
Synthesis and biological effect of halogen substituted phenyl acetic acid derivatives of...
8
Journal of Steroid Biochemistry & Molecular Biology 111 (2008) 232–239 Contents lists available at ScienceDirect Journal of Steroid Biochemistry and Molecular Biology journal homepage: www.elsevier.com/locate/jsbmb Synthesis and biological effect of halogen substituted phenyl acetic acid derivatives of progesterone as potent progesterone receptor antagonists Marisa Cabeza a,∗ , Eugene Bratoeff b , Georgina Gómez a , Ivonne Heuze a , Arely Rojas b , Martha Ochoa b , Miguel Angel Palomino c , Cristina Revilla c a Department of Biological Systems and Animal Production, Metropolitan University-Xochimilco, Mexico, D.F., Mexico b Department of Pharmacy, Faculty of Chemistry, National University of Mexico City, Mexico, D.F., Mexico c Unit of Medical Research in Metabolic Diseases, Cardiology Hospital, National Medical Center, Mexican Institute of Social Security (IMSS), Mexico, D.F., Mexico article info Article history: Received 29 October 2007 Received in revised form 23 May 2008 Accepted 5 June 2008 Keywords: Novel 5-halogen phenyl acidic derivatives of progesterone Progesterone receptor selectivity Anti-progestin effect Inhibitors of ovulation Antagonist of the progesterone receptors abstract In this paper, we report the relative binding affinities to the progesterone receptor (PR) of several pro- gesterone derivatives containing an acetoxyphenyl substituent at C-17 and their structure–bioactivity relationship. The inhibitory effect to ovulation as well as their function as interrupters of endometrial maturation is also described. The biological activity of the novel steroids was determined in vivo and in vitro experiments using female cycling mice, which were synchronized for estrus with luteinizing hormone-releasing hormone (LHRH) and injected with the steroidal compounds. The cytosol used for the determination of the PR, was obtained from the uteri of adult estrogen-primed rabbits and the androgen (AR), mineralocorticoid (MR) and glucocorticoid (GR) receptors were determined in the cytosolic fractions from the prostate of castrated rats and from the kidneys and livers of adenalectomized male rats. We evaluated six related steroidal compounds 8a–8f differing in the nature of the 17 ester side chain for the inhibition of [ 3 H] R5020 binding to the PR. The IC 50 values for the displacement of [ 3 H] R5020 binding to the PR and its relative binding affinities (RBAs) were determined. Progesterone and R5020 had similar IC 50 values; steroids 8a, 8f and 8c bind to the progesterone receptor with RBAs of 100%, whereas 8e, 8b and 8d have RBA values <100%. These data indicate that there is a relationship between the structure of these steroids and their binding activity to the progesterone receptors. Having demonstrated in this study that steroids 8a–8f bind to the PR, we also evaluated the receptor’s selectivity, since some progesterone derivatives bind to AR, MR, GR receptors. We demonstrated that the tested steroids did not bind to the AR, MR, GR, since none of the steroids inhibited the labeled mibolerone, aldosterone or dexamethasone binding to the AR, MR or GR, respectively. These results show that the novel compounds have certain selectivity for the PR. After LHRH treatment, the mice of the control group showed the presence of ova in the oviduct, whereas the animals treated with steroids 8a, 8f, 8e and 8c with RBAs of 92–100%, did not exhibit any ovum in the oviducts. As a result of this study, it is evident that the novel steroids 8a, 8f, 8e and 8c inhibited the ovulation in these animals at dose of 0.22 mg/kg. After the treatment with LHRH, the uterus of the control group showed the typical progestational activity with an enlarged endometrial thickness with secretory activity. However, the endometrium of the mice treated with steroids 8a, 8f, 8e and 8c (with RBAs of 92–100%) neither did show any enlargement of the endometrium, nor a secretory activity could be detected. The diameter of the uterus was also significantly reduced compared to those of the control group, thus indicating that compounds 8a, 8f, 8e and 8c had antagonistic activity in this tissue. The overall data showed that steroids 8a, 8f, 8e and 8c have a high and selective binding activity to the PR. Furthermore there is a relationship between the structure of these steroids and their binding activity, since the presence of fluorine atom in meta position in the acetoxyphenyl substituent at C-17, improved the binding activity as compared to that for the ortho and para positions. These data also demonstrated that 8a–8f have an anti-progestational activity in vivo, and therefore they have better characteristics than the compounds previously reported. © 2008 Elsevier Ltd. All rights reserved. ∗ Corresponding author at: Departamento de Sistemas Biológicos, Universidad Autónoma Metropolitana-Xochimilco, Calzada del Hueso No. 1100, México, D.F, C.P. 04960, Mexico. Tel.: +52 55 5483 72 60; fax: +52 55 5483 72 60. E-mail address: [email protected] (M. Cabeza). 0960-0760/$ – see front matter © 2008 Elsevier Ltd. All rights reserved. doi:10.1016/j.jsbmb.2008.06.011
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Synthesis and biological effect of halogen substituted phenyl acetic acid derivatives of...

Sd
MMa
b
c
a
ARRA
KNoPAIA
M
0d
Journal of Steroid Biochemistry & Molecular Biology 111 (2008) 232–239
Contents lists available at ScienceDirect
Journal of Steroid Biochemistry and Molecular Biology
journa l homepage: www.e lsev ier .com/ locate / j sbmb
ynthesis and biological effect of halogen substituted phenyl acetic aciderivatives of progesterone as potent progesterone receptor antagonists
arisa Cabezaa,∗ , Eugene Bratoeffb , Georgina Gómeza , Ivonne Heuzea , Arely Rojasb , Martha Ochoab ,iguel Angel Palominoc, Cristina Revillac
Department of Biological Systems and Animal Production, Metropolitan University-Xochimilco, Mexico, D.F., MexicoDepartment of Pharmacy, Faculty of Chemistry, National University of Mexico City, Mexico, D.F., MexicoUnit of Medical Research in Metabolic Diseases, Cardiology Hospital, National Medical Center, Mexican Institute of Social Security (IMSS), Mexico, D.F., Mexico
r t i c l e i n f o
rticle history:eceived 29 October 2007eceived in revised form 23 May 2008ccepted 5 June 2008
eywords:ovel 5-halogen phenyl acidic derivativesf progesteronerogesterone receptor selectivitynti-progestin effect
nhibitors of ovulationntagonist of the progesterone receptors
a b s t r a c t
In this paper, we report the relative binding affinities to the progesterone receptor (PR) of several pro-gesterone derivatives containing an acetoxyphenyl substituent at C-17 and their structure–bioactivityrelationship. The inhibitory effect to ovulation as well as their function as interrupters of endometrialmaturation is also described.
The biological activity of the novel steroids was determined in vivo and in vitro experiments usingfemale cycling mice, which were synchronized for estrus with luteinizing hormone-releasing hormone(LHRH) and injected with the steroidal compounds. The cytosol used for the determination of the PR,was obtained from the uteri of adult estrogen-primed rabbits and the androgen (AR), mineralocorticoid(MR) and glucocorticoid (GR) receptors were determined in the cytosolic fractions from the prostate ofcastrated rats and from the kidneys and livers of adenalectomized male rats.
We evaluated six related steroidal compounds 8a–8f differing in the nature of the 17� ester side chainfor the inhibition of [3H] R5020 binding to the PR. The IC50 values for the displacement of [3H] R5020binding to the PR and its relative binding affinities (RBAs) were determined. Progesterone and R5020 hadsimilar IC50 values; steroids 8a, 8f and 8c bind to the progesterone receptor with RBAs of 100%, whereas 8e,8b and 8d have RBA values <100%. These data indicate that there is a relationship between the structureof these steroids and their binding activity to the progesterone receptors.
Having demonstrated in this study that steroids 8a–8f bind to the PR, we also evaluated the receptor’sselectivity, since some progesterone derivatives bind to AR, MR, GR receptors. We demonstrated that thetested steroids did not bind to the AR, MR, GR, since none of the steroids inhibited the labeled mibolerone,aldosterone or dexamethasone binding to the AR, MR or GR, respectively. These results show that thenovel compounds have certain selectivity for the PR.
After LHRH treatment, the mice of the control group showed the presence of ova in the oviduct, whereasthe animals treated with steroids 8a, 8f, 8e and 8c with RBAs of 92–100%, did not exhibit any ovum inthe oviducts. As a result of this study, it is evident that the novel steroids 8a, 8f, 8e and 8c inhibited theovulation in these animals at dose of 0.22 mg/kg.
After the treatment with LHRH, the uterus of the control group showed the typical progestational activitywith an enlarged endometrial thickness with secretory activity. However, the endometrium of the micetreated with steroids 8a, 8f, 8e and 8c (with RBAs of 92–100%) neither did show any enlargement of theendometrium, nor a secretory activity could be detected. The diameter of the uterus was also significantlyreduced compared to those of the control group, thus indicating that compounds 8a, 8f, 8e and 8c hadantagonistic activity in this tissue.
The overall data showed that steroids 8a, 8f, 8e and 8c have a high and selective binding activity to thePR. Furthermore there is a relationship between the structure of these steroids and their binding activity,
since the presence of fluorine athe binding activity as comparethat 8a–8f have an anti-progestthe compounds previously repo∗ Corresponding author at: Departamento de Sistemas Biológicos, Universidad Autónomexico. Tel.: +52 55 5483 72 60; fax: +52 55 5483 72 60.
E-mail address: [email protected] (M. Cabeza).
960-0760/$ – see front matter © 2008 Elsevier Ltd. All rights reserved.oi:10.1016/j.jsbmb.2008.06.011
tom in meta position in the acetoxyphenyl substituent at C-17, improvedd to that for the ortho and para positions. These data also demonstratedational activity in vivo, and therefore they have better characteristics thanrted.
© 2008 Elsevier Ltd. All rights reserved.
a Metropolitana-Xochimilco, Calzada del Hueso No. 1100, México, D.F, C.P. 04960,

istry
1
omosocttalRiott
tbpbtac
attaeaee
taH(
t8wo
2
2
du2rppI
(cMcs
3
bmpStMa(
2
di
26
tdSsidanc0((CsN2((
2d8
papm(bTscpo(ıC(1H, s, H-4), 6.06 (1H, d, J = 7 Hz, H-6), 7.01 (2H, m, H-2′, H-6′,aromatic), 7.25 (2H, m, H-3′, H-5′, aromatic). 13C NMR (CDCl3)
M. Cabeza et al. / Journal of Steroid Biochem
. Introduction
Progesterone plays a critical role in the maintenance and devel-pment of female reproductive functions. Its biological activity isediated by a progesterone receptor (PR), which induces a cascade
f transcriptional events after binding to the hormones [1]. Severalynthetic ligands for the progesterone receptors have been devel-ped to compete for binding with the natural hormone and areapable of inhibiting or stimulating receptor activity. In responseo binding with natural hormone, the receptor dissociates fromhe oligomeric complex and acquires the ability to dimerize [1]nd bind to a hormone response element (HRE) that is usuallyocated in the regulatory region of steroid responsive genes [2].eceptor association with HREs leads to an increase or decrease
n the transcription of DNA, which is regulated by co-activatorsr co-repressors of the gene transcription [3]. A central event inhe activation process appears to effect a ligand induced conforma-ional change in receptor structure [4].
Blocking PR function by using PR antagonists should allowhe modulation of various reproductive processes. On thisasis anti-progestins were developed which disrupt the normalrogesterone-induced signal transduction pathway by competitiveinding to PR. Therefore, anti-progestins have considerable poten-ial as therapeutic drugs for numerous gynecological, obstetricalnd oncological afflictions as well as contraceptives since theseompounds can block ovulation [5] and prevent implantation [6].
Currently available anti-progestins are steroidal analogs withn aromatic substituent at the at C-11� position which appearo be essential for antagonistic activity [7]. However, these struc-ures generated enhanced binding to the glucocorticoid (GR) andndrogen (AR) receptors which could produce important sideffects [8–10]. Therefore, new compounds with anti-progestationalctivity devoid of glucocorticoid, antiglucocorticoid or androgenicffects are highly desirable for both clinical applications and basicndocrine research.
Several new halogen substituted phenyl acetic acid ester deriva-ives of progesterone had been synthesized by our group whichct as inhibitors of 5�-reductase enzyme in human prostate [11].owever, these compounds did not bind to the androgen receptors
unpublished data).The aim of this research was to synthesize six different proges-
erone derivatives containing an acetoxyphenyl substituent at C-17,a–8f with a selective affinity to the progesterone receptor. Also, itas of interest to determine the structure–bioactivity relationshipf these steroids to the PR as well as their pharmacological activity.
. Materials and methods
.1. Chemical and radioactive material
Solvents were laboratory grade or better. Melting points wereetermined on a Fisher Johns melting point apparatus and arencorrected. 1H NMR and 13C NMR were taken on Varian gemini00 and VRX-300, respectively. Chemical shifts are given in ppmelative to that of Me4Si (ı = 0) in CDCl3 (the abbreviations of signalatterns are as follows: s, singlet; d, doublet, t, triplet, m, multi-let). Mass spectra were obtained with a HP5985-B spectrometer.
R spectra were recorded on a PerkinElmer 200s spectrometer.The radioligands: promegestone (17�-methyl-3H) [3H] R5020
synthetic progestin with high affinity for the PR [12]) spe-ific activity of 87 Ci/mmol; mibolerone (17�-methyl-3H) [3H]IB (synthetic androgen with high affinity by the AR) spe-
ific activity of 70–87 Ci/mmol, aldosterone, D-[1,2,6,7-3H (N)]pecific activity of 70–100 Ci/mmol and dexamethasone, [6,7-
ı1(b4
& Molecular Biology 111 (2008) 232–239 233
H (N)] specific activity of 30–50 Ci/mmol were providedy PerkinElmer Life Sciences, Inc. (Boston, MA). Radio inertibolerone, aldosterone, dexamethasone and R5020, were sup-
lied by Steraloids (Wilton NH, U.S.A.) and PerkinElmer Lifeciences, Inc. (Boston, MA), respectively. dl-Dithiothreitol and pro-ease inhibitors were purchased from Sigma–Aldrich (St. Louis,
O, U.S.A.). Activated charcoal (acid washed with hydrochloriccid) and Dextran (Mr-70,000) were supplied by Sigma–AldrichSt. Louis, MO).
.2. Synthesis of steroidal derivatives
The synthesis of the new steroidal derivatives 8a–8f is brieflyescribed below (Fig. 1). The preparation of the intermediates 1–7
s given in Ref. [13].
.2.1. 17˛-Phenylacetoxy-16ˇ-methylpregna-4,-diene-3,20-dione 8a
A mixture of phenyl acetic acid (60 mg, 0.026 mmol), p-oluenesulfonic acid (5 mg, 0.026 mmol) and trifluoroacetic anhy-ride (92 mg, 0.438 mmol) was stirred for 2 h at room temperature.teroid 7 (50 mg, 0.15 mmol) was added; the reaction mixture wastirred for an additional 3 h (nitrogen atmosphere). It was neutral-zed with an aqueous sodium bicarbonate solution to pH 7 andiluted with chloroform (30 ml). The organic phase was separatednd dried over anhydrous sodium sulfate; the solvent was elimi-ated in vacuum and the crude product was purified by silica gelolumn chromatography. Hexane–ethyl acetate (8:2) eluted 33 mg,.072 mmol (49%) of pure product 8a, mp 166–169 ◦C. UV (nm) 282ε, 16,200). IR (KBr) cm−1: 1734, 1710, 1661, 1618, 719. 1H NMRCDCl3) ı: 0.74 (3H, s, H-18), 1.11 (3H, s, H-19), 1.34 (3H, d, J = 7 Hz,H3 at C-16), 1.84 (3H, s, H-21), 3.67 (2H, s, COOCH2-Ph), 5.71 (1H,, H-4), 6.09 (1H, d, J = 8 Hz, H-6), 7.29 (5H, m, (CDCl3 aromatic). 13CMR) ı: 14.79 (CH3 at C-16), 16.45 (C-18), 18.92 (C-19), 28.11 (C-1), 123.88 (C-4), 129.37 (C-2′, C-6′) 130.45 (C-3′, C-4′, C-5′), 171.24ester carbonyl), 199.46 (C-3 carbonyl), 204.10 (C-20 carbonyl). MSm/z): 460 (M+).
.2.2. 17˛-(4-Fluorophenyl)acetoxy-16ˇ-methylpregna-4,6-iene-3,20-dioneb
A mixture of 4-fluorophenylacetic acid (147 mg, 0.95 mmol),-toluenesulfonic acid (11 mg, 0.062 mmol) and trifluoroaceticnhydride (0.21 g, 0.98 mmol) was stirred for 2 h at room tem-erature. Steroid 7 (100 mg, 0.29 mmol) was added; the reactionixture was stirred at room temperature for an additional 3 h
nitrogen atmosphere). It was neutralized with an aqueous sodiumicarbonate solution to pH 7 and diluted with chloroform (30 ml).he organic phase was separated and dried over anhydrousodium sulfate; the solvent was eliminated in vacuum and therude product was purified by silica gel column chromatogra-hy. Hexane–ethyl acetate (8:2) eluted 66.9 mg, 0.12 mmol (47.9%)f pure product 8b, mp 208–210 ◦C. UV (nm) 277 (ε, 15,700). IRKBr) cm−1: 1732, 1705, 1670, 1618, 873, 800. 1H NMR (CDCl3): 0.75 (3H, s, H-18), 1.11 (3H, s, H-19), 1.36 (3H, d, J = 5 Hz,H3 at C-16), 1.83 (3H, s, H-21), 3.64 (2H, s, COOCH2-Ph), 5.72
: 14.8 (CH3 at C-16), 16.31 (C-18), 19.70 (C-19), 28.13 (C-21),23.98 (C-4), 128.27 (C-6), 130.99 (C-2′, C-6′, aromatic), 132.56C-3′, C-5′, aromatic), 134.49 (C–F, aromatic), 171.10 (ester car-onyl), 199.40 (C-3 carbonyl), 203.87 (C-20 carbonyl). MS (m/z):78 (M+).

2 istry
2p
paSwabofwam1
1Cm141
2p
34 M. Cabeza et al. / Journal of Steroid Biochem
.2.3. 17˛-(3-Fluorophenyl)acetoxy-16ˇ-methyl-regna-4,6-diene-3,20-dione 8c
A mixture of 3-fluorophenylacetic acid (68 mg, 0.44 mmol),-toluenesulfonic acid (5 mg, 0.026 mmol), and trifluoroaceticnhydride (0.092 mmol) was stirred for 2 h at room temperature.teroid 7 (50 mg, 0.15 mmol) was added; the reaction mixtureas stirred at room temperature for an additional 3 h (nitrogen
tmosphere). It was neutralized with an aqueous sodium bicar-onate solution to pH 7 and diluted with chloroform (30 ml). Therganic phase was separated and dried over anhydrous sodium sul-
ate; the solvent was eliminated in vacuum and the crude productas purified by silica gel column chromatography. Hexane–ethylcetate (8:2) eluted 32 mg, 0.032 mmol (46%) of pure product 8c,p 180–183 ◦C. UV (nm) 284 (ε, 15,400) IR (KBr) cm−1, 1729, 1710,
675, 1620, 685. 1H NMR (CDCl3) ı: 0.75 (3H, s, H-18), 1.15 (3H, s, H-
tar3s
Fig. 1. Synthesis of steroidal compounds 8a–8f, and their preparation fr
& Molecular Biology 111 (2008) 232–239
9), 1.35 (3H, d, J = 7 Hz, CH3 at C-16), 1.86 (3H, S, H-21), 3.67 (2H, s,OOCH2-Ph), 5.72 (1H, s, H-4), 6.09 (1H, d, J = 9 Hz, H-6), 7.0–7.3 (4H,, aromatic). 13CMR (CDCl3) ı: 14.86 (CH3 at C-16), 16.32 (C-18),
9.80 (C-19), 28.15 (C-21), 123.94 (C-4), 125.09 (C-6), 130.41 (C-′, C-5′, C-6′), 132.54 (C-2′), 134.32 (C–F), 170.61 (ester carbonyl),99.43 (C-3 carbonyl), 203.88 (C-20 carbonyl). MS (m/z): 478 (M+).
.2.4. 17˛-(2-Fluorophenyl)acetoxy-16ˇ-methyl-regna-4,6-diene-3,20-dione 8d
A mixture of 2-fluorophenylacetic acid (68 mg, 0.44 mmol) and
rifluoroacetic anhydride (50 mg, 0.026 mmol) was stirred for 2 ht room temperature, steroid 7 (50 mg, 0.15 mmol) was added; theeaction mixture was stirred at room temperature for an additionalh (nitrogen atmosphere). It was neutralized with an aqueousodium bicarbonate solution to pH 7 and diluted with chloroform
om the commercially available 16-dehydropregnenolone acetate.

istry & Molecular Biology 111 (2008) 232–239 235
(dtppcs(J(4((
2p
t(swabofwau1H(dH1(b5
2d
t(7aItwvbeUNJ5Ha(((
2
v
Table 1Relative binding affinities (RBAs) of the novel steroidal compounds 8a–8f to theprogesterone receptor
Structures IC50 (nM) RBA (%)
0.37 100
0.36 100
0.37 100
0.36 100
0.40 92
1.2 31
1.3 28
0.37 100
RBA of the novel steroidal compounds to the progesterone receptor obtained fromuteri from rabbits, which were previously treated with estrogens. The RBA was
c
NrUiost
M. Cabeza et al. / Journal of Steroid Biochem
30 ml). The organic phase was separated and dried over anhy-rous sodium sulfate; the solvent was eliminated in vacuum andhe crude product was purified by silica gel column chromatogra-hy. Hexane–ethyl acetate (8:2) eluted 32 mg, 0.032 mmol (44%), ofure product 8d, mp 176–178 ◦C. UV (nm) 284 (ε, 15,800). IR (KBr)m−1: 1728, 1705, 1670, 1617, 756. 1H NMR (CDCl3) ı: 0.74 (3H,, H-18), 1.10 (3H, s, H-19), 1.35 (3H, d, J = 7 Hz, CH3 at C-16), 1.933H, s, H-21), 3.73 (2H, s, COOCH2-Ph), 5.72 (1H, s, H-4), 6.10 (1H, d,= 10 Hz, H-6), 7.0–7.3 (4 H, m, aromatic). 13C NMR (CDCl3) ı: 14.79CH3 at C-16), 16.29 (C-18), 19.69 (C-19), 28.17 (C-21), 123.98 (C-), 128.27 (C-6), 131.46 (C-2′, C-3′, C-4′, C-5′), 140.31 (C–F), 170.37ester carbonyl), 199.58 (C-3 carbonyl), 204.15 (C-20 carbonyl). MSm/z): 478 (M+).
.2.5. 17˛-(4-Bromophenyl)acetoxy-16ˇ-methyl-regna-4,6-diene-3,20-dione 8e
A mixture of 4-bromophenylacetic acid (223 mg, 5 mmol), p-oluenesulfonic acid (11 mg, 0.062 mmol), trifluoroacetic anhydride210 mg, 0.98 mmol) was stirred for 2 h at room temperature,teroid 7 (100 mg, 0.29 mmol) was added; the reaction mixtureas stirred at room temperature for an additional 3 h (nitrogen
tmosphere). It was neutralized with an aqueous sodium bicar-onate solution to pH 7 and diluted with chloroform (30 ml). Therganic phase was separated and dried over anhydrous sodium sul-ate; the solvent was eliminated in vacuum and the crude productas purified by silica gel column chromatography. Hexane–ethyl
cetate (8:2) eluted 86.4 mg, 0.161 mmol (54.7%) of pure prod-ct 8e, mp 220–222 ◦C. UV (nm) 281 (ε, 16,400). IR (KBr) cm−1:735, 1700, 1675, 1616, 875, 803. 1H NMR (CDCl3) ı: 0.77 (3H, s,-18), 1.13 (3H, s, H-19), 1.35 (3H, d, J = 6 Hz, CH3 at C-16), 1.88
3H, s, H-21), 3.71 (2H,s, COOCH2-Ph), 5.72 (1H, s, H-4), 6.10 (1H,, J = 8 Hz, H-6), 7.18 (2H, d, J = 10 Hz, H-2′, H-6′), 7.49 (2H, d, J = 9 Hz,-3′, H-5′). 13C NMR (CDCl3) ı: 14.85 (CH3 at C-16), 16.32 (C-8), 19.70 (C-19), 28.19 (C-21), 123.51 (C-4), 128.35 (C-6), 132.35C-2′, C-6′), 136.56 (C-3′, C-5′), 136.66 (C–Br), 171.10 (ester car-onyl), 199.40 (C-3-carbonyl), 203.87 (C-20 carbonyl). MS (m/z):36 (M+).
.2.6. 17˛-(4-Chlorophenyl)acetoxy-16ˇ-methylpregna-4,6,iene-3,20-dione 8f
A mixture of 4-chlorophenylacetic acid (176 mg, 1.03 mmol), p-oluenesulfonic acid (11 mg, 0.62 mmol), trifluoroacetic anhydride0.21 g, 0.99 mmol) was stirred for 2 h at room temperature, steroid(100 mg 0.29 mmol) was added; the reaction mixture was stirred
t room temperature for and additional 3 h (nitrogen atmosphere).t was neutralized with an aqueous sodium bicarbonate solutiono pH 7 and diluted with chloroform (30 ml). The organic phaseas separated and dried over anhydrous sodium sulfate; the sol-
ent was eliminated in vacuum and the crude product was purifiedy silica gel column chromatography. Hexane–ethyl acetate (8:2)luted 77 mg, 0.16 mmol (53.3%) of pure product 8f, mp 223–224 ◦C.V (nm) 285 (ε, 16200). IR (KBr) cm−1: 1727, 1695, 1665, 807. 1HMR (CDCl3) ı: 0.75 (3H, s, H-18), 1.11 (3H, s, H-19), 1.36 (3H, d,
= 6 Hz, CH3 at C-16), 1.89 (3H, s, H-21), 3.73 (2H, s, COOCH2-Ph),.73 (1H, s, H-4), 6.09 (1H, d, J = 8 Hz, H-6), 7.20 (2H, d, J = 9 Hz, H-2′,-6′), 7.41 (2H, d, J = 9 Hz, H-3′, H-5′). 13C NMR (CDCl3) ı: 14.84 (CH3t C-16), 16.31 (C-18), 19.69 (C-19), 28.17 (C-21), 123.98 (C-4), 127.40C-6), 131.47 (C-2′, C-6′), 137.2 (C-3′, C-5′). 138.61 (C–Cl), 170.77ester carbonyl), 199.39 (C-3 carbonyl), 203.81 (C-20-carbonyl). MSm/z): 494 (M+).
.3. Biological activity of the progesterone derivatives
The biological activity of 8a–8f (Table 1) was determined inivo and in vitro experiments using CD1 female mice and adult
Mw31u
alculated according to the following equation: RBA = IC50 of [3H] promegestoneIC50 of inhibitor × 100.
ew Zealand White female rabbits. The animals (mice 20–25 g andabbits 4 kg and rats 400 g) were obtained from the Metropolitanniversity-Xochimilco of Mexico. At the end of the in vivo exper-
ment, the mice were sacrificed by cervical dislocation. In order tobtain the cytosol from the uteri from rabbits, the animals wereacrificed with CO2. This protocol was approved by the Institu-ional Care and Use Committee of the Metropolitan University of
exico (UAM). The uteri of the rabbits were removed, blotted,eighed in ice bath and soaked in cold TEMD (40 mM Tris–HCl,
mM EDTA and 20 mM sodium molybdate, dithiotreitol 0.5 mM,0% glycerol at pH 7.4) and maintained in ice bath prior to theirse.
2 istry
g4
b28
(Timt
2
2
(t[tI
tPm
rMctws
bidfi[aTTpfwactrct
2
1mt[dw(
t
Ub
[4fw
aposttgoTt1mca
makwuaw(t
2
wiofwTvlrt(lb
2
eiawa
2
2
36 M. Cabeza et al. / Journal of Steroid Biochem
In order to determine the binding of steroids 8a–8f to the andro-en receptors, a group of adult rats (500 g) were gonadectomized8 h before the experiment.
The prostate of the rats was removed, blotted, weighed in iceath and soaked in cold TEMD (40 mM Tris–HCl, 3 mM EDTA and0 mM sodium molybdate, dithiotreitol 0.5 mM, 10% glycerol at pH) and maintained in ice bath prior to their use.
Kidneys and livers from adrenalectomized male Wistar rats500 g) were used as source for the mineralocorticoid (MR) or GR.issues were removed, blotted, weighed in ice bath and soakedn cold TEMD (40 mM Tris–HCl, 3 mM EDTA and 20 mM sodium
olybdate, dithiotreitol 0.5 mM, 10% glycerol at pH 7.4) and main-ained in ice bath prior to their use.
.4. Receptor binding assays
.4.1. Androgen receptorRat prostates were homogenized with a tissue homogenizer
Teckmar, Cincinnati, OH), in one volume of buffer TEMD plus pro-ease inhibitors (2 mM PMSF, 10 �g/ml antipain, 5 mM leupeptin14]) in ice bath with a tissue homogenizer. Homogenates were cen-rifuged at 140,000 × g for 60 min [15] in a SW 60 Ti rotor (Beckmannstruments, Palo Alto, CA).
The cytosolic fraction obtained from the supernatant liquid ofhe rat prostate homogenate described above, was stored at −70 ◦C.rostatic cytosol proteins (4 mg of protein in 200 �l) were deter-ined by the Bradford method [16].For competitive studies, tubes containing 1 nM of [3H] MIB plus a
ange of increasing concentrations (1 × 10−10 to 4 × 10−7 M) of coldIB or steroids 8a–8f in ethanol or chloroform, or in absence of the
ompetitor were prepared [15]. Incubates also contained 200 nMriamcinolone, in ethanol, (Sigma) to prevent interaction of MIBith glucocorticoid receptors and progesterone receptors [17], the
olvent was completely eliminated.Aliquots of 200 �l of prostate cytosol were added and incu-
ated in the presence of 300 �l of TEMD buffer containing proteasenhibitors (duplicate) for 18 h at 4 ◦C in the tubes as previouslyescribed [14]. After incubation 0.27 ml saturated ammonium sul-
ate in TEMD buffer (35%) was added [18]. The mixture was furtherncubated for 1 h with occasional shaking for precipitation of the3H] MIB-complex. The precipitate was collected by centrifugationt 10,000 × g, 10 min and the pellet was redissolved in 0.5 ml ofEMD and mixed with 0.5 of 0.1% dextran-coated 1% charcoal inEMD buffer. The mixture was incubated for 10 min at 4 ◦C. To pre-are the dextran-coated charcoal mixture, the dextran was agitatedor 30 min before adding the charcoal to the mixture. The mixtureas agitated two more hours before using. The tubes were vortexed
nd immediately centrifuged at 800 × g for 10 min to pellet theharcoal; aliquots (600 �l) were taken and submitted for radioac-ive counting using Ultima Gold (Packard) as counting solution. Theadioactivity was determined in a scintillation counter (Packard tri-arb 2100 TR). The IC50 of each compound was calculated accordingo the plots of concentration versus percentage of binding.
.4.2. Progesterone receptorUteri were isolated from mature rabbits treated for 7 days with
0 �g/animal/estradiol valerate. The uteri from the rabbits wereinced and homogenized in equal volume of TEMD buffer plus pro-
ease inhibitors (2 mM PMSF, 10 �g/ml antipain, 5 mM leupeptin14]; 40 mM Tris–HCl, 3 mM EDTA and 20 mM sodium molybdate,
ithiotreitol 0.5 mM, 10% glycerol at pH 7.4) [11]. The homogenateas centrifuged at 140,000 × g for 1 h at 0 ◦C in a SW 60 Ti rotorBeckman Instruments, Palo Alto, CA).The cytosolic fraction obtained from the supernatant liquid of
he rabbit uteri homogenate described above, was stored at −70 ◦C.
(v
[
& Molecular Biology 111 (2008) 232–239
teri cytosol proteins (4 mg of protein in 200 �l) were determinedy the Bradford method [16].
For competitive studies, tubes containing 0.4 nM of [3H] R502019]; plus a range of increasing concentrations (1 × 10−10 to× 10−6 M) of cold R5020 or steroids 8a–8f in ethanol or chloro-
orm, or in absence of the competitor were prepared the solventas completely eliminated.
Aliquots of 200 �l of uteri cytosol (4 mg of protein) were addednd incubated in the presence of 300 �l of TEMD buffer containingrotease inhibitors (duplicate) for 18 h at 4 ◦C in the tubes as previ-usly described [14]. After incubation 0.21 ml saturated ammoniumulfate in TEMD buffer (30%) was added [20]. The mixture was fur-her incubated for 1 h with occasional shaking for precipitation ofhe [3H] R5020-complex. The precipitate was collected by centrifu-ation at 10,000 × g, 10 min and the pellet was redissolved in 0.5 mlf TEMD and mixed with 0.5 of 0.1% dextran-coated 1% charcoal inEMD buffer. The mixture was incubated for 10 min at 4 ◦C. Theubes were vortexed and immediately centrifuged at 800 × g for0 min to pellet the charcoal; aliquots (600 �l) were taken and sub-itted for radioactive counting. The IC50 of each compound was
alculated according to the plots of concentration versus percent-ge of binding.
The competition analyses of the synthesized steroids for theineralocorticoid and glucocorticoid receptors were carried out
ccording to the method reported by Juchem et al. [21]. Briefly,idneys and livers from adrenalectomized rats were homogenizedith a tissue homogenizer (Teckmar, Cincinnati, OH), in one vol-me of buffer TEMD plus protease inhibitors (2 mM PMSF, 10 �g/mlntipain, 5 mM leupeptin [14]) in an ice bath. Homogenatesere centrifuged at 140,000 × g for 60 min in a SW 60 Ti rotor
Beckman Instruments, Palo Alto, CA) to obtain the cytosolic frac-ions.
.4.3. Mineralocorticoid receptorAliquots of 200 �l of cytosol (5 mg of protein) from rat kidney
ere incubated with 8nM [3H] aldosterone together with increas-ng concentrations (1 × 10−10 to 4 × 10−6 M) of cold aldosteroner steroids 8a–8f or in absence of the competitor were incubatedor 4 h at 4 ◦C. Unbound steroids were absorbed by incubatingith 0.5 ml of 0.1% dextran-coated 1% charcoal in TEMD buffer.
he mixture was incubated for 10 min at 4 ◦C. The tubes wereortexed and immediately centrifuged at 800 × g for 10 min to pel-et the charcoal; aliquots (600 �l) were taken and submitted foradioactive counting using Ultima Gold (Packard) as counting solu-ion. The radioactivity was determined in a scintillation counterPackard tri-carb 2100 TR). The IC50 of each compound was calcu-ated according to the plots of concentration versus percentage ofinding.
.4.4. Glucocorticoid receptorAliquots of 200 �l of cytosol (5 mg of protein) from rat liv-
rs were incubated with 8 nM [3H] dexamethasone together withncreasing concentrations (1 × 10−10 to 4 × 10−6 M) of cold dex-methasone or steroids 8a–8f or in absence of the competitorere incubated for 4 h at 4 ◦C. Unbound steroids were absorbed
s described above for mineralocorticoids.
.5. In vivo experiments
.5.1. Mouse ovulation inhibition model
The animals were kept in a room with controlled temperature22 ◦C) and light–dark periods of 12 h. Food and water were pro-ided ad libitum.
Ovulation inhibition experiments were carried out as described22]. A brief summary is given below; random cycling mature
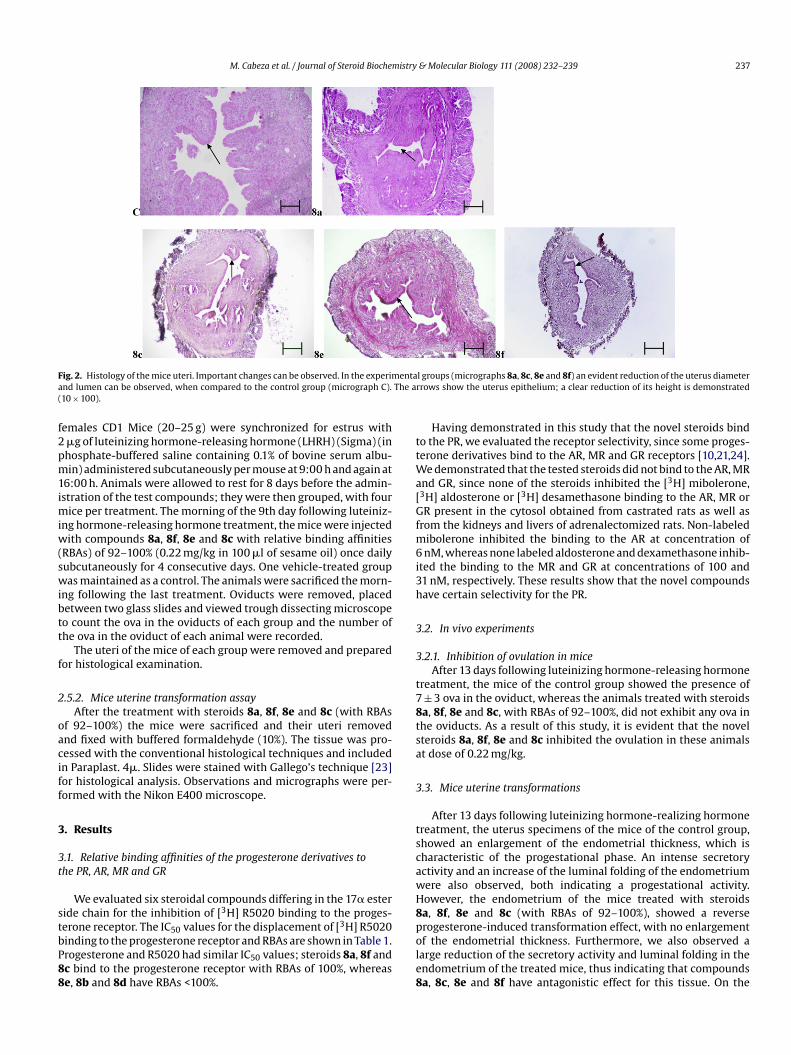
M. Cabeza et al. / Journal of Steroid Biochemistry & Molecular Biology 111 (2008) 232–239 237
F entaa The ar(
f2pm1imiw(swibtt
f
2
oaciff
3
3t
stbP88
ttWa[Gfm6i3h
3
3
t78tsa
3
tscawH8
ig. 2. Histology of the mice uteri. Important changes can be observed. In the experimnd lumen can be observed, when compared to the control group (micrograph C).10 × 100).
emales CD1 Mice (20–25 g) were synchronized for estrus with�g of luteinizing hormone-releasing hormone (LHRH) (Sigma) (inhosphate-buffered saline containing 0.1% of bovine serum albu-in) administered subcutaneously per mouse at 9:00 h and again at
6:00 h. Animals were allowed to rest for 8 days before the admin-stration of the test compounds; they were then grouped, with four
ice per treatment. The morning of the 9th day following luteiniz-ng hormone-releasing hormone treatment, the mice were injected
ith compounds 8a, 8f, 8e and 8c with relative binding affinitiesRBAs) of 92–100% (0.22 mg/kg in 100 �l of sesame oil) once dailyubcutaneously for 4 consecutive days. One vehicle-treated groupas maintained as a control. The animals were sacrificed the morn-
ng following the last treatment. Oviducts were removed, placedetween two glass slides and viewed trough dissecting microscopeo count the ova in the oviducts of each group and the number ofhe ova in the oviduct of each animal were recorded.
The uteri of the mice of each group were removed and preparedor histological examination.
.5.2. Mice uterine transformation assayAfter the treatment with steroids 8a, 8f, 8e and 8c (with RBAs
f 92–100%) the mice were sacrificed and their uteri removednd fixed with buffered formaldehyde (10%). The tissue was pro-essed with the conventional histological techniques and includedn Paraplast. 4�. Slides were stained with Gallego’s technique [23]or histological analysis. Observations and micrographs were per-ormed with the Nikon E400 microscope.
. Results
.1. Relative binding affinities of the progesterone derivatives tohe PR, AR, MR and GR
We evaluated six steroidal compounds differing in the 17� esteride chain for the inhibition of [3H] R5020 binding to the proges-
erone receptor. The IC50 values for the displacement of [3H] R5020inding to the progesterone receptor and RBAs are shown in Table 1.rogesterone and R5020 had similar IC50 values; steroids 8a, 8f andc bind to the progesterone receptor with RBAs of 100%, wherease, 8b and 8d have RBAs <100%.pole8
l groups (micrographs 8a, 8c, 8e and 8f) an evident reduction of the uterus diameterrows show the uterus epithelium; a clear reduction of its height is demonstrated
Having demonstrated in this study that the novel steroids bindo the PR, we evaluated the receptor selectivity, since some proges-erone derivatives bind to the AR, MR and GR receptors [10,21,24].
e demonstrated that the tested steroids did not bind to the AR, MRnd GR, since none of the steroids inhibited the [3H] mibolerone,3H] aldosterone or [3H] desamethasone binding to the AR, MR orR present in the cytosol obtained from castrated rats as well as
rom the kidneys and livers of adrenalectomized rats. Non-labeledibolerone inhibited the binding to the AR at concentration ofnM, whereas none labeled aldosterone and dexamethasone inhib-
ted the binding to the MR and GR at concentrations of 100 and1 nM, respectively. These results show that the novel compoundsave certain selectivity for the PR.
.2. In vivo experiments
.2.1. Inhibition of ovulation in miceAfter 13 days following luteinizing hormone-releasing hormone
reatment, the mice of the control group showed the presence of± 3 ova in the oviduct, whereas the animals treated with steroidsa, 8f, 8e and 8c, with RBAs of 92–100%, did not exhibit any ova inhe oviducts. As a result of this study, it is evident that the novelteroids 8a, 8f, 8e and 8c inhibited the ovulation in these animalst dose of 0.22 mg/kg.
.3. Mice uterine transformations
After 13 days following luteinizing hormone-realizing hormonereatment, the uterus specimens of the mice of the control group,howed an enlargement of the endometrial thickness, which isharacteristic of the progestational phase. An intense secretoryctivity and an increase of the luminal folding of the endometriumere also observed, both indicating a progestational activity.owever, the endometrium of the mice treated with steroidsa, 8f, 8e and 8c (with RBAs of 92–100%), showed a reverse
rogesterone-induced transformation effect, with no enlargementf the endometrial thickness. Furthermore, we also observed aarge reduction of the secretory activity and luminal folding in thendometrium of the treated mice, thus indicating that compoundsa, 8c, 8e and 8f have antagonistic effect for this tissue. On the
238 M. Cabeza et al. / Journal of Steroid Biochemistry & Molecular Biology 111 (2008) 232–239
F ) shows dant sa
ooT
4
cp
saRro8cRtggartsf
Maec[c
ihqThft
epfttotcs
slpr8ae
np
A
ft
R
ig. 3. Histology of the mice uteri. Experimental groups (micrographs 8a, 8c, 8e, 8fize and dilation when compared with the control groups (micrograph C); an abunnd 8f.
ther hand, we also observed a dramatic reduction of the diameterf the uteri of the treated mice compared with the control group.he pictures of the histological sections are shown in Figs. 2 and 3.
. Discussion
In this paper, we demonstrated, that steroids 8a–8f display aertain selectivity for the PR present in the cytosol from estrogen-rimed rabbits.
Progesterone, promegestone as well as steroids 8a, 8f and 8chowed RBA values of 100%; on the other hand compound 8e hadlower RBA value of 92%. Steroid 8b and 8d exhibited much lowerBA values, 31% and 28%, respectively. In view of the fact that theeceptor–steroid complex depends upon the correct conformationf the ligand (steroid) for binding activity, apparently compoundsb and 8d lack the appropriate conformation and as a result of thisannot form a stable complex with the protein receptor, ergo lowBA values. These data indicate that a relationship exists betweenhe structure of these steroids and their binding activity to the pro-esterone receptors. Compound 8a containing an acetoxyphenylroup at C-17 ester side chain had similar potency than that of 8fnd 8c which contain chlorine and fluorine atoms in the molecule,espectively. However, the fluorine atom in para and ortho posi-ions had less potency than in meta position (compound 8c) thusuggesting that this compound has the appropriate configurationor superior binding to the PR.
On the other hand, these compounds did not bind to the AR,R or GR receptors and, as a result of this did not produce an
ndrogenic, mineral corticoid, glucocorticoid or antiglucocorticoidffects. This characteristic could be advantageous for its use in theontrol of fertility (contraception, menses-induction and abortion)8,9] in hormone dependent tumors (e.g. breast cancer) and theontrol of cell growth and differentiation with minor side effects.
Steroids 8a, 8f, 8e and 8c, having RBA values of 92–100% inhib-ted the ovulation, in mice following luteinizing hormone-releasingormone treatment for estrus synchronization and with a subse-
uent treatment with the novel steroids as described in Section 2.his fact could be explained by considering that these compoundsad a marked pituitary inhibitory activity as previously reportedor other anti-progestins. This phenomenon could be useful for thereatment of endometriosis [25,26].
a reduction in the number and activity of endometrial glands, manifested by theecretion can be observed (arrows) (40 × 100). This reduction is more evident in 8e
It had been demonstrated by several authors [8,24,27–30] thatndocrine bioassays using the uterine transformation assay, asarameter to measure progestational activity is a reliable methodor the in vivo evaluation, which gives more real information thathose assays carried out in vitro and can be used as a definiteest. Previously, it had been demonstrated that progesterone antag-nists such as mifepristone have reverse progesterone-inducedransformation in estrogen-primed rabbit uterus [28] and later thisompound was successfully used for the treatment of endometrio-is [26].
The novel steroids (8a, 8f, 8e and 8c) inhibited progesterone-timulated transformation of the mice uterus treated withuteinizing hormone-releasing hormone therefore these com-ounds could be considered as antagonists for the progesteroneeceptors present in the mouse uterus. Based on our results, steroidsa, 8f and 8c showed a high binding capacity as well as highntagonistic activity for the progesterone receptor present in thestrogen-primed-rabbit uterus and had high potency in vivo.
The overall data obtained in this study indicate that PR antago-ists 8a–8f are potent and more selective than the anti-progestinsreviously reported [31].
cknowledgements
We would like to thank Dr. E. Pichardo for technical assistanceor the ovulation assay. This work was supported by CONACYT withhe project no. 54853.
eferences
[1] A.M. DeMarzo, C.A. Beck, S.A. Onate, D.P. Edwars, Dimerization of mammalianprogesterone receptors occurs in the absence of DNA and is related to the releaseof the 90 kDa heat shock protein, Proc. Natl. Acad. Sci. U.S.A. 88 (1991) 72–76.
[2] M.K. Bagchi, M.J. Tsai, B.O. ’Malley, S.Y. Tsai, Analysis of the mechanism of steroidhormone receptor-dependent gene activation in cell-free systems, Endocr. Rev.13 (1992) 525–535.
[3] B.L. Wargner, J.D. Norris, T.A. Notts, N.L. Weigel, D.-P. McDonnell, The nuclear
corepressors NCoR and SMRT are key regulators of both ligand- and 8-bromo-cyclic AMP-dependent transcriptional activity of the human progesteronereceptor, Mol. Cell Biol. 18 (1998) 1369–1378.[4] G.F. Allan, X. Leng, S.Y. Tsai, N.L. Weigel, D.P. Edwards, M.J. Tsai, B.W. O’Malley,Hormone and antihormone induce distinct conformational changes which arecentral to steroid receptor activation, J. Biol. Chem. 267 (1992), 19,513–19,520.

istry
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
M. Cabeza et al. / Journal of Steroid Biochem
[5] T. Luukkainen, O. Heikinheimo, Inhibition of folliculogenesis and ovulation byantiprogesterone RU 486, Fertil. Steril. 49 (1988) 961–963.
[6] M.C. Batista, T.L. Bristol, J. Mathews, W.S. Stokes, D.L. Loriaux, L.K. Niermann,Daily administration of progesterone antagonist RU 486 prevents implantationin the cycling guinea pig, Am. J. Obstet. Gynecol. 165 (1991) 82–86.
[7] T. Garcia, B. Benhamou, D. Golfo, A. Vergezac, D. Philibert, P. Chambon, H. Grone-meyer, Switching agonistic, antagonistic, and mixed transcriptional responsesto 11�-substituted progestins by mutation of progesterone receptor, Mol.Endocr. 6 (1992) 2071–2078.
[8] A. Ulmann, G. Teutsch, D. Philibert, RU 486, Sci. Am. 262 (1990) 38–42.[9] P. Raynauud, T. Ojasoo, The design and use of sex-steroid antagonists, J. Steroid
Biochem. 25 (1986) 811–833.10] E. Bratoeff, E. Ramírez, E. Flores, M. Sánchez, I. Heuze, M. Cabeza, New aro-
matic esters of progesterone as antiandrogens, J. Enz. Inh. Med. Chem. 19 (2004)99–105.
11] M. Cabeza, E. Bratoeff, I. Heuze, A. Rojas, N. Terán, M. Ochoa, M.T. Ramírez-Apan,E. Ramírez, V. Pérez, I. Gracia, New progesterone derivatives as inhibitors of 5�-reductase enzyme and prostate cancer cell growth, J. Enz. Inh. Med. Chem. 21(2006) 371–378.
12] F.Z. Stanczyk, Pharmacokinetics and potency of progestins used for hormonereplacement therapy and contraception, Endocr. Metabol. Disord. 3 (2002)211–224.
13] M. Cabeza, E. Bratoeff, I. Heuze, V. Pérez, D. Valdéz, M. Ochoa, N. Terán, G.Jimenez, E. Ramírez, Synthesis and pharmacological evaluation of new proges-terone esters as 5�-reductase inhibitors, Chem. Pharm. Bull. (Jpn.) 53 (2005)1515–1518.
14] W.J. Hendry, B.J. Danzo, Structural conversion of cytosolic steroids receptors byan age-dependent epididymal protease, J. Steroid Biochem. 23 (1985) 883–893.
15] M. Cabeza, F. Vilchis, A.E. Lemus, L. Diaz de León, G. Pérez-Palacios, Molecu-lar interactions of levonorgestrel and its 5�-reduced derivative with androgenreceptors in hamster flanking organs, Steroids 60 (1995) 630–635.
16] M.M. Bradford, A rapid and sensitive method for the quantization of micro-grams quantities of protein utilizing the principle of protein dye binding, Anal.Biochem. 72 (1986) 248–254.
17] K.E. Carlson, J.A. Katzenellenbogen, A comparative study of the selectivity andefficiency of target tissues uptake five tritium-labeled androgens in the rat, J.Steroid Biochem. 36 (1990) 549–561.
18] T. Liang, E.C. Heiss, Inhibition of 5�-reductase, receptor binding and nuclearuptake of androgens in prostate by a 4-methyl-4-aza-steroid, J. Biol. Chem. 256(1981) 7998–8005.
[
& Molecular Biology 111 (2008) 232–239 239
19] S. Palmer, C.A. Campen, G.F. Allan, P. Rybczynski, D. Haynes-Johnson, et al., Non-steroidal progesterone receptor ligands with unprecedent receptor selectivity,J. Steroid Biochem. Mol. Biol. 75 (2000) 33–42.
20] R.G. Smith, M. d’Istria, N.T. Van, Purification of human progesterone receptor,Biochemistry 20 (1981) 5557–5565.
21] M. Juchem, K. Pollow, W. Elger, G. Hoffman, V. Möbus, Receptor binding ofnorgestimate—a new orally active synthetic progestational compound, Con-traception 47 (1993) 283–294.
22] Z. Zhang, A.M. Olland, Y. Zhu, J. Cohen, T. Berrodin, et al., Molecular and pharma-cological properties of a potent and selective novel nonsteroidal progesteronereceptor agonist tanaproget, J. Biol. Chem. 280 (2005) 468–475.
23] E. Estrada Flores, L. Peralta Zamora, P. Rivas Manzano, Manual de TécnicasHistológicas AGT editor S.A., México, 1982, pp. 65 ork.
24] A.A. Murphy, M. Kettel, A. Morales, V. Roberts, S. Yen, Regression of uterineleiomyomata in response to antiprogesterone RU 486, J. Clin. Endocrinol. Metab.76 (1993) 513–517.
25] J.P. Raynauud, T. Ojasoo, L. Martini (Eds.), Medical Management of endometrio-sis, Raven Press, New York, 1984.
26] L.M. Kettel, A.A. Murphy, A.J. Morales, A. Ulman, E.E. Baulieu, S.S.C. Yen, Treat-ment of endometriosis with the antiprogesterone mifepristone (RU 486), Fertil.Steril. 65 (1996) 23–28.
27] A. Philips, K. Demarest, D.W. Hahn, F. Wong, J.L. Mc Guire, Progestational andandrogenic receptor binding affinities and in vivo activities of norgestimate andother progestins, Contraception 41 (1990) 399–410.
28] J.F.H.M. van Uem, J.G. Hsiu, C.F. Chillik, Contraceptive potential of RU-486by ovulation inhibition. I. Pituitary versus ovarian action with blockadeof estrogen-induced endometrial proliferation, Contraception 40 (1989)171–184.
29] U. Fuhrmann, H. Hess-Stumpp, A. Cleve, G. Neef, W. Schweede, J. Hoffmann,K.H. Fritzemeter, K. Chwalisz, Synthesis and biological activity of a novel,highly potent progesterone receptor antagonist, J. Med. Chem. 43 (2000)5010–5016.
30] D. Philibert, M. Hardy, M. Gaillard Moguilewski, F. Nique, C. Tournemine, L.Nédélec, New analogs or mifepristone with more dissociated antiprogesterone
with more dissociated antiprogesterone activities, J. Steroid Biochem. 34 (1989)413–417.31] F.-A. Kang, J. Guan, N. Jain, G. Allan, O. Linton, P. Tannebaum, X. Chen, J. Xu, P.Zhu, J. Gunnet, K. Damarest, S. Lundeen, Z. Sui, Parallel synthesis and SAR studyof novel oxa-steroids as potent and selective receptor antagonists, Bioorg. Med.Chem. Lett. 17 (2007) 2531–2534.




















