
The electro-oxidation of p-aminodiphenylamine in acetonitrile
10
389 J. Eleztroanal. Chem., 348 (1993) 389-398 Elsevier Sequoia S.A., Lausanne JEC 02488 The electro-oxidation of p-aminodiphenylamine in acetonitrile Nuran Pekmez, Kadir Pekmez and Attila Yildiz l Hacettepe University, Department of Chemistry, Beytepe, Ankara (Turkey) (Received 24 August 1992; in revised form 18 September 1992) Abstract The electro-oxidation behavior of p-aminodiphenylamine (PADPA) in acetonitrile was investigated using cyclic voltammetry and controlled potential electrolysis. PADPA is oxidized in two steps in this medium. When the experiment is carried out in the presence of an equivalent amount of pyridine the second oxidation peak disappears. The first oxidation peak vanishes when the solution contains an equivalent amount of anhydrous perchloric acid. An intermediate oxidation peak which is due to the formation of the cation radical appears under certain conditions. The results are interpreted especially in relation to the role of this compound in the formation of polyaniline and a detailed mechanism is proposed. INTRODUCTION The electro-oxidation of aniline is the usual production method for polyaniline which is one of the most widely studied conducting polymers [1,2]. This electro- oxidation is carried out in an acidic medium such as aqueous sulphuric acid. The electro-oxidation products of aniline in this medium are p-aminodiphenylamine (PADPA) and benzidine. PADPA is also electro-oxidizable at_ the potentials of aniline oxidation [3]. The electro-oxidation of PADPA was also found to lead to the formation of polyaniline in this medium 141.The hydrolysis of aniline and its immediate electro-oxidation product, PADPA, leads to the formation of undesired products of the quinone type [5], thought to be responsible for the decrease in conductivity and the loss of stability of polyaniline [6,7]. In addition it is known that polyaniline itself may hold up to 40% water in its structure, via hydrogen l To whom correspondence should be addressed. 0022-0728/93/$06.00 0 1993 - Elsevier Sequoia S.A. All rights reserved
Transcript of The electro-oxidation of p-aminodiphenylamine in acetonitrile

389
J. Eleztroanal. Chem., 348 (1993) 389-398 Elsevier Sequoia S.A., Lausanne
JEC 02488
The electro-oxidation of p-aminodiphenylamine in acetonitrile
Nuran Pekmez, Kadir Pekmez and Attila Yildiz l
Hacettepe University, Department of Chemistry, Beytepe, Ankara (Turkey)
(Received 24 August 1992; in revised form 18 September 1992)
Abstract
The electro-oxidation behavior of p-aminodiphenylamine (PADPA) in acetonitrile was investigated using cyclic voltammetry and controlled potential electrolysis. PADPA is oxidized in two steps in this medium. When the experiment is carried out in the presence of an equivalent amount of pyridine the second oxidation peak disappears. The first oxidation peak vanishes when the solution contains an equivalent amount of anhydrous perchloric acid. An intermediate oxidation peak which is due to the formation of the cation radical appears under certain conditions. The results are interpreted especially in relation to the role of this compound in the formation of polyaniline and a detailed mechanism is proposed.
INTRODUCTION
The electro-oxidation of aniline is the usual production method for polyaniline which is one of the most widely studied conducting polymers [1,2]. This electro- oxidation is carried out in an acidic medium such as aqueous sulphuric acid. The electro-oxidation products of aniline in this medium are p-aminodiphenylamine (PADPA) and benzidine. PADPA is also electro-oxidizable at_ the potentials of aniline oxidation [3]. The electro-oxidation of PADPA was also found to lead to the formation of polyaniline in this medium 141. The hydrolysis of aniline and its immediate electro-oxidation product, PADPA, leads to the formation of undesired products of the quinone type [5], thought to be responsible for the decrease in conductivity and the loss of stability of polyaniline [6,7]. In addition it is known that polyaniline itself may hold up to 40% water in its structure, via hydrogen
l To whom correspondence should be addressed.
0022-0728/93/$06.00 0 1993 - Elsevier Sequoia S.A. All rights reserved

390
bonding, which may provide a reservoir for the degradative side reactions, even during the use of this material in rechargeable batteries [8-111. Some workers have attempted to use an unusual medium for electropolymerization, such as an NH,F + HF eutectic mixture, in order to avoid the use of aqueous media [l].
The use of the aprotic media for the electro-oxidation of aniline appears to be a good alternative. The preparation of polyaniline was therefore attempted in acetonitrile containing pyridine [l]. The presence of pyridine in acetonitrile caused the formation of azobenzene in substantial amounts which also decreased the yield of polyaniline formation.
An understanding of the electro-oxidation mechanism of PADPA in non-aque- ous acetonitrile is of utmost importance if aniline is electropolymerized in this medium. In this study the electro-oxidation behavior of PADPA was investigated in acetonitrile containing anhydrous perchloric acid and was compared with that obtained in acetonitrile containing pyridine as a base.
EXPERIMENTAL
PADPA (Aldrich) was recrystallized from hexane. The purification of acetoni- trile and tetrabutylammonium perchlorate (TBAP) are described elsewhere [ 121. Anhydrous perchloric acid solutions were prepared by oxidative electrolysis of the saturated solution of molecular hydrogen in acetonitrile containing 0.1 M TBAP at a platinized Pt working electrode (A = 12.5 cm2). The Pt counter electrode was kept in a separate compartment containing HClO, and TBAP in acetonitrile during electrolysis and the two compartments were joined by a salt bridge contain- ing a saturated solution of TBAP in acetonitrile. The pressure in the two compart- ments was balanced by the use of a gas bridge. The electrolysis potential was kept at +OS V vs. Ag/AgCl(sat) reference electrode. The quantity of acid produced was controlled coulometrically; concentrations of the acid solutions were also determined by a potentiometric titration with a standard tetra-n-butylammonium hydroxide solution in isopropyl alcohol.
Electrochemical measurements were carried out under a nitrogen atmosphere in a three-electrode cell. A polished Pt disc electrode with a diameter of 0.13 cm, inlaid in a glass capillary of 0.80 cm, was used in cyclic voltammetric experiments as a working electrode. The counter electrode was a Pt wire (2 cm2> immersed in acetonitrile + 0.1 M TBAP, separated from the electrolysis solution by a sintered glass disc (G4). The reference electrode consisted of an AgCl-coated Ag wire in acetonitrile + 0.1 M TBAP which was also separated from the electrolysis solution by a sintered glass disc. Exhaustive controlled potential electrolysis experiments were carried out in the same cell with a Pt foil as the working macroelectrode (A = 12.5 cm2>. Electrochemical instrumentation consisted of a PAR Model 173 potentiostat coupled to a PAR Model 175 universal programmer and a PAR Model 179 digital coulometer. Current-voltage curves were recorded on a Model SE-790 BBC Goerzt Metrawatt X-Y recorder.
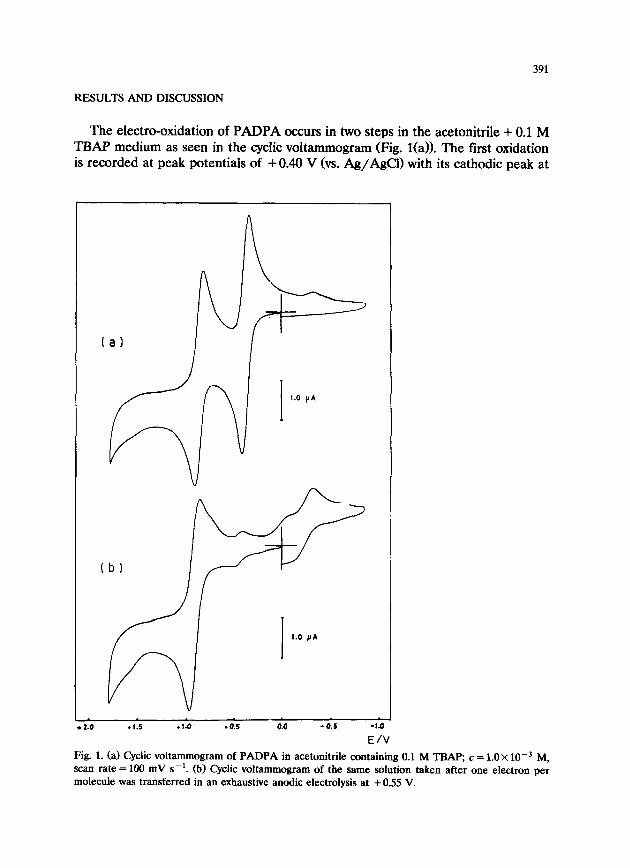
391
RESULTS AND DISCUSSION
The electro-oxidation of PADPA occurs in two steps in the acetonitrile + 0.1 M TBAP medium as seen in the cyclic voltammogram (Fig. l(a)>. The first oxidation is recorded at peak potentials of + 0.40 V (vs. Ag/AgCl) with its cathodic peak at
I 1.0 PA
I 1.0 PA
1.0 .I.5 .I.0 .0.5 0.0 -0.5 -1.0
E/V
Fig. 1. (a) Cyclic voltammogram of PADPA in acetonitrile containing 0.1 M TBm c = 1.0 X 10e3 M, scan rate = 100 mV s-l. (b) Cyclic voltammogram of the same solution taken after one electron per molecule was transferred in an exhaustive anodic electrolysis at +0.55 V.

392
+0.32 V. The second oxidation peak potential is found to be +0.90 V with its corresponding reverse peak at +0.82 V. If the potential is scanned towards more negative values after either the first or the second oxidation peak a cathodic peak appears at -0.40 V. This cathodic peak coincides with the peak obtained when a cyclic voltammogram is taken in a PADPA solution containing an equivalent amount of anhydrous perchloric acid. This means that during the first electro- oxidation process protons are released and the PADPA molecules that are not yet oxidized at the electrode are protonated.
&-(--JNH2 - [eglVNH2].+ + e-
(1)
I ‘f
NH2 + NH,
1
NH;
(2) The neutral PADPA radical that is formed as a product of this protonation reaction is also oxidized at this potential
giving rise to a net one-electron oxidation reaction, as follows
[Q-I&J-NH]’ + (&J--NH; + 2e- (4)
The inverse reaction of eqn. (4) is responsible for the cathodic reverse peak observed at +0.32 V:
(5)
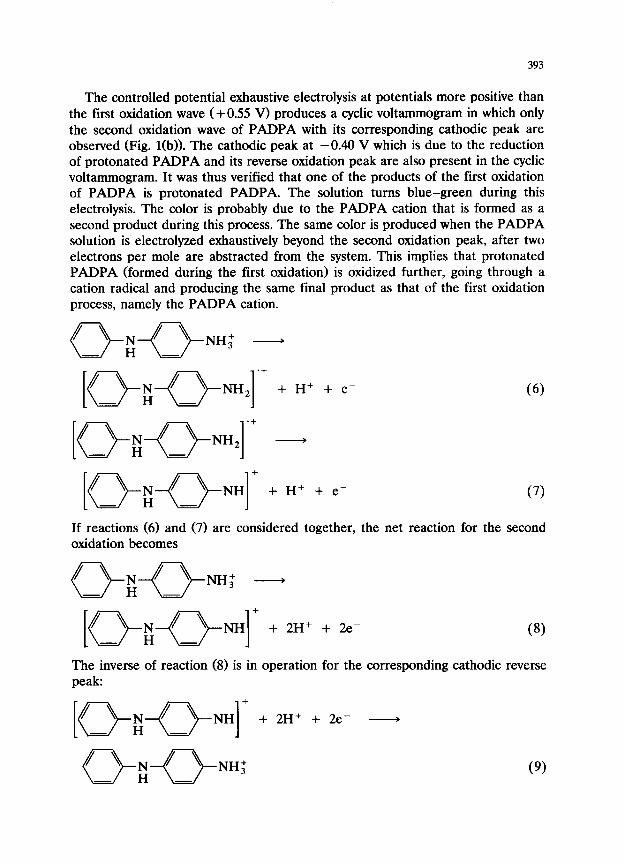
393
The controlled potential exhaustive electrolysis at potentials more positive than the first oxidation wave (+0.55 V) produces a cyclic voltammogram in which only the second oxidation wave of PADPA with its corresponding cathodic peak are observed (Fig. l(b)). The cathodic peak at -0.40 V which is due to the reduction of protonated PADPA and its reverse oxidation peak are also present in the cyclic voltammogram. It was thus verified that one of the products of the first oxidation of PADPA is protonated PADPA. The solution turns blue-green during this electrolysis. The color is probably due to the PADPA cation that is formed as a second product during this process. The same color is produced when the PADPA solution is electrolyzed exhaustively beyond the second oxidation peak, after two electrons per mole are abstracted from the system. This implies that protonated PADPA (formed during the first oxidation) is oxidized further, going through a cation radical and producing the same final product as that of the first oxidation process, namely the PADPA cation.
[~fIl’dlVH2]” + H+ + e-
[@eNHZ].+ -
[~~~_I’ + H+ + e-
(6)
(7)
If reactions (6) and (7) are considered together, the net reaction for the second oxidation becomes
(8)
The inverse of reaction (8) is in operation for the corresponding cathodic reverse peak:
[O~~JNH]’ + 2H+ + 2e- -
(E-NH; (9)
394
7 + I.5 . 1.0 . 0.5 O.!, -0.5 -1.0 E/V -1.5 - I 0
Fig. 2. Cyclic voltammogram of PADPA in acetonitrile containing 0.1 M TBAP and equivalent amount of pyridine: c = 2.0~ 10m3, scan rate = 100 mV SC’.
The ratio of the peak current of the second oxidation to that of the first oxidation was found to decrease as the sweep rate was increased. This suggests that the protonation reaction (2) that produces the reactant of the second electro- oxidation, protonated PADPA, is the rate limiting step in the second oxidation. The same protonated PADPA is also the reactant for the reverse reduction process at + 0.32 V as written in reaction (5). It was found that the intensity of this cathodic peak also decreases with respect to the cathodic peak intensity observed at higher potentials, when faster scan rates were used in cyclic voltammetry.
When increasing amounts of a base (pyridine) are added to PADPA solution, it was found that the peak int’ensities of the first redox system increase and eventu- ally double in intensity, at the expense of those of the second couple which eventually disappears. Figure 2 shows the cyclic voltammogram of a PADPA solution after addition of an equivalent amount of base. The presence of a strong base affects the oxidation process according to the protonation reaction
(10)
In other words the role of the unreacted PADPA is taken over by the more basic species, pyridine, in protonating the product of the first oxidation. Further oxida- tion of the neutral radical is again possible at these potentials, as proposed above in reaction (3). Reactions (11, (10) and (3) give rise to the net oxidation of reaction (11)

395
[OE-Q-NH]’ + OH + 2e- (11)
The doubling of the peak intensities is thus accounted for, since two electrons are abstracted from each PADPA molecule in this process. Since no protonated PADPA is produced, the oxidation wave at more positive potentials disappears, together with its corresponding cathodic reverse peak. It must also be pointed out that the blue-green coloration of the PADPA cation appears here also, if exhaustive oxidative electrolysis is carried out.
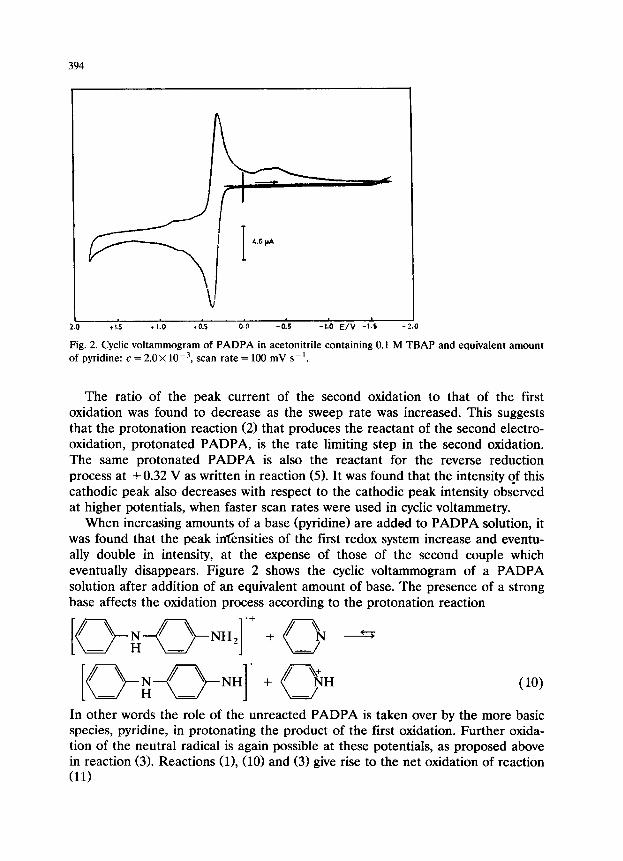
If the electrochemical behavior of PADPA is investigated under acidic condi- tions a new cyclic voltammetric oxidation peak at +0.70 V and corresponding cathodic peak at +0.54 V are observed between the first and the second oxidation peaks (Fig. 3). The same oxidation peak without the corresponding reverse cathodic peak is also observed in solutions of PADPA when no additional acid or base is present, if the cyclic voltammogram is taken at much higher scan rates (50 V s-l). We interpret this peak as being due to the oxidation of the PADPA cation radical, reaction (61, formed as a product of the first oxidation, reaction (2).
It was established experimentally that stability of the PADPA cation radical can be achieved if excess pyridine and anhydrous perchloric acid are added to the PADPA solution together. Figure 4 shows the cyclic voltammogram of a solution containing PADPA, pyridine and HClO,. For each mole of PADPA, three moles
. 2.0 l 1.5 l l:O l Qi 0.0 -0.5 -1.0 - E/v -2.0
Fig. 3. Cyclic voltammogram of PADPA in acetonitrile containing 0.1 M TEtAP +5.0x 10e4 M anhydrous HClO,: c = 2.0 x 10m3 M, scan rate = 100 mV s-l.
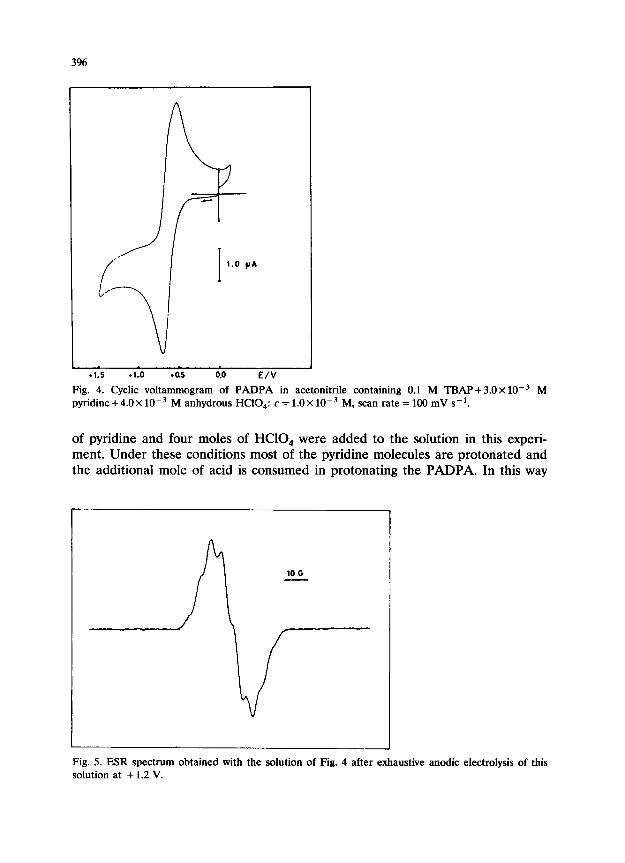
-1.5 .1-o -0.5 0.0 E/V
Fig. 4. Cyclic voltammogram of PADPA in acetonitrile containing 0.1 M TBAP+ 3.0~ 10W3 M pyridine + 4.0 X 10m3 M anhydrous HCIO,: c = 1.0 X 10W3 M, scan rate = 100 mV s-‘.
of pyridine and four moles of HClO, were added to the solution in this experi- ment. Under these conditions most of the pyridine molecules are protonated and the additional mole of acid is consumed in protonating the PADPA. In this way
10 G
Fig. 5. ESR spectrum obtained with the solution of Fig. 4 after exhaustive anodic electrolysis of this solution at + 1.2 V.

397
the solution conditions are so adjusted that there is no substantial amount of basic species in the medium that can abstract protons from the protonated PADPA. The new electro-oxidation wave obtained under these conditions is then due to reaction (6), in which the cation radical is formed. The cathodic reverse peak here is due to the one-electron reduction of the cation radical, the inverse of reaction (6).
It was found that one electron per molecule was transferred during the controlled potential oxidative electrolysis of this solution at + 1.20 V. The final color of the solution was deep blue. The same cyclic voltammogram as in Fig. 4 was obtained after the electrolysis. The production of the cation radical was also verified by the ESR spectrum obtained after this electrolysis (Fig. 5). This solution in which the cation radical is stable was electrolyzed cathodically at +0.20 V and the blue color of the radical disappeared after one electron per molecule was consumed. The cyclic voltammogram taken after this experiment was found to be exactly the same as the starting solution.
CONCLUSION
It is known that cation radicals, in general, are stabilized if they are produced under acidic conditions [13-151. It has been established from earlier studies that the growth of polyaniline passes through reactions involving the cation radicals of aniline and PADPA during the electro-oxidation of aniline [ 1,2]. As is evident from the above results, stability of the cation radical of PADPA can also be achieved under acidic nonaqueous acetonitrile solutions. Conditions which ensure the stability of the PADPA cation radical are of considerable interest since this species plays a key role during the growth of polyaniline film.
This study represents the first attempt towards an understanding of the electro- oxidation mechanism of PADPA in non-aqueous acetonitrile. The only related study in the literature concerns the electro-oxidation of the so-named phenyl-end- capped dimer of aniline [16]. This dimer gave rise to two CV oxidation peaks, the first producing the cation radical and the second the imine of the original amine. Stability of the cation radical was achieved by working in acetonitrile containing perchloric acid.
The electro-oxidation mechanism of PADPA differs considerably from that of the phenyl-capped dimer and tetramer. The first oxidation of PADPA gives rise to the formation of the PADPA cation (not the cation radical) and protonated PADPA. The second step involves the oxidation of the protonated PADPA which is one of the products of the first oxidation process. It must be re-emphasized that the final product of the second oxidation process, the PADPA cation, is the same as that of the first oxidation. The PADPA cation is stable only on a cyclic voltammetric time scale; it decomposes into soluble oligomeric products. The formation of the cation radical of PADPA occurs at a potential between these oxidation potentials, under the special conditions as outlined above. Anhydrous conditions reduce the risk of forming quinone-type products which cause un- desired side effects as far as the eventual polyaniline formation is concerned. As

demonstrated in other studies from this laboratory [17,18] the use of anhydrous perchloric acid in such experiments reveals more detailed mechanistic information.
ACKNOWLEDGMENT
We thank the Alexander von Humboldt Foundation for the donation of instru- ments. This work was partly supported by the Scientific and Technical Research Council of Turkey (TUBITAK), Project No. TBAG 963.
REFERENCES
1 E.M. Genies, A. Boyle, M. Lapkowski and C. Tsintavis, Synth. Met., 36 (1990) 139. 2 A.A. Syed and M.K. Dinesan, Talanta, 38 (1991) 815. 3 J. Bacon and R.N. Adams, J. Am. Chem. Sot., 90 (1968) 6596. 4 E.M. Genies, J.F. Penneau, M. Lapkowski and A. Boyle, J. Electroanal. Chem., 269 (1989) 63. 5 R.L. Hand and R.F. Nelson, J. Am. Chem. Sot., 96 (1974) 850. 6 T. Kobayashi, H. Yoneyama and H. Tamura, J. Electroanal. Chem., 177 (1984) 293. 7 D.F. Stilwell and S.M. Park, J. Electrochem. Sot., 135 (1988) 2497. 8 A. Kitani, J. Izumi, J. Yano, Y. Hiromoto and K. Sasaki, Bull. Chem. Sot. Jpn., 57 (1984) 2254. 9 N.S. SariqifQi, H. Kuzmany, H. Neugebauer and A. Neckel, J. Chem. Phys., 92 (7) (1990) 4530.
10 H.H. Javadi, M. Angelopoulos, A.G. MacDiarmid and A.J. Epstein, Synth. Met., 26 (1988) 1. 11 B.Z. Lubentsov, O.N. Timofeeva and M.L. Khidekel, Synth. Met., 45 (1991) 235. 12 M. Sertel, A. Yildiz and H. Baumgartel, Electrochim. Acta, 31 (1986) 1625. 13 W.F. Forbes and P.D. Sullivan, J. Am. Chem. Sot., 88 (1966) 2862. 14 H. Van Willigen, J. Am. Chem. Sot., 89 (1967) 2229. 15 R.M. Desseau, S. Shih and E.I. Heiba, J. Am. Chem. Sot., 92 (1970) 412. 16 L.W. Shacklatte, J.F. Wolf, S. Gould and R.H. Baughman, J. Chem. Phys., 88 (1988) 3955. 17 K. Pekmez, M. Can and A. Yildiz, Electrochim. Acta, in press. 18 K. Pekmez, H. Gzyiiriik and A. Yildiz, Ber. Bunsenges. Phys. Chem., in press.




















