
The energetics of the interaction of BamHI endonuclease with its recognition site GGATCC1
18
The Energetics of the Interaction of BamHI Endonuclease with its Recognition Site GGATCC Lisa E. Engler 1 , Paul Sapienza 1 , Lydia F. Dorner 2 , Rebecca Kucera 2 , Ira Schildkraut 2 and Linda Jen-Jacobson 1 * 1 Department of Biological Sciences, University of Pittsburgh, Pittsburgh PA 15260, USA 2 New England Biolabs, Beverly MA 01915, USA The interaction of BamHI endonuclease with DNA has been studied crys- tallographically, but has not been characterized rigorously in solution. The enzyme binds in solution as a homodimer to its recognition site GGATCC. Only six base-pairs are directly recognized, but binding affinity (in the absence of the catalytic cofactor Mg 2 ) increases 5400-fold as oligonucleotide length increases from 10 to 14 bp. Binding is modulated by sequence context outside the recognition site, varying about 30-fold from the best (GTG or TAT) to the worst (CGG) flanking triplets. BamHI, EcoRI and EcoRV endonucleases all have different context preferences, suggesting that context affects binding by influencing the free energy levels of the complexes rather than that of the free DNA. Ethylation interference footprinting in the absence of divalent metal shows a localized and symmetrical pattern of phosphate contacts, with strong contacts at NpNpNpGGApTCC. In the presence of Mg 2 , first- order cleavage rate constants are identical in the two GGA half-sites, are the same for the two nicked intermediates and are unaffected by sub- strate length in the range 10-24 bp. DNA binding is strongly enhanced by mutations D94N, E111A or E113K, by binding of Ca 2 at the active site, or by deletion of the scissile phosphate GpGATCC, indicating that a cluster of negative charges at the catalytic site contributes at least 3-4 kcal/mol of unfavorable binding free energy. This electrostatic repul- sion destabilizes the enzyme-DNA complex and favors metal ion binding and progression to the transition state for cleavage. # 2001 Academic Press Keywords: BamHI endonuclease; protein-DNA recognition; flanking sequence; electrostatic repulsion; phosphate contacts *Corresponding author Introduction Restriction endonucleases have generated signifi- cant interest because of their biological function in host-controlled restriction 1 and their well-known uses in recombinant DNA technologies. Their highly specific interactions with their DNA recog- nition sites also present an attractive set of models for site-specific protein-DNA interactions. The endonucleases discriminate against incorrect DNA sites much more stringently than do other site- specific DNA-binding proteins, 2 so that one might expect these enzymes to exemplify the most rigor- ous rules and constraints that govern specificity. The structures of a substantial number of restric- tion endonucleases in complex with their DNA rec- ognition sites have now been solved 3–9 and for EcoRV, BamHI and EcoRI, multiple high-resolution structures (e.g. cognate and non-specific com- plexes, with or without divalent metals) are available 6,10-15 (J. M. Rosenberg, unpublished struc- tures deposited in RCSB Protein Data Bank). Despite these evident advantages, the study of the solution thermodynamics of these protein-DNA interactions has lagged behind the structural characterization. Extensive thermodynamic and pre-steady-state kinetic data were first obtained for EcoRI endonuclease, 16-20 which demonstrates high binding specificity in the absence of the divalent cation (Mg 2 ) required for catalysis, as does its iso- schizomer RsrI. 21 The progress of similar studies on EcoRV endonuclease has generated some con- troversy because some reports claim little or no DNA-binding specificity in the absence of divalent cation, 22-24 whereas other studies suggest that sub- stantial specific binding can be detected, 25 and is E-mail address of the corresponding author: [email protected] doi:10.1006/jmbi.2001.4428 available online at http://www.idealibrary.com on J. Mol. Biol. (2001) 307, 619–636 0022-2836/01/020619–18 $35.00/0 # 2001 Academic Press
Transcript of The energetics of the interaction of BamHI endonuclease with its recognition site GGATCC1

doi:10.1006/jmbi.2001.4428 available online at http://www.idealibrary.com on J. Mol. Biol. (2001) 307, 619±636
The Energetics of the Interaction of BamHIEndonuclease with its Recognition Site GGATCC
Lisa E. Engler1, Paul Sapienza1, Lydia F. Dorner2, Rebecca Kucera2,Ira Schildkraut2 and Linda Jen-Jacobson1*
1Department of BiologicalSciences, University ofPittsburgh, PittsburghPA 15260, USA2New England Biolabs, BeverlyMA 01915, USA
E-mail address of the [email protected]
0022-2836/01/020619±18 $35.00/0
The interaction of BamHI endonuclease with DNA has been studied crys-tallographically, but has not been characterized rigorously in solution.The enzyme binds in solution as a homodimer to its recognition siteGGATCC. Only six base-pairs are directly recognized, but bindingaf®nity (in the absence of the catalytic cofactor Mg2�) increases5400-fold as oligonucleotide length increases from 10 to 14 bp. Binding ismodulated by sequence context outside the recognition site, varyingabout 30-fold from the best (GTG or TAT) to the worst (CGG) ¯ankingtriplets. BamHI, EcoRI and EcoRV endonucleases all have different contextpreferences, suggesting that context affects binding by in¯uencing thefree energy levels of the complexes rather than that of the free DNA.Ethylation interference footprinting in the absence of divalent metalshows a localized and symmetrical pattern of phosphate contacts, withstrong contacts at NpNpNpGGApTCC. In the presence of Mg2�, ®rst-order cleavage rate constants are identical in the two GGA half-sites, arethe same for the two nicked intermediates and are unaffected by sub-strate length in the range 10-24 bp. DNA binding is strongly enhancedby mutations D94N, E111A or E113K, by binding of Ca2� at the activesite, or by deletion of the scissile phosphate GpGATCC, indicating thata cluster of negative charges at the catalytic site contributes at least3-4 kcal/mol of unfavorable binding free energy. This electrostatic repul-sion destabilizes the enzyme-DNA complex and favors metal ion bindingand progression to the transition state for cleavage.
# 2001 Academic Press
Keywords: BamHI endonuclease; protein-DNA recognition; ¯ankingsequence; electrostatic repulsion; phosphate contacts
*Corresponding authorIntroduction
Restriction endonucleases have generated signi®-cant interest because of their biological function inhost-controlled restriction1 and their well-knownuses in recombinant DNA technologies. Theirhighly speci®c interactions with their DNA recog-nition sites also present an attractive set of modelsfor site-speci®c protein-DNA interactions. Theendonucleases discriminate against incorrect DNAsites much more stringently than do other site-speci®c DNA-binding proteins,2 so that one mightexpect these enzymes to exemplify the most rigor-ous rules and constraints that govern speci®city.
The structures of a substantial number of restric-tion endonucleases in complex with their DNA rec-ognition sites have now been solved3 ± 9 and for
ing author:
EcoRV, BamHI and EcoRI, multiple high-resolutionstructures (e.g. cognate and non-speci®c com-plexes, with or without divalent metals) areavailable6,10-15 (J. M. Rosenberg, unpublished struc-tures deposited in RCSB Protein Data Bank).Despite these evident advantages, the study of thesolution thermodynamics of these protein-DNAinteractions has lagged behind the structuralcharacterization. Extensive thermodynamic andpre-steady-state kinetic data were ®rst obtained forEcoRI endonuclease,16-20 which demonstrates highbinding speci®city in the absence of the divalentcation (Mg2�) required for catalysis, as does its iso-schizomer RsrI.21 The progress of similar studieson EcoRV endonuclease has generated some con-troversy because some reports claim little or noDNA-binding speci®city in the absence of divalentcation,22-24 whereas other studies suggest that sub-stantial speci®c binding can be detected,25 and is
# 2001 Academic Press

620 BamHI interaction with its Cognate GGATCC Site
only enhanced25-27 by Ca2�, which does not sup-port catalysis. Similarly, the TaqI,28 Cfr9I,29 PvuII,30
Cfr10I31and MunI32 endonucleases are reported tohave very weak or no binding speci®city in theabsence of divalent cation. Speci®c binding bythese enzymes is enhanced by several factorswhich may be related: the presence of Ca2�
(PvuII,30 MunI,32 Cfr10I31), lower pH (MunI,32
EcoRV25) or mutations that eliminate active-sitecarboxylate residues.33 These are not unique prop-erties of the latter group, however, since EcoRIbinding is also enhanced by the same factors16
(G. Bosco and L.J.-J., unpublished results). It isthus important, in the interest of further thermo-dynamic studies to illuminate structure-functionrelationships, that we understand whether thereare truly two distinct classes of restriction endonu-cleases which differ in their dependence on diva-lent cation to ``trigger'' or ``confer'' speci®city33 or,alternatively, whether these various enzymes rep-resent a continuum of speci®c-binding af®nitieswhich are only enhanced in various degrees by thebinding of divalent cations at their active sites.
The earlier experience with EcoRI and EcoRVendonucleases suggests another potentially obscur-ing issue: it is easy to be misled by arbitrarychoices of DNA substrates, conditions and/ormethods for studying protein-DNA interactions.For example, it was shown for EcoRV endo-nuclease25 that the widely used gel-retardationmethod yielded anomalously low values of theequilibrium association constant under suboptimalconditions, but gave excellent agreement with themembrane-®lter-binding method if scrupulousattention was paid to conditions such as pH andsalt concentration. Furthermore, it has long beenknown34 that EcoRI cleavage is sensitive to DNAbase sequence surrounding the recognition site,and much later shown for both EcoRI35 ± 37 andEcoRV25 endonucleases that this effect represents asurprisingly large in¯uence on protein-DNA bind-ing. This factor should be explored systematicallyrather than preselected for some arbitrary reasonsuch as resemblance to a plasmid sequence ofdubious biological relevance.
Here, we introduce BamHI endonuclease as anew model system. BamHI endonuclease has beenwell studied crystallographically: structures areavailable for the apoenzyme,38 the speci®c recog-nition complex without6 and with11 divalentmetals, for the post-cleavage complex11 and for thenon-speci®c complex.15 These structures demon-strate that BamHI and EcoRI endonucleases have astriking structural homology in the spatial arrange-ment of the a/b-core that includes the recognitiona-helices and the active site residues, even thoughthere is little or no homology between theseenzymes at the level of primary sequence.39 TheBamHI recognition sequence GGATCC is related tothat of EcoRI (GAATTC) by two symmetrical A �Tto G �C changes; both the similarities and thedifferences in these sites provide potentially usefulpoints of comparison. We report here the funda-
mental features of the BamHI endonuclease equili-brium interaction with DNA in solution, as well assome elementary aspects of the pre-steady-statecleavage kinetics. We show that protein-DNAbinding is strongly sensitive to both the length of aDNA oligonucleotide and the base sequence sur-rounding the GGATCC site. We provide datashowing that ¯anking-sequence preferences aredominated by the immediately abutted three base-pairs on both sides of the cognate site, and thatpreferences do not simply correlate with thermalstability of the DNA duplex.40 We also show that acluster of negative charges at the BamHI catalyticsite contributes at least 3-4 kcal/mol of unfavor-able binding free energy.
Results and Discussion
Oligomeric state of BamHI endonucleasein solution
The intracellular concentration of BamHI hasbeen estimated to be slightly less than 1 mM.41 Inthe absence of DNA, the protein demonstrates adimer-tetramer equilibrium at micromolar concen-trations42 forming tetramers below 0.3 M NaCl anddimers at 0.45 M NaCl or higher. Since mostin vitro studies are carried out at relatively low saltconcentrations (0.1-0.2 M), we directly determinedthe oligomeric state of the active (DNA-binding)form of BamHI endonuclease by gel-permeationchromatography in low salt buffer (see Materialsand Methods). The endonuclease-DNA complexeluted at 66 kDa, as predicted for a dimer-DNAcomplex. Neither the DNA nor the endonucleasewere detected at the positions expected for tetra-mer-DNA or monomer-DNA complexes, as deter-mined by analysis of the fractions on SDS-PAGEand non-denaturing gel electrophoresis. In exper-iments at various ratios of DNA to protein, thefraction of DNA in the complex was found to bethat predicted for protein dimer interacting withDNA. Thus, the form of the protein that functionsin DNA binding in solution is the homodimer, as itis in the co-crystalline protein-DNA complex, andall subsequent calculations are based upon thisstoichiometry.
Equilibrium binding of DNA substrates
Prior experience with other restriction endonu-cleases25,35,43 indicates that both substrate lengthand nucleotide sequence ¯anking the recognitionsite may affect protein binding. The oligonucleo-tide in the co-crystalline complex6 is the 12 bppalindrome 50-TATGGATCCATA-30. The central10 bp demonstrate only small deviations fromB-DNA parameters, but large distortion at the50-ends cause the terminal thymine residue in theright half-site to be ¯ipped out of the plane of theDNA helix, while the ®nal A �T base-pair in the lefthalf-site is stacked. This allows base-pairing in thecrystal between successive oligonucleotides,

BamHI interaction with its Cognate GGATCC Site 621
elongating the helix along the dyad axis. This alsohas the effect of placing the phosphate groups at50-TpA in abnormal positions.
We determined equilibrium association constants(KA) for a set of substrates derived from thesequence in the co-crystalline complex. The data(Figure 1) demonstrate a dramatic increase in KA
with increased substrate size from 10 bp to 16 bp,and only minor improvements for sizes greaterthan 16 bp. The most striking improvement occurswhen the 10 bp DNA is increased to 12 bp, wherewe see a 240-fold enhancement of the binding af®-nity (� ÿ3.2 kcal/mol in binding free energy�G�bind). Note that the added phosphate groups areanalogous to those mispositioned by the distor-tions in the crystal.
A smaller improvement (tenfold or � ÿ1.4 kcal/mol in �G�bind) is seen for the addition of a 50-phos-phate group to the 12 bp substrate. This phosphategroup does not exist in the synthetic 50-OH-terminated 12 bp oligonucleotide found in thecrystalline complex.6 In contrast, the addition of a50-phosphate group to the 14 bp substrate had noeffect. These data are entirely consistent withthe protein-phosphate contacts identi®ed byethylation-interference footprinting (see below).Additional base-pairs beyond the 12 bp size(12 � P to 14 and 14 � P to 16) only improve bind-ing approximately twofold with each step; thismay re¯ect either a minor in¯uence of the added
Figure 1. Effect of substrate length on BamHI endonu-clease-DNA binding. Equilibrium association constants(KA) determined by equilibrium competition in bindingbuffer plus 0.12 M potassium acetate (pH 7.30) at 21 �C.Values are means � S.D. for three to seven independentmeasurements. Each smaller oligomer is a symmetrictruncation of the 24 bp oligonucleotideCGCGGGCTATggatccATACGGGC (recognition site inlowercase); the ¯anking triplet TAT is that in the crystalstructure.6 Oligomers labeled as ``P'' were exhaustivelyphosphorylated at the 50-ends. Unless noted as ``P'', theoligomers have 50-OH termini. For example, the ``P''between 10 and 12 denotes a 50-phosphorylated 10 bpoligonucleotide.
base-pair on the context dependence of binding(see below), or increased thermal stability at theoligonucleotide ends, creating a more B-DNA-likesubstrate.
We also tested the ®rst-order strand scissionrates (see below) for the set of substrates inFigure 1, and found that, within experimentalerror, there is no effect of substrate length on clea-vage rate constants. This implies that the same con-tacts between protein and DNA phosphate groupscontribute to the recognition and transition-statecomplexes. A similar inference has been drawnpreviously for EcoRI endonuclease.35
Sequence context and equilibrium binding
To test the effects of sequence context on theendonuclease-DNA interaction, we embedded theBamHI cognate site in 22 and 23 bp oligomeric con-structs with variable ¯anking regions. Ourapproach was conditioned by a large amount ofliterature on sequence tendencies in strongly bentnucleosomal DNA,44 the bending tendencies at YR(purine-pyrimidine steps) in free DNA45 ± 47 andtranscription-factor-DNA complexes48 ± 50 and theinference from both computational and informaticsapproaches51,52 that YR motifs (CA, TA, TG, or CGmodules) are especially ¯exible (i.e. conformation-ally polymorphic) and susceptible to large twist,slide, and roll deformations, whereas RR steps arerelatively in¯exible. We have observed for EcoRIand EcoRV endonucleases that context effects arestrongly dominated by the 3 bp immediately ¯ank-ing the recognition site; the fourth base-pair andbeyond have only minor effects (M. Kurpiewski,D. Chi, R. Varma & L.J.-J., unpublished results).We therefore examined representatives of the eightpossible classes of ¯anking triplets that result fromvarious combinations and permutations of YR, RY,RR, or YY steps. (Note that in free DNA, RR andYY steps can be considered as equivalent, but theyare potentially distinct when a protein interactsasymmetrically with respect to the two DNAstrands.)
The effect of ¯anking triplets on the binding ofBamHI endonuclease was assessed by measuringequilibrium association constants (Tables 1 and 2).We chose to use only symmetrical triplets ¯ankingthe two rotationally symmetrical half sites (e.g.GTGggatccCAC/CACcctaggGTG), because weobserved that oligonucleotides with asymmetriccontext lead to asymmetric adaptations in the com-plexes, as judged by footprinting. The �G�bind valuefor a ``hybrid'' site surrounded by two different¯anking triplets (GTGggatccCCA) was roughly themean of the �G�bind values of the two symmetri-cally ¯anked parents (GTGggatccCAC andTGGggatccCCA; best and near-worst, Table 1).Although the ``sequence space'' of possible ¯ank-ing regions is too great to be explored exhaus-tively, we can draw the following inferences fromTables 1 and 2:

Table 1. Thermodynamics of binding of BamHI endonuclease to cognate sequences
SequenceaKA
b
(Mÿ1)��G�bind
c
(kcal molÿ1)TM (�C)d
ExperimentalTM (�C)e
Predicted�G�duplex
f
(kcal molÿ1)
GGGATGGGTGggatccCACCCAC 1.5 (�0.1) � 109 0 71.5 71.3 ÿ39.8GCGGTTATggatccATAACGCG 1.3 (�0.2) � 109 0.1 � 0.1 65.9 65.4 ÿ36.5
GGATGGGCGggatccCGCCCAC 9.4 (�0.5) � 108 0.3 � 0.1 - 75.3 ÿ40.8GGATGCGGCggatccGCCGCAC 9.1 (�0.6) � 108 0.3 � 0.1 - 76.4 ÿ42.6GGATGGATAggatccTATCCAC 8.9 (�1.0) � 108 0.3 � 0.1 60.1 60.2 ÿ32.6GGATGGCACggatccGTGCCAC 8.5 (�0.7) � 108 0.3 � 0.1 70.1 71.6 ÿ39.5GGATGGGCTggatccAGCCCAC 7.6 (�0.5) � 108 0.4 � 0.1 68.9 70.8 ÿ38.2GGATCGCCCggatccGGGCGAC 7.3 (�0.7) � 108 0.4 � 0.1 - 75.0 ÿ41.4
GGGATGGGGTggatccACCCCAC 6.6 (�0.5) � 108 0.5 � 0.1 69.8 70.3 ÿ39.8GGGATGGTGTggatccACACCAC 6.5 (�0.6) � 108 0.5 � 0.1 67.6 66.6 ÿ38.4GGATCGCGCggatccGCGGGAC 6.0 (�0.5) � 108 0.5 � 0.1 - 75.0 ÿ41.4GGATGGTAAggatccTTACCAC 5.6 (�0.5) � 108 0.6 � 0.1 57.1 61.1 ÿ33.2GGATGATTAggatccTAATCAC 4.1 (�0.6) � 108 0.8 � 0.1 60.1 61.6 ÿ31.1GGATGGCGTggatccACGCCAC 4.0 (�0.3) � 108 0.8 � 0.1 - 71.6 ÿ39.5
GGGATGGTTTggatccAAACCAC 3.1 (�0.4) � 108 0.9 � 0.1 66.0 65.7 ÿ36.6GGGATGGCCAggatccTGGCCAC 3.0 (�0.5) � 108 0.9 � 0.1 72.2 73.0 ÿ40.3GGATGAAAGggatccCTTTCAC 2.1 (�0.3) � 108 1.2 � 0.1 - 63.1 ÿ33.8
GGGATGGCAAggatccTGGCCAC 2.0 (�0.4) � 108 1.2 � 0.1 69.6 69.6 ÿ38.8ATAAAGGGggatccCCCTAAAT 1.8 (�0.2) � 108 1.3 � 0.1 60.2 60.9 ÿ32.5
CGCGGGAAAAggatccTTTTGGGC 1.6 (�0.2) � 108 1.3 � 0.1 70.1 70.8 ÿ41.0GGATGAAGAggatccTCTTCAC 1.4 (�0.1) � 108 1.4 � 0.1 - 62.0 ÿ33.4GGATGGCGAggatccTCGCCAC 1.2 (�0.1) � 108 1.5 � 0.1 69.6 70.7 ÿ38.8
GGGATGGTGGggatccCCACCAC 7.6 (�1.0) � 107 1.7 � 0.1 70.1 70.3 ÿ39.8GGATGGCGGggatccCCGCCAC 4.8 (�0.8) � 107 2.0 � 0.1 - 75.3 ÿ40.8
a Recognition site in lower case; note that the ¯anking triplets are palindromic.b Equilibrium association constants were measured in binding buffer plus 0.18 M potassium acetate (pH 7.3, 21 �C); means � stan-
dard deviation of 53 determinations for each site.c Observed difference in standard binding free energy between each ¯anking sequence variant and the reference sequence (line 1);
��G�bind � ÿ RT ln (KAvar/KA
ref).d Melting temperatures were measured in 10 mM bis-tris-propane plus 0.10 M potassium acetate (pH 7.3); TM values in all cases
represent sharp, cooperative transitions; -, not determined.e Melting temperatures predicted for 2 � 10ÿ6 M total strand concentration, using the uni®ed oligonucleotide �H� and �S� nearest
neighbor parameters of Santa Lucia;71 �S� corrected for salt concentration.71
f Predicted standard free energy for duplex stability at 21 �C, calculated from the uni®ed oligonucleotide �H� and �S� nearestneighbor parameters reported by SantaLucia.71
622 BamHI interaction with its Cognate GGATCC Site
(1) For BamHI, context effects are dominated bythe 3 bp immediately abutting each half site, asthey were for EcoRI and EcoRV. For example,KA values are identical for sequences containingone of the best triplets, TAT (see lines 4 and 5,Table 2), despite the fact that the dinucleotide
Table 2. Thermodynamics of BamHI endonuclease-DNA bind
SequenceaKA
b
(Mÿ1)��G�bind
c
(kcal molÿ1)
GGGATGGGTGggatccCACCCAC 1.5 (�0.1) � 109 0GGGATGGTGGggatccCCACCAC 7.6 (�1.0) � 107 1.8 � 0.1GCGGGGTGggatccCACGCGCG 1.1 (�0.3) � 109 0.2 � 0.1GCGGTTATggatccATAACGCG 1.3 (�0.2) � 109 0.1 � 0.1ATAAATATggatccATATAAAT 1.1 (�0.1) � 109 0.2 � 0.1
a Recognition site ggatcc in lower case; note that ¯anking triplets ab Equilibrium association constants were measured in binding bu
standard deviation of 53 determinations for each site.c Observed difference in standard binding free energy between ea
��G�bind � ÿ RT ln (KAvar/KA
ref).d Melting temperatures were measured in melting buffer (10 mM
strand concentration was 2 � 10ÿ6 M.e Predicted melting temperatures calculated as in Table 1 (footnotef Observed standard free energy (at 37 �C) for duplex stability calc
obtained from a plot of 1/TM versus ln(Ct/4), assuming a two-state tconcentration (Ct) ranged from 0.5 mM to 8 mM.
g Predicted standard free energy for duplex stability, calculated frreported by SantaLucia.71 Note that the values of �G�duplex in Table 1
steps and base composition in the outer regionsbeyond the ¯anking TAT triplet are markedlydifferent. Compare also the data (lines 3, 4 and5) in Table 2, which show that the ¯ankingGTG and TAT triplets (the most favored triplets,Table 1) give similar BamHI binding constants
ing and oligonucleotide stability
TM (�C)
Experimentald Predictede�G�37, van't Hoff
f
(kcal molÿ1)�G�37,duplex
g
(kcal molÿ1)
71 71.3 ÿ31.8 ÿ32.271 70.3 ÿ30.6 ÿ32.278 78.0 ÿ35.5 ÿ35.666 65.4 ÿ27.3 ÿ28.751 49.8 ÿ17.6 ÿ19.6
re palindromic.ffer plus 0.18 M potassium acetate ( pH 7.30, 21 �C); means �
ch ¯anking sequence variant and the reference sequence (line 1);
bis-Tris-propane plus 0.10 M potassium acetate (pH 7.3)), total
e).ulated from the transition enthalpy (�HvH) and entropy (�SvH)
ransition and a temperature-independent enthalpy;70 total strand
om the uni®ed nearest-neighbor free energy parameters at 37 �Care given for 21 �C.
BamHI interaction with its Cognate GGATCC Site 623
regardless of the sequence farther out from the¯anking triplet.
(2) Purportedly ``¯exible'' trinucleotides that arestatistically preferred in ``deformed'' regions ofpromoter sites44 do not guarantee strong bindingby their mere presence. For example, the preferredGTG triplet is found both in the best sequence (line1, Table 1) where it is directly abutted to the recog-nition site and in one of the worst sequences(second from bottom, Table 1), where it is onebase-pair farther from the GGATCC site.
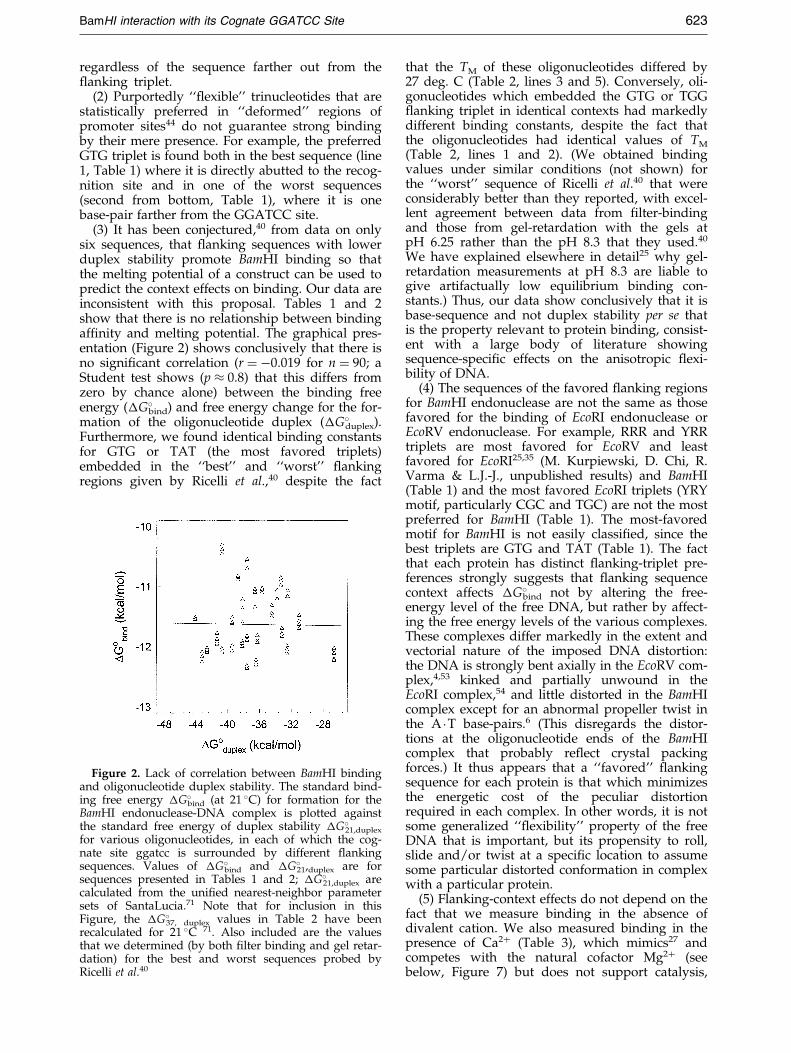
(3) It has been conjectured,40 from data on onlysix sequences, that ¯anking sequences with lowerduplex stability promote BamHI binding so thatthe melting potential of a construct can be used topredict the context effects on binding. Our data areinconsistent with this proposal. Tables 1 and 2show that there is no relationship between bindingaf®nity and melting potential. The graphical pres-entation (Figure 2) shows conclusively that there isno signi®cant correlation (r � ÿ0.019 for n � 90; aStudent test shows (p � 0.8) that this differs fromzero by chance alone) between the binding freeenergy (�G�bind) and free energy change for the for-mation of the oligonucleotide duplex (�G�duplex).Furthermore, we found identical binding constantsfor GTG or TAT (the most favored triplets)embedded in the ``best'' and ``worst'' ¯ankingregions given by Ricelli et al.,40 despite the fact
Figure 2. Lack of correlation between BamHI bindingand oligonucleotide duplex stability. The standard bind-ing free energy �G�bind (at 21 �C) for formation for theBamHI endonuclease-DNA complex is plotted againstthe standard free energy of duplex stability �G�21,duplex
for various oligonucleotides, in each of which the cog-nate site ggatcc is surrounded by different ¯ankingsequences. Values of �G�bind and �G�21,duplex are forsequences presented in Tables 1 and 2; �G�21,duplex arecalculated from the uni®ed nearest-neighbor parametersets of SantaLucia.71 Note that for inclusion in thisFigure, the �G�37, duplex values in Table 2 have beenrecalculated for 21 �C 71. Also included are the valuesthat we determined (by both ®lter binding and gel retar-dation) for the best and worst sequences probed byRicelli et al.40
that the TM of these oligonucleotides differed by27 deg. C (Table 2, lines 3 and 5). Conversely, oli-gonucleotides which embedded the GTG or TGG¯anking triplet in identical contexts had markedlydifferent binding constants, despite the fact thatthe oligonucleotides had identical values of TM
(Table 2, lines 1 and 2). (We obtained bindingvalues under similar conditions (not shown) forthe ``worst'' sequence of Ricelli et al.40 that wereconsiderably better than they reported, with excel-lent agreement between data from ®lter-bindingand those from gel-retardation with the gels atpH 6.25 rather than the pH 8.3 that they used.40
We have explained elsewhere in detail25 why gel-retardation measurements at pH 8.3 are liable togive artifactually low equilibrium binding con-stants.) Thus, our data show conclusively that it isbase-sequence and not duplex stability per se thatis the property relevant to protein binding, consist-ent with a large body of literature showingsequence-speci®c effects on the anisotropic ¯exi-bility of DNA.
(4) The sequences of the favored ¯anking regionsfor BamHI endonuclease are not the same as thosefavored for the binding of EcoRI endonuclease orEcoRV endonuclease. For example, RRR and YRRtriplets are most favored for EcoRV and leastfavored for EcoRI25,35 (M. Kurpiewski, D. Chi, R.Varma & L.J.-J., unpublished results) and BamHI(Table 1) and the most favored EcoRI triplets (YRYmotif, particularly CGC and TGC) are not the mostpreferred for BamHI (Table 1). The most-favoredmotif for BamHI is not easily classi®ed, since thebest triplets are GTG and TAT (Table 1). The factthat each protein has distinct ¯anking-triplet pre-ferences strongly suggests that ¯anking sequencecontext affects �G�bind not by altering the free-energy level of the free DNA, but rather by affect-ing the free energy levels of the various complexes.These complexes differ markedly in the extent andvectorial nature of the imposed DNA distortion:the DNA is strongly bent axially in the EcoRV com-plex,4,53 kinked and partially unwound in theEcoRI complex,54 and little distorted in the BamHIcomplex except for an abnormal propeller twist inthe A �T base-pairs.6 (This disregards the distor-tions at the oligonucleotide ends of the BamHIcomplex that probably re¯ect crystal packingforces.) It thus appears that a ``favored'' ¯ankingsequence for each protein is that which minimizesthe energetic cost of the peculiar distortionrequired in each complex. In other words, it is notsome generalized ``¯exibility'' property of the freeDNA that is important, but its propensity to roll,slide and/or twist at a speci®c location to assumesome particular distorted conformation in complexwith a particular protein.
(5) Flanking-context effects do not depend on thefact that we measure binding in the absence ofdivalent cation. We also measured binding in thepresence of Ca2� (Table 3), which mimics27 andcompetes with the natural cofactor Mg2� (seebelow, Figure 7) but does not support catalysis,

Table 3. The effect of divalent cation on speci®c and non-speci®c BamHI binding
SequenceaKA
b
(Mÿ1)Relative KA
c
(fold decrease)��G�bind
d
(kcal molÿ1)��G�Ca2�
e
(kcal molÿ1)
SpecificGGGATGGGTGggatccCACCCAC
ÿCa2� 4.2 (�0.4) � 108 1.0 0�Ca2� 1.0 (�0.1) � 1011 1.0 0 ÿ3.2 � 0.1
CGCGGGCTATggatccATACGGGCÿCa2� 3.8 (�0.2) � 108 1.1 0.1 � 0.1�Ca2� 6.7 (�0.9) � 1010 1.5 0.2 � 0.1 ÿ3.0 � 0.1
CGCGGGCGGCggatccGGGCGGGCf
ÿCa2� 2.3 (�0.5) � 108 1.8 0.4 � 0.1�Ca2� 8.6 (�0.9) � 1010 1.2 0.1 � 0.1 ÿ3.5 � 0.1
GGGATGGTGGggatccCCACCACÿCa2� 1.8 (�0.2) � 107 23 1.8 � 0.1�Ca2� 6.7 (�0.8) � 109 21 1.6 � 0.1 ÿ3.5 � 0.1
GGGATGGCGGggatccCCGCCACg
ÿCa2� 1.3 (�0.3) � 107 32 2.1 � 0.1�Ca2� 4.0 (�0.3) � 109 25 1.9 � 0.1 ÿ3.4 � 0.1
NonspecificCGCGGGCGGCcctaggGGGCGGGCh
ÿCa2� 1.3 (�0.8) � 105 3200i 4.7 � 0.4i
�Ca2� 1.2 � 105 830,000i 8.0i 0
a Speci®c sequences contain the recognition site (ggatcc) embedded in different ¯anking contexts; reference sequence (line 1) is thebest context (GTG triplet) of Table 1.
b Equilibrium association constants were determined in binding buffer plus 0.24 M potassium acetate (pH 7.3, 21 �C) or bindingbuffer plus 0.225 M potassium acetate and 0.015 M calcium acetate; note that the total cation ion concentration [K�] or [K� plusCa2�] is 0.24 M. Also note that the K� concentration is higher than that in Tables 1 and 2 but the same as that used in Table 5; seeMaterials and Methods.
c Relative KA (fold decrease) is KAref/KA
var. Values ÿCa2� are compared to reference ÿCa2� and values � Ca2� are compared toreference � Ca2�.
d Observed difference in standard binding free energy between each ¯anking sequence variant and the reference sequence;��G�bind � ÿ RT ln (KA
var/KAref).
e ��G�Ca2� � ÿ RT ln (KA (�Ca2�)/KA (ÿCa2�)).f Sequence context used for data in Tables 4, 5 and 6.g Worst cognate context (Table 1).h Non-speci®c sequence contains an inverted BamHI site (cctagg) embedded in the same context as that of footnote f.i Relative KA and ��G� bind for the non-speci®c sequence will vary depending on K� concentration; the slopes of log KA versus
log [K�Acÿ] are ÿ4.4 and ÿ6.1 for formation of the speci®c and non-speci®c complexes, respectively.
Figure 3. Dependence of BamHI endonuclease-DNAcomplex stability on substrate length for three differentsets of ¯anking sequences. Each curve represents sym-metrical truncations of parent 24 bp oligomers:CGCGGGCTATggatccATACGGGC (~), CGCGGGC-GTGggatccCACCGGGC (*), and CGCGGGC-GGCggatccGGGCGGGC (*). Values are means � S.D.for ®ve to ten determinations. All oligonucleotides wereexhaustively phosphorylated at the 50-ends.
624 BamHI interaction with its Cognate GGATCC Site
and which binds as a Mg2� analogue in the BamHIactive site to form a ``pre-reactive complex''11 inwhich the active site negative charge cluster is neu-tralized (see below). The addition of Ca2� does notchange the hierarchy of dependences on ¯ankingcontext; that is, the ratio of KA for each sequence tothe KA for the best sequence remains approxi-mately the same in the presence and absence ofCa2� (Table 3, column 3). For each of the ®vespeci®c sequences (GGATCC in various contexts)in Table 3, Ca2� increases binding an average of300-fold (average ��G� � ÿ3.3 kcal/mol). Thus,¯anking-context effects within the category of``speci®c'' complexes are, to a ®rst approximation,independent of divalent metal.
These ¯anking-context effects raise the questionof whether some of the substrate-length effectsobserved in Figure 1 result from changes in the¯anking sequence motif or module as length isincreased. To test this, we determined the lengthdependence of complex stability for three different¯anking sequences. We found (Figure 3) thatincreasing substrate length caused the same rela-tive increases in complex half-life for each oligonu-cleotide series. This con®rms that increasing DNAlength promotes binding by providing critical

BamHI interaction with its Cognate GGATCC Site 625
DNA phosphate groups rather than through base-sequence effects.
Comparison of specific and non-specificbinding
We determined the equilibrium association con-stant (KA) for binding to non-speci®c DNA using aninverted recognition site embedded in a 24 bp oligo-nucleotide (CGCGGGCGGCcctaggGGGCGGGC).Inversion of the recognition site minimizes protein-DNA recognition by replacing all hydrogen-bonddonors in the GGATCC recognition site with hydro-gen-bond acceptors and acceptors with donors.55
For purposes of calculating the speci®city ratio (i.e.the ratio of speci®c KA to non-speci®c KA), it isimportant to note that non-speci®c binding, unlikespeci®c binding, is insensitive to ¯anking sequencecontext (D. Chi & L.J.-J., unpublished results). Thus,Table 3 shows that in the absence of divalent cationand using the best sequence context (GTG ¯ank; seealso Table 1, line 1) for comparison, speci®c bindingto the GGATCC site is 3200-fold stronger than non-speci®c binding; for the worst context (CGG ¯ank)the ratio is 100-fold. This emphasizes the point thata better sequence context improves speci®city. Fur-thermore, speci®c and non-speci®c binding havedifferent dependences on pH and salt concen-tration,56 (the slope of a log KA versus log [K�Acÿ]plot is ÿ4.4 for speci®c and ÿ6.1 for non-speci®csites) so the speci®city ratio depends on the (arbitra-rily) chosen values of both these environmentalvariables. (The salt concentration and pH used inTable 3 are close to physiological.) We concludethat BamHI endonuclease binds to its recognitionsite with moderate speci®city in the absence of diva-lent metal; the numerical value of ``speci®city'' is sodependent on context and environmental con-ditions that it is only informative to make numericalcomparisons with the speci®city ratios of otherDNA-binding proteins under precisely the samepH, cation and anion conditions.
We also found (Table 3) that non-speci®c bind-ing is unaffected by Ca2�, whereas speci®c bindingis strongly enhanced. As a result, Ca2� dramati-cally increases the speci®city ratio, for the bestsequence context from 3200 (ÿCa2�) to 830,000(�Ca2�), and for the worst context from 100(ÿCa2�) to 33,300 (�Ca2�) We conclude that thedifferentiation between speci®c and non-speci®cbinding depends quantitatively, but not qualitat-ively, on divalent metal (as was observed forEcoRV endonuclease25). Even without divalentmetal, the ``speci®c'' complex is readily distin-guished by the thermodynamic signature of a largenegative heat capacity change upon binding37andthe formation of a localized footprint on the DNA.
Footprinting analysis of protein-phosphatecontacts
Ethylation-interference footprinting57 shows theextent to which ethylation at individual phosphate
groups inhibits binding. This technique mapsfunctionally signi®cant contacts rather than simpleproximity of the protein.19,58 Comparison ofethylation-interference patterns with the crystalstructures of EcoRI-DNA and repressor-DNA com-plexes shows that strong interference occurs onlyat those phosphate groups tightly constrained byhydrogen bonds to short polar side-chains or poly-peptide backbone.35,59 Phosphate groups whichcontact the protein via salt-links or water-mediatedinteractions show only weak ethylation interfer-ence.
Figure 4 shows sample footprints made on thespeci®c site GGATCC (using the same ¯ankingtriplet as that in the BamHI-DNA co-crystalstructure6) and on a ``non-speci®c'' site, CCTAGG.There is no evidence of a localized footprint on thenon-speci®c DNA, indicating that binding is equi-probable at any point along the oligonucleotide,but the GGATCC site supports a strong localizedfootprint which is approximately symmetrical onthe two DNA strands.
Figure 5 shows a quantitative comparison ofthe ethylation interference footprints of BamHIand EcoRI endonucleases. A set of eight strongcontacts are made by BamHI endonuclease atNpNpNpGGApTCC, symmetrically on each strand.The same set of phosphate contacts, with quantitat-ively similar levels of interference, was observed(data not shown) for several other ¯anking con-texts (Table 1). These ``clamp'' contacts appear tobe indispensable for binding to the canonicalsubstrate. Note especially the relatively strong(50-fold) inhibition of binding by single-strandethylation at NpNNGGATCC (Figure 5) and thestrong (240-fold) increase in binding as the samephosphate was added (on both strands) to a 10 bpoligonucleotide (Figure 1). This phosphate, wherethere is no analogous strong interaction in theEcoRI complex, is the one that is mispositioned inthe BamHI-DNA crystalline complex.6
To con®rm the identi®cation of these criticalphosphate contacts, we also performed a deletionanalysis by synthesizing a series of substrates inwhich, in only one DNA strand, each phosphatewas either deleted entirely, or the phosphodiesterbond converted to a 50-phosphomonoester. Theresults (Table 4) show that the energetic cost ofphosphate deletion ranges from �0.6 kcal/mol to�2.4 kcal/mol in �G�bind, corresponding to a pen-alty of 3 to 60-fold in binding. In each case, thesingle-strand ``nick'' leaving a phosphomonoesterwas more deleterious to binding than was com-plete deletion of a phosphate, even though thephosphomonoester still has non-bridging phos-phoryl oxygen atoms to interact with protein whilethe corresponding oxygen atoms are completelyabsent from the deletion oligonucleotide. This indi-cates that introducing the additional charged oxy-gen atom of the phosphomonoester into eachlocally crowded environment in the complexentails steric and/or electrostatic penalties that out-weigh the energetic bene®t of direct protein inter-
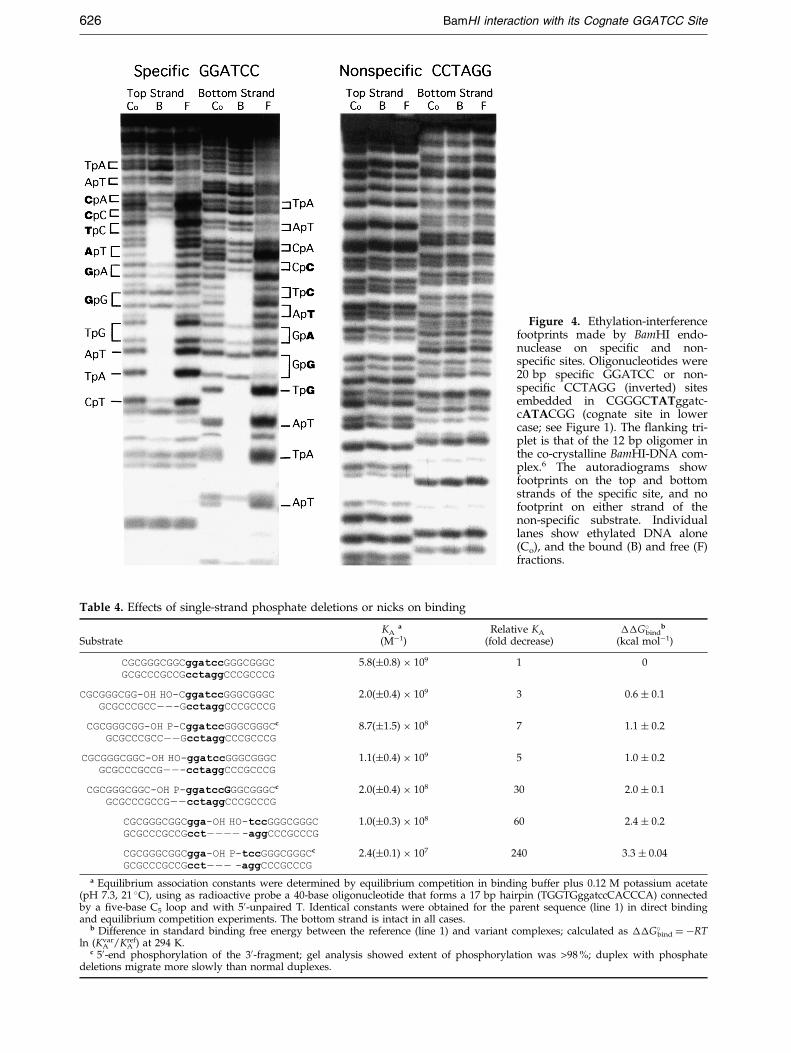
Figure 4. Ethylation-interferencefootprints made by BamHI endo-nuclease on speci®c and non-speci®c sites. Oligonucleotides were20 bp speci®c GGATCC or non-speci®c CCTAGG (inverted) sitesembedded in CGGGCTATggatc-cATACGG (cognate site in lowercase; see Figure 1). The ¯anking tri-plet is that of the 12 bp oligomer inthe co-crystalline BamHI-DNA com-plex.6 The autoradiograms showfootprints on the top and bottomstrands of the speci®c site, and nofootprint on either strand of thenon-speci®c substrate. Individuallanes show ethylated DNA alone(Co), and the bound (B) and free (F)fractions.
Table 4. Effects of single-strand phosphate deletions or nicks on binding
SubstrateKA
a
(Mÿ1)Relative KA
(fold decrease)��G�bind
b
(kcal molÿ1)
CGCGGGCGGCggatccGGGCGGGC 5.8(�0.8) � 109 1 0GCGCCCGCCGcctaggCCCGCCCG
CGCGGGCGG-OH HO-CggatccGGGCGGGC 2.0(�0.4) � 109 3 0.6 � 0.1GCGCCCGCCÐ Ð-GcctaggCCCGCCCG
CGCGGGCGG-OH P-CggatccGGGCGGGCc 8.7(�1.5) � 108 7 1.1 � 0.2GCGCCCGCCÐ ÐGcctaggCCCGCCCG
CGCGGGCGGC-OH HO-ggatccGGGCGGGC 1.1(�0.4) � 109 5 1.0 � 0.2GCGCCCGCCGÐ Ð-cctaggCCCGCCCG
CGCGGGCGGC-OH P-ggatccGGGCGGGCc 2.0(�0.4) � 108 30 2.0 � 0.1GCGCCCGCCGÐ ÐcctaggCCCGCCCG
CGCGGGCGGCgga-OH HO-tccGGGCGGGC 1.0(�0.3) � 108 60 2.4 � 0.2GCGCCCGCCGcctÐ Ð Ð Ð -aggCCCGCCCG
CGCGGGCGGCgga-OH P-tccGGGCGGGCc 2.4(�0.1) � 107 240 3.3 � 0.04GCGCCCGCCGcctÐ Ð Ð -aggCCCGCCCG
a Equilibrium association constants were determined by equilibrium competition in binding buffer plus 0.12 M potassium acetate(pH 7.3, 21 �C), using as radioactive probe a 40-base oligonucleotide that forms a 17 bp hairpin (TGGTGggatccCACCCA) connectedby a ®ve-base C5 loop and with 50-unpaired T. Identical constants were obtained for the parent sequence (line 1) in direct bindingand equilibrium competition experiments. The bottom strand is intact in all cases.
b Difference in standard binding free energy between the reference (line 1) and variant complexes; calculated as ��G�bind � ÿRTln (KA
var/KAref) at 294 K.
c 50-end phosphorylation of the 30-fragment; gel analysis showed extent of phosphorylation was >98 %; duplex with phosphatedeletions migrate more slowly than normal duplexes.
626 BamHI interaction with its Cognate GGATCC Site
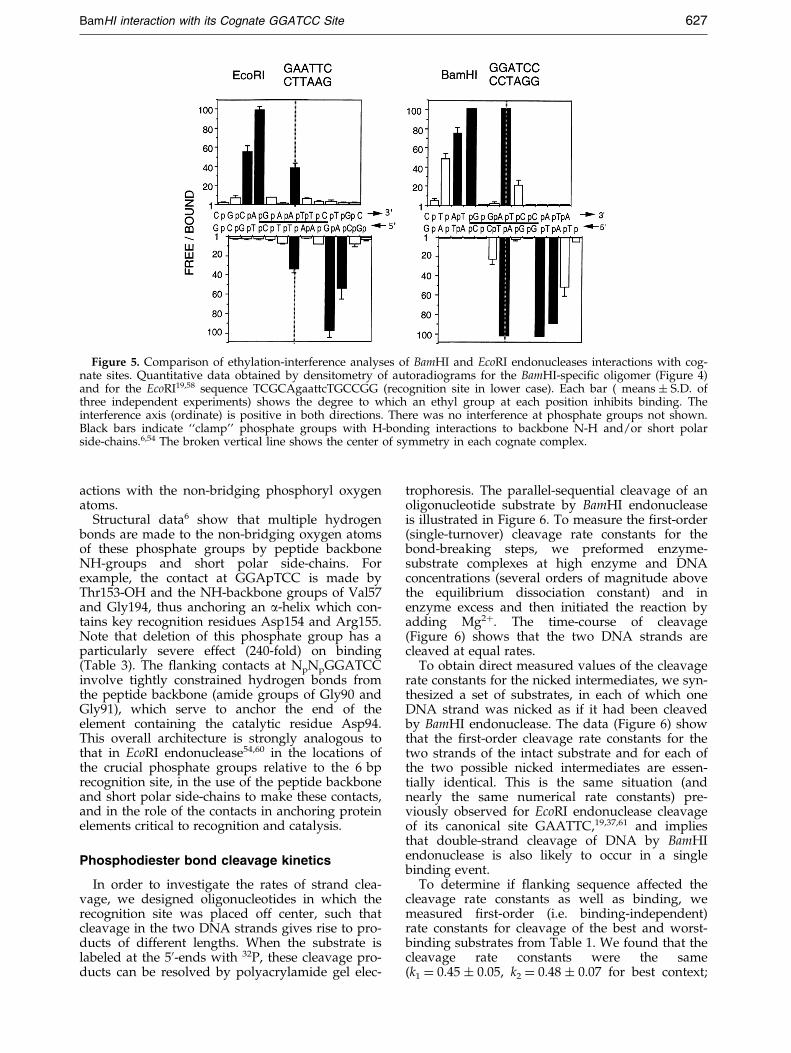
Figure 5. Comparison of ethylation-interference analyses of BamHI and EcoRI endonucleases interactions with cog-nate sites. Quantitative data obtained by densitometry of autoradiograms for the BamHI-speci®c oligomer (Figure 4)and for the EcoRI19,58 sequence TCGCAgaattcTGCCGG (recognition site in lower case). Each bar ( means � S.D. ofthree independent experiments) shows the degree to which an ethyl group at each position inhibits binding. Theinterference axis (ordinate) is positive in both directions. There was no interference at phosphate groups not shown.Black bars indicate ``clamp'' phosphate groups with H-bonding interactions to backbone N-H and/or short polarside-chains.6,54 The broken vertical line shows the center of symmetry in each cognate complex.
BamHI interaction with its Cognate GGATCC Site 627
actions with the non-bridging phosphoryl oxygenatoms.
Structural data6 show that multiple hydrogenbonds are made to the non-bridging oxygen atomsof these phosphate groups by peptide backboneNH-groups and short polar side-chains. Forexample, the contact at GGApTCC is made byThr153-OH and the NH-backbone groups of Val57and Gly194, thus anchoring an a-helix which con-tains key recognition residues Asp154 and Arg155.Note that deletion of this phosphate group has aparticularly severe effect (240-fold) on binding(Table 3). The ¯anking contacts at NpNpGGATCCinvolve tightly constrained hydrogen bonds fromthe peptide backbone (amide groups of Gly90 andGly91), which serve to anchor the end of theelement containing the catalytic residue Asp94.This overall architecture is strongly analogous tothat in EcoRI endonuclease54,60 in the locations ofthe crucial phosphate groups relative to the 6 bprecognition site, in the use of the peptide backboneand short polar side-chains to make these contacts,and in the role of the contacts in anchoring proteinelements critical to recognition and catalysis.
Phosphodiester bond cleavage kinetics
In order to investigate the rates of strand clea-vage, we designed oligonucleotides in which therecognition site was placed off center, such thatcleavage in the two DNA strands gives rise to pro-ducts of different lengths. When the substrate islabeled at the 50-ends with 32P, these cleavage pro-ducts can be resolved by polyacrylamide gel elec-
trophoresis. The parallel-sequential cleavage of anoligonucleotide substrate by BamHI endonucleaseis illustrated in Figure 6. To measure the ®rst-order(single-turnover) cleavage rate constants for thebond-breaking steps, we preformed enzyme-substrate complexes at high enzyme and DNAconcentrations (several orders of magnitude abovethe equilibrium dissociation constant) and inenzyme excess and then initiated the reaction byadding Mg2�. The time-course of cleavage(Figure 6) shows that the two DNA strands arecleaved at equal rates.
To obtain direct measured values of the cleavagerate constants for the nicked intermediates, we syn-thesized a set of substrates, in each of which oneDNA strand was nicked as if it had been cleavedby BamHI endonuclease. The data (Figure 6) showthat the ®rst-order cleavage rate constants for thetwo strands of the intact substrate and for each ofthe two possible nicked intermediates are essen-tially identical. This is the same situation (andnearly the same numerical rate constants) pre-viously observed for EcoRI endonuclease cleavageof its canonical site GAATTC,19,37,61 and impliesthat double-strand cleavage of DNA by BamHIendonuclease is also likely to occur in a singlebinding event.
To determine if ¯anking sequence affected thecleavage rate constants as well as binding, wemeasured ®rst-order (i.e. binding-independent)rate constants for cleavage of the best and worst-binding substrates from Table 1. We found that thecleavage rate constants were the same(k1 � 0.45 � 0.05, k2 � 0.48 � 0.07 for best context;
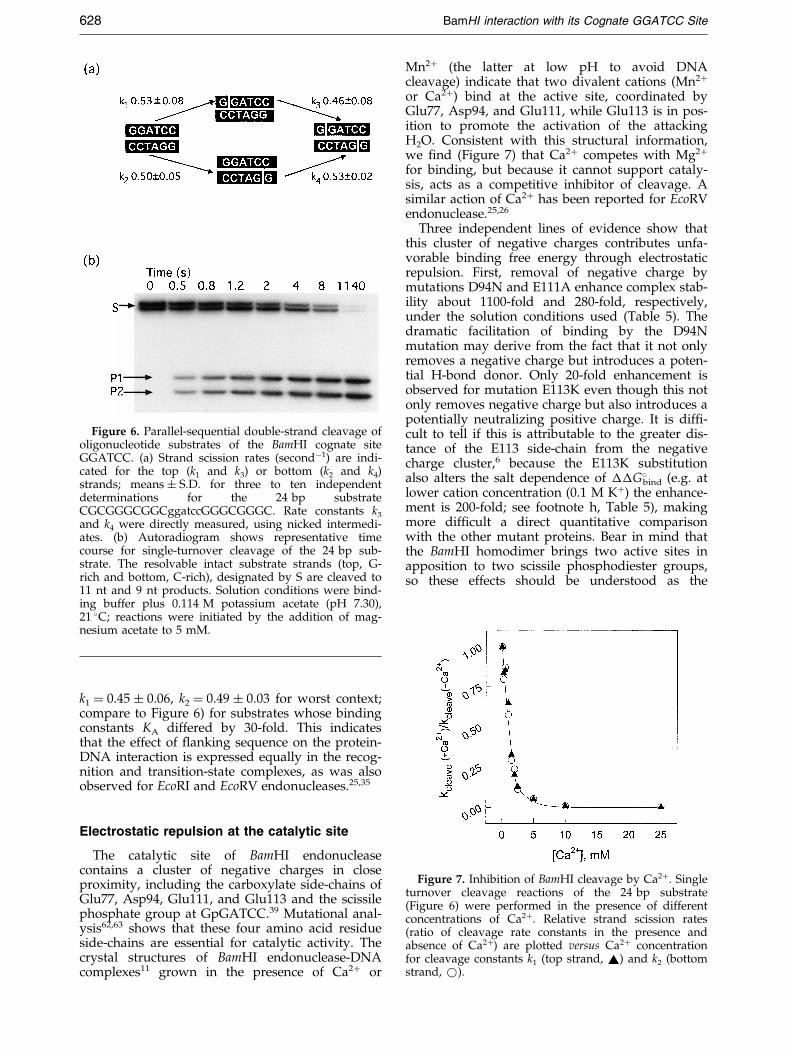
Figure 7. Inhibition of BamHI cleavage by Ca2�. Singleturnover cleavage reactions of the 24 bp substrate(Figure 6) were performed in the presence of differentconcentrations of Ca2�. Relative strand scission rates(ratio of cleavage rate constants in the presence andabsence of Ca2�) are plotted versus Ca2� concentrationfor cleavage constants k1 (top strand, ~) and k2 (bottomstrand, *).
Figure 6. Parallel-sequential double-strand cleavage ofoligonucleotide substrates of the BamHI cognate siteGGATCC. (a) Strand scission rates (secondÿ1) are indi-cated for the top (k1 and k3) or bottom (k2 and k4)strands; means � S.D. for three to ten independentdeterminations for the 24 bp substrateCGCGGGCGGCggatccGGGCGGGC. Rate constants k3
and k4 were directly measured, using nicked intermedi-ates. (b) Autoradiogram shows representative timecourse for single-turnover cleavage of the 24 bp sub-strate. The resolvable intact substrate strands (top, G-rich and bottom, C-rich), designated by S are cleaved to11 nt and 9 nt products. Solution conditions were bind-ing buffer plus 0.114 M potassium acetate (pH 7.30),21 �C; reactions were initiated by the addition of mag-nesium acetate to 5 mM.
628 BamHI interaction with its Cognate GGATCC Site
k1 � 0.45 � 0.06, k2 � 0.49 � 0.03 for worst context;compare to Figure 6) for substrates whose bindingconstants KA differed by 30-fold. This indicatesthat the effect of ¯anking sequence on the protein-DNA interaction is expressed equally in the recog-nition and transition-state complexes, as was alsoobserved for EcoRI and EcoRV endonucleases.25,35
Electrostatic repulsion at the catalytic site
The catalytic site of BamHI endonucleasecontains a cluster of negative charges in closeproximity, including the carboxylate side-chains ofGlu77, Asp94, Glu111, and Glu113 and the scissilephosphate group at GpGATCC.39 Mutational anal-ysis62,63 shows that these four amino acid residueside-chains are essential for catalytic activity. Thecrystal structures of BamHI endonuclease-DNAcomplexes11 grown in the presence of Ca2� or
Mn2� (the latter at low pH to avoid DNAcleavage) indicate that two divalent cations (Mn2�
or Ca2�) bind at the active site, coordinated byGlu77, Asp94, and Glu111, while Glu113 is in pos-ition to promote the activation of the attackingH2O. Consistent with this structural information,we ®nd (Figure 7) that Ca2� competes with Mg2�
for binding, but because it cannot support cataly-sis, acts as a competitive inhibitor of cleavage. Asimilar action of Ca2� has been reported for EcoRVendonuclease.25,26
Three independent lines of evidence show thatthis cluster of negative charges contributes unfa-vorable binding free energy through electrostaticrepulsion. First, removal of negative charge bymutations D94N and E111A enhance complex stab-ility about 1100-fold and 280-fold, respectively,under the solution conditions used (Table 5). Thedramatic facilitation of binding by the D94Nmutation may derive from the fact that it not onlyremoves a negative charge but introduces a poten-tial H-bond donor. Only 20-fold enhancement isobserved for mutation E113K even though this notonly removes negative charge but also introduces apotentially neutralizing positive charge. It is dif®-cult to tell if this is attributable to the greater dis-tance of the E113 side-chain from the negativecharge cluster,6 because the E113K substitutionalso alters the salt dependence of ��G�bind (e.g. atlower cation concentration (0.1 M K�) the enhance-ment is 200-fold; see footnote h, Table 5), makingmore dif®cult a direct quantitative comparisonwith the other mutant proteins. Bear in mind thatthe BamHI homodimer brings two active sites inapposition to two scissile phosphodiester groups,so these effects should be understood as the

Table 5. Probes of electrostatic repulsion in the BamHI endonuclease active site
Protein DNAa AdditionKA
b
(Mÿ1)Relative KA
c
(fold increase)��G�bind
d
(kcal molÿ1)
wt BamHI intact 2.3(�0.5) � 108 1 0D94N intact N.D.e 1100f ÿ4.1 � 0.1g
E111A intact 6.1(�1.0) � 1010 280 ÿ3.3 � 0.1E113K intact 4.9(�0.4) � 109 20 ÿ1.8 � 0.1h
wt BamHI intact Ca2� 8.6(�0.9) � 1010 370 ÿ3.5 � 0.1wt BamHI G-OH P- GATCC 6.1(�0.6) � 108 3 ÿ0.6 � 0.1
CÐ Ð Ð Ð CTAGGi
wt BamHI G-OHHOÐGATCC 1.2(�0.1) � 1011 560 ÿ3.7 � 0.2CÐ Ð-Ð CTAGGi
a The intact oligodeoxynucleotide was CGCGGCGGCggatccGGGCGGGC (recognition site in lower case) except when ``nicked'' asindicated.
b Equilibrium association constants were determined in binding buffer plus 0.24 M potassium acetate (pH 7.3, 21 �C); Note thatthe K� concentration is higher than that in Tables 1 and 2.
c Relative KA (fold increase) is KA/KAref where KA
ref is the equilibrium association constant for wild type BamHI endonuclease withintact oligomer in the absence of Ca2�.
d ��G�bind � ÿRT ln KA/KAref at 294 K.
e KA too large to be determined reliably.f Determined from dissociation kinetics of wild-type and mutant complexes; kd/kd
ref (see footnote g).g ��G�{d � ÿRT ln (kd/kd
ref). It is generally true that ÿ��G�{d � ��G�bind.43
h The slopes of log KA versus log [K�Acÿ] are ÿ4.4 for wild-type protein and ÿ7.1 for E113K; thus at lower salt concentration��G�bind is greater (e.g. at 0.1 M potassium acetate, ��G�bind for E113K is ÿ3.1 kcal/mol).
i The bottom strand is intact.
BamHI interaction with its Cognate GGATCC Site 629
removal of two negative charges in each case, andthe introduction of two positive charges for E113K.Similar, but semi-quantitative observations (no KA
values were reported), have been made formutations of MunI endonuclease33 that removeactive-site carboxylate charges. As might beexpected from this model, we have also found(L.E.E., G. Bosco & L.J.-J., unpublished results) thatprotonation of these active-site residues at low pHalso strongly enhances binding (KA at pH 5.5approximately 160-fold higher than at pH 8.5).
Second, stability of the complex with wild-typeBamHI endonuclease is enhanced about 300-foldby the addition of Ca2� (Tables 3 and 5), presum-ably because Ca2� binding neutralizes negativecharge in the active-site clusters. (Binding of theE111A mutant protein is not enhanced by Ca2�;data not shown.) However, it is not clear howmany calcium ions are binding to produce thiseffect. In BamHI-DNA co-crystals, only one of thetwo active sites is occupied by two metal atoms,while the other contains no metal.11 This single-siteoccupancy may be a function of constraints in thecrystal, and it is possible that as many as fourcalcium ions bind to each BamHI homodimer insolution. Neither cleavage kinetics (Figure 6) northe Ca2�-competition curve (Figure 7) illuminatethis point, and we are investigating further byisothermal titration calorimetry studies. Neverthe-less, because Ca2� does not stimulate binding tonon-speci®c DNA (Table 3), binding speci®city isenhanced by Ca2�. This suggests that the negative-charge clusters at the active sites are not assembledin the complex with non-speci®c DNA (the struc-ture of the non-speci®c BamHI-DNA15 shows thatthe active site residues are displaced away fromthe scissile phosphate group by greater than 6 AÊ ).
A similar differential effect of Ca2� has been pro-posed for MunI endonuclease.32
Third, stability of the complex is enhanced about560-fold (Table 5) by deletion of the scissile phos-phate group at GpGATCC (on one strand only).This effect also results primarily from the removalof negative charge, since nicking of the substratewithout phosphate deletion (i.e. converting the50-30 phosphodiester to a 50-phosphomonoester),enhances binding only threefold (Table 5). The nickalso introduces an additional negative charge, butthe electrostatic disadvantage of this is apparentlymore than offset by the increased freedom of thephosphomonoester to relieve the electrostaticrepulsion by movement away from the negativelycharged protein side-chains. Such a movementaway from Glu77, Asp94, and Glu111 is plainlyvisible in the crystal structure of the post-reactivecomplex.11
These observations reinforce our general view35
that the observed �G�bind for site-speci®c DNA-binding proteins is the resultant of large favorableand unfavorable energetic factors. The electrostaticclusters at the active sites are a particularly inter-esting example of unfavorable forces in the assem-bly of a recognition complex, because the attendantrepulsive energy destabilizes the ground state ofthe protein-DNA complex and strongly favors thebinding of metal ions and progression to the tran-sition state. This mechanism helps solve the essen-tial chemical problem for restriction endonucleases:they must bind their recognition sites very tightlyin order to discriminate between the correct sitesand the vast molar excess of non-speci®c DNAin vivo, yet this strong ground-state binding mustnot be a deep ``energy trap'' that increases theenergy of activation to the point where catalysis isdisfavored. We have argued elsewhere35 that the

630 BamHI interaction with its Cognate GGATCC Site
problem is partly addressed by the formation of``pre-transition-state'' complexes which have veryclose structural resemblance to the transition-statecomplexes over the entire protein-DNA interface,such that binding energy is ef®ciently utilized forcatalysis. In the case of BamHI endonuclease,where the relatively minor DNA distortion is likelynot a factor in destabilizing the pre-transition-statecomplex, we see how an unfavorable electrostaticcomponent of binding energy (a form of ``strainenergy''36) arising directly at the catalytic centercan contribute to catalysis. This active site con-®guration is not assembled in the complex withnon-speci®c DNA and thus there is no comparableelectrostatic drive towards the transition state.Similar but less extensive clusters of negativecharges occur at the active sites of EcoRI andEcoRV endonucleases, in both of which the residuein a position analogous to BamHI-Glu113 is alysine.39,64 In contrast to BamHI, EcoRI and EcoRVendonucleases strongly distort their bound DNAs,such that the strain energy of DNA distortion alsodestabilizes their pre-transition-state complexes.
Are there asymmetric interactions with theDNA minor groove?
The structures of the endonuclease apoenzymeand the protein-DNA complex6,39 suggest thatupon binding to DNA, the a-helices at the C termi-ni of the homodimer unfold to produce extended``arms'', which interact asymmetrically with DNA.
Table 6. Binding of BamHI to sites with inosine substitutions
KAb
(Mÿ1)
Sitea
GGATCC 5.8(�0.8) � 109
CCTAGG
IGATCC 1.6(�0.5) � 109
CCTAGG
GGATCC 1.1(�0.4) � 108
CCTAGI
IGATCC 3.0(�0.2) � 107
CCTAGI
GIATCC 5.2(�1.1) � 108
CCTAGG
GGATCC 4.0(�1.6) � 108
CCTAIG
GIATCC 2.5(�0.9) � 107
CCTAIG
aEach site is embedded in the duplex: CGCGGCGGCggatccGGGCGGGCGCCGCCGcctaggCCCGCC
b Equilibrium association constants were determined by equilibriu(pH 7.3, 21 �C ) using as radioactive probe the same 40 base oligonuobtained for the parent sequence (line 1) in direct binding and equili
c Difference in the observed standard binding free energy betweecalculated as ��G�bind � ÿ RT ln (KA/KA
ref) at 294 K.d Predicted value is the sum of observed ��G�bind values for each
orientation as in the double-analogue site.
One arm makes hydrogen bonds with functionalgroups in the minor groove, whereas the other armmakes a salt bridge and hydrogen bond with phos-phate groups of a ``translationally related DNAfragment''.6 This marked asymmetry is unprece-dented for a protein homodimer interacting with aperfectly palindromic recognition site.
There are no minor-groove contacts proposed forthe ®rst G �C base-pair in either half-site, but forthe second G �C base-pair (in one half-site only) ahydrogen bond is proposed between the 2-NH2
group of G2 and the carboxylate group of Asp196.In the other half-site, only a water molecule isbound to the corresponding 2-NH2 group. To testif this asymmetric interaction occurs in solution,we substituted the second guanine in the BamHIrecognition site with inosine in one or both DNAstrands so as to delete one or both minor-groove 2-NH2 groups. The results (Table 6) show that ino-sine substitution in one strand in the second base-pair (GIATCC) evokes a penalty of �1.4 kcal/mol,and the derivative in which inosine is substitutedin both DNA strands (i.e. GIATCC/CCTAIG) pro-duces nearly twice the penalty for the single substi-tution. As a control we examined inosinesubstitution in the ®rst base-pair (i.e. IGATCC),where no minor-groove interaction is proposed,and also observed a penalty of similar magnitudeand twice the penalty in the double-substitutedderivative. There are three possible interpretationsof these observations:
��G�bindc
(kcal molÿ1)��G�bind
d
(kcal molÿ1)
Observed Predicted
0 0
0.8 � 0.2
1.0 � 0.2
1.7 � 0.1 1.8 � 0.4
1.4 � 0.2
1.5 � 0.2
2.4 � 0.2 2.9 � 0.4
GC
CG
m competition in binding buffer plus 0.12 M potassium acetate,cleotide as that of Table 4 (footnote a). Identical constants were
brium competition experiments.n the unmodifed reference site (line 1) and each analogue site;
of the constituent single-analogue sites, each taken in the same

BamHI interaction with its Cognate GGATCC Site 631
(1) Protein binds to the single-substituted DNAasymmetrically but in random orientation, so thatin half the complexes the ``arm'' makes minor-groove contact with the unmodi®ed half-site andin half the complexes it contacts the modi®ed half-site.
(2) Protein binds symmetrically so the armmakes minor-groove contact in both half-sites.
(3) Protein binds symmetrically, but the armmakes minor-groove contacts in neither half-site.In this case, the penalties for inosine substitutionswould re¯ect effects of the 2-NH2 group and/orminor-groove Watson-Crick hydrogen bond onDNA conformational properties rather than disrup-tion of a minor-groove protein-base contact.
If interpretation (1) were correct we wouldexpect that the single-substituted complex wouldbe an approximately equimolar mixture of stableand unstable complexes, whose stabilities shoulddiffer by 1.5 kcal/mol19,55,65 or about tenfold in dis-sociation rate. To test this possibility, we measuredthe kinetics of dissociation of this complex(Figure 8). There is obviously no sign of suchbiphasic dissociation kinetics, ruling out interpret-ation (1).
To distinguish between interpretations (2) and(3), we studied a mutant BamHI endonucleasein which Asp196 is replaced by alanine.
Figure 8. Kinetics of dissociation of BamHI endonu-clease-DNA complexes with unmodi®ed and inosine-substituted sites. Complexes between BamHI endonu-clease and each radiolabeled oligonucleotide wereformed such that the protein and DNA concentrationswere 500-fold above the equilibrium dissociation con-stant (KD) for the solution conditions of the experiment(binding buffer plus 0.07 M potassium acetate (pH 7.30),21 �C), and dissociation measured as described inMaterials and Methods. Radiolabeled DNA concen-tration was 5 nM for the unmodi®ed site, 20 nM for theIGATCC site and 62.5 nM for the GIATCC site; enzymeconcentration was 90 % that of the respective DNA con-centrations. Curves represent data from three individualexperiments for the cognate site GGATCC (*), IGATCC(~), and GIATCC (!).
This mutation should eliminate the proposedminor-groove contact to G2-NH2. Surprisingly,we saw an improvement in binding(��G�bind � ÿ 1.1(�0.1) kcal/mol, same solutionconditions as those of Table 5) rather than the pen-alty expected if this side-chain was hydrogen-bonding to DNA. This result is consistent with alocation of the Asp196 side-chain near a negativelycharged group, perhaps a DNA phosphate. Themutant enzyme was able to cleave DNA as well aswild-type BamHI endonuclease. These observationsrule out interpretation (2), and are also inconsistentwith interpretation (1).
We therefore infer that at both the ®rst andsecond base-pairs of the recognition site the 2-NH2
groups of guanine and/or the minor-groove Wat-son-Crick hydrogen bonds affect the energetics ofBamHI-DNA binding through effects on the confor-mational properties of the DNA, and that there islikely no protein contact in solution to the 2-NH2
group of guanine in the minor groove.
Conclusions: Principal Features of theBamHI Model System
Our data show that the interaction of BamHIendonuclease with DNA has signi®cant similaritieswith those of EcoRI and EcoRV endonucleases, andalso signi®cant differences. All three enzymes exhi-bit localized, site-speci®c DNA binding in theabsence of divalent metal, although binding isenhanced by divalent metals (Table 3). Divalentmetal ions do not ``trigger'' binding speci®city forEcoRI or BamHI, since site-speci®c complexes in theabsence of divalent metal show ``thermodynamicsignatures'' (strongly negative �C�P) completelydistinct from those of the non-speci®c complexes(�C�P � 0).37 We expect the same will be true ofEcoRV and are currently investigating this.
It is the need to bind divalent metals as catalyticcofactors near the site of phosphodiester bond clea-vage that requires all three enzymes to place nega-tively charged side-chains into homologouspositions39,64 in proximity with each other and thescissile phosphate group. This has the consequenceof producing a large unfavorable electrostatic con-tribution to the binding free energy of the speci®ccomplex in the absence of metals. We have pro-vided data (Table 5) that show how individualnegatively charged side-chains and the DNA phos-phate group contribute to this repulsion energy inthe BamHI active site, and it is reasonable to sup-pose that a similar (albeit perhaps slightly smaller)repulsion term contributes to the net unfavorableelectrostatic energy calculated for EcoRI-DNAbinding. In accord with this prediction, Ca2� alsoenhances EcoRI binding (our unpublished results)but to a lesser extent (140-fold) than for BamHI(300-fold, Tables 3 and 5) under similar pH and K�
solution conditions. Note that BamHI binding inthe absence of divalent metal is signi®cantly weak-er than that of EcoRI, in part because the BamHI

632 BamHI interaction with its Cognate GGATCC Site
active site has a greater number of negativelycharged side-chains clustered around the scissilephosphate group, generating a larger unfavorableelectrostatic contribution to binding free energy.This difference, in turn, re¯ects the fact that BamHI(like EcoRV, which also binds relatively weakly inthe absence of divalent metal13) forms two metalion binding sites in each active site11 whereasEcoRI forms only one (J.M. Rosenberg, personalcommunication).
The overall disposition of the critical protein-phosphate contacts relative to the recognition siteis roughly similar in the BamHI-DNA and EcoRI-DNA interfaces. In both cases, functionally import-ant phosphate contacts (Figure 5) lie well beyondthe boundaries of the six base-pair recognitionsequence in contrast to the EcoRV-DNA interfacewhere most of the functionally important contactslie within the recognition site.25 As a consequence,optimum BamHI binding requires an oligonucleo-tide substrate long enough (about 14-16 base-pairs)to provide the full complement of protein-phos-phate contacts, each of which contributes ÿ0.6 toÿ2.4 kcal/mol to a total net �G�bind of aboutÿ13.5 kcal/mol. Thus, the ``recognition site''should properly be considered to be signi®cantlylarger than the ``recognition sequence'' so as toinclude the crucial DNA phosphate groups.
Like the binding of EcoRI and EcoRV endonu-cleases, the binding of BamHI endonuclease to itsrecognition site (GGATCC) is sensitive to the sur-rounding sequence context, primarily in the three¯anking base-pairs on each side. The quantitativerange over which binding af®nity is modulated bycontext is similar for BamHI and EcoRV, but bothof these show a narrower range than that ofEcoRI35 (M. Kurpiewski, D. Chi, L.J.-J., unpublishedresults). In all cases, context affects binding af®nitywithout affecting the ®rst-order cleavage-rate con-stants, implying that ¯anking context does notalter the basic structure of the pre-transition-statecomplex, but only affects the energetic cost offorming that complex.35 We have shown else-where36,37 that the thermodynamic mechanisms ofthe sequence-context effects are generally similarfor BamHI and EcoRI: a better context improvesbinding free energy by making �H� more negativeand �S� more negative, while �C�P becomes morenegative. Despite these similar thermodynamictrends, the actual sequence preferences of the threeenzymes are pronouncedly different. Noteespecially that these differences do not simplyresult from apposition of the ¯anking region to adifferent ®rst base in the recognition site so as tocreate a new ``module'' or ``motif'', since the threerecognition sites all begin with G (EcoRI GAATTC;BamHI GGATCC; EcoRV GATATC). Thus, thecomparative study of one phenomenon (contexteffects) in several systems has allowed us to dis-cern that better �H� is promoted by differentsequences in each interface, implying that it is theconformational requirements of each complex thatdetermine sequence-context preference. Because
the physical boundaries of these restrictionnucleases in the protein-DNA complexes extendbeyond their six-base recognition sites to make aset of critical phosphate contacts, it should not besurprising that the proteins make use of DNA con-formational information outside each recognitionsite, as they do within the recognition site.19 Weanticipate that future comparative study of theseand other restriction endonucleases will revealother interesting points of contrast, as well as testsof the generality of inferences from experiments oneach system.
Materials and Methods
BamHI endonucleases
Wild-type BamHI and mutant E113K endonucleaseswere overexpressed and puri®ed as described.41 A geneencoding mutant E111A was cloned between the NdeIand SapI sites downstream of the T7 promoter in thepTYB1 vector of the IMPACT-CN system (New EnglandBiolabs) such that the fusion order was BamHI endonu-clease-intein-chitin binding domain. The E111A mutantprotein was then puri®ed as described66 by chitin-af®nitychromatography; cleavage of the intein to release theprotein leaves no extraneous amino acid residues. D94Nand D196A were the kind gifts of Dr Shuan-yong Xuand Richard D. Whitaker, respectively (both of NewEngland Biolabs). Enzyme purity was assessed by SDS15 % (w/v) polyacrylamide gel electrophoresis (SDS-PAGE). Lanes were overloaded with protein and silverstained67 to allow detection of as little as 0.05 % impur-ity. The wild-type, E113K, D196A, and E111A enzymeswere found to be >99 % homogeneous, while the D94Nmutant >92 % pure.
Absolute protein concentration for the wild-type pro-tein was determined by direct amino acid analysis usingnorleucine as an internal standard.16 Protein concen-trations for all the endonuclease mutants were deter-mined by comparison on coomassie-stained SDS-PAGE.
DNA oligonucleotides
All unmodi®ed oligonucleotides were synthesized oneither an Applied Biosystems or Expedite automatedDNA synthesizer by the standard phosphoramiditemethod at the University of Pittsburgh DNA synthesisfacility. Deoxyoligonucleotides containing inosine (GlenResearch) were synthesized by the standard protocolsexcept that the base addition step for the base analogwas increased to ®ve minutes (normal time of 15seconds) at 2 times the standard base concentration(1 mM).
Oligomers were puri®ed as described.25 Concen-trations of single-strand DNA fragments were deter-mined spectroscopically from extinction coef®cientscalculated by the nearest-neighbor method68 as elabo-rated by Senior et al.69 Stoichiometric amounts of comp-lementary single strands were annealed and theproducts con®rmed to be >98 % in duplex form by non-denaturing gel electrophoresis (12 % polyacrylamide,20 mM bis-Tris-propane acetate (pH 7.3), 25 �C). Concen-trations of single-stranded and duplex oligomers werealso checked by quanti®cation of UV-visualized andphotographed bands from ethidium bromide stainedgels in which the 24 nt or base-pair parent was used as

BamHI interaction with its Cognate GGATCC Site 633
standards. DNA concentrations determined by the twomethods were equivalent within 5 %.
Duplex oligonucleotides were end-labeled with 32Pusing T-4 polynucleotide kinase as described.16
DNA duplex stability studies
Absorbance versus temperature pro®les weremeasured at 260 nm on a Cary 100 (Varian) UV/visspectrophotometer interfaced to a Dell Optiplex GXI(Pentium II) computer for data analysis (Cary WinUVv2.0). Melting curves of duplex stocks (in 10 mM bis-Tris-propane plus 0.1 M potassium acetate (pH 7.3))were obtained by increasing the temperature from 15 �Cto 95 �C at a constant rate of 0.5 deg. C/minute with aprogrammable, thermoelectrically controlled cell holder.Oligomer concentrations ranged from 0.5 mM to 8mMtotal strand concentration (Ct). Concentration-dependentmelting temperature data were used to construct 1/Tm
versus ln(Ct/4) plots to evaluate the van't Hoff transitionenthalpy (�H�vH) and entropy (�S�vH; assuming a two-state transition and temperature-independent enthalpy),from which the van't Hoff free energy (�G�vH) was calcu-lated.70 The uni®ed nearest neighbor thermodynamicparameter sets for �G�37, �H� and �S� reported by San-taLucia71 were used to predict values of �G�duplex forduplex stability at 37 � and 21 �C.
Gel permeation chromatography
A column (0.9 cm � 49 cm) of Sephacryl S-200HR(Pharmacia) was equilibrated in buffer (10 mM bis-tris-propane, 7 mM b-mercaptoethanol, 1 mM EDTA) plus0.045 M potassium acetate, adjusted to pH 7.4 at 5 �C.Elution was at a ¯ow rate of 15 ml/hour. The columnwas calibrated with the following markers: blue dextran(void volume), catalase (240 kDa), albumin (86 kDadimer, 43 kDa monomer), b-lactalbumin (36 kDa), carbo-nic anhydrase (29 kDa), trypsin inhibitor (21.5 kDa),myoglobin (17 kDa) and cytochrome C (12.5 kDa).BamHI endonuclease (0.5 mM in a volume of 100 ml) wasmixed with various amounts of radiolabeled 20 bp oligo-nucleotide containing a single recognition site. Com-plexes were formed in the column equilibration buffer at5 �C and equilibrated for 30 minutes prior to loading onthe column. Fractions (0.55 ml each) were collected andassayed for radioactivity, location of the free and boundradioactive DNA species on native PAGE (pH 7.3 at25 �C) and location of the protein on SDS-15 %-PAGE.
Equilibrium binding
Equilibrium binding constants for BamHI endonu-clease-DNA complex formation were probed by eitherdirect binding or equilibrium competition methods onnitrocellulose ®lters, using minor adaptations of themethods described previously.16,18,72 All numericalvalues reported in Figures and Tables were obtained bythese nitrocellulose ®lter-binding methods. For compari-son, some binding constants were also determined bythe gel-retardation method as described;25 the pH of thegel and electrophoresis running buffers was 6.25. Allbinding reactions were carried out in binding buffer(10 mM bis-Tris-propane, 50 mM dithiothreitol, 1 mMEDTA, bovine serum albumin at 100 mg/ml), adjusted tothe concentration of K � (with potassium acetate) andpH indicated in each Table and Figure. KA is steeplydependent on cation concentration,56,73 so ®nal cation
concentrations were carefully selected to place the valuesin a convenient range for each experiment. For BamHIendonuclease,56 acetate and ¯uoride are relatively non-interactive anions (KD,Acetate � 2.0 M) whereas chloride isan interactive anion (KD; cloride � 0.045 M).
Kinetics of DNA cleavage
Single-turnover cleavage rates were determined asdescribed.19,61 Enzyme (1.6 mM) and DNA (0.66 mM)were pre-equilibrated in binding buffer (plus 0.114 Mpotassium acetate), at pH 7.3 for ®ve minutes on ice,then switched to 21 �C for 20 minutes. Reactions wereinitiated by the addition of magnesium acetate (®nal con-centration 5 mM) and quenched at the indicated timeswith stop solution (®nal concentration of 64 % forma-mide, 36 mM EDTA). Products were separated by dena-turing (8 M urea) polyacrylamide gel electrophoresis,and ®rst-order rate constants extracted from themeasured band intensities as described.19
Kinetics of complex dissociation
Dissociation rate constants (kd) for BamHI endonu-clease-DNA complexes were determined as described18,74
except that all pre-incubations were ®ve minutes on ice,15 minutes at 21 �C. This incubation time was deter-mined to be suf®cient to attain binding equilibrium.
Ethylation-interference footprinting
Footprints were obtained as described19,57 with the fol-lowing modi®cations: 22 mg of the 24 bp single strandoligonucleotide in 100 ml of 6.25 mM sodium cacodylatebuffer (pH 8.0) was treated with 100 ml ethyl-nitrosourea.Complexes were formed in binding buffer (0.14 M pot-assium acetate (pH 7.30), 21 �C) using concentrations ofDNA and enzyme that were 20-fold and 60-fold, respect-ively, above the equilibrium dissociation constantbetween the unethylated substrate and BamHI.
Acknowledgments
This work was supported in part by grant GM-29207from the National Institutes of Health to L.J.-J. We aregrateful to Lewis Jacobson for many insightful discus-sions and critical reading of the manuscript.
References
1. Arber, W. (1974). DNA modi®cation and restriction.Prog. Nucl. Acid Res. Mol. Biol. 14, 1-37.
2. Roberts, R. J. & Halford, S. E. (1993). Type II restric-tion endonucleases. In Nucleases (Linn, S. M., Lloyd,R. S. & Roberts, R. J., eds), pp. 35-88, Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY.
3. Kim, Y. C., Grable, J. C., Love, R., Greene, P. J. &Rosenberg, J. M. (1990). Re®nement of EcoRI endo-nuclease crystal structure: a revised protein chaintracing. Science, 249, 1307-1309.
4. Winkler, F. K., Banner, D. W., Oefner, C.,Tsernoglou, D., Brown, R. S., Heathman, S. P.,Bryan, R. K., Martin, P. D., Petratos, K. & Wilson,K. S. (1993). The crystal structure of EcoRV endonu-

634 BamHI interaction with its Cognate GGATCC Site
clease and of its complexes with cognate and non-cognate DNA fragments. EMBO J. 12, 1781-1795.
5. Cheng, X., Balendiran, K., Schildkraut, I. &Anderson, J. E. (1994). Structure of PvuII endo-nuclease with cognate DNA. EMBO J. 13, 3927-3935.
6. Newman, M., Strzelecka, T., Dorner, L. F.,Schildkraut, I. & Aggarwal, A. K. (1995). Structureof BamHI endonuclease bound to DNA: partial fold-ing and unfolding on DNA binding. Science, 269,656-663.
7. Newman, M., Lunnen, K., Wilson, G., Greci, J.,Schildkraut, I. & Phillips, S. E. V. (1998). Crystalstructure of restriction endonuclease BglI bound toits interrupted DNA recognition sequence. EMBO J.17, 5466-5476.
8. Lukacs, C. M., Kucera, R., Schildkraut, I. &Aggarwal, A. K. (2000). Understanding the immut-ability of restriction enzymes: crystal structure ofBglII and its DNA substrate at 1.5 AÊ resolution.Nature Struct. Biol. 7, 134-140.
9. Deibert, M., Grazulis, S., Sasnauskas, G., Siksnys, V.& Huber, R. (2000). Structure of the tetramericrestriction endonuclease NgoMIV in complex withcleaved DNA. Nature Struct. Biol. 7, 792-799.
10. Kostrewa, D. & Winkler, F. K. (1995). Mg2 � bindingto the active site of EcoRV endonuclease: a crystallo-graphic study of complexes with substrate andproduct DNA at 2 AÊ resolution. Biochemistry, 34,683-696.
11. Viadiu, H. & Aggarwal, A. K. (1998). The role ofmetals in catalysis by the restriction endonucleaseBamHI. Nature Struct. Biol. 5, 910-916.
12. Horton, N. C. & Perona, J. J. (1998). Recognition of¯anking DNA sequences by EcoRV endonucleaseinvolves alternative patterns of water-mediated con-tacts. J. Biol. Chem. 273, 21721-21729.
13. Horton, N. C., Newberry, K. J. & Perona, J. J. (1998).Metal ion-mediated substrate-assisted catalysis intype II restriction endonucleases. Proc. Natl Acad.Sci. USA, 95, 13489-13494.
14. Martin, A. M., Sam, M. D., Reich, N. O. & Perona,J. J. (1999). Structural and energetic origins of indir-ect readout in site-speci®c DNA cleavage by arestriction endonuclease. Nature Struct. Biol. 6, 269-277.
15. Viadiu, H. & Aggarwal, A. K. (2000). Structure ofBamHI bound to non-speci®c DNA: a model forDNA sliding. Mol. Cell, 5, 889-895.
16. Jen-Jacobson, L., Kurpiewski, M., Lesser, D., Grable,J. C., Boyer, H. W., Rosenberg, J. M. & Greene, P. J.(1983). Coordinate ion pair formation between EcoRIendonuclease and DNA. J. Biol. Chem. 258, 14638-14646.
17. Terry, B. J., Jack, W. E., Rubin, R. A. & Modrich, P.(1983). Thermodynamic parameters governinginteraction of EcoRI endonuclease with speci®c andnon-speci®c DNA sequences. J. Biol. Chem. 258,9820-9825.
18. Jen-Jacobson, L., Lesser, D. & Kurpiewski, M. (1986).The enfolding arms of EcoRI endonuclease: role inDNA binding and cleavage. Cell, 45, 619-629.
19. Lesser, D. R., Kurpiewski, M. R. & Jen-Jacobson, L.(1990). The energetic basis of speci®city in the EcoRIendonuclease-DNA interaction. Science, 250, 776-786.
20. Thielking, V., Alves, J., Fliess, A., Maass, G. &Pingoud, A. (1990). Accuracy of the EcoRI restrictionendonuclease: binding and cleavage studies with oli-godeoxynucleotide substrates containing degeneraterecognition sequences. Biochemistry, 29, 4682-4691.
21. Aiken, C. R., Fisher, E. W. & Gumport, R. I. (1991).The speci®c binding, bending, and unwinding ofDNA by RsrI endonuclease, an isoschizomer ofEcoRI endonuclease. J. Biol. Chem. 266, 19063-19069.
22. Taylor, J. D., Badcoe, I. G., Clarke, A. R. & Halford,S. E. (1991). EcoRV restriction endonuclease bindsall DNA sequences with equal af®nity. Biochemistry,30, 8743-8753.
23. Vipond, I. B. & Halford, S. E. (1993). Structure-func-tion correlation for the EcoRV restriction enzyme:from non-speci®c binding to speci®c DNA cleavage.Mol. Microbiol. 9, 225-231.
24. Erskine, S. G. & Halford, S. E. (1998). Reactions ofthe EcoRV restriction endonuclease with ¯uorescentoligodeoxynucleotides: identical equilibrium con-stants for binding to speci®c and non-speci®c DNA.J. Mol. Biol. 275, 759-772.
25. Engler, L. E., Welch, K. K. & Jen-Jacobson, L. (1997).Speci®c binding by EcoRV endonuclease to its DNArecognition site GATATC. J. Mol. Biol. 269, 82-101.
26. Vipond, I. B., Baldwin, G. S. & Halford, S. E. (1995).Divalent metal ions at the active sites of the EcoRVand EcoRI restriction endonucleases. Biochemistry, 34,687-704.
27. Martin, A. M., Horton, N. C., Lusetti, S., Reich, N. O.& Perona, J. J. (1999). Divalent metal dependence ofsite-speci®c DNA binding by EcoRV endonuclease.Biochemistry, 38, 8430-8439.
28. Zebala, J., Choi, J. & Barany, F. (1992). Characteriz-ation of steady state, single-turnover, and bindingkinetics of the TaqI restriction endonuclease. J. Biol.Chem. 267, 8097-8105.
29. Siksnys, V. & Pleckaityte, M. (1993). Catalytic andbinding properties of restriction endonuclease Cfr9I.Eur. J. Biochem. 217, 411-419.
30. Nastri, H. G., Evans, P. D., Walker, I. H. & Riggs,P. D. (1997). Catalytic and DNA binding propertiesof PvuII restriction endonuclease mutants. J. Biol.Chem. 272, 25761-25767.
31. Skirgaila, R. & Siksnys, V. (1998). Ca2� ionsstimulate DNA binding speci®city of Cfr10I restric-tion enzyme. Biol. Chem. Hoppe-Seyler, 39, 595-598.
32. Lagunavicius, A., Grazulis, S., Balciunaite, E.,Vainius, D. & Siksnys, V. (1997). DNA bindingspeci®city of MunI restriction endonuclease is con-trolled by pH and calcium ions: involvement ofactive site carboxylate residues. Biochemistry, 36,11093-11099.
33. Lagunavicius, A. & Siksnys, V. (1997). Site-directedmutagenesis of putative active site residues of MunIrestriction endonuclease: replacement of catalyticallyessential carboxylate residues triggers DNA bindingspeci®city. Biochemistry, 36, 11086-11092.
34. Thomas, M. & Davis, R. W. (1975). Studies on thecleavage of bacteriophage lambda DNA with EcoRIrestriction endonuclease. J. Mol. Biol. 91, 315-328.
35. Jen-Jacobson, L. (1997). Protein-DNA recognitioncomplexes: conservation of structure and bindingenergy in the transition state. Biopolymers, 44, 153-180.
36. Jen-Jacobson, L., Engler, L. E. & Jacobson, L. A.(2000). Structural and thermodynamic strategies forsite-speci®c DNA binding proteins. Structure, 8,1015-1023.
37. Jen-Jacobson, L., Engler, L. E., Ames, J. T.,Kurpiewski, M. R. & Grigorescu, A. (2000). Thermo-dynamic parameters of speci®c and non-speci®cprotein-DNA binding. Supramol. Chem. 12, 143-160.

BamHI interaction with its Cognate GGATCC Site 635
38. Newman, M., Strzelecka, T., Dorner, L. F.,Schildkraut, I. & Aggarwal, A. K. (1994). Structureof restriction endonuclease BamHI phased at 1.95 AÊ
resolution by MAD analysis. Structure, 2, 439-452.39. Newman, M., Strzelecka, T., Dorner, L. F.,
Schildkraut, I. & Aggarwal, A. K. (1994). Structureof restriction endonuclease BamHI and its relation-ship to EcoRI. Nature, 368, 660-664.
40. Riccelli, P. V., Vallone, P. M., Kashin, L., Faldasz,B. D., Lane, J. J. & Benight, A. S. (1999). Thermodyn-amic, spectroscopic, and equilibrium binding studiesof DNA sequence context effects in six 22-base pairdeoxyoligonucleotides. Biochemistry, 38, 11197-11208.
41. Jack, W. E., Greenough, L., Dorner, L. F., Xu, S. Y.,Strzelecka, T., Aggarwal, A. K. & Schildkraut, I.(1991). Overexpression, puri®cation and crystalliza-tion of BamHI endonuclease. Nucl. Acids Res. 19,1825-1829.
42. Nardone, G. & Chirikjian, J. G. (1987). The enzymesof the BamHI restriction modi®cation system. GeneAmplif. Anal. 5, 147-184.
43. Cleve, M. D. & Gumport, R. I. (1992). In¯uence ofenzyme-substrate contacts located outside the EcoRIrecognition site on cleavage of duplex oligodeoxy-ribonucleotide substrates by EcoRI endonuclease.Biochemistry, 31, 334-339.
44. Travers, A. A. (1991). DNA bending and kinking-sequence dependence and function. Curr. Opin.Struct. Biol. 1, 114-122.
45. Gorin, A. A., Zhurkin, V. B. & Olson, W. K. (1995).B-DNA twisting correlates with base-pair mor-phology. J. Mol. Biol. 247, 34-48.
46. El Hassan, M. A. & Calladine, C. R. (1996). Propel-ler-twisting of base-pairs and the conformationalmobility of dinucleotide steps in DNA. J. Mol. Biol.259, 95-103.
47. Dickerson, R. E. (1999). Helix structure and molecu-lar recognition by B-DNA. In Oxford Handbook ofNucleic Acid Structure (Neidle, S., ed.), pp. 145-197,Oxford University Press, Oxford.
48. Suzuki, M. & Yagi, N. (1995). Stereochemical basisof DNA bending by transcription factors. Nucl.Acids Res. 23, 2083-2091.
49. Olson, W. K., Gorin, A. A., Lu, X. J., Hock, L. M. &Zhurkin, V. B. (1998). DNA sequence-dependentdeformability deduced from protein-DNA crystalcomplexes. Proc. Natl Acad. Sci. USA, 95, 11163-11168.
50. Dickerson, R. E. & Chiu, T. K. (1998). Helix bendingas a factor in protein/DNA recognition. Biopolymers,44, 361-403.
51. Zhurkin, V. B., Ulyanov, N. B., Gorin, A. A. &Jernigan, R. L. (1991). Static and statistical bendingof DNA evaluated by Monte Carlo simulations.Proc. Natl Acad. Sci. USA, 88, 7046-7050.
52. Packer, M. J., Dauncey, M. P. & Hunter, C. A.(2000). Sequence-dependent DNA structure: dinu-cleotide conformational maps. J. Mol. Biol. 295, 71-83.
53. Horton, N. C. & Perona, J. J. (1998). Role of protein-induced bending in the speci®city of DNArecognition: crystal structure of EcoRV endonucleasecomplexed with d(AAAGAT) � d(ATACTT). J. Mol.Biol. 277, 779-787.
54. Kim, Y., Choi, J., Grable, J. C., Greene, P., Hager, P.& Rosenberg, J. M. (1994). Studies on the canonicalDNA-EcoRI endonuclease complex and the EcoRIkink. In Structural Biology: The State of the Art. Pro-ceedings of the Eighth Conversation, SUNY, Albany
(Sarma, R. & Sarma, M., eds), pp. 225-246, AdeninePress, New York.
55. Jen-Jacobson, L. (1995). Structural-perturbationapproaches to thermodynamics of site-speci®cprotein-DNA interactions. Methods Enzymol. 259,305-344.
56. Engler, L. E. (1998). Speci®city determinants in theBamHI endonuclease-DNA interaction. PhD. thesis,University of Pittsburgh, Pittsburgh, PA.
57. Siebenlist, U. & Gilbert, W. (1980). Contacts betweenEscherichia coli RNA polymerase and an early pro-moter of phage T7. Proc. Natl Acad. Sci. USA, 77,122-126.
58. Becker, M. M., Lesser, D., Kurpiewski, M., Baranger,A. & Jen-Jacobson, L. (1988). ``Ultraviolet footprint-ing'' accurately maps sequence-speci®c contacts andDNA kinking in the EcoRI endonuclease-DNA com-plex. Proc. Natl Acad. Sci. USA, 85, 6247-6251.
59. Pabo, C. O., Aggarwal, A. K., Jordan, S. R., Beamer,L. J., Obeysekare, U. R. & Harrison, S. C. (1990).Conserved residues make similar contacts in tworepressor-operator complexes. Science, 247, 1210-1213.
60. Rosenberg, J. M. (1991). Structure and function ofrestriction endonucleases. Curr. Opin. Struct. Biol. 1,104-113.
61. Lesser, D. R., Grajkowski, A., Kurpiewski, M. R.,Koziolkiewicz, M., Stec, W. J. & Jen-Jacobson, L.(1992). Stereoselective interaction with chiral phos-phorothioates at the central DNA kink of the EcoRIendonuclease-GAATTC complex. J. Biol. Chem. 267,24810-24818.
62. Xu, S. Y. & Schildkraut, I. (1991). Isolation of BamHIvariants with reduced cleavage activities. J. Biol.Chem. 266, 4425-4429.
63. Dorner, L. F. & Schildkraut, I. (1994). Direct selec-tion of binding pro®cient/catalytic de®cient variantsof BamHI endonuclease. Nucl. Acids Res. 22, 1068-1074.
64. Winkler, F. K. (1992). Structure and function ofrestriction endonucleases. Curr. Opin. Struct. Biol. 2,93-99.
65. Lesser, D. R., Kurpiewski, M. R., Waters, T.,Connolly, B. A. & Jen-Jacobson, L. (1993). Facilitateddistortion of the DNA site enhances EcoRI endonu-clease-DNA recognition. Proc. Natl Acad. Sci. USA,90, 7548-7552.
66. Chong, S., Mersha, F. B., Comb, D. G., Scott, M. E.,Landry, D., Vence, L. M., Perler, F. B., Benner, J.,Kucera, R. B., Hirvonen, C. A., Pelletier, J. J., Paulus,H. & Xu, M.-Q. (1997). Single-column puri®cation offree recombinant proteins using a self-cleavable af®-nity tag derived from a protein splicing element.Gene, 192, 277-281.
67. Morrisey, J. H. (1981). Silver stain for proteins inpolyacrylamide gels: a modi®ed procedure withenhanced uniform sensitivity. Anal. Biochem. 117,307-310.
68. Warshaw, M. M. & Cantor, C. R. (1970). Oligonu-cleotide interactions. IV. Conformational differencesbetween deoxy- and ribodinucleoside phosphates.Biopolymers, 9, 1079-1103.
69. Senior, M., Jones, R. A. & Breslauer, K. J. (1988).In¯uence of dangling thymidine residues on thestability and structure of two DNA duplexes.Biochemistry, 27, 3879-3885.
70. Breslauer, K. J. (1995). Extracting thermodynamicdata from equilibrium melting curves for oligonu-

636 BamHI interaction with its Cognate GGATCC Site
cleotide order-disorder transitions. Methods Enzymol.259, 221-242.
71. SantaLucia, J., Jr (1998). A uni®ed view of polymer,dumbell, and oligonucleotide DNA nearest-neighborthermodynamics. Proc. Natl Acad. Sci. USA, 95, 1460-1465.
72. Connolly, B. A., Liu, H., Parry, D., Engler, L. E.,Kurpiewski, M. R., Engler, L. E. & Jen-Jacobson, L.(2001). Assay of restriction endonucleases usingoligonucleotides. In Methods in Molecular Biology:DNA-Protein Interactions: Principles and Protocols
(Kneale, G. G., ed.), pp. 465-490 Humana Press,Totowa, NJ.
73. Record, M. T., Jr, Ha, J. H. & Fisher, M. A. (1991).Analysis of equilibrium and kinetic measurementsto determine thermodynamic origins of stability andspeci®city and mechanism of formation of site-speci®c complexes between protein and helicalDNA. Methods Enzymol. 208, 291-343.
74. Riggs, A. D., Bourgeois, S. & Cohn, M. (1970). Thelac repressor operator interaction. III. Kinetic studies.J. Mol. Biol. 53, 401-417.
Edited by R. Ebright
(Received 4 October 2000; received in revised form 21 December 2000; accepted 21 December 2000)




















