
BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB2...
11
Cell, Vol. 75, 175-185, October8, 1993,Copyright© 1993by Cell Press BCR-ABL-Induced Oncogenesis Is Mediated by Direct Interaction with the SH2 Domain of the GRB-2 Adaptor Protein Ann Marie Pendergast,* Lawrence A. Quilliam,t Larry D. Cripe,*$ Craig H. Bassing,* Zonghan Dai,* Nanxin Li,§ Andreaa Batzer,§ Kelly M. Rabun,t Channing J. Der,t Joseph Schlessinger,§ and Mikhail L. Gishizkyll *Department of Pharmacology Duke University Medical Center Durham, North Carolina 27710 tDepartment of Pharmacology University of North Carolina Chapel Hill, North Carolina 27599 $Division of Hematology-Oncology Department of Medicine Duke University Medical Center Durham, North Carolina 27710 §Department of Pharmacology New York University Medical Center New York, New York 10016 IISugen, Inc. Redwood City, California 94063 Summary BCR-ABL is a chimeric oncoprotein that exhibits de- regulated tyrosine kinase activity and is implicated in the pathogeneele of Philadelphia chromosome (phi) - positive human leukemias. Sequences within the first exon of BCR are required to activate the transforming potential of BCR-ABL. The SH2/SH3 domain-con- taining GRB-2 protein links tyroalne kinases to Ras sig- naling. We demonstrate that BCR-ABL exists in a com- plex with GRB-2 in vivo. Binding of GRB-2 to BCR-ABL is mediated by the direct interaction of the GRB-2 SH2 domain with a phosphorylated tyrosine, ¥177, within the BCR first exon. The BCR-ABL-GRB-2 interaction is required for activation of the Raa signaling pathway. Mutation of Y177 to phenylalanine (Y177F) abolishes GRB-2 binding and abrogates BCR-ABL-induced Ras activation. The BCR-ABL (Y177F) mutant is unable to transform primary bone marrow cultures and is im- paired in its ability to transform Rat1 fibroblasts. These findings Implicate activation of Raa function as an im- portant component in BCR-ABL-mediated transforma- tion and demonstrate that GRB-2 not only functions in normal development and mitogenesis but also plays a role in oncogeneals. Introduction Activation of the oncogenic potential of normal cellular proteins may occur by alteration of their corresponding enzymatic activities, their inappropriate binding to other cellular components, or both. Changes in both enzyme activity and altered noncovalent protein interactions may underlie the mechanism whereby the BCR-ABL oncopro- tein transforms cells. BCR-ABL is a chimeric oncogene implicated in the pathogenesis of Philadelphia chromo- some (Phl)-positive human leukemias. The BCR-ABL on- cogene is generated by the translocation of sequences from the c-ABL protein-tyrosine kinase on chromosome 9 into BCR sequences on chromosome 22 (reviewed by Kurzrock et al., 1988; Rosenberg and Witte, 1988). Alter- native chimeric proteins, p210 BCR-ABL and p185 BCR- ABL, are produced that are characteristic of chronic my- elogenous leukemia and acute lymphocytic leukemia (ALL), respectively. The BCR-ABL proteins exhibit height- ened tyrosine kinase and transforming properties com- pared with the normal c-ABL protein (Konopka et al., 1984; Muller et al., 1991). Previously, we and others have shown that BCR first exon sequences specifically activate the tyrosine kinase and transforming potential of BCR-ABL (Muller et al., 1991; McWhirter and Wang, 1991). A unique property of the BCR first exon is its capacity to bind to the ABL SH2 domain in a phosphotyrosine-independent manner (Pen- dergast et al., 1991a). Deletion of BCR sequences essen- tial for ABL SH2-binding render BCR-ABL nontrans- forming (Pendergast et al., 1991a). In addition to the ABL SH2 domain, BCR binds in vitro to a limited subset of SH2 domains encoded by proteins such as phospholipase C-y and Src (Muller et al., 1992). These results implicate BCR SH2 binding in the oncogenicity of BCR-ABL through in- teraction between BCR and the ABL SH2 domain, through transmediated interactions of BCR with other SH2-con- taining proteins, or through both. In addition to phosphoserine/phosphothreonine-depen- dent binding, BCR sequences in the BCR-ABL chimera may bind to SH2-containing proteins through tyrosine- phosphorylated residues. There are 11 tyrosines within the BCR first exon. Phosphopeptide mapping followed by phosphoamino acid analysis has revealed that several of these tyrosines are phosphorylated in the BCR-ABL chi- mera (Liu et al., 1993; A. M. P., unpublished data). Ras plays a critical role in the transmission of mitogenic signals from receptor tyrosine kinases (Mulcahy et al., 1986; Cai et al., 1990; Thomas et al., 1992; Wood et al., 1992). Activation of receptor tyrosine kinases by ligand binding results in the accumulation of the active, GTP- bound form of Ras (Gibbs et al., 1990; Satoh et al., 1990; Li et al., 1992; Buday and Downward, 1993a; Medema et al., 1993). Ras activation is also required for transforma- tion by oncogenic tyrosine kinases (Smith at al., 1986). Ras activity is regulated by the opposing actions of GTPase-activating proteins (GAPs) and guanine nucleo- tide exchange factors. GAPs stimulate the slow intrinsic rate of GTP hydrolysis on Ras and therefore are negative regulators of Ras function. Exchange factors stimulate the basal rate of exchange of GDP for GTP on Ras and func- tion as Ras activators (Bollag and McCormick, 1991; Down- ward, 1992). A number of exchange factors have been identified in yeast (Broek et al., 1987; Jones et al., 1991) and Drosophila (Simon et al., 1991; Bonfini et al., 1992). Murine homologs of the yeast CDC25 exchange factor
-
Upload
independent -
Category
Documents
-
view
6 -
download
0
Transcript of BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB2...

Cell, Vol. 75, 175-185, October 8, 1993, Copyright © 1993 by Cell Press
BCR-ABL-Induced Oncogenesis Is Mediated by Direct Interaction with the SH2 Domain of the GRB-2 Adaptor Protein
Ann Marie Pendergast,* Lawrence A. Quilliam,t Larry D. Cripe,*$ Craig H. Bassing,* Zonghan Dai,* Nanxin Li,§ Andreaa Batzer,§ Kelly M. Rabun,t Channing J. Der,t Joseph Schlessinger,§ and Mikhail L. Gishizkyll *Department of Pharmacology Duke University Medical Center Durham, North Carolina 27710 tDepartment of Pharmacology University of North Carolina Chapel Hill, North Carolina 27599 $Division of Hematology-Oncology Department of Medicine Duke University Medical Center Durham, North Carolina 27710 §Department of Pharmacology New York University Medical Center New York, New York 10016 IISugen, Inc. Redwood City, California 94063
Summary
BCR-ABL is a chimeric oncoprotein that exhibits de- regulated tyrosine kinase activity and is implicated in the pathogeneele of Philadelphia chromosome (phi) - positive human leukemias. Sequences within the first exon of BCR are required to activate the transforming potential of BCR-ABL. The SH2/SH3 domain-con- taining GRB-2 protein links tyroalne kinases to Ras sig- naling. We demonstrate that BCR-ABL exists in a com- plex with GRB-2 in vivo. Binding of GRB-2 to BCR-ABL is mediated by the direct interaction of the GRB-2 SH2 domain with a phosphorylated tyrosine, ¥177, within the BCR first exon. The BCR-ABL-GRB-2 interaction is required for activation of the Raa signaling pathway. Mutation of Y177 to phenylalanine (Y177F) abolishes GRB-2 binding and abrogates BCR-ABL-induced Ras activation. The BCR-ABL (Y177F) mutant is unable to transform primary bone marrow cultures and is im- paired in its ability to transform Rat1 fibroblasts. These findings Implicate activation of Raa function as an im- portant component in BCR-ABL-mediated transforma- tion and demonstrate that GRB-2 not only functions in normal development and mitogenesis but also plays a role in oncogeneals.
Introduction
Activation of the oncogenic potential of normal cellular proteins may occur by alteration of their corresponding enzymatic activities, their inappropriate binding to other cellular components, or both. Changes in both enzyme activity and altered noncovalent protein interactions may underlie the mechanism whereby the BCR-ABL oncopro- tein transforms cells. BCR-ABL is a chimeric oncogene
implicated in the pathogenesis of Philadelphia chromo- some (Phl)-positive human leukemias. The BCR-ABL on- cogene is generated by the translocation of sequences from the c-ABL protein-tyrosine kinase on chromosome 9 into BCR sequences on chromosome 22 (reviewed by Kurzrock et al., 1988; Rosenberg and Witte, 1988). Alter- native chimeric proteins, p210 BCR-ABL and p185 BCR- ABL, are produced that are characteristic of chronic my- elogenous leukemia and acute lymphocytic leukemia (ALL), respectively. The BCR-ABL proteins exhibit height- ened tyrosine kinase and transforming properties com- pared with the normal c-ABL protein (Konopka et al., 1984; Muller et al., 1991).
Previously, we and others have shown that BCR first exon sequences specifically activate the tyrosine kinase and transforming potential of BCR-ABL (Muller et al., 1991; McWhirter and Wang, 1991). A unique property of the BCR first exon is its capacity to bind to the ABL SH2 domain in a phosphotyrosine-independent manner (Pen- dergast et al., 1991a). Deletion of BCR sequences essen- tial for ABL SH2-binding render BCR-ABL nontrans- forming (Pendergast et al., 1991a). In addition to the ABL SH2 domain, BCR binds in vitro to a limited subset of SH2 domains encoded by proteins such as phospholipase C-y and Src (Muller et al., 1992). These results implicate BCR SH2 binding in the oncogenicity of BCR-ABL through in- teraction between BCR and the ABL SH2 domain, through transmediated interactions of BCR with other SH2-con- taining proteins, or through both.
In addition to phosphoserine/phosphothreonine-depen- dent binding, BCR sequences in the BCR-ABL chimera may bind to SH2-containing proteins through tyrosine- phosphorylated residues. There are 11 tyrosines within the BCR first exon. Phosphopeptide mapping followed by phosphoamino acid analysis has revealed that several of these tyrosines are phosphorylated in the BCR-ABL chi- mera (Liu et al., 1993; A. M. P., unpublished data).
Ras plays a critical role in the transmission of mitogenic signals from receptor tyrosine kinases (Mulcahy et al., 1986; Cai et al., 1990; Thomas et al., 1992; Wood et al., 1992). Activation of receptor tyrosine kinases by ligand binding results in the accumulation of the active, GTP- bound form of Ras (Gibbs et al., 1990; Satoh et al., 1990; Li et al., 1992; Buday and Downward, 1993a; Medema et al., 1993). Ras activation is also required for transforma- tion by oncogenic tyrosine kinases (Smith at al., 1986). Ras activity is regulated by the opposing actions of GTPase-activating proteins (GAPs) and guanine nucleo- tide exchange factors. GAPs stimulate the slow intrinsic rate of GTP hydrolysis on Ras and therefore are negative regulators of Ras function. Exchange factors stimulate the basal rate of exchange of GDP for GTP on Ras and func- tion as Ras activators (Bollag and McCormick, 1991; Down- ward, 1992). A number of exchange factors have been identified in yeast (Broek et al., 1987; Jones et al., 1991) and Drosophila (Simon et al., 1991; Bonfini et al., 1992). Murine homologs of the yeast CDC25 exchange factor
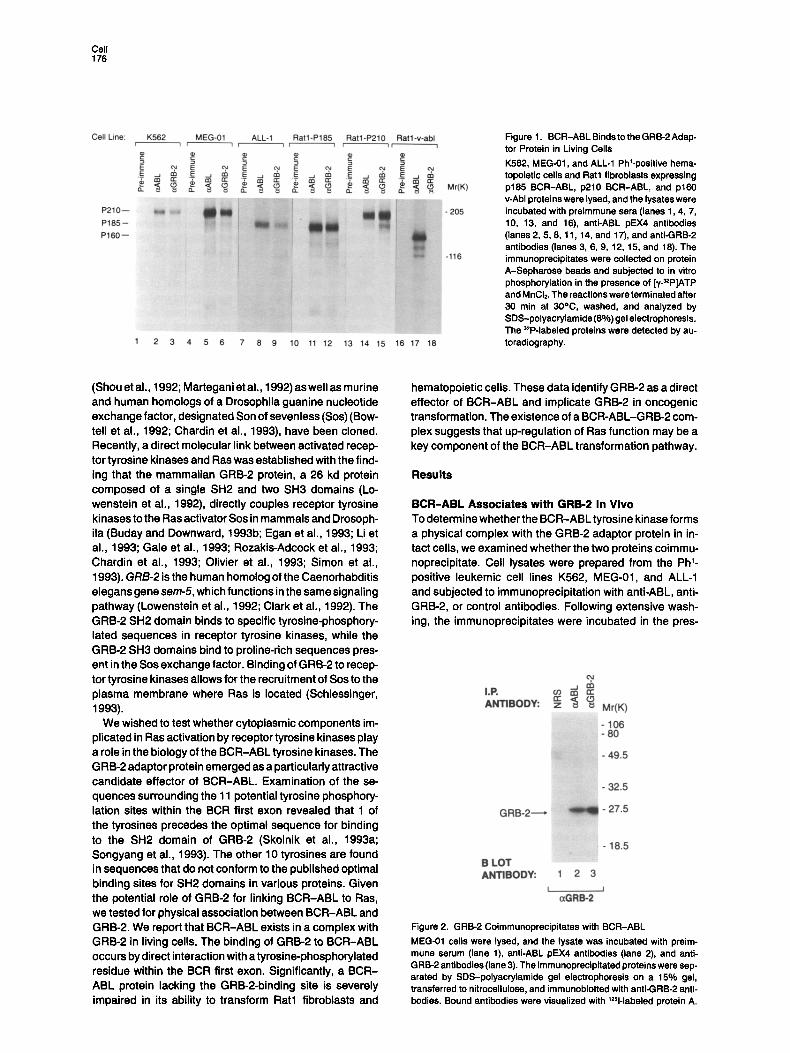
Cell 176
Cell Line: K562 MEG-01 ALL-1 Ratl-P185 n i I - - I i i I - - 1 I - -
; i "+'+ F= +++m+ +++ +..++ o .
P 2 1 0 - -
P185 - -
P 1 6 0 - -
1 2 3 4 5 6 7 8 9 10 11 12
Rat1-P210 Rstl-v-abl
13 14 15 16 17 18
Mr(K)
- 205
-116
Figure 1. BCR-ABL Binds to the GRB-2 Adap- tor Protein in Living Cells K562, MEG-01, and ALL-1 phi-positive hema- topoietic cells and Rat1 fibroblasts expressing p185 BCR-ABL, p210 BCR-ABL, and p160 v-Abl proteins were lysed, and the lysates were incubated with preimmune sera (lanes 1,4, 7, 10, 13, and 16), anti-ABL pEX4 antibodies (lanes 2, 5, 8, 11, 14, and 17), and anti-GRB-2 antibodies (lanes 3, 6, 9, 12, 15, and 18). The immunoprecipitates were collected on protein A-Sepharose beads and subjected to in vitro phosphorylation in the presence of [y-~P]ATP and MnCI2. The reactions were terminated after 30 rain at 30°C, washed, and analyzed by SDS-polyacrylamide (8%) gel electrophoresis. The 32P-labeled proteins were detected by au- toradiography.
(Shou et al., 1992; Martegani et al., 1992) as well as mu rine and human homologs of a Drosophila guanine nucleotide exchange factor, designated Son of sevenless (Sos) (Bow- tell et al., 1992; Chardin et al., 1993), have been cloned. Recently, a direct molecular link between activated recep- tor tyrosine kinases and Ras was established with the find- ing that the mammalian GRB-2 protein, a 26 kd protein composed of a single SH2 and two SH3 domains (Lo- wenstein et al., 1992), directly couples receptor tyrosine kinases to the Ras activator Sos in mammals and Drosoph- ila (Buday and Downward, 1993b; Egan et al., 1993; Liet al., 1993; Gale et al., 1993; Rozakis-Adcock et al., 1993; Chardin et al., 1993; Olivier et al., 1993; Simon et al., 1993). GRB-2 is the human homolog of the Caenorhabditis elegans gene sem-5, which functions in the same signaling pathway (Lowenstein et al., 1992; Clark et al., 1992). The GRB-2 SH2 domain binds to specific tyrosine-phosphory- lated sequences in receptor tyrosine kinases, while the GRB-2 SH3 domains bind to proline-rich sequences pres- ent in the Sos exchange factor. Binding of GRB-2 to recep- tor tyrosine kinases allows for the recruitment of Sos to the plasma membrane where Ras is located (Schlessinger, 1993).
We wished to test whether cytoplasmic components im- plicated in Ras activation by receptor tyrosine kinases play a role in the biology of the BCR-ABL tyrosine kinases. The GRB-2 adaptor protein emerged as a particularly attractive candidate effector of BCR-ABL. Examination of the se- quences surrounding the 11 potential tyrosine phosphory- lation sites within the BCR first exon revealed that 1 of the tyrosines precedes the optimal sequence for binding to the SH2 domain of GRB-2 (Skolnik et al., 1993a; Songyang et al., 1993). The other 10 tyrosines are found in sequences that do not conform to the published optimal binding sites for SH2 domains in various proteins. Given the potential role of GRB-2 for linking BCR-ABL to Ras, we tested for physical association between BCR-ABL and GRB-2. We report that BCR-ABL exists in a complex with GRB-2 in living cells. The binding of GRB-2 to BCR-ABL occurs by direct interaction with a tyrosine-phosphorylated residue within the BCR first exon. Significantly, a BCR- ABL protein lacking the GRB-2-binding site is severely impaired in its ability to transform Rat1 fibroblasts and
hematopoietic cells. These data identify GRB-2 as a direct effector of BCR-ABL and implicate GRB-2 in oncogenic transformation. The existence of a BCR-ABL-GRB-2 com- plex suggests that up-regulation of Ras function may be a key component of the BCR-ABL transformation pathway.
Results
B C R - A B L Associates with GRB-2 In Vivo To determine whether the BCR-ABL tyrosine kinase forms a physical complex with the GRB-2 adaptor protein in in- tact cells, we examined whether the two proteins coimmu- noprecipitate. Cell lysates were prepared from the Ph 1- positive leukemic cell lines K562, MEG-01, and ALL-1 and subjected to immunoprecipitation with anti.ABL, anti- GRB-2, or control antibodies. Following extensive wash- ing, the immunoprecipitates were incubated in the pres-
I.R ANTIBOD~
GRB-2----~
c~
'~ ~ Mr(K)
106 8O
49.5
32.5
27.5
B LOT ANTIBODY: 1 2 3
I
ccGRB-2
18.5
Figure 2. GRB-2 Coimmunoprecipitates with BCR-ABL MEG-01 cells were lysed, and the lysate was incubated with preim- mune serum (lane 1), anti-ABL pEX4 antibodies (lane 2), and anti- GRB-2 antibodies (lane 3). The immunoprecipitated proteins were sep- arated by SDS-polyacrylamide gel electrophoresis on a 15% gel, transferred to nitrocellulose, and immunoblotted with anti-GRB-2 anti- bodies. Bound antibodies were visualized with ~2Sl-labeled protein A.

In Vivo BCR-ABL Chimera-GRB-2 Complex 177
. -
M r ( K )
- 205
: . i 3 ..... " "
1 2 3 4 5
-116
Figure 3. BCR-ABL Binds to GRB-2 In Vitro [~S]methionine-labeled proteins from lysatas of Sf9 insect ceils in- fected with a p185 BCR-ABL recombinant baculovirus were incubated with equal amounts of immobilized GST alone (lane 1), GST-GRB-2 full length (lane 2), GST-amino-terminal (N) GRB-2 SH3 domain (lane 3), GST-GRB-2 SH2 domain (lane 4), GST--carboxy-terminal (C) GRB-2 SH3 domain (lane 5), and anti-ABL antibodies bound to protein A-Sepharose beads (lane 6). After incubation for 90 min at 4°C, the beads were washed four times with incubation buffer and twice with RIPA buffer to remove unbound material. Bound proteins were ana- lyzed by SDS-polyacrylamide gel electrophoresis on a 7% gel and detected by fluorography.
ence of radioactive ATP under conditions that promote autophosphorylation of the BCR-ABL kinase. Compara- ble levels of BCR-ABL protein were precipitated using either anti-GRB-2 or anti-ABL antibodies (Figure 1, lanes 1-9). Both forms of BCR-ABL, p210 and p185, associated with GRB-2 in the Ph ~ leukemic cell lines. Also, p185 and p210 BCR-ABL were found to associate with GRB-2 in Rat1 fibroblasts expressing the corresponding BCR-ABL proteins (Figure 1, lanes 10-15). Similarly, immunoprecipi- tation with anti-GRB-2 or anti-ABL antibodies followed by Western blotting with anti-GRB-2 antibodies showed that GRB-2 can be precipitated by anti-ABL antibodies in cell lines where BCR-ABL is expressed (Figure 2). Metabolic labeling of the BCR-ABL-expressing cells with [~sS]methi- onine followed by immunoprecipitation with anti-ABL or anti-GRB-2 antibodies demonstrated that 50% to 90% of the BCR-ABL kinase available in the cell is complexed with GRB-2, in agreement with the results shown in Figure 1 (data not shown). Interestingly, no association of GRB-2 with the oncoganic v-Abl kinase (Figure 1, lanes 16-18) was observed. These experiments demonstrate that the GRB-2 adaptor protein forms a stable complex with both forms of the BCR-ABL tyrosine kinase but not with the v-Abl kinase and that the BCR-ABL-GRB-2 complexes remain intact following in vitro phosphorylation of BCR-ABL.
BCR-ABL Binds to GRB-2 In Vitro To examine the molecular basis for the association of BCR-ABL with GRB-2, we cloned the full-length GRB-2 into pGEX-2T for expression in bacteria as a giutathione S-transferase (GST) fusion protein, The GST-GRB-2 pro- tein was purified and tested for its ability to bind to BCR- ABL in vitro. We found that p185 BCR-ABL, expressed in baculovirus-infected insect cells, bound to full-length GRB-2 immobilized on glutathione-Sepharose beads (Figure 3,
A. cABL
P 145 cABL'---"
(.g Mr(K)
- 205
- .. . . . : :;'.~" . -116
B. cBCR P16O
c'8CR ~
I I C. cBCR+cABL ~ ~, ~_
P160 cBCR ---" P145 CABL'-'-~ 1
1 2
I I I I I I
I l I l
. . . . . . . .
3 4 5 6
- 205
-116
- 205
-116
Figure 4. SH2 Domain-Mediated Binding of GRB-2 to BCR-ABL Re- quires Tyrosine Phosphorylation of BCR Sequences
Sf9 insect cells were singly infected with c-ABL (A) or c-BCR (B) recom- binant baculoviruses or coinfected with both c-ABL and c-BCR bacu- Ioviruses (C). Three days postinfection, the cells were labeled with [~S]methionine. The labeled cells were lysed, and the lysate proteins were incubated with GST alone (lanes 1), GST-GRB-2 full length (lanes 2), GST-GRB-2 NSH3 (lanes 3), GST-GRB-2 SH2 (lanes 4), GST-GRB-2 CSH3 (lanes 5), and the corresponding anti-ABL (lane 6 in [A] and [C]) and anti-BCR (lane 6 in [B] and [C]) antibodies in the presence of protein A-Sepharose beads. After incubation for 90 min at 4°C, the beads were washed four times with incubation buffer and twice with RIPA buffer. Bound proteins were analyzed by SDS-poly- acrylamide (10%) gel electmphoresis and detected by fluorography.
lane 2). No binding to GST alone was detected (Figure 3, lane 1). The complex of BCR-ABL and GST-GRB-2 remained intact after washing with buffer containing the detergents SDS and deoxycholate (Figure 3, lane 2).
To identify which GRB-2 domain(s) binds to BCR-ABL, we prepared GST fusion proteins that contained the iso- lated GRB-2 SH3 and SH2 domains. BCR-ABL bound to the GST-GRB-2 SH2 fusion protein (Figure 3, lane 4). GST fusion proteins containing the amino- and carboxy- terminal SH3 domains of GRB-2 also bound to the baculo- virus-produced BCR-ABL protein in vitro (Figure 3, lanes 3 and 5). The interaction of BCR-ABL with the SH2 and SH3 domains of GRB-2 was resistant to washing with a buffer containing SDS and deoxycholate (Figure 3, lanes

Cell 178
3-5). Treatment of BCR-ABL with potato acid phospha- tase completely eliminated its ability to associate with the GRB-2 SH2 domain, but did not affect its binding to the GRB-2 SH3 domains (data not shown).
SH2-Mediated Binding of GRB-2 to BCR-ABL Requires Tyrosine Phosphorylation of BCR Sequences To identify which regions in BCR-ABL participate in GRB-2 binding, BCR and ABL sequences were expressed separately in baculovirus-infected insect cells and tested for their ability to bind to full-length GRB-2 as well as to the isolated GRB-2 SH2 and SH3 domains. Baculovirus- produced c-ABL bound to full-length GRB-2 in vitro (Figure 4A, lane 2), but not to the G RB-2 SH2 domain alone (Figu re 4A, lane 4). c-ABL hyperexpressed in insect cells is phos- phorylated on tyrosine residues (Pendergast et al., 1991 b). Thus, the inability of the GRB-2 SH2 domain to bind to the baculovirus-produced c-ABL protein was not due to lack of tyrosine phosphorylation.
The binding of c-ABL to GRB-2 in vitro appears to be mediated exclusively via the amino- and carboxy-terminal SH3 domains of GRB-2 (Figure 4A, lanes 3 and 5). SH3 domains bind to specific proline-rich motifs (Cicchetti et al., 1992; Ren et al., 1993). Recently, it has been shown that GRB-2 binds to the Sos1 guanine nucleotide ex- changer through the direct interaction of the GRB-2 SH3 domains with the proline-rich sequence, PPPVPPR, pres- ent in the carboxy-terminal region of Sos1 (Li et al., 1993; Rozakis-Adcock et al., 1993). The carboxyl terminus of c-ABL contains several proline-rich stretches. However, no PPPVPPR sequence is found in the c-ABL protein. Binding of the GRB-2 SH3 domains to c-ABL in vitro is significantly reduced but not completely eliminated in a c-ABL deletion mutant protein that lacks the majority of the c-ABL carboxy-terminal domain (data not shown). These data, together with the lack of detectable association be- tween c-ABL and GRB-2 following immunoprecipitation
from cell lysates (see Figure 5 below), suggest that the c-ABL-GRB-2 interaction observed in vitro is not specific and may not occur in vivo. However, it is possible that the proline-rich domain in c-ABL serves as a binding site for other SH3 domains.
A similar analysis of c-BCR binding to full-length or the isolated domains of GRB-2 in vitro revealed that only the full-length GRB-2 and to a lesser extent the N-terminal SH3 domain of GRB-2 bound to c-BCR (Figure 4B, lanes 2 and 3). Interestingly, no binding of c-BCR to the SH2 domain of GRB-2 was detected (Figure 4B, lane 4).
The complete lack of in vitro binding of the GRB-2 SH2 domain to c-ABL and c-BCR contrasts with the strong bind- ing of this domain to the chimeric BCR-ABL tyrosine ki- nase (compare Figure 3 with Figures 4A and 4B, lanes 4). The phosphorylation state of c-BCR sequences is dif- ferent in the full-length c-BCR protein than in the BCR- ABL chimera. The c-BCR protein is phosphorylated only on serine/threonine residues in all cell types examined even following hyperexpression in insect cells (Timmons and Witte, 1989; Pendergast et al., 1991a, 1991b). In con- trast, in the BCR-ABL chimera, the activated ABL kinase phosphorylates BCR first exon sequences on tyrosine (Liu et al., 1993; A. M. P., unpublished data). To evaluate whether tyrosine phosphorylation of BCR sequences could uncover binding to the isolated GRB-2 SH2 domain, we coinfected insect cells with baculoviruses encoding for the full-length c-BCR and c-ABL proteins. Transphosphor- ylation of c-BCR by the c-ABL tyrosine kinase resulted in binding of the GRB-2 SH2 domain to c-BCR (Figure 4C, lane 4). Western blotting with anti-phosphotyrosine anti- bodies demonstrated that c-BCR was tyrosine phosphory- lated in Sf9 cells coinfected with c-ABL and c-BCR bacu- Ioviruses (data not shown). A low level of binding of c-BCR to the isolated amino- and carboxy-terminal SH3 domains of GRB-2 in vitro was also detected (Figure 4C, lanes 3 and 5). These results demonstrate that binding of the GRB-2 SH2 domain to BCR sequences requires tyrosine phosphorylation.
A. So
n" ,.n
n-z ~ z ~ Mr(K) z m ~
- 205 BCR/ABL'~'
cABL~ ='~ i l ~
Mr(K)
• 205
-116 BCR._.. mlJ$. -116 ~,(872-1271) •
1 2 3 4 5 6 1 2 3
Figure 5. GRB-2 Forms a Complex with the Chimeric BCR-ABL Tyrosine Kinase but Not with c-ABL and c-BCR Sequences In Vivo (A) COS cells were transfected with p185 wild- type (lanes 1-3) and c-ABL (lanes 4-6) cDNAs cloned into the pSR¢ vector. Three days post- transfection the cells were lysed and the lysates were incubated for 2 hr at 4°C with normal rab- bit serum (NRS) (lanes 1 and 4), anti-ABL 2/3 antibodies (lanes 2 and 5), and anti-GRB-2 anti- bodies (lanes 3 and 6). The immunoprecipi- tates were collected on protein A-Sepharose beads and washed six times with incubation buffer. Bound proteins were analyzed by SDS- polyacrylamide (8%) gel electrophoresis and
transferred to nitrocellulose filters, and the filters were incubated with anti-ABL mouse monoclonal antibody. Immunoreactive bands were visualized with the enhanced chemiluminescence detection system (Amersham). (B) COS ceils were transfected with BCR (&872-1271) cDNA cloned into the pSR(~ vector. The cells were lysed 3 days posttransfection and the lysates were incubated with control serum (NRS) (lane 1), anti-BCR antibodies (lane 2), or anti-GRB-2 antibodies (lane 3) for 2 hr at 4°C. The immunopracipitates were collected with protein A-Sepharose beads, washed four times with incubation buffer and twice with 50 mM Tris-HCI (pH 7.0), and then subjected to in vitro phosphorylation in the presence of [7-~P]ATP and MnCI2. Proteins were analyzed by SDS-polyacrylamide (8%) gel electrophoresis and autoradiography.

In Vivo BCR-ABL Chimera-GRB-2 Complex 179
A. P185 wild type
Or)
c
~ - 205
~ " -116
B. P185 Y177F
I I
- 205
: ~ -116 • =
1 2
Figure 6. Mutation of Tyrosine 177 to Phenylalanine in the BCR First Exon Abolishes Binding of the GRB-2 SH2 Domain to BCR-ABL Sf9 insect cells were cotransfected with wild-type baculovirus DNA and wild-type p 185 BCR-ABL (A) or the p185 BCR-ABL (Y 177F) mutant (13) cDNAs cloned into the pAcC12 vector. Six days posttransfection, the cells were metabolically labeled with [=S]methionine. The cells were lysed, and the lysates were incubated with GST-GRB-2 SH2 (lanes 1) or with anti-ABL pEX4 antibodies with protein A-Sepharose beads (lanes 2). The bound proteins were analyzed by SDS-polyacrylamide gel electrophoresis on a 10% gel followed by fluorography.
GRB-2 Interacts with BCR-ABL but Not c-ABL and c-BCR Sequences In Vivo To examine whether the in vitro binding of GRB-2 to c-ABL and c-BCR also occurred in vivo, we transfected COS cells with expression constructs of the corresponding cDNAs. Transfection of these cDNAs results in an approximately 50- to 200-fold increase in the expression of c-ABL and c-BCR over the corresponding endogenous protein levels (Pendergast et al., 1991b). Under these conditions, no appreciable levels of c-ABL coimmunoprecipitated with GRB-2 (Figure 5A, lane 6). Similarly, no interaction of GRB-2 with BCR sequences could be detected following hyperexpression of the BCR sequences retained in the p210 BCR-ABL chimera in COS cells (Figure 5B, lane 3). In contrast, significant levels of p185 BCR-ABL were precipitated by the anti-GRB-2 antibodies in the same ex- periment (Figure 5A, lane 3).
These data, together with the lack of detectable endoge- nous c-ABL and c-BCR proteins in anti-GRB-2 immunopre- cipitates from normal cells (data not shown), support the contention that no c-ABL-GRB-2 or c-BCR-GRB-2 com- plexes may be found in vivo. These findings demonstrate that the BCR-ABL chimera exhibits novel protein binding properties that are distinct from those of its BCR and ABL protein sequence components.
A Point Mutation in the BCR First Exon Abolishes Binding of the GRB-2 SH2 Domain to BCR Binding of SH2 domains to specific tyrosine-phosphory- lated proteins is dependent on the primary sequence
A. COS B. RAT1
Pill6 P186 P186 P l U wfkl type (Y I?TF ) w i l d t ype (Y I " /TF )
i , i i r l r 1
2 3 4 5 6 1 2 3 4 5 6
Figure 7. Formation of a BCR-ABL-GRB-2 Complex In Vivo Requires the Presence of Tyrosine 177 in the BCR First Exon (A) COS cells were transfected with p185 wild-type (lanes 1-3) and p185 (Y177F) (lanes 4--6) cDNAs cloned into the pSR~x MSVtkneo vector. Three days posttransfection the cells were lysed and the lysates were incubated with normal rabbit serum (NRS) (lanes 1 and 4), anti- ABL 2/3 antibodies (lanes 2 and 5), or anti-GRB-2 antibodies (lanes 3 and 6) for 2 hr at 4°C. The immunoprecipitates were collected on protein A-Sepharose beads and subjected to in vitro phosphorylation in the presence of Iy-=P]ATP and MnCI2. Proteins were analyzed by SDS-polyacrylamide (10%o) gel electrophoresis and autoradiography. (B) Lysates of G418-selected Rat1 cells expressing p185 wild type (lanes 1-3) and p185 (Y177F) (lanes 4-6) were incubated for 2 hr at 4°C with anti-G RB-2 antibodies (lanes 1 and 4), anti-ABL 2/3 antibodies (lanes 2 and 5), or normal rabbit serum (NRS) (lanes 3 and 6). The immunoprecipitates were collected on protein A-Sepharose beads. The bound proteins were analyzed by SDS-polyacrylamide (10%o) gel electrophoresis followed by immunoblotting with anti-ABL mouse monoclonal antibody. Immunoreactive bands were visualized with the enhanced chemiluminescence detection system (Amersham).
C-terminal to the phosphorylated tyrosine (Fantl et al., 1992; Kashishian et al., 1992; Sonyang et al., 1993). Ex- amination of the sequences surrounding the 11 potential tyrosine phosphorylation sites within the first exon of BCR revealed that tyrosine 177 is found within the sequence YVNV, which corresponds to an optimal binding site for the GRB-2 SH2 domain (Sonyang et al., 1993; Skoinik et al., 1993a). Sequences surrounding the other 10 tyrosines in the BCR first exon do not conform to optimal binding sites for the GRB-2 SH2 domain or other SH2 domains examined (Sonyang et al., 1993). To determine whether tyrosine 177 is required for the binding of BCR-ABL to the GRB-2 SH2 domain, we substituted phenylalanine for tyrosine 177 in BCR-ABL by site-directed mutagenesis, expressed the mutated protein in insect cells, and tested for binding to GRB-2. Comparison of the phosphopeptide map patterns of the p185 (Y177F) mutant and p185 wild- type proteins following in vitro autophosphorylation re- vealed the absence of at least one major phosphopeptide in p185 (Y177F) (data not shown). Binding studies demon- strated that in contrast with the wild-type p185 BCR-ABL, the p185 BCR-ABL (Y177F) mutant protein did not interact with the GRB-2 SH2 domain in vitro (Figure 6, lanes 1).
Next, we evaluated the ability of the p185 BCR-ABL (Y177F) mutant to interact with GRB-2 in vivo. p185 (Y177F) did not interact with endogenous GRB-2 when hyperexpressed in COS cells (Figure 7A, lane 6) or in Rat1 fibroblasts that stably express the mutant protein (Figure 7B, lane 4). Similarly, BCR-ABL deletion mutants that lack
Cell 180
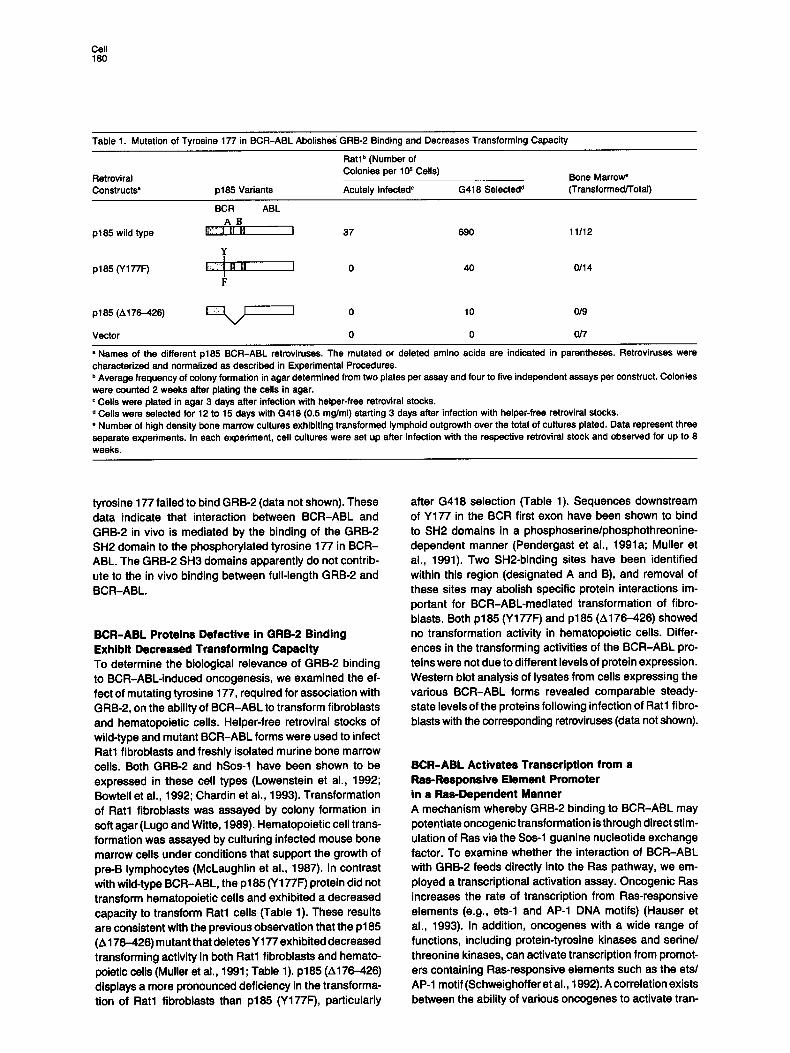
Table 1. Mutation of Tyrosine 177 in BCR-ABL Abolishes" GRB-2 Binding and Decreases Transforming Capacity
Ratlb (Number of Colonies per 10 s Cells)
Retroviral Constructs • p185 Variants Acutely Infected c G418 Selected d
Bone Marrow" (Transformed/Total)
BCR ABL A ] ]
p185 wild type E~I !~1 II I 37
¥ !
p185 (Y177F) I;:iii~1 g B I 0 I F
p185 (A 176-426) ~ , , , ~ 0
Vector 0
690 11/12
40 0114
10 0/9
0 0/7
• Names of the different p185 BCR-ABL retroviruses. The mutated or deleted amino acids are indicated in parentheses. Retrovirusas were characterized and normalized as described in Experimental Procedures. b Average frequency of colony formation in agar determined from two plates per assay and four to five independent assays per construct. Colonies were counted 2 weeks after plating the cells in agar. c Cells were plated in agar 3 days after infection with helper-free retroviral stocks. d Cells were selected for 12 to 15 days with G418 (0.5 mg/ml) starting 3 days after infection with helper-free retroviral stocks. • Number of high density bone marrow cultures exhibiting transformed lymphoid outgrowth over the total of cultures plated. Data represent three separate experiments. In each experiment, cell cultures were set up after infection with the respective retroviral stock and observed for up to 8 weeks.
tyrosine 177 failed to bind GRB-2 (data not shown). These data indicate that interaction between BCR-ABL and GRB-2 in vivo is mediated by the binding of the GRB-2 SH2 domain to the phosphorylated tyrosine 177 in BCR- ABL. The GRB-2 SH3 domains apparently do not contrib- ute to the in vivo binding between full-length GRB-2 and BCR-ABL.
BCR-ABL Proteins Defective in GRB-2 Binding Exhibit Decreased Transforming Capacity To determine the biological relevance of GRB-2 binding to BCR-ABL-induced oncogenesis, we examined the ef- fect of mutating tyrosine 177, required for association with GRB-2, on the ability of BCR-ABL to transform fibroblasts and hematopoietic cells. Helper-free retroviral stocks of wild-type and mutant BCR-ABL forms were used to infect Rat1 fibroblasts and freshly isolated murine bone marrow cells. Both GRB-2 and hSos-1 have been shown to be expressed in these cell types (Lowenstein et al., 1992; Bowtell et al., 1992; Chardin et al., 1993). Transformation of Rat1 flbroblasts was assayed by colony formation in soft agar (Lugo and Witte, 1989). Hematopoietic cell trans- formation was assayed by culturing infected mouse bone marrow cells under conditions that support the growth of pre-B lymphocytas (McLaughlin et al., 1987). In contrast with wild-type BCR-ABL, the p185 (Y177F) protein did not transform hematopoietic cells and exhibited a decreased capacity to transform Rat1 cells (Table 1). These results are consistent with the previous observation that the p185 (A176-426) mutant that deletes Y177 exhibited decreased transforming activity in both Rat1 fibroblasts and hemato- poletic cells (Muller at al., 1991; Table 1). p185 (A176-426) displays a more pronounced deficiency in the transforma- tion of Rat1 fibroblasts than p185 (Y177F), particularly
after G418 selection (Table 1). Sequences downstream of Y177 in the BCR first exon have been shown to bind to SH2 domains in a phosphoserine/phosphothreonine- dependent manner (Pendergast et al., 1991a; Muller et al., 1991). Two SH2-binding sites have been identified within this region (designated A and B), and removal of these sites may abolish specific protein interactions im- portant for BCR-ABL-mediated transformation of fibro- blasts. Both p185 (Y177F) and p185 (4176-426) showed no transformation activity in hematopoietic cells. Differ- ences in the transforming activities of the BCR-ABL pro- teins were not due to different levels of protein expression. Western blot analysis of lysates from cells expressing the various BCR-ABL forms revealed comparable steady- state levels of the proteins following infection of Rat1 fibro- blasts with the corresponding retroviruses (data not shown).
BCR-ABL Activates Transcription from a Ras-Responsive Element Promoter in a Ras-Dependent Manner A mechanism whereby GRB-2 binding to BCR-ABL may potentiate oncogenic transformation is th rough direct sti m- ulation of Ras via the Sos-1 guanine nucleotide exchange factor. To examine whether the interaction of BCR-ABL with GRB-2 feeds directly into the Ras pathway, we em- ployed a transcriptional activation assay. Oncogenic Ras increases the rate of transcription from Ras-responsive elements (e.g., ets-1 and AP-1 DNA motifs) (Hauser et al., 1993). In addition, oncogenas with a wide range of functions, including protein-tyrosine kinases and serine/ threonine kinases, can activate transcription from promot- ers containing Ras-responsive elements such as the ets/ AP-1 motif (Schweighoffer et al., 1992). A correlation exists between the ability of various oncogenas to activate tran-
In Vivo BCR-ABL Chimera-GRB-2 Complex 181
,° 1
< 5
k- <
= re I~
+ co <$
~- ®
==
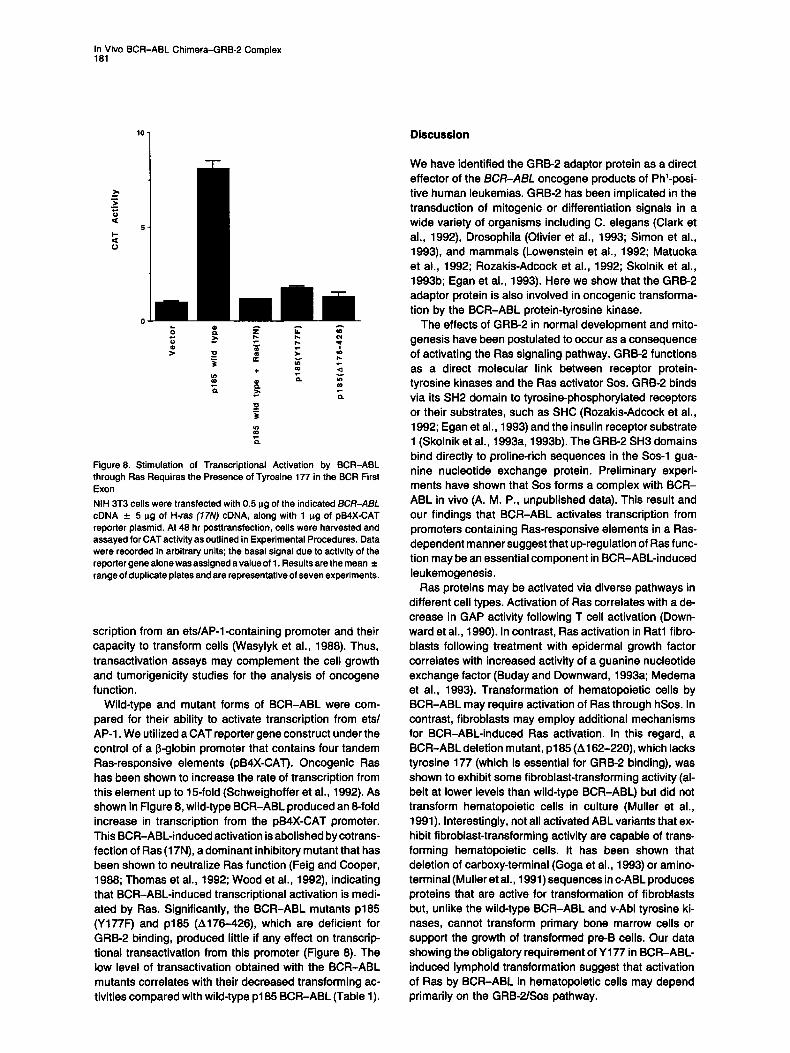
Figure 8. Stimulation of Transcriptional Activation by BCR-ABL through Ras Requires the Presence of Tyrosine 177 in the BCR First Exon NIH 3T3 cells were transfected with 0.5 ~g of the indicated BCR-ABI_ cDNA -4- 5 p.g of H-rss (17N) cDNA, along with 1 p.g of pB4X-CAT reporter plasmid. At 48 hr posttransfection, cells were harvested and assayed for CAT activity as outlined in Experimental Procedures. Data were recorded in arbitrary units; the basal signal due to activity of the reporter gene alone was assigned a value of 1. Results are the mean ± range of duplicate plates and are representative of seven experiments.
scription from an ets/AP-l-containing promoter and their capacity to transform cells (Wasylyk et al., 1988). Thus, transactivation assays may complement the cell growth and tumorigenicity studies for the analysis of oncogene function.
Wild-type and mutant forms of BCR-ABL were com- pared for their ability to activate transcription from ets/ AP-1. We utilized a CAT reporter gene construct under the control of a I~-globin promoter that contains four tandem Ras-responsive elements (pB4X-CAT). Oncogenic Ras has been shown to increase the rate of transcription from this element up to 15-fold (Schweighoffer et al., 1992). As shown in Figure 8, wild-type BCR-ABL produced an 8-fold increase in transcription from the pB4X-CAT promoter. This BCR-ABL-induced activation is abolished by cotrans- fection of Ras (17N), a dominant inhibitory mutant that has been shown to neutralize Ras function (Feig and Cooper, 1988; Thomas et al., 1992; Wood et al., 1992), indicating that BCR-ABL-induced transcriptional activation is medi- ated by Ras. Significantly, the BCR-ABL mutants p185 (Y177F) and p185 (A176-426), which are deficient for GRB-2 binding, produced little if any effect on transcrip- tional transactivation from this promoter (Figure 8). The low level of transactivation obtained with the BCR-ABL mutants correlates with their decreased transforming ac- tivities compared with wild-type p185 BCR-ABL (Table 1).
Discuss ion
We have identified the GRB-2 adaptor protein as a direct effector of the BCR-ABL oncogene products of Phl-posi - tive human leukemias. GRB-2 has been implicated in the transduction of mitogenic or differentiation signals in a wide variety of organisms including C. elegans (Clark et al., 1992), Drosophila (Olivier et al., 1993; Simon et al., 1993), and mammals (Lowenstein et al., 1992; Matuoka et al., 1992; Rozakis-Adcock et al., 1992; Skolnik et al., 1993b; Egan et al., 1993). Here we show that the GRB-2 adaptor protein is also involved in oncogenic transforma- tion by the BCR-ABL protein-tyrosine kinase.
The effects of GRB-2 in normal development and mito- genesis have been postulated to occur as a consequence of activating the Ras signaling pathway. GRB-2 functions as a direct molecular link between receptor protein- tyrosine kinases and the Ras activator Sos. GRB-2 binds via its SH2 domain to tyrosine-phosphorylated receptors or their substrates, such as SHC (Rozakis-Adcock et al., 1992; Egan et al., 1993) and the insulin receptor substrate 1 (Skolnik et al., 1993a, 1993b). The GRB-2 SH3 domains bind directly to proline-rich sequences in the Sos-1 gua- nine nucleotide exchange protein. Preliminary experi- ments have shown that Sos forms a complex with BCR- ABL in vivo (A. M. P., unpublished data). This result and our findings that BCR-ABL activates transcription from promoters containing Ras-responsive elements in a Ras- dependent manner suggest that up-regulation of Ras func- tion may be an essential component in BCR-ABL-induced leukemogenesis.
Ras proteins may be activated via diverse pathways in different cell types. Activation of Ras correlates with a de- crease in GAP activity following T cell activation (Down- ward et al., 1990). In contrast, Ras activation in Rat1 fibro- blasts following treatment with epidermal growth factor correlates with increased activity of a guanine nucleotide exchange factor (Buday and Downward, 1993a; Medema et al., 1993). Transformation of hematopoietic cells by BCR-ABL may require activation of Ras through hSos. In contrast, fibroblasts may employ additional mechanisms for BCR-ABL-induced Ras activation. In this regard, a BCR-ABL deletion mutant, p185 (A162-220), which lacks tyrosine 177 (which is essential for GRB-2 binding), was shown to exhibit some fibroblast-transforming activity (al- beit at lower levels than wild-type BCR-ABL) but did not transform hematopoietic cells in culture (Muller et al., 1991). Interestingly, not all activated ABL variants that ex- hibit fibroblast-transforming activity are capable of trans- forming hematopoietic cells. It has been shown that deletion of carboxy-terminal (Goga et al., 1993) or amino- terminal (Muller et al., 1991) sequences in c-ABL produces proteins that are active for transformation of fibroblasts but, unlike the wild-type BCR-ABL and v-Abl tyrosine ki- nases, cannot transform primary bone marrow cells or support the growth of transformed pre-B cells. Our data showing the obligatory requirement of Y177 in BCR-ABL- induced lymphoid transformation suggest that activation of Ras by BCR-ABL in hematopoietic cells may depend primarily on the GRB-2/Sos pathway.

Cell 182
Different tyrosine kinases appear to employ alternate pathways to activate Ras. The v-Abl tyrosine kinase lacks a GRB-2-binding site and does not bind to GRB-2 directly. Activation of Ras by v-Abl may be achieved by phosphory- lation of the SH2-containing protein SHC (Pelicci et al., 1992; Rozakis-Adcock et al., 1992). Tyrosine phosphoryla- tion of SHC is increased in cells transformed by v-Src and v-Fps (McGlade et al., 1992) and fol lowing insulin stimula- tion of CHO cells overexpressing the human insulin recep- tor (Skolnik et al., 1993a, 1993b). The tyrosine-phos- phorylated SHC protein associates directly with GRB-2 (Rozakis-Adcock et al., 1993; Egan et al., 1993; Skolnik et al., 1993a, 1993b). It is postulated that this association leads to activation of Ras. Similarly, BCR-ABL may em- ploy alternative pathways such as phosphorylat ion of SHC for Ras activation. It appears, however, that the direct bind- ing of GRB-2 to the phosphorylated tyrosine 177 of BCR- ABL provides a critical interaction that is essential for BCR-ABL oncogenicity, particularly in hematopoiet ic cells.
An alternative and nonexclusive possibil i ty for ex- plaining the role of GRB-2 in BCR-ABL transformation is that binding of GRB-2 to BCR-ABL in leukemic cells may result in altered subcellular distribution of GRB-2. It was recently reported that GRB-2 localizes to membrane ruf- fles (Bar-Saul et al., 1993). In contrast, BCR-ABL has been shown to localize to the actin cytoskeleton (McWhirter and Wang, 1991, 1993). The aberrant expression of the BCR- ABL chimera in hematopoiet ic cells may produce the re- crui tment of a large pool of GRB-2 to the actin cytoskeleton and disrupt its interactions with specif ic cellular proteins in the membrane ruffles, leading to altered cellular pro- cesses.
Our data and that of others define at least three distinct structural motifs relevant for transformation within the BCR first exon. First, amino-terminal BCR sequences are required for proper localization of BCR-ABL to the actin cytoskeleton, and deletion of these sequences renders BCR-ABL inactive for transformation (McWhirter and Wang, 1991; Muller et al., 1991). Second, amino acids 192 to 413 are impl icated in phosphoserine/phosphothreo- nine-dependent binding to a subset of SH2 domains, and deletion of this region correlates with a complete loss of transforming activity (Pendergast et al., 1991a). Third, ty- rosins 177 is required for GRB-2 binding and for connect- ing BCR-ABL to the Ras pathway.
Our identif ication of the specific amino acid residues involved in the biologically relevant association of BCR- ABL with GRB-2 makes this interaction a prime candidate for testing the therapeutic utility of pept ide mimetics in disrupting BCR-ABL- induced oncogenesis.
Experimental Procedures
Cells and Viruses Sf9 insect cells were propagated in complete Graces medium. The Phi-positive leukemic cell lines K562 and MEG-01 were derived from patients with chronic myelogenous leukemia and express p210 BCR- ABL. The Phi-positive cell line, ALL-l, was derived from a patient with acute lymphocytic leukemia and express p185 BCR-ABL. The Phi-positive cell lines were cultured in RPM11640 medium with 10% fetal calf serum. COS African green monkey cells and Rat1 fibroblasts
were grown in Dulbocco's modified Eagle's medium with 5% fetal calf serum.
Recombinant baculoviruses for expression of c-BCR, c-ABL, and BCR-ABL were prepared by cotransfecting the corresponding cDNAs cloned into the pAcC12 baculovirus vector in the presence of wild-type baculovirus DNA as described (Pendergast et al., 1991a, 1991 b).
Helper-free retroviral stocks were prepared by transient hyperex- pression in COS cells according to methods previously described (Muller et al., 1991). Retroviral stocks were characterized for their ability to transfer wild-type and mutant forms of the BCR-ABL gene product to Rat1 fibroblasts using immunohistochemical methods (Muller et al., 1991). Endogenous levels of rat c-Abl protein were not detected in these staining procedures. The level of gene transfer was further evaluated by measuring levels of BCR-ABL protein expression (Western blot). Only retroviral stocks showing comparable levels of gene transfer were used in these studies. Titers were in the range of 105 infectious particles per ml as determined by the frequency of G418-resistant Rat1 colonies following exposure to limiting dilutions of the viral stocks.
Antibodies Polyclonal rabbit antibodies directed against the amino terminus of c-BCR and the amino- and carboxy-terminal sequences of c-ABL have been previously described (Pendergast at al., 1991a; Konopka st al., 1984). A mouse monoclonal anti-ABL (21-53) antibody was employed for immunoblotting (Pendergast et al., 1991 b). Polycional rabbit anti- bodies to the C-terminal SH3 domain of GRB-2 (Ab50) and to a syn- thetic peptide derived from the N-terminal SH3 domain of GRB-2 (Ab86) were used for immunoprecipitation and immunoblotting, re- spectively (Lowenstein etal., 1992).
Plssmld Constructions The 650 bp human GRB-2 cDNA (Lowenstein etal., 1992; Matuoka et al., 1992) was cloned from a human placenta cDNA library by poly- merase chain reaction as (5') SH3-SH2 and (3') SH3 fragments. A unique Kpnl site at codon 154 of GRB-2 was employed to generate the full-length GRB-2 cDNA. The entire coding sequence of GRB-2 and the (3') SH3 domain of GRB-2 were subcloned in-frame into the BamHI site of the pGEX-2T vector (Pharmacia). The isolated (5') SH3 and SH2 domains of GRB-2 were prepared as described (Skolnik et al., 1993a).
Preparation of cDNAs for wild-type p185 BCR-ABL (McLaughlin et al., 1989) and p185 (A176-426) (Muller et al., 1991) and cloning of the corresponding cDNAs into pSRa (Pendergast et al., 1991b) and pSRa MSVtkneo (Muller et al., 1991) vectors were performed as pre- viously described. The p185 BCR-ABL (Y177F) mutant was created by oligonucleotide site-directed mutagenesis using the Muta-Gene Phagemid in vitro mutagenesis system (Bio-Rad). Template was gen- erated by subcloning a 1.6 kb EcoRI-Sacl fragment of c-BCR from pGEM41c-BCR into the EcoRI and Sacl sites of the pBlueScript SK(+) vector (Stratagene) and rescuing the single-stranded DNA by coinfect- ing XL-1 Blue bacteria with the helper phage R408 (Stratagene), The mutagenic oligonucleotide, 5'-AAG CCC TTC TIC GTT AAC GTC GAG-3', was employed to create a phenylalanine codon in place of tyrosine, by changing nucleotide 530 from an A to a T. In addition, a silent base change at nucleotide 534 was introduced to create a unique Hpal site. Mutagenized plasmids were selected for the presence of the unique Hpal site. The mutations were verified by dideoxy chain termination sequence analysis in both directions. The mutated BCR sequence was introduced into the wild-type p185 BCR-ABL cDNA. p185 BCR-ABL (Y1771:) was subcloned into the pSRa and pSRaMSV- tkneo mammalian expression vectors and the AcC12 vector for bacu- Iovirus expression. Cloning of the cDNA for wild-type c-ABL into the pSRa vector has been previously described (Pendergast et al., 1991b). BCR (,d872-1271) was also cloned into pSRa as described (Pender- gast et al., 1991a).
Expression and Purification of GST Fusion Proteins GST fusion proteins were expressed and purified using glutathione- Sepharose 4B beads (Pharmacia) as previously described (Pender- gast et al., 1991a). Fusion proteins were left on the resin and stored at 4°C.

In Vivo BCR-ABL Chimera-GRB-2 Complex 183
Metabolic Labeling and ImmunopreclpltaUon Sf9 cells were infected with the indicated recombinant baculoviruses. Three days postinfection the cells were incubated with 0.1 mCi/ml [~S]methionine (ICN) in methionine-frse medium for 4 to 6 hr at 27°C. Labeled cells were lysed with either KLB (10 mM sodium phosphate [pH 7.0], 150 mM NaCI, 1% Triton X-100) or PCLB (50 mM HEPES lpH 7.0], 150 mM NaCI, 1 mM MgCI2, 1% Triton X-100, 10% glycerol) supplemented with 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 50 p.g/ml leupeptin, 25 p,g/ml aprotinin, 25 mM NaF, 1 mM Na3VO4, and 0.1 mM Na2MoO4. The lysates were clarified by centrifugation at 100,000 x g for 1 hr. Lysates were incubated with the indicated antibodies directly or after a 3-fold dilution with incubation buffer (20 mM HEPES [pH 7.0], 150 mM NaCI, 0.1% Triton X-100, 10% glycerol, 0.5 mM Na3VO4, 0.1 mM Na2MoO4, 25 mM NaF, 1 mM phenylmethylsul- fonyl fluoride, 25 i~g/ml leupeptin) as indicated. Immune complexes were collected by incubation with protein A-Sepharose beads (Phar- macia) for 120 min at 4°C. The beads were washed extensively with incubation buffer to remove unbound material. Bound proteins were analyzed by SDS-polyacrylamide gel electrophoresis and visualized by fluorography. Whenever unlabeled lysates were employed, the pro- teins were detected by immunoblotting with the indicated antibodies.
Binding Assays Protein lysates were diluted 3-fold with incubation buffer and incubated with GST or GST fusion proteins attached to glutathione-Sepharose beads. After incubation for 90 rain to 2 hr at 4°C, the beads were washed extensively with incubation buffer or with RIPA buffer (20 mM Tris-HCI [pH 7.4], 137 mM NaCI, 10O/o glycerol, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, and 2 m M EDTA), as indicated. Bound proteins were analyzed by SDS-polyacrylamide followed by fluorogra- phy for radiolabeled proteins.
Immunoblottlng, In Vitro Autophoephoryletlon, and Dephosphorylstlon Reactions Procedures for immunoblotting and in vitro labeling with [7-:~P]ATP were carried out essentially as previously described (Pendergast et al., 1991a, 1991 b). Dephosphorylation with potato acid phosphatase was performed as described (Pendergast st al., 1991a, 1991b).
Transformation Assays Infection of Rat1 fibroblasts was carried out as previously described (Lugo and WiRe, 1989). Titers were in the range of 10 s infectious parti- cles per ml as determined by the frequency of gene transfer to fibro- blasts. Rat1 colony formation in semisolid medium was measured by plating 5 x 104 cells per 6 cm 2 dish in 5 ml of Iscove's medium supple- mented with 15% fetal calf serum and 0.4% Noble agar (Lugo and Witte, 1989). G418-resistant populations were established by culturing the infected cells for 12-15 days in G518 (0.5 mg/ml). Following selec- tion, cells from G418-resistant cultures were plated in agar at a density of 104 cells per ml. The number of colonies formed in the agar was recorded 2 weeks after plating the cells.
Infection and establishment of hematopoietic cell cultures from freshly isolated routine bone marrow were performed as previously described (McLaughlin etal., 1987). In brief, bone marrow elements from 4- to 6-week-old female BALBIc mice were resusbended at 2 x 10 s cells per ml in the appropriate retroviral stock supplemented with 4 p.g of Polybrene per ml. Cells were incubated for 4 hr at 37°C. Following infection, cells were washed and resusbended in RPM11640 medium supplemented with 5% fetal calf serum and 50 p.M ~-mer- captoethanol, at a density of 106 cells per ml. Cell suspension (5 ml) was plated into each 6 cm 2 dish. Cultures were maintained for up to 8 weeks, and fresh medium was added once a week. The cultures were considered transformed when the cell density of the nonadherent hematopoietic cells exceeded 106 cells per ml.
Transcriptional Activation Assay Transcriptional activation of expression from a Ras-responsive ele- ment (ets/AP-1) promoter was done essentially as described previously (Clark st al., 1993; Hauser st al., 1993). In brief, NIH 3T3 cells were transfected with 1 rig of the pB4X-CAT reporter plasmid (Wasylyk et at., 1989), together with 0.5 I~g of pSR(xMSVTkneo containing the indicated BCR-ABL mutants in the presence or absence of 5 p.g of pZIP H.ras (17N) (Feig and Cooper, 1988). Transfections were performed in
duplicate in 60 mm dishes, and the cells were harvested after 48 hr. Following lysis by freeze-thaw in 100 i11 of 250 mM Tris-HCI (pH 7.8), cell debris was removed by centrifugation and the supernatant heated to 62°C to denature endogenous acyl transferases. Following further centrifugation, a 50 I~1 aliquot of each supernatant was assayed for CAT activity by incubation with 0.1 i~Ci of ["C]chloramphenicol (NEN) and 0.34 mM acetyl CoA in 250 mM Tris-HCI in a final reaction volume of 140 pl for 45 min. The reaction was then terminated by extraction with 500 pl of ethyl acetate and evaporated under vacuum, and the resulting pellets were redissolved and subjected to thin layer chroma- tography on silica gel plates, using 5% methanol-95% chloroform (v/v) as solvent. Assays were quantitated using an AMBIS beta scanner.
Acknowledgments
We thank Jonathan Horowitz for critically reading the manuscript, Axel UIIrich for the MEG-01 human megakaryocyte cell line, and Gary Reuther for technical assistance and illustrations. This work was sup- ported by National Institutes of Health (NIH) Training Grant GM07184 (C. H. B.), NIH Physician-Scientist Training Grant DK01965 (L. D. C.), NIH Grants CA42978, CA52072, and CA55008 (C. J. D.), Sugen, Inc. (J. S. and M. L. G.), and the Human Frontier Science Program (J. S.). C. J. D. is the recipient of an American Cancer Society Faculty Research Award. A. M. P. is a Whitehead Scholar.
Received August 6, 1993; revised September 3, 1993.
References
Bar-Sagi, D., Rotin, D., Batzer, A., Mandiyan, V., and Schlessinger, J. (1993). SH3 domains direct cellular localization of signaling molecules. Cell 74, 83-91. Bollag, G., and McCormick, F. (1991). Regulators and effectors of ras proteins. Annu. Rev. Cell Biol. 7, 601-632.
Bonfini, L., Karlovich, C. A., Dasgupta, C., and Banerjes, U. (1992). The Son of sevenless gene product: a putative activator of ras. Science 255, 603-606. Bowtell, D., Fu, P., Simon, M., and Senior, P. (1992). Identification of routine homologues of the Drosophila Son ofsevenless gene: potential activators of ras. Proc. Natl. Acad. Sci. USA 89, 6511-6515.
Broek, D., Toda, T., Michaeli, T., Levin, L., Birchmeier, C., Zoller, M., Powers, S., and Wigler, M. (1987). The S. cerevisiae CDC25 gene product regulates the RASladenylate cyclase pathway. Cell 48, 789- 799. Buday, L., and Downward, J. (1993a). Epidermal growth factor regu- lates the exchange rate of guanine nucleotides on p21r" in fibroblasts. Mol. Cell. Biol. 13, 1903-1910.
Buday, L., and Downward, J. (1993b). Epidermal growth factor regu- lates p21 ~' through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 73, 611- 620.
Cai, H., Szeberenyi, J., and Cooper, G. M. (1990). Effect of a dominant inhibitory Ha-ras mutation on mitogenic signal transduction in NIH3T3 cells. Mol. Cell. Biol. 10, 5314-5323.
Chardin, P., Camonis, J. H., Gale, N. W., Aelst, L. V., Schlessinger, J., Wigler, M. H., and Bar-Sagi, D. (1993). Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260, 1338-1343.
Cicchetti, P., Mayer, B. J., Thiel, G., and Baltimore, D. (1992). Identifi- cation of a protein that binds to the SH3 region of ABL and is similar to BCR and GAP-rho. Science 257, 803-806.
Clark, G. J., Quilliam, L. A., Hisaka, M. M., and Der, C. J. (1993). Differential antagonism of Ras biological activity by catalytic and Src homology domains of Ras GTPase activation protein. Proc. Natl. Acad. Sci. USA 90, 4887-4891.
Clark, S. G., Stern, M. J., and Horvitz, H. R. (1992). C. elegens cell- signaling gene sam-5 encodes a protein with SH2 and SH3 domains. Nature 356, 340-344.

Cell 184
Downward, J. (1992). Regulatory mechanisms for ras proteins. Bioas- says 14, 177-184. Downward, J., Graves, J. D., Warne, P. H., Rayter, S., and Cantrell, D. A. (1990). Stimulation of p21 r" upon T-cell activation. Nature 346, 719-723.
Egan, S. E., Giddings, B. W., Brooks, M. W., Buday, L., Sizeland, A. M., and Weinberg, R. A. (1993). Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 363, 45-51. Fantl, W. J., Escobedo, J. A., Martin, G. A., Turck, C. W., del Rosario, M., McCormick, F., and Williams, L. T. (1992). Distinct phosphotyro- sines on a growth factor receptor bind to specific molecules that medi- ate different signaling pathways. Cell 69, 413-423. Feig, L. A., and Cooper, G. M. (1988). Inhibition of NIH 3T3 cell prolifer- ation by a mutant Ras protein with preferential affinity for GDP. Mol. Cell. Biol. 6, 3235-3243. Gale, N. W., Kaplan, S., Lowenstein, E. J., Schlessinger, J., and Bar- Sagi, D. (1993). Grb2 mediates the EGF-dependent activation of gua- nine nucleotide exchange on Ras. Nature 363, 88-92. Gibbs, J. B., Marshall, M. S., Scolnick, E. M., Dixon, R. F., and Vogel, U. S. (1990). Modulation of guanine nucleotides bound to rats in NIH 3T3 cells by oncogenes, growth factors and the GTPase activating protein (GAP). J. Biol. Chem. 265, 20437-20442. Goga, A., McLaughlin, J., Pendergast, A. M., Parmar, K., Muller, A., Rosenberg, N., and Witte, O. N. (1993). Oncogenic activation of c-ABL by mutation within its last exon. Mol. Cell. Biol. 13, 4967--4975. Hauser, C. A., Der, C. J., and Cox, A. D. (1993). Transcriptional activa- tion analysis of oncogene function. Meth. Enzymol., in press. Jones, S., Vignais, M. L., and Broach, J. R. (1991). The CDC25 protein of Sacoharomyces ceravisiae promotes exchange of guanine nucleo- tides bound to Ras. Mol. Cell. Biol. 11, 2641-2646.
Kashishian, A., Kazlauskas, A., and Cooper, J. A. (1992). Phosphoryla- tion sites in the PDGF receptor with different specificities for binding GAP and PI3 kinase in vivo. EMBO J. 11, 1373-1382. Konopka, J. B., Watanabe, S. M., and Witte, O. N. (1984). An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell 37, 1035-1042. Kurzrock, R., Gutterman, J. U., and Talpaz, M. (1988). The molecular genetics of Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 319, 990-998. Li, B.-Q., Kaplan, D., Kung, H.-F., and Kamata, T. (1992). Nerve growth factor stimulation of the ras-guanine nucleotide exchange factor and GAP activities. Science 256, 1456-1459. Li, N., Batzer, A., Daly, R., Yajnik, V., Skolnik, E., Chardin, P., Bar- Sagi, D., Margolis, B., and Schlessinger, J. (1993). Guanine- nucleotide-releasing factor hSosl binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 363, 85-87. Liu, J., Campbell, M., Guo, J. Q., Lu, C., Xian, Y. M., Anderson, B. S., and Arlinghaus, R. B. (1993). BCR-ABL tyrosine kinase is auto- phosphorylated or transphosphorylates P160 BCR on tyrosine pre- dominantly within the first BCR exon. Oncogene 8, 101-109. Lowenstein, E. J., Daly, R. J., Batzer, A. G., Li, W., Margolis, B., Lammers, R., UIIrich, A., Skolnik, E. Y., Bar-Sagi, D., and Schles- singer, J. (1992). The SH2 and SH3 domain-containing protein GRB-2 links receptor tyrosine kinases to ras signaling. Cell 70, 431-442.
Lugo, T., and Witte, O. N. (1989). The BCR/ABL oncogene transforms Rat1 cells and cooperates with v-myc. Mol. Cell. Biol. 9, 1263-1270.
Martegani, E., Vanoni, M., Zippel, R., Coccetti, P., Brambilla, R., Fer- rari, C., Sturani, E., and Alberghina, L. (1992). Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a Saccharomyces cerevisiae ras activator. EMBO J. 6, 2151-2157.
Matuoka, K., Shibata, M., Yamakawa, A., and Takenawa, T. (1992). Cloning of ASH, a ubiquitous protein composed of one Src homology region (SH) 2 and two SH3 domains, from human and rat cDNA librar- ies. Proc. Natl. Acad. Sci. USA 89, 9015-9019.
McGlade, J., Cheng, A., Pelicci, G., Pelicci, P. G., and Pawson, T. (1992). Shc proteins are phosphorylated and regulated by the v-Src
and v-Fps protein-tyrosine kinases. Proc. Natl. Acad. Sci. USA 89, 8869-8873.
McLaughlin, J., Chianese, E., and Witte, O. N. (1987). In vitro transfor- mation of immature hematopoietic cells by the P210 BCFUABL onco- gene product of the Philadelphia chromosome. Proc. Natl. Acad. Sci. USA 84, 6558-6562. McLaughlin, J., Chianese, E., and Witte, O. N. (1989). Alternative forms of the BCR/ABL oncogene have quantitatively different poten- cies for stimulation of immature lymphoid cells. Mol. Cell. Biol. g, 1866- 1874. McWhirter, J. R., and Wang, J. Y. J. (1991). Activation of tyrosine kinase and microfilament-binding functions of c-abl by bcr sequences in bcr-abl fusion proteins. Mol. Cell. Biol. 11, 1553-1565. McWhirter, J. R., and Wang, J. Y. J. (1993). An actin-binding function contributes to transformation by the BCR-ABL oncoproteins of Phila- delphia chromosome-positive human leukemias. EMBO J. 12, 1533-- 1546. Medema, R. H., de Vries-Smits, A. M. M., van der Zon, G. C. M., Maassen, J. A., and Bos, J. L. (1993). Ras activation by insulin and epidermal growth factor through enhanced exchange of guanine nu- cleotides on p21 '==. MoI. Cell. Biol. 13, 155-162. Mulcahy, L. S., Smith, M. R., and Stacey, D. W. (1985). Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 313, 241-243. Muller, A. J., Young, J. C., Pendergast, A. M., Pondel, M., Littman, D. R., and Witte, O. N. (1991). BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chro- mosome-positive human leukemias. Mol. Cell. Biol. 11, 1785-1792. Muller, A. J., Pendergast, A. M., Havlik, M. H., Pull, L., Pawson, T., and Witte, O. N. (1992). A limited set of SH2 domains binds BCR through a high-affinity phosphotyrosine-independent interaction. Mol. Cell. Biol. 12, 5087-5093. Olivier, J. P., Raabe, T., Henkemeyer, M., Dickson, B., Mbamalu, G., Margolis, B., Schlessinger, J., Hafen, E., and Pawson, T. (1993). A Drosophila SH2-SH3 adaptor protein implicated in coupling the Sev- enless tyrosine kinase to an activator of Ras guanine nucleotide ex- change, Sos. Cell 73, 179-191. Pelicci, G., Lanfrancone, L., Grignani, F., McGlade, J., Cavallo, F., Forni, G., Nicoletti, I., Grignani, F., Pawson, T., and Pelicci, P. G. (1992)~ A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70, 93-104. Pendergast, A. M., Muller, A. J., Havlik, M. H., Maru, Y., and WiRe, O. N. (1991a). BCR sequences essential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non- phosphotyrosine-dependent manner. Cell 66, 161-171. Pendergast, A. M., Muller, A. J., Havlik, M. H., Clark, R., McCormick, F., and Witte, O. N. (1991 b). Evidence for regulation of the ABL tyrosine kinase by a cellular inhibitor. Proc. Natl. Acad. Sci. USA 88, 5927- 5931. Ren, R., Mayer, B. J., Cicchetti, P., and Baltimore, D. (1993). Identifica- tion of a ten-amino acid proline-rich SH3 binding site. Science 259, 1157-1161. Rosenperg, N., and Witte, O. N. (1988). The viral and cellular forms of the Abelson (abl) oncogene. Adv. Virus Res. 35, 39-61. Rozakis-Adcock, M., McGlade, J., Mbamalu, G., Pelicci, G., Daly, R., Li, W., Batzer, A., Thomas, S., Brugge, J., Pelicci, P. G., Schlessinger, J., and Pawson, T. (1992). Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 360, 689-692. Rozakis-Adcock, M., Fernley, R., Wade, J., Pawson, T., and Bowtell, D. (1993). The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSosl. Nature 363, 83-85. Satoh, T., Endo, M., Nakafuku, M., Nakamura, S., and Kaziro, Y. (1990). Platelet-derived growth factor stimulates formation of active p21"=-GTP complex in Swiss mouse 3T3 cells. Proc. Natl. Acad. ScL USA 87, 5993-5997. Schlessinger, J. (1993). How receptor tyrosine kinases activate Ras. Trends Biochem. Sci. 18, 273-275.

In Vivo BCR-ABL Chimera-GRB-2 Complex 185
Schweighoffer, F., Barlat, I., Chevallier-Multon, M.-C., and Tocque, B. (1992). Implication of GAP in Ras-dependent transactivation of a
polyoma enhancer sequence. Science 256, 825-827. Shou, C., Farnsworth, C. L., Neel, B. G., and Feig, L. A. (1992). Molecu- lar cloning of cDNAs encoding a guanine-nucleotide-releasing factor for Ras p21. Nature 358, 351-354. Simon, M. A., Bowtell, D. D. L., Dodson, G. S., Laverty, T. R., and Rubin, G. M. (1991). Rasl and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the Sevenless protein tyro- sine kinase. Cell 67, 701-716. Simon, M. A., Dodson, G. S., and Rubin, G. M. (1993). An SH3-SH2- SH3 protein is required for p21 ~'=1 activation and binds to Sevenless and Sos proteins in vitro. Cell 73, 169-177. Skolnik, E. Y., Lee, C.-H., Batzer, A., Vicentini, L. M., Zhou, M., Daly, R., Myers, M J., Jr., Backer, J. M., UIIrich, A., White, M. F., and Schlassinger, J. (1993a). The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: implica- tions for insulin control of ras signalling. EMBO J. 12, 1929-1936. Skolnik, E. Y., Batzer, A., Li, N., Lee, C.-H., Lowenstein, E., Moharn- madi, M., Margolis, B., and Schlessinger, J. (1993b). The function of GRB-2 in linking the insulin receptor to Ras signaling pathways. Sci- ence 260, 1953-1955. Smith, M. R., DaGudicibus, S. J., and Stacey, D. W. (1986). Require- ment for c-ras proteins during viral oncogene transformation. Nature 320, 540-543. Songyang, Z., Shoelson, S. E., Chaudhuri, M., Gish, G., Pawson, T., Haser, W. G., King, F., Roberts, T., Ratnofsky, S., Lechleider, R. J., Neel, B. G., Birge, R. B., Fajardo, J, E., Chou, M. M., Hanafusa, H., Schaffhausen, B., and Cantley, L. C. (1993). SH2 domains recognize specific phosphopeptide sequences. Cell 72, 767-778. Thomas, S. M., DeMarco, M., D'Arcangelo, G., Halegoua, S., and Brugge, J. S. (1992). Ras is essential for nerve growth factor- and phorbol ester-induced tyrosine phosphorylation of MAP kinases. Cell 68, 1031-1040. Timmons, M. S., and Witte, O. N. (1989). Structural characterization of the BCR gene product. Oncogene 4, 559-567. Wasylyk, C., Imler, J. L., and Wasylyk, B. (1988). Transforming but not immortalizing oncogenes activate the transcription factor PEA1. EMBO J. 7, 2475-2483. Wasylyk, C., Wasylyk, B., Heidecker, G., Huleihel, M., and Rapp, U. R. (1989). Expression of raf oncogenes activates the PEA1 transcription factor motif. Mol. Cell. Biol, 9, 2247-2250. Wood, K. W., Sarnecki, C., Roberts, T. M., and Blenis, J. (1992). ras mediates nerve growth factor receptor modulation of three signal- transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell 68, 1041-1050.




















