
Targeting Bcr–Abl by combining allosteric with ATP-binding-site inhibitors
7
ARTICLES Targeting Bcr–Abl by combining allosteric with ATP-binding-site inhibitors Jianming Zhang 1 *, Francisco J. Adria ´n 2 *, Wolfgang Jahnke 3 , Sandra W. Cowan-Jacob 3 , Allen G. Li 2 , Roxana E. Iacob 4 , Taebo Sim 1,5 , John Powers 6 , Christine Dierks 2 , Fangxian Sun 2 , Gui-Rong Guo 2 , Qiang Ding 2 , Barun Okram 7 , Yongmun Choi 1 , Amy Wojciechowski 1 , Xianming Deng 1 , Guoxun Liu 2 , Gabriele Fendrich 3 , Andre ´ Strauss 3 , Navratna Vajpai 8 , Stephan Grzesiek 8 , Tove Tuntland 2 , Yi Liu 2 , Badry Bursulaya 2 , Mohammad Azam 6 , Paul W. Manley 3 , John R. Engen 4 , George Q. Daley 6 , Markus Warmuth 9 & Nathanael S. Gray 1 In an effort to find new pharmacological modalities to overcome resistance to ATP-binding-site inhibitors of Bcr–Abl, we recently reported the discovery of GNF-2, a selective allosteric Bcr–Abl inhibitor. Here, using solution NMR, X-ray crystallography, mutagenesis and hydrogen exchange mass spectrometry, we show that GNF-2 binds to the myristate-binding site of Abl, leading to changes in the structural dynamics of the ATP-binding site. GNF-5, an analogue of GNF-2 with improved pharmacokinetic properties, when used in combination with the ATP-competitive inhibitors imatinib or nilotinib, suppressed the emergence of resistance mutations in vitro, displayed additive inhibitory activity in biochemical and cellular assays against T315I mutant human Bcr–Abl and displayed in vivo efficacy against this recalcitrant mutant in a murine bone-marrow transplantation model. These results show that therapeutically relevant inhibition of Bcr–Abl activity can be achieved with inhibitors that bind to the myristate-binding site and that combining allosteric and ATP-competitive inhibitors can overcome resistance to either agent alone. Chronic myelogenous leukaemia (CML) is a haematological malig- nancy caused by a chromosomal rearrangement that generates a fusion protein, Bcr–Abl, with deregulated tyrosine kinase activity. Although clinical remission is usually achieved in early-stage disease with the drug imatinib, which targets the ATP-binding site, advanced-stage patients may relapse as a result of the emergence of clones expressing inhibitor-resistant forms of Bcr–Abl. Two recently approved drugs, nilotinib (AMN107) 1 and dasatinib (BMS- 354825) 2,3 , address most of the imatinib resistance mutations except the ‘gatekeeper’ T315I mutation, which is situated in the middle of the ATP-binding cleft 4–6 . GNF-2 is a highly selective non-ATP competitive inhibitor of onco- genic Bcr–Abl activity (half-maximal inhibitory concentration (IC 50 ) 0.14 mM) 7 . Using NMR spectroscopy and X-ray crystallography, we identify the myristoyl pocket located near the carboxy terminus of the Abl kinase domain as the precise binding site of GNF-2 to Bcr–Abl. By selecting for Bcr–Abl alleles resistant to GNF-2 in vitro, we identify residues both within and outside of the myristate cleft that are required for drug efficacy. Simultaneous binding to Bcr–Abl of a myristoyl mimic and an ATP-competitive inhibitor decreases the appearance of resistance-conferring mutations and results in the inhibition of both wild-type and T315I Bcr–Abl kinase activity and cell growth. Hydrogen-exchange mass spectrometry demonstrates that binding of GNF-5 to the myristate pocket results in alterations to the conformational dynamics of the ATP-binding site and provides a possible mechanism for allosteric communication between these sites. GNF-2 binds to the C-terminal myristate pocket of Abl GNF-2 had previously been suggested to bind in the myristate-binding pocket of Abl, on the basis of the observation that engineered mutations located at the entrance (A337N) and rear (A344L) of the myristoyl cleft conferred resistance to GNF-2 but not to imatinib 7 . To establish the GNF-2-binding site by an independent biophysical method, we used solution NMR 8,9 on the Abl/imatinib/GNF-2 complex 10,11 , to demon- strate that GNF-2 induces chemical shift changes that cluster around the myristate-binding pocket (Fig. 1a). No significant perturbations in chemical shift were observed for the ATP pocket, indicating that GNF-2 does not interfere with imatinib for binding at the ATP-binding site. Myristic acid was found to induce qualitatively the same pattern of perturbations in chemical shift (Fig. 1b), providing additional evidence that GNF-2 and myristate share the same binding site. Crystal structure of Abl/imatinib/GNF-2 complex. The binding of GNF-2 to the myristoyl pocket of Abl was further confirmed by X-ray crystallography. The structure of the Abl/imatinib/GNF-2 complex was obtained by soaking crystals of Abl/imatinib/myristate, obtained as described in ref. 10, in an excess of GNF-2. As judged by the shape of the electron density (Supplementary Fig. 1), GNF-2 replaces the myristoylated peptide in the crystals. There are two molecules in the asymmetric unit, and the myristate-binding site is fully occupied by *These authors contributed equally to this work. 1 Dana-Farber Cancer Institute, Harvard Medical School, Department of Cancer Biology and Department of Biological Chemistry and Molecular Pharmacology, 250 Longwood Avenue, Seeley G. Mudd Building 628, Boston, Massachusetts 02115, USA. 2 Genomics Institute of the Novartis Research Foundation, Department of Chemistry, 10675 John Jay Hopkins Drive, San Diego, California 92121, USA. 3 Novartis Institutes for Biomedical Research, CH-4056 Basel, Switzerland. 4 The Barnett Institute of Chemical & Biological Analysis and Department of Chemistry & Chemical Biology, Northeastern University, Boston, Massachusetts 02115, USA. 5 Life Sciences Research Division, Korea Institute of Science and Technology 39-1, Hawolgok-dong, Seongbuk-gu, Seoul, 136-791, Korea. 6 Division of Pediatric Hematology/Oncology, Children’s Hospital and Dana-Farber Cancer Institute; Division of Hematology, Brigham and Women’s Hospital; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School; Howard Hughes Medical Institute; Boston, Massachusetts 02115, USA. 7 Department of Chemistry and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. 8 Biozentrum, University of Basel, Klingelbergstrasse 70, CH-4056 Basel, Switzerland. 9 Novartis Institutes for BioMedical Research, Inc., 250 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. Vol 463 | 28 January 2010 | doi:10.1038/nature08675 501 Macmillan Publishers Limited. All rights reserved ©2010
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Targeting Bcr–Abl by combining allosteric with ATP-binding-site inhibitors

ARTICLES
Targeting Bcr–Abl by combining allostericwith ATP-binding-site inhibitorsJianming Zhang1*, Francisco J. Adrian2*, Wolfgang Jahnke3, Sandra W. Cowan-Jacob3, Allen G. Li2,Roxana E. Iacob4, Taebo Sim1,5, John Powers6, Christine Dierks2, Fangxian Sun2, Gui-Rong Guo2, Qiang Ding2,Barun Okram7, Yongmun Choi1, Amy Wojciechowski1, Xianming Deng1, Guoxun Liu2, Gabriele Fendrich3,Andre Strauss3, Navratna Vajpai8, Stephan Grzesiek8, Tove Tuntland2, Yi Liu2, Badry Bursulaya2,Mohammad Azam6, Paul W. Manley3, John R. Engen4, George Q. Daley6, Markus Warmuth9 & Nathanael S. Gray1
In an effort to find new pharmacological modalities to overcome resistance to ATP-binding-site inhibitors of Bcr–Abl, werecently reported the discovery of GNF-2, a selective allosteric Bcr–Abl inhibitor. Here, using solution NMR, X-raycrystallography, mutagenesis and hydrogen exchange mass spectrometry, we show that GNF-2 binds to themyristate-binding site of Abl, leading to changes in the structural dynamics of the ATP-binding site. GNF-5, an analogue ofGNF-2 with improved pharmacokinetic properties, when used in combination with the ATP-competitive inhibitors imatinibor nilotinib, suppressed the emergence of resistance mutations in vitro, displayed additive inhibitory activity in biochemicaland cellular assays against T315I mutant human Bcr–Abl and displayed in vivo efficacy against this recalcitrant mutant in amurine bone-marrow transplantation model. These results show that therapeutically relevant inhibition of Bcr–Abl activitycan be achieved with inhibitors that bind to the myristate-binding site and that combining allosteric and ATP-competitiveinhibitors can overcome resistance to either agent alone.
Chronic myelogenous leukaemia (CML) is a haematological malig-nancy caused by a chromosomal rearrangement that generates afusion protein, Bcr–Abl, with deregulated tyrosine kinase activity.Although clinical remission is usually achieved in early-stage diseasewith the drug imatinib, which targets the ATP-binding site,advanced-stage patients may relapse as a result of the emergence ofclones expressing inhibitor-resistant forms of Bcr–Abl. Two recentlyapproved drugs, nilotinib (AMN107)1 and dasatinib (BMS-354825)2,3, address most of the imatinib resistance mutations exceptthe ‘gatekeeper’ T315I mutation, which is situated in the middle ofthe ATP-binding cleft4–6.
GNF-2 is a highly selective non-ATP competitive inhibitor of onco-genic Bcr–Abl activity (half-maximal inhibitory concentration (IC50)0.14mM)7. Using NMR spectroscopy and X-ray crystallography, weidentify themyristoyl pocket located near the carboxy terminus of theAbl kinase domain as the precise binding site of GNF-2 to Bcr–Abl. Byselecting for Bcr–Abl alleles resistant to GNF-2 in vitro, we identifyresidues both within and outside of the myristate cleft that arerequired for drug efficacy. Simultaneous binding to Bcr–Abl of amyristoyl mimic and an ATP-competitive inhibitor decreases theappearance of resistance-conferring mutations and results in theinhibition of both wild-type and T315I Bcr–Abl kinase activity andcell growth. Hydrogen-exchange mass spectrometry demonstratesthat binding of GNF-5 to the myristate pocket results in alterationsto the conformational dynamics of the ATP-binding site and provides
a possible mechanism for allosteric communication between thesesites.
GNF-2 binds to the C-terminal myristate pocket of Abl
GNF-2 had previously been suggested to bind in the myristate-bindingpocket ofAbl, on the basis of the observation that engineeredmutationslocated at the entrance (A337N) and rear (A344L) of themyristoyl cleftconferred resistance to GNF-2 but not to imatinib7. To establish theGNF-2-binding site by an independent biophysical method, we usedsolution NMR8,9 on the Abl/imatinib/GNF-2 complex10,11, to demon-strate thatGNF-2 induces chemical shift changes that cluster around themyristate-binding pocket (Fig. 1a). No significant perturbations inchemical shift were observed for the ATP pocket, indicating thatGNF-2 does not interfere with imatinib for binding at the ATP-bindingsite.Myristic acid was found to induce qualitatively the same pattern ofperturbations in chemical shift (Fig. 1b), providing additional evidencethat GNF-2 and myristate share the same binding site.Crystal structure of Abl/imatinib/GNF-2 complex. The binding ofGNF-2 to themyristoyl pocket of Abl was further confirmed by X-raycrystallography. The structure of the Abl/imatinib/GNF-2 complexwas obtained by soaking crystals of Abl/imatinib/myristate, obtainedas described in ref. 10, in an excess of GNF-2. As judged by the shapeof the electron density (Supplementary Fig. 1), GNF-2 replaces themyristoylated peptide in the crystals. There are two molecules in theasymmetric unit, and the myristate-binding site is fully occupied by
*These authors contributed equally to this work.
1Dana-Farber Cancer Institute, HarvardMedical School, Department of Cancer Biology and Department of Biological Chemistry andMolecular Pharmacology, 250 Longwood Avenue,Seeley G.Mudd Building 628, Boston,Massachusetts 02115, USA. 2Genomics Institute of the Novartis Research Foundation, Department of Chemistry, 10675 John Jay Hopkins Drive,San Diego, California 92121, USA. 3Novartis Institutes for Biomedical Research, CH-4056 Basel, Switzerland. 4The Barnett Institute of Chemical & Biological Analysis and Departmentof Chemistry & Chemical Biology, Northeastern University, Boston, Massachusetts 02115, USA. 5Life Sciences Research Division, Korea Institute of Science and Technology 39-1,Hawolgok-dong, Seongbuk-gu, Seoul, 136-791, Korea. 6Division of Pediatric Hematology/Oncology, Children’s Hospital and Dana-Farber Cancer Institute; Division of Hematology,Brigham and Women’s Hospital; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School; Howard Hughes Medical Institute; Boston,Massachusetts 02115, USA. 7Department of Chemistry and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla,California 92037, USA. 8Biozentrum, University of Basel, Klingelbergstrasse 70, CH-4056 Basel, Switzerland. 9Novartis Institutes for BioMedical Research, Inc., 250 MassachusettsAvenue, Cambridge, Massachusetts 02139, USA.
Vol 463 |28 January 2010 |doi:10.1038/nature08675
501Macmillan Publishers Limited. All rights reserved©2010

GNF-2 in one and partly in the other. GNF-2 binds in an extendedconformation in themyristate pocket with theCF3 group buried at thesame depth as the final two carbons of the myristate ligand (Fig. 2).There is a favourable, but probably weak, polar interaction betweenone fluorine atom and the main chain of L340 (similar to thatobserved between nilotinib and D381 of Abl)12, and there are water-mediated hydrogen bonds, but no direct hydrogen bonds with theprotein. As expected, most of the interactions between GNF-2 andthe protein are hydrophobic. As discussed below, mutation of threeresidues near the mouth of the myristate-binding site (C464Y, P465Sand V506L) is found to cause resistance to the binding of GNF-2,presumably for steric reasons. The overall structure of the Abl kinasedomain complexed to GNF-2 is similar to that of the myristate com-plex (Fig. 2b), with some small differences that probably result fromcrystal contacts, but no changes in the ATP-binding site.
GNF-2 analogue structure–activity relations
After the identification of GNF-2 as a lead compound, a systematicevaluation of the structural features necessary to impart function as acellular Bcr–Abl inhibitor were investigated through the synthesis ofmore than 200 analogues and rationalized in the context of bindingto themyristate-binding site (X.D. andN.S.G., unpublished observa-tions, and Supplementary Fig. 2a). The structure–activity relationsare fully consistent with the conformation of GNF-2 observed in thecrystal structure: the trifluoromethoxy group of GNF-2 can only beaccommodated at the para position; the aniline NH is requiredbecause of the formation of a water-mediated hydrogen bond tothe backbone carbonyls of A433 and E462; water-mediated hydrogenbonds between the carboxamide of GNF-2 and Abl confer enhancedinhibitory activity; and the extended compound conformation isrequired to fit the cylindrical binding cavity.
Drug combinations reduce emergence of resistant mutants
We sought to investigate the frequency with which Bcr–Abl-dependentBa/F3 cells would become resistant to combinations of GNF-2 andimatinib in comparison with each compound alone. The number ofresistant clones that emerged as a result of continuous exposure to 1mMimatinib was decreased by at least 90%when cells were treated for up to
21 dayswith 1mMimatinib combinedwith 5 or 10mMGNF-2 (Fig. 3a).These results demonstrate that combinations of GNF-2 and imatinibcan cooperate to suppress the emergence of resistance mutations.Identification of the Bcr–Abl mutants resistant to GNF-2. To dis-cover the full complement of Bcr–Abl mutants that induce resistanceto GNF-2 we performed two types of selection. In the first, Bcr–Abl-transformed Ba/F3 cells were cultured in the presence of increasingconcentrations of GNF-2 to allow cells to evolve drug resistance asdescribed previously for the ATP-competitive inhibitor PD166326(ref. 13). In the second approach, Bcr–Abl was randomly mutated inEscherichia coli and the mutant clones were expressed in Ba/F3 cells,which were then grown in the presence of inhibitor14. These screensresulted in the identification of a total of 306 mutants, 163 (12 sites)from the first and143 (22 sites) from the second (Supplementary Fig. 3).More than 80% of the resistant colonies contained Bcr–Abl mutationsclustered in themyristate-binding pocket or the SH2 and SH3 domains(Supplementary Fig. 3). This is in contrast to ATP-competitive inhibi-tors such as imatinib, PD166321 and AP23464 (refs 13–15), for whichmost resistance mutations cluster adjacent to the kinase catalytic site.
Tovalidate the functional relevanceof thesemutations,we engineeredindividualmutant Bcr–Abl-transformed Ba/F3 cells for nine of themostfrequently isolated GNF-2-resistant mutations and for the T315I ‘gate-keeper’ mutation. These selected mutations located in the SH3 domain(P112S), the SH3–SH2 domain linker (Y128D), the SH2 domain(Y139C), the SH2–kinase-domain linker (S229P), the ATP-binding site(T315I) and adjacent to the myristate-binding site (C464Y, P465S,F497L, E505K and Y506L) were introduced individually into Bcr–Ablby site-directed mutagenesis. Of these, only the T315I substitution haspreviously been reported to confer resistance to imatinib16,17. GNF-2hadan IC50 against all tenmutants thatwas elevated5–50-fold relative to thatagainst wild-type Bcr–Abl-transformed Ba/F3 cells (Fig. 3b). The threemost frequently recovered mutations (60% of the total) were located inclose proximity to themyristate-binding site (C464Y,P465S andE505K)and were shown to confer complete resistance to GNF-2 up to a con-centration of 10mM (Fig. 3c).
To examine how the mutations affected the ability of GNF-2to inhibit Bcr–Abl-mediated signalling, we examined Bcr–Ablautophosphorylation and phosphorylation of a downstream substrate,STAT5, after treatment with inhibitor (Supplementary Fig. 4a). At aconcentration of 10mM, GNF-2 could inhibit the phosphorylation ofBcr–Abl and STAT5 in all mutants except the three myristate-sitemutations (E505K, P465S and C646Y) and the ‘gatekeeper’ T315Imutation.Mutations in the myristate pocket interfere with GNF-2 binding.We tested the ability of mutant Bcr–Abl proteins, obtained fromcrude cell lysates, to bind to a GNF-2 affinity resin (SupplementaryFig. 4b, c)7. These experiments revealed that only the mutationslocated in the myristate-binding site (C464Y, P465S and E505K)
a b122.0
125.0
124.0
123.0
122.0
125.0
124.0
123.0
8.0 7.0 p.p.m. 7.0 p.p.m.8.0
Figure 1 | NMR spectroscopy provides evidence for GNF-2 binding to theC-terminalmyristate pocket ofAbl. a, Top: a heteronuclear single-quantumcoherence spectrum of the Abl/imatinib complex with (red) and without(black) GNF-2, showing chemical-shift changes induced by ligand binding(the y-axis scale is in p.p.m.). Bottom:mapping of the chemical-shift changesto the structure of the Abl/imatinib complex (PDB accession 1OPK; ref. 10)identifies the myristate pocket as the GNF-2-binding site. The size of thespheres is proportional to the magnitude of the chemical shift changes. b, Asin a except that myristic acid (blue in top panel) was used instead of GNF-2.
aa E462
L510
P465
L429
L340L341
HOH
HOH
HOH
!E
!I
!I"
!H
!F
I–I" loop
Y435
C464
E505
V468
V506
F497A337
A344
A413
I432
F493
I502
ba
Figure 2 | Crystal structure of GNF-2 bound to the Abl myristoyl pocket.a, Abl kinase is indicated in green (helices are indicated by transparentcylinders), with the bent part of the I-helix in yellow, GNF-2 resistancemutations in pink, and GNF-2 carbons in cyan. Hydrogen bonding andother polar interactions are indicated by dotted red lines. b, Superposition ofthe Abl/imatinib/myristate (white), Abl/imatinib/GNF-2 (green and yellow)and Abl-imatinib (red) structures. GNF-2 is coloured in cyan, and myristicacid in magenta.
ARTICLES NATURE |Vol 463 |28 January 2010
502Macmillan Publishers Limited. All rights reserved©2010

ablated the binding of Bcr–Abl to GNF-2; all other mutationsretained binding to GNF-2. Using an NMR-based titration, we con-firmed that GNF-2 still binds to the ‘gatekeeper’ mutant, T315I Abl(residues 229–500, not including helix I), albeit with half the affinityof the wild type (Supplementary Fig. 5). These results suggest that themyristate-site mutations directly interfere with drug binding,whereas the non-myristate-site mutants function through a differentmechanism, which may involve disfavouring the inhibited con-formation induced after binding of GNF-2.
GNF-5 and nilotinib combinations inhibit T315I Bcr–Abl
GNF-5, theN-hydroxyethyl carboxamide analogue of GNF-2 posses-sing similar cellular Bcr–Abl inhibitory activity but having morefavourable pharmacokinetic properties, was chosen for further singleand combination studies in vitro and in vivo (Supplementary Fig. 2b).Combinations of GNF-5 and nilotinib inhibited T315I Bcr–Abl-dependent cell growth with a calculated combination index18 of 0.6,indicating moderate synergy (Fig. 4a). For example, at a fixed GNF-5concentration of 2mM, nilotinib inhibits T315I Bcr–Abl-dependentproliferation with an IC50 of 0.86 0.05mM (mean6 s.d.). Flow cyto-metry analysis showed that GNF-5 and nilotinib act additively toinhibit STAT5 phosphorylation; this could be rescued by the additionof interleukin-3 to the medium (Supplementary Fig. 6a). Nilotiniband GNF-5 also exhibited cooperativity for inhibition of wild-typeBcr–Abl-transformedBa/F3 cells with a calculated combination indexof 0.6 (Supplementary Fig. 6b). We confirmed that the cooperativityobserved between GNF-5 and nilotinib is directly mediated by theinhibition of Bcr–Abl, on the basis of the ability of a double mutationof T315I in the ATP-binding site and E505K in the myristate-bindingsite to confer complete resistance to the combination of both inhibi-tors in phosphorylation (Fig. 4b) and proliferation (SupplementaryFig. 6c, d) assays. GNF-5 and nilotinib also acted cooperatively against
p190 Bcr–Abl, a variant commonly found in acute lymphocytic leu-kaemia that typically responds only transiently to imatinib therapy19,with a calculated combination index of 0.5 (Supplementary Fig. 7).Biochemical characterization of GNF-5 in combination withimatinib and nilotinib. To determine whether the additive inter-action between GNF-5 and the ATP-competitive inhibitors imatiniband nilotinib observed in cellular assays could be confirmed at theprotein level, we performed steady-state kinetic analyses of Abl kinaseby using a pyruvate kinase–lactate dehydrogenase detection system20.We first tested the inhibitory activity of imatinib, nilotinib andGNF-5in bacterially expressed wild-type, T315I and E505K Abl kinases(Supplementary Fig. 8). Inhibition of wild-type Abl was observed forall three inhibitors with GNF-5 showing an IC50 of 0.226 0.01mM,imatinib an IC50 of 0.246 0.03mM and nilotinib an IC50 of0.296 0.06mM at an ATP concentration of 20mM, which is close tothe apparent Km under our assay conditions. Although the activity ofrecombinant Abl was previously reported to be insensitive to GNF-2(ref. 7), we subsequently discovered that this was due to the presenceof Brij-35, a common additive to kinase assay buffers, which maskedinhibition by GNF-2 (ref. 21). The myristate-binding-site mutantE505K was inhibited by imatinib with an IC50 of 0.226 0.03mMand nilotinib with an IC50 of 0.206 0.01mM, but not by GNF-5(IC50. 10mM). The T315I mutant was not inhibited by imatinib orGNF-5, but nilotinib did exhibitweak activity against thismutantwithan IC50 of 1.426 0.3mM. GNF-5 was confirmed to inhibit wild-typeAbl in a non-ATP competitive fashion (Supplementary Fig. 9).
We next examined whether combinations of GNF-5 and nilotinibresulted in additive inhibition of wild-type, T315I or E505K recombi-nantAbl proteins. Positive cooperativitywas observed for combinationsof GNF-5 and nilotinib on the wild-type and T315I enzymes withcalculated combination indices of 0.53 and 0.61, respectively (Fig. 4c,d and Supplementary Fig. 10). For example, at a fixed GNF-5 concen-tration of 1mM and an ATP concentration of 20mM, the IC50 values ofnilotinib against T315I and wild-type enzyme were decreased 4.7- and
25 10 5 4 2 1 25 10 5
Day 9
Day 12
Day 210
8491
2 1070
9691
0
7581
2 440
96
72
0
52 59
2 200
96
66
0
50
100
SH3 domainSH3 domainSH3 domain
S229PS229PS229P
P112SP112SP112S
Y128DY128DY128D
SH2 domainSH2 domainSH2 domain
F497LF497LF497L
E505KE505KE505KCOOHCOOHCOOH
V506LV506LV506LC464YC464YC464Y
T315lT315lT315l
Y139CY139CY139C
P465SP465SP465SMyristoylpocketMyristoylpocketMyristoylpocket
Catalytic siteCatalytic siteCatalytic site
Kinase domainKinase domainKinase domain
H2NH2NH2N
MutationNo. of
resistantcoloniesImatinib GNF-2
IC50 (µM)
Bcr–AblT315lP112SY128DY139CS229PC464YP465SF497LE505KV506L
0.4810.00.640.400.410.850.300.240.130.240.23
0.2610.05.284.072.226.0610.010.01.9310.01.25
02413191419242412411
GNF-2 Imatinib GNF-2 + 1 µM imatinibConcentration (µM)
Resistant colonies
a
b c
Figure 3 | Location and cellular IC50 of Bcr–Abl GNF-2 resistancemutations. a, Effect of various concentrations of GNF-2, imatinib, orcombinations of both on the number of emerging Ba/F3.Bcr–Abl-resistantclones (shown on top of each bar). b, Mutations indicated by red spheres onAbl with size proportional to the degree of resistance (PDB accession 1OPK(ref. 10), amino-acid residues numbered as in Abl 1a). c, IC50 for growthinhibition by imatinib or GNF-2 for wild-type and mutant Bcr–Abl-transformedBa/F3 cells. The numbers of colonies that emerged after 12 daysin the presence of 20 mM GNF-2 are indicated.
0 0.5
T315l10 µM nilotinib
T315l/E505K
5 10 0 0.5 5 10GNF-5 (µM)
GNF-5 + nilotinib (1:1)
Nilotinib/GNF-5 (µM)
NilotinibGNF-5
GNF-5 + nilotinib (1:1)NilotinibGNF-5
p-Bcr–AblBcr–Abl
p-STAT5
100
50
0
100
50
00 5 10
0 5 10
Inhi
bito
ry e
ffect
(%)
a b
d
c
Abl T315I
Abl E505KAbl Nilotinib
+ 1 µMGNF-5
NilotinibGNF-5
10
>10>10
2
1 IC50
(µM
)1.42
0.3
0.18
0.03
0.290.2 0.22
Figure 4 | Cellular and enzymatic inhibition of wild-type and mutants bycombination treatments. a, Effects of GNF-5, nilotinib and variousconcentrations of GNF-5 in combination with nilotinib (0.3–10mM) on theproliferation of T315I Bcr–Abl-expressing Ba/F3 cells. The combinationcurve (red, and also in c) contains twice the total drug concentration of thesingle agent curves because both drugswere present. b, Inhibition of Bcr–Ablautophosphorylation was determined by Bcr–Abl immunoprecipitation,followed by an immunoblot for phospho-Tyr (Y412)1, phospho-STAT5(Y694) and total Bcr–Abl (antibody K-12) from cell lystates obtained aftertreatment of T315I Bcr–Abl-expressing Ba/F3 cells with 10 mM of nilotiniband increasing concentrations of GNF-5 (0, 0.5, 5 and 10mM) for 90min.c, Percentage inhibition of T315I Abl kinase by nilotinib and GNF-5 or acombination of the two. d, IC50 for inhibition of wild-type, E505K andT315IAbl kinase activity by GNF-5, nilotinib or a combination of the two at anATP concentration of 20 mM.
NATURE |Vol 463 |28 January 2010 ARTICLES
503Macmillan Publishers Limited. All rights reserved©2010

9.6-fold, respectively, compared with those calculated in the absence ofGNF-5. As expected, no additivity was observed with the E505Kmyristate-binding-site mutant. These results demonstrate that allostericcommunicationbetweenthemyristate-bindingsiteandtheATP-bindingsite can be observed in biochemical kinase assays.GNF-5 binding alters the conformation of the ATP-binding site.To examine how the binding of GNF-5 to the myristate-binding sitemight influence the conformational dynamics of the ATP-bindingsite and other regions of Abl, we performed hydrogen-exchange massspectrometry. This technique allows the dynamics of a protein to beinvestigated by measuring the exchange of backbone amide hydro-gens with the bulk solvent22. Unbound Abl and the GNF-5–Abl com-plex were independently exposed to D2O for periods ranging from10 s to 4 h, as described previously23. Changes in deuterium incor-poration in the presence of GNF-5 were observed in several peptides(Fig. 5) surrounding themyristate-binding cleft. In addition, changesin peptides near the ATP-binding site (for example residues 306–316and 317–324) were also seen, implying that binding of GNF-5 affectedthe ATP-binding site. Hydrogen exchange in these peptides was notaltered in a control experiment with GNF-5 and the non-binding AblE505Kmyristatemutant, indicating that the binding ofGNF-5 iswhatis responsible for the altered conformation and hydrogen exchange inthepeptides near theATP-binding site. These results demonstrate thatligation of themyristate-binding site can cause dynamic perturbationsto residues in the ATP-binding site and provides a mechanism bywhich synergistic interactions between these two sites could occur.
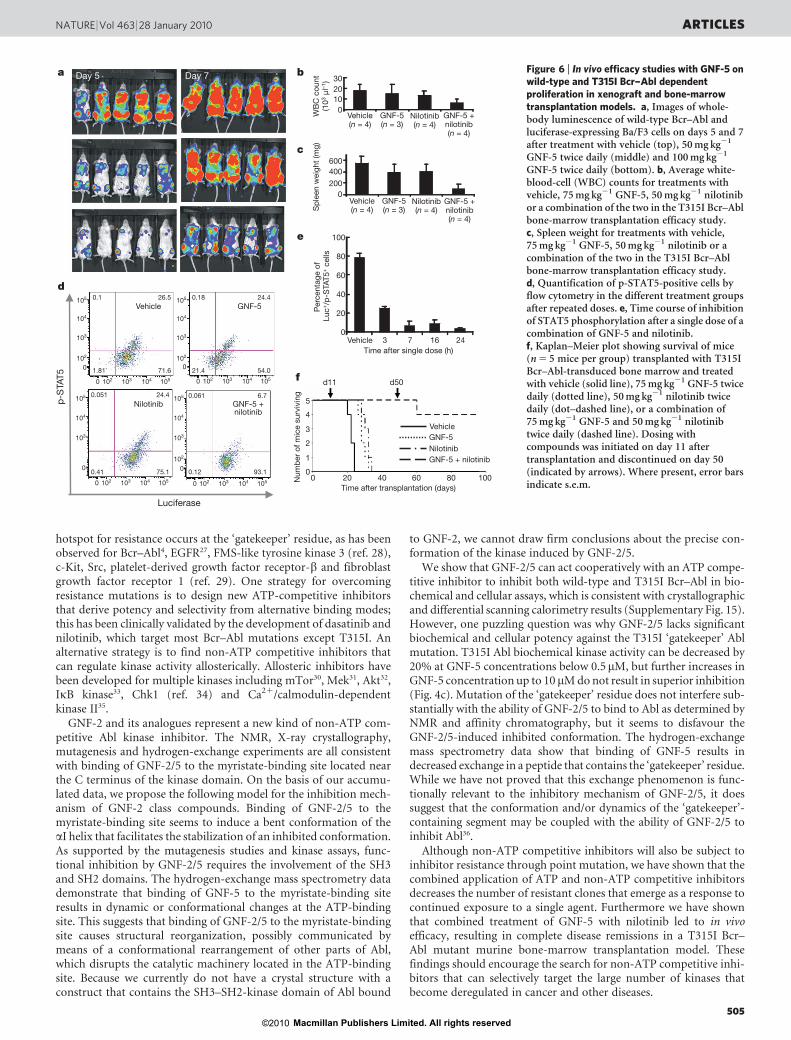
Drug combinations inhibit T315I Bcr–Abl in vivo
The murine pharmacokinetic parameters of GNF-5 were measuredand determined to be suitable for use of the compound in vivo(Supplementary Fig. 11). GNF-5 was shown to be efficacious in vivoat well tolerated doses in a murine xenograft model of p210 Bcr–AblBa/F3-induced leukaemia, but relapses were observed (Fig. 6a and
Supplementary Fig. 12). The target modulation in vivo was furtherconfirmed by examining the phosphorylation of STAT5 (Sup-plementary Fig. 13).GNF-5 and nilotinib combinations against T315I Bcr–Abl. Toevaluate the in vivo efficacy of GNF-5 on wild-type and T315I Bcr–Abl further, we used a bone-marrow transduction/transplantationmouse model that more closely resembles human CML disease24.Initial experiments with p210 Bcr–Abl demonstrated that 50mg kg21
GNF-5 twice daily could normalize blood counts and spleen size(Supplementary Fig. 14). We then addressed whether the combina-tion of GNF-5 with nilotinib would result in efficacy in a T315I Bcr–Abl bone-marrow transduction/transplantation model. Mice treatedtwice daily with either nilotinib (50mg kg21) or GNF-5 (75mg kg21)alone at 15 days after transplantation showed no significant responsecompared with the vehicle group, with twofold to threefold highercell counts and spleens fourfold larger than those of healthy mice. Incontrast, the combination brought about a normalization of bloodcell counts and spleen size without signs of toxicity, suggesting an addi-tive effect of the compounds in a combination treatment (Fig. 6b, c). Toestablish a correlation between efficacy and pharmacodynamic res-ponse, bonemarrow cells from the differentmouse groupswere isolatedat the end of the efficacy study, stained with anti-p-STAT5 and anti-luciferase specific antibodies, and analysed by flow cytometry. The per-centages of p-STAT5-positive Bcr–Abl-expressing bone marrow cellswere similar (approximately 25%) in the groups treated with vehicle,with GNF-5 and with nilotinib. In the combination group, the propor-tion of p-STAT5-positive cells was about 6%, reflecting a correlationbetween the tumour growth inhibition and ablock inBcr–Abl signalling(Fig. 6d). To determine the extent of the inhibition, mice transplantedwith T315I Bcr–Abl-expressing bone marrow cells were treated with asingle dose of the combination (50mgkg21 nilotinib plus 75mgkg21
GNF-5) or vehicle onday 21 after transplantation, and the bonemarrowcells were collected and analysed at 3, 7, 16 and 24h after dose. In thevehicle group, about 80%of the luciferase-positive cells had phosphory-lated STAT5.At 3 h after dosing, STAT5phosphorylationwas decreasedfrom 80% to 25%; from 7–24h the number of p-STAT5-positive cellsremained below10%, showing a strong and sustained inhibition of Bcr–Abl-mediated signalling after administration of the GNF-5/nilotinibcombination (Fig. 6e).
In a third experiment we monitored the survival of the mice trans-planted with T315I Bcr–Abl-transduced bone marrow and treatedwith GNF-5, nilotinib, or both in combination. Mice transplantedwith T315I Bcr–Abl-transduced bone marrow and treated withvehicle control died by day 24 after transplantation, with a mediansurvival of 22 days (Fig. 6f). GNF-5 (75mg kg21 twice daily)extended survival (median 28 days) significantly compared withvehicle-treated controls (P5 0.023). Mice treated with nilotinibalone (50mg kg21 twice daily) also survived longer (median 32 days)than those treated with vehicle (P5 0.023). The overall survival ofmice treatedwith GNF-5 plus nilotinib was improved comparedwiththose treated with either GNF-5 alone (P5 0.002) or nilotinib alone(P5 0.002). All of these mice survived to day 50 after transplanta-tion, after which the treatment was discontinued; 46 days after thecombination treatment was completed, four out of five mice weresurviving without signs of disease. Cumulatively, these results suggestthat a combination of an ATP-competitive inhibitor with an allos-teric inhibitor may be a therapeutically appropriate strategy for tar-geting the T315I Bcr–Abl mutation.
Discussion
The successful development of efficacious inhibitors against kinasessuch as Bcr–Abl, c-Kit and epidermal growth factor receptor (EGFR),which are activated by genetic alterations, has stimulated a massiveeffort to develop new ATP-competitive inhibitors targeted to avariety of kinases25,26. One major problem for all clinically approvedATP-competitive kinase inhibitors is resistance that results from theselection of drug-resistant mutant forms of the kinase target. One
ATP-bindingsite
a
0
2
4
6
8 WT Abl WT Abl + GNF-5E505KE505K + GNF-5
WT Abl WT Abl + GNF-5E505KE505K + GNF-5
0
Time (min)
2
4
6
8
0.1 1 10 100 1,000
Major changes
No changesMinor changes
b306–316
317–324
506–515
306–316
506–515
Rel
ativ
e de
uter
ium
leve
l (D
a)
Figure 5 | Hydrogen-exchange mass spectrometry on binding of GNF-5 toAbl. a, Deuterium uptake curves for the peptides 306–316 and 506–515. They-axis maximum corresponds to the theoretical maximum amount ofdeuterium that could be incorporated into this peptide. No alterations indeuterium incorporationwere seenwith the E505Kmutant, whereas exchangewas affectedbybindingofGNF-5 to thewild-type (WT)protein.b, Location ofthe peptides that showed differences in deuterium incorporation mapped onthe crystal structure of active Abl (PDB accession 2F4J). This crystal structurewas chosen because in the absence of myristoylation (as for the protein usedhere), Abl protein is believed to be in this conformation23. Major changes(coloured magenta) were defined as a difference between exchange curves of1.0Daormore.Minor changes (coloured yellow)were 0.4–1.0Da.Nochanges(coloured grey) were differences of 0–0.4Da.
ARTICLES NATURE |Vol 463 |28 January 2010
504Macmillan Publishers Limited. All rights reserved©2010
hotspot for resistance occurs at the ‘gatekeeper’ residue, as has beenobserved for Bcr–Abl4, EGFR27, FMS-like tyrosine kinase 3 (ref. 28),c-Kit, Src, platelet-derived growth factor receptor-b and fibroblastgrowth factor receptor 1 (ref. 29). One strategy for overcomingresistance mutations is to design new ATP-competitive inhibitorsthat derive potency and selectivity from alternative binding modes;this has been clinically validated by the development of dasatinib andnilotinib, which target most Bcr–Abl mutations except T315I. Analternative strategy is to find non-ATP competitive inhibitors thatcan regulate kinase activity allosterically. Allosteric inhibitors havebeen developed for multiple kinases including mTor30, Mek31, Akt32,IkB kinase33, Chk1 (ref. 34) and Ca21/calmodulin-dependentkinase II35.
GNF-2 and its analogues represent a new kind of non-ATP com-petitive Abl kinase inhibitor. The NMR, X-ray crystallography,mutagenesis and hydrogen-exchange experiments are all consistentwith binding of GNF-2/5 to the myristate-binding site located nearthe C terminus of the kinase domain. On the basis of our accumu-lated data, we propose the following model for the inhibition mech-anism of GNF-2 class compounds. Binding of GNF-2/5 to themyristate-binding site seems to induce a bent conformation of theaI helix that facilitates the stabilization of an inhibited conformation.As supported by the mutagenesis studies and kinase assays, func-tional inhibition by GNF-2/5 requires the involvement of the SH3and SH2 domains. The hydrogen-exchange mass spectrometry datademonstrate that binding of GNF-5 to the myristate-binding siteresults in dynamic or conformational changes at the ATP-bindingsite. This suggests that binding of GNF-2/5 to the myristate-bindingsite causes structural reorganization, possibly communicated bymeans of a conformational rearrangement of other parts of Abl,which disrupts the catalytic machinery located in the ATP-bindingsite. Because we currently do not have a crystal structure with aconstruct that contains the SH3–SH2-kinase domain of Abl bound
to GNF-2, we cannot draw firm conclusions about the precise con-formation of the kinase induced by GNF-2/5.
We show that GNF-2/5 can act cooperatively with an ATP compe-titive inhibitor to inhibit both wild-type and T315I Bcr–Abl in bio-chemical and cellular assays, which is consistent with crystallographicand differential scanning calorimetry results (Supplementary Fig. 15).However, one puzzling question was why GNF-2/5 lacks significantbiochemical and cellular potency against the T315I ‘gatekeeper’ Ablmutation. T315I Abl biochemical kinase activity can be decreased by20% at GNF-5 concentrations below 0.5mM, but further increases inGNF-5 concentration up to 10mMdonot result in superior inhibition(Fig. 4c). Mutation of the ‘gatekeeper’ residue does not interfere sub-stantially with the ability of GNF-2/5 to bind to Abl as determined byNMR and affinity chromatography, but it seems to disfavour theGNF-2/5-induced inhibited conformation. The hydrogen-exchangemass spectrometry data show that binding of GNF-5 results indecreased exchange in a peptide that contains the ‘gatekeeper’ residue.While we have not proved that this exchange phenomenon is func-tionally relevant to the inhibitory mechanism of GNF-2/5, it doessuggest that the conformation and/or dynamics of the ‘gatekeeper’-containing segment may be coupled with the ability of GNF-2/5 toinhibit Abl36.
Although non-ATP competitive inhibitors will also be subject toinhibitor resistance through point mutation, we have shown that thecombined application of ATP and non-ATP competitive inhibitorsdecreases the number of resistant clones that emerge as a response tocontinued exposure to a single agent. Furthermore we have shownthat combined treatment of GNF-5 with nilotinib led to in vivoefficacy, resulting in complete disease remissions in a T315I Bcr–Abl mutant murine bone-marrow transplantation model. Thesefindings should encourage the search for non-ATP competitive inhi-bitors that can selectively target the large number of kinases thatbecome deregulated in cancer and other diseases.
a
d
f
c
b
e
d11 d50
Day 5 Day 7
105
104
103
102
0
105
104
103
102
0
105
104
103
102
0
105
104
103
102 103 104 105
0
0 102 103 104 1050
102 103 104 1050102 103 104 1050
Luciferase
p-S
TAT5
3020100
600400200
0
0
20
40
60
80
100
Vehicle(n = 4)
GNF-5(n = 3)
Nilotinib(n = 4)
GNF-5 +nilotinib(n = 4)
GNF-5 +nilotinib(n = 4)
Vehicle(n = 4)
GNF-5(n = 3)
Nilotinib(n = 4)
Vehicle
Vehicle0.1
0.051 24.4
0.41 75.1
0.061 6.7
0.12 93.1
26.5
1.81 71.6
0.18 24.4
21.4 54.0
GNF-5
Nilotinib GNF-5 +nilotinib
VehicleGNF-5NilotinibGNF-5 + nilotinib
3 7 16 24
WB
C c
ount
(103
µl–1
)S
plee
n w
eigh
t (m
g)P
erce
ntag
e of
Lu
c+ /p-
STA
T5+
cells
Time after single dose (h)
Time after transplantation (days)
5
4
3
2
20 40 60 80 100
1
00N
umbe
r of m
ice
surv
ivin
g
Figure 6 | In vivo efficacy studies with GNF-5 onwild-type and T315I Bcr–Abl dependentproliferation in xenograft and bone-marrowtransplantation models. a, Images of whole-body luminescence of wild-type Bcr–Abl andluciferase-expressing Ba/F3 cells on days 5 and 7after treatment with vehicle (top), 50mg kg21
GNF-5 twice daily (middle) and 100mg kg21
GNF-5 twice daily (bottom). b, Average white-blood-cell (WBC) counts for treatments withvehicle, 75mg kg21 GNF-5, 50mg kg21 nilotinibor a combination of the two in the T315I Bcr–Ablbone-marrow transplantation efficacy study.c, Spleen weight for treatments with vehicle,75mg kg21 GNF-5, 50mg kg21 nilotinib or acombination of the two in the T315I Bcr–Ablbone-marrow transplantation efficacy study.d, Quantification of p-STAT5-positive cells byflow cytometry in the different treatment groupsafter repeated doses. e, Time course of inhibitionof STAT5 phosphorylation after a single dose of acombination of GNF-5 and nilotinib.f, Kaplan–Meier plot showing survival of mice(n5 5 mice per group) transplanted with T315IBcr–Abl-transduced bone marrow and treatedwith vehicle (solid line), 75mg kg21 GNF-5 twicedaily (dotted line), 50mg kg21 nilotinib twicedaily (dot–dashed line), or a combination of75mg kg21 GNF-5 and 50mg kg21 nilotinibtwice daily (dashed line). Dosing withcompounds was initiated on day 11 aftertransplantation and discontinued on day 50(indicated by arrows). Where present, error barsindicate s.e.m.
NATURE |Vol 463 |28 January 2010 ARTICLES
505Macmillan Publishers Limited. All rights reserved©2010

METHODS SUMMARYAll NMR experiments were performed as described previously37. Abl crystalswere grown as described in ref. 10.Wild-type andmutant Bcr–Abl Ba/F3 cellularproliferation assays were performed after incubation for 48 h with a range ofdrug concentrations as described in ref. 7. Selection for clones resistant to GNF-2and imatinib was as described in ref. 13. Abl protein expression in E. coli, andpurification as described in ref. 38. The location of each phosphorylation wasdetermined by liquid chromatography–tandem mass spectrometry. The ATP/NADH-coupled assay system was used to determine inhibition kinetics of Abltyrosine kinase. Hydrogen exchange experiments were performed as described inref. 23. Male Balb/c mice were chosen for GNF-5 pharmacokinetic parameterstudies. The in vivo efficacy study in theBa/F3.p210 xenograftmodelwasperformedin female SCIDbeigemice. The experimental procedures for protein crystallizationand synthetic chemistry are described in Supplementary Information.
Full Methods and any associated references are available in the online version ofthe paper at www.nature.com/nature.
Received 24 March; accepted 11 November 2009.Published online 13 January 2010.
1. Weisberg, E. et al. Characterization of AMN107, a selective inhibitor of native andmutant Bcr-Abl. Cancer Cell 7, 129–141 (2005).
2. Quintas-Cardama, A., Kantarjian, H. & Cortes, J. Flying under the radar: the newwave of BCR-ABL inhibitors. Nature Rev. Drug Discov. 6, 834–848 (2007).
3. Shah, N. P. et al. Overriding imatinib resistance with a novel ABL kinase inhibitor.Science 305, 399–401 (2004).
4. Gorre, M. E. et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880 (2001).
5. Nagar, B. et al. Crystal structures of the kinase domain of c-Abl in complex withthe small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62,4236–4243 (2002).
6. Bradeen, H. A. et al. Comparison of imatinib mesylate, dasatinib (BMS-354825),and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesisscreen: high efficacy of drug combinations. Blood 108, 2332–2338 (2006).
7. Adrian, F. J. et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation.Nature Chem. Biol. 2, 95–102 (2006).
8. Jahnke, W. & Widmer, H. Protein NMR in biomedical research. Cell. Mol. Life Sci.61, 580–599 (2004).
9. Vajpai, N. et al. Solution conformations and dynamics of ABL kinase-inhibitorcomplexes determined by NMR substantiate the different binding modes ofimatinib/nilotinib and dasatinib. J. Biol. Chem. 283, 18292–18302 (2008).
10. Nagar, B. et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell112, 859–871 (2003).
11. Hantschel, O. et al.Amyristoyl/phosphotyrosine switch regulates c-Abl. Cell 112,845–857 (2003).
12. Ray, A. et al. Identification of BCR-ABL point mutations conferring resistance tothe Abl kinase inhibitor AMN107 (nilotinib) by a random mutagenesis study.Blood 109, 5011–5015 (2007).
13. von Bubnoff, N. et al. A cell-based screen for resistance of Bcr-Abl-positiveleukemia identifies the mutation pattern for PD166326, an alternative Abl kinaseinhibitor. Blood 105, 1652–1659 (2005).
14. Azam, M., Latek, R. R. & Daley, G. Q. Mechanisms of autoinhibition and STI-571/imatinib resistance revealedbymutagenesis of BCR-ABL.Cell 112,831–843 (2003).
15. Azam, M. et al. Activity of dual SRC-ABL inhibitors highlights the role of BCR/ABLkinasedynamics indrug resistance.Proc.NatlAcad.Sci.USA 103,9244–9249(2006).
16. Branford, S. et al. High frequency of point mutations clustered within theadenosine triphosphate-binding region of BCR/ABL in patients with chronicmyeloid leukemia or Ph-positive acute lymphoblastic leukemia who developimatinib (STI571) resistance. Blood 99, 3472–3475 (2002).
17. Shah, N. P. et al. Multiple BCR-ABL kinase domain mutations confer polyclonalresistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase andblast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 (2002).
18. Chou, T. C. & Talalay, P. Quantitative analysis of dose–effect relationships: thecombined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22,27–55 (1984).
19. Mishra, S. et al. Resistance to imatinib of bcr/abl p190 lymphoblastic leukemiacells. Cancer Res. 66, 5387–5393 (2006).
20. Kornberg, A. & Pricer, W. E. Jr. Di- and triphosphopyridine nucleotide isocitricdehydrogenases in yeast. J. Biol. Chem. 189, 123–136 (1951).
21. Choi, Y. et al. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmicreticulum upon binding to an allosteric inhibitor. J. Biol. Chem. 284, 29005–29014(2009).
22. Wales, T. E. & Engen, J. R. Hydrogen exchangemass spectrometry for the analysisof protein dynamics. Mass Spectrom. Rev. 25, 158–170 (2006).
23. Iacob, R. E. et al. Conformational disturbance in Abl kinase upon mutation andderegulation. Proc. Natl Acad. Sci. USA 106, 1386–1391 (2009).
24. van Etten, R. A. Disease progression in a murine model of bcr/ablleukemogenesis. Leuk. Lymphoma 11 (suppl. 1), 239–242 (1993).
25. Joensuu, H. et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with ametastatic gastrointestinal stromal tumor.N. Engl. J.Med. 344, 1052–1056 (2001).
26. Lynch, T. J. et al. Activating mutations in the epidermal growth factor receptorunderlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J.Med. 350, 2129–2139 (2004).
27. Kobayashi, S. et al. EGFRmutation and resistance of non-small-cell lung cancer togefitinib. N. Engl. J. Med. 352, 786–792 (2005).
28. Cools, J. et al. Prediction of resistance to small molecule FLT3 inhibitors:implications for molecularly targeted therapy of acute leukemia. Cancer Res. 64,6385–6389 (2004).
29. Blencke, S. et al.Characterization of a conserved structural determinant controllingprotein kinase sensitivity to selective inhibitors. Chem. Biol. 11, 691–701 (2004).
30. Brown, E. J. et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369, 756–758 (1994).
31. Dudley, D. T. et al. A synthetic inhibitor of the mitogen-activated protein kinasecascade. Proc. Natl Acad. Sci. USA 92, 7686–7689 (1995).
32. Barnett, S. F. et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 385,399–408 (2005).
33. Burke, J. R. et al. BMS-345541 is a highly selective inhibitor of IkB kinase that bindsat an allosteric site of the enzyme and blocks NF-kB-dependent transcription inmice. J. Biol. Chem. 278, 1450–1456 (2003).
34. Converso, A. et al. Development of thioquinazolinones, allosteric Chk1 kinaseinhibitors. Bioorg. Med. Chem. Lett. 19, 1240–1244 (2009).
35. Tokumitsu, H. et al. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca21/calmodulin-dependentprotein kinase II. J. Biol. Chem. 265, 4315–4320 (1990).
36. Lee, T. S. et al.Molecular basis explanation for imatinib resistance of BCR-ABL dueto T315I and P-loop mutations from molecular dynamics simulations. Cancer 112,1744–1753 (2008).
37. Strauss, A. et al. Efficient uniform isotope labeling of Abl kinase expressed inBaculovirus-infected insect cells. J. Biomol. NMR 31, 343–349 (2005).
38. Seeliger, M. A. et al. High yield bacterial expression of active c-Abl and c-Srctyrosine kinases. Protein Sci. 14, 3135–3139 (2005).
Supplementary Information is linked to the online version of the paper atwww.nature.com/nature.
Acknowledgements We thank C. Henry and G. Rummel for technical assistance;R. Beigi for help with the bone-marrow transplantation studies; A. Velentza forperforming the DSC experiments; and J. Kuriyan, M. Seeliger, C. Yun, M. Eck,E. Weisberg, D. Fabbro, P. L. Yang, G. Superti-Furga and A. Kung for helpfuldiscussions. We also acknowledge the support of staff at beamline PXII of theSwiss Light Source, Villigen, Switzerland, during X-ray data collection, theICCB-Longwood Screening facility at Harvard Medical School for the cellproliferation and enzyme assay, and Barnet Institute for hydrogen-exchangeexperiments.
Author Contributions F.J.A., J.Z., J.P., Y.C., G.L., M.A. and G.D. designed andperformed cellular and biochemical experiments. J.Z. performed bacterial Ablexpression and enzyme assays. W.J., N.V. and S.G. designed and performed theNMR experiments. S.W.C.-J. designed and performed the crystallographicexperiments. G.F. and A.S. produced the protein for the NMR and X-rayexperiments. T.S., Q.D., B.O., A.W. and X.D. designed and synthesized thecompounds. A.G.L., C.D., F.S., G.-R.G. and T.T. conducted the in vivo studies. Y.L.and B.B. contributed to the design of the compounds. R.E.I. and J.R.E. performed anddesigned the hydrogen-exchange experiments. M.W. contributed to the design ofthe in vivo experiments. F.J.A., M.W. and P.M. provided critical input to the overallresearch direction. N.S.G. directed the research and wrote the paper with inputfrom all co-authors.
Author Information The coordinates and structure factors of the complete Abl/imatinib/GNF-2 complex crystal structure are deposited in the Protein Data Bankunder accession 3K5V. Reprints and permissions information is available atwww.nature.com/reprints. The authors declare competing financial interests:details accompany the full-text HTML version of the paper at www.nature.com/nature. Correspondence and requests for materials should be addressed to F.J.A.([email protected]), M.W. ([email protected]) or N.S.G.([email protected]).
ARTICLES NATURE |Vol 463 |28 January 2010
506Macmillan Publishers Limited. All rights reserved©2010

METHODSNMR spectroscopy. All NMR experiments were performed as described previ-ously37 at 296K on a Bruker AV600 NMR spectrometer at a proton resonancefrequency of 600MHz.Abl crystallography. Crystals were grown as described in ref. 10 using the con-ditions listed in Supplementary Table 1. After a single crystal had been soaked for7 days at 4 uC in an excess of GNF-2, data were collected at beamline PXII of theSwiss Light Source (Supplementary Table 1). Statistics of the refinement anddetails of ligand–protein interactions in the final model are listed inSupplementary Tables 2 and 3.Wild-type and mutant Bcr–Abl-expressing Ba/F3 cellular proliferationassays. Viability of wild-type and mutant Bcr–Abl-expressing Ba/F3 cells aftertreatment for 48 h with various concentrations of single or combined agents wasdetermined by the AlamarBlue (TREK Diagnostic Systems) reduction method.The combination index was calculated as described in ref. 18, using Calcusynsoftware.Selection for clones resistant to GNF-2 and imatinib. Emergence of com-pound-resistant Ba/F3.p210 clones was evaluated as described previously13.One 96-well plate was used for every compound concentration or combination,and the medium was renewed every 3–4 days. The plates were incubated for21 days, and the number of wells with evident cell growth was recorded at days 9,12 and 21.Abl protein expression and purification. Bacterial expression andpurification ofhuman c-Abl kinase (residues 46–515, Abl 1a numbering39) were performed asdescribed previously38. Phosphorylation (180 or 1160Da) was observed in theintact protein spectra (SupplementaryFig. 8).The locationof eachphosphorylationwas determined by digestion with trypsin followed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). For both wild-type Abl and AblE505K, single phosphorylation corresponded to modification at Tyr 412, anddouble phosphorylation involved Tyr 412 and Tyr89. For Abl T315I, single phos-phorylation was on Tyr 89 and only a small quantity of the molecules containedphosphorylation at both Tyr89 and Tyr 412.Kinetic characterization of Abl inhibition. The ATP/NADH-coupled assaysystem in a 96-well format was used to determine the initial velocity of peptidephosphorylation catalysed by Abl tyrosine kinase. The reactionmixture contained20mM Tris-HCl pH8.0, 50mM NaCl, 10mM MgCl2, 2mM 2-(phosphono-oxy)-2-propenoic acid (Sigma-Aldrich) and 20mM Abl peptide substrate(EAIYAAPFAKKK; New England Biolabs), a fixed or varied (to determine inhibi-tor kinetic parameters) concentration of inhibitor applied, 1/50 of the final reac-tionmixture volumeof pyruvate kinase/lactic dehydrogenase enzymes from rabbitmuscle (Sigma-Aldrich), 160mMNADH and 0.16mMAbl; ATP was added last tostart the reaction. Absorbance data were collected every 20 s at 340 nm with aSpectraMaxM5microplate reader. The two-substrate kinase reaction was simpli-fied to two one-substrate reactions to determine ATP kinetic parameters andinhibitor parameters separately.When determiningATPparameters, the inhibitorconcentration was kept constant. When determining inhibition parameters, theATP concentration was kept the same at 20mM. Steady-state initial velocity datawere drawn from the slopes of the A340 curves and fit to the Michaelis–Mentenequation to determine Vmax and Km values. Data were fitted globally withGraphPad Prism (GraphPad Software) and Excel XLfit 4.0 to fit velocity equationsfor competitive and mixed inhibition.Hydrogen exchange experiments. Hydrogen exchange experiments were per-formed essentially as described in ref. 23. Abl protein (38 pmol) was incubatedfor 30min at 25 uC with 45 mM GNF-5 at a protein:drug ratio of 1:7 (4.38mMGNF-5 at labelling solution). For an estimated EC50 of 0.169mM for GNF-5(Supplementary Fig. 9d), 97.41% of the protein was expected to be bound toGNF-5. Deuterium exchange was initiated by dilution of the protein–drug mix-ture 15-fold with 20mMTris-HCl, 100mMNaCl (pD 8.3) in D2O at 21 uC. Thesame procedure was used for the Abl protein alone and for the E505Kmutant. Ateach deuterium exchange time point (from 10 s to 4 h) an aliquot from theexchange reaction was removed and labelling was quenched by adjusting thepH to 2.6 with an equal volume of quench buffer (50mM potassium phosphate
pH2.6 in H2O). Quenched samples were immediately frozen on solid CO2 andstored at 280 uC until analysis. Each frozen sample was thawed rapidly to 0 uCand injected into a customWaters nanoACQUITYUPLC system and analysed asdescribed previously40. The protein sample was digested online with pepsin, andthe resulting peptides were trapped and desalted for 3min at 100ml min21 andthen separated in 6min by an 8–40% acetonitrile:water gradient at 40ml min21.The separation column was a 1.0mm3 100.0mm ACQUITY UPLC C18 BEH(Waters Corp.) containing 1.7-mm particles; the back pressure averaged8,800 lb in22 at 1 uC.Themass spectra were obtainedwith aWatersQTOFPremier equippedwith a
standard electrospray ionization source (Waters Corp.) as described previ-ously23. No correction was made for back-exchange, and all results are reportedas relative deuterium level22. Mass spectra were processed with the software HX-Express41; the deuteration levels were calculated by subtracting the centroid ofthe isotopic distribution for peptide ions of undeuterated protein from thecentroid of the isotopic distribution for peptide ions from the deuterium-labelled sample.In vivo studies of GNF-5 and drug combinations. GNF-5 pharmacokineticparameters in mice: male Balb/c mice were dosed with GNF-5 in PEG400/saline(1:1) at 5mg kg21 intravenously or 20mg kg21 orally. The compound plasmaconcentration at any given time point was determined by LC–MS/MS.Pharmacokinetic parameters were calculated by non-compartmental regressionanalysis with Winnonlin 4.0 software (Pharsight).In vivo efficacy in Ba/F3.p210 xenograft model: female SCID beige mice, 6–8
weeks of age (n5 5 for each GNF-5-treated or vehicle control group) wereinjected into the tail vein with 106 Ba/F3 cells co-expressing Bcr–Abl p210 andluciferase. Three days after injection, mice were dosed orally twice daily with 50or 100mg kg21 GNF-5 for seven days. At days 5 and 7, bioluminescence wasquantified with luciferin and an IVIS imaging system (Xenogen).GNF-5 pharmacodynamics in Ba/F3.p210 xenograft mice: bone marrow
samples were collected at 3 h (n5 3 mice per time point) after a single oraladministration of GNF-5 at 50 and 100mg kg21 at day 7 after cell injection.Fixed and permeabilized bone marrow cells were stained with phycoerythrin-conjugated anti-phospho-Y694 STAT5 antibody (pSTAT5; Becton Dickinson)and subjected to flow cytometry.In vivo efficacy in bone-marrow transduction/transplantation model: bone
marrow cells harvested from 6–8-week-old 5-fluorouracil-injected male Balb/cmice were transduced with a pMSCVBcr–Abl wild-type or T315I Bcr–Abl retro-viral construct and transplanted into irradiated recipient female Balb/c mice(6–8 weeks old). A 7-day treatment with GNF-5, nilotinib or vehicle controlwas started on days 7 (wild-type Bcr–Abl) or 15 (T315I) after transplantation(ten (wild-type Bcr–Abl) or four (T315I Bcr–Abl) mice per treatment group).Blood cell counts and spleen size were determined at treatment day 7. Bonemarrow cells were isolated, fixed, permeabilized, stained with anti-p-STAT5and anti-luciferase antibodies, and analysed by flow cytometry. For survival stud-ies, the treatment with vehicle or with GNF-5 or nilotinib or a combination of thetwo (n5 5 mice per group) was initiated 11 days after transplantation and pro-longed until day 50 after transplantation or until the mice had to be sacrificedbecause they became moribund. Overall survival and time to relapse were deter-mined by the Kaplan–Meier method. Statistical significance was assessed byKaplan–Meier survival analysis, under the assumption of a normal distributionof normalized ratios with an estimate of variance (a5 0.05, two-sided).
39. Oppi, C., Shore, S. K. & Reddy, E. P. Nucleotide sequence of testis-derived c-ablcDNAs: implications for testis-specific transcription and abl oncogene activation.Proc. Natl Acad. Sci. USA 84, 8200–8204 (1987).
40. Wales, T. E., Fadgen, K. E., Gerhardt, G. C. & Engen, J. R. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 80, 6815–6820(2008).
41. Weis, D. D., Engen, J. R. & Kass, I. J. Semi-automated data processing of hydrogenexchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17,1700–1703 (2006).
doi:10.1038/nature08675
Macmillan Publishers Limited. All rights reserved©2010




















