
The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in...
9
Original Contribution The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM +/– /ApoE –/– mice John R. Mercer a , Emma Yu a , Nichola Figg a , Kian-Kai Cheng b , Tracy A. Prime c , Julian L. Griffin b, d , Mojgan Masoodi d , Antonio Vidal-Puig e , Michael P. Murphy c , Martin R. Bennett a, ⁎ a Division of Cardiovascular Medicine, University of Cambridge, Addenbrooke's Hospital, Cambridge CB2 0QQ, UK b Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge, CB2 1GA, UK c MRC Mitochondrial Biology Unit, MRC/Wellcome Trust Building, Hills Road, Cambridge CB2 0XY, UK d MRC Human Nutrition Research, Elsie Widdowson Laboratory, 120 Fulbourn Road, Cambridge, CB1 9NL, UK e Institute of Metabolic Sciences, University of Cambridge, Addenbrooke's Hospital, Cambridge, CB2 0QQ, UK abstract article info Article history: Received 18 February 2011 Revised 14 November 2011 Accepted 22 November 2011 Available online 14 December 2011 Keywords: Metabolic syndrome MitoQ Atherosclerosis Mitochondria A number of recent studies suggest that mitochondrial oxidative damage may be associated with atherosclerosis and the metabolic syndrome. However, much of the evidence linking mitochondrial oxidative damage and excess reactive oxygen species (ROS) with these pathologies is circumstantial. Consequently the importance of mitochondrial ROS in the etiology of these disorders is unclear. Furthermore, the potential of decreasing mitochondrial ROS as a therapy for these indications is not known. We assessed the impact of decreasing mitochondrial oxidative damage and ROS with the mitochondria-targeted antioxidant MitoQ in models of atherosclerosis and the metabolic syndrome (fat-fed ApoE –/– mice and ATM +/– /ApoE –/– mice, which are also haploinsufficient for the protein kinase, ataxia telangiectasia mutated (ATM). MitoQ administered orally for 14 weeks prevented the increased adiposity, hypercholesterolemia, and hypertriglyceridemia associated with the metabolic syndrome. MitoQ also corrected hyperglycemia and hepatic steatosis, induced changes in multiple metabolically relevant lipid species, and decreased DNA oxidative damage (8-oxo-G) in multiple organs. Although MitoQ did not affect overall atherosclerotic plaque area in fat-fed ATM +/+ /ApoE –/– and ATM +/– / ApoE –/– mice, MitoQ reduced the macrophage content and cell proliferation within plaques and 8-oxo-G. MitoQ also significantly reduced mtDNA oxidative damage in the liver. Our data suggest that MitoQ inhibits the development of multiple features of the metabolic syndrome in these mice by affecting redox signaling pathways that depend on mitochondrial ROS such as hydrogen peroxide. These findings strengthen the growing view that elevated mitochondrial ROS contributes to the etiology of the metabolic syndrome and suggest a potential therapeutic role for mitochondria-targeted antioxidants. © 2011 Elsevier Inc. All rights reserved. Introduction The metabolic syndrome describes a combination of obesity, fatty liver, hypertension, hypercholesterolemia, and insulin resistance that is reaching epidemic proportions in the industrialised world [1]. While excessive food intake and associated obesity are clearly major underlying factors, it is thought that elevated mitochondrial oxidative damage contributes to the development and progression of the syndrome, although the mechanistic details are unclear [2–6]. Patients with the metabolic syndrome are particularly susceptible to coronary artery disease due to atherosclerosis and this remains the commonest cause of death in the Western world [7]. The advanced atherosclerotic plaque consists of an accumulation of vascular smooth muscle cells (VSMCs) and their extracellular matrix products, inflammatory cells (macrophages, T lymphocytes, dendritic cells), and intracellular and extracellular lipid and extracellular debris. The risk factors for atherosclerosis are well known, and include increased serum low density lipoprotein cholesterol, hypertension, and diabetes [8]. In previous work to investigate the etiology of the metabolic syndrome and atherosclerosis, we studied mouse models that had features of both of these diseases [9]. Systemic single allelic loss of the gene encoding the highly conserved Ataxia Telangiectasia Mutated (ATM) protein (a member of the phosphoinositide 3-kinase-related kinase family involved in DNA repair) accelerated atherosclerosis and multiple features of the metabolic syndrome in these ApoE –/– mice. ATM +/– /ApoE –/– mice also showed evidence of elevated oxidative damage to mitochondria, including increased mtDNA adducts and deletions, and reduced respiratory complex activity. However, it was unclear whether the mitochondrial oxidative damage directly promoted one or both of the metabolic syndrome and atherosclerosis, or whether the mitochondrial alterations were just a secondary consequence of systemic metabolic effects. Free Radical Biology & Medicine 52 (2012) 841–849 ⁎ Corresponding author. Fax: + 44 1223 331505. E-mail address: [email protected] (M.R. Bennett). 0891-5849/$ – see front matter © 2011 Elsevier Inc. All rights reserved. doi:10.1016/j.freeradbiomed.2011.11.026 Contents lists available at SciVerse ScienceDirect Free Radical Biology & Medicine journal homepage: www.elsevier.com/locate/freeradbiomed
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in...

Free Radical Biology & Medicine 52 (2012) 841–849
Contents lists available at SciVerse ScienceDirect
Free Radical Biology & Medicine
j ourna l homepage: www.e lsev ie r .com/ locate / f reeradb iomed
Original Contribution
The mitochondria-targeted antioxidant MitoQ decreases features of the metabolicsyndrome in ATM+/–/ApoE–/– mice
John R. Mercer a, Emma Yu a, Nichola Figg a, Kian-Kai Cheng b, Tracy A. Prime c, Julian L. Griffin b,d,Mojgan Masoodi d, Antonio Vidal-Puig e, Michael P. Murphy c, Martin R. Bennett a,⁎a Division of Cardiovascular Medicine, University of Cambridge, Addenbrooke's Hospital, Cambridge CB2 0QQ, UKb Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge, CB2 1GA, UKc MRC Mitochondrial Biology Unit, MRC/Wellcome Trust Building, Hills Road, Cambridge CB2 0XY, UKd MRC Human Nutrition Research, Elsie Widdowson Laboratory, 120 Fulbourn Road, Cambridge, CB1 9NL, UKe Institute of Metabolic Sciences, University of Cambridge, Addenbrooke's Hospital, Cambridge, CB2 0QQ, UK
⁎ Corresponding author. Fax: +44 1223 331505.E-mail address: [email protected] (M.R. Benn
0891-5849/$ – see front matter © 2011 Elsevier Inc. Alldoi:10.1016/j.freeradbiomed.2011.11.026
a b s t r a c t
a r t i c l e i n f oArticle history:Received 18 February 2011Revised 14 November 2011Accepted 22 November 2011Available online 14 December 2011
Keywords:Metabolic syndromeMitoQAtherosclerosisMitochondria
A number of recent studies suggest that mitochondrial oxidative damagemay be associatedwith atherosclerosisand the metabolic syndrome. However, much of the evidence linking mitochondrial oxidative damage andexcess reactive oxygen species (ROS) with these pathologies is circumstantial. Consequently the importance ofmitochondrial ROS in the etiology of these disorders is unclear. Furthermore, the potential of decreasingmitochondrial ROS as a therapy for these indications is not known. We assessed the impact of decreasingmitochondrial oxidative damage and ROS with the mitochondria-targeted antioxidant MitoQ in models ofatherosclerosis and the metabolic syndrome (fat-fed ApoE–/– mice and ATM+/–/ApoE–/– mice, which are alsohaploinsufficient for the protein kinase, ataxia telangiectasia mutated (ATM). MitoQ administered orally for14 weeks prevented the increased adiposity, hypercholesterolemia, and hypertriglyceridemia associated withthemetabolic syndrome. MitoQ also corrected hyperglycemia and hepatic steatosis, induced changes inmultiplemetabolically relevant lipid species, and decreased DNA oxidative damage (8-oxo-G) in multiple organs.Although MitoQ did not affect overall atherosclerotic plaque area in fat-fed ATM+/+/ApoE–/– and ATM+/–/ApoE–/– mice, MitoQ reduced the macrophage content and cell proliferation within plaques and 8-oxo-G.MitoQ also significantly reduced mtDNA oxidative damage in the liver. Our data suggest that MitoQ inhibitsthe development of multiple features of the metabolic syndrome in these mice by affecting redox signalingpathways that depend onmitochondrial ROS such as hydrogen peroxide. These findings strengthen the growingview that elevated mitochondrial ROS contributes to the etiology of the metabolic syndrome and suggest apotential therapeutic role for mitochondria-targeted antioxidants.
© 2011 Elsevier Inc. All rights reserved.
Introduction
The metabolic syndrome describes a combination of obesity, fattyliver, hypertension, hypercholesterolemia, and insulin resistance thatis reaching epidemic proportions in the industrialised world [1].While excessive food intake and associated obesity are clearly majorunderlying factors, it is thought that elevated mitochondrial oxidativedamage contributes to the development and progression of thesyndrome, although the mechanistic details are unclear [2–6]. Patientswith the metabolic syndrome are particularly susceptible to coronaryartery disease due to atherosclerosis and this remains the commonestcause of death in the Western world [7]. The advanced atheroscleroticplaque consists of an accumulation of vascular smooth muscle cells(VSMCs) and their extracellular matrix products, inflammatory cells
ett).
rights reserved.
(macrophages, T lymphocytes, dendritic cells), and intracellular andextracellular lipid and extracellular debris. The risk factors foratherosclerosis are well known, and include increased serum lowdensity lipoprotein cholesterol, hypertension, and diabetes [8].
In previous work to investigate the etiology of the metabolicsyndrome and atherosclerosis, we studied mouse models that hadfeatures of both of these diseases [9]. Systemic single allelic loss of thegene encoding the highly conserved Ataxia Telangiectasia Mutated(ATM) protein (a member of the phosphoinositide 3-kinase-relatedkinase family involved in DNA repair) accelerated atherosclerosis andmultiple features of the metabolic syndrome in these ApoE–/– mice.ATM+/–/ApoE–/– mice also showed evidence of elevated oxidativedamage to mitochondria, including increased mtDNA adducts anddeletions, and reduced respiratory complex activity. However, it wasunclear whether the mitochondrial oxidative damage directlypromoted one or both of the metabolic syndrome and atherosclerosis,or whether the mitochondrial alterations were just a secondaryconsequence of systemic metabolic effects.

842 J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
We used the mitochondria-targeted antioxidant MitoQ toinvestigate the role of mitochondrial ROS and oxidative damage in thedevelopment of the metabolic syndrome and atherosclerosis. MitoQconsists of the lipophilic TPP (triphenylphosphonium) cation covalentlylinked to ubiquinone. MitoQ is taken up rapidly and extensively bymitochondria within cells in vivo, where it is reduced to the activeantioxidant ubiquinol form [10]. MitoQ protects mitochondria bydecreasing oxidative damage to mitochondrial proteins, lipids, andDNA [11,12], and has been shown to decrease mitochondrial damageand to lower levels of mitochondrial ROS in a range of animal models(reviewed in [13]). Thus, if MitoQ affects the development of eithermetabolic syndrome or atherosclerosis, this would suggest a role formitochondrial oxidative damage or ROS levels in their etiology. Wefound that MitoQ significantly reduced multiple features of themetabolic syndrome in these models but had more limited effects onatherosclerosis. These findings have significant implications for ourunderstanding of the etiology of the metabolic syndrome and to thedevelopment of potential therapies for this disorder.
Materials and methods
MitoQ administration and animal protocols
All animal experimental procedures conformed to animal ethicalcommittee approval and UK Home Office licensing. The generationand genotyping of ATM+/–/ApoE–/– mice (Mus. musculus) back-crossed >10 times onto a C57BLK6/J background has been describedpreviously [9]. ATM+/–/ApoE–/– and littermate ATM+/+/ApoE–/– maleand female mice were fed a high fat western diet comprising 19.5%Casein milk fat, 0.05% cholesterol (Special Diet Services, UK DietCode 829100) from 6 weeks of age for 14 weeks in an isolatedpathogen-free environment. Mice were administered MitoQ (as a α-cyclodextrin complex of the methanesulfonate salt of MitoQ) from6 weeks of age in their drinking water which was changed biweekly.The initial dose was 100 μM (due to initial taste aversion) and thiswas increased weekly by 100 μM until the maintenance dose of500 μM was reached. These mice consumed ~4.3 ml drinking waterper mouse per day, corresponding to a maintenance dose of approxi-mately 71.6 μmol/kg/day for a 30 g mouse. Previous studies haveshown that feeding MitoQ in this way results in long-term steady-state MitoQ concentrations of approximately ~133 pmol MitoQ/g inheart and ~20 pmol MitoQ/g in liver [12]. Control mice were suppliedwith water alone. Some mice were administered the controlmitochondria-targeted compound decylTPP, which has the sameTPP moiety as MitoQ but lacks antioxidant efficacy, at the samedosing regimen as used for MitoQ. This was done to control for anynonantioxidant effects of the accumulation of MitoQ by mitochondria.Mice were killed by CO2 overdose and the tissues were snap-frozen inliquid nitrogen and stored at -80 °C. Histological samples were fixedwith 5% neutral-buffered formalin (NBF).
Mitochondria assays
Heart and liver tissue were homogenised and mitochondriaextracted by ultracentrifugation. The activities of Complex I, citratesynthase, and the levels of protein carbonyls were assayed asdescribed previously [9].
Serum lipid and glucose analysis
Cholesterol, triglyceride, and low density (LDL) and high densitylipoproteins (HDL) were assayed as previously described [9]. Forglucose tolerance testingmicewere fasted overnight, and blood glucosewas assayed prior to intraperitoneal injection of glucose (1 g/kg).Glucose, insulin, and HbA1cwere assayed using a Dade Behring Dimen-sion RXL analyser using an adaptation of the hexokinase-glucose-6-
phosphate dehydrogenase colorimetric method and a Mercodia Rat/Mouse Insulin ELISA kit.
Histological analysis
Plaquemorphometry and histological analysis including anti-mousemonoclonal 8-oxo-G (Millipore AB3560) adducts were performed asdescribed previously [9].
Metabolomics
Mice were killed after 14 weeks of high fat feeding. Whole bloodwas collected for plasma analysis and tissues were freshly dissectedand snap-frozen in liquid nitrogen before storage at -80 °C. Tissues(100 mg) were extracted using methanol:chloroform:water to sepa-rate aqueous soluble metabolites from lipids. Metabolomic profilingusing 1H NMR for extracts was undertaken using a Bruker AVANCEII+spectrometer. Total fatty acids were examined using GC-FIDfollowing methyl esterification; the fatty acids were identified byretention time matching to known standards and confirmation by GC-MS. Data analysis was undertaken for 1H NMR using 1D NMR processor(version 8, ACD, Toronto, Canada). Chromatograms were analysedusing Xcalibur, (v. 2.0, Thermo Fisher Corp). Identification of majormetabolic perturbations within the pattern recognition models wasachieved by analysis of corresponding loadings plots, and thenconfirmed by standard univariate statistical analysis (Student's t test).
Measurement of mitochondrial DNA damage by quantitative PCR
PCR products were initially generated using generic primers toidentify control and common mutation products. Products were gel-extracted and sequenced and qPCR primers were designed based onthese sequences. A control region of the mitochondrial genome: FWD,GGTCTTGTAAACCTGAAATGAAG, and REV, AAGCTGGTATTCTAAT-TAAACT, primers generated a 101-bp fragment. Mutant primers weredesigned to incorporate the predicted break point: FWD, TCCCAGA-GACTTGGGGATCTAAC, and REV, TCTCATCAATAATTCGCTACC, producinga 251-bp product. Both sequences were screened by BLASTn to eliminatepotential redundancies in the mouse nuclear genome, and against theNCBI SNP database and Japanese Mitochondrial SNP database. PCR wasperformed on a Rotorgene 6000 series using Rotorgene-specific cyber-green in 20 μl reactions for 45 cycles in triplicate. Serial dilutions ofknown positive control DNA were used to generate standard curves.Melt curves confirmed specific generation of a single product per primerpair. Absolute quantification was performed using the standard curvemethod. Mitochondrial mutations were calculated as per the 2-ΔΔCtmethod [14]. Semiquantitiative MtDNA adduct assessment was aspreviously described [9].
Measurement of oxidative stress
To estimate the levels of cellular oxidative stress we used the generaloxidative stress probe DCFDA 2′7′-dichlorodihydrofluorescein (Invitro-gen, D-399) fluorescence as described previously [9].
In vivo measurement of physical activity and oxygen consumption
Experimental mice that had undergone 14 weeks of high fatfeeding and MitoQ treatment or controls were assessed for physicalactivity and oxygen consumption in a Comprehensive Lab AnimalMonitoring System (CLAMS, Columbus Instruments, OH, USA). Micewere individually caged and acclimatized to the monitoring cages at22 °C under an alternating 12 h light:dark cycle for 72 h prior toassessment. Mice were then assessed for a further 72 h duringwhich physical activity, water and food consumption, and respiratoryexchange ratio (RER=CO2 /%O) were measured.

843J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
Statistical analysis
Student's t test was used for data following a Gaussian distributionand the Mann-Whitney rank sum test used under nonbinominalconditions.
Results
Effects of MitoQ on body composition, energy expenditure, and physicalactivity in ApoE–/– mice
We first assessed the effect of MitoQ on fat-fed ApoE–/– mice todetermine whether there were any nonspecific effects on foodconsumption and energy expenditure that might affect subsequentexperiments examining atherosclerosis and the metabolic syndrome.To do this we administered MitoQ or normal drinking water toApoE–/– mice fed a high fat diet for 14 weeks. Dual energy X-rayabsorptiometry (DEXA) scans at the end of this period showed nochange in lean mass or bone density between MitoQ-treated andcontrol mice, but showed that MitoQ led to a loss of adipose tissue(Figs. 1A and B). To determine if there was a selective effect of MitoQon the mass of particular tissues, major organs were weighed and nor-malized as a percentage of total body weight. MitoQ reduced combined
Fig. 1. MitoQ reduces features of the metabolic syndrome in ApoE–/– mice. (A) DEXA scans oof control and MitoQ-treated mice (n=5). (C) Mouse organ weights normalised as a perceintake over 24 h (n=4) at end of protocol in control or MitoQ-treated mice. (E) Respiratime. (F) Mean RER in control or MitoQ–treated mice.
gonadal white adipose tissue (WAT), which accounts for approximate-ly 30% of total WAT deposition in ApoE–/– mice (Fig. 1C), but caused arelative increase in weights of the kidneys, heart, brown adipose tissue(BAT), and brain, which reflects the relative loss of WAT. As decreasedfood intake or increased levels of activity can reduce adiposity, weexamined whether MitoQ altered these parameters. Mice that hadbeen administered MitoQ for 14 weeks or controls were assessed in acomprehensive laboratory animal monitoring system (CLAMS) over72 h. There was no significant difference in food or water consumptionbetween MitoQ-treated and control mice (Fig. 1D). There was also nodifference in physical activity between mice administered MitoQ orcontrols, as measured by counting the number of infrared beam breakscaused by the mice, as total X-axis (XTOT), X-axis during ambulation(XAMB) or Z-axis total (ZTOT) (Supplemental Fig. 1A). Measurementof the total energy expenditure by these mice was also unaffected byMitoQ (Supplemental Figs. 1B and C). These results suggest that thedecrease in adipose content in the fat-fed mice on administration ofMitoQ was not due to changes in food consumption, physical activity.or energy expenditure, but via a specific change in lipid metabolism.
Todetermine ifMitoQ affected overallmetabolism,wenext assessedthe relative use of carbohydrate and fat for energy production, bymeasuring the respiratory exchange ratio (RER=CO2/O2) for controland MitoQ-treated mice. Control mice had a RER=0.827 which is~40% of calories obtained from carbohydrates and ~60% from fat.
f control and MitoQ-treated mice, used to calculate lean/fat mass. (B) Lean and fat massntage of total body weight (n=5), *P=b0.05, **P=b0.01. (D) Mean food and watertory exchange ratio (RER) measured every 18 min over a 72 h period shown in real

844 J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
MitoQ-treated mice had an RER of 0.850, which equates to ~51% ofcalories from carbohydrates vs ~49% from fat (Figs. 1E and F). Thissuggests that MitoQ increases carbohydrate utilisation relative to fatoxidation for energy production. Together these data suggest thatMitoQ might ameliorate some aspects of the metabolic syndrome inthe fat-fed ATM+/+/ApoE–/– mouse model directly, most likely byaffecting mitochondrial oxidative damage and/or ROS, rather than byaffecting the phenotype indirectly by altering food consumption,activity, or energy expenditure.
Fig. 2. MitoQ reduces hepatic steatosis. (A) H+E-stained liver sections from fat-fed
Effect of MitoQ on liver steatosis and inflammation in ATM+/+/ApoE–/–
and ATM+/–/ApoE–/– mice
Having established that MitoQ could be used to investigate therole of mitochondrial oxidative damage in fat-fed ApoE–/– mice, weextended these studies to include ATM+/–/ApoE–/– mice, a mousemodel of the metabolic syndrome and accelerated atherosclerosis.Mice received an escalating dose regime to 500 μM MitoQ or controltreatment for 14 weeks, concurrent with a high fat diet. Both ATM+/–/ApoE–/– mice and ATM+/+/ApoE–/– mice receiving MitoQ gainedless weight over the experimental period vs their respective controls(Supplemental Fig. 1D). Hepatic steatosis, liver inflammation, andincreased serum liver enzymes are prominent features of the meta-bolic syndrome; these features also occur in ATM+/–/ApoE–/– mice,and to a lesser extent in fat-fed ATM+/+/ApoE–/– mice [9]. Wefound that MitoQ markedly reduced hepatic steatosis and triglyceridedeposition in both fat-fed ATM+/–/ApoE–/– and ATM+/+/ApoE–/–
mice, as indicated by a decreased amount of fat accumulation onliver sections (Figs. 2A and B). This decrease in liver steatosis byMitoQ was also reflected in a significant decrease in the serum livertransaminases in ATM+/–/ApoE–/– (Fig. 2C) and ATM+/+/ApoE–/–
mice (not shown), consistent with decreased damage to the liverdue to inflammation. Together these data indicate that MitoQ signifi-cantly decreases liver steatosis and subsequent damage to this organ,a major aspect of the metabolic syndrome.
ATM+/+/ApoE–/– or ATM+/–/ApoE–/– mice treated with MitoQ for 14 weeks or control(scale bar=500 μm). (B) Hepatic lipid content expressed as percentage of liver lipidvoids, n=5. (C) Serum liver enzymes (ALK, alkaline phosphatise; AST, aspartate trans-aminase; ALT, alanine transaminase) in fat-fed ATM+/–/ApoE–/– mice treated withMitoQ or control. Data are means±SEM. *P=b0.05, **P=b0.01, ***Pb0.001.
Effects of MitoQ on lipid and glucose metabolism in ATM+/–/ApoE–/– andATM+/+ApoE–/– mice
Disruption to mitochondrial function can affect carbohydrate and fatmetabolism, both of which are significantly altered in the metabolicsyndrome. Furthermore, our previous work has shown that bothcarbohydrate and fat metabolism are disrupted in ATM+/–/ApoE–/–
mice, with significant changes in specific liver lipid species [9]. Wetherefore assessed whether MitoQ could normalize lipid andcarbohydrate metabolism in fat-fed ATM+/–/ApoE–/– and ATM+/+
ApoE–/– mice.First we assessed changes in lipid metabolism in plasma and in tis-
sue extracts from the liver and pancreas of MitoQ-treated and controlfat-fed ATM+/–/ApoE–/– and ATM+/+ApoE–/– mice. We used a meta-bolomic approach that identifies intermediates of lipid metabolismthat have been shown previously to be disrupted in these mice [9].MitoQ caused a significant decrease in a number of lipid species,including palmitoleic and heptadecenoic acids, and a significantincrease in other lipid species, including oleic, arachidonic, and doco-sahexaenoic acid (Supplemental Figs. 2A and B). This suggests thatdisruption to mitochondrial metabolism affects lipid metabolism inthese mouse models, and that this is altered by MitoQ, consistent withincreased mitochondrial oxidative stress underlying this phenomenon.To determine if these alterations in lipid metabolism led to changes inserum lipid profiles, we next investigated serum total cholesterol andtriglyceride. MitoQ decreased serum total cholesterol and triglyceridein ATM+/+/ApoE–/– and ATM+/–/ApoE–/– mice. MitoQ also reducedLDL cholesterol concentrations in ATM+/+/ApoE–/– but not ATM+/–/
ApoE–/– mice (Figs. 3A and B). HDL levels in ApoE–/– mice are verylow and no change was found with MitoQ treatment.
We have previously shown that ATM+/–/ApoE–/– mice show hyper-glycemia and impaired glucose tolerance in conjunction with relativelynormal insulin tolerance [9]. This combination can occur due to theonset of insulin resistance, to defective insulin release, or both. Wehave previously carried out a detailed histological analysis of thepancreatic islets in these mice [9] and found normal numbers and sizeof α-glucagon-producing and β-insulin-producing cells in bothgenotypes, suggesting that peripheral insulin resistance underlies thephenotype. As insulin resistance is a core feature of the metabolicsyndrome, we next investigated whether MitoQ treatment alteredglucose metabolism in fat-fed ATM+/–/ApoE–/– mice. We found thatMitoQ reduced fasted serum glucose and insulin at 7 weeks after highfat diet feeding (Supplemental Figs. 3A and B) and normalized glucosetolerance (Supplemental Fig. 3C). MitoQ had no effect on pancreatichistology (Supplemental Fig. 3D).
Effects of MitoQ on atherosclerotic plaque burden in ATM+/+/ApoE–/–
and ATM+/–/ApoE–/– mice
As ATM+/–/ApoE–/– mice fed a high fat diet develop extensiveatherosclerotic lesions [9], we next investigated the impact ofadministering MitoQ. First, we examined the effect of MitoQ on the
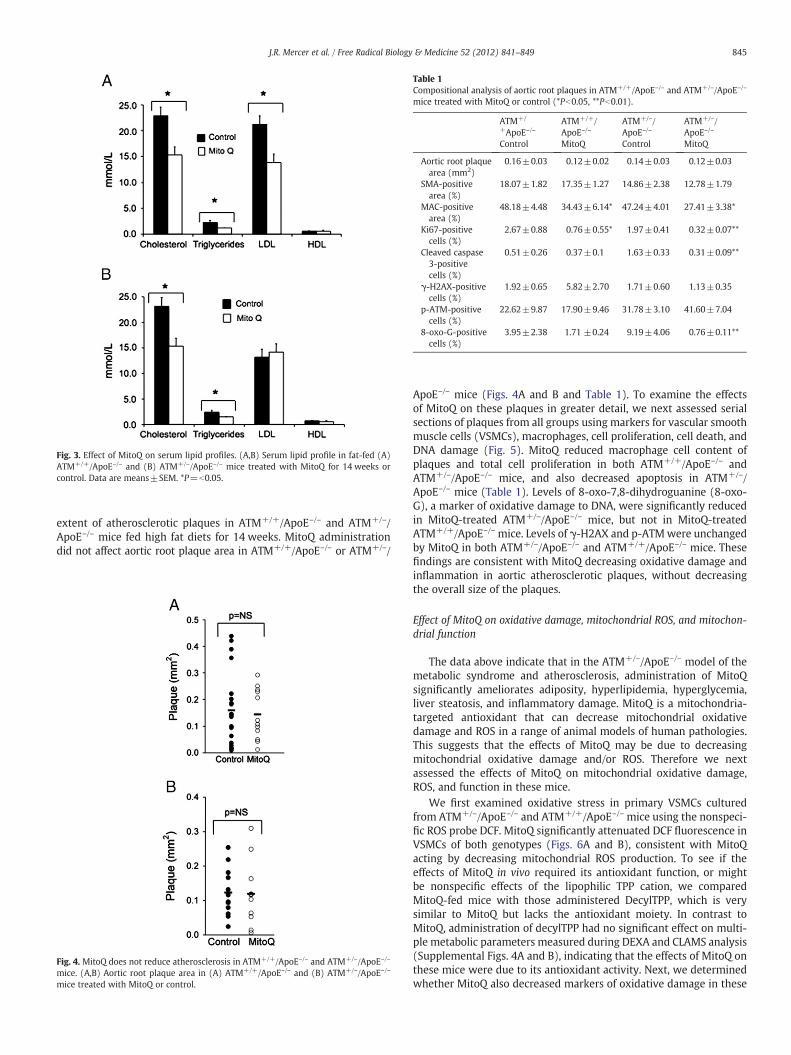
Fig. 3. Effect of MitoQ on serum lipid profiles. (A,B) Serum lipid profile in fat-fed (A)ATM+/+/ApoE–/– and (B) ATM+/–/ApoE–/– mice treated with MitoQ for 14 weeks orcontrol. Data are means±SEM. *P=b0.05.
Table 1Compositional analysis of aortic root plaques in ATM+/+/ApoE–/– and ATM+/–/ApoE–/–
mice treated with MitoQ or control (*Pb0.05, **Pb0.01).
ATM+/
+ApoE–/–
Control
ATM+/+/ApoE–/–
MitoQ
ATM+/–/ApoE–/–
Control
ATM+/–/ApoE–/–
MitoQ
Aortic root plaquearea (mm2)
0.16±0.03 0.12±0.02 0.14±0.03 0.12±0.03
SMA-positivearea (%)
18.07±1.82 17.35±1.27 14.86±2.38 12.78±1.79
MAC-positivearea (%)
48.18±4.48 34.43±6.14* 47.24±4.01 27.41±3.38*
Ki67-positivecells (%)
2.67±0.88 0.76±0.55* 1.97±0.41 0.32±0.07**
Cleaved caspase3-positivecells (%)
0.51±0.26 0.37±0.1 1.63±0.33 0.31±0.09**
γ-H2AX-positivecells (%)
1.92±0.65 5.82±2.70 1.71±0.60 1.13±0.35
p-ATM-positivecells (%)
22.62±9.87 17.90±9.46 31.78±3.10 41.60±7.04
8-oxo-G-positivecells (%)
3.95±2.38 1.71 ±0.24 9.19±4.06 0.76±0.11**
845J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
extent of atherosclerotic plaques in ATM+/+/ApoE–/– and ATM+/–/ApoE–/– mice fed high fat diets for 14 weeks. MitoQ administrationdid not affect aortic root plaque area in ATM+/+/ApoE–/– or ATM+/–/
Fig. 4. MitoQ does not reduce atherosclerosis in ATM+/+/ApoE–/– and ATM+/–/ApoE–/–
mice. (A,B) Aortic root plaque area in (A) ATM+/+/ApoE–/– and (B) ATM+/–/ApoE–/–
mice treated with MitoQ or control.
ApoE–/– mice (Figs. 4A and B and Table 1). To examine the effectsof MitoQ on these plaques in greater detail, we next assessed serialsections of plaques from all groups using markers for vascular smoothmuscle cells (VSMCs), macrophages, cell proliferation, cell death, andDNA damage (Fig. 5). MitoQ reduced macrophage cell content ofplaques and total cell proliferation in both ATM+/+/ApoE–/– andATM+/–/ApoE–/– mice, and also decreased apoptosis in ATM+/–/ApoE–/– mice (Table 1). Levels of 8-oxo-7,8-dihydroguanine (8-oxo-G), a marker of oxidative damage to DNA, were significantly reducedin MitoQ-treated ATM+/–/ApoE–/– mice, but not in MitoQ-treatedATM+/+/ApoE–/– mice. Levels of γ-H2AX and p-ATMwere unchangedby MitoQ in both ATM+/–/ApoE–/– and ATM+/+/ApoE–/– mice. Thesefindings are consistent with MitoQ decreasing oxidative damage andinflammation in aortic atherosclerotic plaques, without decreasingthe overall size of the plaques.
Effect of MitoQ on oxidative damage, mitochondrial ROS, and mitochon-drial function
The data above indicate that in the ATM+/–/ApoE–/– model of themetabolic syndrome and atherosclerosis, administration of MitoQsignificantly ameliorates adiposity, hyperlipidemia, hyperglycemia,liver steatosis, and inflammatory damage. MitoQ is a mitochondria-targeted antioxidant that can decrease mitochondrial oxidativedamage and ROS in a range of animal models of human pathologies.This suggests that the effects of MitoQ may be due to decreasingmitochondrial oxidative damage and/or ROS. Therefore we nextassessed the effects of MitoQ on mitochondrial oxidative damage,ROS, and function in these mice.
We first examined oxidative stress in primary VSMCs culturedfrom ATM+/–/ApoE–/– and ATM+/+/ApoE–/– mice using the nonspeci-fic ROS probe DCF. MitoQ significantly attenuated DCF fluorescence inVSMCs of both genotypes (Figs. 6A and B), consistent with MitoQacting by decreasing mitochondrial ROS production. To see if theeffects of MitoQ in vivo required its antioxidant function, or mightbe nonspecific effects of the lipophilic TPP cation, we comparedMitoQ-fed mice with those administered DecylTPP, which is verysimilar to MitoQ but lacks the antioxidant moiety. In contrast toMitoQ, administration of decylTPP had no significant effect on multi-ple metabolic parameters measured during DEXA and CLAMS analysis(Supplemental Figs. 4A and B), indicating that the effects of MitoQ onthese mice were due to its antioxidant activity. Next, we determinedwhether MitoQ also decreased markers of oxidative damage in these

Fig. 5. Compositional analysis of atherosclerosis in MitoQ-treated or control mice. Aor-tic root plaque composition from ATM+/–/ApoE–/– mice treated with MitoQ or controlstained for VSMCs (SMA), macrophages (Mac-3), proliferation (ki67), apoptosis(cleaved caspase 3), and the DNA damage markers 8-oxoG, γ-H2AX, and p-ATM.Scale bar =50 μm (low power) and 500 μm (high power).
846 J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
mice in vivo. To do this we first examined the accumulation of 8-oxo-G, a marker of oxidative damage to DNA. We found that total 8-oxo-Gadducts were widespread, seen particularly in heart, liver, and pan-creas in ATM+/–/ApoE–/– mice, and were significantly reduced in allthese organs by MitoQ (Figs. 6C and D). We also explored whetherMitoQ affected the accumulation of oxidative damage to mtDNA. Todo this we assessed the accumulation of a large~5-kbp deletion toMtDNA that has previously been shown to increase in ATM+/–/ApoE–/– mice as an indication of mitochondrial oxidative damage[9]. MitoQ reduced the abundance of the mitochondrial deletion inboth ATM+/+/ApoE–/– and ATM+/–/ApoE–/– mouse livers (Figs. 7Aand B, Supplemental Figs. 5A and B, Supplemental Fig. 6A). We nextused a qPCR-based assay of generalised MtDNA damage, includingstrand breaks, abasic sites, and oxidised lesions induced by oxidativedamage. We have previously shown that this assay shows a signifi-cant accumulation of damage to mtDNA in the livers of ATM+/–/ApoE–/– mice [9]. MitoQ significantly reduced MtDNA damage in
livers of ATM+/–/ApoE–/– but not ATM+/+/ApoE–/– mice (Figs. 7Cand D), although no difference was seen in heart and brown adiposetissue (BAT) (not shown).
To further assess a role forMitoQ in protecting againstmitochondrialoxidative damage, we isolated mitochondria from the livers and heartsof fat-fed ATM+/+ApoE–/– and ATM+/–/ApoE–/– mice administeredMitoQ for 14 weeks and assessed the accumulation of protein carbonylsas a marker of oxidative damage. We found no significant difference inthe abundance of these species between genotypes or MitoQ treatment(Figs. 8A and B).
To determine if therewere any changes in the levels ofmitochondrialproteins, we used Western blotting to assess the relative abundance ofrespiratory chain and ATP synthase proteins in MitoQ-treated or controlATM+/+ApoE–/– and ATM+/–/ApoE–/– mice. The relative abundance ofrespiratory complexes Complex I, II, III, and IV and of the ATP synthase(green) was compared to levels of MnSOD (shown in red) and did notchange markedly on treatment with MitoQ (Supplemental Fig. 6B). Toexamine mitochodrial function further, we measured the activity ofrespiratory complex I, as this protein complex is susceptible to oxidativedamage. In addition, we have previously shown that Complex I activityrelative to citrate synthase is reduced in ATM+/–/ApoE–/– mice with nochange in respiratory complex expression [9]. The differences inComplex I activity normalised to citrate synthase were not significantin this smaller group of ATM+/–ApoE–/– and ATM+/+ApoE–/– mice, andsimilarly the effect of MitoQ was not statistically significant (Supple-mentary Figs. 7A and C). Therefore the protective effects of MitoQ arenot a consequence of changing the expression or turnover of mitochon-drial respiratory complexes, but appear to correlate with a change inmitochondrial protein oxidative damage.
Discussion
We and others have previously shown that ATM heterozygosity inApoE null mice promotes atherosclerosis and multiple features of themetabolic syndrome, including hypertension, hypercholesterolemia,hepatic steatosis, glucose intolerance, and alterations in lipidmetabolism [9,15,16]. ATM+/–/ApoE–/– mice also exhibit oxidativeDNA andmitochondrial damage and these features have been hypothe-sised to contribute to the metabolic features seen in these mice [9].However, it was unclear whether the mitochondrial oxidative damageand ROS seen in thesemicewere simply a by-product of the acceleratedatherosclerosis and the metabolic syndrome, or if mitochondrialoxidative damage and/or ROS had a causal role in these pathologies.
To address these issues, we utilised the mitochondria-targetedantioxidant MitoQ to assess the potential role of mitochondrialdamage in accelerating atherosclerosis and the metabolic syndrome.We found that MitoQ affected multiple features of the metabolicsyndrome in these mice. Notably, MitoQ reduced fat accumulation,hyperglycemia, hypercholesterolemia, hypertriglyceridemia, andhepatic steatosis, and altered the abundance of multiple fatty acidsin the liver. MitoQ decreased weight gain, adiposity, and hepaticsteatosis, and improved liver function. However, MitoQ did notreduce atherosclerotic plaque formation, although certain featuresof high-risk plaques were reduced, such as macrophage content.
To understand howMitoQmight preventmany of the features of themetabolic syndrome in these mice, we assessed whole bodymetabolism and found that MitoQ did not affect lean body mass, foodand water intake, or energy expenditure. In contrast, MitoQ increasedRER, suggesting an increase in utilisation of carbohydrate over fat as ametabolic fuel. MitoQ also reduced circulating serum cholesterol andtriglycerides, decreased serum glucose and insulin, and normalisedglucose tolerance. Furthermore, MitoQ altered the metabolomic lipidprofile of ATM+/+/ApoE–/– and ATM+/–ApoE–/– mice.
MitoQ is a mitochondria-targeted antioxidant that has beenshown to decrease oxidative damage and lower mitochondrial ROSin a number of animal models. The metabolic changes following

Fig. 6. MitoQ reduces total DCF fluorescence in vascular smooth muscle cells in vitro and accumulation of 8-oxo-G in tissues. (A,B) DCFDA fluorescence (relative fluorescence units,RFU) in VSMCs in vitro from (A) ATM+/–/ApoE–/– or (B) ATM+/+/ApoE–/– mice (n=3). (C) Immunohistochemistry for 8-oxo-G in heart (nuclear), liver (cytosolic) and pancreas(nuclear) in ATM+/–/ApoE–/– mice fat fed for 14 weeks treated with either MitoQ or control (scale bar=20 μm for heart, 75 μm for pancreas, 50 μm for liver (n=5). (D) Percentageof 8-oxo-G-positive cells in each tissue. Data are means±SEM.
847J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
MitoQ treatment may therefore be due to the prevention of oxidativedamage. This concept is supported by our data showing thatDecylTPP, which is accumulated by mitochondria but which has noantioxidant function, did not mimic the metabolic effects of MitoQin vivo. Furthermore, MitoQ decreased oxidative stress in cells fromthese mice, and reduced the accumulation of 8-oxo-G in the heart,liver, and pancreas of ATM+/–/ApoE–/– mice. While MitoQ decreasedoxidative mtDNA damage and the mtDNA deletion, it had no effecton mitochondrial protein carbonyls. We have previously found thatactivity of Complex I normalised to citrate synthase is reduced inATM+/–/ApoE–/– mice [9]. This difference in Complex I activitybetween mice of different genotypes did not reach statistical signifi-cance in a smaller group of mice, so that the effect of MitoQ couldnot be accurately determined.
These data suggest that the mechanism by which mitochondrialfunction is altered in these models, leading to many of the featuresof the metabolic syndrome, involves elevated mitochondrial ROSthat are MitoQ-sensitive. The increased oxidative mtDNA damageand deletion and reduced Complex I activity found previously inATM+/–/ApoE–/– mice suggest that features of the metabolicsyndrome may also be due to mtDNA damage. In contrast, as therewere no changes in mitochondrial protein carbonyls or in the levelof expression or activity of the respiratory chain on MitoQ treatment,
the effects of MitoQ may not be due to preventing damage to mito-chondrial bioenergetic activity. Instead, more subtle changes in mito-chondrial metabolism may underlie the onset of the metabolicsyndrome in this model and that these changes can then be blockedby MitoQ. One scenario is that the onset of the metabolic syndromeis due to alteration of redox and ROS-sensitive carbohydrate andlipid metabolic pathways within mitochondria that are then reversedby MitoQ treatment. This possibility is supported by the fact thatredox signaling is important for mitochondrial metabolism [17] andthat the activities of a large number of mitochondrial enzymes canbe redox-regulated [18]. Furthermore, recently it has been shownthat adipocyte differentiation requires mitochondrial ROS and thatthis process can be blocked by MitoQ [19]. Together these findingssupport a role for mitochondrial ROS in the development of themetabolic syndrome in these mouse models.
Interestingly, MitoQ did not reduce atherosclerotic plaqueformation, nor did it reduce markers of genomic DNA damage withinthe plaques, although certain features of high-risk plaques werereduced, such asmacrophage content. The lack of effect ofMitoQ on pla-que area may be because MitoQ did not significantly decrease LDL cho-lesterol, which is a major factor leading to atherosclerosis in thesemouse models, or reduce genomic damage, which also exacerbatesatherosclerosis.

Fig. 7.MitoQ reduces the common mitochondrial deletion in ATM+/+/ApoE–/– and ATM+/–/ApoE–/– mice and MtDNA adducts in livers of ATM+/–/ApoE–/– mice. (A,B) Expression ofthe common mitochondrial deletion in liver of MitoQ-treated or control (A) ATM+/+/ApoE–/– or (B) ATM+/–/ApoE–/– mice (n=3). (C,D) Mitochondrial DNA adduct formation inlivers of MitoQ-treated or control (C) ATM+/+/ApoE–/– or (D) ATM+/–/ApoE–/– mice (n=4). Reduced adduct formation is shown by increased amplification of mtDNA in ATM+/–/ApoE–/– mice.
Fig. 8. Protein carbonyls in ATM+/+/ApoE–/– and ATM+/–/ApoE–/– mice livers.Proteincarbonyls in livers from (A) ATM+/+/ApoE–/– or (B) ATM+/–/ApoE–/– mice treatedwith MitoQ (n=3) or control (n=4).
848 J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
Our data are consistent with the following model. ATMhaploinsufficiency is associated with an increase in mitochondrialROS, most likely hydrogen peroxide, that modulates mitochondrialcarbohydrate and lipid metabolism so as to alter whole body glucoseand lipid metabolism and thereby lead to hyperglycemia, hyperlipid-emia, and excess fat storage, all of which are features of the metabolicsyndrome. ATM+/–/ApoE–/– mice also have significant mtDNA damage,although it is not clear whether mtDNA damage contributes to themetabolic syndrome features independent of ROS. MitoQ decreasesthe levels of mitochondrial ROS and thereby reverses the deleteriouseffects on carbohydrate and lipid metabolism, preventing many of thesymptoms of themetabolic syndrome, such as adiposity, hepatic steato-sis, and elevated serum cholesterol and triglycerides. While there arestill significant gaps in our understanding of the origins of the elevatedmitochondrial ROS and how this modifiesmitochondrialmetabolism soas to contribute to the metabolic syndrome, our findings clearly showthat MitoQ can prevent multiple features of the metabolic syndromein fat-fed ATM+/–/ApoE–/–mice. These findings provide further supportfor a role for mitochondrial ROS in the etiology of the metabolicsyndrome and suggest that mitochondria-targeted antioxidants suchas MitoQ may be beneficial in treating these pathologies.
Supplementary materials related to this article can be foundonline at doi:10.1016/j.freeradbiomed.2011.11.026.
Acknowledgments
We acknowledge the British Heart Foundation (BHF) Grant RG08/009/25841 to M.R.B.; Diabetes and Wellness Foundation, BBSRC, BHF(PG/05/08), and Wellcome Trust (072829/Z/03/Z) grants to J.G.; MRCand Hepadip grants to A.V.P.; the Cambridge NIHR BiomedicalResearch Centre. M.P.M. acts as a consultant for and holds stock inAntipodean Pharmaceuticals Inc., who are commercializing MitoQ.

849J.R. Mercer et al. / Free Radical Biology & Medicine 52 (2012) 841–849
References
[1] Bruce, K. D.; Hanson, M. A. The developmental origins, mechanisms, and implicationsof metabolic syndrome. J. Nutr. 140:648–652; 2010.
[2] Nisoli, E.; Clementi, E.; Carruba, M. O.; Moncada, S. Defective mitochondrialbiogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome?Circ. Res. 100:795–806; 2007.
[3] Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J. R. Mitochondrial biogenesis in themetabolic syndrome and cardiovascular disease. J. Mol. Med. 88:993–1001; 2010.
[4] Rolo, A. P.; Gomes, A. P.; Palmeira, C. M. Regulation of mitochondrial biogenesis inmetabolic syndrome. Curr. Drug Targets 12:872–878; 2011.
[5] Ballinger, S. W. Mitochondrial dysfunction in cardiovascular disease. Free Radic.Biol. Med. 38:1278–1295; 2005.
[6] Madamanchi, N. R.; Runge, M. S. Mitochondrial dysfunction in atherosclerosis.Circ. Res. 100:460–473; 2007.
[7] Callow, A. D. Cardiovascular disease 2005—the global picture. Vasc. Pharmacol. 45:302–307; 2006.
[8] Libby, P.; Ridker, P. M.; Hansson, G. K. Progress and challenges in translating thebiology of atherosclerosis. Nature 473:317–325; 2011.
[9] Mercer, J. R.; Cheng, K. K.; Figg, N.; Gorenne, I.;Mahmoudi,M.; Griffin, J.; Vidal-Puig, A.;Logan, A.;Murphy,M. P.; Bennett, M. DNA damage linksmitochondrial dysfunction toatherosclerosis and the metabolic syndrome. Circ. Res. 107:1021–1031; 2010.
[10] Ross, M. F.; Prime, T. A.; Abakumova, I.; James, A. M.; Porteous, C. M.; Smith, R. A.;Murphy, M. P. Rapid and extensive uptake and activation of hydrophobic triphe-nylphosphonium cations within cells. Biochem. J. 411:633–645; 2008.
[11] Kelso, G. F.; Porteous, C. M.; Coulter, C. V.; Hughes, G.; Porteous, W. K.; Ledgerwood,E. C.; Smith, R. A.; Murphy, M. P. Selective targeting of a redox-active ubiquinone to
mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem.276:4588–4596; 2001.
[12] Rodriguez-Cuenca, S.; Cocheme, H. M.; Logan, A.; Abakumova, I.; Prime, T. A.; Rose, C.;Vidal-Puig, A.; Smith, A. C.; Rubinsztein, D. C.; Fearnley, I. M.; Jones, B. A.; Pope, S.;Heales, S. J.; Lam, B. Y.; Neogi, S. G.; McFarlane, I.; James, A. M.; Smith, R. A.; Murphy,M. P. Consequences of long-term oral administration of the mitochondria-targetedantioxidant MitoQ to wild-type mice. Free Radic. Biol. Med. 48:161–172; 2009.
[13] Hobbs, C. E.; Murphy, M. P.; Smith, R. A.; Oorschot, D. E. Neonatal rat hypoxia-ischemia: effect of the anti-oxidant mitoquinol, and S-PBN. Pediatr. Int. 50:481–488; 2008.
[14] Schmittgen, T. D.; Livak, K. J. Analyzing real-time PCR data by the comparativeC(T) method. Nat. Protoc. 3:1101–1108; 2008.
[15] Wu, D.; Yang, H.; Xiang, W.; Zhou, L.; Shi, M.; Julies, G.; Laplante, J. M.; Ballard, B. R.;Guo, Z. Heterozygous mutation of ataxia-telangiectasia mutated gene aggravateshypercholesterolemia in apoE-deficient mice. J. Lipid Res. 46:1380–1387; 2005.
[16] Schneider, J. G.; Finck, B. N.; Ren, J.; Standley, K. N.; Takagi, M.; Maclean, K. H.; Bernal-Mizrachi, C.; Muslin, A. J.; Kastan, M. B.; Semenkovich, C. F. ATM-dependentsuppression of stress signaling reduces vascular disease in metabolic syndrome. CellMetab. 4:377–389; 2006.
[17] Murphy, M. P. How mitochondria produce reactive oxygen species. Biochem. J.417:1–13; 2009.
[18] Hurd, T. R.; Prime, T. A.; Harbour, M. E.; Lilley, K. S.; Murphy, M. P. Detection ofreactive oxygen species-sensitive thiol proteins by redox difference gelelectrophoresis: implications for mitochondrial redox signaling. J. Biol. Chem.282:22040–22051; 2007.
[19] Tormos, K. V.; Anso, E.; Hamanaka, R. B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.;Chandel, N. S. Mitochondrial complex III ROS regulate adipocyte differentiation.Cell Metab. 14:537–544; 2011.




















