition less readily than propene, e.g., chlorination requires a Lewis acid catalyst to polarise the chlorine molecule. However, the reaction with sulphuric acid and other protic acids occurs faster for cyclopropane than for propene. Unlike alkenes, cyclopropane is not hydroxylated with aqueous solution of KMnO4 and also does not add ozone.
CH3 Me X > I I I Me HX Me (b} t M --+ Me-CH-C-X + Me-CH2-CH2-C-Me
H I I X
(a) (b)
Additions to cyclopropanes can take place by electrophilic, nucleophilic or free radical mechanism, but the most important is electrophilic addition mechanism. For substituted cyclopropanes, electrophilic additions usually follows Markovnikov rule, although exceptions are known and the degree of selectivity is often small. The application of Markovnikov rule is illustrated by the following reaction : EB
According to this rule the electrophile (in this case H) goes to the ring carbon with the most hydrogens and the nucleophile goes to the carbon that can best stabilise a positive charge (in this case the tertiary rather than a secondary carbon).
Following three mechanisms have been proposed for electrophilic addition (these mechanisms are shown for HX, but analogous mechanisms can be written for other electrophiles).
Mechanism 1 :
Mechanism 2 :
JL:,:::/¥~ H R'
(A)
R A R R')( Y:x
H R'
JL R RAR R,X®~R' X '-H' R' H R'
(B)

Mechanism 3 :
RAR R A © R Ji R'H fi'\~ R' )j' re' .J:j,/ R' H R'
ADVANCED ORGANIC CHEMISTRY ---R A R R'1 'f,x
M . 2 . echanism I involves a corner protonated cyclopropane (A), i:nechan!sm involves an edge-protonated cyclopropane (B) and the mechanism 3 involves a classical calJon (C). A~ regards the stereQchemistry of the addition at the carbon which becomes attached to the electroph1le 100% retention, 100% inversion, or mixtu;e of retention and inversion has been found. At _the carbon which becomes attached to the nucleophile the result is usually inversion, although ~et~nlI?n has also been found. Elimination, rearrangement and recemisation processes often compete mdicatmg that m many cases a positively charged carbon is generated at this position. . . .
It is known that Br2 and CI2 add to cyclopropanes by a free-radical mechanism m the presence of UV light. The addition follows Markovnikov rule; the initial radical attacks the least substituted ring carbon and the second group goes to the most substituted position. It has been shown that the reaction is stereospecific at one carbon, taking place with inversion there, but non-stereospecific at the other carbon. The mechanism is given below :
x~A-H;/\ R X R
X Hh . 2 + X -.. etc. X X
In some cases conjugate addition has been performed where a double bond is "conjugated" with a cyclopyl ring. For example : .
R
H2C=C~ + CH3COOH H3C-C=CH-CH2-CH-,0COCH I "--1 I - 3
Ar Ar I Pd l HYDROGENATION OF AROMATIC RINGS
Aromatic rings can be 11;duced by catalytic hydrogenation but higher temperatures ( I 00 to 200'C) are reqmred than that for ordmary double bonds. With benzene ri · · • · the reaction after only one or two bonds have b ngs It Is usually 1mposs~ble to stop
een reduced because olefins are more eastly reduced . @ H2/Ni/d 0 . . Cyclohexane than aromalJc rmgs. Thus I mole of benzene treated with
1 . .
or cyclohexene but 1/3 mole of cyclohexane and 213 1 mole of hydrogen, gives no cyclohexad1ene
true for all aromatic systems, for example, with phena:~ e of ~e~overed benzene. However, this is not only the 9, 10-bond has been reduced. rene It ts easy to stop the hydrogenation after
H2 . . cat.
When aromatic rmgs are reduced by lith • are ,known as dissolving metal reductions), ~~:1[
0r _K or Na) in liquid ammonia (such reductions 2-propanol, or t-butyl alcohol), I, 4-addition !f 1~ the presence of an alcohol (e.g., ethano~
Ydrogen takes place and noncon jugate

•
cyclohexadienes d aromatic rin is are pro ~ced. This reaction is called the Birch reduction. By this reaction an g more readlly reduced than an alkene.
0 00 ./.?'
Li/liquid NH3 C2H50H 0
I, 4-dihydrobenzene (I, 4-cyclohexadiene)
C2H50H co I, 4-dihydronaphthalene
When lithium (or K or Na) dissolved in ammonia an intense blue solution is obtained. Blue is the colour of the solvated electrons : e9 (NH3)n- In Birch reductions these blue solutions with solvated electrons
LI. fast EB 9 slow 9 I
Li e (NH ) -- NH2 + -2 H2 nNH3 . 3 n NH3 blue solution 9 colourless solution
are used as reducing agent. The reaction of NH3 to NH2 and H2 is quite slow, and a better electron-acceptor is preferentially gets reduced.
0
~CNHJln -o· 0 .
~H~OEt
Delocalised radical anion
Hor'_!. H
Cyclohexadienyl radical
~e~_00Et
Hy H
Cyclohexadienyl anion
H HOH H
I, 4-cyclohexadicne
Formation of a 1,4-cyclohexadiene is quite general in this type of reduction, but the reason for its formation in preference to the more stable conjugated 1,3-cyclohexadiene is not clearly understood. However, one of the reasons is that kinetically controlled reactions of cyclohexadienyl anions with electrophiles typically take place at this central carbon, i.e., C-4.
When substituted aromatic compounds are subjected to the Birch reduction, the electron-donating groups (e.g., alkyl, alkoxy, etc.) decrease the rate of the reaction and are mainly found on the non-reduced positions of the product, for example :
;:,• Na/NH3(/) • :vt• H l) EtOH /UH Anisole I -methoxy-1, 4-
cyclohexadiene (major product)
OMe H +Q H H
3-methoxy- I, 4-cyclohexadiene (minor product)

o th cooH coNH2, etc.) increase the reacr • n e other hand, electron-withdrawing groups (e.g. , ' 1 . ion
· rate and are mainly found on the reduced position of the product, for examp e · COOH COON a COON a
6 N=t Q +Ho: H H H
Major product Minor product
It is general principle that electron-donating - groups promote ortho, meta reduction while e~ect_ron-_ withdrawing groups promote ipso, para reduction. The explanation for ~his is based on the dtStnbutlon of electron density in the intermediate radical anions. Electron-donating groups stabilise electron density at ortho and meta positions, and protonation occurs at these positions, while electron-withdrawing groups stabilise electron density at the ipso and para positions resulting in
t . I pro onatton a~ these positions.
· OMe 6 N.n<H3(0 EtOH
OMe ~o Na/NH3(/) • EtOH
H
OMe Ho H H H
COONa
HQ COOH 6 N.n<H3(0
COONa COONa
EtOH
COONa
6 EtOH D H H H H
If one wants the conjugated dienes as products it is quite a · l · · using an acid catalyst. ' Simp e matter to isomense them
OMe Ho H H H
Non-conjugated diene
© H
OMe
)-y~ UH H
Conjugated diene
With anilines it is impossible to stop the isomeris f k' reduction always gives conjugated enamines. a ion ta mg place during the reaction and Birch
NMe2
6 Na/NH3(1)
NMe2 ;~ H~
Reduction of aromatic rings with lithium or c 1 . . , . and cyclohexenes are obtained instead of I 4-cycl ha cm~ 10 amines (not ammonia) proceeds further
' 0 exad1enes.

PROBLEMS WITH SOLUTIONS 1. Which one of the following alkenes will give optically active product with Br,/CCl4?
(a) !~Butene (b) Propene (c) cts-2-butene (d) trans-2-butene
2. Which one of the followiing alkenes will give optically active product with Baeyer's reagent? (a) !~Butene (b) Propene (c) cis-2-butene (d) trans-2-butene
3. Which compounds will form mesa-product with Br2/CCI
4?
(1) trans-2-butene (2) trans-2-pentene (3) cis-2-butene (4) trans-3-hexene Select the correct answer from the codes given below : Codes:
(a) Only 3 (b) 1, 2 and 3 ( c) 2 and 3 ( d) 1 and 4
4. Arrange reactivity of •given compounds in decreasing order for electrophilic addition reaction with HX: (1) C6H5-CH=CH2 (2) C6H5-C=CH-CH3
I CH3
(4) CH2=CH-N02
C6Hs Select the correct answer from the codes given below :
Codes: 3 2 1 4 (b) ' , • (a) 4, l, 2, 3 (d) 1, 3, 4, 1
( c) 2, 3, 1, 4 · · dd · · f . th HX ? . ounds will give electroph1hc a 1t10n reac 10n w1 • 5. Which among the following comp (2) CH
3-C==C-H
(I) CH2=CH2 o,N" /No, (4) c=e,
0 2N/ "-N02 (3) CH2=CH-CH=CH2
. . from the codes given below : Select the correct answer
Codes : (b) only 2 (a) I and 3 (d) 1, 2 and 3
(c) only 4
. . dd'tion takes place according to anti-Markovnikov rule? 6. In which compound electroph t_hc a 1
· (2) CH2=CH-CHO .
(1) CH2=CH-N02 (4) CH3-CH=CH2 (3) CH2=CH-CN from the codes given below : Select the correct answer
Codes : (b) 1, 2, 3 and 4 (a) 1, 2 and 3 (d) only I (c) only 4
I

522 ------I matched :
. h air is correct Y -C=CH; vinyl carbocat1on 7. For electrophilic addition with HX w~•c p (2) CH3 - -cH--CH ; Benzyl
(1) CH3-CH=CH2; alkyl carbocatton . n (4) c6H5-CH- 3 carbocation (3) CH2=CH-CH=CH2; alkyl carbocatio .
d iven below . d 4 Select the correct answer from the co es g (b) 1. 2 an (a) 1 and 4 (d) 1. 3 and 4 (c) 2, 3 and 4
8. Consider the following statements : 1 trophilic addition . th lkyne for e ec d" . n (1) Alkene is more reactive an a I philic ad 1t10 .
(2) Alkyne is more reactive than alkene for nuc eo h •lie addition reactmn (3) Alkyne is more reactive than alkene for electrop
1t· e than alkene having CH3 group
• b • more reac 1v (4) Alkene h~ving CF3 at vinylic car on is Of these, correct statements are : (b) 1, 2 and 3 003~4 ~1~2 (c) 1, 2 and 4 . dd"tion with alkenes.
9. Which among the following reagents gives syn-a 1 0
(2) Dil. KMnOJOH (1) Br2 INil6 (3) 0s04 I NaS03H I HOH (4) Hz Select the correct answer from the· codes given below : Codes: (a) only 1 (c) 2, 3 and 4
10. In the given reaction
[X] will be :
(b) 2 and 3 (d) only 4
Hg(OAc)if HOH CH3--CH=CH2 ----""? [X]
OH
(a) CH3-CH2-CH20H OH I
(c) CH3-CH-CH2HgOAc
I (b) CH3-CH-CH3
11. Write short notes on bromonium ions as intermediates in stereoselective synthesis. 12. What is meant by iodolactonisation. Give mechanism of this reactions. 13. Discuss the mechanism of hydration of alkynes. Hydration of alkynes can be performed in highly acidi
medium but this reaction is generally carried out in the presence of dil. H2so iHg ++. Comment on th11
point. 14. Give synthesis of the given compounds starting from methylenecyclohexane :
(5H HCi' OBr (a) (b)
(c)
CH20H CH2-0-CH3 c5~: (d)6 (e) 6 (f)

15. Propose a mechanism for the iollow· . mg reaction.
CH3 @ CH3-CH=CH-CH-CH2-CH=C/ H/HOH
I '--CH 3 CH3
16. There are two diastereomeric dibasic acids (A) and (B) with the general form~la HOOC-CH=CJI-COOH. (A) is Ifialeic acid and (B) is furmaric acid. (A) on treatment with KMn04 I HOH I OH yields meso-tartaric acid. (B) on the other hand gives(±) tartaric acid. Show how this information allow you to write configuration of maleic and fumaric acid.
17. Show final products of the reactions given below. Pay attention to stereochemistry of the reactions.
XY
CH CH e 3 CF3COOOH HOH/NaOH 3 KMnOiHOH/OH
(a) L_Jj (b) LJ C6Hs '-... / C6Hs BrifCCl4
(cl ,....,...c=c........_ H H
18. Complete the following equations, showing the stereochemistry wherever it is known.
(a)O
(i) BD3ffHF
(g) (ii) H202/NaOH
h . for the following reaction. 20. Propose a mec amsm

21.
22.
23.
(a) What piece of information °do you need to kn9w before you can predict whether isobut " I-butene will react faster with HCl? ene Or (b) Knowing result of the above problem predict which of the following will react faster · • . .. Wtth lfC
(1) Ethylene or propylene? Explain. (n) 2-Methyl-I-butene or 3-methyl-1-butene. E _l? Xpla1n When 3,3-dimethyl-l-butene is treated with HBr, a rearrangement takes place. Which alkyl b . · Id r
0tn1d
wou. you expect from the reaction? e Write a reasonable and detailed mechanism for the following transformation :
+HOH
OH
24. ~omplete the following equations, showing first the product of the Michael addition and then the
mtramolecular aldol condensation product.
25.
26.
0
6:CH3 011
( ) KOH/methanol
a O + CH2=CH-C-CH3 Pyrrole/benzene/ ll
0 0
0 11 0 CH C-OC2H5 II I 3
(b) ® e
+ CH3-C-CH2-CH2-N-CH I I 3 CH3
CH3
~o tt (c) u + CH3-CH2-C-CH=CH2
NaH Ethanol
CH3 I e
CH2-N(f) CH3I ?i (d) ttt3 + CH3-C-CH2-COOC H
O 2 5
CH3
CH30Na (2 moles)/ methanol
(ii) HOH/ll
Carcinogen (A} is -Michael acceptor which d ·,1 Glutathione having general structure R-SH ~mage~ the DNA replication process u~seJecU ; inactivated carcinogen. What will be the st · 18 a tripeptide which converts this carcinogen ructure of i • nactivated carcinogen?
0 0
g:CH Draw a detailed mechanism for the given r .
2 ,.p
1
eaction N ·x.tt11"· is base unnecessary? · ote that no base is added to the
011

AOOmON TO CARBO . - N: CARBON MULnPLE BONDS 525
C6HsPH2 +CH i=CH-CN /CH2-CH2-CN
27. Compound (A) . (excess) C6HsP, & I . ts rogleti 'd " io lowmg compound . m1 e which is a d . CH2-CH2-CN is can be prepared in laboratory from the s . se attve. How th'
~COOC2Hs, CH3-CH,-I
Base and CH -CH · 2- -CONH2
0
(A)
28. Complete the give . n reactions and identify th . .
(a) H
3
co~C'_,,,CH2-CH2-C:3 :::::~::;:: _ __..
OCH3 II 0
(C)
CH2=CH-CN
pH4_5 (D) KzCO3 Et3 BnNCI (E) CH3CN/20°C
(A)
base
(i) CH31 (ii) AgOH (iii) ll .
e· HOWH (F)
(B)
{:•·•·••·····•·•······•·••.·.·................... .. . ....... ·•• ijijjjjj§~~m111 .... t·•••hf 1. Addition of HX on alkenes is regioselective. Why? 2. Write note on stereochemistry of addition reactions. 3. Explain why cis-2-butene gives (d[) mixture of 2,3-dibromobutane on addition of bromine. 4. Isobutene reacts with bromine in the presence of aqueous NaCl to give onl
3_b
h 3
hi 2 h I Y romo-2-
c loro-2-methyl propane and not 2-bromo- -c oro- -met Y propane. Explain why?
S. Give mechanism of the following reactions : (a) Oxymercuration-demercuration (b) Boration followed by oxidation
(c) Hydrogenation in the presence of metal catalyst 6. Explain why, cis-2-butene on hydroxylation with Baeyer's reagent gives meso-2,3-butanediol.
7. Give mechanism and some applications of Sharpless asymmetric epoxidation.

I. Complete th , . . e ,ollowmg reaction and give its mechanism :
~H COOH . . . CH -CHB
9• Explain : Addition of HBr on vinyl bromide under polar condiuons give
3 ri ra
ther than
Br-CH2-CH2Br . (IAS 199JJ 10 D ·b . · . , tskY reaction. (IAS 1993 199 · escn e the mechamsm and synthetic uses of Re,orma • 7)
11. Write notes on : (a) Michael addition reaction (IAS 1994) (b) Claisen reaction . (IAS
1995, 1997)
12. Indicate what reagents are used in the following transformation. Explam your choice : (IAS 199S) 0
? II ? C6H5-CH=CH
2 c6H5-1 H-CH2HgO-C-CF3 __:_.. C6Hs-\H-CH3
OCH3
OCH3
13. Using Michael reaction. how would you prepitre the following? Give the mechanism involved. (IAS 1996) 0 II
(ii) CH3-C-CH2-CH2-CH2--C-CH3
II 0
0 II II
(i) CH3-C-CH-CH2-CH2-C-C6Hs
I
0
COOCH3 14. Show ftow Michael addition followed by aldol condensation can transform a mixture of melhyl
vinyl ketone and cyclohexanone into A19 octalone. (/AS 1997) 15. Suggest a method for preparation of the following using the give_n method. Explain your answer:
0 0
CH3, II (a) /C=CH-C-CH3 (using aldol) CHy'
(b) (using aldol)
(c) ov-O (using Michael)
16. When bromine is treated with dihydropyran (A) both cis and t d b · d Why1
rans pro ucts are o tame . ·
0 (A)
17. Propose a mechanism for the following reaction .

~~m~o~N~TO~C~A~R~B~O~N~: ~CAARRBBOONNAMWlUJLLJT1UP;L;E-;;B;;O:N~o=s---------------s-=21 18. Propose a mechanism for the foll . .
owing reaction :
OH (i) Hg(OAc)i
(i i) NaBH4 0 iiiirijij;:l(:t ,~ ,_,_
1. (c) 2. (d) 3. (d) 4. (b) 5. (d) 6. (a) 7. (b) 8. (d) 9. (c) 10. (c)
11. Bromonium ions, intermediates in organic synthesis, are not prepared from bromine. The reagent used for this purpose in laboratory is N-bromosuccinimide (NBS). NBS is an easily handled crystalline solid, and is perfect for electrophilic addition of bromine to alkenes when the bromonium ion is not
e . intended to be opened by Br. It works by providing a very small concentration of bromine in soluuo n : a small amount of hydrobromic acid is enough to get the reacti'on going, and thereafter every addition produces another molecule of HBr which liberates more bromine from NBS. In a sense. NBS is a
E9 source of Br.
0 0 0 ('ie ¢NH + Br2
9 Br H ' N Br ----'
0 0 0
. f Be · I ys low so other nucleophiles whose concentration is more With NBS, the concentration ° r is a wa '
e 1- b monium ion even if they are not the solvent. complete with Br to open t~e c~c ic w~~ks as intermediate i~ organic synthesis are : Examples in which bromomum 100
ci!r c,Hs ~CoHs ~C6H5 NBS C6H5
C6H5 ....._ H20 C6H5 '-•· Br
0 NBS Traces
HBr
DMSO ~H2 (±) isomers
__ ,. CXBr ---
110-CH2-C=CH
.. J OH .. JH2-C ==== CH o;Br
CH3 _...CH3 ,.,.., /
O- CH2- C= C- C~ CH2

528 AIJVM••~--- -· ·-~, •• .., \.,f1f:M ---==--------------------------~ 12.
4-p . . . . . h presence of NaHC03. This reacti . enteno1c acids fonn iodonium ion with iodine in t e e on is an
. . lectrophile is I. In the presence of N example of electrophilic addition reaction m which e h'l of the reaction is ca bo a1-Ico3,
b · Thus nucleop 1 e r xylate ·
car oxylic group converts into carboxylate ion. . dolactone. This type of reactio 10n.
13.
The carboxylate ion attacks intramolecular_ly to form .10
dolactonisation. This reaction is -. the cyclisation of 4-pentenoic acids into lactone is known as
101 ctonisatio~. Inermolecular atta a ~o given
by bromine and in this case reaction is known as bromo a c 0n the
e om lete with the intramolecular cyclisatio bromonium ion by bromide ion ( or by I) does not c P . n step.
f~o ,. 1~0
~OH ,. Br 0 o l.e
Alkynes like alkenes undergo acid catalysed addition of water. The prnduct of the reaction is an enol.
The enol immediately changes to a ketone. 0-H 0 H so I II
R-C==C-R + HOH 2 4 R--C=CH-R R-C-CH2-R Ketone
Terminal alkynes are less reactive than internal alkynes towards addition of water. Terminal alkynes will add water if mercuric ion is added to the acidic mixture. The mercuric ion acts as a catalyst to
O-H Hg++ I ----+ CH3-CH2-C=CHz' ...--
increase the rate of the addition reaction. Mechanism : The first step in the mercuric-ion catalysed hydration of an alkyne is formation °11
cyclic mercurinium ion. Formation of mercurinium ion takes place via formation of 1t-complex. Th:
electron rich pi-bonds make carbon-carbon triple bond a nucleophile capable of donating its electr(I . . 1 . Th 1 · nl~ parr m po ar reactions. e nuc eoph1le, C==C-, reacts with electrophile Hg++ to form a comr
called a 1t-complex.
R-C=CH + Hg++ R-C==CH
ii Hg++
The 1t-bond overlapps head-to-head with the s-orbital f H -++ 1 f ++ • ded as e01be03
in the cloud on one side of the 1t-bond The O •• n act Hg ts ~egar electron ~
of a filled d-orbital of Hg++- with th · broken arrow md1cates the donation of an gtheni1
1t-complex bond even though it 1·n e lempty 1t* MO of-C==C-. This back-bonding st
ren vo ves a MO*
R-C=C-H 6+ H \ I
R-C=C 1',z I /,z
Hg2+ ,./1 Hg 6+ •

- · ,.--'-t::: BONDS In the second step wate 529 I d r attacks th sow an rate determin' e most subsft
addition reaction. Oxy~ng rep. Due to this real Ute~ carb?n of the cyclic intermadiate. This step is mercuric ketone. Los ;n oses a proton to fo son Ydrati~n of alkynes is example 1..;f nucleophilic _
s O
the mercuric ion re I r~ a mercuric enol which immediately rearranges to su ts m form f
\OH a IOn of an enol which rearranges to a ketone . •• 2 (±)
H R-C=CH --. @ 1 9. >._ / R-c1 ~CH-Hg --. @ YI @
(jlg2+ R:-C=CH-Hg R-C-~Hg
Hc.~"-H bH ! Mercuric enol
OH I
Hg2+ + R-C=CH2
y 0 II
R-C-CH3 The acid-catalysed hydration of alkynes is not a good laboratory method for the preparation of ketones because the high acid concentration required for the reaction to proceed at a reasonable rate also promotes polymerisation and other side reactions. Hydration of alkyne in the presence of Hg++ provides milder conditions in which no polymerisation and other side reactions can take place.
6CH2 -s 14.
Methylenecyclohexane
(a) s CHOOH
(i) Hg(OAc)ifHOH _ (ii) NaBH4
(A)
HBr (c) s Peroxide
c5-Br (C)
(d, e) S
(b) s HBr
(B)
(i)Na
c5-0CH3 (E)

asj
pe av Ste nc
pe
I e
fl ti
j
s I
) (
530
(f) s X"oH
(i)Os0-4 9 U e
KMn04'0WHOH or
AO""""' ... .,-··-· ····- -- -- ....... ,HY --(ii) NaS03WOH (F) (I)
15• Alkene gives electrophilic addition with electrophile. Thus, first step is the addition of H which give:
carbocation as reaction intennediate. ®..,.,,CH
-CH2-CH2-CH2-C t"cH Electrophili~ additionl
reaction (lntramolecular)
Et)
Electrophilic centre
:0 p 16. KMnO
4 I HOH I OH gives syn-addition reaction with alkenes. In this addition reaction (Z)-alkenes
the type abC = Cab give meso product whereas (E)-give (di) mixture. Thus. maleic acid h Z-configuration and furmaric acid has £-configuration.
H'-. /COOH c-c H" /H c-c
HOOC / - ". COOH HOOC/ - "H Fumaric acid
~CH3 17. (a) L_Jj
~CH3 (b) L_Jj
Maleic acid
0 KMnO4/l-IOH/OH
Syn addition
0 HOH/OH
Ct.,CH3 OH
11 OH '''H
Syn vie diol
Br I
a ,CH3 OH
l1 H 1110H
trans vie diol
(CJ C6H5 ". / C6H5 BrJCCl4 C6H5-C-H
H/c==e,H --~ I " Anti-addition + Enantiomer
H-C-C6Hs I Br
(±) mixture
U. (a) D Anti addition ''" ,,, B
Anti addition r

TO CWOJW: CA~ IIUll.n,u • •
' OH (•)6
NBS
(+) DET
Ti (OPri)4 TBHP
Ti (OPt),1(-) DET TBHl'
OH
en s>o
Qt ;t.
NBS H20
OH •• 2
OH
A,,,,,,,o x,,,,,,,
H (g) }((CHz)J-OCOCH3
H3C
(h)
H
H'-- /CH3 C II + D2 C
H/ 'CH3
Syn addition
• ex.
OH
(i) BD3/fHF XH (CH2hOCOCH3 (ii) tt2O2/JliaOH Syn addition of H
3C
DandOH H
D
H
+ D
~~1"(CH2lJOCOCH3
H3C;z H
H I
CH3-C-D I . CH3-C-D
I H
Copyright © 2022 FDOKUMEN




















