
The immune response to periodontitis and its relevance to ...
232
The immune response to periodontitis and its relevance to cerebral ischaemia A thesis submitted to The University of Manchester for the degree of Doctor of Philosophy in the Faculty of Biology, Medicine and Health 2019 Conor O’Boyle School of Biological Sciences Division of Neuroscience and Experimental Psychology
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of The immune response to periodontitis and its relevance to ...

The immune response to periodontitis and
its relevance to cerebral ischaemia
A thesis submitted to The University of Manchester for the degree of
Doctor of Philosophy in the Faculty of Biology, Medicine and Health
2019
Conor O’Boyle
School of Biological Sciences
Division of Neuroscience and Experimental Psychology

1
Contents
List of figures ..................................................................................................................... 7
List of tables....................................................................................................................... 9
Abbreviations .................................................................................................................. 10
Abstract...... ...................................................................................................................... 13
Declaration....................................................................................................................... 14
Copyright statement ....................................................................................................... 14
Experimental contributions ........................................................................................... 15
Acknowledgements ......................................................................................................... 16
Chapter 1. Introduction ................................................................................................ 17
1.1. Overview ................................................................................................................ 18
1.2. The importance of the oral barrier ......................................................................... 19
1.2.1. Immunity at the oral barrier .......................................................................... 19
1.3. Periodontal disease ................................................................................................. 21
1.4. Epidemiology of periodontal disease ..................................................................... 22
1.5. Diagnosis and treatment of periodontal disease ..................................................... 24
1.6. The pathogenesis of periodontitis .......................................................................... 25
1.6.1. Microbial dysbiosis ...................................................................................... 25
1.6.2. Aberrant immunity ....................................................................................... 27
1.7. Experimental models of periodontitis .................................................................... 29
1.7.1. Ligature placement ....................................................................................... 30
1.7.2. Oral gavage of periodontal pathogens .......................................................... 30
1.7.3. Ageing .......................................................................................................... 31
1.8. The effects of periodontitis on systemic health...................................................... 31
1.8.1. Periodontitis and rheumatoid arthritis .......................................................... 33
1.8.2. Periodontitis and mucosal disease ................................................................ 33
1.8.3. Periodontitis and cardiovascular disease ...................................................... 35
1.9. Mechanisms by which periodontitis can impact pathology at non-oral sites ......... 36
1.9.1. Bacterial virulence ........................................................................................ 37
1.9.2. Leakage of microbial and host factors .......................................................... 40
1.9.3. Promotion of autoreactivity .......................................................................... 41
1.10. Stroke ................................................................................................................... 43
1.11. The pathophysiology of ischaemic stroke ............................................................ 43
1.12. The immunopathology of ischaemic stroke ......................................................... 44

2
1.13. Experimental models of ischaemic stroke ........................................................... 46
1.14. The impact of co-morbidities on stroke outcome ................................................ 47
1.14.1. Systemic inflammation and infection ........................................................... 47
1.14.2. Metabolic and complex conditions ............................................................... 48
1.15. Periodontitis and stroke: evidence and potential mechanisms ............................. 49
1.15.1. Clinical importance ...................................................................................... 50
1.16. Aims ..................................................................................................................... 50
Chapter 2. Materials and methods ............................................................................... 51
2.1. Media ..................................................................................................................... 52
2.1.1. 3% media ...................................................................................................... 52
2.1.2. Complete media ............................................................................................ 52
2.1.3. Digest media ................................................................................................. 52
2.1.4. FACS buffer ................................................................................................. 53
2.2. Animals .................................................................................................................. 53
2.3. Generation of head-shielded chimeras ................................................................... 54
2.4. Ligature-induced periodontitis ............................................................................... 54
2.4.1. Pre-procedure ............................................................................................... 54
2.4.2. Ligature placement ....................................................................................... 54
2.4.3. Post-procedure .............................................................................................. 55
2.5. Transient middle cerebral artery occlusion ............................................................ 56
2.5.1. Pre-procedure ............................................................................................... 56
2.5.2. Procedure ...................................................................................................... 56
2.5.3. Post-surgical care ......................................................................................... 58
2.5.4. Assessment of neurological deficits ............................................................. 58
2.6. Permanent middle cerebral artery occlusion .......................................................... 58
2.6.1. Pre-procedure ............................................................................................... 58
2.6.2. Procedure ...................................................................................................... 59
2.6.3. Post-operative care ....................................................................................... 60
2.7. Administration of substances ................................................................................. 60
2.8. Euthanasia & tissue harvesting .............................................................................. 60
2.8.1. Carbon dioxide exposure .............................................................................. 60
2.8.2. Terminal anaesthesia & tissue fixation ......................................................... 61
2.9. Histology ................................................................................................................ 62
2.9.1. Brain sectioning ............................................................................................ 62

3
2.9.2. Cresyl violet staining .................................................................................... 62
2.9.3. Immunohistochemistry ................................................................................. 63
2.10. Histological quantification ................................................................................... 64
2.10.1. Quantification of ischaemic damage ............................................................ 64
2.10.2. Quantification of blood-brain barrier breakdown ......................................... 64
2.10.3. Quantification of neutrophils ........................................................................ 64
2.11. Preparation of single cell suspensions ................................................................. 65
2.11.1. Blood ............................................................................................................ 65
2.11.2. Bone marrow ................................................................................................ 65
2.11.3. Lung ............................................................................................................. 66
2.11.4. Spleen ........................................................................................................... 66
2.11.5. Sub-mandibular lymph nodes ....................................................................... 66
2.11.6. Skin ............................................................................................................... 66
2.11.7. Gut ................................................................................................................ 67
2.11.8. Gingiva ......................................................................................................... 67
2.12. Ex vivo re-stimulation .......................................................................................... 68
2.13. Flow cytometry .................................................................................................... 68
2.13.1. Surface staining ............................................................................................ 68
2.13.2. Intracellular and intranuclear staining .......................................................... 68
2.13.3. Sample acquisition ....................................................................................... 69
2.14. Sorting of haematopoietic stem and progenitor cells ........................................... 69
2.15. Bone loss measurements ...................................................................................... 69
2.16. Bacterial growth evaluation ................................................................................. 70
2.17. Cytometric bead array .......................................................................................... 71
2.18. Cerebral microvessel isolation ............................................................................. 71
2.19. qRT-PCR.............................................................................................................. 72
2.19.1. RNA isolation ............................................................................................... 72
2.19.2. cDNA synthesis and qRT-PCR .................................................................... 73
2.20. Bacterial 16S rDNA quantification ...................................................................... 74
2.20.1. DNA isolation ............................................................................................... 74
2.20.2. 16S qPCR ..................................................................................................... 74
2.21. RNA sequencing .................................................................................................. 75
2.21.1. Analysis ........................................................................................................ 76
2.22. Colony-forming assay .......................................................................................... 76

4
2.23. Experimental considerations ................................................................................ 77
2.23.1. Group allocation ........................................................................................... 77
2.23.2. Sample size ................................................................................................... 77
2.23.3. Blinding ........................................................................................................ 77
2.23.4. Exclusion criteria .......................................................................................... 78
2.23.5. Data and statistical analyses ......................................................................... 78
Chapter 3. Immune alterations during experimental periodontitis .......................... 79
3.1. Introduction ............................................................................................................ 80
3.2. Aims ....................................................................................................................... 83
3.3. Results .................................................................................................................... 84
3.3.1. Ligature-induced periodontitis induces inflammatory bone loss, bacterial
outgrowth, and local immune alterations .................................................................. 84
3.3.2. Ligature-induced periodontitis does not lead to increased bacterial load or
altered immunity in the lung ..................................................................................... 91
3.3.3. Ligature-induced periodontitis increases circulating levels of pro-
inflammatory cytokines............................................................................................. 95
3.3.4. Ligature-induced periodontitis differentially modulates the frequency and
functionality of myeloid cells at sites distal from the oral cavity ............................. 98
3.3.5. Ligature-induced periodontitis does not induce vascular or CNS
inflammation ........................................................................................................... 104
3.4. Discussion ............................................................................................................ 107
3.4.1. Ligature-induced periodontitis sufficiently models human disease pathology
in the oral cavity ...................................................................................................... 107
3.4.2. The lung and the bloodstream as gateways to systemic amplification during
periodontitis ............................................................................................................ 108
3.4.3. Periodontitis induces immune alterations at discrete sites in a cell-specific
and tissue-specific manner ...................................................................................... 110
3.4.4. Ligature-induced periodontitis does not induce inflammation of the
vasculature or the brain ........................................................................................... 112
3.4.5. Conclusion .................................................................................................. 113
Chapter 4. The impact of periodontitis on acute outcome after stroke .................. 115
4.1. Introduction .......................................................................................................... 116
4.2. Aims ..................................................................................................................... 120
4.3. Results .................................................................................................................. 121
4.3.1. Ligature-induced periodontitis does not alter acute outcome after transient
MCAo.. ................................................................................................................... 121

5
4.3.2. Systemic challenge of P. gingivalis LPS causes robust inflammatory
responses ................................................................................................................. 128
4.3.3. Ligature-induced periodontitis does not alter acute outcome after permanent
MCAo.. ................................................................................................................... 131
4.4. Discussion ............................................................................................................ 138
4.4.1. Ligature-induced periodontitis causes systemic inflammatory alterations but
does not worsen outcome after stroke ..................................................................... 139
4.4.2. The validity of experimental stroke models ............................................... 141
4.4.3. The validity of experimental periodontitis models ..................................... 142
4.4.4. Conclusion .................................................................................................. 144
Chapter 5. Regulation of the gingival haematopoietic network .............................. 146
5.1. Introduction .......................................................................................................... 147
5.2. Study rationale ..................................................................................................... 152
5.3. Aims ..................................................................................................................... 156
5.4. Results .................................................................................................................. 156
5.4.1. Identification of a haematopoietic niche in the mouse gingiva during steady-
state ..... ................................................................................................................... 156
5.4.2. Ligature-induced periodontitis alters the number and output of gingival
HSPCs.. ................................................................................................................... 158
5.4.3. Ageing leads to a decline in the number of gingival HSPCs ...................... 164
5.4.4. The gingiva undergoes remodelling with age............................................. 171
5.5. Discussion ............................................................................................................ 175
5.5.1. A haematopoietic niche in the gingiva ....................................................... 175
5.5.2. Inflammatory regulation of the gingival haematopoietic niche .................. 176
5.5.3. Ageing and the gingival haematopoietic niche........................................... 178
5.5.4. Factors regulating gingival haematopoietic progenitors ............................ 180
5.5.5. Conclusion .................................................................................................. 182
Chapter 6. General discussion .................................................................................... 183
6.1. Overview .............................................................................................................. 184
6.2. Periodontitis drives myeloid-biased immune alterations ..................................... 185
6.2.1. Haematopoiesis .......................................................................................... 185
6.2.2. Neutrophils ................................................................................................. 186
6.2.3. Monocytes .................................................................................................. 187
6.2.4. Lymphocytes .............................................................................................. 188
6.3. High-grade and low-grade inflammation ............................................................. 189
6.3.1. Comparing periodontitis to infection .......................................................... 189

6
6.3.2. Comparing periodontitis to obesity ............................................................ 190
6.3.3. The impact of specific periodontal pathogens ............................................ 191
6.4. Challenges and future directions .......................................................................... 192
6.5. Concluding remarks ............................................................................................. 194
Bibliography .................................................................................................................. 196
Appendix ... .................................................................................................................... 226

7
List of figures
Chapter 1
Figure 1.1. The pathogenesis of periodontal disease ........................................................ 22
Figure 1.2. Associations and proposed mechanisms between periodontitis and systemic
diseases. ............................................................................................................................ 37
Chapter 2
Figure 2.1. Ligature-induced periodontitis. ...................................................................... 55
Figure 2.2. Transient middle cerebral artery occlusion via intraluminal filament ............ 57
Figure 2.3. Permanent middle cerebral artery occlusion via ferric chloride ..................... 59
Chapter 3
Figure 3.1. Ligature-induced periodontitis causes robust bone loss and an increase in
gingival cellularity within 10 days. ................................................................................... 85
Figure 3.2. Ligature-induced periodontitis induces profound bacterial growth in the oral
cavity within 10 days. ....................................................................................................... 87
Figure 3.3. Gating strategies to identify myeloid and lymphoid cells using flow
cytometry. ......................................................................................................................... 88
Figure 3.4. Ligature-induced periodontitis induces an increase in neutrophils in the
draining sub-mandibular lymph nodes. ............................................................................. 90
Figure 3.5. Ligature-induced periodontitis does not affect lymphoid populations in the
draining sub-mandibular lymph nodes. ............................................................................. 91
Figure 3.6. Ligature-induced periodontitis does not induce robust bacterial growth in the
lungs within 10 days. ........................................................................................................ 93
Figure 3.7. Ligature-induced periodontitis does not affect inflammatory mediators or
myeloid populations in the lung. ....................................................................................... 94
Figure 3.8. Ligature-induced periodontitis does not modulate lymphoid populations in the
lung. .................................................................................................................................. 95
Figure 3.9. Ligature-induced periodontitis increases circulating levels of pro-
inflammatory cytokines without affecting frequencies of myeloid cells. ......................... 97
Figure 3.10. Splenic immune cells are not altered by ligature-induced periodontitis. ...... 99
Figure 3.11. Frequencies of neutrophils in the small intestine are increased post-ligature
placement. ....................................................................................................................... 100
Figure 3.12. Neutrophils and Ly6Chi monocytes in the bone marrow are differentially
modulated during ligature-induced periodontitis. ........................................................... 102
Figure 3.13. Ligature-induced periodontitis increases TNF production by bone marrow
monocytes. ...................................................................................................................... 103
Figure 3.14. Ligature-induced periodontitis does not induce vascular or neuro-
inflammation. .................................................................................................................. 105
Figure 3.15. The spectrum of immune alterations in various tissue sites during ligature-
induced periodontitis. ...................................................................................................... 106

8
Chapter 4
Figure 4.1. Ligature-induced periodontitis does not alter ischaemic damage, neurological
impairment, blood-brain barrier breakdown, or neutrophil infiltration after transient
middle cerebral artery occlusion. .................................................................................... 122
Figure 4.2. Ligature-induced periodontitis does not change the levels of circulating
inflammatory mediators after transient middle cerebral artery occlusion. ...................... 124
Figure 4.3. Ligature-induced periodontitis does not alter the frequency or phenotype of
monocytes and neutrophils in the spleen after transient middle cerebral artery occlusion.
........................................................................................................................................ 125
Figure 4.4. Ligature-induced periodontitis does not alter the balance of T cell subsets in
the bone marrow or spleen after transient middle cerebral artery occlusion. ................. 126
Figure 4.5. Ligature-induced periodontitis does not alter the frequency or phenotype of
monocytes and neutrophils in the bone marrow after transient middle cerebral artery
occlusion. ........................................................................................................................ 127
Figure 4.6. Ligature-induced periodontitis does not alter the functionality of bone marrow
monocytes after transient middle cerebral artery occlusion. ........................................... 128
Figure 4.7. Intravenous challenge with Porphyromonas gingivalis LPS induces robust
systemic inflammatory responses 2 hours after administration. ..................................... 130
Figure 4.8. Periodontitis and systemic P. gingivalis LPS not alter ischaemic damage,
blood-brain barrier breakdown, or neutrophil infiltration after permanent middle cerebral
artery occlusion. .............................................................................................................. 132
Figure 4.9. Gating strategies to identify myeloid and lymphoid cells after permanent
middle cerebral artery occlusion using flow cytometry. ................................................. 133
Figure 4.10. Periodontitis and systemic P. gingivalis LPS do not significantly affect
circulating myeloid cells after permanent middle cerebral artery occlusion. ................. 134
Figure 4.11. Periodontitis and systemic P. gingivalis LPS do not significantly affect
circulating lymphocytes after permanent middle cerebral artery occlusion. .................. 135
Figure 4.12. Periodontitis and systemic P. gingivalis LPS do not significantly affect
splenic myeloid cells after permanent middle cerebral artery occlusion. ....................... 136
Figure 4.13. Periodontitis and systemic P. gingivalis LPS do not significantly affect
splenic lymphocytes after permanent middle cerebral artery occlusion. ........................ 137
Figure 4.14. Periodontitis and systemic P. gingivalis LPS do not significantly affect
myeloid cells in the bone marrow after permanent middle cerebral artery occlusion. ... 138
Chapter 5
Figure 5.1. The haematopoietic tree................................................................................ 150
Figure 5.2. The mouse gingiva contains a unique population of resident monocytes. ... 155
Figure 5.3. The mouse gingiva contains populations of multipotent and oligopotent
haematopoietic stem and progenitor cells during steady-state. ....................................... 157
Figure 5.4. Gingival Ly6Chi monocytes increase in number during ligature-induced
periodontitis. ................................................................................................................... 159
Figure 5.5. Gingival LSKs expand in number during ligature-induced periodontitis. .... 160
Figure 5.6. Discrete populations of multipotent gingival progenitors increase in number
during ligature-induced periodontitis. ............................................................................. 161

9
Figure 5.7. Ligature-induced periodontitis increases the differentiation output of gingival
haematopoietic stem and progenitor cells. ...................................................................... 163
Figure 5.8. Gingival Ly6Chi monocytes are reduced with age. ....................................... 164
Figure 5.9. Gingival and bone marrow haematopoietic stem and progenitor cells are
reduced in aged mice. ..................................................................................................... 166
Figure 5.10. The proliferative capacity of bone marrow, but not gingival, haematopoietic
stem and progenitor cells is reduced in aged mice. ......................................................... 167
Figure 5.11. Lymphoid-biased multipotent progenitors in the gingiva and bone marrow
are reduced in aged mice. ................................................................................................ 168
Figure 5.12. Ageing does not affect the differentiation output of gingival haematopoietic
stem cells. ........................................................................................................................ 169
Figure 5.13. Ageing does not affect the ability of gingival LSKs and MyPs to produce
myeloid cell colonies. ..................................................................................................... 171
Figure 5.14. Haematopoiesis-supporting factors are downregulated in the gingiva of aged
mice. ................................................................................................................................ 173
Figure 5.15. Genes relating to the physical integrity of the gingiva decrease with age. . 174
List of tables
Chapter 2
Table 2.1. Sequences for genes of interest ........................................................................ 73
Appendix
Table A.1. Summary of reagents, chemicals & consumables used for experiments. ..... 226
Table A.2. Details of mouse antibodies used for flow cytometry. .................................. 229
Table A.3. Assessment of neurological impairment (28 points) ..................................... 231
Final word count: 65,423

10
Abbreviations
ABC Alveolar bone crest
ACK Ammonium-chloride-potassium
AD Alzheimer’s disease
ANGPTL Angiopoietin-like protein
ANOVA Analysis of variance
ATP Adenosine triphosphate
BBB Blood-brain barrier
BM Bone marrow
CCA Common carotid artery
CCL2 Chemokine (C-C motif) ligand 2
CCR2 C-C chemokine receptor type 2
CD Cluster of differentiation
cDNA Complementary deoxyribonucleic acid
CEJ Cemento-enamel junction
CFU Colony forming unit
cfu-GM Colony forming units-granulocyte macrophage
CLP Common lymphoid progenitor
cMoP Common monocyte progenitor
CMP Common myeloid progenitor
CNS Central nervous system
CO2 Carbon dioxide
CRP C-reactive protein
CVD Cardiovascular disease
CX3CR1 C-X3-C chemokine receptor type 1
CXCL1 Chemokine (C-X-C motif) ligand 1
DAB 3,3'-diaminobenzidine
DC Dendritic cell
DEG Differentially-expressed genes
DMEM/F-12 Dulbecco's modified eagle medium/nutrient mixture F-12
DNA Deoxyribonucleic acid
DTT Dithiothreitol
Ec-LPS Lipopolysaccharide from Escherichia coli
ECA External carotid artery
ECM Extracellular matrix
EDTA Ethylenediaminetetraacetic acid
EMH Extramedullary haematopoiesis
FACS Fluorescence-activated cell sorting
FDR False discovery rate
FeCl Ferric chloride
FoxP3 Forkhead box P3
GALT Gut-associated lymphoid tissue
G-CSF Granulocyte-colony stimulating factor
GI Gastrointestinal
GM-CSF Granulocyte macrophage-colony stimulating factor
GMP Granulocyte-monocyte progenitor
GO Gene ontology
H2O2 Hydrogen peroxide
HBSS Hank’s balanced salt solution
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HSC Haematopoietic stem cell

11
HSP Heat-shock protein
HSPC Haematopoietic stem and progenitor cell
IBD Inflammatory bowel disease
ICA Internal carotid artery
ICAM-1 Intercellular adhesion molecule-1
IFN Interferon
IGF Insulin-like growth factor
IgG Immunoglobulin G
IL Interleukin
ILC Innate lymphoid cell
IMS Industrial methylated spirit
iNOS Inducible nitric oxide synthase
i.p. Intraperitoneal
i.v. Intravenous
LPS Lipopolysaccharide
LSK Lineage-Sca-1-c-Kit+
LT-HSC Long-term haematopoietic stem cell
Ly6C Lymphocyte antigen 6 complex, locus C1
M-CSF Macrophage-colony stimulating factor
MCA Middle cerebral artery
MCAo Middle cerebral artery occlusion
MDP Monocyte-macrophage-dendritic cell progenitor
MEM Minimal essential medium
MEP Megakaryocyte-erythroid progenitor
MHCII Major histocompatibility complex II
MMP Matrix metalloproteinase
MPP Multipotent progenitor
mRNA Messenger ribonucleic acid
MyP Myeloid progenitor
NK cell Natural killer cell
padj. Adjusted p value
PBS Phosphate-buffered saline
PCR Polymerase chain reaction
PD Periodontitis
PFA Paraformaldehyde
Pg-LPS Lipopolysaccharide from Porphyromonas gingivalis
pMCAo Permanent middle cerebral artery occlusion
PTGS2 Prostaglandin synthase 2
qRT-PCR Quantitative reverse trancription polymerase chain reaction
RA Rheumatoid arthritis
RANKL Receptor activator of nuclear factor kappa-B ligand
rDNA Ribosomal deoxyribonucleic acid
RNA Ribonucleic acid
RORγt Retinoid-related orphan receptor gamma t
RPMI Roswell Park Memorial Institute
Sca-1 Stem cell antigen-1
SEM Standard error of the mean
smLN Sub-mandibular lymph node
SNS Sympathetic nervous system
ST-HSC Short-term haematopoietic stem cell
TGF Transforming growth factor
Th17 T helper-17 cell
TIGIT T-cell immunoreceptor with Ig and ITIM domains

12
TLR Toll-like receptor
TCR T cell receptor
tMCAo Transient middle cerebral artery occlusion
TNFα Tumour necrosis factor-alpha
Treg Regulatory T cell
VEGF Vascular endothelial growth factor
VCAM-1 Vascular cell adhesion molecule-1
wt Wild-type

13
Abstract
Periodontitis is a prevalent chronic inflammatory disease that involves the destruction of
the supporting structures of the teeth. In addition to local tissue damage and bone loss,
periodontitis has been shown to have adverse consequences at sites distant from the oral
cavity. Growing epidemiological and experimental data has associated periodontitis with
the development and/or exacerbation of a myriad of clinically-important diseases, from
rheumatoid arthritis to Alzheimer’s disease to ischaemic stroke. The objective of this
thesis was to provide an insight into the local and systemic immune responses during
experimental periodontitis which could potentially affect peripheral tissue sites, with a
particular focus on the impact on stroke severity.
Using an acute bilateral ligature model in mice, we found that experimental periodontitis
led to bone loss, bacterial growth, and increased local inflammatory cell mobilisation.
Systemically, periodontitis altered the frequencies of monocytes and neutrophils in the
bone marrow and small intestine, increased circulating levels of the pro-inflammatory
cytokines, interleukin (IL)-1β and IL-17A, and increased tumour necrosis factor (TNF)α
production in bone marrow monocytes.
In order to evaluate the impact of periodontitis on stroke outcome, we applied this
ligature-induced model and induced experimental strokes by transient or permanent
occlusion of the middle cerebral artery. In tandem with ligature placement, we
systemically challenged with an oral-specific lipopolysaccharide (LPS) in an effort to
imitate the systemic aspects of the clinical disease. However, periodontitis alone or in
tandem with LPS, did not alter systemic immune trafficking, blood-brain barrier
disruption, or brain damage after stroke.
In addition to the systemic reaches of periodontitis, we also focused on local immune
regulation during disease. In this way, we identified the gingiva as a novel site of
extramedullary haematopoiesis that harbours a population of haematopoietic stem and
progenitor cells in the tissue which can give rise to multiple lineages of myeloid immune
cells, including tissue monocytes. We also describe that these stem cells are differentially
modulated by induced or natural bone loss and provide evidence that stromal cells and the
surrounding matrix may be important in retaining these progenitors in the gingival niche.
Overall, these findings give insight into the fundamental immunological mechanisms
during periodontitis, both in a local context, within the oral cavity, as well as the
implications at distant tissues sites. We specifically provide evidence that periodontitis
does not alter outcome after acute ischaemic stroke, and thus add an important
counterpoint to the growing body of literature associating periodontitis with a negative
impact on stroke.

14
Declaration
No portion of the work referred to in this thesis has been submitted in support of an
application for another degree or qualification of this or any other university or other
institute of learning.
Conor O’Boyle
28/03/2019
Copyright statement
1. The author of this thesis (including any appendices and/or schedules to this thesis)
owns certain copyright or related rights in it (the “Copyright”) and s/he has given The
University of Manchester certain rights to use such Copyright, including for
administrative purposes.
2. Copies of this thesis, either in full or in extracts and whether in hard or electronic
copy, may be made only in accordance with the Copyright, Designs and Patents Act
1988 (as amended) and regulations issued under it or, where appropriate, in
accordance with licensing agreements which the University has from time to time.
This page must form part of any such copies made.
3. The ownership of certain Copyright, patents, designs, trademarks and other
intellectual property (the “Intellectual Property”) and any reproductions of copyright
works in the thesis, for example graphs and tables (“Reproductions”), which may be
described in this thesis, may not be owned by the author and may be owned by third
parties. Such Intellectual Property and Reproductions cannot and must not be made
available for use without the prior written permission of the owner(s) of the relevant
Intellectual Property and/or Reproductions. Further information on the conditions
under which disclosure, publication and commercialisation of this thesis, the
Copyright and any Intellectual Property and/or Reproductions described in it may
take place is available in the University IP Policy (see
http://documents.manchester.ac.uk/DocuInfo.aspx?DocID=24420), in any relevant
Thesis restriction declarations deposited in the University Library, The University
Library’s regulations (see http://www.library.manchester.ac.uk/about/regulations/)
and in The University’s policy on Presentation of Theses.

15
Experimental contributions
All experiments were designed, performed and analysed by myself with the input of my
supervisors, Dr Catherine Lawrence, Dr Joanne Konkel, Prof Stuart Allan, and Prof Craig
Smith. Dr Michael Haley performed the surgeries, neurological scoring, and assisted with
tissue collection in regard to transient strokes (tMCAo). Dr Eloise Lemarchand provided
assistance by teaching permanent stroke surgeries (pMCAo). Dr Siddharth Krishnan and
Hayley Bridgeman assisted with tissue processing for flow cytometry. Dr Siddharth
Krishnan and Dr Joanne Konkel kindly provided the figure for Chapter 5, Figure 5.2., in
which Kelly Wemyss supplied the skin data. Dr Joanne Konkel prepared the gingiva
samples for RNA sequencing, Dr Leo Zeef and Dr Andy Hayes performed the sequencing
and DESeq analysis. Dr Ian Prise performed the gene-set enrichment analysis.

16
Acknowledgements
First and foremost, I would like to thank my supervisors, Cath, Jo, Stuart, and Craig, for
giving me the opportunity to do a PhD and for being great mentors. Cath, thank you for
the constant support, wisdom, and encouragement throughout the last 3.5 years, and for
picking out all the double spaces in my writing, I really couldn’t have asked for a better
supervisor. Jo, thanks for your immunological help, your unwavering work ethic and
scientific knowledge is both terrifying and impressive in equal measure. Stuart, for being
the living embodiment of work hard, play harder, and Craig, for the clinical insight and
the enviable skill of sniffing out the alcohol content of a syrupy beer.
I would also like to thank the BIG characters that have made huge contributions to both
science and life along the way (and tolerating the really terrible jokes), namely Sid,
Siobhan, Matt, Jack B, Pat, Tess, Hannah, and Victor. Thanks for the memorable
conferences in Berlin and Dresden, the brew room chats, and the countless nights in Big
Hands. In particular, I’d like to thank Sid for all his science help and for being a decent
friend by putting up with my nonsense.
I’d also like to thank the rest of the Brain Inflame gang as well as the Grainger lab
members for making it such an enjoyable and productive place to work. In particular,
thank you to Mike H, Eloise, and Jack RA for the surgical and statistical expertise, and
John, Hayley, and Tovah, for the scientific input and hands-on assistance. I would also
like to acknowledge the Flow Cytometry, Genomic Technologies, and Bioimaging
facilities, the Biological Services Facility, as well as the Medical Research Council and
The University of Manchester for funding my research.
Last, but by no means least, Claire, I cannot thank you enough for all you’ve done. A
constant source of encouragement, support, and perspective, thank you for being beside
me to celebrate the highs and to drag me through the lows. You are the silliest and most
wonderful person I’ve ever met.

17
Chapter 1. Introduction

18
1.1. Overview
Periodontitis is an extremely prevalent chronic inflammatory disease of the teeth’s
supporting structures, affecting almost 50% of the global population (Eke et al., 2012). In
addition to localised inflammatory damage and destruction of the underlying bone
anchoring the tooth, periodontitis is also an emerging risk factor for a range of clinically-
important systemic diseases, including Alzheimer’s disease (Ide et al., 2016), rheumatoid
arthritis (Bartold et al., 2005), cancer (Michaud et al., 2018), cardiovascular disease
(Beck and Offenbacher, 2005), and stroke (Grau et al., 2004). In particular, periodontitis
has been frequently associated with increased risk of stroke (Elter et al., 2003; Dorfer et
al., 2004; Grau et al., 2004; Sen et al., 2018), but causal evidence is lacking. Periodontitis
and stroke share a number of risk factors, including old age, male sex, obesity, and
smoking (Allen and Bayraktutan, 2008; Eke et al., 2012). Ischaemic stroke is the second
leading cause of death and the leading cause of disability in the developed world (Feigin
et al., 2010, 2014), but despite a wealth of knowledge about risk factors, co-morbidities,
and the complex immunopathological mechanisms of the disease, current therapeutic
options are severely lacking. Thus, recent efforts have aimed to mitigate the incidence of
stroke, particularly by reducing the impact of common stroke co-morbidities, such as
infection and obesity, in order to reduce the risk of stroke while also improving prognosis
after stroke. Periodontitis is proposed to increase the systemic inflammatory burden
(Tonetti, 2009; de Oliveira et al., 2010), which is known to affect stroke outcome
(McColl et al., 2009). Despite the prevalence of periodontitis, however, there is a lack of
definitive causal evidence tying periodontitis with stroke. Considering the morbidity and
mortality associated with stroke, the impact of periodontitis on stroke risk and outcome
warrants further investigation. Critically, periodontitis may yet be a significant risk factor
for stroke, but it is a modifiable one, as periodontitis is both preventable and treatable
(Kinane et al., 2017).

19
1.2. The importance of the oral barrier
Barrier sites, such as the oral cavity, lung, gut, and skin, are critical interfaces between
the body and external environment. As well as acting as a physical barrier to microbial
invasion and environmental insults, these barrier sites must maintain a tolerance to
harmless commensals while also enforcing immunity to potentially pathogenic microflora
(Moutsopoulos and Konkel, 2018). Thus, immune responses at these sites are
appropriately tuned to the unique requirements of the tissue, driven by tissue-specific and
environmental cues. Although the regulation of immune responses in the gut and skin
have received much attention (Ivanov et al., 2009; Naik et al., 2012; Mortha et al., 2014;
Linehan et al., 2018), by contrast, our understanding of the oral cavity is far more limited.
The oral cavity is unique as a barrier site as it not only harbours a rich and diverse
microbiome, but is the point at which dietary antigens are encountered prior to
gastrointestinal (GI) tract entry (Moutsopoulos and Konkel, 2018) and must also reckon
with regular mechanical damage from ongoing mastication (Dutzan et al., 2017).
Inappropriate or dysregulated responses at barrier sites can result in the development of
disease, and in the context of the oral cavity, periodontitis is a condition that can represent
a deleterious threat to not only the local environment, but also systemic health.
1.2.1. Immunity at the oral barrier
As mentioned, the oral barrier is constantly faced with a number of diverse signals, and
thus even during “steady-state”, it is a site of routine stimulation. In particular, the tooth-
adjacent tissue, the gingiva, is particularly open and vulnerable, as it is exposed to a
diverse biofilm and mechanical trauma from chewing and brushing, and is therefore a site
of constant immune activation (Moutsopoulos and Konkel, 2018). As such, human
gingival tissue is home to a rich immunological network, which comprises of a majority
of recruited neutrophils and resident T cells, minimal B cells, as well as mononuclear

20
phagocytes (monocytes, macrophages and dendritic cells [DCs]), and smaller populations
of mast cells and innate lymphoid cells (ILCs) (Dutzan et al., 2016b). This immune
network is similar in mice, with mucosal sentinels such as Langerhans cells, γδ T cells,
and ILCs, enriched in the gingiva and in the sub-mandibular lymph nodes, which drain
the oral cavity (Capucha et al., 2015; Brown et al., 2018; Krishnan et al., 2018; Wilharm
et al., 2019). Many of these populations exhibit cell-specific variation in ontogeny. For
example, in mice, resident T helper (Th)-17 cells undergo local proliferation (Dutzan et
al., 2017), while Langerhans cells and some γδ T cell populations are seeded after birth
and can be replenished during adulthood from circulating precursors (Capucha et al.,
2015; Krishnan et al., 2018). As a result of this immune diversity, the homeostatic
function of these leukocyte populations is also suitably diverse, and can be either
dependent on (Wilharm et al., 2019), or independent of (Dutzan et al., 2017; Krishnan et
al., 2018), the presence of the microbiota. In particular, gingival-resident Th17 cell
function is tailored by mastication-driven signals. Specifically, regular mechanical
damage from chewing induces interleukin (IL)-6 by epithelial cells which drives
homeostatic Th17 cell expansion and defence (Dutzan et al., 2017). Furthermore, specific
subsets of γδ T cells limit gingival inflammation and bone loss through the production of
IL-17 and amphiregulin respectively (Krishnan et al., 2018; Wilharm et al., 2019).
Neutrophils also function in homeostatic immune surveillance at the space between the
tooth and the gingival epithelium, termed the gingival crevice, as deficiency of tissue-
infiltrating neutrophils leads to immunopathology and bone loss (Eskan et al., 2012;
Moutsopoulos et al., 2014, 2017; Shin et al., 2015). Similarly, T regulatory cells (Tregs)
(Glowacki et al., 2013) and Langerhans cells (Arizon et al., 2012) have crucial roles in
maintaining immune homeostasis. Even though the roles of many gingival leukocyte
populations have been well-characterised, the contributions of others, such as monocytes
and macrophages, to maintenance of oral immune homeostasis have not been elucidated
and require future investigation.

21
1.3. Periodontal disease
Periodontal disease is a dysbiotic inflammatory condition of the hard and soft tissues
supporting the teeth, collectively termed the periodontium. The periodontium comprises
of the superficial epithelium, the gingiva, and deeper sub-gingival structures, including
periodontal ligaments, connective tissue, and alveolar bone (Kinane et al., 2017). Strictly
speaking, periodontal disease is divided into gingivitis and periodontitis. Gingivitis is the
initial phase, where build-up of microbial plaque is met with an acute inflammatory
response, but one that is confined to the gingival tissue (Kinane, 2001). Gingivitis is
reversible with treatment, but if left untreated, can progress to a more severe form,
periodontitis, which is characterised by irreversible destruction of the underlying bone
(Kinane et al., 2017). Gingivitis must precede periodontitis, but not all gingivitis
progresses to periodontitis. Periodontitis is classified based on the severity, extent and
distribution (localised, generalised etc), and rate of progression (Caton et al., 2018).
Periodontitis involves inflammation that extends into the sub-gingival tissues, leading to
loss of contact between tooth and its adjacent supports, and the formation of a deep
“periodontal pocket” that harbours proliferating microbes (Figure 1.1). Persistent and
overwhelming bacterial growth in this pocket leads to breach of the epithelium and
microbial tissue entry, thereby increasing the magnitude of the inflammatory response
and furthering tissue damage. Failure of host immunity to control the microbial threat
leads to chronic inflammatory degradation of tissue and bone, leading to increased tooth
mobility and eventual loss.
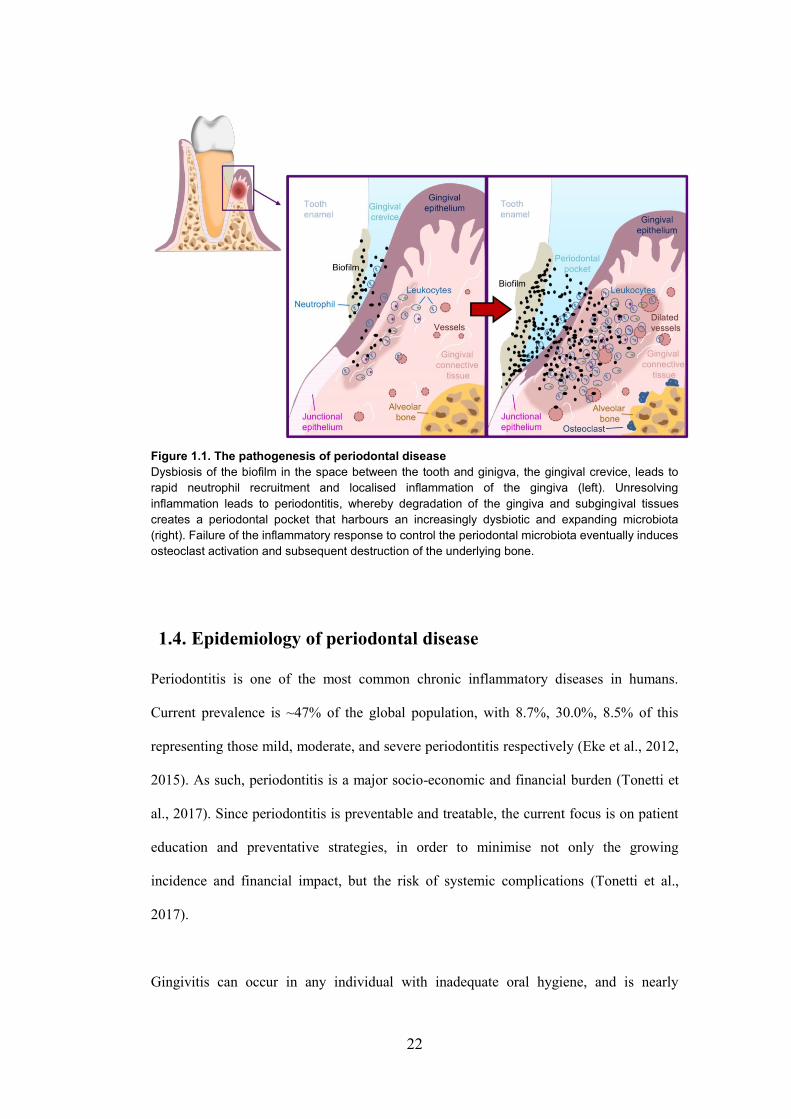
22
Figure 1.1. The pathogenesis of periodontal disease
Dysbiosis of the biofilm in the space between the tooth and ginigva, the gingival crevice, leads to
rapid neutrophil recruitment and localised inflammation of the gingiva (left). Unresolving
inflammation leads to periodontitis, whereby degradation of the gingiva and subgingival tissues
creates a periodontal pocket that harbours an increasingly dysbiotic and expanding microbiota
(right). Failure of the inflammatory response to control the periodontal microbiota eventually induces
osteoclast activation and subsequent destruction of the underlying bone.
1.4. Epidemiology of periodontal disease
Periodontitis is one of the most common chronic inflammatory diseases in humans.
Current prevalence is ~47% of the global population, with 8.7%, 30.0%, 8.5% of this
representing those mild, moderate, and severe periodontitis respectively (Eke et al., 2012,
2015). As such, periodontitis is a major socio-economic and financial burden (Tonetti et
al., 2017). Since periodontitis is preventable and treatable, the current focus is on patient
education and preventative strategies, in order to minimise not only the growing
incidence and financial impact, but the risk of systemic complications (Tonetti et al.,
2017).
Gingivitis can occur in any individual with inadequate oral hygiene, and is nearly

23
pandemic in children and young adults (Craig et al., 2003; Petersen and Ogawa, 2005).
The progression to periodontitis, however, is considerably more complex, as overt disease
only occurs in susceptible individuals (Stabholz et al., 2010). A number of important
factors have been established that contribute to periodontitis risk, some of which are
modifiable. Age, race, and sex are critical non-modifiable risk factors, as chronic
periodontitis is more prevalent amongst Hispanics, African-Americans, males, and in
elderly individuals (aged > 65 years) of either sex (Eke et al., 2012, 2015). Furthermore,
environmental and social factors can increase risk; including stress (Stabholz et al., 2010),
excessive alcohol consumption (Petersen and Ogawa, 2005), cigarette smoking (Eke et
al., 2012; Kumar, 2012; Nociti et al., 2015), low socio-economic status, including lack of
education (Eke et al., 2012), as well as metabolic conditions, such as obesity and diabetes
(Lalla and Papapanou, 2011; Casanova et al., 2014).
Immunoregulatory defects can increase periodontitis incidence, such as in patients with
leukocyte adhesion deficiency type I (LAD-1), who develop severe periodontitis due to a
lack of recruited neutrophils in the tissue and unrestrained IL-17 production
(Moutsopoulos et al., 2014). Similarly, polymorphisms in pro-inflammatory genes that
predispose to a hyper-inflammatory state, such as tumour necrosis factor (TNF)α, IL-1β,
IL-6, IL-10, CD14, and toll-like receptor (TLR)4, can account for an increased incidence
as well as increased severity of chronic and aggressive periodontitis (Stabholz et al.,
2010; Laine et al., 2012). As genome-wide association studies have failed to consistently
identify susceptibility genes (Divaris et al., 2013; Shaffer et al., 2014), the overall
susceptibility to periodontitis is likely a product of multiple genetic polymorphisms, in
addition to social and environmental risk factors.

24
1.5. Diagnosis and treatment of periodontal disease
While gingivitis can be self-diagnosed by bleeding upon brushing (Petersen and Ogawa,
2005), periodontitis is typically diagnosed by an array of clinical measurements; bleeding
upon probing, increased probing depth (of the periodontal pocket, typically ≥ 4 mm),
clinical attachment loss (i.e. loss of tooth contact), and radiographic findings to confirm
loss of bone (Kinane et al., 2017). Early diagnosis and treatment results in an excellent
prognosis, but delays can cause irreversible damage to the bone and tissue, and since
early periodontitis is painless, patients rarely seek early care (Kinane et al., 2017).
Multiple treatment approaches exist, most concerning the physical removal of the
aetiological factors ‒ the microbial biofilm on the teeth and gingival surfaces ‒ and
removal or reduction of risk factors. As such, therapy is often reliant on a combination of
patient self-care, scaling (physical removal of plaque), root planing (deep cleaning under
local anaesthesia), and sometimes topical or oral antibiotics (Kinane et al., 2017; Slots,
2017). Successful management of periodontitis is often the product of patient compliance
and diligence in their post-treatment care. Though most approaches are non-surgical, in
cases of advanced periodontitis, dental implants or surgical intervention may be required,
such as soft tissue grafts or pocket reduction surgery. As treatment may not always lead
to swift resolution in cases of severe periodontitis, emerging anti-inflammatory and
immunotherapies may be useful as adjunct treatment options. Most of these aim to
mitigate the impact of the pro-inflammatory response, and lend credence to the proposal
that dysregulated inflammatory responses precede microbial dysbiosis (Bartold and Van
Dyke, 2017), creating an environment for opportunistic microbes to thrive, which in turn
leads to periodontitis. As such, limiting recruitment of hyper-inflammatory neutrophils to
the periodontal tissues by antagonising lymphocyte function-associated antigen (LFA-1)-
mediated extravasation (Eskan et al., 2012; Shin et al., 2015), or limiting the effects of
pro-inflammatory mediators produced in situ, such as prostaglandin E2 or IL-17 (Bartold

25
and Van Dyke, 2017; Moutsopoulos et al., 2017), may offer benefit to individuals with
periodontitis.
1.6. The pathogenesis of periodontitis
Periodontitis is a complex disease which is initiated in the presence of pathogenic
microbes, progresses in the presence of the entire dysbiotic community, and persists by
disproportionate host immune responses and chronic damage of soft and hard tissues.
1.6.1. Microbial dysbiosis
The human oral cavity is home to a rich and diverse microbial community with as many
as 800 taxa identified, the majority of which are aerobic and Gram-positive in health
(Lourenço et al., 2014). Within the mouth, the composition of the microbiome differs
significantly at distinct surfaces, as palatal (tongue), buccal (cheek), supra-gingival, and
sub-gingival communities all vary in the relative abundance of certain taxa (Marsh et al.,
2011). In regard to periodontitis, an ecological disturbance in sub-gingival communities is
an important initiating factor, leading to a shift in community structure which favours the
growth of anaerobic opportunistic and virulent species (Socransky and Haffajee, 2005;
Moutsopoulos and Madianos, 2006). It was traditionally thought that putative pathogenic
species, such as Porphyromonas gingivalis, Tannerella forsythia, and Treponema
denticola, were causative disease agents, due to their virulent properties and strong
association with diseased sites (Socransky and Haffajee, 2005). However, it has become
clear that periodontitis is not a typical bacterial infection, but rather a disturbance in
microbial homeostasis, insofar as the entire community contributes to the development of
disease (Hajishengallis, 2014a; Kinane et al., 2017). As such, sub-gingival microbial
communities differ markedly in health and periodontitis, with higher diversity and
biomass in periodontitis (Griffen et al., 2012; Abusleme et al., 2013). However, health-

26
associated phyla, such as Actinobacteria, are also present in periodontitis, and conversely,
most disease-associated taxa, such as Firmicutes, Spirochaetes, Synergistetes, and
Bacteroidetes, are also present in health, albeit in low proportions (Abusleme et al.,
2013). Thus, periodontitis leads to a shift in community structure and not membership,
with the emergence of newly dominant taxa without replacement of health-associated
species (Griffen et al., 2012; Abusleme et al., 2013).
Even though the sub-gingival microbiome as a whole is involved in periodontitis, as
mentioned there are a number of virulent periodontal microbes that remodel the
commensal community into a dysbiotic disease-provoking microbiota. These so-called
“periopathogens” include P. gingivalis, Aggregatibacter actinomycetemcomitans, T.
forsythia, T. denticola, and Fusobacterium nucleatum, all of which are routinely detected
in most forms of periodontitis patients (Hernández et al., 2011; Albandar, 2014).
Periopathogens possess a number of virulent characteristics enabling them to thrive in an
inflammatory environment; including invading host cells (Amar et al., 2009) and
subverting immune responses (which will be discussed in later sections) (Hajishengallis,
2015). In particular, the Gram-negative anaerobe, P. gingivalis, has been the subject of
intense scrutiny, given its ability to promote pathology at local and systemic sites. This is
important since P. gingivalis and other periodontal pathogens are often present in the sub-
gingival biofilm at low abundance during disease; P. gingivalis constitutes less than
0.01% of the periodontitis-associated microbiota (Hajishengallis et al., 2011). It also
helps to stress the role of the entire microbial community in disease aetiology and
pathology. In this way, periopathogens facilitate the survival of the entire dysbiotic
community by preventing immune-mediated killing and by orchestrating inflammatory
tissue damage, as tissue products serve as microbial nutrient sources (such as degraded
collagen and haem-rich compounds) (Hajishengallis, 2014a). Indeed, polymicrobial
infections in mice with P. gingivalis and either F. nucleatum or T. forsythia, result in

27
enhanced bone loss which is greater than infection with P. gingivalis alone (Polak et al.,
2009; Orth et al., 2011). Collectively this emphasises the importance of polymicrobial
synergy and dysbiosis in the pathogenesis of periodontitis.
1.6.2. Aberrant immunity
Although microbial dysbiosis commonly instigates disease, tissue destruction and disease
progression are products of an aberrant host immune response. Of note, there is a recent
line of thought that inflammation is the initiating factor, and precedes dysbiosis (Bartold
and Van Dyke, 2017). Regardless of the sequence of initiation, it is clear that an
expanding dysbiotic biofilm accompanies the formation of an inflammatory cell lesion.
Vasodilation of local vasculature allows infiltration of neutrophils which localise at the
gingival crevice, and an increase in gingival crevicular fluid (GCF), an inflammatory
serum exudate that bathes the crevice (Hajishengallis, 2014a). Complement activation
and local chemokine production recruit granulocytes, mononuclear phagocytes, and
lymphocytes, which amplify the cytokine milieu by producing IL-1β, TNFα, IL-6, IL-12,
interferon (IFN)γ, and prostaglandin E2 (Cekici et al., 2014). Furthermore, neutrophils
release cytotoxic reactive oxygen species and matrix metalloproteinases (MMPs) such as
MMP8 and 9, which damage the surrounding soft tissue and weaken structural integrity
(Hernández et al., 2011; Nussbaum and Shapira, 2011). This results in retraction of the
gingival epithelium from the tooth and formation of a periodontal pocket at the gingival
crevice (Cekici et al., 2014). Unresolving inflammation leads to further tissue lysis,
deepening of the pocket, and an increasingly anaerobic biofilm which invades the tissue.
Attempts by responding leukocytes to destroy invading microbes fail to eliminate the
threat (due in part to immune evasion strategies employed by periopathogens), and tissue
breakdown products are swept into the pocket in the GCF and serve as nutrients for the
growing biofilm (Cekici et al., 2014; Hajishengallis, 2014b). This ultimately leads to an

28
advanced inflammatory lesion characterised by the resorption of the alveolar bone by
osteoclasts in a receptor activator of NF-κB ligand (RANKL)-dependent manner (Kawai
et al., 2006; Thorbert-Mros et al., 2015).
The role of dysregulated inflammatory responses in the pathogenesis of human and
experimental periodontitis is well described. Mice deficient in TLR-2/-4 and in
complement receptors C3a/C5aR have reduced bone loss (Hajishengallis et al., 2011; Abe
et al., 2012), highlighting innate immune recognition, complement activation, and
leukocyte recruitment as crucial processes contributing to inflammatory bone loss. In this
way, dysregulated cytokine signalling in the tissue directly promotes the destruction of
bone. For example, IL-33 promotes RANKL expression by B and T cells and
consequently exacerbates periodontal bone loss (Malcolm et al., 2015). Most importantly,
the IL-17-Th17-neutrophil axis has been consistently shown to orchestrate
immunopathology in mice and in humans (Moutsopoulos et al., 2014; Dutzan et al.,
2016b; Dutzan et al., 2017, 2018). Indeed, selective inhibition of IL-17 (Moutsopoulos et
al., 2014, 2017), Th17 differentiation (Dutzan et al., 2018), or neutrophil recruitment
(Eskan et al., 2012), substantially reduces inflammatory bone loss and improves clinical
symptoms. Importantly, while neutrophil accumulation is pathogenic in periodontitis,
defective neutrophil accumulation, such as in LAD-1 patients, also leads to periodontitis,
due to uncontrolled local IL-17 production (Moutsopoulos et al., 2014). It is apparent that
an appropriate balance in neutrophils is required for periodontal tissue homeostasis, as
diminished or excessive recruitment can cause tissue damage and bone loss.
A range of other immune populations are also emerging as mediators of gingival
inflammation and bone destruction during periodontitis. Recently, TNFα-producing mast
cells (Malcolm et al., 2016) and recruited monocytes expressing the chemokine receptor
CX3CR1 (Steinmetz et al., 2016) promote immunopathology in response to P. gingivalis

29
infection in mice. Moreover, B cells promote bone loss in both humans and mice, as
together with T cells, they are major sources of RANKL, which activates osteoclasts (Han
et al., 2006; Kawai et al., 2006; Abe et al., 2015; Oliver-Bell et al., 2015). However, this
is complicated by reports indicating that IL-10-producing (B10) B cells mitigate bone
loss in mice with P. gingivalis-soaked ligatures (Wang et al., 2017). Furthermore, resident
wound-healing γδ T cells and recruited Tregs have been shown in mice to limit gingival
inflammation and bone destruction (Garlet et al., 2009; Krishnan et al., 2018; Wilharm et
al., 2019), with Tregs thought to be recruited to the gingiva through a CCR4-CCL22 axis
(Araujo-Pires et al., 2015).
Thus, a number of key immune populations are at the centre of inflammatory damage
during periodontitis. Immune dysregulation promotes ineffective responses to an
increasingly dysbiotic microbial community, leading to a state of unresolving chronic
inflammation, and with it, an increased risk of systemic involvement.
1.7. Experimental models of periodontitis
Such is the global prevalence of periodontitis that experimental insight into the
pathogenesis is vital for understanding, treating, and preventing disease. A range of
animal model systems have been used, from non-human primates (Emerton et al., 2011),
dogs (Martuscelli et al., 2000), rabbits (Hasturk et al., 2006), to rats (Miyajima et al.,
2015) and mice (Abe and Hajishengallis, 2013; Matsuda et al., 2016). Mice represent a
more tractable model of experimental periodontitis, such is their well-characterised
immune system, ease of genetic manipulation and inexpensive experimental costs (Abe
and Hajishengallis, 2013). Therefore, there are a number of different methods of inducing
periodontitis in mice, ranging from mechanical to infectious. Each offers distinct

30
advantages and disadvantages; selection of the appropriate model must be carefully
considered depending on the experimental objectives.
1.7.1. Ligature placement
This approach involves physical placement of silk ligatures around teeth, which results in
local inflammation, bacterial growth, and robust bone loss (Saadi-Thiers et al., 2013; de
Molon et al., 2014). An important advantage of this model is that bone loss is robust and
reproducible (compared to other models) and occurs in a predictable and acute timeframe
(typically < 1 week) (Abe and Hajishengallis, 2013). Furthermore, this approach allows
dissection of immune pathways as well as periodontal treatments (Graves et al., 2008;
Abe et al., 2012; Dutzan et al., 2018). However, periodontitis in humans is a chronic
condition and often involves specific microbial species, features which are not catered for
using this approach.
1.7.2. Oral gavage of periodontal pathogens
Another method involves oral gavage with human periodontal pathogens, primarily P.
gingivalis (Li et al., 2002; Gibson et al., 2004), but also A. actinomycetemcomitans, and
T. forsythia, and these can be inoculated as single species or as multiple species (Nahid et
al., 2011; Rivera et al., 2013). As periodontal pathogens are not sufficiently virulent,
these approaches often involve frequent and high infection doses over a period of at least
six weeks. Even so, bone loss is generally mild and develops slowly over the time course
(Polak et al., 2009). However, this model does permit the dissection of specific bacteria-
host interactions (Gibson et al., 2004; Maekawa et al., 2014). An important caveat of this
model is that P. gingivalis is not native to mice and requires extensive antibiotic pre-
treatment to successfully colonise, the latter of which is known to disturb commensals at

31
other mucosal sites, leading to profound alterations in host immunity (Ubeda and Pamer,
2012; Saadi-Thiers et al., 2013; Scott et al., 2018).
1.7.3. Ageing
Physiological ageing in mice leads to natural bone loss and this is reported to be
significant from as little as six months of age (Liang et al., 2010; Krishnan et al., 2018).
Since age is one of the most important risk factors for periodontitis (Eke et al., 2015), this
model enables insight into the dysregulated inflammatory networks that occur with age
(Shaw et al., 2013) and the effect of periodontal tissue destruction in a chronic setting.
Aside from the main methods of modelling periodontitis in vivo as detailed above, other
models exist, including P. gingivalis-soaked ligatures (Li and Amar, 2007; Saadi-Thiers
et al., 2013), oral gavages in aged mice (Wu et al., 2015), as well as airpouch and
calvarial models (Graves et al., 2008), all of which can be used to address specific
questions about host-pathogen interactions or mechanisms of inflammatory bone loss.
1.8. The effects of periodontitis on systemic health
Chronic oral inflammation not only results in loss of local barrier integrity but also poses
a major threat to systemic health. Degradation of the sub-gingival tissues alongside
dilation of the periodontal vasculature facilitates increased translocation of bacteria, their
products, and host-derived factors into the systemic circulation. This can amplify the
threat that periodontitis poses to the host, as once in the bloodstream, oral-derived
bacteria can be carried to distal sites and can exacerbate or contribute to disease states.
Furthermore, systemic leakage of soluble host mediators and microbial products can
aggravate the inflammatory response, driving pathology in a number of disease contexts.
There is ample evidence implicating periodontitis in the progression or development of

32
many systemic disease states, including adverse pregnancy outcomes (Schenkein et al.,
2003; Han et al., 2014), non-alcoholic fatty liver disease (Yoneda et al., 2012), lung and
bowel cancer (Michaud et al., 2018), atherosclerosis (Li et al., 2002; Gibson et al., 2004),
and Alzheimer’s disease (AD) (Riviere et al., 2002; Poole et al., 2014; Ide et al., 2016;
Singhrao et al., 2017; Dominy et al., 2019). In many instances, observations in humans
have been corroborated in animal models, which importantly have provided a mechanistic
basis for some of the associations. For example, in humans, periodontitis has been found
to increase the rate of cognitive decline in AD patients (Ide et al., 2016), and this is
supported by experimental reports that have shown that T. denticola and P. gingivalis can
gain access to the brain in genetically-susceptible mice and contribute directly to
pathology by promoting neuroinflammation and the killing of neurons (Foschi et al.,
2006; Poole et al., 2014; Dominy et al., 2019). While each case is evidently distinct,
adverse outcomes as a result of periodontitis are generally attributed to the systemic
dissemination of periopathogens, amplification of systemic inflammation, and/or
dysregulation of immune function. For example, periodontal pathogens F. nucleatum and
P. gingivalis have been shown to colonise the placenta and cause complications of
childbirth, such as low birth weight, premature birth, and miscarriage (Han et al., 2010,
2014; Schenkein et al., 2013). Moreover, F. nucleatum is associated with tumorigenesis
in colorectal cancer by recruiting tumour-infiltrating myeloid cells while preventing
natural killer (NK) cell- and T cell-mediated tumour killing (Kostic et al., 2013; Gur et
al., 2015; Abed et al., 2016). Thus, periodontitis is proposed to adversely affect a wide
spectrum of disease states through a wide spectrum of biologically plausible mechanisms.
In this section the discussion will be restricted to a select few stronger and more relevant
associations.

33
1.8.1. Periodontitis and rheumatoid arthritis
Substantial epidemiological and pre-clinical evidence has indicated a role for
periodontitis in the pathogenesis of rheumatoid arthritis (RA) (Ogrendik et al., 2005;
Ogrendik, 2009, 2013; Bartold et al., 2010). For decades, a link between periodontitis and
RA has been proposed; individuals with chronic RA have a higher incidence of
periodontitis, and similarly, the prevalence of RA is higher in periodontitis patients
(Ogrendik, 2009). Moreover, RA patients with persistent periodontitis are less responsive
to conventional therapeutic interventions (Savioli et al., 2012) and periodontitis treatment
can reduce the severity of RA (Ortiz et al., 2009; Erciyas et al., 2013; Silvestre et al.,
2016). Animal models have also clearly indicated that RA is exacerbated by periodontitis
(Bartold et al., 2010; Cantley et al., 2011). Most research to date has focused on the roles
of periodontal bacteria in driving the inflammatory consequences of periodontitis in RA;
indeed, many periodontally-derived bacteria have been detected in human synovial fluid
during RA (Ogrendik et al., 2005; Moen et al., 2006; Ogrendik, 2013). Importantly, P.
gingivalis and A. actinomycetemcomitans have been shown to drive RA pathology
through antigen mimicry (discussed further in subsequent sections), which promotes self-
reactive T cells that exacerbate disease (Maresz et al., 2013; Konig et al., 2016). Given
the pathogenic potential of anaerobic bacteria such as P. gingivalis, it is unsurprising
therefore that antibacterial treatments (e.g. ornidazole, ievofloxacin, and clarithromycin)
also lessen the severity of RA (Ogrendik, 2006, 2007a, 2007b), highlighting periodontal
bacteria as key drivers of disease pathology.
1.8.2. Periodontitis and mucosal disease
Given that periodontitis is an inflammatory disease of the oral mucosa, it is perhaps not
surprising that periodontitis is associated with driving pathology at other mucosal sites,
most notably the gut (Atarashi et al., 2017), and the lung (Mojon, 2002). Considering that

34
the oral cavity is the access point to both respiratory and gastrointestinal (GI) tracts, it is
conceivable that oral inflammation could affect the lung or GI environments.
Indeed, periodontitis has been associated with lung infections, and although the current
evidence is largely anecdotal, there are clinical and pre-clinical reports of periodontal
bacteria directly contributing to lung pathology; A. actinomycetemcomitans can cause
pulmonary infections of humans either alone or in tandem with Actinomyces species
(Zijlstra et al., 1992; Morris and Sewell, 1994). Furthermore, P. gingivalis has been
isolated with the opportunistic pulmonary pathogen, Pseudomonas aeruginosa, in
tracheal aspirates from patients with chronic obstructive pulmonary disease (Tan et al.,
2014), and in mice, P. gingivalis and T. denticola exacerbate lung pathology more so than
mono-infection with either bacterium (Kimizuka et al., 2003).
Periodontitis has also been proposed to exacerbate inflammatory bowel disease (IBD) and
colorectal cancer. Elevated levels of periodontal bacteria increase the risk of developing
pre-cancerous gastric lesions (Sun et al., 2017), and the oral-derived F. nucleatum has
been shown to directly promote colorectal cancer (Kostic et al., 2012; Rubinstein et al.,
2013; Gur et al., 2015; Abed et al., 2016). In addition to potential oncogenic effects,
periodontal bacteria are proposed to drive inflammatory pathology in IBD (Atarashi et al.,
2017). Patients with IBD are reported to have higher prevalence and extent of
periodontitis compared to healthy individuals (Brito et al., 2008; Habashneh et al., 2012;
Vavricka et al., 2013). However, despite some preclinical evidence indicating a possible
role for periopathogens in the gut (Arimatsu et al., 2015; Atarashi et al., 2017; Blasco-
Baque et al., 2017), a causal association between periodontitis and IBD has not been
established.

35
1.8.3. Periodontitis and cardiovascular disease
To date, perhaps the condition most strongly linked with periodontitis is cardiovascular
disease (CVD). Periodontitis and CVD are intimately tied through common risk factors,
including obesity, hypertension, old age, male sex, and cigarette smoking (Genco and
Van Dyke, 2010). An association between periodontitis and CVD was first reported 20
years ago (Beck et al., 1996). Since, a large number of clinical studies and meta-analyses
have indicated that individuals with periodontitis are at increased risk of developing
coronary artery disease, myocardial infarction, and atherosclerosis, independently of
confounding factors, such as smoking and obesity (Beck et al., 2001; Bahekar et al.,
2007; Gotsman et al., 2007; Humphrey et al., 2008). Indeed, increased sub-gingival
biomass, as well as P. gingivalis, A. actinomycetemcomitans, T. denticola, and T.
forsythia in the sub-gingival plaque, and raised serum antibody levels to periodontal
bacteria, are all associated with increased carotid intima-media thickening (Beck et al.,
2005; Desvarieux et al., 2005). Conversely, an improvement in periodontal clinical and
microbial status reduces carotid intima-media thickening (Desvarieux et al., 2013).
Moreover, interventional studies have found that periodontitis treatments (such as scaling,
root planing, and tooth extractions) can reduce the rate of CVD (Peng et al., 2017), or at
the very least, mitigate some of the risk factors, by improving endothelial function, a sub-
clinical marker of atherosclerosis (Tonetti et al., 2007), or lowering inflammatory
markers, such as IL-6 and C-reactive protein (CRP) (D’Aiuto et al., 2004; Paraskevas et
al., 2008; Offenbacher et al., 2009; Tonetti, 2009). In this way, systemic inflammation is
commonly cited as a biologically plausible mechanism causally linking periodontitis with
CVD; poor oral hygiene with associated low-grade inflammation has been added to the
spectrum of risk factors for CVD (de Oliveira et al., 2010). Specifically, raised CRP, IL-
6, and fibrinogen levels that are associated with poor oral hygiene are also risk factors for
CVD (Paraskevas et al., 2008; Tonetti, 2009; de Oliveira et al., 2010).

36
Even though systemic inflammation is thought to be an important underlying mechanism
linking periodontitis and CVD (Slocum et al., 2016), more frequently it is disseminated
periodontal bacteria that have been proposed to directly drive CVD. Evidence to support
this comes from studies in which bacterial species isolated from human atherosclerotic
plaques are thought to be derived from the oral cavity (Koren et al., 2011). Furthermore,
periodontal bacteria such as P. gingivalis, A. actinomycetemcomitans, T. forsythia,
Prevotella intermedia, and Streptococcus sanguis have been routinely isolated from
atherosclerotic plaques and coronary arteries in humans (Haraszthy et al., 2000; Kozarov
et al., 2005; Gaetti-Jardim et al., 2009; Nakano et al., 2009; Reyes et al., 2013). These
oral microbes are proposed to exert atherogenic effects by accumulating at sites of plaque
development and contributing to local vascular inflammation (Genco and Van Dyke,
2010). Experimental studies have supported the indication that periopathogens, and
specifically P. gingivalis, are causal in the development and progression of
atherosclerosis (Gendron et al., 2000; Kuramitsu et al., 2001; Beck and Offenbacher,
2005). Specifically, a number of studies have demonstrated that P. gingivalis enhances
atherosclerosis progression in pigs (Brodala et al., 2005), rabbits (Jain et al., 2003), and
most frequently, in atherosclerosis-prone apolipoprotein E-deficient mice (Li et al., 2002;
Lalla et al., 2003; Gibson et al., 2004; Brodala et al., 2005; Velsko et al., 2014).
1.9. Mechanisms by which periodontitis can impact pathology at
non-oral sites
As described in the previous section, chronic oral inflammation can exacerbate or
contribute to a multitude of non-oral conditions. Most of the evidence linking oral and
systemic compartments centres on the dissemination of pathogenic periodontal bacteria
and inflammatory molecules from host and microbe to extra-oral sites via the circulation
(Geerts et al., 2002; Kinane et al., 2005; Forner et al., 2006) (Figure 1.2).

37
Figure 1.2. Associations and proposed mechanisms between periodontitis and systemic
diseases.
Chronic inflammatory damage to the hard and soft periodontal tissues can lead to leakage of
bacterial products, host inflammatory factors and pathogenic microbes into the bloodstream where
they are transported to distal sites. Once in the systemic circulation, these factors and microbes
have the potential to adversely affect a multitude of systemic diseases, which is chiefly proposed to
occur either directly in situ or indirectly via amplification of the systemic inflammatory response.
LPS = lipopolysaccharide. IL = interleukin. TNF-α = Tumour necrosis factor-alpha.
1.9.1. Bacterial virulence
Since oral pathogens thrive within anaerobic periodontal pockets (Hajishengallis et al.,
2011), the virulence factors that enable successful oral colonisation may also enable
colonisation at other tissue sites. Indeed, prominent periodontal bacteria including F.
nucleatum, P. gingivalis, and A. actinomycetemcomitans have been detected in a
multitude of extra-oral tissue sites, including the lung, heart, gut, placenta, and inflamed
joints (Meyer and Fives-Taylor, 1997; Mojon, 2002; Foschi et al., 2006; Liu et al., 2007;
Han et al., 2010; Ogrendik, 2013; Dominy et al., 2019). Furthermore, all aforementioned
species are capable of invading epithelial and endothelial cells (Chiu, 1999; Madrigal et
al., 2012) and specifically, A. actinomycetemcomitans can spread to neighbouring cells,
disseminating via membrane-bound vacuoles endocytosed by adjacent host cells (Meyer
and Fives-Taylor, 1997). Even a site like the brain, which possesses a highly restrictive
blood-brain barrier (BBB) to limit entry of molecules, is not immune to invasion by
periodontal bacteria; oral Treponema species have been found in human AD brains and in

38
branches of the trigeminal nerves, indicating a potential route of access to the demented
brain (Riviere et al., 2002). In addition to a remarkable invasiveness potential, periodontal
bacteria possess an extensive suite of virulence factors which facilitate colonisation and
persistence at extra-oral sites. These include hair-like protein structures called fimbriae
which aid binding, colonisation, and immune subversion (Gibson et al., 2004; Jotwani
and Cutler, 2004); adhesion molecules, which enable adherence and invasion of host cells
(Liu et al., 2007; Rubinstein et al., 2013); and capsules, which protect bacteria from
phagocytosis, aid in cell attachment, and subsequent biofilm formation (Singh et al.,
2011).
Periodontal bacteria also utilise sophisticated immune subversion mechanisms to enhance
survival in the oral cavity and these strategies are proposed to also facilitate colonisation
at extra-oral sites (Hajishengallis, 2015). Evasion of complement-mediated killing
(Popadiak et al., 2007; Potempa et al., 2009; Jusko et al., 2012), disarming leukocyte
responses (Wang et al., 2010; Taxman et al., 2012; Maekawa et al., 2014), and
modulation of TLR signalling (Maekawa et al., 2014) are part of a sophisticated arsenal
that can prevent immune-mediated clearance. While most evidence to date has been
carried out in the context of the oral cavity, some studies have demonstrated that these
subversive strategies can be employed at extra-oral sites, mostly in the context of
atherosclerosis (Hayashi et al., 2010; Delbosc et al., 2011; Madrigal et al., 2012; Slocum
et al., 2014). There is also emerging evidence to suggest that oral bacteria can “hijack”
immune cells, such as macrophages and DCs (Wang et al., 2007; Carrion et al., 2012;
Miles et al., 2014), thereby enabling dissemination from the initial oral focus while
simultaneously evading immune-mediated killing.
Independently of direct immune evasion strategies, periodontal bacteria are capable of
disrupting homeostatic biological processes, which has the potential to adversely impact
systemic health. In this regard, P. gingivalis and S. sanguis can induce platelet

39
aggregation (Sharma et al., 2000; Nakayama, 2010), which may contribute to an
increased risk of thrombosis in cardiac or cerebral vessels. P. gingivalis is also capable of
modifying low-density lipoprotein which promotes foam cell formation (Qi et al., 2003;
Miyakawa et al., 2004), a pathogenic feature of atherosclerosis. In the context of
metabolic disease, P. gingivalis can promote glucose intolerance and insulin resistance in
mice fed a diabetogenic diet (Blasco-Baque et al., 2017). And, in the gut environment, F.
nucleatum promotes colorectal cancer through binding to oncogenic cells via its Fap2
protein (Kostic et al., 2013; Abed et al., 2016).
Another potential means by which periodontal bacteria adapt to extra-oral colonisation is
through inter-species microbial synergy. As this has been well documented in the oral
cavity (Polak et al., 2009; Daep et al., 2011; Orth et al., 2011; Settem et al., 2012; Shin et
al., 2013; Zhu et al., 2013), it is plausible that this is a potential means of facilitating
microbial survival in sites outside the periodontal tissues. In particular, P. gingivalis has
been shown to promote the ability of P. aeruginosa to invade and persist in respiratory
epithelial cells in vitro (Pan et al., 2009; Li et al., 2014), and the temporal colonisation of
F. nucleatum and then P. gingivalis which mutually benefits survival in the oral cavity is
also suggested to occur in the gut and contributes to colorectal tumorigenesis (Flynn et
al., 2016).
It is important to note that although viable periopathogens are capable of persisting at
extra-oral sites, presence of bacteria in the circulation will provoke immune responses,
regardless of viability. This undoubtedly escalates the burden that host defences are
tasked with, as large numbers of oral commensals are transported into the bloodstream
with pathogenic microbes. This may explain why chronic bacteraemias as a result of
periodontitis pose an important threat, especially when periodontal pathogens themselves
are usually found in low numbers (Hajishengallis et al., 2011).

40
1.9.2. Leakage of microbial and host factors
Continued degradation of the periodontal tissues not only increases microbial leakage into
the systemic circulation, but also increases the flushing of locally-produced host and
microbial factors. Furthermore, dental procedures (scaling, extractions, root planing etc.)
as well as habitual brushing, flossing, and mastication all increase the rate of blood
contamination (Geerts et al., 2002; Kinane et al., 2005; Forner et al., 2006; Crasta et al.,
2009) and this in turn aggravates the systemic inflammatory response. In patients with
severe periodontitis, even gentle mastication can lead to elevated lipopolysaccharide
(LPS) in the circulation (Geerts et al., 2002). In particular, LPS from P. gingivalis has
been given the most attention (Pulendran et al., 2001; Darveau et al., 2004; DeLeon-
Pennell et al., 2014). Although more weakly immunogenic than its E. coli counterpart
(Liu et al., 2008), elevated circulating levels of P. gingivalis LPS (Pg-LPS) can induce
inflammation in vivo in mice (Pulendran et al., 2001; Bian et al., 2016; Lyu et al., 2017).
Systemic Pg-LPS has also been shown to directly contribute to the pathogenesis of
murine atherosclerosis (Stoll et al., 2004; DeLeon-Pennell et al., 2013, 2014).
Furthermore, chronic systemic challenge with Pg-LPS (1mg/kg; daily for 5 weeks) can
induce neuroinflammation and cognitive dysfunction in mice (Wu et al., 2017). Taken
together, this suggests that chronic leakage of LPS from the oral cavity could amplify
systemic inflammation and impact distal sites.
In addition to microbial factors, chronic periodontal inflammation can also result in
sustained and increased levels of host inflammatory mediators in the circulation, which is
a trigger for the acute-phase response. Indeed, the acute-phase reactants, IL-6, CRP,
haptoglobin, fibrinogen, serum amyloid A, and serum amyloid P, are elevated in
periodontitis patients (Ebersole et al., 1997; Loos et al., 2000; de Maat and Kluft, 2001;
Noack et al., 2001; Sahingur et al., 2003). Moreover, there are reports of serum increases

41
in levels of the pro-inflammatory cytokines, interferon (IFN)-γ (Andrukhov et al., 2011),
as well as TNFα and IL-17, in patients with aggressive periodontitis (Duarte et al., 2010;
Schenkein et al., 2010). Findings from animal models of periodontitis also support a
heightened systemic inflammatory profile (Brito et al., 2013; Arimatsu et al., 2015;
Miyajima et al., 2015; Matsuda et al., 2016). However, not all pre-clinical and clinical
studies report increased serum levels of pro-inflammatory mediators (Yamazaki et al.,
2005), or at least, do not report the same cytokines. As a result, there is a lack of
consensus on the inflammatory profile during periodontitis, and this may be due to
inherent heterogeneity of individuals as well as differences in oral bacterial load and
composition (Andrukhov et al., 2011).
1.9.3. Promotion of autoreactivity
In general, periodontal bacteria can actively modulate the innate immune response and
their products can incite a typical inflammatory reaction, whether this is at oral or extra-
oral sites. These are commonly proposed mechanisms that offer an explanation for the
adverse effects of periodontitis on systemic diseases. However, perhaps the most robust
evidence implicating periodontal bacteria directly in the context of systemic disease is the
ability of P. gingivalis and, recently A. actinomycetemcomitans, to promote RA pathology
via antigen mimicry (Lundberg et al., 2008; Maresz et al., 2013; Gully et al., 2014; Konig
et al., 2016). Citrullination of peptides (the conversion of residues from an arginine to
citrulline) is a pathogenic hallmark of RA as it promotes autoantibody generation, and
importantly, this occurs prior to disease onset as well as correlating strongly with disease
severity (Suwannalai et al., 2012). P. gingivalis is unique in that it can citrullinate
proteins via its enzyme peptidylarginine-deiminase, and therefore, is a direct contributor
to the production of pathogenic antibodies; driving autoreactive T cells to promote
inflammatory synovial destruction (Lundberg et al., 2008; Maresz et al., 2013; Gully et

42
al., 2014). More recently, A. actinomycetemcomitans has also been shown to
hypercitrullinate proteins in host neutrophils in joints (Konig et al., 2016). Specifically,
the pore-forming toxin, leukotoxin A (LtxA), induces hypercitrullination of proteins
within neutrophils, and causes subsequent cell lysis and extracellular release of these
proteins, which provokes autoantibody generation. Taken together, this highlights a direct
deleterious role for periodontal pathogens in systemic disease, by exacerbating specific
pathogenic features.
Similarly, P. gingivalis is also reported to elicit antigen mimicry in the context of CVD
and pregnancy complications. Auto-antibodies to cardiolipin are associated with risk of
atherosclerotic thrombosis (Schenkein et al., 2003) and adverse pregnancy outcomes
(Schenkein et al., 2013), and elevated levels of anti-cardiolipin antibodies are found in the
gingival crevicular fluid and serum of periodontitis patients (Schenkein et al., 2003; Chen
et al., 2009). Importantly, cross-reactive bacterial epitopes from P. gingivalis can mimic
cardiolipin, thereby promoting the generation of pathogenic autoantibodies (Schenkein et
al., 2003, 2013; Han et al., 2014).
Antigen mimicry is also posited to link periodontal and atherosclerotic diseases in the
context of cross-reactivity of heat-shock proteins (HSPs) with components from
periodontal bacteria (Perschinka et al., 2003). HSPs are associated with enhanced
atherosclerosis development (Foteinos et al., 2005), and P. gingivalis possesses GroEL
proteins that are highly homologous to the heat-shock protein HSP60 (Yamazaki et al.,
2002; Perschinka et al., 2003; Ford et al., 2005a, 2005b). As the arterial endothelium
expresses highly immunogenic HSPs (Amberger et al., 1997; Yamazaki et al., 2002),
cross-reactivity with GroEL from disseminated oral bacteria can lead to endothelial
dysfunction which is an early contributor to the development of atherosclerosis.

43
Thus, there is ample clinical and pre-clinical evidence causally linking periodontitis and
specifically, periopathogens, to the development and/or progression of a range of chronic
systemic diseases, by enhancing clinical disease features, subverting immune responses,
and promoting deleterious systemic inflammation. Although current evidence is limited,
given that stroke shares a number of similarities with CVD, it is therefore biologically
plausible that periodontitis may be an independent risk factor for stroke and could also
have a profound impact on pathology and prognosis.
1.10. Stroke
Stroke defines a heterogeneous group of cerebrovascular diseases in which blood supply
to the brain is interrupted. Stroke is a significant cause of mortality and morbidity; it is
the second leading global cause of death and the predominant cause of disability (Feigin
et al., 2010, 2014). Ischaemic stroke is the most common type, accounting for 85% of all
stroke cases, and is caused by obstruction of a cerebral blood vessel (by embolus, formed
elsewhere, or thrombus, formed in situ). In contrast, haemorrhagic stroke, which accounts
for 15% of strokes, is induced by rupture of a cerebral vessel and leakage of blood into
the brain, and is further classified as sub-arachnoid or intracerebral based on the location
of the bleed (An et al., 2017). For the purposes of this thesis, the discussion will be
limited to ischaemic stroke. The terms stroke, ischaemic stroke, cerebral ischaemia, and
ischaemic event are all used interchangeably throughout.
1.11. The pathophysiology of ischaemic stroke
Obstructed cerebral blood flow results in the formation of an “ischaemic core” or
“infarct”, a region in which blood supply is most restricted. Oxygen and glucose
deprivation in the core causes rapid necrotic cell death of neurons and glia, and
consequently, brain injury and neurological deficits (Moskowitz et al., 2010). The core is

44
surrounded by the penumbra, a region in which adjacent collateral vessels limit
immediate neuronal loss, and thus is functionally impaired but potentially salvageable.
Loss of cells in the penumbra (known as secondary damage) and the extent of
neurological dysfunction is dependent on a multitude of factors, including the duration of
ischaemia, extent of inflammation, as well as the presence of co-morbidities (Sommer,
2017). In the core, cellular bioenergetic failure is the major mechanism of cell death, a
cascade of biochemical disturbances that leads to excitotoxicity and oxidative stress
(Iadecola and Anrather, 2011). This drives inflammatory signalling in the brain,
subsequent peripheral immune cell invasion, and BBB breakdown, which together
contribute to further loss of neurons, glia, and endothelial cells.
1.12. The immunopathology of ischaemic stroke
Maturation of the infarct during stroke is intimately dependent on deleterious
inflammatory signalling and dysregulated immune responses (Dénes et al., 2010b).
Inflammation during cerebral ischaemia occurs in sequential order, involving the brain,
the vasculature, blood and peripheral immune tissues (Iadecola and Anrather, 2011).
Initially, “danger signals” (such as adenosine triphosphate [ATP]) released by dying and
dead neurons activate nearby microglia, brain-resident macrophages, astrocytes, and
endothelial cells, through stimulation of pattern-recognition receptors (such as TLRs)
(Iadecola and Anrather, 2011). Activated glial cells, in turn, propagate local damage by
releasing neurotoxic factors (such as inducible nitric oxide [iNOS]), as well as cytokines
(such as IL-1β, TNFα, and IL-6) and chemokines (such as CCL2 and CXCL1), which
feedback on the vascular and perivascular compartments, activating the BBB and
facilitating rapid entry of neutrophils and monocytes into the brain, which occurs in the
first 24–48 hours after the initial ischaemic insult (Iadecola and Anrather, 2011). These
myeloid cells amplify the pro-inflammatory environment within the brain, further disrupt

45
the BBB by degrading tight junction proteins (McColl et al., 2008), and induce
microvascular obstruction through leukocyte-platelet interactions (Burrows et al., 2015).
Lymphocyte infiltration follows and peaks later, usually between 3–7 days, and can either
contribute to damage or repair depending on the inflammatory status and phase of injury.
An extensive body of evidence has implicated a diverse range of immune cell populations
in propagating brain damage post-stroke. Indeed, resident microglia (Tang et al., 2014),
as well as the central nervous system (CNS) infiltration of neutrophils (Gelderblom et al.,
2012) and dendritic cells (Gelderblom et al., 2018) all promote brain damage in the sub-
acute and acute stages post-stroke. However, the role of monocytes is controversial, as
they have been shown to acutely potentiate brain injury (Dimitrijevic et al., 2007), but
promote tissue repair and resolution of inflammation in the chronic stages after stroke
(Gliem et al., 2012; Chu et al., 2015; Wattananit et al., 2016; Ge et al., 2017).
Contradictory observations also extend to lymphocytes, as a number of leukocyte
populations have positive and negative impacts on the ischaemic brain. T cells (Yilmaz et
al., 2006; Kleinschnitz et al., 2010), NK cells (Gan et al., 2014), and IL-17-producing γδ
T cells (Shichita et al., 2009; Arunachalam et al., 2017) have been shown to exacerbate
brain injury by infiltrating the brain and promoting neuroinflammation. However, while B
cells are reportedly neuroprotective in stroke (Ren et al., 2011a; Bodhankar et al., 2013),
not all studies support such a role (Kleinschnitz et al., 2010). Similarly, Tregs are
generally considered protective in the later stages of stroke (Liesz et al., 2009b; Na et al.,
2015; Ito et al., 2019) but can also be detrimental (Kleinschnitz et al., 2013) or non-
essential (Ren et al., 2011b). Despite conflicting findings, acute recruitment of peripheral
leukocytes to the ischaemic brain is a major contributor to neuronal injury and represents
an important potential therapeutic option.

46
1.13. Experimental models of ischaemic stroke
While it is apparent that experimental stroke models cannot sufficiently recapitulate all
aspects of such an immensely complex disease, detailed exploration of therapeutic targets
and pathological mechanisms has benefitted enormously from the use of rodent models.
Although the location of infarcts in humans can vary, a large proportion involve blockage
of the middle cerebral artery (MCA) (Ng et al., 2007). Therefore, in animals the MCA is
principally the focus of surgical manipulation. Broadly, there are two main approaches to
induce focal cerebral ischaemia, either by obstructing the MCA at its origin or at a distal
branch (Howells et al., 2010).
The first approach uses a suture or filament to prevent blood flow through the MCA.
While there are many variations on this method, most are based upon a technique by
Longa et al. (1989), in which a filament is advanced through the extracranial internal
carotid artery to occlude the MCA at its origin. The filament can be left in place
permanently but is commonly withdrawn after a defined time period (usually from 15 to
90 minutes in mice) to allow reperfusion (Howells et al., 2010). This method induces
striatal or striato-cortical infarcts and can yield robust neurological deficits. However, an
important caveat is that natural anatomical variation in collateral vessels of inbred mice
can profoundly and unpredictably alter the size of the infarct, and the rate of mortality is
high (McColl et al., 2004; Howells et al., 2010). To counteract this, the other approach
involves accessing the distal branches of the MCA via craniotomy. The MCA then can be
electrocoagulated (Llovera et al., 2014), photothrombosed (Labat-gest and Tomasi,
2013), or occluded chemically by application of ferric chloride (FeCl3) (Karatas et al.,
2011) or the vasoconstrictor endothelin-1 (Ansari et al., 2013). In this manner, infarcts are
typically smaller and more reproducible, confined to the cortex, and do not produce
robust neurological dysfunction. Although distal and proximal methods vary

47
considerably, for example, vascular-thrombotic inflammation (distal) versus
neuroinflammation (proximal), together they represent valid approaches for studying
mechanisms as well as the impact of co-morbidities, such is their similarities to the
spectrum of human strokes (Dirnagl and Endres, 2014; Llovera et al., 2014).
1.14. The impact of co-morbidities on stroke outcome
Despite a vast amount of knowledge of risk factors and the complex pathobiological
events occurring before, during, and after stroke, treatments are severely lacking (Dirnagl
and Endres, 2014). Mechanical or chemical removal of clots are the only approved
treatments and are severely limited by a short time window which prevents widespread
applicability (Hacke et al., 2008; Vogelgesang et al., 2017). Thus, efforts to mitigate
modifiable risk factors, such as hypertension, smoking, infection, and obesity (Allen and
Bayraktutan, 2008; Goldstein et al., 2011), are now central to minimising stroke
incidence and also minimising the extent of stroke-induced damage. Indeed, concurrent
conditions or co-morbidities are major determinants of both increased risk and poorer
prognosis after stroke (Drake et al., 2011; Maysami et al., 2015), and, since inflammation
is critically involved in determining the fate of the brain during stroke, any condition that
increases the inflammatory burden can be deleterious in the context of stroke-induced
injury.
1.14.1. Systemic inflammation and infection
Systemic inflammation, which is central to all known stroke co-morbidities, amplifies
neuroinflammation and worsens brain injury after stroke (McColl et al., 2009; Dénes et
al., 2010b; Burrows et al., 2015). At the molecular level, inflammatory mediators from
host, such as IL-1β (McColl et al., 2007, 2008), and from non-host, such as bacterial LPS
(McColl et al., 2007; Dénes et al., 2011a), are key drivers of ischaemic brain injury.

48
Furthermore, many infectious diseases, from acute pneumonia (Dénes et al., 2014) to
chronic helminth burden (Dénes et al., 2010a) can exacerbate ischaemic brain damage,
primarily by promoting microvascular injury, thrombosis, and inflammation (Fugate et
al., 2014). Not only is infection a risk factor and a modifier of stroke, but also a
consequence, as stroke leads to profound and acute immunosuppression, which is itself a
major contributor to increased risk of infection after stroke (Prass et al., 2003; Engel et
al., 2015; McCulloch et al., 2017).
1.14.2. Metabolic and complex conditions
It is also appreciated that a range of conditions and complex disease states influence the
extent of ischaemic damage, often by promoting deleterious inflammatory responses. For
example, hypertension increases infarct volumes (O’Collins et al., 2013), and
atherosclerosis promotes stroke-induced inflammation (Drake et al., 2011). In turn, stroke
can accelerate the progression of atherosclerosis (Roth et al., 2018). Moreover, diabetes
increases haemorrhagic transformation and worsens post-stroke recovery without
increasing infarct size (Vannucci et al., 2001; Sweetnam et al., 2012; Mishiro et al.,
2014). In contrast, hypercholesterolaemia (Hayakawa et al., 2007), and obesity (McColl
et al., 2010; Maysami et al., 2015; Haley et al., 2017a), exacerbate acute brain damage in
experimental settings, with obesity specifically exacerbating BBB disruption. However,
somewhat paradoxically, obese stroke patients often fare better than their healthy-weight
counterparts (Haley and Lawrence, 2016), which raises questions about the translational
validity of experimental stroke studies (Dirnagl and Endres, 2014).

49
1.15. Periodontitis and stroke: evidence and potential
mechanisms
Given the associations between periodontitis and CVD, it is not surprising that
periodontitis is also often associated with cerebrovascular disease, as stroke and CVD
share similar aetiologies and risk factors, such as hypertension, obesity, and cigarette
smoking (Elter et al., 2003; Joshipura et al., 2003; Dorfer et al., 2004; Grau et al., 2004).
A recent systematic review and meta-analysis of five case-control and three cohort
studies found that there was a statistically significant association between periodontitis
and ischaemic stroke (Leira et al., 2017). Further, dental infections, including
periodontitis, have been proposed to increase the risk of stroke (Syrjanen et al., 1989;
Grau et al., 1997), although not all indicate an increased risk (Howell et al., 2001).
Furthermore, there is minimal experimental data on the role of periodontitis in stroke
outcome, and as such, definitive causal evidence is not available.
However, much can be inferred from studies on experimental periodontitis and systemic
effects (as previously discussed). Chiefly, pathogenic periodontal bacteria and their
products are typically associated with promoting systemic inflammation (Hajishengallis,
2015) and platelet aggregation (Nakayama, 2010), which are known contributors of
increased brain injury post-stroke (Dénes et al., 2011a). Taking evidence from recent
studies in Alzheimer’s disease, periodontitis is proposed to directly lead to neuronal
damage; P. gingivalis, and T. denticola have been detected in brains in humans and in
mice (Riviere et al., 2002; Foschi et al., 2006; Poole et al., 2014; Dominy et al., 2019),
and proteases from P. gingivalis can kill neurons in vitro and in vivo (Dominy et al.,
2019). Also, chronic leakage of cytokines and microbial products into the circulation
from the oral cavity can induce systemic inflammation (Loos, 2005). Coupled with the
extensive BBB disruption that occurs during stroke, it is plausible that a raised pro-

50
inflammatory profile and blood-borne periodontal pathogens could have devastating
consequences for the ischaemic brain.
1.15.1. Clinical importance
Periodontitis and ischaemic stroke share risk factors (such as age, smoking, stress, and
obesity) and inflammation is central to the aetiology and pathogenesis of both diseases
(Grau et al., 2004; Sfyroeras et al., 2012). A heightened systemic inflammatory profile
pre- and post-stroke is an important determinant of poor outcome in both humans and
mice (McColl et al., 2007, 2008; Dénes et al., 2010a; Dénes et al., 2014). Stroke is
associated with significant morbidity and mitigating the impact of prominent stroke risk
factors is therefore critical for diminishing the incidence of stroke. Periodontitis is
extremely prevalent, but is preventable and treatable, and therefore it is of great
importance to determine the extent that periodontitis influences stroke outcome.
1.16. Aims
The overall aims of this thesis were to characterise the systemic immune landscape during
experimental periodontitis. In this way, we aimed to determine the potential impact of
inflammatory and immune mediators on peripheral tissue sites during periodontitis, and
in particular, the impact of periodontitis-induced immune alterations on acute outcome
after ischaemic stroke. To achieve this, we predominantly employed multiparameter flow
cytometry and relevant in vivo mouse models of periodontitis and stroke.

51
Chapter 2. Materials and methods

52
Details of reagents and flow cytometry antibodies can be found in the Appendix.
2.1. Media
2.1.1. 3% media
RPMI 1640 medium was supplemented with 10 mM 4-(2-hydroxyethyl)-1-
piperazineethanesulfonic acid (HEPES) and 3% heat-inactivated foetal bovine serum
(FBS).
2.1.2. Complete media
RPMI 1640 medium was supplemented with 10 mM HEPES, 100 U/ml penicillin, 100
μg/ml streptomycin, 2 mM L-glutamine, 1X minimum essential medium (MEM) non-
essential amino acids, and 10% FBS.
2.1.3. Digest media
For lung digests, RPMI 1640 medium was supplemented with 10 mM HEPES, 2 mM L-
glutamine, with 0.1 mg/ml liberase TL and 0.25 mg/ml DNase I. For gingiva digests,
RPMI 1640 medium was supplemented with 10 mM HEPES, 2 mM L-glutamine, 3.2
mg/ml collagenase IV and 75 μg/ml DNase I. For gut digest ‘A’, RPMI 1640 medium
was supplemented with 20 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin,
100 U/ml polymyxin B, 5 mM ethylenediaminetetraacetic acid (EDTA), 1 mM freshly-
thawed dithiothreitol, and 3% FBS. For gut digest ‘B’, RPMI 1640 medium was
supplemented with 20 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM
L-glutamine, 1 mM sodium pyruvate, 1X MEM non-essential amino acids, 0.1 mg/ml
Liberase TL, and 0.5 mg/ml DNase I.

53
2.1.4. FACS buffer
Phosphate buffered saline (PBS) containing 2% FBS and 2 mM (EDTA) was sterile-
filtered through a 0.22 µm filter.
2.2. Animals
All C57BL/6 (CD45.2) mice were purchased from Envigo (UK) except for “aged”
C57BL/6 mice (Chapter 5) which were provided by the University of Manchester
Biological Services Facility. In all studies, mice were 8–14 weeks old, except for “aged”
mice which were 6–9 months old. Ccr2-/- mice (available from the Jackson laboratory)
and congenic CD45.1 mice (serially backcrossed from SJL/J onto C57BL/6) were bred
in-house and backcrossed to a C57BL/6 background for at least 10 generations. Male
mice were used for all experiments except in Chapter 5, where female mice were used
for the ligature-induced periodontitis experiments. Animals were maintained in the
University of Manchester Biological Services Facility. Animals were housed in
individually-ventilated cages, with an ambient temperature 21±5°C, relative humidity of
55±10%, with a 12-hour light/dark cycle, and given access to food and water ad libitum
under specific pathogen-free conditions. Mice were allowed one week to acclimatise to
the animal unit before commencing experimental procedures. All scientific procedures
were performed in accordance with the Animals (Scientific Procedures) Act (1986) under
the regulation of the Home Office project licences 40/03617 and P28AA2253 and
relevant personal licences in accordance with the local Animal Welfare and Ethical
Review Board (University of Manchester, UK). All reporting of animal experiments
complied with the ARRIVE guidelines (Animal Research: Reporting In Vivo
Experiments).

54
2.3. Generation of head-shielded chimeras
Procedures in this section were performed by Dr Siddharth Krishnan and Dr Joanne
Konkel. Wild-type (wt) CD45.2+ host mice aged 8 weeks were anaesthetised by
intraperitoneal (i.p.) injection of ketamine (80 mg/kg; Vetoquinol) and xylazine (8 mg/kg;
Bayer). Anaesthetised mice were positioned beneath a lead sheet shielding the head,
including the gingiva, from a split dose of irradiation (2 × 5.5 Gy). Mice therefore
received partial body irradiation with only the head unexposed. After recovery from
anesthaesia, mice were reconstituted by intravenous (i.v.) injection with 7 × 106 CD90.2+
T cell-depleted donor bone marrow cells from congenic CD45.1+ wt donor animals. T
cells were depleted using CD90.2 MicroBeads (Miltenyi Biotec). Mice were maintained
on 0.03% enrofloxacin in drinking water for up to 1 week before and for 2 weeks after
irradiation and then were housed in autoclaved cages with sterile water, diet, and bedding.
Reconstitution was allowed to occur for a minimum of 3 weeks before analysis.
2.4. Ligature-induced periodontitis
2.4.1. Pre-procedure
Animals were anaesthetised with an i.p. injection of with a mix of ketamine (50 mg/kg;
Vetoquinol) and medetomidine (650 μg/kg; Vetoquinol) in sterile PBS at a concentration
of 6.6 μl/g for male mice, and 10 μl/g for female mice. Anaesthetised mice prior to
surgeries were kept briefly in a warm cabinet at 30–32 °C.
2.4.2. Ligature placement
Procedures described here are adapted from Abe and Hajishengallis (2013). Lubrithal™
eye gel was applied, and mice were laid in a supine position on a polysytrene board.
Ligature restraints were placed around upper and lower incisors to retract the jaws and
open the mouth in order to access the upper molar teeth. Straight forceps were used to

55
create spaces between second and third molars and second and first molars in order to
facilitate subsequent ligature placement. A 5/0 silk ligature was passed through the space
between the second and third molar and looped around the second molar using straight
and angled forceps (Figure 2.1). The suture was tied firmly with a triple knot on the
palatal side and excess suture cut using spring scissors. Mice received either unilateral or
bilateral ligatures depending on the study, and this stated in the figures and figure
legends.
2.4.3. Post-procedure
Each mouse was revived with 100 µl atipamezole (1 mg/kg; Vetoquinol) administered
subcutaneously. Animals were kept at 30–32°C, monitored until fully recovered from the
effects of anaesthesia, and subsequently placed back in general housing. Mash was
provided post-surgery and general behaviour and appearance were monitored daily. Body
weights were taken every second day at the same time of day. “Control” animals were
anaesthesised only and not subjected to creation of interdental spaces or ligature
placement (as to do so would cause damage that is associated with periodontitis).
Figure 2.1. Ligature-induced periodontitis. Experimental periodontitis was induced in mice by tying 5/0 ligatures around the upper second molar teeth. Ligatures were left in place for 10 days.

56
2.5. Transient middle cerebral artery occlusion
2.5.1. Pre-procedure
All procedures described in this section were performed by Dr Michael Haley. Transient
focal cerebral ischaemia was induced in mice using a modified version of the middle
cerebral artery occlusion method by intraluminal filament (tMCAo) (Longa et al. 1989).
One day before surgery, animals were acclimatised to mash (standard rodent chow mixed
with sterile water). For surgery, anaesthesia was induced using 3‒4% isoflurane and
maintained with 1.5‒2.0% isoflurane in a N2O:O2 mixture (70:30) via face mask. The
depth of anaesthesia was monitored before and throughout surgical procedures by
respiratory rate and toe pinch reflex. Core body temperature was monitored and
maintained homeothermically at 37±1°C throughout surgery using a rectal probe and
heating pad (Harvard Apparatus, USA). In preparation for surgery, the chest and the area
between the animal’s left eye and ear were shaved and disinfected with Videne®
antiseptic solution with topical analgesia provided by EMLA™ cream.
2.5.2. Procedure
Under an operating microscope (Leica Microsystems, UK), the skin between the eye and
ear was incised and the underlying muscle was bluntly dissected to reveal the skull. A
doppler probe connected to an OxyFlo system (Oxford Optronic, UK) was secured on the
skull, roughly in between the eye and ear, using Vetbond® tissue adhesive to dynamically
assess changes in cerebral perfusion (Beretta et al., 2013). In the supine position, the
midline was incised between the manubrium and mandible to expose the submandibular
glands. The glands and underlying fascia were dissected to expose the common carotid
artery (CCA). Further dissection was performed to free the CCA from the vagus nerve
and visualise the internal carotid artery (ICA), external carotid artery (ECA) and the
occipital branch of the CCA. To manipulate the ICA with tension, a loop of 6/0 braided

57
silk suture was placed around it, proximal to the bifurcation. The ECA was ligated distal
to the bifurcation with a silk suture and a loose ligature was placed proximal to the
bifurcation. The occipital and ECA were both electrocoagulated and severed. The ECA
was caudally reflected, tension was applied on the ICA suture and a microvascular clip
was placed on the CCA. Via an arteriotomy on the distal ECA stump, a 6/0 silicon-coated
nylon filament suture was inserted and advanced along the ICA until it was met with
resistance (~10‒12 mm) and/or a > 70% drop in doppler signal was obtained, effectively
occluding the MCA at its origin (Figure 2.2). The ligature around the filament was
tightened and was left in place for 20 minutes. Post-occlusion, the filament was
withdrawn, ligature tightened and the ECA cauterised at the arteriotomy site. All other
sutures and clips were removed; the submandibular glands were returned to their original
positions; the wound was sutured using non-absorbable 6/0 sutures (Ethicon) and the
doppler probe was removed.
Figure 2.2. Transient middle cerebral artery occlusion via intraluminal filament Experimental stroke was induced by advancing a silicon-coated filament into the ECA, through the ICA, to occlude the MCA at its origin. MCA = middle cerebral artery. CCA = common carotid artery. ICA = internal carotid artery. ECA = external carotid artery. PCA = posterior communicating artery

58
2.5.3. Post-surgical care
Animals received a subcutaneous injection of 50 μg/kg buprenorphine (Vetergesic®) and
topical EMLA™ cream at the wound site for analgesia. They were also given a
subcutaneous injection of 500 μl saline for fluid replacement and a choice of moist
mashed food and pellets for feeding. Post-surgery, animals were transferred and group-
housed in recovery cages in an incubator maintained at 27‒28°C and monitored for 6‒8
hours, and afterwards returned to their holding cages which were placed on a thermal
heating blanket. The body weight of animals was assessed daily, and animals received
500 μl saline for fluid replacement at 24 hours post-surgery.
2.5.4. Assessment of neurological deficits
Neurological dysfunction post-stroke were assessed using the criteria adapted from Clark
et al. (1998). The parameters used to assess neurological deficits were body and front
limb asymmetry, gait, limb weakness, circling behaviour, and contralateral whisker
response (Appendix). Neurological assessment was performed prior to culling.
2.6. Permanent middle cerebral artery occlusion
2.6.1. Pre-procedure
Permanent occlusion of the distal MCA (pMCAo) was induced in mice as described
previously (Le Behot et al., 2014). Anaesthesia was induced using 5% isoflurane and
maintained with 2.0‒2.5% isoflurane in a N2O:O2 mixture (70:30). The area between the
right eye and ear was shaved, and the mouse was placed in a sterotactic frame. A rectal
probe was used to monitor core body temperature throughout surgery at 37±1°C using a
heating pad (Harvard Apparatus, USA).

59
2.6.2. Procedure
Under an operating microscope (Leica Microsystems, UK), the skin between the animal’s
right eye and ear was disinfected with Videne® solution and topical analgesia was
provided using EMLA™ cream. A longitudinal incision was made and the underlying
temporal muscle was dissected and detached to reveal the skull. A small cranial window
was drilled directly above the visible MCA at a bifurication in the rostral part of the
temporal area, below the zygomatic arch. A doppler probe (Moor Instruments) connected
to an OxyFlo system (Oxford Optronic, UK) was secured on the skull at a location dorsal
to the cranial window, above the underlying MCA, to dynamically assess changes in
cerebral perfusion (Beretta et al., 2013). A triangle of filter paper soaked in freshly-
prepared 30% ferric chloride (FeCl3) was applied to the MCA and left in place for 5
minutes (Figure 2.3). Saline was applied after removal of filter paper and the site was
monitored for formation of a platelet-rich thrombus for a few minutes. Confirmation of
successful thrombus formation was achieved by visual cessation of blood flow and a
stable cerebral blood flow reduction of ≥ 70% via doppler probe. The thrombus was
allowed to develop for 45 minutes with regular administrations of saline until it fully and
stably occluded the vessel. After this period, the doppler probe was removed, the
temporal muscle was returned to its original position, and the wound was sutured using
non-absorbable 6/0 sutures (Ethicon).
Figure 2.3. Permanent middle cerebral artery occlusion via ferric chloride Experimental stroke was induced by applying filter paper soaked in 30% ferric chloride (FeCl3) to a distal portion of the middle cerebral artery (MCA) at the bifurcation.

60
2.6.3. Post-operative care
Animals received a subcutaneous injection of 50 μg/kg buprenorphine (Vetergesic®) and
topical EMLA™ cream at the wound site for analgesia. They were also given a
subcutaneous injection of 300 μl saline for fluid replacement and a choice of moist
mashed food and pellets for feeding. Post-surgery, animals were transferred and group-
housed in recovery cages in an incubator maintained at 27‒28°C and monitored for 6‒8
hours, and afterwards returned to their holding cages.
2.7. Administration of substances
The following substances were administered in Chapter 4; 100 μg/kg LPS from E. coli
(Ec-LPS; 0127:B8) or 4 mg/kg LPS from P. gingivalis (Pg-LPS) were injected (i.v.) in a
single bolus and mice were euthanised at 2 hours post-administration. In the repeated Pg-
LPS experiment, Pg-LPS (1 mg/kg) was administered i.v. at days 1, 3, 5, 7, 9 post-
periodontitis or control surgery. The same volume of PBS was used as a control.
2.8. Euthanasia & tissue harvesting
Animals were euthanised and their tissues harvested in different ways depending on the
study; in all experiments regarding stroke (Chapter 4), mice were euthanised by terminal
isoflurane anaesthesia (Section 2.8.2). In all other instances, mice were euthanised via
carbon dioxide (CO2) exposure. Both routes are detailed below.
2.8.1. Carbon dioxide exposure
Animals were euthanised by exposure to a rising concentration of CO2. Blood was
withdrawn by cardiac puncture and collected into tubes with 4 ml 3% media with EDTA
(6.25 mM) to prevent coagulation. Plasma was isolated from blood by centrifugation at
1500 xg for 10 minutes. Depending on the particular study, the animal was briefly

61
intracardially perfused with ~20 ml sterile PBS until the liver was visibly perfused,
indicated by a paling in colour. Sub-mandibular lymph nodes, femurs, lungs, spleen,
aorta, guts, and brain were either collected in 3% media for flow cytometry or snap-
frozen on dry ice for molecular analyses. For 16S rDNA PCR purposes, upper lung lobes
were excised using sterile tools and swabs of the oral cavity were taken using a sterile
cotton applicator which was placed inside a sterile eppendorf and snap-frozen. For bone
loss measurements, heads were removed and frozen at -20°C until use.
2.8.2. Terminal anaesthesia & tissue fixation
Animals were intracardially perfused at 48 hours after tMCAo or pMCAo surgeries. Mice
were anaesthetised with 4% isoflurane in a N2O:O2 mixture (70:30). Prior to perfusion,
blood was withdrawn by cardiac puncture and collected into tubes with 4 ml 3% media
with EDTA (6.25 mM) to prevent coagulation. The chest cavity was then opened to
expose the heart. A 25-gauge butterfly needle was inserted into the left ventricle and held
in place with a haemostat and an incision was made in the right atrium. Animals were
perfused with ~50 ml PBS supplemented with heparin (10 I.U./ml) followed by ~50 ml
4% paraformaldehyde (PFA; in PBS) using a peristaltic pump (Watson Marlow, UK).
Tissues not requiring fixation were removed after PBS perfusion and either collected in
3% media for flow cytometry or snap-frozen on dry ice for molecular analyses. Brains
were excised post-fixation and immersed in 4% PFA overnight, transferred to 30%
sucrose solution for a further 24 hours, snap-frozen in isopentane, and stored at -80°C
until use.

62
2.9. Histology
2.9.1. Brain sectioning
Brains were mounted on a stage using dry ice and sectioned coronally into 8 sets at 30
µm thickness using a freezing sledge microtome (Bright Instruments, UK) and kept at -
20°C in cryoprotectant solution (30% ethylene glycol and 20% glycerol in PBS) until
required. For pMCAo, the brains were sectioned into 8 sets of 30 µm thickness and the
following 8 sections were discarded before continuing with the next 8 sets (to yield 480
µm between sections). Sections were mounted on gelatine-coated slides. Slides were
coated previously by washing in industrial methylated spirit (IMS), dipping in gelatine
solution (0.5% gelatine, 0.05% chromium (III) potassium sulphate in distilled water), and
drying overnight.
2.9.2. Cresyl violet staining
Cresyl violet stains the Nissl bodies of the rough endoplasmic reticulum of neurons and
cell nuclei. Cell death causes neuronal nuclei to shrink, a change visible at the
macroscopic level as a paling of the infarcted tissue. PFA-fixed sections mounted on
gelatine-coated slides were stained with cresyl violet before dehydrating in graded
concentrations of alcohols. In brief, slides were consecutively immersed in 95% industrial
methylated spirit (IMS; 2 minutes), running distilled water (2 minutes) and 1% cresyl
violet (~5 minutes). Excess stain was removed from the sections by briefly washing them
in running distilled water followed by dehydration in increasing concentrations of IMS
(95%, 99%, 99%) for 2 minutes each. Sections were finally immersed in xylene (2
minutes) before mounting and coverslipping onto slides using DPX mounting medium.

63
2.9.3. Immunohistochemistry
Immunohistochemistry was performed on free-floating coronal sections from PFA-fixed
brains. Sections were initially incubated in 1% H2O2 in distilled water for 10 minutes to
quench endogenous peroxidase activity. Sections were then incubated with 5% normal
goat serum (Vector Labs) for 1 hour to block non-specific antibody binding. Primary
antibodies were diluted in reagent diluent (2% normal goat serum and 0.3% Triton X-100
in PBS) and applied overnight at 4°C: rabbit anti-mouse SJC-4 (1:10,000) (kindly
provided by Professor D. Anthony, University of Oxford, UK). After washing, a goat
anti-rabbit biotinylated secondary antibody (1:500 in reagent diluent; Vector Labs) was
added for 2 hours at room temperature. Amplification of signal was achieved by
incubating sections with avidin-biotin complex (ABC) solution tagged to a horseradish
peroxidase (Vectastain ABC HRP Elite, Vector Labs) at dilutions of 1:750 of each of
reagent A and B in PBS. Visualisation of positive peroxidase labelling was determined by
adding the chromogen 3,3'-diaminobenzidine (DAB) (0.05% DAB and 0.005% hydrogen
peroxide in distilled water), which forms an insoluble brown precipitate when in the
presence of hydrogen peroxide. Upon colour change, the reaction was stopped by
washing and removal of the DAB solution (usually > 5 minutes). Sections were washed
in PBS, mounted on gelatine-coated slides, and dehydrated in graded alcohols (70%
ethanol for 2 minutes, 90% ethanol for 2 minutes, and 100% ethanol for 5 minutes) and
finally cleared in xylene (10 minutes) before coverslipping with DPX mounting medium.
For determining blood-brain barrier breakdown, indicated by plasma IgG leakage into the
brain parenchyma, immunohistochemical staining was performed as above but with the
omission of the primary antibody step.

64
2.10. Histological quantification
2.10.1. Quantification of ischaemic damage
The volume of ischaemic damage was calculated as described by Osborne et al. (1987) by
measuring the area of neuronal death at eight defined coronal levels (2.22, 1.54, 0.86,
0.14, -0.58, -1.22, -1.94, -2.7 mm according to Bregma) (Paxinos and Franklin, 2001).
For studies involving tMCAo, the infarcted areas were manually drawn onto
corresponding brain maps (in order to correct for oedema) and scanned digitally. A
custom-made macro was used to calculate the area of each level using ImageJ. The total
volume of damage was then found by plotting the area of damage at each level against its
coordinates and calculating the area under the curve using GraphPad Prism (v7)
(GraphPad, USA). For studies involving pMCAo, cresyl violet-stained slides were
scanned and the infarcted area of each section were determined by manually drawing
around the border of cell death (denoted by paling of tissue) in Image J/FIJI. Total
volume of ischaemic damage was calculated as: total infarcted area × thickness of
section.
2.10.2. Quantification of blood-brain barrier breakdown
Images of brain sections stained for IgG were scanned on a Pannoramic 250 Flash III
slide scanner (3DHISTECH, Hungary). IgG staining intensity in the infarcted hemisphere
was calculated as the change in pixel contrast from the contralateral side and averaged
across three coronal levels (0.14, -0.58, -1.94 mm according to Bregma) (Paxinos and
Franklin, 2001) using Image J/FIJI.
2.10.3. Quantification of neutrophils
Images of brain sections stained for SJC-4 were scanned on a slide scanner
(3DHISTECH, Hungary). SJC-4+ neutrophils were quantified at 5X magnification in the

65
entire area of striatum and/or cortex with Image J/FIJI. Counts were averaged over three
coronal levels (0.86, 0.14, 0.58 mm according to Bregma) (Paxinos and Franklin, 2001)
and expressed as number of neutrophils/section.
2.11. Preparation of single cell suspensions
For all procedures in this section, fresh non-fixed tissue was used. Single cell suspensions
from blood, bone marrow, lung, spleen, sub-mandibular lymph nodes, gut, and gingiva
were obtained as detailed below.
2.11.1. Blood
Bloods were centrifuged (500 xg, 5 minutes, 4°C) and the supernatant was carefully
removed and discarded. Pellets were resuspended in 3 ml ACK lysing buffer for 3
minutes at room temperature to lyse red blood cells. Cells were washed with PBS,
centrifuged and the lysis step was repeated. Cells were finally suspended in 500 µl
complete media.
2.11.2. Bone marrow
Femur bones were cut open at the epiphysis at the knee joint end, placed in a 0.5 ml
eppendorf tube in which a hole had been made in the bottom with a 21-gauge needle. This
tube was placed inside an intact 1.5 ml eppendorf and this was centrifuged to maximum
speed for ~10‒15 seconds in a microcentrifuge so that the marrow was flushed out of the
bone into the collecting tube. Cells were resuspended in 3 ml ACK lysing buffer for 3
minutes at room temperature to lyse red blood cells, washed in PBS, centrifuged (500 xg,
5 minutes, 4°C), the supernatant discarded, and the cells resuspended in 1 ml complete
media.

66
2.11.3. Lung
Lung tissue was finely minced with scissors and incubated in 2 ml lung digest buffer
(Section 2.1.3) for 30 minutes at 37°C with agitation. EDTA (5 mM) was added for the
final 5 minutes. Lung tissue was mashed through a 70 µm cell strainer, centrifuged (500
xg, 5 minutes, 4°C) and the supernatant was discarded. Cells were resuspended in 3 ml
ACK lysing buffer for 3 minutes at room temperature to lyse red blood cells, washed in
PBS, centrifuged, the supernatant discarded, and the cells resuspended in 1 ml complete
media.
2.11.4. Spleen
Spleens were mashed through a 70 µm cell strainer and centrifuged (500 xg, 5 minutes,
4°C). The supernatant was discarded, and red blood cells were lysed with 3 ml ACK
lysing buffer for 3 minutes at room temperature. Cells were washed in PBS, centrifuged,
the supernatant discarded, and the cells resuspended in 3 ml complete media.
2.11.5. Sub-mandibular lymph nodes
Lymph nodes were mashed through a 70 µm cell strainer and centrifuged (500 xg, 5
minutes, 4°C). The supernatant discarded, and the cells resuspended in 500 µl complete
media.
2.11.6. Skin
Skin processing was performed by Kelly Wemyss (Figure 5.2A). Briefly, the back was
shaved, and hair removal cream was used to remove skin. Fat was removed with a scalpel
and the skin cut into small pieces. The tissue was transferred to a gentleMACS C tube
(Miltenyi Biotec) containing 3ml skin digest media (complete media with 0.25 mg/ml
Liberase TL and 0.5 mg/ml DNase I). Tubes were incubated on their sides for 1 hour 45
minutes at 37°C with agitation. After incubating, tissue was homogenised in a

67
gentleMACS dissociator (Miltenyi). EDTA (5 mM) was added in the final 5 minutes and
the suspension was passed over a 100 μm MACS smart strainer (Miltenyi) and washed
with 5ml PBS before centrifuging and resuspending in 500 μl complete media.
2.11.7. Gut
Gut processing was performed by Hayley Bridgeman. Guts were harvested and all fat and
Peyer’s patches were removed. Small intestines were isolated, cut longitudinally and the
contents washed thoroughly with PBS on ice. To remove intestinal epithelial cells and
leukocytes, the gut was cut into segments (2–3 cm) and incubated in pre-warmed gut
digest media ‘A’ (Section 2.1.3) and incubated for 20 minutes at 37°C with agitation.
Tissue was transferred to 10 ml pre-warmed serum-free shake solution (RPMI 1640
medium with 100 U/ml penicillin, 100 μg/ml streptomycin, 20 mM HEPES, and 2 mM
EDTA) and shaken for 30 seconds, repeating this process three times in total (sieving the
guts after each new shake) to ensure optimal dissociation of intestinal epithelial cells and
leukocytes. Remaining tissue (lamina propria and muscularis) was finely minced and
incubated in 10 ml gut digest media ‘B’ (Section 2.1.3) for 30 minutes at 37°C with
agitation. Gut tissue was mashed through a 70 μm cell strainer, centrifuged (500 xg, 5
minutes) and the supernatant discarded. The pellet was resuspended in 3% media and
passed through a 40 µm cell strainer, centrifuged, and the supernatant discarded before
finally resuspending in 1 ml complete media supplemented with polymxyin B.
2.11.8. Gingiva
Mouse gingiva was obtained as previously described (Dutzan et al., 2016a). Gingival
blocks were dissected from the mouth and incubated in 2 ml gingival digest media
(Section 2.1.3) per sample for 25–30 minutes at 37°C with agitation. EDTA (50 mM)
was added in the final 5 minutes. Following digestion, tissue was removed from jaw
bones (maxillae/mandibles) using a #10 scalpel and then mashed through a 70 μm cell

68
strainer and washed through with collagenase-free RPMI 1640 medium (with 10 mM
HEPES, 2 mM L-glutamine, and 75 μg/ml DNase I). The tissue was pelleted at 500 xg (5
minutes) and resuspended in 0.2 ml complete media per sample.
2.12. Ex vivo re-stimulation
Cell suspensions from bone marrow and blood were plated in 96-well U-bottom plates
and stimulated with or without LPS (10 ng; Ultrapure LPS from E.coli O111:B4) in the
presence of brefeldin A (5 µg/ml) in complete media for 2.5 hours at 37°C with 5% CO2
without agitation and then stained for flow cytometry.
2.13. Flow cytometry
2.13.1. Surface staining
Single cell suspensions were first stained with Fc receptor block (anti-CD16/32) along
with the appropriate anti-mouse antibodies (Table A.2) in PBS or FACS buffer for 15‒20
minutes at 4°C. Secondary antibodies and/or a cell viability stain (Live/Dead Blue,
Zombie Aqua, Zombie UV) in PBS were incubated with cells for 15‒20 minutes at 4°C.
Cells were subsequently fixed with 2% PFA for 10 minutes at room temperature.
2.13.2. Intracellular and intranuclear staining
For intracellular cytokine staining, cells were first stained for surface markers, fixed in
2% PFA, then permeabilised in 0.5% Saponin (Sigma) for 15 minutes and stained for
intracellular cytokines in Saponin solution for 1 hour at 4°C. For intranuclear staining
(e.g. FoxP3, Ki67), the Foxp3/Transcription Factor buffer set (eBioscience) was used
according to the manufacturer’s instructions. Briefly, cells were stained and fixed in
Fixation/Permeabilisation buffer for 30 minutes, permeabilised in Permeabilisation buffer

69
for 15 minutes and then incubated with the appropriate intranuclear antibodies in
permeabilisation buffer for 1 hour at 4°C.
2.13.3. Sample acquisition
Gingival suspensions were first filtered through a 35 μm strainer prior to acquisition.
Samples were acquired on a BD Fortessa™ flow cytometer (BD Biosciences) and
analysed using FlowJo software v10 (FlowJo LLC, USA). Compensation was performed
using UltraComp eBeads (eBioscience) for all antibodies and ArC Kit (Fisher Scientific)
for cell viability stains. Fluorescence-minus-one controls were used to validate positive
staining. A “Lineage” gate was used to exclude multiple cell populations.
2.14. Sorting of haematopoietic stem and progenitor cells
Haematopoietic stem and progenitor cells (HSPCs) were isolated from single-cell
suspensions of gingiva (Section 2.11.8) of young and aged mice. For each sample, tissue
from two mice was pooled in order to obtain adequate cell numbers. Cells were stained
for surface markers and cell viability (Zombie Aqua), then resuspended in FACS buffer
prior to acquisition. HSPCs were first defined based on surface marker expression: Live,
CD45+, Lineage (CD3, TCR-β, CD19, B220, NK1.1, Ly6G, Siglec F, Ter119, CD11c,
FcεR1)-, CD11b-, Ly6C-. HSPCs were then divided into four populations based on
differential expression of c-Kit and Sca-1. Cells were sorted on a BD FACSAria™
Fusion and collected in 1 ml complete media before plating on MethoCult™ medium
(Section 2.22).
2.15. Bone loss measurements
For bone loss, mice were euthanised with a rising concentration of CO2. Heads were
removed and frozen at -80°C until use. All skin and fur were manually removed while the

70
head was still frozen, and the head was wrapped in aluminium foil and autoclaved at
121°C for 15 minutes. The mandible (lower jaw) was removed by blunt dissection. The
left and right maxillae (upper jaws) were carefully isolated and the flesh was gently
removed, taking care not to dislodge the teeth, and each maxilla left overnight in 1 ml 3%
H2O2 and subsequently in 1 ml PBS overnight. Maxillae were immersed in 1% methylene
blue for 30 seconds, which stains the underlying bone but not the teeth, followed by a 10-
second rinse in bleach, and finally washed twice in deionised water for 2 minutes each to
remove excess dye. Maxillae were left to dry overnight, separate to one another, in a 12-
well cell culture plate. For imaging, maxillae were fixed in moldable putty (“Play-Doh”)
and aligned accordingly. Images of the palatal side of the maxillae were taken using a
Leica Stereo Fluorescence M205 FA (Leica Microsystems, UK). Bone loss was then
calculated using 6 different sites across all 3 molar teeth by measuring the distance from
cemento-enamel junction (CEJ) and alveolar bone crest (ABC) using Image J (NIH,
USA).
2.16. Bacterial growth evaluation
Following euthanasia, lungs were excised and placed in 2 ml sterile PBS in a
gentleMACS M tube (Miltenyi Biotec) and kept on ice. The oral cavity was swabbed with
a sterile cotton applicator (10 seconds on each of the left buccal and right buccal sides
and 10 seconds on the palate) and the applicator placed in 1 ml sterile PBS and briefly
vortexed. Lungs were homogenised using a gentleMACS dissociator (Miltenyi Biotec).
For lungs, 100 μl homogenate was plated neat or at a 1:10 dilution on tryptone soy agar
plates (Oxoid) and incubated at 37°C for 24 hours to detect non-selective growth of
aerobic bacterial species. For oral swabs, 50 μl solution was plated neat or at serial 1:10
dilutions and incubated at the same conditions. To detect anaerobic growth, oral swab
dilutions were spread on plates containing Wilken-Chalgrens anaerobe agar (Oxoid) and

71
placed in a hypoxic chamber at 37°C for 72 hours. Colony forming units (CFUs) were
determined by manually counting bacterial colonies.
2.17. Cytometric bead array
Alterations in cytokines in the plasma cytokine levels were determined using
LEGENDplex™ Mouse Inflammation Panel (BioLegend), according to the
manufacturer’s instructions but with a 5-fold reduction in the recommended volumes.
This dilution has been validated in the laboratory, giving comparable results to the
undiluted assay. Samples were acquired using a BD FACSVerse™ flow cytometer and
the FCS files were analysed using the proprietary LEGENDplex™ software (v8;
BioLegend) to obtain cytokine concentrations. To permit statistical analyses, samples
with no detectable cytokine levels were assigned the lowest level of detection which was
specified for each analyte in the manual.
2.18. Cerebral microvessel isolation
For cerebral microvessel isolation, mice were briefly intracardially perfused with ~10 ml
PBS, the brains were removed and snap-frozen on dry ice and stored at -80°C until use.
Once defrosted, the cerebella and as much of the white matter were removed with
tweezers, and the brains transferred into 1 ml ice-cold Dulbecco’s Modified Eagle
Medium (DMEM/F-12) in a 7 ml Dounce homogeniser (Glass Precision Engineeing Ltd).
A #10 scalpel was then used to mince the tissue until pieces no greater than 1 mm3
remained. Another 1 ml DMEM/F-12 was added and the tissue homogenised using the
large clearance pestle in ~80 strokes until no large pieces were left, followed by
homogenisation with the small clearance pestle for ~15 strokes. The homogenate was
transferred to a 15 ml falcon along with 3 × 2 ml washes of the Dounce homogeniser and
centrifuged at 200 xg for 5 minutes at 4°C. The supernatant was then removed, and the

72
pellet resuspended in 5 ml 18% dextran solution (in DMEM/F-12) which was then
centrifuged at 4000 xg for 10 minutes at 4°C. The white myelin layer and dextran
supernatant were carefully discarded, and the pellet resuspended in 1 ml Hank’s Balanced
Salt Solution. This suspension was then passed through a 70 µm cell strainer on top of a
50 ml falcon three times so that microvessels caught on the strainer but other cellular
debris passed through. Each time the suspension was passed through the strainer, the
mesh was inverted and the collected microvessels washed off into a fresh 50 ml falcon.
The purified microvessels were pelleted by centrifuging at 1000 xg for 10 minutes at 4°C
and frozen at -80°C in Qiazol lysis reagent (Qiagen, UK).
2.19. qRT-PCR
2.19.1. RNA isolation
Tissue previously stored at -80°C was transferred to Qiazol lysis reagent (in a 10%
[weight/volume]), in Matrix D Lysing tubes. Samples were homogenised using a
FastPrep-24 machine (MP Biomedicals, UK) at 5m/s-1 in 30 second bursts until fully
disaggregated. Samples were transferred to sterile eppendorf tubes, 200 µl chloroform
was added to each sample, and tubes were centrifuged at 12,000 xg for 15 minutes.
Following centrifugation, the mixture separated into a lower, red phenol-chloroform
phase, an interphase, and a colourless upper aqueous phase. Without disturbing the
interphase or lower phase, the upper phase was carefully transferred to fresh eppendorfs.
Iso-2-propanol (500 µl/sample) was added to precipitate the RNA and incubated for 10
minutes at room temperature and then centrifuged at 10,000 xg for 10 minutes. The
supernatants were discarded, the pellets resuspended in 1ml 75% ethanol to wash and
then centrifuged at 5000 xg for 5 minutes. Supernatants were discarded, and the tubes
inverted on tissue paper with lids open to air-dry for ~1 hour. Once dry, each sample was

73
resuspended in 30 µl nuclease-free water and RNA concentration was determined using a
NanoDrop 2000 Spectrophotometer. Samples were stored at -80°C until further use.
2.19.2. cDNA synthesis and qRT-PCR
RNA concentration was equalised amongst samples in nuclease-free water and
complementary DNA (cDNA) was generated using a High Capacity cDNA Reverse
Transcription Kit (Thermo Fisher). The reaction was carried out in a Veriti 96-Well
Thermal Cycler (Applied Biosystems, UK) according to the following parameters: 10
minutes at 25°C, 120 minutes at 37°C and 5 minutes at 85°C. cDNA was kept at 4°C until
collection. Reactions were set up as follows: 5 µl Fast SYBR Green Master Mix, 0.5 µl
(10 µM) forward primers, 0.5 µl (10 µM) reverse primers, 0.5 µl cDNA, 3.5 µl nuclease-
free water for a 10 µl total reaction volume per sample. Samples were plated in triplicate
in a 384-well plate and acquired on a Quant Studio 12K Flex Real-Time PCR System
(Applied Biosystems). The expression of inflammatory genes was determined using the
∆∆-CT method as described by Livak and Schmittgen (2001). All genes were expressed
relative to the housekeeping genes gapdh or hprt. Primer sequences for genes of interest
were purchased from Sigma-Aldrich using validated sequences obtained on PrimerBank
(https://pga.mgh.harvard.edu/primerbank/) (Table 2.1).
Table 2.1. Sequences for genes of interest
Gene Forward Primer Reverse Primer
gapdh 5’ – AGGTCGGTGTGAACGGATTTG – 3’ 5’ – TGTAGACCATGTAGTTGAGGTCA – 3’
hprt 5’ – TCCTCCTCAGACCGCTTTT – 3’ 5’ – CCTGGTTCATCATCGCTAAT – 3’
cxcl1 5’ – CTGGGATTCACCTCAAGAACATC – 3’ 5’ – CAGGGTCAAGGCAAGCCTC – 3’
il-17 5’ – TTTAACTCCCTTGGCGCAAAA – 3’ 5’ – CTTTCCCTCCGCATTGACAC – 3’
il-6 5’ – CCAAGAGGTGAGTGCTTCCC – 3’ 5’ – CTGTTGTTCAGACTCTCTCCCT – 3’
il-1β 5’ – TGTAATGAAAGACGGCACACC – 3’ 5’ – TCTTCTTTGGGTATTGCTTGG – 3’
tnfα 5’ – CCCTCACACTCAGATCATCTTCT – 3’ 5’ – GCTACGACGTGGGCTACAG – 3’
icam-1 5’ – GTGATGCTCAGGTATCCATCCA – 3’ 5’ – CACAGTTCTCAAAGCACAGCG – 3’

74
vcam-1 5’ – AGTTGGGGATTCGGTTGTTCT – 3’ 5’ – CCCCTCATTCCTTACCACCC – 3’
iba-1 5’ – ATCAACAAGCAATTCCTCG – 3’ 5’ – CAGCATTCGCTTCAAGGACATA – 3’
2.20. Bacterial 16S rDNA quantification
2.20.1. DNA isolation
DNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen, UK). Briefly, lung
tissue or oral swabs were suspended in 180 µl enzymatic lysis buffer (30 µl 20 mg/ml
lysozyme and 150 µl 1X TE buffer [20 mM Tris pH 7.4, 2 mM EDTA]) and incubated at
37°C for ~1 hour. Proteinase K (25 µl) and Buffer AL (200 µl) were then added to each
sample and tubes incubated at 56°C for ~2 hours until the contents became translucent.
200 µl ethanol was added to each sample and the mixture then pipetted onto a DNeasy
Mini spin column placed in a 2 ml collection tube. Samples were centrifuged at 5000 xg
for 1 minute and the collection tubes discarded. Spin columns were placed in new
collection tubes and 500 µl Buffer AW1 was added to each sample. Samples were
centrifuged at 5000 xg for 1 minute and the collection tubes discarded. Spin columns
were placed in new collection tubes and 500 µl Buffer AW2 was added to each sample.
Samples were centrifuged at 13,000 xg for 3 minutes and the collection tubes discarded.
Spin columns were placed in clean 2 ml microcentrifuge tubes and 100 µl Buffer AE was
added directly to the DNeasy membrane. Samples were briefly incubated for 1 minute
and then centrifuged at 5000 xg for 1 minute to elute the DNA. DNA concentrations were
determined using a NanoDrop™ 2000 Spectrophotometer (Thermo Fisher).
2.20.2. 16S qPCR
Bacterial rDNA was amplified using universal 16S rDNA primers as described previously
(Nadkarni et al., 2002): Forward primer = 5′-TCCTACGGGAGGCAGCAGT-3′ (Tm,
59·4 °C); Reverse primer = 5′-GGACTACCAGGGTATCTAATCCTGTT-3′ (Tm, 58·1

75
°C) and probe = (6-FAM)-5′-CGTATTACCGCGGCTGCTGGCAC-3′-(TAMRA) (Tm,
69·9 °C). Total genomic DNA from E. coli (ECAT1177T) was used to construct standard
curves of known number of 16S rRNA gene copies (ranging from 102 to 108). Sample
DNA was equalised to the lowest sample concentration before running. Reactions were
set up as follows: 5 µl TaqMan Gene Expression Master Mix, 0.5 µl 16S rDNA
primer/probe mix (20X), 1 µl equalised DNA, 3.5 µl nuclease-free water for a 10 µl total
reaction volume per sample. Samples and standards were plated in triplicate and acquired
on a Quant Studio 12K Flex Real-Time PCR System. Thermal cycle conditions were
50°C for 2 minutes, 95°C for 10 minutes and 40 cycles of: 95°C for 15 seconds followed
by 60°C for 1 minute.
2.21. RNA sequencing
Dr Joanne Konkel processed samples and Dr Leo Zeef and Dr. Andy Hayes performed
the sequencing, read mapping, and DESeq analysis in this section. Dr Ian Prise performed
gene-set enrichment analysis (GSEA). Bulk RNA sequencing on gingiva from young (8
weeks) and “aged” (6 months) mice was performed in-house at the University of
Manchester Genomics Facility. Total RNA from samples was isolated using the phenol-
chloroform method (Section 2.19.1) and the concentration was assessed using the
Qubit™ RNA HS Assay Kit according to the manufacturer’s protocol. The quality of the
isolated RNA was tested using the RNA ScreenTape assay on the 4200 TapeStation
(Agilent Technologies) as per the manufacturer’s protocol. Subsequently, RNA samples
were precipitated adding 0.1 volume of 3 M sodium acetate and 2 volumes of 100%
ethanol for sequencing. Briefly, cDNA libraries were generated using the SMART-Seq
V3 Ultra Low Input RNA Kit (CloneTech) and samples were sequenced on an Illumina
HiSeq4000 platform.

76
2.21.1. Analysis
Unmapped paired-end sequences were tested by FastQC
(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequence adapters were
removed and reads were quality-trimmed and filtered using Trimmomatic (v0.36) (Bolger
et al., 2014). Reads were mapped against the reference mouse genome (mm10/GRCm38)
and counts per gene were calculated using annotation from GENCODE M12 using STAR
(v2.5.3) (Dobin et al., 2013). Filtered reads were then normalized and compared using
DESeq2_1.16.1 (Love et al., 2014). Differentially expressed genes (DEG) were identified
by an adjusted p value (p adj.)/false discovery rate (FDR) < 0.05 and fold change (FC) >
1 or < -1. For functional analysis, gene ontology (GO) enrichment of DEGs was
performed using AmiGO 2 that employs PANTHER as the search engine (Boyle et al.,
2004). Volcano plots using pair-wise FDR and FC, and heatmaps of selected genes using
GO analyses were created using GraphPad Prism (v7). GSEA was performed using
defined gene sets obtained from the Molecular Signatures Database (MSigDB) hallmark
gene set collection (Mootha et al., 2003; Subramanian et al., 2005).
2.22. Colony-forming assay
MethoCult™ medium is a semi-solid methylcellulose medium containing IL-3, IL-6,
stem cell factor, and erythropoietin, which supports the differentiation of multipotent
haematopoietic progenitors into multiple lineages, including granulocyte-macrophage
colonies (cfu-GM). Single cell suspensions from bone marrow or gingiva (Section 2.11),
or FACS-sorted progenitor populations were first washed twice with PBS (supplemented
with 100 U/ml penicillin, 100 μg/ml streptomycin), and then suspended in MethoCult™
medium at a 1:10 dilution. Suspensions were plated directly into the centre of a well of a
6-well plate using a 20-gauge needle attached to a 5 ml syringe. Cultures were plated in
duplicate where possible. Plates were incubated at 37°C with 5% CO2 for 10–13 days and

77
then the quantity of cfu-GM in each well was determined by manual counting of colonies
and collated to give a total cfu-GM count per sample. For subsequent flow cytometry to
determine the identities of the cell progeny, colonies were resuspended in PBS and
stained as usual with an appropriate antibody cocktail.
2.23. Experimental considerations
2.23.1. Group allocation
Where appropriate, animals were allocated treatments or surgical procedures in a
randomised order across cages. The order of surgeries was also randomised. Each cage
was designated as an experimental block, and within each cage, mice were randomly
assigned a number from 1‒5 by an online random number generator.
2.23.2. Sample size
In all animal experiments, sample sizes were determined to allow adequate statistical
power, and this was determined based on a combination of the literature and past
experience. Specifically, for stroke studies we calculated sample sizes by power
calculations of previous data (α=0.05, β=0.2); this data was from previous co-morbidity
experiments on obesity and stroke in the lab, using the group with the highest standard
deviation and an outcome of a 50% change in infarct volume as a guide. In this way, we
accounted for variability in infarct volumes and exclusionary events and used sufficient
numbers to allow for drop outs. Numbers in each group are stated in the figure legends.
2.23.3. Blinding
During in vivo experiments the same experimenter performed surgeries and the tissue
processing, so blinding of surgery status was not possible. However, the investigator was

78
blinded to the treatments administered where appropriate and was blinded downstream
before any histological quantification (such as calculating infarct volumes).
2.23.4. Exclusion criteria
In the fMCAo study, animals were excluded from analysis due to mortality during or after
surgery (n = 1 out of 12, periodontitis + stroke group), presence of sub-arachnoid
haemorrhage (n = 1 out of 12, stroke group) or if they did not have both ligatures in place
at time of sacrifice (n = 1 out of 12, periodontitis + stroke group). In the pMCAo study,
animals were excluded due to absence of stroke (validated by cresyl violet staining) (n =
1 out of 7, stroke PBS group; n = 1 out of 8, stroke LPS group; n = 2 out of 7,
periodontitis + stroke PBS group;).
2.23.5. Data and statistical analyses
All statistical analyses were performed using GraphPad Prism (v7) using the appropriate
test (stated in the figure legends). In instances where variances were significantly
different between groups (determined by Welch’s test for 2 groups, Brown-
Forysthe/Bartlett’s test for 3 groups, and Shapiro-Wilks normality test for >3 groups in
GraphPad Prism), the data was transformed prior to performing the statistical test until
the variance tests were not significant (details are provided in the figure legends). All
figures show untransformed data even if data was transformed. All data are presented as
mean ± standard error (SEM). p < 0.05 was considered statistically significant.

79
Chapter 3. Immune alterations during
experimental periodontitis

80
3.1. Introduction
Periodontitis has emerged as an important risk factor for a multitude of systemic diseases,
which range from cancerous (Abed et al., 2016) to inflammatory (Ogrendik, 2009) to
neurodegenerative (Dominy et al., 2019). Despite persistent epidemiological links, the
means by which oral pathology contributes to extra-oral pathology in many of these
disease settings are largely ill-defined. That being said, a number of shared mechanisms
have been suggested. Principally, these relate to the dissemination of bacteria or oral-
derived products (from host or bacteria) through the bloodstream, and/or the systemic
activation of inflammatory responses, leading to adverse consequences at distal tissue
sites (Geerts et al., 2002; Kinane et al., 2005; Loos, 2005). Recently, a number of other
more speculative mechanisms have been proposed in experimental studies to link
periodontitis with effects at extra-oral sites, as neuronal signals (Klose et al., 2017;
Wallrapp et al., 2017), innate immune training (Askenase et al., 2015; Novakovic et al.,
2016), and commensal bacterial metabolites (Trompette et al., 2014, 2018; Rothhammer
et al., 2018), are instances whereby events at mucosal sites can modulate long-range
immunity. Even though these are mechanisms involved in inter-tissue crosstalk, they have
yet to be explicitly shown in the context of experimental periodontitis. Nevertheless, it is
prudent to appreciate that the mechanisms by which periodontitis impacts certain distal
sites could be a product of one, two, or many of the aforementioned mechanisms, such is
the diverse nature of the diseases in which it is proposed to contribute. What is apparent,
however, is that dysregulated immunity is key not only in periodontitis pathology but is
also central to the aetiology and progression of many of the diseases it is associated with,
and as such, it is likely an important element that links oral disease to extra-oral
complications.

81
Thus, given the clinical relevance of the diseases in which periodontitis is proposed to
impact, it is critical to fully appreciate the dynamics of the host response during
periodontitis. This means locally; in the gingiva and associated lymphoid tissues, as well
as systemically; in the circulation, the bone marrow, and other peripheral sites distant
from the primary disease focus. While our knowledge of systemic immune alterations is
still limited, our understanding of the oral immune network during periodontitis, and
indeed, in health, is quite well-defined (Cekici et al., 2014; Moutsopoulos and Konkel,
2018). Furthermore, our knowledge of oral immunopathology has been recently enhanced
through elegant demonstrations of the pathogenic roles of Th17 cells and neutrophils in
promoting tissue and bone damage in mice (Eskan et al., 2012; Moutsopoulos et al., 2014,
2017, Dutzan et al., 2017, 2018), and conversely, the protective roles of Tregs (Garlet et
al., 2009) and amphiregulin-producing γδ T cells (Krishnan et al., 2018), highlighting the
complex interplay between the cellular mediators that promote bone destruction and those
that mitigate it. Though it is evident that the neutrophil-Th17 axis is key in disease
pathogenesis (Dutzan et al., 2018), recent reports have implicated other immune
populations, such as B cells and mast cells, in contributing to inflammatory bone loss in
mouse models (Abe et al., 2015; Oliver-Bell et al., 2015; Malcolm et al., 2016). Further,
there is a specific pathogenic role for locally-produced soluble mediators, such as TNFα
and IL-1β (Graves and Cochran, 2003), prostaglandin E2 (Noguchi and Ishikawa, 2007),
and recently, the IL-1 family alarmin, IL-33 (Malcolm et al., 2015). All promote tissue
damage and bone destruction, primarily by increasing the expression of RANKL on
immune cells, which leads to osteoclast-mediated resorption of bone. Thus, there exists a
large body of pre-clinical evidence that defines the local contributions of immune cells
and their associated cytokines to inflammatory bone loss during periodontitis.
As mentioned above, far less is known about the immune landscape outside the
immediate oral environment during periodontitis, the alteration of which may be an

82
important predictor of downstream adverse complications. In patients, periodontitis leads
to elevated circulating levels of a range of soluble inflammatory mediators, including
CRP, IL-6, IL-1β, and TNFα amongst others (Noack et al., 2001; Loos, 2005; Duarte et
al., 2010). This can potentially lead to deleterious effects on systemic health, since it is
well-established in other disease contexts that a heightened chronic inflammatory state
exacerbates disease progression and worsens outcome, including rheumatoid arthritis
(Sattar et al., 2003), neurodegeneration (Perry et al., 2007) and stroke (McColl et al.,
2007). However, regarding periodontitis specifically, it is not known how raised levels of
circulating pro-inflammatory molecules affect systemic health or disease progression,
what distant tissues they impact, or which mediators in particular are important for
proposed adverse effects. Little is known about the nature of the systemic leukocyte
response, which remains poorly defined in both mice and humans. Addressing the role of
systemic immunity in humans is severely limited by the fact that the blood is often the
only point of sampling and overlooks how immune responsiveness at peripheral tissue
sites are shaped by oral inflammation. When data is available for changes in the
circulating immune profile, these lack detail about phenotype and functionality and have
only been broadly characterised as white blood cells, lymphocytes, monocytes, and
neutrophils (Loos et al., 2000; Nagasawa et al., 2004; Zekonis et al., 2014).
Given the drawbacks in trying to evaluate systemic immune responses in patients, rodents
represent amenable tools and can be effectively used to dissect microbial-host interactions
and define the humoral and cellular immune responses in a range of tissues. As
previously discussed, the majority of rodent periodontitis studies are centred on
immunopathology in the oral compartment only, with few studies exploring how extra-
oral sites are affected during periodontitis. For example, there are reports of increased
vascular inflammation and endothelial dysfunction during periodontitis, which may
precede cardiovascular complications (Qi et al., 2003; Brito et al., 2013; Miyajima et al.,

83
2015). Nevertheless, the majority of studies are centred around how oral bacteria manage
to evade and subvert immune defences at distant sites (Miyakawa et al., 2004;
Hajishengallis et al., 2011; Maekawa et al., 2014), rather than how the host mounts its
defences in the first place.
In this way, the current experimental focus is largely on the virulence factors that allow
periodontal bacteria (such as P. gingivalis and F. nucleatum) to successfully colonise at
distinct extra-oral niches (Hajishengallis, 2015), or the specific qualities of the microbes
that enhance pathogenic features of the disease, by increasing plaque load during
atherosclerosis (Li et al., 2002), killing neurons during AD (Dominy et al., 2019),
inhibiting tumour killing during colorectal cancer (Abed et al., 2016), or the citrullination
of proteins during RA (Konig et al., 2016). By contrast, the nature of how immune cells
at these sites respond and how they carry out their effector function is comparably ill-
defined. This being said, some studies have addressed immune involvement, for example,
F. nucleatum in the colon binds to the inhibitory receptor TIGIT (T-cell immunoreceptor
with Ig and ITIM domain) on NK cells and T cells via its Fap2 protein and subsequently
prevents immune cell-mediated tumour killing in colorectal cancer (Gur et al., 2015).
Despite this example, in most other contexts, analyses of immune involvement is limited
only to generalised hallmarks of inflammation, such as levels of pro-inflammatory
cytokines like IL-6 (Lalla et al., 2003; Arimatsu et al., 2015) or TNFα (Dominy et al.,
2019), and accordingly, details about the other critical aspects of host immunity are often
overlooked or not addressed fully.
3.2. Aims
Thus, there is a gap in the knowledge of the exact dynamics of systemic host immunity to
periodontitis. We therefore set out to investigate compartmentalised alterations in

84
immune cells in response to experimental periodontitis and to also determine whether
dissemination of bacteria and elevation of systemic inflammatory mediators were a
feature of this disease model, which are proposed complications of the clinical disease.
The main aim of this study therefore was to fully characterise both the local and systemic
immune consequences of ligature-induced periodontitis in a multitude of tissue sites to
define the extent by which periodontitis alters host immunity. As a secondary aim, we
compared the effects of a single or double ligature, in order to establish whether systemic
effects of periodontitis are dependent on the number of afflicted teeth, as the severity of
the disease may be correlated with impact on the systemic compartment.
3.3. Results
3.3.1. Ligature-induced periodontitis induces inflammatory bone
loss, bacterial outgrowth, and local immune alterations
As a number of different periodontitis models exist in mice (Section 1.7), each with
inherent advantages and limitations, we aimed to use a well-established model that would
best mimic the clinical disease hallmarks. Infection-based approaches require chronic
antibiotic pre-treatment known to causes disturbances in immune function (Scott et al.,
2018), and was thus unsuitable for our purposes. Therefore, we employed a mechanical
approach by tying ligatures around the molar teeth, which is reported to drive bone
destruction and local inflammation in a predictably acute timeframe (Abe and
Hajishengallis, 2013). Indeed, within ten days of ligature placement we observed robust
bone destruction, marked by significant increases in the distance from the cemento-
enamel junction to the alveolar bone crest across multiple tooth sites (Figure 3.1A-C).
Moreover, the diseased gingiva exhibited a doubling in overall cell number (Figure
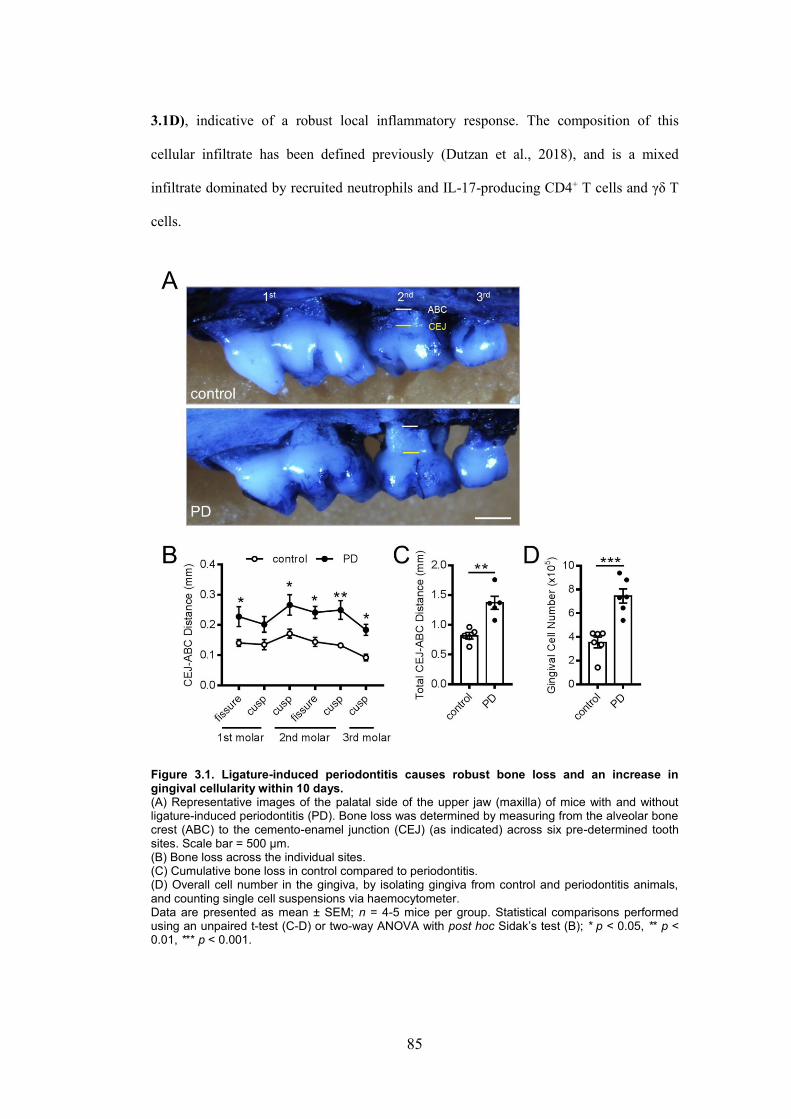
85
3.1D), indicative of a robust local inflammatory response. The composition of this
cellular infiltrate has been defined previously (Dutzan et al., 2018), and is a mixed
infiltrate dominated by recruited neutrophils and IL-17-producing CD4+ T cells and γδ T
cells.
Figure 3.1. Ligature-induced periodontitis causes robust bone loss and an increase in gingival cellularity within 10 days. (A) Representative images of the palatal side of the upper jaw (maxilla) of mice with and without ligature-induced periodontitis (PD). Bone loss was determined by measuring from the alveolar bone crest (ABC) to the cemento-enamel junction (CEJ) (as indicated) across six pre-determined tooth sites. Scale bar = 500 μm. (B) Bone loss across the individual sites. (C) Cumulative bone loss in control compared to periodontitis. (D) Overall cell number in the gingiva, by isolating gingiva from control and periodontitis animals, and counting single cell suspensions via haemocytometer. Data are presented as mean ± SEM; n = 4-5 mice per group. Statistical comparisons performed using an unpaired t-test (C-D) or two-way ANOVA with post hoc Sidak’s test (B); * p < 0.05, ** p < 0.01, *** p < 0.001.

86
In addition to a destructive host inflammatory response, periodontitis pathology is
intimately dependent on the microbial community (as antibiotic-treated rodents exhibit
diminished bone loss (Bezerra et al., 2002)). Thus, we sought to determine if there was a
shift in oral bacterial burden in ligated animals. To achieve this, swabs of the oral cavity
were cultured in either aerobic or anaerobic conditions and we found a marked increase in
both aerobic (Figure 3.2A) and anaerobic (Figure 3.2B) communities. In tandem, we
evaluated bacterial load by quantification of DNA from the bacterial 16S ribosome in the
oral cavity (Figure 3.2C), which appeared to trend towards an increase (p = 0.06) with a
single ligature and further with double ligature placement. Taken together, these data
indicate that the ligature model induces robust microbial growth, in line with the clinical
disease.

87
Figure 3.2. Ligature-induced periodontitis induces profound bacterial growth in the oral cavity within 10 days. (A) Representative images of agar plates grown under aerobic conditions. Suspensions from oral swabs were plated on tryptone soy agar and incubated for 24 hours at 37°C. Colonies were manually counted and expressed as Log10 CFU/ml. CFU = colony forming units. (B) As in (A) except suspensions plated on anaerobic agar and incubated for 72 hours in the absence of oxygen. (C) Number of copies of bacterial 16S rDNA in oral swabs. Total rDNA was amplified using universal 16S rDNA primers using genomic DNA from E. coli as reference standard. Data are presented as mean ± SEM; n = 4-5 mice per group. Data in (C) was square root transformed to permit statistical analysis. Statistical comparisons performed using an unpaired t-test (A-B) or one-way ANOVA with post hoc Tukey’s test (C); *** p < 0.001. Lig. = ligature.
Since the ligature model recapitulated the fundamental aspects of human periodontitis, we
next moved from the oral cavity and focused our attention on the spectrum of immune

88
alterations in other tissue sites. In this manner, we undertook a broad approach to
immunophenotype a number of immune populations, which are enriched in the oral
mucosa (Dutzan et al., 2016b; Brown et al., 2018), in order to shed light on the role of
monocytes, neutrophils, T cells, and ILCs at extra-oral sites (Figure 3.3). We also chose
to specifically investigate monocyte phenotype, using the surface markers CD62L,
CX3CR1, and MHCII to probe activation, trafficking, and differentiation state, as
CX3CR1hi monocytes and F4/80+ macrophages are reported to contribute to inflammatory
bone loss during oral infection with P. gingivalis (Lam et al., 2014; Steinmetz et al.,
2016), but their role in ligature-induced periodontitis is ill-defined.
Figure 3.3. Gating strategies to identify myeloid and lymphoid cells using flow cytometry.

89
(A) Gating strategy to identify neutrophils and Ly6Chi monocytes, as well as surface markers CD62L, CX3CR1, and MHCII.
(B) Gating strategy to identify conventional T cells (TCR+), T cells (TCR+), innate lymphoid
cells (ILCs), CD4+ T cells, CD8+ T cells, as well as RORt+ and FoxP3+ CD4+ T cells.
Beginning at the sub-mandibular lymph nodes (smLNs), we observed increased cell
numbers, in line with the inflammatory response in the gingiva, and this was significantly
increased in animals with double ligatures compared to those with a single ligature
(Figure 3.4A), highlighting that increased severity drives appropriate increases in
immune responsiveness in this model. Interestingly, neutrophils in the smLNs were
significantly increased in animals with two ligatures compared to single ligature and
control mice (Figure 3.4B), although this was not extended to monocytes, as frequencies
and phenotype of the inflammatory Ly6Chi subset remained unaltered (Figure 3.4C-D).
While neutrophils were altered during experimental periodontitis, frequencies of T cells,
γδ T cells, and ILCs in the smLNs were not similarly modulated (Figure 3.5), indicating
the compartmentalised nature of the immune response.

90
Figure 3.4. Ligature-induced periodontitis induces an increase in neutrophils in the draining sub-mandibular lymph nodes. (A) Overall cell numbers in the sub-mandibular lymph nodes (smLN) 10 days post-ligature placement. (B-C) Representative FACS plots and frequencies of neutrophils and Ly6Chi monocytes in the smLNs. Populations were gated as shown in Figure 3.3A. (D) Frequencies of smLN Ly6Chi monocytes expressing CD62L, CX3CR1 and MHCII. Data are presented as mean ± SEM; n = 4-5 mice per group. Data in (A) was log transformed to permit statistical analysis. Statistical comparisons performed using a one-way ANOVA with post hoc Tukey’s test; * p < 0.05, ** p < 0.01, *** p < 0.001. Lig. = ligature.

91
Figure 3.5. Ligature-induced periodontitis does not affect lymphoid populations in the draining sub-mandibular lymph nodes.
(A-B) Frequencies of TCR+, TCR+, ILCs (innate lymphoid cells), CD4+ T cells, and CD8+ T cells in the sub-mandibular lymph nodes (smLNs) 10 days post-ligature placement.
(C) Frequencies of CD4+ T cells expressing FoxP3 and RORt in the smLNs. Data are presented as mean ± SEM; n = 4-5 mice per group. Statistical comparisons performed using a one-way ANOVA with post hoc Tukey’s test. Lig. = ligature.
3.3.2. Ligature-induced periodontitis does not lead to increased
bacterial load or altered immunity in the lung
Despite some evidence implicating specific periodontal bacteria to an exacerbation of
pre-existing lung pathology (Kimizuka et al., 2003; Tan et al., 2014), there is limited
knowledge about the inflammatory effects of periodontitis in a non-diseased lung

92
environment. Given the anatomical continuity of the lung and oral cavity (Mojon, 2002),
it is feasible that oral inflammatory pathology, immune trafficking, and bacterial growth
may lead to adverse pulmonary effects, and possibly further systemic consequences.
Thus, we investigated if periodontitis-induced oral pathology could induce inflammation
and bacterial translocation in the lung.
Since we observed a dramatic increase in bacterial load in the oral cavity (Figure 3.2), we
reasoned that a higher oral bacterial burden could lead to increased dissemination of
microbes into the airways. Thus, we cultured whole lung homogenates from control and
double-ligatured mice under aerobic conditions (Figure 3.6A). However, we did not
observe any discernible bacterial growth in either group, although one mouse with
periodontitis exhibited noticeable colony formation (Figure 3.6B). On a molecular level,
bacterial 16S rDNA copy number in the lung was not increased (Figure 3.6C), which,
when taken together, suggests that experimental periodontitis does not induce robust
translocation of microbes from oral to pulmonary compartments.

93
Figure 3.6. Ligature-induced periodontitis does not induce robust bacterial growth in the lungs within 10 days. (A) Representative images of agar plates grown under aerobic conditions. Lung homogenates were plated on tryptone soy agar and incubated for 24 hours at 37°C. Scale bar = 0.5 mm. (B) One mouse with periodontitis displayed bacterial growth (1/5). (C) Number of copies of bacterial 16S rDNA in the lung. Total rDNA in the lung was amplified using universal 16S rDNA primers using genomic DNA from E. coli as reference standard. Data are presented as mean ± SEM; (A-B) n = 5 mice per group, (C) n = 3 mice per group. Statistical comparisons performed using an unpaired t-test. Lig. = ligature.
While bacterial dissemination is one proposed mechanism linking oral and pulmonary
sites, there may be other factors at play, and as such we sought to fully characterise the
inflammatory and immune landscape in the lung. Nevertheless, we did not observe
alterations in the gene expression of hallmark pro-inflammatory and chemotactic factors
(Figure 3.7A), and populations of myeloid (Figures 3.7B-C) and lymphoid (Figures 3.8)
cells were largely unchanged. Taken together, these data indicate that the lung is
unaffected by acute oral pathology induced by experimental periodontitis.

94
Figure 3.7. Ligature-induced periodontitis does not affect inflammatory mediators or myeloid populations in the lung. (A) Relative expression (RE) of pro-inflammatory genes in the lung 10 days after periodontitis induction. (B) Frequencies of neutrophils, Ly6Chi monocytes, and macrophages in the lung. (C) Frequencies of Ly6Chi monocytes expressing CD62L, CX3CR1, and MHCII. Data are presented as mean ± SEM; n = 5 mice per group. Statistical comparisons performed using a one-way ANOVA with post hoc Tukey’s test. Lig. = ligature.

95
Figure 3.8. Ligature-induced periodontitis does not modulate lymphoid populations in the lung.
(A-B) Frequencies of TCR+, TCR+, ILCs (innate lymphoid cells), CD4+ T cells, and CD8+ T cells in the lung 10 days post-ligature placement.
(B) Frequencies of regulatory (FoxP3+) and helper (RORt+) CD4+ T cell subsets in the lung. Data are presented as mean ± SEM; n = 5 mice per group. Statistical comparisons performed using a one-way ANOVA with post hoc Tukey’s test; * p < 0.05. Lig. = ligature.
3.3.3. Ligature-induced periodontitis increases circulating levels of
pro-inflammatory cytokines
Even though our data indicated that periodontitis did not lead to robust lung inflammation
or bacterial translocation, we hypothesised that the oral pathology could independently
lead to induction of an inflammatory response in the bloodstream. Certainly, clinical and
pre-clinical evidence indicates that chronic periodontitis can lead to bacteraemia (Kinane
et al., 2005), and increases in circulating pro-inflammatory cytokines, such as IL-6, IL-

96
17A, and IL-1β (Duarte et al., 2010; Schenkein et al., 2010; Saadi-Thiers et al., 2013)
which can activate the acute-phase response and thereby heighten the overall
inflammatory profile. Since we did not find evidence of any 16S DNA presence in the
blood of control or periodontitis mice (data not shown), we turned to host mediators, and
determined the concentrations of a range of cytokines by multiplex cytokine bead array.
Interestingly, mice with periodontitis exhibited significant increases in the levels of IL-
1β, IL-17A, and granulocyte-macrophage colony stimulating factor (GM-CSF) compared
to control animals (Figure 3.9A). IL-1β is a driver of inflammatory damage and disease
exacerbation in a number of contexts, including inflammatory bowel disease (Coccia et
al., 2012), stroke (McColl et al., 2007), and atherosclerosis (Kirii et al., 2003). Moreover,
elevated GM-CSF can drive inflammation in experimental autoimmune myocarditis
(Sonderegger et al., 2008), and in the context of periodontitis, production of IL-17A in
the gingiva is associated with immunopathology, through the recruitment of neutrophils
(Dutzan et al., 2018). However, in the present study, elevated plasma cytokines were not
accompanied by altered trafficking or phenotype of neutrophils and Ly6Chi monocytes
(Figure 3.9B-C), indicating that these cytokines may not be actively secreted into the
bloodstream and are more likely to be produced in the inflamed gingiva and leaking into
the circulation.

97
Figure 3.9. Ligature-induced periodontitis increases circulating levels of pro-inflammatory cytokines without affecting frequencies of myeloid cells. (A) Levels of plasma cytokines were measured 10 days after periodontitis induction by cytometric bead array. Grey dotted lines indicate the lower limit of detection for each analyte. (B) Frequencies of neutrophils and Ly6Chi monocytes in the blood. (C) Frequencies of Ly6Chi monocytes expressing CD62L, CX3CR1, and MHCII. Data are presented as mean ± SEM; (A) n = 9-10 mice per group, (B-C) n = 5 mice per group. Data in (A) was log transformed to permit statistical analysis. Statistical comparisons performed using an unpaired t-test (A) or one-way ANOVA with post hoc Tukey’s test (B-C); * p < 0.05, ** p < 0.01. Lig. = ligature.

98
3.3.4. Ligature-induced periodontitis differentially modulates the
frequency and functionality of myeloid cells at sites distal from the
oral cavity
We have demonstrated that ligature-induced periodontitis leads to alterations in blood-
borne cytokines, we next investigated whether there were consequent immune alterations
at peripheral sites. Since the spleen is a major site of immunological function in response
to blood monitoring, and periodontitis is linked to inflammation in the gut (Vavricka et
al., 2013), we examined both tissues for immune changes. We did not observe alterations
in lymphoid populations in the spleen or gut (Figures 3.10B-C, 3.11B-C), again
highlighting the compartmentalisation of immune responses and a lack of profound
inflammation at tissues distant from the oral cavity. Splenic myeloid cells were also
unaffected (Figure 3.10A), although neutrophils in the small intestine were significantly
increased (Figure 3.11). This contrasts with the lung, where neutrophils were unaltered,
indicating that periodontitis exerts differential immune regulation at mucosal barrier sites
outside the local oral tissues.

99
Figure 3.10. Splenic immune cells are not altered by ligature-induced periodontitis. (A) Frequencies of neutrophils and Ly6Chi monocytes in the spleen 10 days post-ligature placement.
(B) Frequencies of TCR+, TCR+, ILCs (innate lymphoid cells), CD4+ T cells, and CD8+ T cells in the spleen 10 days post-ligature placement.
(C) Frequencies of regulatory (FoxP3+) and helper (RORt+) CD4+ T cell subsets in the spleen. Data are presented as mean ± SEM; n = 4-5 mice per group. Statistical comparisons performed using a one-way ANOVA with post hoc Tukey’s test. Lig. = ligature.

100
Figure 3.11. Frequencies of neutrophils in the small intestine are increased post-ligature placement. (A) Frequencies of neutrophils, Ly6Chi monocytes, and macrophages in the small intestine 10 days post-ligature placement.
(B) Frequencies of TCR+, TCR+, ILCs (innate lymphoid cells), CD4+ T cells, and CD8+ T cells in the small intestine 10 days post-ligature placement.
(C) Frequencies of regulatory (FoxP3+) and helper (RORt+) CD4+ T cell subsets in the small intestine. Data are presented as mean ± SEM; n = 4-5 mice per group. Statistical comparisons performed using an unpaired t-test. Lig. = ligature.

101
Since the bone marrow is the primary site responsible for the generation and mobilisation
of mature immune cells (Takizawa et al., 2012), we next determined if bone marrow
Ly6Chi monocytes and neutrophils were altered due to periodontitis. We found that these
leukocytes were differentially modulated; Ly6Chi monocytes were increased and
neutrophils decreased (Figure 3.12A-B). However, the surface phenotype of Ly6Chi
monocytes was unchanged (Figure 3.12C), which was in line with the observations at
other tissue sites. Since Ly6Chi monocytes were altered, we next sought to investigate if
the functionality of these cells were altered by periodontitis. We evaluated the capacity
for cytokine production in Ly6Chi monocytes from both the blood and bone marrow in
response to ex vivo LPS stimulation (Figure 3.13A). In keeping with the unchanged
blood monocyte frequencies, so too was their cytokine production unaffected (Figure
3.13B). However, in the bone marrow there was an increased frequency of TNFα-
producing monocytes, indicating that periodontitis can modulate monocyte phenotype
prior to bone marrow egress, a possible “educating” feature which has also been
described in the setting of acute gastrointestinal infection (Askenase et al., 2015).
Collectively these data show that, outside the oral cavity, experimental periodontitis can
lead to alterations in the function and frequency of myeloid cell populations without
similarly affecting lymphocytes.

102
Figure 3.12. Neutrophils and Ly6Chi monocytes in the bone marrow are differentially modulated during ligature-induced periodontitis. (A) Representative FACS plots and frequencies of neutrophils in the bone marrow 10 days after ligature placement. (B) Representative FACS plots and frequencies of Ly6Chi monocytes in the bone marrow. (C) Frequencies of Ly6Chi monocytes expressing CD62L, CX3CR1, and MHCII. Data are presented as mean ± SEM; n = 4-5 mice per group. Statistical comparisons performed using an unpaired t-test; ** p < 0.01. Lig. = ligature. BM = bone marrow.

103
Figure 3.13. Ligature-induced periodontitis increases TNF production by bone marrow monocytes.
(A) Gating strategy to identify IL-1, IL-1, and TNF expression on Ly6Chi monocytes in blood and bone marrow 10 days post-ligature placement.
(B) Frequencies of circulating Ly6Chi monocytes expressing IL-1, IL-1, and TNF after 2.5 hours ex vivo LPS stimulation.
(C) Frequencies of bone marrow Ly6Chi monocytes expressing IL-1, IL-1, and TNF after 2.5 hours ex vivo LPS stimulation.
(D) Representative FACS plots of bone marrow Ly6Chi monocytes expressing TNF Data are presented as mean ± SEM; n = 4-5 mice per group. Statistical comparisons performed using a two-way ANOVA with post hoc Sidak’s test; *** p < 0.001. Lig. = ligature. BM = bone marrow.

104
3.3.5. Ligature-induced periodontitis does not induce vascular or
CNS inflammation
Since periodontitis is a major risk factor for vascular disease, including atherosclerosis
and ischaemic stroke (Chistiakov et al., 2016), and our observation that levels of
circulating IL-1β were elevated in periodontitis animals, a known driver of vascular
pathology (Dénes et al., 2011b) (Figure 3.9A), we asked if ligature-induced periodontitis
leads to inflammation in peripheral and cerebral vessels. Even though, ligature-induced
periodontitis is reported to induce vascular activation (Brito et al., 2013; Miyajima et al.,
2015), here, expression of pro-inflammatory genes and vascular activation markers were
not significantly affected by experimental periodontitis (Figure 3.14A) and this was
mirrored in microvessels isolated from the brain (microvessel enrichment was confirmed
by endothelial marker CD31 expression) (Figure 3.14B-C). As the vasculature only
comprises a small component of the total brain and ligature placement could be inducing
inflammation in the brain without causing vascular activation, we also determined the
inflammatory status in the whole brain. There were no alterations in il1β expression, nor
did we find expansion of microglia/macrophages (iba1) or activation of the blood-brain
barrier (icam1, vcam1) (Figure 3.14D), suggesting that the brain is unperturbed by the
presence of periodontitis.

105
Figure 3.14. Ligature-induced periodontitis does not induce vascular or neuro-inflammation. (A) Relative expression (RE) of pro-inflammatory and vascular activation genes in the aorta 10 days after periodontitis induction. (B) Experimental workflow for isolating cerebral microvessels from whole brain tissue. (C) Relative expression (RE) of pro-inflammatory and vascular activation genes in cerebral microvessels 10 days after periodontitis induction.

106
(D) Relative expression (RE) of pro-inflammatory and vascular activation genes in whole brain tissue 10 days after periodontitis induction. Data are presented as mean ± SEM; (A-C) n = 8-10 mice per group, (D) n = 5 mice per group. Statistical comparisons performed using an unpaired t-test (A-C) or one-way ANOVA with post hoc Tukey’s test (D). Lig. = ligature.
Taken together, these data suggest that, despite links with cardio- and cerebro-vascular
disease, ligature-induced periodontitis does not induce a robust inflammatory response in
the aorta or the brain. However, despite these findings, as well as a lack of major immune
alterations in many sites (Figure 3.15), we have demonstrated that periodontitis leads to
tissue-specific modulation of neutrophils and monocytes and marked increases in
circulating pro-inflammatory cytokines, indicating that experimental periodontitis
mediates compartmentalised inflammatory effects at non-oral sites which could
contribute to adverse disease outcomes.
Figure 3.15. The spectrum of immune alterations in various tissue sites during ligature-induced periodontitis. (A) Changes in the frequencies of global immune cell populations across peripheral tissue sites.

107
(B) Changes in the phenotype of Ly6Chi monocytes across peripheral tissue sites. Heatmaps show changes in periodontitis (either double or single ligature) compared to control. Blacked-out boxes indicate absence of data. smLNs = sub-mandibular lymph nodes.
3.4. Discussion
Periodontitis has been consistently linked with adverse effects on systemic health
(Hajishengallis, 2015). However, since there is a paucity of experimental data regarding
the spatial dynamics of systemic immune involvement during periodontitis, and
specifically, in ligature-induced periodontitis, we sought to thoroughly characterise the
local and systemic responses in a range of peripheral murine tissues. To this end, we
defined the role of systemic inflammatory mediators and phenotyped a broad range of
immune populations, such as conventional T cells, monocytes, and neutrophils, as well as
smaller subsets such as γδ T cells and Tregs, all of which are involved in
immunopathology in the local oral environment in mice (Garlet et al., 2009; Steinmetz et
al., 2016; Dutzan et al., 2018; Krishnan et al., 2018), and were therefore hypothesised to
contribute to effects at sites outside the oral mucosa. Even so, it is important to appreciate
that in humans, B cells and plasma cells constitute the majority (~70%) of the cellular
infiltrate in periodontal lesions (Berglundh et al., 2007; Thorbert-Mros et al., 2015),
highlighting a fundamental difference in the cellular mediators of human and rodent
disease.
3.4.1. Ligature-induced periodontitis sufficiently models human
disease pathology in the oral cavity
Before attempting to determine the spectrum of immune alterations in non-oral sites, we
first characterised the local response to periodontitis pathology at ten days post-ligature
placement. We have demonstrated that the local pathology of this model mimics that of
the human disease, and encompassed significant bone loss, increased gingival cell
number, as well as marked growth of aerobic and anaerobic bacterial communities, in an

108
acute and predictable timeframe, which is in line with previous studies using this
approach (Abe and Hajishengallis, 2013; Campi et al., 2016; Matsuda et al., 2016).
Furthermore, the oral-draining lymph nodes exhibited increased cellular mobilisation,
which appeared to be a result of a global increase in immune cell populations, as T cells,
ILCs, and monocytes were not preferentially affected over one another. However, we did
observe a specific increase in neutrophils in the smLNs. Neutrophils constitute the
majority of phagocytes in the inflamed periodontal tissues and are crucial mediators of
inflammatory damage (Hajishengallis et al., 2016). Furthermore, neutrophils can migrate
to lymph nodes in other infection settings, such as Toxoplasma gondii, and
Mycobacterium bovis (Abadie et al., 2005; Chtanova et al., 2008). Therefore, it is at least
plausible that neutrophils may be transporting microbial antigens or phagocytosed oral
bacteria to the vicinity of T cells, as neutrophils are reported to shuttle M. bovis to lymph
nodes (Abadie et al., 2005). However, this suggestion may be contraindicated since we
did not detect changes in T cell frequencies or in specific subsets of Tregs (FoxP3+) or
Th17 (RORγt+) cells, and as such, the exact nature of neutrophils in the lymph nodes
during periodontitis is currently unclear.
3.4.2. The lung and the bloodstream as gateways to systemic
amplification during periodontitis
Despite most significant disease pathology being confined to the oral mucosa, it is clear
that in certain cases systemic health can be adversely affected by periodontitis (Kuramitsu
et al., 2001; Beck and Offenbacher, 2005; Moutsopoulos and Madianos, 2006). The
bloodstream is a critical access point for oral bacteria and immunogenic factors to be
transported away from the initial disease focus. Similarly, the lung is also proposed to be
a site of systemic amplification, given its anatomical continuity and proximity to the oral
cavity (Mojon, 2002). Isolated clinical case studies suggest that during periodontitis,

109
aspiration of certain oral bacteria into the lung, such as A. actinomycetemcomitans, can
initiate and contribute to infection in tandem with pulmonary pathogens (Zijlstra et al.,
1992; Morris and Sewell, 1994; Venkataramani et al., 1994; Matzumura-Kuan and
Jennings, 2014). We thus hypothesised that events in the oral cavity could lead to an
altered inflammatory profile in the lung. However, there was no clear distinction between
the status of mice with or without periodontitis in terms of pulmonary inflammation,
bacterial load, and immune cell landscape. The status of the mice used in this study, i.e.
young and otherwise healthy, may explain the lack of prominent lung inflammation,
especially as the evidence associating oral and lung disease is typically observed in
elderly individuals and in those with already-compromised lungs or compromised health
in general (Mojon, 2002). This is an important point, as periodontitis might yet represent
a threat to lung health in older individuals or in the context of concomitant pulmonary
disease.
Intriguingly, in contrast to the absence of overt consequences in the lung, we found that
experimental periodontitis increased circulating inflammatory mediators, IL-1β, IL-17A,
and GM-CSF, which are reported drivers of local tissue damage in the oral cavity (Abe et
al., 2012). Critically, there is currently no consensus on the exact nature of the systemic
inflammatory profile during periodontitis, in humans or in mouse models. Therefore, it is
perhaps not surprising that our findings both support and contradict previous reports.
Whereas unaltered plasma levels of TNFα is in agreement with a report by Miyajima et
al. (2015), we did not observe increased IL-6, in contrast to other mouse studies (Saadi-
Thiers et al., 2013; Matsuda et al., 2016). Furthermore, elevated plasma IL-1β here
corroborates previous data in studies with ligature-induced periodontitis (Saadi-Thiers et
al., 2013), while refuting another (Ma et al., 2011). To our knowledge, however, increases
in IL-17A and GM-CSF have not been previously reported in the circulation in animal
models, although serum IL-17A is elevated in patients with aggressive periodontitis

110
(Schenkein et al., 2010). In the oral tissue, local production of GM-CSF drives P.
gingivalis-induced pathology (Lam et al., 2015) and IL-17A-producing Th17 cells drive
neutrophil-mediated bone loss (Moutsopoulos et al., 2014; Dutzan et al., 2018),
highlighting a pathogenic role for both cytokines during periodontitis in oral and
potentially extra-oral compartments. Even though we did not determine the source of
these cytokines, Th17 cells are major producers of both IL-17A and GM-CSF (Lee et al.,
2012), and are involved in orchestrating periodontitis pathology in the oral mucosa
(Dutzan et al., 2018). Further, in the context of other inflammatory disease states, both
cytokines are deleterious (Sonderegger et al., 2008), and when taken together, our
findings indicate that systemic cytokine presence as a result of periodontitis may lead to
adverse complications.
3.4.3. Periodontitis induces immune alterations at discrete sites in a
cell-specific and tissue-specific manner
While there has been some insight into the soluble inflammatory mediators present in the
bloodstream during periodontitis, the cellular players in extra-oral tissues are poorly
defined. In the present study, we describe alterations in myeloid cell function and
composition in spatially-distinct extra-oral sites. Interestingly, periodontitis did not
significantly modulate lymphocytes outside the oral cavity, despite their well-defined
roles in the diseased oral mucosa of humans and mice (Teng et al., 2000; Garlet et al.,
2009; Dutzan et al., 2016b; Dutzan et al., 2018). However, we show that periodontitis
regulated myeloid cells, which could be a product of increased levels of circulating IL-1β,
IL-17A, and GM-CSF. Specifically, neutrophil frequency was differentially altered;
increased in the small intestine, but decreased in the bone marrow. Neutrophil egress
from the bone marrow and recruitment to inflammatory foci is often pathogenic in the

111
context of inflammatory bowel disease (Zhou and Liu, 2017), and this assigns a possible
deleterious extra-oral role for neutrophils during periodontitis.
By contrast, bone marrow Ly6Chi monocytes increased in frequency and were further
primed to produce TNFα. In ligated rats, TNFα signalling in adherent monocytes and
macrophages is reported to promote aortic inflammation (Miyajima et al., 2015).
Interestingly, this occurred without a corresponding increase in serum TNFα, which
mirrors our data, and suggests that these inflammatory changes are not global but cell-
and site-specific. Tailoring monocyte effector function has been described previously in
other contexts, such as sepsis (Bomans et al., 2018), and specifically during acute T.
gondii infection in the gut, monocytes can be educated prior to bone marrow egress; local
NK cell-derived IFNγ signals to Ly6Chi monocytes to switch to a regulatory phenotype
during systemic inflammation after bone marrow exit (Askenase et al., 2015). Although
the signal is unknown in the current study, systemic IL-12 and local IFNγ have been
shown to synergise to prime the regulatory capacity of monocytes (Askenase et al., 2015),
and bacterial components such as LPS and β-glucan can tolerise or stimulate monocytes
respectively, which is a mechanism of innate immune training (Quintin et al., 2012;
Novakovic et al., 2016; Mitroulis et al., 2018). Thus, it is plausible that systemic host or
bacterial factors actively or passively released from the oral environment during
periodontitis could be exerting immune regulation in the bone marrow. Although reported
in other disease settings, this is a novel finding in the context of periodontitis and a
potentially important mechanism by which oral inflammation can lead to adverse
systemic effects, by indicating that long-range crosstalk can tailor immune responsiveness
at sites distant from local pathology in the oral cavity.
Our observations have demonstrated that during periodontitis communication between
distal sites may potentially alter immune responsiveness. This may be a future predictor

112
of deleterious inflammatory responses, and with it, potential complications for systemic
health.
3.4.4. Ligature-induced periodontitis does not induce inflammation
of the vasculature or the brain
Vascular inflammation and endothelial dysfunction are intimately associated with the
development of atherosclerosis (Chistiakov et al., 2016). Here, ligature-induced
periodontitis did not induce inflammation in the aorta, which contrasts with previous
evidence suggesting that this model leads to aortic upregulation of elements of the TNFα
signalling cascade, il-1β, vcam-1, as well as inos (inducible nitric oxide) and ptgs2 (which
encodes cyclooxygenase-2) (Ma et al., 2011; Miyajima et al., 2015; Campi et al., 2016).
Furthermore, Brito et al. (2013) found that ligature-induced periodontitis in rats reduced
endothelial NOS, which is a hallmark of endothelial dysfunction. Thus, there is a
discrepancy between the findings of the present study and previous work, which suggests
that while ligature-induced periodontitis can lead to mild vascular dysfunction and
inflammation, these effects are relatively mild and are not always consistent from study to
study, leading to inherent variation in the reported findings. Indeed, aortic inflammatory
gene expression was only moderately increased (Miyajima et al., 2015) and the observed
endothelial dysfunction was reportedly transient (Brito et al., 2013), highlighting that
ligature placement causes robust local pathology but outside the oral cavity the impact is
considerably more subtle.
Although there is some evidence for peripheral vascular effects of periodontitis, there are
no reports about how ligature-induced periodontitis impacts the brain or the cerebral
vasculature. Indeed, most studies that have reported adverse CNS effects are those that
employ oral infection models and responses in the brain are specifically attributed to the

113
pathogenicity of the bacterium itself, such as P. gingivalis (Poole et al., 2014; Dominy et
al., 2019). Therefore, even though experimental periodontitis did not induce brain or
cerebrovascular inflammation in our study, we cannot exclude the possibility that specific
periodontal microbes could lead to inflammatory consequences in the CNS.
In general, a practical reason for the discrepancies between this work and previous studies
that try to address distal effects of ligature-induced periodontitis could be due to the
extensive variation in experimental parameters observed in most ligature-based rodent
models. For example, rats and mice are both used, ligatures can be tied around one, two
or bilateral molars, and ligatures can be left in place for two to as long as six weeks (Ma
et al., 2011; Miyajima et al., 2015; Matsuda et al., 2016). Variation of these parameters
may be an incentive to either dampen or drive inflammation at vascular sites, and also,
systemically. This is an issue with experimental periodontitis studies in general, as even
oral infection models vary in dose and bacterial species being inoculated (Graves et al.,
2008), and may explain the contrasting results amongst the literature and indeed, in our
findings, as experimental parameters are rarely conserved from study to study. Thus,
there is a need for more rigorous in-depth study across multiple models, which is
imperative to properly assess the exact systemic inflammatory profile during
periodontitis.
3.4.5. Conclusion
In conclusion, we have uncovered previously undescribed immunomodulatory effects of
ligature-induced periodontitis that occur in spatially-distinct sites, providing evidence to
suggest that periodontitis may lead to adverse effects at distal sites. Specifically, we
substantiate current literature by demonstrating that ligature placement leads to increased
levels of circulating pro-inflammatory mediators in an acute timeframe. We also propose

114
novel findings, in that circulating levels of IL-17A and GM-CSF are augmented, and that
bone marrow monocytes can be educated prior to bone marrow egress to enhance their
pro-inflammatory potential, and these observations have not been reported in the context
of experimental periodontitis. Specifically how these monocytes become primed and how
they respond after bone marrow exit is still unclear, but it helps to emphasise that events
in the oral mucosa can alter distal immune effector function, adding a potential novel
mechanism by which periodontitis can lead to systemic complications by priming
immune cells in the bone marrow.
Our findings here add to the growing body of evidence tying periodontitis with adverse
systemic effects. Though we report that the spectrum of immune alterations is relatively
modest, this actually supports the current opinion, helping to reiterate the point that
periodontitis is not a typical high-grade infection, but a milder and perhaps more
insidious disease in which subtle changes over a long period of time eventually lead to a
deleterious impact.

115
Chapter 4. The impact of periodontitis on
acute outcome after stroke

116
4.1. Introduction
Periodontitis is an emerging risk factor for ischaemic stroke. Originally proposed in the
late 1980s (Syrjanen et al., 1989), there has been a large quantity of observational studies
since then, indicating that individuals with periodontitis have a greater risk of stroke
compared to their healthy counterparts (Kweider et al., 1993; Grau et al., 1997, 2004; Wu
et al., 2000; Elter et al., 2003; Joshipura et al., 2003; Dorfer et al., 2004; Sen et al., 2018).
While most studies suggest a generalised increased risk, Grau et al. (2004) report that the
risk increases in cases of severe periodontitis, but only amongst young men, but not
women or older individuals (> 60 years) of either sex (Grau et al., 2004). Even though the
majority of these studies have assigned a significant role for periodontitis in increasing
stroke incidence, these associations are often moderate and not all studies report
significance (Howell et al., 2001). Critically, even though cumulative evidence seems to
support periodontitis as a risk factor for stroke, there is not enough direct proof that
periodontitis is causal for cerebrovascular disease (Beck and Offenbacher, 2005), and as
such, further investigation is needed. Since stroke and periodontitis share common risk
factors, such as age, inflammatory profile, hypertension, and obesity, defining the exact
impact of periodontitis on stroke on its own, independently of confounders, is often
difficult and presents a challenge for investigators in both clinical and pre-clinical realms.
In general, most of the evidence associating other prominent stroke risk factors (such as
infection and obesity) with worse neuropathology is centred on the inflammatory
response. Extensive preclinical and clinical evidence supports the concept of unresolving
or dysregulated immunity as a major driver of both increased risk and worse outcome (i.e.
the extent of brain damage) post-stroke (Emsley and Hopkins, 2008). For example, in
experimental rodent models, lung and gut infections worsen stroke-induced injury by
driving early inflammatory pathology. Specifically, severe Streptococcus pneumoniae

117
infection augments microvascular injury and brain damage by promoting platelet
activation and IL-1α production in microglia (Dénes et al., 2014). Similarly, chronic low-
dose Trichuris muris infection increases microvascular obstruction and platelet
aggregation in the brain but through a CCL5-dependent Th1-type response (Dénes et al.,
2010a). Furthermore, pro-inflammatory stimuli, such as bacterial LPS and host IL-1β, are
major systemic drivers of post-stroke brain damage. These factors exert detrimental
effects by promoting neutrophil infiltration into the brain, which contributes to
pathological damage by increasing oedema, neurological dysfunction, and neuronal injury
by aggravating blood-brain barrier (BBB) disruption in a matrix metalloproteinase
(MMP)-9-dependent manner (Caso et al., 2007; McColl et al., 2007, 2008).
It is apparent that systemic inflammation has a major impact on post-stroke brain injury.
However, unlike scenarios where infection is a product of a single species such as
Streptococcus pneumoniae or Trichuris muris (Dénes et al., 2010a; Dénes et al., 2014),
periodontitis is not a typical infectious or “high-grade” systemic inflammatory disease
(Moutsopoulos and Madianos, 2006), and therefore it is perhaps not prudent to include it
in the same category. A more suitable comparison may be obesity, a metabolic disorder
associated with “low-grade” chronic inflammation (Haley et al., 2017b), in a manner
similar to that of periodontitis. In rodents, diet-induced or genetic obesity moderately
increases plasma levels of CCL2 and CXCL1, but profoundly worsens stroke outcome,
promoting haemorrhagic transformation (i.e. microbleeds), BBB damage, neurological
dysfunction, and exacerbated the extent of brain damage (McColl et al., 2010; Deng et al.,
2014; Maysami et al., 2015; Haley et al., 2017b). As demonstrated in Chapter 3,
periodontitis induces comparable low-level inflammation, marked by moderate increases
in circulating levels of IL-1β, IL-17A, and GM-CSF. A large body of evidence has
implicated IL-1β and IL-17A in particular with worse outcome after experimental stroke
(McColl et al., 2007; Shichita et al., 2009; Gelderblom et al., 2012, 2018; Burrows et al.,

118
2016; Arunachalam et al., 2017). Thus, even a low-grade systemic inflammatory
environment as observed during periodontitis may have a profound impact on brain injury
after stroke.
Although epidemiological evidence suggests that periodontitis worsens stroke risk and
inflammatory conditions are known to aggravate stroke-induced injury, there is a lack of
direct causal data tying periodontitis to altered outcome after stroke. To our knowledge,
there are only two studies that specifically address this question, with opposing outcomes.
The first and most recent study found that experimental periodontitis, induced by repeated
gingival LPS injections, worsened outcome after stroke in mice, through increased IL-1β
production and microglial and astrocyte activation (Chi et al., 2019). This provides
evidence that periodontitis can contribute to stroke pathology by aggravating deleterious
inflammatory responses in the brain. By contrast, the second study found that ligature-
induced periodontitis in rats was neuroprotective after stroke by inducing a mild systemic
inflammation (Petcu et al., 2008). Since these studies report conflicting findings, it
remains difficult to definitively attribute a positive or negative role for periodontitis in
stroke outcome. However, the results of the latter study indicate that periodontitis can
mitigate brain injury by tolerising host immunity, thus dampening the acute pro-
inflammatory response to stroke. This is indicative of a phenomenon termed “ischaemic
tolerance”, whereby sub-injurious insults can confer neuroprotection during subsequent
ischaemic events (Rosenzweig et al., 2004; Marsh et al., 2009; Wendeln et al., 2018).
This can be induced in experimental settings by a low-dose LPS challenge prior to stroke
which dampens neuroinflammation, by limiting neutrophil infiltration into the CNS and
suppressing microglial activation (Rosenzweig et al., 2004) while at the same time
promoting spleen-derived monocytes to infiltrate and protect the ischaemic brain (Garcia-
Bonilla et al., 2018). While this is a plausible mechanism linking oral inflammation to
neuroprotection after stroke, extrapolating from this is contradictory to recent clinical and

119
preclinical evidence regarding the detrimental role of periodontal bacteria in stroke and
other diseases (Li et al., 2002; Lalla et al., 2003; Gibson et al., 2004; Kostic et al., 2013;
Dominy et al., 2019). Specifically in the context of human stroke, Streptococcus mutans,
a causal agent of dental caries, is associated with haemorrhagic stroke and deep
microbleeds (Tonomura et al., 2016), and increased oral and systemic levels of P.
gingivalis are associated with increased incidence of stroke (Pussinen et al., 2007;
Ghizoni et al., 2012). Furthermore, periodontal bacteria are capable of colonising sites
outside the oral cavity and can directly contribute to pathogenesis of vascular disease. For
example, P. gingivalis can invade endothelial cells (Madrigal et al., 2012), can cause
platelet aggregation (Sharma et al., 2000; Nakayama, 2010), and can modify low-density
lipoprotein which is taken up by inflammatory macrophages in vessel walls, a pathogenic
feature of atherosclerosis (Qi et al., 2003; Miyakawa et al., 2004). Thus, oral bacteria can
not only survive at distal sites but also actively contribute to vascular dysfunction and
predispose to increased risk of thrombosis.
In addition, there is recent evidence to suggest that periodontal bacteria can impact
neuronal survival directly. Studies of Alzheimer’s disease in mice infected with oral
bacteria such as P. gingivalis and T. denticola have demonstrated that these species can
gain access to the brain parenchyma and cause neurotoxicity (Riviere et al., 2002; Foschi
et al., 2006; Dominy et al., 2019). Specifically, P. gingivalis induces complement
activation and produces proteases called gingipains which directly kill neurons and
induce neuroinflammation (Poole et al., 2014; Dominy et al., 2019). Collectively, these
studies highlight the invasive capacity of oral bacteria. Therefore, it is reasonable to
suggest that the profound disruption to the BBB that accompanies stroke could facilitate
enhanced entry of blood-borne oral bacteria into the brain, which could directly potentiate
injury in the CNS.

120
Taken together, most evidence supports a detrimental role for periodontitis, and
specifically, periodontal bacteria, in the progression of systemic diseases, even though
there is a lack of direct mechanistic evidence with respect to ischaemic stroke. Alterations
in inflammatory responses and host immunity are cited as major factors that underpin the
associations between periodontitis and stroke, and as such, require a more thorough
exploration in pre-clinical studies.
4.2. Aims
The primary aim of this study was therefore to determine the impact of periodontitis on
acute stroke outcome. Specifically, we used ligature-induced periodontitis in conjunction
with two well-defined murine stroke models to evaluate ischaemic damage and BBB
breakdown. We also sought to extensively characterise the peripheral inflammatory and
immune responses, with a particular focus on myeloid cell populations, as we have shown
in Chapter 3 that experimental periodontitis affected neutrophil and monocyte dynamics.
Many reports have stressed the importance of dysregulated immunity outside the brain, as
altered peripheral leukocyte responses post-stroke can contribute to delayed ischaemic
injury (Liesz et al., 2009b; Shichita et al., 2009) or increase the risk of infection (Prass et
al., 2003; Liesz et al., 2009a; McCulloch et al., 2017).

121
4.3. Results
4.3.1. Ligature-induced periodontitis does not alter acute outcome
after transient MCAo
In order to evaluate the impact of periodontitis on outcome after stroke, bilateral ligatures
were placed around second molar teeth and left in place for 10 days, upon which mice
were subjected to transient middle cerebral artery occlusion (tMCAo) and sacrificed 48
hours post-stroke (Figure 4.1A). After a 20-minute tMCAo, most mice exhibited small
(<20 mm3) infarcts restricted to the striatum, which was expected and in line with
previous data from our laboratory (Haley and Lawrence, 2017) (Figure 4.1B). Some mice
displayed infarcts that extended into the cortex (>30 mm3) but this was evenly distributed
amongst the groups (36% in the stroke group, 40% in the periodontitis + stroke group).
However, total infarct volumes were not significantly different between groups,
indicating that experimental periodontitis does not modulate ischaemic damage in this
experimental paradigm. This was reflected in the neurological scores, which were not
different between groups (Figure 4.1C). Sustained disruption of the BBB during stroke
results in leakage of plasma proteins into the brain parenchyma (Haley and Lawrence,
2017). We found that periodontitis did not alter the amount of IgG in the infarcted
hemisphere (Figure 4.1D). Since there was variation in infarct volumes within the
periodontitis + stroke group, we wondered if the extent of bone loss was correlated with
size of infarct. However, bone loss values were consistent and exhibited low deviation
within the group and were not significantly correlated with ischaemic damage (Figure
4.1E).

122
Figure 4.1. Ligature-induced periodontitis does not alter ischaemic damage, neurological impairment, blood-brain barrier breakdown, or neutrophil infiltration after transient middle cerebral artery occlusion. (A) Experimental outline. tMCAo = transient middle cerebral artery occlusion. (B) Representative image of cresyl violet staining showing typical striatal infarct (dotted line) and quantification of total infarct volumes. Scale bar = 1 mm. (C) Functional assessment of neurological impairment was performed on the morning of sacrifice and each mouse scored on a 28-point scale (Table A.3).

123
(D) Representative image of IgG staining and quantification of IgG leakage into the brain. Image shows the area of blood-brain barrier breakdown (dotted line). Total IgG staining was quantified as increase in pixel density from the contralateral hemisphere. Scale bar = 1 mm. AU = arbitrary units. (E) The range of bone loss in the periodontitis + stroke group (left) and the relation between bone loss and infarct size (right). (F) Representative image and quantification of SJC-4+ neutrophils in the brain 48 hours after stroke. Inset and arrowheads show brain-infiltrating neutrophils present in the striatum (Str.) and cortex (Ctx.). Scale bar = 0.5 mm. Data are presented as mean ± SEM; n = 10-11 mice per group. Statistical comparisons performed using an unpaired t-test. PD = periodontitis (double ligature).
We have shown that periodontitis modulates peripheral neutrophil frequencies (Figure
3.15) and in the context of stroke, neutrophils are critical cellular mediators of BBB
breakdown and exacerbation of ischaemic brain damage (McColl et al., 2007, 2008;
Gelderblom et al., 2012; Neumann et al., 2015), and are rapidly recruited to the ischaemic
hemisphere in this model within 48 hours after the initial ischaemic event (Stevens et al.,
2002). Thus, we determined the number of brain-infiltrating neutrophils by
immunohistochemical staining with a neutrophil-specific marker, SJC-4 (McColl et al.,
2008; Maysami et al., 2015). However, we did not find differences in neutrophil numbers
across the striatum or cortex in mice with or without periodontitis post-stroke, despite
some individual biological variation within the periodontitis + stroke group (Figure
4.1F).

124
Figure 4.2. Ligature-induced periodontitis does not change the levels of circulating inflammatory mediators after transient middle cerebral artery occlusion. Levels of plasma cytokines were measured 48 hours after stroke by cytometric bead array. Grey dotted lines indicate the lower limit of detection for each analyte. Data are presented as mean ± SEM; n = 10-11 mice per group. Data in was log transformed to permit statistical analysis. Statistical comparisons performed using an unpaired t-test. PD = periodontitis (double ligature).
Since the inflammatory status in the brain was not affected by periodontitis, we turned to
events in the periphery in order to determine if periodontitis may be affecting distinct
tissue sites or certain immune cells, as dysregulated host immunity is known to increase
the risk of infection, a common complication of stroke in mice and humans (Prass et al.,
2003; Liesz et al., 2009a; McCulloch et al., 2017). In this regard, circulating levels of
inflammatory cytokines and chemokines were also unchanged between the groups
(Figure 4.2). Supporting these findings, we did not observe alterations in myeloid
(Figure 4.3) or lymphoid (Figure 4.4B) immune cells in the spleen between ligated and
non-ligated animals after stroke. This was also mirrored in the bone marrow, where

125
frequencies of naïve (CD62L+), CD4+, and CD8+ T cell subsets (Figure 4.4C) or
neutrophils and Ly6Chi monocytes frequencies (Figure 4.5) were unaffected by
periodontitis. Additionally, the effector function of bone marrow Ly6Chi monocytes was
not significantly affected by periodontitis post-stroke (Figure 4.6). Taken together, this
indicates that experimental periodontitis does not alter acute brain damage, BBB
disruption or peripheral leukocyte responses 48 hours after transient MCAo.
Figure 4.3. Ligature-induced periodontitis does not alter the frequency or phenotype of monocytes and neutrophils in the spleen after transient middle cerebral artery occlusion. (A-B) Representative FACS plots and frequencies of neutrophils and Ly6Chi monocytes in the spleen 48 hours after stroke. Monocytes and neutrophils were gated as shown in Figure 3.3. (C) Frequencies of splenic Ly6Chi monocytes expressing CD62L, CX3CR1, MHCII, and Sca-1. (D) The frequency of splenic neutrophils expressing CD62L. Data are presented as mean ± SEM; n = 10-11 mice per group. Statistical comparisons performed using an unpaired t-test. PD = periodontitis (double ligature).

126
Figure 4.4. Ligature-induced periodontitis does not alter the balance of T cell subsets in the bone marrow or spleen after transient middle cerebral artery occlusion. (A) Gating strategy to identify CD4+, CD8+ T cells, and naïve/central memory T cell (CD62L+) subsets in the bone marrow and spleen 48 hours after stroke. (B-C) Frequencies of T cells expressing CD4+ or CD8+, and CD62L+ in the spleen and bone marrow post-stroke.
Data are presented as mean ± SEM; n = 10-11 mice per group. Statistical comparisons performed using an unpaired t-test. PD = periodontitis (double ligature).

127
Figure 4.5. Ligature-induced periodontitis does not alter the frequency or phenotype of monocytes and neutrophils in the bone marrow after transient middle cerebral artery occlusion. (A-B) Representative FACS plots and frequencies of neutrophils and Ly6Chi monocytes in the bone marrow 48 hours after stroke. Monocytes and neutrophils were gated as shown in Figure 3.3. (C) Frequencies of bone marrow Ly6Chi monocytes expressing CD62L, CX3CR1, MHCII, and Sca-1. (D) The frequency of bone marrow neutrophils expressing CD62L. Data are presented as mean ± SEM; n = 10-11 mice per group. Statistical comparisons performed using an unpaired t-test. PD = periodontitis (double ligature).

128
Figure 4.6. Ligature-induced periodontitis does not alter the functionality of bone marrow monocytes after transient middle cerebral artery occlusion.
(A) Gating strategy to identify IL-1, TNF, and IL-6 expression on Ly6Chi monocytes in the bone marrow 48 hours after stroke.
(B) Frequencies of bone marrow Ly6Chi monocytes expressing IL-1, TNF, and IL-6 after 2.5 hours ex vivo LPS stimulation. Data are presented as mean ± SEM; n = 10-11 mice per group. Statistical comparisons performed using a two-way ANOVA with post hoc Sidak’s test. PD = periodontitis (double ligature).
4.3.2. Systemic challenge of P. gingivalis LPS causes robust
inflammatory responses
Since we found that experimental periodontitis did not appear to modulate stroke
outcome after transient MCAo, we wondered whether this was finding was supported
using a different model to induce stroke. A caveat of the tMCAo model is that it involves
major surgery which can alter systemic immunity independent of the stroke itself (Denes
et al., 2011). We reasoned that the relatively modest inflammatory effects of periodontitis

129
could have been masked in this model. We therefore altered the experimental setup to
drive systemic involvement during the course of ligature-induced periodontitis (by
systemic LPS administration), and to mitigate the detrimental impacts of tMCAo surgery
we employed a thrombotic pMCAo model, which results in less physiological stress,
smaller infarcts (which can still be modulated) and more pronounced leukocyte
infiltration into the brain (Liesz et al., 2009a; Zhou et al., 2013).
Individuals with periodontitis are reported to have raised LPS levels in the circulation
(Geerts et al., 2002; DeLeon-Pennell et al., 2013). Thus, we sought to better imitate the
clinical condition in vivo by systemically administering a relevant oral-specific LPS from
P. gingivalis (Pg-LPS). In this sense, Pg-LPS is reported to activate both TLR2 and
TLR4, in contrast to E. coli LPS (Ec-LPS), which activates TLR4 (Hirschfeld et al., 2001;
Le Sage et al., 2017). When injected intravenously in a single bolus, we found that high-
dose Pg-LPS was less immunogenic than Ec-LPS, but nonetheless capable of inducing
peripheral and central inflammation within 2 hours (Figure 4.7). Specifically, Pg-LPS
increased plasma levels of IL-6, and CCL2, but not TNFα and IFNγ, showing that each
molecule has separate and distinct effects (Figure 4.7B). Furthermore, Pg-LPS also
increased the frequencies of Ly6Chi monocytes in blood and bone marrow (Figure 4.7C),
and induced upregulation of inflammatory genes in the hypothalamus, a region that is
sensitive to peripheral inflammatory stimuli since it contains regions with absent blood-
brain barriers (Rodríguez et al., 2010) (Figure 4.7D).

130
Figure 4.7. Intravenous challenge with Porphyromonas gingivalis LPS induces robust systemic inflammatory responses 2 hours after administration. (A) Experimental outline. Mice were injected intravenously (i.v.) with PBS, P. gingivalis LPS (Pg-LPS; 4 mg/kg), or E. coli LPS (Ec-LPS; 100 μg/kg) as a positive control and sacrificed 2 hours post-challenge. (B) Levels of inflammatory cytokines in the blood determined by cytometric bead array. (C) Representative FACS plots and frequencies of Ly6Chi monocytes in the blood and bone marrow post-challenge. Ly6Chi monocytes were gated as shown in Figure 4.9. (D) Relative expression (RE) of pro-inflammatory genes in the hypothalamus post-challenge. Data are presented as mean ± SEM; n = 4 mice per group. Statistical comparisons performed using a one-way ANOVA with post hoc Dunnett’s test; * p < 0.05, ** p < 0.01, *** p < 0.001.

131
4.3.3. Ligature-induced periodontitis does not alter acute outcome
after permanent MCAo
As a single bolus of high-dose Pg-LPS would not reflect the sustained low-level flushing
of bacterial products into the bloodstream reported in periodontitis patients (Geerts et al.,
2002; Kinane et al., 2005; DeLeon-Pennell et al., 2013) we used a lower dose (1 mg/kg)
which was administered intravenously every two days alongside with ligature-induced
periodontitis (Figure 4.8A). After inducing stroke by pMCAo, we observed smaller
infarcts limited to the cortex at 48 hours (Figure 4.8B), as described previously (Llovera
et al., 2014; Cisbani et al., 2018). However, despite repeatedly administering Pg-LPS for
five total doses over a 10-day period, ischaemic brain damage and blood-brain barrier
breakdown after pMCAo (Figure 4.8B-C) were not robustly affected by Pg-LPS or
periodontitis, either alone or in combination. In accordance with these observations,
neutrophil infiltration into the infarcted region in the cortex was also unchanged (Figure
4.8D).

132
Figure 4.8. Periodontitis and systemic P. gingivalis LPS not alter ischaemic damage, blood-brain barrier breakdown, or neutrophil infiltration after permanent middle cerebral artery occlusion. (A) Experimental outline. Before stroke, P. gingivalis LPS (Pg-LPS; 1mg/kg) or PBS were injected intravenously (i.v.) every two days in tandem with ligature-induced periodontitis. pMCAo = permanent middle cerebral artery occlusion. (B) Representative image of cresyl violet staining showing typical cortical infarct (dotted line) and quantification of total infarct volumes. Scale bar = 1 mm. (C) Representative image of IgG staining and quantification of IgG leakage into the brain. Image shows the area of blood-brain barrier breakdown (dotted line). Total IgG staining was quantified as increase in pixel density from the contralateral hemisphere. Scale bar = 1 mm. AU = arbitrary units. (D) Representative image and quantification of SJC-4+ neutrophils in the 48 hours after stroke. arrowheads show brain-infiltrating neutrophils present in cortex (Ctx.). Scale bar = 200 μm. Data are presented as mean ± SEM; Stroke PBS (n = 6), Stroke LPS (n = 7), PD+Stroke PBS (n = 5), PD+Stroke LPS (n = 9). PD = periodontitis (double ligature).

133
Figure 4.9. Gating strategies to identify myeloid and lymphoid cells after permanent middle cerebral artery occlusion using flow cytometry. (A) Gating strategy to identify neutrophils, monocytes, Ly6Chi monocytes, Ly6Clo monocytes, as well as surface markers CCR2, CX3CR1, MHCII, and Sca-1.
(B) Gating strategy to identify B cells, TCR+ cells, TCR+ cells, CD4+ T cells, CD8+ T cells, as
well as FoxP3+ CD4+ T cells.
In the periphery, myeloid and lymphoid populations (gated as shown in Figure 4.9) were
not significantly altered. Specifically, the frequencies of neutrophils and Ly6Chi
monocytes as well as the phenotype of Ly6Chi monocytes were unaltered in the blood,
spleen, and bone marrow (Figures 4.10, 4.12, 4.14). Moreover, there were no differences
in frequencies of B cells, T cells, and T cell subsets in the blood and spleen (Figures

134
4.11, 4.13). Taken together with the findings from the tMCAo model, these data
demonstrate that ligature-induced periodontitis does not modulate acute outcome after
stroke in mice.
Figure 4.10. Periodontitis and systemic P. gingivalis LPS do not significantly affect circulating myeloid cells after permanent middle cerebral artery occlusion. (A) Frequencies of monocytes, Ly6Chi, Ly6Clo, and neutrophils in the blood 48 hours post-stroke. (B) Frequencies of blood Ly6Chi monocytes expressing CCR2, CX3CR1, MHCII, and Sca-1. Data are presented as mean ± SEM; Stroke PBS (n = 6), Stroke LPS (n = 7), PD+Stroke PBS (n = 5), PD+Stroke LPS (n = 9). Statistical comparisons performed using a two-way ANOVA with post hoc Tukey’s test. PD = periodontitis (double ligature).

135
Figure 4.11. Periodontitis and systemic P. gingivalis LPS do not significantly affect circulating lymphocytes after permanent middle cerebral artery occlusion.
(A-B) Frequencies of B cells, TCR+ cells, TCR+ cells, CD4+ T cells, CD8+ T cells in the blood 48 hours post-stroke. (B) The frequency of blood CD4+ T cells expressing FoxP3. Data are presented as mean ± SEM; Stroke PBS (n = 6), Stroke LPS (n = 7), PD+Stroke PBS (n = 5), PD+Stroke LPS (n = 9). Statistical comparisons performed using a two-way ANOVA with post hoc Tukey’s test. PD = periodontitis (double ligature).

136
Figure 4.12. Periodontitis and systemic P. gingivalis LPS do not significantly affect splenic myeloid cells after permanent middle cerebral artery occlusion. (A) Frequencies of monocytes, Ly6Chi, Ly6Clo, and neutrophils in the spleen 48 hours post-stroke. (B) Frequencies of splenic Ly6Chi monocytes expressing CCR2, CX3CR1, MHCII, and Sca-1. Data are presented as mean ± SEM; Stroke PBS (n = 6), Stroke LPS (n = 7), PD+Stroke PBS (n = 5), PD+Stroke LPS (n = 9). Statistical comparisons performed using a two-way ANOVA with post hoc Tukey’s test. PD = periodontitis (double ligature).

137
Figure 4.13. Periodontitis and systemic P. gingivalis LPS do not significantly affect splenic lymphocytes after permanent middle cerebral artery occlusion.
(A-B) Frequencies of B cells, TCR+ cells, TCR+ cells, CD4+ T cells, CD8+ T cells in the spleen 48 hours post-stroke. (B) The frequency of splenic CD4+ T cells expressing FoxP3. Data are presented as mean ± SEM; Stroke PBS (n = 6), Stroke LPS (n = 7), PD+Stroke PBS (n = 5), PD+Stroke LPS (n = 9). Statistical comparisons performed using a two-way ANOVA with post hoc Tukey’s test. PD = periodontitis (double ligature).

138
Figure 4.14. Periodontitis and systemic P. gingivalis LPS do not significantly affect myeloid cells in the bone marrow after permanent middle cerebral artery occlusion. (A) Frequencies of monocytes, Ly6Chi, and neutrophils in the bone marrow 48 hours post-stroke. (B) Frequencies of bone marrow Ly6Chi monocytes expressing CCR2, CX3CR1, MHCII, and Sca-1. Data are presented as mean ± SEM; Stroke PBS (n = 6), Stroke LPS (n = 7), PD+Stroke PBS (n = 5), PD+Stroke LPS (n = 9). Statistical comparisons performed using a two-way ANOVA with post hoc Tukey’s test. PD = periodontitis (double ligature).
4.4. Discussion
From a large body of clinical and pre-clinical evidence, it is apparent that infections and
inflammatory diseases can increase the risk of stroke, and substantially contribute to
worse brain damage and impaired recovery after stroke (Ergul et al., 2007, 2016; McColl
et al., 2010; Dénes et al., 2014; Fugate et al., 2014). In humans, periodontitis has long
been implicated as a contributing factor to increased incidence of stroke, but it is not
currently known if periodontitis leads to altered outcome after stroke. Therefore, in the
present study we sought to address if periodontitis could modulate ischaemic brain
damage in mice. Since amplification of the systemic inflammatory response and
mobilisation of peripheral immune cells are crucial determinants of outcome post-stroke
(Yilmaz et al., 2006; Liesz et al., 2009b; Dénes et al., 2011a; Gelderblom et al., 2012;

139
Neumann et al., 2015), we focused specifically on the nature of the immune landscape in
peripheral and central compartments in the acute phase post-stroke.
4.4.1. Ligature-induced periodontitis causes systemic inflammatory
alterations but does not worsen outcome after stroke
We showed in Chapter 3 that the bilateral ligature model of periodontitis is an attractive
tool for experimental periodontal research. The model successfully imitates the clinical
disease hallmarks, including bone destruction, inflammation, and microbial overgrowth,
and these occur within an acute and predictable timeframe. We also reported that ligature-
induced periodontitis caused a specific increase in plasma IL-1β, and IL-17A, as well as
differentially modulating peripheral neutrophils and monocytes. IL-1β has a well-
documented detrimental role in orchestrating inflammatory pathology in the acute phases
post-stroke (McColl et al., 2009; Dénes et al., 2011b), and trafficking of IL-17A-
producing γδ T cells to the brain exacerbates ischaemic damage (Shichita et al., 2009;
Gelderblom et al., 2012; Benakis et al., 2016; Arunachalam et al., 2017). Furthermore,
neutrophils and monocytes are intimately involved with stroke pathology; neutrophils are
detrimental (Gelderblom et al., 2012; Neumann et al., 2015) but the role of monocytes is
controversial since early brain infiltration promotes damage (Dimitrijevic et al., 2007),
but in later stages they promote tissue repair programs (Gliem et al., 2012). We
hypothesised that periodontitis would worsen outcome after stroke by promoting
leukocyte-mediated inflammatory pathology. However, unlike other experimental co-
morbidities such as infection or obesity, periodontitis did not exacerbate ischaemic brain
damage in two mouse models of stroke.
Though high-grade systemic inflammation is deleterious in the context of stroke (McColl
et al., 2009), there are contrasting reports on whether low-grade inflammation is harmful

140
or protective after cerebral ischaemia. Indeed, obesity in mice (which is associated with
low-grade inflammation) leads to an increase in ischaemic brain damage, blood-brain
barrier breakdown, and neutrophil infiltration into the ischaemic territory (McColl et al.,
2010; Maysami et al., 2015). This is also true in periodontitis induced by gingival LPS
injections, which increases brain injury after photothrombotic stroke (Chi et al., 2019).
Conversely, ligature-induced periodontitis in rats induces mild systemic inflammation
(marked by increased TGFβ, IL-10, and IFNγ mRNA levels in the circulation) which
reduces infarct volume and neuronal loss (Petcu et al., 2008). In our study, even though
ligature-induced periodontitis alone induced circulating pro-inflammatory cytokines and
modulated innate immune cells within the bone marrow (Chapter 3), it is apparent that
these changes were not sufficient to worsen stroke pathology. This is an interesting
finding because it is apparent that periodontitis is not the severe systemic stressor in the
same manner as other stroke modifiers, such as obesity and infection. Furthermore, our
results challenge the studies by Petcu et al. (2008) and Chi et al. (2019) which reported
protective and deleterious effects on brain damage post-stroke, respectively. Addressing
the study by Petcu et al. (2008), their use of a different species (rat), longer periodontitis
timeframe (15–21 days), and a different stroke model as well as longer reperfusion time
(7 days) could account for differing results. However, their study has a number of
important caveats: the authors did not extensively characterise peripheral immune
alterations to evaluate the risk of a delayed-phase injury or future infection susceptibility;
they only used one measure, mRNA levels, to define “mild systemic inflammation”; and
quantified infarct volumes using a neuron-specific marker, when in reality, multiple CNS
cells comprise the infarct (Liu et al., 2009), and they could therefore be misrepresenting
the overall volume of ischaemic damage. In the context of the study by Chi et al. (2019),
the authors induced periodontitis in mice by high-dose LPS (800 µg/kg) injection into the
gingiva three times per week over a one month period, used a photothrombotic stroke
model which resulted in small infarcts (0.5–1 mm3), and assessed long-term (14 days),

141
but not acute, outcome post-stroke. Such profound differences in the experimental
parameters renders their findings and ours impossible to compare and difficult to
interpret. Furthermore, the authors do not specify if their LPS is from a relevant oral
species and do not reveal when the last injection took place prior to stroke as it is likely
that their model of periodontitis was more akin to a high-dose LPS-induced systemic
inflammatory response, which is already known to worsen stroke outcome (McColl et al.,
2007), and not an accurate representation of periodontitis. Therefore, our study has many
advantages over these reports by using a well-established ligature model in mice, two
different stroke models, a clinically-relevant systemic LPS challenge, and extensive and
detailed analyses of immune and inflammatory responses.
4.4.2. The validity of experimental stroke models
In the event of results that do not disprove the null hypothesis, the question of model
suitability is inevitably raised. However, nearly 65% of 2852 experimental stroke studies
on neuroprotection used either filament or coagulation approaches to induce stroke
(Howells et al., 2010), both of which used in the present study. Transient MCAo is a
well-characterised model of severe cerebral ischaemia in humans (Engel et al., 2011;
Sommer, 2017). Importantly, ischaemic damage and the inflammatory response can be
modulated by co-morbidities, allowing dissection of the pathophysiological mechanisms
after stroke (Ergul et al., 2007; Maysami et al., 2015; Crapser et al., 2016). However, it is
prudent to be mindful that spontaneous infections (Prass et al., 2003; McCulloch et al.,
2017), intestinal dysfunction (Singh et al., 2016b), and surgery-induced immune
dysregulation (Denes et al., 2011) are common features of this model, variables that may
occur in stroked mice at random which may mask or confound any potential impact of the
co-morbidity or perturbation in question. Furthermore, infarct volumes in our study were
variable within experimental groups, consistent with previous studies (Liesz et al., 2009b;

142
Zhou et al., 2013). This is an important caveat of this model, as the extent of damage is
affected by collateral vessels which feed the MCA territory and these vessels in inbred
C57BL/6 mice are notoriously variable (McColl et al., 2004). Thus, in our study, the
relatively modest periodontitis-induced systemic effects could have been masked by
stroke- or surgery-induced systemic immune dysfunction, physiological stress and
inherent variation in infarct sizes.
With this in mind, we used the permanent thrombotic stroke model in an attempt to detect
more nuanced changes in infarct volumes, as this model does not cause marked immune
or physiological dysfunction (Liesz et al., 2009a), infarct size is less variable (Zhou et al.,
2013; Llovera et al., 2014), and neuroinflammation is more pronounced (Zhou et al.,
2013). In addition, we sought to enhance the systemic inflammatory response by
administering systemic Pg-LPS to better reflect human disease, as over 50% of
periodontitis patients are reported to contain oral-derived LPS in the circulation (DeLeon-
Pennell et al., 2013), which is distinct from LPS from enterobacteria like E. coli
(Hirschfeld et al., 2001). Since periodontitis and Pg-LPS did not affect ischaemic brain
damage in our study, tolerance to low-dose LPS may be cited as a viable explanation.
However, in studies of ischaemic tolerance, repeated challenge with sub-injurious doses
are neuroprotective (Rosenzweig et al., 2004; Marsh et al., 2009; Garcia-Bonilla et al.,
2018; Wendeln et al., 2018), whereas in our study, we did not find increased protection or
increased injury with Pg-LPS.
4.4.3. The validity of experimental periodontitis models
A valid explanation for our results may be due to the limitations of the experimental
periodontitis approach, although, as discussed previously, mechanical ligature placement
is a well-accepted model of human oral pathology (Graves et al., 2008; Hajishengallis et

143
al., 2015; de Molon et al., 2016). In fact, it is possibly superior than other established
periodontitis models, such as oral P. gingivalis inoculation, as this does not yield
pronounced bone loss (de Molon et al., 2014, 2016). Furthermore, P. gingivalis is a
human-specific pathogen and is found in extremely low numbers (<0.01% of total
bacterial counts) in the human oral cavity (Hajishengallis et al., 2011). This shows a huge
disparity between human periodontitis and experimental P. gingivalis inoculation in mice,
as the bacterium requires frequent and high inoculating doses, as well as antibiotic pre-
treatment, to successfully colonise the murine oral cavity (Graves et al., 2008). Given the
commensal disturbances and immune dysfunction that occur with antibiotic treatment
(Ekmekciu et al., 2017; Scott et al., 2018), which has been shown to affect stroke
outcome (Benakis et al., 2016), this raises the question of the suitability of infection
models for studying the immune response or effects on systemic conditions. It is therefore
interesting that most of the murine studies that associate periodontitis with adverse
systemic effects are typically carried out using oral P. gingivalis infections and not
ligatures (Gibson et al., 2004; Ogrendik, 2013; Dominy et al., 2019), suggesting that
periopathogens are vital for adverse disease-modifying effects and an explanation for the
lack of impact with ligature placement in the present work. While use of P. gingivalis is
directly translatable to human disease, this highlights the fundamental differences in the
pathogenicity of native human and mouse microbial species during periodontitis. Thus,
there are both advantages and limitations of using indigenous or foreign species in rodent
periodontitis studies, and that the decision of which model to use must be carefully
considered when aiming to assess the disease-modifying effects of periodontitis.
The acute timeframe of the periodontitis model used in this study must also be
considered, as this may not lead to sufficiently chronic inflammatory changes to affect
stroke immunopathology. By contrast, human periodontitis is a chronic condition
associated with transient bacteraemias and low-level inflammation that occur over a long

144
period, from years to decades (Kinane et al., 2005; Moutsopoulos and Madianos, 2006).
Indeed, chronic systemic Pg-LPS challenge (1 mg/kg daily for 5 weeks) has been shown
to induce neuroinflammation and cognitive deficits in aged mice (Wu et al., 2017),
suggesting that chronicity is an important component in the impact of periodontitis on
systemic diseases. However, in our study in mice, ligatures left in place for longer would
risk tooth loss, and would be defined as “edentulism” (toothlessness) and not
“periodontitis” per se. Thus, there is a need to develop a more suitable periodontitis
model that encapsulates local and systemic pathology as well as bacterial virulence in a
chronic manner, such as bacteria-soaked ligatures with chronic systemic Pg-LPS
administration. This study was also limited by the absence of a periodontitis-only group,
and thus a suitable comparison between periodontitis and periodontitis + stroke groups
could not be made. Furthermore, the mice used in this study were young and otherwise
healthy, and most individuals with periodontitis have other confounding factors
(smoking, hypertension, old age, obesity) which can independently account for increased
stroke risk (Neuhaus et al., 2014). While no model can sufficiently replicate all aspects of
the human disease, the animal models used in this study are well-characterised, well-
accepted, and as close to human pathology as is possible in rodents, and as such, the
results presented here should be considered valid and act as a basis to inform future
studies.
4.4.4. Conclusion
In conclusion, we have shown that after experimental stroke, ligature-induced
periodontitis does not modulate inflammation or worsen acute outcome. This indicates
that periodontitis alone is not sufficient to aggravate brain damage associated with stroke,
at least in the experimental paradigm of our study. However, we cannot exclude the
possibility that periodontitis causes adverse effects which were not accounted for in this

145
study, such as altered blood-flow, which is known to be altered by systemic inflammation
(Burrows et al., 2016), and this could be a predictor for risk of repeat stroke or
cardiovascular event. Similarly, it is not known if periodontitis affects long-term outcome
after stroke, delaying recovery by affecting tissue repair and angiogenic processes or by
impairing cognition, the latter of which can occur with chronic systemic Pg-LPS
challenge (Wu et al., 2017). Given the impact of P. gingivalis oral infections on systemic
disease progression, it is also feasible that use of a similar methodology could worsen
stroke outcome. Finally, there is also the possibility that in tandem with other co-
morbidities, such as age and obesity, periodontitis can exaggerate stroke-induced
pathology in a synergistic manner. While some of these questions may be difficult to
address in experimental animal models, the high prevalence of periodontitis and the
morbidity of stroke in humans mean that these are important avenues that warrant further
investigation.

146
Chapter 5. Regulation of the gingival
haematopoietic network

147
5.1. Introduction
Mononuclear phagocytes, such as monocytes, macrophages, and dendritic cells (DCs),
are critical responders to sites of injury, infection, and inflammation, and function in
maintaining tissue homeostasis (Guilliams et al., 2014). Monocytes are found mainly in
the bone marrow, blood, and spleen (Swirski et al., 2009) and are functionally plastic
cells that give rise to macrophages and DCs under both steady-state and inflammatory
conditions (Cheong et al., 2010). In mice, monocytes are divided into two subsets, Ly6Chi
monocytes and Ly6Clo monocytes (Geissmann et al., 2003; Auffray et al., 2007). During
pathological states, inflammatory Ly6Chi monocytes exit the bone marrow, circulate via
the blood and rapidly infiltrate the tissue where, in addition to differentiating, they can
take on diverse monocytic functions in line with specific environmental cues, such as
tissue repair (Ikeda et al., 2018), angiogenesis (Avraham-Davidi et al., 2013), and
limiting of pathologic responses to commensals during infection (Grainger et al., 2013;
Askenase et al., 2015). By contrast, Ly6Clo monocytes are derived from blood Ly6Chi
monocytes (Yona et al., 2013), and are not thought to be involved in the response to
inflammation and infection, as they predominantly function in maintaining vascular
health (Geissmann et al., 2003; Auffray et al., 2007; Carlin et al., 2013).
While monocytes are short-lived and actively home to inflammatory sites where they
differentiate into monocyte-derived macrophages and monocyte-derived DCs (Ginhoux
and Jung, 2014), resident macrophages, in contrast, are long-lived self-renewing
populations that inhabit distinct tissue niches and are maintained independently of bone
marrow monocyte input (Hashimoto et al., 2013). These tissue-resident macrophage
populations are seeded either from the embryonic yolk sac, such as brain microglia
(Ginhoux et al., 2010; Sheng et al., 2015), or from monocytes and haematopoietic stem
and progenitor cells (HSPCs) from the foetal liver, which occurs in sites such as the lung

148
(Guilliams et al., 2013), skin (Tamoutounour et al., 2013), intestine (Bain et al., 2014),
and heart (Epelman et al., 2014). However, the macrophage pool in sites like the heart
and intestine is constantly replenished by circulating monocytes after birth (Bain et al.,
2014; Molawi et al., 2014; Shaw et al., 2018). Moreover, circulating monocytes are
capable of re-populating niches in the absence (Scott et al., 2016; Lund et al., 2018), and
even in the presence of (Cronk et al., 2018; Shemer et al., 2018), a tissue-resident
macrophage population, although they maintain a distinct transcriptional identity (Cronk
et al., 2018). Thus, there are potentially distinct macrophage ontogenies present within
the same tissue, wherein resident macrophages can be established from single or multiple
origins, before and after birth (Yona et al., 2013).
Given their phenotypic and functional diversity in both homeostatic and inflammatory
conditions, the factors that shape the monocyte compartment is the subject of recent
study. In this context, monocytes are directed by local tissue-specific cues upon tissue
entry, such as vascular endothelial growth factor (VEGF) (Avraham-Davidi et al., 2013),
and oxidised low-density lipoprotein (Christ et al., 2018), thereby instructing their
effector function. Moreover, there is emerging evidence that monocytes are key mediators
of “trained innate immunity”, whereby metabolites, such as mevalonate (Bekkering et al.,
2018), as well as microbes and their products, such as the Bacille Calmette-Guerin
vaccine (Kleinnijenhuis et al., 2012) and β-glucan from Candida albicans (Quintin et al.,
2012; Saeed et al., 2014; Mitroulis et al., 2018), can reprogram subsequent monocyte
effector function, conferred through epigenetic modifications (Quintin et al., 2012; Saeed
et al., 2014). Training of monocytes can be exerted from within the bone marrow,
allowing monocyte function to be modulated distally from the site of inflammation and
prior to egress. For example, Toxoplasma gondii infection promotes IFNγ release by NK
cells in the bone marrow, which instructs monocytes to assume a regulatory phenotype
upon exit (Askenase et al., 2015). Similarly, a population of regulatory Ym1+ Ly6Chi

149
monocytes are trained in the bone marrow to initiate tissue repair programs in the
inflamed gut during colitis (Ikeda et al., 2018).
While monocytes integrate a diverse range of inputs that tailor their function directly in
the bone marrow or in the tissue, the monocyte compartment can be shaped indirectly by
changes at the progenitor level (Mitroulis et al., 2018). Mature bone marrow monocytes
are terminally derived via sequential intermediate progenitors from rare, self-renewing,
multipotent haematopoietic stem cells (HSCs). These so-called HSCs are responsible for
the generation and maintenance of all blood cell lineages, a process termed
haematopoiesis (Figure 5.1). Downstream of the HSCs is a linear differentiation pathway
where cell fates become more lineage-committed, from common myeloid progenitors
(CMPs), which give rise to myeloid and erythrocyte lineages, to the most recent lineage-
committed common monocyte progenitors (cMoPs) (Hettinger et al., 2013). While it is
thought that monocyte development follows a sequential hierarchy, this has recently been
challenged by demonstrations that Ly6Chi monocytes can be independently generated
from two branches in the haematopoietic tree, the macrophage-monocyte-dendritic cell
progenitors (MDPs) and granulocyte-monocyte progenitors (GMPs), which both diverge
from CMPs. In particular, LPS induces “neutrophil-like” GMP-derived monocytes
whereas CpG motifs drive monocytes derived from MDPs in vivo (Yáñez et al., 2017). As
such, committed monocyte progenitors can modulate their cellular differentiation patterns
based on environmental cues.

150
Figure 5.1. The haematopoietic tree. HSCs are rare self-renewing populations with the capacity to differentiate into all blood cells. MPPs are also multipotent but lose the ability to self-renew. Further downstream, CMPs and CLPs give rise to myeloid and lymphoid lineages respectively, via a hierarchical sequence of intermediate progenitors that become progressively more lineage-committed. The terminal differentiation of mature blood cells, such as monocytes, occurs from recent progenitors, such as cMoPs. LT-HSC = Long-term haematopoietic stem cell. ST-HSC = short-term haematopoietic stem cell. MPP = multipotent progenitor. CLP = common lymphoid progenitor. CMP = common myeloid progenitor. MEP = megakaryocyte-erythroid progenitor. GMP = granulocyte-monocyte progenitor. MDP = monocyte-macrophage-dendritic cell progenitor. cMoP = common monocyte progenitor. CDP = common dendritic cell progenitor.
Similarly, it has recently become appreciated that the upstream progenitor and stem cell
populations (collectively referred to as HSPCs) that give rise to monocytic lineages are
themselves directly influenced by intrinsic and environmental cues, which includes IFNα,
IFNγ, M-CSF, G-CSF, TLR ligands such as LPS, and β-glucan (Nagai et al., 2006;
Baldridge et al., 2010; Griseri et al., 2012; Mossadegh-Keller et al., 2013; Mitroulis et al.,
2018). HSPCs normally reside in specialised microenvironments, or niches, created by
surrounding stromal cells. Bone-forming osteoblasts (Calvi et al., 2003), nestin+
mesenchymal stem cells (Méndez-Ferrer et al., 2010), as well as CD169+ macrophages
(Chow et al., 2011) and endothelial cells (Ding et al., 2012) comprise this support
network, producing many adhesion molecules and soluble factors, including CXCL12
and stem cell factor, which tether and maintain HSPCs within the bone marrow niche
(Ding et al., 2012). As such, there are varied inputs to the HSPC niche, and physiological

151
stressors, such as old age (Mann et al., 2018), infection (Baldridge et al., 2010), or
inflammation (Griseri et al., 2012), can override these intrinsic retention signals,
increasing the rate of haematopoiesis and shaping the overall output of immune effector
populations.
Further adding to this complexity is the concept that haematopoiesis is not confined
solely to the bone marrow and can occur at sites outside this primary haematopoietic site,
a process termed extramedullary haematopoiesis (EMH). During steady-state, HSPCs are
constitutively mobilised from the bone marrow in low numbers into the blood and cycle
through peripheral tissues, but in inflammation are capable of giving rise to myeloid cells
in situ (Wright et al., 2001; McKinney-Freeman et al., 2002; Massberg et al., 2007).
Moreover, the spleen and liver contain significant reservoirs of undifferentiated
monocytes and HSPCs, including cMoPs, which are poised to expand and respond to
infectious and inflammatory stress, which can complement the bone marrow response or
function independently, depending on the context (Swirski et al., 2009; Leuschner et al.,
2012; Robbins et al., 2012; Siracusa et al., 2013). Some emerging evidence has indicated
that under inflammatory conditions, other tissue sites can also provide a niche for
extramedullary haematopoietic activity, such as the colon (Griseri et al., 2012) and gut-
associated lymphoid tissue (GALT) (Saenz et al., 2010). Specifically, during IL-23-
dependent colitis, IFNγ and GM-CSF drive increases in HSCs in the bone marrow and
GMPs in the colon and spleen, which in turn aggravates disease pathology (Griseri et al.,
2012). On the other hand, IL-25 drives an MPPtype2 population in the GALT, and
subsequently promotes Th2-type responses to control helminth infection. Thus, these
reports highlight that EMH can occur at mucosal sites under inflammatory conditions,
and that HSPCs can modulate their progeny depending on the nature of inflammatory
stimulus.

152
Whether other mucosal sites, such as the gingiva, can exhibit a similar independent
haematopoietic capacity under inflammatory conditions is not known. In the context of
periodontitis, gingival Th17 cells exhibit local proliferation (Dutzan et al., 2017, 2018),
and inflammatory myeloid cells are actively recruited from the bloodstream
(Moutsopoulos et al., 2014), but in situ production of the latter has not yet been described.
In addition, while the role of lymphocytes in instructing local immune networks has been
recently demonstrated during steady-state (Dutzan et al., 2017; Krishnan et al., 2018;
Wilharm et al., 2019) and in disease (Dutzan et al., 2018), there is limited knowledge
about the ontogeny of gingival monocytes and macrophage populations and their function
in maintaining oral tissue and immune homeostasis. Given that the gingiva is a site of
constant mechanical damage from mastication (Dutzan et al., 2017), requiring reparative
responses by amphiregulin-producing γδ T cells (Krishnan et al., 2018; Wilharm et al.,
2019), monocytes and macrophages may participate in wound-healing. However, during
oral P. gingivalis infection, CX3CR1+ monocyte/macrophages have been found to
promote bone destruction (Steinmetz et al., 2016), suggesting that the beneficial or
detrimental role of these leukocytes is context-dependent. Critically, the oral
mononuclear phagocyte network has been understudied in comparison to other immune
populations and considering their diverse functions at other barrier sites, from limiting to
promoting inflammation (Grainger et al., 2013; Askenase et al., 2015; Singh et al.,
2016a), they require more thorough exploration to determine their importance in oral
immunity.
5.2. Study rationale
Dr Siddharth Krishnan (SK) and Dr Joanne Konkel (JEK) sought to determine the local
instruction and maintenance of the gingival mononuclear phagocyte network. They have
provided evidence for existence of a tissue-resident monocyte within the gingiva during

153
steady-state (Figure 5.2). Monocytes and macrophages in barrier sites were identified by
their varying expression of Ly6C and MHCII, with a “waterfall” distribution as described
previously (Tamoutounour et al., 2012, 2013), which describes the spectrum of monocyte
to macrophage differentiation. Ly6Chi monocytes dominated in gingival tissue (~80% of
waterfall), in contrast to the skin and small intestine, where macrophages dominated
(Figure 5.2A). Further, gingival Ly6Chi monocytes were not labelled by anti-CD45
antibody, which marks circulating CD45+ cells, in contrast to their blood monocyte
counterparts (Figure 5.2B). These gingival monocytes were also not recruited from the
bone marrow via CCR2, as Ccr2-/- mice, which constitutively lack circulating monocytes
(Serbina and Pamer, 2006), had unaltered levels of Ly6Chi monocytes in the gingiva
compared to wild-type mice (Figure 5.2C). Using head-shielded bone marrow chimeras
(which protected the gingival leukocyte reservoir) (Figure 5.2D), Ly6Chi monocytes in
the gingiva were primarily host-derived (~55–60%), in comparison to blood monocytes
(Figure 5.2E). Taken together these data confirm that gingival monocytes are maintained
locally, independently of the bone marrow, and are infrequently replenished from the
circulating monocyte pool in steady-state.
This work proposes that, in contrast to other barrier sites and indeed other tissues in
general, the gingiva is a tissue dominated by monocytes, raising the possibility that
monocytes can reside in gingival tissue without obligatory differentiation, given the ratio
of monocytes to macrophages. There is emerging evidence that monocytes found in the
skin, lung, and lymph nodes during steady-state lack overt DC or macrophage signatures
and retain a monocytic gene expression profile (Ly6Chi, MHCII+) that resemble that of
blood monocytes (Jakubzick et al., 2013). However, monocytes in the gingiva lack
MHCII expression and are thus distinct from the authors’ proposed definition of a “tissue
monocyte”. Another important consideration is that the lung and skin contain large
populations of resident macrophages in addition to smaller proportions of tissue

154
monocytes (Tamoutounour et al., 2013; Shaw et al., 2018), contrasting with the monocyte
majority and macrophage minority observed in the gingiva. In addition, the authors
propose that these monocytes persist only for a limited time in the tissue (Jakubzick et al.,
2013). By contrast, SK and JEK have shown using multiple approaches that monocytes
are constitutively present in the gingiva and independent of circulating bone marrow-
derived monocytes. Further clarification is needed in regard to how the status of
“monocyte” is maintained, as well as outstanding questions of half-life, ontogeny, and
function, in health and indeed, in inflammation. Comparing the transcriptional signatures
of monocytes from the gingiva, blood, and bone marrow etc. will be key in elucidating
how these gingival monocytes are distinct from or like other monocytes, and also whether
the term “monocyte” is still appropriate.

155
Figure 5.2. The mouse gingiva contains a unique population of resident monocytes. (A) Representative FACS plots and frequencies of Ly6Chi monocytes and macrophages (Ly6C-) in various barrier sites during steady-state. Populations were gated as shown in Figure 3.3A. (B) Representative FACS plots of blood and gingival monocytes labelled with anti-CD45 antibody. CD45 was injected intravenously (i.v.) 5 minutes before sacrifice. (C) Frequencies of monocytes and macrophages in the gingiva of wild-type (wt) and Ccr2-/- mice during steady-state. (D) Generation of head-shielded chimeras. CD45.2 host mice were irradiated and reconstituted with CD45.1 donor bone marrow. (E) Representative FACS plots and frequencies show the contribution of host and donor to the blood and gingival monocyte pool, determined at 12- and 20-weeks post-transfer. Data are presented as mean ± SEM; n = 7-11 mice per group (A), n = 12 mice per group (C), n = 4-5 mice per group (B, D, E). Data is collated from 2 independent experiments.

156
5.3. Aims
The work by SK and JEK indicates that the gingiva represents a previously unrecognised
and unique site that harbours monocytes during steady-state that do not undergo
obligatory differentiation to macrophages, opposing the current paradigm for other barrier
sites such as the intestine (Bain et al., 2014; Hoeffel et al., 2015). However, without bone
marrow input, the question is raised about how these monocytes are generated and
maintained, which could suggest that there are reservoirs of monocyte progenitors which
give rise to local monocytes. Furthermore, it is not known how local inflammation affects
these monocytes. The primary aim of this study therefore was to establish the ontogeny
and local instruction of gingival monocytes and determine the impact of inflammation on
these processes. We used models of acute periodontitis (ligature-induced) and chronic
periodontitis (age-induced) in order to assess the response of the local monocytic and
haematopoietic niche.
5.4. Results
5.4.1. Identification of a haematopoietic niche in the mouse gingiva
during steady-state
In order to define the ontogeny of tissue monocytes, we adopted a flow cytometry-based
approach to identify by surface marker expression (CD45+Lin-CD11b-CD11c-Ly6C-) if
there were reservoirs of HSPCs in the gingiva during steady-state. Indeed, we identified
multiple populations of HSPCs in the gingiva during steady-state (Figure 5.3A), which
were comparable in phenotype to those in the bone marrow (Figure 5.3B), and were in
line with current gating strategies (Griseri et al., 2012; Pietras et al., 2015; Mitroulis et al.,
2018). This included the LSK (Lin-Sca-1+cKit+) fraction, a heterogeneous group loosely
defined as “HSPCs” that contains self-renewing HSCs as well as non-renewing

157
multipotent progenitors (MPPs). Within the LSK compartment, we identified MPPs,
which are multipotent but preferentially give rise to certain lineages; the lymphoid-biased
MPP4s and myeloid-biased MPP2/3s, based on differential expression of Flt3 (CD135)
and CD48 (Pietras et al., 2015; Mitroulis et al., 2018). Further, we identified populations
of oligopotent myeloid progenitors (MyPs; c-Kit+ Sca-1-), and within this population,
CMPs and GMPs, based on the strength of CD16/32 expression. Thus, we have identified
a novel HSPC niche within the adult mouse gingiva during steady-state, which is capable
of giving rise to monocytes locally within the gingival tissue.
Figure 5.3. The mouse gingiva contains populations of multipotent and oligopotent haematopoietic stem and progenitor cells during steady-state. (A-B) The gingiva contains populations of haematopoietic stem and progenitor cells (HSPCs) similar to that of the bone marrow and these HSPCs are present in health. HSPCs were identified
as Lineage(CD3, TCR, B220, CD19, Ly6G, Ter119, NK1.1, FcεR1)- CD11b- Ly6C- CD11c-. MyPs = myeloid progenitors. LSK = Lin- Sca-1+ c-Kit+. MPP = multipotent progenitor. GMP = granulocyte monocyte progenitor. CMP = common myeloid progenitor.

158
5.4.2. Ligature-induced periodontitis alters the number and output of
gingival HSPCs
Thus far we have demonstrated that the gingiva harbours populations of resident
monocytes and HSPCs during “steady-state” (the gingiva is a site of constant stimulation
even in the absence of disease or inflammation). Since EMH has so far been reported to
occur only under inflammatory conditions (Saenz et al., 2010; Griseri et al., 2012;
Leuschner et al., 2012), this is a potentially pivotal observation that the gingiva is a site of
EMH in the absence of overt inflammation. Therefore, we next sought to define the
contribution of these populations in the context of inflammation, and also to what extent
the primary bone marrow niche was involved during oral inflammation. To this end, we
induced periodontitis in 9-10-week-old mice by bilateral ligatures for 10 days and
subsequently determined the impact of oral pathology on the mononuclear phagocyte and
progenitor network in the gingiva and bone marrow. In line with an overall increase in
gingival cell number during periodontitis (Chapter 3, Figure 3.1), Ly6Chi monocytes
increased in number (Figure 5.4A), and this was not observed in the bone marrow
(Figure 5.4B), indicating differential regulation of the niches during experimental
periodontitis. In addition, gingival MyPs and LSK frequencies were not preferentially
altered in animals with periodontitis, but significantly increased in number (Figure 5.5A).
By contrast, bone marrow MyPs and LSKs did not change in frequency or number
(Figure 5.5B), and HSPCs were barely detectable in the blood in both groups (Figure
5.5C). This would indicate that HSPCs are likely not mobilised from the bone marrow in
these mice, although we cannot exclude the possibility that they may not be easily
detectable with the current methodology, as a small but steady population of HSPCs are
reported to constantly migrate via the bloodstream (Wright et al., 2001; Massberg et al.,
2007). Furthermore, we observed increases in numbers of MPP2/3s and MPP4s in the
gingiva but not in the bone marrow during periodontitis (Figure 5.6A-D), in agreement

159
with reports describing these MPPs as emergency responders during inflammation
(Pietras et al., 2015). However, CMPs and GMPs were not significantly affected at either
site (Figure 5.6E-H), suggesting that periodontitis induces selective expansion of
multipotent HSCs over lineage-committed myeloid progenitors. Taken together, this
suggests that local inflammatory pathology results in marked increases in numbers of
primitive HSPCs and mature effector cells, and that this expansion is likely regulated
locally without recruitment from the circulation.
Figure 5.4. Gingival Ly6Chi monocytes increase in number during ligature-induced periodontitis. (A) Representative FACS plots, frequencies, and numbers of Ly6Chi monocytes in the gingiva 10 days after periodontitis induction (double ligatures). Ly6Chi monocytes were gated: CD45+
Lineage(CD3, TCR, B220, CD19, Ly6G, Ter119, NK1.1, FcεR1)- CD11b+ Ly6C+. (B) Frequencies and numbers of Ly6Chi monocytes in the bone marrow 10 days after periodontitis induction. Data are presented as mean ± SEM; n = 6 mice per group. Data in (A) was log transformed to permit statistical analysis. Data is collated from 2 independent experiments. Statistical comparisons performed using an unpaired t-test; ** p < 0.01. PD = periodontitis (double ligature).

160
Figure 5.5. Gingival LSKs expand in number during ligature-induced periodontitis. (A) Representative FACS plots, frequencies, and cell numbers of LSKs and MyPs in the gingiva 10 days after periodontitis induction. (B) Representative FACS plots, frequencies, and cell numbers of LSKs and MyPs in the bone marrow 10 days after periodontitis induction. (C) Representative FACS plots showing absence of LSKs and MyPs in the blood 10 days after periodontitis induction. Data are presented as mean ± SEM; n = 6 mice per group. Data is collated from 2 independent experiments. Data in (A) was log transformed to permit statistical analysis. Statistical comparisons performed using an unpaired t-test; ** p < 0.01. PD = periodontitis (double ligature). LSK = Lin- Sca-1+ c-Kit+. MyP = myeloid progenitor.

161
Figure 5.6. Discrete populations of multipotent gingival progenitors increase in number during ligature-induced periodontitis. (A-H) Frequencies and numbers of MPP2/3, MPP4, CMPs, and GMPs in the gingiva and bone marrow 10 days after periodontitis induction. Data are presented as mean ± SEM; n = 6 mice per group. Data is collated from 2 independent experiments. Data in (A), (C), (E), and (G) was log transformed to permit statistical analysis. Statistical comparisons performed using an unpaired t-test; ** p < 0.01. PD = periodontitis (double ligature). MPP = multipotent progenitor. GMP = granulocyte monocyte progenitor. CMP = common myeloid progenitor.
As gingival HSPCs expanded during periodontitis, we investigated if their differentiation
ability was altered. To achieve this, we isolated gingiva from mice with and without
periodontitis and plated single cell suspensions on a semi-solid methylcellulose medium

162
(MethoCult), which contains growth factors and cytokines (including IL-3, IL-6, and
erythropoietin) to support the differentiation of HSCs into multiple progenitor lineages,
including erythroid progenitors and granulocyte/macrophage progenitors (cfu-GM)
(Figure 5.7A). We observed growth of cfu-GM in both groups, indicating that gingival
HSPCs display a capacity for multilineage differentiation (Figure 5.7B). HSPCs from
periodontitis mice displayed a significantly increased rate of colony formation, as
expected, since periodontitis drives increases in the numbers of gingival progenitors and
this would translate as a greater overall rate of myelopoiesis. Interestingly, periodontitis
skewed HSPC differentiation to granulocytes (Figure 5.7C), suggesting that
inflammatory signals in the oral mucosa tailor the local haematopoietic compartment to
alter their output. Furthermore, we confirmed using this approach that these resident
HSPCs are localised in the gingival tissue itself and not present in the maxillae/mandible
(jaw) bones, as the latter did not exhibit formation of cfu-GM colonies (Figure 5.7D).

163
Figure 5.7. Ligature-induced periodontitis increases the differentiation output of gingival haematopoietic stem and progenitor cells. (A) Experimental outline. (B) Representative images and quantification of granulocyte-macrophage colonies (cfu-GM) that have differentiated from gingival haematopoietic stem cells after 13 days on MethoCult medium. Scale bar = 0.5 mm. (C) Representative FACS plots and frequencies of monocytic/macrophage cells (CD45+ CD11b+
CD115+) and neutrophils (CD45+ CD11b+ Ly6G+) grown on MethoCult plates in (A). (D) Representative images and quantification of granulocyte-macrophage colonies from the femur and maxillae/mandible bones after 13 days on MethoCult medium. Scale bar = 0.5 mm. Data are presented as mean ± SEM; n = 7 mice per group (B, C). n = 1-2 per group (D). Data is collated from 2 independent experiments. Statistical comparisons performed using an unpaired t-test; ** p < 0.01. PD = periodontitis (double ligature). BM = bone marrow.

164
5.4.3. Ageing leads to a decline in the number of gingival HSPCs
Thus far we have shown that acute ligature-induced periodontitis modulates gingival
HSPCs. Since mice naturally develop bone loss as they age (Liang et al., 2010), we next
investigated if ageing, a model of chronic periodontitis, would evoke similar changes in
the haematopoietic compartment in the gingiva. We used young (8–12 weeks) and “aged”
(6–9 months) mice to determine how the gingival landscape is tailored in age. In contrast
to acute periodontitis, aged mice displayed a reduction in gingival Ly6Chi monocytes
(Figure 5.8). Further, Ki67+ staining revealed that bone marrow monocytes were less
proliferative, but gingival monocytes maintained a similar proliferation rate to their
young counterparts even though there were fewer overall, thus demonstrating that ageing
results in different HSPC effects in the gingiva and bone marrow.
Figure 5.8. Gingival Ly6Chi monocytes are reduced with age. (A-B) Frequencies and numbers of Ly6Chi monocytes and frequencies of Ly6Chi monocytes expressing Ki67+ in the gingiva and bone marrow (BM) of young (8-12 weeks) and aged (6-9 months) mice. Data are presented as mean ± SEM; n = 9 mice per group. Data is collated from 3 independent experiments except Ki67+ staining: one experiment, n = 3 mice per group. Statistical comparisons performed using an unpaired t-test; ** p < 0.01.

165
As we identified alterations in local monocyte numbers we next wanted to determine if
these changes were likely due to alterations in the gingival resident HPSC network. There
was a marked decrease in both LSKs and MyPs in the aged gingiva (Figure 5.9A), and
this was largely mirrored in the bone marrow (Figure 5.9B), suggesting that the
haematopoietic compartment as a whole is likely affected by increasing age. However,
the age-dependent proliferative capacity of gingival and bone marrow LSKs appeared to
be differentially modulated (Figure 5.10), indicating that advancing age constrains
gingival HSPC numbers but does not affect the proliferative competency of existing cells.
Furthermore, ageing significantly reduced MPP4s in the gingiva and bone marrow
(Figure 5.11A-B), and also led to a decline in the number of MPP2/3s only in the gingiva
(Figure 5.11C-D). Indeed, this is largely in agreement with previous studies
demonstrating that ageing leads to declines in numbers of lymphoid-biased MPP4s in the
bone marrow (Young et al., 2016). CMPs and GMPs were unchanged in the gingiva
(Figure 5.11E), but GMPs in the bone marrow were increased (Figure 5.11F) indicating
that multipotent and oligopotent progenitors are differently affected in ageing. Thus, both
bone marrow and gingival HSPC niches are similarly affected during physiological
ageing but display a degree of independent regulation, as different subsets of HSPCs are
affected in either niche.
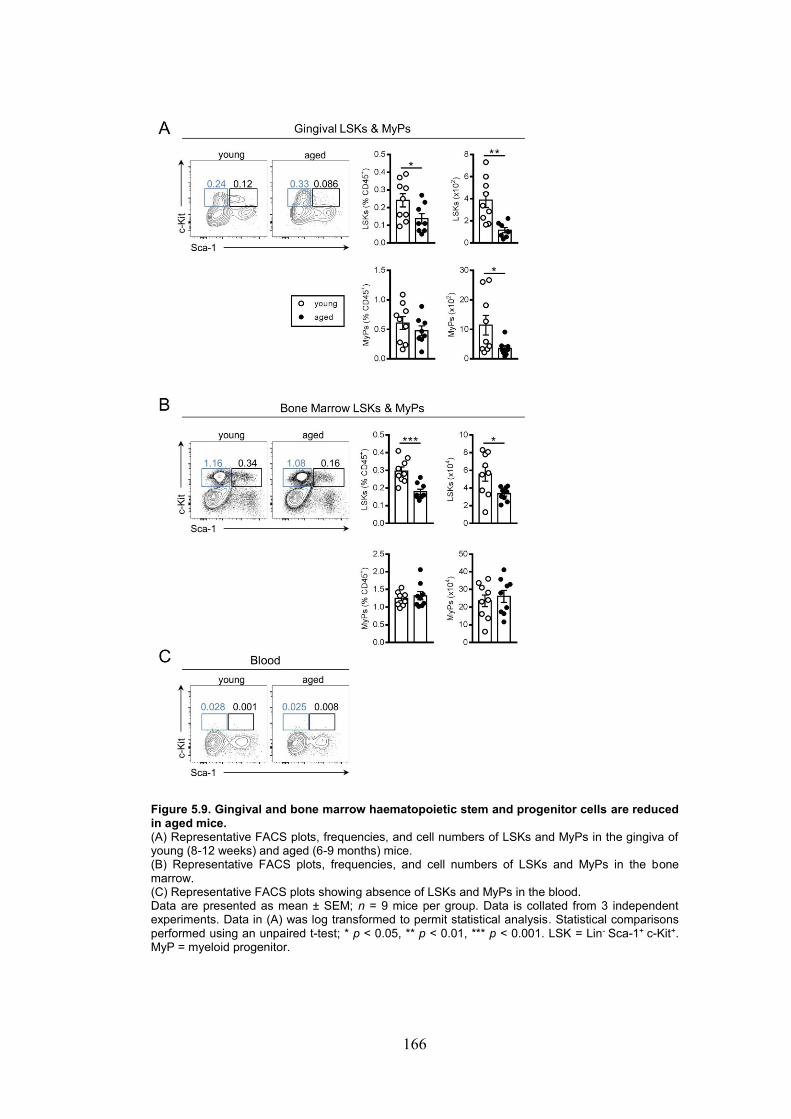
166
Figure 5.9. Gingival and bone marrow haematopoietic stem and progenitor cells are reduced in aged mice. (A) Representative FACS plots, frequencies, and cell numbers of LSKs and MyPs in the gingiva of young (8-12 weeks) and aged (6-9 months) mice. (B) Representative FACS plots, frequencies, and cell numbers of LSKs and MyPs in the bone marrow. (C) Representative FACS plots showing absence of LSKs and MyPs in the blood. Data are presented as mean ± SEM; n = 9 mice per group. Data is collated from 3 independent experiments. Data in (A) was log transformed to permit statistical analysis. Statistical comparisons performed using an unpaired t-test; * p < 0.05, ** p < 0.01, *** p < 0.001. LSK = Lin- Sca-1+ c-Kit+. MyP = myeloid progenitor.

167
Figure 5.10. The proliferative capacity of bone marrow, but not gingival, haematopoietic stem and progenitor cells is reduced in aged mice. (A-B) Representative FACS plots and frequencies of LSKs expressing Ki67+ in the gingiva and bone marrow of young (8-12 weeks) and aged (6-9 months) mice. Data are presented as mean ± SEM; n = 3 mice per group. Statistical comparisons performed using an unpaired t-test; * p < 0.05. LSK = Lin- Sca-1+ c-Kit+.

168
Figure 5.11. Lymphoid-biased multipotent progenitors in the gingiva and bone marrow are reduced in aged mice. (A-B) Representative FACS plots, frequencies, and numbers of lymphoid-biased MPP4s in the gingiva and bone marrow of young (8-12 weeks) and aged (6-9 months) mice. (C-F) Frequencies and numbers of myeloid-biased MPP2/3s and MyPs in the gingiva and bone marrow of young (8-12 weeks) and aged (6-9 months) mice. Data are presented as mean ± SEM; n = 9 mice per group. Data is collated from 3 independent experiments. Data in (A), (C), and (E) was log transformed to permit statistical analysis. Statistical comparisons performed using an unpaired t-test; * p < 0.05, ** p < 0.01, *** p < 0.001. MPP = multipotent progenitor. MyP = myeloid progenitor. BM = bone marrow.
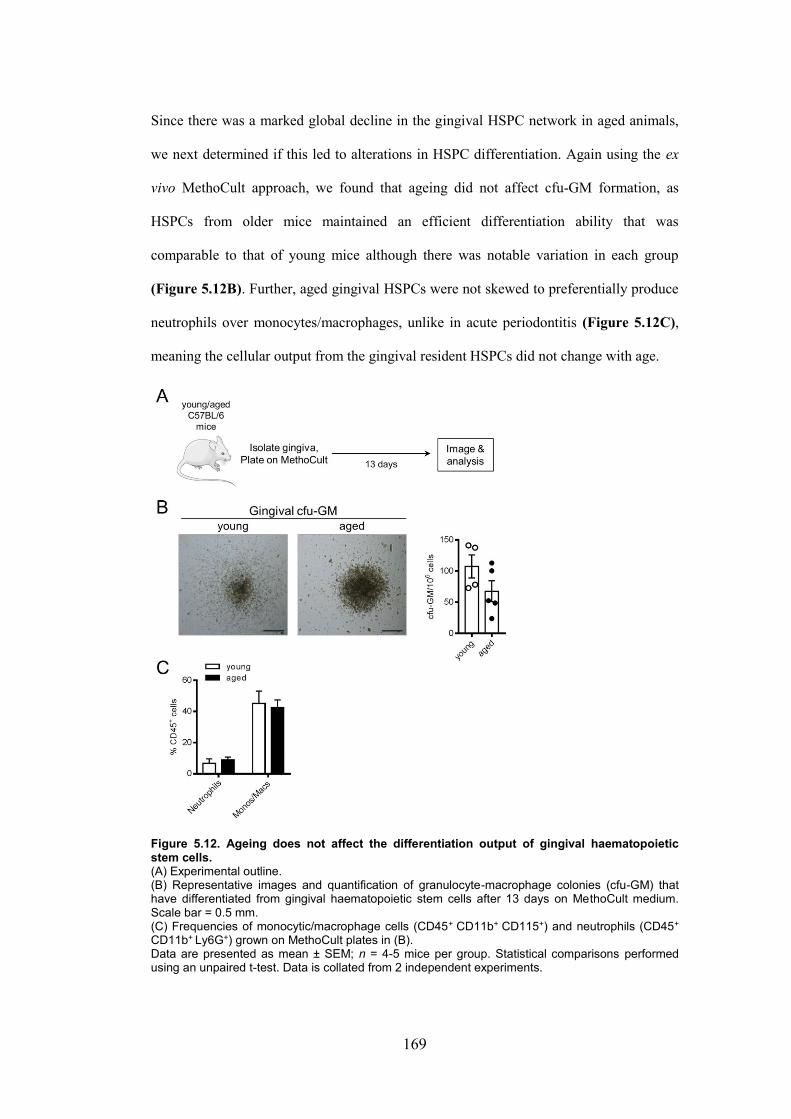
169
Since there was a marked global decline in the gingival HSPC network in aged animals,
we next determined if this led to alterations in HSPC differentiation. Again using the ex
vivo MethoCult approach, we found that ageing did not affect cfu-GM formation, as
HSPCs from older mice maintained an efficient differentiation ability that was
comparable to that of young mice although there was notable variation in each group
(Figure 5.12B). Further, aged gingival HSPCs were not skewed to preferentially produce
neutrophils over monocytes/macrophages, unlike in acute periodontitis (Figure 5.12C),
meaning the cellular output from the gingival resident HSPCs did not change with age.
Figure 5.12. Ageing does not affect the differentiation output of gingival haematopoietic stem cells. (A) Experimental outline. (B) Representative images and quantification of granulocyte-macrophage colonies (cfu-GM) that have differentiated from gingival haematopoietic stem cells after 13 days on MethoCult medium. Scale bar = 0.5 mm. (C) Frequencies of monocytic/macrophage cells (CD45+ CD11b+ CD115+) and neutrophils (CD45+
CD11b+ Ly6G+) grown on MethoCult plates in (B). Data are presented as mean ± SEM; n = 4-5 mice per group. Statistical comparisons performed using an unpaired t-test. Data is collated from 2 independent experiments.

170
Since there was apparent variability in the capacity of young and aged gingival HSPCs to
differentiate in vitro (when isolated from whole tissue) (Figure 5.12B), we further
explored if young and aged HSPCs were indeed displaying a similar capacity to form
colonies. To demonstrate this, we sorted progenitor populations and plated a defined
number of each cell population on MethoCult medium (Figure 5.13A). By taking this
approach, we could control for the observed differences in HSPC number in the tissue of
young and aged mice. In this way, we found that the MyP and LSK populations were
responsible for colony formation, as cKit-Sca-1- and cKit-Sca-1+ were unable to form cfu-
GM, in agreement with previous findings in the inflamed colon (Griseri et al., 2012).
However, although there were significantly fewer HSPCs in the aged gingiva, at a cellular
level, MyPs and LSKs from aged mice could produce colonies at an equivalent rate as
their young counterparts (Figure 5.13B). Together, these data suggest that while the
gingival HSPC pool declines over age, the HSPCs that are still present retain an adequate
capacity to produce progeny and the types of immune effectors produced are not altered.

171
Figure 5.13. Ageing does not affect the ability of gingival LSKs and MyPs to produce myeloid cell colonies. (A) Gingival haematopoietic stem and progenitor cells (HSPCs) were sorted from gingival tissue from young (8-12 weeks) and aged (6-9 months) mice and plated on MethoCult medium for 13 days. (B) Representative images and quantification of granulocyte-macrophage colonies (cfu-GM) that have differentiated from gingival haematopoietic stem cells after 13 days on MethoCult medium. Scale bar = 0.5 mm. Data are presented as mean ± SEM; n = 4 pooled mice per group. Statistical comparisons performed using a one-way ANOVA with post hoc Sidak’s test. Data is collated from 2 independent experiments.
5.4.4. The gingiva undergoes remodelling with age
Physiological stressors such as inflammation, infection, and ageing, modulate bone
marrow HSPCs by skewing their differentiation potential and promoting mobilisation
(King and Goodell, 2011; Mann et al., 2018). Since we identified advancing age as a
negative regulator of the gingival HSPC niche, we wanted to determine whether we could
identify mechanisms by which ageing specifically affects HSPCs. To begin to address
this, we employed a transcriptomic approach by performing bulk RNA sequencing of
gingival tissues of young (8 weeks) and “aged” (6 months) mice at steady-state (Figure
5.14A). Results of this sequencing analysis indicated 1243 differentially-expressed genes
(DEGs; padj <0.05, log2 fold change >1 or <-1) between aged and young gingiva in

172
steady-state, and that the majority (88%) of genes were reduced in aged mice (Figure
5.14B). Moreover, gene-set enrichment analysis using the Molecular Signatures Database
(MSigDB) hallmark gene set collection, showed a significant negative correlation with
haematopoiesis gene sets in the gingiva of aged mice (Figure 5.14C), indicating that the
aged gingiva loses a HSPC-supporting gene signature, supporting our findings by flow
cytometry. To dissect this finding further, we performed gene ontology (GO) analysis
(via AmigGO 2) of the positively- and negatively-regulated DEGs separately, to
determine the biological processes altered in ageing. Lymphocyte and T cell-mediated
responses were increased (Figure 5.14D), and this is in line with reports that gingival
Th17 cells increase in age, as a direct response to mastication-dependent increases in IL-6
production from gingival epithelial cells (Dutzan et al., 2017). By contrast, a striking
number of tissue homeostatic processes were downregulated, including cell adhesion,
extracellular matrix (ECM) organisation, bone maintenance, and haematopoietic
progenitor support (Figure 5.14E). GO analysis specifically revealed that within the
related ECM and cell adhesion modules, ageing downregulated a number of integrins
(itg) and protocadherins (pcdh), as well as an array of collagen genes (col), with matrix
metalloproteinases (mmp, adamst) and their inhibitors (timp) also displaying reductions in
gene expression (Figure 5.15B, C). Specifically in relation to known HSPC-supporting
factors, angiopoietins (angpt), insulin growth factors (igf), and members of the TGFβ
family (tgf, bmp) were all downregulated (Figure 5.15A), factors known to regulate
haematopoietic progenitor migration and activity in vivo and ex vivo (Zhang et al., 2003,
2006). Additionally, the osteoblast-related genes, osteopontin (spp1),
osteomodulin/osteoadherin (omd) and osteoglycin (ogn), were downregulated (Figure
5.15D); osteoblasts are intimately involved in regulating HSPC retention and
mobilisation in the bone marrow (Calvi et al., 2003). These large-scale shifts in the
transcriptional landscape of the aged gingiva suggest that bone remodelling and ECM
reorganisation are possible candidate pathways that could be contributing to a lack of
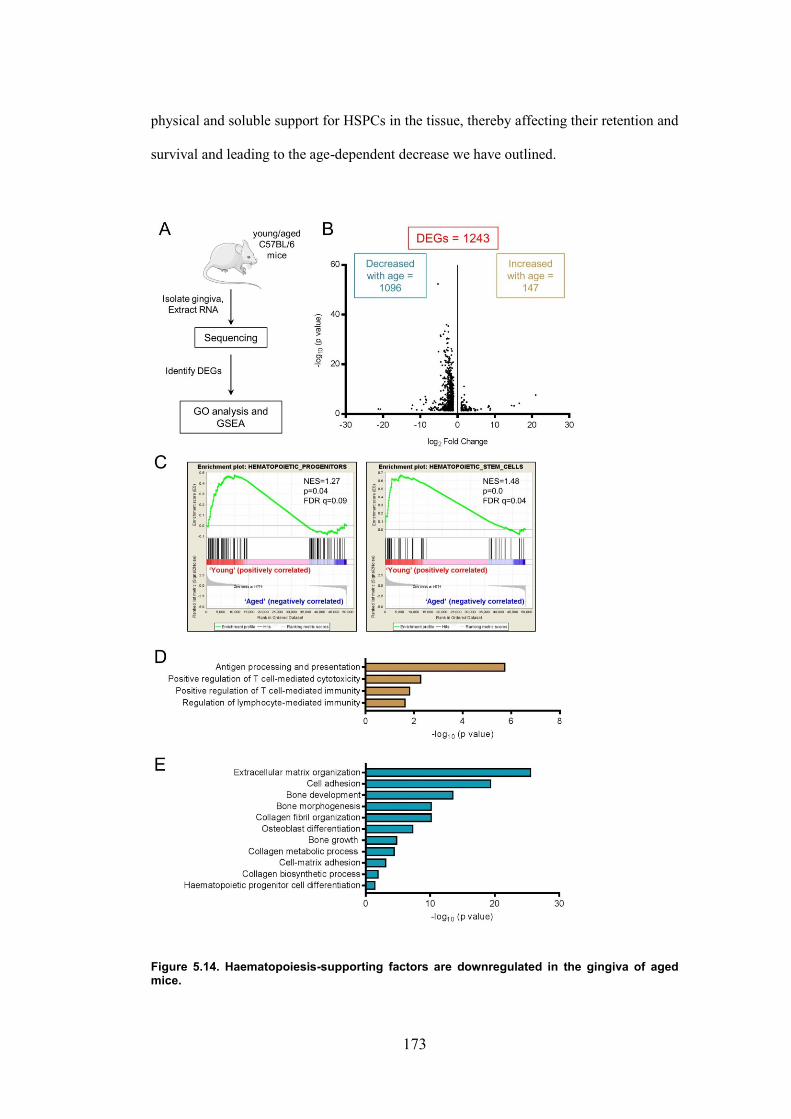
173
physical and soluble support for HSPCs in the tissue, thereby affecting their retention and
survival and leading to the age-dependent decrease we have outlined.
Figure 5.14. Haematopoiesis-supporting factors are downregulated in the gingiva of aged mice.

174
(A) Workflow of RNA sequencing in young (8 weeks) and aged (6 months) mice. GO = gene ontology. GSEA = gene-set enrichment analysis. (B) Volcano plot showing differentially-expressed genes (DEGs) in young and aged gingiva. All DEGs shown have padj < 0.05 and log2 fold change < -1 or > 1. (C) Gene-set enrichment analysis of gene sets associated with haematopoiesis. Gene sets are positively enriched in young compared to aged mice. NES = normalised enrichment score. FDR = false discovery rate. (D-E) Gene ontology analysis of positive and negative DEGs showing differentially-regulated biological processes in aged compared to young gingiva. n = 3-4 mice per group.
Figure 5.15. Genes relating to the physical integrity of the gingiva decrease with age. (A-D) Heatmaps showing individual differentially-expressed genes in young versus aged mice which were identified by GO analysis outlined in Figure 5.14D. n = 3-4 mice per group. GO = gene ontology. HSPC = haematopoietic stem and progenitor cell. ECM = extracellular matrix.

175
5.5. Discussion
5.5.1. A haematopoietic niche in the gingiva
In the present study, we describe a novel HSPC niche within the gingival tissue of adult
mice that expands during periodontitis and contracts with advancing age, and appears to
be differentially regulated from the bone marrow pool. We propose that gingival HSPCs,
like their bone marrow counterparts, have the capacity for multi-lineage differentiation,
and specifically, these gingival progenitors are capable of giving rise to monocytes and
thus may maintain the tissue-resident monocyte population that patrol the oral barrier in
steady-state and in inflammatory settings. The gingiva has not been previously described
as a reservoir for HSPCs, although peripheral tissues can be sites of extramedullary
haematopoiesis, such as muscle (McKinney-Freeman et al., 2002), liver (Cardier and
Barberá-Guillem, 1997), colon (Griseri et al., 2012), and spleen (Leuschner et al., 2012;
Siracusa et al., 2013). Importantly, many of these only undergo EMH in inflammatory or
stressed conditions, in contrast to the gingiva, in which we report presence of HSPCs in
steady-state. HSPCs are reported to constantly traffic between the bone marrow, blood,
lymph and peripheral tissues in steady-state (Wright et al., 2001; Massberg et al., 2007),
and as such, may also be trafficking to the gingiva. Furthermore, these migratory HSPCs
possess the capacity to differentiate in peripheral tissues, giving rise to tissue-resident
myeloid cells (Massberg et al., 2007). Importantly, however, this is only triggered under
inflammatory conditions, such as TLR stimulation (Massberg et al., 2007), which
highlights the unique nature of the gingiva as a HSPC reservoir in steady-state. As we did
not detect overt HSPC presence in the blood, it may be that this haematopoietic niche in
the gingiva is self-maintaining, independently of migrating HSPCs from the bone
marrow. However, we cannot rule out the possibility of low-level mobilisation from the
bone marrow, which may not have been detectable without the use of multiple
experimental time points. Certainly, it is estimated that 100–400 HSCs are thought to be

176
circulating at any one time in the peripheral blood of a mouse (Wright et al., 2001).
Further detailed analyses are therefore required to assess the contribution of the bone
marrow to the gingival niche.
5.5.2. Inflammatory regulation of the gingival haematopoietic niche
As mentioned, during inflammatory stress, sites other than the bone marrow and the main
extramedullary site, the spleen (Swirski et al., 2009; Robbins et al., 2012; Siracusa et al.,
2013), have been reported to undergo EMH. Specifically, IL-25 promotes the
colonisation of MPPtype2 in the gut-associated lymphoid tissue, and a subsequent Th2-type
myeloid output during helminth infection (Saenz et al., 2010). Furthermore, in a murine
model of IL-23-driven colitis, EMH occurs in the inflamed colon, driving myelopoiesis
(Griseri et al., 2012). In agreement, we report increased numbers of LSKs, and
specifically the MPP populations, in the inflamed gingiva, although, notably, these
HSPCs were present in the gingiva even during steady-state. Interestingly, gingival
HSPCs appeared to be regulated independently of the bone marrow, unlike in the report
by Griseri et al. (2012), in which increased progenitor activity occurred simultaneously in
bone marrow, spleen, and colon. This helps to reinforce the point that the gingiva is a site
for EMH under inflammatory conditions but is regulated differently from the
haematopoietic niche in the bone marrow.
The local inflammatory cues regulating the expansion of gingival HSPCs during
periodontitis are not clear. However, mouse bone marrow HSPCs possess a range of
pattern-recognition receptors, such as TLR-2/-4 (Nagai et al., 2006), and a range of
bacterial infections have been shown to expand HSPCs directly via LPS signalling
(Zhang et al., 2008; Rodriguez et al., 2009; Baldridge et al., 2010). Thus, this may also be
true of gingival HSPCs, as barrier breach during periodontitis can lead to increases in

177
TLR-activating bacterial products, such as LPS, which may directly instigate local HSPC
expansion and differentiation. In addition, endogenous cytokines may regulate gingival
HSPCs. Indeed, IL-6, TNFα, and IL-1β are increased in the gingival tissues during
periodontitis (Graves and Cochran, 2003), and these pro-inflammatory cytokines have
been shown to directly act on bone marrow HSPCs to augment proliferation and
differentiation during inflammatory stress (Ueda et al., 2004; Chen et al., 2010), which
could suggest that local cytokine production during periodontitis may affect the gingival
resident population of HSPCs in a similar regard. In particular, TNFα promotes B cell
mobilisation from the bone marrow and IL-1β promotes granulopoiesis (Ueda et al.,
2004), suggesting that the inflammatory milieu can not only augment HSPC output, but
skew towards particular lineages too. Furthermore, IFNγ is a potent factor regulating
HSPCs. IFNγ activates quiescent HSCs during chronic bacterial infection (Baldridge et
al., 2010), and both IFNγ and GM-CSF signalling mediate increased myelopoiesis in the
inflamed colon during colitis (Griseri et al., 2012). Thus, it appears that IFNγ drives
inflammatory haematopoiesis via direct effects on HSPCs. Even though IFNγ is
reportedly increased in patients (Dutzan et al., 2009) and mice (Garlet et al., 2008) during
periodontitis, it is unclear in our study whether periodontitis-induced increases in
numbers of monocytes and HSPCs are specifically mediated by IFNγ. In fact,
periodontitis is primarily considered a IL-17-mediated disease, with neutrophils central to
pathological tissue destruction (Dutzan et al., 2018). Thus, both these mediators may be
important for modulating gingival HSPCs, as IL-17 is known to stimulate granulopoiesis
in vivo (Schwarzenberger et al., 2000). Further, we found that gingival HSPCs isolated
from mice with periodontitis produced more neutrophils when cultured ex vivo. Together,
this indicates that mediators in the inflamed gingiva could directly modulate the HSPC
niche in order to mount efficient immune responses to the microbial invasion.

178
Of note, we observed that periodontitis increased the numbers of a range of stem and
progenitor populations as well as monocytes. This parallels studies in mice whereby
activation of the sympathetic nervous system (SNS), due to either chronic stress (Heidt et
al., 2014) or stroke (Courties et al., 2015), increases numbers of LSKs, CMPs, and GMPs
in the bone marrow, leading to increased myelopoiesis. Since the SNS innervates stromal
cells and HSPCs in the bone marrow, and SNS signalling contributes to HSPC
mobilisation (Mazo et al., 2011), sympathetic activation may be a possible mechanism by
which periodontitis drives local output of gingival HSPCs, especially considering that
chronic stress during experimental periodontitis enhances inflammatory responses (Lu et
al., 2014, 2016).
5.5.3. Ageing and the gingival haematopoietic niche
Physiological ageing leads to a heightened basal inflammatory state and alterations in
haematopoietic output that promotes myeloid over lymphoid lineages, thereby leading to
a decline in immune competence with age (Rossi et al., 2005; King and Goodell, 2011;
Young et al., 2016; Mann et al., 2018). We have observed that even moderate ageing in
mice reduces the output of the gingival HSPC niche. Supporting previous observations of
age-related declines in lymphopoiesis (Mann et al., 2018), and specifically in lymphoid-
biased MPP4s (Young et al., 2016), we observed decreases in overall LSKs, and in
particular, MPP4s, in both the bone marrow and gingiva from six months of age, which
resembles “mature adult” in humans (Flurkey et al., 2007). In addition, the gingiva
exhibited further declines in numbers of MPP2/3s, MyPs, and the downstream progeny,
Ly6Chi monocytes. This demonstrates that the overall HSPC niche begins to fail at an
early age. Importantly, while the bone marrow and gingiva displayed comparable
reductions in the same cell types, there were also gingiva-specific changes, suggesting
that age leads to widespread changes that affect the niche as a whole but also specifically

179
and separately affects the gingiva. Older mice exhibit a CD4+ T cell-dependent decrease
in IFNγ (and increase in IL-17) in the gingival tissues in response to mastication-driven
signals (Dutzan et al., 2017). Since IFNγ is a known HSPC-supporting factor (Baldridge
et al., 2010) and may also be important for supporting HSPCs in the gingiva, decreases in
local cytokine levels may lead to a reduction in HSPC numbers in the gingiva, and this
may also be true of the bone marrow.
Furthermore, ageing in mice leads to natural bone loss (Liang et al., 2010; Krishnan et al.,
2018). Both ageing-induced and periodontitis-induced bone loss are Th17-dependent
processes (Dutzan et al., 2017, 2018). However, despite prominent Th17 involvement in
both settings, we have shown that gingival HSPCs are differentially affected in age and
periodontitis, suggesting that the underlying mechanisms leading to bone loss are
apparently distinct. In the context of ageing, it is feasible that gingival HSPC reduction
may cause loss of oral immune homeostasis and immunosurveillance through decreased
numbers of mature immune cells, and thereby lead to inflammation and bone loss. How
these HSPCs are lost with age is still unclear, although the gingiva undergoes a number of
inherent age-dependent structural changes (discussed more below) which could be
responsible for HSPC decline.
A number of studies have reported that long-term HSCs, a subset of LSKs that have long-
lived self-renewal ability, expand with age (Rossi et al., 2005; Young et al., 2016; Mann
et al., 2018). Specifically, CD61hi long-term HSCs are proposed to drive myelopoiesis
during ageing, which results in inadequate immune responses to infectious challenge
(Mann et al., 2018). In contrast, though we did not subset LSKs into long- and short-term
HSCs, we observed age-dependent decreases in overall numbers of gingival and bone
marrow LSKs. However, it is perhaps difficult to draw a comparison between our
findings and these studies as the advanced ages of their mice (~20–28 months) are known

180
to lead to pronounced inflammation (Franceschi et al., 2018) that is likely not present at
middle age. Thus, age is likely a dynamic regulator of HSCs; constraining numbers
during adulthood, but increasingly becoming dysregulated and promoting a myeloid bias
in older ages.
5.5.4. Factors regulating gingival haematopoietic progenitors
Within the bone marrow HSPC niche, osteoblasts and supporting stromal cells express
key haematopoietic and adhesion factors, primarily stem cell factor (a.k.a. c-Kit ligand),
CXCL12, thrombopoietin, angiopoietin-1, and VCAM-1, which synergise to regulate the
quiescence, proliferation, differentiation, and mobilisation of HSPCs (Arai et al., 2004;
Sugiyama et al., 2006; Yoshihara et al., 2007; Chou and Lodish, 2010). However, our
transcriptomic data examining young and aged gingivae at steady state did not indicate a
role for these specific factors, suggesting that that the gingival HSPC niche is regulated
by different mechanisms. Certainly, an array of additional factors expand and maintain
HSPCs in the bone marrow, including members of the TGFβ superfamily (Zhang et al.,
2003), insulin-like growth factors (IGFs) (Chou and Lodish, 2010), and several
angiopoietin-like proteins (ANGPTLs) (Zhang et al., 2006; Broxmeyer et al., 2012). We
found that genes encoding IGF-2, and ANGPTL-1,-2,-7 were significantly downregulated
in aged mice, factors that are specifically known to promote the expansion and self-
renewal of mouse HSPCs ex vivo (Zhang et al., 2006). In addition, there were extensive
changes to the extracellular matrix and decreases in the expression of adhesion molecules.
Collectively, this could suggest a loss of physical support, as bone marrow HSPCs are
physically anchored in the niche by cell-matrix adhesion via adhesion factors such as
integrins (Janowska-Wieczorek et al., 2012; Kräter et al., 2017).

181
Since osteoblasts are critical supports for HSPCs in the bone marrow (Calvi et al., 2003),
and the gingival tissue is in close proximity to the supporting bone, HSPCs in the gingiva
may be associated with osteoblasts. We found that ageing reduced gingival expression of
osteoblast-related genes, such as osteoglycin (ogn) and osteopontin (spp1) (Stier et al.,
2005; Méndez-Ferrer et al., 2010), and N-cadherin (cdh2), the latter of which is expressed
on both HSCs and HSPC-supporting osteoblasts in the bone marrow (Zhang et al., 2003).
Collectively, these data suggest that the integrity of the HSPC support network declines
with increasing age, and consequently results in the loss of HSPCs, and osteoblasts may
be important in this regard. As the aged gingiva displayed large-scale reductions in
families of trophic genes for haematopoiesis, the relative impact of each factor, and
indeed, each cell type, in maintaining the gingival HSPC niche has yet to be elucidated.
Even though it is likely that a decrease in gingival HSPC numbers is due to age-related
declines in their capacity to self-renew and repopulate (Rando, 2006), there is also the
possibility that age-dependent bone loss and matrix breakdown may render the gingival
tissue unable to support HSPCs, thereby leading to apoptosis or mobilisation into the
blood. Evidence to support the latter comes from observations of patients with primary
myelofibrosis, a fibrotic condition of the bone marrow which renders it untenable, leading
to HSPC egress to extramedullary sites as a compensatory mechanism (Tefferi, 2018).
Experimentally, depletion of osteoblasts reduces cellularity in the bone marrow and leads
to mobilisation of HSPCs to the blood, spleen, and liver in mice (Visnjic et al., 2004).
Thus, an age-mediated reduction in HSPC supporting factors in the gingiva may lead to
their egress. Although we did not observe HSPCs in the blood in either young or aged
mice, they may not have been detectable at the single time-point used in this study and
therefore this possibility cannot be excluded. Adding to this potential complexity, it is
proposed that monocytes can lead to the mobilisation of HSPCs via G-CSF (Christopher
et al., 2011). Specifically, signalling through the G-CSF receptor in monocytic cells

182
inhibits osteoblasts and CXCL12, thereby leading to increased HSPC mobilisation from
the bone marrow. This raises the intriguing possibility that resident gingival monocytes
themselves may be instrumental in regulating the progenitor and stem cell niche.
5.5.5. Conclusion
Although a complete description of the potential function, ontogeny, and regulation of
these multipotent HPSCs and tissue-resident monocytes will require further investigation,
it is clear from our data that the mouse gingiva represents a novel niche that can support
and maintain populations of HSPCs and myeloid progenitors capable of giving rise to a
number of mature immune cell lineages. Our results demonstrate that tissue monocytes
and haematopoietic progenitors exist in the gingiva during steady-state, and these
populations expand during periodontitis, and are reduced with advancing age. While we
can only speculate on the factors that maintain these tissue monocytes and the HSPC
niche, our findings suggest the importance of the supporting matrix and osteoblasts in
physically retaining HSPCs, and also possibly monocytes within the gingival tissue.
Evidently, this highlights the intricate and dynamic immune regulation within the gingiva
during steady-state, physiological ageing, and inflammatory pathology. The existence of a
putative HSPC niche only adds to the complexity governing the oral immune network
during these states, but further exploration is necessary as this has potentially major
implications for emerging periodontitis immunotherapies and may also present novel
targets to better treat the disease.

183
Chapter 6. General discussion

184
6.1. Overview
Even though recent demonstrations of oral immune responses during periodontitis have
enhanced our understanding of some key cellular mediators that contribute to local
disease pathology (Abe et al., 2015; Oliver-Bell et al., 2015; Malcolm et al., 2016;
Dutzan et al., 2018), the immune populations that are involved in adverse systemic effects
are comparably ill-documented. This is in spite of the fact that periodontitis represents a
major threat to systemic health, consistently associated with a myriad of diseases with
diverse pathogeneses, including rheumatoid arthritis (Ogrendik, 2009), bowel cancer
(Abed et al., 2016), atherosclerosis (Li et al., 2002), and stroke (Grau et al., 2004).
Though each case is evidently distinct, it is clear that aberrant immunity underpins many
of the associations between periodontitis and systemic diseases.
Therefore, the overarching aims of this PhD thesis were to extensively characterise the
peripheral immune landscape during experimental periodontitis, evaluating the potential
impact of inflammatory and immune mediators on peripheral tissue sites, and in
particular, the impact on acute outcome after ischaemic stroke. We have demonstrated
that periodontitis leads to systemic changes, by increasing circulating pro-inflammatory
cytokine levels, modulating peripheral monocyte and neutrophil frequencies, and in
particular, altering bone marrow monocytes to take on a pro-inflammatory effector
function. Despite these changes, periodontitis did not alter stroke severity in two different
experimental stroke models. Additionally, by focusing on the role of myeloid cells in
periodontitis, we identified within the gingival tissue leukocytes arising from tissue-
resident haematopoietic stem and progenitor cells (HSPCs) in steady-state and that the
progenitor niche appears to be differentially remodelled in periodontitis and ageing. Thus,
this work enhances our fundamental understanding of the local and systemic mechanisms

185
that underlie oral immunopathology and how oral immunopathology can impact other
clinically-relevant diseases.
6.2. Periodontitis drives myeloid-biased immune alterations
Studies have implicated a number of immune effectors involved within the oral mucosa
during periodontitis, including neutrophils and Th17 cells (Dutzan et al., 2018), B cells
(Abe et al., 2015; Oliver-Bell et al., 2015), macrophages (Lam et al., 2014; Steinmetz et
al., 2016), and mast cells (Malcolm et al., 2016). Our findings add to this expanding body
of knowledge, as we report increases in hitherto unreported populations of resident
Ly6Chi monocytes and multipotent HSPCs, both of which could be propagating local
pathology. Using multiple approaches, we have shown that these populations are
primarily instructed and maintained locally within the oral cavity and that chronic and
acute bone loss evoke differential effects on these populations, as older mice with natural
bone loss had reduced numbers, while mechanically-induced bone loss increased
numbers, suggesting that the mechanisms of bone loss in induced and natural models of
periodontitis are inherently distinct.
6.2.1. Haematopoiesis
Although it remains to be seen if the observed increases in gingival monocytes and
HSPCs have consequences for local or even systemic pathology, altered haematopoietic
output and myeloid cell mobilisation are pathogenic features of many systemic diseases
with which periodontitis is associated. In particular, ischaemic stroke leads to
dysregulated sympathetic and glucocorticoid signalling that alters the balance of
haematopoietic progenitors in the bone marrow, promoting myelopoiesis and
subsequently contributing to an immunosuppressed state, specifically through reduced B
cell output (Denes et al., 2011; Courties et al., 2015, 2019; McCulloch et al., 2017).

186
Moreover, myocardial infarction drives increases in HSPC and monocyte mobilisation
from the bone marrow, aggravating existing atherosclerosis (Dutta et al., 2012), and in the
colon, dysregulated haematopoiesis exacerbates colitis pathology (Griseri et al., 2012).
While the implications of this oral haematopoietic niche have yet to be elucidated, altered
haematopoietic output in the gingiva may be a novel mechanism by which periodontitis
leads to further local pathology and possibly systemic complications, by altering the
balance of immune effectors. Indeed, we found that gingival HSPCs in periodontitis were
skewed towards a neutrophil-dominant output. Since neutrophils are intimately associated
with mediating bone loss (Eskan et al., 2012; Moutsopoulos et al., 2014), this could be a
novel pathway by which neutrophils are generated locally. This is a potentially important
finding, given that some candidate periodontitis treatments, such as the leukocyte integrin
antagonist, Del-1 (Eskan et al., 2012; Shin et al., 2015), function by limiting neutrophil
recruitment to the inflamed gingiva. If neutrophils can be produced locally by resident
HSPCs then this may compromise the efficacy of these targeted immunotherapies.
This work also has implications for mechanisms of bone loss in older individuals.
Physiological ageing, associated with bone loss in mice, skews bone marrow HSPC
output to promote myeloid cells at the expense of lymphoid effectors, resulting in a
decline in immune competence (Rossi et al., 2005; Mann et al., 2018). This could suggest
that older individuals may develop worse bone loss due to dysregulated local
haematopoiesis and inadequate control of microbial growth.
6.2.2. Neutrophils
As an extension of the local myeloid changes in the gingiva during periodontitis, we
observed a distinct myeloid-bias in the immune cell alterations outside the oral cavity;
neutrophil and monocyte frequencies were differentially regulated in sub-mandibular

187
lymph nodes, small intestine, and bone marrow. Neutrophil mobilisation to the site of
injury is pathogenic in a number of disease contexts, including stroke (McColl et al.,
2007; Gelderblom et al., 2012), although it has emerged that brain-invading neutrophils
can shift to a regulatory phenotype (Ym1+CD206+) akin to alternatively activated
macrophages, and subsequently lessen the extent of ischaemic brain damage (Cuartero et
al., 2013). In our work, although periodontitis discretely modulated local and systemic
frequencies of neutrophils, this did not manifest as an increase in brain-infiltrating
neutrophils after stroke, and consequently, brain injury was neither exacerbated nor
mitigated. This contrasts with obesity, where neutrophils are recruited to the ischaemic
hemisphere and release MMP9, enhancing BBB disruption and worsening damage
(McColl et al., 2010; Maysami et al., 2015). Despite altering neutrophils in the periphery,
periodontitis failed to increase neutrophil recruitment to the brain, a hallmark of other co-
morbidities that worsen stroke outcome.
6.2.3. Monocytes
Monocyte effector function is often context- and tissue-dependent; promoting tissue
repair and the resolution of inflammation in one instance (Askenase et al., 2015; Ikeda et
al., 2018) or driving pathology in another, like in atherosclerosis (Swirski et al., 2007;
Dutta et al., 2012). Specifically in stroke, monocyte infiltration into the brain can limit
(Gliem et al., 2012; Garcia-Bonilla et al., 2018) or promote (Chen et al., 2003;
Dimitrijevic et al., 2007) ischaemic injury depending on the phase in question, as
monocytes transition from an early pro-inflammatory phenotype to a reparative one in the
later stages after stroke (Gliem et al., 2016). With this in mind, we sought to define the
contribution of monocyte plasticity in periodontitis. We have demonstrated novel insights
into monocyte regulation; local tissue monocytes increase in the gingiva in line with
inflammatory bone loss and bone marrow monocytes are primed for pro-inflammatory

188
effector function. Though these findings have not been previously reported, the latter is
somewhat supported by the observation of increased TNFα+ circulating monocytes during
ligature-induced periodontitis in rats (Miyajima et al., 2015). In contrast, monocytes in
the blood were unchanged in our study, indicating that the primed bone marrow
monocytes had not been released into the circulation, and this may offer an explanation as
to why periodontitis-primed monocytes did not aggravate brain injury. This finding helps
to re-emphasise the point of chronicity, in that ligature-induced periodontitis in rodents
may prime systemic immune changes but is ultimately too acute as a model to lead to
robust consequences at distal tissue sites. Indeed, in humans with periodontitis,
inflammatory monocytes in peripheral blood are increased (Nagasawa et al., 2004), and
this could promote, or ameliorate pathology at distal sites, including the ischaemic brain.
6.2.4. Lymphocytes
Although critical in the context of local pathology, we found that lymphocytes were not
largely altered at distal peripheral tissue sites. Specifically, Th17 cells, mediators of oral
tissue homeostasis during steady-state (Dutzan et al., 2017) and orchestrating neutrophil-
mediated bone loss during periodontitis (Dutzan et al., 2018), were unaltered outside the
oral cavity. Despite the role of γδ T cells in tissue homeostasis and repair (Krishnan et al.,
2018; Wilharm et al., 2019) and an emerging role for gingival ILCs during steady-state
(Brown et al., 2018), both populations did not undergo systemic changes in ligature-
induced periodontitis. This indicates a disconnect between important cellular mediators at
local and systemic levels, but is potentially important as the peripheral mobilisation of
Th17 and γδ T cells is known to drive worse brain injury in both acute and sub-acute
phases post-stroke (Shichita et al., 2009; Gelderblom et al., 2014; Benakis et al., 2016;
Arunachalam et al., 2017), and could explain why periodontitis did not alter outcome in
our study.

189
Collectively, this work demonstrates that locally, experimental periodontitis can drive
leukocyte production through resident HSPCs, and systemically, preferentially modulates
myeloid but not lymphoid cells. Importantly in the context of stroke, the leukocyte
populations that are pathogenic in periodontitis are not sufficiently mobilised to the
ischaemic brain to promote damage.
6.3. High-grade and low-grade inflammation
6.3.1. Comparing periodontitis to infection
Extensive clinical and pre-clinical evidence demonstrates that severe infections and high-
grade systemic inflammation worsen outcome after stroke and compromise survival
(McColl et al., 2007; Dénes et al., 2011a; Dénes et al., 2014; Fugate et al., 2014).
Mechanistically, in experimental stroke models, concurrent infections promote a
profound pro-inflammatory and pro-thrombotic state, driving deleterious Th1- and
neutrophil-driven responses that promote damage to the cerebral microvasculature and
potentiate brain injury, leading to profound neurological deficits (Dénes et al., 2010a;
Dénes et al., 2014). By contrast, our results presented indicate that periodontitis
represents a scenario whereby substantial pathological changes are largely confined to the
oral cavity. Systemic inflammatory effects are modest or low-grade in nature, and thereby
insufficient to aggravate ischaemic brain damage in a manner analogous to high-grade
infections. Periodontitis did induce increases in circulating IL-1β, IL-17A, and GM-CSF,
which are implicated as soluble mediators promoting brain damage after stroke (McColl
et al., 2009; Shichita et al., 2009; Gelderblom et al., 2012) with the latter shown to
promote pathogenic Th17 cells in experimental autoimmune myocarditis (Sonderegger et
al., 2008). Intriguingly, there was a notable absence of inflammatory and immune
changes in the lung, spleen, blood, and at the cardiac or cerebral vasculature. This is in
stark contrast to a S. pneumoniae infection, for example, which results in elevated pro-

190
inflammatory cytokines and chemokines in the plasma, spleen, lung, liver, and brain, as
well as increased granulocytosis and aortic plaque formation (Dénes et al., 2014).
Furthermore, periodontitis is an atypical “infection” scenario, in that the invading unit is
comprised of aerobic and anaerobic species of commensals, opportunistic microbes and
“pathogens”, of which the latter are in low abundance (Hajishengallis et al., 2011). By
contrast, typical bacterial, viral and worm infections are often caused by a single,
abundant, virulent species, and commonly elicit profound system-wide responses,
including fever, peripheral blood leukocytosis, multi-tissue involvement, and emergency
myelopoiesis (Takizawa et al., 2012). Thus, in spite of some peripheral immune
alterations, it is apparent that the systemic reaches of periodontitis are minimal (in this
model), indicative of a low-level inflammatory state.
6.3.2. Comparing periodontitis to obesity
Since the systemic inflammatory consequences of ligature-induced periodontitis are
relatively modest, obesity is perhaps a more suitable comparison than typical infection.
Diet-induced or transgenic models of obesity in mice are associated with chronic, low-
level inflammation (marked by serum increases in CCL2 and CXCL1) and consistently
worse stroke outcome, by aggravating BBB disruption, promoting haemorrhagic
transformation and increasing infarct volumes (McColl et al., 2010; Deng et al., 2014;
Maysami et al., 2015; Haley et al., 2017b). Thus, obesity and periodontitis both result in
moderately increased serum concentrations of some pro-inflammatory cytokines and
chemokines (Haley et al., 2017b). It is intriguing therefore that periodontitis does not
exacerbate brain damage in a manner similar to obesity, even with the presence of
systemic Pg-LPS as an additional pro-inflammatory stimulus. This is likely attributed to
the fact that obesity-associated inflammation is just one facet of an extended obesity
phenotype, or metabolic syndrome, which involves systemic increases in blood glucose,

191
insulin, free fatty acids, and adipokines, amongst a number of other dysregulated
metabolic pathways and adipose changes (Deng et al., 2014; Haley et al., 2017b). Despite
the fact that periodontitis and obesity share low-grade systemic inflammatory changes
(although the mediators of these changes are different), the additional metabolic stress
imposed by obesity cannot be disregarded as an important contributor to worse stroke
outcome.
At its simplest, it seems that the detrimental effect of a condition on stroke outcome
comes down to whether it reaches a certain threshold. For example, obesity in mice
worsens stroke outcome only after 4 months, but not 3 months on a high-fat diet
(Maysami et al., 2015). Thus, severe or chronic inflammatory, infectious, or metabolic
states cause sufficient cumulative stress to reach this threshold and consequently worsen
stroke outcome, in a manner that periodontitis or localised inflammatory conditions
cannot.
6.3.3. The impact of specific periodontal pathogens
While we have shown that mechanically-induced inflammatory bone loss does not
worsen acute stroke outcome, a number of recent studies have highlighted the critical role
that specific oral microbes play (in other disease contexts), indicating that oral infection
models may induce adverse consequences in a way that ligature placement does not.
These studies also highlight that oral periodontal species may be more pathogenic than
previously thought: P. gingivalis (Pg) is proposed to contribute to the pathophysiology of
Alzheimer’s disease (AD) (Dominy et al., 2019), rheumatoid arthritis (Ogrendik, 2013),
and atherosclerosis (Gibson et al., 2004); A. actinomycetemcomitans to rheumatoid
arthritis (RA) (Konig et al., 2016); and F. nucleatum to bowel cancer (Abed et al., 2016).
These are instances of a particular periodontal bacterial species complicating events at

192
distal sites, often not by inciting a “classical” inflammatory response, but by insidiously
enhancing pathogenic features of the disease, such as citrullinating proteins in RA, which
indirectly contributes to immunopathology (Maresz et al., 2013; Konig et al., 2016). In
light of this, the composition and burden of the individuals’ dysbiotic oral flora becomes
as important as the overt inflammatory symptoms. Indeed, microbial dysbiosis in general
is known to contribute to an array of diseases at distal sites, from multiple sclerosis to
asthma to atherosclerosis (Levy et al., 2017). With this in mind, we can hypothesise that
periodontitis can adversely affect systemic disease in two main ways: local inflammatory
pathology induces sustained low-level systemic inflammation, but the oral microbes
themselves also contribute separately in other ways; increasing the risk of ischaemia by
promoting platelet aggregation (Nakayama, 2010), or the risk of metastasis by inhibiting
tumour killing (Gur et al., 2015). Ideally, experimental studies should endeavour to
address both components in order to adequately evaluate the systemic risks of
periodontitis. Since current models prioritise either host (ligature) or bacteria (oral
inoculation), each is distinctly lacking in a piece of the overall disease picture.
6.4. Challenges and future directions
Since oral microbes and their products, as well as immune effectors and their products are
all involved in varying degrees in oral inflammation, a major challenge is assessing the
relative importance of each aspect in adverse systemic events. With this in mind, we have
provided evidence of the effects of Pg-LPS and ligature induction on acute stroke
outcome, but the impact of oral microbes on stroke outcome needs to be assessed.
Chronic oral P. gingivalis infections have been used in the context of AD (Dominy et al.,
2019) and polymicrobial infection models have been employed in the context of
atherosclerosis (Nahid et al., 2011; Rivera et al., 2013). Using these approaches would

193
help to emphasise the importance of periopathogens and the entire oral microbial
community in causing systemic complications.
Moving forward, a chronic periodontitis model is required, especially one that
incorporates the pathological host and microbial responses over a long period. Chronicity
is a fundamental aspect of the human disease not replicated in the rodent ligature model
and is likely part of the reason why peripheral tissue sites and the brain were unaffected
in this work, because chronic low-level inflammation, as in obesity, likely represents a
slowly-increasing burden that eventually becomes a tangible threat over time.
Even though periodontitis did not alter acute stroke outcome, a number of other key
questions must be considered. It is unclear if periodontitis increases the risk of stroke, or
the long-term recovery after stroke. The former could be evaluated using stroke-prone
mice, to determine the impact of periodontitis on likelihood of stroke, since observational
studies have associated periodontitis with increased risk of stroke (Grau et al., 2004), and
thus it may have an impact in this manner. Regarding the latter, long-term outcome post-
stroke could be affected, in terms of cognitive dysfunction or infection susceptibility.
Certainly, chronic Pg-LPS administration has been shown to cause neuroinflammation
and cognitive deficits (Wu et al., 2017). Equally, it is unknown if dysbiotic oral bacterial
species could contribute to post-stroke infections in the lungs or blood (Prass et al., 2003,
2006). Enhanced dissemination of oral species during post-stroke immunosuppression
(Engel et al., 2015) could promote infection, given that periodontitis in humans is
associated with worsening lung pathology in those with already compromised health
(Mojon, 2002).
As experimental periodontitis did not alter acute stroke outcome in this work it indicates
that periodontitis alone may be insufficient to drive brain injury associated with stroke.

194
That said, it is unknown if periodontitis contributes to poorer prognosis after stroke in
tandem with other co-morbidities. Most individuals with periodontitis present with
confounding factors that can independently account for increased stroke risk, such as
hypertension, age, obesity, and smoking (Neuhaus et al., 2014). Ideally, to model human
stroke and periodontitis more closely, an emphasis should be placed on use of animals of
varying ages and with other co-morbidities (such as obesity and hypertension) as several
co-morbidities may co-exist in the same individual. Most stroke and periodontitis patients
are not young and healthy, and therefore using young healthy rodents is disregarding a
vital aspect of the disease aetiology. Thus, our work has begun to explore the associations
between periodontitis and stroke but there remain a number of unanswered questions that
warrant further investigation.
6.5. Concluding remarks
The oral barrier maintains a physical interface between the host and external
environment, and we have shown that the breakdown of this barrier can lead to
inflammatory and immune changes in the local environment as well as distant sites, with
the potential for adverse systemic complications. The work contained in this thesis
provides novel insights into fundamental oral immunology during periodontitis by
indicating that a novel population of resident HSPCs expand in the tissue, can give rise to
multiple immune lineages, and could be intimately involved in mediating bone loss.
Furthermore, this work provides insight into the impact of periodontitis on peripheral
immunity and specifically on the ischaemic brain. Importantly, we have shown that
experimental periodontitis does not alter acute outcome after stroke in mice, and with this
work we provide a counterpoint to the current hypothesis that periodontitis detrimentally
affects stroke outcome. This being said, periodontitis may remain an important but under-
recognised risk factor for cerebral ischaemia and other systemic diseases. Given the

195
widespread prevalence of periodontitis, it is imperative to define the immunological
mechanisms in the oral cavity, and the extent that oral microbial species impact disease
pathophysiology, to not only better treat periodontitis, but to also mitigate any potential
adverse effects on other clinically-important conditions.

196
Bibliography
Abadie, V., Badell, E., Douillard, P., Ensergueix, D., Leenen, P. J., Tanguy, M., Fiette, L., Saeland, S.,
Gicquel, B. and Winter, N. (2005) ‘Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis
BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes.’ Blood. 2005/05/12, 106(5)
pp. 1843–1850.
Abe, T., AlSarhan, M., Benakanakere, M. R., Maekawa, T., Kinane, D. F., Cancro, M. P., Korostoff, J. M.
and Hajishengallis, G. (2015) ‘The B Cell–Stimulatory Cytokines BLyS and APRIL Are Elevated in Human
Periodontitis and Are Required for B Cell–Dependent Bone Loss in Experimental Murine Periodontitis.’ The
Journal of Immunology. 2015/07/08, 195(4) pp. 1427–1435.
Abe, T. and Hajishengallis, G. (2013) ‘Optimization of the ligature-induced periodontitis model in mice.’
Journal of Immunological Methods, 394(1–2) pp. 49–54.
Abe, T., Hosur, K. B., Hajishengallis, E., Reis, E. S., Ricklin, D., Lambris, J. D. and Hajishengallis, G. (2012)
‘Local Complement-Targeted Intervention in Periodontitis: Proof-of-Concept Using a C5a Receptor (CD88)
Antagonist.’ The Journal of Immunology, 189(11) pp. 5442–5448.
Abed, J., Emgård, J. E. M., Zamir, G., Faroja, M., Almogy, G., Grenov, A., Sol, A., Naor, R., Pikarsky, E.,
Atlan, K. A., Mellul, A., Chaushu, S., Manson, A. L., Earl, A. M., Ou, N., Brennan, C. A., Garrett, W. S. and
Bachrach, G. (2016) ‘Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by
Binding to Tumor-Expressed Gal-GalNAc.’ Cell Host & Microbe, 20(2) pp. 215–225.
Abusleme, L., Dupuy, A. K., Dutzan, N., Silva, N., Burleson, J. A., Strausbaugh, L. D., Gamonal, J. and
Diaz, P. I. (2013) ‘The subgingival microbiome in health and periodontitis and its relationship with
community biomass and inflammation.’ The ISME Journal, 7(5) pp. 1016–1025.
Albandar, J. M. (2014) ‘Aggressive and acute periodontal diseases.’ Periodontology 2000, 65(1) pp. 7–12.
Allen, C. L. and Bayraktutan, U. (2008) ‘Risk Factors for Ischaemic Stroke.’ International Journal of Stroke,
3(2) pp. 105–116.
Amar, S., Wu, S. C. and Madan, M. (2009) ‘Is Porphyromonas gingivalis cell invasion required for
atherogenesis? Pharmacotherapeutic implications.’ The Journal of Immunology, 182(3) pp. 1584–1592.
Amberger, A., Maczek, C., Jürgens, G., Michaelis, D., Schett, G., Trieb, K., Eberl, T., Jindal, S., Xu, Q. and
Wick, G. (1997) ‘Co-expression of ICAM-1, VCAM-1, ELAM-1 and Hsp60 in human arterial and venous
endothelial cells in response to cytokines and oxidized low-density lipoproteins.’ Cell Stress & Chaperones,
2(2) p. 94.
An, S. J., Kim, T. J. and Yoon, B.-W. (2017) ‘Epidemiology, Risk Factors, and Clinical Features of
Intracerebral Hemorrhage: An Update.’ Journal of Stroke, 19(1) pp. 3–10.
Andrukhov, O., Ulm, C., Reischl, H., Nguyen, P. Q., Matejka, M. and Rausch-Fan, X. (2011) ‘Serum
Cytokine Levels in Periodontitis Patients in Relation to the Bacterial Load.’ Journal of Periodontology, 82(6)
pp. 885–892.
Ansari, S., Azari, H., Caldwell, K. J., Regenhardt, R. W., Hedna, V. S., Waters, M. F., Hoh, B. L. and Mecca,
A. P. (2013) ‘Endothelin-1 Induced Middle Cerebral Artery Occlusion Model for Ischemic Stroke with Laser
Doppler Flowmetry Guidance in Rat.’ Journal of Visualized Experiments, (72) February.
Arai, F., Hirao, A., Ohmura, M., Sato, H., Matsuoka, S., Takubo, K., Ito, K., Koh, G. Y. and Suda, T. (2004)
‘Tie2/Angiopoietin-1 Signaling Regulates Hematopoietic Stem Cell Quiescence in the Bone Marrow Niche.’
Cell, 118(2) pp. 149–161.
Araujo-Pires, A. C., Vieira, A. E., Francisconi, C. F., Biguetti, C. C., Glowacki, A., Yoshizawa, S.,
Campanelli, A. P., Trombone, A. P. F., Sfeir, C. S., Little, S. R. and Garlet, G. P. (2015) ‘IL-4/CCL22/CCR4
Axis Controls Regulatory T-Cell Migration That Suppresses Inflammatory Bone Loss in Murine
Experimental Periodontitis.’ Journal of Bone and Mineral Research, 30(3) pp. 412–422.
Arimatsu, K., Yamada, H., Miyazawa, H., Minagawa, T., Nakajima, M., Ryder, M. I., Gotoh, K., Motooka,
D., Nakamura, S., Iida, T. and Yamazaki, K. (2015) ‘Oral pathobiont induces systemic inflammation and

197
metabolic changes associated with alteration of gut microbiota.’ Scientific Reports, 4(1) p. 4828.
Arizon, M., Nudel, I., Segev, H., Mizraji, G., Elnekave, M., Furmanov, K., Eli-Berchoer, L., Clausen, B. E.,
Shapira, L., Wilensky, A. and Hovav, A.-H. (2012) ‘Langerhans cells down-regulate inflammation-driven
alveolar bone loss.’ Proceedings of the National Academy of Sciences, 109(18) pp. 7043–7048.
Arunachalam, P., Ludewig, P., Melich, P., Arumugam, T. V., Gerloff, C., Prinz, I., Magnus, T. and
Gelderblom, M. (2017) ‘CCR6 (CC Chemokine Receptor 6) Is Essential for the Migration of Detrimental
Natural Interleukin-17–Producing γδ T Cells in Stroke.’ Stroke, 48(7) pp. 1957–1965.
Askenase, M. H., Han, S.-J., Byrd, A. L., Morais da Fonseca, D., Bouladoux, N., Wilhelm, C., Konkel, J. E.,
Hand, T. W., Lacerda-Queiroz, N., Su, X., Trinchieri, G., Grainger, J. R. and Belkaid, Y. (2015) ‘Bone-
Marrow-Resident NK Cells Prime Monocytes for Regulatory Function during Infection.’ Immunity.
2015/06/14, 42(6) pp. 1130–1142.
Atarashi, K., Suda, W., Luo, C., Kawaguchi, T., Motoo, I., Narushima, S., Kiguchi, Y., Yasuma, K.,
Watanabe, E., Tanoue, T., Thaiss, C. A., Sato, M., Toyooka, K., Said, H. S., Yamagami, H., Rice, S. A.,
Gevers, D., Johnson, R. C., Segre, J. A., Chen, K., Kolls, J. K., Elinav, E., Morita, H., Xavier, R. J., Hattori,
M. and Honda, K. (2017) ‘Ectopic colonization of oral bacteria in the intestine drives T H 1 cell induction
and inflammation.’ Science, 358(6361) pp. 359–365.
Auffray, C., Fogg, D., Garfa, M., Elain, G., Join-Lambert, O., Kayal, S., Sarnacki, S., Cumano, A., Lauvau,
G. and Geissmann, F. (2007) ‘Monitoring of Blood Vessels and Tissues by a Population of Monocytes with
Patrolling Behavior.’ Science, 317(5838) pp. 666–670.
Avraham-Davidi, I., Yona, S., Grunewald, M., Landsman, L., Cochain, C., Silvestre, J. S., Mizrahi, H.,
Faroja, M., Strauss-Ayali, D., Mack, M., Jung, S. and Keshet, E. (2013) ‘On-site education of VEGF-
recruited monocytes improves their performance as angiogenic and arteriogenic accessory cells.’ The Journal
of Experimental Medicine, 210(12) pp. 2611–2625.
Bahekar, A. A., Singh, S., Saha, S., Molnar, J. and Arora, R. (2007) ‘The prevalence and incidence of
coronary heart disease is significantly increased in periodontitis: A meta-analysis.’ American Heart Journal,
154(5) pp. 830–837.
Bain, C. C., Bravo-Blas, A., Scott, C. L., Gomez Perdiguero, E., Geissmann, F., Henri, S., Malissen, B.,
Osborne, L. C., Artis, D. and Mowat, A. M. (2014) ‘Constant replenishment from circulating monocytes
maintains the macrophage pool in the intestine of adult mice.’ Nature Immunology. Nature Publishing Group,
15(10) pp. 929–937.
Baldridge, M. T., King, K. Y., Boles, N. C., Weksberg, D. C. and Goodell, M. A. (2010) ‘Quiescent
haematopoietic stem cells are activated by IFN-γ in response to chronic infection.’ Nature, 465(7299) pp.
793–797.
Bartold, P. M. and Van Dyke, T. E. (2017) ‘Host modulation: controlling the inflammation to control the
infection.’ Periodontology 2000, 75(1) pp. 317–329.
Bartold, P. M., Marino, V., Cantley, M. and Haynes, D. R. (2010) ‘Effect of Porphyromonas gingivalis-
induced inflammation on the development of rheumatoid arthritis.’ Journal of Clinical Periodontology, 37(5)
pp. 405–411.
Bartold, P. M., Marshall, R. I. and Haynes, D. R. (2005) ‘Periodontitis and Rheumatoid Arthritis: A Review.’
Journal of Periodontology. 2005/11/10, 76(11–s) pp. 2066–2074.
Beck, J. D., Elter, J. R., Heiss, G., Couper, D., Mauriello, S. M. and Offenbacher, S. (2001) ‘Relationship of
Periodontal Disease to Carotid Artery Intima-Media Wall Thickness.’ Arteriosclerosis, Thrombosis, and
Vascular Biology, 21(11) pp. 1816–1822.
Beck, J. D. and Offenbacher, S. (2005) ‘Systemic Effects of Periodontitis: Epidemiology of Periodontal
Disease and Cardiovascular Disease.’ Journal of Periodontology, 76(11–s) pp. 2089–2100.
Beck, J., Eke, P., Lin, D., Madianos, P., Couper, D., Moss, K., Elter, J., Heiss, G. and Offenbacher, S. (2005)
‘Associations between IgG antibody to oral organisms and carotid intima–medial thickness in community-
dwelling adults.’ Atherosclerosis, 183(2) pp. 342–348.
Beck, J., Garcia, R., Heiss, G., Vokonas, P. S. and Offenbacher, S. (1996) ‘Periodontal Disease and

198
Cardiovascular Disease.’ Journal of Periodontology. 1996/10/01, 67(10s) pp. 1123–1137.
Le Behot, A., Gauberti, M., De Lizarrondo, S. M., Montagne, A., Lemarchand, E., Repesse, Y., Guillou, S.,
Denis, C. V., Maubert, E., Orset, C. and Vivien, D. (2014) ‘GpIbα-VWF blockade restores vessel patency by
dissolving platelet aggregates formed under very high shear rate in mice.’ Blood, 123(21) pp. 3354–3363.
Bekkering, S., Arts, R. J. W., Novakovic, B., Kourtzelis, I., van der Heijden, C. D. C. C., Li, Y., Popa, C. D.,
ter Horst, R., van Tuijl, J., Netea-Maier, R. T., van de Veerdonk, F. L., Chavakis, T., Joosten, L. A. B., van
der Meer, J. W. M., Stunnenberg, H., Riksen, N. P. and Netea, M. G. (2018) ‘Metabolic Induction of Trained
Immunity through the Mevalonate Pathway.’ Cell. Cell Press, 172(1–2) p. 135–146.e9.
Benakis, C., Brea, D., Caballero, S., Faraco, G., Moore, J., Murphy, M., Sita, G., Racchumi, G., Ling, L.,
Pamer, E. G., Iadecola, C. and Anrather, J. (2016) ‘Commensal microbiota affects ischemic stroke outcome
by regulating intestinal γδ T cells.’ Nature Medicine, 13(10) pp. 720–726.
Beretta, S., Riva, M., Carone, D., Cuccione, E., Padovano, G., Rodriguez Menendez, V., Pappadá, G. B.,
Versace, A., Giussani, C., Sganzerla, E. P. and Ferrarese, C. (2013) ‘Optimized system for cerebral perfusion
monitoring in the rat stroke model of intraluminal middle cerebral artery occlusion.’ Journal of visualized
experiments : JoVE, (February) January, pp. 1–7.
Berglundh, T., Donati, M. and Zitzmann, N. (2007) ‘B cells in periodontitis ? friends or enemies?’
Periodontology 2000, 45(1) pp. 51–66.
Bezerra, M. M., Brito, G. A., Ribeiro, R. A. and Rocha, F. A. (2002) ‘Low-dose doxycycline prevents
inflammatory bone resorption in rats.’ Braz J Med Biol Res. 2002/05/16, 35(5) pp. 613–616.
Bian, T., Li, L., Lyu, J., Cui, D., Lei, L. and Yan, F. (2016) ‘Human β-defensin 3 suppresses Porphyromonas
gingivalis lipopolysaccharide-induced inflammation in RAW 264.7 cells and aortas of ApoE-deficient mice.’
Peptides, 82, August, pp. 92–100.
Blasco-Baque, V., Garidou, L., Pomié, C., Escoula, Q., Loubieres, P., Le Gall-David, S., Lemaitre, M.,
Nicolas, S., Klopp, P., Waget, A., Azalbert, V., Colom, A., Bonnaure-Mallet, M., Kemoun, P., Serino, M. and
Burcelin, R. (2017) ‘Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota
dysbiosis and insulin resistance via an impaired adaptive immune response.’ Gut, 66(5) pp. 872–885.
Bodhankar, S., Chen, Y., Vandenbark, A. A., Murphy, S. J. and Offner, H. (2013) ‘IL-10-producing B-cells
limit CNS inflammation and infarct volume in experimental stroke.’ Metabolic Brain Disease, 28(3) pp. 375–
386.
Bolger, A. M., Lohse, M. and Usadel, B. (2014) ‘Trimmomatic: a flexible trimmer for Illumina sequence
data.’ Bioinformatics, 30(15) pp. 2114–2120.
Bomans, K., Schenz, J., Sztwiertnia, I., Schaack, D., Weigand, M. A. and Uhle, F. (2018) ‘Sepsis Induces a
Long-Lasting State of Trained Immunity in Bone Marrow Monocytes.’ Frontiers in Immunology. Frontiers
Media SA, 9, November, p. 2685.
Boyle, E. I., Weng, S., Gollub, J., Jin, H., Botstein, D., Cherry, J. M. and Sherlock, G. (2004)
‘GO::TermFinder - Open source software for accessing Gene Ontology information and finding significantly
enriched Gene Ontology terms associated with a list of genes.’ Bioinformatics, 20(18) pp. 3710–3715.
Brito, F., Barros, F. C. De, Zaltman, C., Pugas Carvalho, A. T., de Vasconcellos Carneiro, A. J., Fischer, R.
G., Gustafsson, A. and de Silva Figueredo, C. M. (2008) ‘Prevalence of periodontitis and DMFT index in
patients with Crohn’s disease and ulcerative colitis.’ Journal of Clinical Periodontology, 35(6) pp. 555–560.
Brito, L. C. W., DalBó, S., Striechen, T. M., Farias, J. M., Olchanheski, L. R., Mendes, R. T., Vellosa, J. C.
R., Fávero, G. M., Sordi, R., Assreuy, J., Santos, F. A. and Fernandes, D. (2013) ‘Experimental periodontitis
promotes transient vascular inflammation and endothelial dysfunction.’ Archives of Oral Biology, 58(9) pp.
1187–1198.
Brodala, N., Merricks, E. P., Bellinger, D. A., Damrongsri, D., Offenbacher, S., Beck, J., Madianos, P.,
Sotres, D., Chang, Y.-L., Koch, G. and Nichols, T. C. (2005) ‘Porphyromonas gingivalis Bacteremia Induces
Coronary and Aortic Atherosclerosis in Normocholesterolemic and Hypercholesterolemic Pigs.’
Arteriosclerosis, Thrombosis, and Vascular Biology, 25(7) pp. 1446–1451.
Brown, J. L., Campbell, L., Malcolm, J., Adrados Planell, A., Butcher, J. P. and Culshaw, S. (2018)

199
‘Enrichment of Innate Lymphoid Cell Populations in Gingival Tissue.’ Journal of Dental Research. SAGE
PublicationsSage CA: Los Angeles, CA, 97(12) pp. 1399–1405.
Broxmeyer, H. E., Srour, E. F., Cooper, S., Wallace, C. T., Hangoc, G. and Youn, B.-S. (2012)
‘Angiopoietin-like-2 and -3 act through their coiled-coil domains to enhance survival and replating capacity
of human cord blood hematopoietic progenitors.’ Blood Cells, Molecules, and Diseases, 48(1) pp. 25–29.
Burrows, F. E., Bray, N., Denes, A., Allan, S. M. and Schiessl, I. (2015) ‘Delayed Reperfusion Deficits after
Experimental Stroke Account for Increased Pathophysiology.’ Journal of Cerebral Blood Flow &
Metabolism, 35(2) pp. 277–284.
Burrows, F., Haley, M. J., Scott, E., Coutts, G., Lawrence, C. B., Allan, S. M. and Schiessl, I. (2016)
‘Systemic inflammation affects reperfusion following transient cerebral ischaemia.’ Experimental Neurology,
277 pp. 252–260.
Calvi, L. M., Adams, G. B., Weibrecht, K. W., Weber, J. M., Olson, D. P., Knight, M. C., Martin, R. P.,
Schipani, E., Divieti, P., Bringhurst, F. R., Milner, L. A., Kronenberg, H. M. and Scadden, D. T. (2003)
‘Osteoblastic cells regulate the haematopoietic stem cell niche.’ Nature, 425(6960) pp. 841–846.
Campi, P., Herrera, B. S., de Jesus, F. N., Napolitano, M., Teixeira, S. A., Maia-Dantas, A., Spolidorio, L. C.,
Akamine, E. H., Mayer, M. P. A., de Carvalho, M. H. C., Costa, S. K. P. and Muscara, M. N. (2016)
‘Endothelial dysfunction in rats with ligature-induced periodontitis: Participation of nitric oxide and
cycloxygenase-2-derived products.’ Archives of Oral Biology. Elsevier Ltd, 63, March, pp. 66–74.
Cantley, M. D., Haynes, D. R., Marino, V. and Bartold, P. M. (2011) ‘Pre-existing periodontitis exacerbates
experimental arthritis in a mouse model.’ Journal of Clinical Periodontology, 38(6) pp. 532–541.
Capucha, T., Mizraji, G., Segev, H., Blecher-Gonen, R., Winter, D., Khalaileh, A., Tabib, Y., Attal, T.,
Nassar, M., Zelentsova, K., Kisos, H., Zenke, M., Seré, K., Hieronymus, T., Burstyn-Cohen, T., Amit, I.,
Wilensky, A. and Hovav, A.-H. (2015) ‘Distinct Murine Mucosal Langerhans Cell Subsets Develop from
Pre-dendritic Cells and Monocytes.’ Immunity, 43(2) pp. 369–381.
Cardier, J. E. and Barberá-Guillem, E. (1997) ‘Extramedullary hematopoiesis in the adult mouse liver is
associated with specific hepatic sinusoidal endothelial cells.’ Hepatology, 26(1) pp. 165–75.
Carlin, L. M., Stamatiades, E. G., Auffray, C., Hanna, R. N., Glover, L., Vizcay-Barrena, G., Hedrick, C. C.,
Cook, H. T., Diebold, S. and Geissmann, F. (2013) ‘Nr4a1-Dependent Ly6Clow Monocytes Monitor
Endothelial Cells and Orchestrate Their Disposal.’ Cell, 153(2) pp. 362–375.
Carrion, J., Scisci, E., Miles, B., Sabino, G. J., Zeituni, A. E., Gu, Y., Bear, A., Genco, C. a, Brown, D. L. and
Cutler, C. W. (2012) ‘Microbial Carriage State of Peripheral Blood Dendritic Cells (DCs) in Chronic
Periodontitis Influences DC Differentiation, Atherogenic Potential.’ The Journal of Immunology, 189(6) pp.
3178–3187.
Casanova, L., Hughes, F. J. and Preshaw, P. M. (2014) ‘Diabetes and periodontal disease: a two-way
relationship.’ British Dental Journal, 217(8) pp. 433–437.
Caso, J. R., Pradillo, J. M., Hurtado, O., Lorenzo, P., Moro, M. A. and Lizasoain, I. (2007) ‘Toll-Like
Receptor 4 Is Involved in Brain Damage and Inflammation After Experimental Stroke.’ Circulation, 115(12)
pp. 1599–1608.
Caton, J. G., Armitage, G., Berglundh, T., Chapple, I. L. C., Jepsen, S., Kornman, K. S., Mealey, B. L.,
Papapanou, P. N., Sanz, M. and Tonetti, M. S. (2018) ‘A new classification scheme for periodontal and peri-
implant diseases and conditions - Introduction and key changes from the 1999 classification.’ Journal of
Clinical Periodontology, 45, June, pp. S1–S8.
Cekici, A., Kantarci, A., Hasturk, H. and Van Dyke, T. E. (2014) ‘Inflammatory and immune pathways in the
pathogenesis of periodontal disease.’ Periodontology 2000. NIH Public Access, 64(1) pp. 57–80.
Chen, C., Liu, Y., Liu, Y. and Zheng, P. (2010) ‘Mammalian target of rapamycin activation underlies HSC
defects in autoimmune disease and inflammation in mice.’ Journal of Clinical Investigation, 120(11) pp.
4091–4101.
Chen, Y.-W., Nagasawa, T., Wara-Aswapati, N., Ushida, Y., Wang, D., Takeuchi, Y., Kobayashi, H., Umeda,
M., Inoue, Y., Iwai, T., Ishikawa, I. and Izumi, Y. (2009) ‘Association between periodontitis and anti-

200
cardiolipin antibodies in Buerger disease.’ Journal of Clinical Periodontology, 36(10) pp. 830–835.
Chen, Y., Hallenbeck, J. M., Ruetzler, C., Bol, D., Thomas, K., Berman, N. E. J. and Vogel, S. N. (2003)
‘Overexpression of Monocyte Chemoattractant Protein 1 in the Brain Exacerbates Ischemic Brain Injury and
is Associated with Recruitment of Inflammatory Cells.’ Journal of Cerebral Blood Flow & Metabolism,
23(6) pp. 748–755.
Cheong, C., Matos, I., Choi, J.-H., Dandamudi, D. B., Shrestha, E., Longhi, M. P., Jeffrey, K. L., Anthony, R.
M., Kluger, C., Nchinda, G., Koh, H., Rodriguez, A., Idoyaga, J., Pack, M., Velinzon, K., Park, C. G. and
Steinman, R. M. (2010) ‘Microbial Stimulation Fully Differentiates Monocytes to DC-SIGN/CD209+
Dendritic Cells for Immune T Cell Areas.’ Cell, 143(3) pp. 416–429.
Chi, L., Cheng, X., He, X., Sun, J., Liang, F., Pei, Z. and Teng, W. (2019) ‘Increased cortical infarction and
neuroinflammation in ischemic stroke mice with experimental periodontitis.’ NeuroReport, March, p. 1.
Chistiakov, D. A., Orekhov, A. N. and Bobryshev, Y. V. (2016) ‘Links between atherosclerotic and
periodontal disease.’ Experimental and Molecular Pathology. Elsevier Inc., 100(1) pp. 220–235.
Chiu, B. (1999) ‘Multiple infections in carotid atherosclerotic plaques.’ Am Heart J. 1999/10/28, 138(5 Pt 2)
pp. S534-6.
Chou, S. and Lodish, H. F. (2010) ‘Fetal liver hepatic progenitors are supportive stromal cells for
hematopoietic stem cells.’ Proceedings of the National Academy of Sciences, 107(17) pp. 7799–7804.
Chow, A., Lucas, D., Hidalgo, A., Méndez-Ferrer, S., Hashimoto, D., Scheiermann, C., Battista, M., Leboeuf,
M., Prophete, C., van Rooijen, N., Tanaka, M., Merad, M. and Frenette, P. S. (2011) ‘Bone marrow CD169 +
macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell
niche.’ The Journal of Experimental Medicine, 208(2) pp. 261–271.
Christ, A., Günther, P., Lauterbach, M. A. R., Duewell, P., Biswas, D., Pelka, K., Scholz, C. J., Oosting, M.,
Haendler, K., Baßler, K., Klee, K., Schulte-Schrepping, J., Ulas, T., Moorlag, S. J. C. F. M., Kumar, V., Park,
M. H., Joosten, L. A. B., Groh, L. A., Riksen, N. P., Espevik, T., Schlitzer, A., Li, Y., Fitzgerald, M. L.,
Netea, M. G., Schultze, J. L. and Latz, E. (2018) ‘Western Diet Triggers NLRP3-Dependent Innate Immune
Reprogramming.’ Cell. Cell Press, 172(1–2) p. 162–175.e14.
Christopher, M. J., Rao, M., Liu, F., Woloszynek, J. R. and Link, D. C. (2011) ‘Expression of the G-CSF
receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice.’
The Journal of Experimental Medicine, 208(2) pp. 251–260.
Chtanova, T., Schaeffer, M., Han, S.-J., Van Dooren, G. G., Nollmann, M., Herzmark, P., Chan, S. W., Satija,
H., Camfield, K., Aaron, H., Striepen, B. and Robey, E. A. (2008) ‘Dynamics of Neutrophil Migration in
Lymph Nodes during Infection.’ Immunity, 29(3) pp. 487–496.
Chu, H. X., Broughton, B. R. S., Ah Kim, H., Lee, S., Drummond, G. R. and Sobey, C. G. (2015) ‘Evidence
That Ly6Chi Monocytes Are Protective in Acute Ischemic Stroke by Promoting M2 Macrophage
Polarization.’ Stroke, 46(7) pp. 1929–1937.
Cisbani, G., Le Behot, A., Plante, M.-M., Préfontaine, P., Lecordier, M. and Rivest, S. (2018) ‘Role of the
chemokine receptors CCR2 and CX3CR1 in an experimental model of thrombotic stroke.’ Brain, Behavior,
and Immunity, 70, May, pp. 280–292.
Clark, W., Gunion-Rinker, L., Lessov, N. and Hazel, K. (1998) ‘Citicoline Treatment for Experimental
Intracerebral Hemorrhage in Mice.’ Stroke, 29(10) pp. 2136–2140.
Coccia, M., Harrison, O. J., Schiering, C., Asquith, M. J., Becher, B., Powrie, F. and Maloy, K. J. (2012) ‘IL-
1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate
lymphoid cells and CD4 + Th17 cells.’ The Journal of Experimental Medicine, 209(9) pp. 1595–1609.
Courties, G., Frodermann, V., Honold, L., Zheng, Y., Herisson, F. E., Schloss, M. J., Sun, Y., Presumey, J.,
Severe, N., Engblom, C., Hulsmans, M., Cremer, S., Rohde, D., Pittet, M. J., Scadden, D., Swirski, F. K.,
Kim, D.-E., Moskowitz, M. A. and Nahrendorf, M. (2019) ‘Glucocorticoids Regulate Bone Marrow B
Lymphopoiesis After Stroke.’ Circulation Research, February, p. CIRCRESAHA.118.314518.
Courties, G., Herisson, F., Sager, H. B., Heidt, T., Ye, Y., Wei, Y., Sun, Y., Severe, N., Dutta, P., Scharff, J.,
Scadden, D. T., Weissleder, R., Swirski, F. K., Moskowitz, M. A. and Nahrendorf, M. (2015) ‘Ischemic

201
Stroke Activates Hematopoietic Bone Marrow Stem Cells.’ Circulation Research. American Heart
Association, Inc., 116(3) pp. 407–417.
Craig, R. G., Yip, J. K., So, M. K., Boylan, R. J., Socransky, S. S. and Haffajee, A. D. (2003) ‘Relationship of
destructive periodontal disease to the acute-phase response.’ J Periodontol, 74(7) pp. 1007–1016.
Crapser, J., Ritzel, R., Verma, R., Venna, V. R., Liu, F., Chauhan, A., Koellhoffer, E., Patel, A., Ricker, A.,
Maas, K., Graf, J. and McCullough, L. D. (2016) ‘Ischemic stroke induces gut permeability and enhances
bacterial translocation leading to sepsis in aged mice.’ Aging, 8(5) pp. 1049–1063.
Crasta, K., Daly, C. G., Mitchell, D., Curtis, B., Stewart, D. and Heitz-Mayfield, L. J. A. (2009) ‘Bacteraemia
due to dental flossing.’ Journal of Clinical Periodontology, 36(4) pp. 323–332.
Cronk, J. C., Filiano, A. J., Louveau, A., Marin, I., Marsh, R., Ji, E., Goldman, D. H., Smirnov, I., Geraci, N.,
Acton, S., Overall, C. C. and Kipnis, J. (2018) ‘Peripherally derived macrophages can engraft the brain
independent of irradiation and maintain an identity distinct from microglia.’ The Journal of Experimental
Medicine. Rockefeller University Press, April, p. jem.20180247.
Cuartero, M. I., Ballesteros, I., Moraga, A., Nombela, F., Vivancos, J., Hamilton, J. A., Corbí, Á. L.,
Lizasoain, I. and Moro, M. A. (2013) ‘N2 Neutrophils, Novel Players in Brain Inflammation After Stroke.’
Stroke, 44(12) pp. 3498–3508.
D’Aiuto, F., Ready, D. and Tonetti, M. S. (2004) ‘Periodontal disease and C-reactive protein-associated
cardiovascular risk.’ Journal of Periodontal Research, 39(4) pp. 236–241.
Daep, C. A., Novak, E. A., Lamont, R. J. and Demuth, D. R. (2011) ‘Structural Dissection and In Vivo
Effectiveness of a Peptide Inhibitor of Porphyromonas gingivalis Adherence to Streptococcus gordonii.’
Camilli, A. (ed.) Infection and Immunity, 79(1) pp. 67–74.
Darveau, R. P., Pham, T.-T. T., Lemley, K., Reife, R. A., Bainbridge, B. W., Coats, S. R., Howald, W. N.,
Way, S. S. and Hajjar, A. M. (2004) ‘Porphyromonas gingivalis Lipopolysaccharide Contains Multiple Lipid
A Species That Functionally Interact with Both Toll-Like Receptors 2 and 4.’ Infection and Immunity, 72(9)
pp. 5041–5051.
Delbosc, S., Alsac, J.-M., Journe, C., Louedec, L., Castier, Y., Bonnaure-Mallet, M., Ruimy, R., Rossignol,
P., Bouchard, P., Michel, J.-B. and Meilhac, O. (2011) ‘Porphyromonas gingivalis Participates in
Pathogenesis of Human Abdominal Aortic Aneurysm by Neutrophil Activation. Proof of Concept in Rats.’
Reitsma, P. H. (ed.) PLoS ONE, 6(4) p. e18679.
DeLeon-Pennell, K. Y., de Castro Brás, L. E., Iyer, R. P., Bratton, D. R., Jin, Y.-F., Ripplinger, C. M. and
Lindsey, M. L. (2014) ‘P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction
through matrix metalloproteinase-9.’ Journal of Molecular and Cellular Cardiology, 76, November, pp. 218–
226.
DeLeon-Pennell, K. Y., de Castro Brás, L. E. and Lindsey, M. L. (2013) ‘Circulating Porphyromonas
gingivalis lipopolysaccharide resets cardiac homeostasis in mice through a matrix metalloproteinase-9-
dependent mechanism.’ Physiological Reports, 1(5).
Dénes, Á., Ferenczi, S. and Kovács, K. J. (2011a) ‘Systemic inflammatory challenges compromise survival
after experimental stroke via augmenting brain inflammation, blood- brain barrier damage and brain oedema
independently of infarct size.’ Journal of Neuroinflammation, 8(1) p. 164.
Dénes, Á., Humphreys, N., Lane, T. E., Grencis, R. and Rothwell, N. (2010a) ‘Chronic Systemic Infection
Exacerbates Ischemic Brain Damage via a CCL5 (Regulated on Activation, Normal T-Cell Expressed and
Secreted)-Mediated Proinflammatory Response in Mice.’ Journal of Neuroscience, 30(30) pp. 10086–10095.
Denes, Á., McColl, B. W., Leow-Dyke, S. F., Chapman, K. Z., Humphreys, N. E., Grencis, R. K., Allan, S.
M. and Rothwell, N. J. (2011) ‘Experimental Stroke-Induced Changes in the Bone Marrow Reveal Complex
Regulation of Leukocyte Responses.’ Journal of Cerebral Blood Flow & Metabolism, 31(4) pp. 1036–1050.
Dénes, Á., Pinteaux, E., Rothwell, N. J. and Allan, S. M. (2011b) ‘Interleukin-1 and Stroke: Biomarker,
Harbinger of Damage, and Therapeutic Target.’ Cerebrovascular Diseases. 2011/11/23, 32(6) pp. 517–527.
Dénes, Á., Pradillo, J. M., Drake, C., Sharp, A., Warn, P., Murray, K. N., Rohit, B., Dockrell, D. H.,
Chamberlain, J., Casbolt, H., Francis, S., Martinecz, B., Nieswandt, B., Rothwell, N. J. and Allan, S. M.

202
(2014) ‘Streptococcus pneumoniae worsens cerebral ischemia via interleukin 1 and platelet glycoprotein Ibα.’
Annals of Neurology, 75(5) pp. 670–683.
Dénes, Á., Thornton, P., Rothwell, N. J. and Allan, S. M. (2010b) ‘Inflammation and brain injury: Acute
cerebral ischaemia, peripheral and central inflammation.’ Brain, Behavior, and Immunity, 24(5) pp. 708–723.
Deng, J., Zhang, J., Feng, C., Xiong, L. and Zuo, Z. (2014) ‘Critical role of matrix metalloprotease-9 in
chronic high fat diet-induced cerebral vascular remodelling and increase of ischaemic brain injury in mice.’
Cardiovascular Research, 103(4) pp. 473–484.
Desvarieux, M., Demmer, R. T., Jacobs, D. R., Papapanou, P. N., Sacco, R. L. and Rundek, T. (2013)
‘Changes in Clinical and Microbiological Periodontal Profiles Relate to Progression of Carotid Intima‐Media
Thickness: The Oral Infections and Vascular Disease Epidemiology Study.’ Journal of the American Heart
Association, 2(6) p. e000254.
Desvarieux, M., Demmer, R. T., Rundek, T., Boden-Albala, B., Jacobs, D. R., Sacco, R. L. and Papapanou, P.
N. (2005) ‘Periodontal Microbiota and Carotid Intima-Media Thickness.’ Circulation, 111(5) pp. 576–582.
Dimitrijevic, O. B., Stamatovic, S. M., Keep, R. F. and Andjelkovic, A. V. (2007) ‘Absence of the chemokine
receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice.’ Stroke, 38(4) pp. 1345–1353.
Ding, L., Saunders, T. L., Enikolopov, G. and Morrison, S. J. (2012) ‘Endothelial and perivascular cells
maintain haematopoietic stem cells.’ Nature, 481(7382) pp. 457–462.
Dirnagl, U. and Endres, M. (2014) ‘Found in Translation: Preclinical Stroke Research Predicts Human
Pathophysiology, Clinical Phenotypes, and Therapeutic Outcomes.’ Stroke, 45(5) pp. 1510–1518.
Divaris, K., Monda, K. L., North, K. E., Olshan, A. F., Reynolds, L. M., Hsueh, W.-C., Lange, E. M., Moss,
K., Barros, S. P., Weyant, R. J., Liu, Y., Newman, A. B., Beck, J. D. and Offenbacher, S. (2013) ‘Exploring
the genetic basis of chronic periodontitis: a genome-wide association study.’ Human Molecular Genetics,
22(11) pp. 2312–2324.
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and
Gingeras, T. R. (2013) ‘STAR: ultrafast universal RNA-seq aligner.’ Bioinformatics, 29(1) pp. 15–21.
Dominy, S. S., Lynch, C., Ermini, F., Benedyk, M., Marczyk, A., Konradi, A., Nguyen, M., Haditsch, U.,
Raha, D., Griffin, C., Holsinger, L. J., Arastu-Kapur, S., Kaba, S., Lee, A., Ryder, M. I., Potempa, B., Mydel,
P., Hellvard, A., Adamowicz, K., Hasturk, H., Walker, G. D., Reynolds, E. C., Faull, R. L. M., Curtis, M. A.,
Dragunow, M. and Potempa, J. (2019) ‘Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence
for disease causation and treatment with small-molecule inhibitors.’ Science Advances. American Association
for the Advancement of Science, 5(1) p. eaau3333.
Dorfer, C. E., Becher, H., Ziegler, C. M., Kaiser, C., Lutz, R., Jorss, D., Lichy, C., Buggle, F., Bultmann, S.,
Preusch, M. and Grau, A. J. (2004) ‘The association of gingivitis and periodontitis with ischemic stroke.’
Journal of Clinical Periodontology. 2004/04/17, 31(5) pp. 396–401.
Drake, C., Boutin, H., Jones, M. S., Denes, A., McColl, B. W., Selvarajah, J. R., Hulme, S., Georgiou, R. F.,
Hinz, R., Gerhard, A., Vail, A., Prenant, C., Julyan, P., Maroy, R., Brown, G., Smigova, A., Herholz, K.,
Kassiou, M., Crossman, D., Francis, S., Proctor, S. D., Russell, J. C., Hopkins, S. J., Tyrrell, P. J., Rothwell,
N. J. and Allan, S. M. (2011) ‘Brain inflammation is induced by co-morbidities and risk factors for stroke.’
Brain, Behavior, and Immunity. 2011/03/02, 25(6) pp. 1113–1122.
Duarte, P. M., da Rocha, M., Sampaio, E., Mestnik, M. J., Feres, M., Figueiredo, L. C., Bastos, M. F. and
Faveri, M. (2010) ‘Serum Levels of Cytokines in Subjects With Generalized Chronic and Aggressive
Periodontitis Before and After Non-Surgical Periodontal Therapy: A Pilot Study.’ Journal of Periodontology,
81(7) pp. 1056–1063.
Dutta, P., Courties, G., Wei, Y., Leuschner, F., Gorbatov, R., Robbins, C. S., Iwamoto, Y., Thompson, B.,
Carlson, A. L., Heidt, T., Majmudar, M. D., Lasitschka, F., Etzrodt, M., Waterman, P., Waring, M. T.,
Chicoine, A. T., van der Laan, A. M., Niessen, H. W. M., Piek, J. J., Rubin, B. B., Butany, J., Stone, J. R.,
Katus, H. A., Murphy, S. A., Morrow, D. A., Sabatine, M. S., Vinegoni, C., Moskowitz, M. A., Pittet, M. J.,
Libby, P., Lin, C. P., Swirski, F. K., Weissleder, R. and Nahrendorf, M. (2012) ‘Myocardial infarction
accelerates atherosclerosis.’ Nature, 487(7407) pp. 325–329.
Dutzan, N., Abusleme, L., Bridgeman, H., Greenwell-Wild, T., Zangerle-Murray, T., Fife, M. E., Bouladoux,

203
N., Linley, H., Brenchley, L., Wemyss, K., Calderon, G., Hong, B.-Y., Break, T. J., Bowdish, D. M. E.,
Lionakis, M. S., Jones, S. A., Trinchieri, G., Diaz, P. I., Belkaid, Y., Konkel, J. E. and Moutsopoulos, N. M.
(2017) ‘On-going Mechanical Damage from Mastication Drives Homeostatic Th17 Cell Responses at the
Oral Barrier.’ Immunity, 46(1) pp. 133–147.
Dutzan, N., Abusleme, L., Konkel, J. E. and Moutsopoulos, N. M. (2016a) ‘Isolation, Characterization and
Functional Examination of the Gingival Immune Cell Network.’ Journal of Visualized Experiments, (108)
February.
Dutzan, N., Kajikawa, T., Abusleme, L., Greenwell-Wild, T., Zuazo, C. E., Ikeuchi, T., Brenchley, L., Abe,
T., Hurabielle, C., Martin, D., Morell, R. J., Freeman, A. F., Lazarevic, V., Trinchieri, G., Diaz, P. I.,
Holland, S. M., Belkaid, Y., Hajishengallis, G. and Moutsopoulos, N. M. (2018) ‘A dysbiotic microbiome
triggers Th17 cells to mediate oral mucosal immunopathology in mice and humans.’ Science Translational
Medicine, 10(463) p. eaat0797.
Dutzan, N., Konkel, J. E., Greenwell-Wild, T. and Moutsopoulos, N. M. (2016b) ‘Characterization of the
human immune cell network at the gingival barrier.’ Mucosal Immunology. Nature Publishing Group, 9(5)
pp. 1163–1172.
Dutzan, N., Vernal, R., Hernandez, M., Dezerega, A., Rivera, O., Silva, N., Aguillon, J. C., Puente, J., Pozo,
P. and Gamonal, J. (2009) ‘Levels of Interferon-Gamma and Transcription Factor T-Bet in Progressive
Periodontal Lesions in Patients With Chronic Periodontitis.’ Journal of Periodontology, 80(2) pp. 290–296.
Ebersole, J. L., Machen, R. L., Steffen, M. J. and Willman, D. E. (1997) ‘Systemic acute-phase reactants, C-
reactive protein and haptoglobin, in adult periodontitis.’ Clinical and Experimental Immunology, 107(2) pp.
347–352.
Eke, P. I., Dye, B. A., Wei, L., Slade, G. D., Thornton-Evans, G. O., Borgnakke, W. S., Taylor, G. W., Page,
R. C., Beck, J. D. and Genco, R. J. (2015) ‘Update on Prevalence of Periodontitis in Adults in the United
States: NHANES 2009 to 2012.’ Journal of Periodontology. 2015/02/18, 86(5) pp. 611–622.
Eke, P. I., Dye, B. A., Wei, L., Thornton-Evans, G. O. and Genco, R. J. (2012) ‘Prevalence of Periodontitis in
Adults in the United States: 2009 and 2010.’ Journal of Dental Research, 91(10) pp. 914–920.
Ekmekciu, I., von Klitzing, E., Fiebiger, U., Escher, U., Neumann, C., Bacher, P., Scheffold, A., Kühl, A. A.,
Bereswill, S. and Heimesaat, M. M. (2017) ‘Immune Responses to Broad-Spectrum Antibiotic Treatment and
Fecal Microbiota Transplantation in Mice.’ Frontiers in Immunology, 8, April.
Elter, J. R., Offenbacher, S., Toole, J. F. and Beck, J. D. (2003) ‘Relationship of periodontal disease and
edentulism to stroke/TIA.’ Journal of Dental Research. 2003/11/25, 82(12) pp. 998–1001.
Emerton, K. B., Drapeau, S. J., Prasad, H., Rohrer, M., Roffe, P., Hopper, K., Schoolfield, J., Jones, A. and
Cochran, D. L. (2011) ‘Regeneration of Periodontal Tissues in Non-human Primates with rhGDF-5 and Beta-
Tricalcium Phosphate.’ Journal of Dental Research, 90(12) pp. 1416–1421.
Emsley, H. C. and Hopkins, S. J. (2008) ‘Acute ischaemic stroke and infection: recent and emerging
concepts.’ The Lancet Neurology, 7(4) pp. 341–353.
Engel, O., Akyüz, L., da Costa Goncalves, A. C., Winek, K., Dames, C., Thielke, M., Herold, S., Böttcher,
C., Priller, J., Volk, H. D., Dirnagl, U., Meisel, C. and Meisel, A. (2015) ‘Cholinergic Pathway Suppresses
Pulmonary Innate Immunity Facilitating Pneumonia After Stroke.’ Stroke, 46(11) pp. 3232–3240.
Engel, O., Kolodziej, S., Dirnagl, U. and Prinz, V. (2011) ‘Modeling Stroke in Mice - Middle Cerebral Artery
Occlusion with the Filament Model.’ Journal of Visualized Experiments, (47) January.
Epelman, S., Lavine, K. J., Beaudin, A. E., Sojka, D. K., Carrero, J. A., Calderon, B., Brija, T., Gautier, E. L.,
Ivanov, S., Satpathy, A. T., Schilling, J. D., Schwendener, R., Sergin, I., Razani, B., Forsberg, E. C.,
Yokoyama, W. M., Unanue, E. R., Colonna, M., Randolph, G. J. and Mann, D. L. (2014) ‘Embryonic and
Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State
and during Inflammation.’ Immunity, 40(1) pp. 91–104.
Erciyas, K., Sezer, U., Üstün, K., Pehlivan, Y., Kısacık, B., Şenyurt, S., Tarakçıoğlu, M. and Onat, A. (2013)
‘Effects of periodontal therapy on disease activity and systemic inflammation in rheumatoid arthritis
patients.’ Oral Diseases, 19(4) pp. 394–400.

204
Ergul, A., Elgebaly, M. M., Middlemore, M.-L., Li, W., Elewa, H., Switzer, J. A., Hall, C., Kozak, A. and
Fagan, S. C. (2007) ‘Increased hemorrhagic transformation and altered infarct size and localization after
experimental stroke in a rat model type 2 diabetes.’ BMC Neurology, 7(1) p. 33.
Ergul, A., Hafez, S., Fouda, A. and Fagan, S. C. (2016) ‘Impact of Comorbidities on Acute Injury and
Recovery in Preclinical Stroke Research: Focus on Hypertension and Diabetes.’ Translational Stroke
Research, 7(4) pp. 248–260.
Eskan, M. A., Jotwani, R., Abe, T., Chmelar, J., Lim, J.-H., Liang, S., Ciero, P. A., Krauss, J. L., Li, F.,
Rauner, M., Hofbauer, L. C., Choi, E. Y., Chung, K.-J., Hashim, A., Curtis, M. A., Chavakis, T. and
Hajishengallis, G. (2012) ‘The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory
bone loss.’ Nature Immunology. Nature Publishing Group, 13(5) pp. 465–473.
Feigin, V. L., Barker-Collo, S., Krishnamurthi, R., Theadom, A. and Starkey, N. (2010) ‘Epidemiology of
ischaemic stroke and traumatic brain injury.’ Best Practice & Research Clinical Anaesthesiology.
2011/05/31, 24(4) pp. 485–494.
Feigin, V. L., Forouzanfar, M. H., Krishnamurthi, R., Mensah, G. A., Connor, M., Bennett, D. A., Moran, A.
E., Sacco, R. L., Anderson, L., Truelsen, T., O’Donnell, M., Venketasubramanian, N., Barker-Collo, S.,
Lawes, C. M. M., Wang, W., Shinohara, Y., Witt, E., Ezzati, M., Naghavi, M. and Murray, C. (2014) ‘Global
and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010.’
The Lancet, 383(9913) pp. 245–255.
Flurkey, K., Currer, J. M. and Harrison, D. E. (2007) ‘Mouse Models in Aging Research.’ In The Mouse in
Biomedical Research. Second Edi, New York: Elsevier, pp. 637–672.
Flynn, K. J., Baxter, N. T. and Schloss, P. D. (2016) ‘Metabolic and Community Synergy of Oral Bacteria in
Colorectal Cancer.’ McMahon, K. (ed.) mSphere, 1(3).
Ford, P., Gemmell, E., Hamlet, S. M., Hasan, A., Walker, P. J., West, M. J., Cullinan, M. P. and Seymour, G.
J. (2005a) ‘Cross-reactivity of GroEL antibodies with human heat shock protein 60 and quantification of
pathogens in atherosclerosis.’ Oral Microbiology and Immunology, 20(5) pp. 296–302.
Ford, P. J., Gemmell, E., Walker, P., West, M., Cullinan, M. and Seymour, G. J. (2005b) ‘Characterization of
Heat Shock Protein-Specific T Cells in Atherosclerosis.’ Clinical and Vaccine Immunology, 12(2) pp. 259–
267.
Forner, L., Larsen, T., Kilian, M. and Holmstrup, P. (2006) ‘Incidence of bacteremia after chewing, tooth
brushing and scaling in individuals with periodontal inflammation.’ Journal of Clinical Periodontology, 33(6)
pp. 401–407.
Foschi, F., Izard, J., Sasaki, H., Sambri, V., Prati, C., Müller, R. and Stashenko, P. (2006) ‘Treponema
denticola in Disseminating Endodontic Infections.’ Journal of Dental Research, 85(8) pp. 761–765.
Foteinos, G., Afzal, A. R., Mandal, K., Jahangiri, M. and Xu, Q. (2005) ‘Anti–Heat Shock Protein 60
Autoantibodies Induce Atherosclerosis in Apolipoprotein E–Deficient Mice via Endothelial Damage.’
Circulation, 112(8) pp. 1206–1213.
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. and Santoro, A. (2018) ‘Inflammaging: a new immune–
metabolic viewpoint for age-related diseases.’ Nature Reviews Endocrinology, 14(10) pp. 576–590.
Fugate, J. E., Lyons, J. L., Thakur, K. T., Smith, B. R., Hedley-Whyte, E. T. and Mateen, F. J. (2014)
‘Infectious causes of stroke.’ The Lancet Infectious Diseases, 14(9) pp. 869–880.
Gaetti-Jardim, E., Marcelino, S. L., Feitosa, A. C. R., Romito, G. A. and Avila-Campos, M. J. (2009)
‘Quantitative detection of periodontopathic bacteria in atherosclerotic plaques from coronary arteries.’
Journal of Medical Microbiology, 58(12) pp. 1568–1575.
Gan, Y., Liu, Q., Wu, W., Yin, J.-X., Bai, X.-F., Shen, R., Wang, Y., Chen, J., La Cava, A., Poursine-
Laurent, J., Yokoyama, W. and Shi, F.-D. (2014) ‘Ischemic neurons recruit natural killer cells that accelerate
brain infarction.’ Proceedings of the National Academy of Sciences. National Academy of Sciences, 111(7)
pp. 2704–2709.
Garcia-Bonilla, L., Brea, D., Benakis, C., Lane, D. A., Murphy, M., Moore, J., Racchumi, G., Jiang, X.,
Iadecola, C. and Anrather, J. (2018) ‘Endogenous Protection from Ischemic Brain Injury by Preconditioned

205
Monocytes.’ Journal of Neuroscience, 38(30) pp. 6722–6736.
Garlet, G. P., Cardoso, C. R. B., Campanelli, A. P., Garlet, T. P., Avila-Campos, M. J., Cunha, F. Q. and
Silva, J. S. (2008) ‘The essential role of IFN-γ in the control of lethal Aggregatibacter
actinomycetemcomitans infection in mice.’ Microbes and Infection.
Garlet, G. P., Cardoso, C. R., Mariano, F. S., Claudino, M., De Assis, G. F., Campanelli, A. P., Ávila-
Campos, M. J. and Silva, J. S. (2009) ‘Regulatory T cells attenuate experimental periodontitis progression in
mice.’ Journal of Clinical Periodontology, 37(7) pp. 591–600.
Ge, R., Tornero, D., Hirota, M., Monni, E., Laterza, C., Lindvall, O. and Kokaia, Z. (2017) ‘Choroid plexus-
cerebrospinal fluid route for monocyte-derived macrophages after stroke.’ Journal of Neuroinflammation,
14(1) p. 153.
Geerts, S. O., Nys, M., Mol, P. De, Charpentier, J., Albert, A., Legrand, V. and Rompen, E. H. (2002)
‘Systemic Release of Endotoxins Induced by Gentle Mastication: Association With Periodontitis Severity.’
Journal of Periodontology, 73(1) pp. 73–78.
Geissmann, F., Jung, S. and Littman, D. R. (2003) ‘Blood Monocytes Consist of Two Principal Subsets with
Distinct Migratory Properties.’ Immunity, 19(1) pp. 71–82.
Gelderblom, M., Arunachalam, P. and Magnus, T. (2014) ‘γδ T cells as early sensors of tissue damage and
mediators of secondary neurodegeneration.’ Frontiers in Cellular Neuroscience, 8, November.
Gelderblom, M., Gallizioli, M., Ludewig, P., Thom, V., Arunachalam, P., Rissiek, B., Bernreuther, C.,
Glatzel, M., Korn, T., Arumugam, T. V., Sedlacik, J., Gerloff, C., Tolosa, E., Planas, A. M. and Magnus, T.
(2018) ‘IL-23 (Interleukin-23)-Producing Conventional Dendritic Cells Control the Detrimental IL-17
(Interleukin-17) Response in Stroke.’ Stroke, 49(1) pp. 155–164.
Gelderblom, M., Weymar, A., Bernreuther, C., Velden, J., Arunachalam, P., Steinbach, K., Orthey, E.,
Arumugam, T. V, Leypoldt, F., Simova, O., Thom, V., Friese, M. A., Prinz, I., Holscher, C., Glatzel, M.,
Korn, T., Gerloff, C., Tolosa, E. and Magnus, T. (2012) ‘Neutralization of the IL-17 axis diminishes
neutrophil invasion and protects from ischemic stroke.’ Blood. American Society of Hematology, 120(18) pp.
3793–3802.
Genco, R. J. and Van Dyke, T. E. (2010) ‘Reducing the risk of CVD in patients with periodontitis.’ Nature
Reviews Cardiology, 7(9) pp. 479–480.
Gendron, R., Grenier, D. and Maheu-Robert, L.-F. (2000) ‘The oral cavity as a reservoir of bacterial
pathogens for focal infections.’ Microbes and Infection, 2(8) pp. 897–906.
Ghizoni, J. S., Taveira, L. A. de A., Garlet, G. P., Ghizoni, M. F., Pereira, J. R., Dionísio, T. J., Brozoski, D.
T., Santos, C. F. and Sant’Ana, A. C. P. (2012) ‘Increased levels of Porphyromonas gingivalis are associated
with ischemic and hemorrhagic cerebrovascular disease in humans: an in vivo study.’ Journal of Applied
Oral Science, 20(1) pp. 104–112.
Gibson, F. C., Hong, C., Chou, H.-H., Yumoto, H., Chen, J., Lien, E., Wong, J. and Genco, C. A. (2004)
‘Innate Immune Recognition of Invasive Bacteria Accelerates Atherosclerosis in Apolipoprotein E-Deficient
Mice.’ Circulation, 109(22) pp. 2801–2806.
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., Mehler, M. F., Conway, S. J., Ng, L.
G., Stanley, E. R., Samokhvalov, I. M. and Merad, M. (2010) ‘Fate Mapping Analysis Reveals That Adult
Microglia Derive from Primitive Macrophages.’ Science, 330(6005) pp. 841–845.
Ginhoux, F. and Jung, S. (2014) ‘Monocytes and macrophages: developmental pathways and tissue
homeostasis.’ Nature Reviews Immunology, 14(6) pp. 392–404.
Gliem, M., Mausberg, A. K., Lee, J.-I., Simiantonakis, I., van Rooijen, N., Hartung, H.-P. and Jander, S.
(2012) ‘Macrophages prevent hemorrhagic infarct transformation in murine stroke models.’ Annals of
Neurology. 2012/06/22, 71(6) pp. 743–752.
Gliem, M., Schwaninger, M. and Jander, S. (2016) ‘Protective features of peripheral monocytes/macrophages
in stroke.’ Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1862(3) pp. 329–338.
Glowacki, A. J., Yoshizawa, S., Jhunjhunwala, S., Vieira, A. E., Garlet, G. P., Sfeir, C. and Little, S. R.

206
(2013) ‘Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via
recruitment of regulatory lymphocytes.’ Proceedings of the National Academy of Sciences, 110(46) pp.
18525–18530.
Goldstein, L. B., Bushnell, C. D., Adams, R. J., Appel, L. J., Braun, L. T., Chaturvedi, S., Creager, M. A.,
Culebras, A., Eckel, R. H., Hart, R. G., Hinchey, J. A., Howard, V. J., Jauch, E. C., Levine, S. R., Meschia, J.
F., Moore, W. S., Nixon, J. V. (Ian) and Pearson, T. A. (2011) ‘Guidelines for the Primary Prevention of
Stroke.’ Stroke, 42(2) pp. 517–584.
Gotsman, I., Lotan, C., Soskolne, W. A., Rassovsky, S., Pugatsch, T., Lapidus, L., Novikov, Y., Masrawa, S.
and Stabholz, A. (2007) ‘Periodontal Destruction Is Associated With Coronary Artery Disease and
Periodontal Infection With Acute Coronary Syndrome.’ Journal of Periodontology, 78(5) pp. 849–858.
Grainger, J. R., Wohlfert, E. A., Fuss, I. J., Bouladoux, N., Askenase, M. H., Legrand, F., Koo, L. Y.,
Brenchley, J. M., Fraser, I. D. C. and Belkaid, Y. (2013) ‘Inflammatory monocytes regulate pathologic
responses to commensals during acute gastrointestinal infection.’ Nature Medicine, 19(6) pp. 713–721.
Grau, A. J., Becher, H., Ziegler, C. M., Lichy, C., Buggle, F., Kaiser, C., Lutz, R., Bültmann, S., Preusch, M.
and Dorfer, C. E. (2004) ‘Periodontal disease as a risk factor for ischemic stroke.’ Stroke. 2004/01/07, 35(2)
pp. 496–501.
Grau, A. J., Buggle, F., Ziegler, C., Schwarz, W., Meuser, J., Tasman, A. J., Buhler, A., Benesch, C., Becher,
H. and Hacke, W. (1997) ‘Association between acute cerebrovascular ischemia and chronic and recurrent
infection.’ Stroke. 1997/09/26, 28(9) pp. 1724–1729.
Graves, D. T. and Cochran, D. (2003) ‘The Contribution of Interleukin-1 and Tumor Necrosis Factor to
Periodontal Tissue Destruction.’ Journal of Periodontology, 74(3) pp. 391–401.
Graves, D. T., Fine, D., Teng, Y.-T. A., Van Dyke, T. E. and Hajishengallis, G. (2008) ‘The use of rodent
models to investigate host-bacteria interactions related to periodontal diseases.’ Journal of Clinical
Periodontology, 35(2) pp. 89–105.
Griffen, A. L., Beall, C. J., Campbell, J. H., Firestone, N. D., Kumar, P. S., Yang, Z. K., Podar, M. and Leys,
E. J. (2012) ‘Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S
pyrosequencing.’ The ISME Journal, 6(6) pp. 1176–1185.
Griseri, T., McKenzie, B. S., Schiering, C. and Powrie, F. (2012) ‘Dysregulated Hematopoietic Stem and
Progenitor Cell Activity Promotes Interleukin-23-Driven Chronic Intestinal Inflammation.’ Immunity, 37(6)
pp. 1116–1129.
Guilliams, M., Ginhoux, F., Jakubzick, C., Naik, S. H., Onai, N., Schraml, B. U., Segura, E., Tussiwand, R.
and Yona, S. (2014) ‘Dendritic cells, monocytes and macrophages: a unified nomenclature based on
ontogeny.’ Nature Reviews Immunology, 14(8) pp. 571–578.
Guilliams, M., De Kleer, I., Henri, S., Post, S., Vanhoutte, L., De Prijck, S., Deswarte, K., Malissen, B.,
Hammad, H. and Lambrecht, B. N. (2013) ‘Alveolar macrophages develop from fetal monocytes that
differentiate into long-lived cells in the first week of life via GM-CSF.’ The Journal of Experimental
Medicine, 210(10) pp. 1977–1992.
Gully, N., Bright, R., Marino, V., Marchant, C., Cantley, M., Haynes, D., Butler, C., Dashper, S., Reynolds,
E. and Bartold, M. (2014) ‘Porphyromonas gingivalis Peptidylarginine Deiminase, a Key Contributor in the
Pathogenesis of Experimental Periodontal Disease and Experimental Arthritis.’ Yilmaz, Ö. (ed.) PLoS ONE,
9(6) p. e100838.
Gur, C., Ibrahim, Y., Isaacson, B., Yamin, R., Abed, J., Gamliel, M., Enk, J., Bar-On, Y., Stanietsky-Kaynan,
N., Coppenhagen-Glazer, S., Shussman, N., Almogy, G., Cuapio, A., Hofer, E., Mevorach, D., Tabib, A.,
Ortenberg, R., Markel, G., Miklić, K., Jonjic, S., Brennan, C. A., Garrett, W. S., Bachrach, G. and
Mandelboim, O. (2015) ‘Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory
Receptor TIGIT Protects Tumors from Immune Cell Attack.’ Immunity, 42(2) pp. 344–355.
Habashneh, R. A., Khader, Y. S., Alhumouz, M. K., Jadallah, K. and Ajlouni, Y. (2012) ‘The association
between inflammatory bowel disease and periodontitis among Jordanians: a case-control study.’ Journal of
Periodontal Research, 47(3) pp. 293–298.
Hacke, W., Kaste, M., Bluhmki, E., Brozman, M., Dávalos, A., Guidetti, D., Larrue, V., Lees, K. R.,

207
Medeghri, Z., Machnig, T., Schneider, D., von Kummer, R., Wahlgren, N. and Toni, D. (2008)
‘Thrombolysis with Alteplase 3 to 4.5 Hours after Acute Ischemic Stroke.’ New England Journal of
Medicine, 359(13) pp. 1317–1329.
Hajishengallis, G. (2014a) ‘Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host
response.’ Trends in Immunology. 2013/11/26, 35(1) pp. 3–11.
Hajishengallis, G. (2014b) ‘The inflammophilic character of the periodontitis-associated microbiota.’
Molecular Oral Microbiology, 29(6) pp. 248–257.
Hajishengallis, G. (2015) ‘Periodontitis: from microbial immune subversion to systemic inflammation.’
Nature Reviews Immunology, 15(1) pp. 30–44.
Hajishengallis, G., Lamont, R. J. and Graves, D. T. (2015) ‘The enduring importance of animal models in
understanding periodontal disease.’ Virulence, 6(3) pp. 229–35.
Hajishengallis, G., Liang, S., Payne, M. A., Hashim, A., Jotwani, R., Eskan, M. A., McIntosh, M. L., Alsam,
A., Kirkwood, K. L., Lambris, J. D., Darveau, R. P. and Curtis, M. A. (2011) ‘Low-Abundance Biofilm
Species Orchestrates Inflammatory Periodontal Disease through the Commensal Microbiota and
Complement.’ Cell Host & Microbe. 2011/11/01, 10(5) pp. 497–506.
Hajishengallis, G., Moutsopoulos, N. M., Hajishengallis, E. and Chavakis, T. (2016) ‘Immune and regulatory
functions of neutrophils in inflammatory bone loss.’ Seminars in Immunology. 2016/03/05, 28(2) pp. 146–
158.
Haley, M. J., Krishnan, S., Burrows, D., de Hoog, L., Thakrar, J., Schiessl, I., Allan, S. M. and Lawrence, C.
B. (2017a) ‘Acute high-fat feeding leads to disruptions in glucose homeostasis and worsens stroke outcome.’
Journal of Cerebral Blood Flow & Metabolism, November, pp. 1–12.
Haley, M. J. and Lawrence, C. B. (2016) ‘Obesity and stroke: Can we translate from rodents to patients?’
Journal of Cerebral Blood Flow & Metabolism, 36(12) pp. 2007–2021.
Haley, M. J. and Lawrence, C. B. (2017) ‘The blood–brain barrier after stroke: Structural studies and the role
of transcytotic vesicles.’ Journal of Cerebral Blood Flow & Metabolism, 37(2) pp. 456–470.
Haley, M. J., Mullard, G., Hollywood, K. A., Cooper, G. J., Dunn, W. B. and Lawrence, C. B. (2017b)
‘Adipose tissue and metabolic and inflammatory responses to stroke are altered in obese mice.’ Disease
Models & Mechanisms, 10(10) pp. 1229–1243.
Han, X., Kawai, T., Eastcott, J. W. and Taubman, M. A. (2006) ‘Bacterial-Responsive B Lymphocytes
Induce Periodontal Bone Resorption.’ The Journal of Immunology, 176(1) pp. 625–631.
Han, Y. W., Fardini, Y., Chen, C., Iacampo, K. G., Peraino, V. A., Shamonki, J. M. and Redline, R. W.
(2010) ‘Term Stillbirth Caused by Oral Fusobacterium nucleatum.’ Obstetrics & Gynecology,
115(Supplement) pp. 442–445.
Han, Y. W., Houcken, W., Loos, B. G., Schenkein, H. A. and Tezal, M. (2014) ‘Periodontal Disease,
Atherosclerosis, Adverse Pregnancy Outcomes, and Head-and-Neck Cancer.’ Advances in Dental Research,
26(1) pp. 47–55.
Haraszthy, V. I., Zambon, J. J., Trevisan, M., Zeid, M. and Genco, R. J. (2000) ‘Identification of Periodontal
Pathogens in Atheromatous Plaques.’ Journal of Periodontology. 2000/11/04, 71(10) pp. 1554–1560.
Hashimoto, D., Chow, A., Noizat, C., Teo, P., Beasley, M. B., Leboeuf, M., Becker, C. D., See, P., Price, J.,
Lucas, D., Greter, M., Mortha, A., Boyer, S. W., Forsberg, E. C., Tanaka, M., van Rooijen, N., García-Sastre,
A., Stanley, E. R., Ginhoux, F., Frenette, P. S. and Merad, M. (2013) ‘Tissue-Resident Macrophages Self-
Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes.’ Immunity.
Cell Press, 38(4) pp. 792–804.
Hasturk, H., Kantarci, A., Ebrahimi, N., Andry, C., Holick, M., Jones, V. L. and Van Dyke, T. E. (2006)
‘Topical H2 Antagonist Prevents Periodontitis in a Rabbit Model.’ Infection and Immunity, 74(4) pp. 2402–
2414.
Hayakawa, K., Mishima, K., Nozako, M., Hazekawa, M., Aoyama, Y., Ogata, A., Harada, K., Fujioka, M.,
Abe, K., Egashira, N., Iwasaki, K. and Fujiwara, M. (2007) ‘High-cholesterol feeding aggravates cerebral

208
infarction via decreasing the CB1 receptor.’ Neuroscience Letters.
Hayashi, C., Madrigal, A. G., Liu, X., Ukai, T., Goswami, S., Gudino, C. V., Gibson, III, F. C. and Genco, C.
A. (2010) ‘Pathogen-Mediated Inflammatory Atherosclerosis Is Mediated in Part via Toll-Like Receptor 2-
Induced Inflammatory Responses.’ Journal of Innate Immunity, 2(4) pp. 334–343.
Heidt, T., Sager, H. B., Courties, G., Dutta, P., Iwamoto, Y., Zaltsman, A., von zur Muhlen, C., Bode, C.,
Fricchione, G. L., Denninger, J., Lin, C. P., Vinegoni, C., Libby, P., Swirski, F. K., Weissleder, R. and
Nahrendorf, M. (2014) ‘Chronic variable stress activates hematopoietic stem cells.’ Nature Medicine, 20(7)
pp. 754–758.
Hernández, M., Dutzan, N., García-Sesnich, J., Abusleme, L., Dezerega, A., Silva, N., González, F. E.,
Vernal, R., Sorsa, T. and Gamonal, J. (2011) ‘Host-Pathogen Interactions in Progressive Chronic
Periodontitis.’ Journal of Dental Research, 90(10) pp. 1164–1170.
Hettinger, J., Richards, D. M., Hansson, J., Barra, M. M., Joschko, A.-C., Krijgsveld, J. and Feuerer, M.
(2013) ‘Origin of monocytes and macrophages in a committed progenitor.’ Nature Immunology. Nature
Publishing Group, 14(8) pp. 821–830.
Hirschfeld, M., Weis, J. J., Toshchakov, V., Salkowski, C. A., Cody, M. J., Ward, D. C., Qureshi, N.,
Michalek, S. M. and Vogel, S. N. (2001) ‘Signaling by Toll-Like Receptor 2 and 4 Agonists Results in
Differential Gene Expression in Murine Macrophages.’ Infection and Immunity. American Society for
Microbiology Journals, 69(3) pp. 1477–1482.
Hoeffel, G., Chen, J., Lavin, Y., Low, D., Almeida, F. F., See, P., Beaudin, A. E., Lum, J., Low, I., Forsberg,
E. C., Poidinger, M., Zolezzi, F., Larbi, A., Ng, L. G., Chan, J. K. Y., Greter, M., Becher, B., Samokhvalov, I.
M., Merad, M. and Ginhoux, F. (2015) ‘C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give
Rise to Adult Tissue-Resident Macrophages.’ Immunity, 42(4) pp. 665–678.
Howell, T. H., Ridker, P. M., Ajani, U. A., Christen, W. G. and Hennekens, C. H. (2001) ‘Periodontal disease
and risk of subsequent cardiovascular disease in U.S. male physicians.’ Journal of the American College of
Cardiology. 2001/02/24, 37(2) pp. 445–450.
Howells, D. W., Porritt, M. J., Rewell, S. S. J., O’Collins, V., Sena, E. S., van der Worp, H. B., Traystman, R.
J. and Macleod, M. R. (2010) ‘Different Strokes for Different Folks: The Rich Diversity of Animal Models of
Focal Cerebral Ischemia.’ Journal of Cerebral Blood Flow & Metabolism, 30(8) pp. 1412–1431.
Humphrey, L. L., Fu, R., Buckley, D. I., Freeman, M. and Helfand, M. (2008) ‘Periodontal Disease and
Coronary Heart Disease Incidence: A Systematic Review and Meta-analysis.’ Journal of General Internal
Medicine, 23(12) pp. 2079–2086.
Iadecola, C. and Anrather, J. (2011) ‘The immunology of stroke: from mechanisms to translation.’ Nature
Medicine, 17(7) pp. 796–808.
Ide, M., Harris, M., Stevens, A., Sussams, R., Hopkins, V., Culliford, D., Fuller, J., Ibbett, P., Raybould, R.,
Thomas, R., Puenter, U., Teeling, J., Perry, V. H. and Holmes, C. (2016) ‘Periodontitis and Cognitive Decline
in Alzheimer’s Disease.’ Garg, P. (ed.) PLOS ONE, 11(3) p. e0151081.
Ikeda, N., Asano, K., Kikuchi, K., Uchida, Y., Ikegami, H., Takagi, R., Yotsumoto, S., Shibuya, T., Makino-
Okamura, C., Fukuyama, H., Watanabe, T., Ohmuraya, M., Araki, K., Nishitai, G. and Tanaka, M. (2018)
‘Emergence of immunoregulatory Ym1+ Ly6Chi monocytes during recovery phase of tissue injury.’ Science
Immunology, 3(28) p. eaat0207.
Ito, M., Komai, K., Mise-Omata, S., Iizuka-Koga, M., Noguchi, Y., Kondo, T., Sakai, R., Matsuo, K.,
Nakayama, T., Yoshie, O., Nakatsukasa, H., Chikuma, S., Shichita, T. and Yoshimura, A. (2019) ‘Brain
regulatory T cells suppress astrogliosis and potentiate neurological recovery.’ Nature. Nature Publishing
Group, 565(7738) pp. 246–250.
Ivanov, I. I., Atarashi, K., Manel, N., Brodie, E. L., Shima, T., Karaoz, U., Wei, D., Goldfarb, K. C., Santee,
C. A., Lynch, S. V., Tanoue, T., Imaoka, A., Itoh, K., Takeda, K., Umesaki, Y., Honda, K. and Littman, D. R.
(2009) ‘Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria.’ Cell, 139(3) pp. 485–498.
Jain, A., Batista, E. L., Serhan, C., Stahl, G. L. and Dyke, T. E. Van (2003) ‘Role for Periodontitis in the
Progression of Lipid Deposition in an Animal Model.’ Infection and Immunity, 71(10) pp. 6012–6018.

209
Jakubzick, C., Gautier, E. L., Gibbings, S. L., Sojka, D. K., Schlitzer, A., Johnson, T. E., Ivanov, S., Duan,
Q., Bala, S., Condon, T., Van Rooijen, N., Grainger, J. R., Belkaid, Y., Ma’, A., Riches, D. W. H.,
Yokoyama, W. M., Ginhoux, F., Henson, P. M. and Randolph, G. J. (2013) ‘Minimal Differentiation of
Classical Monocytes as They Survey Steady-State Tissues and Transport Antigen to Lymph Nodes.’
Janowska-Wieczorek, A., Marquez-Curtis, L. A., Shirvaikar, N. and Ratajczak, M. Z. (2012) ‘The role of
complement in the trafficking of hematopoietic stem/progenitor cells.’ Transfusion, 52(12) pp. 2706–2716.
Joshipura, K. J., Hung, H. C., Rimm, E. B., Willett, W. C. and Ascherio, A. (2003) ‘Periodontal disease, tooth
loss, and incidence of ischemic stroke.’ Stroke. 2003/01/04, 34(1) pp. 47–52.
Jotwani, R. and Cutler, C. W. (2004) ‘Fimbriated Porphyromonas gingivalis Is More Efficient than Fimbria-
Deficient P. gingivalis in Entering Human Dendritic Cells In Vitro and Induces an Inflammatory Th1 Effector
Response.’ Infection and Immunity, 72(3) pp. 1725–1732.
Jusko, M., Potempa, J., Karim, A. Y., Ksiazek, M., Riesbeck, K., Garred, P., Eick, S. and Blom, A. M. (2012)
‘A Metalloproteinase Karilysin Present in the Majority of Tannerella forsythia Isolates Inhibits All Pathways
of the Complement System.’ The Journal of Immunology, 188(5) pp. 2338–2349.
Karatas, H., Erdener, S. E., Gursoy-Ozdemir, Y., Gurer, G., Soylemezoglu, F., Dunn, A. K. and Dalkara, T.
(2011) ‘Thrombotic distal middle cerebral artery occlusion produced by topical FeCl 3 application: A novel
model suitable for intravital microscopy and thrombolysis studies.’ Journal of Cerebral Blood Flow &
Metabolism, 31(6) pp. 1452–1460.
Kawai, T., Matsuyama, T., Hosokawa, Y., Makihira, S., Seki, M., Karimbux, N. Y., Goncalves, R. B.,
Valverde, P., Dibart, S., Li, Y.-P., Miranda, L. A., Ernst, C. W. O., Izumi, Y. and Taubman, M. A. (2006) ‘B
and T Lymphocytes Are the Primary Sources of RANKL in the Bone Resorptive Lesion of Periodontal
Disease.’ The American Journal of Pathology, 169(3) pp. 987–998.
Kimizuka, R., Kato, T., Ishihara, K. and Okuda, K. (2003) ‘Mixed infections with Porphyromonas gingivalis
and Treponema denticola cause excessive inflammatory responses in a mouse pneumonia model compared
with monoinfections.’ Microbes and Infection, 5(15) pp. 1357–1362.
Kinane, D. F. (2001) ‘Causation and pathogenesis of periodontal disease.’ Periodontology 2000. 2001/01/13,
25(1) pp. 8–20.
Kinane, D. F., Riggio, M. P., Walker, K. F., MacKenzie, D. and Shearer, B. (2005) ‘Bacteraemia following
periodontal procedures.’ Journal of Clinical Periodontology, 32(7) pp. 708–713.
Kinane, D. F., Stathopoulou, P. G. and Papapanou, P. N. (2017) ‘Periodontal diseases.’ Nature Reviews
Disease Primers, 3, June, p. 17038.
King, K. Y. and Goodell, M. A. (2011) ‘Inflammatory modulation of HSCs: viewing the HSC as a foundation
for the immune response.’ Nature Reviews Immunology, 11(10) pp. 685–692.
Kirii, H., Niwa, T., Yamada, Y., Wada, H., Saito, K., Iwakura, Y., Asano, M., Moriwaki, H. and Seishima,
M. (2003) ‘Lack of Interleukin-1β Decreases the Severity of Atherosclerosis in ApoE-Deficient Mice.’
Arteriosclerosis, Thrombosis, and Vascular Biology, 23(4) pp. 656–660.
Kleinnijenhuis, J., Quintin, J., Preijers, F., Joosten, L. A. B., Ifrim, D. C., Saeed, S., Jacobs, C., van Loenhout,
J., de Jong, D., Stunnenberg, H. G., Xavier, R. J., van der Meer, J. W. M., van Crevel, R. and Netea, M. G.
(2012) ‘Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via
epigenetic reprogramming of monocytes.’ Proceedings of the National Academy of Sciences, 109(43) pp.
17537–17542.
Kleinschnitz, C., Kraft, P., Dreykluft, A., Hagedorn, I., Gobel, K., Schuhmann, M. K., Langhauser, F.,
Helluy, X., Schwarz, T., Bittner, S., Mayer, C. T., Brede, M., Varallyay, C., Pham, M., Bendszus, M., Jakob,
P., Magnus, T., Meuth, S. G., Iwakura, Y., Zernecke, A., Sparwasser, T., Nieswandt, B., Stoll, G. and Wiendl,
H. (2013) ‘Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction
of the cerebral microvasculature.’ Blood. American Society of Hematology, 121(4) pp. 679–691.
Kleinschnitz, C., Schwab, N., Kraft, P., Hagedorn, I., Dreykluft, A., Schwarz, T., Austinat, M., Nieswandt,
B., Wiendl, H. and Stoll, G. (2010) ‘Early detrimental T-cell effects in experimental cerebral ischemia are
neither related to adaptive immunity nor thrombus formation.’ Blood. American Society of Hematology,
115(18) pp. 3835–3842.

210
Klose, C. S. N., Mahlakõiv, T., Moeller, J. B., Rankin, L. C., Flamar, A.-L., Kabata, H., Monticelli, L. A.,
Moriyama, S., Putzel, G. G., Rakhilin, N., Shen, X., Kostenis, E., König, G. M., Senda, T., Carpenter, D.,
Farber, D. L. and Artis, D. (2017) ‘The neuropeptide neuromedin U stimulates innate lymphoid cells and type
2 inflammation.’ Nature. Nature Publishing Group, 549(7671) pp. 282–286.
Konig, M. F., Abusleme, L., Reinholdt, J., Palmer, R. J., Teles, R. P., Sampson, K., Rosen, A., Nigrovic, P.
A., Sokolove, J., Giles, J. T., Moutsopoulos, N. M. and Andrade, F. (2016) ‘Aggregatibacter
actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in
rheumatoid arthritis.’ Science Translational Medicine, 8(369) p. 369ra176-369ra176.
Koren, O., Spor, A., Felin, J., Fak, F., Stombaugh, J., Tremaroli, V., Behre, C. J., Knight, R., Fagerberg, B.,
Ley, R. E. and Backhed, F. (2011) ‘Human oral, gut, and plaque microbiota in patients with atherosclerosis.’
Proceedings of the National Academy of Sciences, 108(Supplement_1) pp. 4592–4598.
Kostic, A. D., Chun, E., Robertson, L., Glickman, J. N., Gallini, C. A., Michaud, M., Clancy, T. E., Chung,
D. C., Lochhead, P., Hold, G. L., El-Omar, E. M., Brenner, D., Fuchs, C. S., Meyerson, M. and Garrett, W. S.
(2013) ‘Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune
Microenvironment.’ Cell Host & Microbe, 14(2) pp. 207–215.
Kostic, A. D., Gevers, D., Pedamallu, C. S., Michaud, M., Duke, F., Earl, A. M., Ojesina, A. I., Jung, J., Bass,
A. J., Tabernero, J., Baselga, J., Liu, C., Shivdasani, R. A., Ogino, S., Birren, B. W., Huttenhower, C.,
Garrett, W. S. and Meyerson, M. (2012) ‘Genomic analysis identifies association of Fusobacterium with
colorectal carcinoma.’ Genome Research, 22(2) pp. 292–298.
Kozarov, E. V., Dorn, B. R., Shelburne, C. E., Dunn, W. A. and Progulske-Fox, A. (2005) ‘Human
Atherosclerotic Plaque Contains Viable Invasive Actinobacillus actinomycetemcomitans and Porphyromonas
gingivalis.’ Arteriosclerosis, Thrombosis, and Vascular Biology, 25(3).
Kräter, M., Jacobi, A., Otto, O., Tietze, S., Müller, K., Poitz, D. M., Palm, S., Zinna, V. M., Biehain, U.,
Wobus, M., Chavakis, T., Werner, C., Guck, J. and Bornhauser, M. (2017) ‘Bone marrow niche-mimetics
modulate HSPC function via integrin signaling.’ Scientific Reports, 7(1) p. 2549.
Krishnan, S., Prise, I. E., Wemyss, K., Schenck, L. P., Bridgeman, H. M., McClure, F. A., Zangerle-Murray,
T., O’Boyle, C., Barbera, T. A., Mahmood, F., Bowdish, D. M. E., Zaiss, D. M. W., Grainger, J. R. and
Konkel, J. E. (2018) ‘Amphiregulin-producing γδ T cells are vital for safeguarding oral barrier immune
homeostasis.’ Proceedings of the National Academy of Sciences, 115(42) pp. 10738–10743.
Kumar, P. S. (2012) ‘Smoking and the subgingival ecosystem: a pathogen-enriched community.’ Future
Microbiology, 7(8) pp. 917–919.
Kuramitsu, H. K., Qi, M., Kang, I. and Chen, W. (2001) ‘Role for Periodontal Bacteria in Cardiovascular
Diseases.’ Annals of Periodontology. 2002/03/13, 6(1) pp. 41–47.
Kweider, M., Lowe, G. D., Murray, G. D., Kinane, D. F. and McGowan, D. A. (1993) ‘Dental disease,
fibrinogen and white cell count; links with myocardial infarction?’ Scott Med J. 1993/06/01, 38(3) pp. 73–74.
Labat-gest, V. and Tomasi, S. (2013) ‘Photothrombotic Ischemia: A Minimally Invasive and Reproducible
Photochemical Cortical Lesion Model for Mouse Stroke Studies.’ Journal of Visualized Experiments, (76)
June.
Laine, M. L., Crielaard, W. and Loos, B. G. (2012) ‘Genetic susceptibility to periodontitis.’ Periodontology
2000, 58(1) pp. 37–68.
Lalla, E., Lamster, I. B., Hofmann, M. A., Bucciarelli, L., Jerud, A. P., Tucker, S., Lu, Y., Papapanou, P. N.
and Schmidt, A. M. (2003) ‘Oral Infection With a Periodontal Pathogen Accelerates Early Atherosclerosis in
Apolipoprotein E–Null Mice.’ Arteriosclerosis, Thrombosis, and Vascular Biology, 23(8) pp. 1405–1411.
Lalla, E. and Papapanou, P. N. (2011) ‘Diabetes mellitus and periodontitis: a tale of two common interrelated
diseases.’ Nature Reviews Endocrinology, 7(12) pp. 738–748.
Lam, R. S., O’Brien-Simpson, N. M., Hamilton, J. A., Lenzo, J. C., Holden, J. A., Brammar, G. C., Orth, R.
K., Tan, Y., Walsh, K. A., Fleetwood, A. J. and Reynolds, E. C. (2015) ‘GM-CSF and uPA are required for
Porphyromonas gingivalis -induced alveolar bone loss in a mouse periodontitis model.’ Immunology and Cell
Biology, 93(8) pp. 705–715.

211
Lam, R. S., O’Brien-Simpson, N. M., Lenzo, J. C., Holden, J. A., Brammar, G. C., Walsh, K. A.,
McNaughtan, J. E., Rowler, D. K., Van Rooijen, N. and Reynolds, E. C. (2014) ‘Macrophage Depletion
Abates Porphyromonas gingivalis –Induced Alveolar Bone Resorption in Mice.’ The Journal of Immunology,
193(5) pp. 2349–2362.
Lee, Y., Awasthi, A., Yosef, N., Quintana, F. J., Xiao, S., Peters, A., Wu, C., Kleinewietfeld, M., Kunder, S.,
Hafler, D. A., Sobel, R. A., Regev, A. and Kuchroo, V. K. (2012) ‘Induction and molecular signature of
pathogenic TH17 cells.’ Nature Immunology, 13(10) pp. 991–999.
Leira, Y., Seoane, J., Blanco, M., Rodríguez-Yáñez, M., Takkouche, B., Blanco, J. and Castillo, J. (2017)
‘Association between periodontitis and ischemic stroke: a systematic review and meta-analysis.’ European
Journal of Epidemiology, 32(1) pp. 43–53.
Leuschner, F., Rauch, P. J., Ueno, T., Gorbatov, R., Marinelli, B., Lee, W. W., Dutta, P., Wei, Y., Robbins,
C., Iwamoto, Y., Sena, B., Chudnovskiy, A., Panizzi, P., Keliher, E., Higgins, J. M., Libby, P., Moskowitz,
M. A., Pittet, M. J., Swirski, F. K., Weissleder, R. and Nahrendorf, M. (2012) ‘Rapid monocyte kinetics in
acute myocardial infarction are sustained by extramedullary monocytopoiesis.’ The Journal of Experimental
Medicine, 209(1) pp. 123–137.
Levy, M., Kolodziejczyk, A. A., Thaiss, C. A. and Elinav, E. (2017) ‘Dysbiosis and the immune system.’
Nature Reviews Immunology, 17(4) pp. 219–232.
Li, C. H. and Amar, S. (2007) ‘Morphometric, Histomorphometric, and Microcomputed Tomographic
Analysis of Periodontal Inflammatory Lesions in a Murine Model.’ Journal of Periodontology, 78(6) pp.
1120–1128.
Li, L., Messas, E., Batista, E. L., Levine, R. A. and Amar, S. (2002) ‘Porphyromonas gingivalis Infection
Accelerates the Progression of Atherosclerosis in a Heterozygous Apolipoprotein E–Deficient Murine
Model.’ Circulation. 2002/02/21, 105(7) pp. 861–867.
Li, Q., Pan, C., Teng, D., Lin, L., Kou, Y., Haase, E. M., Scannapieco, F. A. and Pan, Y. (2014)
‘Porphyromonas gingivalis modulates Pseudomonas aeruginosa-induced apoptosis of respiratory epithelial
cells through the STAT3 signaling pathway.’ Microbes and Infection.
Liang, S., Hosur, K. B., Domon, H. and Hajishengallis, G. (2010) ‘Periodontal inflammation and bone loss in
aged mice.’ Journal of Periodontal Research. 2010/03/27, 45(4) pp. 574–578.
Liesz, A., Hagmann, S., Zschoche, C., Adamek, J., Zhou, W., Sun, L., Hug, A., Zorn, M., Dalpke, A.,
Nawroth, P. and Veltkamp, R. (2009a) ‘The Spectrum of Systemic Immune Alterations After Murine Focal
Ischemia.’ Stroke, 40(8) pp. 2849–2858.
Liesz, A., Suri-Payer, E., Veltkamp, C., Doerr, H., Sommer, C., Rivest, S., Giese, T. and Veltkamp, R.
(2009b) ‘Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke.’
Nature Medicine, 15(2) pp. 192–199.
Linehan, J. L., Harrison, O. J., Han, S.-J., Byrd, A. L., Vujkovic-Cvijin, I., Villarino, A. V., Sen, S. K., Shaik,
J., Smelkinson, M., Tamoutounour, S., Collins, N., Bouladoux, N., Dzutsev, A., Rosshart, S. P., Arbuckle, J.
H., Wang, C.-R., Kristie, T. M., Rehermann, B., Trinchieri, G., Brenchley, J. M., O’Shea, J. J. and Belkaid,
Y. (2018) ‘Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair.’ Cell,
172(4) p. 784–796.e18.
Liu, F., Schafer, D. P. and McCullough, L. D. (2009) ‘TTC, Fluoro-Jade B and NeuN staining confirm
evolving phases of infarction induced by middle cerebral artery occlusion.’ Journal of Neuroscience Methods,
179(1) pp. 1–8.
Liu, H., Redline, R. W. and Han, Y. W. (2007) ‘Fusobacterium nucleatum Induces Fetal Death in Mice via
Stimulation of TLR4-Mediated Placental Inflammatory Response.’ The Journal of Immunology, 179(4) pp.
2501–2508.
Liu, R., Desta, T., Raptis, M., Darveau, R. P. and Graves, D. T. (2008) ‘P. gingivalis and E. coli
Lipopolysaccharides Exhibit Different Systemic but Similar Local Induction of Inflammatory Markers.’
Journal of Periodontology, 79(7) pp. 1241–1247.
Livak, K. J. and Schmittgen, T. D. (2001) ‘Analysis of Relative Gene Expression Data Using Real-Time
Quantitative PCR and the 2−ΔΔCT Method.’ Methods, 25(4) pp. 402–408.

212
Llovera, G., Roth, S., Plesnila, N., Veltkamp, R. and Liesz, A. (2014) ‘Modeling Stroke in Mice: Permanent
Coagulation of the Distal Middle Cerebral Artery Video Link.’ J. Vis. Exp, (8910) pp. 517293791–51729.
Longa, E. Z., Weinstein, P. R., Carlson, S. and Cummins, R. (1989) ‘Reversible middle cerebral artery
occlusion without craniectomy in rats.’ Stroke. 1989/01/01, 20(1) pp. 84–91.
Loos, B. G. (2005) ‘Systemic Markers of Inflammation in Periodontitis.’ Journal of Periodontology.
2005/11/10, 76(11–s) pp. 2106–2115.
Loos, B. G., Craandijk, J., Hoek, F. J., Dillen, P. M. E. W. and Velden, U. Van Der (2000) ‘Elevation of
Systemic Markers Related to Cardiovascular Diseases in the Peripheral Blood of Periodontitis Patients.’
Journal of Periodontology, 71(10) pp. 1528–1534.
Lourenço, T. G. B., Heller, D., Silva-Boghossian, C. M., Cotton, S. L., Paster, B. J. and Colombo, A. P. V.
(2014) ‘Microbial signature profiles of periodontally healthy and diseased patients.’ Journal of Clinical
Periodontology, 41(11) pp. 1027–1036.
Love, M. I., Huber, W. and Anders, S. (2014) ‘Moderated estimation of fold change and dispersion for RNA-
seq data with DESeq2.’ Genome Biology, 15(12) p. 550.
Lu, H., Xu, M., Wang, F., Liu, S., Gu, J. and Lin, S. (2014) ‘Chronic stress enhances progression of
periodontitis via α1-adrenergic signaling: a potential target for periodontal disease therapy.’ Experimental &
Molecular Medicine, 46(10) pp. e118–e118.
Lu, H., Xu, M., Wang, F., Liu, S., Gu, J., Lin, S. and Zhao, L. (2016) ‘Chronic stress accelerates ligature-
induced periodontitis by suppressing glucocorticoid receptor-α signaling.’ Experimental & Molecular
Medicine. Nature Publishing Group, 48(3) p. e223.
Lund, H., Pieber, M., Parsa, R., Han, J., Grommisch, D., Ewing, E., Kular, L., Needhamsen, M., Espinosa,
A., Nilsson, E., Överby, A. K., Butovsky, O., Jagodic, M., Zhang, X.-M. and Harris, R. A. (2018)
‘Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like
cells.’ Nature Communications, 9(1) p. 4845.
Lundberg, K., Kinloch, A., Fisher, B. A., Wegner, N., Wait, R., Charles, P., Mikuls, T. R. and Venables, P. J.
(2008) ‘Antibodies to citrullinated α-enolase peptide 1 are specific for rheumatoid arthritis and cross-react
with bacterial enolase.’ Arthritis & Rheumatism, 58(10) pp. 3009–3019.
Lyu, J., Bian, T., Chen, B., Cui, D., Li, L., Gong, L. and Yan, F. (2017) ‘β-defensin 3 modulates macrophage
activation and orientation during acute inflammatory response to Porphyromonas gingivalis
lipopolysaccharide.’ Cytokine, 92, April, pp. 48–54.
Ma, S., Guo, J., You, X., Xia, W. and Yan, F. (2011) ‘Expressions of interleukin-1beta and interleukin-6
within aortas and uteri of rats with various severities of ligature-induced periodontitis.’ Inflammation.
2010/07/27, 34(4) pp. 260–268.
de Maat, M. P. and Kluft, C. (2001) ‘Determinants of C-reactive protein concentration in blood.’ Italian
Heart Journal. 2001/04/18, 2(3) pp. 189–195.
Madrigal, A. G., Barth, K., Papadopoulos, G. and Genco, C. A. (2012) ‘Pathogen-Mediated Proteolysis of the
Cell Death Regulator RIPK1 and the Host Defense Modulator RIPK2 in Human Aortic Endothelial Cells.’
Isberg, R. R. (ed.) PLoS Pathogens, 8(6) p. e1002723.
Maekawa, T., Krauss, J. L., Abe, T., Jotwani, R., Triantafilou, M., Triantafilou, K., Hashim, A., Hoch, S.,
Curtis, M. A., Nussbaum, G., Lambris, J. D. and Hajishengallis, G. (2014) ‘Porphyromonas gingivalis
Manipulates Complement and TLR Signaling to Uncouple Bacterial Clearance from Inflammation and
Promote Dysbiosis.’ Cell Host & Microbe. 2014/06/13, 15(6) pp. 768–778.
Malcolm, J., Awang, R. A., Oliver-Bell, J., Butcher, J. P., Campbell, L., Adrados Planell, A., Lappin, D. F.,
Fukada, S. Y., Nile, C. J., Liew, F. Y. and Culshaw, S. (2015) ‘IL-33 Exacerbates Periodontal Disease
through Induction of RANKL.’ Journal of Dental Research, 94(7) pp. 968–975.
Malcolm, J., Millington, O., Millhouse, E., Campbell, L., Adrados Planell, A., Butcher, J. P., Lawrence, C.,
Ross, K., Ramage, G., McInnes, I. B. and Culshaw, S. (2016) ‘Mast Cells Contribute to Porphyromonas
gingivalis– induced Bone Loss.’ Journal of Dental Research, 95(6) pp. 704–710.

213
Mann, M., Mehta, A., de Boer, C. G., Kowalczyk, M. S., Lee, K., Haldeman, P., Rogel, N., Knecht, A. R.,
Farouq, D., Regev, A. and Baltimore, D. (2018) ‘Heterogeneous Responses of Hematopoietic Stem Cells to
Inflammatory Stimuli Are Altered with Age.’ Cell Reports, 25(11) p. 2992–3005.e5.
Maresz, K. J., Hellvard, A., Sroka, A., Adamowicz, K., Bielecka, E., Koziel, J., Gawron, K., Mizgalska, D.,
Marcinska, K. A., Benedyk, M., Pyrc, K., Quirke, A.-M., Jonsson, R., Alzabin, S., Venables, P. J., Nguyen,
K.-A., Mydel, P. and Potempa, J. (2013) ‘Porphyromonas gingivalis Facilitates the Development and
Progression of Destructive Arthritis through Its Unique Bacterial Peptidylarginine Deiminase (PAD).’
Kazmierczak, B. I. (ed.) PLoS Pathogens, 9(9) p. e1003627.
Marsh, B., Stevens, S. L., Packard, A. E. B., Gopalan, B., Hunter, B., Leung, P. Y., Harrington, C. A. and
Stenzel-Poore, M. P. (2009) ‘Systemic Lipopolysaccharide Protects the Brain from Ischemic Injury by
Reprogramming the Response of the Brain to Stroke: A Critical Role for IRF3.’ Journal of Neuroscience,
29(31) pp. 9839–9849.
Marsh, P. D., Moter, A. and Devine, D. A. (2011) ‘Dental plaque biofilms: communities, conflict and
control.’ Periodontology 2000, 55(1) pp. 16–35.
Martuscelli, G., Fiorellini, J. P., Crohin, C. C. and Howard Howell, T. (2000) ‘The Effect of Interleukin-11 on
the Progression of Ligature-Induced Periodontal Disease in the Beagle Dog.’ Journal of Periodontology,
71(4) pp. 573–578.
Massberg, S., Schaerli, P., Knezevic-Maramica, I., Köllnberger, M., Tubo, N., Moseman, E. A., Huff, I. V.,
Junt, T., Wagers, A. J., Mazo, I. B. and von Andrian, U. H. (2007) ‘Immunosurveillance by Hematopoietic
Progenitor Cells Trafficking through Blood, Lymph, and Peripheral Tissues.’ Cell. Cell Press, 131(5) pp.
994–1008.
Matsuda, Y., Kato, T., Takahashi, N., Nakajima, M., Arimatsu, K., Minagawa, T., Sato, K., Ohno, H. and
Yamazaki, K. (2016) ‘Ligature-induced periodontitis in mice induces elevated levels of circulating
interleukin-6 but shows only weak effects on adipose and liver tissues.’ Journal of Periodontal Research,
51(5) pp. 639–646.
Matzumura-Kuan, M. and Jennings, J. (2014) ‘Aggregatibacter actinomycetemcomitans infection mimicking
lung cancer: A case report.’ Scandinavian Journal of Infectious Diseases. 2014/06/10, 46(9) pp. 669–672.
Maysami, S., Haley, M. J., Gorenkova, N., Krishnan, S., McColl, B. W. and Lawrence, C. B. (2015)
‘Prolonged diet-induced obesity in mice modifies the inflammatory response and leads to worse outcome
after stroke.’ Journal of neuroinflammation. Journal of Neuroinflammation, 12 p. 140.
Mazo, I. B., Massberg, S. and von Andrian, U. H. (2011) ‘Hematopoietic stem and progenitor cell
trafficking.’ Trends in Immunology. Elsevier Current Trends, 32(10) pp. 493–503.
McColl, B. W., Allan, S. M. and Rothwell, N. J. (2009) ‘Systemic infection, inflammation and acute ischemic
stroke.’ Neuroscience, 158(3) pp. 1049–1061.
McColl, B. W., Carswell, H. V., McCulloch, J. and Horsburgh, K. (2004) ‘Extension of cerebral
hypoperfusion and ischaemic pathology beyond MCA territory after intraluminal filament occlusion in
C57Bl/6J mice.’ Brain Research. Elsevier, 997(1) pp. 15–23.
McColl, B. W., Rose, N., Robson, F. H., Rothwell, N. J. and Lawrence, C. B. (2010) ‘Increased Brain
Microvascular MMP-9 and Incidence of Haemorrhagic Transformation in Obese Mice after Experimental
Stroke.’ Journal of Cerebral Blood Flow & Metabolism, 30(2) pp. 267–272.
McColl, B. W., Rothwell, N. J. and Allan, S. M. (2007) ‘Systemic Inflammatory Stimulus Potentiates the
Acute Phase and CXC Chemokine Responses to Experimental Stroke and Exacerbates Brain Damage via
Interleukin-1- and Neutrophil-Dependent Mechanisms.’ Journal of Neuroscience. 2007/04/20, 27(16) pp.
4403–4412.
McColl, B. W., Rothwell, N. J. and Allan, S. M. (2008) ‘Systemic inflammation alters the kinetics of
cerebrovascular tight junction disruption after experimental stroke in mice.’ Journal of Neuroscience, 28(38)
pp. 9451–9462.
McCulloch, L., Smith, C. J. and McColl, B. W. (2017) ‘Adrenergic-mediated loss of splenic marginal zone B
cells contributes to infection susceptibility after stroke.’ Nature Communications. Nature Publishing Group,
8(D) p. 15051.

214
McKinney-Freeman, S. L., Jackson, K. A., Camargo, F. D., Ferrari, G., Mavilio, F. and Goodell, M. A.
(2002) ‘Muscle-derived hematopoietic stem cells are hematopoietic in origin.’ Proceedings of the National
Academy of Sciences, 99(3) pp. 1341–1346.
Méndez-Ferrer, S., Michurina, T. V., Ferraro, F., Mazloom, A. R., MacArthur, B. D., Lira, S. A., Scadden, D.
T., Ma’ayan, A., Enikolopov, G. N. and Frenette, P. S. (2010) ‘Mesenchymal and haematopoietic stem cells
form a unique bone marrow niche.’ Nature, 466(7308) pp. 829–834.
Meyer, D. H. and Fives-Taylor, P. M. (1997) ‘The role of Actinobacillus actinomycetemcomitans in the
pathogenesis of periodontal disease.’ Trends in Microbiology. 1997/06/01, 5(6) pp. 224–228.
Michaud, D. S., Lu, J., Peacock-Villada, A. Y., Barber, J. R., Joshu, C. E., Prizment, A. E., Beck, J. D.,
Offenbacher, S. and Platz, E. A. (2018) ‘Periodontal Disease Assessed Using Clinical Dental Measurements
and Cancer Risk in the ARIC Study.’ JNCI: Journal of the National Cancer Institute, 110(8) pp. 843–854.
Miles, B., Zakhary, I., El-Awady, A., Scisci, E., Carrion, J., O’Neill, J. C., Rawlings, A., Stern, J. K., Susin,
C. and Cutler, C. W. (2014) ‘Secondary Lymphoid Organ Homing Phenotype of Human Myeloid Dendritic
Cells Disrupted by an Intracellular Oral Pathogen.’ Blanke, S. R. (ed.) Infection and Immunity, 82(1) pp. 101–
111.
Mishiro, K., Imai, T., Sugitani, S., Kitashoji, A., Suzuki, Y., Takagi, T., Chen, H., Oumi, Y., Tsuruma, K.,
Shimazawa, M. and Hara, H. (2014) ‘Diabetes Mellitus Aggravates Hemorrhagic Transformation after
Ischemic Stroke via Mitochondrial Defects Leading to Endothelial Apoptosis.’ Drossopoulou, G. (ed.) PLoS
ONE, 9(8) p. e103818.
Mitroulis, I., Ruppova, K., Wang, B., Chen, L.-S., Grzybek, M., Grinenko, T., Eugster, A., Troullinaki, M.,
Palladini, A., Kourtzelis, I., Chatzigeorgiou, A., Schlitzer, A., Beyer, M., Joosten, L. A. B., Isermann, B.,
Lesche, M., Petzold, A., Simons, K., Henry, I., Dahl, A., Schultze, J. L., Wielockx, B., Zamboni, N.,
Mirtschink, P., Coskun, Ü., Hajishengallis, G., Netea, M. G. and Chavakis, T. (2018) ‘Modulation of
Myelopoiesis Progenitors Is an Integral Component of Trained Immunity.’ Cell. Cell Press, 172(1–2) p. 147–
161.e12.
Miyajima, S., Naruse, K., Kobayashi, Y., Nakamura, N., Nishikawa, T., Adachi, K., Suzuki, Y., Kikuchi, T.,
Mitani, A., Mizutani, M., Ohno, N., Noguchi, T. and Matsubara, T. (2015) ‘Periodontitis-activated
monocytes/macrophages cause aortic inflammation.’ Scientific Reports. 2014/06/05, 4(1) p. 5171.
Miyakawa, H., Honma, K., Qi, M. and Kuramitsu, H. K. (2004) ‘Interaction of Porphyromonas gingivalis
with low-density lipoproteins: implications for a role for periodontitis in atherosclerosis.’ Journal of
Periodontal Research, 39(1) pp. 1–9.
Moen, K., Brun, J. G., Valen, M., Skartveit, L., Eribe, E. K. R., Olsen, I. and Jonsson, R. (2006) ‘Synovial
inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral
bacterial DNAs.’ Clinical and Experimental Rheumatology, 24(6) pp. 656–63.
Mojon, P. (2002) ‘Oral health and respiratory infection.’ Journal of the Canadian Dental Association.
2002/05/30, 68(6) pp. 340–345.
Molawi, K., Wolf, Y., Kandalla, P. K., Favret, J., Hagemeyer, N., Frenzel, K., Pinto, A. R., Klapproth, K.,
Henri, S., Malissen, B., Rodewald, H.-R., Rosenthal, N. A., Bajenoff, M., Prinz, M., Jung, S. and Sieweke,
M. H. (2014) ‘Progressive replacement of embryo-derived cardiac macrophages with age.’ The Journal of
Experimental Medicine, 211(11) pp. 2151–2158.
de Molon, R. S., de Avila, E. D., Boas Nogueira, A. V., Chaves de Souza, J. A., Avila-Campos, M. J., de
Andrade, C. R. and Cirelli, J. A. (2014) ‘Evaluation of the Host Response in Various Models of Induced
Periodontal Disease in Mice.’ Journal of Periodontology. 2013/06/29, 85(3) pp. 465–477.
de Molon, R. S., Mascarenhas, V. I., de Avila, E. D., Finoti, L. S., Toffoli, G. B., Spolidorio, D. M. P.,
Scarel-Caminaga, R. M., Tetradis, S. and Cirelli, J. A. (2016) ‘Long-term evaluation of oral gavage with
periodontopathogens or ligature induction of experimental periodontal disease in mice.’ Clinical Oral
Investigations, 20(6) pp. 1203–1216.
Mootha, V. K., Lindgren, C. M., Eriksson, K.-F., Subramanian, A., Sihag, S., Lehar, J., Puigserver, P.,
Carlsson, E., Ridderstråle, M., Laurila, E., Houstis, N., Daly, M. J., Patterson, N., Mesirov, J. P., Golub, T.
R., Tamayo, P., Spiegelman, B., Lander, E. S., Hirschhorn, J. N., Altshuler, D. and Groop, L. C. (2003)
‘PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human

215
diabetes.’ Nature Genetics, 34(3) pp. 267–273.
Morris, J. F. and Sewell, D. L. (1994) ‘Necrotizing pneumonia caused by mixed infection with Actinobacillus
actinomycetemcomitans and Actinomyces israelii: case report and review.’ Clinical Infectious Diseases.
1994/03/01, 18(3) pp. 450–452.
Mortha, A., Chudnovskiy, A., Hashimoto, D., Bogunovic, M., Spencer, S. P., Belkaid, Y. and Merad, M.
(2014) ‘Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis.’
Science (New York, N.Y.), 343(6178) p. 1249288.
Moskowitz, M. A., Lo, E. H. and Iadecola, C. (2010) ‘The Science of Stroke: Mechanisms in Search of
Treatments.’ Neuron, 67(2) pp. 181–198.
Mossadegh-Keller, N., Sarrazin, S., Kandalla, P. K., Espinosa, L., Stanley, E. R., Nutt, S. L., Moore, J. and
Sieweke, M. H. (2013) ‘M-CSF instructs myeloid lineage fate in single haematopoietic stem cells.’ Nature.
Nature Publishing Group, 497(7448) pp. 239–243.
Moutsopoulos, N. M. and Konkel, J. E. (2018) ‘Tissue-Specific Immunity at the Oral Mucosal Barrier.’
Trends in Immunology, 39(4) pp. 276–287.
Moutsopoulos, N. M., Konkel, J., Sarmadi, M., Eskan, M. A., Wild, T., Dutzan, N., Abusleme, L., Zenobia,
C., Hosur, K. B., Abe, T., Uzel, G., Chen, W., Chavakis, T., Holland, S. M. and Hajishengallis, G. (2014)
‘Defective Neutrophil Recruitment in Leukocyte Adhesion Deficiency Type I Disease Causes Local IL-17-
Driven Inflammatory Bone Loss.’ Science Translational Medicine. American Association for the
Advancement of Science, 6(229) p. 229ra40-229ra40.
Moutsopoulos, N. M. and Madianos, P. N. (2006) ‘Low-Grade Inflammation in Chronic Infectious Diseases:
Paradigm of Periodontal Infections.’ Annals of the New York Academy of Sciences. 2006/12/29, 1088(1) pp.
251–264.
Moutsopoulos, N. M., Zerbe, C. S., Wild, T., Dutzan, N., Brenchley, L., DiPasquale, G., Uzel, G., Axelrod,
K. C., Lisco, A., Notarangelo, L. D., Hajishengallis, G., Notarangelo, L. D. and Holland, S. M. (2017)
‘Interleukin-12 and Interleukin-23 Blockade in Leukocyte Adhesion Deficiency Type 1.’ New England
Journal of Medicine, 376(12) pp. 1141–1146.
Na, S. Y., Mracsko, E., Liesz, A., Hünig, T. and Veltkamp, R. (2015) ‘Amplification of regulatory T cells
using a CD28 superagonist reduces brain damage after ischemic stroke in mice.’ Stroke, 46(1) pp. 212–220.
Nadkarni, M. A., Jacques, N. A., Martin, F. E. and Hunter, N. (2002) ‘Determination of bacterial load by real-
time PCR using a broad-range (universal) probe and primers set.’ Microbiology, 148(1) pp. 257–266.
Nagai, Y., Garrett, K. P., Ohta, S., Bahrun, U., Kouro, T., Akira, S., Takatsu, K. and Kincade, P. W. (2006)
‘Toll-like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment.’
Immunity, 24(6) pp. 801–812.
Nagasawa, T., Kobayashi, H., Aramaki, M., Kiji, M., Oda, S. and Izumi, Y. (2004) ‘Expression of CD14,
CD16 and CD45RA on monocytes from periodontitis patients.’ Journal of Periodontal Research, 39(1) pp.
72–78.
Nahid, M. A., Rivera, M., Lucas, A., Chan, E. K. L. and Kesavalu, L. (2011) ‘Polymicrobial Infection with
Periodontal Pathogens Specifically Enhances MicroRNA miR-146a in ApoE −/− Mice during Experimental
Periodontal Disease.’ Blanke, S. R. (ed.) Infection and Immunity, 79(4) pp. 1597–1605.
Naik, S., Bouladoux, N., Wilhelm, C., Molloy, M. J., Salcedo, R., Kastenmuller, W., Deming, C., Quinones,
M., Koo, L., Conlan, S., Spencer, S., Hall, J. A., Dzutsev, A., Kong, H., Campbell, D. J., Trinchieri, G.,
Segre, J. A. and Belkaid, Y. (2012) ‘Compartmentalized Control of Skin Immunity by Resident
Commensals.’ Science, 337(6098) pp. 1115–1119.
Nakano, K., Nemoto, H., Nomura, R., Inaba, H., Yoshioka, H., Taniguchi, K., Amano, A. and Ooshima, T.
(2009) ‘Detection of oral bacteria in cardiovascular specimens.’ Oral Microbiology and Immunology, 24(1)
pp. 64–68.
Nakayama, K. (2010) ‘Porphyromonas gingivalis cell-induced hemagglutination and platelet aggregation.’
Periodontology 2000, 54(1) pp. 45–52.

216
Neuhaus, A. A., Rabie, T., Sutherland, B. A., Papadakis, M., Hadley, G., Cai, R. and Buchan, A. M. (2014)
‘Importance of Preclinical Research in the Development of Neuroprotective Strategies for Ischemic Stroke.’
JAMA Neurology. American Medical Association, 71(5) p. 634.
Neumann, J., Riek-Burchardt, M., Herz, J., Doeppner, T. R., König, R., Hütten, H., Etemire, E., Männ, L.,
Klingberg, A., Fischer, T., Görtler, M. W., Heinze, H.-J., Reichardt, P., Schraven, B., Hermann, D. M.,
Reymann, K. G. and Gunzer, M. (2015) ‘Very-late-antigen-4 (VLA-4)-mediated brain invasion by
neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in
experimental stroke.’ Acta Neuropathologica, 129(2) pp. 259–277.
Ng, Y. S., Stein, J., Ning, M. and Black-Schaffer, R. M. (2007) ‘Comparison of Clinical Characteristics and
Functional Outcomes of Ischemic Stroke in Different Vascular Territories.’ Stroke, 38(8) pp. 2309–2314.
Noack, B., Genco, R. J., Trevisan, M., Grossi, S., Zambon, J. J. and Nardin, E. De (2001) ‘Periodontal
Infections Contribute to Elevated Systemic C-Reactive Protein Level.’ Journal of Periodontology, 72(9) pp.
1221–1227.
Nociti, F. H., Casati, M. Z. and Duarte, P. M. (2015) ‘Current perspective of the impact of smoking on the
progression and treatment of periodontitis.’ Periodontology 2000, 67(1) pp. 187–210.
Noguchi, K. and Ishikawa, I. (2007) ‘The roles of cyclooxygenase-2 and prostaglandin E2 in periodontal
disease.’ Periodontology 2000. John Wiley & Sons, Ltd (10.1111), 43(1) pp. 85–101.
Novakovic, B., Habibi, E., Wang, S.-Y., Arts, R. J. W., Davar, R., Megchelenbrink, W., Kim, B.,
Kuznetsova, T., Kox, M., Zwaag, J., Matarese, F., van Heeringen, S. J., Janssen-Megens, E. M., Sharifi, N.,
Wang, C., Keramati, F., Schoonenberg, V., Flicek, P., Clarke, L., Pickkers, P., Heath, S., Gut, I., Netea, M.
G., Martens, J. H. A., Logie, C. and Stunnenberg, H. G. (2016) ‘β-Glucan Reverses the Epigenetic State of
LPS-Induced Immunological Tolerance.’ Cell. Elsevier, 167(5) p. 1354–1368.e14.
Nussbaum, G. and Shapira, L. (2011) ‘How has neutrophil research improved our understanding of
periodontal pathogenesis?’ Journal of Clinical Periodontology. 2011/02/26, 38, March, pp. 49–59.
O’Collins, V. E., Donnan, G. A., Macleod, M. R. and Howells, D. W. (2013) ‘Hypertension and
Experimental Stroke Therapies.’ Journal of Cerebral Blood Flow & Metabolism, 33(8) pp. 1141–1147.
Offenbacher, S., Beck, J. D., Moss, K., Mendoza, L., Paquette, D. W., Barrow, D. A., Couper, D. J., Stewart,
D. D., Falkner, K. L., Graham, S. P., Grossi, S., Gunsolley, J. C., Madden, T., Maupome, G., Trevisan, M.,
Van Dyke, T. E. and Genco, R. J. (2009) ‘Results From the Periodontitis and Vascular Events (PAVE) Study:
A Pilot Multicentered, Randomized, Controlled Trial to Study Effects of Periodontal Therapy in a Secondary
Prevention Model of Cardiovascular Disease.’ Journal of Periodontology. Wiley-Blackwell, 80(2) pp. 190–
201.
Ogrendik, M. (2006) ‘Treatment of rheumatoid arthritis with ornidazole: a randomized, double-blind,
placebo-controlled study.’ Rheumatology International, 26(12) pp. 1132–1137.
Ogrendik, M. (2007a) ‘Effects of clarithromycin in patients with active rheumatoid arthritis.’ Current
Medical Research and Opinion, 23(3) pp. 515–522.
Ogrendik, M. (2007b) ‘Levofloxacin Treatment in Patients with Rheumatoid Arthritis Receiving
Methotrexate.’ Southern Medical Journal, 100(2) pp. 135–139.
Ogrendik, M. (2009) ‘Rheumatoid arthritis is linked to oral bacteria: etiological association.’ Modern
Rheumatology, 19(5) pp. 453–456.
Ogrendik, M. (2013) ‘Rheumatoid arthritis is an autoimmune disease caused by periodontal pathogens.’
International Journal of General Medicine, 6, May, p. 383.
Ogrendik, M., Kokino, S., Ozdemir, F., Bird, P. S. and Hamlet, S. (2005) ‘Serum antibodies to oral anaerobic
bacteria in patients with rheumatoid arthritis.’ Medscape General Medicine, 7(2) p. 2.
de Oliveira, C., Watt, R. and Hamer, M. (2010) ‘Toothbrushing, inflammation, and risk of cardiovascular
disease: results from Scottish Health Survey.’ BMJ, 340(may27 1) pp. c2451–c2451.
Oliver-Bell, J., Butcher, J. P., Malcolm, J., MacLeod, M. K. L., Adrados Planell, A., Campbell, L., Nibbs, R.
J. B., Garside, P., McInnes, I. B. and Culshaw, S. (2015) ‘Periodontitis in the absence of B cells and specific

217
anti-bacterial antibody.’ Molecular Oral Microbiology, 30(2) pp. 160–169.
Orth, R. .-H., O’Brien-Simpson, N. M., Dashper, S. G. and Reynolds, E. C. (2011) ‘Synergistic virulence of
Porphyromonas gingivalis and Treponema denticola in a murine periodontitis model.’ Molecular Oral
Microbiology, 26(4) pp. 229–240.
Ortiz, P., Bissada, N. F., Palomo, L., Han, Y. W., Al-Zahrani, M. S., Panneerselvam, A. and Askari, A.
(2009) ‘Periodontal Therapy Reduces the Severity of Active Rheumatoid Arthritis in Patients Treated With or
Without Tumor Necrosis Factor Inhibitors.’ Journal of Periodontology, 80(4) pp. 535–540.
Osborne, K. A., Shigeno, T., Balarsky, A. M., Ford, I., McCulloch, J., Teasdale, G. M. and Graham, D. I.
(1987) ‘Quantitative assessment of early brain damage in a rat model of focal cerebral ischaemia.’ Journal of
Neurology, Neurosurgery & Psychiatry, 50(4) pp. 402–410.
Pan, Y., Teng, D., Burke, A. C., Haase, E. M. and Scannapieco, F. A. (2009) ‘Oral bacteria modulate
invasion and induction of apoptosis in HEp-2 cells by Pseudomonas aeruginosa.’ Microbial Pathogenesis,
46(2) pp. 73–79.
Paraskevas, S., Huizinga, J. D. and Loos, B. G. (2008) ‘A systematic review and meta-analyses on C-reactive
protein in relation to periodontitis.’ Journal of Clinical Periodontology, 35(4) pp. 277–290.
Paxinos, G. and Franklin, K. B. J. (2001) The mouse brain in stereotaxic coordinates. Academic Press.
Second Edi, New York: Academic Press.
Peng, C.-H., Yang, Y.-S., Chan, K.-C., Kornelius, E., Chiou, J.-Y. and Huang, C.-N. (2017) ‘Periodontal
Treatment and the Risks of Cardiovascular Disease in Patients with Type 2 Diabetes: A Retrospective Cohort
Study.’ Internal Medicine, 56(9) pp. 1015–1021.
Perry, V. H., Cunningham, C. and Holmes, C. (2007) ‘Systemic infections and inflammation affect chronic
neurodegeneration.’ Nature Reviews Immunology. 2007/01/16, 7(2) pp. 161–167.
Perschinka, H., Mayr, M., Millonig, G., Mayerl, C., van der Zee, R., Morrison, S. G., Morrison, R. P., Xu, Q.
and Wick, G. (2003) ‘Cross-Reactive B-Cell Epitopes of Microbial and Human Heat Shock Protein 60/65 in
Atherosclerosis.’ Arteriosclerosis, Thrombosis, and Vascular Biology, 23(6) pp. 1060–1065.
Petcu, E., Kocher, T., Kuhr, A., Buga, A.-M., Kloting, I., Herndon, J., Kessler, C. and Popa-Wagner, A.
(2008) ‘Mild Systemic Inflammation has a Neuroprotective Effect After Stroke in Rats.’ Current
Neurovascular Research. 2008/11/11, 5(4) pp. 214–223.
Petersen, P. E. and Ogawa, H. (2005) ‘Strengthening the Prevention of Periodontal Disease: The WHO
Approach.’ Journal of Periodontology, 76(12) pp. 2187–2193.
Pietras, E. M., Reynaud, D., Kang, Y.-A., Carlin, D., Calero-Nieto, F. J., Leavitt, A. D., Stuart, J. M.,
Gottgens, B. and Passegué, E. (2015) ‘Functionally Distinct Subsets of Lineage-Biased Multipotent
Progenitors Control Blood Production in Normal and Regenerative Conditions.’ Cell Stem Cell, 17(1) pp. 35–
46.
Polak, D., Wilensky, A., Shapira, L., Halabi, A., Goldstein, D., Weiss, E. I. and Houri-Haddad, Y. (2009)
‘Mouse model of experimental periodontitis induced by Porphyromonas gingivalis / Fusobacterium
nucleatum infection: bone loss and host response.’ Journal of Clinical Periodontology, 36(5) pp. 406–410.
Poole, S., Singhrao, S. K., Chukkapalli, S., Rivera, M., Velsko, I., Kesavalu, L. and Crean, S. (2014) ‘Active
invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains.’
Journal of Alzheimer’s Disease. 2014/07/26, 43(1) pp. 67–80.
Popadiak, K., Potempa, J., Riesbeck, K. and Blom, A. M. (2007) ‘Biphasic Effect of Gingipains from
Porphyromonas gingivalis on the Human Complement System.’ The Journal of Immunology, 178(11) pp.
7242–7250.
Potempa, M., Potempa, J., Kantyka, T., Nguyen, K.-A., Wawrzonek, K., Manandhar, S. P., Popadiak, K.,
Riesbeck, K., Eick, S. and Blom, A. M. (2009) ‘Interpain A, a Cysteine Proteinase from Prevotella
intermedia, Inhibits Complement by Degrading Complement Factor C3.’ von Pawel-Rammingen, U. (ed.)
PLoS Pathogens, 5(2) p. e1000316.
Prass, K., Braun, J. S., Dirnagl, U., Meisel, C. and Meisel, A. (2006) ‘Stroke Propagates Bacterial Aspiration

218
to Pneumonia in a Model of Cerebral Ischemia.’
Prass, K., Meisel, C., Höflich, C., Braun, J., Halle, E., Wolf, T., Ruscher, K., Victorov, I. V, Priller, J.,
Dirnagl, U., Volk, H.-D. and Meisel, A. (2003) ‘Stroke-induced Immunodeficiency Promotes Spontaneous
Bacterial Infections and Is Mediated by Sympathetic Activation Reversal by Poststroke T Helper Cell Type
1–like Immunostimulation.’ The Journal of Experimental Medicine, 198(5) pp. 725–736.
Pulendran, B., Kumar, P., Cutler, C. W., Mohamadzadeh, M., Van Dyke, T. and Banchereau, J. (2001)
‘Lipopolysaccharides from Distinct Pathogens Induce Different Classes of Immune Responses In Vivo.’ The
Journal of Immunology, 167(9) pp. 5067–5076.
Pussinen, P. J., Alfthan, G., Jousilahti, P., Paju, S. and Tuomilehto, J. (2007) ‘Systemic exposure to
Porphyromonas gingivalis predicts incident stroke.’ Atherosclerosis, 193(1) pp. 222–228.
Qi, M., Miyakawa, H. and Kuramitsu, H. K. (2003) ‘Porphyromonas gingivalis induces murine macrophage
foam cell formation.’ Microbial Pathogenesis, 35(6) pp. 259–267.
Quintin, J., Saeed, S., Martens, J. H. A., Giamarellos-Bourboulis, E. J., Ifrim, D. C., Logie, C., Jacobs, L.,
Jansen, T., Kullberg, B.-J., Wijmenga, C., Joosten, L. A. B., Xavier, R. J., van der Meer, J. W. M.,
Stunnenberg, H. G. and Netea, M. G. (2012) ‘Candida albicans Infection Affords Protection against
Reinfection via Functional Reprogramming of Monocytes.’ Cell Host & Microbe, 12(2) pp. 223–232.
Rando, T. A. (2006) ‘Stem cells, ageing and the quest for immortality.’ Nature, 441(7097) pp. 1080–1086.
Ren, X., Akiyoshi, K., Dziennis, S., Vandenbark, A. A., Herson, P. S., Hurn, P. D. and Offner, H. (2011a)
‘Regulatory B Cells Limit CNS Inflammation and Neurologic Deficits in Murine Experimental Stroke.’
Journal of Neuroscience, 31(23) pp. 8556–8563.
Ren, X., Akiyoshi, K., Vandenbark, A. A., Hurn, P. D. and Offner, H. (2011b) ‘CD4+FoxP3+ regulatory T-
cells in cerebral ischemic stroke.’ Metabolic Brain Disease, 26(1) pp. 87–90.
Reyes, L., Herrera, D., Kozarov, E., Roldán, S. and Progulske-Fox, A. (2013) ‘Periodontal bacterial invasion
and infection: contribution to atherosclerotic pathology.’ Journal of Clinical Periodontology, 40, April, pp.
S30–S50.
Rivera, M. F., Lee, J.-Y., Aneja, M., Goswami, V., Liu, L., Velsko, I. M., Chukkapalli, S. S., Bhattacharyya,
I., Chen, H., Lucas, A. R. and Kesavalu, L. N. (2013) ‘Polymicrobial Infection with Major Periodontal
Pathogens Induced Periodontal Disease and Aortic Atherosclerosis in Hyperlipidemic ApoEnull Mice.’
Seshu, J. (ed.) PLoS ONE, 8(2) p. e57178.
Riviere, G. R., Riviere, K. H. and Smith, K. S. (2002) ‘Molecular and immunological evidence of oral
Treponema in the human brain and their association with Alzheimer’s disease.’ Oral Microbiology and
Immunology. 2002/04/04, 17(2) pp. 113–118.
Robbins, C. S., Chudnovskiy, A., Rauch, P. J., Figueiredo, J.-L., Iwamoto, Y., Gorbatov, R., Etzrodt, M.,
Weber, G. F., Ueno, T., van Rooijen, N., Mulligan-Kehoe, M. J., Libby, P., Nahrendorf, M., Pittet, M. J.,
Weissleder, R. and Swirski, F. K. (2012) ‘Extramedullary Hematopoiesis Generates Ly-6C high Monocytes
That Infiltrate Atherosclerotic Lesions.’ Circulation, 125(2) pp. 364–374.
Rodríguez, E. M., Blázquez, J. L. and Guerra, M. (2010) ‘The design of barriers in the hypothalamus allows
the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood
and the latter to the cerebrospinal fluid.’ Peptides, 31(4) pp. 757–776.
Rodriguez, S., Chora, A., Goumnerov, B., Mumaw, C., Goebel, W. S., Fernandez, L., Baydoun, H.,
HogenEsch, H., Dombkowski, D. M., Karlewicz, C. A., Rice, S., Rahme, L. G. and Carlesso, N. (2009)
‘Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis.’
Blood, 114(19) pp. 4064–4076.
Rosenzweig, H. L., Lessov, N. S., Henshall, D. C., Minami, M., Simon, R. P. and Stenzel-Poore, M. P. (2004)
‘Endotoxin Preconditioning Prevents Cellular Inflammatory Response During Ischemic Neuroprotection in
Mice.’ Stroke, 35(11) pp. 2576–2581.
Rossi, D. J., Bryder, D., Zahn, J. M., Ahlenius, H., Sonu, R., Wagers, A. J. and Weissman, I. L. (2005) ‘Cell
intrinsic alterations underlie hematopoietic stem cell aging.’ Proceedings of the National Academy of
Sciences, 102(26) pp. 9194–9199.

219
Roth, S., Singh, V., Tiedt, S., Schindler, L., Huber, G., Geerlof, A., Antoine, D. J., Anfray, A., Orset, C.,
Gauberti, M., Fournier, A., Holdt, L. M., Harris, H. E., Engelhardt, B., Bianchi, M. E., Vivien, D., Haffner,
C., Bernhagen, J., Dichgans, M. and Liesz, A. (2018) ‘Brain-released alarmins and stress response synergize
in accelerating atherosclerosis progression after stroke.’ Science Translational Medicine, 10(14).
Rothhammer, V., Borucki, D. M., Tjon, E. C., Takenaka, M. C., Chao, C.-C., Ardura-Fabregat, A., de Lima,
K. A., Gutiérrez-Vázquez, C., Hewson, P., Staszewski, O., Blain, M., Healy, L., Neziraj, T., Borio, M.,
Wheeler, M., Dragin, L. L., Laplaud, D. A., Antel, J., Alvarez, J. I., Prinz, M. and Quintana, F. J. (2018)
‘Microglial control of astrocytes in response to microbial metabolites.’ Nature, 557(7707) pp. 724–728.
Rubinstein, M. R., Wang, X., Liu, W., Hao, Y., Cai, G. and Han, Y. W. (2013) ‘Fusobacterium nucleatum
Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin.’
Cell Host & Microbe, 14(2) pp. 195–206.
Saadi-Thiers, K., Huck, O., Simonis, P., Tilly, P., Fabre, J.-E., Tenenbaum, H. and Davideau, J.-L. (2013)
‘Periodontal and Systemic Responses in Various Mice Models of Experimental Periodontitis: Respective
Roles of Inflammation Duration and Porphyromonas gingivalis Infection.’ Journal of Periodontology.
2012/06/05, 84(3) pp. 396–406.
Saeed, S., Quintin, J., Kerstens, H. H. D., Rao, N. A., Aghajanirefah, A., Matarese, F., Cheng, S.-C., Ratter,
J., Berentsen, K., van der Ent, M. A., Sharifi, N., Janssen-Megens, E. M., Ter Huurne, M., Mandoli, A., van
Schaik, T., Ng, A., Burden, F., Downes, K., Frontini, M., Kumar, V., Giamarellos-Bourboulis, E. J.,
Ouwehand, W. H., van der Meer, J. W. M., Joosten, L. A. B., Wijmenga, C., Martens, J. H. A., Xavier, R. J.,
Logie, C., Netea, M. G. and Stunnenberg, H. G. (2014) ‘Epigenetic programming of monocyte-to-
macrophage differentiation and trained innate immunity.’ Science, 345(6204) pp. 1251086–1251086.
Saenz, S. A., Siracusa, M. C., Perrigoue, J. G., Spencer, S. P., Urban Jr, J. F., Tocker, J. E., Budelsky, A. L.,
Kleinschek, M. A., Kastelein, R. A., Kambayashi, T., Bhandoola, A. and Artis, D. (2010) ‘IL25 elicits a
multipotent progenitor cell population that promotes TH2 cytokine responses.’ Nature, 464(7293) pp. 1362–
1366.
Le Sage, F., Meilhac, O. and Gonthier, M.-P. (2017) ‘Porphyromonas gingivalis lipopolysaccharide induces
pro-inflammatory adipokine secretion and oxidative stress by regulating Toll-like receptor-mediated signaling
pathways and redox enzymes in adipocytes.’ Molecular and Cellular Endocrinology, 446, May, pp. 102–110.
Sahingur, S. E., Sharma, A., Genco, R. J. and De Nardin, E. (2003) ‘Association of Increased Levels of
Fibrinogen and the –455G/A Fibrinogen Gene Polymorphism with Chronic Periodontitis.’ Journal of
Periodontology, 74(3) pp. 329–337.
Sattar, N., McCarey, D. W., Capell, H. and McInnes, I. B. (2003) ‘Explaining How “High-Grade” Systemic
Inflammation Accelerates Vascular Risk in Rheumatoid Arthritis.’ Circulation, 108(24) pp. 2957–2963.
Savioli, C., Ribeiro, A. C. M., Fabri, G. M. C., Calich, A. L., Carvalho, J., Silva, C. a, Viana, V. S. T., Bonfá,
E. and Siqueira, J. T. T. (2012) ‘Persistent Periodontal Disease Hampers Anti–Tumor Necrosis Factor
Treatment Response in Rheumatoid Arthritis.’ JCR: Journal of Clinical Rheumatology, May, p. 1.
Schenkein, H. A., Berry, C. R., Burmeister, J. A., Brooks, C. N., Barbour, S. E., Best, A. M. and Tew, J. G.
(2003) ‘Anti-cardiolipin Antibodies in Sera from Patients with Periodontitis.’ Journal of Dental Research,
82(11) pp. 919–922.
Schenkein, H. A., Bradley, J. L. and Purkall, D. B. (2013) ‘Anticardiolipin in Porphyromonas gingivalis
Antisera Causes Fetal Loss in Mice.’ Journal of Dental Research, 92(9) pp. 814–818.
Schenkein, H. A., Koertge, T. E., Brooks, C. N., Sabatini, R., Purkall, D. E. and Tew, J. G. (2010) ‘IL-17 in
Sera from Patients with Aggressive Periodontitis.’ Journal of Dental Research, 89(9) pp. 943–947.
Schwarzenberger, P., Huang, W., Ye, P., Oliver, P., Manuel, M., Zhang, Z., Bagby, G., Nelson, S. and Kolls,
J. K. (2000) ‘Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-
17-mediated granulopoiesis.’ Journal of Immunology, 164(9) pp. 4783–9.
Scott, C. L., Zheng, F., De Baetselier, P., Martens, L., Saeys, Y., De Prijck, S., Lippens, S., Abels, C.,
Schoonooghe, S., Raes, G., Devoogdt, N., Lambrecht, B. N., Beschin, A. and Guilliams, M. (2016) ‘Bone
marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells.’ Nature
Communications, 7(1) p. 10321.

220
Scott, N. A., Andrusaite, A., Andersen, P., Lawson, M., Alcon-Giner, C., Leclaire, C., Caim, S., Le Gall, G.,
Shaw, T., Connolly, J. P. R., Roe, A. J., Wessel, H., Bravo-Blas, A., Thomson, C. A., Kästele, V., Wang, P.,
Peterson, D. A., Bancroft, A., Li, X., Grencis, R., Mowat, A. M., Hall, L. J., Travis, M. A., Milling, S. W. F.
and Mann, E. R. (2018) ‘Antibiotics induce sustained dysregulation of intestinal T cell immunity by
perturbing macrophage homeostasis.’ Science Translational Medicine, 10(464) p. eaao4755.
Sen, S., Giamberardino, L. D., Moss, K., Morelli, T., Rosamond, W. D., Gottesman, R. F., Beck, J. and
Offenbacher, S. (2018) ‘Periodontal Disease, Regular Dental Care Use, and Incident Ischemic Stroke.’
Stroke. American Heart Association, Inc., 49(2) pp. 355–362.
Serbina, N. V and Pamer, E. G. (2006) ‘Monocyte emigration from bone marrow during bacterial infection
requires signals mediated by chemokine receptor CCR2.’ Nature Immunology, 7(3) pp. 311–317.
Settem, R. P., El-Hassan, A. T., Honma, K., Stafford, G. P. and Sharma, A. (2012) ‘Fusobacterium nucleatum
and Tannerella forsythia Induce Synergistic Alveolar Bone Loss in a Mouse Periodontitis Model.’ Blanke, S.
R. (ed.) Infection and Immunity, 80(7) pp. 2436–2443.
Sfyroeras, G. S., Roussas, N., Saleptsis, V. G., Argyriou, C. and Giannoukas, A. D. (2012) ‘Association
between periodontal disease and stroke.’ Journal of Vascular Surgery. Elsevier Inc., 55(4) pp. 1178–1184.
Shaffer, J. R., Polk, D. E., Wang, X., Feingold, E., Weeks, D. E., Lee, M.-K., Cuenco, K. T., Weyant, R. J.,
Crout, R. J., McNeil, D. W. and Marazita, M. L. (2014) ‘Genome-Wide Association Study of Periodontal
Health Measured by Probing Depth in Adults Ages 18−49 years.’ G3 Bethesda, 4(2) pp. 307–314.
Sharma, A., Novak, E. K., Sojar, H. T., Swank, R. T., Kuramitsu, H. K. and Genco, R. J. (2000)
‘Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of
murine platelets.’ Oral Microbiology and Immunology. Wiley/Blackwell (10.1111), 15(6) pp. 393–396.
Shaw, A. C., Goldstein, D. R. and Montgomery, R. R. (2013) ‘Age-dependent dysregulation of innate
immunity.’ Nature Reviews Immunology, 13(12) pp. 875–887.
Shaw, T. N., Houston, S. A., Wemyss, K., Bridgeman, H. M., Barbera, T. A., Zangerle-Murray, T.,
Strangward, P., Ridley, A. J. L., Wang, P., Tamoutounour, S., Allen, J. E., Konkel, J. E. and Grainger, J. R.
(2018) ‘Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4
expression.’ The Journal of Experimental Medicine. Rockefeller University Press, May, p. jem.20180019.
Shemer, A., Grozovski, J., Tay, T. L., Tao, J., Volaski, A., Süß, P., Ardura-Fabregat, A., Gross-Vered, M.,
Kim, J.-S., David, E., Chappell-Maor, L., Thielecke, L., Glass, C. K., Cornils, K., Prinz, M. and Jung, S.
(2018) ‘Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin
landscape and response to challenge.’ Nature Communications, 9(1) p. 5206.
Sheng, J., Ruedl, C. and Karjalainen, K. (2015) ‘Most Tissue-Resident Macrophages Except Microglia Are
Derived from Fetal Hematopoietic Stem Cells.’ Immunity, 43(2) pp. 382–393.
Shichita, T., Sugiyama, Y., Ooboshi, H., Sugimori, H., Nakagawa, R., Takada, I., Iwaki, T., Okada, Y., Iida,
M., Cua, D. J., Iwakura, Y. and Yoshimura, A. (2009) ‘Pivotal role of cerebral interleukin-17–producing γδT
cells in the delayed phase of ischemic brain injury.’ Nature Medicine. Nature Publishing Group, 15(8) pp.
946–950.
Shin, J. E., Baek, K. J., Choi, Y. S. and Choi, Y. (2013) ‘A periodontal pathogen Treponema denticola hijacks
the Fusobacterium nucleatum -driven host response.’ Immunology and Cell Biology, 91(8) pp. 503–510.
Shin, J., Maekawa, T., Abe, T., Hajishengallis, E., Hosur, K., Pyaram, K., Mitroulis, I., Chavakis, T. and
Hajishengallis, G. (2015) ‘DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in
nonhuman primates.’ Science Translational Medicine, 7(307) p. 307ra155-307ra155.
Silvestre, F., Silvestre-Rangil, J., Bagan, L. and Bagan, J. (2016) ‘Effect of nonsurgical periodontal treatment
in patients with periodontitis and rheumatoid arthritis: A systematic review.’ Medicina Oral Patología Oral y
Cirugia Bucal pp. e349–e354.
Singh, A., Wyant, T., Anaya-Bergman, C., Aduse-Opoku, J., Brunner, J., Laine, M. L., Curtis, M. A. and
Lewis, J. P. (2011) ‘The Capsule of Porphyromonas gingivalis Leads to a Reduction in the Host
Inflammatory Response, Evasion of Phagocytosis, and Increase in Virulence.’ Bliska, J. B. (ed.) Infection and
Immunity, 79(11) pp. 4533–4542.

221
Singh, T. P., Zhang, H. H., Borek, I., Wolf, P., Hedrick, M. N., Singh, S. P., Kelsall, B. L., Clausen, B. E. and
Farber, J. M. (2016a) ‘Monocyte-derived inflammatory Langerhans cells and dermal dendritic cells mediate
psoriasis-like inflammation.’ Nature Communications.
Singh, V., Roth, S., Llovera, G., Sadler, R., Garzetti, D., Stecher, B., Dichgans, M. and Liesz, A. (2016b)
‘Microbiota Dysbiosis Controls the Neuroinflammatory Response after Stroke.’ Journal of Neuroscience,
36(28) pp. 7428–7440.
Singhrao, S. K., Chukkapalli, S., Poole, S., Velsko, I., Crean, S. J. and Kesavalu, L. (2017) ‘Chronic
Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE – / – mice
brains.’ Journal of Oral Microbiology. Taylor & Francis, 9(1) p. 1270602.
Siracusa, M. C., Saenz, S. A., Tait Wojno, E. D., Kim, B. S., Osborne, L. C., Ziegler, C. G., Benitez, A. J.,
Ruymann, K. R., Farber, D. L., Sleiman, P. M., Hakonarson, H., Cianferoni, A., Wang, M.-L., Spergel, J. M.,
Comeau, M. R. and Artis, D. (2013) ‘Thymic Stromal Lymphopoietin-Mediated Extramedullary
Hematopoiesis Promotes Allergic Inflammation.’ Immunity. Elsevier Inc., 39(6) pp. 1158–1170.
Slocum, C., Coats, S. R., Hua, N., Kramer, C., Papadopoulos, G., Weinberg, E. O., Gudino, C. V., Hamilton,
J. A., Darveau, R. P. and Genco, C. A. (2014) ‘Distinct Lipid A Moieties Contribute to Pathogen-Induced
Site-Specific Vascular Inflammation.’ PLoS Pathogens.
Slocum, C., Kramer, C. and Genco, C. A. (2016) ‘Immune dysregulation mediated by the oral microbiome:
potential link to chronic inflammation and atherosclerosis.’ Journal of Internal Medicine, 280(1) pp. 114–
128.
Slots, J. (2017) ‘Periodontitis: facts, fallacies and the future.’ Periodontology 2000, 75(1) pp. 7–23.
Socransky, S. S. and Haffajee, A. D. (2005) ‘Periodontal microbial ecology.’ Periodontology 2000, 38(1) pp.
135–187.
Sommer, C. J. (2017) ‘Ischemic stroke: experimental models and reality.’ Acta Neuropathologica, 133(2) pp.
245–261.
Sonderegger, I., Iezzi, G., Maier, R., Schmitz, N., Kurrer, M. and Kopf, M. (2008) ‘GM-CSF mediates
autoimmunity by enhancing IL-6–dependent Th17 cell development and survival.’ The Journal of
Experimental Medicine, 205(10) pp. 2281–2294.
Stabholz, A., Soskolne, W. A. and Shapira, L. (2010) ‘Genetic and environmental risk factors for chronic
periodontitis and aggressive periodontitis.’ Periodontology 2000, 53(1) pp. 138–153.
Steinmetz, O., Hoch, S., Avniel-Polak, S., Gavish, K., Eli-Berchoer, L., Wilensky, A. and Nussbaum, G.
(2016) ‘CX3CR1hi Monocyte/Macrophages Support Bacterial Survival and Experimental Infection–Driven
Bone Resorption.’ Journal of Infectious Diseases, 213(9) pp. 1505–1515.
Stevens, S. L., Bao, J., Hollis, J., Lessov, N. S., Clark, W. M. and Stenzel-Poore, M. P. (2002) ‘The use of
flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in
mice.’ Brain Research, 932(1–2) pp. 110–119.
Stier, S., Ko, Y., Forkert, R., Lutz, C., Neuhaus, T., Grünewald, E., Cheng, T., Dombkowski, D., Calvi, L.
M., Rittling, S. R. and Scadden, D. T. (2005) ‘Osteopontin is a hematopoietic stem cell niche component that
negatively regulates stem cell pool size.’ The Journal of Experimental Medicine, 201(11) pp. 1781–1791.
Stoll, L. L., Denning, G. M. and Weintraub, N. L. (2004) ‘Potential Role of Endotoxin as a Proinflammatory
Mediator of Atherosclerosis.’ Arteriosclerosis, Thrombosis, and Vascular Biology, 24(12) pp. 2227–2236.
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A.,
Pomeroy, S. L., Golub, T. R., Lander, E. S. and Mesirov, J. P. (2005) ‘Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression profiles.’ Proceedings of the National
Academy of Sciences, 102(43) pp. 15545–15550.
Sugiyama, T., Kohara, H., Noda, M. and Nagasawa, T. (2006) ‘Maintenance of the Hematopoietic Stem Cell
Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches.’ Immunity, 25(6) pp.
977–988.
Sun, J., Zhou, M., Salazar, C. R., Hays, R., Bedi, S., Chen, Y. and Li, Y. (2017) ‘Chronic Periodontal

222
Disease, Periodontal Pathogen Colonization, and Increased Risk of Precancerous Gastric Lesions.’ Journal of
Periodontology, 88(11) pp. 1124–1134.
Suwannalai, P., Trouw, L. a, Toes, R. E. M. and Huizinga, T. W. J. (2012) ‘Anti-citrullinated protein
antibodies (ACPA) in early rheumatoid arthritis.’ Modern Rheumatology, 22(1) pp. 15–20.
Sweetnam, D., Holmes, A., Tennant, K. A., Zamani, A., Walle, M., Jones, P., Wong, C. and Brown, C. E.
(2012) ‘Diabetes Impairs Cortical Plasticity and Functional Recovery Following Ischemic Stroke.’ Journal of
Neuroscience, 32(15) pp. 5132–5143.
Swirski, F. K., Libby, P., Aikawa, E., Alcaide, P., Luscinskas, F. W., Weissleder, R. and Pittet, M. J. (2007)
‘Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in
atheromata.’ Journal of Clinical Investigation, 117(1) pp. 195–205.
Swirski, F. K., Nahrendorf, M., Etzrodt, M., Wildgruber, M., Cortez-Retamozo, V., Panizzi, P., Figueiredo,
J.-L., Kohler, R. H., Chudnovskiy, A., Waterman, P., Aikawa, E., Mempel, T. R., Libby, P., Weissleder, R.
and Pittet, M. J. (2009) ‘Identification of Splenic Reservoir Monocytes and Their Deployment to
Inflammatory Sites.’ Science, 325(5940) pp. 612–616.
Syrjanen, J., Peltola, J., Valtonen, V., Iivanainen, M., Kaste, M. and Huttunen, J. K. (1989) ‘Dental infections
in association with cerebral infarction in young and middle-aged men.’ Journal of Internal Medicine.
1989/03/01, 225(3) pp. 179–184.
Takizawa, H., Boettcher, S. and Manz, M. G. (2012) ‘Demand-adapted regulation of early hematopoiesis in
infection and inflammation.’ Blood, 119(13) pp. 2991–3002.
Tamoutounour, S., Guilliams, M., Montanana Sanchis, F., Liu, H., Terhorst, D., Malosse, C., Pollet, E.,
Ardouin, L., Luche, H., Sanchez, C., Dalod, M., Malissen, B. and Henri, S. (2013) ‘Origins and Functional
Specialization of Macrophages and of Conventional and Monocyte-Derived Dendritic Cells in Mouse Skin.’
Immunity, 39(5) pp. 925–938.
Tamoutounour, S., Henri, S., Lelouard, H., de Bovis, B., de Haar, C., van der Woude, C. J., Woltman, A. M.,
Reyal, Y., Bonnet, D., Sichien, D., Bain, C. C., Mowat, A. M., Reis e Sousa, C., Poulin, L. F., Malissen, B.
and Guilliams, M. (2012) ‘CD64 distinguishes macrophages from dendritic cells in the gut and reveals the
Th1-inducing role of mesenteric lymph node macrophages during colitis.’ European Journal of Immunology,
42(12) pp. 3150–3166.
Tan, L., Wang, H., Li, C. and Pan, Y. (2014) ‘16S rDNA-based metagenomic analysis of dental plaque and
lung bacteria in patients with severe acute exacerbations of chronic obstructive pulmonary disease.’ Journal
of Periodontal Research, 49(6) pp. 760–769.
Tang, Z., Gan, Y., Liu, Q., Yin, J.-X., Liu, Q., Shi, J. and Shi, F.-D. (2014) ‘CX3CR1 deficiency suppresses
activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke.’ Journal of
Neuroinflammation, 11(1) p. 26.
Taxman, D. J., Swanson, K. V., Broglie, P. M., Wen, H., Holley-Guthrie, E., Huang, M. T.-H., Callaway, J.
B., Eitas, T. K., Duncan, J. A. and Ting, J. P. Y. (2012) ‘Porphyromonas gingivalis Mediates Inflammasome
Repression in Polymicrobial Cultures through a Novel Mechanism Involving Reduced Endocytosis.’ Journal
of Biological Chemistry, 287(39) pp. 32791–32799.
Tefferi, A. (2018) ‘Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management.’
American Journal of Hematology, 93(12) pp. 1551–1560.
Teng, Y. T., Nguyen, H., Gao, X., Kong, Y. Y., Gorczynski, R. M., Singh, B., Ellen, R. P. and Penninger, J.
M. (2000) ‘Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in
periodontal infection.’ The Journal of Clinical Investigation. American Society for Clinical Investigation,
106(6) pp. 59–67.
Thorbert-Mros, S., Larsson, L. and Berglundh, T. (2015) ‘Cellular composition of long-standing gingivitis
and periodontitis lesions.’ Journal of Periodontal Research, 50(4) pp. 535–543.
Tonetti, M. S. (2009) ‘Periodontitis and risk for atherosclerosis: an update on intervention trials.’ Journal of
Clinical Periodontology, 36(SUPPL. 10) pp. 15–19.
Tonetti, M. S., D’Aiuto, F., Nibali, L., Donald, A., Storry, C., Parkar, M., Suvan, J., Hingorani, A. D.,

223
Vallance, P. and Deanfield, J. (2007) ‘Treatment of periodontitis and endothelial function.’ The New England
journal of medicine.
Tonetti, M. S., Jepsen, S., Jin, L. and Otomo-Corgel, J. (2017) ‘Impact of the global burden of periodontal
diseases on health, nutrition and wellbeing of mankind: A call for global action.’ Journal of Clinical
Periodontology, 44(5) pp. 456–462.
Tonomura, S., Ihara, M., Kawano, T., Tanaka, T., Okuno, Y., Saito, S., Friedland, R. P., Kuriyama, N.,
Nomura, R., Watanabe, Y., Nakano, K., Toyoda, K. and Nagatsuka, K. (2016) ‘Intracerebral hemorrhage and
deep microbleeds associated with cnm-positive Streptococcus mutans; a hospital cohort study.’ Scientific
Reports. 2016/02/06, 6(1) p. 20074.
Trompette, A., Gollwitzer, E. S., Pattaroni, C., Lopez-Mejia, I. C., Riva, E., Pernot, J., Ubags, N., Fajas, L.,
Nicod, L. P. and Marsland, B. J. (2018) ‘Dietary Fiber Confers Protection against Flu by Shaping Ly6c−
Patrolling Monocyte Hematopoiesis and CD8+ T Cell Metabolism.’ Immunity, 48(5) p. 992–1005.e8.
Trompette, A., Gollwitzer, E. S., Yadava, K., Sichelstiel, A. K., Sprenger, N., Ngom-Bru, C., Blanchard, C.,
Junt, T., Nicod, L. P., Harris, N. L. and Marsland, B. J. (2014) ‘Gut microbiota metabolism of dietary fiber
influences allergic airway disease and hematopoiesis.’ Nature Medicine, 20(2) pp. 159–166.
Ubeda, C. and Pamer, E. G. (2012) ‘Antibiotics, microbiota, and immune defense.’ Trends in Immunology,
33(9) pp. 459–466.
Ueda, Y., Yang, K., Foster, S. J., Kondo, M. and Kelsoe, G. (2004) ‘Inflammation Controls B Lymphopoiesis
by Regulating Chemokine CXCL12 Expression.’ The Journal of Experimental Medicine, 199(1) pp. 47–58.
Vannucci, S. J., Willing, L. B., Goto, S., Alkayed, N. J., Brucklacher, R. M., Wood, T. L., Towfighi, J., Hurn,
P. D. and Simpson, I. A. (2001) ‘Experimental Stroke in the Female Diabetic, db/db , Mouse.’ Journal of
Cerebral Blood Flow & Metabolism, 21(1) pp. 52–60.
Vavricka, S. R., Manser, C. N., Hediger, S., Vögelin, M., Scharl, M., Biedermann, L., Rogler, S., Seibold, F.,
Sanderink, R., Attin, T., Schoepfer, A., Fried, M., Rogler, G. and Frei, P. (2013) ‘Periodontitis and Gingivitis
in Inflammatory Bowel Disease.’ Inflammatory Bowel Diseases, 19(13) pp. 2768–2777.
Velsko, I. M., Chukkapalli, S. S., Rivera, M. F., Lee, J.-Y., Chen, H., Zheng, D., Bhattacharyya, I., Gangula,
P. R., Lucas, A. R. and Kesavalu, L. (2014) ‘Active Invasion of Oral and Aortic Tissues by Porphyromonas
gingivalis in Mice Causally Links Periodontitis and Atherosclerosis.’ Glogauer, M. (ed.) PLoS ONE, 9(5) p.
e97811.
Venkataramani, A., Santo-Domingo, N. E. and Main, D. M. (1994) ‘Actinobacillus actinomycetemcomitans
pneumonia with possible septic embolization.’ Chest. 1994/02/01, 105(2) pp. 645–646.
Visnjic, D., Kalajzic, Z., Rowe, D. W., Katavic, V., Lorenzo, J. and Aguila, H. L. (2004) ‘Hematopoiesis is
severely altered in mice with an induced osteoblast deficiency.’ Blood, 103(9) pp. 3258–64.
Vogelgesang, A., Lange, C., Blümke, L., Laage, G., Rümpel, S., Langner, S., Bröker, B. M., Dressel, A. and
Ruhnau, J. (2017) ‘Ischaemic stroke and the recanalization drug tissue plasminogen activator interfere with
antibacterial phagocyte function.’ Journal of Neuroinflammation, 14(1) p. 140.
Wallrapp, A., Riesenfeld, S. J., Burkett, P. R., Abdulnour, R.-E. E., Nyman, J., Dionne, D., Hofree, M.,
Cuoco, M. S., Rodman, C., Farouq, D., Haas, B. J., Tickle, T. L., Trombetta, J. J., Baral, P., Klose, C. S. N.,
Mahlakõiv, T., Artis, D., Rozenblatt-Rosen, O., Chiu, I. M., Levy, B. D., Kowalczyk, M. S., Regev, A. and
Kuchroo, V. K. (2017) ‘The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation.’ Nature.
Nature Publishing Group, 549(7672) pp. 351–356.
Wang, M., Krauss, J. L., Domon, H., Hosur, K. B., Liang, S., Magotti, P., Triantafilou, M., Triantafilou, K.,
Lambris, J. D. and Hajishengallis, G. (2010) ‘Microbial Hijacking of Complement-Toll-Like Receptor
Crosstalk.’ Science Signaling, 3(109) pp. ra11-ra11.
Wang, M., Shakhatreh, M.-A. K., James, D., Liang, S., Nishiyama, S. -i., Yoshimura, F., Demuth, D. R. and
Hajishengallis, G. (2007) ‘Fimbrial Proteins of Porphyromonas gingivalis Mediate In Vivo Virulence and
Exploit TLR2 and Complement Receptor 3 to Persist in Macrophages.’ The Journal of Immunology, 179(4)
pp. 2349–2358.
Wang, Y., Yu, X., Lin, J., Hu, Y., Zhao, Q., Kawai, T., Taubman, M. A. and Han, X. (2017) ‘B10 Cells

224
Alleviate Periodontal Bone Loss in Experimental Periodontitis.’ Blanke, S. R. (ed.) Infection and Immunity,
85(9).
Wattananit, S., Tornero, D., Graubardt, N., Memanishvili, T., Monni, E., Tatarishvili, J., Miskinyte, G., Ge,
R., Ahlenius, H., Lindvall, O., Schwartz, M. and Kokaia, Z. (2016) ‘Monocyte-Derived Macrophages
Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice.’ Journal of Neuroscience.
2016/04/15, 36(15) pp. 4182–4195.
Wendeln, A., Degenhardt, K., Kaurani, L., Gertig, M., Ulas, T., Jain, G., Wagner, J., Häsler, lisa M., Wild,
K., Skodras, A., Blank, T., Staszewski, O., Datta, M., Centeno, T. P., Capece, V., Islam, M. R., Kerimoglu,
C., Staufenbiel, M., Schultze, J. L., Beyer, M., Prinz, M., Jucker, M., Fischer, A. and Neher, J. J. (2018)
‘Innate immune memory in the brain shapes neurological disease hallmarks.’ Nature, 556(7701) pp. 332–338.
Wilharm, A., Tabib, Y., Nassar, M., Reinhardt, A., Mizraji, G., Sandrock, I., Heyman, O., Barros-Martins, J.,
Aizenbud, Y., Khalaileh, A., Eli-Berchoer, L., Elinav, E., Wilensky, A., Förster, R., Bercovier, H., Prinz, I.
and Hovav, A.-H. (2019) ‘Mutual interplay between IL-17–producing γδT cells and microbiota orchestrates
oral mucosal homeostasis.’ Proceedings of the National Academy of Sciences, 116(7) pp. 2652–2661.
Wright, D. E., Wagers, A. J., Gulati, A. P., Johnson, F. L. and Weissman, I. L. (2001) ‘Physiological
migration of hematopoietic stem and progenitor cells.’ Science, 294(5548) pp. 1933–6.
Wu, T., Trevisan, M., Genco, R. J., Dorn, J. P., Falkner, K. L. and Sempos, C. T. (2000) ‘Periodontal Disease
and Risk of Cerebrovascular Disease.’ Archives of Internal Medicine, 160(18) p. 2749.
Wu, Y., Dong, G., Xiao, W., Xiao, E., Miao, F., Syverson, A., Missaghian, N., Vafa, R., Cabrera-Ortega, A.
A., Rossa, C. and Graves, D. T. (2015) ‘Effect of aging on periodontal inflammation, microbial colonization,
and disease susceptibility.’ Journal of Dental Research.
Wu, Z., Ni, J., Liu, Y., Teeling, J. L., Takayama, F., Collcutt, A., Ibbett, P. and Nakanishi, H. (2017)
‘Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic
exposure to lipopolysaccharide from Porphyromonas gingivalis in mice.’ Brain, Behavior, and Immunity, 65,
October, pp. 350–361.
Yamazaki, K., Honda, T., Oda, T., Ueki-Maruyama, K., Nakajima, T., Yoshie, H. and Seymour, G. J. (2005)
‘Effect of periodontal treatment on the C-reactive protein and proinflammatory cytokine levels in Japanese
periodontitis patients.’ Journal of Periodontal Research.
Yamazaki, K., Ohsawa, Y., Tabeta, K., Ito, H., Ueki, K., Oda, T., Yoshie, H. and Seymour, G. J. (2002)
‘Accumulation of human heat shock protein 60-reactive T cells in the gingival tissues of periodontitis
patients.’ Infection and Immunity, 70(5) pp. 2492–501.
Yáñez, A., Coetzee, S. G., Olsson, A., Muench, D. E., Berman, B. P., Hazelett, D. J., Salomonis, N., Grimes,
H. L. and Goodridge, H. S. (2017) ‘Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell
Progenitors Independently Produce Functionally Distinct Monocytes.’ Immunity, 47(5) p. 890–902.e4.
Yilmaz, G., Arumugam, T. V, Stokes, K. Y. and Granger, D. N. (2006) ‘Role of T Lymphocytes and
Interferon-γ in Ischemic Stroke.’ Circulation. American Heart Association, Inc., 113(17) pp. 2105–2112.
Yona, S., Kim, K. W., Wolf, Y., Mildner, A., Varol, D., Breker, M., Strauss-Ayali, D., Viukov, S., Guilliams,
M., Misharin, A., Hume, D. A., Perlman, H., Malissen, B., Zelzer, E. and Jung, S. (2013) ‘Fate Mapping
Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis.’ Immunity.
Elsevier, 38(1) pp. 79–91.
Yoneda, M., Naka, S., Nakano, K., Wada, K., Endo, H., Mawatari, H., Imajo, K., Nomura, R., Hokamura, K.,
Ono, M., Murata, S., Tohnai, I., Sumida, Y., Shima, T., Kuboniwa, M., Umemura, K., Kamisaki, Y., Amano,
A., Okanoue, T., Ooshima, T. and Nakajima, A. (2012) ‘Involvement of a periodontal pathogen,
Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease.’ BMC Gastroenterology,
12(1) p. 16.
Yoshihara, H., Arai, F., Hosokawa, K., Hagiwara, T., Takubo, K., Nakamura, Y., Gomei, Y., Iwasaki, H.,
Matsuoka, S., Miyamoto, K., Miyazaki, H., Takahashi, T. and Suda, T. (2007) ‘Thrombopoietin/MPL
Signaling Regulates Hematopoietic Stem Cell Quiescence and Interaction with the Osteoblastic Niche.’ Cell
Stem Cell, 1(6) pp. 685–697.
Young, K., Borikar, S., Bell, R., Kuffler, L., Philip, V. and Trowbridge, J. J. (2016) ‘Progressive alterations

225
in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging.’ The Journal of Experimental
Medicine, 213(11) pp. 2259–2267.
Zekonis, G., Barzdziukaite, I., Zekonis, J., Sadzeviciene, R., Simonyte, S. and Zilinskas, J. (2014) ‘Local and
systemic immune responses in gingivitis and periodontitis.’ Open Medicine, 9(5).
Zhang, C. C., Kaba, M., Ge, G., Xie, K., Tong, W., Hug, C. and Lodish, H. F. (2006) ‘Angiopoietin-like
proteins stimulate ex vivo expansion of hematopoietic stem cells.’ Nature Medicine, 12(2) pp. 240–245.
Zhang, J., Niu, C., Ye, L., Huang, H., He, X., Tong, W.-G., Ross, J., Haug, J., Johnson, T., Feng, J. Q.,
Harris, S., Wiedemann, L. M., Mishina, Y. and Li, L. (2003) ‘Identification of the haematopoietic stem cell
niche and control of the niche size.’ Nature, 425(6960) pp. 836–841.
Zhang, P., Nelson, S., Bagby, G. J., Siggins, R., Shellito, J. E. and Welsh, D. A. (2008) ‘The Lineage−c-
Kit+Sca-1+ Cell Response to Escherichia coli Bacteremia in Balb/c Mice.’ Stem Cells, 26(7) pp. 1778–1786.
Zhou, G. X. and Liu, Z. J. (2017) ‘Potential roles of neutrophils in regulating intestinal mucosal inflammation
of inflammatory bowel disease.’ Journal of Digestive Diseases, 18(9) pp. 495–503.
Zhou, W., Liesz, A., Bauer, H., Sommer, C., Lahrmann, B., Valous, N., Grabe, N. and Veltkamp, R. (2013)
‘Postischemic Brain Infiltration of Leukocyte Subpopulations Differs among Murine Permanent and
Transient Focal Cerebral Ischemia Models.’ Brain Pathology, 23(1) pp. 34–44.
Zhu, Y., Dashper, S. G., Chen, Y. Y., Crawford, S., Slakeski, N. and Reynolds, E. C. (2013) ‘Porphyromonas
gingivalis and Treponema denticola Synergistic Polymicrobial Biofilm Development.’ PLoS ONE.
Zijlstra, E. E., Swart, G. R., Godfroy, F. J. M. and Degener, J. E. (1992) ‘Pericarditis, pneumonia and brain
abscess due to a combined Actinomyces—Actinobacillus actinomycetemcomitans infection.’ Journal of
Infection, 25(1) pp. 83–87.

226
Appendix
Table A.1. Summary of reagents, chemicals & consumables used for experiments.
Reagent Catalogue# Supplier
0.22 µm Sterile Filter (PES Membrane Millex -
GP) 10268401 EMD Millipore
1 ml Syringe 15489199 BD Biosciences
100 μm MACS Smart Strainer 130-098-463 Miltenyi Biotec
12-well Cell Culture Plate BC011 Corning
16% Formaldehyde (w/v), Methanol-free (used
at 1:8) 28908 Fisher Scientific
16S rDNA Primer/Probe Mix NA Applied Biosystems
20-Gauge Microlance Needles 10201211 BD Biosciences
21-Gauge Butterfly Needles 12977706 Fisher Scientific
21-Gauge Microlance Needles 10775295 BD Biosciences
25-Gauge Microlance Needles 10442204 BD Biosciences
3,3'-Diaminobenzidine (DAB) D8001 Sigma-Aldrich
35 μm Cell Strainer Caps 352235 BD Biosciences
384-well Plate 4309849 Thermo Fisher
40 μm Cell Strainer 11587522 Fisher Scientific
5/0 Silk Suture 18020-50 Fine Science Tools,
Germany
5 ml Syringe SYR6162 BD Biosciences
6/0 Sutures ETW812 Ethicon
6-well Cell Culture Plate BC010 Corning
70 µm Cell Strainer 11597522 Fisher Scientific
96-well U-bottom Plate 3799 Corning
96-well V-bottom Plate 611V96 Fisher Scientific
ACK Lysing Buffer 10-548E Lonza
ArC™ Amine Reactive Compensation Bead Kit A10346 Fisher Scientific
Atipamezole (Antisedan®) Vm 06043/4004 Vetoquinol, UK
Biotinylated goat anti-rabbit IgG BA-1000 Vector Labs
Biotinylated horse anti-mouse IgG BA-2000 Vector Labs
Brefeldin A/GolgiPlug™ Solution (1000x) 555029 BD Biosciences
Buprenorphine (Vetergesic®) Vm 15052/4080 Reckitt Benckiser
Healthcare UK Ltd.
CD90.2 MicroBeads 130-121-278 Miltenyi Biotec
cDNA Reverse Transcription Kit 4368814 Thermo Fisher
Chloroform 288306 Sigma-Aldrich
Chromium (III) potassium sulphate 1010360250 Sigma-Aldrich
Collagenase, type IV 17104019 Thermo Fisher

227
Cresyl Violet Acetate AC229630250 Fisher Scientific
Dextran (MW: 60k–90k) 14495 USB Corporation
DL-Dithiothreitol D9779 Sigma-Aldrich
DNeasy Blood and Tissue Kit 69506 Qiagen
Dumont 5 Angled Forceps 11251-35 Fine Science Tools,
Germany
Dumont 5 Straight Forceps 11251-10 Fine Science Tools,
Germany
DNase I DN25 Sigma-Aldrich
DPX Mounting Medium 10050080 Fisher Scientific
Dulbeccos’s Modified Eagle Medium Nutrient
Mixture (F-12) 21331-020 Invitrogen
Dulbecco's Phosphate-Buffered Saline (1x) D8537 Sigma-Aldrich
EDTA solution (0.5 M) 3690 Sigma-Aldrich
EMLA™ PL 39699/0088 AstraZeneca
Ethanol 10644795 Fisher Scientific
Ethylene glycol 85978 Sigma-Aldrich
Fast SYBR™ Green Master Mix 4385618 Thermo Fisher
Foxp3/Transcription Factor Staining Buffer Set 00-5523-00 eBioscience
Foetal Bovine Serum F9665 Sigma-Aldrich
Gelatine G9391 Sigma-Aldrich
GentleMACS C Tubes 130-093-237 Miltenyi Biotec
GentleMACS M Tubes 130-093-236 Miltenyi Biotec
Glycerol G5516 Sigma-Aldrich
Hank’s Balanced Salt Solution (Ca2+, Mg2+
free) 14170-070 Invitrogen
Heparin sodium (1000 I.U./ml) PL 29831/0109 Wockhardt
HEPES Solution (1M) H0887 Sigma-Aldrich
Hydrogen peroxide solution (30% w/v) H1009 Sigma-Aldrich
Industrial Methylated Spirit 11412884 Fisher Scientific
Iso-2-propanol I9516 Sigma-Aldrich
Isoflurane PL 41042/0002 Abbvie Ltd
Isopentane 277258 Sigma-Aldrich
Ketamine (Narketan® 10) Vm 08007/4090 Vetoquinol, UK
LEGENDplex™ Mouse Inflammation Panel 740150 BioLegend
L-Glutamine Solution G7513 Sigma-Aldrich
Liberase™ TL 5401020001 Roche
Lipopolysaccharide from P. gingivalis tlrl-pglps InvivoGen
Lipopolysaccharide from E. coli 0111:B4 tlrl-3pelps InvivoGen
Lipopolysaccharide from E. coli 0127:B8 L4516-1MG Sigma-Aldrich
LIVE/DEAD™ Blue L23105 Thermo Fisher

228
Lubrithal™ eye gel Dechra
Lysing Matrix D Tubes 116913050-CF MP Biochemicals
Lysozyme L6876 Sigma-Aldrich
Medetomidine (Domitor®) Vm 06043/4003 Vetoquinol, UK
MEM Non-essential Amino Acid Solution
(100x) M7145 Sigma-Aldrich
MethoCult™ GF M3434 Methylcellulose
Medium M3434 Stem Cell Technologies
Methylene Blue M9140 Sigma-Aldrich
Normal Goat Serum S-1000 Vector Labs
Normal Horse Serum S-2000 Vector Labs
Nylon Filament Suture 6021PK10 Doccol Corporation
Optical Adhesive Film 4360954 Fisher Scientific
Paraformaldehyde, prilled 441244 Sigma-Aldrich
Penicillin-Streptomycin P4333 Sigma-Aldrich
Petri Dishes 15370366 Fisher Scientific
Polymyxin B sulfate P4932 Sigma-Aldrich
QIAzol® Lysis Reagent 79306 Qiagen
RPMI 1640 Medium R0883 Sigma-Aldrich
Saline 5/436464/0217 B. Braun Melsungen AG
Saponin 47036-250G-F Sigma-Aldrich
Scalpels, disposable 05XX Swann-Morton, UK
Scalpel blades, #10 INS4670 Scientific Laboratory
Supplies Ltd
Sodium pyruvate 11360070 Thermo Fisher
Sterile Water W3500 Sigma-Aldrich
Sucrose S9378 Sigma-Aldrich
TaqMan™ Gene Expression Master Mix 4369016 Thermo Fisher
Triton™ X-100 T8787 Sigma-Aldrich
Tryptone Soy Agar CM0131 Oxoid
UltraComp eBeads 01-2222-42 eBioscience
Vectastain® Elite® ABC HRP Kit (Peroxidase,
Standard) PK-6100 Vector Labs
Vetbond® 1469SB 3M
Videne® 3030440 Ecolab Ltd
Wilkins-Chalgren Anaerobe Agar CM0619 Oxoid
Xylazine (Rompun®) Vm 00010/4093 Bayer
Xylene 10385910 Fisher Scientific
Zombie Aqua (used at 1:500) 423102 BioLegend
Zombie UV (used at 1:500) 423108 BioLegend
229
Table A.2. Details of mouse antibodies used for flow cytometry.
Target Fluorochrome Dilution Clone Catalogue # Supplier
B220 APC-eFluor® 780 1:200 RA3-6B2 47-0452-82 eBioscience
B220 Brilliant Violet 605™ 1:400 RA3-6B2 103243 BioLegend
B220 NA (Biotin) 1:400 RA3-6B2 103204 BioLegend
CCR2 PE 1:50 475301 FAB5538P-
100
R&D
Systems
CD115 APC 1:200 AFS98 135509 BioLegend
CD115 PE-Cy7 1:200-400 AFS98 25-1152-82 eBioscience
CD117 (c-Kit) PerCP-Cy5.5 1:200 2B8 105824 BioLegend
CD11b Brilliant Violet 605™ 1:400 M1/70 101237 BioLegend
CD11b Brilliant Violet 650™ 1:400 M1/70 101239 BioLegend
CD11c NA (Biotin) 1:400 N418 117304 BioLegend
CD11c Brilliant Violet 785™ 1:200 N418 117336 BioLegend
CD16/32 PE-Cy7 1:100 93 101317 BioLegend
CD135 (Flt3) PE 1:100 A2F10 12-1351-83 eBioscience
CD19 APC-eFluor® 780 1:200 1D3 47-0193 eBioscience
CD19 NA (Biotin) 1:400 6D5 115504 BioLegend
CD3 NA (Biotin) 1:400 145-2C11 100304 BioLegend
CD3 APC-eFluor® 780 1:200 17A2 47-0032 eBioscience
CD4 Brilliant Violet 605™ 1:400 RM4-5 100548 BioLegend
CD4 Brilliant Violet 650™ 1:400 RM4-5 100546 BioLegend
CD44 Brilliant Violet 785™ 1:200 IM7 103059 BioLegend
CD45 Brilliant Violet 510™ 1:400 30-F11 103138 Biolegend
CD45 Alexa Fluor® 700 1:200 30-F11 103128 BioLegend
CD45.1 Alexa Fluor® 700 1:200 A20 110723 BioLegend
CD45.2 PE-Cy7 1:200 104 25-0454-82 eBioscience
CD48 Brilliant Violet 711™ 1:100 HM48-1 103439 BioLegend
CD62L APC 1:200 MEL-14 17-0621-82 eBioscience
CD68 PE 1:200 FA-11 137013 BioLegend
CD68 PerCP-Cy5.5 1:400 FA-11 137009 BioLegend
CD8a Brilliant Violet 711™ 1:400 53-6.7 100759 BioLegend
CD8a PE-Cy7 1:400 53-6.7 25-0081 eBioscience
CD90.2 PerCP-Cy5.5 1:200 53-2.1 140322 BioLegend
CD90.2 APC-eFluor® 780 1:200 53-2.1 46-0902-82 eBioscience
CX3CR1 PerCP-Cy5.5 1:200 SA011F11 149009 BioLegend
F4/80 eFluor® 450 1:400 BM8 48-4801-82 eBioscience
FcεR1 APC-Cy7 1:200 MAR-1 134325 BioLegend
FcεR1 NA (Biotin) 1:200 MAR-1 134303 BioLegend
FoxP3 FITC 1:100 FJK-16s 11-5773-80 eBioscience
FoxP3 eFluor® 450 1:100 FJK-16s 48-5773-82 eBioscience

230
I-A/I-E (MHCII) eFluor® 450 1:200 M5/114.15.2 48-5321-82 eBioscience
I-A/I-E (MHCII) Brilliant Violet 510™ 1:200-400 M5/114.15.2 107635 BioLegend
I-A/I-E (MHCII) FITC 1:400 M5/114.15.2 107605 BioLegend
IFN-γ PE 1:100 XMG1.2 505808 BioLegend
IL-1α PE 1:300 ALF-161 12-7011-81 eBioscience
IL-1β (pro-form) FITC 1:300 NJTEN3 11-7114-82 eBioscience
IL-6 APC 1:200 MP5-20F3 504508 BioLegend
Ki67 eFluor® 450 1:200 SolA15 48-5698-80 eBioscience
Ly6A/E (Sca-1) Alexa Fluor® 488 1:200-400 D7 108115 BioLegend
Ly6C Brilliant Violet 711™ 1:400 HK1.4 128037 BioLegend
Ly6C eFluor® 450 1:200-400 HK1.4 48-5932-80 eBioscience
Ly6G APC/Fire 750™ 1:200-400 1A8 127652 BioLegend
Ly6G NA (Biotin) 1:200-400 1A8 127604 BioLegend
NK-1.1 APC-eFluor® 780 1:200 PK136 47-5941 eBioscience
NK-1.1 NA (Biotin) 1:200-400 PK136 108704 BioLegend
ROR-γt PE 1:100 B2D 12-6981-80 eBioscience
Siglec F PE-CF594 1:400 E50-2440 562757 BD
Biosciences
Streptavidin PE-CF594 1:1000 None 562284 BD
Biosciences
TCR-β NA (Biotin) 1:200-400 H57-597 109203 BioLegend
TCR-β PerCP-Cy5.5 1:200 H57-597 109228 BioLegend
TCR-β Brilliant Violet 605™ 1:200-400 H57-597 109241 BioLegend
TCR-β APC-eFluor® 780 1:200-400 H57-597 47-5961-82 eBioscience
TCR-γδ PE-Cy7 1:200-400 eBioGL3 25-5711-82 eBioscience
Ter-119 APC-eFluor® 780 1:200 TER-119 47-5921-82 eBioscience
Ter-119 NA (Biotin) 1:200-400 TER-119 116204 BioLegend
TNF-α Alexa Fluor® 700 1:400 MP6-XT22 506338 BioLegend
TruStain fcX™
(CD16/32) None 1:100 93 101320 BioLegend

231
Table A.3. Assessment of neurological impairment (28 points)
0 1 2 3 4
(1) Body symmetry (open bench top)
Normal Slight asymmetry
Moderate asymmetry
Prominent asymmetry
Extreme asymmetry
(2) Gait (open bench top)
Normal Stiff, inflexible Limping Trembling, drifting, falling
Does not walk
(3) Climbing (gripping surface, 45° angle)
Normal Climbs with strain, limb weakness present
Holds onto slope, does not slip or climb
Slides down slope, unsuccesful effort to prevent slope
Slides Immediately, no effort to prevent fall
(4) Circling behaviour (open bench top)
Not Present Predominantly one-sided turns
Circles to one side
Circles contantly to one side
Pivoting, swaying or no movement (not constantly)
(5) Front limb symmetry (mouse suspended by its tail)
Normal Light asymmetry
Marked asymmetry
Prominent asymmetry
Slight asymmetry, no body/limb movement
(6) Compulsory circling (front limbs on bench, rear suspended by tail)
Not Present Tendency to turn to one side
Circles to one side
Pivots to one side sluggishly
Does not advance
(7) Whisker response (light touch from behind)
Symetrical Response
Light asymmetry
Prominent asymmetry
Absent response ipsilaterally, diminished contralaterally
Absent proprioceptive response bilaterally




















