
The differential effects of single or repeated restraint stress on kainic acid-induced neuronal...
12
The differential effects of single or repeated restraint stress on kainic acid-induced neuronal death in the hippocampal CA3 region: the role of glucocorticoid and various signal molecules Min-Soo Kwon, Young-Jun Seo, Seung-Min Choi, Hee-Woo Choi, Jun-Sub Jung, Soo-Hyun Park and Hong-Won Suh Department of Pharmacology, Institute of Natural Medicine, College of Medicine, Hallym University, Gangwon-do, South Korea Abstract The effect of stress mediators following the stress period and addition time is a controversial issue until now. Thus, we aim to clarify the differential effects of single restraint stress (SS) or repeated restraint stress (RS) on kainic acid (KA)-induced neuronal death especially as addressing not only the role of glucocorticoid (Gc) and its receptor but also the signal path- way leading to cAMP response element binding protein phosphorylation (pCREB) and its functional role during stress. In the present study, we found that although RS did not show any difference on serum Gc level and hippocampal Gc receptor level compared to SS, SS exacerbated KA-induced neuronal death in hippocampal CA3 region, but RS did not. Moreover, pre-treatment with RU 38486 (Gc receptor antag- onist) abolished the effect of SS on KA-induced neuronal death without an effect on KA toxicity itself. Furthermore, RS aggravates KA-induced neuronal death when CREB phos- phorylation was deprived by KN-93 (calcium/calmodulin- dependent protein kinase II inhibitor). However, other signal molecules inhibitors such as PD98059 (MEK1/2 inhibitor) and SP600125 (p-p38 inhibitor) have no effect on KA-induced neuronal death after RS although these signal molecule were increased during SS or RS. These findings suggest that pCREB expression via calcium/calmodulin-dependent protein kinase II phosphorylation during RS comprise one of the balancers against Gc induced by stress. Keywords: hippocampus, kainic acid, neuronal death, phosphorylated calcium/calmodulin dependent protein kinase II, phosphorylated cyclic-AMP response element binding protein, stress. J. Neurochem. (2007) 103, 1530–1541. A series of studies have demonstrated that stress causes changes in the body which lead to disease, suggesting that stress mediators have damaging effects following the stress period and time (McEwen 2002). In addition, stress elicits longer-lasting effects that include atrophy of neurons in the hippocampus, evoking cognitive function disorder (Dellu et al. 1994). There are abundant evidences that glucocortic- oids (Gc) secreted in the adrenal gland during stress can augment the brain damage induced by trauma, stroke, and seizure. Within CNS, Gc improves infiltration of inflamma- tory cells such as granulocyte, macrophage and microglia (Dinkel et al. 2003). Under restraint stress, which is one of the most commonly employed models to mimic psychological stress (Glavin et al. 1994), rats showed increased extracellular levels of glutamate in the hippocampus, and adrenalectomy markedly attenuates this elevation (Lowy et al. 1993). In addition, previous studies have demonstrated that Gc not only appear to be involved in potentiating the increased extracellular levels of excitatory amino acids during stress (Moghaddam et al. 1994) but also enhance kainic acid (KA)-induced calcium and glutamate elevation in cultured hippocampal neurons (Elliott and Sapolsky 1992; Stein- Behrens et al. 1992). Recently, it has been known that exposure of rats to immobilization stress causes differential (protective or damage) outcomes against the ischemic model following stress duration (Madrigal et al. 2003). Thus, it is speculated that single (SS) or repeated restraint stress (RS) may have an effect on the outcome of in vivo KA-induced neuronal death. Received May 11, 2007; revised manuscript received July 4, 2007; accepted July 5, 2007. Address correspondence and reprint requests to Hong-Won Suh, Department of Pharmacology, College of Medicine, Hallym University, 1 Okchun-Dong, Chuncheon, Gangwon-do 200-702, South Korea. E-mail: [email protected] Abbreviations used: BCL, B cell lymphoma; CaMK, calcium/cal- modulin-dependent protein kinase; CREB, cyclic-AMP response element binding protein; ERK, extracellular signal-regulated kinase; Gc, glucocorticoid; GR, glucocorticoid receptor; i.c.v., intracerebroventric- ular; JNK, c-jun NH2-terminal protein kinase; MAPK, mitogen-activated protein kinase; PBS, phosphate-buffered saline; pCaMKII, phosphory- lated calcium/calmodulin-dependent protein kinase II; pCREB, phos- phorylated cyclic-AMP response element binding protein; pERK, phosphorylated extracellular cell-regulated protein kinase; pJNK, phos- phorylated c-jun NH2-terminal protein kinase; RS, repeated restraint stress; SS, single restraint stress. Journal of Neurochemistry , 2007, 103, 1530–1541 doi:10.1111/j.1471-4159.2007.04865.x 1530 Journal Compilation ȑ 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541 ȑ 2007 The Authors
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of The differential effects of single or repeated restraint stress on kainic acid-induced neuronal...

The differential effects of single or repeated restraint stress onkainic acid-induced neuronal death in the hippocampal CA3region: the role of glucocorticoid and various signal molecules
Min-Soo Kwon, Young-Jun Seo, Seung-Min Choi, Hee-Woo Choi, Jun-Sub Jung, Soo-Hyun Parkand Hong-Won Suh
Department of Pharmacology, Institute of Natural Medicine, College of Medicine, Hallym University, Gangwon-do, South Korea
Abstract
The effect of stress mediators following the stress period and
addition time is a controversial issue until now. Thus, we aim
to clarify the differential effects of single restraint stress (SS)
or repeated restraint stress (RS) on kainic acid (KA)-induced
neuronal death especially as addressing not only the role of
glucocorticoid (Gc) and its receptor but also the signal path-
way leading to cAMP response element binding protein
phosphorylation (pCREB) and its functional role during stress.
In the present study, we found that although RS did not show
any difference on serum Gc level and hippocampal Gc
receptor level compared to SS, SS exacerbated KA-induced
neuronal death in hippocampal CA3 region, but RS did not.
Moreover, pre-treatment with RU 38486 (Gc receptor antag-
onist) abolished the effect of SS on KA-induced neuronal
death without an effect on KA toxicity itself. Furthermore, RS
aggravates KA-induced neuronal death when CREB phos-
phorylation was deprived by KN-93 (calcium/calmodulin-
dependent protein kinase II inhibitor). However, other signal
molecules inhibitors such as PD98059 (MEK1/2 inhibitor) and
SP600125 (p-p38 inhibitor) have no effect on KA-induced
neuronal death after RS although these signal molecule were
increased during SS or RS. These findings suggest that
pCREB expression via calcium/calmodulin-dependent protein
kinase II phosphorylation during RS comprise one of the
balancers against Gc induced by stress.
Keywords: hippocampus, kainic acid, neuronal death,
phosphorylated calcium/calmodulin dependent protein kinase
II, phosphorylated cyclic-AMP response element binding
protein, stress.
J. Neurochem. (2007) 103, 1530–1541.
A series of studies have demonstrated that stress causeschanges in the body which lead to disease, suggesting thatstress mediators have damaging effects following the stressperiod and time (McEwen 2002). In addition, stress elicitslonger-lasting effects that include atrophy of neurons in thehippocampus, evoking cognitive function disorder (Delluet al. 1994). There are abundant evidences that glucocortic-oids (Gc) secreted in the adrenal gland during stress canaugment the brain damage induced by trauma, stroke, andseizure. Within CNS, Gc improves infiltration of inflamma-tory cells such as granulocyte, macrophage and microglia(Dinkel et al. 2003).
Under restraint stress, which is one of the mostcommonly employed models to mimic psychological stress(Glavin et al. 1994), rats showed increased extracellularlevels of glutamate in the hippocampus, and adrenalectomymarkedly attenuates this elevation (Lowy et al. 1993). Inaddition, previous studies have demonstrated that Gc notonly appear to be involved in potentiating the increasedextracellular levels of excitatory amino acids during stress(Moghaddam et al. 1994) but also enhance kainic acid(KA)-induced calcium and glutamate elevation in culturedhippocampal neurons (Elliott and Sapolsky 1992; Stein-
Behrens et al. 1992). Recently, it has been known thatexposure of rats to immobilization stress causes differential(protective or damage) outcomes against the ischemic modelfollowing stress duration (Madrigal et al. 2003). Thus, it isspeculated that single (SS) or repeated restraint stress (RS)may have an effect on the outcome of in vivo KA-inducedneuronal death.
Received May 11, 2007; revised manuscript received July 4, 2007;accepted July 5, 2007.Address correspondence and reprint requests to Hong-Won Suh,
Department of Pharmacology, College of Medicine, Hallym University,1 Okchun-Dong, Chuncheon, Gangwon-do 200-702, South Korea.E-mail: [email protected] used: BCL, B cell lymphoma; CaMK, calcium/cal-
modulin-dependent protein kinase; CREB, cyclic-AMP responseelement binding protein; ERK, extracellular signal-regulated kinase; Gc,glucocorticoid; GR, glucocorticoid receptor; i.c.v., intracerebroventric-ular; JNK, c-jun NH2-terminal protein kinase; MAPK, mitogen-activatedprotein kinase; PBS, phosphate-buffered saline; pCaMKII, phosphory-lated calcium/calmodulin-dependent protein kinase II; pCREB, phos-phorylated cyclic-AMP response element binding protein; pERK,phosphorylated extracellular cell-regulated protein kinase; pJNK, phos-phorylated c-jun NH2-terminal protein kinase; RS, repeated restraintstress; SS, single restraint stress.
Journal of Neurochemistry, 2007, 103, 1530–1541 doi:10.1111/j.1471-4159.2007.04865.x
1530 Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541� 2007 The Authors

Kainic acid, which is an analog of the excitatory aminoacid L-glutamate, elicits neuronal cell death followed bysevere status epilepticus in the pyramidal layer of thehippocampal CA3 region (Sperk 1994) when KA is admin-istered intracerebroventricularly (i.c.v.). It may be because ofexcessive activation of neurons by excitatory neurotransmit-ters (e.g. glutamate), which are massively released as aconsequence of energy depletion and which result inexcitotoxic neuron death (Rothman and Olney 1986; Beal1992).
It is important to note that the brain is normally resilient inthe face of stress, suggesting that there are factors that help inthe resilience of brain cells in the presence of stressfulstimuli. The CREB phosphorylation, which mediates notonly synaptic plasticity but also survival of certain neurons(Mabuchi et al. 2001; Ma et al. 2006), can be one of thosefactors. The CREB is also phosphorylated by mitogen-activated protein kinase (MAPK) (Deak et al. 1998; Daviset al. 2000) and calcium/calmodulin-dependent proteinkinase (CaMK) (Finkbeiner 2000; Kwon et al. 2006) whendiverse extracellular stimuli such as hypoxia (Goldbart et al.2003), oxidative stress (Deak et al. 1998), glutamate(Mabuchi et al. 2001), swim stress (Liu et al. 2004; Shenet al. 2004), and restraint stress (Kwon et al. 2006) aredelivered.
A series of studies have demonstrated that various stressstimuli appear to show controversial effects in the in vivo/invitro cell death model (Moghaddam et al. 1994; DeVrieset al. 2001; May et al. 2002; Madrigal et al. 2003). How-ever, the differential effects of SS or RS on in vivo KA-induced neuronal death have not been fully elucidated untilnow. Therefore, we aim to clarify the differential effects ofSS or RS on KA-induced neuronal death especially toaddress not only the role of Gc and its receptor but also thesignal pathway leading to CREB phosphorylation (pCREB)and its functional role during stress.
Materials and Methods
These experiments were approved by the Hallym University Animal
Care and Use Committee. All procedures were conducted in
accordance with the ‘Guide for Care and Use of Laboratory
Animals’ published by the National Institutes of Health.
Experimental animals and stress procedure
Male ICR mice (MJ Co., Seoul, Korea) weighing 25–30 g and
aging 6 weeks were used for all the experiments. Animals were
housed three per cage in a room maintained at 22 ± 0.5�C with
an alternating 12 h light–dark cycle. Food and water were
available ad libitum. Animals were allowed to acclimate to the
laboratory 1 week before the beginning of the experiments. In
addition, the animals were allowed to adapt to the laboratory for
at least 2 h before testing and were only used once. All
experiments were performed during the light phase of the cycle
(10:00–17:00 h).
Single restraint stress was conducted for 2 h by placing the
mouse in 50-mL Corning tubes from 9:00 to 11:00 h. Adequate
ventilation was provided by means of holes at the sides of the tubes
(Kwon et al. 2006). In repeated stress model, the mice were
repeatedly exposed to restraint stress 2 h daily for four consecutive
days. The control group was submitted to the same handling at the
same time except for the restraint procedure. The restraint stress was
conducted in a procedure room next to the housing room to avoid
transportation.
Following the last period of restraint stress, the mice were
sacrificed by cervical dislocation, and then immediately the brain
was dissected for immunoblot. For immunohistochemistry, the mice
were prepared for perfusion after restraint stress procedure. For
cresyl violet staining, the mice were immediately administered
vehicle or KA (i.c.v.) after stress, and then the mice were prepared
for perfusion after 24 h.
Cresyl violet staining method and histological analysis
Animals were sacrificed for the brain sample by perfusion at 24 h
after phosphate-buffered saline (PBS) or KA administration. All
perfusion procedures were worked in the fume hood. For perfusion,
all mice were first deeply anesthetized with sodium pentobarbital
(100 mg/kg, i.p., Hanlim Pharm Co., Seoul, Korea) and perfused
intracardially with physiological saline followed with ice-cold
phosphate-buffered 4% paraformaldehyde (pH 7.4). Whole brain
was removed from the skull and post-fixed in the same fixative for
4 h at 4�C. Then the brains were cryoprotected in 30% sucrose for
24 h at 4�C and sectioned coronally (45 lm) on a freezing
microtome and collected in cryoprotectant for storage at )20�Cuntil processed. Prepared sections were rinsed 3 · 10 min in PBS to
remove cryoprotectant. Sections were mounted on microscope slides
(Fisher, Fair Lawn, NJ, USA) and dried on air. The slides were
soaked in cresyl violet working solution (0.02% buffer solution;
0.2% sodium acetate, 0.3% acetic acid) for 30 min. Then the
sections were dehydrated through alcohol and xylene and covers-
lipped using Permount (Fisher).
Histological analysis method in pyramidal layer of hippocampal
CA3 region was performed following under procedures. The number
of cresyl violet-positive neurons was counted by two blinded
observers at the same time using an image analyzing system
equipped with a computer-based CCD camera (Olympus AX70;
Center Valley, PA, USA). The number of cresyl violet-positive
neurons in the CA3 region of the hippocampus was counted in three
sections in reference to the mouse atlas (Franklin 1997) for each
animal (Fig. 1a). Starting from the first section (interaural 2.10 mm,
bregma )1.70 mm), counts were taken from at least three coronal
sections at 0.135 mm increments. Thus, we could always perform
neuronal counting of the same brain region and minimize any
counting bias. The number of cresyl violet-positive neurons was
compared to that of the control group of the same brain area from all
animals. All experiments were conducted independently twice. The
neuronal counting of the same group was combined for final analysis.
Glucocorticoid (corticosterone in mice) assay and blood
sampling
The plasma corticosterone level was determined by the fluorometric
determination method (Glick et al. 1964). Four hundred microliters
of blood were collected by puncturing the retro-orbital venous
The differential effect of SS or RS on KA-induced neuronal death 1531
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541
plexus. Plasma was separated by centrifugation and stored at )80�Cuntil assayed.
Total cellular protein extraction and western blot analysis
After dissecting the hippocampus, tissue was washed two times with
cold Tris-buffered saline (20 mmol/L Trizma base and 137 mmol/L
NaCl, pH 7.5). Immediately after washing, cells were lysed with
sodium dodecyl sulfate lysis buffer (62.5 mmol/L Trizma base, 2%
w/v sodium dodecyl sulfate, 10% glycerol) containing 0.1 mmol/L
Na3VO4, 3 mg/mL aprotonin, and 20 mmol/L NaF. After brief
sonication to shear DNA and reduce viscosity, the concentration of
protein was determined with a detergent-compatible protein assay
reagent (Bio-Rad Laboratories, Hercules, CA, USA) using bovine
serum albumin as the standard. After adding dithiothreitol (5 mmol/
L) and bromophenol blue (0.1% w/v), the proteins were boiled,
separated by electrophoresis in 10–12% polyacrylamide gels, and
transferred onto a polyvinylidene difluoride membrane (Amersham
Pharmacia Biotech, Little Chalfont, Buckinghamshire, UK). The
membranes were immunoblotted with antibodies against gluco-
corticoid receptor (GR) (1 : 1000; Santa Cruz Biotechnology, Santa
Cruz, CA, USA), phosphorylated c-jun NH2-terminal protein kinase
(pJNK) (1 : 1000; Cell Signaling Technology, Danvers, MA, USA),
phosphorylated extracellular cell-regulated protein kinase (pERK)
(1 : 2000; Cell Signaling Technology), p-p38 (1 : 500; Cell Signal-
ing Technology), phosphorylated calcium/calmodulin-dependent
protein kinase II (pCaMKII) a phosphorylated at thr-286
(1 : 1000; Santa Cruz Biotechnology), and pCREB phosphorylated
at ser-133 (1 : 1000; Cell Signaling Technology) and then visual-
ized with ECL-plus solution (Amersham Pharmacia Biotech). Then,
the membranes were exposed to Hyperfilm-MP (Amersham Phar-
macia Biotech) for detection of light emission. The specific signal
was quantified with the use of laser scanning densitometry with a
Bio-profil Bio-1D application (Vilber–Lourmat, Maine-la Vallee,
France) and plotted.
Immunohistochemistry
For perfusion, all mice were first deeply anesthetized with sodium
pentobarbital (100 mg/kg), i.p., and perfused intracardially with
physiological saline followed with ice-cold phosphate-buffered 4%
paraformaldehyde (pH 7.4). Whole brain was dissected and post-
fixed in the same fixative for 4 h at 4�C. Then the brain blocks
were cryoprotected in 30% sucrose for 24 h at 4�C. Sections werecut with a cryostat at a thickness of 45 lm. Immuno-histochemical
staining was performed with the Elite ABC Kit (Vector Labora-
tories, Burlingame, CA, USA). Sections were first rinsed with
0.1 mol/L bovine serum albumin three times for 10 min each, then
pre-incubated in 0.1 mol/L PBS containing 1% bovine serum
albumin and 0.2% Triton X-100 for 30 min. After rinsing twice
with 0.1 mol/L PBS containing 0.5% BSA for 10 to 15 min each,
sections were incubated with polyclonal anti-rabbit pCaMKIIa IgG
phosphorylated at thr-286 (1 : 1000; Santa Cruz biotechnology),
and polyclonal anti-rabbit pCREB IgG phosphorylated at ser-133
(1 : 300; Cell Signaling Technology) diluted with 0.1 mol/L PBS
containing 0.5% BSA and 0.05% sodium azide at 20–25�C. Afterovernight incubation, sections were rinsed and incubated with
biotinylated anti-rabbit IgG secondary antibody 1 : 200 diluted
with 0.1 mol/L PBS containing 0.5% BSA for 1 h at 20–25�C.After rinsing, the sections were incubated with ABC reagent
1 : 200 diluted with PBS for 1 h at 20–25�C and then rinsed with
PBS followed with 0.1 mol/L phosphate buffer. Finally, sections
were incubated in SIGMA FAST DAB kit (Sigma, St Louis, MO,
USA) until the desired stain intensity developed. Sections were
rinsed with 0.05 mol/L phosphate buffer, and then the sections
were dehydrated through graded ethanols, cleared in histoclear
(Fisher), and coverslipped using Permount (Fisher).
Iimmunofluorescence study
For the immunofluorescence studies, floating sections were rinsed
3 · 15 min in PBS to remove cryoprotectant, pre-incubated for
30 min in 0.1 mol/L PBS with 1% bovine serum albumin and 0.2%
Triton X-100, and incubated overnight with rabbit polyclonal anti-
pCREB IgG (1 : 300; Cell Signaling Technology) in the hippo-
campus. On the following day, after washing free-floating sections
two times with 0.1 mol/L PBS with 0.5% BSA for immunofluores-
cence labeling, we incubated them for 1 h with the purified anti-
rabbit IgG conjugated to Rhodamine (tetramethyl rhodamine
isothiocyanate) secondary antibody (Jackson Immunoresearch, Bar
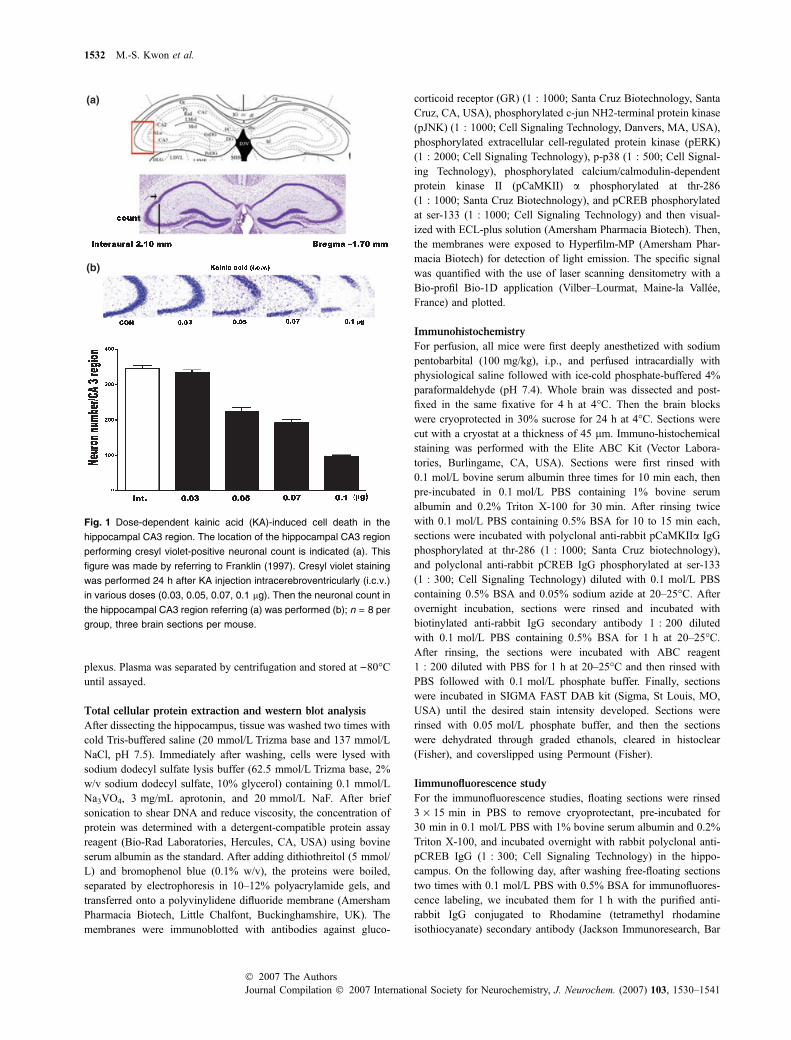
Fig. 1 Dose-dependent kainic acid (KA)-induced cell death in the
hippocampal CA3 region. The location of the hippocampal CA3 region
performing cresyl violet-positive neuronal count is indicated (a). This
figure was made by referring to Franklin (1997). Cresyl violet staining
was performed 24 h after KA injection intracerebroventricularly (i.c.v.)
in various doses (0.03, 0.05, 0.07, 0.1 lg). Then the neuronal count in
the hippocampal CA3 region referring (a) was performed (b); n = 8 per
group, three brain sections per mouse.
1532 M.-S. Kwon et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541� 2007 The Authors
Harbor, ME, USA). After incubation sections were washed and
mounted, air-dried, and coverslipped using cover glass (Fisher) and
Vectashield Mounting Medium (Vector Laboratories). Then the
slides were observed under a microscope (Bio-Rad confocal product
1024; Bio-Rad Laboratories) and photographed.
Drug treatment and intracerebroventricular kainic acid
injection
KN-93 (CaMK II inhibitor; Calbiochem-Novabiochem San Diego,
CA, USA) and PD98059 (MEK1/2 inhibitor; Sigma) were dissolved
in a phosphate buffer solution. SP600125 (JNK inhibitor; Sigma)
was dissolved in 1% dimethyl sulfoxide. RU38486 (GR antagonist;
Sigma) was dissolved in propylene glycol. The KA (0.03, 0.05,
0.07, 0.1 lg/5 lL; Sigma) was dissolved in a phosphate buffer
solution. RU 38486 (GR antagonist, 40 mg/kg) was injected
intraperitonearly (i.p.) 30 min prior to SS. KN-93, PD98059, or
SP600125 (i.c.v. 15 lg) was pre-treated 15 min prior RS on the last
day. The i.c.v. administrations of each drug such as KN-93,
PD98059, SP600125, and KA were performed following the
procedure established by Laursen and Belknap (1986). Briefly,
each mouse was injected at bregma with a 50 lL Hamilton
microsyringe (Reno, NV, USA) fitted with a 26-gauge needle that
was inserted to a depth of 2.4 mm.
Statistical analysis
Statistical analysis was carried out by t-test or one-way ANOVA
with a Bonferroni post hoc test using GRAPHPAD Prism version
4.0 for Windows (GraphPad Software, San Diego, CA, USA); p-values < 0.05 were considered to indicate statistical significance.
All values were expressed as the mean ± SEM.
Results
Dose-dependent KA-induced neuronal death in the
hippocampal CA3 region
The dose-dependent effect of i.c.v. KA injection on theneuronal death was observed in the hippocampal CA3region. In the previous study, we demonstrated that an i.c.v.KA injection (0.1 lg/5 lL) induced neuronal death in mostof the hippocampal CA3 region (Lee et al. 2004). In thepresent study, because the purpose of this experiment is toinvestigate whether the SS or RS affects KA-inducedneuronal death, the dose (0.1 lg/5 lL) used in previousstudy is a high dose to observe the effect. Thus, weperformed a dose-dependent experiment to find what theadequate dose is for this experiment. As shown in Fig. 1bi.c.v. KA injection showed dose-dependent effects onneuronal death in the hippocampal CA3 region. We chosea dose of 0.05, 0.07 lg/5 lL for the following experiment toexamine the effects of SS or RS on KA-induced neuronaldeath. In addition, we chose a dose of 0.03 lg/5 lL as themaximum dose that did not cause neuronal death in thehippocampal CA3 region.
The effect of SS or RS on KA-induced neuronal death in
the hippocampal CA3 region
To investigate the effect of SS or RS on KA-inducedneuronal death, the mice were administered KA (i.c.v. 0.05,
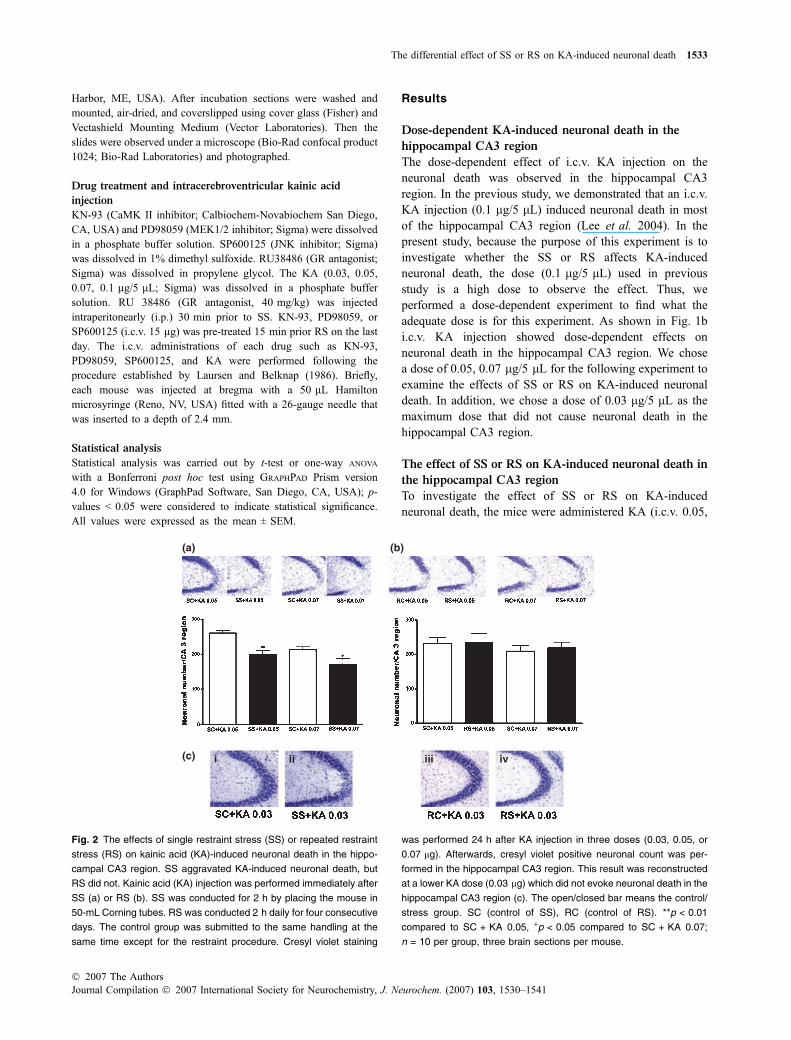
Fig. 2 The effects of single restraint stress (SS) or repeated restraint
stress (RS) on kainic acid (KA)-induced neuronal death in the hippo-
campal CA3 region. SS aggravated KA-induced neuronal death, but
RS did not. Kainic acid (KA) injection was performed immediately after
SS (a) or RS (b). SS was conducted for 2 h by placing the mouse in
50-mL Corning tubes. RS was conducted 2 h daily for four consecutive
days. The control group was submitted to the same handling at the
same time except for the restraint procedure. Cresyl violet staining
was performed 24 h after KA injection in three doses (0.03, 0.05, or
0.07 lg). Afterwards, cresyl violet positive neuronal count was per-
formed in the hippocampal CA3 region. This result was reconstructed
at a lower KA dose (0.03 lg) which did not evoke neuronal death in the
hippocampal CA3 region (c). The open/closed bar means the control/
stress group. SC (control of SS), RC (control of RS). **p < 0.01
compared to SC + KA 0.05, +p < 0.05 compared to SC + KA 0.07;
n = 10 per group, three brain sections per mouse.
The differential effect of SS or RS on KA-induced neuronal death 1533
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541
0.07 lg/5 lL) after SS or RS. As shown in Fig. 2a, the SSaggravates KA-induced neuronal death in two KA doses(0.05, 0.07 lg/5 lL). However, RS did not show any effectson the KA-induced neuronal death when compared to thecontrol group (Fig. 2b). In addition, we performed thisexperiment again at a dosage of 0.03 lg/5 lL to exclude thepossibility of neuronal count error. As shown in Fig. 2c, SSevoked neuronal death in the hippocampal CA3 region, butRS did not when following KA (0.03 lg/5 lL) injection(SC + KA 0.03 vs. SS + KA0.03, p-value = 0.0048,t = 3.611).
The alteration of the serum Gc level and hippocampal
GR expression during SS or RS
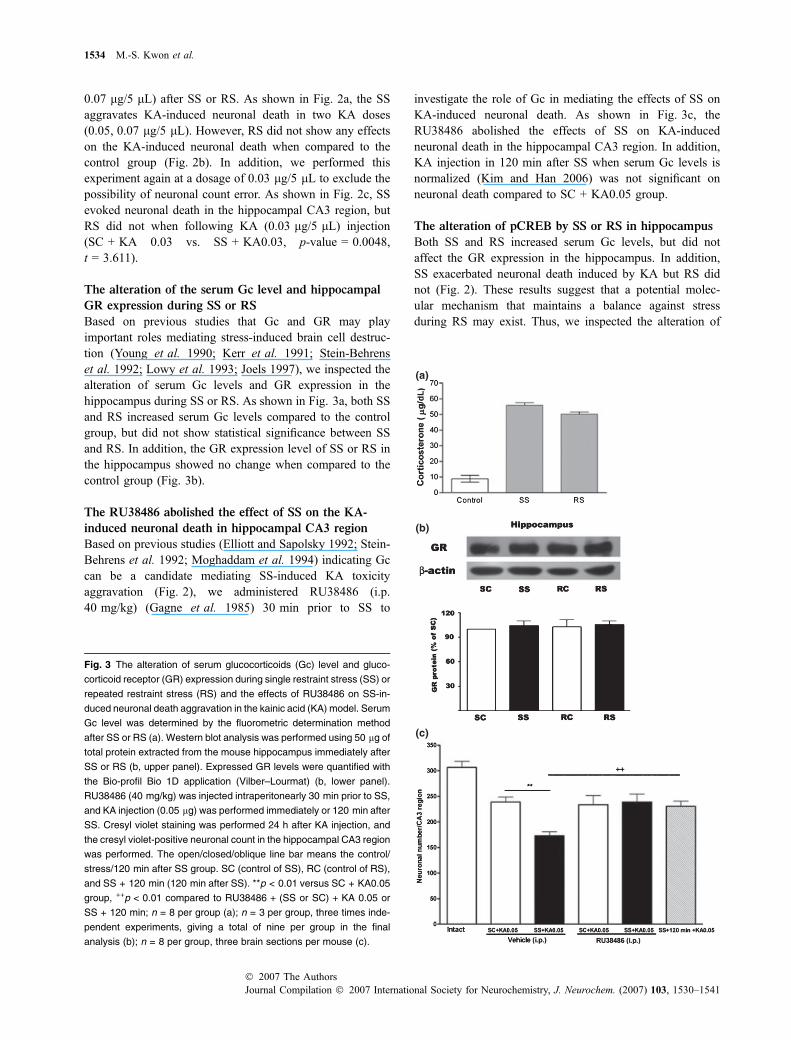
Based on previous studies that Gc and GR may playimportant roles mediating stress-induced brain cell destruc-tion (Young et al. 1990; Kerr et al. 1991; Stein-Behrenset al. 1992; Lowy et al. 1993; Joels 1997), we inspected thealteration of serum Gc levels and GR expression in thehippocampus during SS or RS. As shown in Fig. 3a, both SSand RS increased serum Gc levels compared to the controlgroup, but did not show statistical significance between SSand RS. In addition, the GR expression level of SS or RS inthe hippocampus showed no change when compared to thecontrol group (Fig. 3b).
The RU38486 abolished the effect of SS on the KA-
induced neuronal death in hippocampal CA3 region
Based on previous studies (Elliott and Sapolsky 1992; Stein-Behrens et al. 1992; Moghaddam et al. 1994) indicating Gccan be a candidate mediating SS-induced KA toxicityaggravation (Fig. 2), we administered RU38486 (i.p.40 mg/kg) (Gagne et al. 1985) 30 min prior to SS to
investigate the role of Gc in mediating the effects of SS onKA-induced neuronal death. As shown in Fig. 3c, theRU38486 abolished the effects of SS on KA-inducedneuronal death in the hippocampal CA3 region. In addition,KA injection in 120 min after SS when serum Gc levels isnormalized (Kim and Han 2006) was not significant onneuronal death compared to SC + KA0.05 group.
The alteration of pCREB by SS or RS in hippocampus
Both SS and RS increased serum Gc levels, but did notaffect the GR expression in the hippocampus. In addition,SS exacerbated neuronal death induced by KA but RS didnot (Fig. 2). These results suggest that a potential molec-ular mechanism that maintains a balance against stressduring RS may exist. Thus, we inspected the alteration of
Fig. 3 The alteration of serum glucocorticoids (Gc) level and gluco-
corticoid receptor (GR) expression during single restraint stress (SS) or
repeated restraint stress (RS) and the effects of RU38486 on SS-in-
duced neuronal death aggravation in the kainic acid (KA) model. Serum
Gc level was determined by the fluorometric determination method
after SS or RS (a). Western blot analysis was performed using 50 lg of
total protein extracted from the mouse hippocampus immediately after
SS or RS (b, upper panel). Expressed GR levels were quantified with
the Bio-profil Bio 1D application (Vilber–Lourmat) (b, lower panel).
RU38486 (40 mg/kg) was injected intraperitonearly 30 min prior to SS,
and KA injection (0.05 lg) was performed immediately or 120 min after
SS. Cresyl violet staining was performed 24 h after KA injection, and
the cresyl violet-positive neuronal count in the hippocampal CA3 region
was performed. The open/closed/oblique line bar means the control/
stress/120 min after SS group. SC (control of SS), RC (control of RS),
and SS + 120 min (120 min after SS). **p < 0.01 versus SC + KA0.05
group, ++p < 0.01 compared to RU38486 + (SS or SC) + KA 0.05 or
SS + 120 min; n = 8 per group (a); n = 3 per group, three times inde-
pendent experiments, giving a total of nine per group in the final
analysis (b); n = 8 per group, three brain sections per mouse (c).
1534 M.-S. Kwon et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541� 2007 The Authors
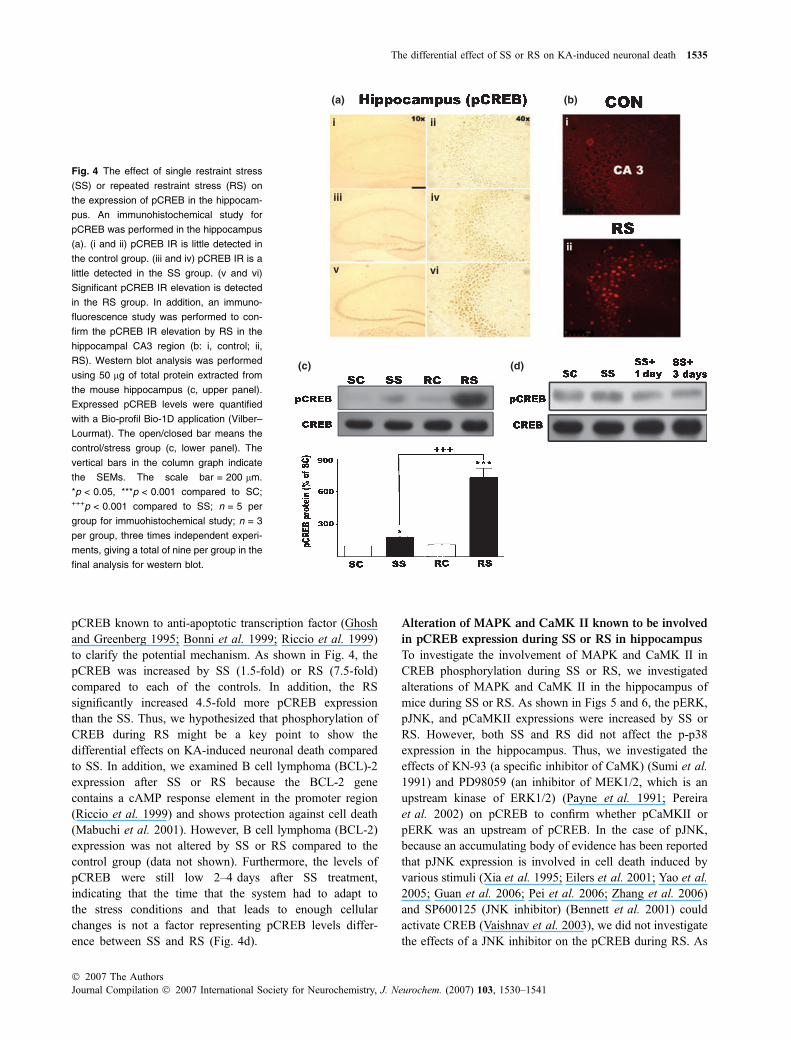
pCREB known to anti-apoptotic transcription factor (Ghoshand Greenberg 1995; Bonni et al. 1999; Riccio et al. 1999)to clarify the potential mechanism. As shown in Fig. 4, thepCREB was increased by SS (1.5-fold) or RS (7.5-fold)compared to each of the controls. In addition, the RSsignificantly increased 4.5-fold more pCREB expressionthan the SS. Thus, we hypothesized that phosphorylation ofCREB during RS might be a key point to show thedifferential effects on KA-induced neuronal death comparedto SS. In addition, we examined B cell lymphoma (BCL)-2expression after SS or RS because the BCL-2 genecontains a cAMP response element in the promoter region(Riccio et al. 1999) and shows protection against cell death(Mabuchi et al. 2001). However, B cell lymphoma (BCL-2)expression was not altered by SS or RS compared to thecontrol group (data not shown). Furthermore, the levels ofpCREB were still low 2–4 days after SS treatment,indicating that the time that the system had to adapt tothe stress conditions and that leads to enough cellularchanges is not a factor representing pCREB levels differ-ence between SS and RS (Fig. 4d).
Alteration of MAPK and CaMK II known to be involved
in pCREB expression during SS or RS in hippocampus
To investigate the involvement of MAPK and CaMK II inCREB phosphorylation during SS or RS, we investigatedalterations of MAPK and CaMK II in the hippocampus ofmice during SS or RS. As shown in Figs 5 and 6, the pERK,pJNK, and pCaMKII expressions were increased by SS orRS. However, both SS and RS did not affect the p-p38expression in the hippocampus. Thus, we investigated theeffects of KN-93 (a specific inhibitor of CaMK) (Sumi et al.1991) and PD98059 (an inhibitor of MEK1/2, which is anupstream kinase of ERK1/2) (Payne et al. 1991; Pereiraet al. 2002) on pCREB to confirm whether pCaMKII orpERK was an upstream of pCREB. In the case of pJNK,because an accumulating body of evidence has been reportedthat pJNK expression is involved in cell death induced byvarious stimuli (Xia et al. 1995; Eilers et al. 2001; Yao et al.2005; Guan et al. 2006; Pei et al. 2006; Zhang et al. 2006)and SP600125 (JNK inhibitor) (Bennett et al. 2001) couldactivate CREB (Vaishnav et al. 2003), we did not investigatethe effects of a JNK inhibitor on the pCREB during RS. As
Fig. 4 The effect of single restraint stress
(SS) or repeated restraint stress (RS) on
the expression of pCREB in the hippocam-
pus. An immunohistochemical study for
pCREB was performed in the hippocampus
(a). (i and ii) pCREB IR is little detected in
the control group. (iii and iv) pCREB IR is a
little detected in the SS group. (v and vi)
Significant pCREB IR elevation is detected
in the RS group. In addition, an immuno-
fluorescence study was performed to con-
firm the pCREB IR elevation by RS in the
hippocampal CA3 region (b: i, control; ii,
RS). Western blot analysis was performed
using 50 lg of total protein extracted from
the mouse hippocampus (c, upper panel).
Expressed pCREB levels were quantified
with a Bio-profil Bio-1D application (Vilber–
Lourmat). The open/closed bar means the
control/stress group (c, lower panel). The
vertical bars in the column graph indicate
the SEMs. The scale bar = 200 lm.
*p < 0.05, ***p < 0.001 compared to SC;+++p < 0.001 compared to SS; n = 5 per
group for immuohistochemical study; n = 3
per group, three times independent experi-
ments, giving a total of nine per group in the
final analysis for western blot.
The differential effect of SS or RS on KA-induced neuronal death 1535
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541

shown in Fig. 7a and b, the increased pCaMKII expressionduring RS was inhibited by KN-93 (i.c.v. 15 lg) adminis-tered 15 min prior to RS on the fourth day. In addition, thepCREB expression during RS was effectively blocked by the
same dose of KN-93. However, both pCaMKII and pCREBexpressions increased by RS were not inhibited by the lowerdose of KN-93 (5 and 10 lg, data not shown). In contrast toKN-93, PD98059 did not inhibit pCREB, suggesting that
Fig. 5 The effects of single restraint stress
(SS) or repeated restraint stress (RS) on
the expression of MAPK (pERK, pJNK,
p-p38) in the hippocampus. The pERK and
pJNK expressions were increased by SS or
RS. Western blot analysis was performed
using 50 lg of total protein extracted from
the mouse hippocampus. Representative
immunoblot for pERK/ERK, pJNK/JNK, and
p-p38/p38 expression during SS or RS (a).
Expressed pERK (b), pJNK (c) and p-p38
(d) levels were quantified with a Bio-profil
Bio-1D application (Vilber–Lourmat). The
open/closed bar means the control/stress
group. The vertical bars in the column graph
indicate the SEMs. *p < 0.05, ***p < 0.001
compared to SC; ++p < 0.01 compared to
SS; n = 3 per group, three times indepen-
dent experiments, giving a total of nine per
group in the final analysis for western blot.
Fig. 6 The effects of single restraint stress (SS) or repeated restraint
stress (RS) on the expression of pCaMKII in the hippocampus.
Phosphorylation of CaMK II was increased in similar pattern to pCREB
expression. An immunohistochemical study for pCaMKII was per-
formed in hippocampus (a). (i and ii) pCaMKII IR in the control group.
(iii and iv) pCaMKII IR in the SS group. (v and vi) pCaMK II IR in the
RS group. Western blot analysis was performed using 50 lg of total
protein extracted from the mouse hippocampus after SS or RS
(b, upper panel). Expressed pCaMK II levels were quantified with a
Bio-profil Bio-1D application (Vilber–Lourmat). The open/closed bar
means the control/stress group (b, lower panel). The vertical bars in
the column graph indicate the SEMs. The scale bar = 200 lm.
***p < 0.001 compared to SC; +++p < 0.001 compared to SS; n = 5 per
group for the immuohistochemical study; n = 3 per group, three times
independent experiments, giving a total of nine per group in the final
analysis for western blot.
1536 M.-S. Kwon et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541� 2007 The Authors

pERK was not an upstream signal molecule of pCREBduring RS (Fig. 7c and d). The KN-93 or PD98059 alone hadno effect on CREB activation.
The role of pCaMKII, pERK and pJNK, and GR
activation during RS in KA-induced neuronal death
As mentioned above, we observed that KN-93 pre-treatmentinhibited the pCREB expression during RS in the hippo-campus. To investigate the role of pCREB expression viapCaMKII during RS on KA-induced neuronal death, weinhibited phosphorylation of CREB by using KN-93 in theRS model, and then observed the effects of RS on KA-induced neuronal death. As shown in Fig. 8a, the RS, whichshowed no effect on KA-induced neuronal death, aggra-vated neuronal death when pCREB expression during RSwas inhibited by pre-treatment with KN-93. Therefore, wespeculate that the pCREB expression via pCaMKII duringRS may play a role in inhibiting the effects of stress, whichaggravated KA-induced neuronal death. Indeed, we couldconfirm this result again at the lower dose (KA 0.03 lg)that did not evoke neuronal death (Fig. 8d, Veh + RS +KA0.03 vs. KN-93 + RS + KA0.03, p-value = 0.0107,t = 2.845). Intracerebroventricular KN-93 injection alonedid not cause neuronal death in the hippocampal CA3
region at a lower dose (KA 0.03 lg). As shown in Fig. 8band c, other signal molecule inhibitors in the mice appliedto RS did not show any effects on KA-induced neuronaldeath. Each drug alone had no effect on KA-inducedneuronal death. Finally, we observed the effect of RU38486on KA-induced neuronal death after RS. As shown inFig. 8d, RU38486 exerted no effect on KA toxicity afterRS.
Discussion
The major findings of this study are as follows: (i) Gc can bea factor mediating SS induced neuronal death aggravation;(ii) Although RS did not show any differences in serum Gclevels and GR levels in the hippocampus compared to SS, SSaggravated KA-induced neuronal death in the hippocampalCA3 region, but RS did not. In addition, pre-treatment withRU38486 (GR antangonist) reversed the effects of SS onKA-induced neuronal death; (iii) pCREB expression viapCaMKII during RS may be associated with making thedifferential effects between SS and RS in KA-inducedneuronal death, although further experiments showing thatinduction of pCREB without prior stress is able to affordprotection would be required. Taken together, it is speculated
Fig. 7 The effects of KN-93 (CaMK II inhibitor) and PD98059 (MEK1/2
inhibitor) on the expression of pCREB during repeated stress (RS) in
the hippocampus. The pretreatment with KN-93 not PD98059 prior to
RS inhibits CREB activation. KA-induced neuronal death. RS was
conducted 2 h daily for four consecutive days. The control group was
submitted to the same handling at the same time except for the
restraint procedure. KN-93 (a and b) or PD98059 (c) was administered
15 min prior to RS on the last day. Western blot analysis was per-
formed using 50 lg of total protein extracted from the hippocampus of
the mice after RC or RS. Expressed pCaMK II and pCREB levels were
quantified with Bio-profil Bio-1D application (Vilber–Lourmat). The
open/closed bar means the control/stress group (a and b, lower
panel). The vertical bars in the column graph indicate the SEMs.
***p < 0.001 versus all other RC group; +++p < 0.001 compared to
Veh + RS; n = 3 per group, three times independent experiments,
giving a total of nine per group in the final analysis for western blot.
The differential effect of SS or RS on KA-induced neuronal death 1537
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541

that pCREB expression via pCaMKII during RS plays animportant role as a balancer against Gc, which aggravatesKA-induced neuronal death.
A series of studies have demonstrated that stress can leadto stroke or aggravate its outcome. This is reconstructed inthe transient forebrain ischemia model, which evokesneuronal death in the hippocampal CA1 region (Simonset al. 1998; McEwen 1999; Everson et al. 2001). The effectsof stress on the outcome of ischemia may be determined bystress duration (Madrigal et al. 2003). In the present study,we examined that KA-induced neuronal death was exacer-bated by SS, but not RS. First of all, it is well known thatserum Gc levels were increased during stress (Etches 1976;Siegel et al. 1982; Schlatter and Dokas 1989; Kim et al.2000), and many studies have demonstrated that Gc can leadto cell death or exacerbate cell death in various in vivo/vitrocell death models (Kerr et al. 1991; Elliott and Sapolsky1992; Stein-Behrens et al. 1992; Lowy et al. 1993; Mog-haddam et al. 1994). Thus, we observed serum Gc levels inmice during SS or RS. Although RS displayed slightly
decreased Gc level compared with SS, the differencebetween these two groups was not statistically significant,suggesting that serum Gc is not a key point in showing thedifferential effect between SS and RS on KA-inducedneuronal death. However, it is evident that significant Gclevel elevation was even observed in stress groups comparedto control groups. In addition, Moghaddam (1993) reportedthat restraint stress preferentially increased extracellularlevels of excitatory amino acid in the hippocampus, andadrenalectomy attenuated stress-induced elevations of extra-cellular glutamate concentrations in the hippocampus (Lowyet al. 1993), suggesting that Gc mediates extracellularglutamate concentration elevation during stress (Moghaddamet al. 1994). Taken together, it is speculated that extracellularamino acid increased by Gc during SS may play a synergisticrole in neuronal death with KA. To confirm whether Gcmediates SS-induced neuronal death aggravation, we per-formed RU38486 (i.p. 40 mg/kg) pre-treatment 30 min priorto SS. RU 38486 counter-balanced SS-induced neuronaldeath aggravation, which suggested that Gc was an important
Fig. 8 The effects of KN-93 (CaMK II
inhibitor), PD98059 (MEK1/2 inhibitor),
SP600125 (JNK inhibitor), or RU38486 (GR
antagonist) on kainic acid (KA)-induced
neuronal death in mice applied to repeated
stress (RS). The pre-treatment with KN-93
prior to RS aggravates KA-induced neuro-
nal death. RS was conducted 2 h daily for
four consecutive days. The control group
was submitted to the same handling at the
same time except for the restraint proce-
dure. KN-93 (a), PD98059 (b), or SP600125
(c) was administered 15 min prior to RS on
the last day. RU38486 (e) was injected
intraperitoneally 30 min prior to RS. After
RS, KA (0.5 lg) was immediately injected
intracerebroventricularly (i.c.v.). Cresyl vio-
let staining was performed 24 h after KA
injection, and the cresyl violet-positive
neuronal count in the hippocampal CA3
region was performed. The experiment of
(a) was reconstructed again at the lower KA
dose (0.03 lg) which did not evoke neuro-
nal death in the hippocampal CA3 region
(d). (i) Veh + RC, (ii) Veh + RS, (iii) KN-
93 + RC, (iv) KN-93 + RS. The open/
closed bar means the control/stress
group; *p < 0.05, compared to Veh (i.c.v.) +
RS + KA0.05; n = 10 per group, three brain
sections per mouse. RC, control of RS.
1538 M.-S. Kwon et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541� 2007 The Authors

factor mediating SS-induced neuronal death aggravation in aGR-dependent manner. However, KA0.05 toxicity itself wasnot inhibited by RU38486 pre-treatment.
Another candidate exerting a differential effect betweenSS and RS on KA-induced neuronal death may be a GR.An accumulating body of evidence has demonstrated thatGR are down-regulated by repeated stress (Young et al.1990; Wright et al. 2006), which can be an example of aresistant response to Gc. In addition, a biphasic regulationof receptor mRNA expressions for Gc in the rat hippocam-pus during acute stress was investigated (Fujikawa et al.2000). However, we examined in the present study that bothSS and RS did not show any differences on the GR level inthe hippocampus compared to the control group. Thedecreased extracellular level of glutamate also can beanother factor making the differential effect. The previousstudy (Bagley and Moghaddam 1997) reported that extra-cellular levels of glutamate increased by a single stress inthe hippocampus was not inhibited by repeated stressalthough we did not confirm this in the present study. Takentogether, these results suggest that a potential molecularmechanism that maintains a balance against stress duringRS may exist. To find this molecular mechanism, wefocused on CREB, which may be one of the candidatemolecules in explaining these results. CREB plays pivotalroles on not only synaptic plasticity but also neurotropin-mediated neuronal survival (Bonni et al. 1999; Riccio et al.1999). In addition, it has been recently demonstrated thatthe ‘preconditioning’ phenomenon (Yellon et al. 1998)happens as an inducing of phosphorylation of CREB inhippocampus (Ma et al. 2006). Furthermore, the BCL-2gene, which mediates cell survival by neurotropin-inducedCREB phosphorylation in sympathetic and cortical neurons(Riccio et al. 1999), was found to have a cAMP responseelement in the 5¢ promotor region. Thus, we observed thealteration of pCREB expression in the hippocampus duringSS or RS. Although both SS and RS increased pCREBexpression in the hippocampus, the expression level of RSis 4.5-fold more than SS, suggesting that repeated stressstimuli can induce accumulated pCREB expression in thehippocampus. In addition, we explored the alteration ofsome signal molecules (Davis et al. 2000; Mabuchi et al.2001; Kwon et al. 2006) known to be involved inphosphorylation of CREB during SS or RS. The signalmolecules (pERK, pJNK, and pCaMKII) were increased insimilar pattern to pCREB expression pattern and pCaMKIIwas found to be associated with phosphorylation of CREBin agreement with a previous study (Mabuchi et al. 2001).This result suggests that we can investigate the role ofpCREB via pCaMKII during RS by using the CaMK IIinhibitor because CREB activation during RS can beinhibited by KN-93 pre-treatment. As expected, the RSwith KN-93 pre-treatment aggravated KA-induced neuronaldeath in a similar fashion to SS. These results suggest that
phosphorylation of CREB via pCaMKII during RS plays apivotal role as a balancer against stress. However, the effectof pCaMKII itself should be addressed because it is clearthat, besides an increase in pCaMKII in the cell bodies ofCA3 pyramidal cells there is a huge increase in pCaMKII inthe mossy fiber pathway, suggesting pre-synaptic changesmay also play some role in the protection against exacer-bated toxicity, for example by somehow reducing the levelsof extracellular glutamate.
A growing body of evidence has demonstrated that CREBis activated not only in response to pro-growth and pro-survival stimuli, but also in response to stressful stimuli suchas hypoxia (Goldbart et al. 2003), oxidative stress (Deaket al. 1998), glutamate (Mabuchi et al. 2001), swim stress(Liu et al. 2004; Shen et al. 2004), and restraint stress(Kwon et al. 2006), suggesting that the activation of aCREB-dependent survival program in response to harmfulstimuli might represent a cellular form of defense. Mabuchiet al. (2001) reported that exposure to low glutamateconcentration in vitro or glutamate exposure by pre-condi-tioning ischemia in vivo in hippocampal neurons could showa protective effect via CREB activation in a gerbil globalischemia model. Based on this report, it is speculated thatrepeated extracellular glutamate level increase by RS (Mog-haddam et al. 1994) leads to enough CREB activationagainst GR activation. In addition, pCREB during RS mayinduce a survival program in response to Gc that is increasedby restraint stress. However, BCL-2 appears not to beinvolved in these survival programs during RS because analteration of BCL-2 expression was not significant during RScompare to the control group (data not shown), suggestingthat other survival programs may be involved via CREBphosphorylation by pCaMKII. The phosphorylation ofCREB also may interfere with GR-mediated transcriptionalactivation within nucleus to modulate both DNA-binding andgene transcription during RS (Stauber et al. 1992) althoughthe phenomenon happens in either a positive or inhibitorymanner depending on cell type (Adcock et al. 1996). IfpCREB expression during RS induces survival program, Gcinhibition with pCREB expression during RS may showprotective effect against KA toxicity. However, we found thatRU38486 exerted no effect on KA toxicity after RS.Therefore, the latter hypothesis appears to be more appro-priate in explaining our results, indicating that CREBactivation during RS only may play a major role ininterfering with the GR-mediated transcriptional activationinduced by RS. In the event, Gc-induced excitatory aminoacid elevation (Elliott and Sapolsky 1992; Stein-Behrenset al. 1992) in hippocampus during RS may be inhibited byCREB activation. However, further study will be needed forcharacterizing this assumption.
In conclusion, our results suggest that RS are capable ofinitiating two reciprocal pathways: (i) serum Gc levelelevation that encodes the nature of the injury as increasing
The differential effect of SS or RS on KA-induced neuronal death 1539
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541

extracellular glutamate level and (ii) a CREB-directedpro-survival program activation via CaMK II activation orGR-mediated transcription interference by CREB activation.In contrast to RS, SS, which did not lead enough CREBactivation while induced serumGc level elevation, aggravatedKA-induced neuronal death. Therefore, the fate of a cellappears to be determined by the relative strengths of these twosignals (Lonze and Ginty 2002). In addition, the lack ofinvolvement of both pERK and pJNK in phosphorylation ofCREB during RS may not be associated with KA-inducedneuronal death although these signal molecules were increasedby RS. Further study will be needed to determine the roles ofthese signal molecules that are increased by SS or RS.
Acknowledgements
This research was supported by the Research Grants from the
Korean Ministry of Science and Technology under the Brain
Frontier (M103KV010014-06K2201-01410) and Medical Research
Center program of MOST/KOSEF (R13-2005-022-01001-0).
References
Adcock I. M., Stevens D. A. and Barnes P. J. (1996) Interactions ofglucocorticoids and beta 2-agonists. Eur. Respir. J. 9, 160–168.
Bagley J. and Moghaddam B. (1997) Temporal dynamics of glutamateefflux in the prefrontal cortex and in the hippocampus followingrepeated stress: effects of pretreatment with saline or diazepam.Neuroscience 77, 65–73.
Beal M. F. (1992) Mechanisms of excitotoxicity in neurologic diseases.FASEB J. 6, 3338–3344.
Bennett B. L., Sasaki D. T., Murray B. W. et al. (2001) SP600125, ananthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. NatlAcad. Sci. USA 98, 13681–13686.
Bonni A., Brunet A., West A. E., Datta S. R., Takasu M. A. andGreenberg M. E. (1999) Cell survival promoted by the Ras-MAPKsignaling pathway by transcription-dependent and -independentmechanisms. Science 286, 1358–1362.
Davis S., Vanhoutte P., Pages C., Caboche J. and Laroche S. (2000) TheMAPK/ERK cascade targets both Elk-1 and cAMP response ele-ment-binding protein to control long-term potentiation-dependentgene expression in the dentate gyrus in vivo. J. Neurosci. 20,4563–4572.
Deak M., Clifton A. D., Lucocq L. M. and Alessi D. R. (1998) Mitogen-and stress-activated protein kinase-1 (MSK1) is directly activatedby MAPK and SAPK2/p38, and may mediate activation of CREB.EMBO J. 17, 4426–4441.
Dellu F., Mayo W., Vallee M., Le Moal M. and Simon H. (1994)Reactivity to novelty during youth as a predictive factor of cog-nitive impairment in the elderly: a longitudinal study in rats. BrainRes. 653, 51–56.
DeVries A. C., Joh H. D., Bernard O., Hattori K., Hurn P. D., TraystmanR. J. and Alkayed N. J. (2001) Social stress exacerbates strokeoutcome by suppressing Bcl-2 expression. Proc. Natl Acad. Sci.USA 98, 11824–11828.
Dinkel K., MacPherson A. and Sapolsky R. M. (2003) Novel gluco-corticoid effects on acute inflammation in the CNS. J. Neurochem.84, 705–716.
Eilers A., Whitfield J., Shah B., Spadoni C., Desmond H. and Ham J.(2001) Direct inhibition of c-Jun N-terminal kinase in sympathetic
neurones prevents c-jun promoter activation and NGF withdrawal-induced death. J. Neurochem. 76, 1439–1454.
Elliott E. M. and Sapolsky R. M. (1992) Corticosterone enhances kainicacid-induced calcium elevation in cultured hippocampal neurons.J. Neurochem. 59, 1033–1040.
Etches R. J. (1976) A radioimmunoassay for corticosterone and itsapplication to the measurement of stress in poultry. Steroids 28,763–773.
Everson S. A., Lynch J. W., Kaplan G. A., Lakka T. A., Sivenius J.and Salonen J. T. (2001) Stress-induced blood pressure reac-tivity and incident stroke in middle-aged men. Stroke 32, 1263–1270.
Finkbeiner S. (2000) CREB couples neurotrophin signals to survivalmessages. Neuron 25, 11–14.
Franklin K. B. J. P. G. (1997) The Mouse Brain in Stereotaxic Coordi-nates, 2nd edn. Academic Press, San Diego.
Fujikawa T., Soya H., Fukuoka H., Alam K. S., Yoshizato H., McEwenB. S. and Nakashima K. (2000) A biphasic regulation of receptormRNA expressions for growth hormone, glucocorticoid and min-eralocorticoid in the rat dentate gyrus during acute stress. BrainRes. 874, 186–193.
Gagne D., Pons M. and Philibert D. (1985) RU 38486: a potent anti-glucocorticoid in vitro and in vivo. J. Steroid Biochem. 23, 247–251.
Ghosh A. and Greenberg M. E. (1995) Calcium signaling in neurons:molecular mechanisms and cellular consequences. Science 268,239–247.
Glavin G. B., Pare W. P., Sandbak T., Bakke H. K. and Murison R.(1994) Restraint stress in biomedical research: an update. Neurosci.Biobehav. Rev. 18, 223–249.
Glick D., Vonredlich D. and Levine S. (1964) Fluorometric determina-tion of corticosterone and cortisol in 0.02–0.05 milliliters of plasmaor submilligram samples of adrenal tissue. Endocrinology 74, 653–655.
Goldbart A., Row B. W., Kheirandish L., Schurr A., Gozal E., Guo S. Z.,Payne R. S., Cheng Z., Brittian K. R. and Gozal D. (2003) Inter-mittent hypoxic exposure during light phase induces changes incAMP response element binding protein activity in the rat CA1hippocampal region: water maze performance correlates. Neuro-science 122, 585–590.
Guan Q. H., Pei D. S., Liu X. M., Wang X. T., Xu T. L. and Zhang G. Y.(2006) Neuroprotection against ischemic brain injury by SP600125via suppressing the extrinsic and intrinsic pathways of apoptosis.Brain Res. 1092, 36–46.
Joels M. (1997) Steroid hormones and excitability in the mammalianbrain. Front. Neuroendocrinol. 18, 2–48.
Kerr D. S., Campbell L. W., Applegate M. D., Brodish A. and LandfieldP. W. (1991) Chronic stress-induced acceleration of electrophysi-ologic and morphometric biomarkers of hippocampal aging.J. Neurosci. 11, 1316–1324.
Kim K. S. and Han P. L. (2006) Optimization of chronic stress paradigmsusing anxiety- and depression-like behavioral parameters. J. Neu-rosci. Res. 83, 497–507.
Kim D. H., Jung J. S., Kim H. S., Suh H. W., Son B. K., Kim Y. H. andSong D. K. (2000) Inhibition of brain protein kinase C attenuatesimmobilization stress-induced plasma corticosterone levels inmice. Neurosci. Lett. 291, 69–72.
Kwon M. S., Seo Y. J., Shim E. J., Choi S. S., Lee J. Y. and Suh H. W.(2006) The effect of single or repeated restraint stress on severalsignal molecules in paraventricular nucleus, arcuate nucleus andlocus coeruleus. Neuroscience 142, 1281–1292.
Laursen S. E. and Belknap J. K. (1986) Intracerebroventricular injectionsin mice. Some methodological refinements. J. Pharmacol. Methods16, 355–357.
1540 M.-S. Kwon et al.
Journal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541� 2007 The Authors

Lee H. K., Choi S. S., Han E. J., Lee J. Y., Kwon M. S., Shim E. J., SeoY. J. and Suh H. W. (2004) Role of nicotinic acetylcholinereceptors in the regulation of kainic acid-induced hippocampal celldeath in mice. Brain Res. Bull. 64, 309–317.
Liu Y. F., Bertram K., Perides G., McEwen B. S. and Wang D. (2004)Stress induces activation of stress-activated kinases in the mousebrain. J. Neurochem. 89, 1034–1043.
Lonze B. E. and Ginty D. D. (2002) Function and regulation of CREBfamily transcription factors in the nervous system. Neuron 35, 605–623.
Lowy M. T., Gault L. and Yamamoto B. K. (1993) Adrenalectomyattenuates stress-induced elevations in extracellular glutamateconcentrations in the hippocampus. J. Neurochem. 61, 1957–1960.
Ma D., Hossain M., Pettet G. K., Luo Y., Lim T., Akimov S., SandersR. D., Franks N. P. and Maze M. (2006) Xenon preconditioningreduces brain damage from neonatal asphyxia in rats. J. Cereb.Blood Flow Metab. 26, 199–208.
Mabuchi T., Kitagawa K., Kuwabara K., Takasawa K., Ohtsuki T., XiaZ., Storm D., Yanagihara T., Hori M. and Matsumoto M. (2001)Phosphorylation of cAMP response element-binding protein inhippocampal neurons as a protective response after exposure toglutamate in vitro and ischemia in vivo. J. Neurosci. 21, 9204–9213.
Madrigal J. L., Caso J. R., de Cristobal J., Cardenas A., Leza J. C.,Lizasoain I., Lorenzo P. and Moro M. A. (2003) Effect of subacuteand chronic immobilisation stress on the outcome of permanentfocal cerebral ischaemia in rats. Brain Res. 979, 137–145.
May M., McCarron P., Stansfeld S., Ben-Shlomo Y., Gallacher J., Yar-nell J., Davey Smith G., Elwood P. and Ebrahim S. (2002) Doespsychological distress predict the risk of ischemic stroke andtransient ischemic attack? The Caerphilly Study Stroke 33, 7–12.
McEwen B. S. (1999) Stress and hippocampal plasticity. Annu. Rev.Neurosci. 22, 105–122.
McEwen B. S. (2002) Protective and damaging effects of stress medi-ators: the good and bad sides of the response to stress. Metabolism51, 2–4.
Moghaddam B. (1993) Stress preferentially increases extraneuronallevels of excitatory amino acids in the prefrontal cortex: compar-ison to hippocampus and basal ganglia. J. Neurochem. 60, 1650–1657.
Moghaddam B., Bolinao M. L., Stein-Behrens B. and Sapolsky R.(1994) Glucocorticoids mediate the stress-induced extracellularaccumulation of glutamate. Brain Res. 655, 251–254.
Payne D. M., Rossomando A. J., Martino P., Erickson A. K., Her J. H.,Shabanowitz J., Hunt D. F., Weber M. J. and Sturgill T. W. (1991)Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 10, 885–892.
Pei D. S., Wang X. T., Liu Y., Sun Y. F., Guan Q. H., Wang W., Yan J. Z.,Zong Y. Y., Xu T. L. and Zhang G. Y. (2006) Neuroprotectionagainst ischaemic brain injury by a GluR6-9c peptide containingthe TAT protein transduction sequence. Brain 129, 465–479.
Pereira D. B., Carvalho A. P. and Duarte C. B. (2002) Non-specificeffects of the MEK inhibitors PD098, 059 and U0126 on glutamaterelease from hippocampal synaptosomes. Neuropharmacology 42,9–19.
Riccio A., Ahn S., Davenport C. M., Blendy J. A. and Ginty D. D.(1999) Mediation by a CREB family transcription factor of
NGF-dependent survival of sympathetic neurons. Science 286,2358–2361.
Rothman S. M. and Olney J. W. (1986) Glutamate and the pathophysi-ology of hypoxic-ischemic brain damage. Ann. Neurol. 19, 105–111.
Schlatter L. K. and Dokas L. A. (1989) Receptor specificity of a glu-cocorticoid- and stress-induced hippocampal protein. J. Neurosci.9, 1134–1140.
Shen C. P., Tsimberg Y., Salvadore C. and Meller E. (2004) Activation ofErk and JNK MAPK pathways by acute swim stress in rat brainregions. BMC Neurosci. 5, 36.
Siegel R. A., Chowers I., Conforti N., Feldman S. and Weidenfeld J.(1982) Effects of naloxone on basal and stress-induced ACTH andcorticosterone secretion in the male rat-site and mechanism ofaction. Brain Res. 249, 103–109.
Simons L. A., McCallum J., Friedlander Y. and Simons J. (1998) Riskfactors for ischemic stroke: Dubbo Study of the elderly. Stroke 29,1341–1346.
Sperk G. (1994) Kainic acid seizures in the rat. Prog. Neurobiol. 42,1–32.
Stauber C., Altschmied J., Akerblom I. E., Marron J. L. and Mellon P. L.(1992) Mutual cross-interference between glucocorticoid receptorand CREB inhibits transactivation in placental cells. New Biol. 4,527–540.
Stein-Behrens B. A., Elliott E. M., Miller C. A., Schilling J. W., New-combe R. and Sapolsky R. M. (1992) Glucocorticoids exacerbatekainic acid-induced extracellular accumulation of excitatory aminoacids in the rat hippocampus. J. Neurochem. 58, 1730–1735.
Sumi M., Kiuchi K., Ishikawa T., Ishii A., Hagiwara M., Nagatsu T. andHidaka H. (1991) The newly synthesized selective Ca2+/calmod-ulin dependent protein kinase II inhibitor KN-93 reduces dopaminecontents in PC12h cells. Biochem. Biophys. Res. Commun. 181,968–975.
Vaishnav D., Jambal P., Reusch J. E. and Pugazhenthi S. (2003)SP600125, an inhibitor of c-jun N-terminal kinase, activates CREBby a p38 MAPK-mediated pathway. Biochem. Biophys. Res.Commun. 307, 855–860.
Wright R. L., Lightner E. N., Harman J. S., Meijer O. C. and ConradC. D. (2006) Attenuating corticosterone levels on the day ofmemory assessment prevents chronic stress-induced impairmentsin spatial memory. Eur. J. Neurosci. 24, 595–605.
Xia Z., Dickens M., Raingeaud J., Davis R. J. and Greenberg M. E.(1995) Opposing effects of ERK and JNK-p38 MAP kinases onapoptosis. Science 270, 1326–1331.
Yao M., Nguyen T. V. and Pike C. J. (2005) Beta-amyloid-inducedneuronal apoptosis involves c-Jun N-terminal kinase-dependentdownregulation of Bcl-w. J. Neurosci. 25, 1149–1158.
Yellon D. M., Baxter G. F., Garcia-Dorado D., Heusch G. and SumerayM. S. (1998) Ischaemic preconditioning: present position andfuture directions. Cardiovasc. Res. 37, 21–33.
Young E. A., Akana S. and Dallman M. F. (1990) Decreased sensitivityto glucocorticoid fast feedback in chronically stressed rats.Neuroendocrinology 51, 536–542.
Zhang Q. X., Pei D. S., Guan Q. H., Sun Y. F., Liu X. M. and ZhangG. Y. (2006) Blockade of the translocation and activation ofmitogen-activated protein kinase kinase 4 (MKK4) signalingattenuates neuronal damage during later ischemia-reperfusion.J. Neurochem. 98, 170–179.
The differential effect of SS or RS on KA-induced neuronal death 1541
� 2007 The AuthorsJournal Compilation � 2007 International Society for Neurochemistry, J. Neurochem. (2007) 103, 1530–1541
![On Justiciability, Judicial Review, and Judicial Restraint: Steps to Restoring Trust in The Israel Supreme Court [Hebrew]](https://static.fdokumen.com/doc/165x107/631a79a9fd704e1d390a2561/on-justiciability-judicial-review-and-judicial-restraint-steps-to-restoring-trust.jpg)



















