
Deletion analysis of BMI1 oncoprotein identifies its negative regulatory domain
Upload
independentCategory
view
3download
0
The FASEB Journal • Research Communication
BCR-ABL residues interacting with ponatinib are criticalto preserve the tumorigenic potential ofthe oncoprotein
Pietro Buffa,*,1 Chiara Romano,†,1 Alessandro Pandini,*,1 Michele Massimino,†
Elena Tirrò,† Francesco Di Raimondo,† Livia Manzella,† Franca Fraternali,*,2
and Paolo G. Vigneri†,‡,2
*Randall Division of Cell and Molecular Biophysics, King’s College London, London, UK;†Department of Clinical and Molecular Biomedicine, University of Catania, Catania, Italy; and‡Department of Hematology, Oncology, and Molecular Medicine, Istituto Superiore di Sanita,Rome, Italy
ABSTRACT Patients with chronic myeloid leukemiain whom tyrosine kinase inhibitors (TKIs) fail oftenpresent mutations in the BCR-ABL catalytic domain.We noticed a lack of substitutions involving 4 aminoacids (E286, M318, I360, and D381) that form hydro-gen bonds with ponatinib. We therefore introducedmutations in each of these residues, either preservingor altering their physicochemical properties. We foundthat E286, M318, I360, and D381 are dispensable forABL and BCR-ABL protein stability but are critical forpreserving catalytic activity. Indeed, only a “conserva-tive” I360T substitution retained kinase proficiency andtransforming potential. Molecular dynamics simula-tions of BCR-ABLI360T revealed differences in bothhelix �C dynamics and protein-correlated motions,consistent with a modified ATP-binding pocket. Never-theless, this mutant remained sensitive to ponatinib,imatinib, and dasatinib. These results suggest thatchanges in the 4 BCR-ABL residues described herewould be selected against by a lack of kinase activity orby maintained responsiveness to TKIs. Notably, aminoacids equivalent to those identified in BCR-ABL areconserved in 51% of human tyrosine kinases. Hence,these residues may represent an appealing target forthe design of pharmacological compounds that wouldinhibit additional oncogenic tyrosine kinases whileavoiding the emergence of resistance due to pointmutations.—Buffa, P., Romano, C., Pandini. A., Mas-simino, M., Tirrò, E., Di Raimondo, F., Manzella, L.,Fraternali. F., Vigneri, P. G. BCR-ABL residues inter-acting with ponatinib are critical to preserve the tumor-
igenic potential of the oncoprotein. FASEB J. 28,000–000 (2014). www.fasebj.org
Key Words: CML � molecular dynamics � mutations � resis-tance � TKIs
Chronic myeloid leukemia (CML) has historicallyrepresented one of the few “paradigm diseases” in thefield of cancer research (1–3). Indeed, CML was one ofthe first malignancies associated with a known cytoge-netic alteration (i.e., the Philadelphia chromosome)and a specific molecular hallmark (i.e., the BCR-ABLoncoprotein; refs. 4, 5). Furthermore, specific BCR-ABL characteristics (e.g., constitutive tyrosine kinaseactivity and exclusive cytoplasmic localization) havebeen ingeniously targeted to devise innovative thera-peutic strategies that ushered in the era of tailoredcancer treatment (6, 7).
Suppression of BCR-ABL catalytic activity by thetyrosine kinase inhibitor (TKI) imatinib mesylate (IM)has dramatically improved the natural history of CML(5). Despite the unparalleled results achieved with thisdrug, IM treatment fails in �30% of patients with CML(3). To address this critical issue, second- and third-generation TKIs [e.g., dasatinib (DAS) and ponatinib(PON)] were generated and tested on IM-resistantindividuals and showed early clinical success (8, 9).However, more mature data suggest that up to 50% ofpatients receiving either DAS or PON will not have
1 These authors contributed equally to this work.2 Correspondence: P.G.V., Department of Clinical and Mo-
lecular Biomedicine, University of Catania, Via Androne, 85,95124 Catania, Italy. E-mail: [email protected]; F.F., RandallDivision of Cell and Molecular Biophysics, King’s CollegeLondon, SE1 1UL London, UK. E-mail: [email protected]
doi: 10.1096/fj.13-236992This article includes supplemental data. Please visit http://
www.fasebj.org to obtain this information.
Abbreviations: CML, chronic myeloid leukemia; DAS, da-satinib; FBS, fetal bovine serum; HA, hemagglutinin; IL-3,interleukin 3; IM, imatinib mesylate; KD, kinase domain; KI,kinase inactive; MD, molecular dynamics; MOE, MolecularOperating Environment; Ni2�-NTA, Ni-nitrilotriacetic acid;PDB, Protein Data Bank; PMSF, phenylmethylsulfonyl fluo-ride; PON, ponatinib; TKI, tyrosine kinase inhibitor; WT, wildtype
10892-6638/14/0028-0001 © FASEB
The FASEB Journal article fj.13-236992. Published online December 2, 2013.

long-term benefits from these drugs and will progressto the advanced phases of the disease (10, 11).
Multiple mechanisms have been proposed to explainthe failure of TKI treatment. Although this phenome-non can be associated with different causes, pointmutations in the BCR-ABL kinase domain (KD) repre-sent one of the most common reasons underlyingresistance to TKIs (12–15). Substitution of threonine315 with an isoleucine (T315I) represents the mostproblematic BCR-ABL mutation because alterations ofthis gatekeeper residue preserve the kinase activity ofthe oncoprotein while inducing resistance to first- andsecond-generation TKIs (16, 17). Furthermore, recentevidence suggests that the T315I substitution is associ-ated with inferior overall and failure-free survival (18).
Because the mechanisms allowing different BCR-ABLmutants to avoid catalytic inhibition by first-, second-,and third-generation inhibitors are still partially unre-solved, there is a clear need for a structural character-ization of the interactions occurring between the BCR-ABL KD and different TKIs. Experimental measuresand quantification of the delicate balance of differentphysicochemical components are very difficult to ob-tain, and a complete overview of the molecular deter-minants in the inhibitor binding modes is challengingfor this class of flexible proteins. Molecular dynamics
(MD) investigations represent an ideal tool to extractenergetic and dynamic features in binding processesand conformational equilibria. In this respect, compu-tational studies addressing BCR-ABL binding mecha-nisms can be grouped into 3 types of approaches:studies addressing the BCR-ABL KD alone, aimed tocharacterize the equilibrium between the active andinactive conformations (19–21); studies addressingbinding to various inhibitors (22–24); and studies eval-uating the impact of mutations on BCR-ABL kinaseactivity (25–30). However, a comparative investigationof the binding impact modes and dynamics character-izing the interactions between the BCR-ABL kinase anddifferent TKIs is still lacking.
By analyzing all reported BCR-ABL point mutationsassociated with TKI resistance (12–15), we noticed alack of amino acid substitutions involving 4 residues(E286, M318, I360, and D381) that are known togenerate hydrogen bond interactions with PON (ref. 31and Fig. 1A). The same residues (with the addition ofT315) also generate hydrogen bonds with IM (ref. 32and Fig. 1B), whereas T315 and M318 are the onlyamino acids forming hydrogen bonds with DAS (ref. 33and Fig. 1C). We therefore used a combined experi-mental and computational approach to investigate theeffects of different mutations on these amino acids.
Figure 1. Cartoon showing the amino acids within the BCR-ABL catalytic domain that generate hydrogen bonds with differentTKIs. Top panels: structural representation of PON (brown; PDB accession number 3OXZ; A), IM (orange; PDB 2HYY; B) andDAS (blue; PDB 2GQG; C) fitted inside the BCR-ABL KD. Bottom panels: amino acid residues generating hydrogen bonds witheach drug are highlighted with the corresponding color.
2 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org

Our results shed light on the molecular details under-lying the essential role of E286, M318, I360, and D381in preserving BCR-ABL kinase activity and oncogenicpotential. We also outline some of the fingerprints ofthe inhibition mechanisms of PON, IM, and DAS,offering a rationale for PON-dependent inhibition ofall described BCR-ABL point mutations, includingT315I. Because we report that 51% of human tyrosinekinases retain residues equivalent to those forminghydrogen bonds between BCR-ABL and PON, ourresults may aid in the design of inhibitory compoundstargeting additional kinases involved in the develop-ment of other malignancies.
MATERIALS AND METHODS
Mutagenesis and constructs
The BCR-ABL sequence, cloned in the expression vectorpcDNA3.1 (Life Technologies, Grand Island, NY, USA), wasmutagenized using a QuikChange II XL Site-Direct Mutagen-esis Kit (Agilent Technologies, Santa Clara, CA, USA). Incor-poration of each mutation was verified by direct sequencing.Every BCR-ABL construct was then amplified by PCR usingthe indicated forward (FLAG tag underlined) 5=-GACTAGTGCCACCATGGATTACAAGGATGACGAC-GATAAGATGGTGGACCCGGTGGGC-3= and reverse 5=-CGACGCGTCTACCTCTGCACTATGTCACT-3= primers andcloned in the SpeI and MluI unique sites of the pLEXlentiviral vector (Open Biosystems, Lafayette, CO, USA). Togenerate ABL constructs corresponding to the reported na-tive and mutated BCR-ABL, we amplified the first 425 bp ofthe ABL1A coding sequence from HL-60 immortalized cells(DSMZ, Braunschweig, Germany) and substituted this am-plimer for the BCR sequences located upstream of the ABLbreakpoint. PCRs were performed with the indicated forward[hemagglutinin (HA) tag underlined] 5=-CTAGCTAG-CGGATCCATGTACCCATACGATGTTCCAGATTACGCT-ATGTTGGAGATCTGCCTGAAGC-3= and reverse 5=-CTCAG-CAGATACTCAGCGGCAT-3= primers. The resultant PCRproduct was initially cloned in the pcDNA 3.1 vector using theNheI (on the forward primer) and KpnI (in the ABL1Asequence) restriction sites. Each ABL construct was subse-quently subcloned in the BamHI and NotI unique sites of thepLEX vector.
Cell culture, transfection, and lentiviral transduction
Ba/F3 cells (DSMZ) were maintained in RPMI 1640 medium(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 4mM l-glutamine, 50 U/ml penicillin, 50 �g/ml streptomycin(all from Sigma-Aldrich), 10% fetal bovine serum (FBS;Lonza, Basel, Switzerland), and 10% WEHI-3B conditionedmedium, as a source of interleukin 3 (IL-3). WEHI-3B cells(DSMZ) were grown in Iscove’s modified Dulbecco’s medium(Sigma-Aldrich), supplemented with 2 mM l-glutamine, 50U/ml penicillin, 50 �g/ml streptomycin, 10% FBS, and 25�M 2-mercaptoethanol (Sigma-Aldrich). All cells were grownat 37°C in 5% CO2. For the production of the lentiviralsupernatant, transfer vectors (empty vector, native and mu-tant ABL, and BCR-ABL constructs) were cotransfected withthe packaging plasmids in the HEK293T cell line, using thecalcium phosphate transfection method (Trans-LentiviralORF Packaging Kit, TLP4617; Open Biosystems). Viral super-natants were harvested 48 h after transfection. Ba/F3 cells
were then transduced by a double round of spin infection(1200 g at 32°C for 90 min) in the presence of 1 ml of viralsupernatant and 8 �g/ml Polybrene (Sigma-Aldrich). Trans-duced cells were finally selected with 3 �g/ml puromycin(Sigma-Aldrich) for 72 h.
Proliferation assays and drug treatments
Ba/F3 cells (1�104) transduced with empty vector, wild-type(WT) or mutant ABL, or BCR-ABL constructs were plated inquadruplicate in a 96-well plate in the absence of IL-3. Cellproliferation was assessed after 24, 48, and 72 h using anATPlite Luminescence Kit (PerkinElmer, Waltham, MA,USA), following the manufacturer’s instructions. To calculatethe IC50 values for cellular proliferation of different TKIs,Ba/F3 cells transduced with empty vector and WT, T315I, andI360T BCR-ABL were plated in triplicate and incubated for 24h with logarithmic dilutions of PON (Santa Cruz Biotechnol-ogy, Santa Cruz, CA, USA), IM (a gift from Novartis, Basel,Switzerland), and DAS (a gift from Bristol-Myers Squibb,Princeton, NJ, USA). IL-3 was maintained in the culturemedium of Ba/F3 cells transduced with the empty vector.Proliferation was measured using CellTiter 96 AQueous OneSolution Reagent (Promega, Madison, WI, USA). IC50 valueswere calculated using the logistic nonlinear regression in-cluded in GraphPad Prism 4.0a (GraphPad Software Inc., SanDiego, CA, USA) and are reported as the means of 3independent experiments. For evaluation of total tyrosinekinase activity by immunoblots, Ba/F3 cells transduced withBCR-ABLWT, BCR-ABLT315I, or BCR-ABLI360T were treated for24 h with the BCR-ABLWT IC50 concentrations determinedfor PON, IM, and DAS; 70 nM PON was used for experimentsincluding the T315I construct.
Immunoblots
Ba/F3 cells expressing empty vector, native or mutant ABL,or BCR-ABL proteins were starved of IL-3 for 4 h (or 24 hwhen exposed to TKIs) before centrifugation and suspensionin isotonic buffer containing 100 mM NaCl, 25 mM Trizmabase (pH 8.5), 2 mM phenylmethylsulfonyl fluoride (PMSF),and 1� protease inhibitor cocktail (Roche, Mannheim, Ger-many). Proteins were extracted by sonication and analyzed bySDS-PAGE, loading 60 �g of cell lysate/lane. Blots wereprobed using anti-HA (Covance, Princeton, NJ, USA), anti-FLAG (Sigma-Aldrich), anti-phosphotyrosine (clone 4G10;Millipore, Billerica, MA, USA), and anti-actin (Sigma-Aldrich) monoclonal antibodies.
Purification of BCR-ABL proteins and in vitro kinase assays
BCR-ABL constructs cloned in the pLEX vector were modifiedto include 6 histidine codons at the N terminus to allowprotein purification. As a negative control, we generated akinase-inactive (KI) BCR-ABL mutant carrying the K271Hsubstitution (ABL1A numbering) in the ABL KD as describedpreviously (34). The Ni2�-nitrilotriacetic acid (Ni-NTA) Puri-fication System (Life Technologies) was used for proteinpurification under native conditions. BCR-ABL constructswere expressed by transient transfection in HEK293T cellsusing the calcium phosphate method. After 48 h, 1 � 107 cellswere lysed in 8 ml of 1� native binding buffer, supplementedwith 0.5% Triton X-100, 5% glycerol, 0.1 mM PMSF, and 2�g/ml aprotinin. Lysates were loaded onto preequilibratedNi-NTA agarose columns and purified, following the manu-facturer’s protocol. Proteins were then eluted in 6 ml ofnative elution buffer and concentrated up to 100-fold bySpin-X UF concentrators (Corning Life Sciences, Corning,
3CRITICAL BCR-ABL RESIDUES DETERMINE TKI EFFICACY

NY, USA). Concentrations of BCR-ABL proteins were deter-mined by SDS-PAGE analysis and Coomassie blue stainingusing purified bovine serum albumin (Sigma-Aldrich) as acomparative standard, with quantification performed by den-sitometry using ImageJ 1.42q software (U.S. National Insti-tutes of Health, Bethesda, MD, USA).
Kinase activity was detected using the ADP-Glo KinaseAssay (Promega) with an ABL peptide substrate (Abltide;SignalChem, Richmond, ON, Canada). Assays were per-formed in triplicate at room temperature for 1 h, using 12 nMenzyme, 50 �M ATP, and peptide concentrations of 1.56,3.12, 6.25, 12.5, 25, 50, 100, and 200 �M. Luminescencevalues were measured using a Victor Wallac microplate reader(PerkinElmer) and normalized for background ATP to ADPconversion by omitting the peptide substrate from the kinasereaction. Nonlinear regression fits (GraphPad Prism 4.0a)were used to determine maximum rate (Vmax), substratebinding (Km), and Michaelis-Menten kinetics by plottingvelocity as a function of peptide concentration for eachmutant. Velocity values are reported as signal/backgroundratios. The catalytic constant rate (Kcat) was obtained fromthe equation Kcat � Vmax/[enzyme].
Structure preparation, MD simulations, and energeticanalysis
MD simulations were performed on the human WT ABL KDand the T315I, I360T, and I360R mutants. For the ABLWT,ABLT315I, and ABLI360T systems, simulations were also per-formed in complex with PON, IM, and DAS. Starting struc-tures for the mutants were derived by in silico mutagenesiswith MODELLER 9.8 (35). The following structures weredownloaded from the Protein Data Bank (PDB; ref. 36):inactive or “closed” conformation with PON (PDB accessionnumber 3OXZ; ref. 31), inactive conformation with IM (PDB2HYY; ref. 37), and active or “open” conformation with DAS(PDB 2GQG; ref. 33). Unbound forms were generated bymanual removal of the inhibitors. Missing residues wererebuilt with MODELLER 9.8. The GROMACS 4.0 program(38) was used to prepare the initial system coordinates, runthe MD simulations, and analyze the resulting trajectories.Simulations were performed using the ffamber99sb (39)porting of the AMBER parm99 force field (40), the TIP3Pmodel for water (41), and the general AMBER force field(42) with the AM1-BCC charges (43) for the inhibitors. Thesystems were minimized and then gradually heated to 300 Kwith potential restraints on the solute, performing severalsteps of equilibration (44). After equilibration, the resultingstructures were used as a starting point for production of50-ns MD simulations. Secondary structure analysis was per-formed with DSSP (45). The extent of correlated motions wasestimated from the correlation matrices on C� positions, and
the correlations are represented as correlation webs (46).The energetic analysis was performed with the MolecularOperating Environment (MOE) 2012.10. The Pose Rescoringmethod of the Dock module was used to calculate the freeenergy of binding for each simulated system in complex withthe 3 TKIs. Representative structures were extracted by clus-ter analysis (47) from each MD trajectory and validated withthe MOE Structure Preparation module. Average values cal-culated over the representative structures of the 3 largestclusters are reported. The GBVI/WSA dG scoring functionwith the Amber 12EHT force field was used (48). In silicomutagenesis scanning was performed with the Residue Scanmethod of the Protein Design module. The representativestructure of the largest cluster in the MD trajectory of ABLWT
was used. Sequence-based predictions of the effect of eachmutation were performed with the web servers PROVEAN(49), SIFT (50), and PolyPhen-2 (51). Images were generatedwith VMD 1.8.5 (52) and PyMOL 1.2 (Schrödinger Inc.,Portland, OR, USA).
Multiple sequence alignment of tyrosine kinase proteins
KD sequences were downloaded from the Kinase.Comwebsite (http://kinase.com). The human tyrosine kinasesubset (90 sequences) was extracted and aligned usingT-Coffee (53).
RESULTS
PON binds to the inactive conformation of the BCR-ABL catalytic pocket by generating hydrogen bondswith 4 amino acids: E286, M318, I360, and D381 (Fig.1A and ref. 31). Notably, these residues do not undergosubstitutions in patients in whom TKI therapy has failed(12–15). We therefore decided to generate point mu-tations in these sites and investigate their effect on theexpression and kinase activity of both ABL and BCR-ABL. To this end, we engineered 8 ABL and 8 BCR-ABLconstructs displaying 2 alternative substitutions inE286, M318, I360, and D381. The inserted mutationswere deemed conservative if they preserved the physi-cochemical properties of the amino acid found in theWT sequence or nonconservative if they altered thesecharacteristics (Table 1). ABL and BCR-ABL constructsdisplaying the WT sequence or bearing the T315Isubstitution were used as controls.
TABLE 1. Conservative and nonconservative substitutions inserted in the BCR-ABL catalyticdomain
Amino acid ABL position C mutation NC mutation
Glutamic acid (E) 286 Aspartic acid (D) Arginine (R)Methionine (M) 318 Threonine (T) Lysine (K)Isoleucine (I) 360 Threonine (T) Arginine (R)Aspartic acid (D) 381 Glutamic acid (E) Arginine (R)
Letters in parentheses refer to the corresponding single-letter code for each amino acidic residue.Amino acid positions specified above are derived from the human ABL1A protein sequence (NationalCenter for Biotechnology Information accession number NP_005148.2). However, in the article, weattribute these residues to the BCR-ABL sequence maintaining the ABL numbering. C, conservative, NC,nonconservative.
4 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org

E286, M318, I360, and D381 are dispensable for ABLand BCR-ABL protein stability but are critical topreserve BCR-ABL catalytic activity
Because point mutations in E286, M318, I360, andD381 have never been reported in patients with CML inwhom TKIs have failed, we hypothesized that substitu-tions in any of these residues might alter proteinstability. To address this issue, we initially used theabove-indicated ABL constructs that were lentivirallytransduced in the Ba/F3 cell line. We found that allABL mutants were expressed at levels comparable tothose of the WT protein and were devoid of catalyticactivity (Fig. 2A) and transforming potential (Fig. 2B).As expected, the ABLT315I control represented the onlyexception to these findings because this proteinshowed high kinase proficiency, thereby allowingBa/F3 proliferation and survival in the absence of IL-3(ref. 54 and Fig. 2A, B).
Because ABLWT displays very low tyrosine phos-phorylation, we could not investigate the influence ofeach substitution in modulating catalytic activity. Wetherefore repeated the same experiments using thecorresponding BCR-ABL constructs, exploiting theconstitutive kinase activity of the oncoprotein (2).Although again all mutants were expressed at levelscomparable to those of BCR-ABLWT, any substitution
involving residues E286, M318, and D381 completelyabrogated their kinase proficiency. The conservativeI360T substitution was the only variation tested thatwas capable of maintaining BCR-ABL tyrosine phos-phorylation (Fig. 2C). Accordingly, only Ba/F3 cellsexpressing BCR-ABLI360T survived and proliferatedafter IL-3 withdrawal, albeit at lower levels thanthose reported for BCR-ABLWT and BCR-ABLT315I
(Fig. 2D).To further confirm these results, we also performed
an in vitro kinase assay using purified BCR-ABL mutantsand a previously reported synthetic peptide substrate(55). The Michaelis-Menten parameters Km, Vmax, andKcat were determined using nonlinear regression fits. AKI BCR-ABL was included in the experiments as anegative control (34). As expected, 7 of 8 BCR-ABLmutants generated Vmax values similar to those for theKI protein, indicating the complete lack of phosphor-ylation of the synthetic substrate (Fig. 2E and Table 2).On the contrary, BCR-ABLI360T maintained its kinaseactivity, although at lower levels than BCR-ABLWT.However, the low Km value displayed by this mutantsuggests that minimal substrate concentrations are suf-ficient to saturate its catalytic activity (Table 2).
To ascertain the expected effects of additional muta-tions on E286, M318, I360, and D381, we also per-
Figure 2. I360T is the only substitutionthat maintains BCR-ABL catalyticactivity and transforming potential.A) Cell lysates from Ba/F3 cells lenti-virally transduced with HA-tagged ABLconstructs displaying the specified mu-tations were blotted using anti-HA(top panel), anti-phosphotyrosine(middle panel), or anti-actin (bottompanel) antibodies. WT and T315I con-structs were used as controls. Actinlevels confirmed equal protein load-ing in each lane. EV, empty vector. B)Same cells were also used to performproliferation assays for 24, 48, or 72 h.
Numbers represent averages � sd of 3 separate experiments performed in quadruplicate. Results are expressed as relativeluminescence fold increase with cells plated at baseline arbitrarily set at 1. C, D) Identical experiments as in A and B,respectively, were repeated on Ba/F3 cells transduced with FLAG-tagged BCR-ABL mutants that reproduce the amino acidsubstitutions generated for ABL. Anti-FLAG immunoblots confirmed protein expression of each construct. E) In vitro kinasereactions of BCR-ABL mutants were performed in the presence of increasing concentrations of the Abltide syntheticsubstrate. Velocity is plotted vs. peptide concentrations in a Michaelis-Menten graph. KI BCR-ABL was used as a negativecontrol. Velocity values are expressed as relative luminescence fold increase, with background calculated by omittingsubstrate addition in the reaction. Results shown represent averages � sd of 3 separate experiments performed in triplicate.Table 2 shows Km and Vmax values of the different BCR-ABL mutants.
5CRITICAL BCR-ABL RESIDUES DETERMINE TKI EFFICACY

formed in silico screening using 3 sequence-based meth-ods (PROVEAN, SIFT, and PolyPhen-2). The 3predictors consistently classified the majority of thesubstitutions as deleterious (labeled “not tolerated” inSIFT or “damaging” in PolyPhen-2). A few cases werepredicted as deleterious by 2 of 3 methods, and onlyselected substitutions in I360 were suggested as possiblytolerated. Sequence-based methods predict the ex-pected effect on both stability and function. To isolatethe contribution from protein structure destabilizationwe performed an analysis of the mutants using MOE.The differences in free energy of folding (��G) of themutants vs. the WT are also reported in Table 3.Substitutions on position M318 and I360 are generallydestabilizing (��G values of 1 kcal/mol are under-lined in Table 3), whereas changes on E286 and D381do not affect stability, suggesting that they are delete-rious for their effect on function.
Overall, these results offer a solid rationale for theobserved lack of mutations on residues E286, M318,and D381 in patients with CML in whom TKI treatmenthas failed. Indeed, both conservative and nonconserva-tive substitutions of these amino acids would result inloss of BCR-ABL catalytic activity, thus compromising itstransforming potential. In contrast, a conservative sub-stitution in position 360 appeared to preserve BCR-ABLkinase functionality.
MD simulations of BCR-ABL mutants I360T andI360R reveal differences in the helix �C dynamicsand in the protein-correlated motions
In an effort to better understand the structural anddynamic differences between the KDs of the kinase-proficient I360T and the kinase-deficient I360R, weanalyzed the MD trajectories of both BCR-ABL mu-tants and compared them with the WT oncoprotein.In particular, we investigated the correlation betweenthe motions of the different parts of the protein. Thiswas then mapped onto the protein structures usingcorrelation webs, in which residues that move to-gether are connected by a line (Fig. 3, top panels).Correlated motions were previously shown to becritical for the activation of the ABL tyrosine kinasedomain (26).
We found that the correlated motions of the BCR-ABLI360T KD were mostly located at the bottom of thecatalytic pocket. On the contrary, the N terminus ofhelix �C is not coupled in its motion to the N lobe as inthe WT domain. This provides more freedom of mo-tion to helix �C that shifts upward and toward theoutside (Fig. 3A, B, top panels). We then analyzed theconformational changes and stability of helix �C byplotting its time evolution along the sequence. Weobserved that in BCR-ABLI360T helix �C maintained itsstructure despite retaining the modified orientation forthe rest of the simulated time (Fig. 3B, bottom panel,red arrow). BCR-ABLI360R presented a strong decreasein correlated motions compared with those for both theWT oncoprotein and the I360T variant (Fig. 3C, toppanel). The I360R mutant also showed an upward shiftof helix �C that appeared to partially unwind for a 6-nsinterval in the middle of the simulation (Fig. 3C,bottom panel, red arrow).
Overall, our results detected differences in the dy-namics of BCR-ABLI360T and BCR-ABLI360R that couldbe related to the diverse catalytic activity of thesemutants. However, it remained to be establishedwhether different TKIs would successfully abrogate thecatalytic activity of BCR-ABLI360T.
Persistence of some hydrogen bond interactions iscritical for successful inhibition of BCR-ABLI360T
kinase activity by TKIs
To investigate whether PON would suppress BCR-ABLI360T kinase activity, we simulated the KD of thismutant in complex with the drug and analyzed theaverage distance of the residues involved in hydrogenbond interactions during the simulations. BecauseBCR-ABLI360T has never been reported in the litera-ture, we also analyzed this mutant in complex with IMand DAS. Additional simulations were performed withthe KDs of BCR-ABLWT and BCR-ABLT315I used ascontrols. The time evolution of the root mean squaredeviation on the C� atoms indicated that all systemsrapidly reached equilibrium and remained stable dur-ing the simulated time (Supplemental Fig. S1).
PON binds the inactive conformation of the BCR-ABLWT kinase, establishing 4 hydrogen bonds with
TABLE 2. Km and Vmax values of different BCR-ABL mutants obtained from the Michaelis-Men-ten equation by plotting the velocity of the reactions as a function of the substrate concentration
Mutant Km (�M) Vmax (pmol/min) Kcat (min1)
BCR-ABLWT 5.650 � 0.959 2.736 � 0.106 0.228 � 0.008BCR-ABLKI 0.147 � 0.092 1.152 � 0.024 0.096 � 0.002BCR-ABLE286D 0.025 � 0.102 1.094 � 0.030 0.091 � 0.002BCR-ABLE286R 0.00 0.956 � 0.022 0.079 � 0.002BCR-ABLM318T 0.110 � 0.089 1.051 � 0.022 0.087 � 0.002BCR-ABLM318K 0.00 1.079 � 0.028 0.089 � 0.003BCR-ABLI360T 0.039 � 0.083 1.448 � 0.030 0.120 � 0.003BCR-ABLI360R 0.123 � 0.090 1.080 � 0.022 0.090 � 0.001BCR-ABLD381E 0.00 1.005 � 0.025 0.083 � 0.002BCR-ABLD381R 0.041 � 0.095 1.072 � 0.025 0.089 � 0.002
Kcat values were generated from the equation Kcat � Vmax/[enzyme].
6 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org

TABLE 3. In silico prediction of the effects of different mutations on amino acid positions 286, 318, 360, and 381
Position PROVEAN SIFT PolyPhen-2 MOE ��G (kcal/mol)
E286A Deleterious Not tolerated Damaging 2.49E286C Deleterious Not tolerated Damaging 5.72E286D Deleterious Not tolerated Damaging 1.90E286F Deleterious Not tolerated Damaging 3.55E286G Deleterious Not tolerated Damaging 1.39E286H Deleterious Not tolerated Damaging 0.62E286I Deleterious Not tolerated Damaging 3.73E286K Deleterious Not tolerated Damaging 1.13E286L Deleterious Not tolerated Damaging 4.52E286M Deleterious Not tolerated Damaging 4.19E286N Deleterious Not tolerated Damaging 2.47E286P Deleterious Not tolerated Damaging 2.65E286Q Deleterious Not tolerated Damaging 1.88E286R Deleterious Not tolerated Damaging 3.40E286S Deleterious Not tolerated Damaging 3.00E286T Deleterious Not tolerated Possibly damaging 2.40E286V Deleterious Not tolerated Damaging 3.93E286W Deleterious Not tolerated Damaging 2.64E286Y Deleterious Not tolerated Damaging 5.21M318A Deleterious Not tolerated Damaging 1.26M318C Deleterious Not tolerated Damaging 1.45M318D Deleterious Not tolerated Damaging 4.73M318E Deleterious Not tolerated Damaging 5.55M318F Deleterious Not tolerated Damaging 4.01M318G Deleterious Not tolerated Damaging 3.90M318H Deleterious Not tolerated Damaging 9.37M318I Deleterious Not tolerated Damaging 0.26M318K Deleterious Not tolerated Damaging 2.79M318L Deleterious Not tolerated Possibly damaging 0.01M318N Deleterious Not tolerated Damaging 1.38M318P Deleterious Not tolerated Damaging 0.92M318Q Deleterious Not tolerated Damaging 3.55M318R Deleterious Not tolerated Damaging 7.68M318S Deleterious Not tolerated Damaging 2.31M318T Deleterious Not tolerated Damaging 1.88M318V Deleterious Not tolerated Possibly damaging 0.09M318W Deleterious Not tolerated Damaging 5.32M318Y Deleterious Not tolerated Damaging 2.96I360A Deleterious Not tolerated Damaging 0.50I360C Deleterious Not tolerated Damaging 1.60I360D Deleterious Not tolerated Damaging 3.05I360E Deleterious Not tolerated Damaging 1.85I360F Deleterious Not tolerated Damaging 1.18I360G Deleterious Not tolerated Damaging 2.71I360H Deleterious Not tolerated Damaging 3.30I360K Deleterious Not tolerated Damaging 1.49I360L Neutral Not tolerated Possibly damaging 0.03I360M Deleterious Not tolerated Damaging 0.38I360N Deleterious Not tolerated Damaging 1.13I360P Deleterious Not tolerated Damaging 0.38I360Q Deleterious Not tolerated Damaging 2.00I360R Deleterious Not tolerated Damaging 1.83I360S Deleterious Not tolerated Damaging 0.76I360T Deleterious Not tolerated Damaging 1.65I360V Neutral Tolerated Possibly damaging 0.10I360W Deleterious Not tolerated Damaging 1.51I360Y Deleterious Not tolerated Damaging 0.26D381A Deleterious Not tolerated Damaging 2.34D381C Deleterious Not tolerated Damaging 4.91D381E Deleterious Not tolerated Damaging 0.36D381F Deleterious Not tolerated Damaging 5.21D381G Deleterious Not tolerated Damaging 1.47D381H Deleterious Not tolerated Damaging 1.51
(continued on next page)
7CRITICAL BCR-ABL RESIDUES DETERMINE TKI EFFICACY

E286, M318, I360, and D381 (ref. 31 and Fig. 4A).These interactions remained stable during the simula-tion with the exception of the transient binding withE286 (Fig. 4A). Because PON does not form hydrogen
bonds with threonine 315, its mutation to isoleucineshould not affect drug binding (56). Indeed, in ourBCR-ABLT315I-PON system, the drug maintained persis-tent interactions with M318, I360, and D381, while
TABLE 3. (continued)
Position PROVEAN SIFT PolyPhen-2 MOE ��G (kcal/mol)
D381I Deleterious Not tolerated Damaging 4.73D381K Deleterious Not tolerated Damaging 0.91D381L Deleterious Not tolerated Damaging 4.72D381M Deleterious Not tolerated Damaging 4.16D381N Deleterious Not tolerated Damaging 2.58D381P Deleterious Not tolerated Damaging 6.07D381Q Deleterious Not tolerated Damaging 2.33D381R Deleterious Not tolerated Damaging 1.69D381S Deleterious Not tolerated Damaging 3.11D381T Deleterious Not tolerated Damaging 2.68D381V Deleterious Not tolerated Damaging 4.50D381W Deleterious Not tolerated Damaging 3.26D381Y Deleterious Not tolerated Damaging 4.66
Values of ��G 1 kcal/mol are underlined.
Figure 3. I360T and I360R kinase domains display differences in correlated motions and helix �C dynamics compared with those forABL WT. Correlation webs calculated from 50-ns production runs for ABLWT (A), ABLI360T (B), and ABLI360R (C) KDs are shownas a cartoon representation with the DFG motif indicated in red and the activation loop in blue. Residues with correlated motions areconnected by a line. Only correlations 0.6 were reported. The time evolution of the secondary structure elements during the MDsimulations is shown below each cartoon. The simulation time is reported on the x axis, and the protein residue index is specified onthe y axis. Residues belonging to helix �C are designated by a red arrow. Secondary structures were attributed with DSSP (45):�-helixes (blue), �-sheets (red), turns (yellow), bends (green), B-bridges (black), coils (white), and 3-helixes (gray).
8 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org
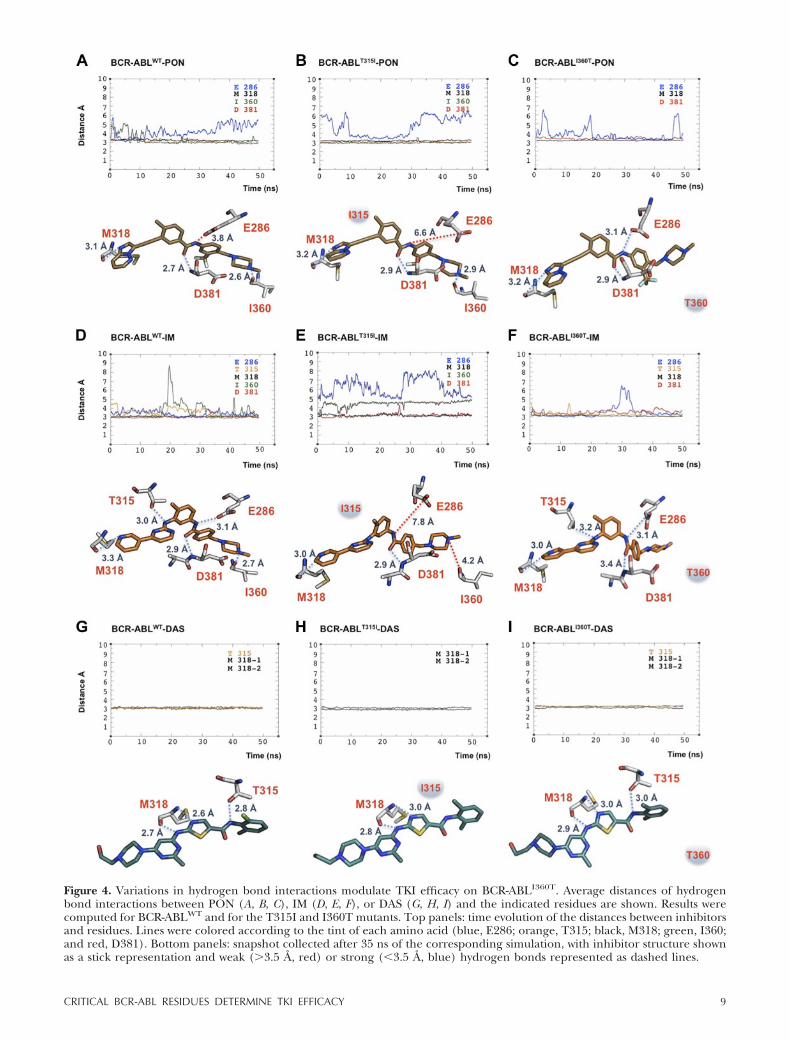
Figure 4. Variations in hydrogen bond interactions modulate TKI efficacy on BCR-ABLI360T. Average distances of hydrogenbond interactions between PON (A, B, C), IM (D, E, F), or DAS (G, H, I) and the indicated residues are shown. Results werecomputed for BCR-ABLWT and for the T315I and I360T mutants. Top panels: time evolution of the distances between inhibitorsand residues. Lines were colored according to the tint of each amino acid (blue, E286; orange, T315; black, M318; green, I360;and red, D381). Bottom panels: snapshot collected after 35 ns of the corresponding simulation, with inhibitor structure shownas a stick representation and weak (3.5 Å, red) or strong (�3.5 Å, blue) hydrogen bonds represented as dashed lines.
9CRITICAL BCR-ABL RESIDUES DETERMINE TKI EFFICACY

again binding to E286 was transient (Fig. 4B). More-over, the linear ethynyl linker of PON helped thecompound to skirt the steric hindrance phenomenoncaused by I315. In the BCR-ABLI360T-PON system, lossof the hydrogen bond with I360 coupled with the weakinteraction with E286 did not affect the ability of thedrug to bind this mutant (Fig. 4C). Hence, our simula-tions suggested that PON should effectively suppressthe catalytic activity of the I360T mutant.
Like PON, IM binds the BCR-ABL KD in the inactiveconformation, generating hydrogen bond interactionswith the 4 residues described above (E286, M318, I360,and D381) plus the gatekeeper threonine in position315 (32, 57; Fig. 4D). Our analysis showed that theseinteractions persisted during the entire BCR-ABLWT
simulation, with small oscillations of the tail of the drugthat caused one short increase of the distance fromI360 in the middle of the simulated time (Fig. 4D). Inthe BCR-ABLT315I-IM system, the drug adapted to themutagenized catalytic pocket owing to a slightly out-ward displacement, thus avoiding the increased sterichindrance caused by the isoleucine side chain. Thesechanges can be recognized by comparing the differ-ences in IM structure depicted in Fig. 4D, E. This spatialrearrangement alleviated the clash of bulky residues.However, in the simulation, the drug lost 3 of 5hydrogen bond interactions, compromising optimalprotein binding. Indeed, aside from the loss of thehydrogen bond with threonine 315 because of itssubstitution with isoleucine, we observed displacementsof the interactions with E286 and I360 (Fig. 4E).Analysis of BCR-ABLI360T in complex with IM revealedthat all hydrogen bond interactions previously shownfor the WT system were maintained over time, with theexception of the interaction with the mutagenized I360residue. Insertion of a threonine at this site caused aslight change in the conformation of the activationloop, resulting in a missing hydrogen bond with the tailof the drug (Fig. 4F). However, IM binding appearedstable despite this alteration.
Unlike PON, DAS binds the BCR-ABL catalyticpocket in the active conformation, forming 3 hydrogenbond interactions: 1 with T315 and 2 with M318 (ref. 33and Fig. 4G). Hydrogen bond analysis of BCR-ABLWT incomplex with DAS showed that these 3 points ofcontact were preserved over the entire simulation (Fig.4G). Both biological and clinical evidence has demon-strated that DAS is unable to inhibit the T315I gate-keeper mutation (58, 59). Simulation of BCR-ABLT315I
in complex with DAS showed the expected loss of thecritical interaction with I315 (ref. 33 and Fig. 4H). Inthis case, the hydrophobic pocket of the BCR-ABL KDplays a key role in anchoring the phenyl ring of DASthrough hydrophobic interactions with several aminoacids. Four residues (M290, V299, I313, and F382), alllocated in this region (33), showed an increased dis-tance from the inhibitor in the BCR-ABLT315I-DASsystem, becoming unavailable for hydrophobic interac-tions (Supplemental Fig. S2). Thus, the loss of DASbinding to BCR-ABLT315I can mainly be attributed to
changes in the hydrophobic anchoring of the drug.However, DAS did not lose any hydrogen bond inter-actions with BCR-ABLI360T, implying effectiveness ofthis drug in inhibiting the kinase activity of the I360Tmutant (Fig. 4I).
In addition to the MD simulations, we also estimatedthe free energy of binding (�G) for each of the TKIswith the simulated systems. The �G values were calcu-lated with MOE and are reported in Table 4, togetherwith their corresponding pKi values. The results aregenerally consistent with our simulations: PON showseffective binding in all 3 systems, DAS has decreasedability to bind BCR-ABLT315I, and IM maintains strongbinding to BCR-ABLI360T. The apparent inconsistencyof the values for T315I:IM and I360T:DAS can beexplained, considering that the approximate free en-ergy calculation included in MOE cannot accuratelydescribe contributions from significant conformationalchanges in the oncoprotein. From previous studies(54), we know that this is the case for the destabilizationof the interaction of the gatekeeper mutant with IM. Itis possible that conformational changes also play a rolein modulating DAS binding affinity for the previouslyuncharacterized I360T mutant.
In summary, our computational analyses suggest thatBCR-ABLI360T should be responsive to PON, IM, andDAS.
PON, IM, and DAS inhibit BCR-ABLI360T kinaseactivity, suppressing its transforming potential
To ascertain the validity of our computational analysison BCR-ABLI360T, we initially exposed Ba/F3 cellsexpressing this construct to increasing concentrationsof PON, IM, and DAS after IL-3 deprivation. Calcula-tion of the IC50 values for cellular proliferation inresponse to each TKI confirmed our computationalpredictions, because the I360T mutant was sensitive toeach drug, displaying IC50 rates inferior to those for theWT oncoprotein (Fig. 5, left panels, and Table 5). Wenext analyzed total tyrosine phosphorylation levels aftera 24-h exposure to the 3 inhibitors and found thatBCR-ABLI360T catalytic activity was efficiently sup-pressed by each TKI (Fig. 5, right panels). Finally, ourexperiments also confirmed that PON was the onlyeffective compound for the treatment of BCR-ABLT315I
(Fig. 5A), whereas the WT oncoprotein was responsiveto all TKIs.
TABLE 4. Binding free energies and inhibition constants for thedifferent TKIs bound to the simulated mutants
Mutant
�G (kcal/mol) pKi
PON IM DAS PON IM DAS
BCR-ABLWT 13.8 13.5 12.5 10.0 9.8 9.1BCR-ABLT315I 13.4 14.0 10.0 9.8 10.2 7.3BCR-ABLI360T 13.1 16.0 10.6 9.5 11.7 7.7
pKi is calculated as log Kd, where Kd is the dissociation constantderived from the corresponding �G of binding.
10 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org

Thus, PON, IM, and DAS all successfully inhibitedBCR-ABLI360T oncogenic activity, as predicted by thehydrogen bond interactions analysis (Fig. 4). Thesedata also explain why the I360T substitution has neverbeen observed in patients with CML unresponsive toTKIs because, despite the transforming potential of thismutant, BCR-ABLI360T remains sensitive to multiplekinase inhibitors.
DISCUSSION
More than 100 point mutations involving more than 60amino acid residues have been associated with TKIresistance in patients with CML (12–15). However, nostudy has ever reported amino acid substitutions involv-ing E286, M318, I360, and D381, 4 residues of BCR-
ABL kinase that generate hydrogen bond interactionswith PON (31). In this article, we present biological andcomputational data demonstrating that these 4 aminoacids play a pivotal role in preserving BCR-ABL trans-forming activity. Indeed, our results suggest that, withthe exception of the I360T substitution, both conserva-tive and nonconservative mutations in E286, M318,I360, and D381 abolish BCR-ABL kinase activity. Thesefindings explain the lack of point mutations in these 4amino acid residues, because any change in these siteswould be selected against by either the lack of catalyticactivity or maintained responsiveness to TKI treatment,as we observed with BCR-ABLI360T. These findings arealso corroborated by sequencing data derived from 95patients with CML in whom IM treatment either failedor achieved suboptimal responses according to the2009 European Leukemia Net criteria (60). In theseanalyses, we detected the E286K mutation in 1 subject,3 substitutions at the M318 residue (M318T-V-I) in 3patients, and 3 amino acid replacements at the I360residue (I360T-L-S) in 5 additional patients. All substi-tutions were found in a small percentage of clones(�10%), and none were detected when the samepatients were sequenced at a later time (unpublishedresults).
Because the I360T mutation preserved BCR-ABLcatalytic activity, we investigated this amino acid substi-tution from a computational standpoint, comparing itwith the corresponding nonconservative I360R muta-tion devoid of kinase activity. MD simulations of these 2mutants revealed major differences in correlated mo-tions and in the dynamics of helix �C compared withthose for WT BCR-ABL. I360T displayed strong corre-lations in the region between the N and C lobes of theBCR-ABL catalytic region. We also detected a signifi-cant upward shift of helix �C, which is allowed morefreedom and is partially displaced from the core of thedomain, with an ensuing tendency to widen the accessto the binding cavity. These results suggest a modifiedarrangement of the ATP pocket that could compromiseATP binding and would be compatible with the re-duced kinase activity of this mutant. More dramaticchanges were detected in the dynamics of helix �C inBCR-ABLI360R because in this mutant the helix is looserand has a tendency to unwind. In addition, the domainhas lost the interlobe communication known to becritical for activation (26). This is in agreement with thedrastic reduction in kinase activity observed for thismutant. In addition, the hydrogen bond interaction
TABLE 5. Average IC50 values derived from 3 independent exper-iments performed in triplicate
Mutant
IC50 (nM)
PON IM DAS
Empty vector 1131.00 8392.00 1000.00BCR-ABLWT 3.13 1439.00 5.83BCR-ABLT315I 72.23 6475.00 1000.00BCR-ABLI360T 0.53 21.15 0.69
Figure 5. BCR-ABLI360T catalytic activity is suppressed bymultiple TKIs. To calculate IC50 values for PON (A), IM (B),and DAS (C), Ba/F3 cells transduced with empty vector (EV;diamonds), BCR-ABLWT (squares), BCR-ABLT315I (triangles)or BCR-ABLI360T (circles) were treated for 24 h with increas-ing concentrations of the specified TKIs. Values shown areexpressed as fold decrease, with untreated cells arbitrarily setat 100 (left panels). To verify BCR-ABL inhibition, cell lysatesfrom Ba/F3 cells expressing the indicated mutants wereblotted using an anti-phosphotyrosine (p-Tyr) antibody (rightpanels). Table 5 shows average IC50 values.
11CRITICAL BCR-ABL RESIDUES DETERMINE TKI EFFICACY

analyses followed by biochemical and survival assaysdemonstrated that PON, IM, and DAS successfullyinhibit BCR-ABLI360T.
Hydrogen bond analyses also contributed to explain-ing the structural reasons underlying the resistance ofthe T315I gatekeeper mutation to IM and DAS. Thecommon hypotheses advocated to explain the failure ofIM and DAS to suppress BCR-ABLT315I is that thissubstitution mediates drug resistance through a sterichindrance mechanism (61). However, by studying thedynamics of the inhibitor bound systems in silico, wefound that a slightly outward displacement of both IMand DAS allowed these drugs to adapt to the T315Icatalytic pocket, thus circumventing the increasedsteric hindrance caused by the bulky isoleucine sidechain (Fig. 4D, E). Indeed, the steric hindrance gener-ated by the T315I mutation does not lead to completeobstruction of the binding cavity, and the flexibility ofthe drug structure allows for a suitable rearrangementaround the bulkier side chain, unlike cases in whichmore rigid molecules cannot fit into partially (or to-tally) occluded pockets. This is generally not visiblefrom a single experimental structure, and simulation ofthe system is required to estimate the degree of flexi-bility of both drug and protein. We therefore investi-gated other mechanisms contributing to the lack ofefficacy of IM and DAS on BCR-ABLT315I. In accor-dance with previous reports, we found that loss ofhydrogen bonds between IM and E286, T315 (replacedby an isoleucine), and I360 in the BCR-ABLT315I KDplayed a pivotal role in IM failure against the T315Imutation (30). However, loss of hydrogen bonds wasnot sufficient to explain BCR-ABLT315I resistance toDAS because only the interaction between T315 andDAS was compromised in the T315I mutant simula-tions. On further investigation, we detected a perturba-tion in the dynamics of amino acid residues that formhydrophobic interactions with the drug. These altera-tions are likely to factor heavily in the lack of DASbinding to the T315I mutant.
Further computational investigations of BCR-ABLT315I revealed a net enhancement of correlatedmotions within the N and C lobes compared with thosefor the WT oncoprotein (Fig. 6A, B). The mutationaffects the dynamics of the 2 lobes modifying the accessto and the shape of the binding site. The correlationsalso involve helix �C (which undergoes a partial un-winding) and the activation loop, suggesting that theT315I mutation dynamically perturbs several structuralelements crucially involved in drug binding (Fig. 6B).This observation is in line with previous suggestionsabout the main role of gatekeeper mutations in orches-trating effective communication between the 2 lobes(26) and in stabilizing the hydrophobic spine (17, 54).Simulation of the BCR-ABLT315I-PON system showedthat, aside from the effects ascribed to the ethynyllinker in bypassing the steric hindrance (31), 3 pivotalfeatures enabled this drug to inhibit BCR-ABLT315I:PON showed intermittent interactions with E286,which is often dislocated from its natural position whenhelix �C is destabilized, as in the WT domain; the drugmaintained the same hydrogen bond interactions andthe same average distance with all residues involved inhydrophobic contacts in the BCR-ABLWT-PON simula-tion; and there were only minor differences betweenthe BCR-ABLWT-PON and BCR-ABLT315I-PON systemsin terms of correlated motions (Fig. 6C, D), implyingthat PON seems to “revert” the dynamics of the T315Imutant to those of BCR-ABLWT.
The critical issue stemming from our results con-cerns the implications of the reported findings forfuture therapeutic developments. Patients with CMLunresponsive to TKI treatment sometimes developpoint mutations involving multiple residues within theBCR-ABL KD (12–15, 62). It should be emphasized thatamino acid substitutions engaging different residues ofa catalytic domain are not a prerogative of the ABLkinase, because identical phenomena (including muta-tions of the gatekeeper threonines) have been reportedin patients with lung cancer or gastrointestinal stromal
Figure 6. Ponatinib reverts the dynamics of the BCR-ABLT315I catalytic domain to those of the WT oncoprotein. Correlationwebs were calculated over 50-ns production runs and are reported on the cartoon representation of BCR-ABLWT (A) andBCR-ABLT315I (B), alone or complexed (C, D) with PON. Only correlations 0.6 were included.
12 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org

tumors in whom treatment with epidermal growthfactor receptor or c-KIT inhibitors, respectively, hasfailed (63, 64). Two different approaches have beenpursued to address these problems. Several companieshave synthesized irreversible TKIs that, unlike revers-ible inhibitors (e.g., IM), generate covalent bonds withspecific amino acid residues (usually cysteines but alsoserines, threonines, or lysines) of their substrates. Al-though highly effective, irreversible TKIs have oftenbeen associated with increased (sometimes limiting)toxicities that have hindered their clinical development(65). An alternative approach has been to designcompounds that do not interact with the critical gate-keeper residue of the substrate kinase (66). The caveatof this method is that, theoretically, amino acid substi-tutions in residues other than the gatekeeper couldinduce resistance to these drugs. However, to date, nosingle point mutations have been associated with resis-tance to PON. Our data suggest that the lack ofmutations causing PON failure derives from the factthat this inhibitor forms hydrogen bonds with 4 criticalresidues (E286, M318, I360, and D381) of the BCR-ABLKD that, if mutated, would compromise the transform-ing ability of the oncoprotein. Hence, changes in any ofthe above-mentioned 4 residues would be selectedagainst by a lack of kinase activity or by maintainedresponsiveness to TKIs (BCR-ABLI360T). Notably, whenwe analyzed the 12 known PON biological targets (9,67), we found that these tyrosine kinases conserved theABL E, I/V, and D residues but displayed differentsubstitutions in place of M318 (Table 6). These resultssuggest a structural scenario in which E286, M318, I360,and D381 are “untouchable” residues for the ABLkinase, but only E, I, and D are critical for the remain-ing PON targets because the methionine in position318 can be replaced by either an alanine or a cysteine.
A remarkably high number (46 of 90, 51%) of
human tyrosine kinases including both receptor andnonreceptor proteins display residues equivalent toABL E286, M318, I360, and D381 (Table 7). Of these46 tyrosine kinases, 42 were tested for PON responsive-ness in a previously published in vitro kinase assay and25 of 42 (59%) were efficiently inhibited by the drug(IC50 �20 nM; ref. 9). Although we cannot offer a
TABLE 7. List of 46 human tyrosine kinases displaying residuesequivalent to E286, M318, I360, and D381 identified in theBCR-ABL KD
Kinase
Amino acid and ABL
E: 286 M: 318 IV: 1360 D: 381
ALK 1167 1199 I1246 1270ARG 332 364 I406 427BLK 284 315 I357 378BRK 235 267 I309 330CSK 236 269 V311 332DDR1 672 704 V763 784DDR2 625 657 V707 728EGFR 762 793 V834 855EphA1 673 705 V746 767EphA2 663 695 V736 757EphA3 670 702 V743 764EphA4 670 702 V743 764EphA5 724 756 V797 818EphA6 679 753 V794 815EphA7 682 714 V755 776EphA8 684 716 V757 778EphB1 668 700 V741 762EphB2 670 702 V743 764EphB3 682 714 V755 776EphB4 664 696 V737 758ErbB2 770 801 V842 863ErbB4 768 799 V840 861FGR 306 337 I379 400FRK 277 309 I351 372FYN 314 345 I387 408HCK 305 336 I378 399IGF1R 1050 1082 V1132 1153INSR 1074 1106 V1156 1176IRR 1030 1062 V1112 1133ITK 406 438 I479 500LCK 288 319 I361 382LTK 561 593 I640 664LYN 290 322 I364 385MET 1127 1160 V1201 1222MUSK 626 658 V722 743RON 1131 1164 V1205 1226ROS 1997 2029 I2076 2102SRC 312 343 V385 406TEC 413 445 I486 507TRKA 560 592 V647 668TRKB 588 620 V673 694TRKC 588 620 V673 694TXK 314 346 I387 408TYR03 568 606 I652 673YES 320 351 I393 414Zap70 386 416 V458 479
The isoleucine in position 360 is replaced by a valine in 29 of 46tyrosine kinases. The reported proteins were efficiently inhibited byPON (IC50 �20 nM) in a previously published in vitro kinase assay,with the exception of the underlined kinases (IC50 20 nM). Proteinsin italics have not been tested.
TABLE 6. List of the 12 known PON targets, summarizing theposition of the conserved residues in the specified protein kinases
Kinase
Amino acid and ABL
E: 286 M: 318 I/V: 1360 D: 381
FGFR1 531 A564 I620 641FGFR2 534 A567 I623 644FGFR3 525 A558 I614 635FGFR4 520 A553 I609 630FLT3 661 C694 V808 829KIT 640 C673 I789 810PDGFRa 644 C677 V815 836PDGFRb 651 C684 V823 844RET 775 A807 V87 892VEGFR1 878 C912 I1019 1040VEGFR2 885 C919 I1025 1046VEGFR3 896 C930 I1034 1055
An alanine or a cysteine is always found in place of BCR-ABLM318. The isoleucine is replaced by a valine in 4 of 12 proteins. Thisconservative substitution does not influence the interaction with theinhibitor, because the hydrogen bond generated with PON involvesthe backbone oxygen of I360 and can be reproduced by the equiva-lent oxygen atom in the valine backbone.
13CRITICAL BCR-ABL RESIDUES DETERMINE TKI EFFICACY

detailed explanation as to why PON lacks efficacy onthe remaining 17 kinases, several observations shouldbe kept in mind: although a sequence alignment sug-gests that E, M, I, and D residues are preserved in thesetyrosine kinases, a large-scale MD study is necessary toconfirm that these amino acids conserve a role inprotein flexibility similar to that for the ABL KD, in linewith recent evidence that selective inhibition of ty-rosine kinases could also be connected to differences intheir intrinsic dynamics (19, 22); additional residueswithin the ABL KD are likely to play pivotal roles inregulating both BCR-ABL kinase activity and interac-tion with different TKIs (e.g., our analyses suggest amajor role for alterations in hydrophobic interactionsin negatively modulating DAS binding to BCR-ABLT315I); because our current structural informationis mostly limited to the catalytic domain of multipletyrosine kinases, we cannot account for the significantmodifications that other portions of the protein mayexert on the catalytic pocket, heavily affecting PONbinding; and likewise, the shape and orientation of thedrug-binding pocket of each tyrosine kinase may pres-ent subtle differences that could partially or completelyabrogate its interaction with PON. Hence, preservationof E, M, I, and D residues in positions equivalent tothose of the ABL KD appears to be necessary but notsufficient to predict PON binding to other tyrosinekinases.
In summary, we report here the identification of 4amino acid residues (E286, M318, I360, and D381)that are critical for BCR-ABL catalytic activity andleukemogenic potential. We also find that theseamino acids are conserved in several human tyrosinekinases and are likely to represent unmodifiableresidues that contribute to the proper structuralfolding and enzymatic activity of these proteins.Small molecule inhibitors targeting these amino ac-ids in other tyrosine kinases might be of great clinicalinterest to avoid the emergence of resistance becauseof point mutations.
The authors thank Maria Stella Pennisi, Stefania Stella, andSilvia Rita Vitale for critical reading of the manuscript. Thiswork was supported by an investigator grant to P.G.V. fromthe Associazione Italiana per la Ricerca sul Cancro (AIRC)and by funding from the Biotechnology and Biological Sci-ences Research Council (BB/I023291/1 and BB/H018409/1to A.P. and F.F.). P.B. is the recipient of an AIRC-Marie Curiefellowship.
REFERENCES
1. Goldman, J. M., and Melo, J. V. (2003) Chronic myeloidleukemia—advances in biology and new approaches to treat-ment. N. Engl. J. Med. 349, 1451–1464
2. Melo, J. V., and Barnes, D. J. (2007) Chronic myeloid leukaemiaas a model of disease evolution in human cancer. Nat. Rev.Cancer 7, 441–453
3. Deininger, M., O’Brien, S. G., Guilhot, F., Goldman, J. M.,Hochhaus, A., Hughes, T. P., Radich, J. P., Hatfield, A. K.,Mone, M., Filian, J., Reynolds, J., Gathmann, I., Larson, R. A.,and Druker, B. J. (2009) International Randomized Study of
Interferon vs STI571 (IRIS) 8-year follow up: sustained survivaland low risk for progression or events in patients with newlydiagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood (ASH Annu. Meet. Abstr.) 114,1126
4. Kantarjian, H. M., Talpaz, M., Giles, F., O’Brien, S., and Cortes,J. (2006) New insights into the pathophysiology of chronicmyeloid leukemia and imatinib resistance. Ann. Intern. Med. 145,913–923
5. Druker, B. J. (2008) Translation of the Philadelphia chromo-some into therapy for CML. Blood 112, 4808–4817
6. Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal,G. M., Fanning, S., Zimmermann, J., and Lydon, N. B. (1996)Effects of a selective inhibitor of the Abl tyrosine kinase on thegrowth of Bcr-Abl positive cells. Nat. Med. 2, 561–566
7. Aloisi, A., Di Gregorio, S., Stagno, F., Guglielmo, P., Mannino,F., Sormani, M. P., Bruzzi, P., Gambacorti-Passerini, C., Saglio,G., Venuta, S., Giustolisi, R., Messina, A., and Vigneri, P. (2006)BCR-ABL nuclear entrapment kills human CML cells: ex vivostudy on 35 patients with the combination of imatinib mesylateand leptomycin B. Blood 107, 1591–1598
8. Shah, N. P., Tran, C., Lee, F. Y., Chen, P., Norris, D., andSawyers, C. L. (2004) Overriding imatinib resistance with anovel ABL kinase inhibitor. Science 305, 399–401
9. O’Hare, T., Shakespeare, W. C., Zhu, X., Eide, C. A., Rivera,V. M., Wang, F., Adrian, L. T., Zhou, T., Huang, W.-S., Xu, Q.,Metcalf, C. A., Tyner, J. W., Loriaux, M. M., Corbin, A. S.,Wardwell, S., Ning, Y., Keats, J. A., Wang, Y., Sundaramoorthi,R., Thomas, M., Zhou, D., Snodgrass, J., Commodore, L.,Sawyer, T. K., Dalgarno, D. C., Deininger, M. W. N., Druker,B. J., and Clackson, T. (2009) AP24534, a pan-BCR-ABL inhib-itor for chronic myeloid leukemia, potently inhibits the T315Imutant and overcomes mutation-based resistance. Cancer Cell16, 401–412
10. Jabbour, E., Jones, D., Kantarjian, H. M., O’Brien, S., Tam, C.,Koller, C., Burger, J. A., Borthakur, G., Wierda, W. G., andCortes, J. E. (2009) Long-term outcome of patients with chronicmyeloid leukemia treated with second-generation tyrosine ki-nase inhibitors after imatinib failure is predicted by the in vitrosensitivity of BCR-ABL kinase domain mutations. Blood 114,2037–2043
11. Cortes, J. E., Kantarjian, H., Shah, N. P., Bixby, D., Mauro, M. J.,Flinn, I., O’Hare, T., Hu, S., Narasimhan, N. I., Rivera, V. M.,Clackson, T., Turner, C. D., Haluska, F. G., Druker, B. J.,Deininger, M. W., and Talpaz, M. (2012) Ponatinib in refractoryPhiladelphia chromosome-positive leukemias. N. Engl. J. Med.367, 2075–2088
12. Apperley, J. F. (2007) Part I: mechanisms of resistance toimatinib in chronic myeloid leukaemia. Lancet Oncol. 8, 1018–1029
13. Jabbour, E., Branford, S., Saglio, G., Jones, D., Cortes, J. E., andKantarjian, H. M. (2011) Practical advice for determining therole of BCR-ABL mutations in guiding tyrosine kinase inhibitortherapy in patients with chronic myeloid leukemia. Cancer 117,1800–1811
14. Quintas-Cardama, A., Kantarjian, H., and Cortes, J. (2009)Imatinib and beyond—exploring the full potential of targetedtherapy for CML. Nat. Rev. Clin. Oncol. 6, 535–543
15. Ernst, T., and Hochhaus, A. (2012) Chronic myeloid leukemia:clinical impact of BCR-ABL1 mutations and other lesions asso-ciated with disease progression. Semin. Oncol. 39, 58–66
16. Gorre, M. E., Mohammed, M., Ellwood, K., Hsu, N., Paquette,R., Rao, P. N., and Sawyers, C. L. (2001) Clinical resistance toSTI-571 cancer therapy caused by BCR-ABL gene mutationor amplification. Science 293, 876–880
17. Gibbons, D. L., Pricl, S., Kantarjian, H., Cortes, J., and Quintás-Cardama, A. (2012) The rise and fall of gatekeeper mutations?The BCR-ABL1 T315I paradigm. Cancer 118, 293–299
18. Nicolini, F. E., Ibrahim, A. R., Soverini, S., Martinelli, G., Muller,M. C., Hochhaus, A., Dufva, I. H., Kim, D. W., Cortes, J., Mauro,M. J., Chuah, C., Labussiere, H., Morisset, S., Roche-Lestienne,C., Lippert, E., Hayette, S., Peter, S., Zhou, W., Maguer-Satta, V.,Michallet, M., Goldman, J., Apperley, J. F., Mahon, F. O., Marin,D., and Etienne, G. (2013) The BCR-ABLT315I mutation com-promises survival in chronic phase chronic myelogenous leuke-mia patients resistant to tyrosine kinase inhibitors, in a matchedpair analysis. Haematologica 98, 1510–1516
14 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org

19. Lovera, S., Sutto, L., Boubeva, R., Scapozza, L., Dölker, N., andGervasio, F. L. (2012) The different flexibility of c-Src and c-Ablkinases regulates the accessibility of a druggable inactive con-formation. J. Am. Chem. Soc. 134, 2496–2499
20. Shan, Y., Seeliger, M. A., Eastwood, M. P., Frank, F., Xu, H.,Jensen, M. Ø., Dror, R. O., Kuriyan, J., and Shaw, D. E. (2009)A conserved protonation-dependent switch controls drug bind-ing in the Abl kinase. Proc. Natl. Acad. Sci. U. S. A. 106, 139–144
21. Levinson, N. M., Kuchment, O., Shen, K., Young, M. A.,Koldobskiy, M., Karplus, M., Cole, P. A., and Kuriyan, J. (2006)A Src-like inactive conformation in the abl tyrosine kinase do-main. PLoS Biol. 4, e144
22. Aleksandrov, A., and Simonson, T. (2010) Molecular dynamicssimulations show that conformational selection governs thebinding preferences of imatinib for several tyrosine kinases. J.Biol. Chem. 285, 13807–13815
23. Verkhivker, G. M. (2007) In silico profiling of tyrosine kinasesbinding specificity and drug resistance using Monte Carlosimulations with the ensembles of protein kinase crystal struc-tures. Biopolymers 85, 333–348
24. Lin, Y. L., Meng, Y., Jiang, W., and Roux, B. (2013) Explainingwhy Gleevec is a specific and potent inhibitor of Abl kinase. Proc.Natl. Acad. Sci. U. S. A. 110, 1664–1669
25. Dixit, A., and Verkhivker, G. M. (2009) Hierarchical modelingof activation mechanisms in the ABL and EGFR kinase domains:thermodynamic and mechanistic catalysts of kinase activation bycancer mutations. PLoS Comput. Biol. 5, e1000487
26. Dixit, A., and Verkhivker, G. M. (2011) Computational model-ing of allosteric communication reveals organizing principles ofmutation-induced signaling in ABL and EGFR kinases. PLoSComput. Biol. 7, e1002179
27. Lee, T.-S., Ma, W., Zhang, X., Giles, F., Cortes, J., Kantarjian,H., and Albitar, M. (2008) BCR-ABL alternative splicing as acommon mechanism for imatinib resistance: evidence frommolecular dynamics simulations. Mol. Cancer Ther. 7, 3834 –3841
28. Lee, T.-S., Potts, S. J., and Albitar, M. (2009) Basis forresistance to imatinib in 16 BCR-ABL mutants as determinedusing molecular dynamics. Recent Pat. Anticancer Drug Discov.4, 164 –173
29. Lee, T.-S., Potts, S. J., Kantarjian, H., Cortes, J., Giles, F., andAlbitar, M. (2008) Molecular basis explanation for imatinibresistance of BCR-ABL due to T315I and P-loop mutationsfrom molecular dynamics simulations. Cancer 112, 1744 –1753
30. Pricl, S., Fermeglia, M., Ferrone, M., and Tamborini, E. (2005)T315I-mutated Bcr-Abl in chronic myeloid leukemia and ima-tinib: insights from a computational study. Mol. Cancer Ther. 4,1167–1174
31. Zhou, T., Commodore, L., Huang, W.-S., Wang, Y., Thomas, M.,Keats, J., Xu, Q., Rivera, V. M., Shakespeare, W. C., Clackson, T.,Dalgarno, D. C., and Zhu, X. (2011) Structural mechanism ofthe Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons forovercoming kinase inhibitor resistance. Chem. Biol. Drug Des. 77,1–11
32. Nagar, B., Bornmann, W. G., Pellicena, P., Schindler, T., Veach,D. R., Miller, W. T., Clarkson, B., and Kuriyan, J. (2002) Crystalstructures of the kinase domain of c-Abl in complex with thesmall molecule inhibitors PD173955 and imatinib (STI-571).Cancer Res. 62, 4236–4243
33. Tokarski, J. S., Newitt, J. A., Chang, C. Y. J., Cheng, J. D.,Wittekind, M., Kiefer, S. E., Kish, K., Lee, F. Y. F., Borzillerri,R., Lombardo, L. J., Xie, D., Zhang, Y., and Klei, H. E. (2006)The structure of dasatinib (BMS-354825) bound to activatedABL kinase domain elucidates its inhibitory activity againstimatinib-resistant ABL mutants. Cancer Res. 66, 5790 –5797
34. Preyer, M., Vigneri, P., and Wang, J. Y. (2011) Interplay betweenkinase domain autophosphorylation and F-actin binding do-main in regulating imatinib sensitivity and nuclear import ofBCR-ABL. PLoS One 6, e17020
35. Sali, A., and Blundell, T. L. (1993) Comparative protein mod-elling by satisfaction of spatial restraints. J. Mol. Biol. 234,779–815
36. Bernstein, F. C., Koetzle, T. F., Williams, G. J., Meyer, E. F.,Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T., andTasumi, M. (1977) The Protein Data Bank: a computer-basedarchival file for macromolecular structures. J. Mol. Biol. 112,535–542
37. Cowan-Jacob, S. W., Fendrich, G., Floersheimer, A., Furet, P.,Liebetanz, J., Rummel, G., Rheinberger, P., Centeleghe, M.,Fabbro, D., and Manley, P. W. (2007) Structural biologycontributions to the discovery of drugs to treat chronicmyelogenous leukaemia. Acta Crystallogr. D Biol. Crystallogr.63, 80 –93
38. Van der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark,A. E., and Berendsen, H. J. C. (2005) GROMACS: fast, flexible,and free. J. Comput. Chem. 26, 1701–1718
39. Sorin, E. J., and Pande, V. S. (2005) Exploring the helix-coiltransition via all-atom equilibrium ensemble simulations. Bio-phys. J. 88, 2472–2493
40. Hornak, V., Abel, R., Okur, A., Strockbine, B., Roitberg, A., andSimmerling, C. (2006) Comparison of multiple Amber forcefields and development of improved protein backbone param-eters. Proteins 65, 712–725
41. Jorgensen, W., Chandrasekhar, J., Madura, J., Impey, R., andKlein, M. (1983) Comparison of simple potential functions forsimulating liquid water. J. Chem. Phys. 79, 926
42. Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case,D. A. (2004) Development and testing of a general amberforce field. J. Comput. Chem. 25, 1157–1174
43. Jakalian, A., Jack, D. B., and Bayly, C. I. (2002) Fast, efficientgeneration of high-quality atomic charges. AM1-BCC model:II. Parameterization and validation. J. Comput. Chem. 231623–1641
44. Pandini, A., Fornili, A., Fraternali, F., and Kleinjung, J.(2012) Detection of allosteric signal transmission by informa-tion-theoretic analysis of protein dynamics. FASEB J. 26,868 –881
45. Kabsch, W., and Sander, C. (1983) Dictionary of protein sec-ondary structure: pattern recognition of hydrogen-bonded andgeometrical features. Biopolymers 22, 2577–2637
46. Barrett, C. P., Hall, B. A., and Noble, M. E. M. (2004) Dynamite:a simple way to gain insight into protein motions. Acta Crystal-logr. D Biol. Crystallogr. 60, 2280–2287
47. Daura, X., Gademann, K., Jaun, B., Seebach, D., van Gunsteren,W. F., and Mark, A. E. (1999) Peptide folding: when simulationmeets experiment. Angew. Chem. Int. Ed. 38, 236–240
48. Steinbrecher, T., Latzer, J., and Case, D. A. (2012) RevisedAMBER parameters for bioorganic phosphates. J. Chem. TheoryComput. 8, 4405–4412
49. Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P.(2012) Predicting the functional effect of amino acid substitu-tions and indels. PLoS One 7, e46688
50. Kumar, P., Henikoff, S., and Ng, P. C. (2009) Predicting theeffects of coding non-synonymous variants on protein functionusing the SIFT algorithm. Nat. Protoc. 4, 1073–1081
51. Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E.,Gerasimova, A., Bork, P., Kondrashov, A. S., and Sunyaev, S. R.(2010) A method and server for predicting damaging mis-sense mutations. Nat. Methods 7, 248–249
52. Humphrey, W., Dalke, A., and Schulten, K. (1996) VMD: visualmolecular dynamics. J. Mol. Graph. 14, 33–38
53. Notredame, C., Higgins, D. G., and Heringa, J. (2000) T-Coffee:a novel method for fast and accurate multiple sequence align-ment. J. Mol. Biol. 302, 205–217
54. Azam, M., Seeliger, M. A., Gray, N. S., Kuriyan, J., and Daley,G. Q. (2008) Activation of tyrosine kinases by mutation of thegatekeeper threonine. Nat. Struct. Mol. Biol. 15, 1109 –1118
55. Griswold, I. J., MacPartlin, M., Bumm, T., Goss, V. L., O’Hare,T., Lee, K. A., Corbin, A. S., Stoffregen, E. P., Smith, C.,Johnson, K., Moseson, E. M., Wood, L. J., Polakiewicz, R. D.,Druker, B. J., and Deininger, M. W. (2006) Kinase domainmutants of Bcr-Abl exhibit altered transformation potency,kinase activity, and substrate utilization, irrespective of sensitiv-ity to imatinib. Mol. Cell. Biol. 26, 6082–6093
56. Shah, N. P. (2011) Ponatinib: targeting the T315I mutation inchronic myelogenous leukemia. Clin. Adv. Hematol. Oncol. 9,925–926
57. Manley, P. W., Cowan-Jacob, S. W., Buchdunger, E., Fabbro, D.,Fendrich, G., Furet, P., Meyer, T., and Zimmermann, J. (2002)Imatinib: a selective tyrosine kinase inhibitor. Eur. J. Cancer 38(Suppl. 5), S19–S27
58. Bradeen, H. A., Eide, C. A., O’Hare, T., Johnson, K. J., Willis,S. G., Lee, F. Y., Druker, B. J., and Deininger, M. W. (2006)Comparison of imatinib mesylate, dasatinib (BMS-354825), and
15CRITICAL BCR-ABL RESIDUES DETERMINE TKI EFFICACY

nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-basedmutagenesis screen: high efficacy of drug combinations. Blood108, 2332–2338
59. Jabbour, E., and Kantarjian, H. (2012) Chronic myeloid leuke-mia: 2012 update on diagnosis, monitoring, and management.Am. J. Hematol. 87, 1037–1045
60. Baccarani, M., Cortes, J., Pane, F., Niederwieser, D., Saglio, G.,Apperley, J., Cervantes, F., Deininger, M., Gratwohl, A., Guilhot,F., Hochhaus, A., Horowitz, M., Hughes, T., Kantarjian, H.,Larson, R., Radich, J., Simonsson, B., Silver, R. T., Goldman, J.,Hehlmann, R., and European, L. (2009) Chronic myeloidleukemia: an update of concepts and management recommen-dations of European LeukemiaNet. J. Clin. Oncol. 27, 6041–6051
61. Zhou, T., Parillon, L., Li, F., Wang, Y., Keats, J., Lamore, S., Xu,Q., Shakespeare, W., Dalgarno, D., and Zhu, X. (2007) Crystalstructure of the T315I mutant of AbI kinase. Chem. Biol. Drug.Des. 70, 171–181
62. Quintas-Cardama, A., and Cortes, J. (2008) Therapeutic optionsagainst BCR-ABL1 T315I-positive chronic myelogenous leuke-mia. Clin. Cancer Res. 14, 4392–4399
63. Wheeler, D. L., Dunn, E. F., and Harari, P. M. (2010) Under-standing resistance to EGFR inhibitors-impact on future treat-ment strategies. Nat. Rev. Clin. Oncol. 7, 493–507
64. Guo, T., Agaram, N. P., Wong, G. C., Hom, G., D’Adamo, D.,Maki, R. G., Schwartz, G. K., Veach, D., Clarkson, B. D., Singer,S., DeMatteo, R. P., Besmer, P., and Antonescu, C. R. (2007)
Sorafenib inhibits the imatinib-resistant KITT670I gatekeepermutation in gastrointestinal stromal tumor. Clin. Cancer Res. 13,4874–4881
65. Barf, T., and Kaptein, A. (2012) Irreversible protein kinaseinhibitors: balancing the benefits and risks. J. Med. Chem. 55,6243–6262
66. Huang, W. S., Metcalf, C. A., Sundaramoorthi, R., Wang, Y., Zou, D.,Thomas, R. M., Zhu, X., Cai, L., Wen, D., Liu, S., Romero, J., Qi, J.,Chen, I., Banda, G., Lentini, S. P., Das, S., Xu, Q., Keats, J., Wang, F.,Wardwell, S., Ning, Y., Snodgrass, J. T., Broudy, M. I., Russian, K.,Zhou, T., Commodore, L., Narasimhan, N. I., Mohemmad, Q. K.,Iuliucci, J., Rivera, V. M., Dalgarno, D. C., Sawyer, T. K., Clackson, T.,and Shakespeare, W. C. (2010) Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a po-tent, orally active pan-inhibitor of breakpoint cluster region-Abelson(BCR-ABL) kinase including the T315I gatekeeper mutant. J. Med.Chem. 53, 4701–4719
67. De Falco, V., Buonocore, P., Muthu, M., Torregrossa, L., Basolo,F., Billaud, M., Gozgit, J. M., Carlomagno, F., and Santoro, M.(2013) Ponatinib (AP24534) is a novel potent inhibitor ofoncogenic RET mutants associated with thyroid cancer. J. Clin.Endocrinol. Metab. 98, E811–E819
Received for publication June 24, 2013.Accepted for publication November 18, 2013.
16 Vol. 28 March 2014 BUFFA ET AL.The FASEB Journal � www.fasebj.org
Copyright © 2022 FDOKUMEN




















