
Synthesis and Antitumor Activity of Potentially Tumor ...
375
City University of New York (CUNY) City University of New York (CUNY) CUNY Academic Works CUNY Academic Works Dissertations, Theses, and Capstone Projects CUNY Graduate Center 2-2020 Synthesis and Antitumor Activity of Potentially Tumor Targeting Synthesis and Antitumor Activity of Potentially Tumor Targeting Analogues of the Tetrahydrofuran Containing Acetogenins Analogues of the Tetrahydrofuran Containing Acetogenins Patricia Gonzalez Periche The Graduate Center, City University of New York How does access to this work benefit you? Let us know! More information about this work at: https://academicworks.cuny.edu/gc_etds/3641 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected]
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Synthesis and Antitumor Activity of Potentially Tumor ...

City University of New York (CUNY) City University of New York (CUNY)
CUNY Academic Works CUNY Academic Works
Dissertations, Theses, and Capstone Projects CUNY Graduate Center
2-2020
Synthesis and Antitumor Activity of Potentially Tumor Targeting Synthesis and Antitumor Activity of Potentially Tumor Targeting
Analogues of the Tetrahydrofuran Containing Acetogenins Analogues of the Tetrahydrofuran Containing Acetogenins
Patricia Gonzalez Periche The Graduate Center, City University of New York
How does access to this work benefit you? Let us know!
More information about this work at: https://academicworks.cuny.edu/gc_etds/3641
Discover additional works at: https://academicworks.cuny.edu
This work is made publicly available by the City University of New York (CUNY). Contact: [email protected]

i
SYNTHESIS AND ANTITUMOR ACTIVITY OF POTENTIALLY TUMOR TARGETING ANALOGUES OF
THE TETRAHYDROFURAN CONTAINING ACETOGENINS
by
PATRICIA GONZALEZ PERICHE
A dissertation submitted to the Graduate Faculty in Chemistry in partial fulfilment of the requirements for the degree of Doctor of Philosophy, The City University of New York
2020

ii
© 2020
PATRICIA GONZALEZ PERICHE
All rights reserved

iii
SYNTHESIS AND ANTITUMOR ACTIVITY OF POTENTIALLY TUMOR TARGETING ANALOGUES OF
THE TETRAHYDROFURAN CONTAINING ACETOGENINS
by
Patricia Gonzalez Periche
This manuscript has been read and accepted for the Graduate Faculty in Chemistry
In satisfaction of the dissertation requirement for the degree of
Doctor of Philosophy
________________________
Date
________________________
Date
______________________________________David R. Mootoo
Chair of Examining Committee
______________________________________Brian R. Gibney
Executive office
Supervisory Committee:
Ryan P. Murelli
Akira Kawamura
Naga Vara Kishore Pillarsetty
THE CITY UNIVERSITY OF NEW YORK

iv
ABSTRACT
Synthesis and antitumor activity of potentially tumor targeting analogues of the tetrahydrofuran containing
acetogenins
by
Patricia Gonzalez Periche
Advisor: Professor David R. Mootoo
The tetrahydrofuran containing acetogenins (THF-ACGs) are a naturally occurring class of compounds
with potent toxicity against a broad range of tumors, including multidrug resistant (MDR) strains. However,
the equally high toxicity to normal cells presents a hurdle for their clinical advancement. This study entails
synthetic and tumor targeting strategies that are relevant to their therapeutic development.
Chapter 1 presents a review of the synthesis and biological activity of the THF-ACGs. Chapter 2 covers a
modular approach for the synthesis of libraries of THF-ACGs. This strategy is illustrated in the synthesis
of C-10 epimers of 4-deoxyannonomontacin (4-DAN), one of the more potent monotetrahydrofuran
acetogenins. Towards the discovery of synthetically more accessible and tumor-selective analogues,
Chapter 3 describes the application of this methodology to analogues of 4-DAN where the THF ring is
substituted by mannose with different chain lengths and/or the butenolide is substituted by simpler
heterocycles.
To address the issue of systemic toxicity, Chapter 4 presents tumor-targeting strategies for the THF-
ACGs and their mimics. In the ‘chameleon’ approach the THF moiety was replaced with the 3-O-
carbamoyl-mannose residue, present in bleomycin, a family of clinically used anti-tumor agents and
believed to be responsible for their tumor selectivity. Therefore, the carbamoyl containing mannose
residue could confer both drug potency and selectivity. A prodrug strategy in which a naturally occurring
THF-ACG or synthetic analogue is conjugated via a cleavable linker to a tumor vector, is also described.
The guiding hypothesis is that following uptake by the tumor cell, the prodrug will be degraded to release

v
the active cytotoxic agent. As proof of principle, prodrugs comprising a sugar analogue of 4-DAN or the
naturally occurring acetogenin annonacin, 2-[3-(1,3-dicarboxypropyl)ureido] pentanedioic acid (DUPA), a
highly specific vector for prostate specific membrane antigen (PSMA) in prostate cancer (PCa), and a
traceless disulfide-based linker were synthesized.
Chapter 5 presents the cytotoxicity data for the new THF-ACGs and structure activity conclusions. Using
an MTT assay at both 16 and 48 hours, the cell viability of the THF-ACGs and their mimetics were
evaluated against five cancer cell lines: MDA-MD-231 (triple negative breast), MCF-7 (breast), HCT116
(colon), PC-3 (PSMA negative prostate cancer) and LNCaP (PSMA positive prostate). THF containing
analogues generally showed a lower IC50 (ca 0.5-5 micromolar) across the panel of cell lines than ones in
which the THF is replaced by a mannose. For the analogues with butenolide replacements, the
thiophene-2-carboxamide residue was the most active substitute and their derivatives showed
comparable or higher activity than their butenolide parents. The cytotoxicity of sugar mimetics with alkyl
ether chains of different lengths and positions was also determined. While individual analogues displayed
low micromolar potency and selectivity across the five cell lines tested, no clear structure activity trends
emerged. An ACG mimetic in which the THF and butenolide segments were replaced with 3-O-
carbamoyl-mannose and thiophene-2-carboxamide residues respectively, showed potent activity in the
0.1-10 M range against LNCaP and PC-3. Although not evaluated, the tumor selectivity of this mimetic is
particularly interesting given its structural similarity to the bleomycins.
The cytotoxicity activity of the DUPA-linked prodrugs derived from a mannose based ACG mimetic and
the naturally occurring ACG annonacin was also evaluated. In the 16 h MTT assay against LNCaP
(PSMA positive), the two prodrugs were more active than their parent drugs. However, against the PSMA
negative cell lines, MDA-MD-231, MCF-7, HCT116 and PC-3 both prodrugs and their parent drugs
showed similar activity and were appreciably less active than for LNCaP. The higher toxicity for LNCaP
over the PSMA negative cell lines, albeit modest (ca 2:1), supports the notion that this selectivity is
connected to DUPA-PSMA binding, and that PSMA is an internalizing receptor. However, in a 48 h MTT
assay, while the cytotoxicity of the prodrugs and parent drugs against MDA-MD-231, MCF-7, HCT116
was similar to that observed at 16 h, the activity of the prodrugs (but not the parent drugs), against PC-3

vi
increased markedly. This unexpected trend for PC-3 does not align with our prodrug hypothesis and may
be an indication that these DUPA-linked ACGs have a different mechanism of action than their parent
drugs. More robust studies are needed to resolve this issue, as well as to improve prodrug selectivity.

vii
“Our greatest weakness lies in giving up. The most certain
way to succeed is always to try just one more time.”
Thomas Edison

viii
ACKNOWLEDGEMENTS
First, I would like to thank my mentor, Professor Mootoo, for his constant support, patience and for
helping me grow both as a scientist and as a person. Thank you, Professor. The opportunity that you
provided for undergraduate research inspired me to pursue a Ph.D.
I would also like to thank my committee members, Professor Akira Kawamura, Professor Ryan Murelli
and Professor Naga Vara Kishore Pillarsetty for their support, guidance and stimulating conversations
over the years.
To the other members of the Mootoo lab, both past and present. Particularly, Dr. Stewart Bachan, Dr.
Ahmad Altiti and Dr. Clayton Mattis, Steven Truong, Amanda Ramdular, Dayanni Bhagwandin, Avelyn de
los Reyes, I am forever thankful for your help, encouragement and support over the years.
Thanks to everyone in the Chemistry Department at Hunter College. In particular, to Dr. Matthew Devany
for his help and patience with NMR characterization. To Professor Donna McGregor, thank you for
helping me come from Spain and for being a role model as a strong and successful woman in science.
Thank you for constant guidance. I would also like to acknowledge the academic and technical staff in the
department.
I am very grateful to Graduate Center for creating such a supportive work environment and for allowing
me the opportunity to earn a Ph.D. In particular, I would like to acknowledge my classmates for their
support.
To my friends in New York, my family away from home, thank you for the good times, the support and the
endless encouragement.
To my friends and family back home, thank you for always being there, even with an ocean in between
us.
To my mom and my sister, thank you for being there every step of the way and for helping me get where I
am today.

ix

x
TABLE OF CONTENTS
ABSTRACT.................................................................................................................................................. iv
ACKNOWLEDGEMENTS .......................................................................................................................... viii
TABLE OF CONTENTS ............................................................................................................................... x
LIST OF SYMBOLS AND ABBREVIATIONS ........................................................................................... xiii
LIST OF CANCER CELL LINES .............................................................................................................. xvii
LIST OF FIGURES .................................................................................................................................. xviii
LIST OF SCHEMES .................................................................................................................................... xx
LIST OF TABLES ...................................................................................................................................... xxi
Chapter 1. Tetrahydrofuran containing acetogenins: structure, in vivo and in vitro activity
1.1. Introduction and background ................................................................................................ 2
1.2. Tetrahydrofuran containing acetogenins ............................................................................. 2
1.3. Synthesis of THF-ACGs .......................................................................................................... 3
1.4. Mode of action ....................................................................................................................... 13
1.5. Activity studies ...................................................................................................................... 15
1.5.1. In vitro .............................................................................................................................. 16
1.5.2. In vivo .............................................................................................................................. 17
1.6. SAR studies ........................................................................................................................... 20
Chapter 2. Synthesis of 4-Deoxyannonmontacin
2.1. Introduction and background .................................................................................................. 24
2.2. Synthetic strategy ..................................................................................................................... 26
2.3. Synthesis ................................................................................................................................... 30
2.3.1. THF alkene, 2.16.................................................................................................................. 30
2.3.2. Butenolide alkene, 2.30 ....................................................................................................... 31
2.3.3. CM of THF and butenolide alkenes ..................................................................................... 32
2.3.4. Comparison of naturally obtained 4-DAN and synthetic C-10 epimers ............................... 33
2.4. Experimental .............................................................................................................................. 36

xi
Chapter 3. Analogues of 4-Deoxyannomontacin
3.1. Introduction and background .................................................................................................. 50
3.2. Butenolide substitutes ............................................................................................................. 51
3.2.1. Piperazine ............................................................................................................................ 51
3.2.2. Thiophene ............................................................................................................................ 52
3.2.3. O-butyl .................................................................................................................................. 53
3.3. Tetrahydrofuran substitutes .................................................................................................... 53
3.3.1. Mannose replacement ......................................................................................................... 53
3.4. Hybrid structures ...................................................................................................................... 55
3.5. Modifications on the hydrocarbon spacer ............................................................................. 56
3.6. Synthetic strategy ..................................................................................................................... 56
3.7. Synthesis ................................................................................................................................... 58
3.7.1. Mannose alkenes ................................................................................................................. 58
3.7.2. Butenolide replacements ..................................................................................................... 59
3.7.3. Cross metathesis and final products .................................................................................... 60
3.8. Summary ……………………………………………………………………………………………….62
3.9. Experimental .............................................................................................................................. 64
Chapter 4. Design and synthesis of potential tumor-selective cytotoxic agents
4.1. Introduction and background .................................................................................................. 92
4.2. The ‘chameleon’ approach ....................................................................................................... 92
4.2.1. Design .................................................................................................................................. 93
4.2.2. Synthesis .............................................................................................................................. 94
4.3. The prodrug approach .............................................................................................................. 95
4.3.1. Design .................................................................................................................................. 95
4.3.2. Drugs .................................................................................................................................... 96
4.3.2. Receptor .............................................................................................................................. 97
4.3.3. Vector ................................................................................................................................... 97
4.3.4. Linker ................................................................................................................................... 98
4.4. C11 mannose derivative .......................................................................................................... 100
4.4.1. Sugar-butenolide Prodrug Synthesis ................................................................................. 100
4.4.2. Sugar-thiophene Prodrug Synthesis .................................................................................. 103
4.5. Annonacin derivative .............................................................................................................. 104
4.5.1. Isolation, purification and characterization of annonacin ................................................... 104

xii
4.5.2. Annonacin prodrug synthesis ............................................................................................. 108
4.6. Conclusions ............................................................................................................................. 112
4.7. Experimental ............................................................................................................................ 114
Chapter 5. Antitumor activity of 4-Deoxyannomontacin and analogues
5.1. Introduction ............................................................................................................................. 131
5.2. Results ..................................................................................................................................... 131
5.2.1. THF with butenolide or butenolide substitutes (1, 2, 3, 4, 5) ............................................. 132
5.2.2. Butenolide analogues with sugar substitutes for the THF (6, 7, 8, 9, 10) .......................... 134
5.2.3. Hybrid analogues with THF and butenolide substitutes (12, 13, 14, 15) ........................... 135
5.3. Discussion ............................................................................................................................... 137
5.4. Prodrug studies ....................................................................................................................... 138
5.4.1. Cytotoxicity data for 6 and 15 against LNCaP……………………………………………...…139
5.4.2. Cytotoxicity data for 6 and 15 against PSMA negative cells………….……………………..140
5.4.3. Annonacin prodrugs…………………………………………………………..……………….…142
5.5. Summary and future directions ............................................................................................. 143
5.6. Experimental ............................................................................................................................ 145
APPENDIX ................................................................................................................................................ 146
REFERENCES .......................................................................................................................................... 344

xiii
LIST OF SYMBOLS AND ABBREVIATIONS
Ac
Ac2O
AgOTf
ATP
Bn
brine
br
brsm
BST
Bu
oC
ca
calcd
cat
CM
13C NMR
CSA
COSY
δ
d
DEAD
DCC
DCM
DIBAL-H
DMAP
DMF
acetyl
acetic anhydride
silver trifluoromethanesulfonate
adenosine triphosphate
benzyl
saturated aqueous sodium chloride solution
broad
based on recovering started material
brine shrimp letality test
butyl
degree Celsius
about
calculated
catalytic
cross metathesis
carbon-13 nuclear magnetic resonance spectrometry
camphorsulfonic acid
correlation spectroscopy
chemical shift in ppm
doublet
diethyl azodicarboxylate
dicyclohexylcarbodiimide
dichloromethane
diisobutylaluminum hydride
4-dimethylaminopyridine
N,N-dimethylformamide

xiv
2,2-DMP
DMSO
ED50
ee
Et2O
EtOAc
EtOH
eq
EVE
FCC
g
h
1H NMR
hmn
HRMS
HSQC
Hz
IC50
J
L
m
M
mCPBA
Me
MeOH
mg
MHz
min
2,2-dimetoxypropane
dimethyl sulfoxide
effective dose
enantiomeric excess
diethyl ether
ethyl acetae
ethanol
equivalent
ethyl vinyl ether
flash column chromatography
gram
hour
proton nuclear magnetic resonance spectrometry
human
high resolution mass spectrometry
heteronuclear single quantum correlation spectroscopy
hertz
50% inhibitory concentration
coupling constant
liter
multiplet
molar
meta-chloroperoxybenzoic acid
methyl
methanol
milligram
megahertz
minute

xv
mL
mmol
MOM
MP
MTPA
MS
ND
NIS
PE
PCC
Ph
Piv
ppm
PPTS
pTSOH
q
rt
s
SAR
t
TBAF
TBAI
TBDPS
TFA
THF
THP
TLC
Tr
milliliter
millimole
chloromethyl methyl
melting point
α-methoxy-α-(trifluoromethyl)phenyl acetic acid
molecular sieves
Not determined
N-Iodosuccinimide
petroleum ether
pyridinium chlorochromate
phenol
pivaloyl or trimethylacetyl)
parts per million
para-pyridinium toluenesulfonate
para-toluene sulphonic acid
quartet
room temperature
singlet
structure activity relationship
triplet
tetra-n-butylammonium fluoride
tetra-n-butylammonium iodide
tert-butyldiphenylsilyl
trifluoroacetic acid
tetrahydrofuran
tetrahydropyran
thin layer chromatography
trityl or triphenylmethyl

xvi
Ts
vs
toluenesulfonyl
versus

xvii
LIST OF CANCER CELL LINES1
3PS
9PS
A-2780
A-549
HeLa
Hep G2
HepS
L1210
LNCaP
MCF-7
MDA-MB-231
MKN-45
OC-194
OC-222
OVCAR-3
PACA-2
PC-3
S180
SMMC-7721
mouse leukemia
mouse lymphocytic leukemia
hmn ovarian
hmn non-small lung
hmn cervical epithetial
hmn liver
liver
lymphocytic leukemia
hmn prostate
hmn breast
mn breast
hmn gastric
hmn ovarian epithelial
hmn ovarian epithelial
ovarian adenocarcinoma
hmn pancreas
hmn prostate
murine sarcoma
hmn liver

xviii
LIST OF FIGURES
Chapter 1. Tetrahydrofuran containing acetogenins: structure, in vivo and in vitro activity
Figure 1.1. Structure of uvaricin .................................................................................................................... 2
Figure 1.2. Structural features of the THF containing acetogenins .............................................................. 3 Figure 1.3. Proposed interaction of the THF-ACGs with mitochondria ....................................................... 13
Figure 1.4. Takada studies on the modification on the butenolide ............................................................. 21
Chapter 2. Synthesis of 4-Deoxyannonmontacin
Figure 2.1. Structure of 4-deoxyannomontacin .......................................................................................... 24
Figure 2.2. 4-deoxyannoreticuin (2.10 and 2.11) and 4-deoxyannomotacin (2.12) .................................... 26 Figure 2.3. Previous THF-ACGs by the Mootoo laboratory ........................................................................ 27
Figure 2.4. Konno’s CM approach to solamin type structures ................................................................... 28 Figure 2.5. CM approach to the synthesis of the C-10 epimeric mixture of 4-DAN .................................... 29
Figure 2.6. X-ray structure of compound 2.39 ............................................................................................ 30 Figure 2.7. 1H chemical shifts of 4-DAN both naturally obtained and synthesized ..................................... 34
Figure 2.8. 13C chemical shifts of 4-DAN both naturally obtained and synthesized ................................... 35
Chapter 3. Analogues of 4-Deoxyannomontacin
Figure 3.1. Structural characteristics of the THF-AGEs and proposed replacements ................................ 50
Figure 3.2. Piperazine analogues ............................................................................................................... 51 Figure 3.3. Thiophene derivative ................................................................................................................ 52
Figure 3.4. Simple n-alkyl derivative ........................................................................................................... 53 Figure 3.5. Activity of DAR 3.8 and mannose analog 3.9 ........................................................................... 54
Figure 3.6. THF replacement analogues .................................................................................................... 55
Figure 3.7. Hybrid compounds .................................................................................................................... 55
Figure 3.8. Deoxygenated analog ............................................................................................................... 56
Figure 3.9. Coupling alkene partners for the synthesis of analogues of 4-DAN ......................................... 57
Chapter 4. Design and synthesis of potential tumor-selective cytotoxic agents
Figure 4.1. Bleomycin and carbamoylmannose derivative 4.2 ................................................................... 93
Figure 4.2. Recognition mechanism of prodrugs ....................................................................................... 95 Figure 4.3. Example of a prodrug construct using 4-DAN as the parent drug ............................................ 95
Figure 4.4. Proposed drug and prodrug pairs 4.9-4.10, 4.11-4.12, 4.13-4.14 ............................................ 96 Figure 4.5. DUPA derived vector ................................................................................................................ 97
Figure 4.6. Structure of the linker (left) and release mechanism of the prodrug (right) .............................. 99
Figure 4.7. 1H of the C11-butenolide prodrug 4.10 ................................................................................... 102 Figure 4.8. Annona muricata ..................................................................................................................... 104 Figure 4.9. Extraction of annonacin .......................................................................................................... 105 Figure 4.10. Purification of annonacin ...................................................................................................... 105 Figure 4.11. 1H NMR of annonacin reported in the literature .................................................................... 106
Figure 4.12. 1H NMR isolated annonacin .................................................................................................. 106
Figure 4.13. Linker-annonacin purification. ............................................................................................... 108 Figure 4.14. Fraction 1.1 (4.40 or 4.41) .................................................................................................... 110 Figure 4.15. Fraction 1.2 (4.38) ................................................................................................................ 110

xix
Chapter 5. Antitumor activity of 4-Deoxyannomontacin and analogues
Figure 5.1. SAR conclusion based on cytotoxicity assays ........................................................................ 137 Figure 5.2. Structure of compound 6 and its prodrug, 15 ......................................................................... 138 Figure 5.3. Cytotoxicity at 16 and 48 hours of compounds 6 and 15 in PSMA positive cells ................... 139
Figure 5.4. Cytotoxicity at 16 and 48 hours of compounds 6 and 15 in PSMA negative cells ................. 140 Figure 5.5. Structure of annonacin, 2, and annonacin prodrug, 16 .......................................................... 142 Figure 5.6. Cytotoxicity at 16 and 48 hours of compounds 2 and 16 in prostate cancer cells ................. 143

xx
LIST OF SCHEMES
Chapter 2. Synthesis of 4-Deoxyannonmontacin
Scheme 2.1. The Wu approach to 4-DAN .................................................................................................. 25
Scheme 2.2. Synthesis of the THF-alkene 2.16 ......................................................................................... 31 Scheme 2.3. Synthesis of butenolide 2.30.................................................................................................. 32
Scheme 2.4. CM reaction and synthesis of C-10 epimers of 4-DAN .......................................................... 33
Chapter 3. Analogues of 4-Deoxyannomontacin
Scheme 3.1. Synthesis of mannose alkenes .............................................................................................. 58
Scheme 3.2. Synthesis of butenolide replacements ................................................................................... 59 Scheme 3.3. Synthesis of the compounds with butenolide replacements .................................................. 60
Scheme 3.4. Synthesis of mannose containing compounds ...................................................................... 61 Scheme 3.5. Route to hybrid analogues ..................................................................................................... 62
Chapter 4. Design and synthesis of potential tumor-selective cytotoxic agents
Scheme 4.1. Synthesis of “chameleon” compound 4.7 .............................................................................. 94
Scheme 4.2. Synthetic route to the synthesis of the DUPA derived vector 4.15 ........................................ 98 Scheme 4.3. Synthesis of linker 4.24 .......................................................................................................... 99
Scheme 4.4. Synthesis of the C11-butenolide prodrug .............................................................................. 101 Scheme 4.5. Synthesis of the C11-thiophene prodrug .............................................................................. 103
Scheme 4.6. Synthesis of the annonacin based prodrug ........................................................................ 109
Scheme 4.7. En route to the synthesis of 4-DAN prodrug ........................................................................ 112

xxi
LIST OF TABLES
Chapter 1. Tetrahydrofuran containing acetogenins: structure, in vivo and in vitro activity
Table 1.1. Strategies for the thetrahydrofuran ring synthesis ....................................................................... 4
Table 1.2. Synthetic approaches to the butenolide ....................................................................................... 7
Table 1.3. Coupling strategies of the THF and butenolide segments ........................................................... 8
Table 1.4. Recent publications on THF-ACGs syntheses .......................................................................... 11
Table 1.5. Example of the data disparity among different cell assasys in the THF-ACGs ......................... 17
Table 1.6. Summary of in vivo studies of THF-AGCs ................................................................................. 19
Chapter 2. Synthesis of 4-Deoxyannonmontacin
Table 2.1. Naturally obtained vs synthetic 4-DAN ...................................................................................... 34
Chapter 4. Design and synthesis of potential tumor-selective cytotoxic agents
Table 4.1.13C of literature reported and isolated annonacin ..................................................................... 107
Chapter 5. Antitumor activity of 4-Deoxyannomontacin and analogues
Table 5.1. Cell viability (%) over 16 h at 12.5 and 25 µM ......................................................................... 132
Table 5.2. THF with butenolide or butenolide substitutes IC50s (µM) in 16 and 48 h assays ................... 133 Table 5.3. Cell viability (%) over 16 h at 12.5 and 25 µM ......................................................................... 134
Table 5.4. Cell viability (%) over 16 h at 12.5 and 25 µM ......................................................................... 135 Table 5.5. Hybrid compounds IC50s (µM) (16 and 48 h assays) .............................................................. 136
Table 5.6. Comparison of activity (% cell viability) of 15 for LNCaP vs PSMA negative cell lines at 5 M
.................................................................................................................................................................. 141

1
TETRAHYDROFURAN CONTAINING ACETOGENINS:
STRUCTURE, IN VIVO AND IN VITRO ACTIVITY

2
1.1. Introduction and background
Cancer remains a major health challenge despite major advances in early detection, diagnosis and
treatment. The current lines of treatment involve chemotherapy, surgery, radiation or most commonly, a
combination of the three. Small organic molecules are a primary line of treatment and usually those have
been identified from natural sources. Plants have been used since ancient times to treat many diseases:
between the 1940s and 2014, 175 molecules were approved for the treatment of cancer and 49% of
those are natural products or modified natural compounds.2 To this end, the naturally occurring
tetrahydrofuran containing acetogenins (THF-ACGs) are of interest because of their high potencies
against a broad panel of human cancer cell lines, including several multidrug resistant (MDR) ones and
their relatively simple structures. However, while certain THF-ACGs show selectivity for certain cancer
cells, challenges for their clinical use are systemic toxicity and a lack of understanding of their
mechanisms of action.
1.2. Tetrahydrofuran containing acetogenins
The tetrahydrofuran containing annonaceous acetogenins (THF-ACGs) are a relatively untested family of
cytotoxic agents isolated from the Annona species. These evergreen trees are usually found in tropical
and subtropical areas. ACGs can be found in the leaves, roots, seeds, pulp of fruits like graviola, soursop,
pawpaw, guanabanana, Brazilian pawpaw and
custard apple. The first compound of this class,
uvaricin3 1.1, was isolated in 1982 via an activity-
directed fractionalization using 3PS cells in vivo
(murine leukemia) testing of the extracts. Its
unusual bioactivity attracted the attention of many,
hoping to isolate and/or synthesize the future
generation of antitumor drugs based on this new Figure 1.1. Structure of uvaricin

3
class of compounds. Since then, more than 400 compounds have been isolated from natural sources.4,5,6
Many of them have been synthesized and analogues have been designed and synthesized seeking for
the “magic bullet”. Many of them have antitumoral effects, as well as antimicrobial, antimalarial,
antifeedant, antiviral, anthelminthic, antiprotozoal, pesticidal and immunosuppressive.7,2,8 Unfortunately,
as of 2019, this class of compounds are still strong candidates for the development of new drugs, but the
lack of understanding of their mode of action makes targeting of specific tumors and designing active and
selective analogues a tremendous challenge.
The highly lipophilic THF-ACGs derive from the polyketide pathway and have a C35 or C37 aliphatic chain
that contains: (i) one, two or three THF tetrahydrofuran rings that can be adjacent or not; (ii) a terminal α,
β-unsaturated δ-lactone; (iii) a series of oxygenated groups (hydroxyls, acetoxyls, ketones, epoxides)
and/or double bonds (Figure 1.2).
Figure 1.2. Structural features of the THF containing acetogenins
1.3. Synthesis of THF-ACGs
The potent biological activities of the THF-ACGs and their stereochemical complexity have motivated
considerable interest in their synthesis. Since 1996, over 100 total syntheses have been reported.9, 10, 11
This topic has been extensively reviewed.11c,12

4
Convergent, linear and biomimetic strategies for naturally occurring THF-ACGs, derivates, as well as non-
natural analogues have been described. A major emphasis has been the synthesis of the
stereochemically complex tetrahydrofuran segment which has employed inexpensive chiral starting
materials, such as carbohydrates and amino acids, and a variety of asymmetric reactions including: (i)
oxidative cyclization of hydroxy alkenes, (ii) epoxidation-epoxide opening sequence on 4-alkene-1-ol
precursor, (iii) iodoetherification of hydroxy alkenes, (iv) ring closing metathesis, (v) Sharpless
dihydroxylation followed by acid catalyzed cyclization; (vi) base catalyzed cyclization;11d (vii)
diastereoselective Williamson reaction; (viii) chiral ligand-controlled cyclization; (ix) radical cyclization.
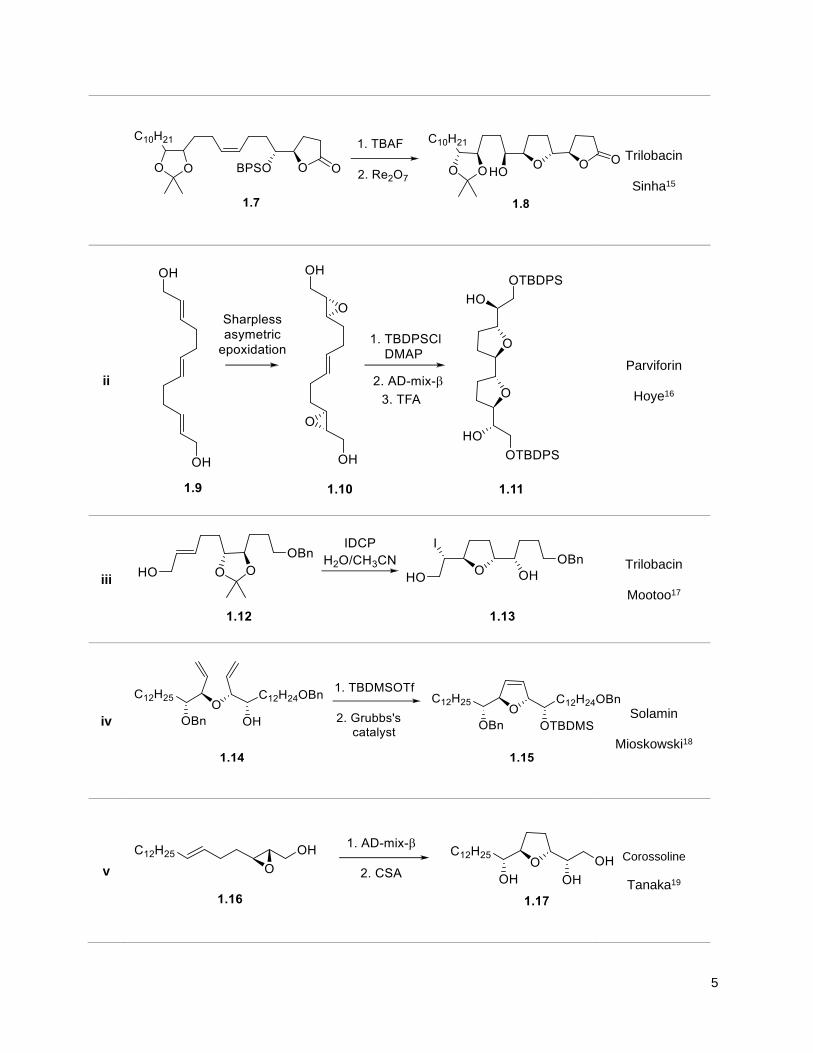
Table 1.1 exemplifies the more popular strategies.
Table 1.1. Strategies for the tetrahydrofuran ring synthesis
Examples Target / Group
i
cis- solamin
Brown13
(+)-cis-solamin
Donohoe14
5
Trilobacin
Sinha15
ii
Parviforin
Hoye16
iii
Trilobacin
Mootoo17
iv
Solamin
Mioskowski18
v
Corossoline
Tanaka19
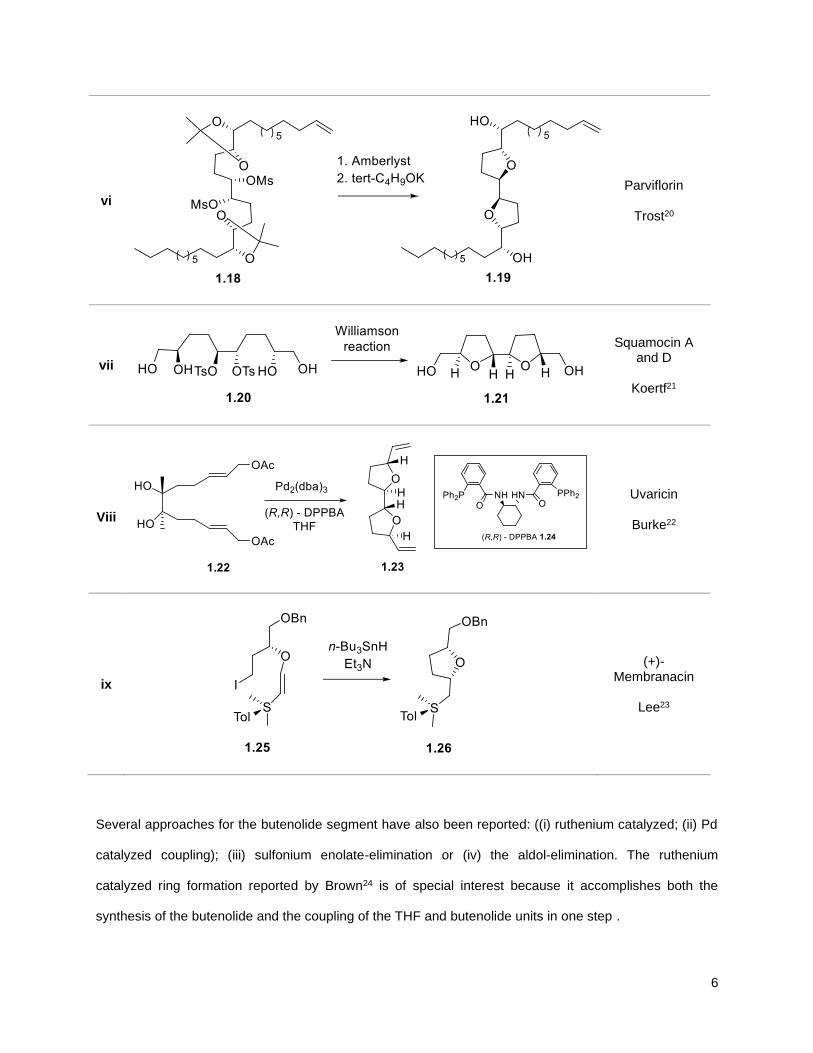
6
vi
Parviflorin
Trost20
vii
Squamocin A and D
Koertf21
Viii
Uvaricin
Burke22
ix
(+)- Membranacin
Lee23
Several approaches for the butenolide segment have also been reported: ((i) ruthenium catalyzed; (ii) Pd
catalyzed coupling); (iii) sulfonium enolate-elimination or (iv) the aldol-elimination. The ruthenium
catalyzed ring formation reported by Brown24 is of special interest because it accomplishes both the
synthesis of the butenolide and the coupling of the THF and butenolide units in one step .
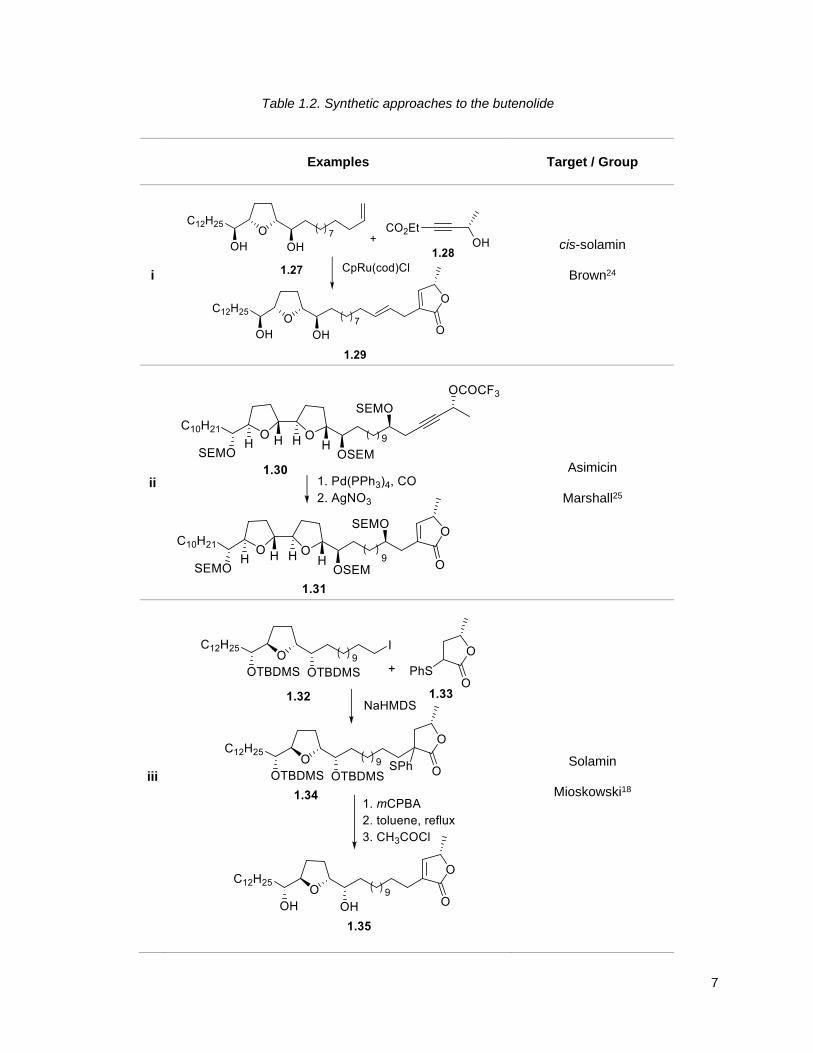
7
Table 1.2. Synthetic approaches to the butenolide
Examples
Target / Group
i
cis-solamin
Brown24
ii
Asimicin
Marshall25
iii
Solamin
Mioskowski18
8
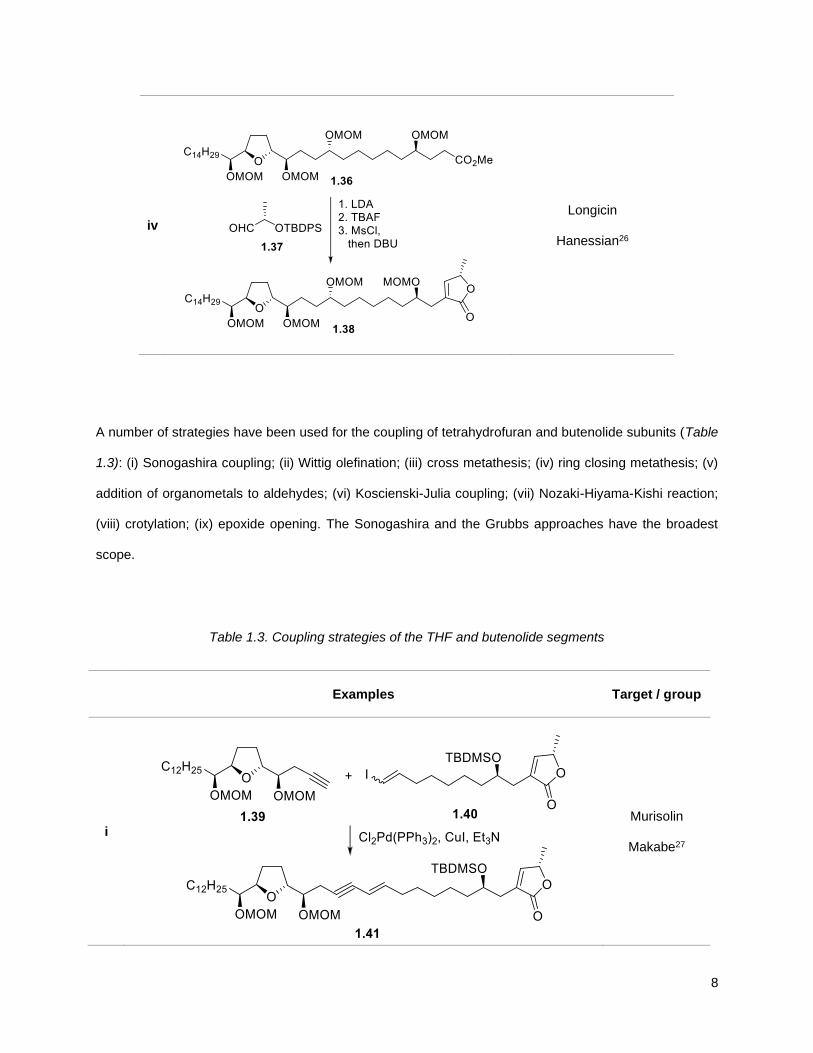
iv
Longicin
Hanessian26
A number of strategies have been used for the coupling of tetrahydrofuran and butenolide subunits (Table
1.3): (i) Sonogashira coupling; (ii) Wittig olefination; (iii) cross metathesis; (iv) ring closing metathesis; (v)
addition of organometals to aldehydes; (vi) Koscienski-Julia coupling; (vii) Nozaki-Hiyama-Kishi reaction;
(viii) crotylation; (ix) epoxide opening. The Sonogashira and the Grubbs approaches have the broadest
scope.
Table 1.3. Coupling strategies of the THF and butenolide segments
Examples
Target / group
i
Murisolin
Makabe27
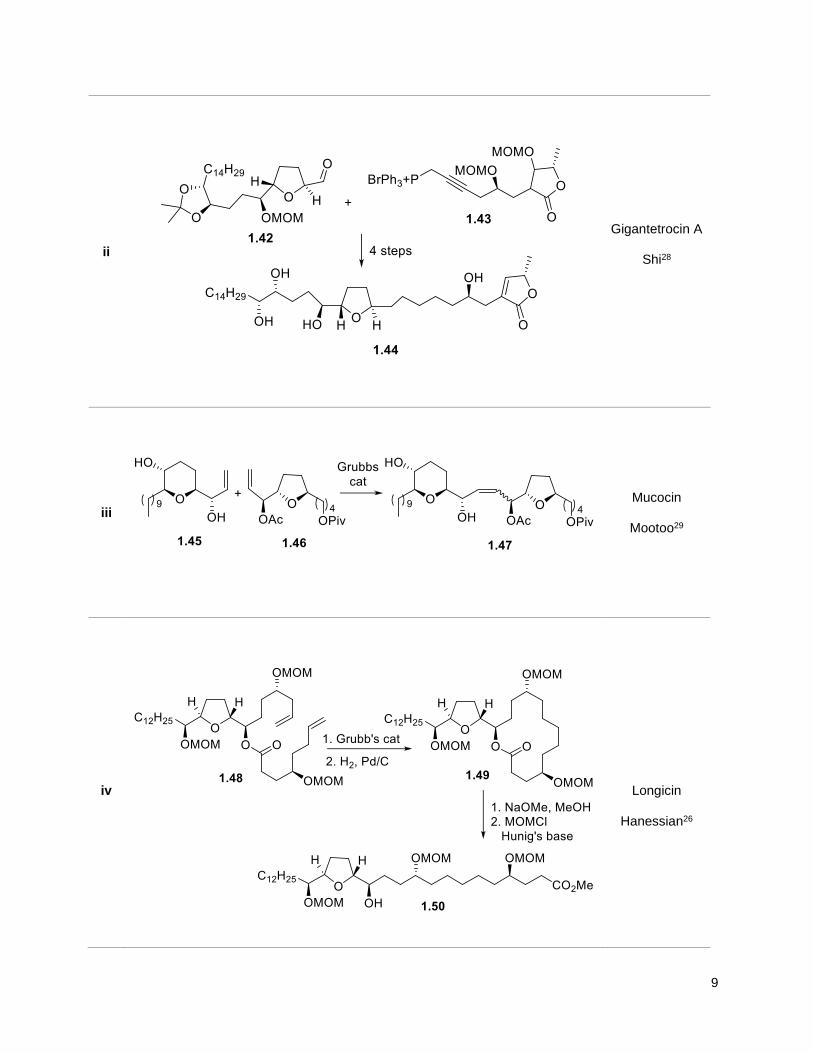
9
ii
Gigantetrocin A
Shi28
iii
Mucocin
Mootoo29
iv
Longicin
Hanessian26
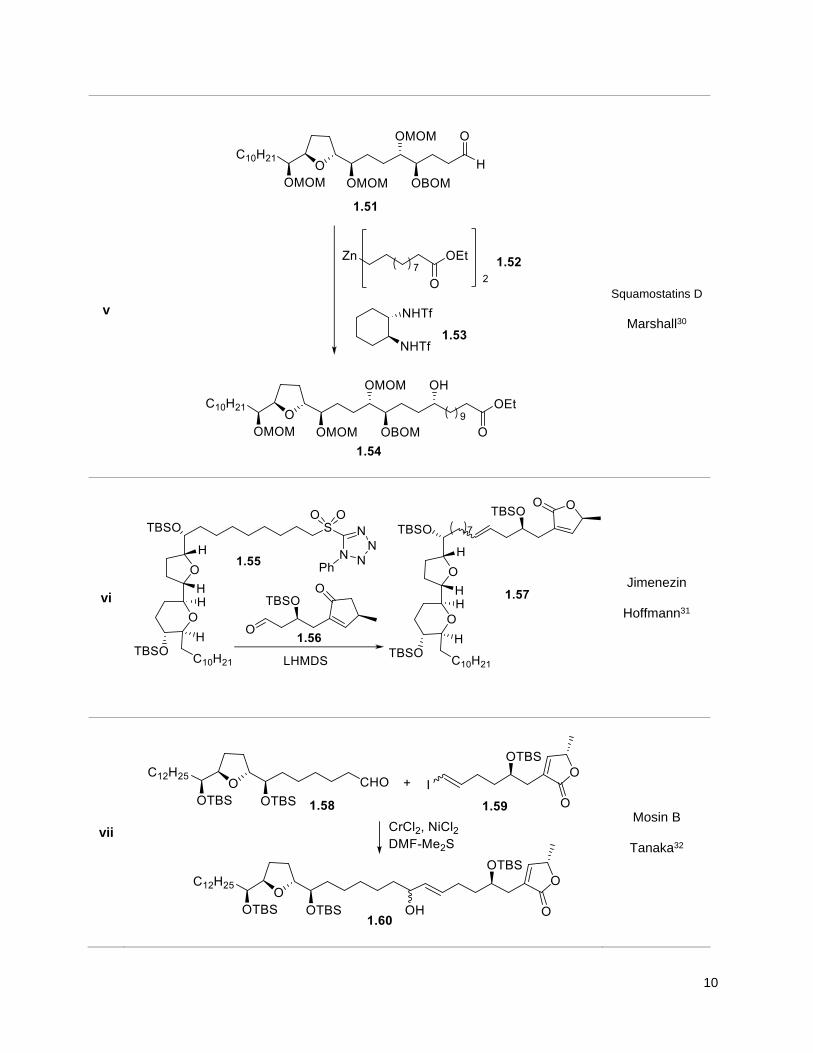
10
v
Squamostatins D
Marshall30
vi
Jimenezin
Hoffmann31
vii
Mosin B
Tanaka32
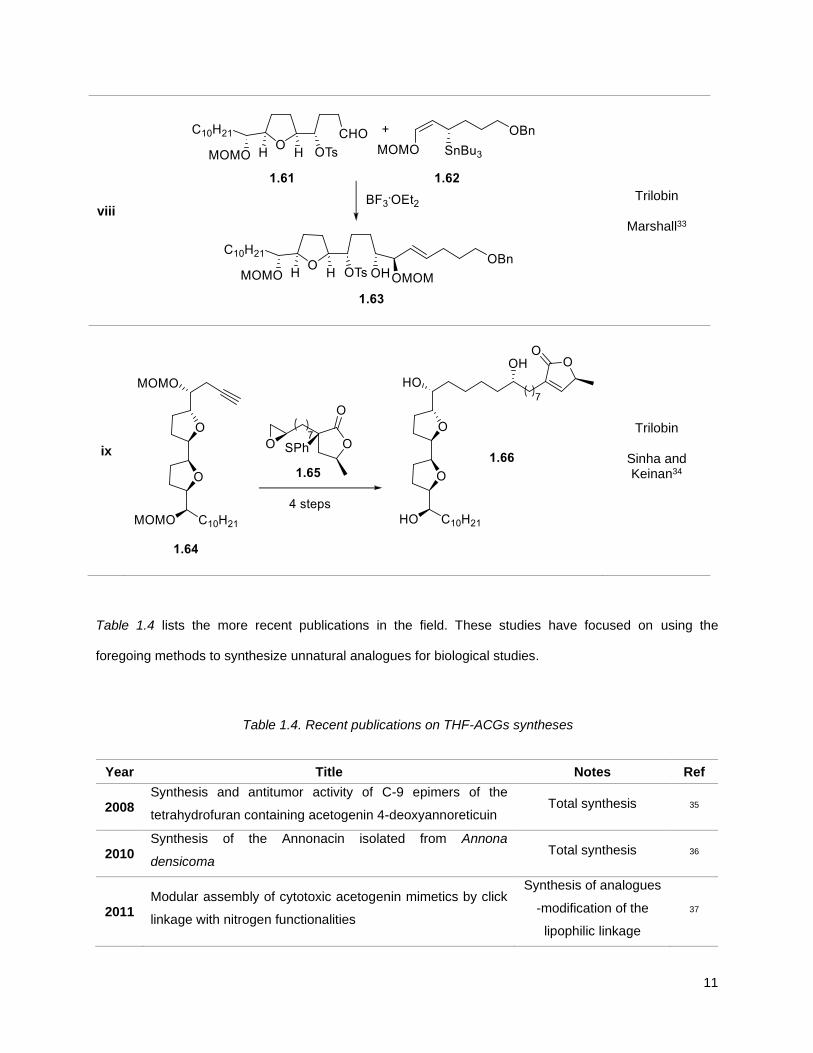
11
viii
Trilobin
Marshall33
ix
Trilobin
Sinha and Keinan34
Table 1.4 lists the more recent publications in the field. These studies have focused on using the
foregoing methods to synthesize unnatural analogues for biological studies.
Table 1.4. Recent publications on THF-ACGs syntheses
Year Title Notes Ref
2008 Synthesis and antitumor activity of C-9 epimers of the
tetrahydrofuran containing acetogenin 4-deoxyannoreticuin Total synthesis 35
2010 Synthesis of the Annonacin isolated from Annona
densicoma Total synthesis 36
2011 Modular assembly of cytotoxic acetogenin mimetics by click
linkage with nitrogen functionalities
Synthesis of analogues
-modification of the
lipophilic linkage
37

12
2013
Critical role of a methyl group on the δ-lactones ring of
annonaceous acetogenins in the potent inhibition of
mitochondrial complex I
Development of
analogues for SAR
studies
38
2013
Structure-activity relationships of hybrid annonaceous
acetogenins: powerful growth inhibitory effects of their
connecting groups between heterocycle and hydrophobic
carbon chain bearing THF ring on human cancer cell lines
Solamin analogues
using as butenolide
replacement N-pyrazole
39
2013 Total synthesis of 14,21-diepi-squamocin-K Total synthesis for SAR
studies 40
2014
New cytotoxic annonaceous acetogenin mimetics having a
nitrogen-heterocyclic terminal and their application to cell
imaging
Analogues
development -
substitution of the
butenolide
41
2014 Thiophene-3-carboxamide analogue of annonaceous
acetogenins as antitumor drug lead
Substitution of the
butenolide by a
thiophene delivers new
potent analogues
42
2014 Total synthesis of Muricadienin, the Putative Key precursor
in the Solamin Biosynthesis Total synthesis 43
2015
Synthesis of dansyl-labeled probe of thiophene analogues of
annonaceous acetogenins for visualization of cell
distribution and growth inhibitory activity toward human
cancer cell lines
Synthesis of a probe for
imaging studies 44
2016
Total synthesis of (+)- Goniodecin Total synthesis
45
2017
Covergent synthesis of stereoisomers of THF ring moiety of
acetogenin thiophene analogue and their antiproliferative
activities against human cell lines
Synthesis of analogues
– replacement of
butenolide by a
thiophene moiety
46
2017
Total synthesis of two possible diastereomers of natural 6-
chlorotetrahydrofuran acetogenin and its stereostructural
elucidation
Total synthesis 47
2018 Convergent total synthesis of asimicin via decarbonylative
radical dimerization Total synthesis 48
2018
A general diastereoselective strategy for both cis- and trans-
2,6- disubstituted tetrahydropyrans: formal total synthesis of
(+)-muconin
Total synthesis 49
13
It is evident that organic chemists have been fascinated with this class of complex structures since their
discovery and are excited and intrigued by their wide range of biological activities and potential use in the
clinic.
1.4. Mode of action
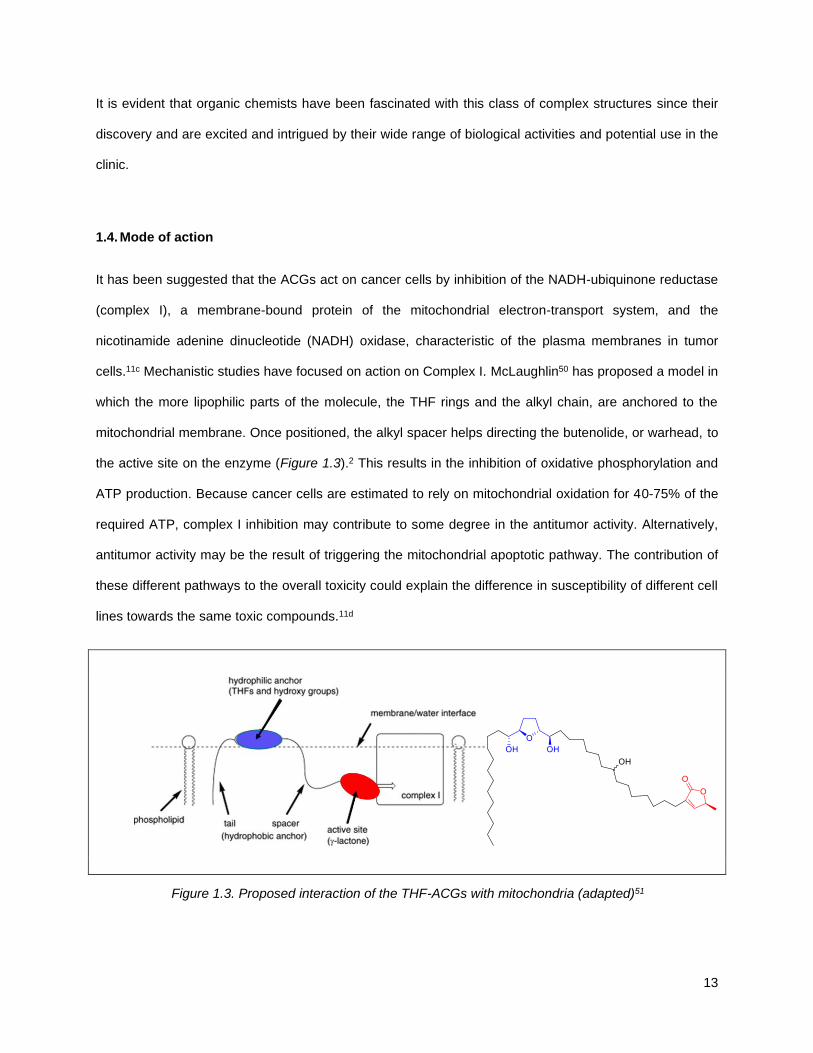
It has been suggested that the ACGs act on cancer cells by inhibition of the NADH-ubiquinone reductase
(complex I), a membrane-bound protein of the mitochondrial electron-transport system, and the
nicotinamide adenine dinucleotide (NADH) oxidase, characteristic of the plasma membranes in tumor
cells.11c Mechanistic studies have focused on action on Complex I. McLaughlin50 has proposed a model in
which the more lipophilic parts of the molecule, the THF rings and the alkyl chain, are anchored to the
mitochondrial membrane. Once positioned, the alkyl spacer helps directing the butenolide, or warhead, to
the active site on the enzyme (Figure 1.3).2 This results in the inhibition of oxidative phosphorylation and
ATP production. Because cancer cells are estimated to rely on mitochondrial oxidation for 40-75% of the
required ATP, complex I inhibition may contribute to some degree in the antitumor activity. Alternatively,
antitumor activity may be the result of triggering the mitochondrial apoptotic pathway. The contribution of
these different pathways to the overall toxicity could explain the difference in susceptibility of different cell
lines towards the same toxic compounds.11d
Figure 1.3. Proposed interaction of the THF-ACGs with mitochondria (adapted)51

14
The mitochondrial apoptotic pathway entails the efflux of mitochondrial contents into the cytosol as a
result of the disruption of the mitochondrial membrane, which in turn may be promoted by depletion of
mitochondrial ATP. In this context, different apoptotic mechanisms have been studied.
Alteration of the cytosol would affect the endoplasmic reticulum causing release of Ca2+. The inability of
the cell to restore Ca2+ homeostasis, will trigger caspase activation an eventually cell death. This
sequence of events that are part of the endoplasmic reticulum stress disorder (ERS) were observed by
Wu’s group52 when treating the human nasopharyngeal carcinoma (NPC) cells with THF-ACGs.
Other indicators of the apoptotic pathway are the alteration of the Bcl-2/Bax protein ratio, the activation of
caspases 3 and 9 and the production superoxide anion and hydrogen peroxide (ROS). All of which were
reported by Tormo53 when treating Hep2G cells with THF-AGEs. Tormo also reported a dramatic
decrease in the mitochondrial transmembrane potential (MTP) – a measurement of the mitochondrial
function, as it is generated by the pumping of electrons across the membrane- and in the mitochondrial
membrane permeability transition pore (MPTP) – which opening allows calcium homeostasis.
Other signals/targets that have been reported to be altered by the THF-AGEs related to the mitochondrial
apoptotic pathway are: (i) Notch signaling54. Treatment of gastric cancer cells with THF-ACGs resulted in
Notch2 up-regulation in a dose dependent manner, which resulted in cell growth inhibition; (ii) The
hypoxia-inducible factor-1 (HIF-1)55 activates the expression of more than 100 genes involved in
adaptation and cell survival under hypoxia conditions. It is suggested that hypoxia increases the levels of
ROS, which in turn inactivate HIF-1α protein, resulting in activation of HIF-1. T47D cells treated with THF-
ACGs inhibited HIF-1 activation with IC50 values from 0.02-31 µM; (iii) Estrogen receptor-α (ERα).
Annonacin decreased cell survival in ERα positive cells but not on ERα negative cells, suggesting that the
receptor may play a key role in the mode of action of THF-AGEs. Moreover, annonacin also inhibited
expression of D1 and Bcl-2 receptors. These in vitro results were confirmed in in vivo assays using cell-
grafted nude mice.
It has also been observed that ACGs act against certain multidrug resistant (MDR) tumors.8 MDR is

15
attributed to an increased expression of the P-170 glycoprotein, drug efflux pump in the plasma
membrane. ACGs may act against MDR cells by their inhibition of ATP production, which is required for
active efflux.
The inhibition of NADH oxidase in the plasma membrane of certain tumor cells, involved in ATP
production for glycolysis, may lead to both necrotic and apoptotic pathways. This may explain the
selectivity observed against certain cell lines. Another basis for selectivity, could be that the mitochondria
for certain tumor cells seems to be different from their healthy counterparts. They exhibit comprehensive
metabolic differences that makes them more sensitive to mitochondrial perturbation than normal cells.56
In this context, the ACGs are an interesting group of potential clinical agents.
1.5. Activity studies
In the early stages of extraction and isolation of THF-AGEs from seeds, pulp, leaves or other parts of the
Annonaceae plants, the potential as cytotoxic agents of this class of compounds was recognized. In order
to evaluate the extracts, each one of the partition fractions from the extractions of the naturally obtained
samples were tested using the brine shrimp lethality assay. If the fraction contained the cytotoxic
compound, shrimp would not survive, and therefore the extract would be further purified to isolate
individual compounds. That could be considered the first cytotoxicity assessment of THF-ACGs.
Some research has been focused on the biological evaluation in vitro and in vivo of the crude extracts
from the plants57,58 and other researchers have isolated compounds from the extracts and studied their
biological activities. Even though the data collected for the extracts is valuable and seems to be in
concordance with the one of individual compounds, it is beyond the scope of this discussion and we will
center our attention in studies of isolated compounds.
When first discovered, the tetrahydrofuran containing annonaceous acetogenins (THF-ACGs) showed
potent cytotoxicity against a wide range of human tumor cell lines. The first in vivo study carried out by
the Upjohn company59 showed great potential against L1210 murine leukemia and A2780 ovarian

16
xenografts. Thereafter, the interest in this family of compounds, grew exponentially, resulting in the
isolation of 400 compounds in 27 years in the search of the ‘magic bullet’ against tumor cells. Even
though in vitro studies have proven useful to identify potential cytotoxic compounds or “magic bullets”
have not been so successful when carried out in vivo. In solid tumors, other factors need to be considered
such as pH differences or gradient of nutrition concentrations. Those have a great impact in the cell
viability, the metabolism and the sensitivity to treatment60 and may be the reason why these compounds
have not yet been advanced to clinical studies.
Although these compounds showed potential in the cell studies, there has been a gradual loss of interest
in this class of compounds due to the challenges with systemic toxicity. In particular there is a concern in
neuronal toxicity. It has been reported that in regions where there is a high consumption of herbal tea and
fruits from plants of the Annonaceae family61 there is a high rate of parkinsonism. Targeting compounds
to cancer cells could address these issues.
1.5.1. In vitro
The lack of a stablished protocol towards the evaluation of the compounds, makes the makes the
reproducibility and comparison of newly acquired data challenging. Based on the comparison of different
reports, the activity seems to vary with incubation time. Holschneider62 treated different cell lines with
bullatacin for 48 and 72 h observing ED50s of 10 vs 10-7 µg/mL for OC-194 cells and 50 vs 4 µg/mL for
OVCAR-3 cells, respectively. Cytotoxicities vary with incubation time and across different cell lines. To
this date there is not an explanation as to why some cell lines are more affected by THF-ACGs than
others. Elucidation of the mode of action could bring some insight to this matter. The disparity of data
collected by different groups on bullatacin is exemplified in Table 1.5.63, 64, 65
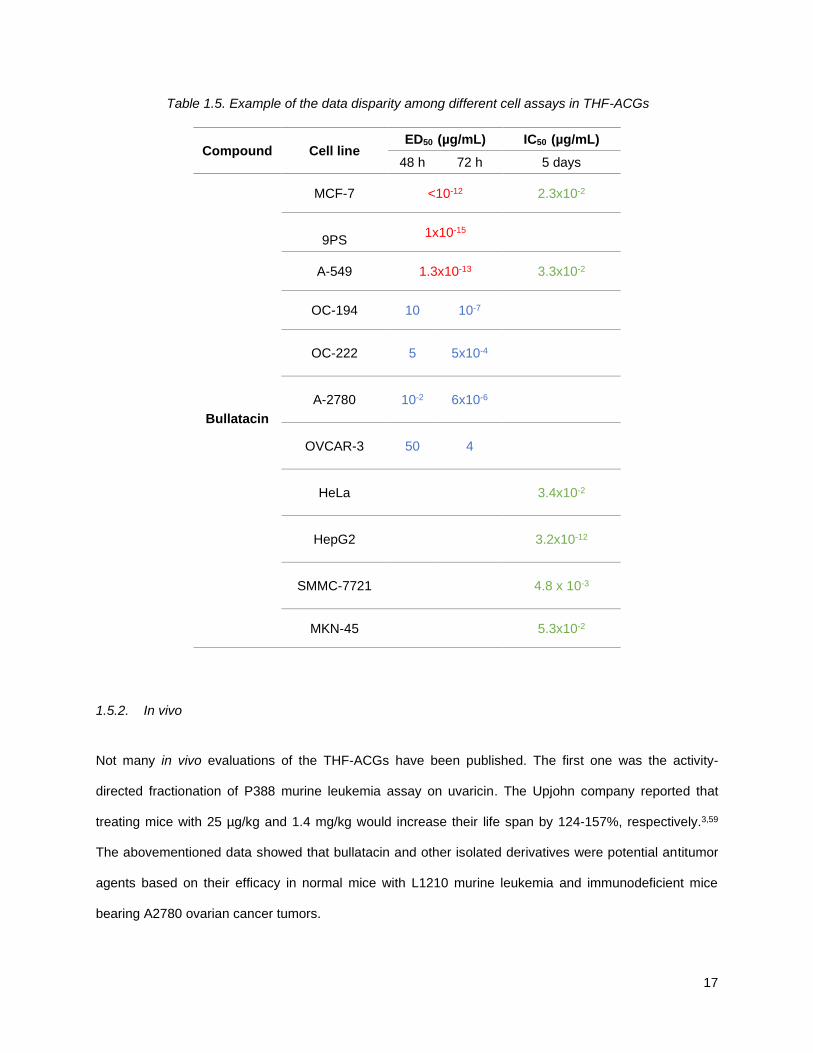
17
Table 1.5. Example of the data disparity among different cell assays in THF-ACGs
Compound Cell line ED50 (µg/mL) IC50 (µg/mL)
48 h 72 h 5 days
Bullatacin
MCF-7 <10-12 2.3x10-2
9PS
1x10-15
A-549 1.3x10-13 3.3x10-2
OC-194 10 10-7
OC-222
5 5x10-4
A-2780
10-2 6x10-6
OVCAR-3
50 4
HeLa
3.4x10-2
HepG2
3.2x10-12
SMMC-7721
4.8 x 10-3
MKN-45 5.3x10-2
1.5.2. In vivo
Not many in vivo evaluations of the THF-ACGs have been published. The first one was the activity-
directed fractionation of P388 murine leukemia assay on uvaricin. The Upjohn company reported that
treating mice with 25 µg/kg and 1.4 mg/kg would increase their life span by 124-157%, respectively.3,59
The abovementioned data showed that bullatacin and other isolated derivatives were potential antitumor
agents based on their efficacy in normal mice with L1210 murine leukemia and immunodeficient mice
bearing A2780 ovarian cancer tumors.

18
In another study, the in vivo activity of annonacin was examined using mouse lung carcinoma (LLC)
tumor cells in BDF-1 mice.66 The difference of this study with the ones that had been published before is
that the ratio of activity towards the tumor vs activity towards the host was much higher than in previous
cases: at a dose of 10 mg/kg of annonacin no mice died while three and five out of six died in the
controls. This seemed to suggest that annonacin was less toxic towards the host than other compounds.
In a separate study also on annonacin, mice bearing MCF-7 xenografts were treated with 50 mg/kg/day
for 21 days and the tumor size was measured every 3 to 4 days. It was observed that the size of the
tumor decreased in 7 to 22 days. In 2012, Fan et al67 evaluated 5 compounds: squamostatiin A,
squamostatiin E, 4-deoxyannoreticuin, desacetyluvaricin, bullatacin were tested the against S180 and
Hep9 xenograpft bearing mice. Their findings agreed with their in vitro results: (i) the two bis-THF
compounds, showed the highest antitumor inhibition of between 64 and 71% at a dose of 60 µg/kg; (ii) out
of the two bis-THF compounds, the one with adjancent THF showed higher activity and (iii) bullatacin at
higher doses showed the highest activity but also more adverse effects.
The above examples suggest that treatment of mice with THF-ACGs in vivo reduced tumor size without
affecting the host. However, in the studies below, treatment of mice with THF-AGEs would not stop tumor
growth (at low dosages) and/or would cause host death (at high dosages).
The data published in 1994 by Montz and coworkers63 proved that using a murine ovarian
terotocarcinoma (MOT) model in C3HeB/Fej mice, the injection of bullatacin made no difference in the
MOT-related animal death at non-lethal dose ranges for both, single dose administration or if the dosage
was divided over time. At low dosages the tumor growth was not inhibited and at higher dosages
bullatacin killed the host.
In the most recent in vivo study, in 2013, Li60 injected H22 hepatoma cells bearing mice with
annosquamin B, bullatacin and annosquatin B. They established that: (i) a single injection of 100 µg/kg
did not influence healthy mice; (ii) treatment of tumor bearing mice with 5 doses of 25-50 µg/kg produced
61% reduction of the tumor size, whereas the other two compounds reduced the tumor size by 53.7 and
58.7% This study agrees with Fan’s67. Additionally, they found that bullatacin resulted in severe liver and
19
kidney tissue damage proved by an increase in calcium concentration, ROS production and Bax/Bcl-2
ratio in rats.
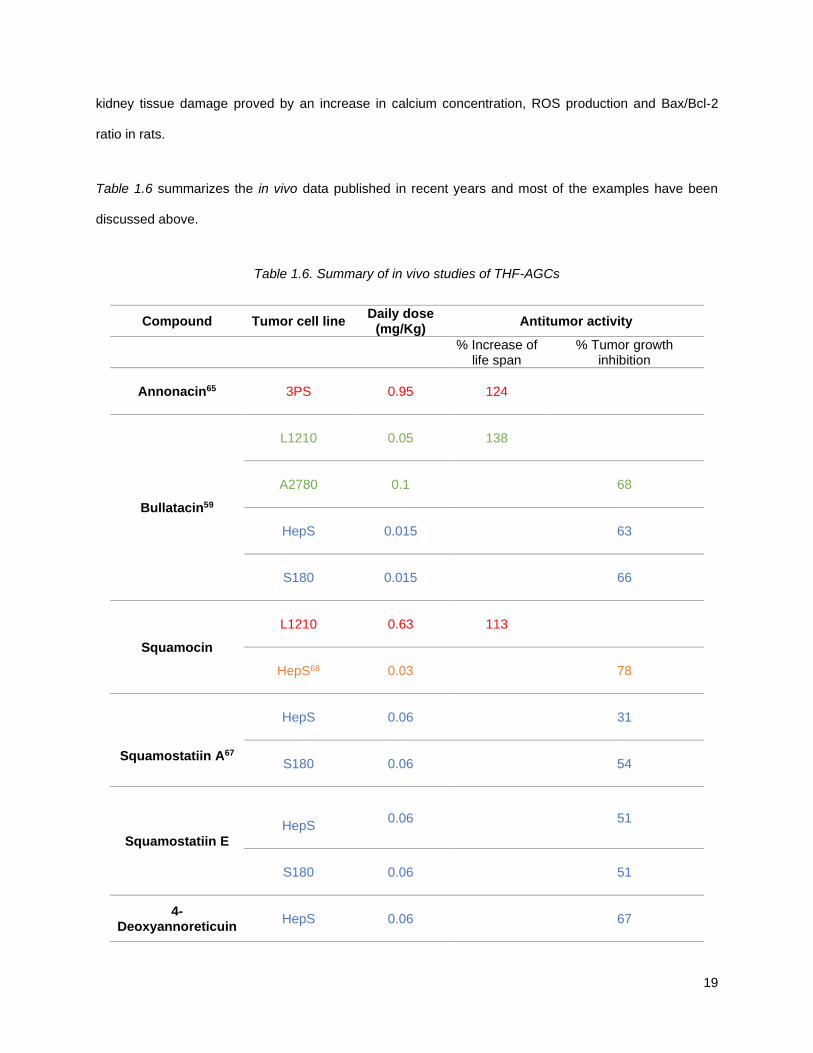
Table 1.6 summarizes the in vivo data published in recent years and most of the examples have been
discussed above.
Table 1.6. Summary of in vivo studies of THF-AGCs
Compound Tumor cell line Daily dose
(mg/Kg) Antitumor activity
% Increase of
life span % Tumor growth
inhibition
Annonacin65
3PS
0.95 124
Bullatacin59
L1210
0.05 138
A2780
0.1 68
HepS
0.015 63
S180
0.015 66
Squamocin
L1210
0.63 113
HepS68
0.03 78
Squamostatiin A67
HepS
0.06 31
S180
0.06 54
Squamostatiin E
HepS
0.06 51
S180
0.06 51
4-Deoxyannoreticuin
HepS
0.06 67
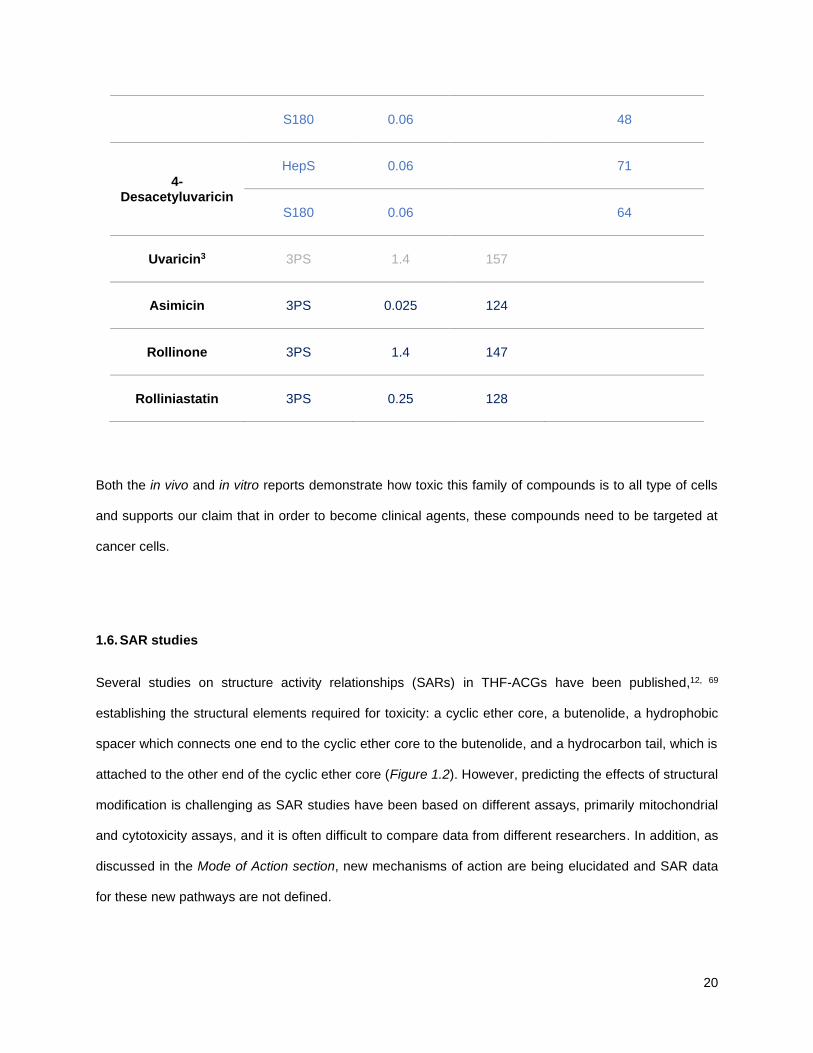
20
S180
0.06 48
4-Desacetyluvaricin
HepS
0.06 71
S180
0.06 64
Uvaricin3
3PS
1.4 157
Asimicin
3PS
0.025 124
Rollinone
3PS
1.4 147
Rolliniastatin
3PS
0.25 128
Both the in vivo and in vitro reports demonstrate how toxic this family of compounds is to all type of cells
and supports our claim that in order to become clinical agents, these compounds need to be targeted at
cancer cells.
1.6. SAR studies
Several studies on structure activity relationships (SARs) in THF-ACGs have been published,12, 69
establishing the structural elements required for toxicity: a cyclic ether core, a butenolide, a hydrophobic
spacer which connects one end to the cyclic ether core to the butenolide, and a hydrocarbon tail, which is
attached to the other end of the cyclic ether core (Figure 1.2). However, predicting the effects of structural
modification is challenging as SAR studies have been based on different assays, primarily mitochondrial
and cytotoxicity assays, and it is often difficult to compare data from different researchers. In addition, as
discussed in the Mode of Action section, new mechanisms of action are being elucidated and SAR data
for these new pathways are not defined.

21
On the mitochondrial and cytotoxic assays, the role of the butenolide is the more extensively studied.
However, it is still uncertain if its presence is necessary for the compounds to remain active. In an early
study, Takada and coworkers70 used a bis THF model to evaluate whether inverting the (S) configuration
of the lactone found would affect the inhibitory potency of complex I. Their study concluded that the R-
configuration analog was as potent as the natural occurring one. Based on those findings, Takada
expanded the study using a bullatacin-like structure (Figure 1.4).70 He concluded that: (i) removing the
alcohol from position 4 in bullatacin 1.67 did not affect inhibitory potency, see 1.68; (ii) removing the
methyl 1.69 had no effect in the potency; (iii) the bulkier n-butyl derivative, 1.70 retained an activity in the
same order of magnitude as the parent drug.
Figure 1.4. Takada studies on the modification of the butenolide
Based on those results, it was suggested that the recognition of the butenolide (or any butenolide
substituent) by the enzyme is weak and that could be a consequence of a large cavity binding domain in
complex I.
Hoppen71 substituted the butenolide by the natural target of the mitochondrial complex I (NADH-
ubiquinone oxidoreductase), quinone. In the inhibitory studies, it was found that the lactone could be
substituted by the quinone ring and the compound would still be active. These findings led to the idea that
both ubiquinone and the anonnaceous acetogenins are competitive inhibitors of the same binding site of
complex I. Hocquemiller72 explored the possible replacements of the butenolide by synthesizing a library
of bis THF squamocin-heterocycle analogues using known complex I inhibitors, such as pyrimidines,

22
quinazoles or benzimidazoles. Some of the modifications resulted in analogues with high activities,
however, it must be noted that alteration of the structure may result in a completely different mechanism
of action.
In more recent years, other groups have substituted the butenolide for simpler heterocycles such as
thiophene,39, 42, 44, 46 piperazine41 or N-methylpyrazole39 and have obtained analogues with maintained or
increased activities. The thiophene and piperazine cases will be discussed in Chapter 3. These studies
suggest that a butenolide-like structure may be necessary for complex I targeting.
Other SAR studies propose the middle of the hydrophobic spacer as the ideal position for conjugation for
tumor targeting or for introduction of a fluorescent group or radiolabel for imaging. Others12 have
evaluated the length of the hydrophilic spacers as well as the polarity of the tetrahydrofuran and its
surroundings and was determined that it was a fundamental feature to maintain activity.
There is controversy regarding the hydroxyl groups in the hydrophilic spacer. Some claim that activity
varies depending on the position of the hydroxyls,73 Chen67 suggested that two hydroxyls flanking the
THF and a third one somewhere else are necessary, others proposed that hydroxylation at position 4
could be involved in a unique intramolecular interaction with the butenolide,74 and Takada’s70 results (see
Figure 1.3) seem to indicate that removal of the hydroxyl at position 4 does not compromise the activity of
the compound.
Even though much progress has been made in elucidating the role of the structural elements of the THF-
ACGs there is a need for a wider library of molecules to expand SAR trends and elucidate new pathways.
To this end simple, high-throughput, synthetic strategies that can deliver novel structures are of interest.

23
SYNTHESIS OF 4-DEOXYANNOMONTACIN

24
2.1. Introduction and background
The tetrahydrofuran containing acetogenins (THF-AGEs) exhibit high activities and selectivities for certain
tumors, but they are also too potent to normal cells. Therefore, there is an interest in developing novel
synthetic analogues that may present higher selectivity towards tumors. THF-AGEs can be obtained from
natural resources, but it is challenging because of the difficult purification that results in low yields. In
order to easily access a library of compounds for structure-activity relationship studies and clinical
development is important to develop a synthetic method. That method should be convergent to allow
synthetic modification at late stages and tolerate a broad range of functional groups.
The mono THF-AGE 4-deoxyannomontacin (4-DAN), 2.1 was selected as a model to develop new
synthetic strategies as its structure is relatively simple and it shows high potency against a wide range of
human tumor cell lines in an MTT 7-day assay (Figure 2.1).
Figure 2.1. Structure of 4-deoxyannomontacin (7-day MTT assays)
4-DAN was first isolated from Goniothalamus giganteus in 1997 by McLaughin75 and coworkers. The only
total synthesis was reported in 1999 by the Wu group.76 The key reaction in this synthesis was the
Sonogashira coupling of vinyl iodide 2.5 and alkyne 2.9, in which diimide reduction25 on the enyne
product gave 4-DAN.
Iodide 2.5 was obtained in 13 steps starting from compound 2.3 in a 22% overall yield. Aldehyde 2.3 was
obtained from D-glucose 2.2. After elongation of the side chain using a Wittig-Horner reaction and
reduction of the alkene, the resulting ester was treated with propane-1,3-dithiol in the presence of
BF3.Et2O to give the lactone-diol. Then, it was protected in situ to afford a lactone that was reduced with
DIBAL-H and treated with (bromotridecylidene)triphenylphosphorane to yield a mixture of the E and Z

25
alkenes. After photoisomerization of the alkene, only Z was obtained. The alcohol was converted to the
tosylester and the alkene was subjected to asymmetric dihydroxylation using AD-mix-β. With the diol in
hand the THF ring was constructed using an intramolecular Williamson etherification on substrate 2.4.
Scheme 2.1. The Wu approach to 4-DAN
The free alcohol was protected with MOM. After deprotection of the isopropylidene, epoxide formation
and epoxide opening using lithium trimethylsilylacetylide, the resulting alkyne was converted into the vinyl

26
iodide 2.5 using tributyltin hydride followed by iodine. Alkyne 2.9 was obtained from methyl ester 2.6 and
ethyl (S)-(-)-lactate using an aldol-cyclization tandem previously reported.77 Then, the epoxide was
introduced using the terminal alkene as substrate and upon elimination, the racemic mixture was
obtained. The CoIII-Salen catalyzed hydrolytic kinetic resolution of epoxide 2.7 delivered the resolved
oxirane that was treated with n-butyl lithium and trimethylsilylacetylene to deliver silyl 2.8. Desylation of
the alkyne yielded lactone 2.9. Overall 4-DAN was synthesized in an overall yield of 15% from over 17
steps starting from 2.3 and 2.8.
In earlier studies in our laboratory, 4-deoxyannoreticuin (4-DAR), 2.10 and 2.11, a very closely related
analog of 4-DAN, showed that the configuration at the position corresponding to C-10 in 4-DAN did not
impact on cytotoxicity against several cell lines. Therefore we decided, for synthetic convenience, to
prepare 4-DAN as a mixture of C-10 epimers 2.12.35 (Figure 2.2).
Figure 2.2. 4-deoxyannorecticuin (2.10 and 2.11) and 4-deoxyannomontacin (2.12)
2.2. Synthetic strategy
Our synthetic plan follows a modular approach, involving a cross metathesis (CM) strategy that was first
developed for the synthesis of tetrahydrofuran containing acetogenins such as bullatanocin, 2.13, both
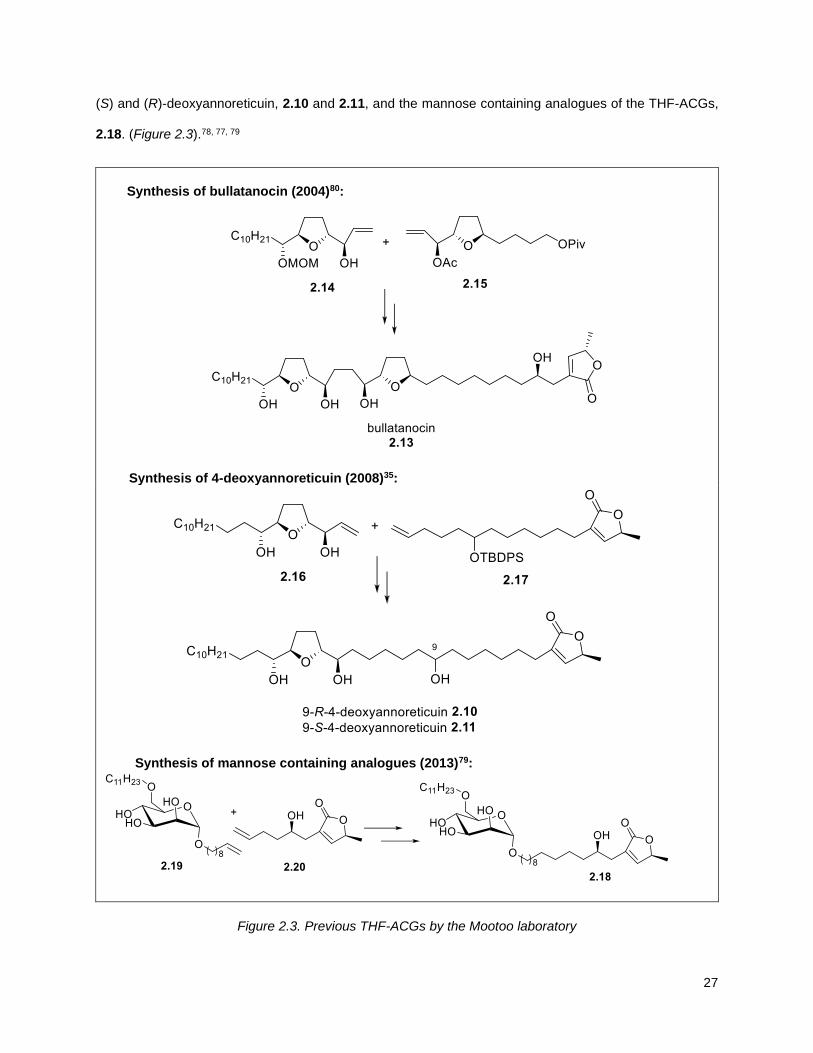
27
(S) and (R)-deoxyannoreticuin, 2.10 and 2.11, and the mannose containing analogues of the THF-ACGs,
2.18. (Figure 2.3).78, 77, 79
Synthesis of bullatanocin (2004)80:
Synthesis of 4-deoxyannoreticuin (2008)35:
Synthesis of mannose containing analogues (2013)79:
Figure 2.3. Previous THF-ACGs by the Mootoo laboratory
28
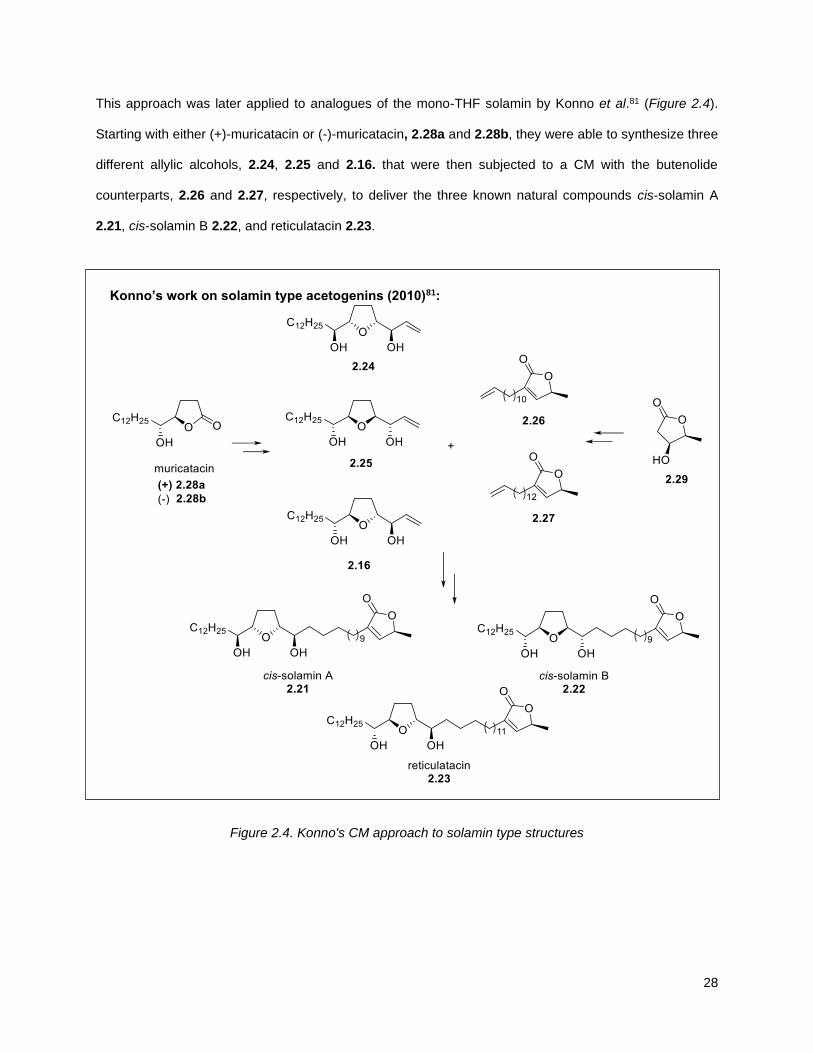
This approach was later applied to analogues of the mono-THF solamin by Konno et al.81 (Figure 2.4).
Starting with either (+)-muricatacin or (-)-muricatacin, 2.28a and 2.28b, they were able to synthesize three
different allylic alcohols, 2.24, 2.25 and 2.16. that were then subjected to a CM with the butenolide
counterparts, 2.26 and 2.27, respectively, to deliver the three known natural compounds cis-solamin A
2.21, cis-solamin B 2.22, and reticulatacin 2.23.
Konno’s work on solamin type acetogenins (2010)81:
Figure 2.4. Konno's CM approach to solamin type structures

29
Current work:
Figure 2.5. CM approach to the synthesis of the C-10 epimeric mixture of 4-DAN
For their synthesis of the THF-alkene, 2.16, (-)-muricatacin 2.28b was converted into the α,β-unsaturated
ester and then cyclized back to yield the allylic alcohol. Then, it was converted to the epoxide, that was
subsequently converted into the diol and subjected to oxidative cleavage. The resulting aldehyde was
then treated with vinyl magnesium bromide and after removal of the protecting group, compound 2.16
was obtained in 8 steps with an overall yield of 4%.
Konno’s group obtained the butenolide partners, 2.26 and 2.27 by subjecting trans-3-pentenenitrile to an
osmium catalyzed dihydroxylation followed by acid catalyzed hydrolysis-lactonization that yielded the
racemic mixture of the hydroxylactone 2.29. The mixture was then separated by using a lipase-mediated
kinetic transesterification. Our approach to the butenolide 2.30 segment must be different because of the
presence of a hydroxyl group in the hydrophobic spacer. In both cases, the convergent nature of this CM
strategy is particularly attractive because of its synthetic efficiency and flexibility for later modification in
the synthesis of analogues.
Towards the synthesis of the epimeric mixture of 4-DAN, 2.12, we applied the same strategy developed in
our lab and then expanded by Konno’s group using the alkene partners 2.16 and 2.30 (Figure 2.5).
30
2.3. Synthesis
2.3.1. THF alkene, 2.16
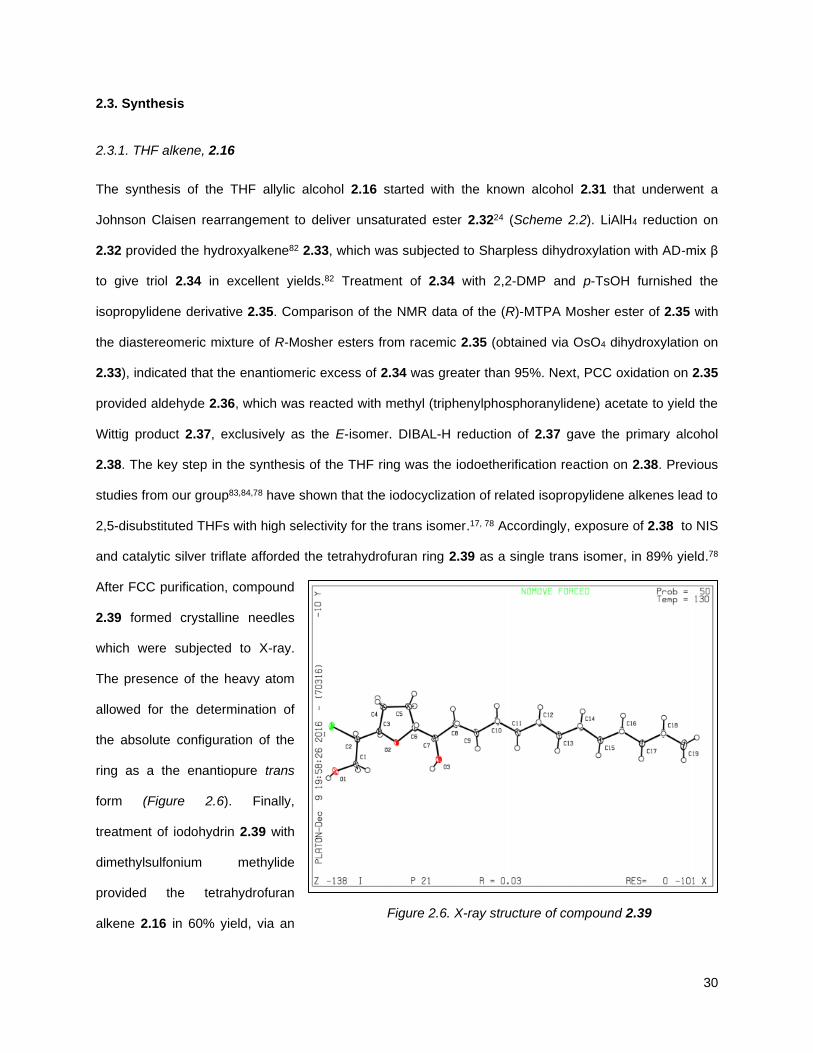
The synthesis of the THF allylic alcohol 2.16 started with the known alcohol 2.31 that underwent a
Johnson Claisen rearrangement to deliver unsaturated ester 2.3224 (Scheme 2.2). LiAlH4 reduction on
2.32 provided the hydroxyalkene82 2.33, which was subjected to Sharpless dihydroxylation with AD-mix β
to give triol 2.34 in excellent yields.82 Treatment of 2.34 with 2,2-DMP and p-TsOH furnished the
isopropylidene derivative 2.35. Comparison of the NMR data of the (R)-MTPA Mosher ester of 2.35 with
the diastereomeric mixture of R-Mosher esters from racemic 2.35 (obtained via OsO4 dihydroxylation on
2.33), indicated that the enantiomeric excess of 2.34 was greater than 95%. Next, PCC oxidation on 2.35
provided aldehyde 2.36, which was reacted with methyl (triphenylphosphoranylidene) acetate to yield the
Wittig product 2.37, exclusively as the E-isomer. DIBAL-H reduction of 2.37 gave the primary alcohol
2.38. The key step in the synthesis of the THF ring was the iodoetherification reaction on 2.38. Previous
studies from our group83,84,78 have shown that the iodocyclization of related isopropylidene alkenes lead to
2,5-disubstituted THFs with high selectivity for the trans isomer.17, 78 Accordingly, exposure of 2.38 to NIS
and catalytic silver triflate afforded the tetrahydrofuran ring 2.39 as a single trans isomer, in 89% yield.78
After FCC purification, compound
2.39 formed crystalline needles
which were subjected to X-ray.
The presence of the heavy atom
allowed for the determination of
the absolute configuration of the
ring as a the enantiopure trans
form (Figure 2.6). Finally,
treatment of iodohydrin 2.39 with
dimethylsulfonium methylide
provided the tetrahydrofuran
alkene 2.16 in 60% yield, via an Figure 2.6. X-ray structure of compound 2.39

31
in situ, tandem epoxide formation-epoxide opening-elimination sequence of reactions. Compound 2.16
was obtained in 8 steps from 2.32 with a 6% overall yield. Protection of the diol using MOMCl afforded
compound 2.40.
Scheme 2.2. Synthesis of THF alkene 2.16
2.3.2. Butenolide alkene, 2.30
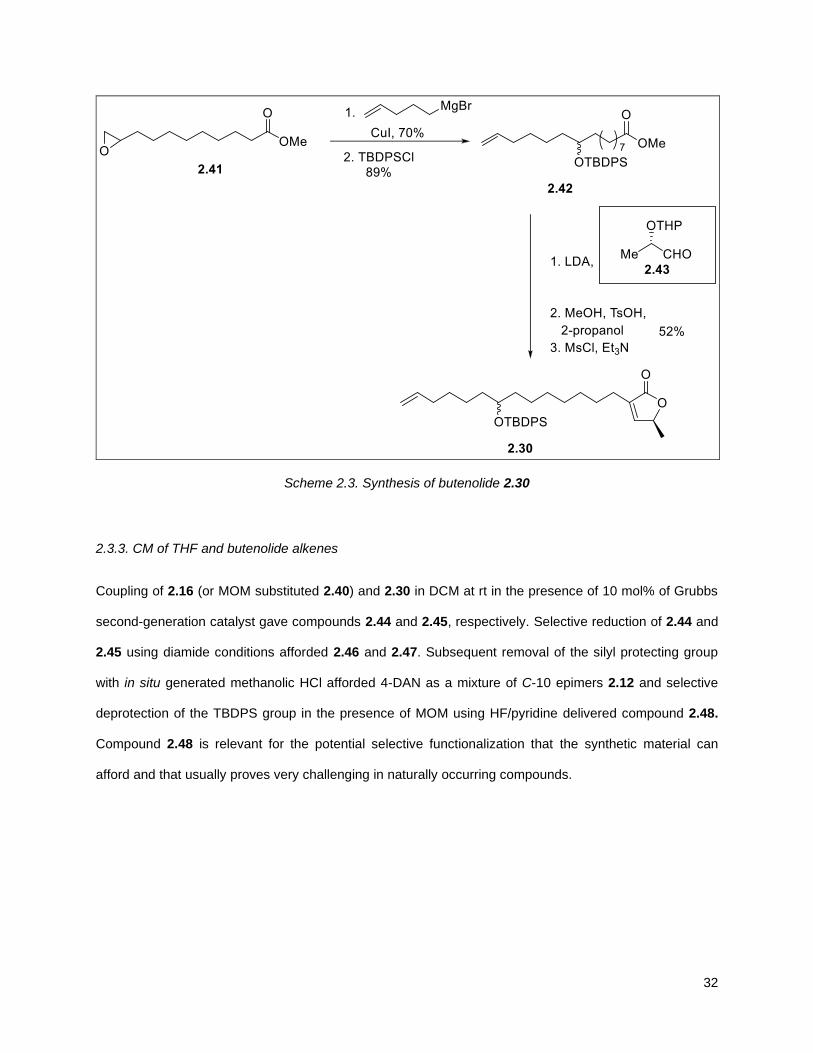
The synthesis of the diastereomeric mixture of butenolide 2.30 started with known compound 2.41.85 This
compound was prepared as the racemic mixture via the esterification of undecenoic acid with methanol
and the treatment of the resulting methyl 10-undecenoate ester with mCPBA. Copper (I) iodide assisted
epoxide opening with 4-pentenylmagnesium bromide and protection of the resulting alkenol as the
TBDPS ether, yielded 2.42. An stablished two step aldol-elimination sequence on 2.42 and the known
lactic acid 2.43 afforded the desired butenolide 2.3035 in 5 steps from 2.41 in 32% yield.
32
Scheme 2.3. Synthesis of butenolide 2.30
2.3.3. CM of THF and butenolide alkenes
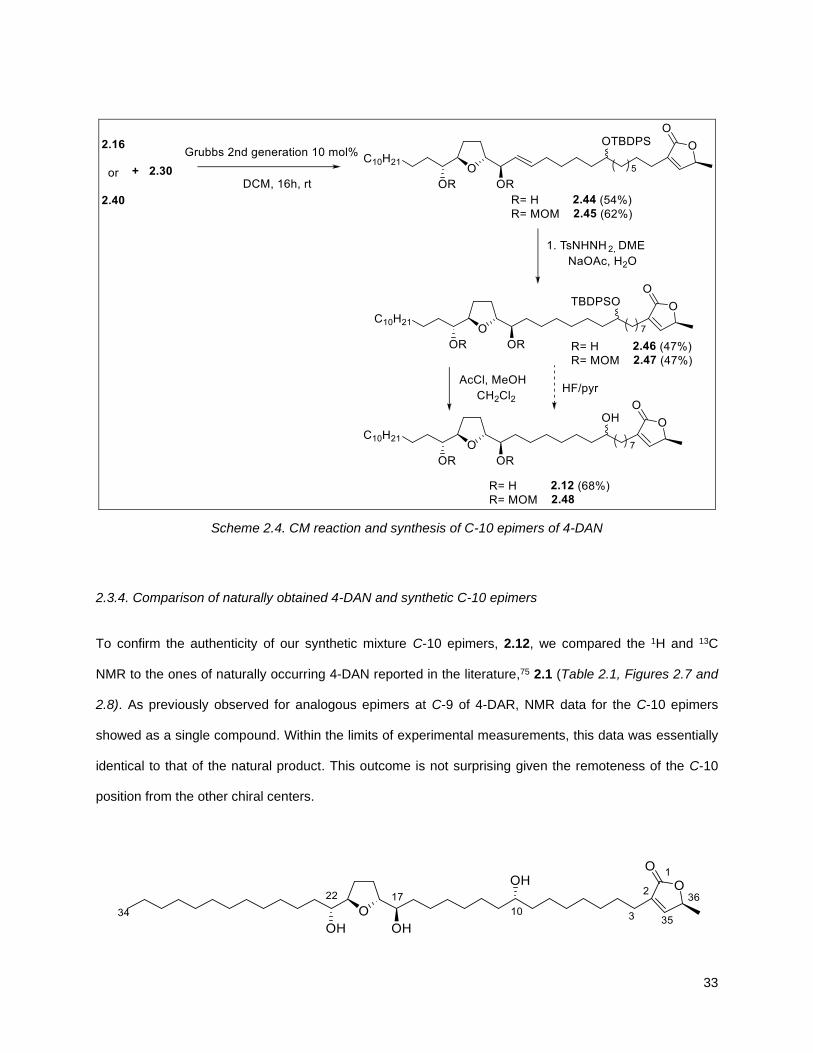
Coupling of 2.16 (or MOM substituted 2.40) and 2.30 in DCM at rt in the presence of 10 mol% of Grubbs
second-generation catalyst gave compounds 2.44 and 2.45, respectively. Selective reduction of 2.44 and
2.45 using diamide conditions afforded 2.46 and 2.47. Subsequent removal of the silyl protecting group
with in situ generated methanolic HCl afforded 4-DAN as a mixture of C-10 epimers 2.12 and selective
deprotection of the TBDPS group in the presence of MOM using HF/pyridine delivered compound 2.48.
Compound 2.48 is relevant for the potential selective functionalization that the synthetic material can
afford and that usually proves very challenging in naturally occurring compounds.
33
Scheme 2.4. CM reaction and synthesis of C-10 epimers of 4-DAN
2.3.4. Comparison of naturally obtained 4-DAN and synthetic C-10 epimers
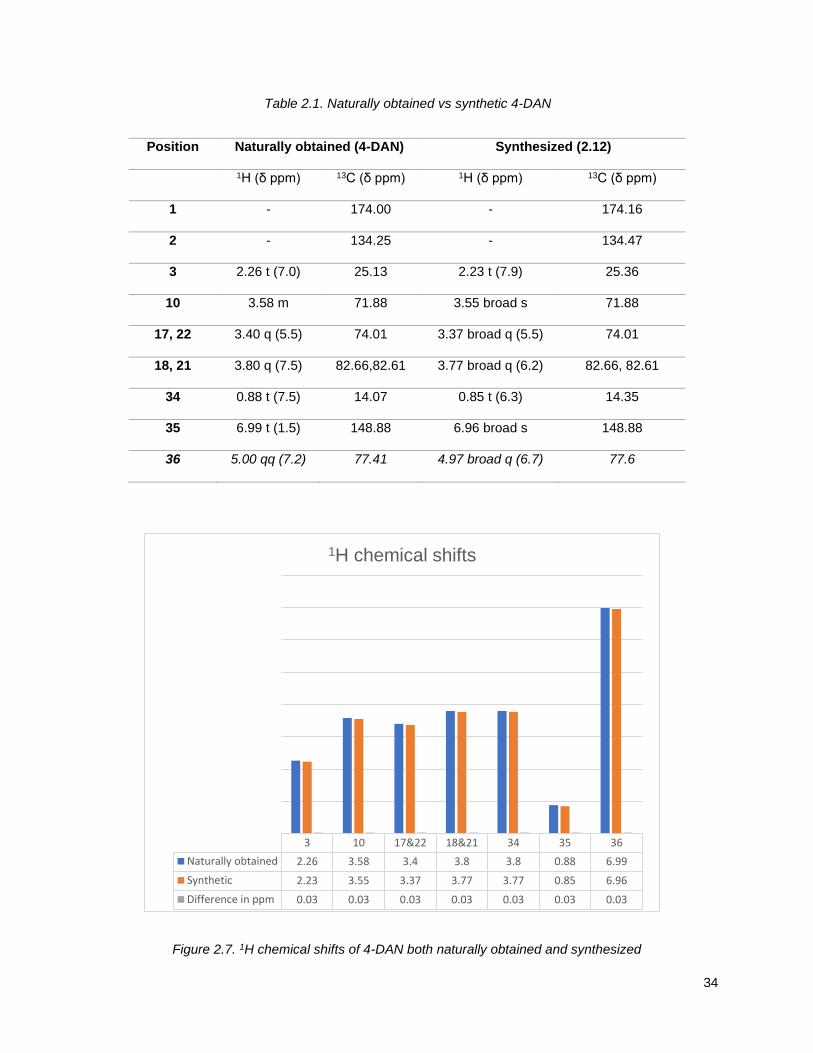
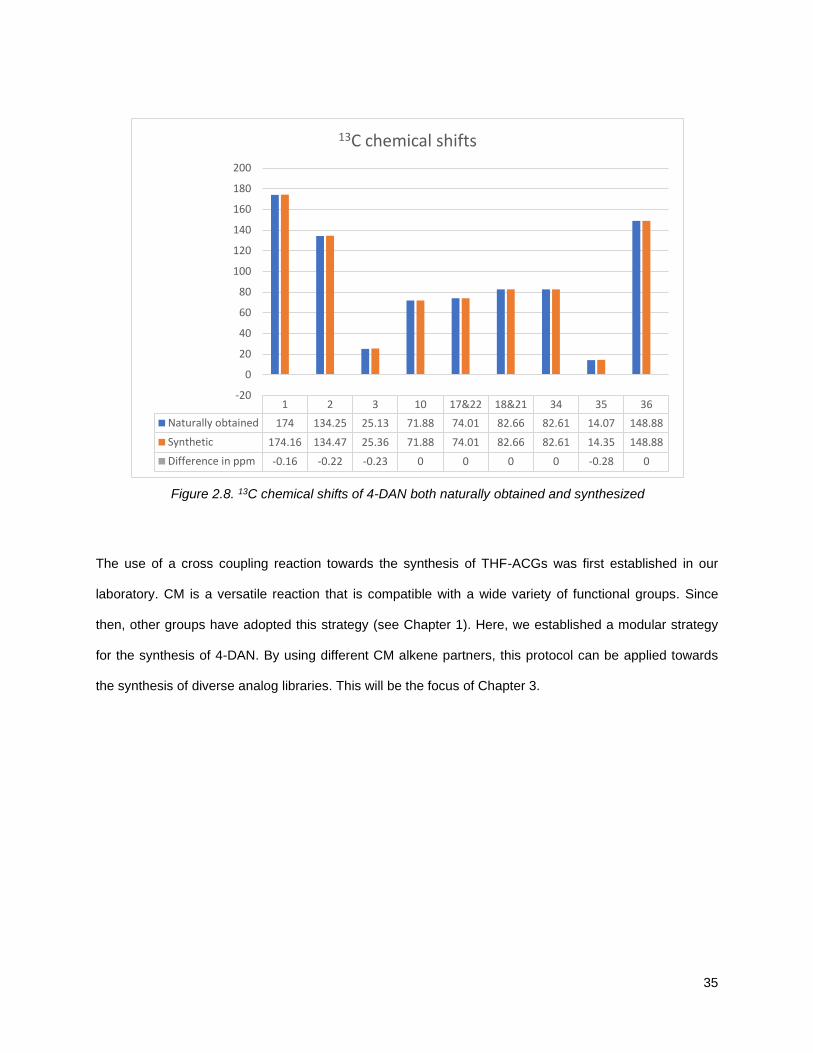
To confirm the authenticity of our synthetic mixture C-10 epimers, 2.12, we compared the 1H and 13C
NMR to the ones of naturally occurring 4-DAN reported in the literature,75 2.1 (Table 2.1, Figures 2.7 and
2.8). As previously observed for analogous epimers at C-9 of 4-DAR, NMR data for the C-10 epimers
showed as a single compound. Within the limits of experimental measurements, this data was essentially
identical to that of the natural product. This outcome is not surprising given the remoteness of the C-10
position from the other chiral centers.
34
Table 2.1. Naturally obtained vs synthetic 4-DAN
Position Naturally obtained (4-DAN) Synthesized (2.12)
1H (δ ppm) 13C (δ ppm) 1H (δ ppm) 13C (δ ppm)
1 - 174.00 - 174.16
2 - 134.25 - 134.47
3 2.26 t (7.0) 25.13 2.23 t (7.9) 25.36
10 3.58 m 71.88 3.55 broad s 71.88
17, 22 3.40 q (5.5) 74.01 3.37 broad q (5.5) 74.01
18, 21 3.80 q (7.5) 82.66,82.61 3.77 broad q (6.2) 82.66, 82.61
34 0.88 t (7.5) 14.07 0.85 t (6.3) 14.35
35 6.99 t (1.5) 148.88 6.96 broad s 148.88
36 5.00 qq (7.2) 77.41 4.97 broad q (6.7) 77.6
Figure 2.7. 1H chemical shifts of 4-DAN both naturally obtained and synthesized
3 10 17&22 18&21 34 35 36
Naturally obtained 2.26 3.58 3.4 3.8 3.8 0.88 6.99
Synthetic 2.23 3.55 3.37 3.77 3.77 0.85 6.96
Difference in ppm 0.03 0.03 0.03 0.03 0.03 0.03 0.03
1H chemical shifts
35
Figure 2.8. 13C chemical shifts of 4-DAN both naturally obtained and synthesized
The use of a cross coupling reaction towards the synthesis of THF-ACGs was first established in our
laboratory. CM is a versatile reaction that is compatible with a wide variety of functional groups. Since
then, other groups have adopted this strategy (see Chapter 1). Here, we established a modular strategy
for the synthesis of 4-DAN. By using different CM alkene partners, this protocol can be applied towards
the synthesis of diverse analog libraries. This will be the focus of Chapter 3.
1 2 3 10 17&22 18&21 34 35 36
Naturally obtained 174 134.25 25.13 71.88 74.01 82.66 82.61 14.07 148.88
Synthetic 174.16 134.47 25.36 71.88 74.01 82.66 82.61 14.35 148.88
Difference in ppm -0.16 -0.22 -0.23 0 0 0 0 -0.28 0
-20
0
20
40
60
80
100
120
140
160
180
200
13C chemical shifts

36
2.4. Experimental
General methods
Moisture and oxygen sensitive reactions were performed under an argon atmosphere. Solvents were
purified by standard procedures or used directly from commercial sources. Thin layer chromatography
(TLC) was done on 0.25 mm thick precoated silica gel HF254 aluminum sheets. Chromatograms were
observed under UV (short and long wavelength) light and were visualized by heating plates that were
dipped in a solution of ammonium (VI) molybdate tetrahydrate (12.5 g) and cerium (IV) sulfate
tetrahydrate (5.0 g) in 10 % aqueous sulfuric acid (500 mL). Flash column chromatography (FCC) was
performed using silica gel 60 (230-400 mesh) and employed a stepwise solvent polarity gradient,
correlated with TLC mobility. 1H and 13C NMR spectra were obtained on a Bruker 400, 500 and 600 MHz
spectrometer. Chemicals shifts are relative to the deuterated solvent peak and are in parts per million
(ppm). X-ray diffraction data were collected on a Bruker X8 Kappa Apex II diffractometer. Crystal data,
data collection and refinement parameters are summarized in Table S1-I. The structure was solved using
direct methods and standard difference map techniques and was refined by full-matrix least-squares
procedures on F2 with SHELXTL86 (Version 2014/7). The absolute configuration was established by
anomalous dispersion effects.87
THF-butenolide (2.12)
A mixture of 5% Ac2Cl in MeOH (0.5 mL) was added at rt to a solution of 2.46 (30 mg, 0.035 mmol) in
CH2Cl2 (1 mL). The mixture was stirred at this temperature for 4 h, diluted with CH2Cl2 and washed with
saturated aqueous NaHCO3. The organic layer was dried (Na2SO4), filtered, and concentrated under
reduced pressure. FCC of the residue afforded 2.12 (15 mg, 68%). Rf = 0.2 (60% EtOAc/hexanes). 1H
NMR (CDCl3, 500 MHz) δ 6.96 (s, 1H), 4.97 (m, 1H), 3.77 (m, 2H), 3.55 (m, 1H), 3.37 (m, 2H), 2.23 (m,

37
3H), 1.95 (m, 3H), 1.67 (m, 4H), 1.43-1.38 (m, 7H), 1.39 (d, partially buried, J = 6.8 Hz, 3H), 1.38-1.20 (s,
38H), 0.88 (t, J = 6.3 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 174.1, 149.1, 134.5, 82.8, 82.8, 77.7, 74.3,
74.2, 72.2, 37.7, 37.6, 33.7, 33.6, 32.1, 29.9 (4 signals), 29.8 (2 signals), 29.6, 29.5, 29.3, 29.0, 27.6,
25.8, 25.7, 25.4, 22.9, 19.4, 14.4. HRMS (ESI) calcd for C37H69O6 (M+H)+ 609.5094, found 609.5087.
THF-alkene (2.16)
To a solution of Me3SI (2.23 g, 10.4 mmol) in THF (11 mL) at -78 °C was added n-BuLi (3.6 mL, 9.1
mmol) under N2. The mixture was warmed to rt, maintained at this temperature for 1 h, then recooled to -
78 °C. A solution of compound 2.39 (0.5 g, 1.2 mmol) in THF (12 mL) was then introduced and the
mixture stirred at -78 °C for 2 h. A solution of sulfonium ylide in THF (11 mL), Me3SI (1.88 g, 9.1 mmol)
and n-BuLi (2.9 mL, 7.3 mmol) was prepared at -78 oC and then warmed to rt. Then, that portion was
added to the main reaction at -78 °C, let warm up to rt, and stirred for 2 h. The reaction was then
quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic phase was dried
(Na2SO4) filtered and evaporated under reduced pressure. The residue was subjected to FCC to give
2.16 (220 mg, 60%) as a clear oil. Rf = 0.57 (50% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.76 (m,
1H), 5.31 (d, J = 18.6 Hz, 1H), 5.17 (m, 1H), 3.92 (m, 1H), 3.82 (m, 2H), 3.38 (m, 1H), 2.93 (br s, 1H),
2.67 (br s, 1H), 1.93 (m, 2H), 1.64 (m, 2H), 1.34 (m, 2H), 1.21 (s, 20H), 0.74 (t, J = 6.9 Hz, 3H); 13C NMR
(CDCl3, 500 MHz) δ 136.8, 117.4, 83.1, 82.4, 75.8, 74.2, 33.5, 32.1, 29.9 (2 signals), 29.8 (4 signals),
29.5, 28.7, 28.6, 25.8, 22.9, 14.3. HRMS (ESI) calcd for C20H38O3Na (M+Na)+ 349.2719, found 349.2715.
Butenolide-alkene (2.30)

38
To a solution of diisopropylamine (23.2 mL, 0.16 mmol) in anhydrous THF (290 mL) at -78 oC was added
n-BuLi (46.3 mL, 2.5 M in hexane, 0.12 mol). The mixture was warmed to 0 oC and stirred at this
temperature for 10 min, then re-cooled to -78 oC. A solution of 2.42 (11.6 g, 0.02 mol) in anhydrous THF
(115 mL) was then added and the mixture maintained at -78 oC for 1 h, at which time a solution of 2.43
(11.07 g, 0.07 mmol) in anhydrous THF (115 mL) was slowly added. After 30 min, the reaction was
diluted with saturated aqueous NH4Cl and extracted with ethyl ether. The organic phase was dried
(Na2SO4) and concentrated under reduced pressure. FCC of the residue (20% EtOAc/hexanes) afforded
mixture of products (11.3 g). To the solution of this material in a mixture of MeOH (28 mL) and 2-propanol
(280 mL) was added TsOH.H2O (0.34 g, 1.79 mmol) at 0 oC. The reaction mixture was stirred at rt for 20
h, then concentrated under reduced pressure. The residue was purified by FCC (15-20 %
EtOAc/hexanes) to afford the derived lactone 2.30 as a yellow oil (6.4 g). To a solution of the material
obtained in the previous step (6.4 g) in CH2Cl2 (280 mL) was added Et3N (8.46 mL, 0.06 mol) and MsCl (2
mL, 0.02 mol) at 0 oC. After stirring at rt for 14 h, the reaction was diluted with saturated aqueous NH4Cl
and extracted with ethyl ether. The organic phase was dried (Na2SO4) and concentrated under reduced
pressure. The residue was purified by FCC to afford 2.30 (3.21 g, 52% after two steps from 2.42). 1H
NMR (CDCl3, 500 MHz) δ 7.65-7.63 (m, 4H), 7.42-7.31 (m, 6H), 6.94 (s, 1H), 5.76-5.67 (m, 1H), 4.96 (m,
1H), 4.87 (m, 1H), 4.83 (m, 1H), 3.67 (m, 1H), 2.22 (m, 2H), 1.91 (m, 2H), 1.16 (m, 3H), 1.15-1.07 (m,
18H), 1.03 (s, (H); 13C NMR (CDCl3, 500 MHz) δ 149.0, 139.2, 136.1, 135.0, 130.0, 129.5, 127.9, 127.5,
114.3, 77.3, 36.4, 36.3, 33.9, 29.7, 29.4, 29.3, 29.1, 27.5, 27.3, 26.7, 25.3, 25.0, 24.6, 19.6, 19.4. HRMS
(ESI) calcd C35H51O3Si for (M+H)+ 546.3529, found 546.3529.
Ethyl (E)-heptadec-4-enoate (2.32)
A mixture of compound 2.3182 (7.7 g, 34.0 mmol) in toluene (80 mL), triethyl orthoacetate (56.09 mL, 0.3
mol) and propionic acid (0.51 mL, 6.8 mmol), was heated at 125 °C under nitrogen for 4 h. The reaction
was then quenched with saturated aqueous NaHCO3 and extracted with EtOAc. The organic phase was
dried (Na2SO4) and evaporated under reduced pressure. The residue was subjected to FCC (2%

39
EtOAc/hexanes) to give product 2.32 (9.86 g, 98%) as a yellow oil. 1H NMR (CDCl3, 500 MHz) δ 5.37-
5.21 (m, 1H), 5.46-5.38 (m, 1H), 4.12-4.08 (m, 2H), 2.34-2.29 (d, J = 25.0 Hz, 2H), 2.29-2.27 (t, J = 11.6
Hz, 2H), 1.95-1.91 (q, J = 20.4 Hz, 2H), 1.22 (s, 20H), 0.86-0.84 (t, J = 13.7 Hz, 3H); 13C NMR (CDCl3,
500 MHz) δ 59.2, 33.4, 31.5, 30.9, 28.7, 28.1, 26.9, 21.7. HRMS (ESI) calcd for C19H37O2 (M+H)+
297.2794, found 297.2787. The NMR data was essentially identical to that reported in the literature.24
(E)-heptadec-4-en-1-ol (2.33)
LAH (2.63 g, 69.3 mmol) was added over 30 min to a solution of 2.32 (10.3 g, 34.7 mmol) in THF (400
mL) at 0 °C. The mixture was stirred at 0 °C under nitrogen for 1.5 h. Then, water (20 mL) and 1 M
aqueous NaOH (20 mL) was slowly added, sequentially, to the reaction mixture, after which anhydrous
Na2SO4 was added until a granular precipitate was obtained. The suspension was filtered, and the filtrate
was concentrated under reduced pressure. The residue was subjected to FCC (5% EtOAc/hexanes) to
give 2.33 (8.08 g, 91%) as a white solid (MP: 31-33⁰C). Rf = 0.4 (10% EtOAc/hexanes). 1H NMR (CDCl3,
500 MHz) δ 5.45-5.34 (m, 2H), 3.64 (m, 2H), 2.05 (m, 2H), 1.95 (m, 2H), 1.62 (m, 2H), 1.30-1.23 (br s,
20H), 0.86 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 131.5, 129.5, 62.8, 32.8, 32.7, 32.1, 29.9 (3
signals), 29.8, 29.7, 29.6, 29.4, 29.2, 22.9, 14.4. HRMS (ESI) calcd for C17H35O3 (M+H)+ 255.2688, found
255.2676.
(4R,5R)-heptadecane-1,4,5-triol (2.34)
Methanesulfonamide (2.0 g, 45.3 mmol), AD-mix β (18.5 g, 0.023 mol) in 1:1 (v/v) water:t-BuOH (100 mL)
was stirred for 1 h until the mixture was homogeneous. The mixture was cooled to 0 °C for 30 min at
which time an orange precipitate was formed. A solution of compound 2.33 (6.16 g, 0.17 mmol) in t-BuOH
(17 mL) was next added and the mixture was stirred at 0 °C for 48 h. The reaction was then quenched

40
with Na2SO3, filtered, extracted with EtOAc. The organic phase was concentrated in vacuo and the solid
residue recrystallized from ethyl acetate to give 2.34 (6.71 g, 95%) as white crystals (MP: 66-69⁰C). Rf =
0.15 (75% EtOAc/hexanes). 𝛼𝐷25 = 15.3. 1H NMR (CDCl3, 500 MHz) δ 3.72 (m, 2H), 3.47 (m, 2H), 3.08 (br
s, 1H), 2.33 (br s, 1H), 2.30 (br s, 1H), 1.77 (m, 2H), 1.58-1.42 (m, 4H), 1.28 (br s, 20H), 0.90 (t, J = 7.1
Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 74.9, 74.6, 63.2, 33.8, 32.1, 31.1, 29.9 (2 signals), 29.8 (2
signals), 29.6, 29.2, 25.9, 22.9, 14.3. HRMS (ESI) calcd for C17H36O3Na (M+Na)+ 311.2555, found
311.2562. The enantiomeric purity of 2.34 was determined to be greater than 95% by Mosher ester
analysis of its isopropylidene derivative 2.35 (vide infra).
(4R,5R)-4,5-O-isopropylidene-heptadecan-1-ol (2.35)
To a solution of 2.34 (8.55 g, 29.3 mmol) in CH2Cl2 (58 mL) was added 2,2-dimethoxypropane (4.32 mL,
35.1 mmol) and p-TsOH (0.56 g, 2.93 mmol). The mixture was stirred for 1 h, then quenched with
saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic phase was dried (Na2SO4), filtered
and concentrated under reduced pressure. FCC of the residue (10-15% EtOAc/hexanes) afforded 2.35
(9.23 g, 96%) as a clear oil. Rf = 0.36 (35% acetone/hexanes). 1H NMR (CDCl3, 500 MHz) δ 3.65 (m, 2H),
3.59 (m, 2H), 2.24 (br s, 1H), 1.71 (m, 2H), 1.56-1.20 (m, 22H), 1.37 (s, partially buried, 6H), 0.86 (t, J =
7.1 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 108.2, 81.2, 81.1, 62.9, 32.9, 32.1, 29.9 (2 signals), 29.8 (3
signals), 29.7 (2 signals), 29.5, 27.5, 27.4, 26.3, 22.8, 14.3. HRMS (ESI) calcd for C20H40O3Na (M+Na)+
351.2875, found 351.2869.

41
(R)-MTPA ester of 2.35 (2.35a)
To a stirred solution of 2.35 (50 mg, 0.15 mmol) and (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl
chloride (77 mg, 0.30 mmol) in CH2Cl2 (10 mL) at 0 oC, DMAP (ca 2 mg) and DCC (63 mg, 0.30 mmol)
were added. The reaction was brought to rt and stirred for an additional 1 h at which time the mixture was
diluted with ethyl ether. The resulting suspension was filtered over Celite and the filtrate was washed with
brine and water. The organic phase was dried (Na2SO4), filtered and concentrated in vacuo. The crude
mixture was purified by FCC to give the R-MTPA-ester 2.35a as a white solid. Rf = 0.78 (30%
acetone/hexanes). 1H NMR (C6D6, 500 MHz) δ 7.68 (m, 2H), 7.09-6.96 (m, 3H), 4.10 (m, 1H), 4.05 (m,
1H), 3.49 (m 1H), 3.42 (s, 3H), 3.41 (partially buried m, 1H), 1.65-1.55 (m, 6H), 1.29 (s, 3H), 1.28 (s, 3H),
1.26 (m, 20H), 0.91 (t, J = 5.7 Hz, 3H); 13C NMR (C6D6, 500 MHz) δ 166.9, 133.5, 130.1, 130.0, 129.2,
129.0, 128.8, 128.7, 127.9, 125.6, 123.7, 108.5, 81.6, 81.0, 66.4, 55.7, 33.5, 32.7, 30.6, 30.5 (2 signals),
30.4 (2 signals), 30.2, 29.5, 28.0, 27.9, 27.1, 26.1 (2 signals), 26.0, 23.5, 14.7, 1.7. HRMS (ESI) calcd for
C30H47F3O5Na (M+Na)+ 567.3268, found 567.3249.
(R)-MTPA Ester of (+/-) 2.35 (2.35b)
2.33 (0.1 g, 0.3 mmol) was dissolved in a mixture of water and acetone (1:1, 2 mL). The reaction was
cooled down to 0 oC and osmium tetroxide in t-butanol (2.5% wt, 0.7 mL, 0.06 mmol) was added. Then,
N-methylmorpholine (0.04 mL, 0.4 mmol) was added and the reaction was stirred for 2 h at 0 oC. The
reaction was quenched with Na2SO3 (17 mg, 0.06 mmol) and stirred for 2 h. The product was extracted
using ethyl acetate and purified over FCC (70% EtOAc/hexanes) to afford a racemic mixture of the diol.

42
Rf = 0.2 (70% EtOAc/hexanes). The isopropylidene was installed using the same conditions as for the
chiral compound 2.35 and the primary alcohol was then converted to the R-MTPA-ester, 2.35b, was
purified using FCC (30-40% EtOAc/hexanes). Rf = 0.8 (35% acetone/hexanes). 1H NMR (C6D6, 400 MHz)
δ 7.66 (m, 2H), 7.05 (m, 3H), 4.07 (m, 2H), 3.89 (m, 1H), 3.50 (m, 1H), 3.47 (m, 1H), 3.38 (m, 3H), 3.33
(m, 3H), 1.55 (m, 2H), 1.40 (br s, 16H), 0.8 (m, 2H), 0.7 (m, 2H), 0.4 (m, 3H); 13C NMR (C6D6, 400 MHz) δ
165.4, 165.3, 132.0, 128.6, 128.5, 128.5, 127.7, 127.5, 127.4, 127.1, 127.0, 126.9, 126.7, 126.6, 126.1,
124.5, 121.7, 107.0, 84.1, 83.8, 80.1, 79.5, 64.9, 64.9, 60.9, 55.7, 54.1 (2 signals), 51.1, 48.9, 32.0, 31.3,
31.2, 30.2, 29.0 (2 signals), 28.9 (3 signals), 28.6, 28.0, 26.5, 26.4, 25.5, 25.3, 25.2, 24.6 (3 signals),
24.5, 23.6 (2 signals), 21.9, 13.2, 12.6.
(4R,5R)-4,5-O-isopropylidene-heptadecanal (2.36)
A suspension of powdered, freshly activated molecular sieves (7 g), Fluorosil (7 g), Celite (7 g),
CH3COONa (2.54 g, 31.2 mmol), PCC (6.61 g, 31 mmol) and 2.35 (6.84 g, 20.8 mmol) in CH2Cl2 (19 mL)
was stirred under nitrogen for 1 h. At that time the mixture was diluted with ether and filtered over
Fluorosil. The filtrate was concentrated under reduced pressure and the residue subjected to FCC to give
2.36 (4.42 g, 64%) as a yellow oil. Rf = 0.45 (15% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 9.74 (s,
1H), 3.54 (m, 2H), 2.57 (m, 2H), 1.90 (m, 1H), 1.68 (m, 1H), 1.46 (m, 2H), 1.34-1.19 (m, 20H), 1.34 (s,
partially buried, 6H), 0.86 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 202.1, 108.4, 81.0, 80.0, 40.7,
33.0, 32.1, 29.9 (2 signals), 29.8, 29.7, 27.5, 27.4, 26.3, 25.3, 22.9, 14.3.
Methyl (6R,7R,E)-6,7-O-isopropylidenenonadec-2-enoate (2.37)

43
To a solution of compound 2.36 (5.06 g, 15.5 mmol) in CH3CN (150 mL) was added methyl
(triphenylphosphoranylidene) acetate (15.5 g, 46.5 mmol). The mixture was heated at reflux for 1.5 h,
then filtered over Celite. The filtrate was concentrated under reduced pressure and the residue purified
by FCC to give 2.37 (4.67 g, 76%) as a clear oil. Rf = 0.85 (15% EtOAc/hexanes). 1H NMR (CDCl3, 500
MHz) δ 6.94 (m, 1H), 5.81 (m, 1H), 3.68 (s, 3H), 3.55 (m, 2H), 2.36 (m, 1H), 2.25 (m, 1H), 1.76 (m, 2H),
1.45 (m, 2H), 1.32-1.10 (m, 20H), 1.32 (s, partially buried 6H), 0.82 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3,
500 MHz) δ 167.2, 148.8, 121.4, 108.2, 81.0, 80.2, 51.6, 33.1, 32.1, 31.4, 29.9, 29.8 (3 signals), 29.7,
29.5, 29.0, 27.5, 27.4, 26.3, 22.9, 14.3. HRMS (ESI) calcd for C23H42O4Na (M+Na)+ 405.2981, found
405.2985.
(6R,7R,E)-6,7-O-isopropylidenenonadec-2-en-1-ol (2.38)
To a solution of compound 2.37 (1.4 g, 3.54 mmol) in CH2Cl2 (70 mL) was added a solution of 1 M
DIBAL-H in hexane (2.16 mL, 10.6 mmol) at -78 °C under nitrogen. The mixture was warmed to rt and
stirred at this temperature for an additional 1.5 h. Then water (10 mL) and 1 M aqueous NaOH (1 mL)
was slowly added, sequentially, to the reaction mixture after which anhydrous Na2SO4 was added until a
granular precipitate was obtained. The mixture was filtered over Celite and the filtrate was concentrated
under reduced pressure. FCC of the residue (10% EtOAc/hexanes) afforded 2.38 (0.7 g, 56%) as a clear
oil. Rf = 0.53 (25% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.66 (m, 2H), 4.06 (m, 2H), 3.57 (m,
2H), 2.23 (m, 1H), 2.10 (m, 1H), 1.69 (s, 1H), 1.56 (m, 4H), 1.32 (s, partially buried, 6H), 1.18 (m, 20H),
0.81 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 132.5, 129.6, 108.1, 81.2, 81.1, 63.9, 33.1, 32.6,
32.1, 30.0, 29.9, 29.8 (2 signals), 29.7, 29.5, 29.0, 27.5 (2 signals), 26.4, 22.9, 14.3. HRMS (ESI) calcd
for C22H42O3Na (M+Na)+ 377.3032, found 377.3026.

44
Iodohydrin (2.39)
To a solution of compound 2.38 (390 mg, 1.10 mmol) in CH3CN (100 mL) was added NIS (100 mg, 4.40
mmol) and AgOTf (6 mg, 0.02 mmol). The mixture was stirred at rt for 3 h, then diluted with a saturated
solution of Na2S2O3 and extracted with EtOAc. The organic phase was dried (Na2SO4), filtered and
evaporated in vacuo. FCC of the residue gave 2.39 (430 mg, 89%) as a white solid (MP: 47-49⁰C). Rf =
0.33 (25% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 4.23 (m, 1H), 4.11 (m, 1H), 4.00 (m, 1H), 3.95
(m, 2H), 3.46 (m, 1H), 2.85 (t, J = 13.1 Hz, 1H), 2.41 (m, 1H), 2.10 (m, 2H), 1.99-1.73 (m, 2H), 1.42 (m,
2H), 1.28 (s, 20H), 0.85 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 84.1, 83.1, 74.0, 68.0, 39.3,
34.3, 33.9, 32.1, 29.9 (2 signals), 29.8 (2 signals), 29.6, 28.2, 25.8, 22.9, 14.4 HRMS (ESI) calcd for
C19H37IO3Na (M+Na)+ 463.1685, found 463.1681.
THF alkene MOM protected (2.40)
2.16 (0.150 g, 0.4 mmol) were dissolved in 8 mL CH2Cl2. Then, MOMCl (0.14 mL,1.84 mmol) and DIPEA
(0.41 mL, 2.3 mmol) were added at 0 oC and the solution was stirred overnight. The reaction was then
quenched with water and extracted with DCM (3x). The organic phase was dried (NaSO4), concentrated
in vacuo and purified via FCC (10% EtOAc/hexanes) to afford compound 2.40 (0.18 g, quant) as a white
solid. Rf = 0.5 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.71 (m, 1H), 5.28 (m, 1H), 5.24 (m,
1H), 4.82 (m, 1H), 4.65 (m, 4H), 3.99 (m, 3H), 3.37 (d, J = 2.8 Hz, 6H), 1.89 (m, 2H), 1.66 (m, 2H), 1.39
(m, 4H), 1.23 (m, 20H), 0.86 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 135.0, 118.9, 96.9, 94.2,
81.8, 81.1, 79.7, 79.4, 55.9, 55.5, 32.1, 31.3, 30.0, 29.9 (2 signals), 29.8, 29.6, 28.3, 28.2, 25.8, 22.9,
14.3. HRMS (ESI) calcd for C24H46O5Na (M+Na)+ 437.3243, found 437.3238.

45
Methyl 10-hydroxyhexadec-15-enoate (2.41a)
To a suspension of CuBr (I) (3.70 g, 0.26 mol) in THF (300 mL) at 0 oC was added 4-pentenylmagnesium
bromide (24 mL, 1M, 0.13 mol) over 5 min. A solution of 2.4185 (10.6 g, 43 mmol) in anhydrous THF (60
mL) was then introduced, dropwise over 5 min. The reaction was stirred 0 oC for 10 min and saturated
aqueous NH4Cl was added and the mixture extracted with ethyl ether. The organic phase was washed
with brine, dried (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified to
give the derived alcohol 2.41a (10.2 g, 70%). Rf = 0.15 (10% EtOAc /hexanes). 1H NMR (CDCl3, 500
MHz) δ 5.77 (m, 1H), 4.92 (m, 1H), 4.91 (m, 1H), 3.63 (s, 3H), 3.53 (br s, 1H), 2.26 (m, 2H), 2.03 (m, 2H),
1.34-1.26 (m, 2H); 13C NMR (CDCl3, 500 MHz) δ 174.6, 139.1, 114.6, 72.1, 51.7, 37.6, 37.5, 34.3, 29.8,
29.6, 29.4, 29.3 (2 signals), 29.1, 25.8, 25.3, 25.1.
Methyl 10-((tert-butyldiphenylsilyl)oxy)hexadec-15-enoate (2.42)
A portion of the material from the previous step (7.31 g, 0.024 mol), TBDPSCl (6.24 mL, 0.024 mol) and
imidazole (8.33 g, 0.12 mol) in anhydrous THF (200 mL) were heated at reflux under nitrogen for 4 h. The
mixture was then diluted with water, extracted with ethyl ether and washed with brine. The organic phase
was dried (Na2SO4), filtered, and evaporated under reduced pressure. The residue was purified by FCC
to afford 2.42 (9.37 g, 89%) as a clear oil. Rf = 0.75 (10% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ
7.64 (m, J = 7.6 Hz, 4H), 7.36 (m, 6H), 5.72 (m, 1H), 4.89 (m, 1H), 4.87 (m, 1H), 3.67 (m, partially buried,
1H), 3.64 (s, 3H), 2.27 (t, J = 7.5 Hz, 2H), 1.91 (m, 2H), 1.08-1.25 (m, 20H), 1.04 (s, 9H); 13C NMR
(CDCl3, 500 MHz) δ 174.4, 139.1, 136.0, 134.8, 129.4, 127.4, 114.2, 73.2, 51.5, 36.3, 36.1, 34.1, 33.7,
29.6, 29.4, 29.2, 29.1, 29.0, 27.1, 25.0, 24.8, 24.4, 19.4. HRMS (ESI) calcd for C33H51O3Si (M+H)+
523.3602, found 523.3585.

46
THF-butenolide alkene (2.44)
A mixture of 2.16 (0.1 g, 0.3 mmol) and 2.30 (0.28 g, 0.51 mmol) in CH2Cl2 (2 mL) was purged with
nitrogen for 30 min. Grubbs 2nd generation catalyst (25 mg, 0.03 mmol) was then introduced and the
reaction stirred for 16 h at rt, after which DMSO (0.05 mL) was added and stirring continued for 30 min.
The mixture was then concentrated in vacuo. FCC of the residue (40% EtOAc/hexanes) afforded 2.44
(136 mg, 54%) as a clear oil. Rf = 0.65 (40% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 7.63 (m, 4H),
7.40-7.30 (m, 6H), 6.94 (s, 1H), 5.67 (m, 1H), 5.30 (m, 1H) 4.96 (m, 1H), 3.84-3.77 (m, 3H), 3.80 (m, 1H),
3.38 (m, 1H), 2.47 (br s, 1H), 2.28 (br s, partially buried, 1H), 2.21 (m, 2H), 1.90 (m, 4H), 1.63 (m, 2H),
1.45 (m, 3H), 1.33 (m, 10H), 1.22-1.14 (m, 30H), 1.00 (s, 9H), 0.85 (t, J = 6.6 Hz, 3H); 13C NMR (CDCl3,
500 MHz) δ 174.1, 149.0, 136.1, 134.9, 134.5, 129.6, 128.3, 127.6, 82.9, 82.7, 77.7, 75.9, 74.2, 73.3,
36.4, 36.3, 33.8, 32.5, 32.5, 32.1, 29.9 (4 signals), 29.8 (3 signals), 29.6, 29.4, 29.3, 28.7, 27.6, 27.3,
25.8, 25.4, 25.0, 24.6, 22.9, 19.6, 19.4, 14.3. HRMS (ESI) calcd C53H84O6SiNa (M+Na)+ 867.5935, found
867.5909.
THF-MOM-butenolide-alkene (2.45)
A mixture of 2.40 (0.180 g, 0.45 mmol) and 2.30 (0.4 g, 0.73 mmol) in CH2Cl2 (4 mL) was purged with
nitrogen for 30 min. Grubbs 2nd generation catalyst (31 mg, 0.04 mmol) was then introduced and the
reaction stirred for 16 h at rt, after which DMSO (0.05 mL) was added and stirring continued for 30 min.
The mixture was then concentrated in vacuo. FCC of the residue in 40% EtOAc/hexanes afforded 2.45
(0.26 g, 62%). Rf = 0.4 (20% EtOAc/hexanes).

47
THF-butenolide (2.46)
A solution of sodium acetate (0.62 g, 3 mmol) in water (12 mL) was added via a syringe pump, over 4 h,
to a mixture of 2.44 (85 mg, 0.10 mmol), p-toluenesulfonylhydrazide (1.25 g, 3 mmol) and DME (12 mL) at
reflux. After cooling to rt, the reaction mixture was poured into water and extracted with EtOAc. The
organic extract was washed with 2M HCl, water and brine, dried (Na2SO4), filtered and concentrated
under reduced pressure. FCC of the residue (1-5% acetone/DCM) gave 2.46 (0.04 g, 47%). Rf = 0.68 (2%
acetone/dichloromethane). 1H NMR (CDCl3, 500 MHz) δ 7.63 (m, 4H), 7.45-7.29 (m, 6H), 6.94 (s, 1H),
4.95 (m, 1H), 3.76 (m, 2H), 3.66 (m, 1H), 3.36 (m, 2H), 2.21 (m, 2H), 1.96 (m, 4H), 1.85 (br s, 2H), 1.36
(d, partially buried, J = 6.8 Hz, 3H), 1.34 (m, 10H), 1.25 (m, 36H), 1.00 (s, 9H), 0.85 (t, J = 7.1, 3H); 13C
NMR (CDCl3, 500 MHz) δ 174.1, 149.0, 136.1, 135.0, 134.5, 129.5, 127.5, 82.8, 74.2, 73.4, 36.5, 36.4,
33.7 (2 signals), 32.1, 31.8, 29.9 (2 signals), 29.8 (3 signals), 29.6, 29.4, 29.3, 28.9, 27.5, 27.3, 25.8,
25.8, 25.4, 25.0, 22.9 (2 signals), 19.6, 19.4, 14.3, 11.7. HRMS (ESI) calcd for C53H86O6SiNa (M+Na)+
869.6091, found 869.6068.
THF-MOM-butenolide(2.47)
A solution of sodium acetate (1.89 g, 0.023 mol) in water (57 mL) was added via a syringe pump, over 4
h, to a mixture of 2.45 (0.2 g, 0.21 mmol), p-toluenesulfonylhydrazide (1.7 g, 9.28 mmol) and DME (57
mL) at reflux. After cooling to rt, the reaction mixture was poured into water and extracted with EtOAc.
The organic extract was washed with 2 M HCl, water and brine, dried (Na2SO4), filtered and concentrated
under reduced pressure. FCC of the residue gave 2.47 (4 mg, 47%). Rf = 0.4 (20% EtOAc/hexanes). 1H
NMR (CDCl3, 500 MHz) δ 7.68 (m, 4H), 7.45-7.28 (m, 6H), 7.01 (s, 1H), 4.99 (m, 1H), 4.86 (m, 2H), 4.68

48
(m, 2H), 3.99 (m, 2H), 3.70 (m, 1H), 3.51 (m, 2H), 3.39 (m, 6H), 2.20 (m, 2H), 1.94 (m, 2H), 1.45 (m, 6H),
1.41 (m, 9H), 1.38 (m, 36H), 1.06 (s, 9H), 0.90 (t, J = 7.1 Hz, 3H). HRMS (ESI) calcd for C57H94O8SiNa
(M+Na)+ 957.6616, found 957.6601.

49
ANALOGUES OF 4-DEOXYANNOMONTACIN

50
3.1. Introduction and background
THF-AGCs show potent activity against human cancer cell lines and therapeutic potential in animal
studies. However, their equally high toxicity to normal cells has limited their clinical development. Towards
more clinically viable analogues, we are interested in THF-ACGs that are easily accessed and more
selective to tumor cells. Accordingly, we designed a series of compounds based on the SAR trends for
the THF-ACGs discussed in Chapter 1. The potently active mono-THF-ACG 4-DAN was used as a
template for these structures. The modifications that we propose are: (i) replacement of the butenolide by
simpler non-chiral heterocycles or alkyl chains; (ii) substitution of the THF ring with a carbohydrate
moiety; (iii) variations in the hydrophobic spacer that connects the THF core or its replacement to the
butenolide or butenolide substitute (Figure 3.1). While certain of these individual modifications have been
previously shown to deliver compounds with similar activity to naturally occurring THF-ACGs, hybrid
structures that combine two or more such changes have not been widely tested. For instance, we have
shown that the THF core could be replaced with a cyclic sugar and other researchers have replaced the
butenolide with simple heterocycles, but the activity of analogues that combine both of these
modifications is unknown. Such hybrids are of interest in that they may be more easily prepared than less
modified analogues.
Figure 3.1. Structural characteristics of THF-AGEs and proposed replacements

51
As discussed earlier, previous studies from this laboratory on 4-deoxyannoreticuin,35 showed that both C-
10 epimers were similarly active. Based on this result, for synthetic simplicity we decided to prepare our
new analogues of DAN as an epimeric mixture at the corresponding C-9 carbinol in 4-DAN.
3.2. Butenolide substitutes
3.2.1. Piperazine
Zen and others88 have evaluated ACG mimetics like AA005 3.1, in which the bis-THF ring is replaced with
an ethylene glycol ether moiety (Figure 3.2). These simplified mimetics presented activities comparable to
the natural products. More recently, the same group examined even simpler mimics in which the
butenolide in these acyclic analogues was replaced with simple achiral heterocycles (cf 3.2).41 These
compounds still showed low micromolar activity. We are particularly interested in the analog containing a
terminal piperazine, 3.3, not just because it exhibited the most potent antiproliferation activity but also
because the secondary amine provides a handle for prodrugs.
Figure 3.2. Piperazine analogues

52
3.2.2. Thiophene
Kojima also investigated small heterocycles as butenolide substitutes. In the initial study, an amide linked
N-methylpyrazole, showed selectivity towards the same cell lines as the alkyl-linked equivalent, but with
higher potencies (Figure 3.3).39 In a later study, other amide-linked heterocycles, including furan or
thiophene were evaluated.42, 44, 46 Both the thiophene-3-carboxamide and thiophene-2-carboxamide
showed high potencies across a panel of 39 cell lines. The 3-carboxamide derivative was used in vivo
studies and inhibited tumor growth of the NCI-H23 xenografts implanted in mice. Following this study, we
decided to replace the butenolide in 4-DAN with a thiophene-2-carboxamide moiety. Our analogues differ
from Kojima’s with respect to the length of the hydrophobic chain that connects the THF and the
heterocycle, and the presence of the hydroxyl group in the middle of the chain.
Figure 3.3. Thiophene derivative

53
3.2.3. O-butyl
Other groups have examined simpler replacements, such as a n-butyl ester in the case of squamocin
(Figure 3.4).89 This analog was several orders of magnitude less active than squamocin but was still in
the low micromolar range. Interestingly, its activity against complex I was comparable to the natural
product. To probe whether the carbonyl group of the ester was important for activity, we targeted the n-
butyl ether derivative of 4-DAN, 3.7.
Figure 3.4. Simple n-alkyl derivative
3.3. Tetrahydrofuran substitutes
3.3.1. Mannose replacement
Several reviews have been published on the modification of the THF ring.90,91,92 Our group has showed
that replacing the THF core with a sugar residue delivered compounds with low micromolar level activity
that was comparable to the mono-THF-ACG, (9S)-4-deoxyannonereticuin (DAR), 3.8 (Figure 3.5).79
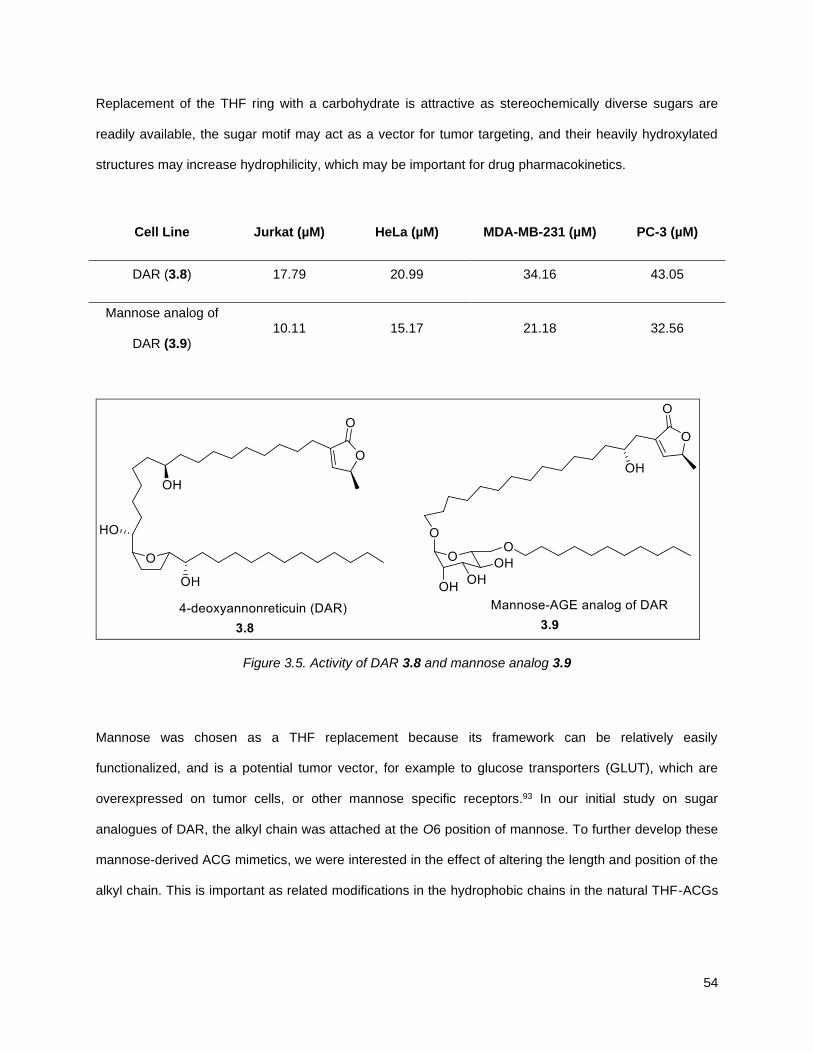
54
Replacement of the THF ring with a carbohydrate is attractive as stereochemically diverse sugars are
readily available, the sugar motif may act as a vector for tumor targeting, and their heavily hydroxylated
structures may increase hydrophilicity, which may be important for drug pharmacokinetics.
Figure 3.5. Activity of DAR 3.8 and mannose analog 3.9
Mannose was chosen as a THF replacement because its framework can be relatively easily
functionalized, and is a potential tumor vector, for example to glucose transporters (GLUT), which are
overexpressed on tumor cells, or other mannose specific receptors.93 In our initial study on sugar
analogues of DAR, the alkyl chain was attached at the O6 position of mannose. To further develop these
mannose-derived ACG mimetics, we were interested in the effect of altering the length and position of the
alkyl chain. This is important as related modifications in the hydrophobic chains in the natural THF-ACGs
Cell Line Jurkat (µM) HeLa (µM) MDA-MB-231 (µM) PC-3 (µM)
DAR (3.8) 17.79 20.99 34.16 43.05
Mannose analog of
DAR (3.9) 10.11 15.17 21.18 32.56
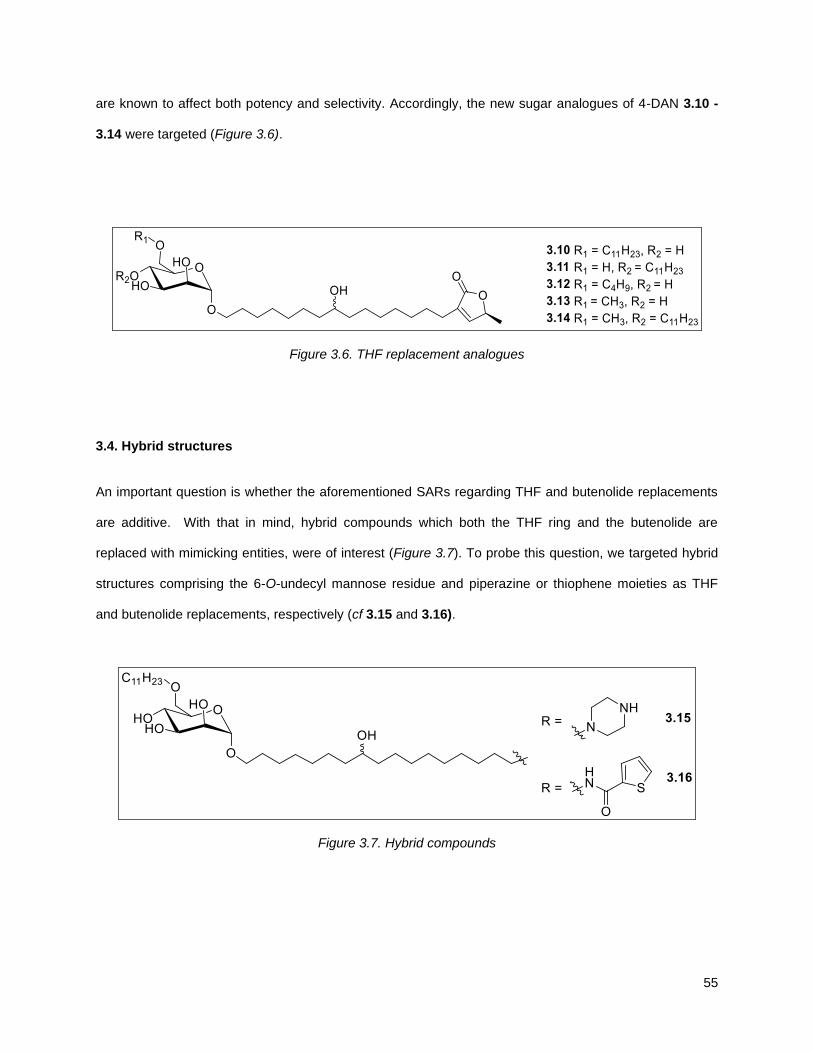
55
are known to affect both potency and selectivity. Accordingly, the new sugar analogues of 4-DAN 3.10 -
3.14 were targeted (Figure 3.6).
Figure 3.6. THF replacement analogues
3.4. Hybrid structures
An important question is whether the aforementioned SARs regarding THF and butenolide replacements
are additive. With that in mind, hybrid compounds which both the THF ring and the butenolide are
replaced with mimicking entities, were of interest (Figure 3.7). To probe this question, we targeted hybrid
structures comprising the 6-O-undecyl mannose residue and piperazine or thiophene moieties as THF
and butenolide replacements, respectively (cf 3.15 and 3.16).
Figure 3.7. Hybrid compounds

56
3.5. Modification of the hydrocarbon spacer
It has been proposed that the hydrophobic spacer connecting the THF and the butenolide controls the
active spatial arrangement between the THF and the butenolide.91 Consistent with this hypothesis, 14-
and 12- carbon chains appear to be optimal for the mono and bis-THF ACGs, respectively.11c The position
and number of hydroxyl groups also impacts on activity. Some studies suggest that having too many
alcohols in the middle chain can lower the activity of the compounds as an increase in hydrophilicity may
decrease mitochondrial uptake. Against this background we targeted the deoxygenated sugar-piperazine
hybrid structure 3.17 (Figure 3.8).
Figure 3.8. Deoxygenated analog
3.6. Synthetic strategy
The overall synthetic plan for these new analogues follows the CM approach described in Chapter 2 for 4-
DAN. Thus, the THF-alkene 3.18 (2.16 in Chapter 2), or one of its mannose substitutes 3.19-3.23, were
combined with either the butenolide alkene 3.24 (2.32 in Chapter 2) or its replacements 3.25-3.28.
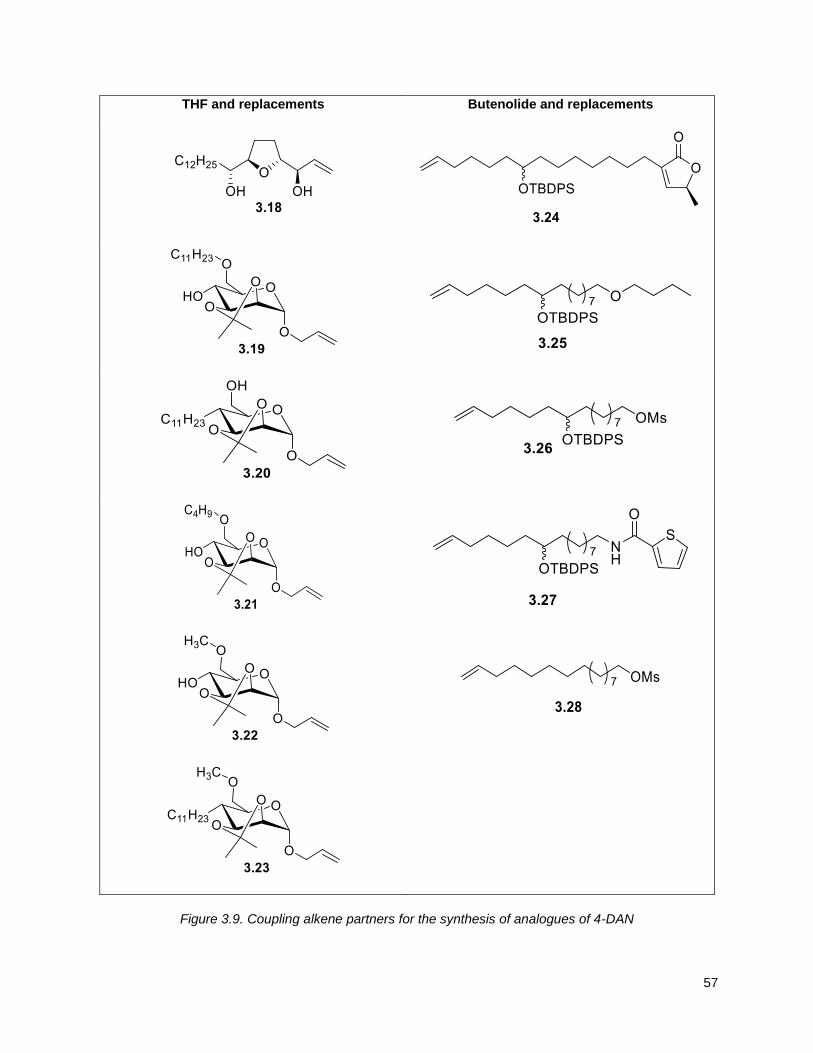
57
THF and replacements Butenolide and replacements
Figure 3.9. Coupling alkene partners for the synthesis of analogues of 4-DAN

58
3.7. Synthesis
3.7.1. Mannose alkenes
For efficiency, the synthesis of the different mannose alkenes was conducted in a divergent fashion from
a known precursor, 1-propenyl 2,3-O-isopropylidene mannopyranoside 3.32. This material can be
obtained in multi-gram quantity, in a straightforward fashion, over three steps from D-mannose: (i) acid
promoted glycosidayion of mannose with allyl alcohol, (ii) isopropylidenation of the product 3.30 to give
the bis-O-isopropylidene 3.31, and (iii) selective removal of the 4,6-O-isopropylidene (Scheme 3.1). The
yield over these three steps was 35%.
Scheme 3.1. Synthesis of mannose alkenes

59
Alkylation of 3.32 using one equivalent of base in THF provided a separable mixture of 3.19 (27%) and
3.20 (13%). For a streamlined synthesis of 3.19, the primary alcohol on 3.32 was first selectively
protected as the its TBDPS ether. Then, protection of the secondary alcohol by treatment with ethyl vinyl
ether and PPTS and desilylation of the resulting product provided 3.33. Alkylation of 3.33 with undecyl
bromide and treatment of the product under mildly acidic conditions yielded 3.19 (65%). Similarly,
butylation on 3.33 afforded 3.21 (58%) and methylation on 3.20 and 3.33 afforded 3.23 (64%) and 3.22
(90%), respectively.
3.7.2. Butenolide replacements
The precursor for the butenolide replacements was alkenol 3.35, which was obtained from the LAH
reduction of ester 3.34 (Scheme 3.2). Alkenes 3.25 and 3.26, the CM precursors for the piperazine and
butyl ether analogues, were obtained from butylation and mesylation on 3.35, respectively.
Scheme 3.2. Synthesis of butenolide replacements

60
Reaction of 3.26 with ethanolic ammonia and amidation of the resulting amine 3.36 with thiophene 2-
carboxylic acid provided 3.27, the CM precursor for the thiophene analogues.
3.7.3. Cross metathesis and final products
The CM reactions for the THF analogues were performed under similar conditions to those used in the
synthesis of 4-DAN in Chapter 2. 77, 80 Thus, reaction of THF alkene 3.18 with 2 equivalents of alkenes
3.25-2.37, in the presence of 10 mol% of 2nd generation Grubbs catalyst, in DCM at room temperature
for 20 h afforded CM products 3.37, 3.38 and 3.39 in 40, 27, 23% yields, respectively. Hydrogenation of
these products provided the reduced derivatives, which were transformed via straightforward substitution,
alkylation and desilylation procedures to give the final products 3.3, 3.4 and 3.7 respectively (Scheme
3.3).
Scheme 3.3. Synthesis of the compounds with butenolide replacements

61
For the synthesis of the mannose containing analogs, 3.10-3.14, modified CM conditions using Grubbs I
catalyst at 40 oC were needed (Scheme 3.4).94 The mannose butenolide metathesis products 3.40-3.44
were obtained in 25-96% yields. Subsequently, the alkene in the CM product was selectively reduced with
diimide as described in the earlier synthesis of 4-DAN, and the isopropylidene and silyl protecting groups
on the sugar were removed using acidic conditions to afford 3.10-3.14. The yields for the deprotection
steps were low as these reactions were not optimized. In the later compounds, after optimization, these
yields were higher.
Scheme 3.4. Synthesis of mannose containing compounds
CM between the 6-O-undecenyl-mannose alkene 3.19 and the mesylate containing alkenes, 3.26 and
3.28, and the thiophene alkene 3.27 provided 3.45, 3.46 and 3.47 respectively (Scheme 3.5)
Hydrogenation of 3.45 and 3.46, treatment of the reduced derivatives with piperazine, and acid promoted
removal of isopropylidene and TBDPS protecting groups provided the mannose-piperazine hybrid
compounds 3.15 and 3.17 respectively. Similarly, hydrogenation of 3.47 and removal of protecting groups
afforded the mannose-thiophene hybrid 3.16.

62
Scheme 3.5. Route to hybrid analogues
3.8 Summary
In Chapter 3, the CM methodology that was used for the synthesis of the 4-DAN in Chapter 2, was
applied to THF analogues of 4-DAN with butenolide substitutes, 3.3, 3.4 and 3.7. It has been shown that
the butenolide residue in other ACGs or mimetics thereof, can be replaced with heterocycles such as
thiophene or piperazine without loss in activity. Biological evaluation of such heterocyclic analogues of
DAN is needed to determine the generality of this trend. The CM methodology was also applied to ACG
analogues in which the THF moiety is replaced with a mannose residue, and the butenolide is preserved
or replaced with piperazine or thiophene residues, i.e. 3.10-3.17. These carbohydrate-derived structures
which follow from earlier studies in this laboratory and the aforementioned observations on butenolide
replacement, are attractive for their relatively easy accessibility and for the potential of the sugar motif to
act as a vector for tumor targeting. Biological evaluation of this wider set of sugar-derived analogues

63
would expand the SARs for these less well-known carbohydrate analogues and pave the way to their
possible development as therapeutic agents. The development of potentially more tumor selective ACGs
and antitumor evaluation are the foci of Chapters 4 and 5.

64
3.9. Experimental
THF-piperazine (3.3)
Compound 3.37b (29 mg, 0.033 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in
MeOH (0.5 mL). The reaction was stirred for 24 h and then quenched with a saturated solution of
NaHCO3 in MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (10%
MeOH/DCM) afforded 3.3 (3 mg, 14%). Rf = 0.6 (10% MeOH/DCM). 1H NMR (CD3OD, 600 MHz) δ 3.72
(m, 2H), 3.51 (m, 1H), 3.33 (m, 2H), 3.16 (m, 4H), 2.72 (m, 4H), 2.35 (m, 2H), 1.98 (m, 2H), 1.66 (m, 2H),
1.50-1.28 (m, 50H), 0.98 (t, J = 8.7 Hz, 3H); 13C NMR (CD3OD, 600 MHz) δ 84.0, 75.3, 72.6, 59.4, 50.3,
45.0, 38.6, 34.4, 33.3, 31.0, 30.8, 30.7 (2 signals), 29.8, 26.9, 23.9, 14.6. HRMS (ESI) calcd for
C38H77N2O4 (M+H)+ 625.5883, found 625.5874.
THF-thiophene (3.4)
Compound 3.38b (31 mg, 0.034 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in
MeOH (0.5 mL). The reaction was stirred for 20 h and then quenched with a saturated solution of
NaHCO3 in MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (10%
MeOH/DCM) afforded 3.4 (7 mg, 33%). Rf = 0.4 (60% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ
7.40 (m, 1H), 7.38 (m, 1H), 7.00 (m, 1H), 5.88 (br s, 1H), 3.73 (m, 2H), 3.60 (m, 1H), 3.34 (m, 4H), 3.28
(m, 2H), 2.24 (m, 2H), 1.91 (m, 3H), 1.55-1.00 (m, 50H), 0.95 (t, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 600
MHz) δ 161.8, 139.1, 129.6, 127.8, 127.6, 82.6, 74.0 (2 signals), 72.0 (2 signals), 59.0, 40.0, 37.5, 37.1,
33.5 (2 signals), 32.8, 31.9, 30.0, 29.7 (2 signals), 29.6 (2 signals), 29.5, 29.4 (2 signals), 29.2, 28.7,

65
28.0, 27.1, 26.9, 25.6 (3 signals), 25.5, 24.9, 24.5, 24.1, 22.7, 21.9, 19.8, 14.1, 13.6, 12.6, 11.4. HRMS
(ESI) calcd for C39H72NO5S (M+H)+ 666.5131, found 666.5127.
THF-butyl ether (3.7)
Compound 3.39 (50 mg, 0.047 mmol) was dissolved in EtOAc (4 mL). The solution was degassed for 30
min and Pd/C (10%, 5 mg) was introduced. Then, it was stirred under H2 atmosphere for 20 h. The
solution was filtered over Celite, concentrated in vacuo. FCC of the residue (30-35% EtOAc/hexanes)
yielded 3.39a (42 mg, 84%). Rf = 0.4 (40% EtOAc/hexanes).
Compound 3.39a (42 mg, 0.049 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in
MeOH (0.5 mL). The reaction was stirred overnight and then quenched with a saturated solution of
NaHCO3 in MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (5%
MeOH/DCM) afforded 3.7 (15 mg, 51%). Rf = 0.4 (60% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ
3.76 (m, 2H), 3.37 (m, 1H), 3.37 (m, 6H), 1.95 (m, 2H), 1.66 (m, 2H), 1.56-1.39 (m, 54H), 0.88 (t, J = 7.4
Hz, 3H), 0.84 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 82.6, 74.0 (2 signals), 72.0, 71.0, 70.6 (2
signals), 37.5 (3 signals), 33.5 (2 signals), 31.9 (2 signals), 29.8, 29.7 (4 signals), 29.6 (3 signals), 29.5 (2
signals), 29.4, 28.7, 26.2, 25.7, 25.6 (3 signals), 25.5, 22.7, 19.4, 19.2, 14.1. HRMS (ESI) calcd for
C38H77O5 (M+H)+ 613.5771, found 613.5760.
6-O-Undecyl mannoside-butenolide (3.10)

66
Compound 3.40a (35 mg, 0.036 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in
MeOH (0.5 mL). The reaction was stirred for 24 h and then quenched with a saturated solution NaHCO3
in MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (5-10% MeOH/DCM)
afforded 3.10 (5 mg, 20%). Rf = 0.5 (5% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ 5.96 (s, 1H), 4.97 (m,
1H), 4.80 (s, 1H), 3.90 (s, 1H), 3.81 (m, 1H), 3.76-3.36 (m, 13H), 2.24 (m, 2H), 1.45-1.20 (m, 42H), 1.37
(partially buried d, J = 6.8 Hz, 3H), 0.86 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 600 MHz) 148.9, 134.3, 99.5,
72.2, 71.9, 71.8, 71.7, 70.8, 70.5, 69.1, 67.8, 37.5, 31.9, 29.7, 29.6 (2 signals), 29.5 (2 signals), 29.3 (3
signals), 29.2, 29.1, 27.4, 26.1, 26.0, 25.6, 25.5, 25.2, 22.7, 19.2, 14.1. HRMS (ESI) calcd for C37H69O9
(M+H)+ 657.4942, found 657.4930.
4-O-Undecyl mannoside-butenolide (3.11)
Compound 3.41 (60 mg, 0.064 mmol) was transformed to 3.41a following the reduction protocol
described for preparation of 3.40a. FCC in 1-5% (acetone/DCM) delivered 3.41a (35 mg, 58%). Rf = 0.3
(5% acetone/DCM). 1H NMR (CDCl3, 600 MHz) δ 7.59 (m, 4H), 7.31 (m, 6H), 6.90 (s, 1H), 4.99 (m, 2H),
4.18 (m, 1H), 4.10 (m, 1H), 3.77 (m, 2H), 3.65-3.42 (m, 4H), 3.31 (m, 1H), 2.17 (m, 2H), 2.03 (br s, 1H),
1.61-1.03 (m, 51H), 0.97 (s, 9H), 3.43 (t, J = 6.7 Hz, 3H.
Compound 3.41a (27 mg, 0.028 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in
MeOH (0.5 mL). The reaction was stirred overnight and then quenched with saturated solution of
NaHCO3 in MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (5-10%
MeOH/DCM) afforded 3.11 (4 mg, 17%). Rf = 0.5 (5% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ 6.95 (s,
1H), 5.00 (m, 1H), 4.78 (s, 1H), 3.90 (m, 1H), 3.80 (m, 1H), 3.75-3.48 (m, 8H) 3.37 (m, 1H), 2.23 (m, 2H),
1.54-1.22 (m, 42H), 1.28 (partially buried d, J = 5.0 Hz, 3H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3,
600 MHz) δ 174.1, 148.8, 134.4, 99.6, 76.2, 73.6, 72.2, 71.8, 71.6, 71.3, 68.0, 62.4, 37.6, 32.1, 30.6,

67
29.9, 29.8 (2 signals), 29.7 (2 signals), 29.6, 29.5, 29.4, 29.3, 27.6, 26.3, 26.2, 25.8, 25.7, 25.4, 22.9,
19.4, 14.3. HRMS (ESI) calcd for C37H68O9Na (M+Na)+ 679.4761, found 679.4754.
6-O-Butyl mannoside-butenolide (3.12)
Compound 3.42a (60 mg, 0.07 mmol) was dissolved in CH2Cl2 (5 mL) and treated with 5% AcCl in MeOH
(1 mL). The reaction was stirred for 24 h and then quenched with a saturated solution of NaHCO3 in
MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (5-10% MeOH/DCM)
afforded 3.12 (5 mg, 14%). Rf = 0.6 (5% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ 6.95 (s, 1H), 4.97 (m,
1H), 4.78 (s, 1H), 3.87 (m, 1H), 3.90-3.60 (m, 5H), 3.50 (m, 4H), 3.38 (m, 1H), 2.35 (m, 2H), 2.22 (m, 3H),
1.65-1.10 (m, 28H), 0.87 (m, 3H); 13C NMR (CDCl3, 600 MHz) δ 174.1, 149.1, 136.1, 134.4, 99.7, 72.1,
72.0, 71.9, 71.6 (2 signals), 70.7, 70.3, 69.7, 67.9, 37.6, 32.1, 31.7, 29.8 (2 signals), 29.6, 29.5, 29.4, 29.3
(2 signals), 29.2, 29.0, 28.7, 27.5 (2 signals), 27.2, 27.1, 26.1 (2 signals), 25.7, 25.6, 25.3, 25.0, 23.9 (2
signals), 22.8 (2 signals), 19.4, 19.3, 14.0. HRMS (ESI) calcd C30H55O9 for (M+H)+ 559.3846, found
559.3837.
6-O-Methyl mannoside-butenolide (3.13)
Compound 3.43a (54 mg, 0.06 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in MeOH
(0.5 mL). The reaction was stirred for 24 h and then quenched with a saturated solution of NaHCO3 in
MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (5-10% MeOH/DCM)
afforded 3.13 (3 mg, 7%). Rf = 0.7 (5% MeOH/DCM). 1H NMR (CD3OD, 500 MHz) δ 7.79 (s, 1H), 7.14 (s,

68
1H), 4.94 (m, 1H), 4.57 (s, 1H), 3.64-3.18 (m, 11H), 3.28 (partially buried s, 3H), 2.10 (m, 2H), 1.26 (m,
27H). HRMS (ESI) calcd for C27H48O9Na (M+Na)+ 539.3196, found 539.3190.
6-O-Methyl-4-O-undecyl mannoside-butenolide (3.14)
Compound 3.44a (27 mg, 0.027 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in
MeOH (0.5 mL). The reaction was stirred for 24 h and then quenched with a saturated solution of
NaHCO3 in MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (60-80%
EtOAc/hexanes) afforded 3.14 (3 mg, 16%). Rf = 0.5 (60% EtOAc/hexanes). 1H NMR (CD3OD, 500 MHz)
δ 7.13 (s, 1H), 4.93 (m, 1H), 4.54 (s, 1H), 3.70 (m, 1H), 3.55 (m, 3H), 3.50-3.30 (m, 5H), 3.25 (m, 2H),
3.24 (partially buried s, 3H), 2.11 (m, 2H), 1.44 (m, 6H), 1.16 (m, 42H), 0.77 (t, J = 7.5 Hz, 3H). HRMS
(ESI) calcd for C38H70O9Na (M+Na)+ 693.4918, found 693.4910.
6-O-Undecyl mannoside-piperazine (3.15)
Compound 3.45b (62 mg, 0.063 mmol) was dissolved in CH2Cl2 (5 mL) and treated with 5% AcCl in
MeOH (1 mL). The reaction was stirred for 24 h and then quenched with a saturated solution of NaHCO3
in MeOH. The mixture was filtered and concentrated in vacuo. FCC of the residue (10% MeOH/DCM)
afforded 3.15 (7 mg, 18%). Rf = 0.3 (20% MeOH/DCM). 1H NMR (CD3OD, 600 MHz) δ 4.58 (s, 1H), 3.65-
3.24 (m, 11H), 3.18 (m, 4H), 2.88 (m, 4H), 2.48 (m, 2H), 1.64-1.04 (m, 46H), 0.88 (m, J = 7.5 Hz, 3H); 13C
NMR (CD3OD, 600 MHz) δ 101.7, 73.5, 72.9, 72.8, 72.6, 72.3, 71.9, 69.1, 68.8, 59.3, 51.4, 45.0, 38.6 (2

69
signals), 33.2, 31.0 (2 signals), 30.9 (3 signals), 30.8, 30.7 (2 signals), 30.6, 27.5 (2 signals), 26.9 (2
signals), 23.9, 14.6. HRMS (ESI) calcd for C38H77N2O7 (M+H)+ 673.5731, found 673.5724.
6-O-Undecyl mannoside-thiophene (3.16)
3.46 (70 mg, 0.07 mmol) was dissolved in EtOH (2 mL). Then 10% wt (0.7 mg) Pd/C was added and the
mixture was maintained under H2 for 16 h (2x). The mixture was filtered over Celite and the filtrate
evaporated in vacuo. The residue was dissolved in DCM (2 mL) and treated with 5% AcCl in MeOH (0.4
mL) for 4 h. Additional 5% AcCl in MeOH (0.2 mL) was added over 9 h. The mixture was neutralized with
K2CO3 in MeOH, filtered through Celite and purified by FCC (60-100% EtOAC/hexanes) to yield 3.16 (14
mg, 65%).1H NMR (CDCl3, 600 MHz) δ 7.46 (d, J = 3.6 Hz, 1H), 7.42 (d, J = 4.9 Hz, 1H), 7.04 (dd, J1 =
4.9 Hz, J2 = 3.8 Hz, 1H), 5.99 (br s, 1H), 4.79 (s, 1H), 3.88 (br s, 1H), 3.79-3.63 (m, 6H), 3.54 (m, 1H),
3.45 (m, 2H), 3.37 (m, 3H), 1.56 (m, 6H), 1.36 (m, 6H), 1.38-1.22 (m, 37H), 0.85 (t, J = 6.8 Hz, 3H); 13C
NMR (CDCl3, 600 MHz) δ 162.1, 139.3, 129.8, 128.1, 127.8, 99.7, 72.3, 72.1, 71.9, 70.7, 67.9, 40.2, 37.6
(2 signals), 32.1, 29.8 (4 signals), 29.7 (2 signals), 29.6 (2 signals), 29.5, 29.4 (2 signals), 27.1, 26.2,
25.8, 25.6, 22.9, 14.3. HRMS (ESI) calcd for C39H71NO8SNa (M+Na)+ 736.4793, found 736.4782.
6-O-Undecyl mannoside-deoxypiperazine (3.17)
Compound 3.47b (44 mg, 0.067 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in
MeOH (0.5 mL). The reaction was stirred for 2 h and then quenched with a solution of methanol and

70
NaHCO3. The mixture was filtered and concentrated in vacuo. FCC of the residue (5% MeOH/DCM)
afforded 3.17 (15 mg, 38%). Rf = 0.2 (10% MeOH/DCM). 1H NMR (CD3OD, 600 MHz) δ 4.62 (s, 1H), 3.66
(m, 3H), 3.58 (m, 3H), 3.44 (m, 3H), 3.33 (m, 1H), 3.12 (m, 4H), 2.65 (br s, 4H), 2.41 (br s, 2H), 1.53-1.44
(m, 6H), 1.21 (m, 42H), 0.82 (t, J = 6.8 Hz, 3H); 13C NMR (CD3OD, 600 MHz) δ 101.7, 73.5, 72.9, 72.8,
72.3, 72.0, 69.0, 68.8, 59.3, 51.2, 44.9, 33.3, 31.0, 31.0 (3 signals), 30.9 (4 signals), 30.8, 30.7 (3
signals), 28.5, 27.6, 27.5, 23.9, 14.6. HRMS (ESI) calcd for C38H77N2O6 (M+H)+ 657.5782, found
657.5775.
Propan-1-yl-2,3-O-isopropylidene-6-O-undecyl-α-D-mannopyranoside (3.19)
3.32 (1.30 g, 5 mmol) and TBAI (0.32 g, 1 mmol) were dissolved in THF (36 mL) under a N2 atmosphere.
The reaction was cooled to 0 oC and NaH (0.22 g, 5.5 mmol) was added. The reaction was warmed
slowly to rt, then recooled to 0 oC and bromoundecane (1.23 mL, 5.5 mmol) added. The mixture was
stirred for 2 h, quenched and purified via FCC (40-50% EtOAc/hexanes), to give: 0.2 g of dialkylated
material, 3.19 (0.16 g, 13%), 3.20 (0.34 g, 26%), 3.32 (0.50 g, 38% bsrm) and 4,6-di-O-undecenyl
derivative (0.20 g, 7%).
Compound 3.19 was also prepared from 3.33. Compound 3.33 (1.51 g, 4.5 mmol) and TBAI (0.29 g, 0.9
mmol) were dissolved in DMF (22 mL) under a N2 atmosphere. The mixture was cooled to 0 oC and NaH
(0.2 g, 5.0 mmol) was added. The reaction was warmed to rt over 30 min. Then, it was cooled down to 0
oC and undecenyl bromide (1.18 mL, 5 mmol) was added. The reaction was stirred overnight at rt, then
cooled to 0 oC and quenched slowly with water. The mixture was extracted with ethyl ether and the
organic phase washed with brine, dried (NaSO4) and evaporated in vacuo. The residue was resuspended
in MeOH and treated with PPTS (ca 0.5 g, pH 3-4). The mixture was stirred for 1 h, then, quenched with
saturated NaHCO3 in MeOH. The solution was filtrated and concentrated under pressure. FCC of the
residue (40-50% EtOAc/hexanes) afforded 3.19 (1.22 g, 65%). 1H NMR (CDCl3, 500 MHz) δ 5.89 (m, 1H),

71
5.28 (m, 1H), 5.20 (m, 1H), 5.03 (s, 1H), 4.20-4.10 (m, 3H), 3.99 (m, 1H), 3.73-3.60 (m, 4H), 3.47 (m, 2H),
2.93 (br s, 1H), 1.57 (m, 2H), 1.50 (s, 3H), 1.33 (s, 3H), 1.33-1.23 (m, 16 H), 0.85 (t, J = 6.0 Hz, 3H); 13C
NMR (CDCl3, 500 MHz) δ 133.7, 118.2, 109.7, 96.5, 78.2, 75.5, 72.3, 71.8, 71.7, 68.3, 68.0, 32.1, 29.8 (3
signals), 29.7, 29.5, 28.1, 26.3 (2 signals), 22.9, 14.34. HRMS (ESI) calcd for C23H42O6Na (M+Na)+
437.2879, found 437.2879.
Propan-1-yl-2,3-O-isopropylidene-4-O-undecyl-α-D-mannopyranoside (3.20)
1H NMR (CDCl3, 500 MHz) δ 5.86 (m, 1H), 5.26 (m, 1H), 5.19 (m, 1H), 5.04 (s, 1H), 4.20-4.11 (m, 3H),
3.94 (m, 1H), 3.74 (m, 2H), 3.71 (m, 1H), 3.60 (m, 1H), 3.44 (m, 1H), 3.36 (m, 1H), 1.52 (m, 5H), 1.51
(partially buried s, 3H), 1.33 (s, 3H), 1.23 (m, 14H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ
133.6, 118.2, 109.4, 96.4, 78.8 (2 signals), 75.9, 71.8, 68.6, 68.3, 62.9, 32.1, 30.3, 29.8, 29.7, 29.6, 28.2,
26.5, 26.3, 22.9, 14.3. HRMS (ESI) calcd for C23H42O6Na (M+Na)+ 437.2862, found 437.2874.
Propan-1-yl-2,3-O-isopropylidene-6-O-butyl-α-D-mannopyranoside (3.21)
Compound 3.21 was obtained from compound 3.33 (0.33 g, 0.99 mmol) following the procedure for 3.19
with 1-bromobutane (0.16 mL, 1.48 mmol) as the alkylating agent. After purification via FCC (20-30%
EtOAc/hexanes) 3.21 (0.18 g, 58%) was obtained. Rf = 0.75 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500
MHz) δ 5.88 (m, 1H), 5.28 (m, 1H), 5.19 (m, 1H), 5.03 (s, 1H), 4.18 (m, 1H), 4.13 (s, 2H), 3.98 (m, 1H),
3.72-3.63 (m, 3H), 3.61 (m, 1H), 3.46 (m, 2H), 3.00 (br s, 1H), 1.54 (m, 2H), 1.50 (s, 3H), 1.34 (m, 2H),

72
1.32 (partially buried s, 3H), 0.89 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 133.6, 118.1, 109.7,
96.5, 78.2, 75.5, 71.9, 71.7, 71.6, 68.3, 68.0, 31.8, 28.1 (2 signals), 26.3, 19.4, 14.1. HRMS (ESI) calcd
for C16H28O6Na (M+Na)+ 339.1784, found 339.1780.
Propan-1-yl-2,3-O-isopropylidene-6-O-methyl-α-D-mannopyranoside (3.22)
3.33 (0.74 g, 2.2 mmol) and TBAI (0.24 g, 0.74 mmol) were dissolved in DMF (20 mL) under a N2
atmosphere. The reaction was cooled to 0 oC and NaH (0.13 g, 0.01 mmol) was added. The reaction was
warmed up slowly over 30 min. Then, it was cooled down to 0 oC and CH3I (0.3 mL, 4.8 mmol) was
added. The mixture was stirred overnight. The solution was brought to 0 oC, quenched slowly with water.
Then, it was extracted using ethyl ether (3x100 mL) and washed with brine. The crude residue was
resuspended in MeOH and treated with PPTS (ca 0.5 g, pH 3-4). The mixture was stirred for 1 h. Then,
quenched with saturated NaHCO3 in methanol and the solvent was evaporated in vacuo. FCC yielded
3.22 (0.55 g, 90%). Rf = 0.38 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.88 (m, 1H), 5.28 (m,
1H), 5.19 (m, 1H), 5.04 (s, 1H), 4.19 (m, 1H), 4.12 (m, 2H), 3.67 (m, 3H), 3.59 (m, 1H), 3.38 (s, 3H), 2.68
(br s, 1H), 1.49 (s, 3H), 1.32 (s, 3H); 13C NMR (CDCl3, 500 MHz) δ 133.5, 118.0, 96.5, 78.0, 75.4, 72.9,
70.6, 68.3, 68.1, 59.6, 27.9, 26.1. HRMS (ESI) calcd for C13H23O6 (M+H)+ 275.1489, found 275.1470.
Propan-1-yl-2,3-O-isopropylidene-6-O-methyl-4-O-undecyl-α-D-mannopyranoside (3.23)

73
NaH (58 mg, 1.45 mmol) was added at 0 oC to a solution of 3.20 (0.28 g, 0.9 mmol), TBAI (66 mg, 0.18
mmol) in DMF (15 mL). The mixture was stirred for 30 min, and CH3I (0.09 mL, 1.45 mmol) was added.
The reaction was stirred for 3-4 h at rt, then cooled to 0 oC and quenched with water. The mixture was
extracted with ethyl ether (3x25 mL) and the organic phase was dried (Na2SO4) and evaporated under
reduced pressure. The residue was purified via FCC (10-15% EtOAc/hexanes) to give 3.23 (0.27 g, 64%).
Rf = 0.9 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.86 (m, 1H), 5.25 (m, 1H), 5.16 (m, 1H),
5.05 (s, 1H), 4.17 (m, 2H), 4.10 (m, 1H), 3.96 (m, 1H), 3.76 (m, 1H), 3.64-3.53 (m, 3H), 3.44 (m, 1H), 3.36
(partially buried s, 3H), 3.34 (m, 1H), 1.52 (m, 2H), 1.50 (partially buried s, 3H), 1.31 (partially buried s,
3H), 1.22 (br s, 16H), 0.84 (t, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 133.8, 117.8, 109.3, 96.6,
79.0, 76.3, 76.0, 71.7 (2 signals), 68.4, 68.0 (2 signals), 67.9, 59.4, 32.1, 30.2, 29.8, 29.6, 29.5, 28.2,
26.4, 26.3, 22.8, 14.3. HRMS (ESI) calcd for C24H44O6Na (M+Na)+ 451.3036, found 451.3028.
((1-Butoxydec-9-en-4-yl)oxy)(tert-butyl)diphenylsilane (3.25)
Compound 3.25 was prepared following the procedure described for 3.21. For 3.25: 1H NMR (CDCl3, 500
MHz) δ 7.67 (m, 4H), 7.42-7.34 (m, 6H), 5.73 (m, 1H), 4.93 (m, 1H), 4.90 (m, 1H), 3.69 (m, 1H), 3.40 (m,
4H), 1.94 (m, 2H), 1.56 (m, 4H), 1.40-1.07 (m, 22H), 1.05 (br s, 9H), 0.90 (t, J = 7.4 Hz, 3H); 13C NMR
(CDCl3, 500 MHz) δ 139.3, 136.2, 135.8, 135.0, 129.7, 129.6, 127.8, 127.6, 114.4, 73.4, 71.2, 70.9, 36.5,
36.3, 33.9, 32.1, 30.0, 29.9, 29.7 (3 signals), 29.2, 27.3, 27.1, 26.4, 25.1, 24.6, 19.6 (2 signals), 14.2.
HRMS (ESI) calcd for C36H59O2Si (M+H)+ 551.4279, found 551.4263.
10-((tert-Butyldiphenylsilyl)oxy)hexadec-15-en-1-yl methanesulfonate (3.26)
To a solution of compound 3.35 (1.0 g, 2.14 mmol) at 0 oC in CH2Cl2 (30 mL) was added Et3N (0.35 mL,
2.57 mmol) and MsCl (0.19 mL, 2.57 mmol). The reaction was warmed to rt and stirred overnight. The

74
reaction was diluted with H2O and extracted with ethyl ether (3x100 mL). The organic phase was washed
with brine, dried (Na2SO4) and evaporated in vacuo. FCC of the residue afforded 3.26 (0.84 g, 69%) as a
yellow oil. Rf = 0.5 (20% EtOAc/hexanes).1H NMR (CDCl3, 500 MHz) δ 7.65 (m, 4H), 7.41-7.32 (m, 6H),
5.72 (m, 1H), 4.91 (m, 1H), 4.88 (m, 1H), 4.20 (t, J = 6.6 Hz, 2H), 3.67 (m, 1H), 2.98 (s, 3H), 1.91 (m, 2H),
1.72 (m, 2H), 1.40-1.06 (m, 20H), 1.02 (br s, 9H); 13C NMR (CDCl3, 500 MHz) δ 139.3, 136.1, 129.6,
127.6, 114.4, 73.4, 70.4, 37.6, 36.5, 36.3, 34.0, 29.9, 29.8, 29.6, 29.5, 29.3, 29.2, 29.1, 27.3, 25.6, 25.1,
24.6, 19.6. HRMS (ESI) calcd for C33H52O4SSiNa (M+Na)+ 595.3253, found 595.3246.
10-Hydroxyhexadec-15-en-1-yl methanesulfonate (3.26a)
A 5% solution of acetyl chloride in MeOH (1.7 mL) was added to a mixture of 3.26 (1.0 g, 1.75 mmol) and
DCM (17 mL). The mixture was stirred for 16 h at rt, then neutralized with methanolic sodium bicarbonate.
The mixture was filtered and concentrated in vacuo. FCC of the residue (20-30% EtOAc/hexanes) yielded
3.26a (0.39 g, 66%). Rf = 0.5 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.77 (m, 1H), 4.97 (m,
1H), 4.93 (m, 1H), 4.20 (t, J = 6.0 Hz, 2H), 3.56 (m, 1H), 2.99 (s, 3H), 2.06 (m, 2H), 1.70 (m, 2H), 1.39-
1.34 (m, 20H); 13C NMR (CDCl3, 500 MHz) δ 139.1, 114.5, 72.0, 70.4, 37.6, 37.5, 37.4, 33.9, 29.7, 29.6,
29.5, 29.2, 29.1 (2 signals), 25.7, 25.5, 25.3. HRMS (ESI) calcd for C17H34O4SiNa (M+Na)+ 357.2070,
found 357.2064.
10-Iodohexadec-15-en-1-yl methanesulfonate (3.26b)
A mixture of 3.26a (0.38 g, 1.15 mmol), Ph3P (0.33 g, 1.26 mmol) and imidazole (90 mg, 1.38 mmol)
benzene (12 mL) was stirred for 15 min at rt. Then, iodine (0.15 g, 1.26 mmol) was introduced and stirring
continued for 2 h. The reaction was quenched with aqueous Na2S2O3 and extracted with ethyl acetate.

75
The organic phase was dried (Na2SO4) and evaporated in vacuo. The residue was purified via FCC (15%
EtOAc/hexanes) to yield 3.26b (0.31 g, 61%). Rf = 0.6 (20% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz)
δ 5.78 (m, 1H), 4.98 (m, 1H), 4.92 (m, 1H), 4.20 (t, J = 6.0 Hz, 2H), 4.08 (m, 1H), 2.98 (s, 3H), 2.05 (m,
2H), 1.84 (m, 2H), 1.75-1.63 (m, 4H), 1.52 (s, 2H), 1.43 (m, 6H), 1.31 (m, 8H); 13C NMR (CDCl3, 600
MHz) δ 138.7, 114.6, 70.2, 40.6, 40.5, 37.4, 33.6, 29.5, 29.3, 29.1, 29.0 (2 signals), 28.8, 28.1 25.4.
N-(10-((tert-Butyldiphenylsilyl)oxy)hexadec-15-en-1-yl)thiophene-2-carboxamide (3.27)
Amine 3.36 (0.26 g, 0.52 mmol) and 2-thiophene carboxylic acid (0.10 g, 0.78 mmol) were azeotroped
and dissolved in DCM (26 mL) Then, EDCI (0.30 g, 1.58 mmol), dry Et3N (0.29 mL, 2.10 mmol) and
catalytic DMAP (13 mg, 0.10 mmol) were added. The reaction was stirred for 16 h. Then, it was extracted
using more DCM (3x10 mL) and it was washed with water (3x). The organic phase was dried (Na2SO4)
and evaporated and FCC of the residue yielded 3.27 (0.22 g, 69%). Rf = 0.7 (20% EtOAc/hexanes). 1H
NMR (CDCl3, 600 MHz) δ 7.65 (m, 4H), 7.45 (m, 2H), 7.42 (m, 6H), 7.04 (d, J = 6.0 Hz, 1H) 5.90 (br s,
1H), 5.71 (m, 1H), 4.90 (m, 1H), 4.88 (m, 1H), 3.67 (m, 1H), 3.40 (m, 2H), 1.90 (m, 2H), 1.57 (m, 4H),
1.36 (m, 6H), 1.15 (m, 12H), 1.08 (s, 9H); 13C NMR (CDCl3, 500 MHz) δ 139.3, 136.2, 135.0, 129.8,
129.6, 128.0, 127.8, 127.6, 114.4, 73.4, 40.3, 36.5, 36.3, 33.9, 30.0, 29.9, 29.7, 29.7, 29.5, 29.2, 27.3,
27.2, 25.0, 24.6, 19.6.
Hexadec-15-en-1-yl methanesulfonate (3.28)
Compound 3.26b (0.31 g, 0.07 mmol) was stirred with zinc (0.26 g, 4.05 mmol) and methanol (15 mL).
The reaction was quenched with water, extracted with ethyl ether (3x10 mL) and purified via FCC to
obtain 3.28 (0.25 g, quantitative yield). Rf = 0.46 (20% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ

76
5.80 (m, 1H), 4.98 (m, 1H), 4.91 (m, 1H), 4.21 (t, J = 6.0 Hz, 2H), 2.99 (s, 3H), 2.02 (m, 2H), 1.73 (m, 2H),
1.39-1.30 (m, 4H), (br s, 18H). 13C NMR (CDCl3, 600 MHz) δ 139.5, 114.3, 70.4, 37.6, 34.0, 29.8, 29.8,
29.7, 29.6, 29.4, 29.3, 29.2 (2 signals), 25.6.
Propen-1-yl-2,3-O-isopropylidene-α-D-mannopyranoside (3.32)
To a solution of 3.34 (3.3 g, 0.01 mmol) in MeOH (50 mL) was added PPTS (0.24 g, 0.96 mmol). The
reaction mixture was stirred for 4 h and then quenched with a saturated solution of NaHCO3 in MeOH.
The mixture was concentrated in vacuo. FCC of the residue in 50-60% EtOAc/hexanes gave 3.35 (2.19 g,
95% brsm). Rf = 0.34 (50% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.86 (m, 1H), 5.30 (m, 1H),
5.22 (m, 1H), 5.05 (s, 1H), 4.17 (m, 3H), 3.98 (m, 1H), 3.83 (m, 2H), 3.74 (m, 1H), 3.64 (m 1H), 2.51 (br s,
1H), 2.04 (m, 1H), 1.51 (s, 3H), 1.34 (s, 3H); 13C NMR (CDCl3, 500 MHz) δ 133.5, 128.6, 118.4, 109.9,
96.7, 75.7, 70.1, 69.8, 68.5, 62.8, 28.1, 26.3.
Propen-1-yl-6-O-tert-butyldiphenylsilyl-2,3-O-isopropylidene-α-D-mannopyranoside (3.32a)
3.32 (2.19 g, 8.41 mmol), TBDPSCl (2.37 mL, 9.25 mmol) and imidazole (1.14 g, 16.82 mmol) were
dissolved in THF (84 mL) and stirred overnight. The solvent was evaporated in vacuo and the mixture
was redissolved in ethyl ether and water and extracted (3x100 mL). The combined organic phase was
washed with brine, dried (Na2SO4) and the solvent was evaporated in vacuo. FCC of the residue afforded
product 3.32a (1.7 g, 56% brsm). Rf = 0.4 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 7.72 (m,
4H), 7.44 (m, 6H), 5.90 (m, 1H), 5.28 (m, 1H), 5.22 (m, 1H), 5.06 (s, 1H), 4.18 (m, 3H), 3.99 (m, 1H), 3.95
(m, 2H), 3.91 (m, 1H), 3.88 (m, 1H), 2.75 (m, 1H), 1.53 (s, 3H), 1.27 (s, 3H), 1.08 (s, 9H); 13C NMR

77
(CDCl3, 500 MHz) δ 135.8, 135.7, 133.6, 133.0, 130.0 (2 signals), 127.9 (2 signals), 118.1, 109.7, 96.3,
78.3, 75.5, 71.0, 69.6, 68.0, 64.8, 28.1, 27.0, 26.3, 19.4. HRMS (ESI) calcd for C28H38O6SiNa (M+Na)+
521.2335, found 521.2329.
Propen-1-yl-6-O-tert-butyldiphenylsilyl-4-O-[1-ethoxyethyl]-2,3-O-isopropylidene-α-D-
mannopyranoside (3.32b)
PPTS (ca 1 g) was added to a mixture of 3.32a (1.7 g, 3.4 mmol), ethyl vinyl ether and DCM solution (1:1,
100 mL) until a pH of 4. The reaction was stirred for 2 h and quenched with methanolic NaHCO3. The
solution was filtered and evaporated to give crude 3.32b (ca 1.42 g). Rf = 0.69 (20% EtOAc/hexanes). 1H
NMR (CDCl3, 500 MHz) δ 7.71 (m, 4H), 7.38 (m, 6H), 5.90 (m, 1H), 5.26 (m, 1H), 5.21 (m, 1H), 5.08 (m,
1H), 4.90 (m, 1H), 4.24-3.99 (m, 3H), 3.96-3.75 (m, 3H), 3.74 (m, 2H), 3.74-3.68 (m, 2H), 1.52 (s, 3H),
1.26 (s, 3H), 1.20 (m, 4H), 1.04 (s, 9H), 0.80 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 136.1,
135.8 (2 signals), 134.0, 133.9, 133.8 (2 signals), 133.5, 133.3, 129.8 (2 signals), 129.7 (2 signals), 127.8
(2 signals), 127.7, 118.1, 118.0, 109.4, 109.3, 100.9, 100.0, 96.1, 95.9, 79.1, 78.7, 76.0 (2 signals), 73.3,
73.2, 69.8 (2 signals), 67.7, 67.6, 63.6, 62.9, 61.8, 61.0, 28.2, 28.1, 27.0, 26.6 (2 signals), 22.9, 21.2,
20.4, 19.5, 19.4 (2 signals), 15.3, 14.3.
Propen-1-yl-4-O-[1-ethoxyethyl]-2,3-O-isopropylidene-α-D-mannopyranoside (3.33)
A mixture of crude 3.35b (ca 1.42 g), TBAF (0.1 M in THF, 50 mL, 5 mmol) and THF (100 mL) was stirred
overnight at rt. The solvent was then evaporated in vacuo. FCC of the residue yielded 3.36 (0.7 g, 85%).
Rf = 0.28 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 5.98 (m, 1H), 5.34 (m, 1H), 5.22 (m, 1H),

78
5.10 (m, 1H), 4.95 (m, 1H), 4.29 (m, 1H), 4.14 (m, 2H), 3.95 (m, 1H), 3.78 (m, 3H), 3.64 (m, 3H), 1.47 (s,
3H), 1.26 (m, 6H), 1.23 (m, 3H); 13C NMR (CDCl3, 500 MHz) δ 133.7, 133.6, 118.2 (2 signals), 109.5,
109.4, 101.3, 100.9, 78.9, 78.5, 76.3, 76.0, 73.5, 72.8, 69.0, 68.7, 68.3, 68.2, 62.2, 62.1, 62.0, 28.3, 28.2,
26.5 (2 signals), 20.7, 20.4, 15.4, 15.3.
10-((tert-Butyldiphenylsilyl)oxy)hexadec-15-en-1-ol (3.35)
LAH (0.1 g, 2.7 mmol) was slowly added to a solution of compound 3.34 (0.92 g, 1.8 mmol) in CH2Cl2 (18
mL) at 0 oC. The mixture was warmed to rt and stirred for an additional 1 h. Then, water (10 mL) and
NaOH (1 mL, 1M), were added, subsequently. Na2SO4 was added to dry the H2O and the mixture was
filtered over Celite. The filtrated was concentrated in vacuo and then was subjected to FCC to afford
compound 3.35 (0.71 g, 85%). Rf = 0.2 (10% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ 7.65 (m,
4H), 7.34 (m, 6H), 5.72 (m, 1H), 4.92 (m, 1H), 4.88 (m, 1H), 4.10 (m, 1H), 3.68 (m, 1H), 3.62 (m, 2H),
1.92 (m, 2H), 1.54 (m, 4H), 1.38-1.17 (m, 8H), 1.02 (s, 9H). HRMS (ESI) calcd for C32H50O2SiNa (M+Na)+
517.3478, found 517.3472.
10-((tert-Butyldiphenylsilyl)oxy)hexadec-15-en-1-amine (3.36)
Ethanol (ca 200 mL) was saturated with ammonia gas at 0 oC by bubbling for 30 minutes. 3.26 (0.65 g,
1.13 mmol) was placed in a high-pressure tube and the saturated ammonia solution (150 mL) was added.
The vessel was closed, and the reaction was heated at approximately 80 oC for 4 h. The reaction was
brought to rt and the solvent was concentrated in vacuo. The residue was purified via FCC (10%
MeOH/DCM) to afford 3.36 (0.48 g, 94%). Rf = 0.32 (20% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ
7.63 (m, 4H), 7.36 (m, 6H), 5.70 (m, 1H), 4.88 (m, 1H), 4.85 (m, 1H), 4.10 (m, 1H), 3.10 (m, 2H), 1.90 (m,
2H), 1.45 (m, 2H), 1.35 (m, 4H), 1.16 (m, 16H), 1.06 (s, 9H); 13C NMR (CDCl3, 500 MHz) δ 139.2, 136.1,

79
134.9, 129.5, 127.5, 114.3, 73.3, 40.1, 39.4, 36.4, 36.3, 33.9, 29.8, 29.7, 29.6, 29.2, 29.1, 27.8, 27.2,
26.7, 25.0, 24.5, 19.6.
THF-mesylate-alkene (3.37)
Nitrogen was bubbled through a solution of 3.18 (96 mg, 0.29 mmol) and 3.26 (0.33 g, 0.58 mmol) in dry
CH2Cl2 (3 mL) for 30 min and at rt. At that point Grubbs 2nd generation catalyst (25 mg, 10 mol%) was
added and the mixture stirred for 20 h. Then, the reaction was quenched with DMSO (0.05 mL). After
stirring for an additional 30 in the solution was evaporated in vacuo. FCC of the residue afforded 3.37 (45
mg, 40%). Rf = 0.35 (40% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 7.64 (m, 4H), 7.34 (m, 6H),
5.67 (m, 1H), 5.30 (m, 1H), 4.19 (t, J = 6.7 Hz, 2H), 3.79 (m, 3H), 3.66 (m, 1H), 3.38 (m, 1H), 2.97 (s, 3H),
2.40 (br s, 1H), 2.24 (br s, 1H), 1.90 (m, 4H), 1.72-1.06 (m, 46H), 1.06 (s, partially buried, 9H), 0.85 (t, J =
7.1 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 135.9, 129.4, 127.4, 82.7, 82.5, 74.0, 73.1, 70.2, 37.4, 36.3,
33.6, 31.9, 29.6 (3 signals), 29.4, 29.3 (2 signals), 29.1, 29.0, 28.4, 27.1, 25.6, 25.4, 24.8, 22.7,19.4, 14.1.
HRMS (ESI) calcd for C51H86O7SSiNa (M+Na)+ 893.5761, found 893.5743.
THF-mesylate (3.37a)
A solution of compound 3.37 (45 mg, 0.05 mmol) in EtOAc (2 mL) and Pd/C (20 mg, 10% wt) was purged
with N2 for 30 min. Then, the mixture was stirred under a hydrogen atmosphere (balloon) for 20 h. The
reaction was then purged with N2 and filtered through a bed of Celite to yield 3.37a (45 mg, quantitative).
1H NMR (CDCl3, 600 MHz) δ 7.64 (m, 4H), 7.36 (m, 6H), 4.19 (t, J = 6.6 Hz, 2H), 3.76 (m, 2H), 3.67 (m,
1H), 3.36 (m, 2H), 2.98 (s, 3H), 1.95 (m, 2H), 1.74–1.07 (m, 22H), 1.01 (s, 9H), 0.85 (t, J = 6.5 Hz, 3H);

80
13C NMR (CDCl3, 600 MHz) δ 136.1, 135.0, 129.5, 127.5, 82.8, 74.2 (2 signals), 73.4, 70.4, 37.5, 36.5,
36.4, 33.7, 33.6, 32.1, 29.9 (2 signals), 29.8 (3 signals), 29.6 (2 signals), 29.5, 29.3, 29.2, 28.9, 27.3, 25.8
(2 signals), 25.6, 25.0, 22.9, 19.6, 14.3. HRMS (ESI) calcd for C51H88O7SSiNa (M+Na)+ 895.5918, found
895.5904.
THF-piperazine (3.37b)
To a solution of compound 3.37a (45 mg, 0.05 mmol) in dry CH3CN (2 mL) was added piperazine (0.22 g,
0.26 mmol) and the mixture was refluxed for 4 h. The mixture was evaporated under reduced pressure.
FCC of the residue yielded 3.37b (29 mg, 64%). Rf = 0.57 (10% MeOH/DCM). 1H NMR (CDCl3, 500 MHz)
δ 7.63 (m, 4 H), 7.34 (m, 6H), 3.76 (m, 2H), 3.66 (m, 1H), 3.36 (m, 2H), 3.11 (br s, 4H), 2.64 (br s, 4H),
2.39 (m, 2H), 1.96 (m, 2H), 1.62-1.22 (m, 18H), 1.01 (br s, partially buried), 1.00 (s, partially buried, 9H),
0.84 (t, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 136.1, 135.0, 129.6, 127.6, 82.8, 74.4, 73.4, 58.5,
50.7, 44.0, 36.7, 36.4, 33.6, 32.1, 29.9, 29.9, 29.9, 29.8, 29.7, 29.7, 29.7, 29.6, 29.6, 29.1, 29.0, 27.5,
27.3, 26.6, 25.8, 25.7, 25.0, 24.9, 22.9, 19.6, 14.4. HRMS (ESI) calcd for C54H95N2O4Si (M+H)+ 862.6983,
found 862.6940.
THF-thiophene-alkene (3.38)
Following the procedure described for 3.37, CM of 3.18 (60 mg, 0.18 mmol) and 3.27 (0.22 g, 0.36 mmol)
yielded 3.38 (43 mg, 27%). Rf = 0.47 (40% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ 7.42 (m, 4H),
7.39 (m, 1H), 7.37 (m, 1H), 7.32 (m, 6H), 7.04 (m, 1H), 6,02 (br s, 1H), 5.67 (m, 1H), 5.30 (m, 1H), 3.80
(m, 3H), 3.66 (m, 1H), 3.39 (m, 3H), 2.59 (br s, 1H), 2.43 (br s, 1H), 1.90 (m, 4H), 1.54-1.15 (m, 46H),
0.86 (t, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 161.9, 139.3, 136.0, 134.9 (2 signals), 129.7,

81
129.5, 128.1, 127.9, 127.7, 127.5, 82.8, 82.6, 74.1, 73.2, 40.2, 36.4, 36.2, 33.62, 32.4 (2 signals), 32.0,
29.8 (5 signals), 29.7 (2 signals), 29.6, 29.5 (2 signals), 29.4, 29.2, 28.6, 27.2, 27.1, 25.8, 25.0, 24.5,
22.8, 19.5, 14.2.
THF-thiophene (3.38a)
Compound 3.38 (43 mg, 0.047 mmol) was dissolved in EtOAc (2 mL). The solution was degassed for 30
min and Pd/C (40 mg, 10%) was introduced. Then, the reaction was stirred under H2 atmosphere for 20 h.
The solution was filtered over Celite and concentrated in vacuo. NMR indicated the presence of alkene,
so the hydrogenation procedure was repeated. Then, FCC (40-50% EtOAc/hexanes) of the crude product
gave 3.38a (31 mg, 72%). Rf = 0.5 (40% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ 7.64 (m, 4H),
7.46 (d, J = 1.1 Hz, 1H), 7.45 (d, J = 1.1 Hz, 1H), 7.35 (m, 6H), 7.04 (d, J = 1.3 Hz ,1H), 5.98 (br s, 1H),
3.76 (m, 2H), 3.66 (m, 1H), 3.39 (m, 4H), 1.96 (m, 3H), 1.65 (m, 3H), 1.56 (m, 3H), 1.35-1.12 (m, 47H),
1.09 (s, 9H), 0.86 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 162.0, 139.3, 136.1, 135.0, 134.9 (2
signals), 129.8, 129.6, 129.5, 128.2, 128.0, 127.7, 127.6, 127.5, 82.9, 72.2 (2 signals), 73.4, 73.3, 40.2,
36.4, 32.1, 29.9 (2 signals), 29.8 (2 signals), 29.6 (3 signals), 29.5, 28.9, 28.6, 27.3, 27.1, 25.8, 25.0,
22.9, 19.6, 14.3.
THF-butyl ether-alkene (3.39)
Following the procedure for 3.37, CM of alkenes 3.18 (86 mg, 0.26 mmol) and 3.25 (0.13 g, 0.52 mmol)
yielded 3.39 (50 mg, 23%). Rf = 0.51 (40% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 7.63 (m, 4H),
7.35 (m, 6H), 5.65 (m, 1H), 5.30 (m, 1H), 3.80 (m, 3H), 3.67 (m, 1H), 3.37 (m, 4H),1.8 (m, 5H), 1.32 (m,

82
8H), 1.23 (br s, 44H), 1.01 (s, 9H), 0.89 (t, J = 7.3 Hz, 3H), 0.84 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 500
MHz) δ 139.3, 136.1, 135.0, 129.5, 127.6, 114.3, 73.4, 71.2, 70.9, 36.5, 36.3, 33.9, 32.1, 30.0, 29.9, 29.7
(3 signals), 29.1, 27.3, 26.4, 25.0, 24.7, 19.6 (2 signals), 14.2.
6-O-Undecyl mannoside-butenolide-alkene (3.40)
Nitrogen was bubbled through a solution of 3.19 (57 mg, 0.14 mmol) and butenolide 3.24 (39 mg g, 0.07
mmol) in dry CH2Cl2 (1 mL) for 30 min and at rt. At that point Grubbs 1st generation catalyst (25 mg, 10
mol%) was added and the mixture refluxed for 6 h. Then, the reaction was quenched with DMSO (0.05
mL). After stirring for an additional 30 min the solution was evaporated in vacuo. FCC of the residue
afforded 3.40 (54 mg, 96%) as an E/Z mixture (E/Z~3/1). Rf = 0.38 (20% EtOAc/hexanes x 2). 1H NMR
(CDCl3, 500 MHz) δ 7.59 (m, 4H), 7.31 (m, 6H), 6.90 (m, 1H), 5.60-5.41 (m, 2H), 4.97 (s, 1H), 4.92 (m,
1H), 4.10-3.84 (m, 4H), 3.61-3.56 (m, 5H), 3.39 (m, 2H), 2.93 (m, 1H), 2.17 (t, J = 7.7 Hz, 2H), 1.85 (m,
2H), 1.52-1.18 (bm, 45H), (s, 9H), 0.80 (t, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 174.1, 149.0,
136.3 (2 signals), 136.1, 135.1, 134.9 (2 signals), 134.5, 129.6, 127.6, 124.9, 124.6, 109.7, 96.3, 96.0,
75.6, 75.5, 73.3, 72.3, 71.8, 71.7, 71.6, 68.0 (2 signals), 36.5, 36.3, 32.4, 32.1, 29.9, 29.8 (2 signals), 29.7
(2 signals), 29.5, 29.4, 29.3, 29.2, 28.1 (2 signals), 27.5, 27.3, 26.3, 25.4, 25.0, 24.7 (2 signals), 22.9,
19.6, 19.4, 14.3. HRMS (ESI) calcd for C56H88O9SiNa (M+Na)+ 955.6090, found 955.6088.

83
6-O-Undecyl mannoside-butenolide (3.40a)
To a solution of 3.40 (70 mg, 0.078 mmol) and TsNHNH2 (0.98 g, 5.26 mmol) in DME (7.8 mL) at reflux,
was added a solution of NaOAc (0.53 g, 6.51 mmol) in H2O (10 mL) over 4 h. The mixture was then
cooled, diluted with EtOAc, and washed with water. The organic phase was dried (Na2SO4), filtered and
concentrated in vacuo. FCC in 1-5% (acetone/DCM) delivered 3.40a (35 mg, 48%). Rf = 0.42 (5%
acetone/DCM). 1H NMR (CDCl3, 500 MHz) δ 7.64 (m, 4H), 7.34 (m, 6H), 6.94 (s, 1H), 4.96 (m, 2H), 4.12
(m, 2H), 3.66 (m, 6H), 3.37 (m, 3H), 2.93 (m, 1H), 2.21 (m, 2H), 1.49-1.09 (bm, 51H), 1.08 (s, 9H), 0.85
(t, J = 7.5 Hz, 3H). HRMS (ESI) calcd for C56H90O9SiNa (M+Na)+ 957.6252, found 957.6225.
4-O-Undecyl mannoside-butenolide-alkene (3.41)
Following the procedure described for 3.40, CM for 3.20 (0.21 g, 0.52 mmol) and 3.24 (0.150 g, 0.26
mmol) yielded 3.41 (6 mg, 25%) as an E/Z mixture (E/Z~4/1). Rf = 0.4 (30% EtOAc/hexanes). 1H NMR
(CDCl3, 500 MHz) δ 7.59 (m, 4H), 7.28 (m, 6H), 6.90 (s, 1H), 5.56-5.40 (m, 2H), 5.40 (m, 1H), 4.99 (s,
1H), 4.92 (m, 1H), 4.16 (m, 1H), 4.05 (m, 2H), 3.82-3.75 (m, 5H), 3.56 (m, 1H), 3.43 (m, 1H), 3.31 (m,
1H), 2.16 (m, 2H), 2.05-1.80 (m, 2H), 1.60-1.10 (bm, 45H), 1.10 (s, 9H), 0.82 (t, J = 6.7 Hz, 3H); 13C NMR
(CDCl3, 500 MHz) δ 174.1, 149.0, 136.3 (2 signals), 136.1, 134.9 (2 signals), 134.4, 129.5, 127.5, 124.8,
109.3, 96,2, 95.9, 78.8, 76.0, 71.7, 68.5, 68.0, 62.9, 36.4, 36.3, 32.4, 32.1, 30.2, 28.8, 29.7 (2 signals),
29.6, 29.5, 29.4, 29.3, 29.1, 28.2, 27.5, 27.2, 26.5, 26.2, 25.3, 25.0, 22.8, 19.6, 19.4.

84
6-O-Butyl mannoside-butenolide-alkene (3.42)
Following the procedure described for 3.40, the CM of 3.21 (0.12 g, 0.37 mmol) and 3.24 (80 mg, 0.18
mmol) yielded 3.42 (60 mg, 80%) as an E/Z mixture (E/Z~4/1). Rf = 0.2 (30% EtOAc/hexanes). 1H NMR
(CDCl3, 500 MHz) δ 7.63 (m, 4H), 7.37 (m, 6H), 6.94 (s, 1H), 5.61-5.44 (m, 2H), 5.00 (s, 1H), 4.96 (m,
1H), 4.12-3.88 (m, 4H), 3.71-3.60 (m, 5H), 3.50 (m, 2H), 2.91 (br s, 1H), 2.20 (m, 2H), 1.90 (m, 2H), 1.55-
1.10 (m, 33H), 1.00 (s, 9H), 0.86 (m, 3H); 13C NMR (CDCl3, 500 MHz) 149.1, 136.1, 129.6, 127.6, 124.9,
109.7, 96.1, 75.6, 73.3, 72.0, 71.9, 71.8, 68.1, 68.0, 36.5, 36.3, 32.5, 31.9, 29.8, 29.5, 29.4, 29.2, 28.1,
27.6, 27.4, 27.3, 26.3, 25.4, 25.1, 24.8 (2 signals), 19.7, 19.5 (2 signals), 14.1.
6-O-Butyl mannoside-butenolide (3.42a)
Compound 3.42 (0.1 g, 0.12 mmol) was transformed to 3.42a following the reduction protocol described
for preparation of 3.40a. FCC in 1-5% (acetone/DCM) delivered 3.42a (60 mg, 61%). Rf = 0.42 (5%
acetone/DCM). 1H NMR (CDCl3, 500 MHz) δ 7.68 (m, 4H), 7.28 (m, 6H), 6.90 (s, 1H), 4.91 (m, 2H), 4.08
(m, 2H), 3.63 (m, 6H), 3.45 (m, 2H), 3.35 (m, 1H), 3.00 (br s, 1H), 2.15 (m, 2H), 1.49-1.10 (m, 37H), 1.02
(s, 9H), 0.79 (t, J = 7.7 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ 174.1, 149.1, 136.1, 135.0, 134.9, 134.5,
129.9, 129.6, 127.5, 126.7, 109.7, 97.3, 78.1, 75.6, 73.4, 71.9, 71.8, 71.5,68.0, 36.5 (2 signals), 31.8,
29.8 (2 signals), 29.5, 29.4, 29.3, 28.1, 27.5, 27.3, 26.3 (2 signals), 25.4, 25.0, 19.6, 19.5, 19.4, 14.1.

85
6-O-Methyl mannoside-butenolide-alkene (3.43)
Following the procedure described for 3.40, the CM of 3.22 (91 mg, 0.33 mmol) and 3.24 (90 mg, 0.17
mmol) yielded 3.43 (0.12 g, 92%) as an E/Z mixture (E/Z~3/1). Rf = 0.2 (30% EtOAc/hexanes). 1H NMR
(CDCl3, 500 MHz) δ 7.64 (m, 4H), 7.34 (m, 6H), 6.94 (s, 1H), 5.65-5.45 (m, 2H), 5.10 (s, 1H), 5.02 (m,
1H), 4.08-3.91 (m, 4H), 3.68 (m, 4H), 3.58 (m, 1H), 3.39 (m, 3H), 2.67 (br s, 1H), 2.22 (m, 2H), 1.89-1.13
(m, 27H), 1.01 (s, 9H); 13C NMR (CDCl3, 600 MHz) δ 174.1, 149.0, 136.3, 136.2 (3 signals), 136.1, 134.9
(2 signals), 134.5, 129.6, 129.5, 127.6, 127.5 (2 signals), 125.2, 125.0, 109.4, 98.3, 96.4, 96.0. 73.3, 72.0
(2 signals), 70.4, 70.3, 69.9, 69.8, 69.5, 68.3, 68.0, 67.8, 56.1, 36.5, 36.3, 32.4 (2 signals), 32.1, 31.1,
29.9 (2 signals), 29.8, 29.7 (2 signals), 29.5, 29.4, 29.3, 29.2, 28.0, 27.5, 27.2, 26.6, 26.3 (2 signals),
25.3, 25.0, 24.7, 22.9, 19.6, 19.4, 14.3. HRMS (ESI) calcd for C46H68O9SiNa (M+Na)+ 815.4525, found
815.4514.
6-O-Methyl mannoside-butenolide (3.43a)
Compound 3.43 (66 mg, 0.084 mmol) was transformed to 3.43a following the reduction protocol
described for preparation of 3.40a. FCC in 1-5% (acetone/DCM) delivered 3.43a (60 mg, 91%). Rf = 0.28
(5% acetone/DCM). 1H NMR (CDCl3, 500 MHz) δ 7.59 (m, 4H), 7.30 (m, 6H), 6.90 (s, 1H), 4.92 (m, 2H),
4.09 (m, 2H), 3.61 (m, 6H), 3.33 (m, 1H), 3.32 ( partially buried s, 3H), 2.16 (m, 2H), 1.46-1.10 (m, 33H),
1.03 (s, 9H); 13C NMR (CDCl3, 500 MHz) δ 174.1, 149, 136.1, 134.9 (2 signals), 134.5, 129.5, 127.5,
109.7, 97.3, 78.2, 75.6, 73.3, 73.0, 70.5, 68.5, 68.0, 59.7, 36.4, 29.8, 29.7, 29.5, 29.4, 29.3, 28.0, 27.5,
27.2, 26.2 (2 signals), 25.3, 25.0, 19.6, 19.4.

86
6-O-Methyl-4-O-undecyl mannoside-butenolide-alkene (3.44)
Following the procedure described for 3.40, CM of 3.23 (0.12 g, 0.29 mmol) and 3.24 (80 g, 0.14 mmol)
yielded 3.44 (60 mg, 44%) as an E/Z mixture (E/Z~3/1). Rf = 0.8 (20% EtOAc/hexanes). 1H NMR (CDCl3,
500 MHz) δ 7.63 (m, 4H), 7.35 (m, 6H), 6.94 (s, 1H), 5.60-5.45 (m, 2H), 5.03 (s, 1H), 4.96 (m, 1H), 4.15
(m, 1H), 4.08 (m, 2H), 3.86 (m, 1H), 3.78 (m, 1H), 3.59 (m, 4H), 3.46 (1H), 3.41 (s, 3H), 3.36 (m, 4H),
2.22 (m, 2H), 1.89 (m, 2H), 1.57-1.49 (m, 45H), 1.08 (s, 9H), 0.86 (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3,
600 MHz) δ 171.2, 149.1, 146.6 (2 signals), 139.3, 136.7, 136.2, 135.0, 134.8, 129.6, 129.0, 127.6, 124.9,
114.4, 109.3, 104.2, 96.7, 96.6, 96.2, 79.1, 73.4, 73.3, 71.8, 62.8, 59.5, 36.5, 36.4, 33.9, 32.1, 30.2, 29.9,
29.7 (2 signals), 29.6, 29.4, 29.3 (2 signals), 29.2, 28.2, 27.4, 27.3, 26.5, 26.3, 25.1 (2 signals), 25.0,
24.6, 22.9, 19.6, 19.4, 14.3. HRMS (ESI) calcd for C57H90O9SiNa (M+Na)+ 969.6246, found 969.6237.
6-O-Methyl-4-O-undecyl mannoside-butenolide (3.44a)
Compound 3.44 (60 mg, 0.063 mmol) was transformed to 3.44a following the reduction protocol
described for preparation of 3.40a. FCC in 1-5% (acetone/DCM) delivered 3.44a (27 mg, 45%). Rf = 0.5
(2% acetone/DCM). 1H NMR (CDCl3, 500 MHz) δ 7.64 (m, 4H), 7.34 (m, 6H), 6.94 (s, 1H), 4.98 (s, 1H),
4.97 (m, 1H), 4.15 (m, 1H), 4.07 (m, 1H), 3.78 (m, 1H), 3.62 (m, 5H), 3.46 (m, 1H), 3.36 (partially buried s,
3H), 3.34 (m, 2H), 2.22 (m, 2H), 1.50-1.08 (m, 51H), 1.01 (s, 9H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR
(CDCl3, 500 MHz) δ 174.1, 149.0, 136.1, 135.0 (2 signals), 130.0 (2 signals), 128.0, 109.3, 97.3, 79.1,
77.6, 76.4, 76.2, 73.4, 71.8, 71.7, 68.2, 67.7, 59.5, 36.5 (2 signals), 32.1, 30.3, 29.9, 29.8 (3 signals),
29.7, 29.6, 29.4, 29.3, 28.2, 27.5, 27.3, 26.5, 26.3 (2 signals), 25.4, 25.1, 25.0, 22.9, 19.6.

87
6-O-Undecyl mannoside-mesylate-alkene (3.45)
Following the procedure described for 3.40, CM of 3.19 (0.3 g, 0.72 mmol) and 3.26 (0.2 g, 0.35 mmol)
yielded 3.45 (0.160 g, 48%) as an E/Z mixture (E/Z~4/1). 1H NMR (CDCl3, 600 MHz) δ 7.65 (m, 4H), 7.35
(m, 6H), 5.65-5.46 (m, 2H), 5.02 (s, 1H), 4.21 (t, J = 5.0 Hz, 2H), 4.13 (m, 3H), 3.90 (m, 1H), 3.69 (m, 4H),
3.63 (m, 1H), 3.49 (m, 2H), 2.99 (s, 3H), 2.91 (br s, 1H), 1.9 (m, 2H), 1.73 (m, 2H), 1.57-1.24 (m, 46H),
1.05 (s, 9H), 0.87 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 136.3, 136.2, 135.0 (2 signals), 129.6,
128.6, 127.6 (2 signals), 125.0, 109.7, 96.2, 75.6, 73.4, 72.3, 71.9, 71.7, 70.4, 68.1, 37.6, 36.6, 36.4,
32.5, 32.1, 31.2, 29.8, 29.7(2 signals), 29.6, 29.4, 29.3, 29.2, 28.1, 27.3, 26.3, 25.6, 25.1, 24.8, 22.9,
19.7, 14.3; HRMS (ESI) calcd for C54H90O10SSiNa (M+Na)+ 958.5916, found 958.5917.
6-O-Undecyl mannoside-mesylate (3.45a)
Compound 3.45 (0.16 g, 1 mmol) was transformed to 3.45a following the reduction protocol described for
preparation of 3.37a (0.16 g, quant). 1H NMR (CDCl3, 600 MHz) δ 7.65 (m, 4H), 7.38 (m, 6H), 4.96 (s,
1H), 4.21 (t, J = 6.0 Hz, 2H), 4.13 (m, 2H), 3.68 (m, 6H), 3.45 (m, 3H), 2.99 (s, 3H), 2.94 (s, 1H), 1.73 (m,
2H), 1.55-1.13 (m, 50H), 1.01 (s, 9H), 0.88 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 136.1, 135.0,
129.6, 127.6, 109.7, 97.3, 78.1, 75.6,73.4, 72.3,71.9, 71.6, 70.4, 68.1, 68.0, 37.6, 36.5, 32.1, 29.8 (3
signals), 29.7, 29.6 (2 signals), 29.5, 29.3, 29.2, 28.1, 27.3, 26.3, 25.6, 25.1, 22.9, 19.6, 14.3.

88
6-O-Undecyl mannoside-piperazine (3.45b)
Following the procedure for the synthesis of 3.37b, 3.45a (0.16 g, 0.16 mmol) was converted to
piperazine 3.45b (62 mg, 40%). 1H NMR (CDCl3, 600 MHz) δ 7.64 (m, 4H), 7.35 (m, 6H), 4.95 (s, 1H),
4.10 (m, 2H), 3.65 (m, 5H), 3.47 (m, 4H), 3.19 (br s, 4H), 2.72 (br s, 4H), 2.39 (m, 2H), 1.50-1.09 (m,
52H), 1.01 (s, 9H), 0.86 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 136.1, 135.0 (2 signals), 129.6,
127.6, 109.7, 97.3, 78.2, 75.6, 73.4, 72.3, 71.7, 71.4, 68.1, 68.0, 58.4, 50.3, 43.8, 36.5 (2 signals), 32.1,
29.9 (2 signals), 29.8 (2 signals), 29.7 (2 signals), 29.6, 28.1, 27.5, 27.30, 26.6, 26.3 (2 signals), 25.1 (2
signals), 22.9, 19.6, 14.4.
6-O-Undecyl mannoside-thiophene-alkene (3.46)
Following the procedure described for 3.40, CM for 3.19 (0.1 g, 0.24 mmol) and 3.27 (0.29 g, 0.48 mmol)
yielded 3.41 (6 mg, 25%). FCC gave a mixture of CM product and thiophene dimer (~300 mg), which was
treated with 5% AcCl in MeOH (1.5 mL) in DCM (15 mL). The reaction was stirred for 1 h at rt and then
quenched with a solution of methanolic NaHCO3. The mixture was filtered and concentrated in vacuo.
FCC of the residue (5% MeOH/DCM) afforded 3.46 (100 mg, 44% after two steps). 1H NMR (CDCl3, 600
MHz) δ 7.59 (m, 4H), 7.41 (d, J = 3.7 Hz, 1H), 7.38 (d, J = 0.9 Hz, 1H), 7.33 (m, 2H), 7.28 (m, 4H), 6.99
(m, 1H), 5.88 (br s, 1H), 5.59-5.36 (m, 2H), 4.91 (m, 1H), 4.03 (m, 1H), 3.94-3.53 (m, 8H), 3.46-3.29 (m,
4H), 3.10 (m, 1H), 2.78 (br s, 1H), 2.50 (br s, 1H), 1.87 (m, 2H), 1.33-1.03 (m, 40H), 0.97 (s, 9H), 0.81 (t,
J = 7.5 Hz, 3H).

89
6-O-Undecyl mannoside-10-deoxy-mesylate-alkene (3.47)
Following the procedure described for 3.40, CM for 3.19 (0.28 g, 0.67 mmol) and 3.28 (0.11 g, 0.33
mmol) yielded 3.47 (0.12 g, 48%) as an E/Z mixture (E/Z~4/1). Rf = 0.4 (20% EtOAc/hexanes). 1H NMR
(CDCl3, 600 MHz) δ 5.71-5.68 (m, 2H), 5.01 (m, 1H), 4.19 (t, J = 7.5 Hz, 2H), 4.12 (m, 3H), 3.92 (m, 1H),
3.68 (m, 3H), 3.60 (m, 1H), 3.45 (m, 2H), 2.97 (s, 3H), 2.95 (m, 1H), 2.01 (m, 2H), 1.72 (m, 2H), 1.59-1.26
(m, 44H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 136.5, 124.9, 109.7, 96.1, 78.2, 75.6,
72.3, 71.8, 71.7, 70.4, 68.1, 68.0, 37.6, 32.5, 32.1, 29.9 (2 signals), 29.8 (2 signals), 29.7, 29.7 (2
signals), 29.6, 29.5 (2 signals), 29.3, 29.4, 29.2, 28.1, 26.3, 25.6, 22.9, 14.3.
6-O-Undecyl mannoside-10-deoxy-mesylate (3.47a)
3.47 (0.117 g, 0.16 mmol) was dissolved in ethyl acetate (3 mL) and degassed for 30 minutes. At that
time, Pd/C (11 mg, 10% wt) was added and the reaction was stirred overnight under hydrogen
atmosphere. The solution was filtered over Celite and evaporated to deliver 3.47a (85 mg, 48%).

90
6-O-Undecylmannoside-10-deoxy-piperazine (3.47b)
Following the procedure for 3.37b, compound 3.47a (85 mg, 0.011 mmol) was converted into piperazine
3.47b (44 mg, 30%). 1H NMR (CDCl3, 500 MHz) δ 4.96 (s, 1H), 4.12 (m, 1H), 4.10 (s, 1H), 3.66 (m, 4H),
3.61 (m, 1H), 3.47 (m, 2H), 3.40 (m, 1H), 2.89 (t, J = 5.0 Hz, 4H), 2.40 (br s, 4H), 2.28 (m, 1H), 1.70 (br s,
4H), 1.55 (m, 4H), 1.50 (s, 3H), 1.33 (s, 3H), 1.23 (br s, 33H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3,
500 MHz) δ 109.6, 97.3, 78.2, 75.6, 72.3, 71.8, 71.5, 68.1, 68.0, 59.7, 54.7, 46.2, 32.1, 29.9 (2 signals),
29.8 (4 signals), 29.7, 29.6 (3 signals), 28.1, 27.8, 26.8, 26.4, 26.3, 22.9, 14.3.

91
DESIGN AND SYNTHESIS OF POTENTIAL
TUMOR-SELECTIVE CYTOTOXIC AGENTS

92
4.1. Introduction and background
Traditional cancer therapies are designed to interfere with cell pathways involved in proliferation or cell
survival.95 Those therapies are effective against cancer cells, but invariably. Therefore, tumor-specific
cytotoxic agents are desirable.96 Herein, we describe targeting strategies for THF-ACGs. For reasons
stated earlier, 4-DAN was used for proof of principle.
Two studies on the targeting of THF-AGEs to cancer cells have been reported. In one, the authors
explore folate targeted, β-cyclodextrin derived, nano-suspensions, (a biphasic system of pure drug
particles of less than 1 nanometer in an aqueous vehicle) in vivo.97 The conjugates showed enhanced
cytotoxicity and cell uptake for folate receptor-positive 4T1 cells. In 4T1 tumor bearing mice, the
acetogenin nanosuspensions aggregated in tumors and enhanced drug uptake only in the tumor site. In
the other study, biotin-conjugated acetogenins showed higher potency and selectivity in a cellular assay,
than the parent drugs.68
We have designed two strategies for tumor-targeted THF-ACGs. The first is a prodrug approach in which
an ACG or an ACG mimetic is attached to a vector that binds to a tumor specific receptor. Upon cellular
internalization the cytotoxic agent will be released. The other strategy is a ‘chameleon’ approach, in which
the sugar ring of a sugar containing ACG mimetic is “disguised” as a tumor vector. Advantages of the
chameleon strategy over the prodrug approach are (i) simpler synthetic chemistry; (ii) the drug and the
vector entity are one in the same and potential problems with release of the active drug before delivery to
the cellular target do not arise. However, designing a chameleon molecule can be more challenging than
a prodrug as precise site modification may be needed (vide infra).
4.2. The ‘chameleon’ approach
The mannose structures described in earlier chapters are relevant to the chameleon approach. As
discussed previously, mannose replacements are not only of interest for their easy synthetic accessibility,
but also because they are potentially tumor vectors. For example, certain GLUTs that are overexpressed
on tumor cells also recognize mannose motifs and could help internalize mannose containing

93
molecules.98 In addition, the 3-O-carbamoyl mannose subunit of the bleomycin family of clinical agents,
has been shown to be primarily responsible for the tumor selectivity of the bleomycins (Figure 4.1).
93c,99,100
4.2.1. Design
The design of our chameleon analogues is based on the following hypothesis: replacement of the THF
moiety in 4-DAN with a 3-O-carbamoylmannose residue will confer tumor selectivity but will not abrogate
potency. This design is challenging because the domain responsible for selectivity essentially overlaps
with a segment of the molecule that impacts on potency. In this regard, questions are: (i) will the presence
of the 3-O-carbomyl moiety reduce cytotoxicity; (ii) will the 6-O-alkyl chain on the mannose residue lower
tumor selectivity. In comparison, the prodrug constructs, while involving more complex synthetic
chemistry, are simpler to design as the domains for potency and selectively are remote and therefore
likely to function independently .
Figure 4.1. Bleomicyn and carbamoylmannose derivative 4.2
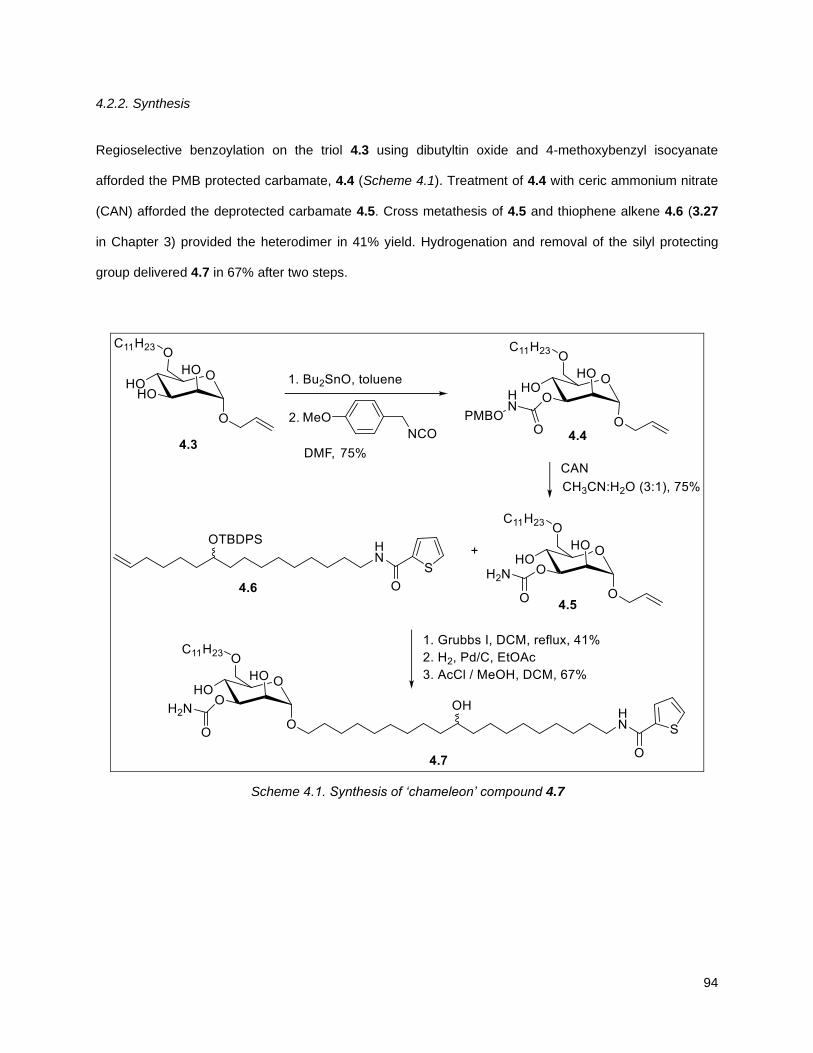
94
4.2.2. Synthesis
Regioselective benzoylation on the triol 4.3 using dibutyltin oxide and 4-methoxybenzyl isocyanate
afforded the PMB protected carbamate, 4.4 (Scheme 4.1). Treatment of 4.4 with ceric ammonium nitrate
(CAN) afforded the deprotected carbamate 4.5. Cross metathesis of 4.5 and thiophene alkene 4.6 (3.27
in Chapter 3) provided the heterodimer in 41% yield. Hydrogenation and removal of the silyl protecting
group delivered 4.7 in 67% after two steps.
Scheme 4.1. Synthesis of ‘chameleon’ compound 4.7

95
4.3. The prodrug approach
This strategy uses a prodrug that is
comprised of an ACG or an ACG mimetic
that is conjugated via a cleavable linker to a
vector for a tumor-specific cell surface
receptor (Figure 4.2). Upon recognition by
the tumor cell, the prodrug is internalized and
the drug is released by an intracellular
mechanism.
4.3.1. Design
The underlying hypothesis is that the tumor-specific vector on the prodrug will facilitate delivery to tumor
but not to normal cells. Once internalized, the linkage will be cleaved, and the liberated drug will kill the
cell. The mechanism for drug release is critical as this should be a pathway that is tumor specific to
prevent drug release before delivery to the drug target. The use of a traceless linker is important as the
released drug entity is identical to the parent drug, thus assuring activity.
Figure 4.3. Example of a prodrug construct using 4-DAN as the parent drug
Figure 4.2. Recognition mechanism of prodrugs

96
For proof of principle, our vector target is PSMA which is overexpressed on prostate cancer cells and the
linker is a previously validated disulfide-based construct (Figure 4.3). Details of the choice and synthesis
of the specific drug, tumor receptor, vector and linker are presented in the following sections.
4.3.2. Drugs
One of the analogues of 4-DAN, 4.9 (3.10 in Chapter 3) was selected as the drug entity as it is easily
prepared and was active against several important tumor cell lines (Figure 4.4). The synthesis of the
derived prodrug 4.10 lays the groundwork for prodrugs from other drugs like ACG mimetic 4.11 (3.16 in
Chapter 3), and the naturally occurring ACG annonacin 4.13 (Figure 4.4).
Figure 4.4. Proposed drug and prodrug pairs 4.9-4.10, 4.11-4.12, 4.13-4.14

97
Figure 4.5. DUPA derived vector
4.3.2. Receptor
Prostate-specific membrane antigen (PSMA) is a type II cell surface glycoprotein and is the most well
established highly restricted prostate epithelial cell membrane antigen.101 This highly restricted prostate-
related antigen is an integral cell membrane protein. Moreover, the expression level of PSMA has been
shown to increase as the stage and the grade of the tumor progresses. There is a high interest in PSMA
as a target for prostate-specific imaging and antitumor agents.102 Furthermore, recent studies have
demonstrated that PSMA is also expressed in nonprostatic tumor neovasculature and it is almost absent
in the vasculature of healthy tissues.101 Accordingly much effort has been made in developing PSMA-
targeted agents.103
4.3.3. Vector
2-[3-(1,3-dicarboxypropyl)ureido] pentanedioic acid (DUPA), is a
PSMA-specific ligand and enters PSMA expressing cells via
endocytosis.103b DUPA has shown promise for targeting human
lymph node prostate cancer (LNCaP) cells with many different
therapeutic warheads.103a The specificity of DUPA conjugates
for prostate cancers was evaluated by studying the uptake of
DUPA-targeted 99mTc radioimaging agents by human prostate
cancer (LNCaP) tumors in athymic nude mice. It was found that the retention of the agent was limited to
PCa xenograft and kidneys.104 Unlike murine kidneys, human kidneys express low levels of PSMA, These
data suggested that DUPA might constitute an ideal ligand for prostate cancer-targeted therapy.103a
Following a known procedure,105 glutamate salts 4.16 and 4.17 were coupled using triphosgene to deliver
urea 4.18. Selective removal of the benzyl protecting group provided 4.19, which was treated with 4.20 to
deliver 4.21. Chloride present under these conditions apparently led to substitution of the tosylate.
Treatment of 4.21 with potassium thioacetate yielded 4.22. Exposure of 4.22 to trifluoroacetic acid in the
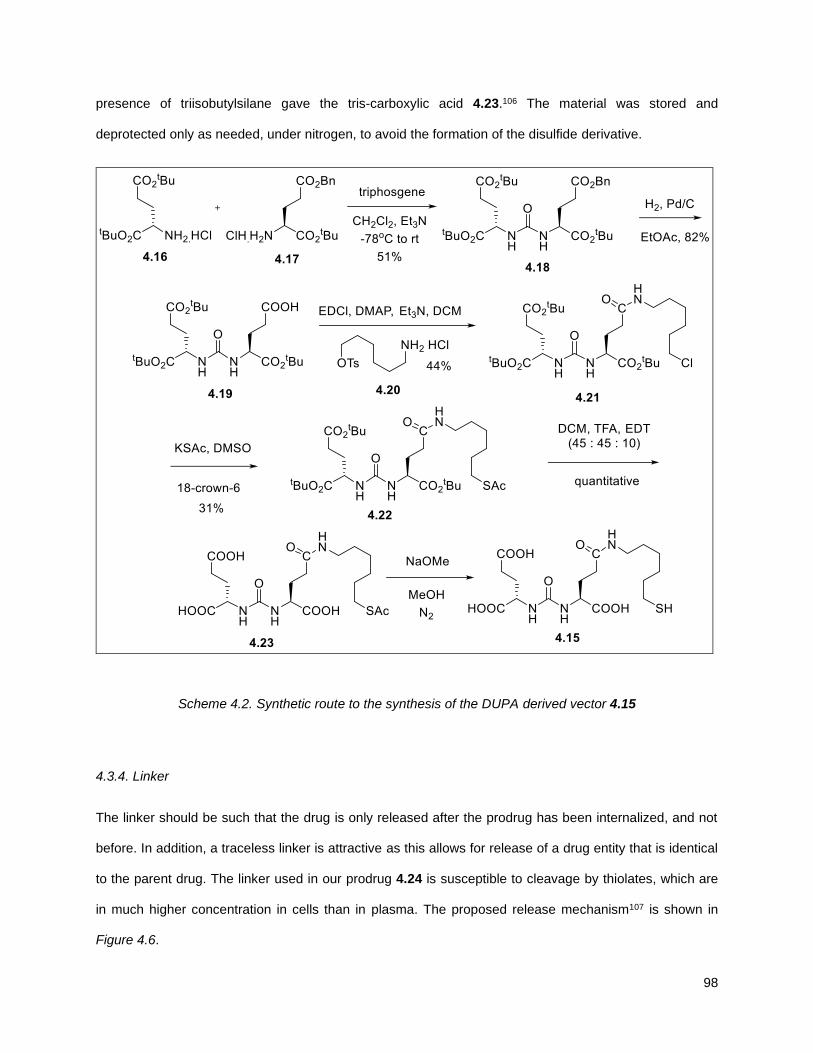
98
presence of triisobutylsilane gave the tris-carboxylic acid 4.23.106 The material was stored and
deprotected only as needed, under nitrogen, to avoid the formation of the disulfide derivative.
Scheme 4.2. Synthetic route to the synthesis of the DUPA derived vector 4.15
4.3.4. Linker
The linker should be such that the drug is only released after the prodrug has been internalized, and not
before. In addition, a traceless linker is attractive as this allows for release of a drug entity that is identical
to the parent drug. The linker used in our prodrug 4.24 is susceptible to cleavage by thiolates, which are
in much higher concentration in cells than in plasma. The proposed release mechanism107 is shown in
Figure 4.6.
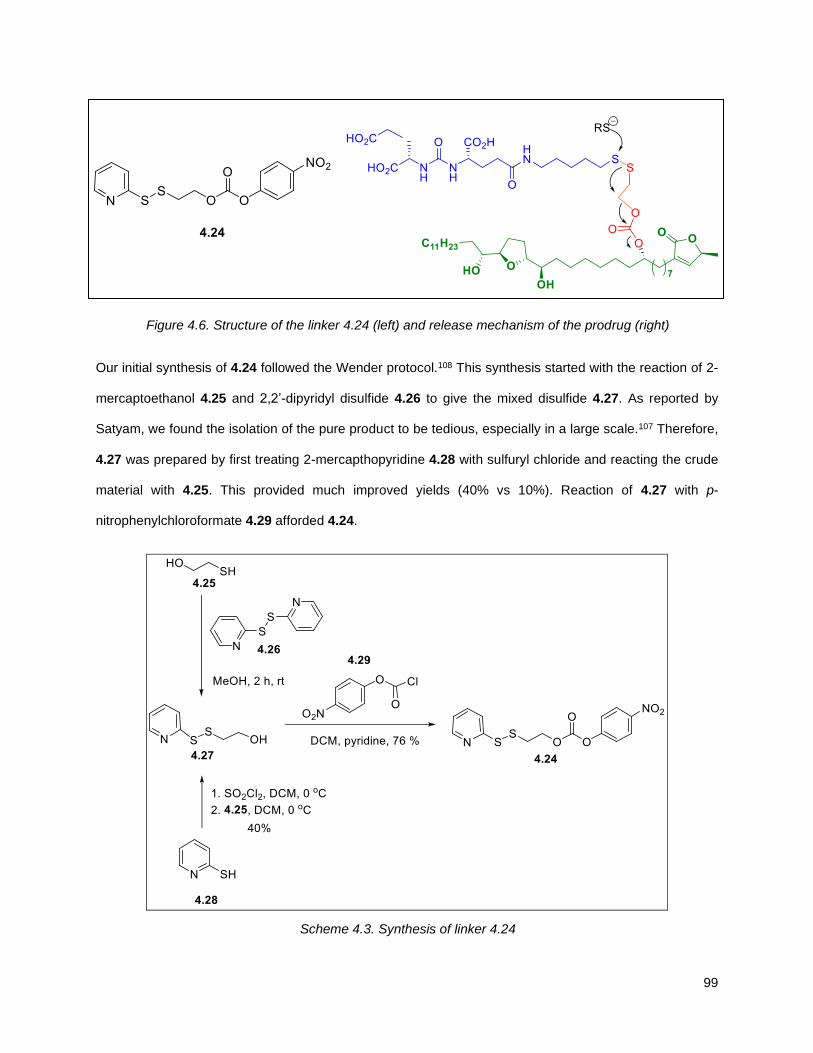
99
Figure 4.6. Structure of the linker 4.24 (left) and release mechanism of the prodrug (right)
Our initial synthesis of 4.24 followed the Wender protocol.108 This synthesis started with the reaction of 2-
mercaptoethanol 4.25 and 2,2’-dipyridyl disulfide 4.26 to give the mixed disulfide 4.27. As reported by
Satyam, we found the isolation of the pure product to be tedious, especially in a large scale.107 Therefore,
4.27 was prepared by first treating 2-mercapthopyridine 4.28 with sulfuryl chloride and reacting the crude
material with 4.25. This provided much improved yields (40% vs 10%). Reaction of 4.27 with p-
nitrophenylchloroformate 4.29 afforded 4.24.
Scheme 4.3. Synthesis of linker 4.24

100
4.4. C11 mannose derivatives
4.4.1. Sugar-butenolide Prodrug Synthesis
The free alcohol in alkene 4.30 was protected as the MOM derivative 4.31, which was then subjected to
CM with butenolide alkene 4.32 under the previously described conditions to afford 4.33. Reduction of the
double bond and selective deprotection of the TBDPS ether, provided alcohol 4.34 for vector conjugation.
Treatment of 4.34 with nitrophenylcarbonate 4.24 in the presence of DMAP gave 4.35. Next, removal of
the isopropylidene and MOM protecting groups by treatment of 4.35 with methanolic HCl in
dichloromethane yielded the derived triol. Disulfide exchange on this material with in situ generated
DUPA-derived thiol 4.15 delivered prodrug 4.10 in 80% yield from the sugar disulfide precursor (Scheme
4.4). This product was characterized by NMR and HRMS (Figure 4.7).
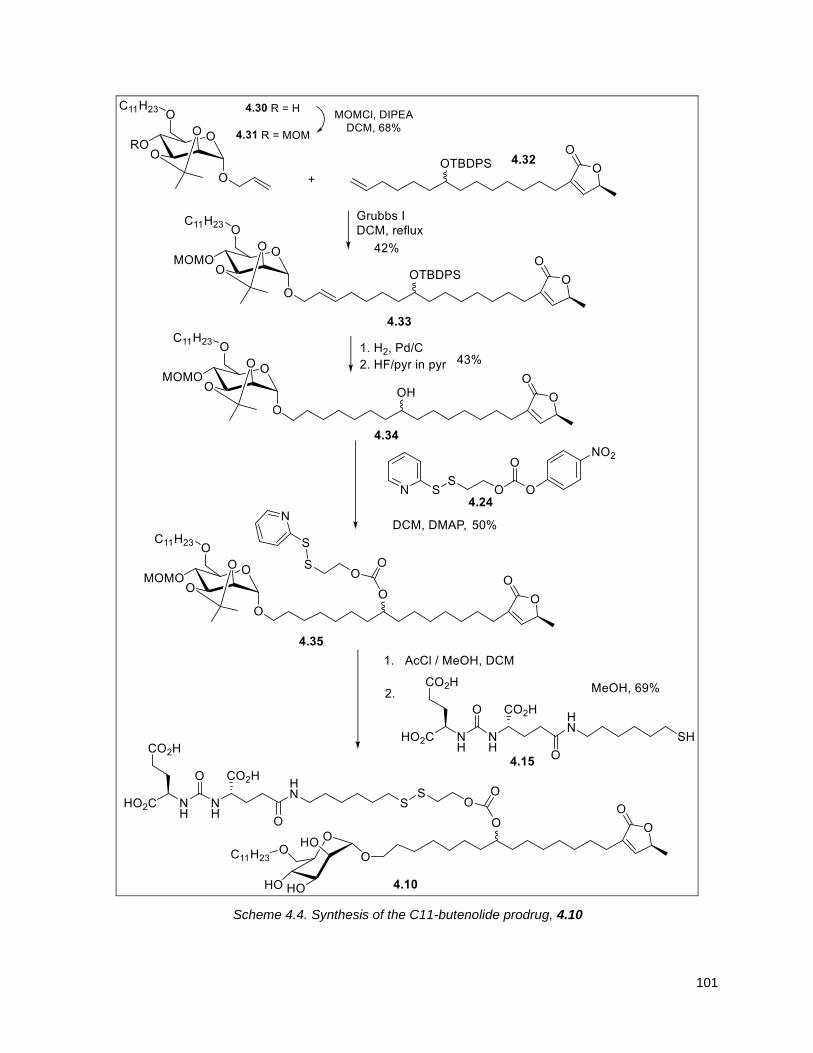
101
Scheme 4.4. Synthesis of the C11-butenolide prodrug, 4.10

102
Figure 4.7. 1H of C11-butenolide prodrug 4.10
11’
m, o q, k
l, p
20
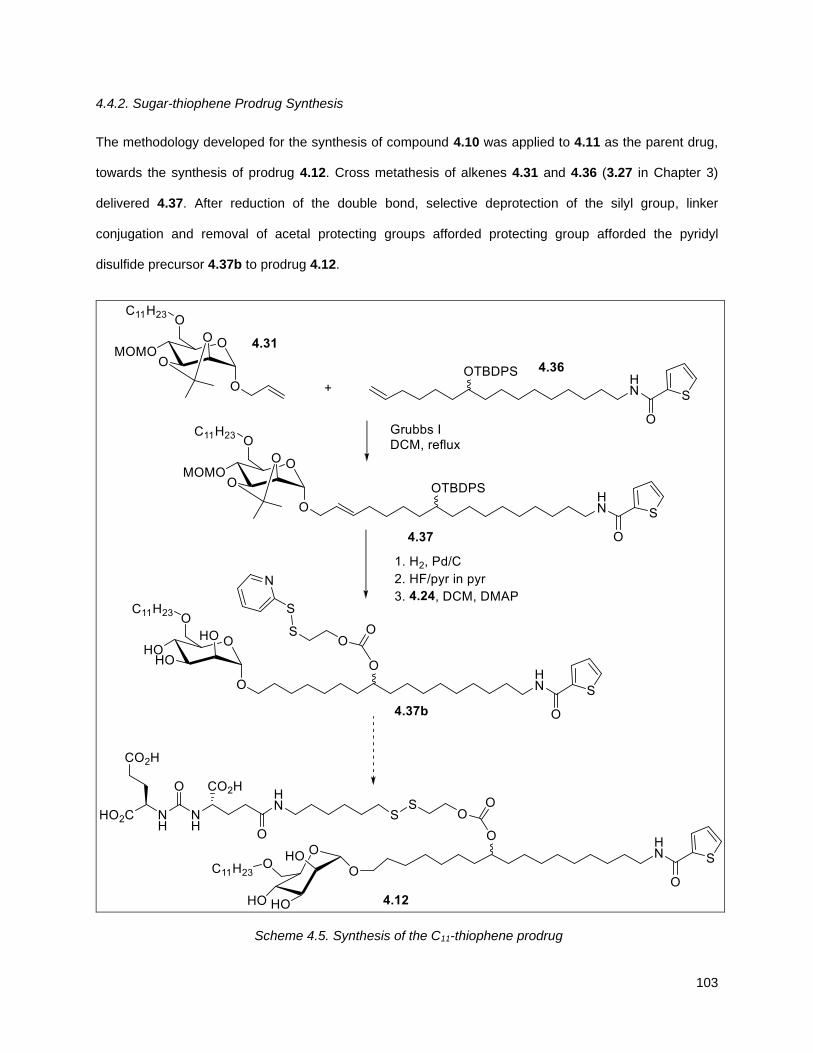
103
4.4.2. Sugar-thiophene Prodrug Synthesis
The methodology developed for the synthesis of compound 4.10 was applied to 4.11 as the parent drug,
towards the synthesis of prodrug 4.12. Cross metathesis of alkenes 4.31 and 4.36 (3.27 in Chapter 3)
delivered 4.37. After reduction of the double bond, selective deprotection of the silyl group, linker
conjugation and removal of acetal protecting groups afforded protecting group afforded the pyridyl
disulfide precursor 4.37b to prodrug 4.12.
Scheme 4.5. Synthesis of the C11-thiophene prodrug

104
4.5. Annonacin prodrugs
Using the naturally obtained compound avoids a challenging
synthesis. However, the purification of the natural extract may not be
trivial. Furthermore, executed complex reactions on the natural
product may also be problematic. In this context, total synthesis is
advantageous as such reactions can be more straightforwardly
performed on synthetic precursors.
A crude sample of annonacin from Annona muricata was provided by
Professor Mohindra Seepersaud of the University of The West Indies, St Augustine. Details for extraction
of this material and our protocol for the further purification of annonacin are described in the following
section.
4.5.1. Isolation, purification and characterization of annonacin
Approximately 1.5 kg of mature and semi-mature fruits’ seeds were blended and extracted with ethanol
(Figure 4.9). The extract was then partitioned in DCM and water and using the brine lethality test (BST)
the organic extract was found to be more active.
The extraction process was repeated two more times on the crude initial extract and crude compound
was extracted every time. Each of the extracts were purified using dichloromethane and increasing
amounts of methanol. We performed two subsequent purifications on 25 g of this sample (Figure 4.10).
The first purification was done using 60-100% EtOAc/hexanes to 20% methanol/EtOAc. The second
column involved only increasing amounts of methanol (up to 6%) in dichloromethane. This sequence
provided 1.22 g of pure annonacin, 4.13, whose NMR data was essentially identical to that reported
(Figure 4.9, 4.10, Table 4.1).109
Figure 4.8. Annona muricata
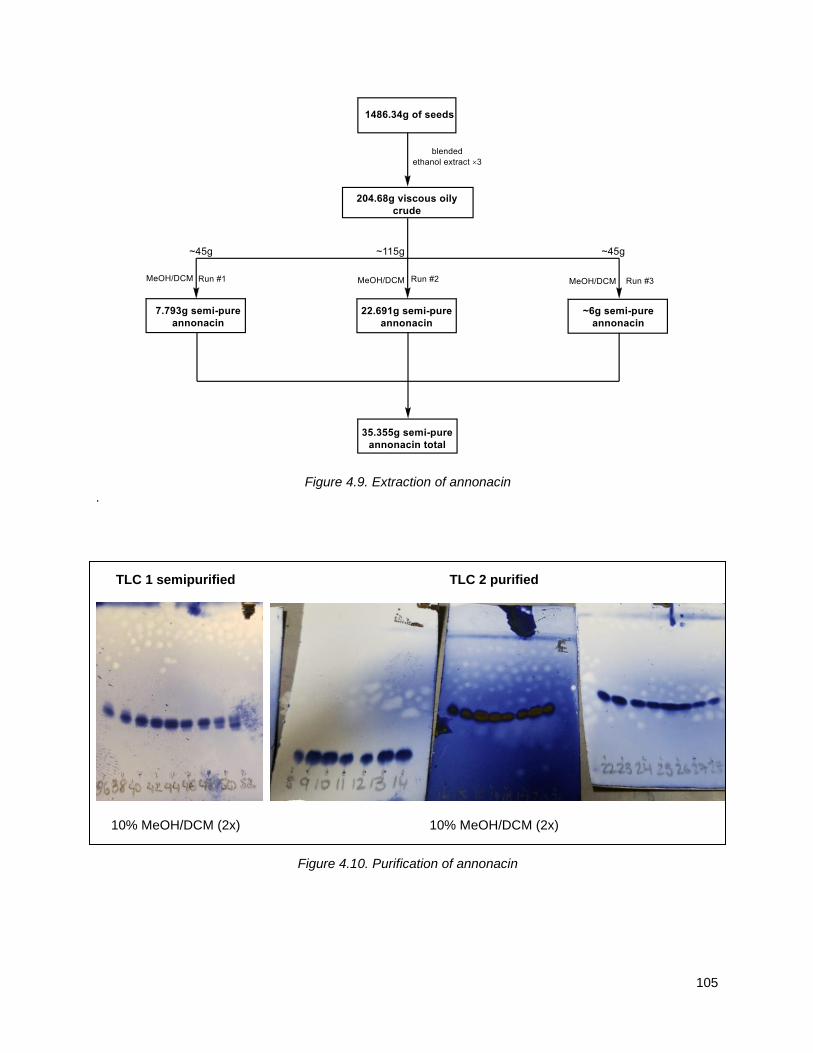
105
Figure 4.9. Extraction of annonacin .
TLC 1 semipurified TLC 2 purified
10% MeOH/DCM (2x) 10% MeOH/DCM (2x)
Figure 4.10. Purification of annonacin
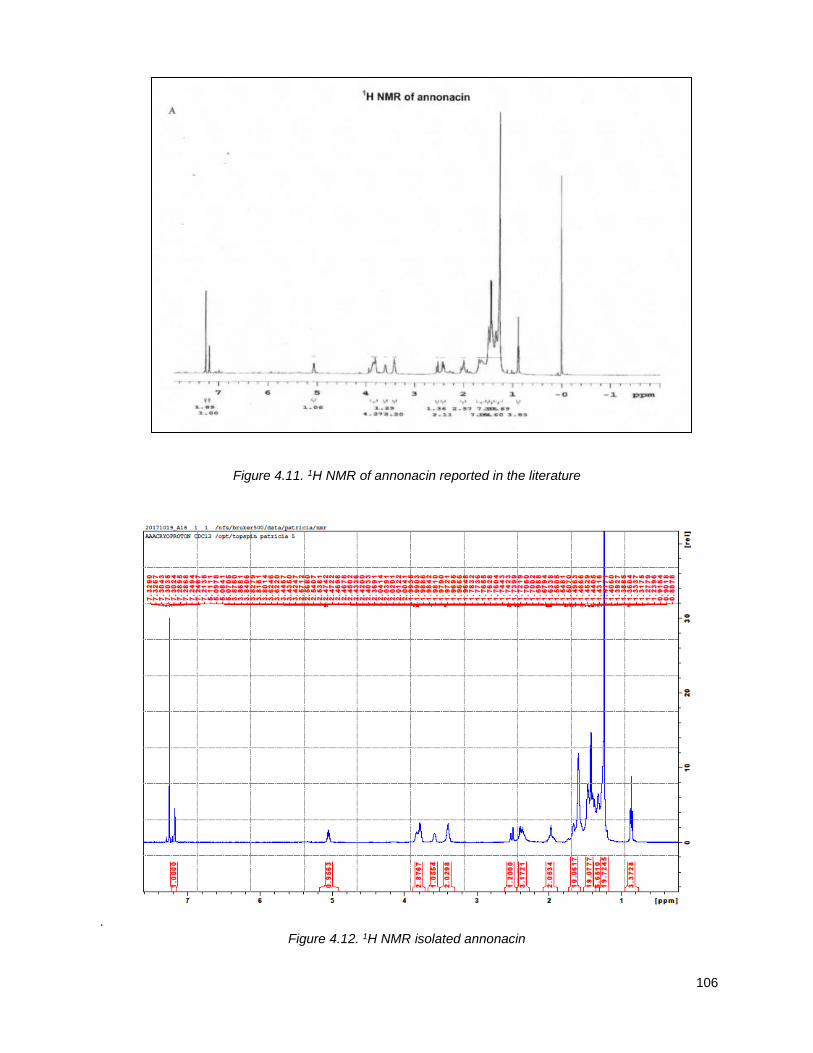
106
Figure 4.11. 1H NMR of annonacin reported in the literature
.
Figure 4.12. 1H NMR isolated annonacin

107
The 13C of the extracted annonacin and the one reported in the literature are comparable. See table
below:
Table 4.1. 13C of literature reported
and isolated annonacin
Carbon Literature annonacin
Isolated annonacin
1 174.6 174.32
2 131.1 131.05
3 22-38 22.58-37.18
4 71.6 70.25
5 29.5 29.77-29.12
6 29.5 29.77-29.13
7 29.5 29.77-29.14
8 29.5 29.77-29.15
9 29.5 29.77-29.16
10 69.8 69.85
11 29.5 29.77-29.12
12 29.5 29.77-29.13
13 29.5 29.77-29.14
14 29.5 29.77-29.15
15 73.9 74.17
16 82.6 82.72
17 22-38 22.58-37.18
18 22-38 22.58-37.18
19 82.7 82.63
20 74.1 74.85
21 29.5 29.77-29.12
22-29 29.5 29.77-29.13
30 29.5 29.77-29.14
31 29.5 29.77-29.15
32 14.1 14.13
33 151.8 152.09
34 77.9 78.14
35 19.0 19.05
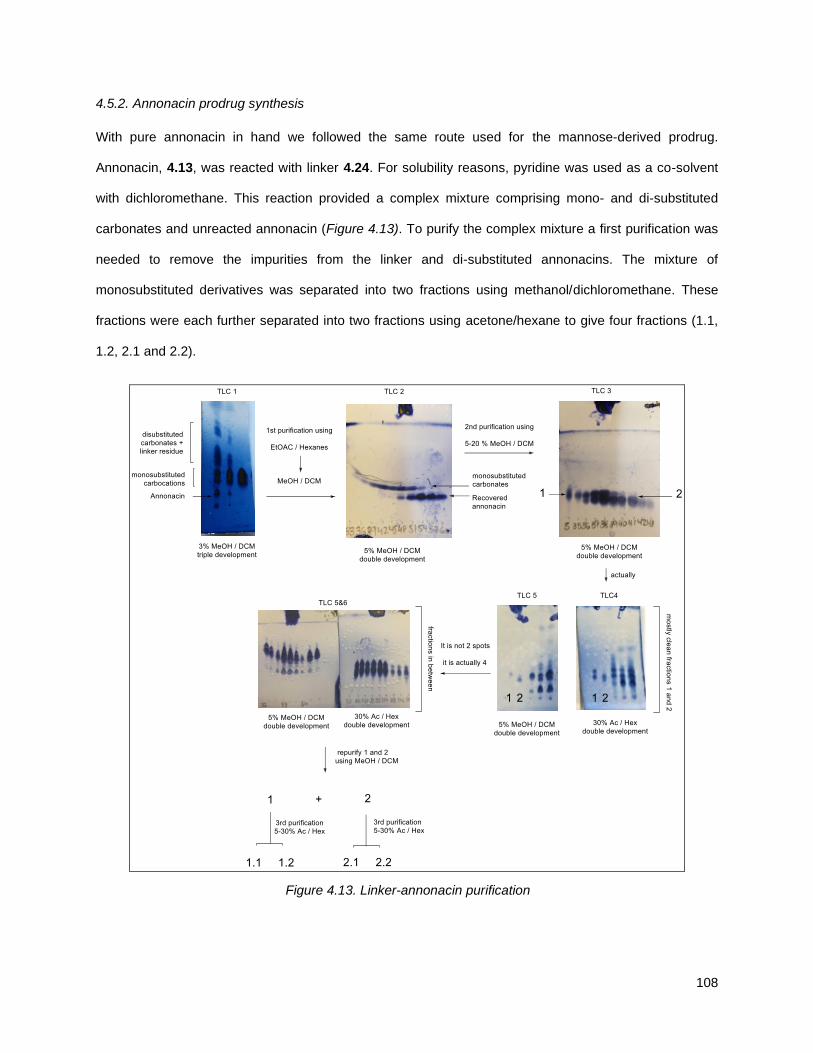
108
4.5.2. Annonacin prodrug synthesis
With pure annonacin in hand we followed the same route used for the mannose-derived prodrug.
Annonacin, 4.13, was reacted with linker 4.24. For solubility reasons, pyridine was used as a co-solvent
with dichloromethane. This reaction provided a complex mixture comprising mono- and di-substituted
carbonates and unreacted annonacin (Figure 4.13). To purify the complex mixture a first purification was
needed to remove the impurities from the linker and di-substituted annonacins. The mixture of
monosubstituted derivatives was separated into two fractions using methanol/dichloromethane. These
fractions were each further separated into two fractions using acetone/hexane to give four fractions (1.1,
1.2, 2.1 and 2.2).
Figure 4.13. Linker-annonacin purification

109
Fractions 1.2 and 2.2 were assigned the structures 4.38 and 4.39, respectively from the 1HNMR and
HRMS data. Similarly, the compounds in fractions 1.1 and 2.1 were determined to be the other two mono-
carbonate regioisomers, 4.40 and 4.41. Unfortunately, it was not possible to distinguish between these
two structures. Nevertheless, as fraction 1.1 was the only material that was obtained in significant
amounts, it was subjected to the disulfide exchange reaction with 4.15. NMR analysis indicated that the
product was either 4.14 which is derived from 4.41, or the regioisomer that comes from 4.40 (Figures
4.14-4.16 and Appendix).
Scheme 4.6. Synthesis of the annonacin based prodrug

110
Figure 4.14. Fraction 1.1 (4.40 or 4.41)
Figure 4.15. Fraction 1.2 (4.38)
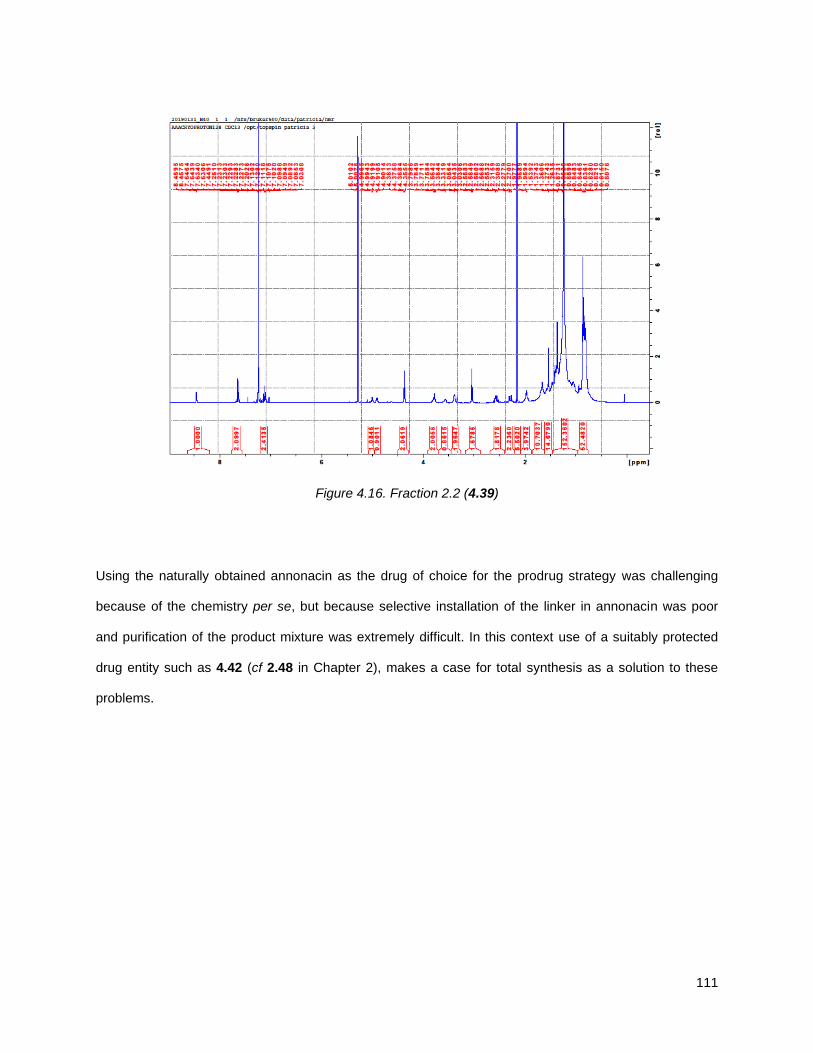
111
Figure 4.16. Fraction 2.2 (4.39)
Using the naturally obtained annonacin as the drug of choice for the prodrug strategy was challenging
because of the chemistry per se, but because selective installation of the linker in annonacin was poor
and purification of the product mixture was extremely difficult. In this context use of a suitably protected
drug entity such as 4.42 (cf 2.48 in Chapter 2), makes a case for total synthesis as a solution to these
problems.
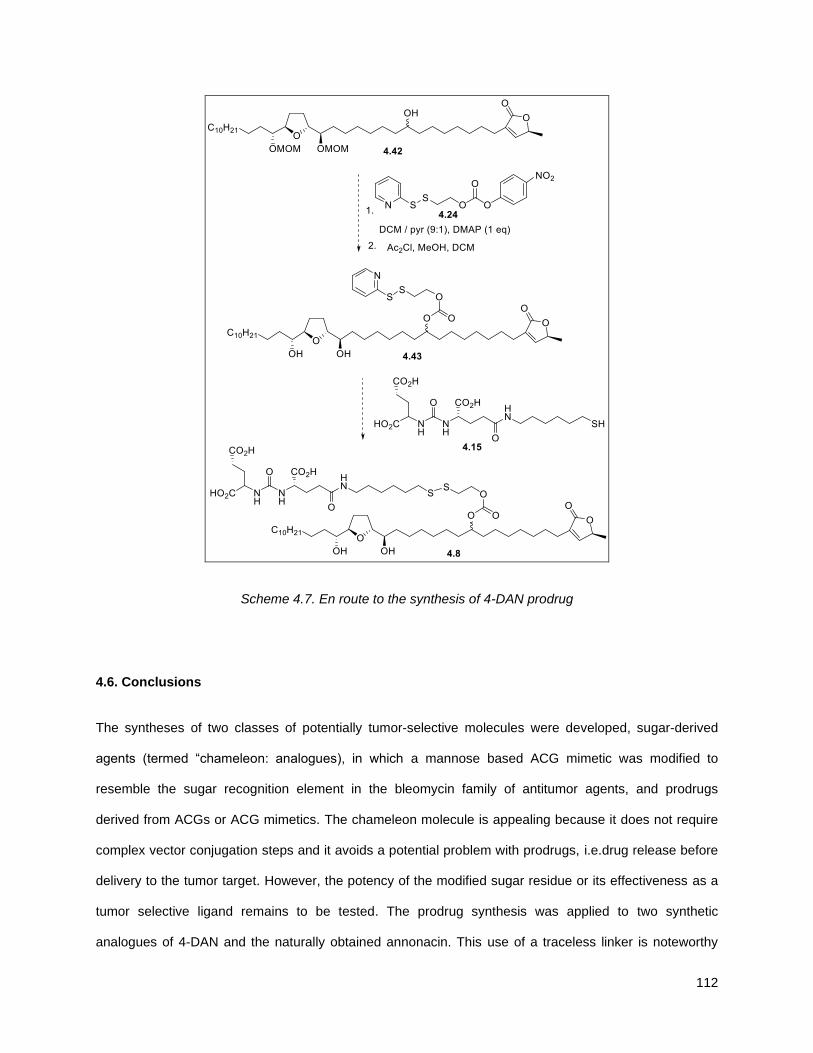
112
Scheme 4.7. En route to the synthesis of 4-DAN prodrug
4.6. Conclusions
The syntheses of two classes of potentially tumor-selective molecules were developed, sugar-derived
agents (termed “chameleon: analogues), in which a mannose based ACG mimetic was modified to
resemble the sugar recognition element in the bleomycin family of antitumor agents, and prodrugs
derived from ACGs or ACG mimetics. The chameleon molecule is appealing because it does not require
complex vector conjugation steps and it avoids a potential problem with prodrugs, i.e.drug release before
delivery to the tumor target. However, the potency of the modified sugar residue or its effectiveness as a
tumor selective ligand remains to be tested. The prodrug synthesis was applied to two synthetic
analogues of 4-DAN and the naturally obtained annonacin. This use of a traceless linker is noteworthy

113
because it facilitates release of the original unmodified drug. Therefore, retention of drug potency is very
likely, but as mentioned above, drug release before delivery to the tumor target could be problematic. For
both classes of these ACG-based drugs, their high degree of hydrophobicity could compromise selectivity
because of non-specific cellular uptake. The cytotoxic properties of these potential tumor selective
molecules and their forerunners from Chapter 3 will be discussed in Chapter 5.

114
4.7. Experimental
Propen-1-yl-4-O-(4-methoxybenzyl)carbamoyl-6-O-undecyl-α-D-mannopyrannoside (4.4)
Triol 4.3 (53 mg, 0.14 mmol) and Bu2SnO (45 mg, 0.18 mmol) were heated in toluene at reflux for 1 h,
then the solvent was evaporated. The residue was dissolved in DMF (1 mL) and PMBNCO (22 µL, 0.15
mmol) was added. The reaction mixture was stirred at rt for 1 h and quenched with MeOH (0.05 mL) and
stirred for additional 30 min. The solvent was removed in vacuo and the residue was purified via FCC to
deliver 4.4 (57 mg, 75%). Rf = 0.55 (5% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ 7.23 (m, 2H), 6.88
(m, 2H), 5.91 (m, 1H), 5.33 (m, 1H), 5.23 (m, 1H), 5.03 (m, 1H), 4.87 (br s, 1H), 4.33 (m, 2H), 4.22 (m,
1H), 4.02 (m, 2H), 3.94 (m, 1H), 3.78 (m, 4H), 3.73 (m, 2H), 3.48 (m, 2H), 1.60 (m, 4H), 1.30 (m, 14H),
0.91 (t, J = 7.5 Hz, 3H).
Propen-1-yl-4-O-carbamoyl-6-O-undecyl-α-D-mannopyranoside (4.5)
Compound 4.4 (57 mg, 0.11 mmol) and CAN (0.27 g, 0.49 mmol) were dissolved in a 3:1 mixture of
CH3CN:H2O (3 mL) and stirred for 30 min. Then, it was diluted with saturated NaHCO3 and stirred for
additional 30 min. The mixture was then filtered through Celite, extracted with EtOAc and dried (Na2SO4).
FCC of the residue afforded 4.5 (33 mg, 75%). Rf = 0.2 (5% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ
5.83 (m, 1H), 5.23 (m, 1H), 5.13 (m, 1H), 4.85 (m, 1H), 4.78 (br s, 1H), 4.12 (m, 1H), 3.91 (m, 3H), 3.69
(m, 3H), 3.41 (m, 2H), 1.51 (m, 2H), 1.19 (m, 16H), 0.81 (t, J = 6.7 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ
157.6, 133.5, 117.8, 98.9, 75.3, 72.1, 71.1, 70.4, 69.5, 68.2, 66.9, 31.9, 29.6, 29.5, 29.5, 29.4, 26.0, 22.7,
14.1.

115
4-O-Carbamoyl-6-O-undecylmannose-thiophene-alkene (4.5a)
Nitrogen was bubbled through a solution of 4.5 (33 mg, 0.08 mmol) and 4.6 (0.1 g, 0.17 mmol) in dry
CH2Cl2 (3 mL) for 30 min and at rt. At that point Grubbs 1st generation catalyst (12 mg, 0.015 mmol) was
added and the mixture heated for 2.5 h at 40 oC. Then, the reaction was quenched with DMSO (0.05 mL).
After stirring for an additional 30 min the solution was evaporated in vacuo. FCC of the residue afforded
0.312 g Rf = 0.25 (30% EtOAc/hexanes). FCC afforded heterodimer 4.5a (0.44 g, 41%). Rf = 0.15 (5%
MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ 7.58 (m, 4H), 7.40 (m, 1H), 7.32 (m, 1H), 7.28 (m, 6H), 6.99
(m, 1H), 5.94 (br s, 1H), 5.55-5.36 (m, 2H), 4.91 (m, 1H), 4.78 (br s, 1H), 4.03 (m, 1H), 3.94-3.82 (m, 2H),
3.63 (m, 4H), 3.43 (m, 3H), 3.35 (m, 2H), 1.86 (m, 2H), 1.51 (m, 4H), 1.30 (m, 4H), 1.29-1.26 (m, 34H),
0.97 (s, 9H), 0.81 (t, J = 6.9 Hz, 3H).
4-O-Carbamoyl-6-O-undecylmannose-8’-tert-butyldiphenylsiloxy-thiophene (4.5b)
A mixture of 4.5a (0.44 g, 0.44 mmol) and 10% Pd/C (0.05 g) in EtOH (2 mL) was stirred for 16 h under a
H2 atmosphere, then filtered over Celite. The filtrate was then evaporated in vacuo and the residue
subjected to FCC to give 4.5b (0.44 g, quantitative). Rf = 0.15 (5% MeOH/DCM). 1H NMR (CDCl3, 600
MHz) δ 7.69 (m, 4H), 7.50 (m, 1H), 7.47 (m, 1H), 7.38 (m, 6H), 7.09 (m, 1H), 6.01 (br s, 1H), 4.98 (m,
1H), 4.83 (s, 1H), 4.04 (br s, 1H), 3.99 (t, 1H), 3.77-3.67 (m, 4H), 3.50 (m, 3H), 3.44 (m, 3H), 1.61 (m,
4H), 1.33-1.10 (m, 38H), 1.07 (s, 9H), 0.89 (t, J = 6.8 Hz, 3H).
116
8’-Hydroxy-17’-(thiophene-2-carboxamido)heptadecyl-3-O-carbamoyl-6-O-undecyl-α-D-
mannopyranoside (4.7)
A crude sample of 4.5b (40 mg, 0.052 mmol) was dissolved in DCM (2 mL) and treated with 5% AcCl in
MeOH (0.6 mL) for 4 h. Additional 5% AcCl in MeOH (0.3 mL) was then introduced and stirring continued
for 4 h The reaction was then quenched with saturated K2CO3 in methanol, filtered and evaporated in
vacuo. FCC of the residue (80-100% EtOAc/hexanes and 5-10% MeOH/EtOAc) afforded 4.7 (20 mg,
67%). Rf = 0.3 (5% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ 7.51 (dd, J1 = 3.66 Hz , J2 = 1.08 Hz, 1H),
7.47 (dd, J1 = 4.98 Hz, J2 = 1.14 Hz, 1H), 7.08 (dd, J1 = 4.98 Hz , J2 = 3.72 Hz, 1H), 6.04 (br s, 1H), 4.98
(m, 1H), 4.83 (s, 1H), 4.03 (br s, 1H), 3.96 (t, J = 2.58 Hz, 1H), 3.79-3.69 (m, 4H), 3.59 (m, 1H), 3.45 (m,
2H), 3.41 (m, 3H), 1.60 (m, 6H), 1.30-1.10 (m, 40H), 0.88 (t. J = 6.8 Hz, 3H); 13C NMR (CDCl3, 600 MHz)
162.1, 157.5, 139.3, 136.1, 129.8, 128.1, 127.8, 99.8, 75.8, 72.3, 72.1, 70.9, 69.9, 68.1, 40.3, 37.6 (3
signals), 37.5, 32.1, 29.8 (3 signals), 29.7 (2 signals), 29.6 (2 signals), 29.4 (2 signals), 29.3, 29.2, 27.1,
26.3, 26.2, 26.1, 25.8, 25.6 (2 signals), 22.9, 14.3. HRMS (ESI) calcd for C40H72N2O9SNa (M+Na)+
779.4851, found 779.4841.
6-O-Undecyl mannose-butenolide-DUPA conjugate (4.10)
Under a N2 atmosphere, 4.35a (16 mg, 0.021 mmol) was dissolved in MeOH (0.5 mL) and NaOMe (0.1
mL, 0.83 M) was added until a pH 8-10. After 10 min, 5 M HCl in MeOH was added until pH 6-7. Next, a

117
solution of 4.35a (18 mg, 0.031 mmol) in methanol (0.5 mL) was added to the reaction mixture. The
reaction was stirred for 18 h, then, the solvent was removed in vacuo. FCC of the residue (15-20%
MeOH/DCM) afforded 4.10 (20 mg, 80%). 1H NMR (MeOD, 600 MHz) δ 7.20 (s, 1H), 4.97 (m, 1H), 4.61
(partially buried s, 1H), 4.60 (m, 1H), 4.25 (t, J = 6.3 Hz, 2H), 4.06 (m, 2H), 3.67-3.20 (m, 9H), 3.11 (m,
3H), 2.84 (t, J = 6.3 Hz, 2H), 2.64 (t, J = 7.3 Hz, 2H), 2.26 (m, 4H), 2.15 (m, 2H), 2.13 (m, 2H), 1.88 (m,
2H), 1.40 (m, 14H), 1.28 (partially buried d, J = 6.7 Hz, 3H), 1.24 (m, 18H), 1.31-1.19 (m, 25H), 0.80 (t, J
= 6.7 Hz, 3H); 13C NMR (MeOD, 600 MHz) δ 176.4, 156.7, 152.4, 134.7, 106.8, 101.7, 80.2, 70.8, 73.5,
72.9, 72.8, 72.3, 71.9, 69.1, 68.8, 66.7, 40.5, 39.8, 38.4, 35.4 (2 signals), 33.3, 31.5, 31.0 (2 signals),
30.8, 30.7 (2 signals), 30.6, 30.5, 30.4 (2 signals), 30.2 (2 signals), 29.3, 28.7, 27.7, 27.5, 27.4, 26.5,
26.4, 23.9, 23.1, 19.4, 14.6. HRMS (ESI) calcd for C57H100N3O19S2 (M+H)+ 1194.6387, found 1194.6299.
Annonacin-DUPA conjugate (4.14)
Following the procedure for the synthesis of 4.10, 4.40/4.41 (9 mg, 0.01 mmol) was transformed to 4.14
(5 mg, 41%). 1H NMR (CDCl3, 600 MHz) δ 7.16 (s, 1H), 4.86 (m, 1H), 4.45 (m, 3H), 4.15 (t, J = 6.2 Hz,
2H), 3.93 (m, 5H), 3.59 (m, 2H), 3.45 (m, 2H), 3.21 (m, 2H), 3.03 (m, 8H), 2.75 (t, J = 6.2 Hz, 2H), 2.54 (t,
J = 7.1 Hz, 2H), 2.49 (m, 2H), 2.34 (m, 4H), 2.23-2.10 (m, 16H), 1.87-1.06 (m, 44H), 0.71 (t, J = 6.8 Hz,
3H). HRMS (ESI) calcd for C57H100N3O17S2 (M+H)+ 1162.6489, found 1162.6495.
5-Benzyl 1-(tert-butyl) (((S)-1,5-di-tert-butoxy-1,5-dioxopentan-2-yl)carbamoyl)-L-glutamate (4.18)

118
A solution of L-glutamic acid di-tertbutyl ester hydrochloride 4.16 (1.0 g, 3.38 mmol) and Et3N (1.54 mL,
11.09 mmol) in DCM (30 mL) was cooled to -78 oC. A solution of triphosgene (0.34 g, 0.34 mmol) in DCM
(10 mL) was added dropwise via a syringe over 30 min. The reaction was stirred and allowed to warm to
rt for 1 h. Then, 4.17 (0.7 g, 2.03 mmol) and Et3N (3 mL, 2.03 mmol) were succesively added and the
reaction was stirred at rt for 16 h. The reaction mixture was then diluted with DCM and washed with H2O,
dried (Na2SO4) and concentrated under reduced pressure. FCC (80% EtOAc/hexanes) delivered 4.18
(1.0 g, 51%). Rf = 0.67 (50% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ 7.33 (br s, 5H), 5.23 (s, 2H),
5.09 (m, 1H), 4.98 (m, 1H), 4.43 (m, 1H), 4.26 (m, 1H), 2.24 (m, 4H), 2.11 (m, 2H), 1.80 (m, 2H), 1.51 (s,
9H), 1.40 (s, 9H), 1.36 (s, 9H); 13C NMR (CDCl3, 600 MHz) δ 172.9, 172.7, 172.5, 172.1, 156.9, 135.6,
128.8, 128.6, 128.5, 82.3, 82.3, 80.9, 80.8 (2 signals), 67.4, 53.3, 52.9, 31.8, 31.7, 28.3, 28.3, 28.2.
HRMS (ESI) calcd for C30H46N2O9Na (M+Na)+ 601.3096, found 601.3091. The NMR data was essentially
identical to that reported in the literature.110
(S)-5-(tert-butoxy)-4-(3-((S)-1,5-di-tert-butoxy-1,5-dioxopentan-2-yl)ureido)-5-oxopentanoic acid
(4.19)
Compound 4.18 (1.0 g, 1.7 mmol) was dissolved in ethyl acetate (30 mL). The mixture was purged with
N2 and Pd/C catalyst (0.1 g, 10%) was added. Then, the reaction was stirred under H2 overnight. The
mixture was then purged with N2 and filtered over Celite, and the filtrate concentrated under reduced
pressure. FCC of the residue yielded 4.19 (0.7 g, 82%). Compound did not show well on TLC, so the
reaction was followed by NMR. 1H NMR (CDCl3, 600 MHz) δ 5.29 (br s, 1H), 5.22 (br s, 1H), 4.43 (m, 1H),
4.29 (m, 1H), 2.28 (m, 4H), 2.02 (m, 2H), 1.84 (m, 2H), 1.43 (s, 9H), 1.40 (m, 18H); 13C NMR (CDCl3, 500
MHz) δ 172.5, 172.0, 156.8, 82.0, 80.5, 53.0, 31.6, 28.4, 28.1, 28.0. HRMS (ESI) calcd for C23H40N2O9Na
(M+Na)+ 511.2626, found 511.2619. The NMR data was essentially identical to that reported in the
literature.110

119
6-Aminohexyl-4-methylbenzenesulfonate hydrochloride (4.20)
6-((tert-butoxycarbonyl)amino)hexyl 4-methylbenzenesulfonate111 (5.0 g, 0.013 mol) was stirred in a
solution of 4 M HCl in dioxane at rt for 16 h. After that, it was concentrated in vacuo to provide 4.20 as a
solid (4.13 g, quantitative). 1H NMR (C5H5N, 500 MHz) δ 8.01 (d, J = 8.1 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H),
4.07 (t, J = 6.4 Hz, 2H), 3.18 (t, J = 7.5 Hz, 2H), 2.24 (s. 3H), 1.90 (m, 2H), 1.50 (m, 2H), 1.23 (m, 2H),
1.17 (m, 2H); 13C NMR (C5H5N, 500 MHz) δ 145.6, 134.2, 129.5, 128.8, 71.5, 40.0, 29.3, 28.4, 26.7, 25.5,
21.8.
Di-tert-butyl (((S)-1-(tert-butoxy)-5-((6-chlorohexyl)amino)-1,5-dioxopentan-2-yl)carbamoyl)-L-
glutamate (4.21)
To a mixture of 4.19 (0.7 g, 1.43 mmol) and DCM (70 mL) at 0 oC under N2, 4.20 (0.64 g, 2.11 mmol),
EDCI (0.81 g, 4.24 mmol), Et3N (0.73 mL, 5.72 mmol) and DMAP (5 mg, 0.043 mmol) were added. The
reaction was warmed slowly to rt and then stirred for 16 h. FCC (70% EtOAc/hexanes) delivered 4.19
(0.38 g, 44%). Rf = 0.3 (30 % EtOAC/hexanes). 1H NMR (CDCl3, 500 MHz) δ 6.61 (br s, 1H), 5.90 (m,
1H), 5.45 (m, 1H), 4.27 (m, 1H), 4.24 (m, 1H), 3.45 (m, 2H), 3.19 (m, 2H), 2.34 (m, 1H), 2.20 (m, 3H),
1.98 (m, 2H), 1.80 (m, 2H), 1.70-1.27 (m, 33H); 13C NMR (CDCl3, 500 MHz) δ 173.5, 172.6, 172.3, 172.1,
157.3, 82.2, 81.1, 80.8, 53.7, 53.2, 45.2, 39.6, 32.6, 32.1, 31.7, 29.5, 28.5, 26.7, 26.3.
Tri-tert-butyl (14S,18S)-2,11,16-trioxo-3-thia-10,15,17-triazaicosane-14,18,20-tricarboxylate (4.22)

120
Compound 4.21 (0.38 g, 0.65 mmol) was dissolved in dry DMSO (2 mL). KSAc (0.22 g, 1.9 mmol) and
18-crown-6 ether (17 mg, 0.064 mmol) were added and the mixture stirred at rt for 2 h. The solvent was
evaporated in vacuo. FCC of the residue provided 4.22 (0.13 g, 31%). Rf = 0.3 (20 % EtOAC/hexanes).
1H NMR (CDCl3, 600 MHz) δ 6.48 (m, 1H), 5.50 (br s, 1H), 5.05 (m, 1H), 4.30 (m, 1H), 4.19 (m, 1H), 3.22
(m, 2H), 2.82 (t, J = 7.3 Hz, 2H), 2.40 (m, 1H), 2.29 (s, 3H), 2.40 (m, 1H), 2.24 (m, 1H), 2.03 (m, 1H), 1.84
(m, 1H), 1.48-1.20 (m, 35H); 13C NMR (CDCl3, 600 MHz) δ 195.5, 173.5, 172.6, 172.2, 172.1, 157.3, 82.2,
81.1 (2 signals), 53.7, 53.2, 39.5, 32.1, 31.7, 30.9, 29.5, 29.4, 29.1, 28.5, 28.4 (2 signals), 28.3 (2
signals), 28.2, 26.4. HRMS (ESI) calcd for C31H55N3O9SNa (M+Na)+ 668.3551, found 668.3543.
(14S,18S)-2,11,16-Trioxo-3-thia-10,15,17-triazaicosane-14,18,20-tricarboxylic acid (4.23)
4.22 (25 mg, 0.037 mmol) was stirred in TFA (0.25 mL), DCM (0.25 mL) and TIBS (0.02 mL) at rt for 1.5
h. The solution was then concentrated in vacuo. FCC of the residue (25% MeOH/DCM to
HOAc:MeOH:DCM, 0.1:2:8) yielded 4.23 (20 mg, quantitatve). Rf = 0.3 (30% MeOH/DCM). 1H NMR
(MeOD, 400 MHz) δ 4.30 (m, 1H), 4.20 (m, 1H), 3.21 (m, 2H), 2.88 (t, J = 7.2 Hz, 2H), 2.38 (m, 4H), 2.32
(s, 3H), 2.16 (m, 1H), 2.04 (m, 1H), 1.81 (m, 1H), 1.77 (m, 2H), 1.52 (m, 4H), 1.39 (m, 4H); 13C NMR
(CDCl3, 400 MHz) δ 197.8, 176.8, 174.7, 160.0, 54.7, 54.0, 40.4, 31.4, 30.8, 30.6, 30.3, 29.9, 29.6, 29.5,
29.0, 27.5. HRMS (ESI) calcd for C19H32N3O9S (M+H)+ 478.1854, found 478.1843.
4-Nitrophenyl (2-(pyridin-2-yldisulfaneyl)ethyl) carbonate (4.24)
To a solution of 4.27 (0.50 g, 2.7 mmol), pyridine (0.3 mL, 4 mmol) and DCM (10 mL) at rt was added
dropwise over 20 min a solution of 4-nitrophenylchloroformate (0.75 g, 4.0 mmol) in DCM (10 mL). The

121
mixture was stirred overnight, then was washed with water (3x). The organic phase was dried (Na2SO4),
filtered and evaporated under reduced pressure. FCC (15% EtOAc/hexanes) of the residue yielded 4.24
(0.71 g, 76%). Rf = 0.45 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 8.47 (m, 1H), 8.34 (m, 2H),
8.14 (m, 2H), 7.55 (m, 2H), 7.32 (m, 2H), 7.14 (m, 1H), 4.58 (t, J = 1.7 Hz, 2H), 3.10 (t, J = 6.2 Hz, 2H);
13C NMR (CDCl3, 500 MHz) δ 159.1, 155.4, 152.2, 149.9, 137.2, 126.3, 125.0, 123.2, 122.1, 120.2, 115.7,
66.7, 36.7. HRMS (ESI) calcd for C14H13N2O5S2 (M+H)+ 352.0188, found 352.0187. The NMR data
reported is essentially identical to the one found in literature.96
2-(Pyridin-2-yldisulfaneyl)ethan-1-ol (4.27)
Sulfuryl chloride (50 mL, 1M solution in DCM) was added over a period of 20 min to a stirred solution of 2-
mercaptopyridine, 4.28 (5.0 g, 0.045 mol) in dry DCM (50 mL) at 0-5 oC under a nitrogen atmosphere.
The mixture was stirred for 2 h and concentrated in vacuo. The yellow granular solid was taken up in dry
DCM (200 mL) and cooled to 0 oC. To this stirred suspension at 0-5 oC was added a solution of 2-
mercaptoethanol 4.25 (3.4 mL, 0.048 mol) in DCM (10 mL) dropwise over 5 min. Within 15-20 min a
yellow solid started to separate. The mixture was stirred at rt overnight. The product was filtered, washed
with DCM, and dried under high vacuum. The residue was resuspended in DCM and DMAP (5.50 g,
0.045 mmol) was added. The suspension was filtered and concentrated in vacuo. FCC of the residue
(10% EtOAc/hexanes) yielded 4.27 (3.4 g, 40%). Rf = 0.16 (20% EtOAc/hexanes). 1H NMR (CDCl3,
500MHz) δ 8.53 (d, J = 3.8 Hz, 1H), 7.41 (m, 1H), 7.32 (m, 1H), 7.19 (m, 1H), 3.78 (m, 2H), 2.93 (m, 2H);
13C NMR (CDCl3, 500MHz) δ 159.1, 149.8, 136.9, 122.0, 121.6, 58.2, 42.8; HRMS (ESI) calcd for
C7H10NOS2 (M+H)+ 187.0126, found 187.0130.

122
Propen-1-yl-3,4-O-isopropyledene-4-O-methoxymethyl-6-O-undecyl-α-D-mannopyranoside (4.31)
Alcohol 4.30 (0.40 g, 0.96 mmol) was dissolved in DCM (16 mL), MOMCl (0.29 mL, 3.81 mmol), DIPEA
(0.86 mL, 0.006 mmol) were added at 0 oC and the solution was stirred overnight. The reaction was then
quenched with water and extracted with DCM. The organic phase was dried Na2SO4, concentrated in
vacuo and purified via FCC (25% EtOAc/hexanes) to afford compound 4.31 (0.3 g, 68%). Rf = 0.62 (20%
EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ 5.87 (m, 1H), 5.26 (m, 1H), 5.17 (m, 1H), 5.65 (s, 1H),
4.86 (d, J = 5.3 Hz, 1H), 4.65 (d, J = 5.2 Hz, 1H), 4.19 (m, 1H), 4.11 (m, 1H), 3.98 (m, 1H), 3.66 (m, 3H),
3.57 (m, 1H), 3.48 (m, 1H), 3.42 (m, 4H), 1.55 (m, 4H), 1.50 (s, 3H), 1.31 (partially buried s, 3H), 1.30-
1.22 (m, 21H), 0.86 (t, J = 5.8 Hz, 3H).; 13C NMR δ 133.6, 117.8, 109.3, 96.3, 96.2, 78.4, 75.9, 73.2, 71.8,
69.7, 68.3, 67.9, 56.0, 31.9, 29.8, 29.7, 29.6, 29.5, 29.3, 27.9, 26.4, 26.2, 22.7, 14.2. HRMS (ESI) calcd
for C25H47O7 (M+H)+ 458.3244, found 458.3243.
4-O-Methoxymethyl-6-O-undecyl mannoside-butenolide-alkene (4.33)
A mixture of 4.31 (0.3 g, 0.65 mmol) and 4.32 (0.18 g, 0.33 mmol) in dry CH2Cl2 (3.3 mL) was purged with
nitrogen. Grubbs I catalyst (26 mg, 0.03 mmol) was then added and the mixture purged with nitrogen,
then heated at reflux for 6 h. At that time the mixture was cooled to rt and DMSO (0.05 mL) was added.
After 1 h the mixture was concentrated in vacuo. FCC (30% EtOAc/hexanes) of the residue afforded 4.33
(0.27 g, 42%). Rf = 0.53 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz) δ 7.37 (m, 4H), 7.33 (m, 6H),
6.95 (s, 1H), 5.62 (m, 1H), 5.48 (m, 1H), 5.05 (s, 1H), 4.97 (m, 1H), 4.86 (m, 1H), 4.66 (d, J = 6.3 Hz, 1H),
4.18-4.10 (m, 3H), 3.80 (m, 1H), 3.67 (m, 4H), 3.57 (m, 1H), 3.50 (m, 1H), 3.38 (m, 4H), 2.22 (m, 2H),

123
1.90 (m, 2H), 1.50-1.20 (m, 45H), 1.00 (s, 9H), 0.85 (t, J = 7.2 Hz, 3H). HRMS (ESI) calcd for C58H93O10SI
(M+H)+ 976.6460, found 976.6454.
4-O-Methoxymethyl-6-O-undecyl mannoside-butenolide (4.33a)
Compound 4.33 (0.27 g, 0.27 mmol) and TsNHNH2 (1.8 g, 9.67 mmol) were dissolved in DME (60 mL). At
reflux, a solution of NaOAc (2.1 g, 25.6 mmol) in H2O (63 mL) was added dropwise over a 4 h period. The
mixture was then cooled, diluted with ethyl acetate, and washed with water. The organic phase was dried
(Na2SO4), filtered and concentrated in vacuo. FCC in 1-5% (acetone/DCM) delivered 4.33a. Rf = 0.5 (2%
acetone/DCM). 1H NMR (CDCl3, 600 MHz) δ 7.64 (m, 4H), 7.33 (m, 6H), 6.93 (m, 1H), 4.99 (s, 1H), 4.97
(m, 1H), 4.86 (d, J = 6.2 Hz, 1H), 4.66 (d, J = 6.2 Hz, 1H), 4.17 (m, 1H), 4.07 (m, 1H), 3.66 (m, 5H), 3.58
(m, 1H), 3.46 (m, 1H), 3.40 (m, 2H), 3.38 (s, 3H), 2.40 (s, 1H), 2.22 (m, 2H), 1.50 (m, 17H), 1.24 (m, 6H),
1.21 (m, 18H), 1.19 (m, 8H), 1.01 (s, 9H), 0.87 (m, 3H). HRMS (ESI) calcd for C58H92O10Si (M+Na)+
1001.6514, found 1001.6500.
10-Hydroxy-4-O-methoxymethyl-6-O-undecylmannoside-butenolide-alkene (4.34)
Compound 4.33a was then dissolved in a mixture of pyridine (1 mL) and HF/pyridine (1 mL) at 0 oC. The
mixture was stirred for 16 h, then diluted with water and extracted with ethyl ether. FCC of the residue
afforded 4.33b (85 mg, 43% after two steps). 1H NMR (CDCl3, 500 MHz) δ 6.96 (s, 1H), 4.99 (s, 1H), 4.97

124
(m, 1H), 4.86 (d, J = 6.2 Hz, 1H), 4.66 (d, J = 6.3 Hz, 1H), 4.17 (m, 1H), 4.07 (d, J = 5.6 Hz, 1H), 3.68 (m,
5H), 3.56 (m, 2H), 3.47 (m, 1H), 3.38 (m, 4H), 2.41 (m, 1H), 1.60-1.32 (m, 51H), 0.86 (t, J = 6.9 Hz, 3H);
13C NMR (CDCl3, 600 MHz) δ 174.1, 149.1, 134.5, 109.4, 97.1, 95.5, 78.7, 76.2, 73.4, 72.1, 72.0, 69.9,
68.4, 67.6, 56.2, 37.7 (2 signals), 32.1, 29.9, 29.8, 29.7 (2 signals), 29.6, 29.5 ( 2 signals), 29.3, 28.0,
27.6, 26.6, 26.3 (2 signals), 25.8, 25.3, 22.9, 19.4, 14.3.
[1-(5-Methyl-2-oxo-2,5-dihydrofuran-3-yl)-pentadecan-8-yl-(2-(pyridin-2-yldisulfaneyl)ethyl)
carbonate]-15-yl-2,3-O-isopropylidene-4-O-methoxymethyl-6-O-undecyl-α-D-mannopyranoside
(4.35)
DMAP (12 mg, 0.11 mmol) was added to a solution of 4.34 (50 mg, 0.07 mmol) and linker 4.24 (40 mg,
0.11 mmol) in DCM (1 mL). The reaction was monitored by TLC, and quenched with water after 16 h, on
disappearance of 4.24. The solvent was evaporated and FCC of the residue (20% EtOAc/hexanes)
yielded 4.35 (30 mg, 50%). Rf = 0.3 (20% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ 8.45 (d, J = 4.7
Hz, 1H), 7.63 m, 2H), 7.07 (m, 1H), 6.95 (m, 1H), 4.98 (s, 1H), 4.96 (m, 1H), 4.85 (d, J = 6.2 Hz, 2H), 4.35
(m, 2H), 4.15 (t, J = 6.1 Hz, 1H), 4.10 (m, 1H), 4.05 (m, 1H), 3.56 (m, 4H), 3.55 (m, 1H), 3.48 (m, 1H),
3.45 (m, 1H), 3.37 (m, 4H), 3.04 (t, J = 6.1 Hz, 2H), 2.22 (m, 2H), 1.52 (m, 10H), 1.49 (s, 3H), 1.37 (d, J =
6.8 Hz, 3H), 1.29 (s, 3H), 1.27-1.21 (m, 26H), 0.84 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 500 MHz) δ
174.1, 159.7, 155.0, 149.9, 149.1, 137.3, 134.4, 121.1, 120.0, 109.4, 97.1, 96.5, 79.6, 78.6, 76.2, 73.3,
72.0, 69.8, 68.3, 67.6, 65.2, 56.2, 37.2, 34.2, 32.1, 29.9, 29.8, 29.7, 29.5, 29.4, 29.3 (2 signals), 28.0,
27.5, 26.6, 26.3, 25.3, 22.9, 19.4, 14.3.

125
[1-(5-Methyl-2-oxo-2,5-dihydrofuran-3-yl)-pentadecan-8-yl-(2-(pyridin-2-
yldisulfaneyl)ethyl)carbonate]-15-yl-6-O-undecyl-α-D-mannopyranoside (4.35a)
Compound 4.35 (30 mg, 0.03 mmol) was dissolved in CH2Cl2 (2 mL) and treated with 5% AcCl in MeOH
(0.5 mL). The reaction was stirred for 16 h and then quenched with a saturated solution of NaHCO3 in
methanol. The mixture was filtered, and the filtrate concentrated in vacuo. FCC of the residue (5%
MeOH/DCM) afforded 4.35a (18 mg, 69%). 1H NMR (CDCl3, 500 MHz) δ 8.51 (d, J = 4.5 Hz, 1H), 7.69
(m, 2H), 7.14 (m, 1H), 7.01 (s, 1H), 5.02 (m, 1H), 4.85 (s, 1H), 4.60 (m, 1H), 4.41 (t, J = 6.5 Hz, 2H), 3.94
(br s, 1H), 3.85-3.45 (m, 6H), 3.54 (m, 2H), 3.43 (m, 1H), 3.10 (t, J = 6.5 Hz, 2H), 2.28 (t, J = 8.2 Hz, 2H),
1.59 (m, 18H), 1.40 (d, J = 5.8 Hz, 3H), 1.28 (m, 27H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 500
MHz) δ 174.2, 160.0, 155.0, 149.9, 149.2, 137.3, 134.4, 121.1, 120.1, 99.7 (2 signals), 79.6, 72.3, 71.9,
71.8, 70.8, 70.6, 69.0 (2 signals), 68.0 (2 signals), 65.2, 53.6, 51.1, 37.2, 34.2, 32.1, 29.9, 29.8, 29.7,
29.6, 29.5 (2 signals), 29.4 (2 signals), 29.3 (2 signals), 27.5, 26.2 (3 signals), 25.3 (2 signals), 22.9, 19.4,
14.3. HRMS (ESI) calcd for C45H76NO11S2 (M+H)+ 870.4860, found 870.4850.
4-O-Methoxymethyl-6-O-undecyl mannoside-thiophene alkene (4.37)
A mixture of 4.31 (160 mg, 0.35 mmol) and 4.36 (0.56 g, 0.93 mmol) in dry CH2Cl2 (10 mL) was purged
with nitrogen. Grubbs I catalyst (43 mg, 0.05 mmol) was then introduced and the mixture heated at reflux
for 6 h. At that time the mixture was cooled to rt and DMSO (0.05 mL) was added. After 1 h the mixture

126
was concentrated in vacuo. FCC (40% EtOAc/hexanes) of the residue afforded compound 4.37 (245 mg,
68%). Rf = 0.53 (20% EtOAc/hexanes). 1H NMR (CDCl3, 600 MHz) δ 7.64 (m, 4H), 7.46 (m, 1H), 7.44 (m,
1H), 7.34 (m, 6H), 7.04 (m, 1H), 5.05 (br s, 1H), 4.86 (d, J = 6.3 Hz, 1H), 4.66 (d, J = 6.2 Hz, 1H), 4.18
(m, 1H), 4.10 (m, 2H), 3.86 (m, 1H), 3.66 (m, 3H), 3.58 (m, 1H), 3.48 (m, 1H), 3.39 (m, 6H), 1.91 (m, 2H),
1.55 (m, 6H), 1.37 (s, 3H), 1.37-1.12 (m, 41H), 1.02 (s, 9H), 0.86 (t, J = 5.1 Hz, 3H); 13C NMR (CDCl3,
600 MHz) δ 162.0, 139.3, 136.1 (2 signals), 135.0, 134.9, 129.8, 129.6 (2 signals), 128.0, 127.7, 127.6,
125.1, 109.4, 96.5, 96.0, 78.6, 76.2, 73.4, 73.3, 72.0, 69.9, 68.4, 67.8, 56.1, 40.2, 36.5, 35.3, 32.4, 32.1,
29.9 (2 signals), 29.8, 29.7 (2 signals), 29.6, 29.5 (2 signals), 29.2, 28.0, 27.3, 27.1, 26.6, 26.4, 25.0,
24.7, 22.9, 19.6, 14.3. HRMS (ESI) calcd for C60H95NO9SSiNa (M+Na)+ 1056.6389, found 1056.6389.
4-O-Methoxymethyl-6-O-undecyl mannoside-thiophene (4.37a)
A solution of compound 4.37 (245 mg, 0.23 mmol) in EtOH (10 mL) and Pd/C (150 mg, 10% wt) was
purged with N2 for 30 min. Then, the mixture was stirred under a hydrogen atmosphere (balloon) for 12 h.
Additional Pd/C was then added (50 mg, 10% wt) and stirring continued for additional 24 h. The reaction
was then purged with N2 and filtered through a bed of Celite to yield 4.37a (0.2 g, 82 %). 1H NMR (CDCl3,
600 MHz) δ 7.64 (m, 4H), 7.43 (m, 1H), 7.38 (m, 1H), 7.34 (m, 6H), 7.04 (m, 1H), 5.92 (br s, 1H), 5.00 (s,
1H), 4.86 (d, J = 6.3 Hz, 1H), 4.66 (d, J = 6.2 Hz, 1H), 4.18 (m, 1H), 4.07 (m, 1H), 3.68 (m, 6H), 3.64 (m,
1H), 3.47 (m, 1H), 3.37 (m, 7H), 1.56 (m, 6H), 1.55 (s, 3H), 1.37 (m, 4H), 1.32 (partially buried s, 3H),
1.09-1.26 (m, 35H), 1.02 (s, 9H), 0.85 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 162.0, 139.4,
136.2, 135.0, 129.9, 129.8, 129.6, 128.0, 127.9, 127.8, 127.6, 109.5, 97.1, 96.5, 78.7, 76.3, 73.4, 72.0,
69.9, 68.4, 67.8, 56.2, 40.3, 36.5, 32.1, 30.0, 29.9 (2 signals), 29.8 (2 signals), 29.7 (2 signals), 29.6 (2
signals), 29.5, 28.1, 27.3, 27.2, 26.8, 26.6, 26.4 (2 signals), 25.1, 22.9, 22.6, 19.6, 14.3.

127
6-O-undecyl mannoside-2-(pyridin-2-yldisulfaneyl)ethyl)carbonate-thiophene (4.37b)
4.37a (200 mg, 0.19 mmol) was dissolved in pyridine (2 mL) and HF/pyridine (10%, 1 mL) was added.
The reaction was stirred at 45 oC for 24 h and then at rt for additional 24 h. The product was isolated and
purified following the procedure used for 4.33b to afford the derived alcohol (91 mg, 65%). Rf = 0.4 (40%
EtOAc/hexanes). A mixture of this material (90 mg, 0.11 mmol), linker 4.24 (80 mg, 0.23 mmol) and
DMAP (30 mg, 0.25 mmol) in DCM (1 mL) was stirred at rt for 16 h. The solvent was then evaporated,
and the residue subject to FCC. The partially purified product (Rf = 0.4 (40% EtOAc/hexanes). was
dissolved in DCM (4 mL) and 5% AcCl in MeOH (0.6 mL) was added. The reaction mixture was then
evaporated under reduced pressure at rt. FCC of the residue afforded 4.37b (76 mg, 73% after two
steps) Rf = 0.42 (8% MeOH/DCM). 1H NMR (CDCl3, 600 MHz) δ 8.41 (m, 1H), 7.61 (m, 1H), 7.59 (m,
1H), 7.41 (m, 1H), 7.38 (m, 1H), 7.19 (m, 1H), 7.05 (m, 1H), 5.99 (br s, 1H), 4.75 (s, 1H), 4.62 (m, 1H),
4.31 (t, J = 6.5 Hz, 2H), 3.84-3.58 (m, 7H), 3.44 (m, 2H), 3.33 (m, 3H), 3.01 (t, J = 6.3 Hz, 2H), 1.56 (m,
12H), 1.27 (m, 37H), 0.81 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 600 MHz) δ 162.1, 159.7, 155.0, 149.9,
139.3, 137.3, 129.8, 128.0, 127.7, 121.1, 120.1 (2 signals), 99.7, 79.6, 72.3, 71.9, 71.8 (2 signals), 70.7
(2 signals), 69.4 (2 signals), 68.0, 67.9, 65.2, 40.2, 37.3, 34.1 (2 signals), 32.1, 29.8 (3 signals), 29.7,
29.6, 29.5 (3 signals), 29.4 (2 signals), 27.1, 26.2 (2 signals), 25.3, 22.9, 14.3. HRMS (ESI) calcd for
C47H79N2O10S3 (M+H)+ 927.4890, found 927.4891.
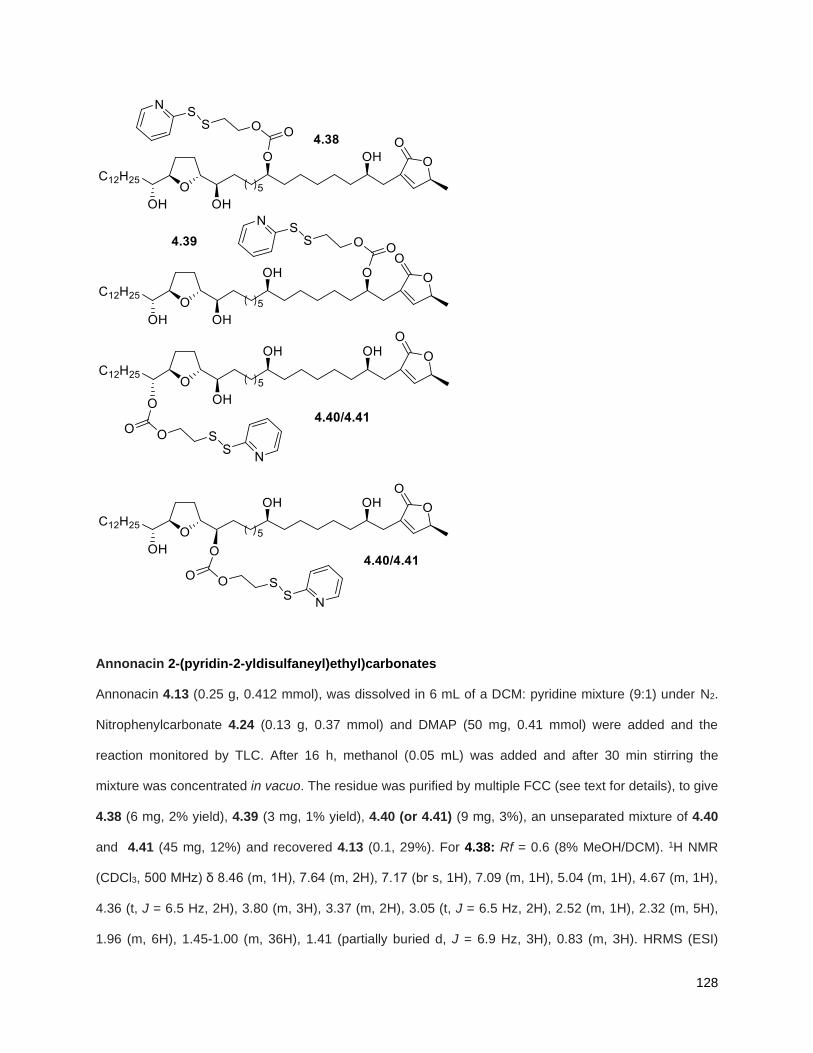
128
Annonacin 2-(pyridin-2-yldisulfaneyl)ethyl)carbonates
Annonacin 4.13 (0.25 g, 0.412 mmol), was dissolved in 6 mL of a DCM: pyridine mixture (9:1) under N2.
Nitrophenylcarbonate 4.24 (0.13 g, 0.37 mmol) and DMAP (50 mg, 0.41 mmol) were added and the
reaction monitored by TLC. After 16 h, methanol (0.05 mL) was added and after 30 min stirring the
mixture was concentrated in vacuo. The residue was purified by multiple FCC (see text for details), to give
4.38 (6 mg, 2% yield), 4.39 (3 mg, 1% yield), 4.40 (or 4.41) (9 mg, 3%), an unseparated mixture of 4.40
and 4.41 (45 mg, 12%) and recovered 4.13 (0.1, 29%). For 4.38: Rf = 0.6 (8% MeOH/DCM). 1H NMR
(CDCl3, 500 MHz) δ 8.46 (m, 1H), 7.64 (m, 2H), 7.17 (br s, 1H), 7.09 (m, 1H), 5.04 (m, 1H), 4.67 (m, 1H),
4.36 (t, J = 6.5 Hz, 2H), 3.80 (m, 3H), 3.37 (m, 2H), 3.05 (t, J = 6.5 Hz, 2H), 2.52 (m, 1H), 2.32 (m, 5H),
1.96 (m, 6H), 1.45-1.00 (m, 36H), 1.41 (partially buried d, J = 6.9 Hz, 3H), 0.83 (m, 3H). HRMS (ESI)

129
calcd for C45H76NO9S2 (M+H)+ 838.4956, found 838.4927. For 4.39: Rf = 0.5 (8% MeOH/DCM). 1H NMR
(CDCl3, 500 MHz) δ 8.46 (m, 1H), 7.64 (m, 2H), 7.11 (br s, 1H), 7.03 (m, 1H), 4.99 (m, 1H), 4.91 (m. 1H),
4.37 (t , J = 6.7 Hz, 2H), 3.78 (m, 2H), 3.56 (br s, 1H), 3.38 (br s, 2H), 3.04 (t, J = 6.5 Hz, 2H), 2.57 (m,
2H), 2.31 (m, 1H), 2.27 (m, 1H), 1.97 (m 4H), 1.54-1.36 (m, 8H), 1.37 (partially buried d, J = 6.9 Hz, 3H),
0.34 (m, 3H). For 4.40 (or 4.41): Rf = 0.6 (8% MeOH/DCM). 1H NMR (CDCl3, 500 MHz) δ 8.40 (m, 1H),
7.60 (m, 2H), 7.12 (s, 1H), 7.06 (m, 1H), 4.99 (m, 1H), 4.6 (m, 1H), 4.32 (m, 2H), 3.92 (m, 1H), 3.75 (m,
2H), 3.51 (br s, 1H), 3.28 (m, 1H), 2.99 (m, 2H), 2.47 (m, 1H), 2.34 (m, 2H), 2.28 (m ,1H), 1.91 (m, 3H),
1.45-1.29 (m, 10H), 1.39 (partially buried d, J = 6.9 Hz, 3H), 1.19 (m, 34H), 0.82 (m, 3H).

130
ANTITUMOR ACTIVITY OF 4-DEOXYANNOMONTACIN AND ANALOGUES

131
5.1. Introduction
The cytotoxicity of naturally occurring THF-ACGs and unnatural analogues against a broad range of
human tumor cell lines has been widely studied.12, 63 The MTT assay is the most common assay used.
However, because studies have been performed at different timelines and against different cell lines, it is
often difficult to compare the data. This is particularly important as the activity of certain analogues has
been shown to vary with the length of time that the cells are incubated with the drug.62 Herein, the activity
of our synthetic compounds in the MTT assay at 16 and 48 h is presented.
The cell lines evaluated were the prostate cancer (PCa) cells, PC-3 and LNCaP, MCF-7 (MDR breast),
MDA-MB-231 (breast) and HTC116 (colon). This panel was chosen to test activity against different tumor
types and to evaluate activity of parent drugs and their DUPA-linked conjugates against PSMA (prostate
specific membrane antigen) positive and negative cells. LNCaP is known to overexpress PSMA which
binds DUPA with high specificity and affinity, whereas wild-type PC-3, and the non-PCa lines do not
present PSMA.103a, b, 110 Therefore, the DUPA-drug conjugate of a given parent drug is expected to be
more active than the parent drug against LNCaP cells, and other PSMA positive cells compared to PSMA
negative cells.
5.2. Results
Activities are reported as the half maximal inhibitory concentration (IC50). As it was not possible to
calculate an IC50 for analogues with low activity, the % cell viability at 12.5 µM and 25 µM is also reported
to give a measure of the relative activity of all test compounds. Compounds were first assayed at 16 h at
1-25 µM against MCF-7, MDA-MB-231, HTC116, PC-3 and LNCaP. Analogues with significant activity or
of structure activity significance were advanced to a 48 h assay starting at a wider range of
concentrations.
For comparison of the data, the test compounds are grouped according to structural similarity: (a) THF
with butenolide or butenolide substitutes (1, 2, 3, 4, 5); (b) Sugar analogues with butenolide (6, 7, 8, 9,

132
10); (c) Hybrid analogues with both THF and butenolide substitutes (11, 12, 13, 14), including the
‘chameleon’ analog, 14. The results for each subgroup are discussed in the following sections.
5.2.1. THF with butenolide or butenolide substitutes (1, 2, 3, 4, 5)
This group comprises the naturally occurring THF-butenolides 4-DAN, 1 and annonacin, 2, and 3, 4 and
5, THF analogues in which the butenolide is replaced with O-n-butyl, N-linked piperazine and thiophene-
2-carboxamide moieties, respectively (Table 5.1). In the 16 h MTT assay, all analogues except 3 showed
less than 50% cell viability at concentrations lower than 25 M against at least four of the five cell lines.
The four active analogues 1, 2, 4 and 5 were further evaluated at lower concentrations over 48 h (Table
5.2, Figure S5-I).
Table 5.1. Cell viability (%) over 16 h at 12.5 and 25 µM (ND = Not determined, *data collected in an independent study)
Compound
MBA-MD-231
HTC116 LNCaP PC-3 MCF-7
12.5 25 12.5 25 12.5 25 12.5 25 12.5 25
75 60 40 30 10 10 45 40 45 35
80 70 55 45 30 30 40 30 50 50
80 75 ND ND 70 55 80 75 ND ND
75 60 60 40 20 20 40 30 35 25
30 25 30 20 10 0 30 30 45 40
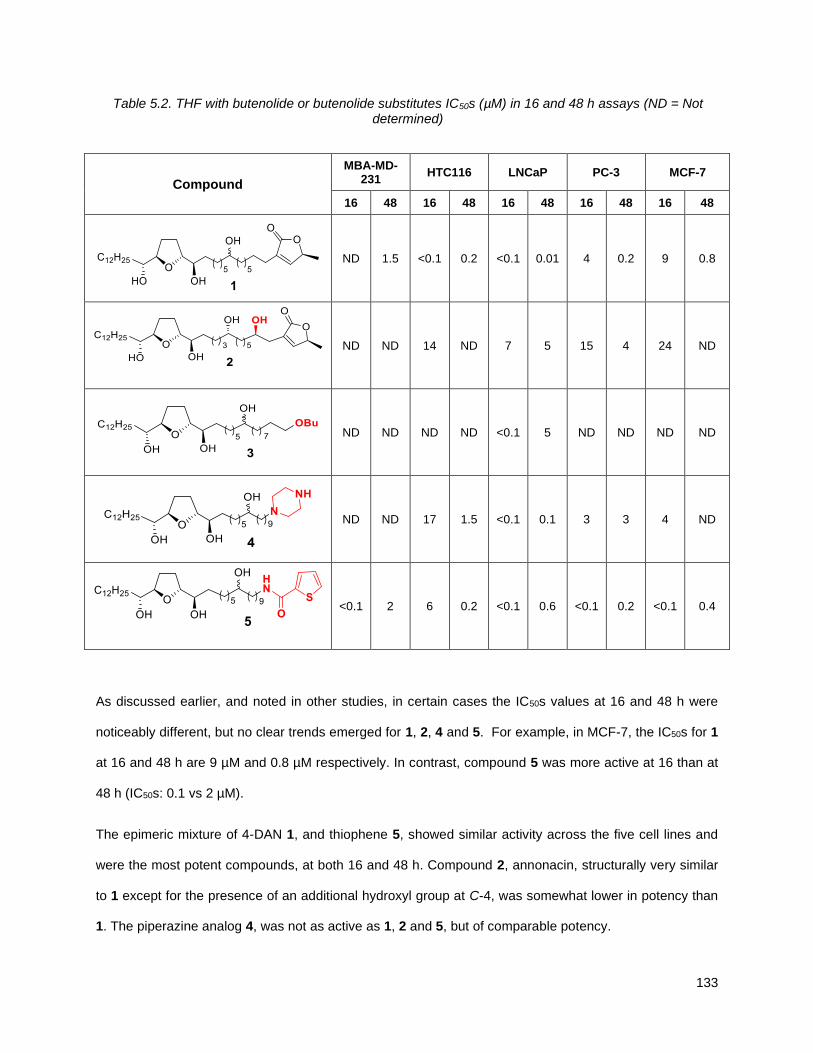
133
Table 5.2. THF with butenolide or butenolide substitutes IC50s (µM) in 16 and 48 h assays (ND = Not determined)
Compound
MBA-MD-231
HTC116 LNCaP PC-3 MCF-7
16 48 16 48 16 48 16 48 16 48
ND 1.5 <0.1 0.2 <0.1 0.01 4 0.2 9 0.8
ND ND 14 ND 7 5 15 4 24 ND
ND ND ND ND <0.1 5 ND ND ND ND
ND ND 17 1.5 <0.1 0.1 3 3 4 ND
<0.1 2 6 0.2 <0.1 0.6 <0.1 0.2 <0.1 0.4
As discussed earlier, and noted in other studies, in certain cases the IC50s values at 16 and 48 h were
noticeably different, but no clear trends emerged for 1, 2, 4 and 5. For example, in MCF-7, the IC50s for 1
at 16 and 48 h are 9 µM and 0.8 µM respectively. In contrast, compound 5 was more active at 16 than at
48 h (IC50s: 0.1 vs 2 µM).
The epimeric mixture of 4-DAN 1, and thiophene 5, showed similar activity across the five cell lines and
were the most potent compounds, at both 16 and 48 h. Compound 2, annonacin, structurally very similar
to 1 except for the presence of an additional hydroxyl group at C-4, was somewhat lower in potency than
1. The piperazine analog 4, was not as active as 1, 2 and 5, but of comparable potency.
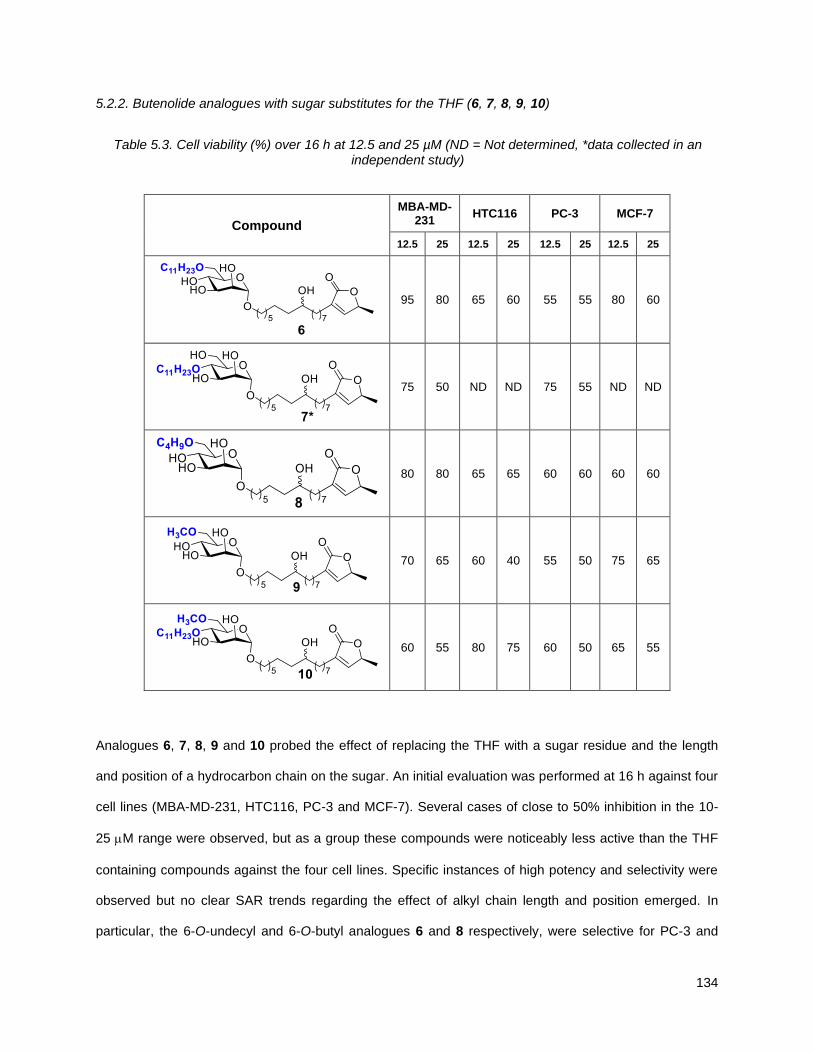
134
5.2.2. Butenolide analogues with sugar substitutes for the THF (6, 7, 8, 9, 10)
Table 5.3. Cell viability (%) over 16 h at 12.5 and 25 µM (ND = Not determined, *data collected in an independent study)
Compound
MBA-MD-231
HTC116 PC-3 MCF-7
12.5 25 12.5 25 12.5 25 12.5 25
95 80 65 60 55 55 80 60
75 50 ND ND 75 55 ND ND
80 80 65 65 60 60 60 60
70 65 60 40 55 50 75 65
60 55 80 75 60 50 65 55
Analogues 6, 7, 8, 9 and 10 probed the effect of replacing the THF with a sugar residue and the length
and position of a hydrocarbon chain on the sugar. An initial evaluation was performed at 16 h against four
cell lines (MBA-MD-231, HTC116, PC-3 and MCF-7). Several cases of close to 50% inhibition in the 10-
25 M range were observed, but as a group these compounds were noticeably less active than the THF
containing compounds against the four cell lines. Specific instances of high potency and selectivity were
observed but no clear SAR trends regarding the effect of alkyl chain length and position emerged. In
particular, the 6-O-undecyl and 6-O-butyl analogues 6 and 8 respectively, were selective for PC-3 and
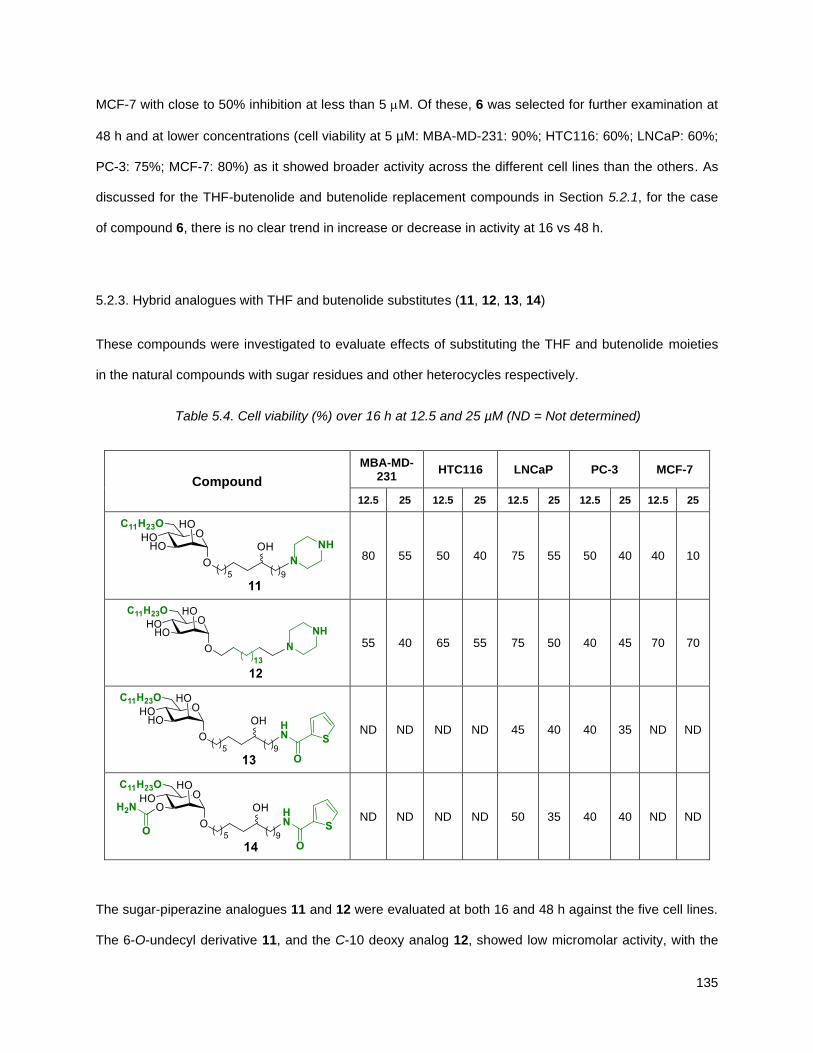
135
MCF-7 with close to 50% inhibition at less than 5 M. Of these, 6 was selected for further examination at
48 h and at lower concentrations (cell viability at 5 µM: MBA-MD-231: 90%; HTC116: 60%; LNCaP: 60%;
PC-3: 75%; MCF-7: 80%) as it showed broader activity across the different cell lines than the others. As
discussed for the THF-butenolide and butenolide replacement compounds in Section 5.2.1, for the case
of compound 6, there is no clear trend in increase or decrease in activity at 16 vs 48 h.
5.2.3. Hybrid analogues with THF and butenolide substitutes (11, 12, 13, 14)
These compounds were investigated to evaluate effects of substituting the THF and butenolide moieties
in the natural compounds with sugar residues and other heterocycles respectively.
Table 5.4. Cell viability (%) over 16 h at 12.5 and 25 µM (ND = Not determined)
Compound
MBA-MD-231
HTC116 LNCaP PC-3 MCF-7
12.5 25 12.5 25 12.5 25 12.5 25 12.5 25
80 55 50 40 75 55 50 40 40 10
55 40 65 55 75 50 40 45 70 70
ND ND ND ND 45 40 40 35 ND ND
ND ND ND ND 50 35 40 40 ND ND
The sugar-piperazine analogues 11 and 12 were evaluated at both 16 and 48 h against the five cell lines.
The 6-O-undecyl derivative 11, and the C-10 deoxy analog 12, showed low micromolar activity, with the
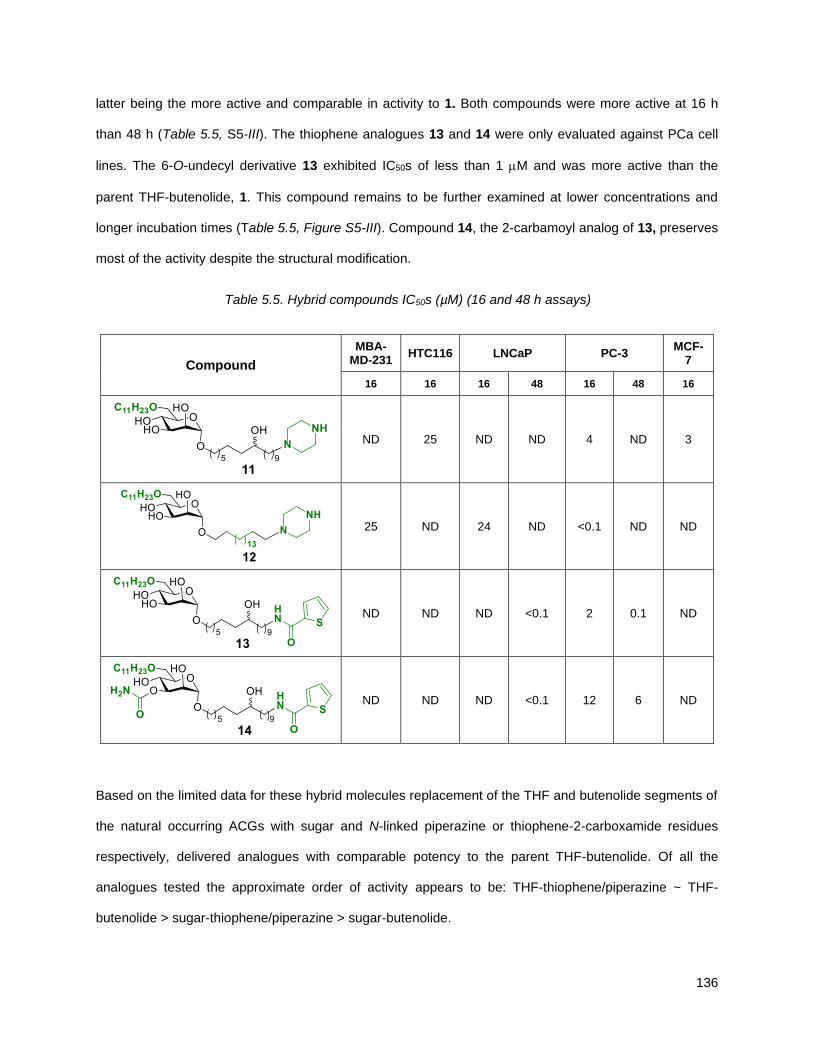
136
latter being the more active and comparable in activity to 1. Both compounds were more active at 16 h
than 48 h (Table 5.5, S5-III). The thiophene analogues 13 and 14 were only evaluated against PCa cell
lines. The 6-O-undecyl derivative 13 exhibited IC50s of less than 1 M and was more active than the
parent THF-butenolide, 1. This compound remains to be further examined at lower concentrations and
longer incubation times (Table 5.5, Figure S5-III). Compound 14, the 2-carbamoyl analog of 13, preserves
most of the activity despite the structural modification.
Table 5.5. Hybrid compounds IC50s (µM) (16 and 48 h assays)
Compound
MBA-MD-231
HTC116 LNCaP PC-3 MCF-
7
16 16 16 48 16 48 16
ND 25 ND ND 4 ND 3
25 ND 24 ND <0.1 ND ND
ND ND ND <0.1 2 0.1 ND
ND ND ND <0.1 12 6 ND
Based on the limited data for these hybrid molecules replacement of the THF and butenolide segments of
the natural occurring ACGs with sugar and N-linked piperazine or thiophene-2-carboxamide residues
respectively, delivered analogues with comparable potency to the parent THF-butenolide. Of all the
analogues tested the approximate order of activity appears to be: THF-thiophene/piperazine ~ THF-
butenolide > sugar-thiophene/piperazine > sugar-butenolide.

137
5.3. Discussion
As determination of IC50s was not possible for the entire panel of test compounds, to allow for a more
complete comparison of activities, Table S5-II summarizes activities at 12.5 and 25 mM for all analogues.
Our SAR observations are generally consistent with previous studies (Figure 5.1).12,69 Thus, the
observation that replacement of the butenolide with thiophene or a piperazine leads to analogues with
comparable activity to the THF-butenolide parent, agrees with earlier results by Kojima42, 44, 56, 112 and
Yao.41 However, in contrast to other studies, substitution of the butenolide with a simple n-butyl chain led
to considerable loss in activity.113
Figure 5.1. SAR conclusion based on cytotoxicity assays
Substitution of the THF ring by a mannose delivered compounds with activity in the 25 µM range, which
was somewhat lower than the THF-butenolide parent, and in line with earlier results from this laboratory.79
No clear trend emerged regarding the effects of the length and location of the hydrocarbon chain on the
sugar, as individual analogues with specific chain lengths and positions all displayed significant activity.
However, these SAR effects merit closer evaluation as several notable cases of selectivity and potency
were observed.

138
There have been contradicting reports of the effect that the hydroxyl group in the hydrocarbon spacer has
on activity.67, 70,70 In this context, 11, the C6-undecyl-mannose-piperazine and 12, its C10-deoxygenated
counterpart both showed high potency, but their relative toxicity was dependent on the cell line studied. In
a similar vein, we also found that annonacin 2, was less active than 4-DAN, 1, the 4-deoxy congener of 2.
The most interesting compounds resulted from replacing both the THF and butenolide segments of the
naturally occurring ACGs with sugar and N-piperazine or thiophene-2-carboxamide residues. These
hybrid structures are appealing in that they are of comparable potency to the parent THF-butenolide
parents but are considerably more synthetically accessible. The piperazine analogues are of additional
interest in that the secondary amine allows for straightforward conjugation to imaging entities or tumor
vectors. In the latter context, sugar hybrids such as the 3-O-carbamoyl derivative, 14 have additional
appeal in that the sugar residue, following from the tumor selectivity of the bleomycins, is an “in-built”
potential tumor vector.93c, 99-100
5.4. Prodrug studies
Figure 5.2. Structure of compound 6 and its prodrug 15
Our hypothesis is that conjugation of a cytotoxic entity to a tumor-specific vector leads to a drug that is
more toxic to tumor cells that overexpress the receptor than cells that do not. To test this hypothesis for
our ACG mimetics, the sugar-butenolide 6 and its DUPA prodrug analog 15, were assayed against the
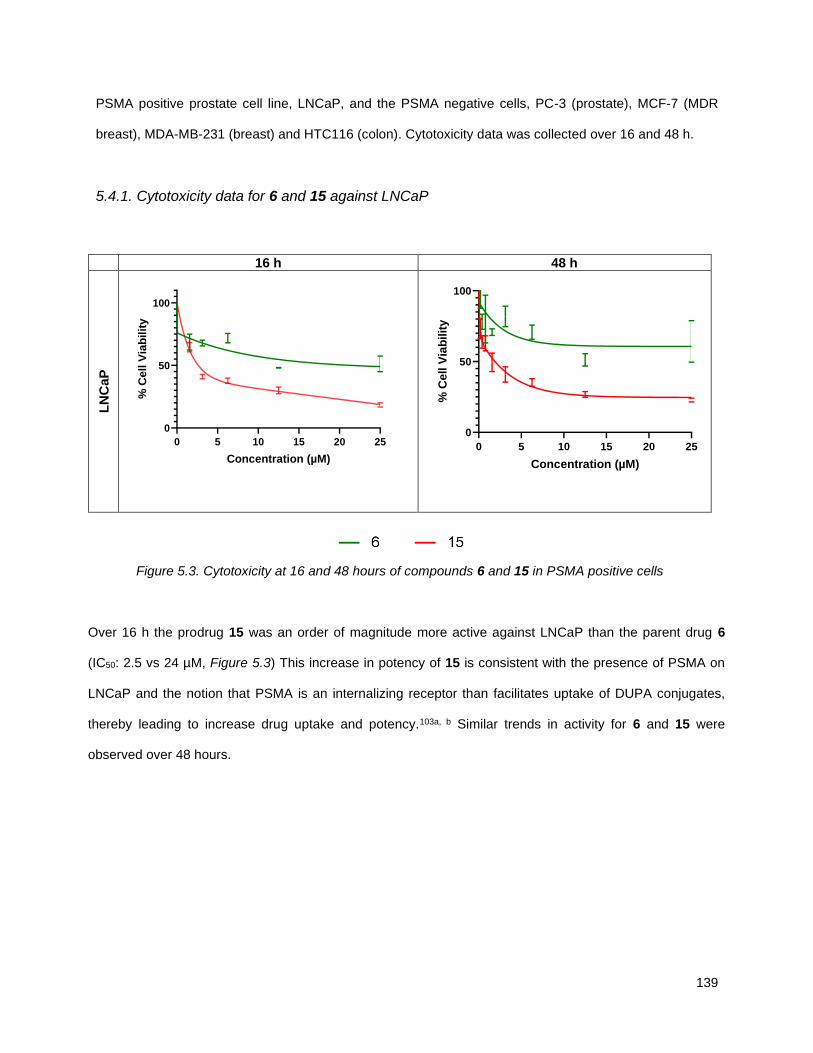
139
PSMA positive prostate cell line, LNCaP, and the PSMA negative cells, PC-3 (prostate), MCF-7 (MDR
breast), MDA-MB-231 (breast) and HTC116 (colon). Cytotoxicity data was collected over 16 and 48 h.
5.4.1. Cytotoxicity data for 6 and 15 against LNCaP
Over 16 h the prodrug 15 was an order of magnitude more active against LNCaP than the parent drug 6
(IC50: 2.5 vs 24 µM, Figure 5.3) This increase in potency of 15 is consistent with the presence of PSMA on
LNCaP and the notion that PSMA is an internalizing receptor than facilitates uptake of DUPA conjugates,
thereby leading to increase drug uptake and potency.103a, b Similar trends in activity for 6 and 15 were
observed over 48 hours.
16 h 48 h
LN
CaP
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
Figure 5.3. Cytotoxicity at 16 and 48 hours of compounds 6 and 15 in PSMA positive cells
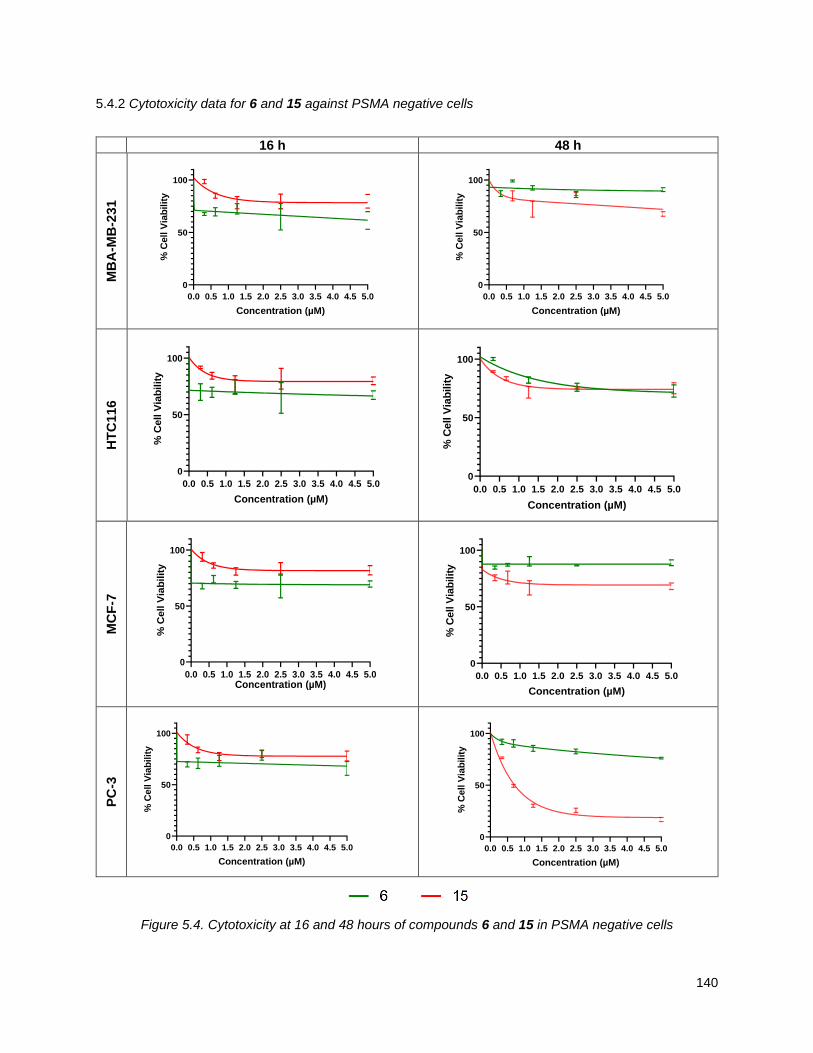
140
5.4.2 Cytotoxicity data for 6 and 15 against PSMA negative cells
16 h 48 h
MB
A-M
B-2
31
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
HT
C116
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
MC
F-7
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
PC
-3
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
Figure 5.4. Cytotoxicity at 16 and 48 hours of compounds 6 and 15 in PSMA negative cells

141
Compounds 6 and 15 showed a similar activity profile against the PSMA negative cells MBA-MB-231,
HTC116 and MCF-7 (Figure 5.4). At 16 h, whereas the prodrug 15 was significantly more active than 6
against LNCaP (ca 20 vs 50% cell viability at 25 M), the activity of 6 and 15 against the three PSMA
negative cell lines was similar (ca 70-90% cell viability at 25 M).
As a crude measure of selectivity, the activity at 5 M of 15 at 16 h against LNCaP vs the PSMA negative
cell lines are compared in Table 5.6. The relatively higher toxicity against LNCaP vs the PSMA negative
cells, albeit modest, supports our hypothesis that the DUPA conjugated prodrug 15 should be more
selective for PSMA than PSMA negative cell lines.
Table 5.6. Comparison of activity (% cell viability) of 15 for LNCaP vs PSMA negative cell lines at 5 M
MBA-MB-231 HTC116 PC-3 LNCaP MCF-7
% viability at
5 M and 16 h 80 80 80 40 80
% viability PSMA (–)
% viability LNCaP 2 2 2 1 2
It is also noteworthy that 15 was subtly but consistently lower in activity than 6 against the three PSMA
negative cell lines (ca 90 vs 70% cell viability at 5 M). This may be an indication that in the absence of
the PSMA transporter, the more polar DUPA-conjugate 15 is internalized more slowly than 6. For the 48 h
assay, the activities of 6 and 15 against these three PSMA negative cell lines, were essentially the same
compared to the data at 16 h.
In contrast to data for the three previously discussed PSMA negative cell lines, the 16 and 48 h data
against PC-3, which is also PSMA negative, is conflicting. The 16 h data for PC-3 is in line with our
hypothesis and the observations for the other PSMA negative cell lines in that both 6 and 15 show
similarly modest activity (ca 70 - 90% cell viability at 5 M). However, the activity of 6 and 15 in the 48 h
assay seemingly disagrees with our hypothesis in that the prodrug 15 is significantly more active than the
parent drug 6. This was unexpected as PC-3 is PSMA negative, and therefore the activity of 6 and 15

142
was predicted to be similar, as was the case for the other three PSMA negative cell lines at 48 h. In
comparison, the activity of 6 in the 16 and 48 h assay was relatively unchanged.
5.4.3. Annonacin prodrugs
To evaluate the generality of the behavior of ACGs and their DUPA conjugates against LNCaP and PC-3
cells, the cytotoxicity of annonacin 2 and its prodrug 16 was evaluated (Figure 5.5). These data indicate
similar trends in activity against LNCaP and PC-3 as for 6 and 15.
Figure 5.5. Structure of annonacin, 2, and annonacin prodrug, 16
At 16 h, the prodrug 16 was more active against LNCaP than PC-3 (40 vs 60% viability at 5 M, Figure
5.6). However, it should be noted that the difference in selectivity against the two cell lines is somewhat
lower than for 6 and 15, which may be a consequence of the higher intrinsic toxicity of parent drug 2
compared to 6. Similar to the behavior of 6 and 15, at 48 h, prodrug 16 is significantly more active than 2
against PC-3. This unexpected activity of DUPA-conjugated prodrugs 15 and 16 against PC-3 suggest
that they may have a different mechanism of action against PC-3 compared to the other cell lines.
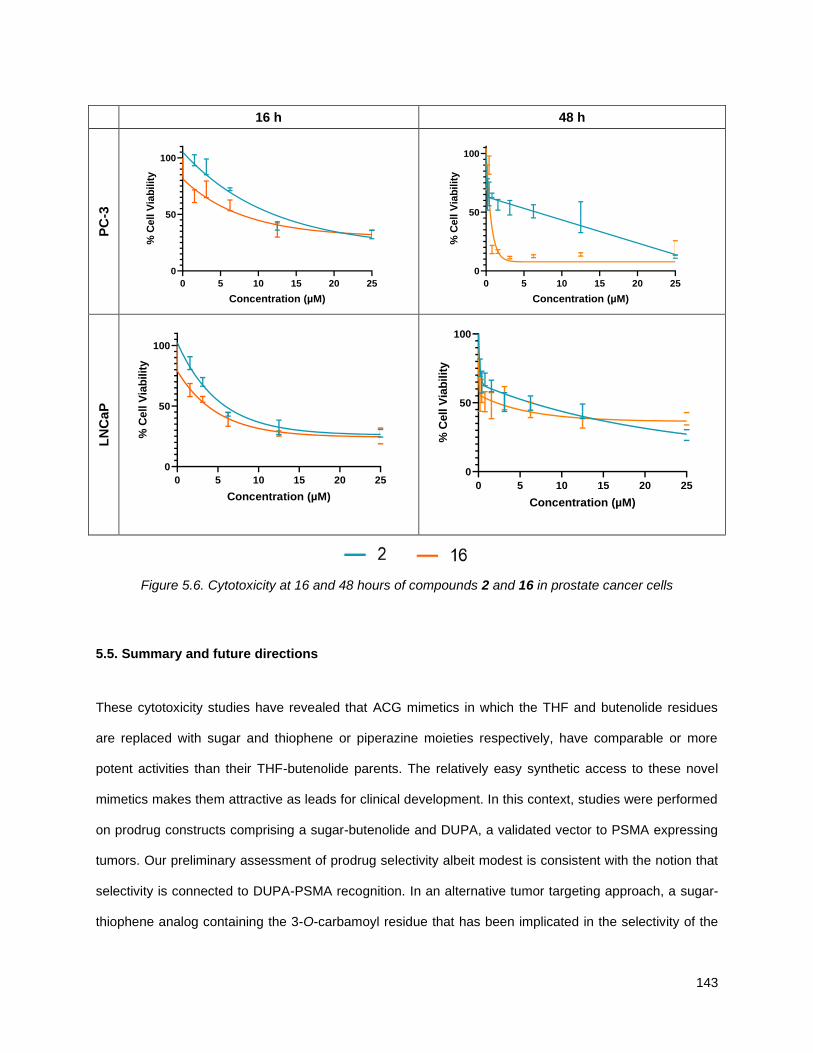
143
Figure 5.6. Cytotoxicity at 16 and 48 hours of compounds 2 and 16 in prostate cancer cells
5.5. Summary and future directions
These cytotoxicity studies have revealed that ACG mimetics in which the THF and butenolide residues
are replaced with sugar and thiophene or piperazine moieties respectively, have comparable or more
potent activities than their THF-butenolide parents. The relatively easy synthetic access to these novel
mimetics makes them attractive as leads for clinical development. In this context, studies were performed
on prodrug constructs comprising a sugar-butenolide and DUPA, a validated vector to PSMA expressing
tumors. Our preliminary assessment of prodrug selectivity albeit modest is consistent with the notion that
selectivity is connected to DUPA-PSMA recognition. In an alternative tumor targeting approach, a sugar-
thiophene analog containing the 3-O-carbamoyl residue that has been implicated in the selectivity of the
16 h 48 h
PC
-3
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
LN
CaP
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y

144
bleomycin family of antitumor agents, was also synthesized. This molecule showed activities in the 0.1-10
M range against the prostate tumor cell lines LNCaP and PC-3; its tumor selectivity remains to be
investigated.
More detailed studies will be performed to probe the selectivity observed for our ACG-DUPA prodrugs.
First, to confirm that the toxicity of the prodrug is due primarily to the drug entity and not the DUPA vector,
activity of the disulfide of the vector (i.e the DUPA ligand), and prodrugs with inactive “drug” entities To
improve selectivity prodrugs with drug entities of varying potencies such as the parent THF-butenolide,
THF-thiophene and sugar-thiophene analogues will be screened. Along these lines, it will be also
interesting to evaluate sugar residues with shorter hydrocarbon chains, as this may decrease non-specific
drug uptake. To determine whether the selectivity of these conjugates is in fact due to binding to PSMA,
competitive inhibitory experiments using 2-(phosphonomethyl)pentanedioic acid (PMPA) - an inhibitor of
DUPA – can be performed.103a These studies will lay the groundwork for ACG derived prodrugs with other
established tumor vectors, such as folate and less explored entities as the 3-O-carbamoyl subunit of the
bleomycins.
The activity against a wider range of tumor cells will further inform on selectivity. The prostate cell lines
MDA-PCA-2b and RWPE-1, are of particular interest. The first is highly prevalent among African
Americans and is relevant to health disparities; the second is a normal prostate epithelial cell line and is
needed for evaluating selectivity. More detailed mechanistic studies will require fluorescently labeled
analogues to determine surface binding, and the uptake and intracellular distribution of the compounds. In
this context, the C-10 alcohol, the amine in piperazine analogues and the 6-O-alkyl chain are practical
positions for incorporation of these labels. Animal experiments using xenografts will be the prelude to
more long-term drug development.

145
5.6. Experimental
The MTT assay was used to determine metabolic activity of cells growing in monolayers by measuring
using a microplate reader (PowerWave HT Microplate Spectrophotometer) their ability to reduce the
yellow- colored MTT to a colored formazan. The data is expressed in percentage of control (i.e. optical
density of formazan from control cells).
HTC-116 colon cancer cell line, MCF-7 breast cancer, MDA-MB-231 triple negative breast cancer, PC-3,
and LNCaP prostate cancer were obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA). All batches of culture media were supplemented with 10% (v/v) of fetal bovine serum (Life
Technologies, Carlsbad, CA, USA), 100 U/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL
amphotericin B (Sigma). HCT116 colon cancer cells, MCF-7, and MDA-MB-231 breast cancers were
cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma), LNCaP was cultured in RPMI-1640,
and PC-3 in F-12K Medium (Kaighn's Modification of Ham's F-12 Medium). All cells were maintained at
37 oC in a 5% CO2 humidified atmosphere. Briefly, 20000 cells were plated on sterile 96-well plates (100
µL of medium per well) and left undisturbed for 48 h. Then, cells were treated with different
concentrations of compounds and incubated for 16 and 48 hours, respectively. After the incubation, cell
viability was evaluated and quantified.
The values of percentage of cell viability presented correspond to the mean of 3 independent experiments
with error bars corresponding to the standard deviation of the mean. The IC50s were estimated using
GraphPad Prism v8.3.0.

146
APPENDIX
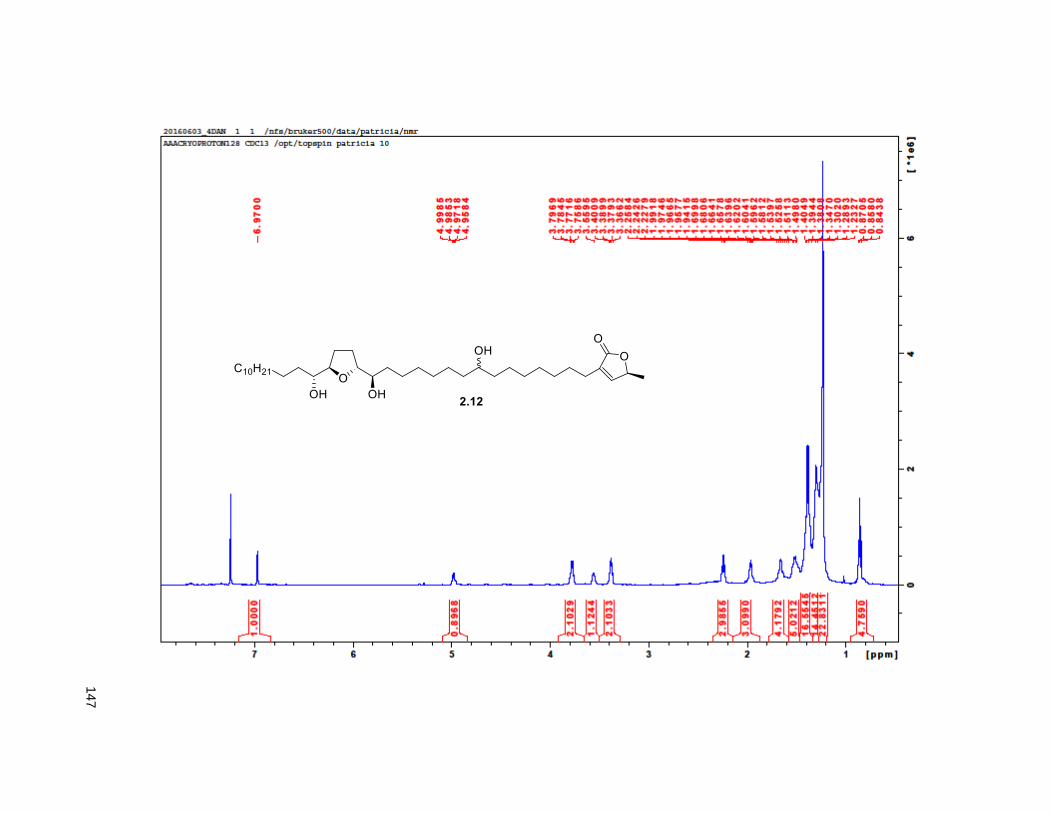
147
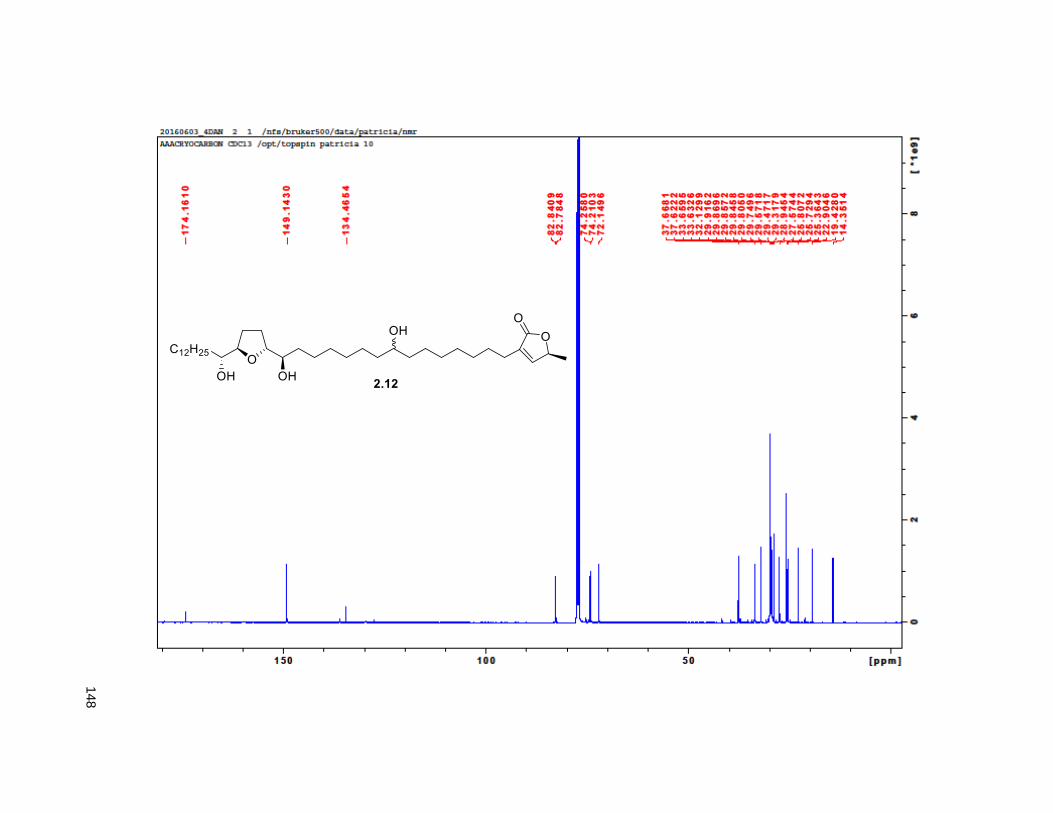
148
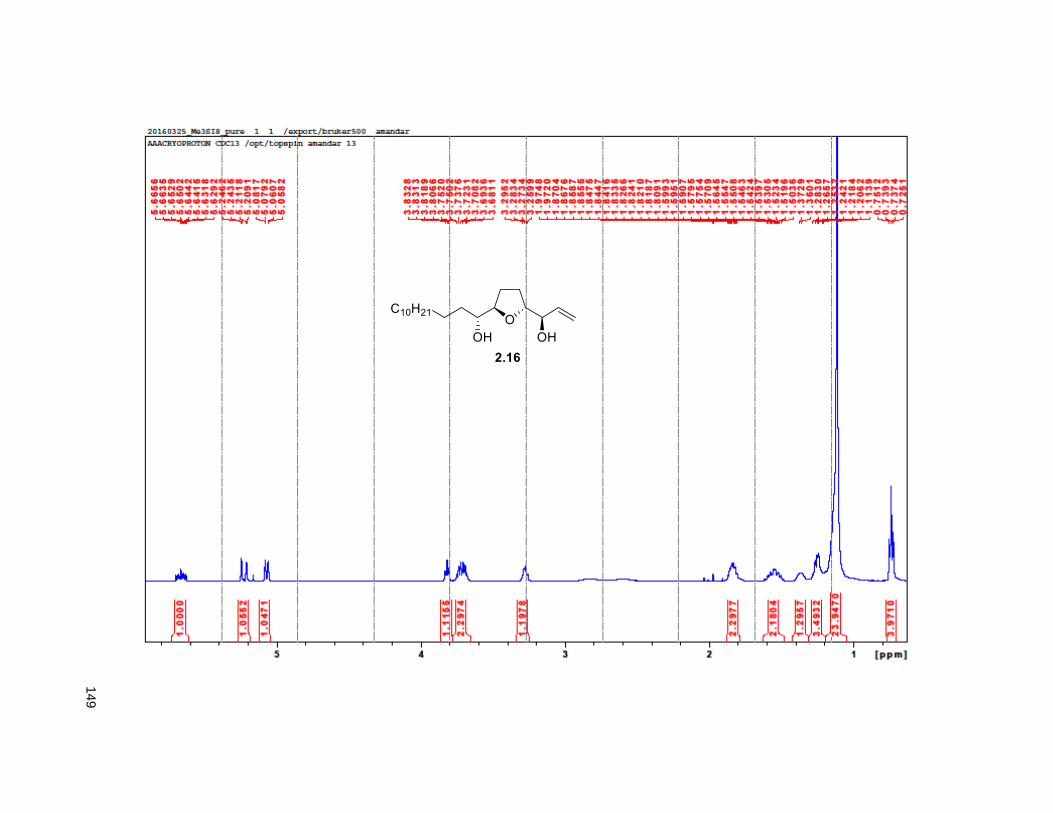
149
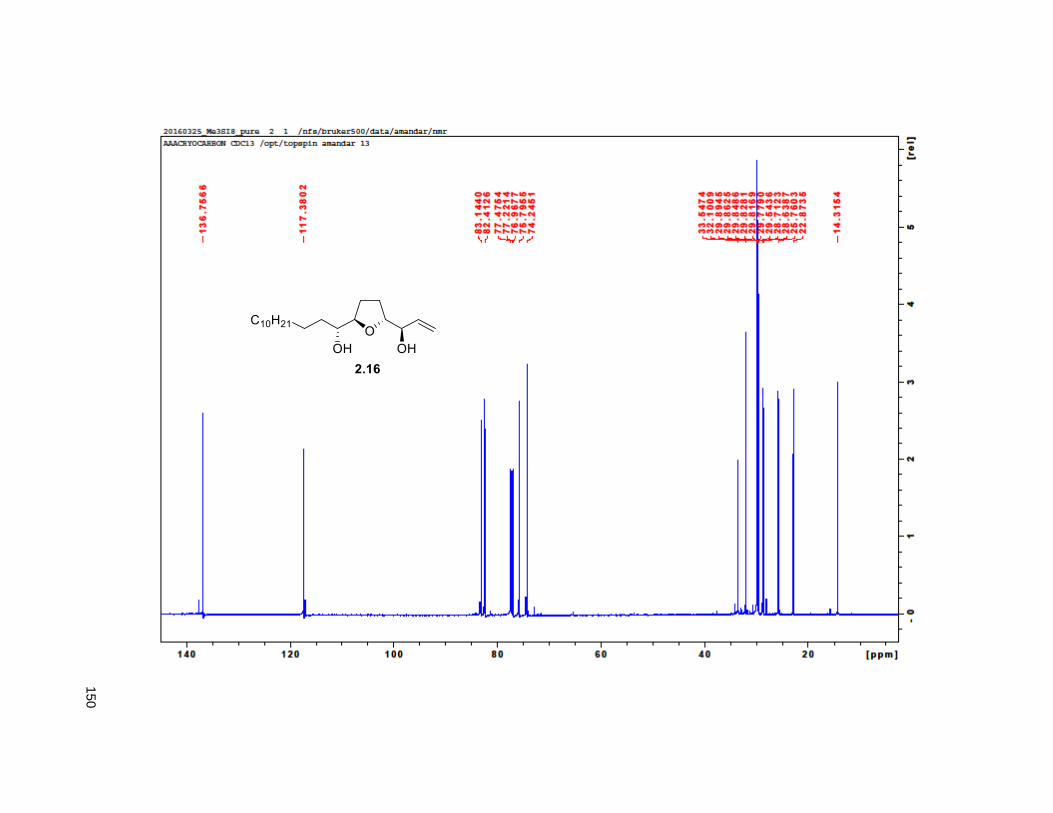
150
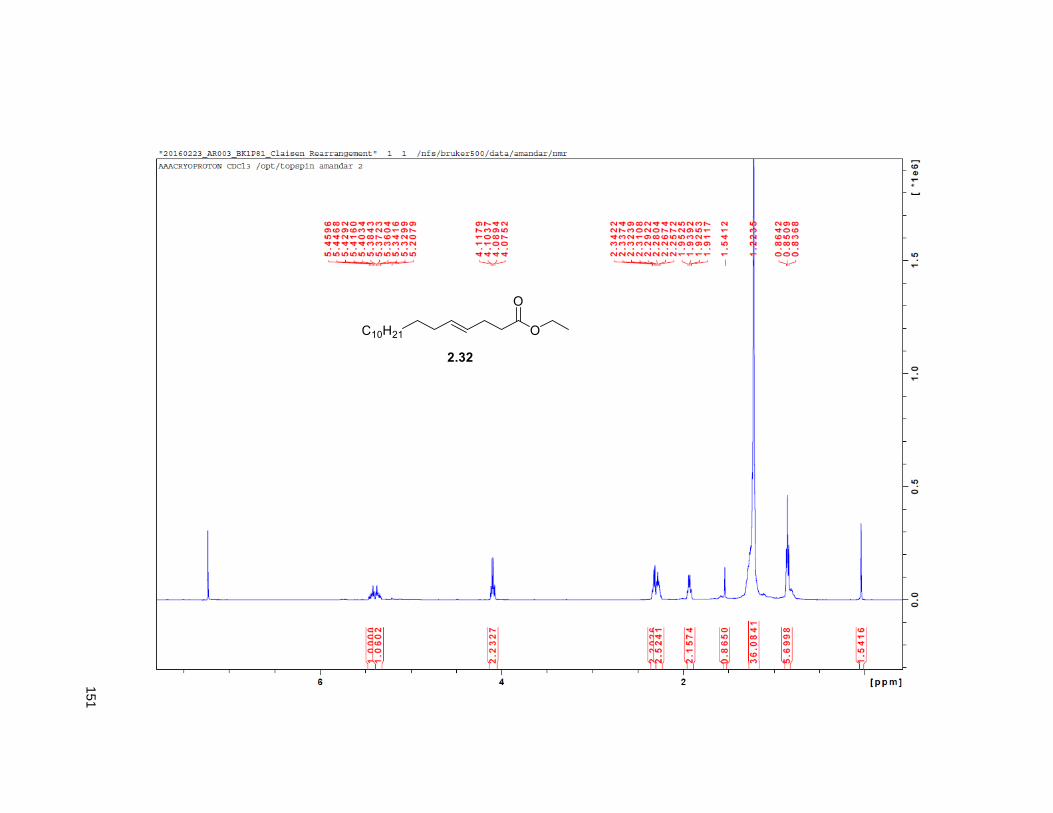
151

152

153
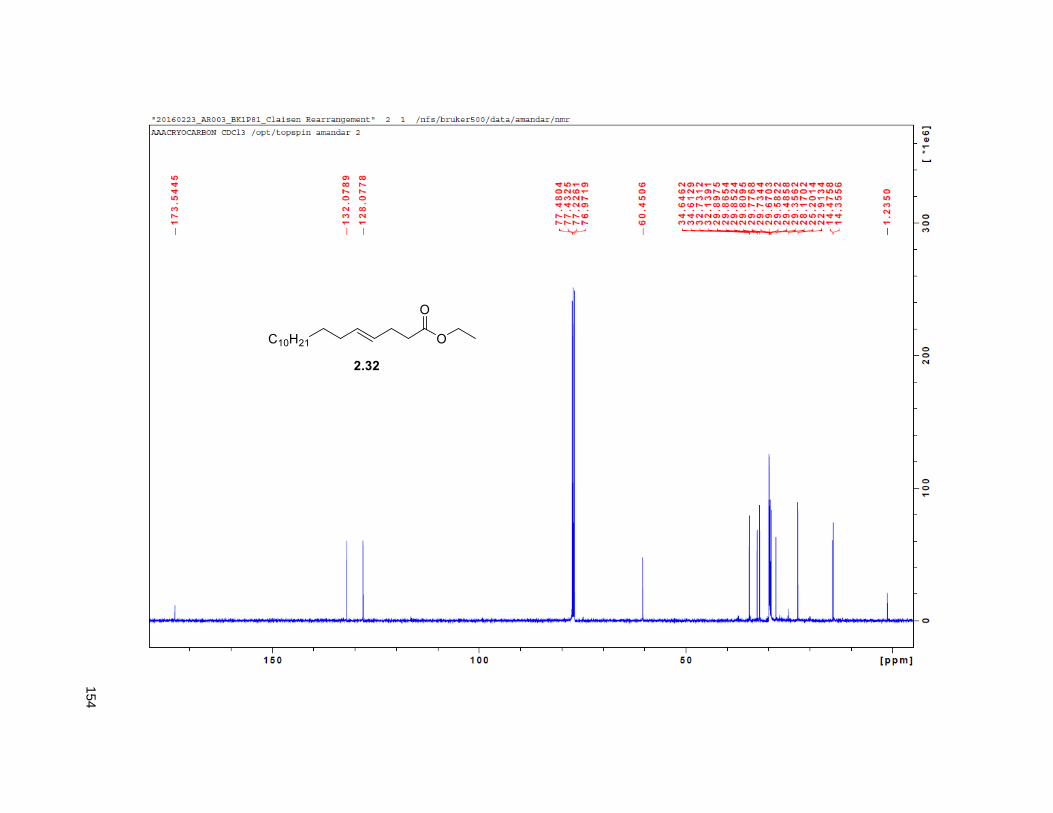
154
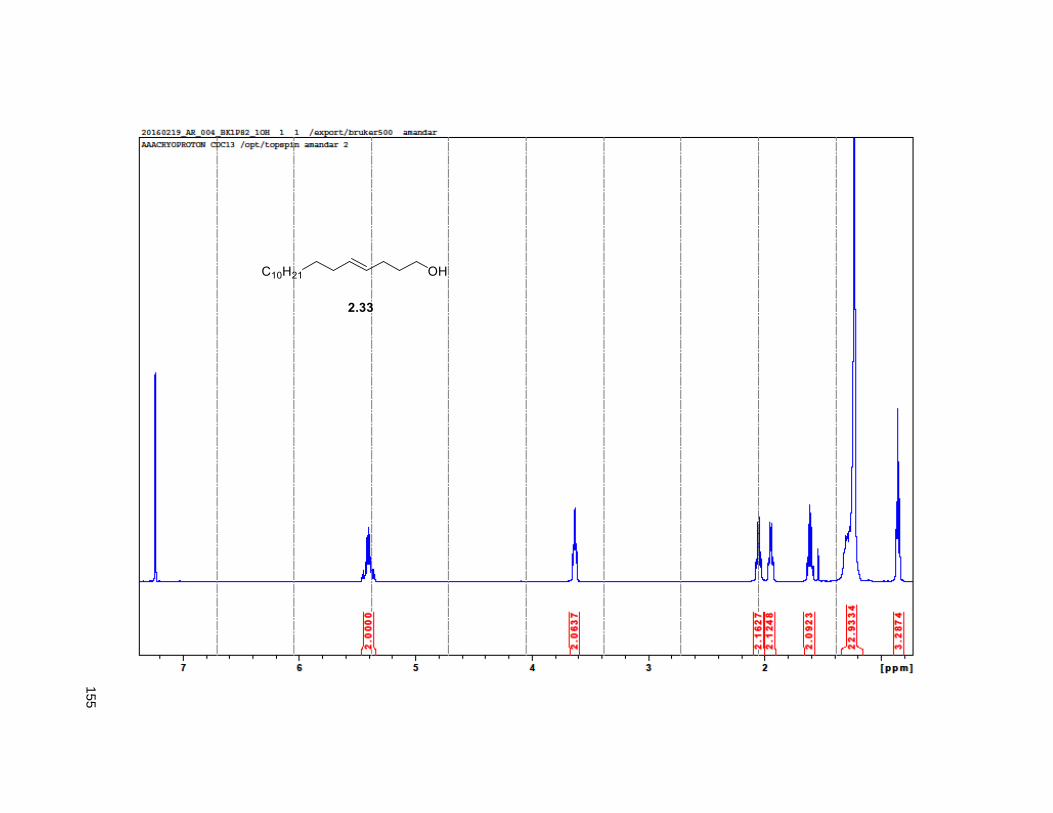
155

156
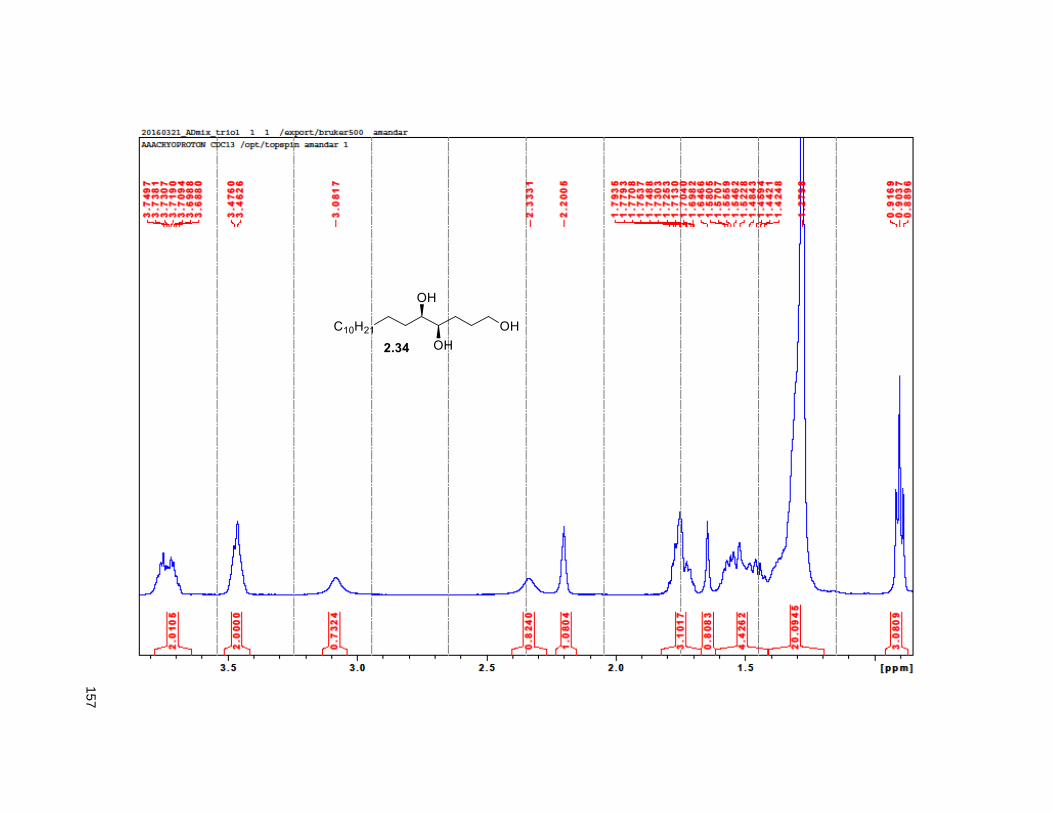
157
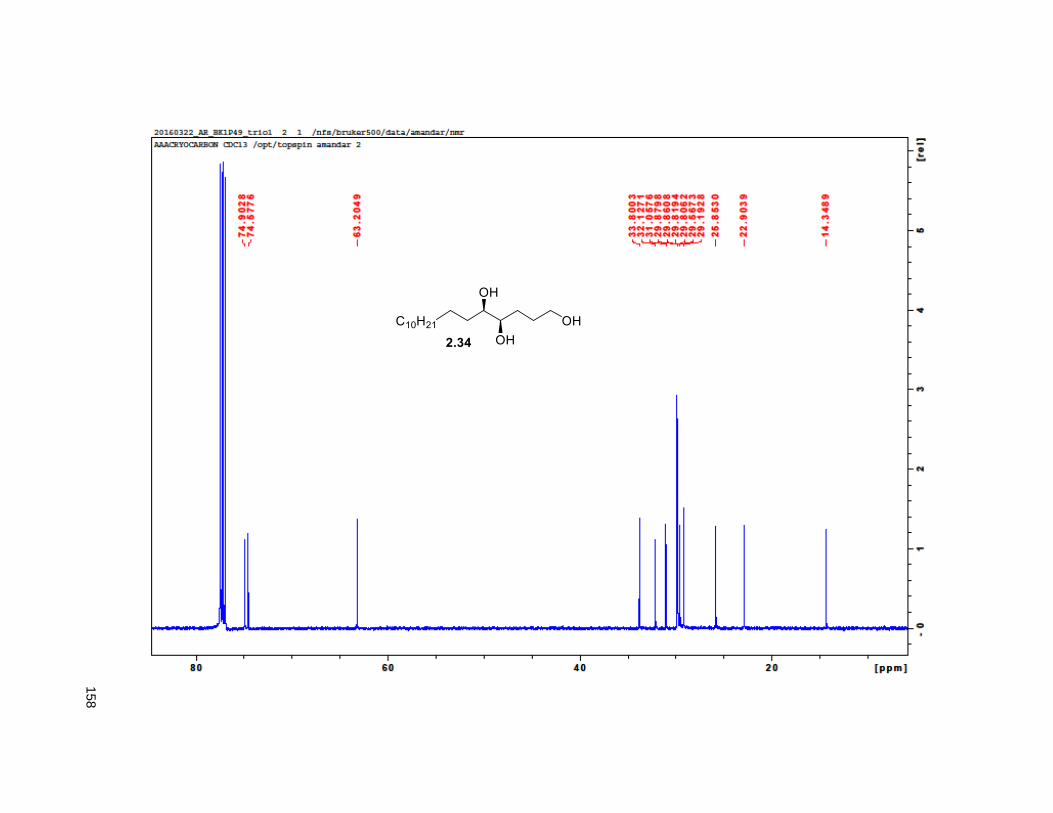
158

159
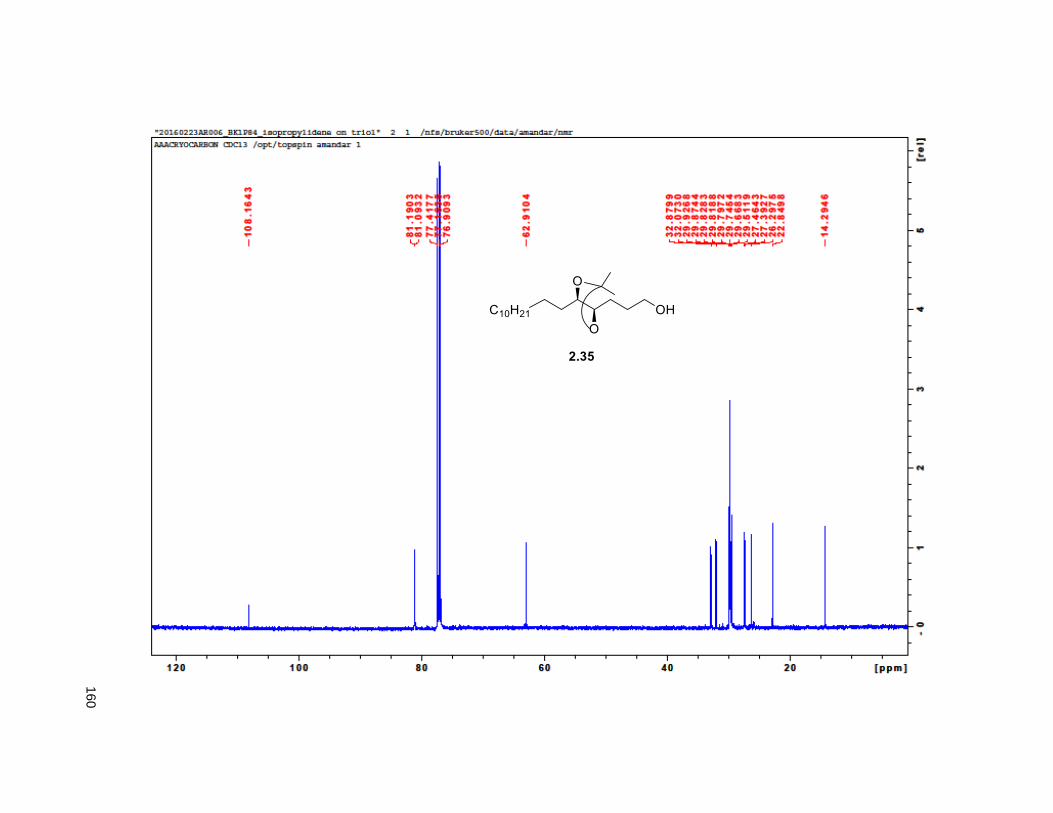
160

161
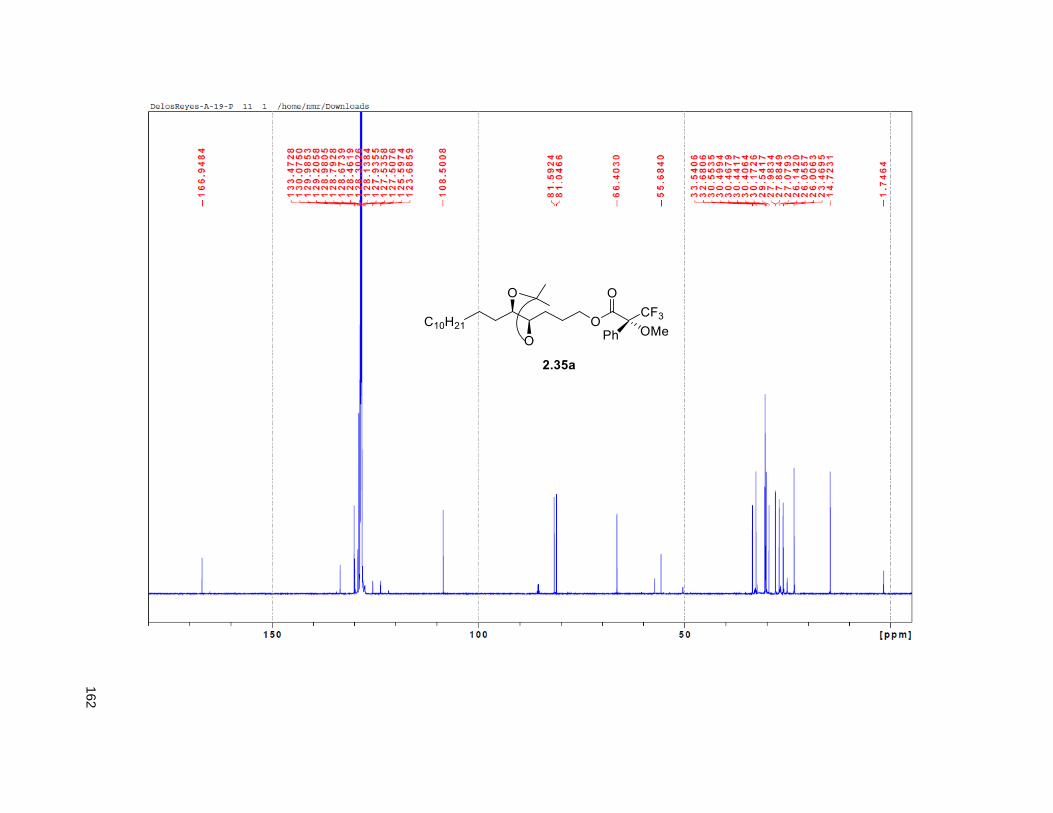
162
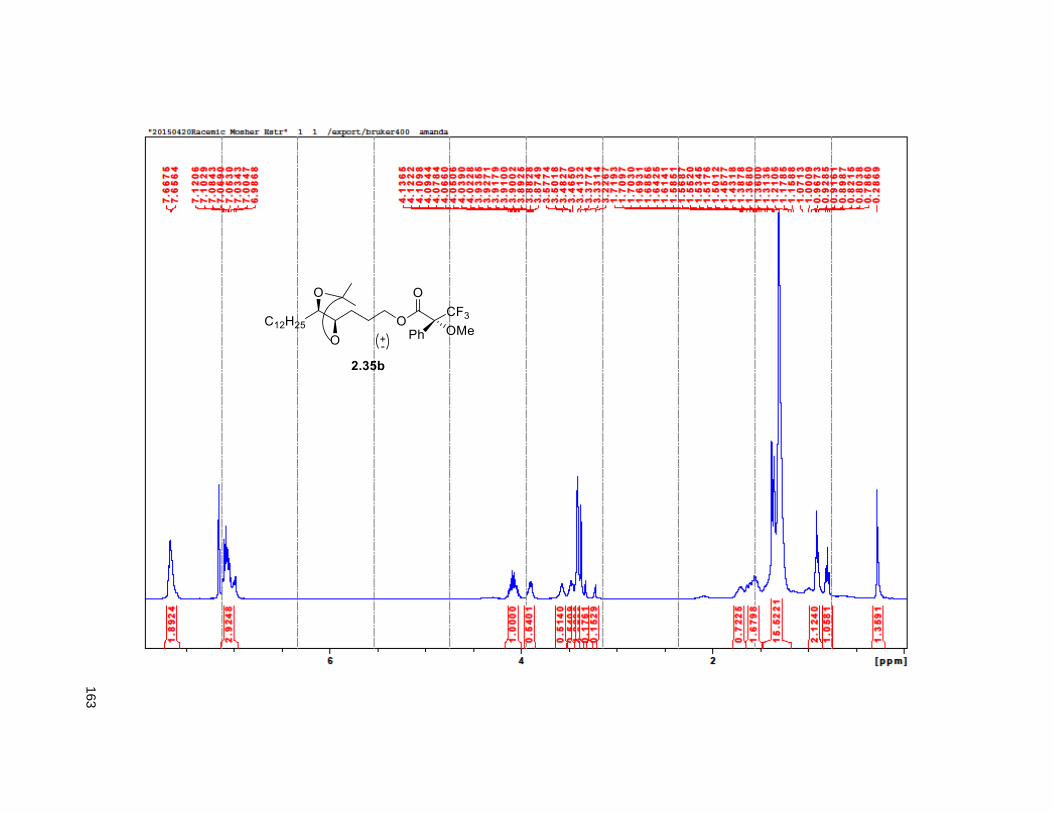
163
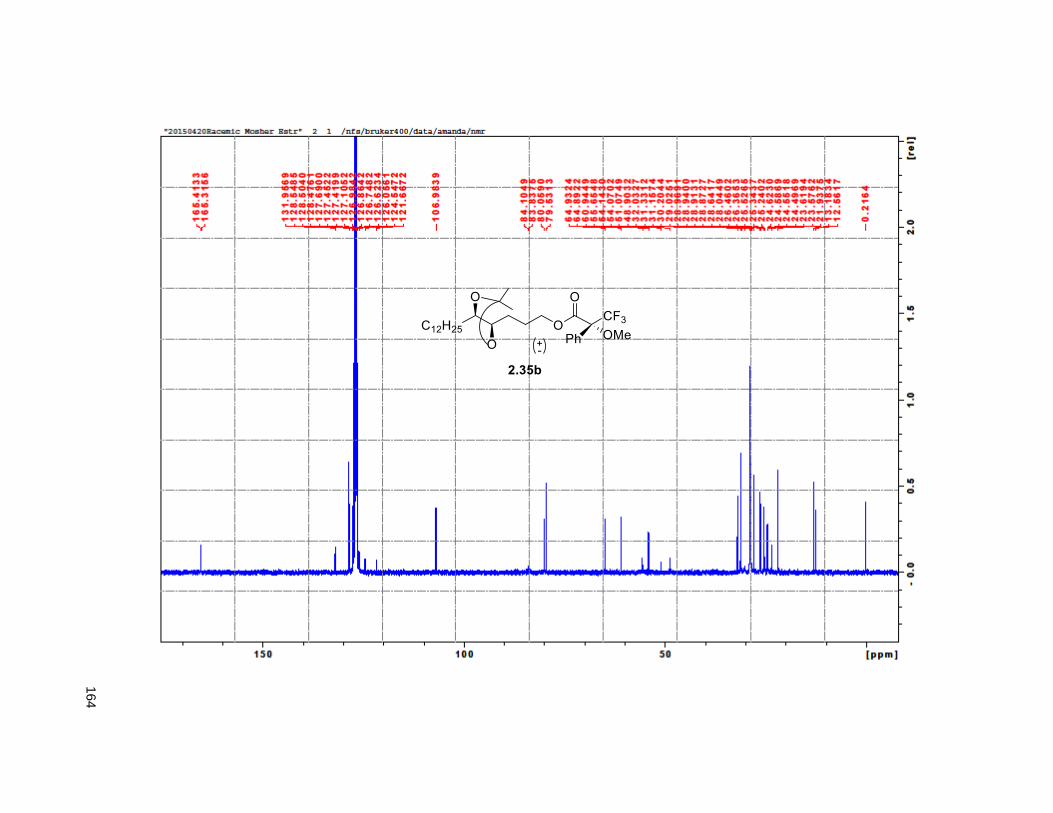
164
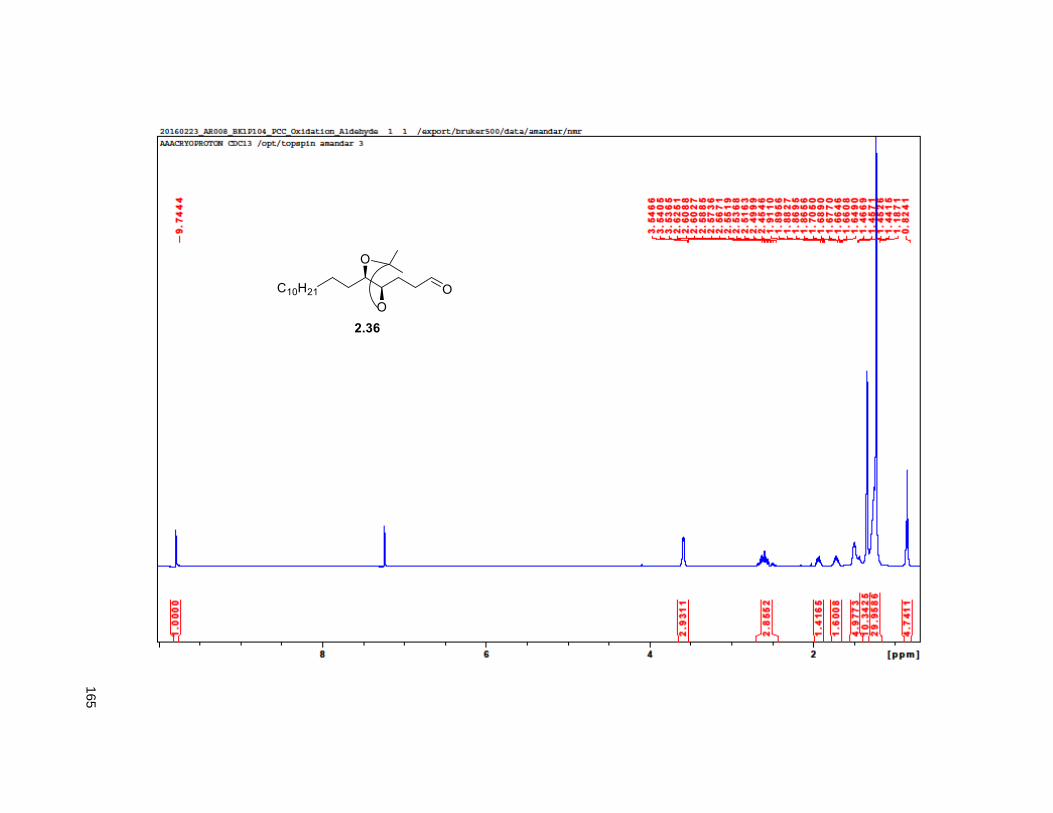
165
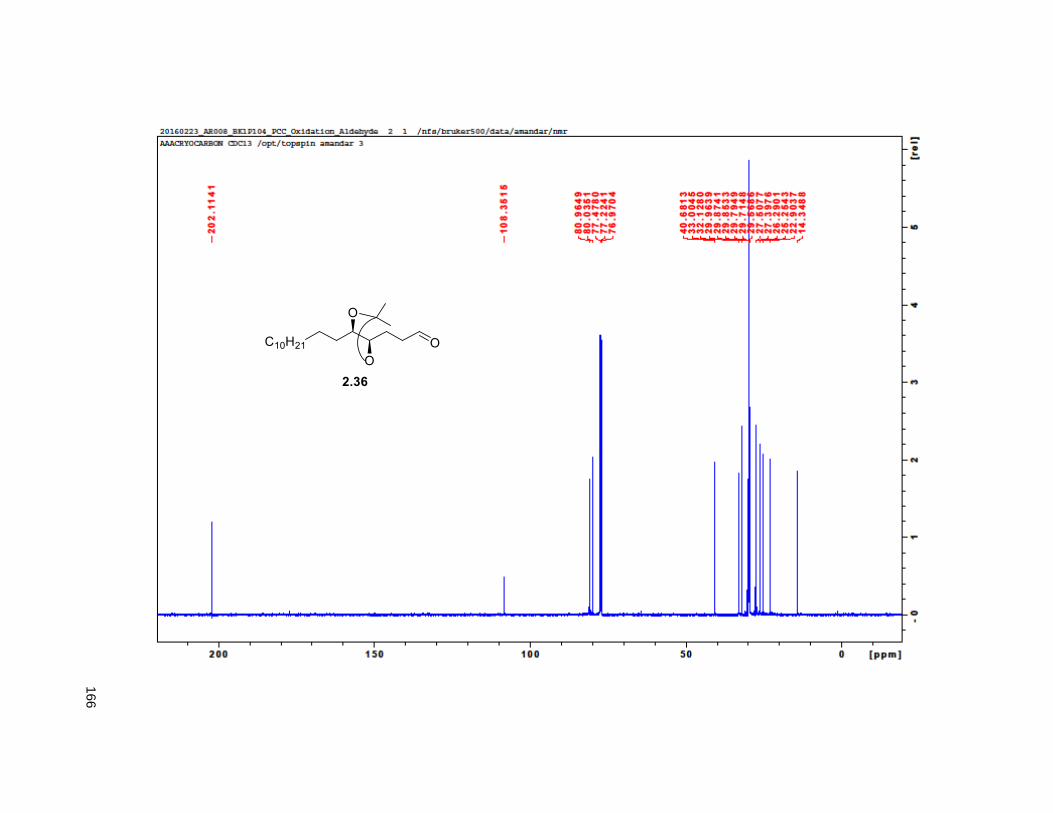
166
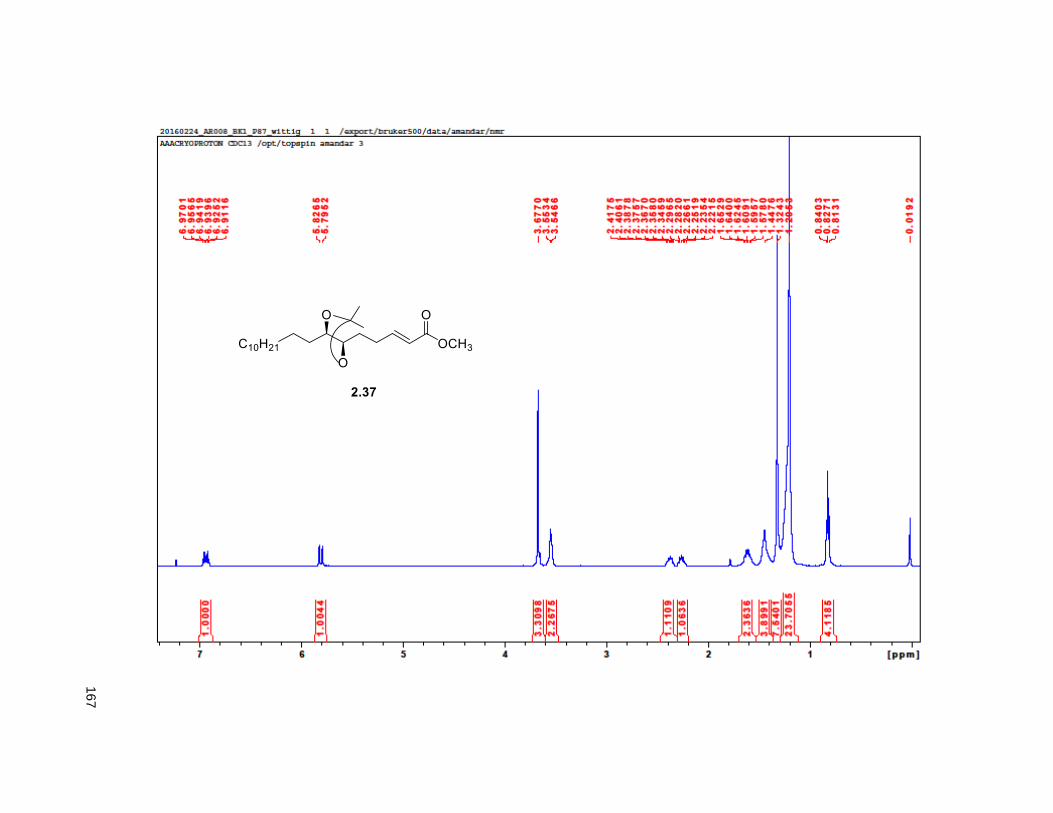
167
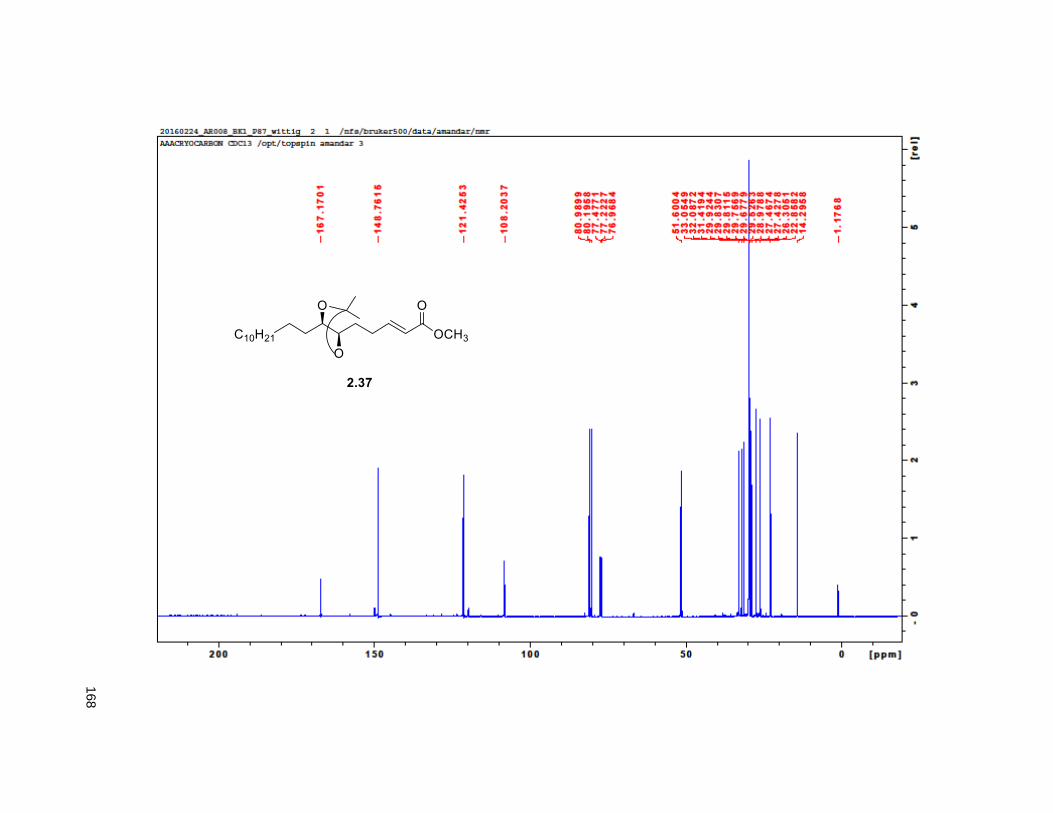
168
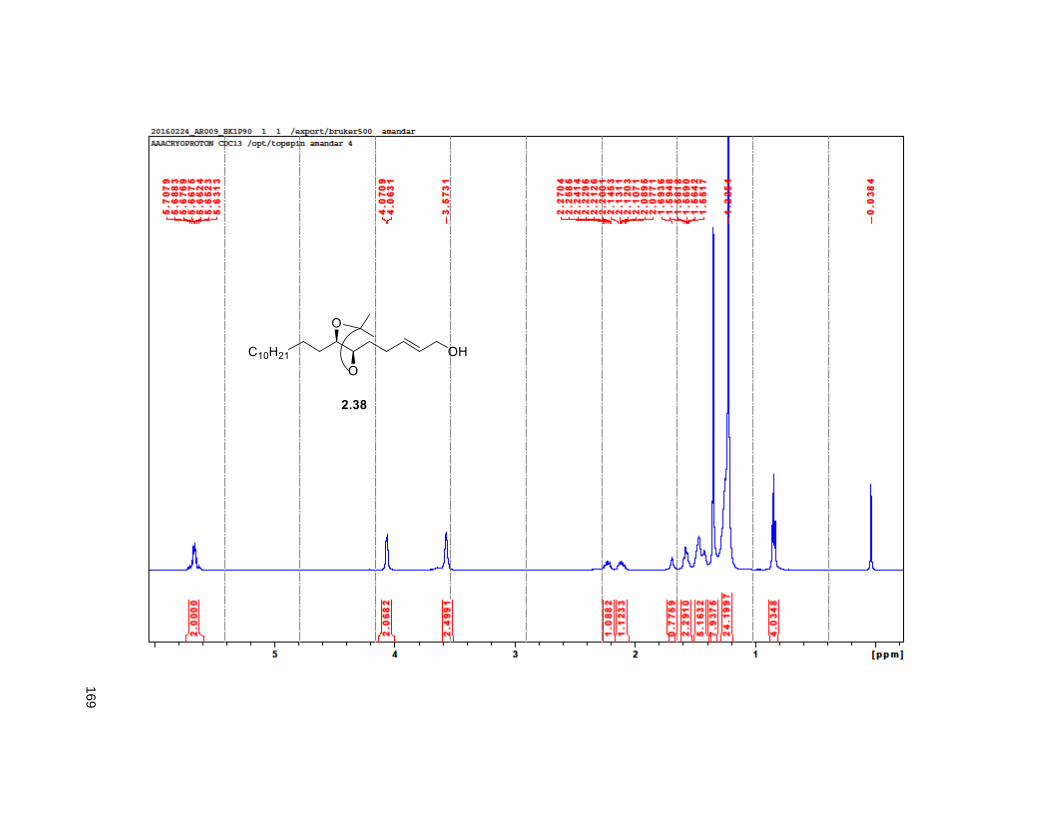
169
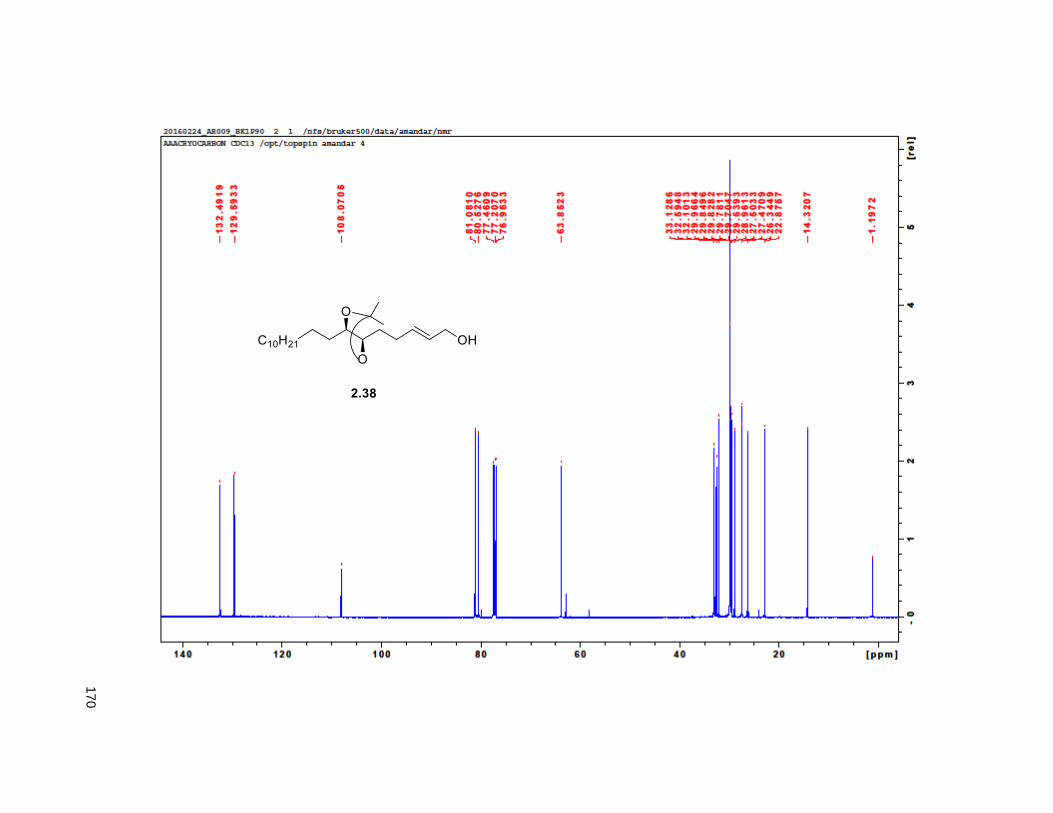
170

171

172
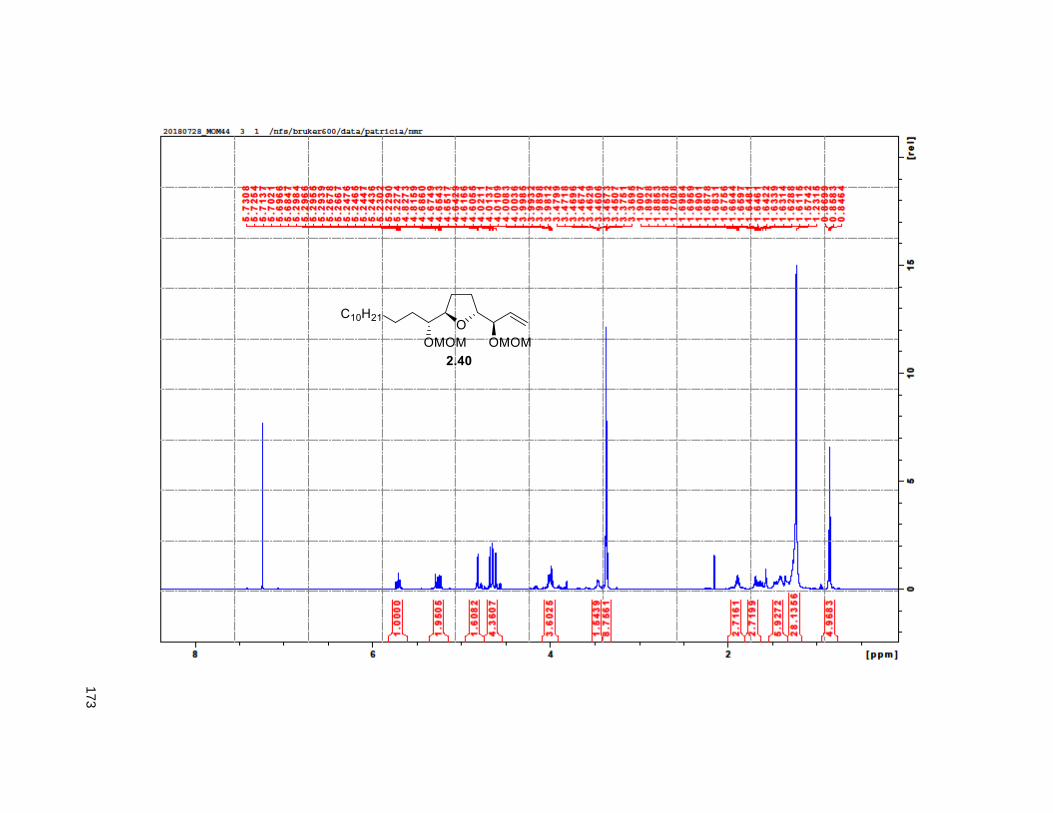
173

174
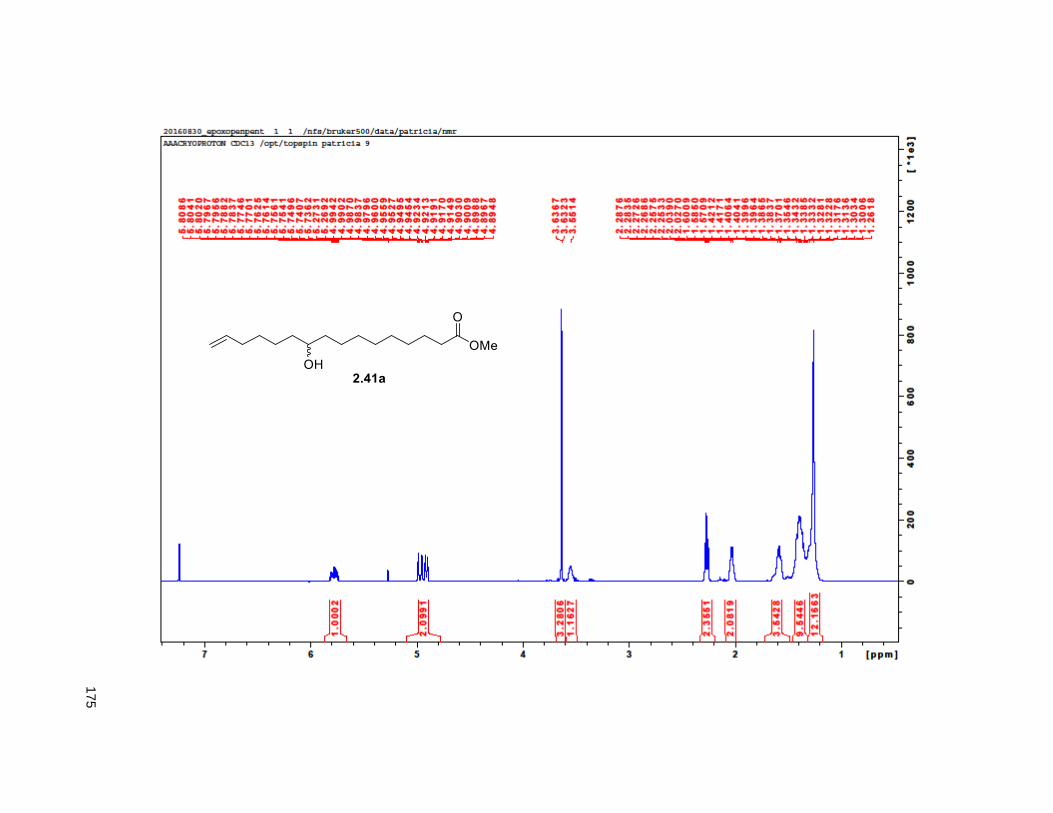
175
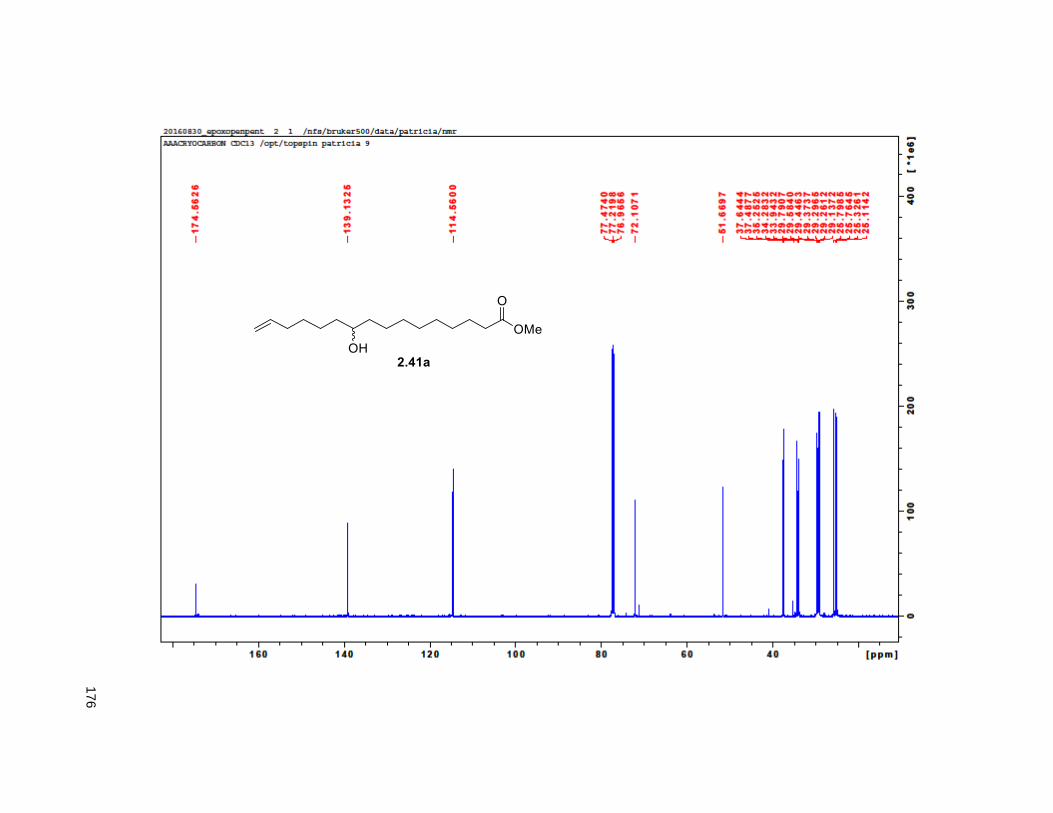
176

177
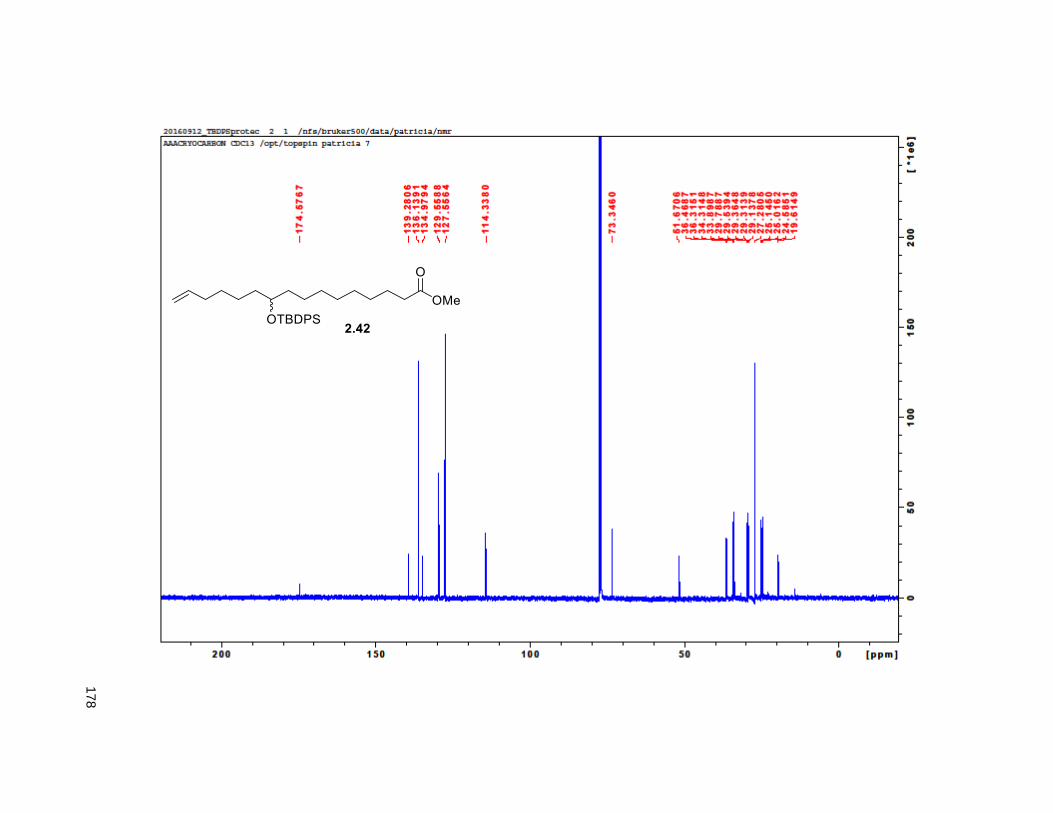
178

179
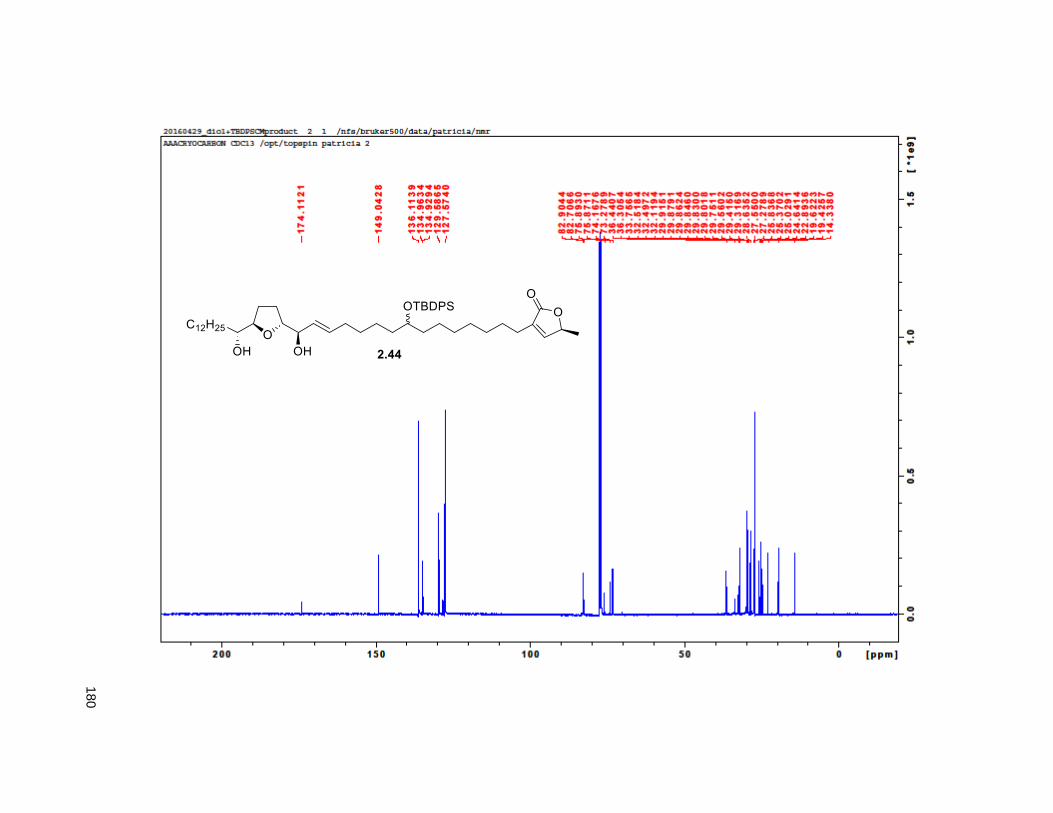
180
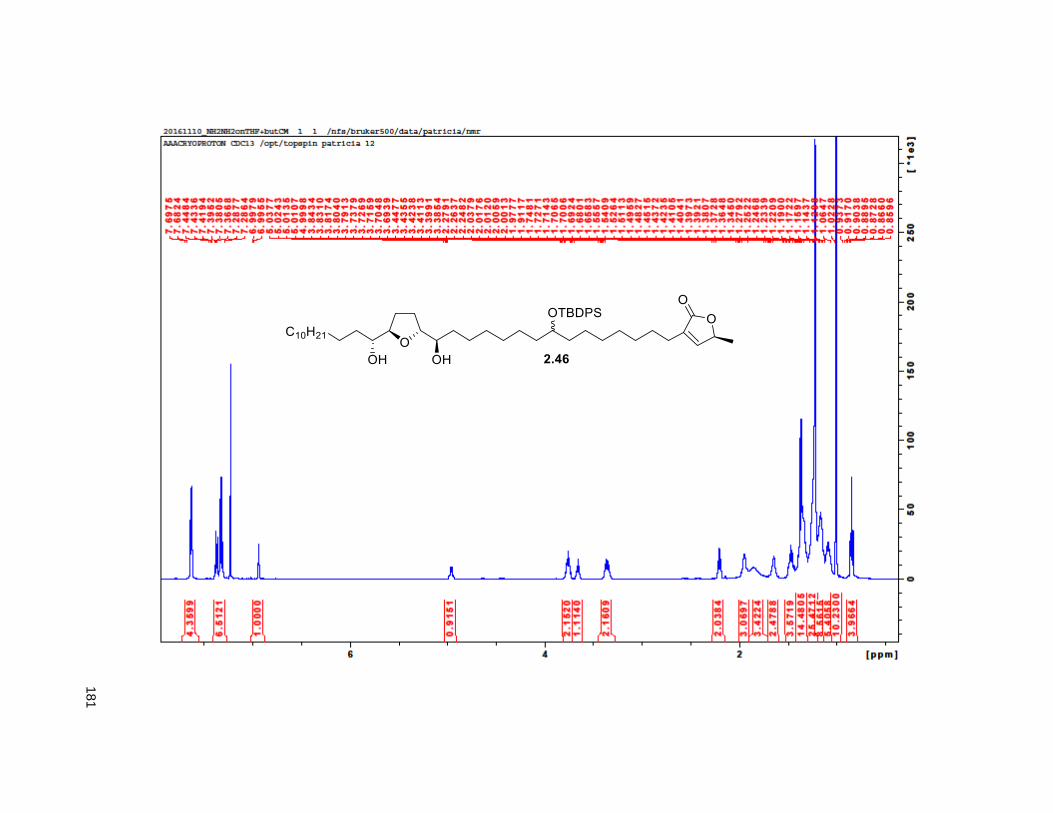
181
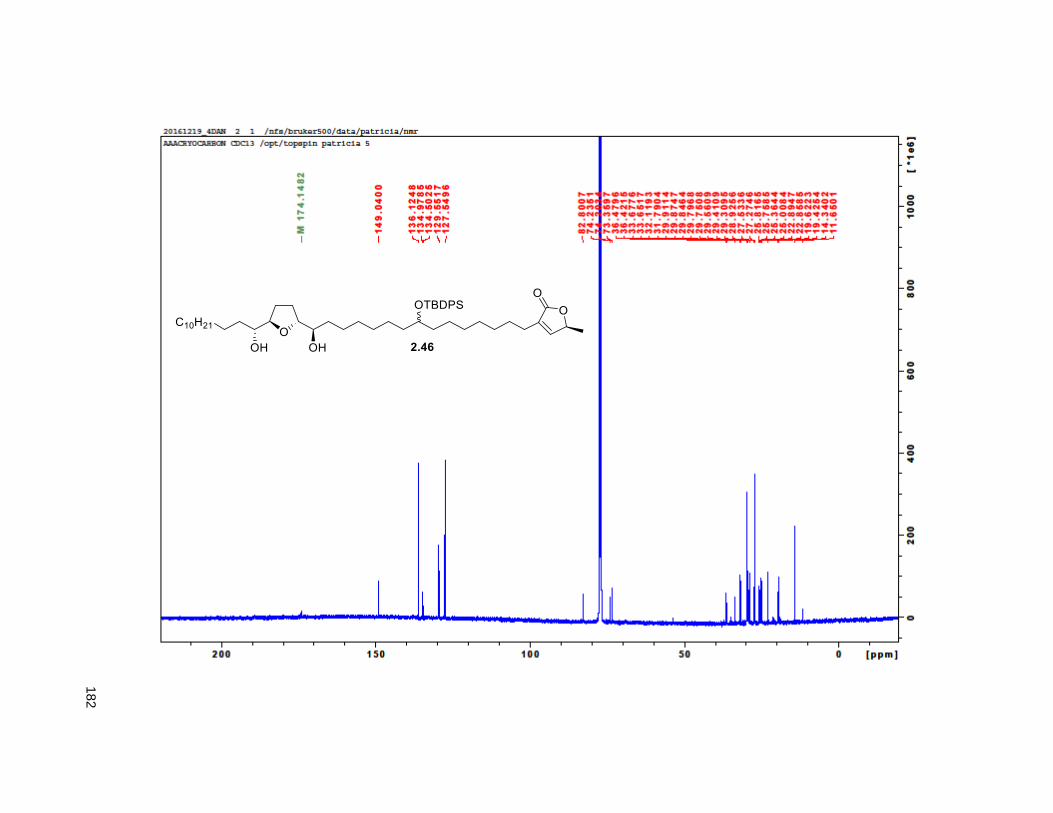
182

183
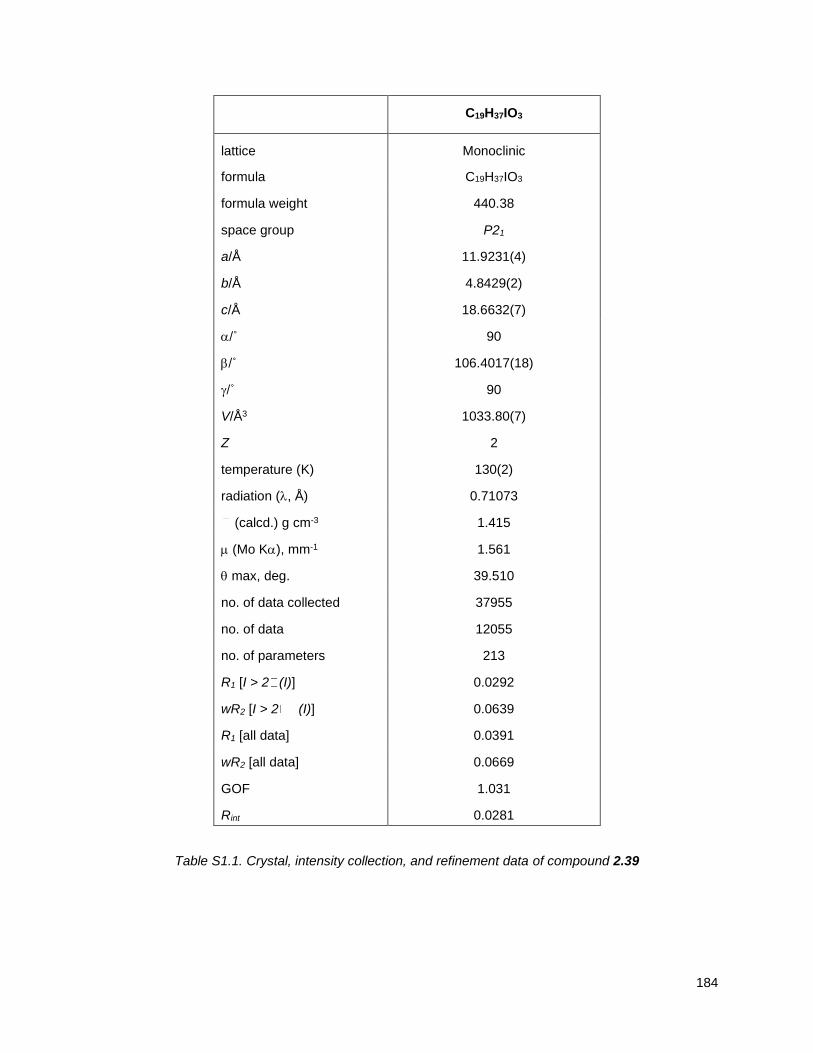
184
C19H37IO3
lattice Monoclinic
formula C19H37IO3
formula weight 440.38
space group P21
a/Å 11.9231(4)
b/Å 4.8429(2)
c/Å 18.6632(7)
/˚ 90
/˚ 106.4017(18)
/˚ 90
V/Å3 1033.80(7)
Z 2
temperature (K) 130(2)
radiation (, Å) 0.71073
(calcd.) g cm-3 1.415
(Mo K), mm-1 1.561
max, deg. 39.510
no. of data collected 37955
no. of data 12055
no. of parameters 213
R1 [I > 2 (I)] 0.0292
wR2 [I > 2 (I)] 0.0639
R1 [all data] 0.0391
wR2 [all data] 0.0669
GOF
1.031
Rint 0.0281
Table S1.1. Crystal, intensity collection, and refinement data of compound 2.39
185
185
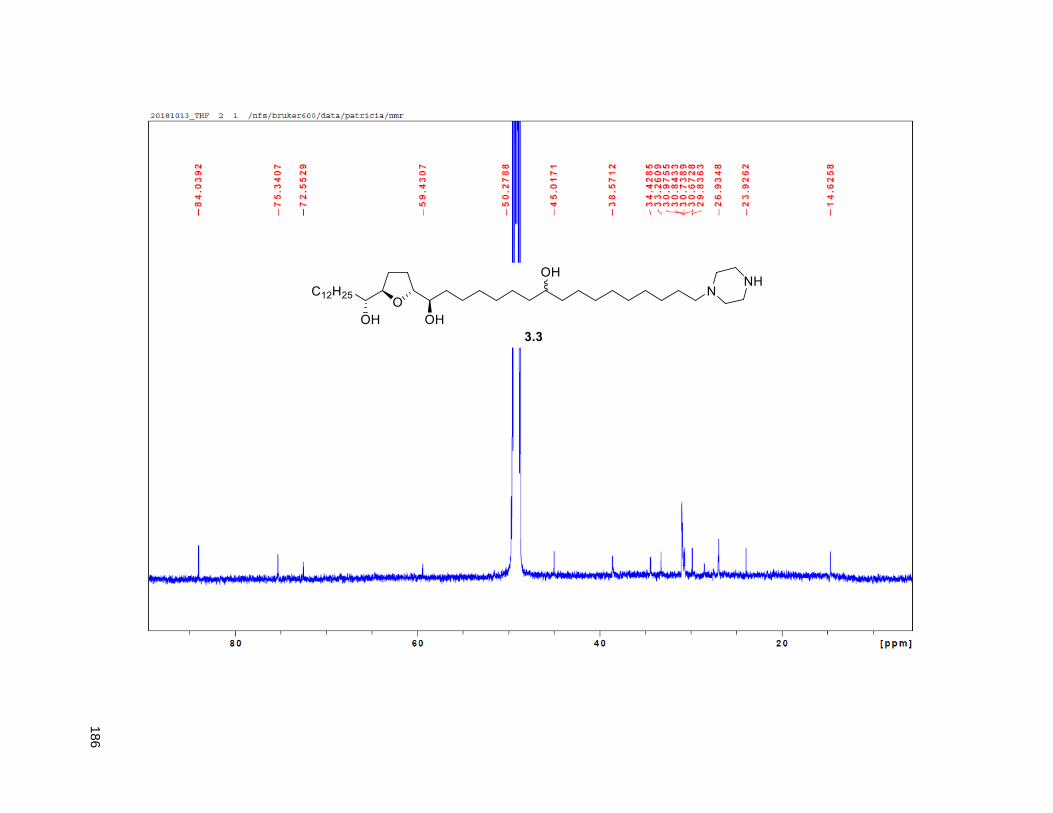
186
186
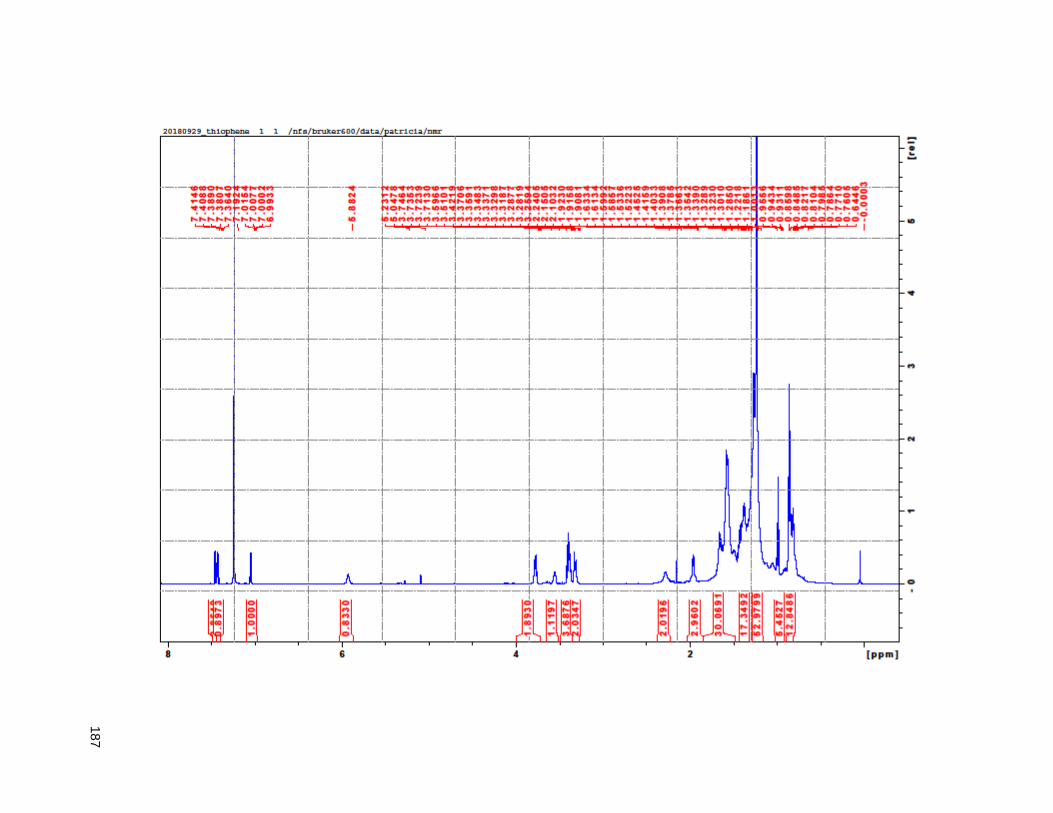
187
187
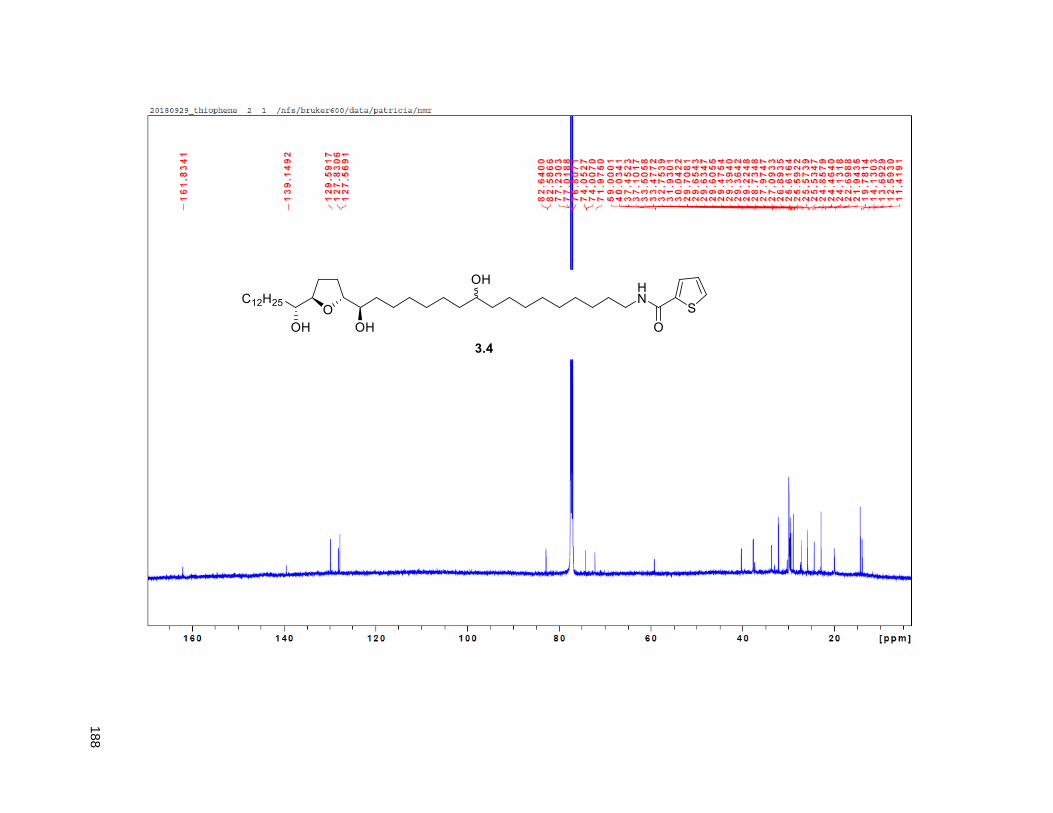
188
188
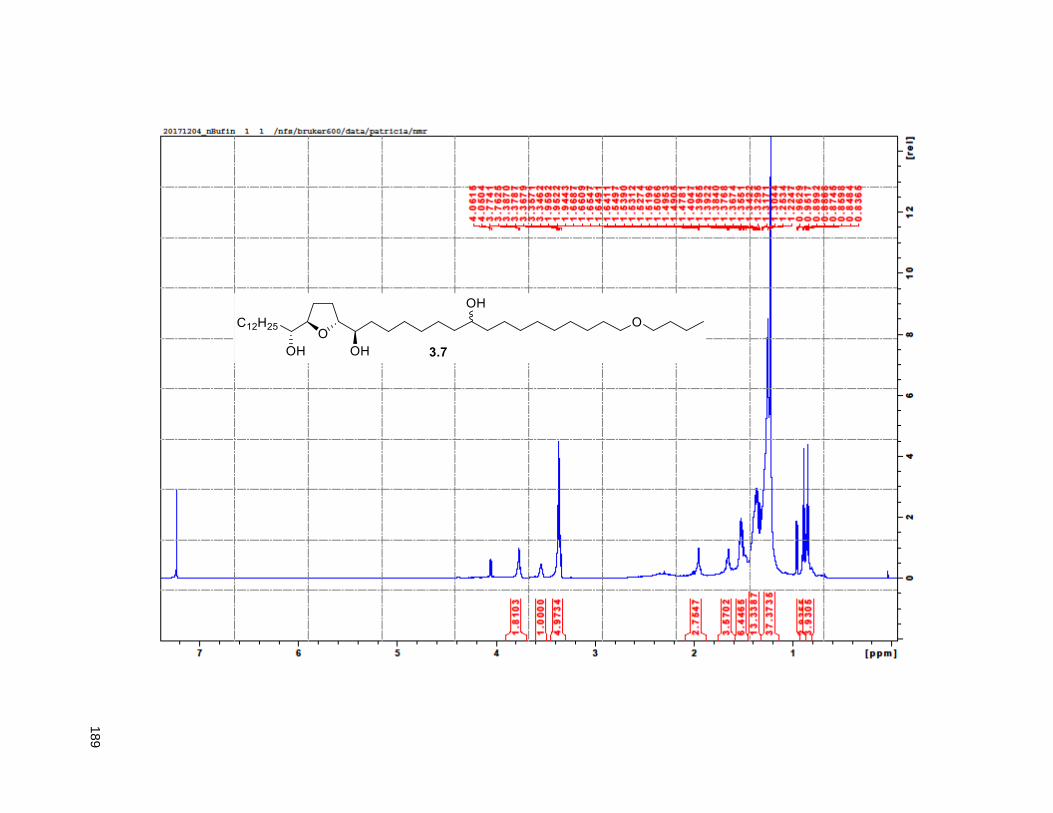
189
189
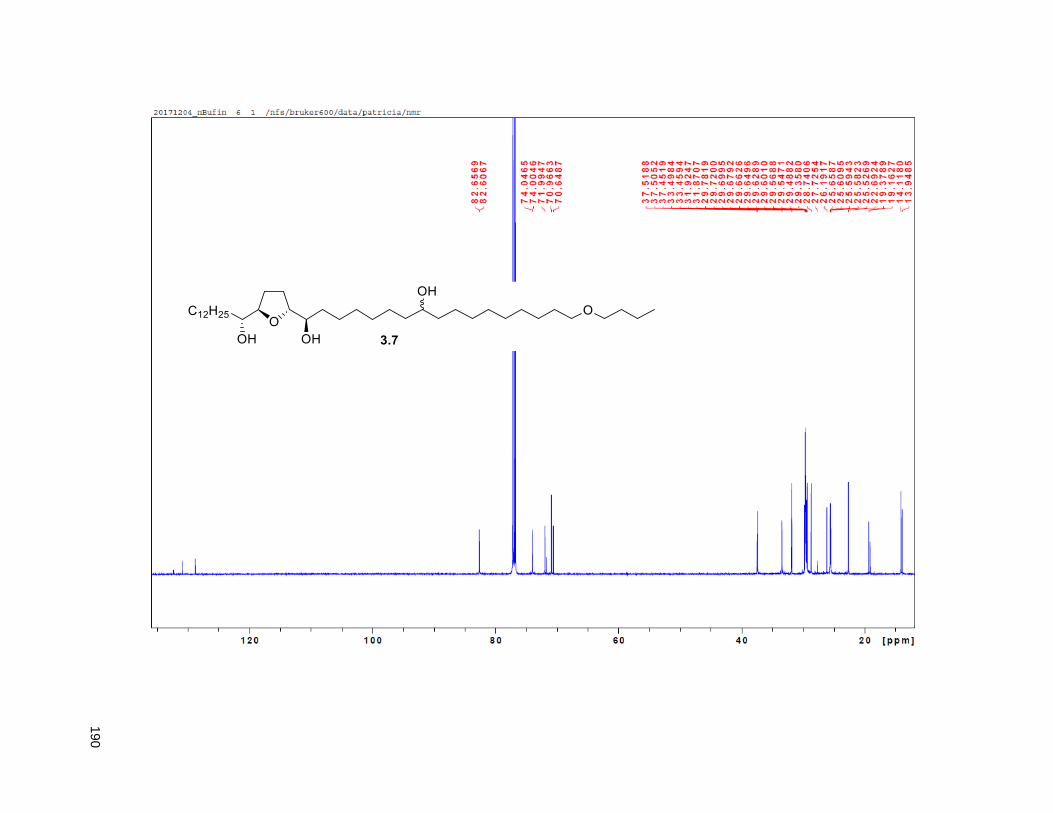
190
190

191
191
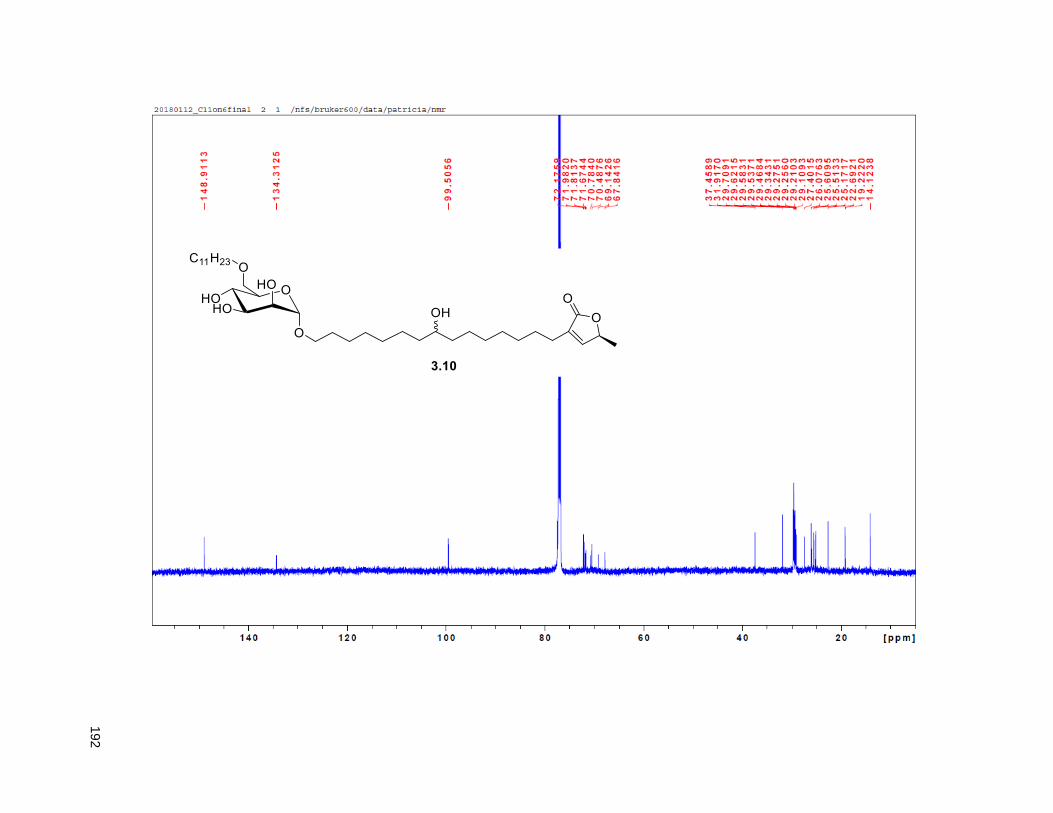
192
192
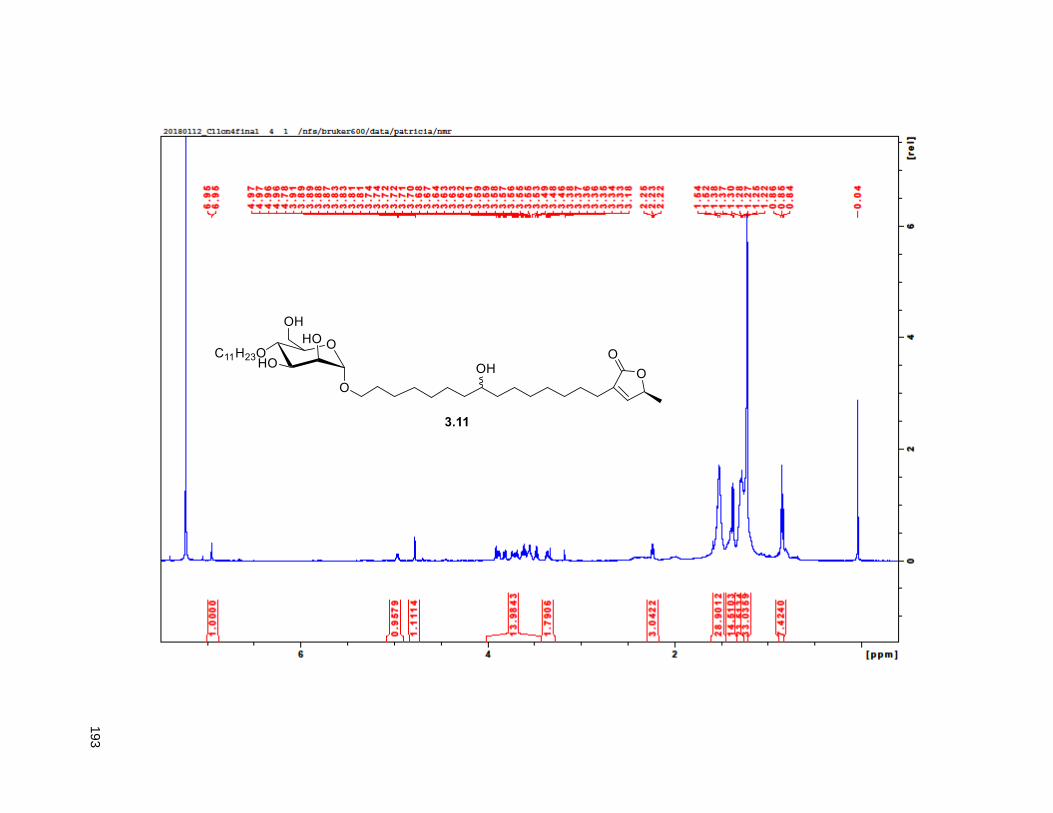
193
193
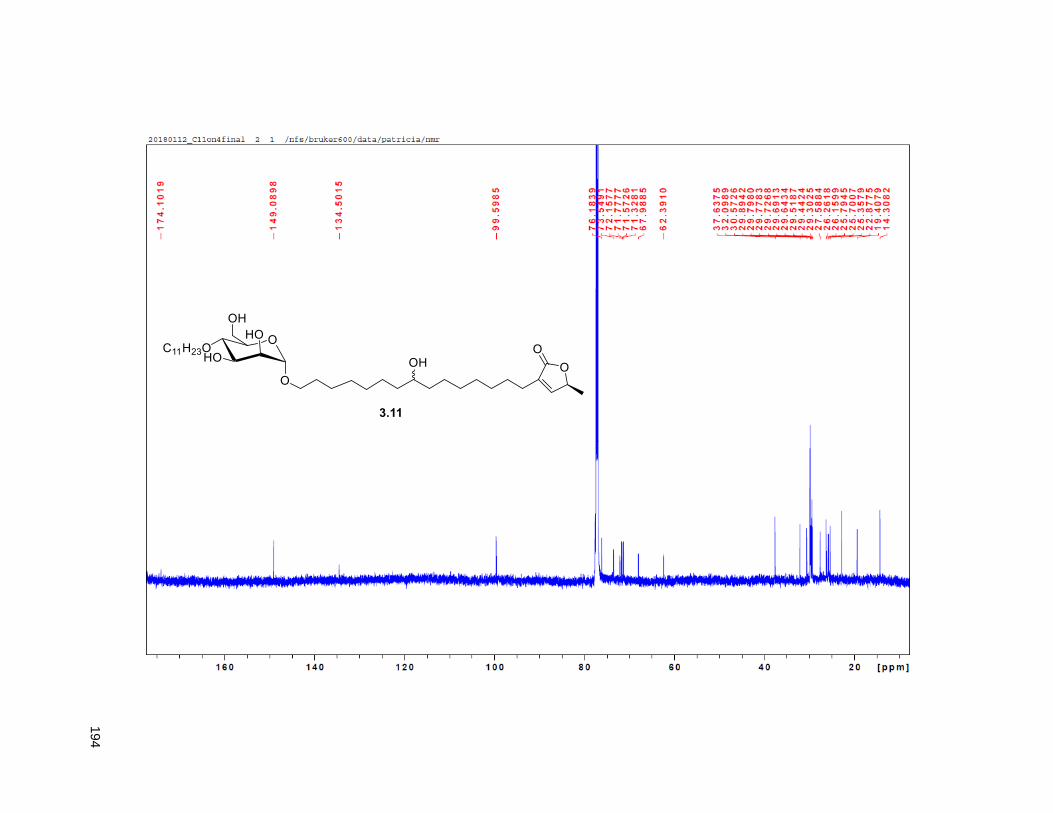
194
194
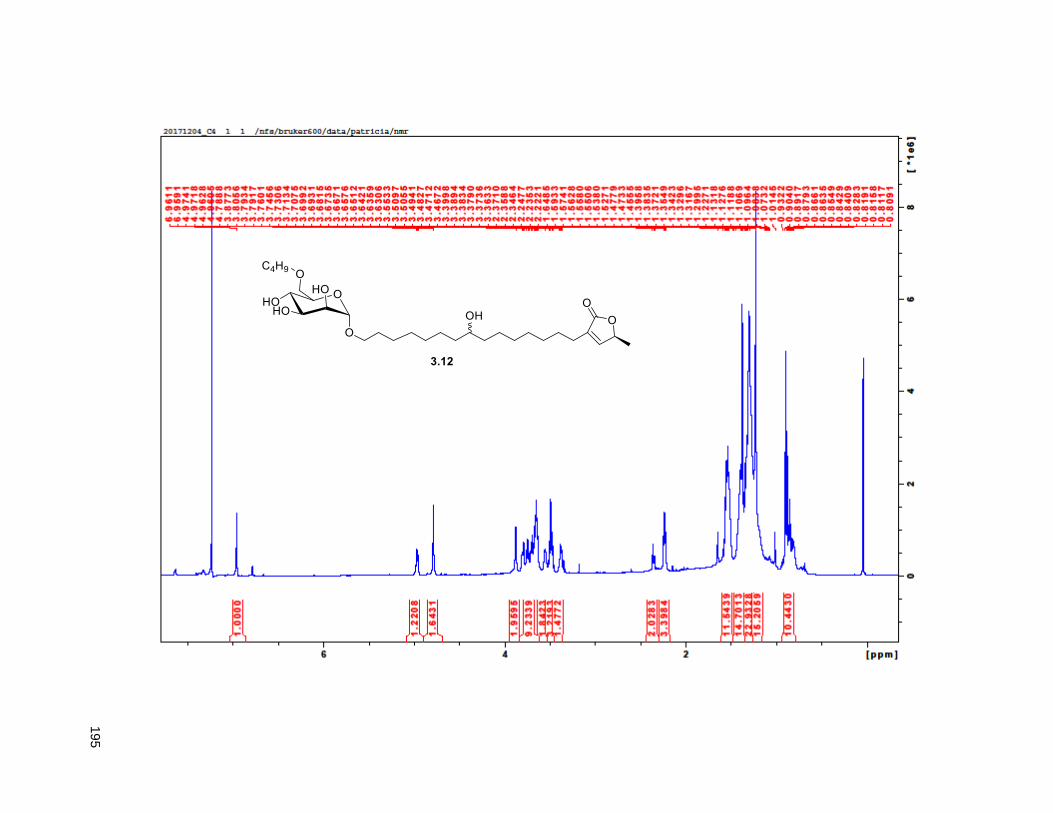
195
195
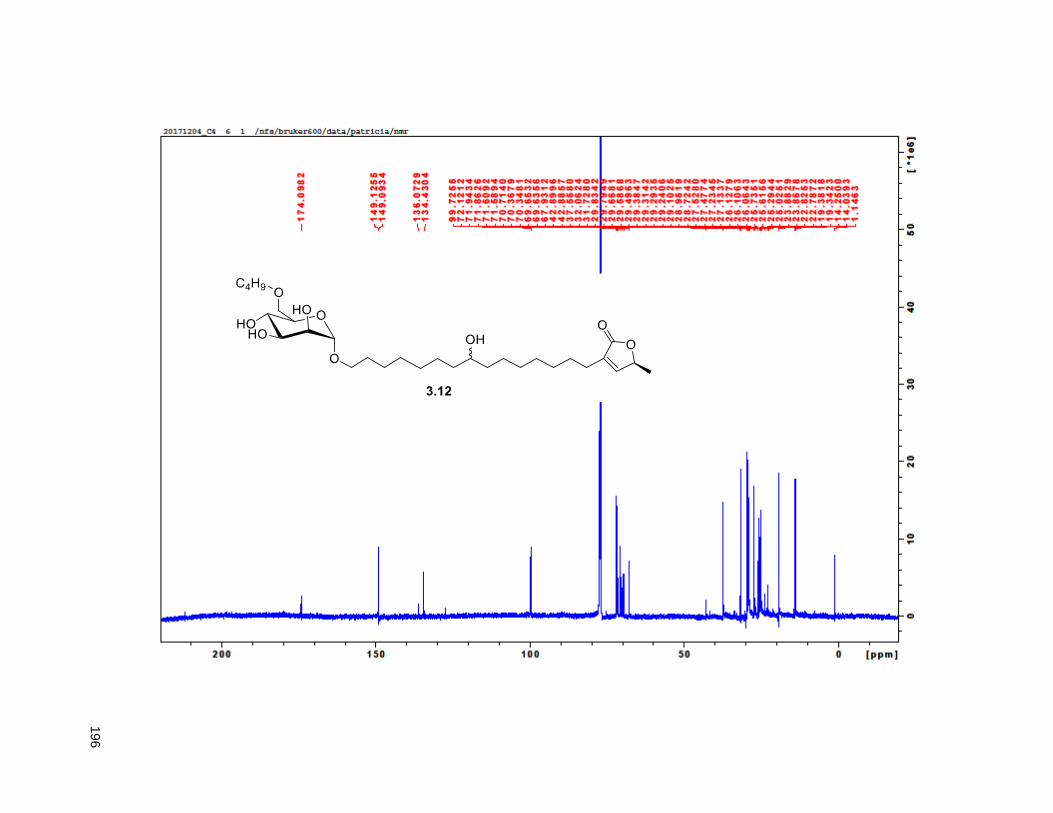
196
196
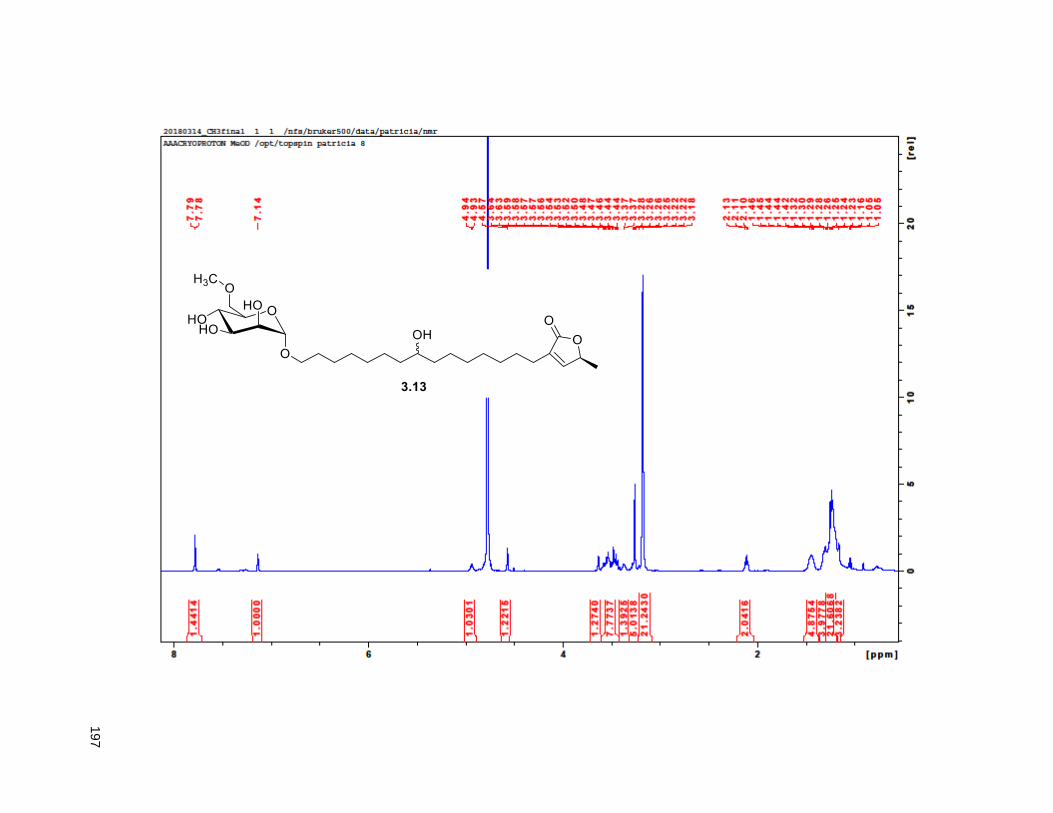
197
197
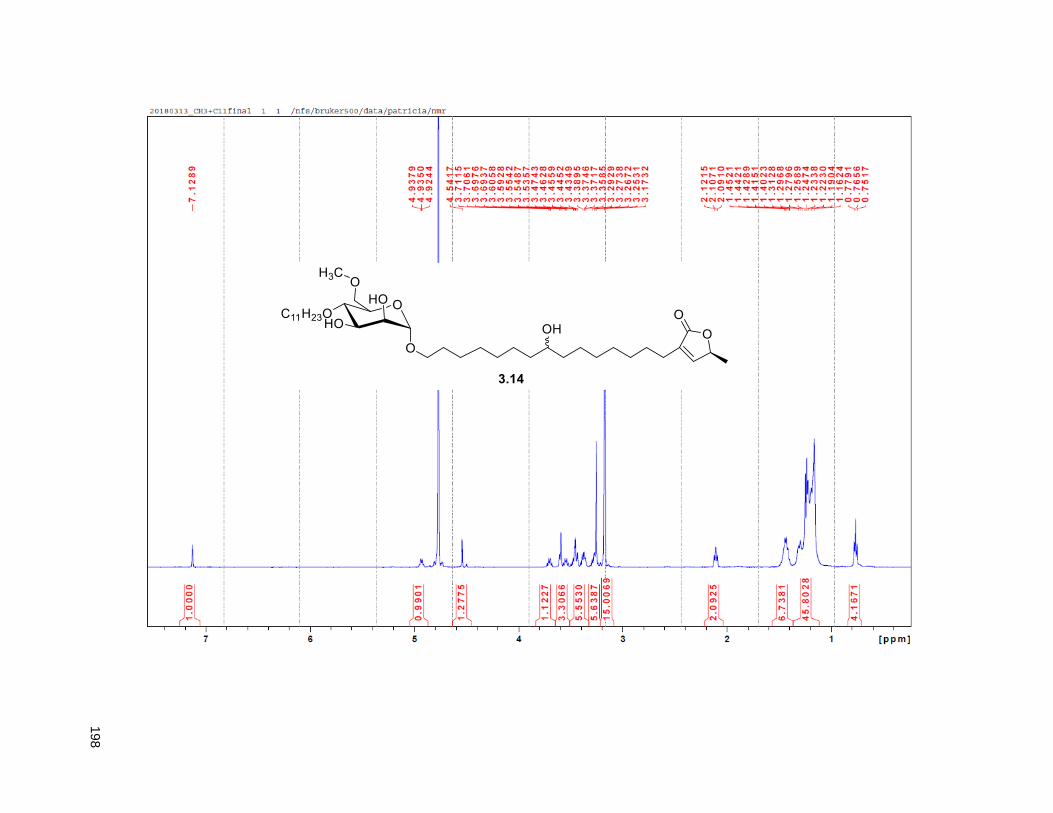
198
198
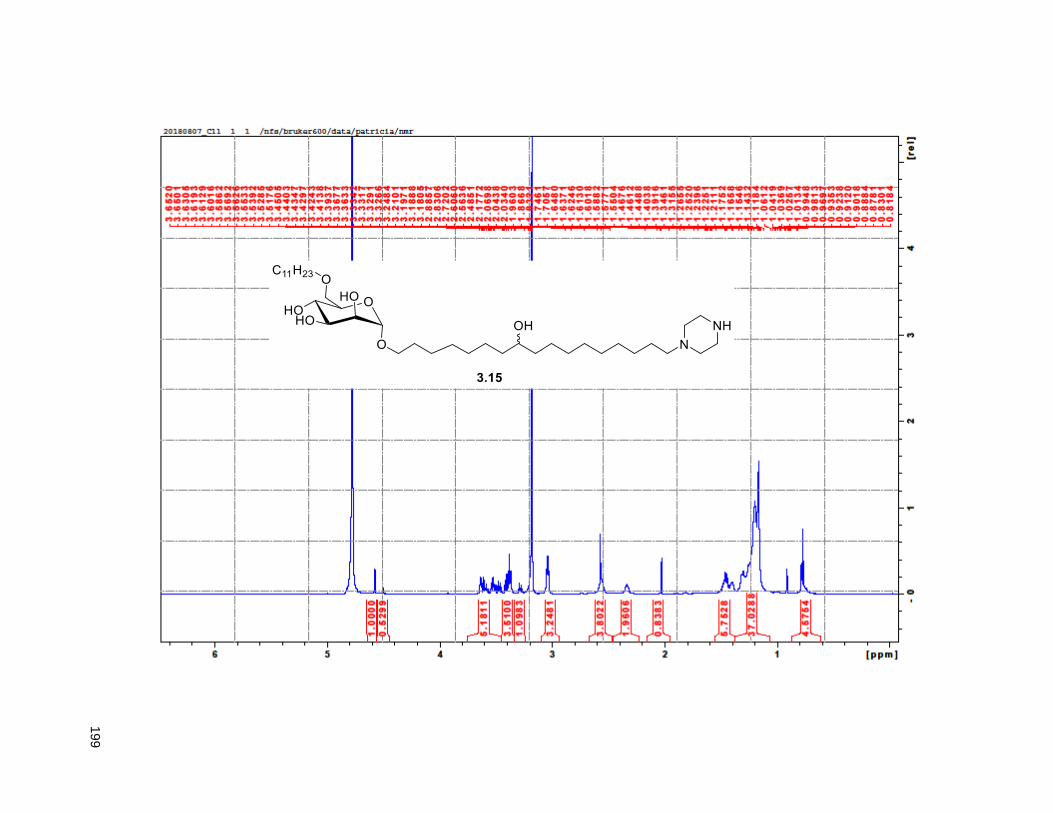
199
199
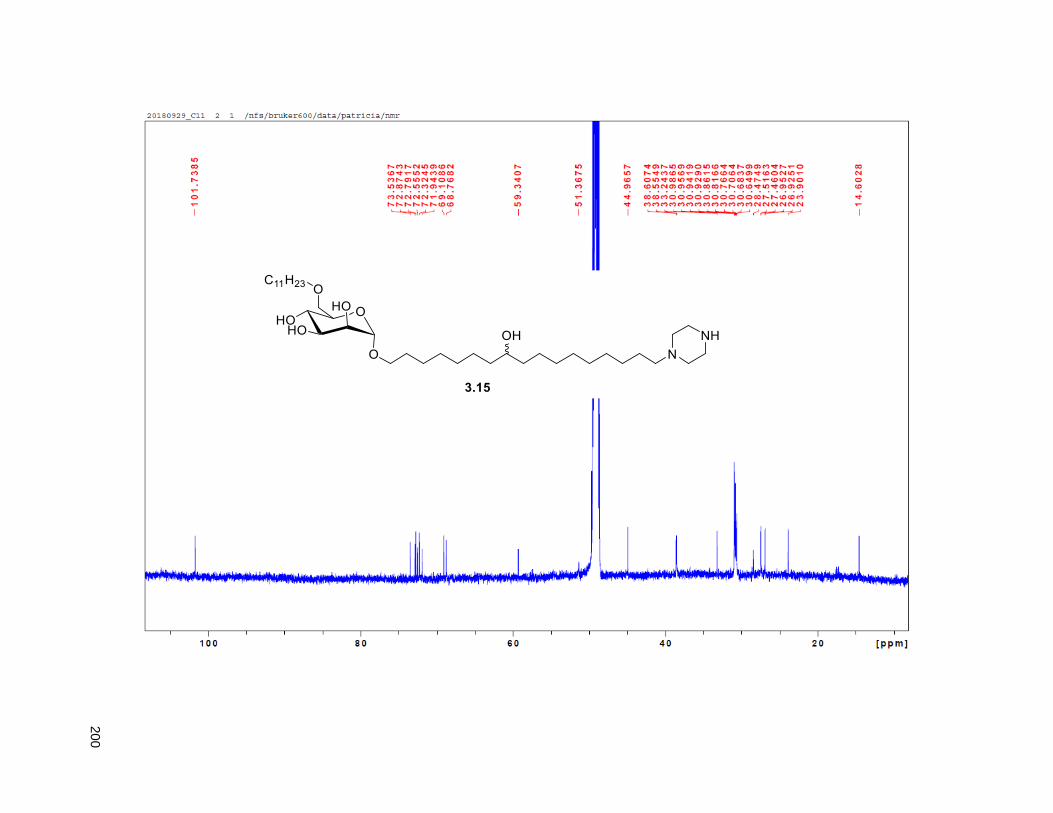
200
200
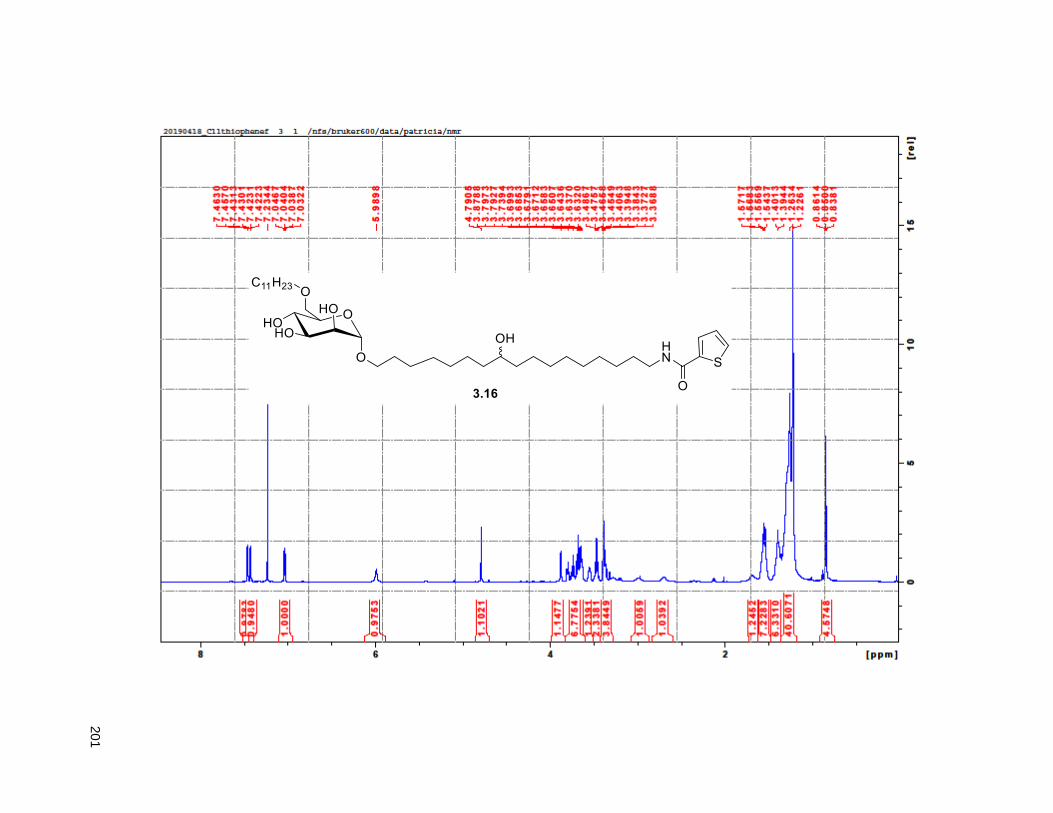
201
201

202
202
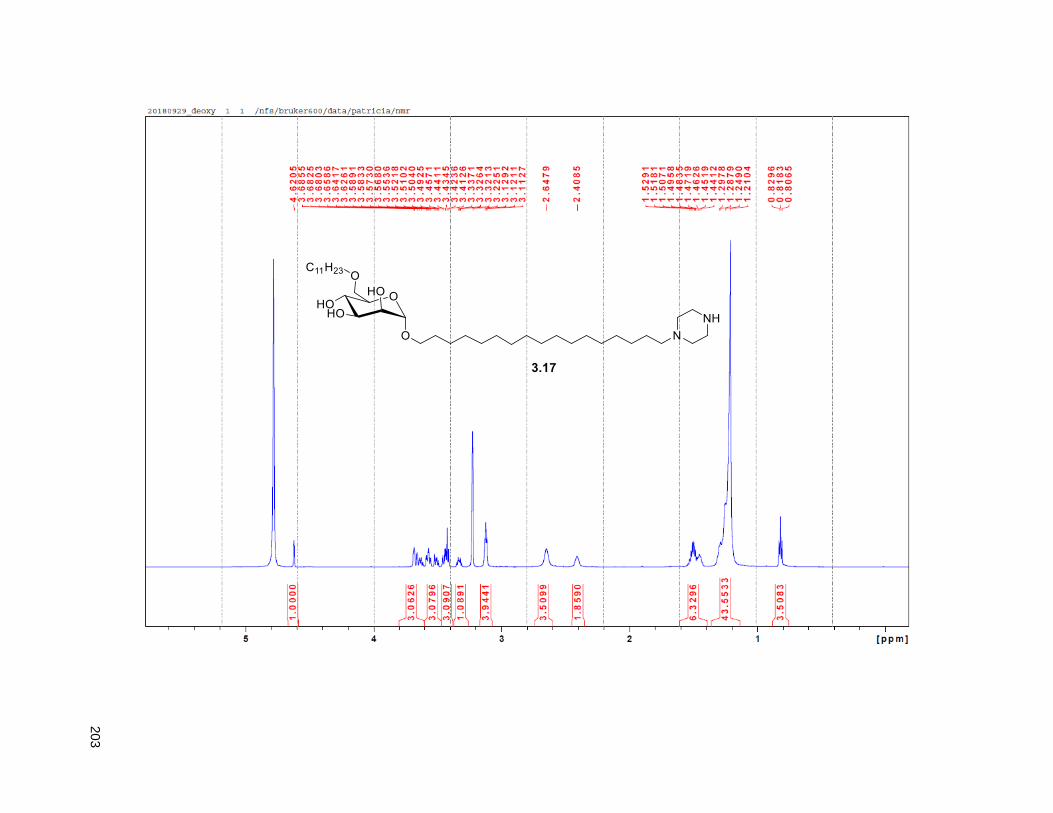
203
203
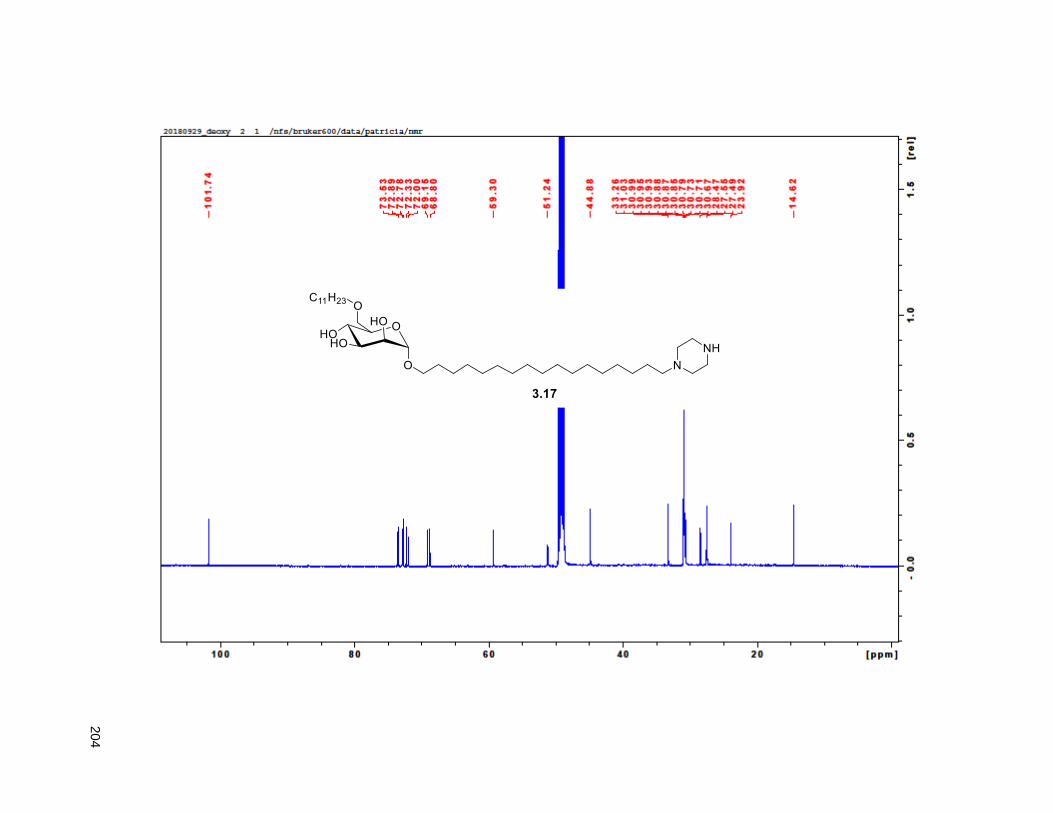
204
204
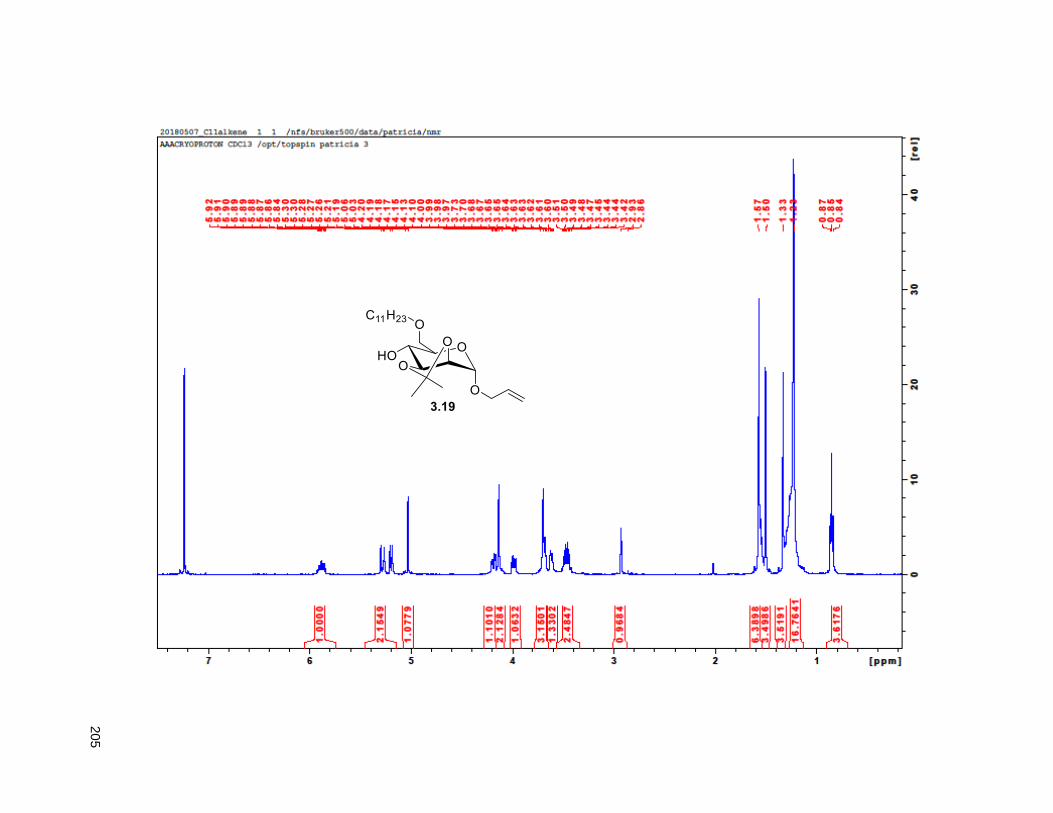
205
205
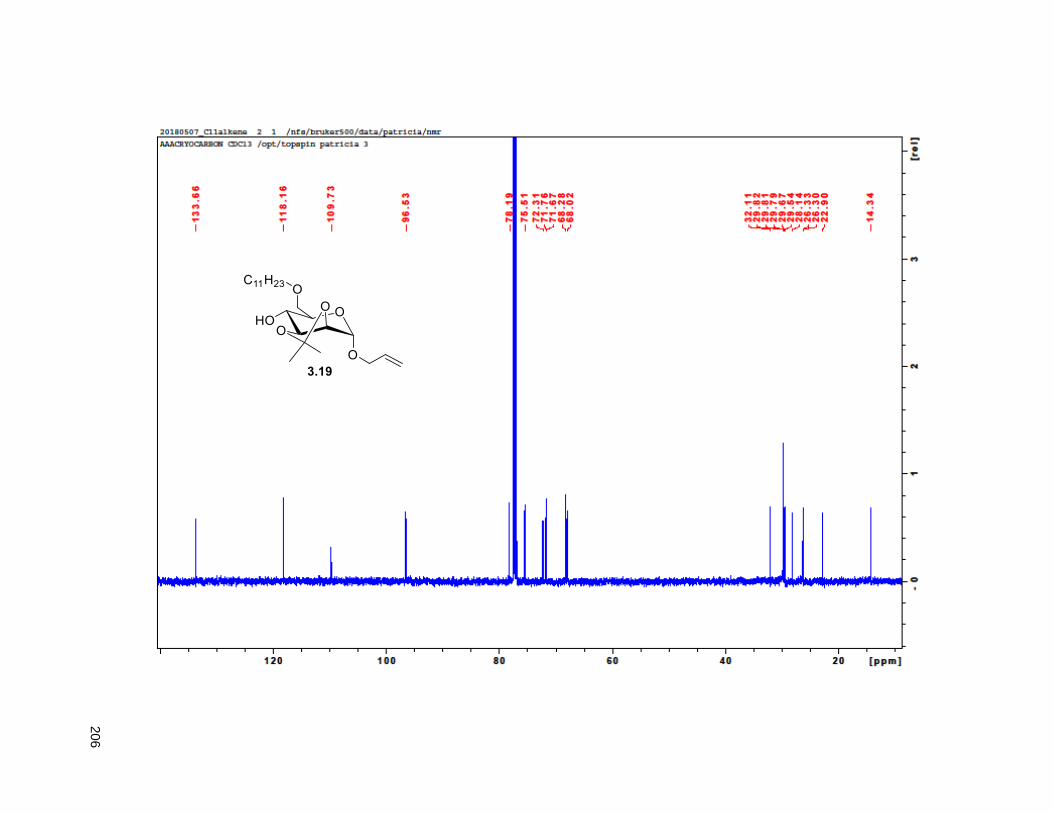
206
206
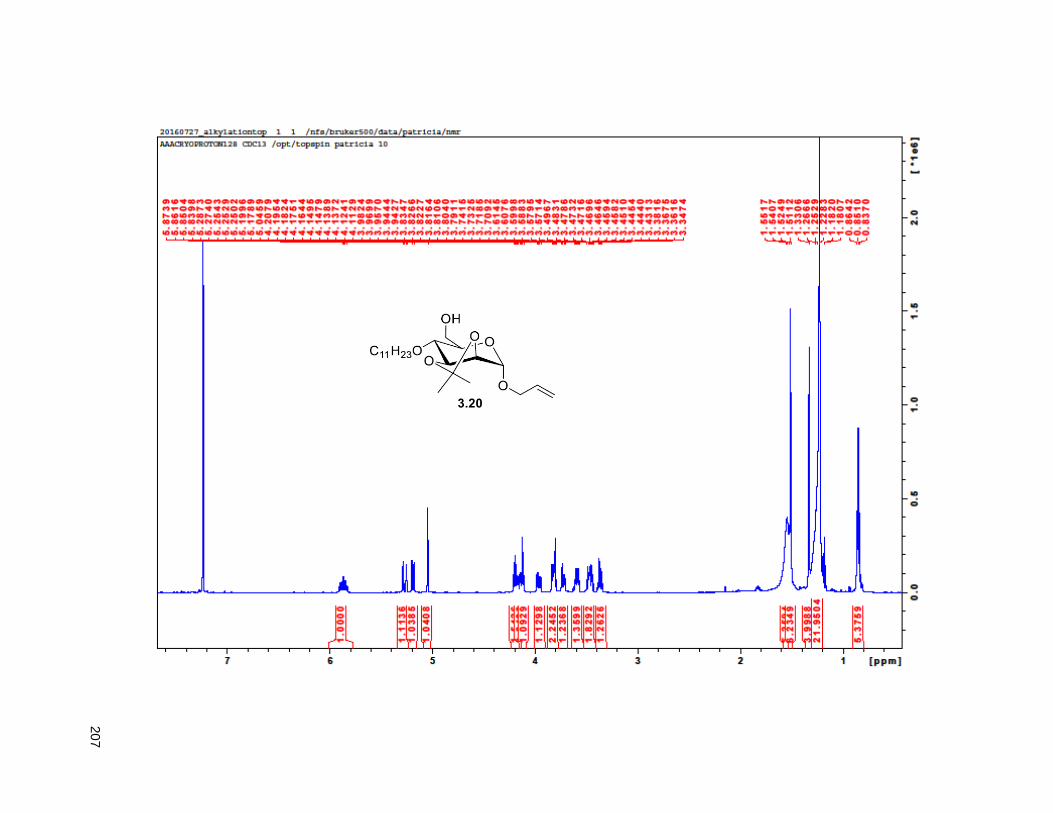
207
207
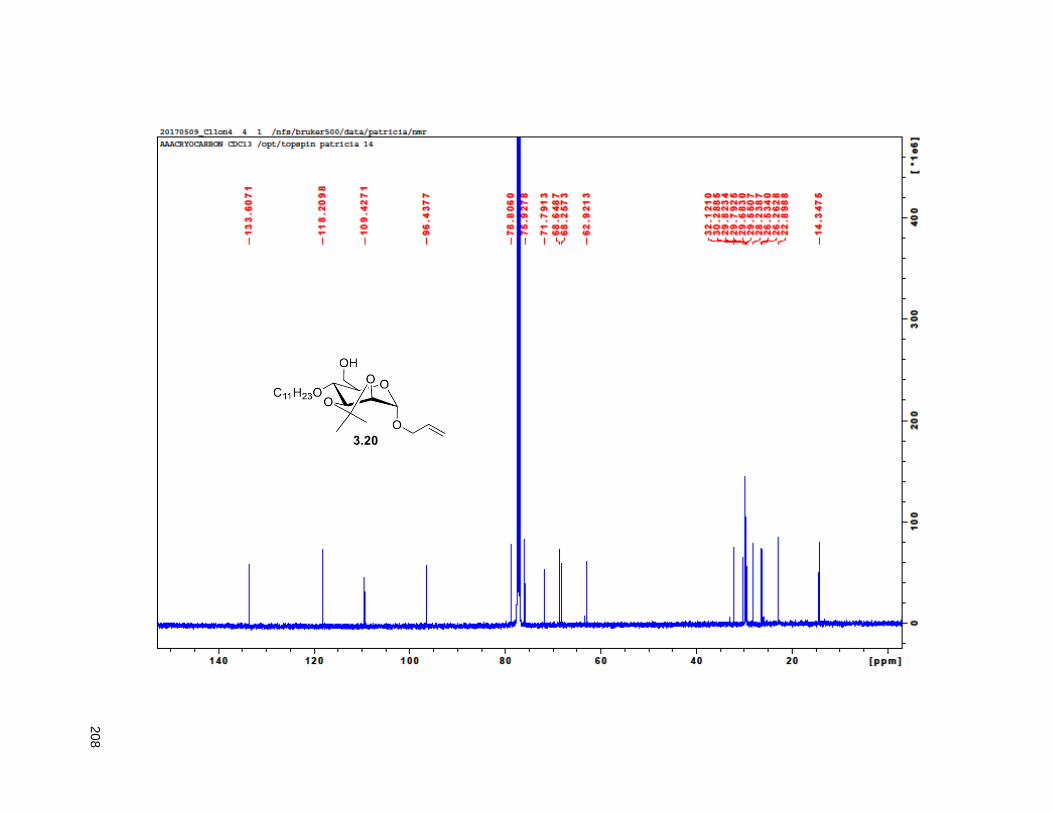
208
208
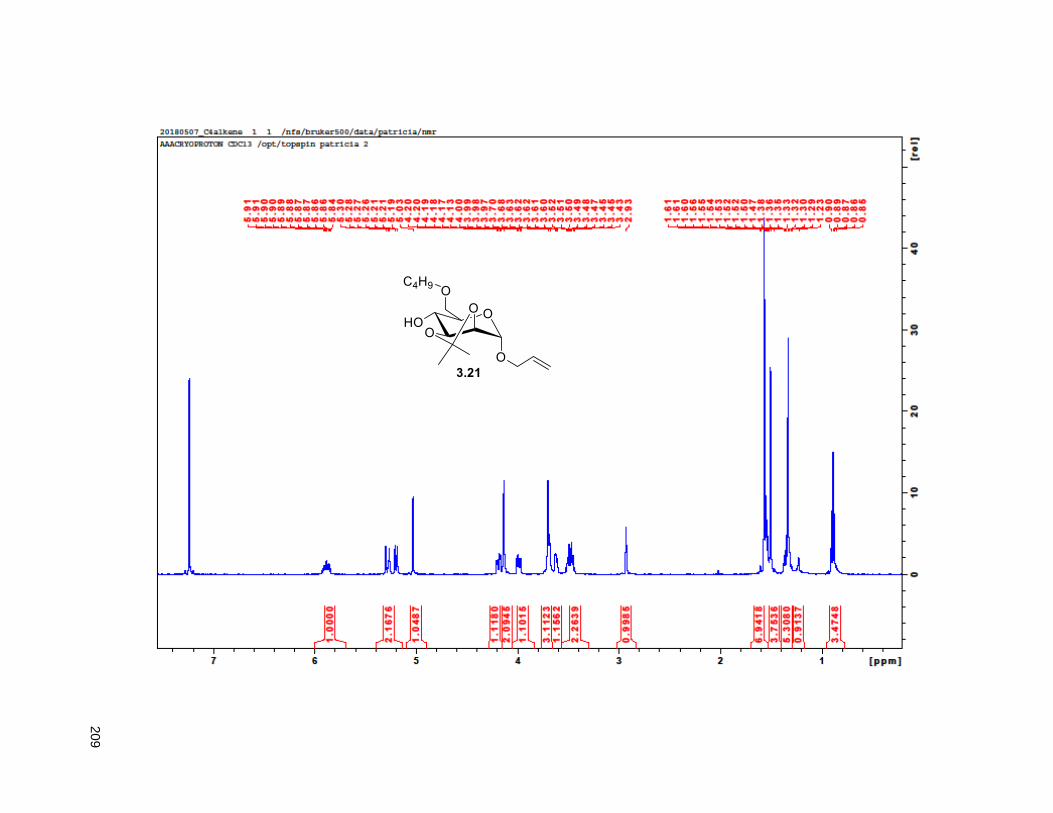
209
209
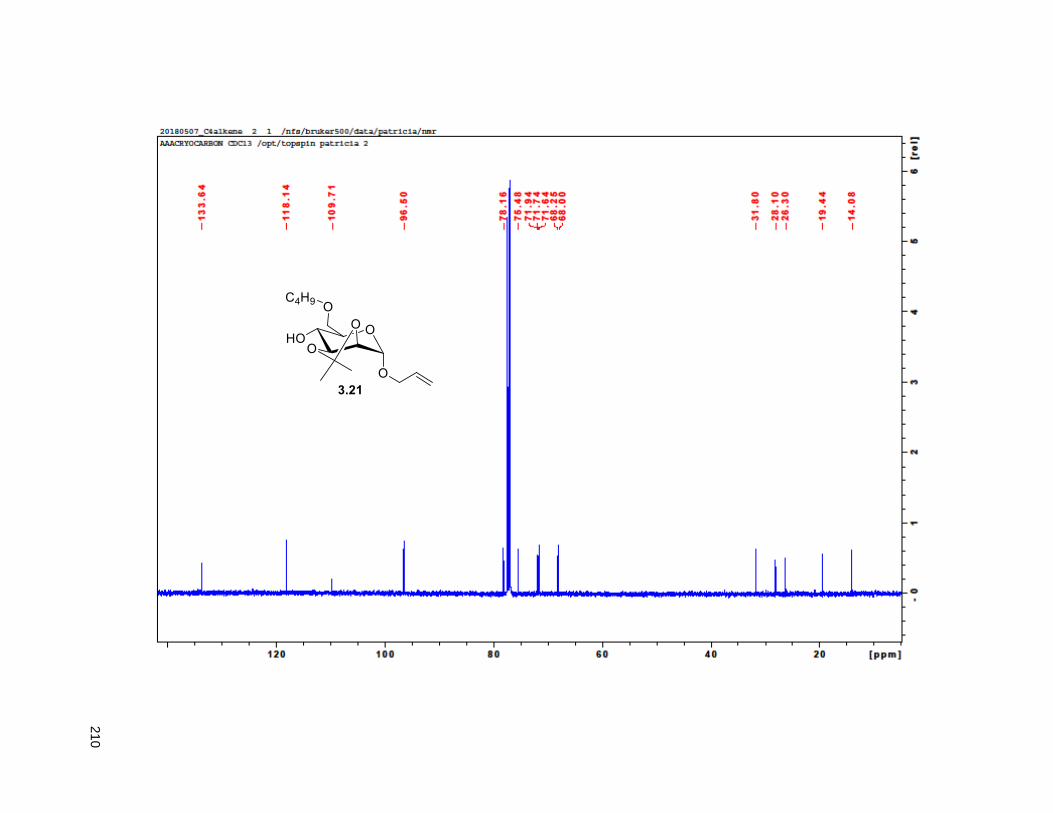
210
210
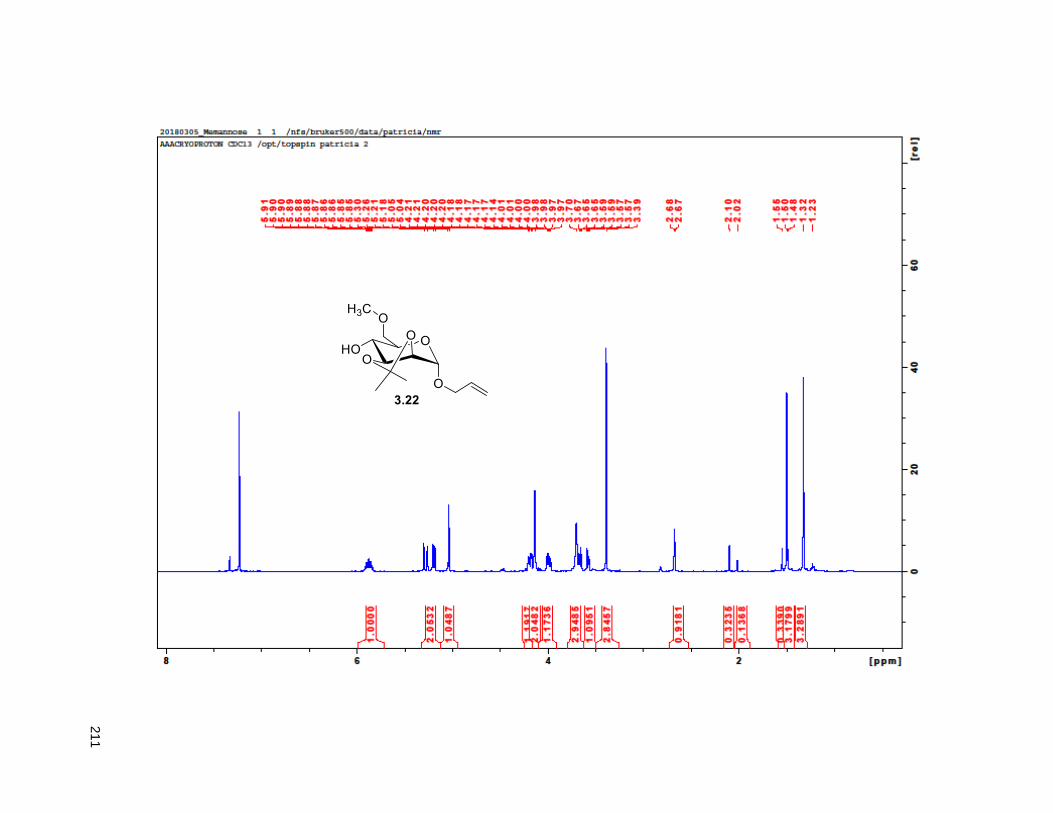
211
211
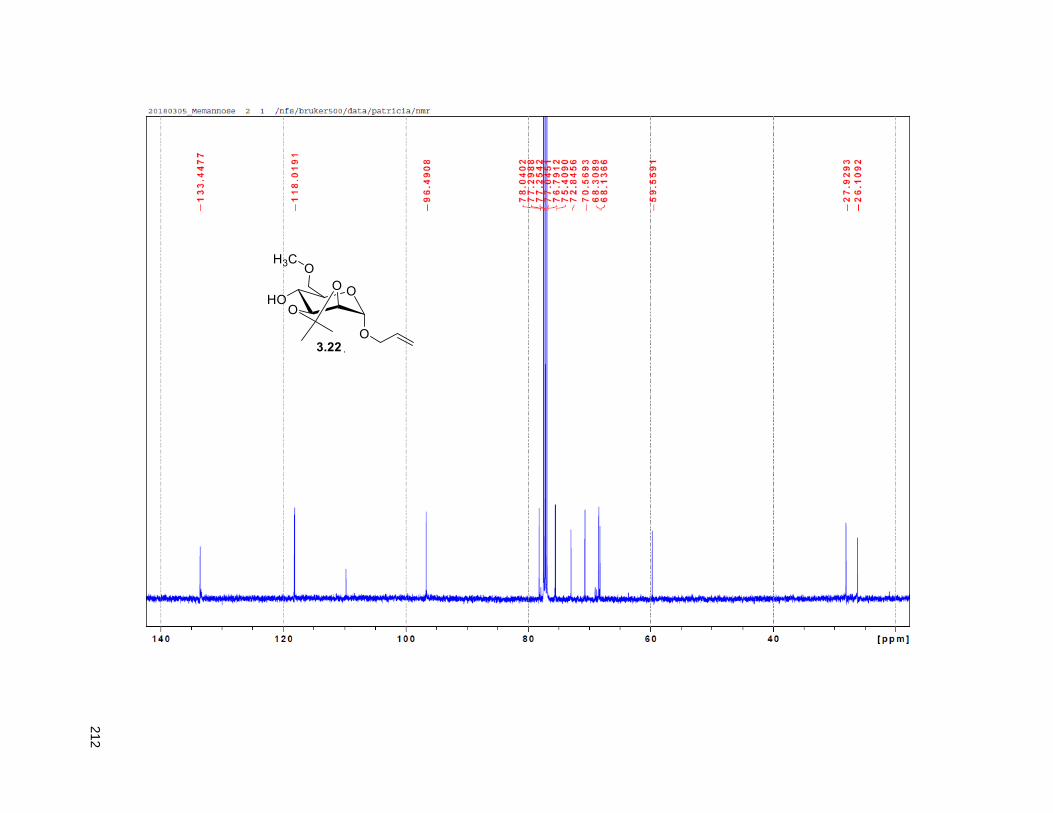
212
212
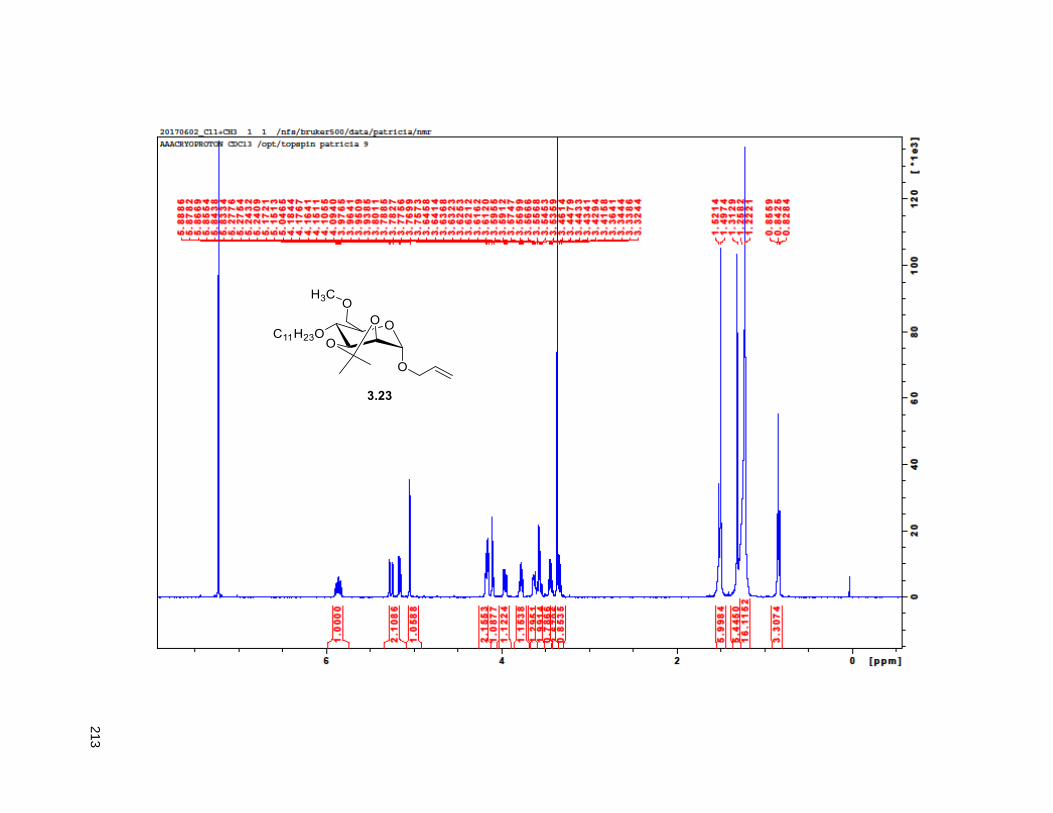
213
213

214
214
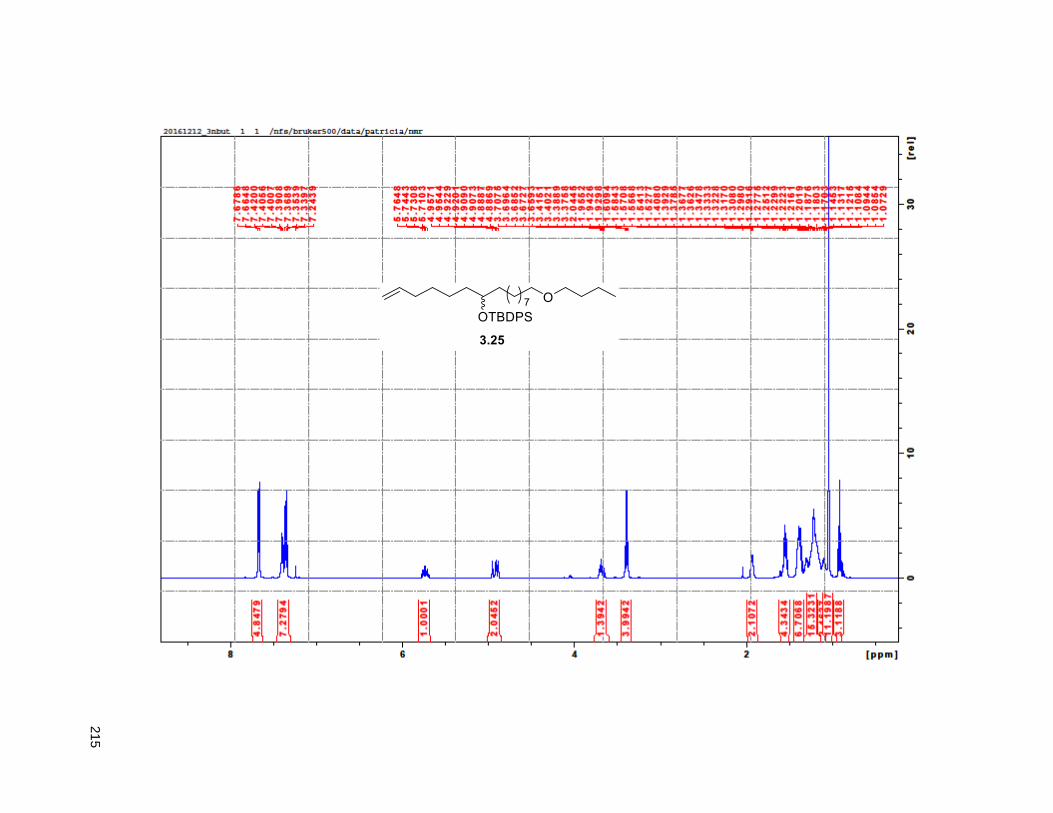
215
215
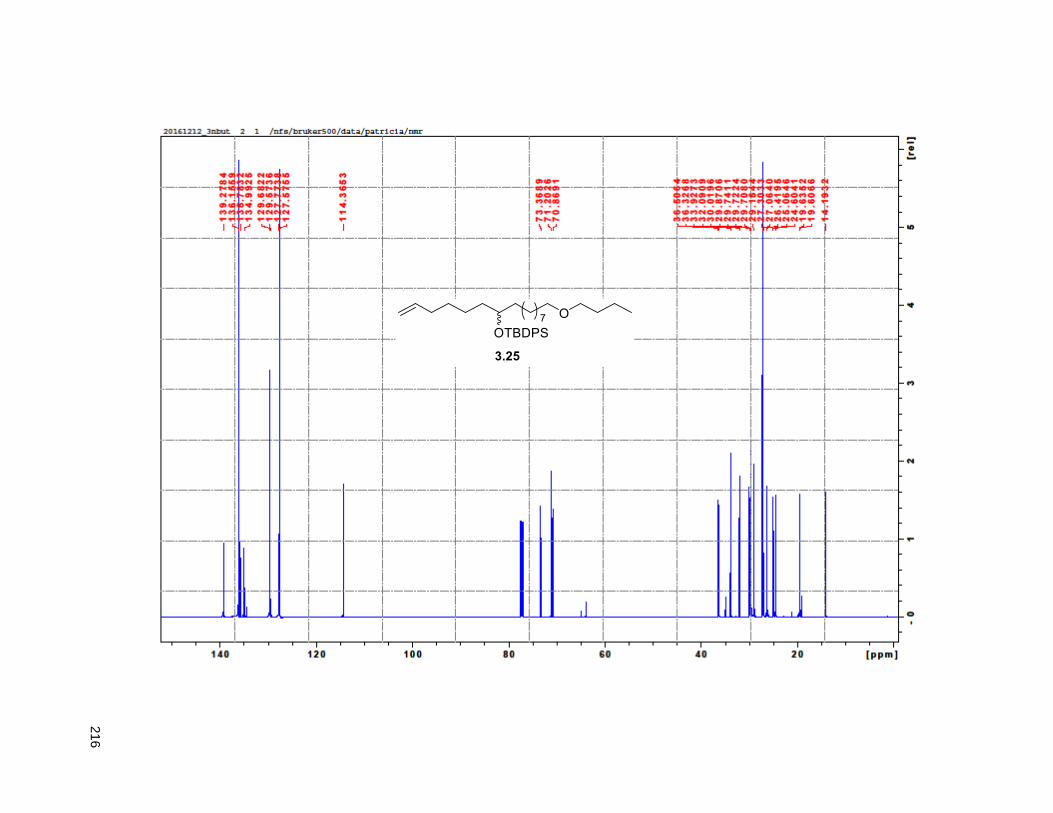
216
216
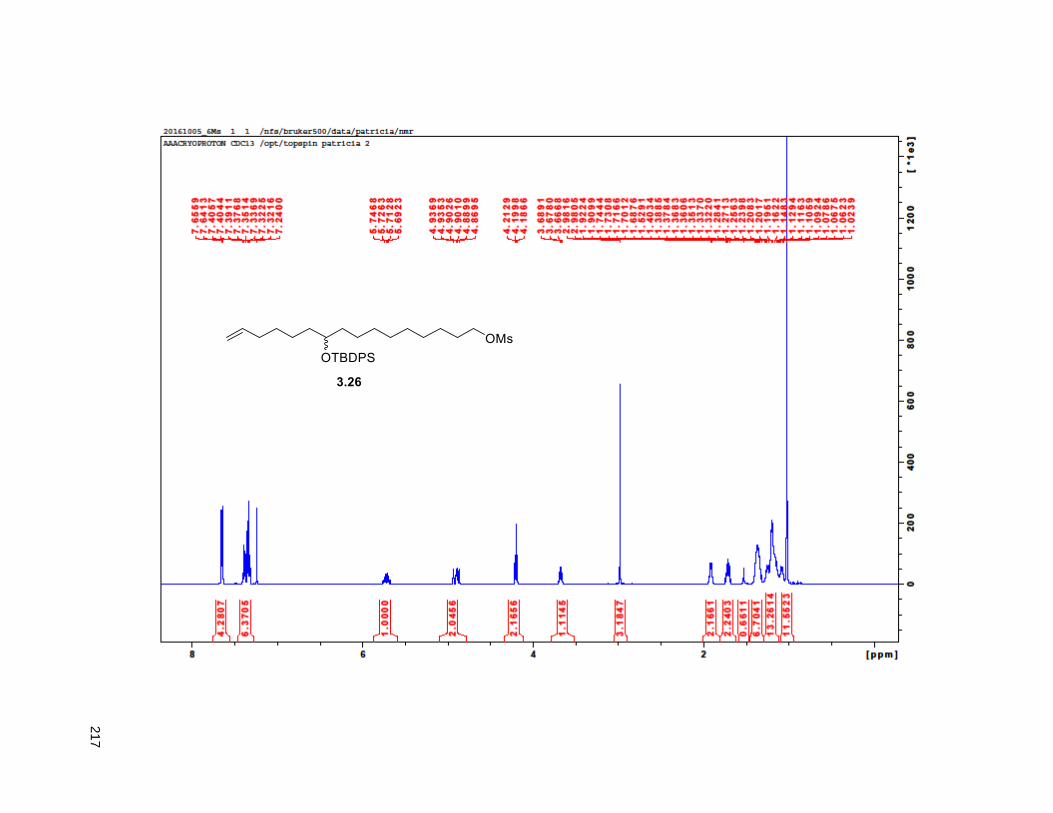
217
217
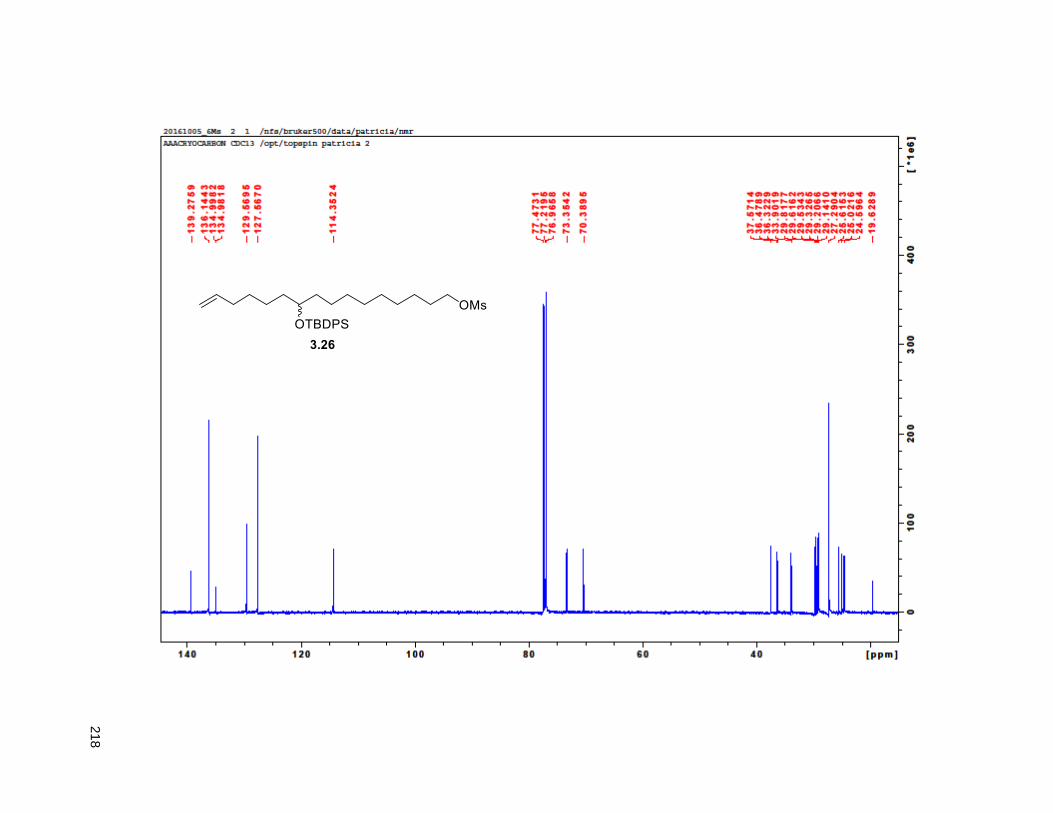
218
218
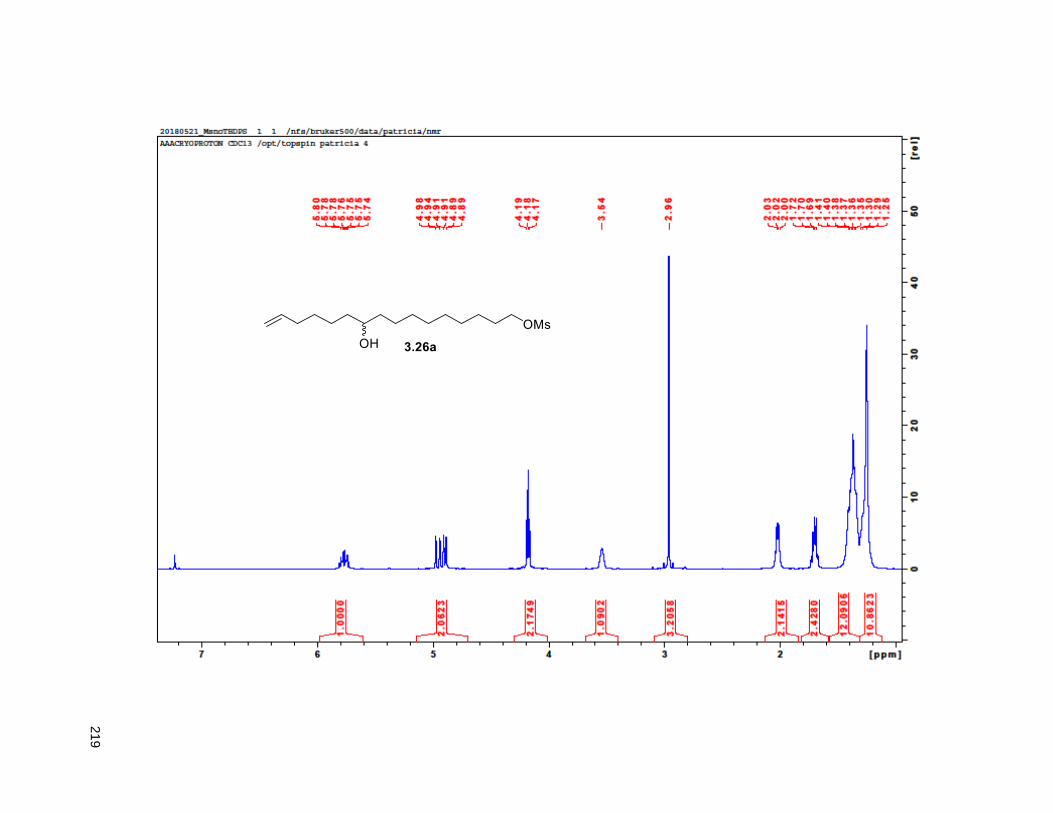
219
219
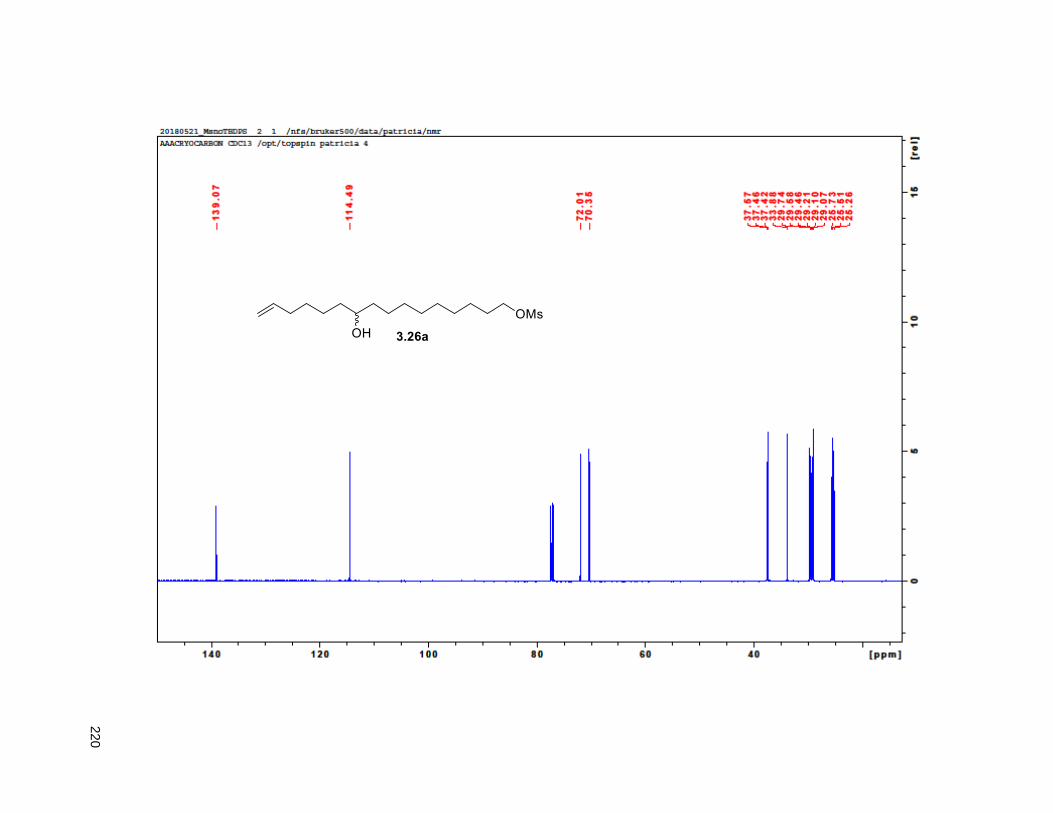
220
220

221
221
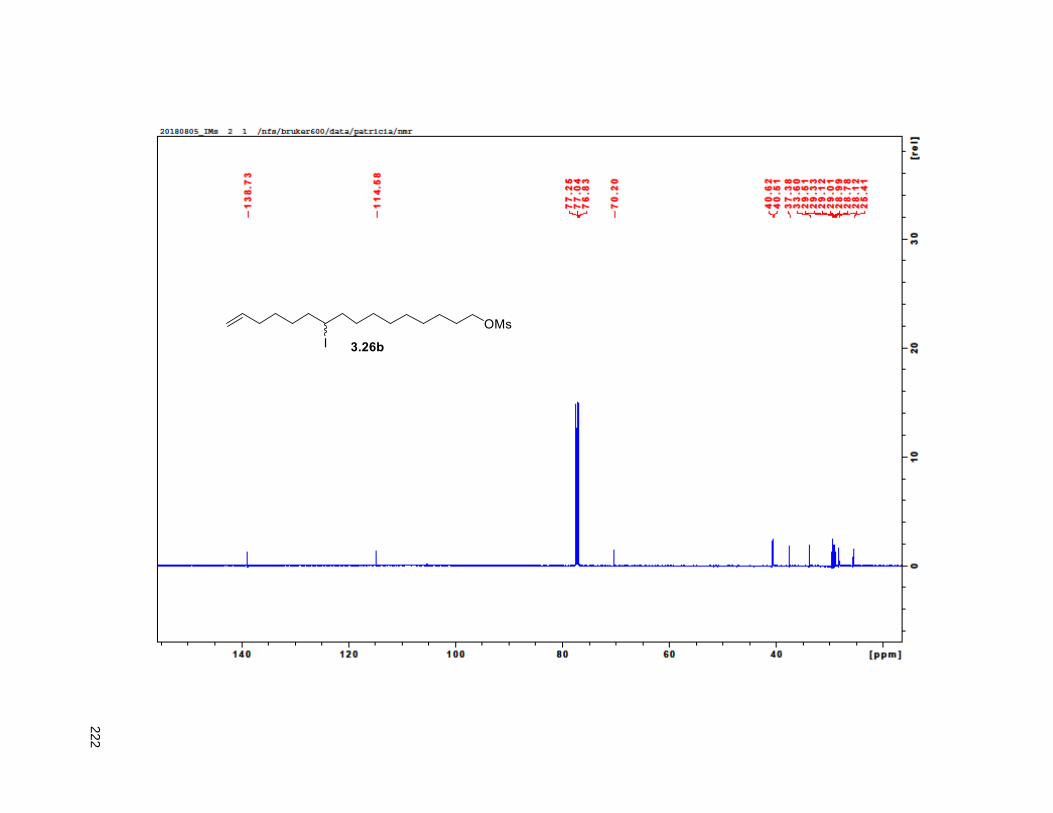
222
222
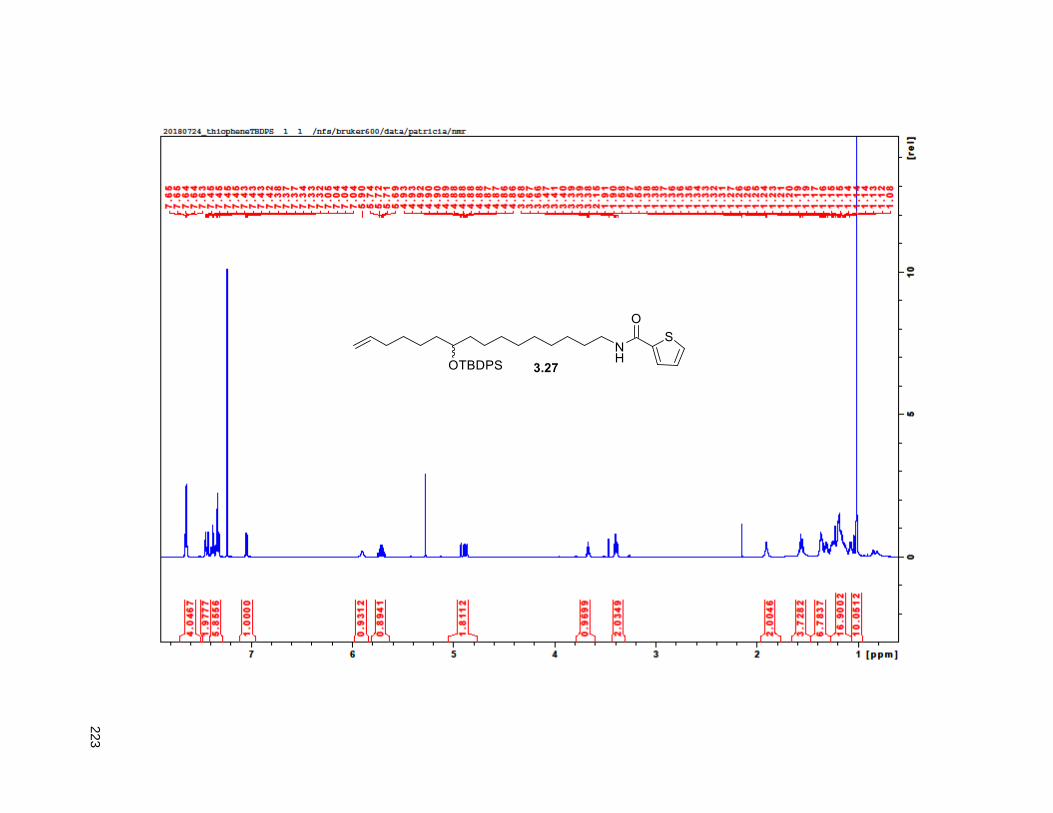
223
223
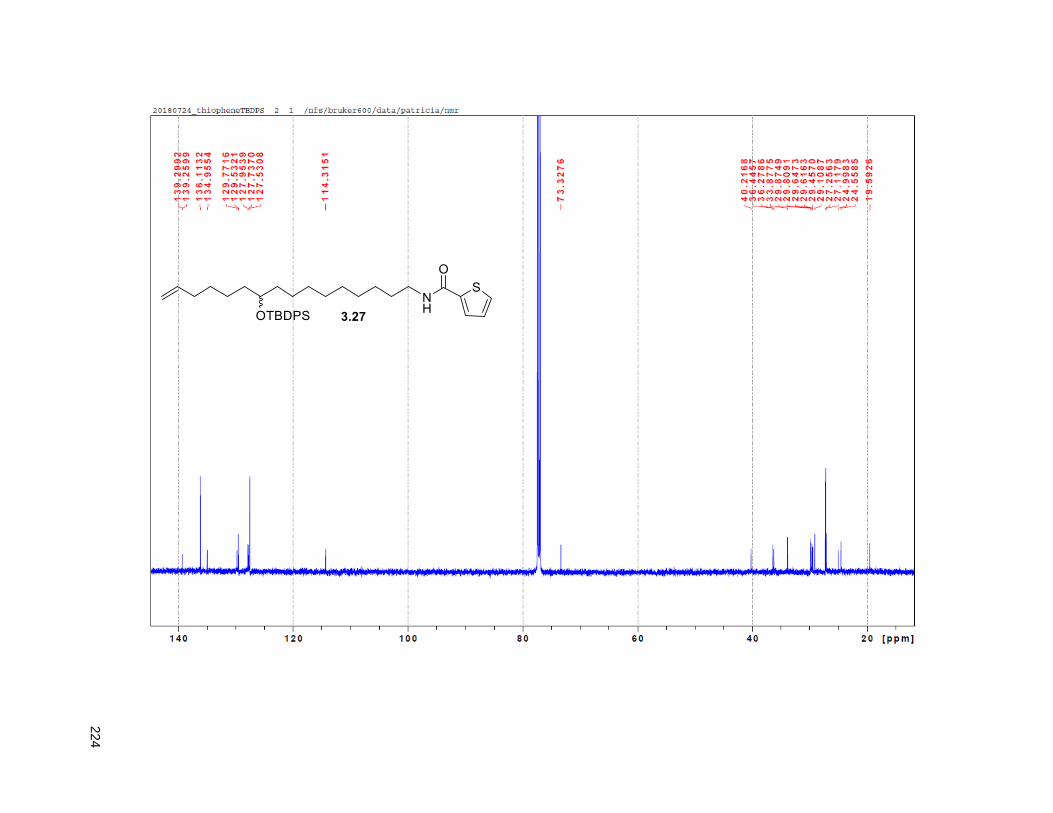
224
224
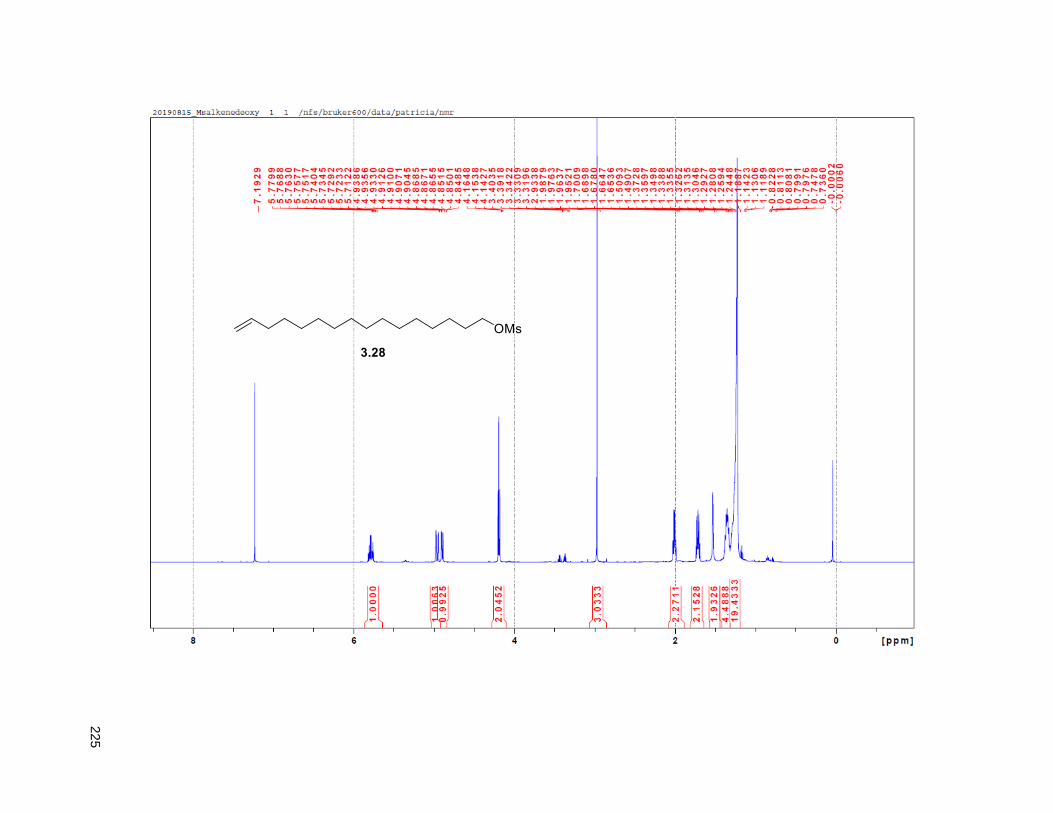
225
225
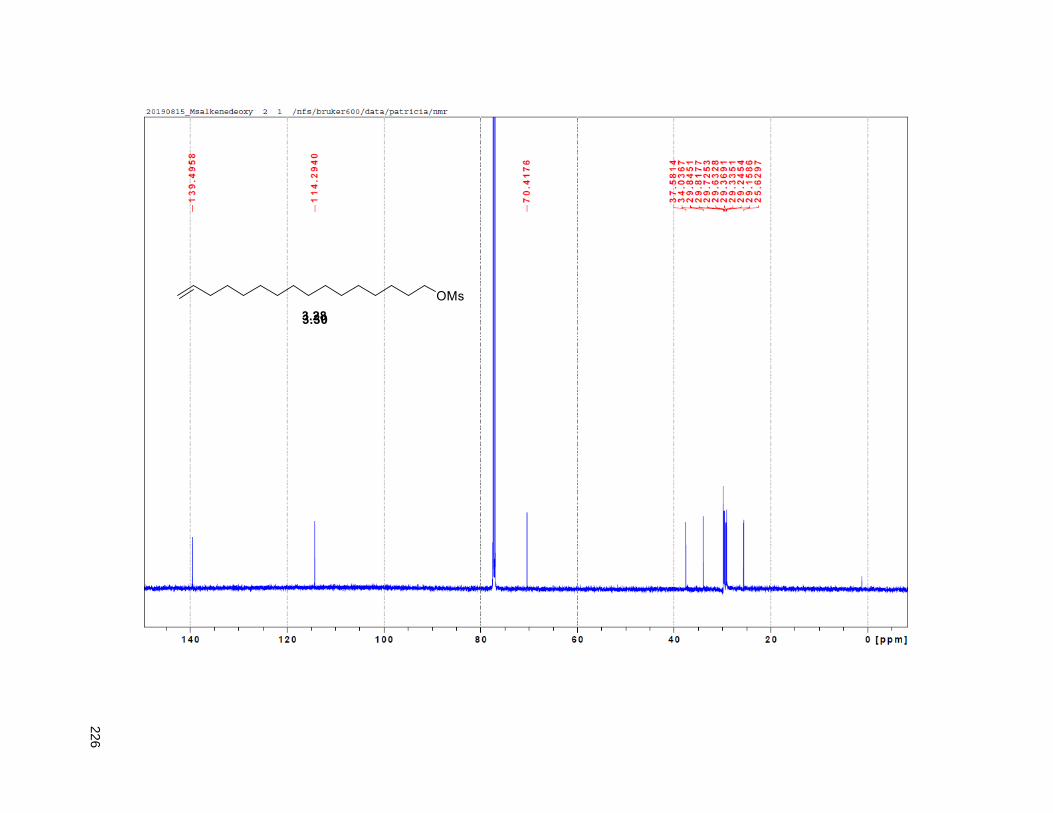
226
226
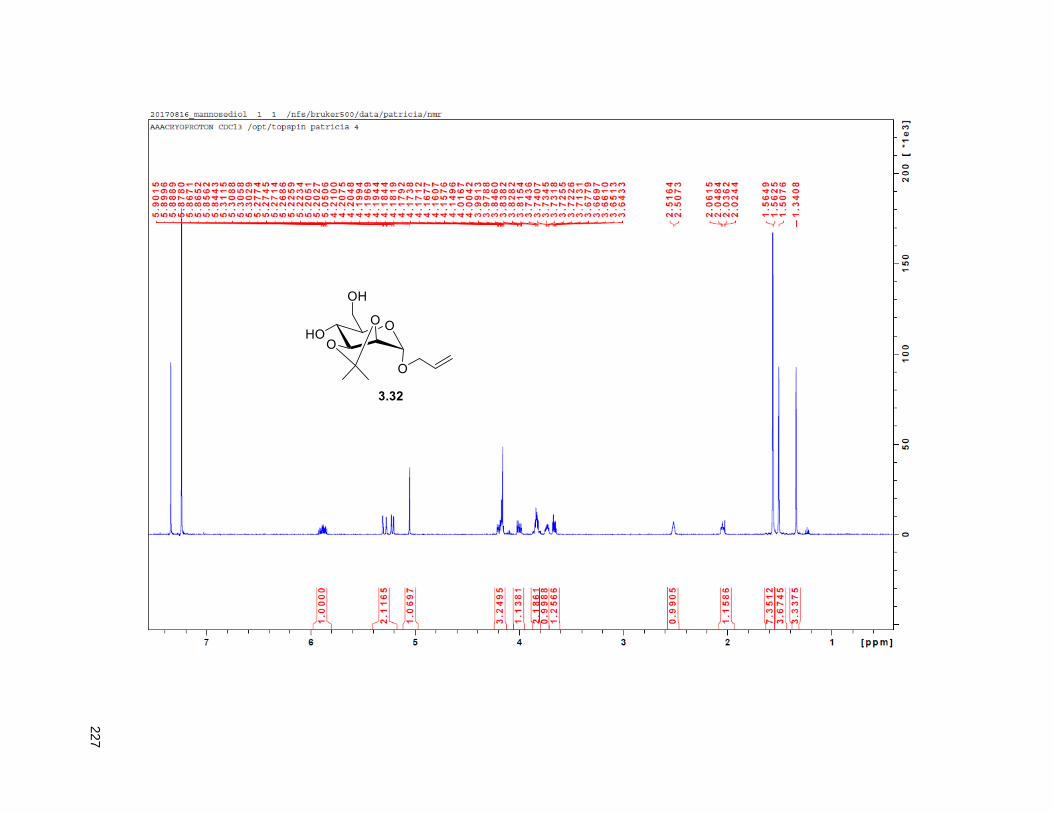
227
227
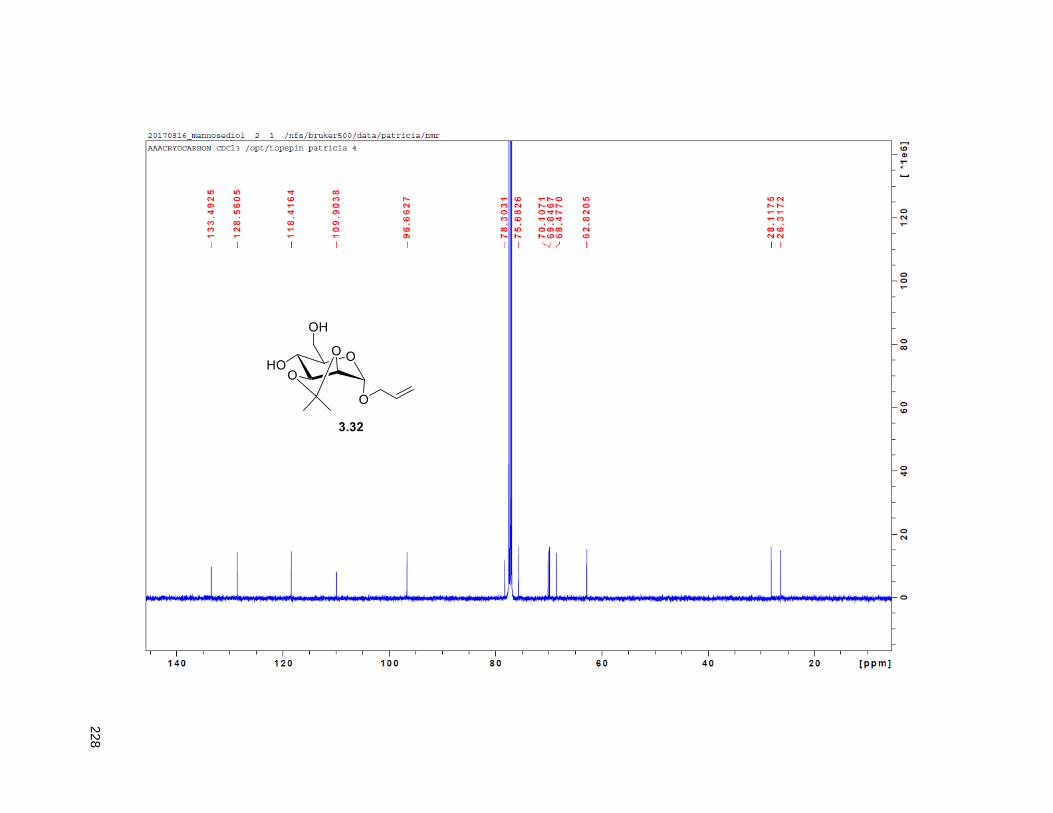
228
228
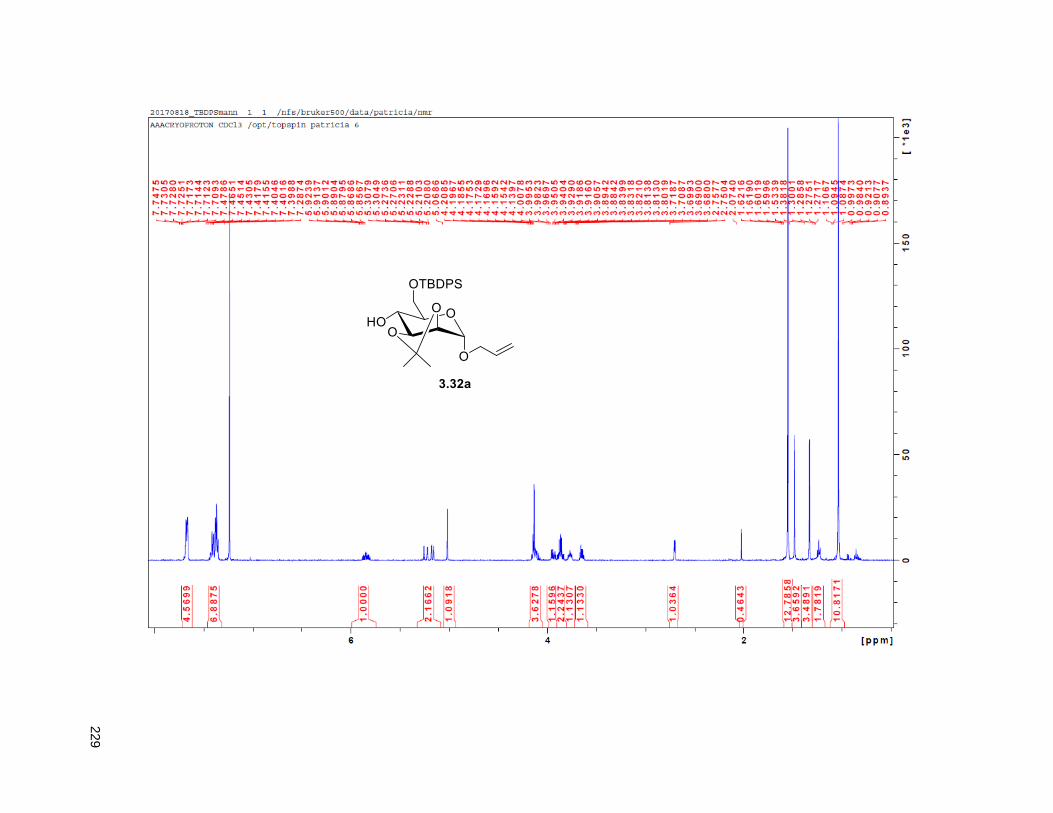
229
229
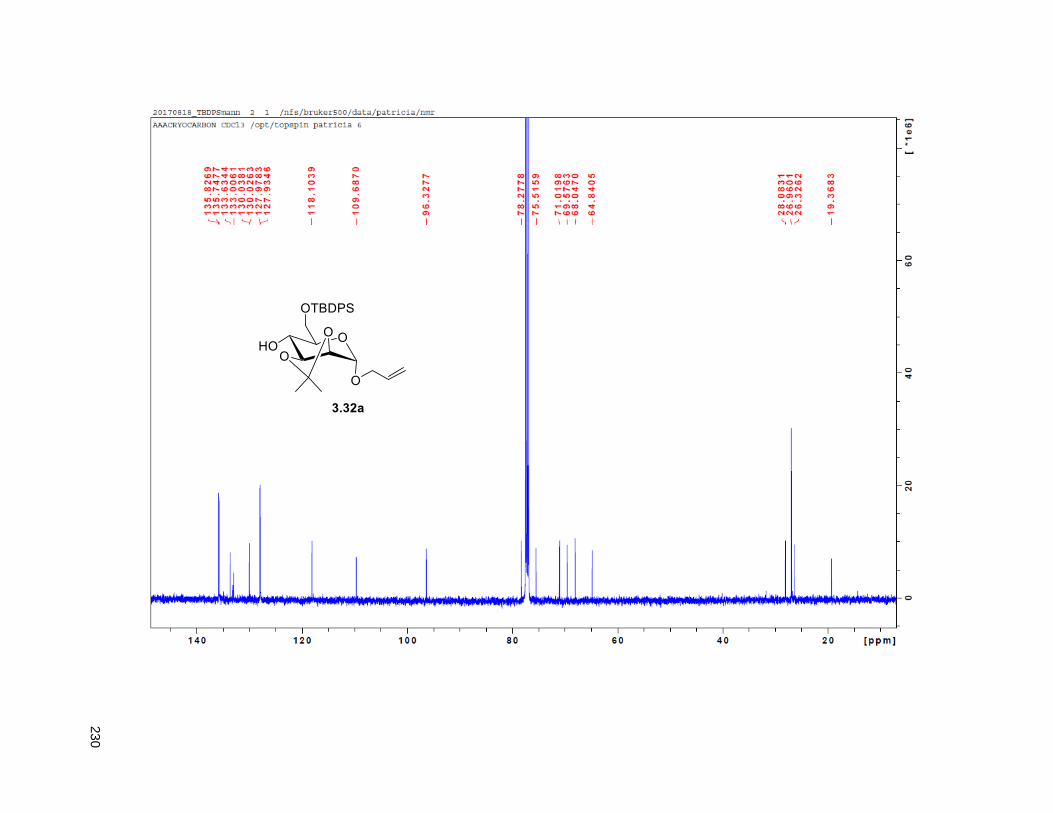
230
230
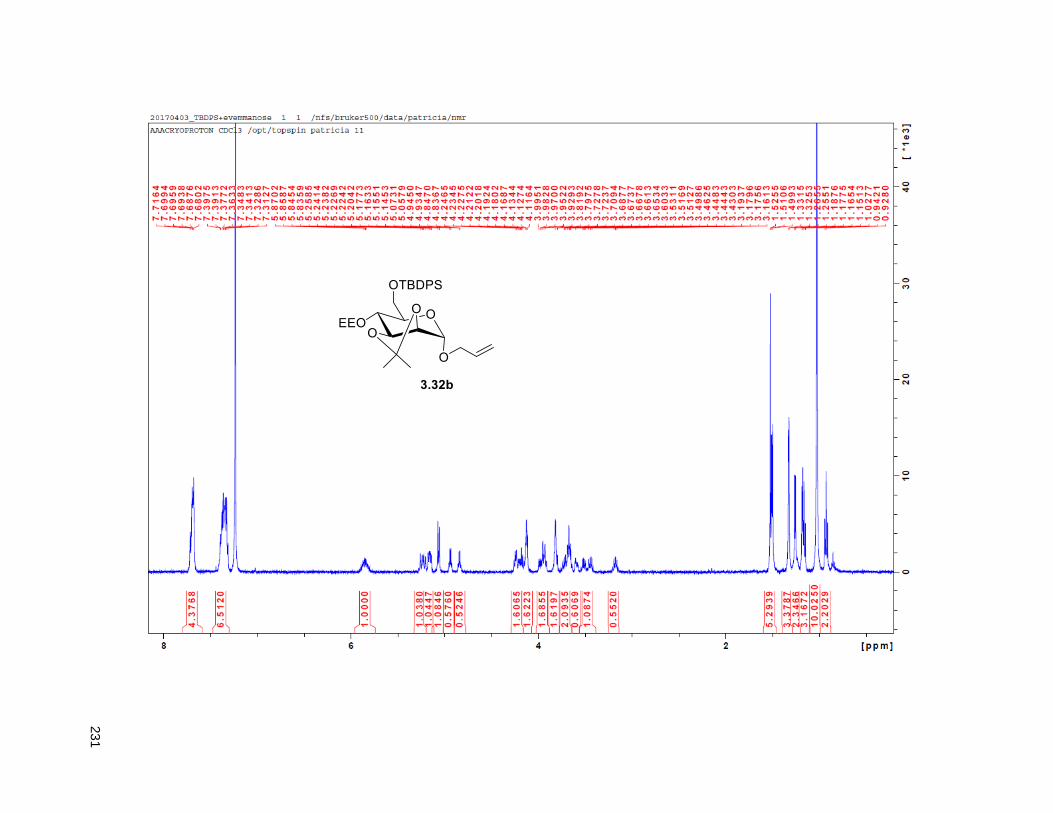
231
231
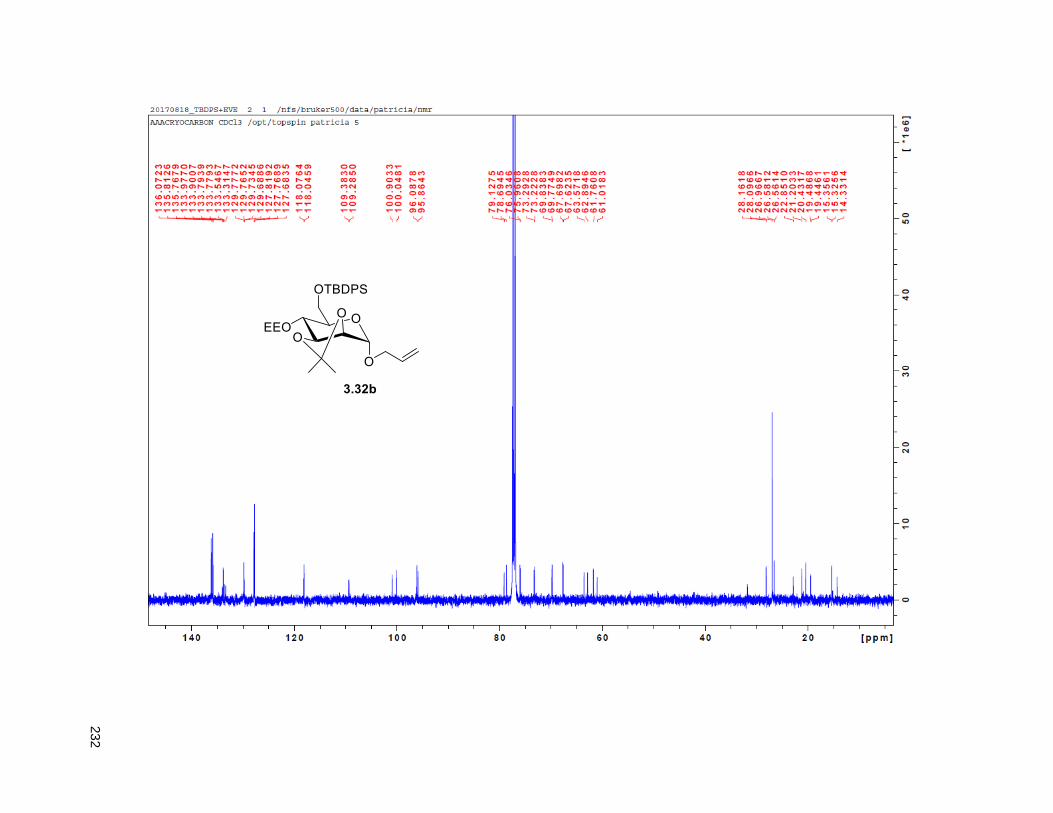
232
232
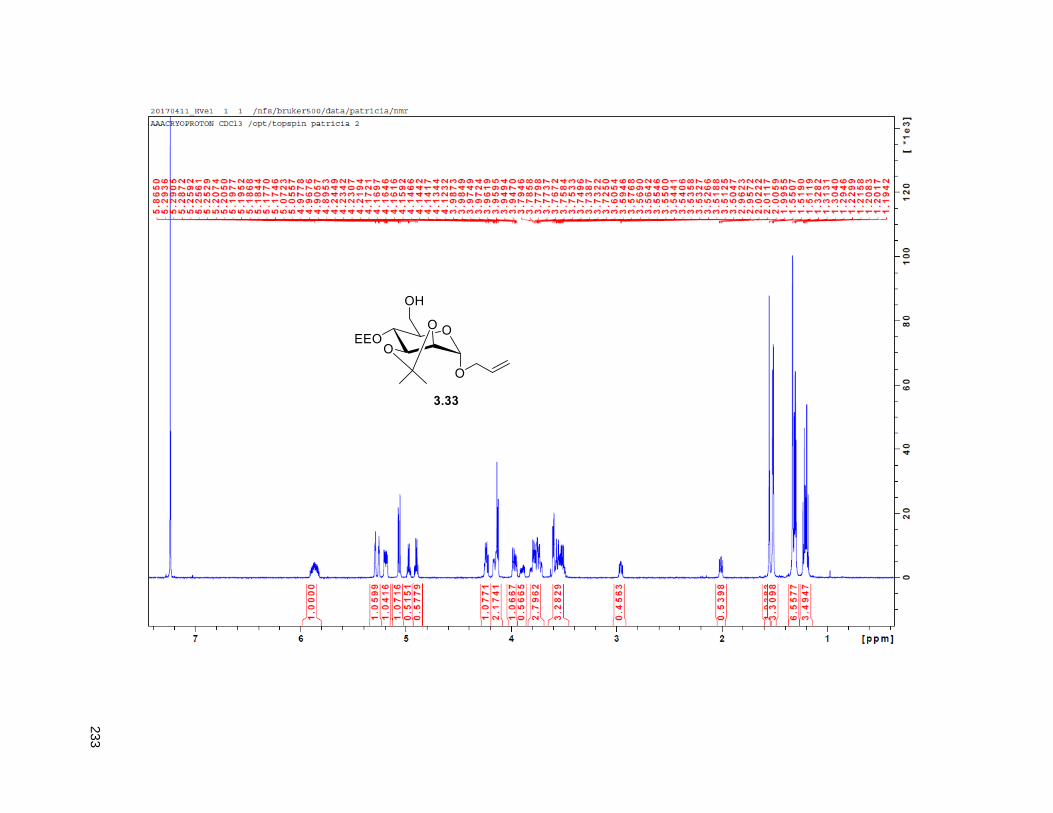
233
233
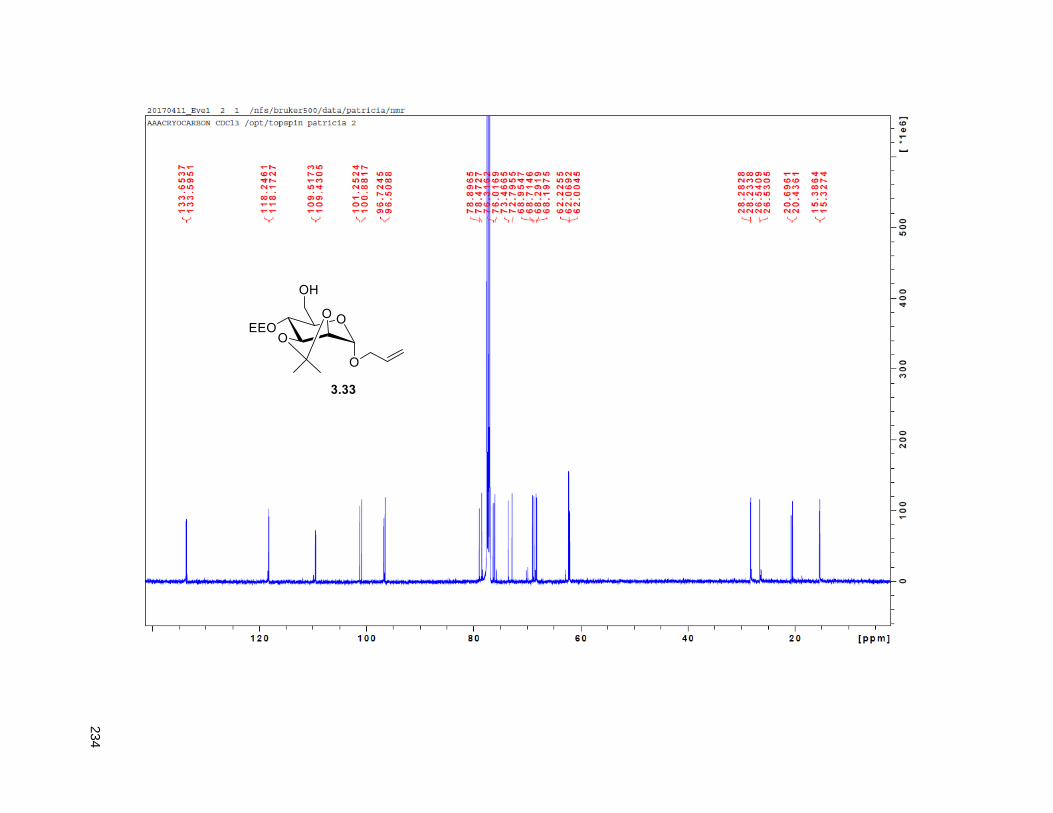
234
234
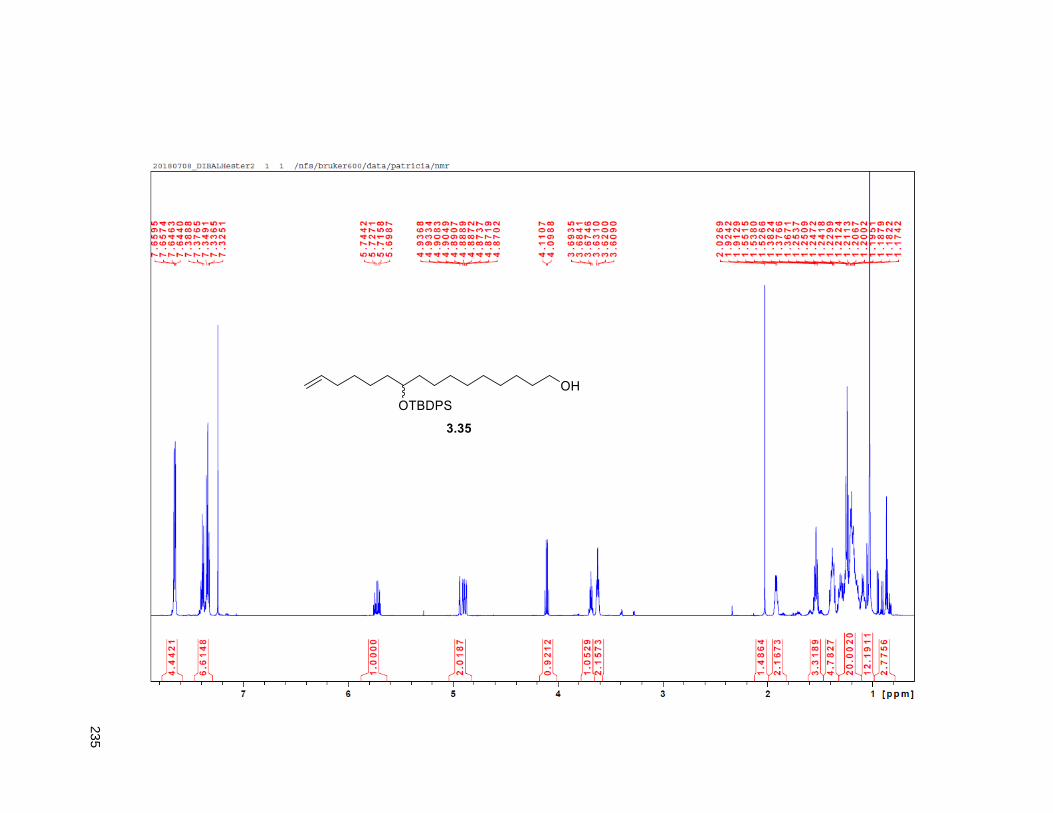
235
235
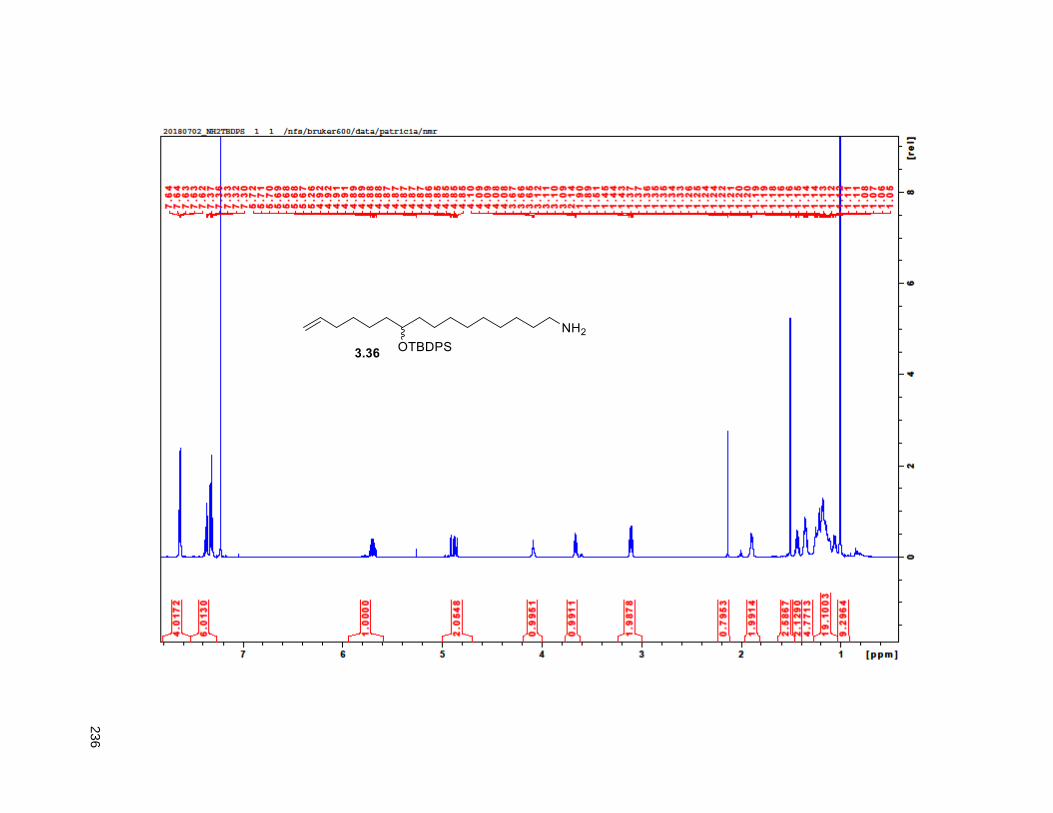
236
236

237
237
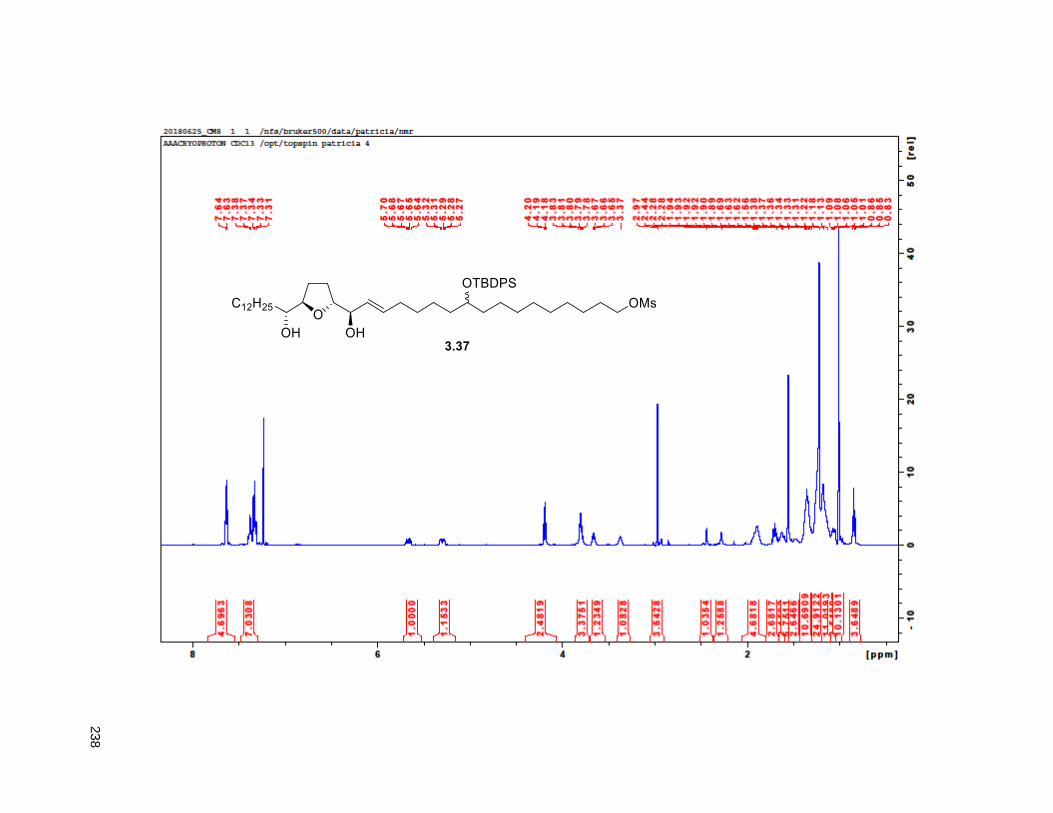
238
238
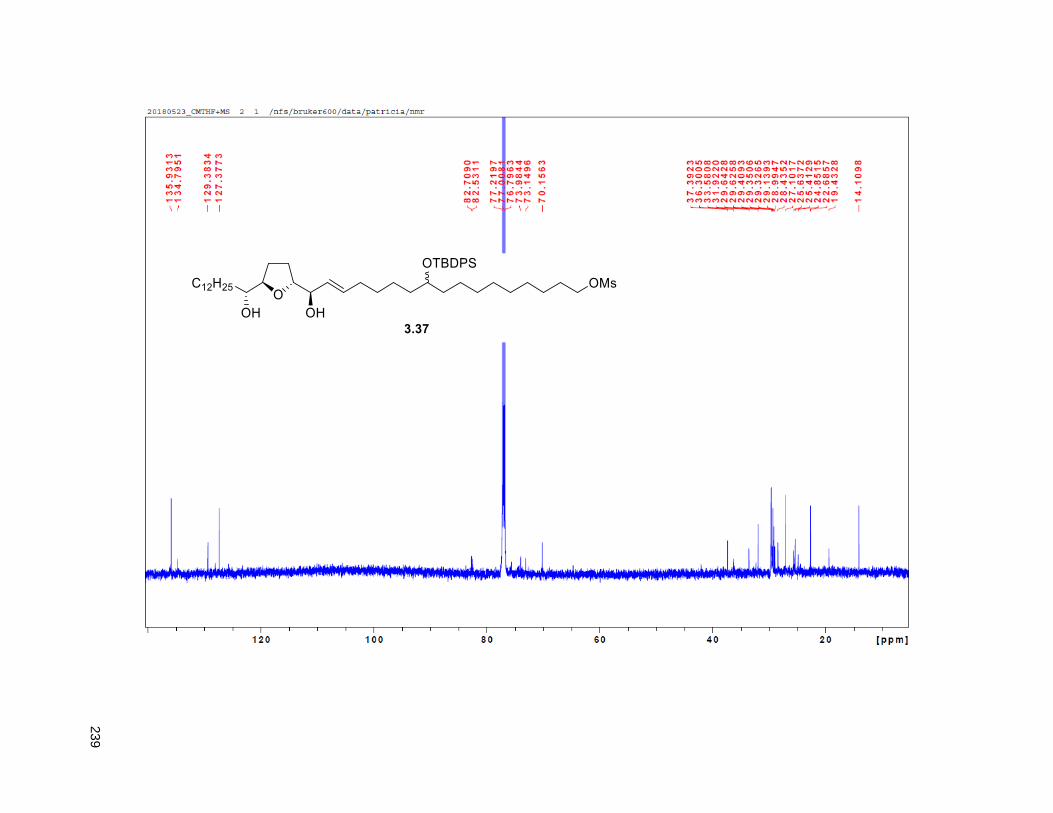
239
239
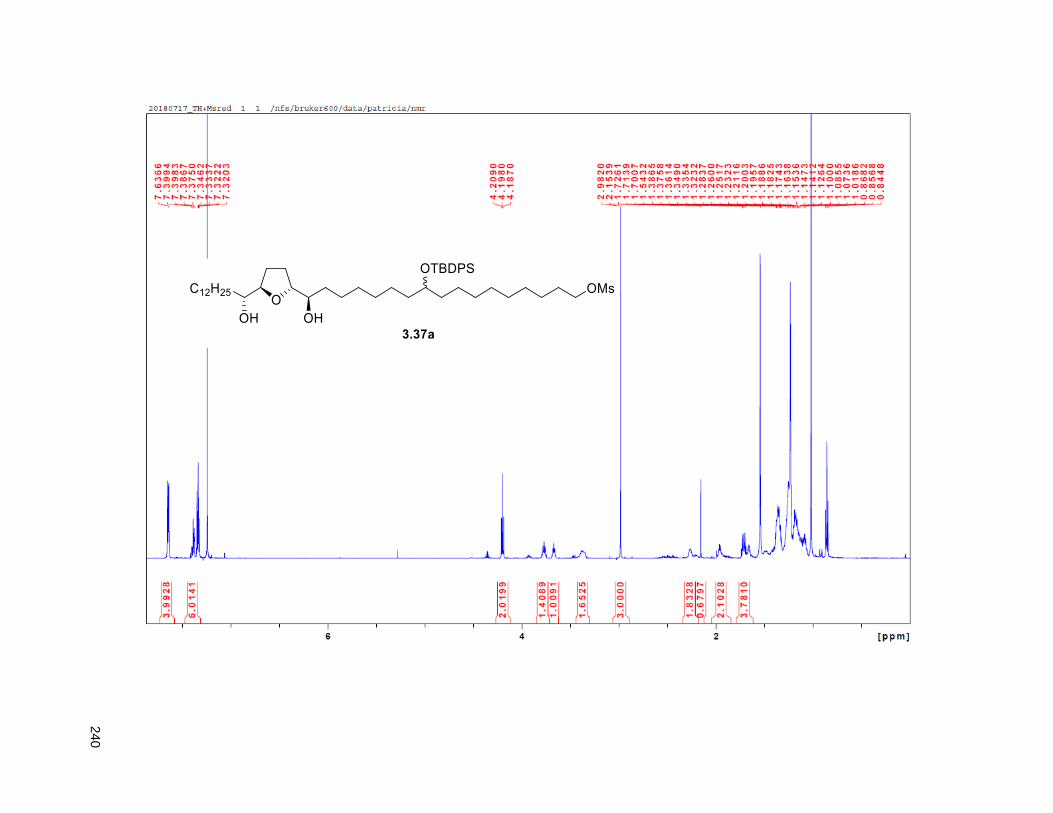
240
240
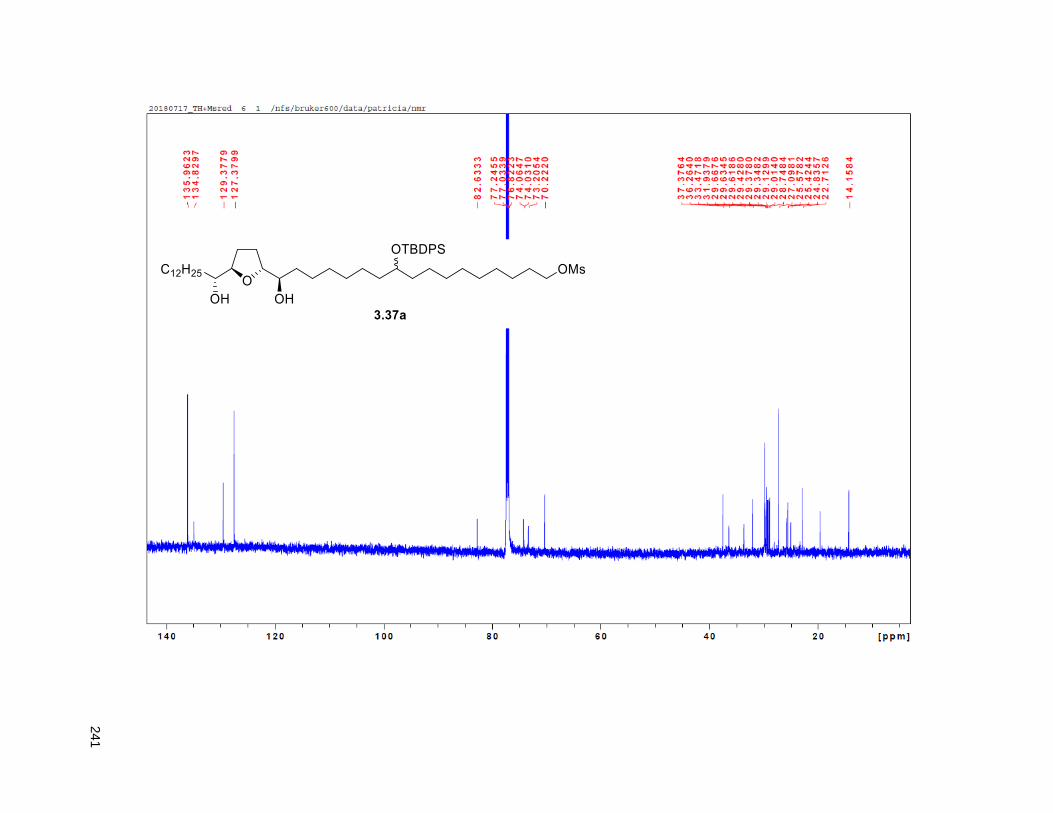
241
241

242
242
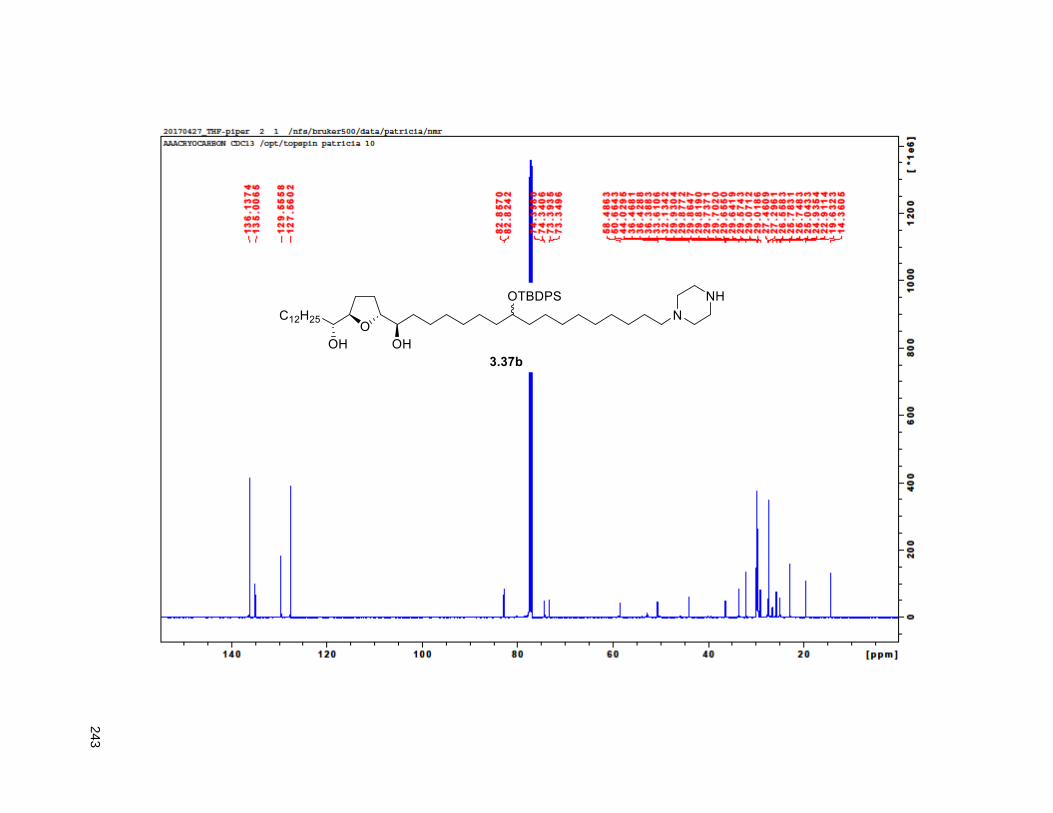
243
243

244
244
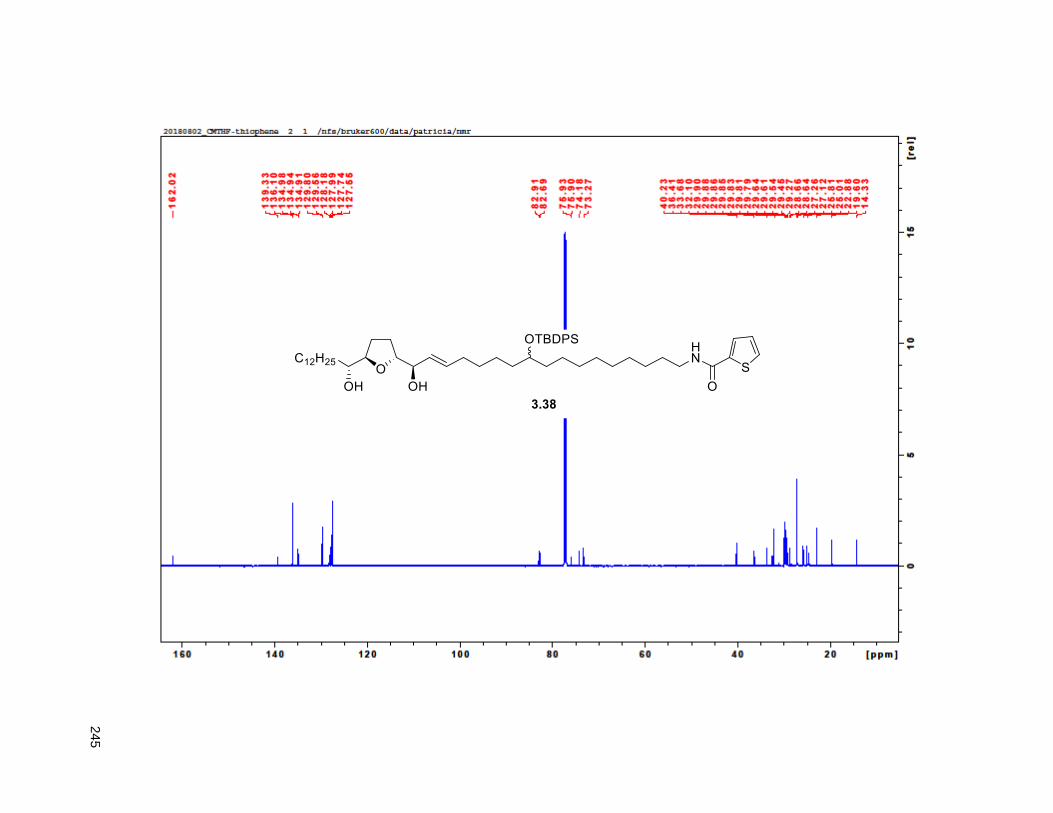
245
245
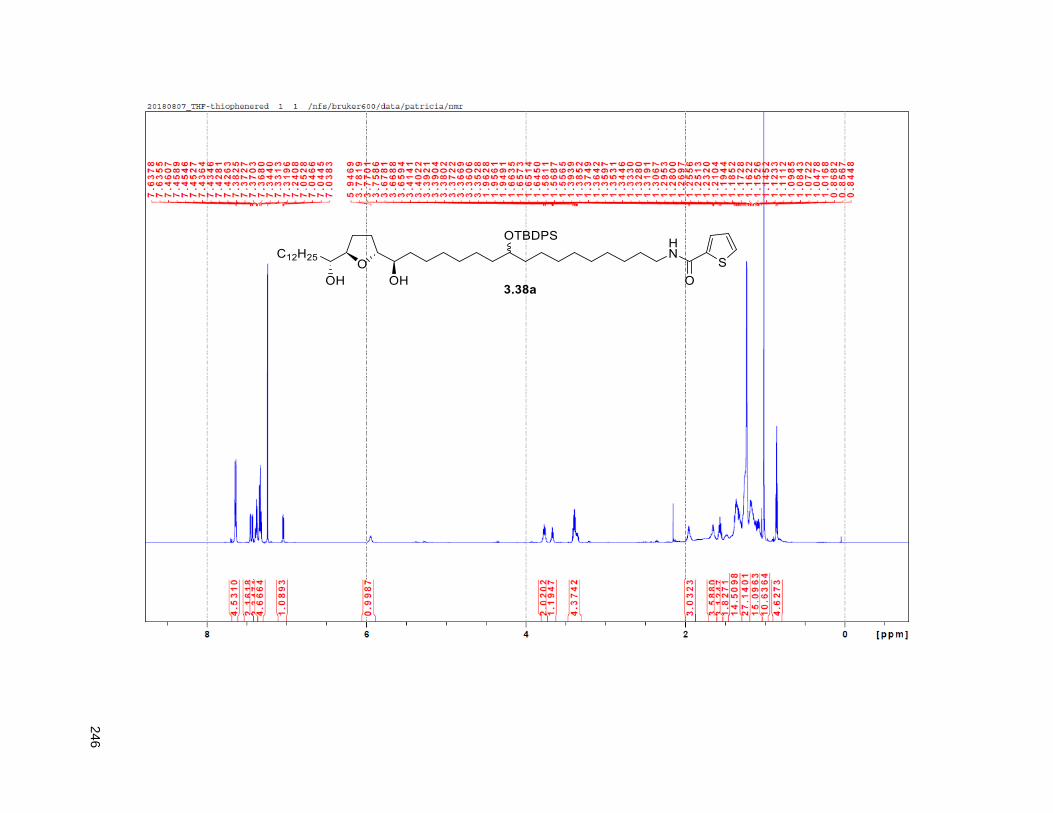
246
246

247
247
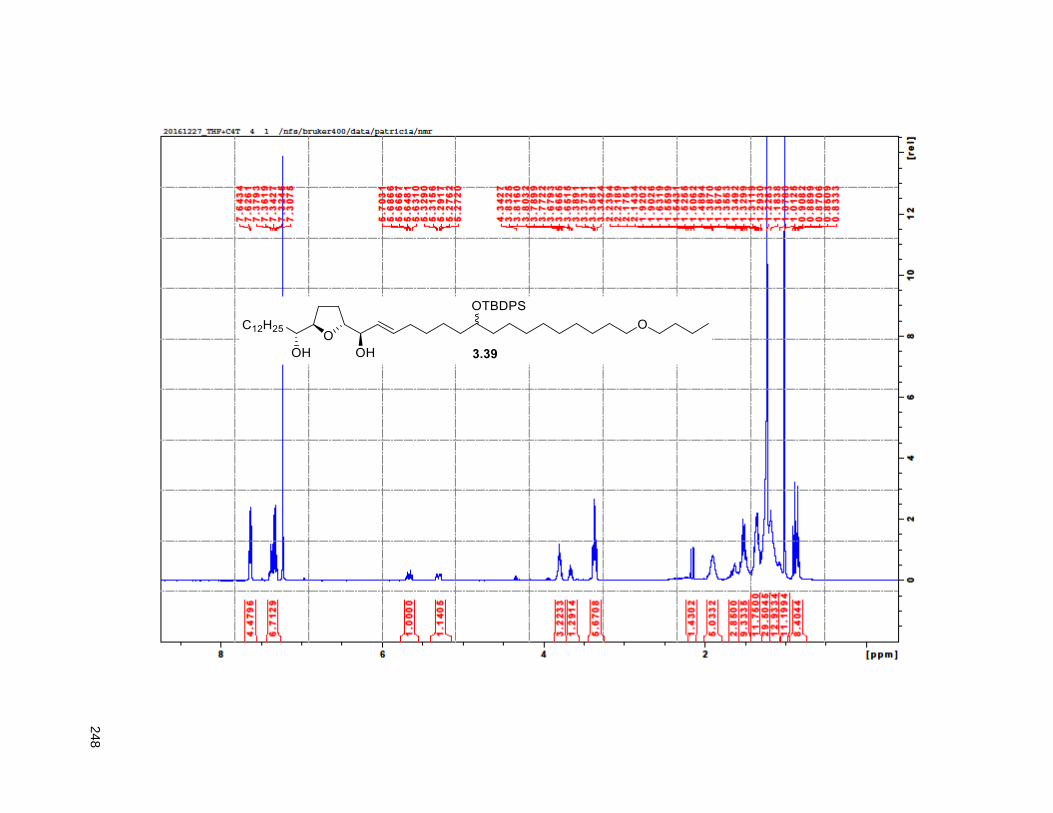
248
248
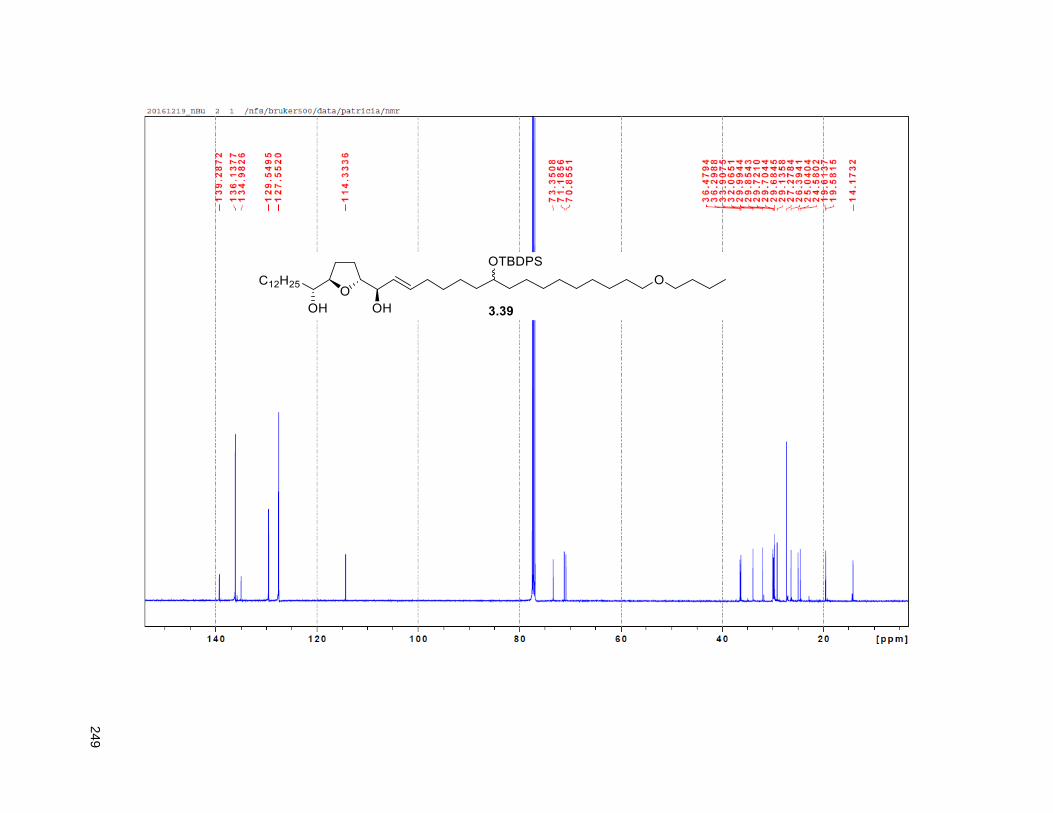
249
249
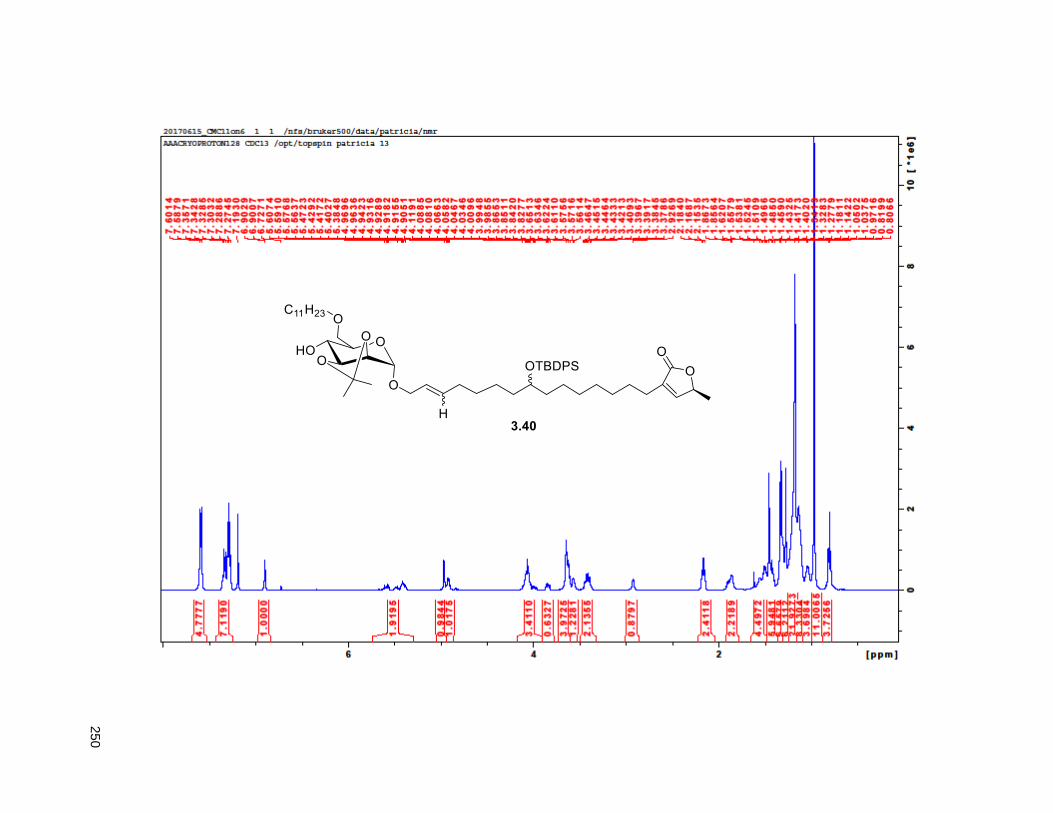
250
250
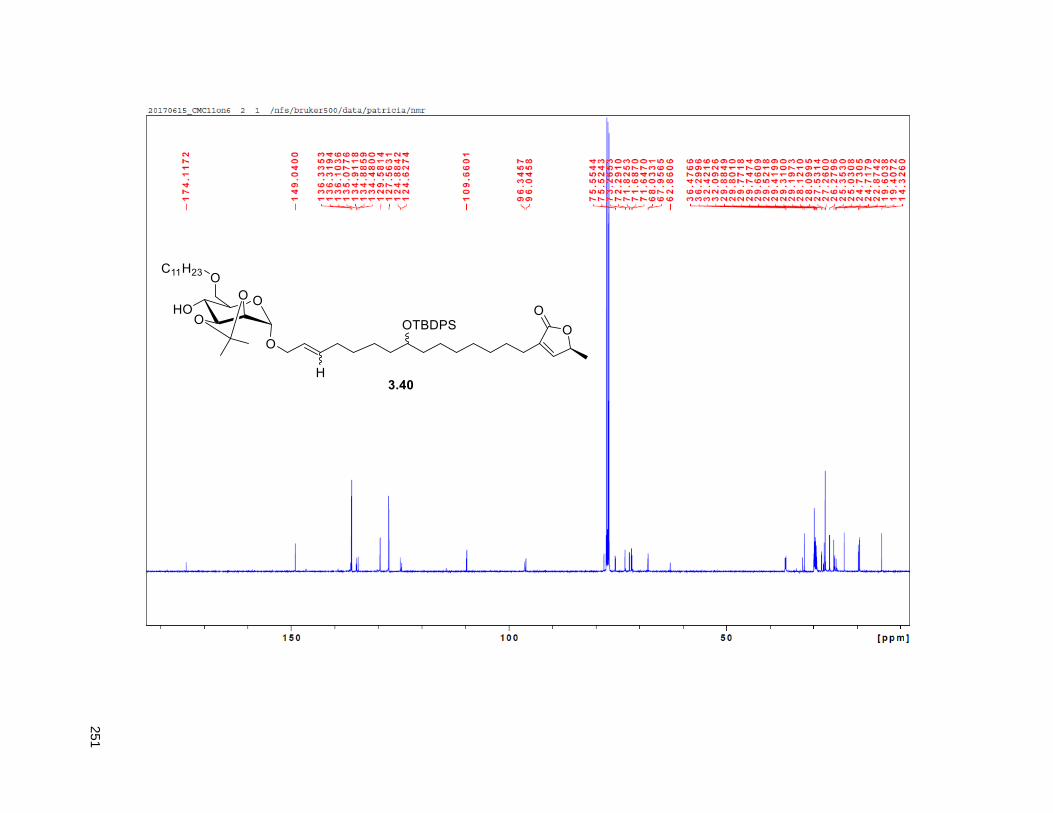
251
251
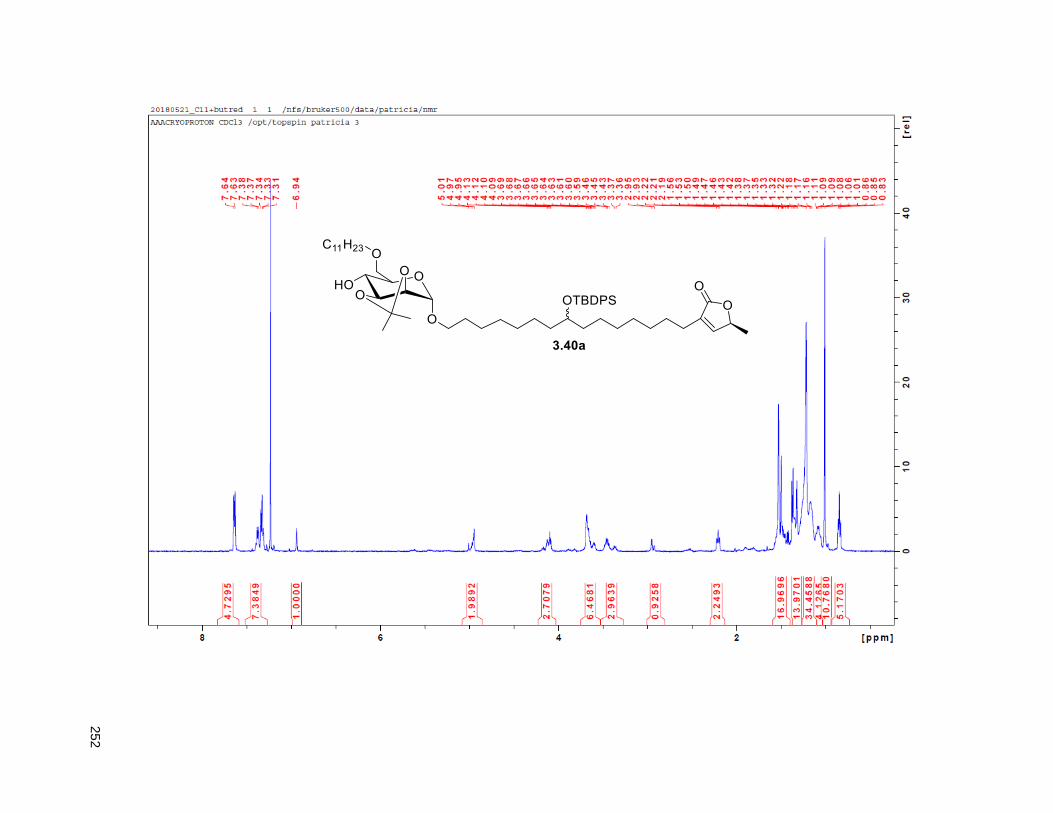
252
252
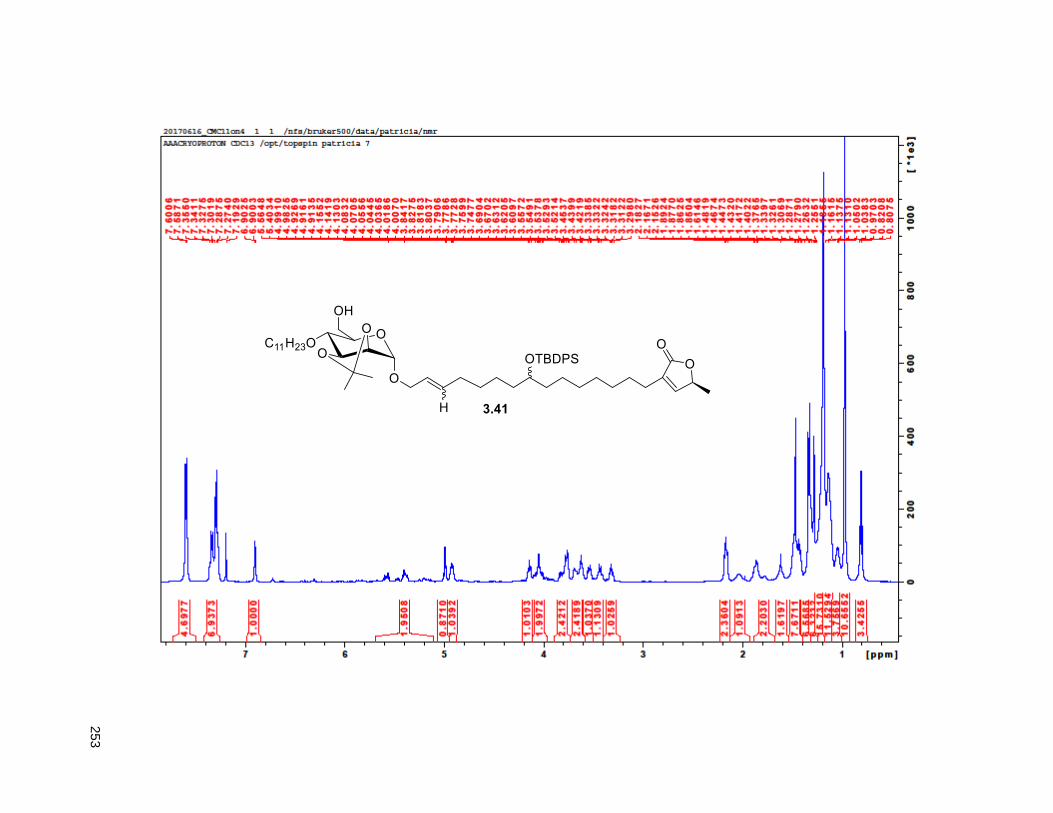
253
253
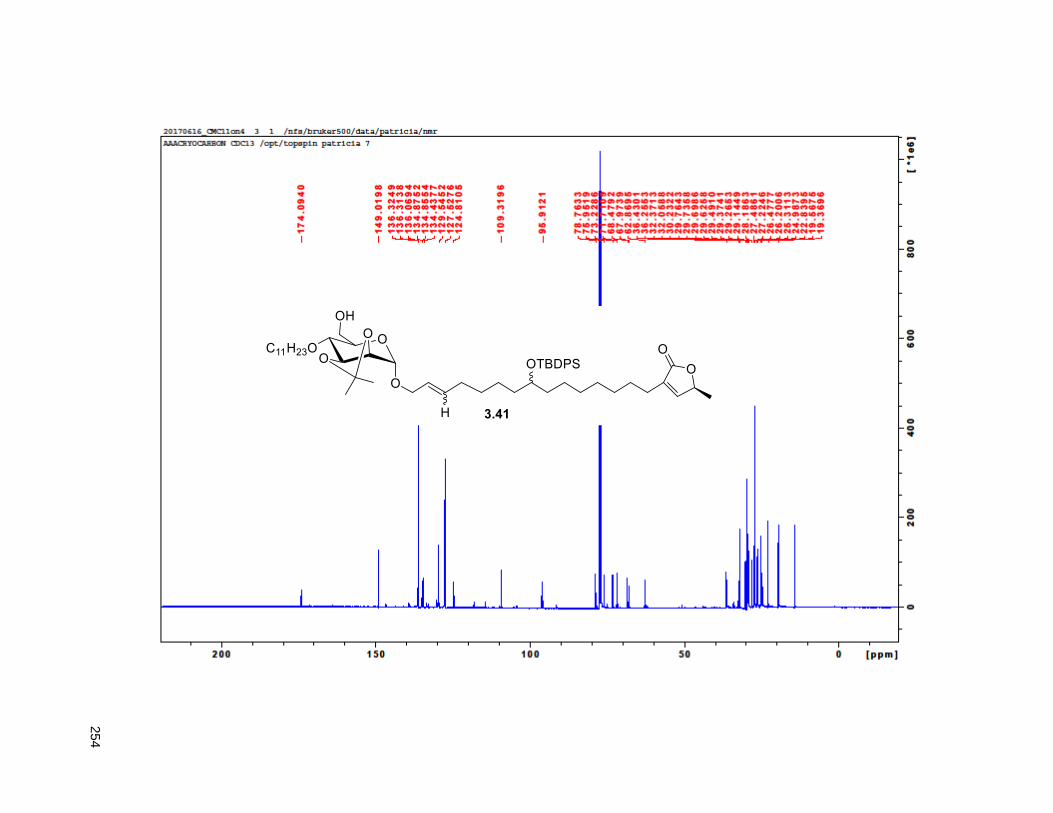
254
254
255
255
256
256
257
257
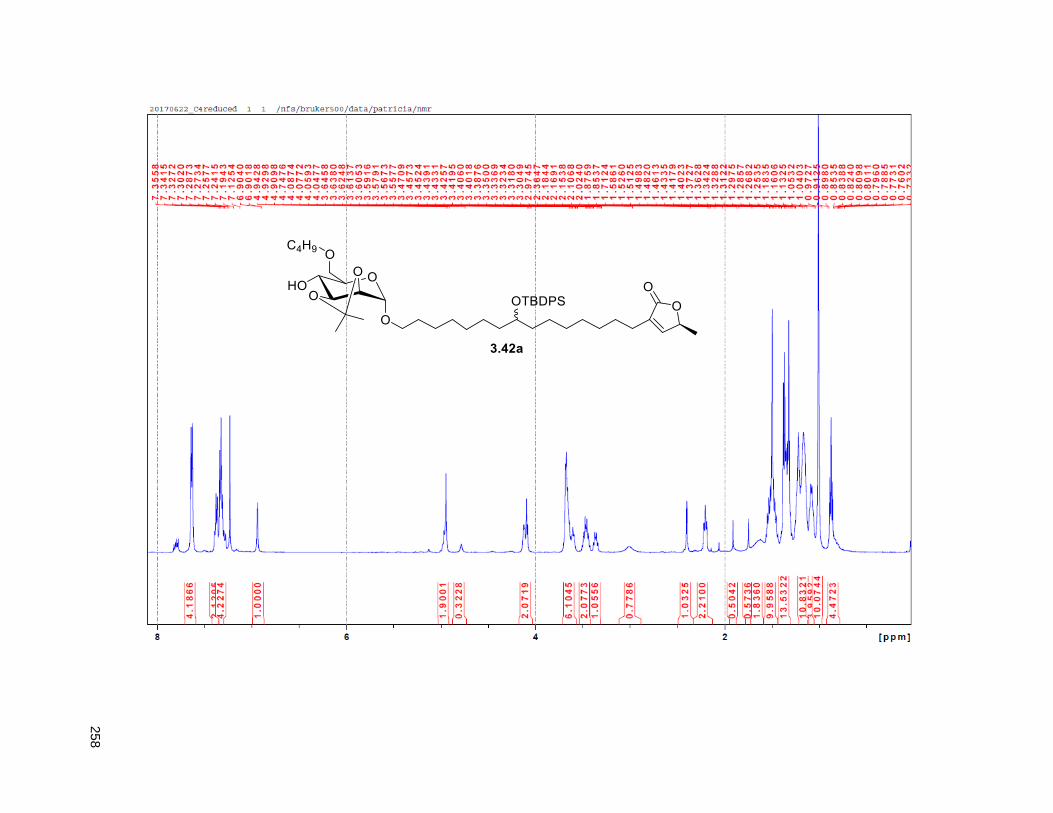
258
258
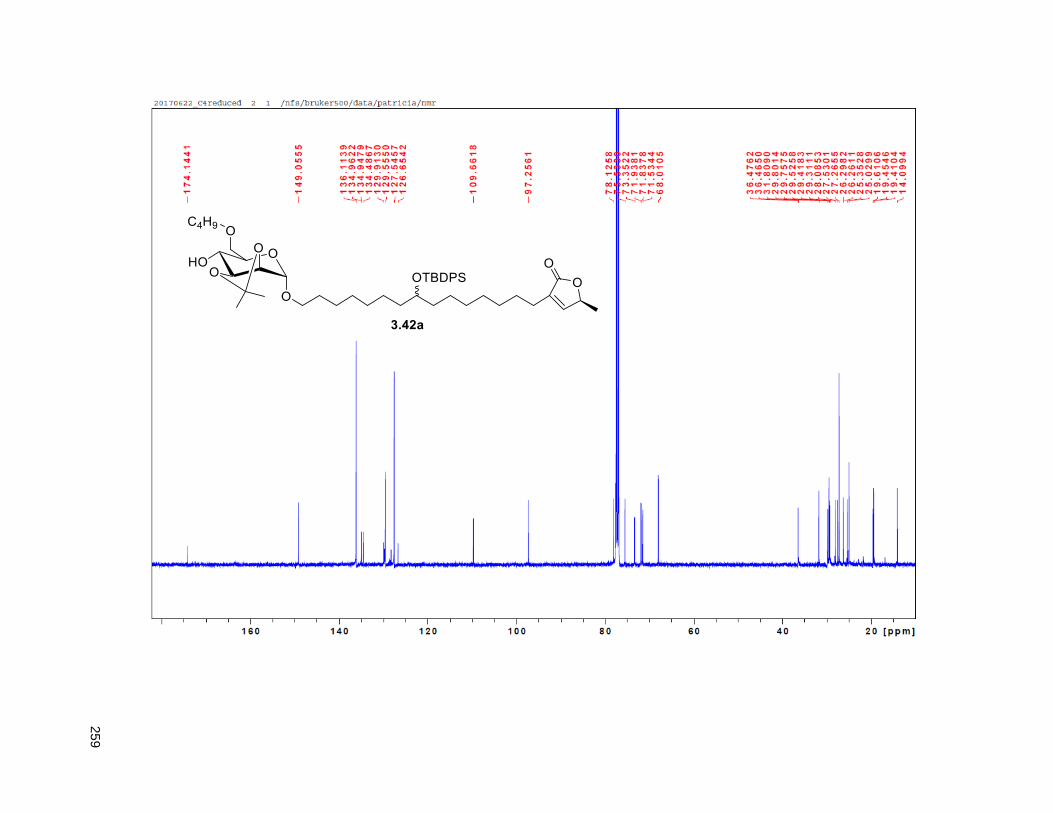
259
259

260
260
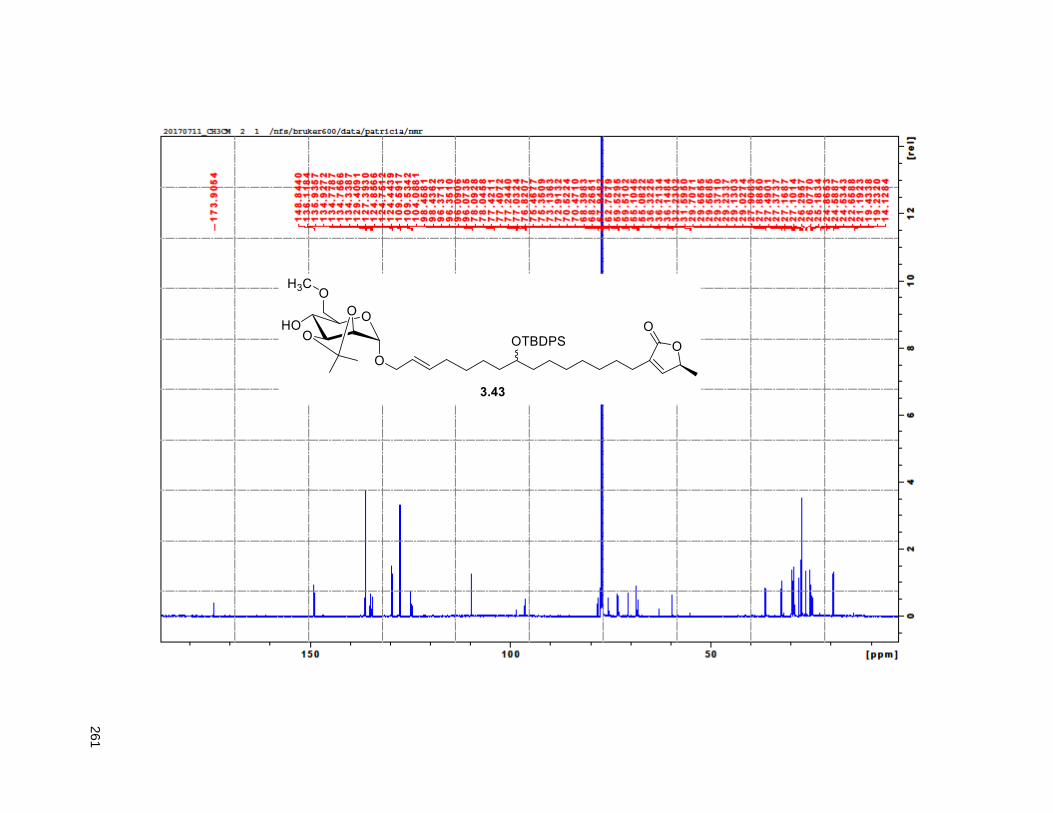
261
261
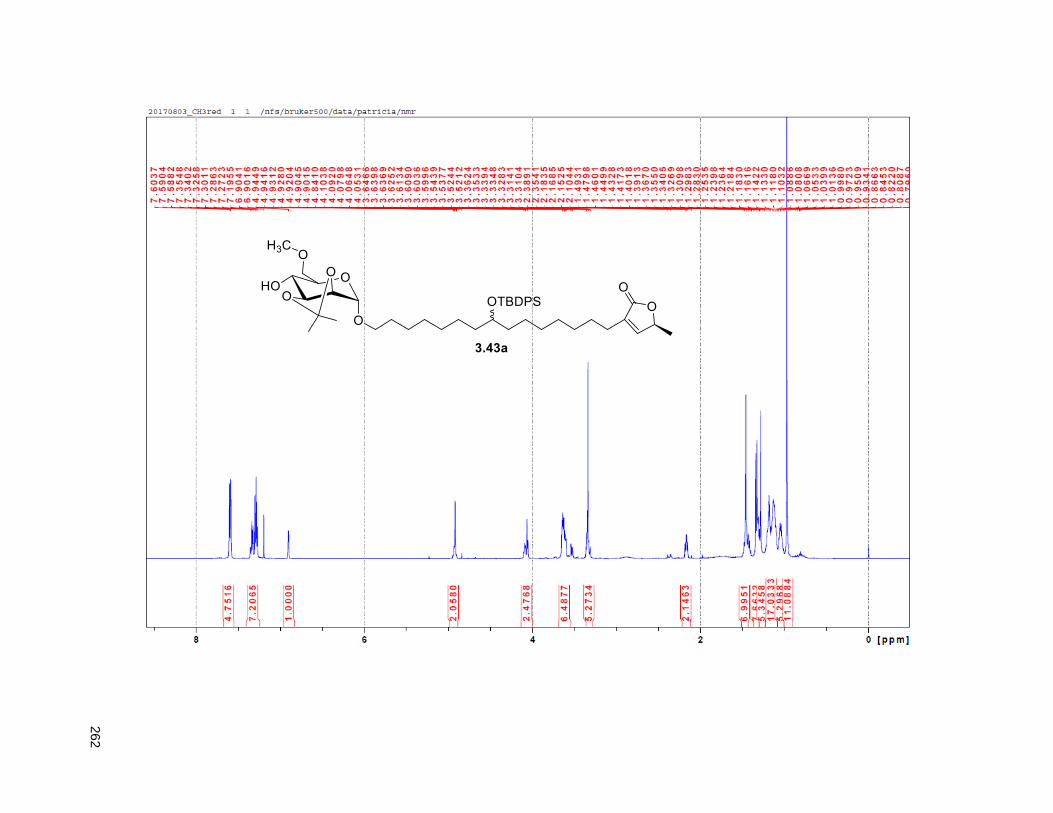
262
262
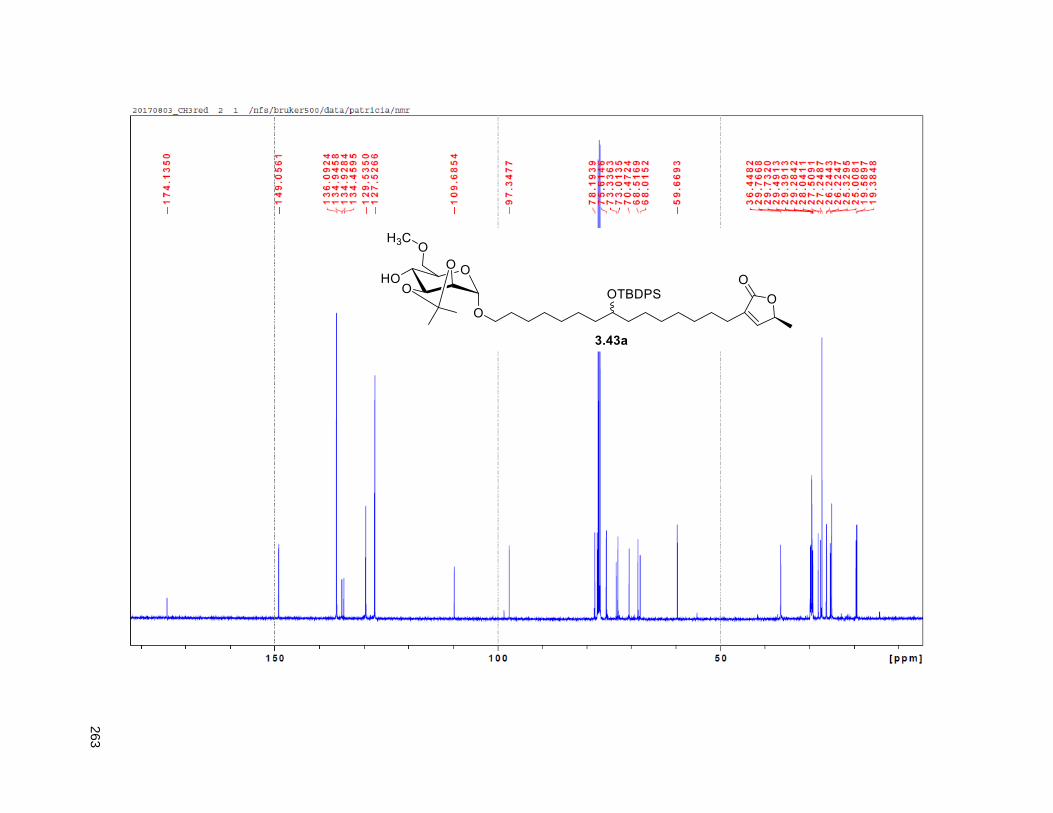
263
263

264
264
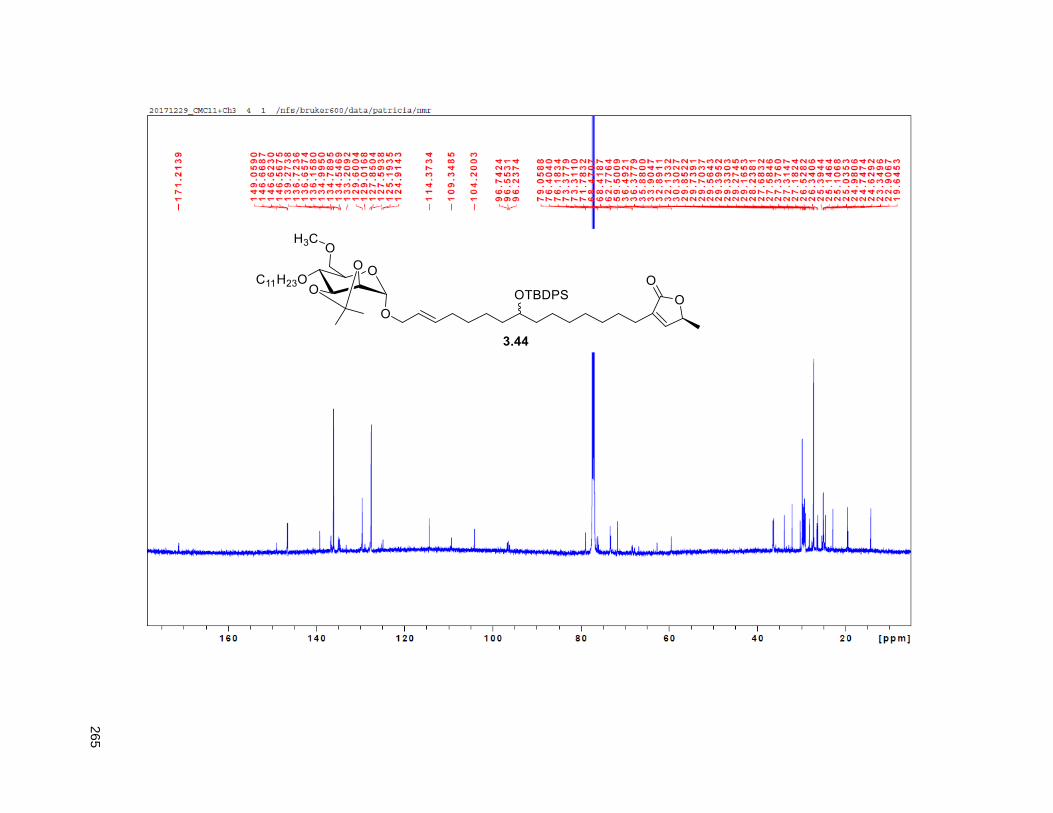
265
265

266
266
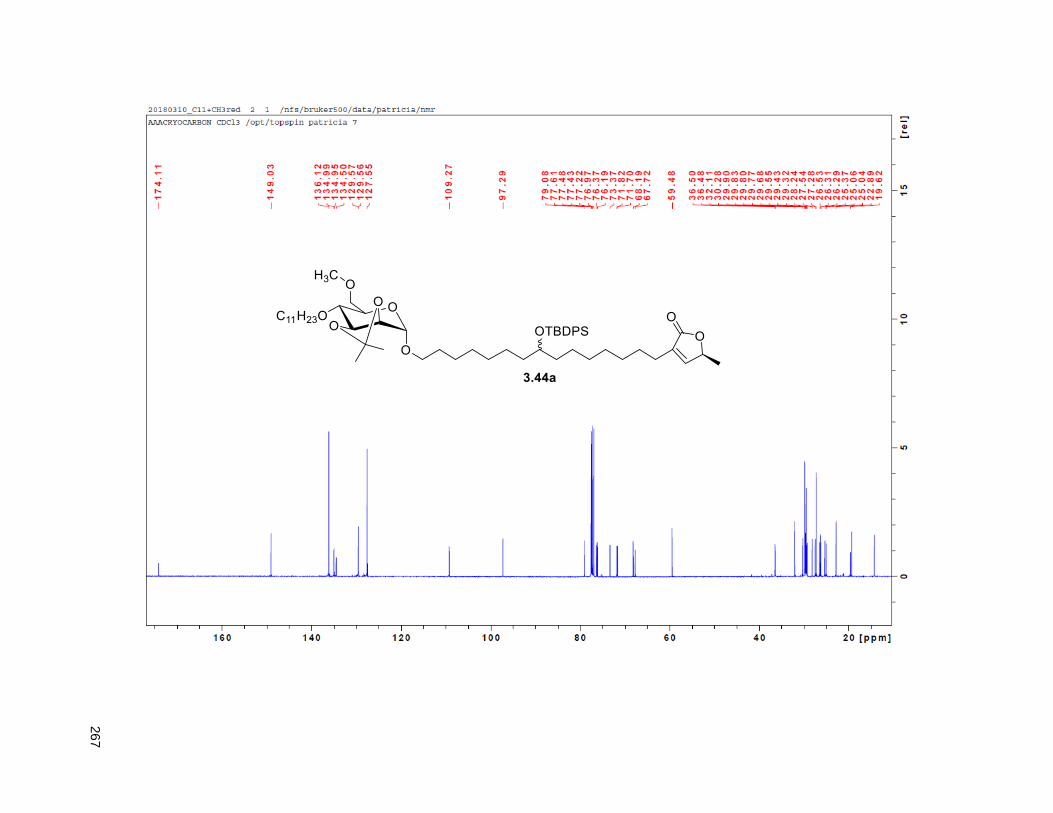
267
267
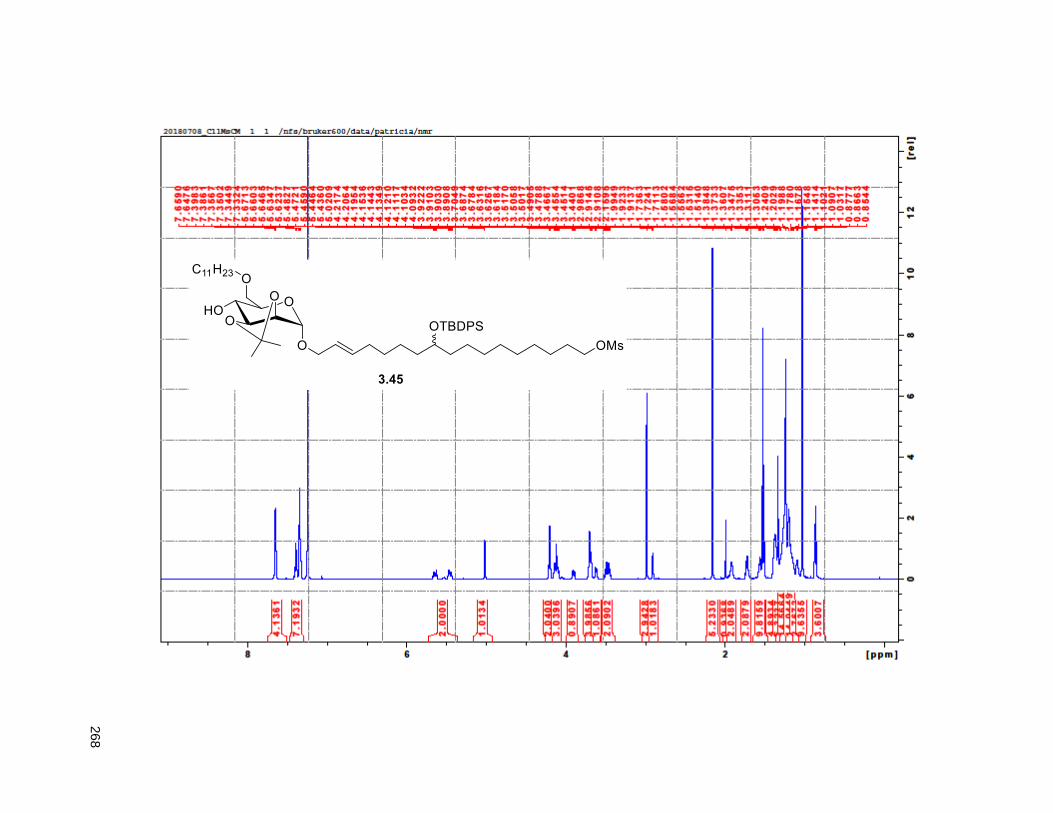
268
268
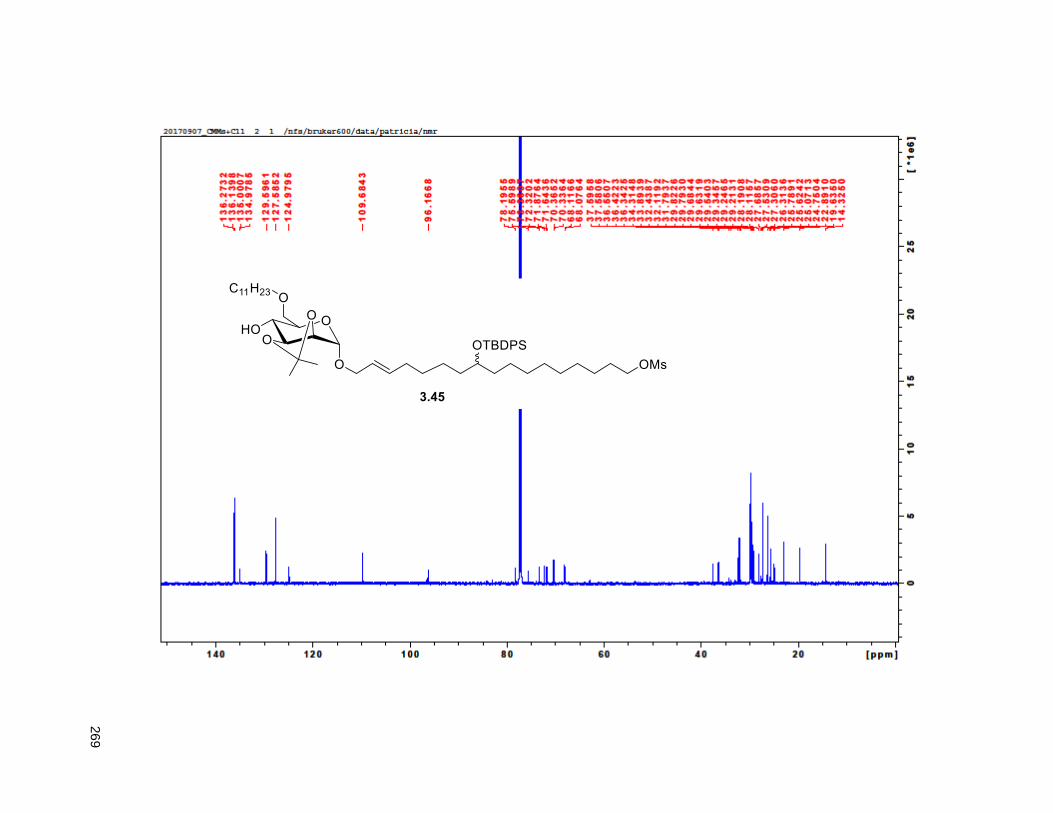
269
269
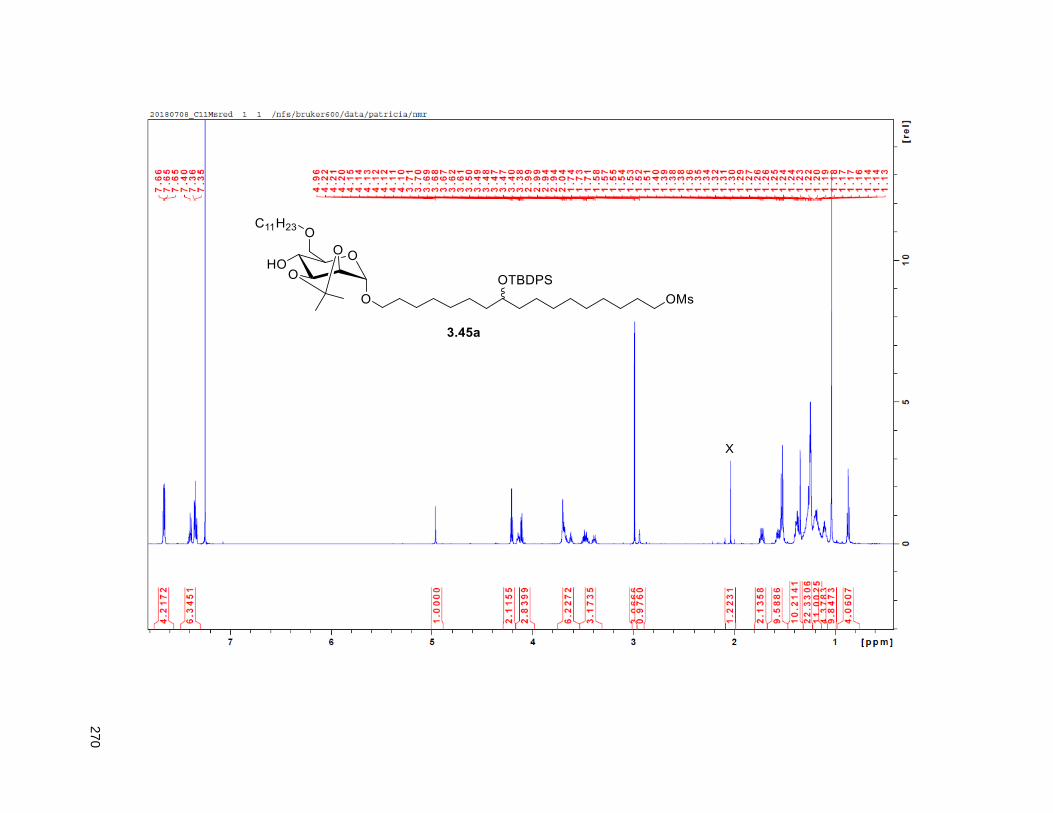
270
270
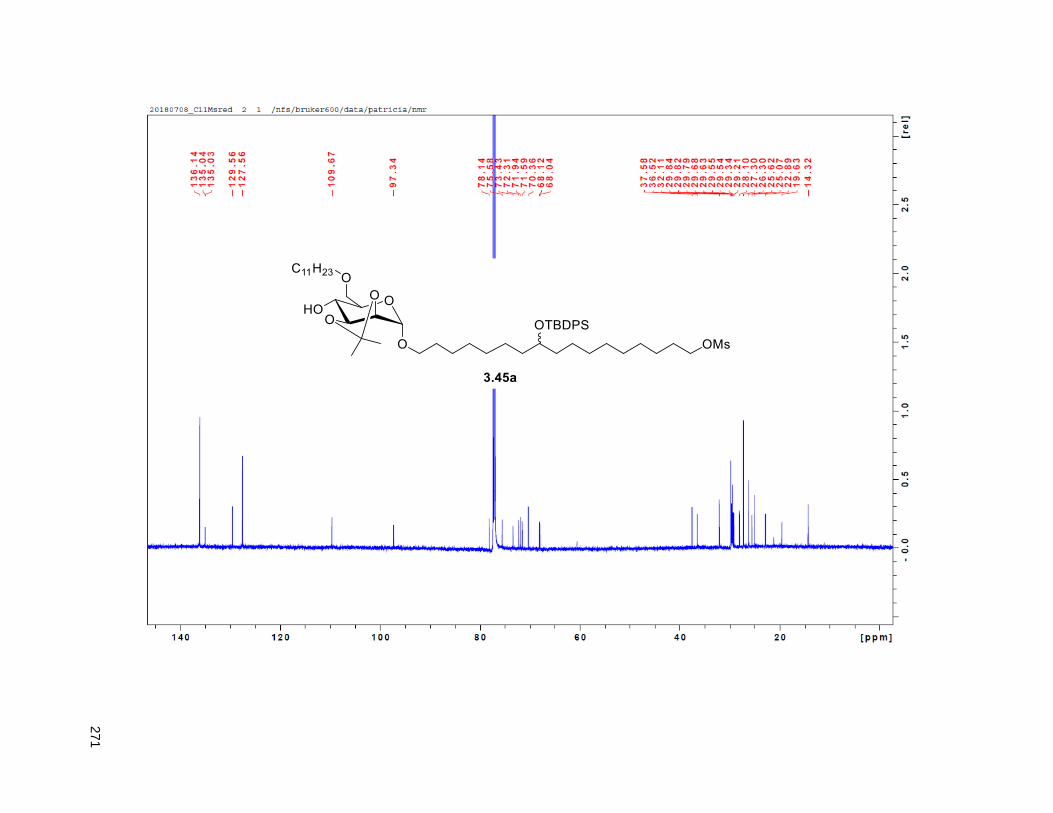
271
271
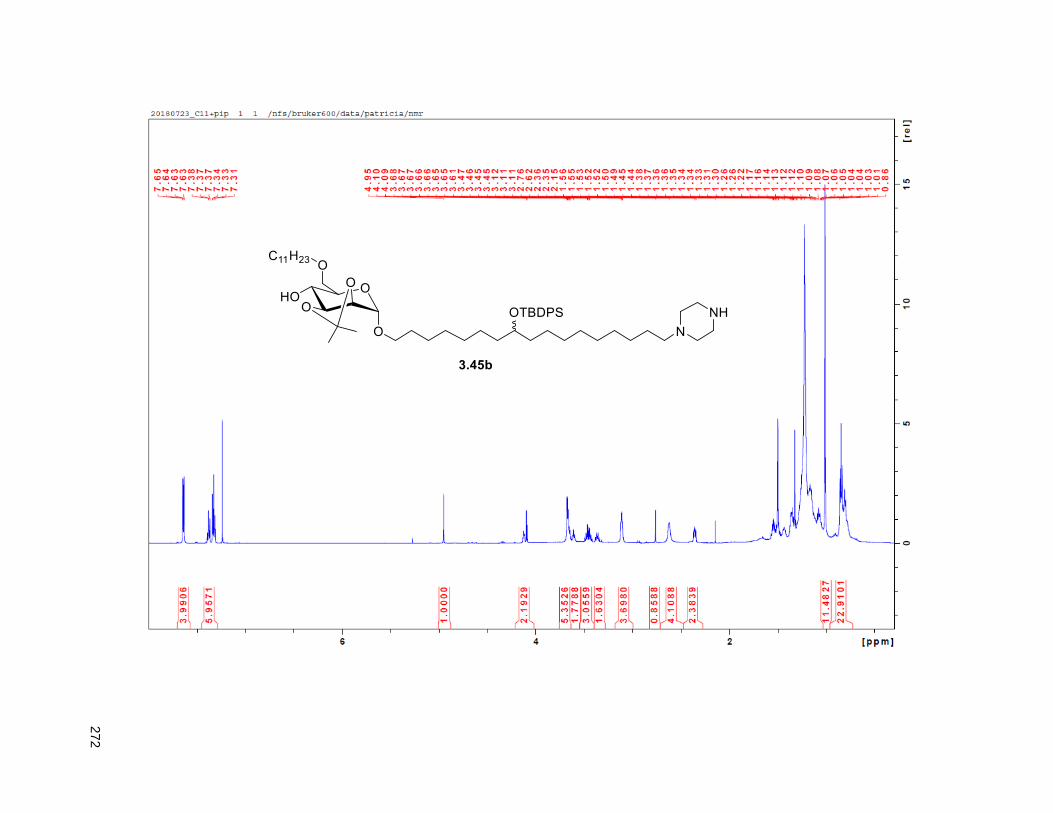
272
272
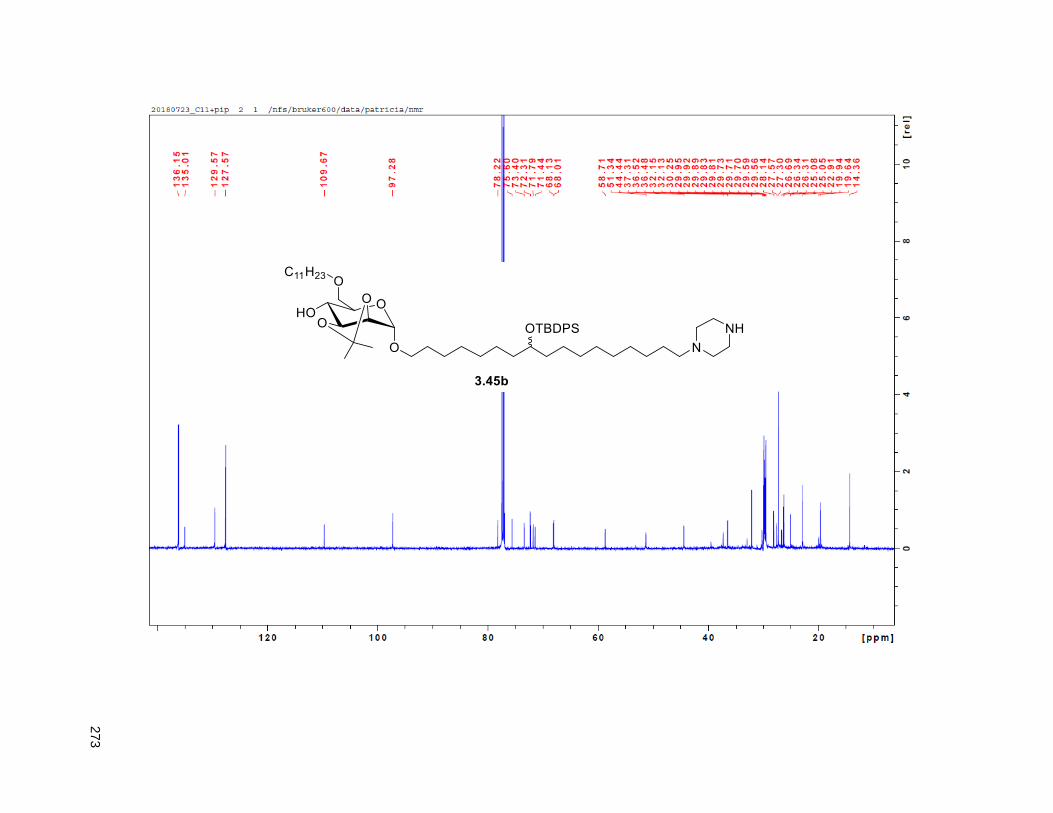
273
273

274
274
275
275
276
276
277
277
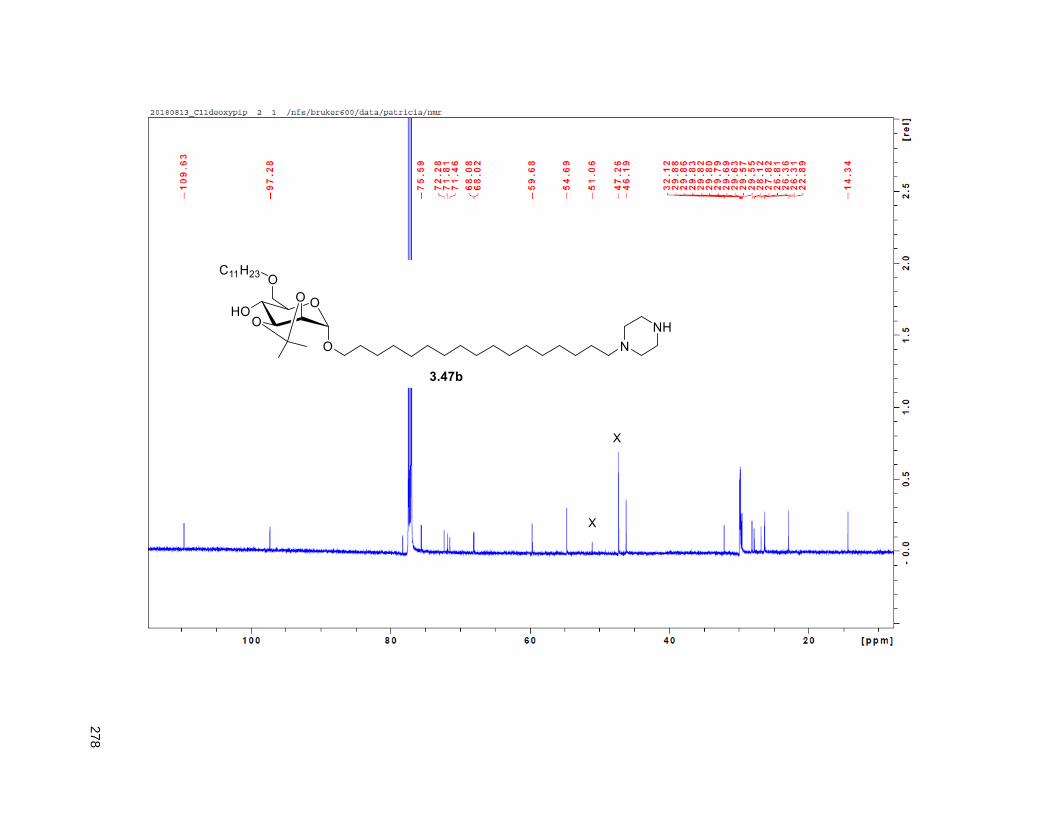
278
278
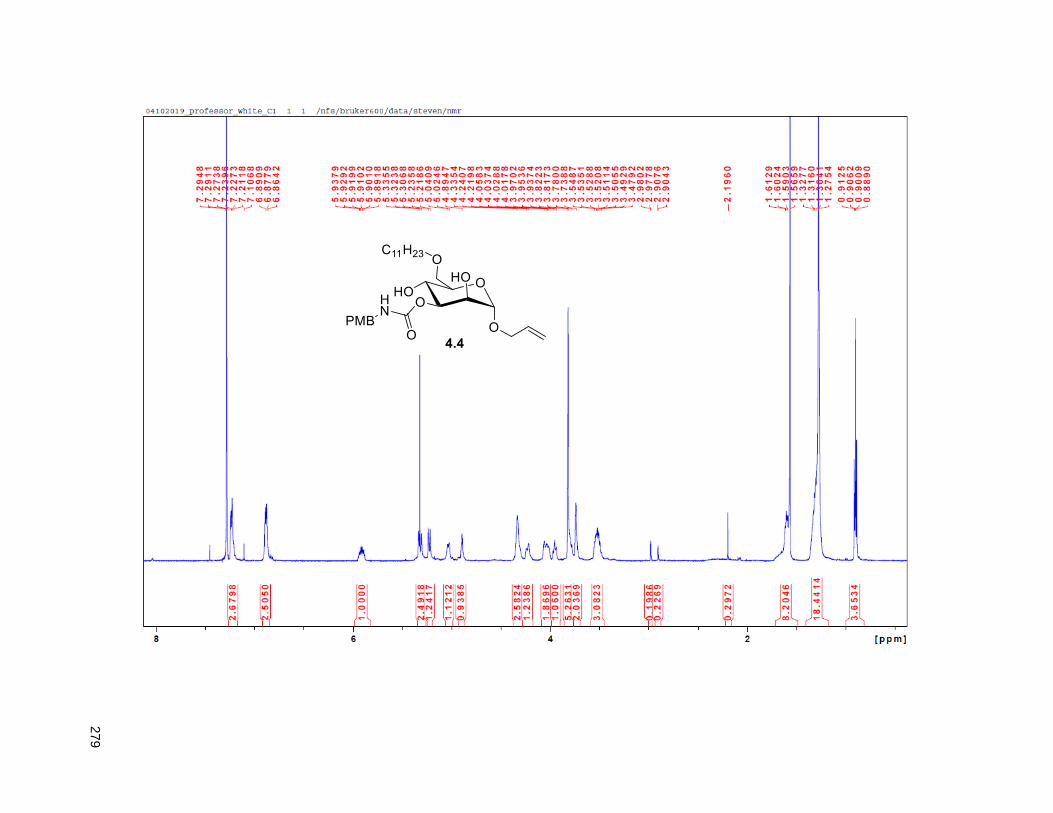
279
279
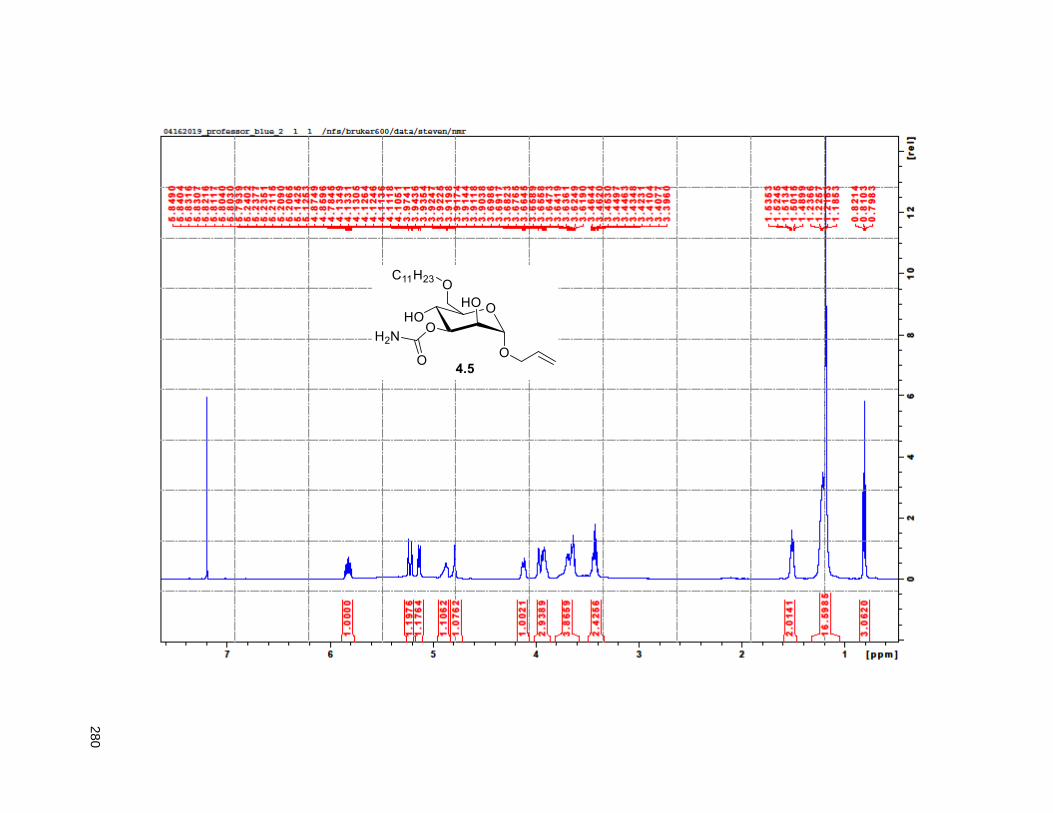
280
280
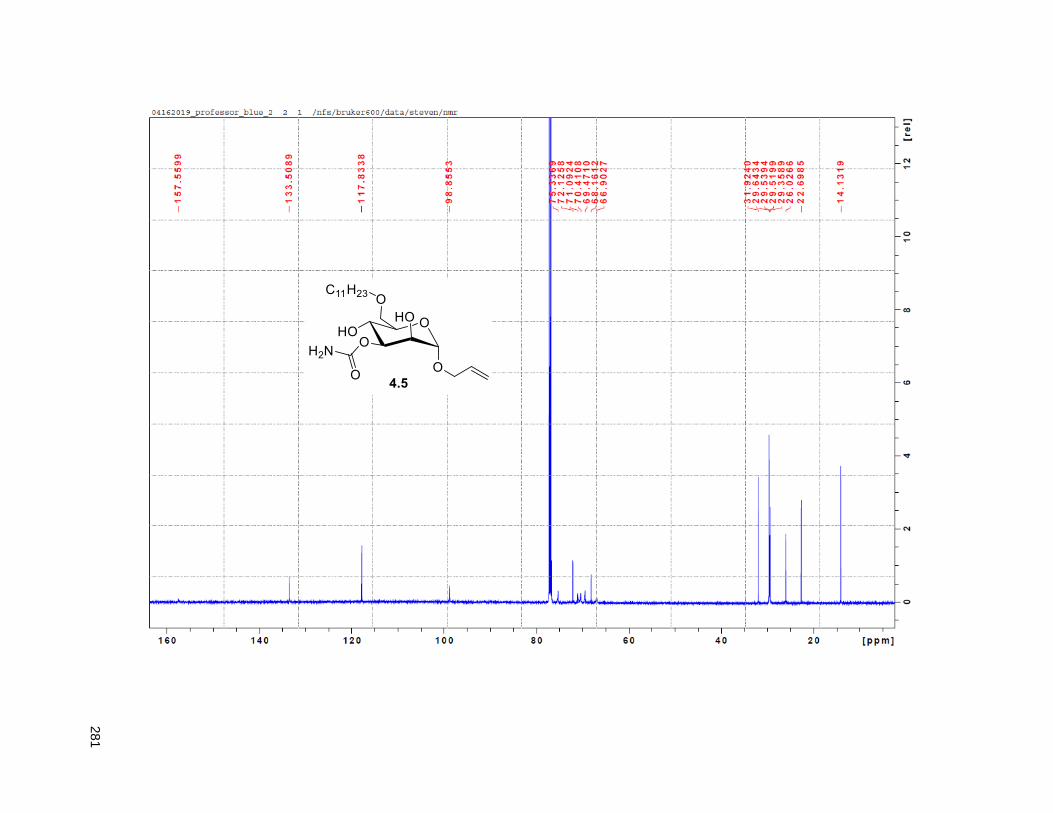
281
281

282
282
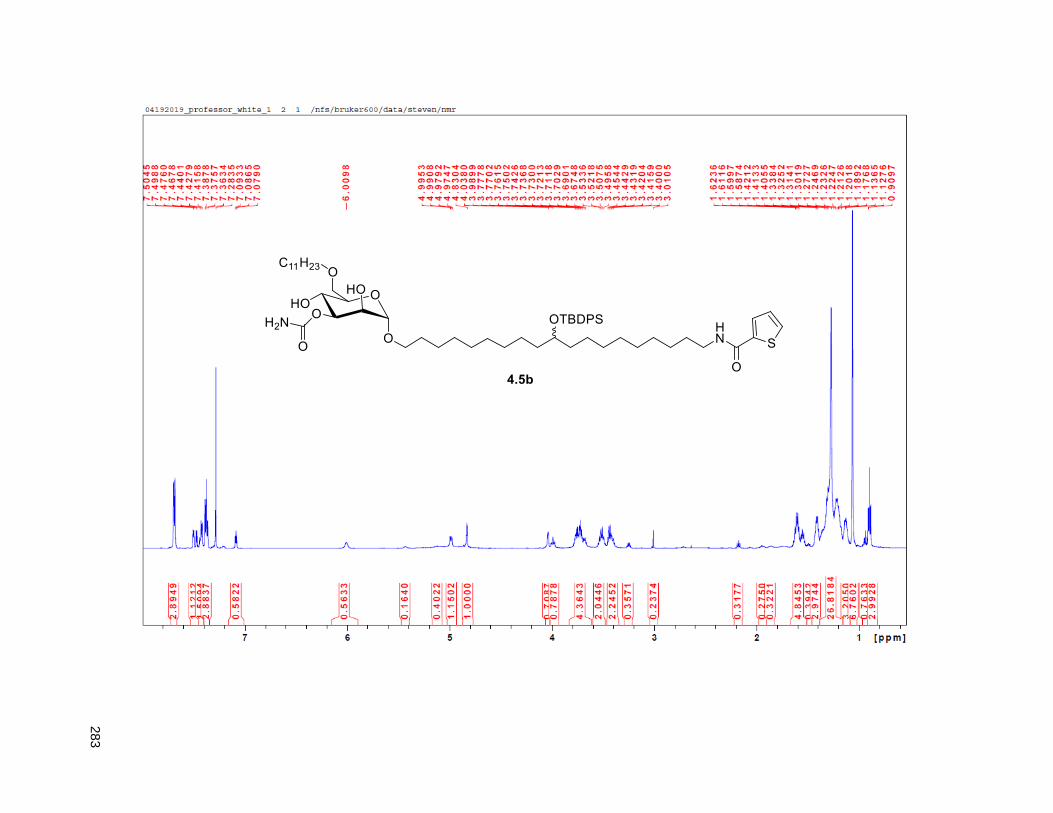
283
283
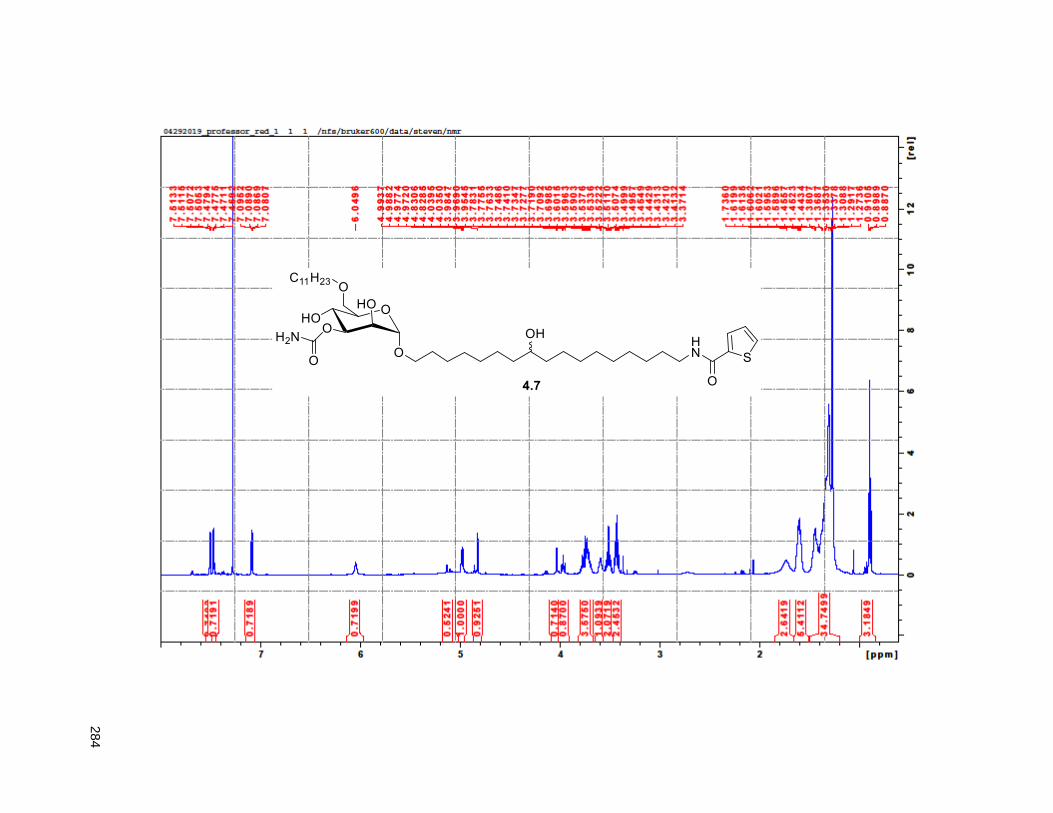
284
284
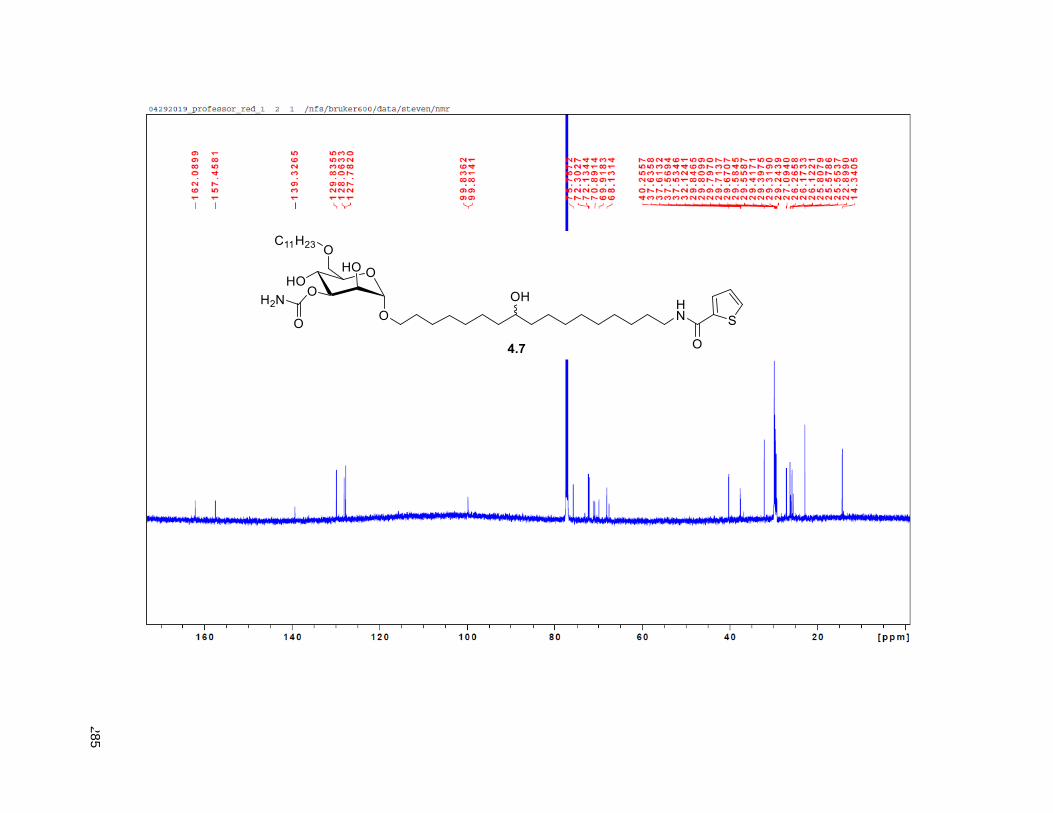
285
285
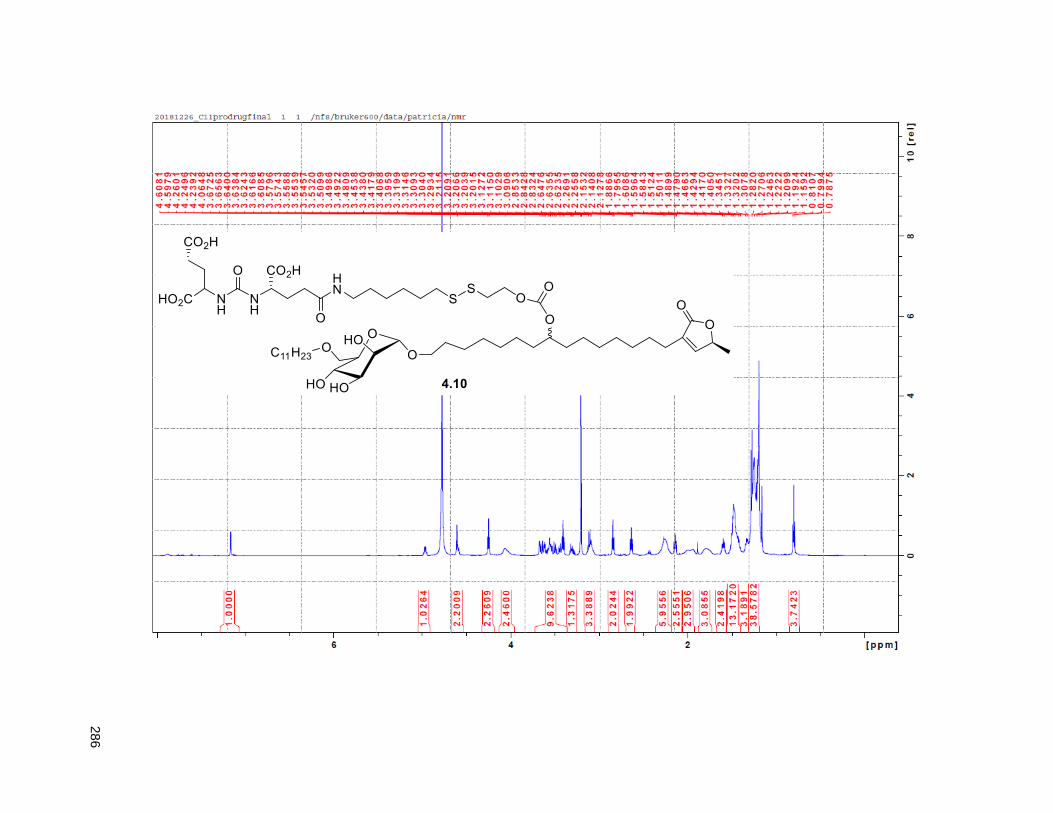
286
286
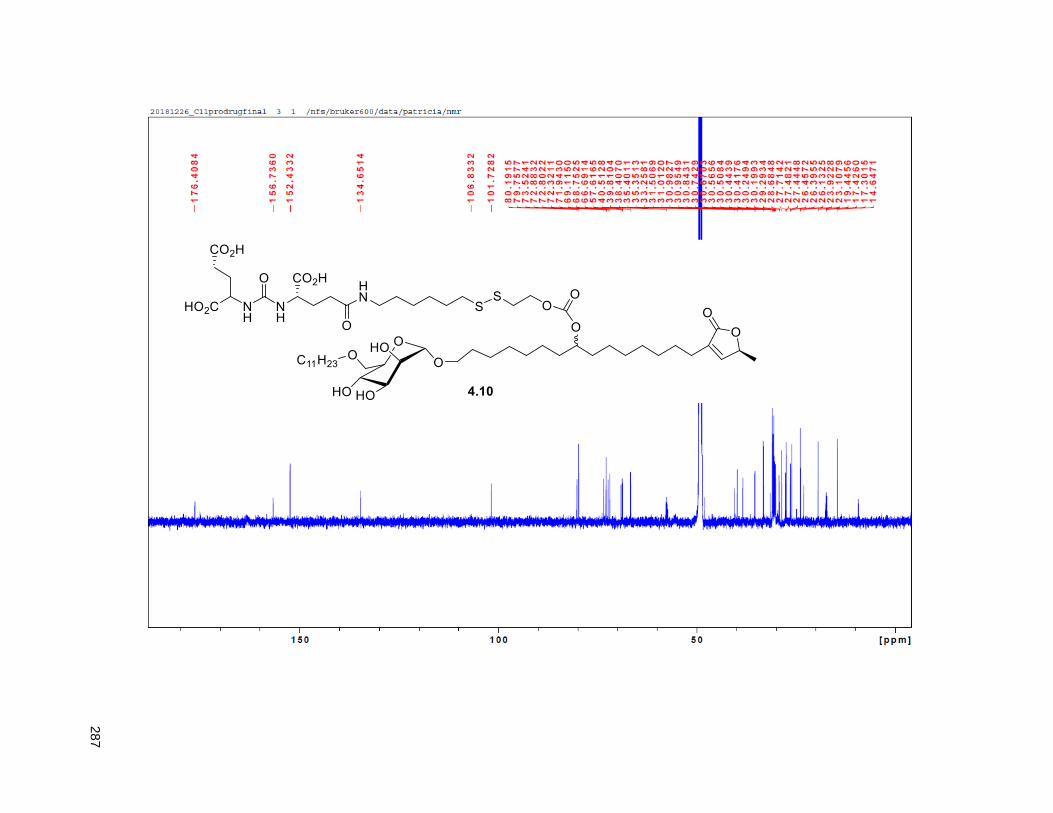
287
287
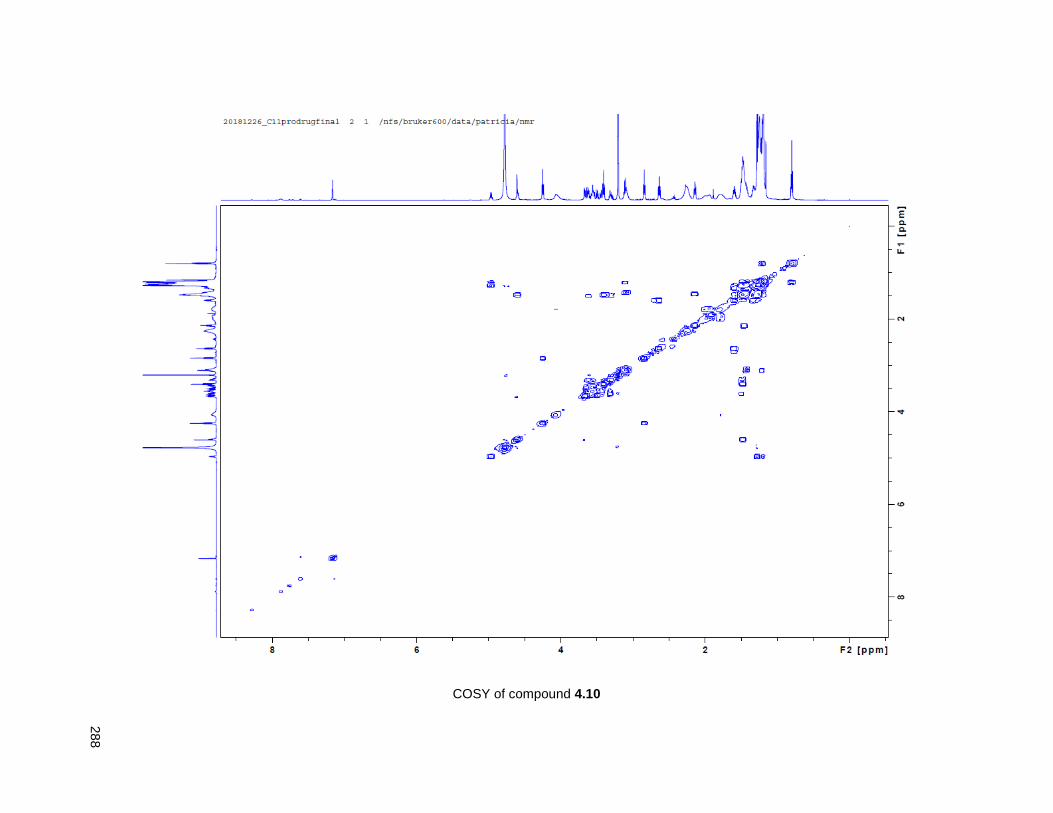
288
288
COSY of compound 4.10
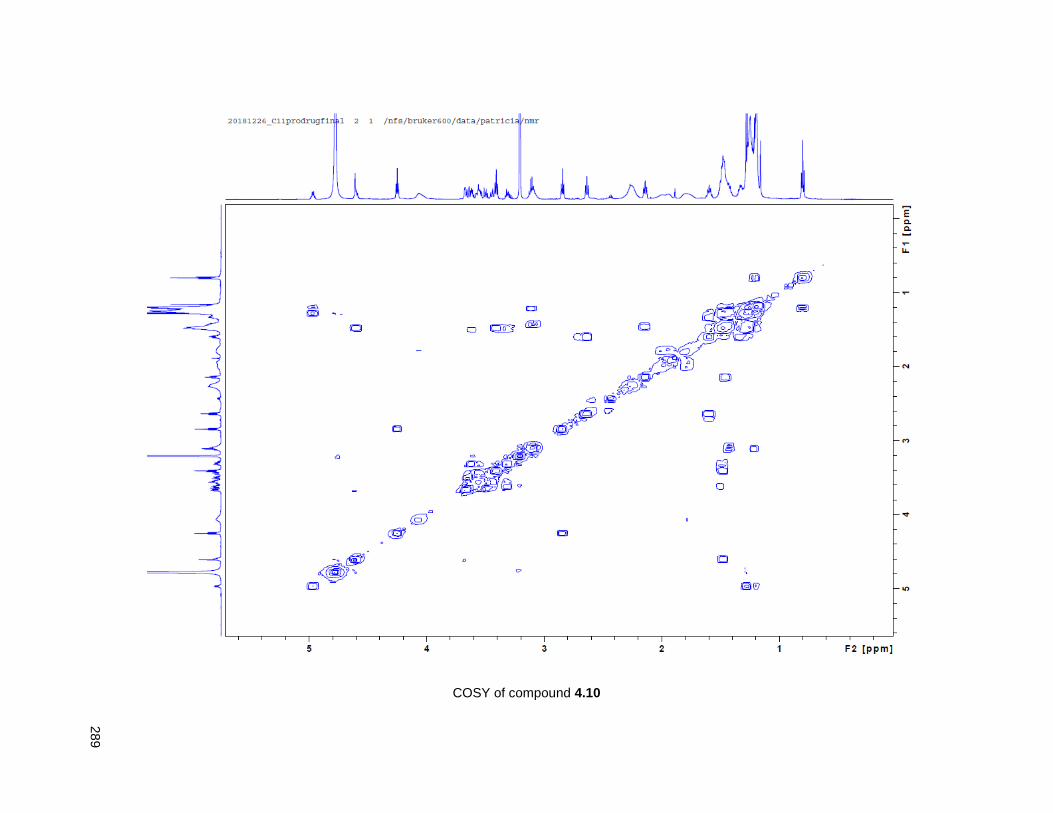
289
289
COSY of compound 4.10
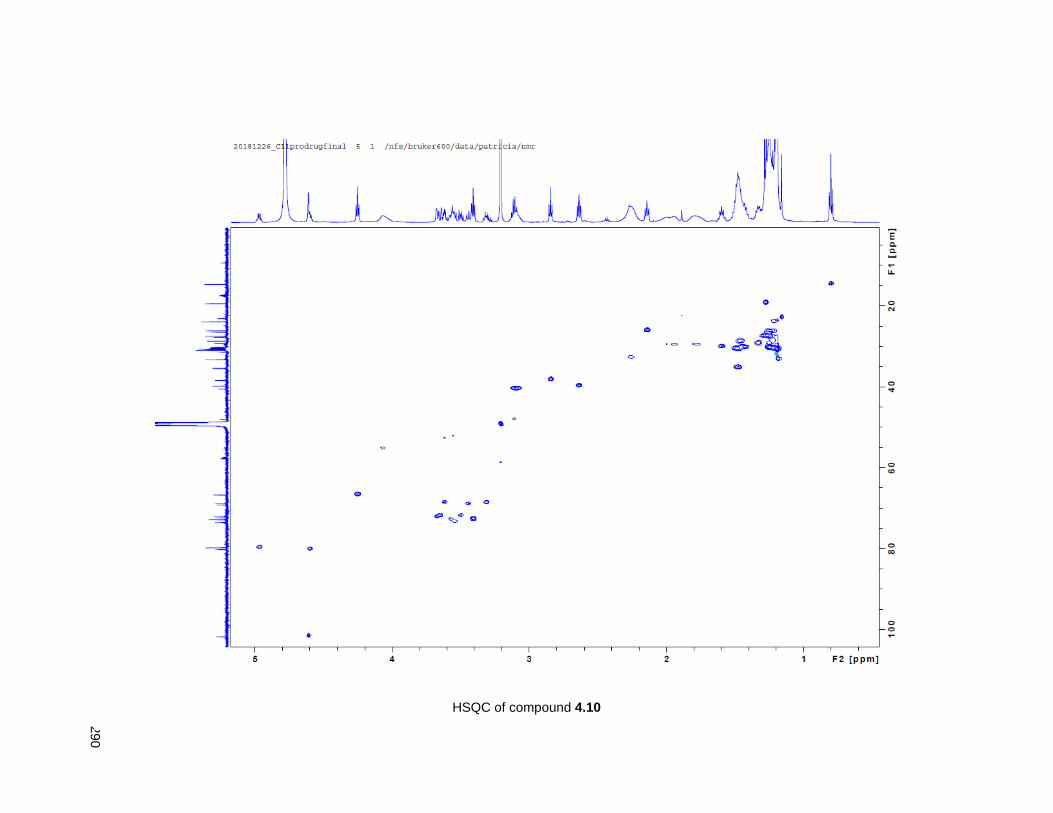
290
290
HSQC of compound 4.10
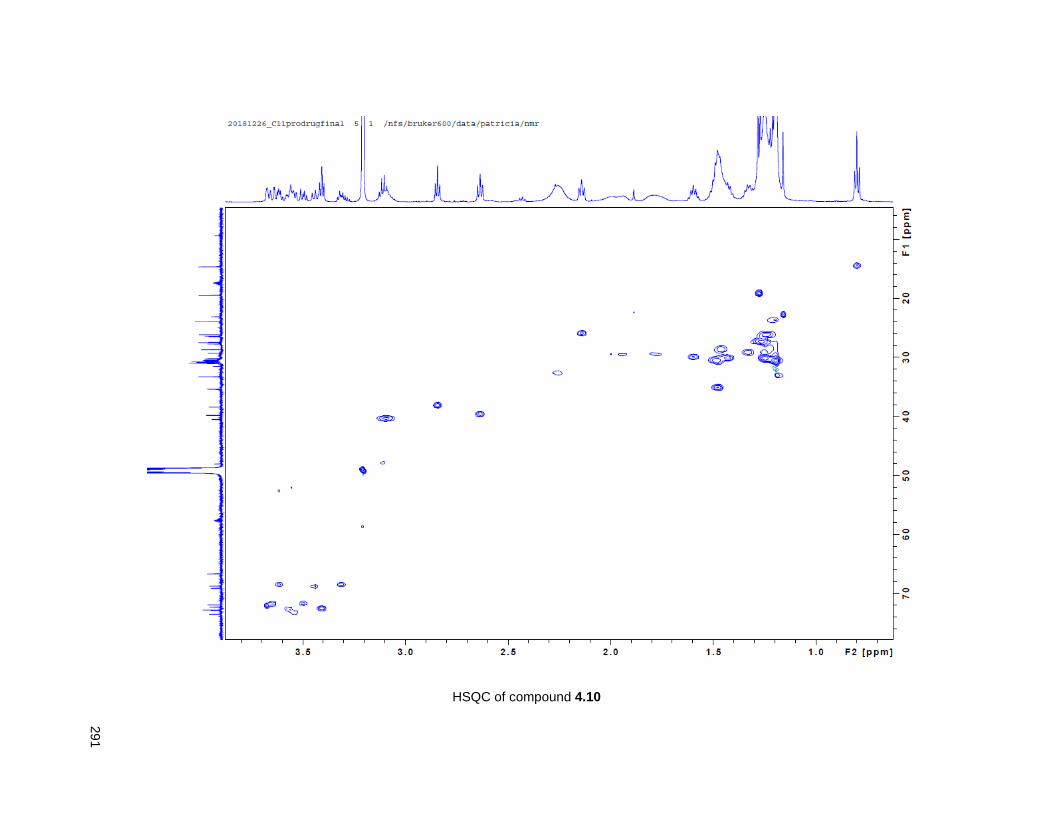
291
291
HSQC of compound 4.10
292
292
HSQC of compound 4.10
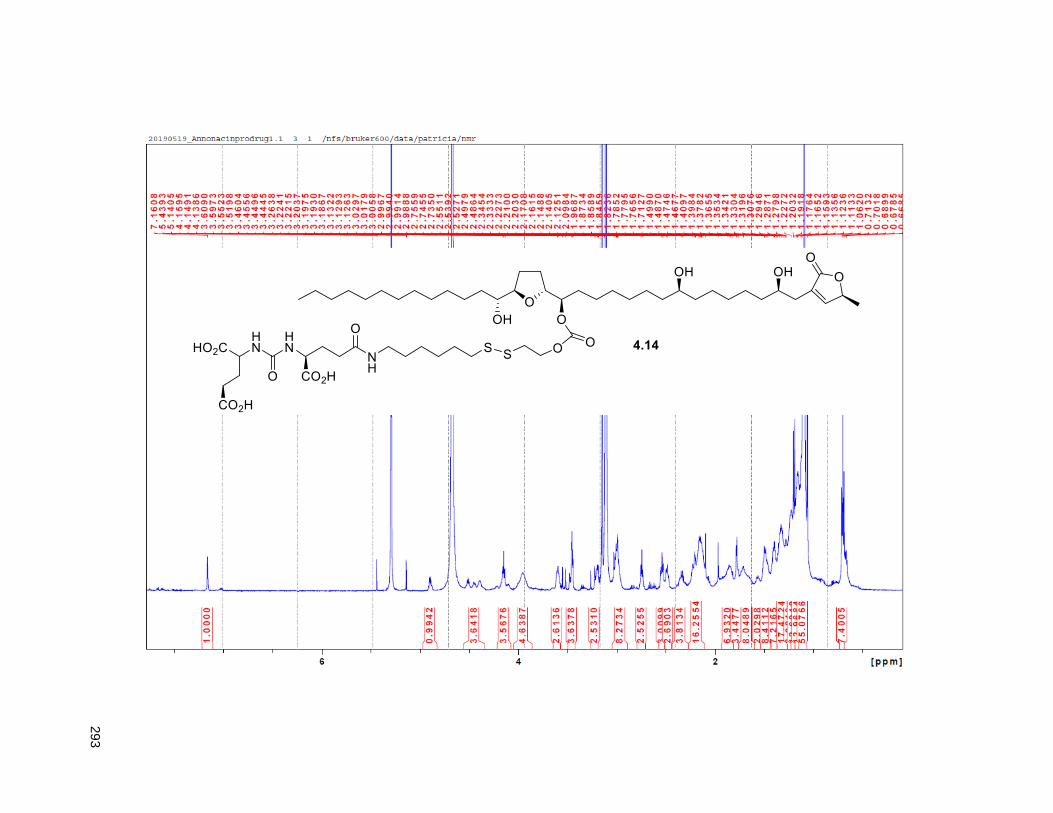
293
293
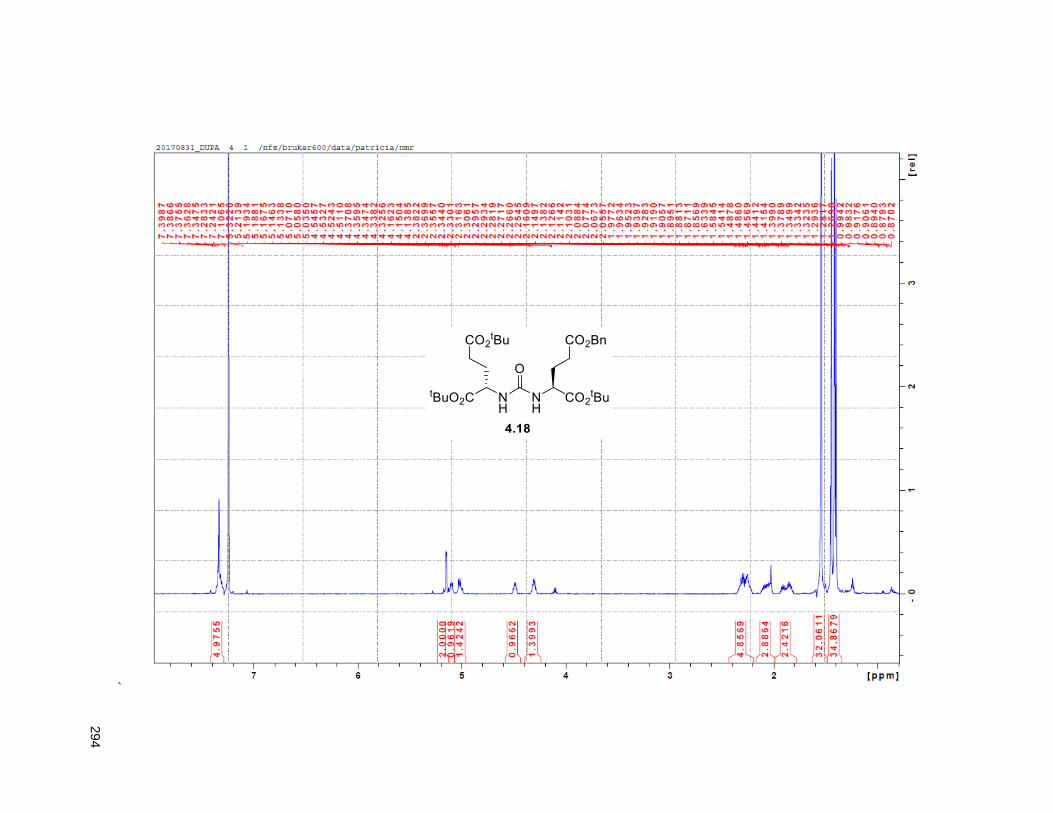
294
294
`
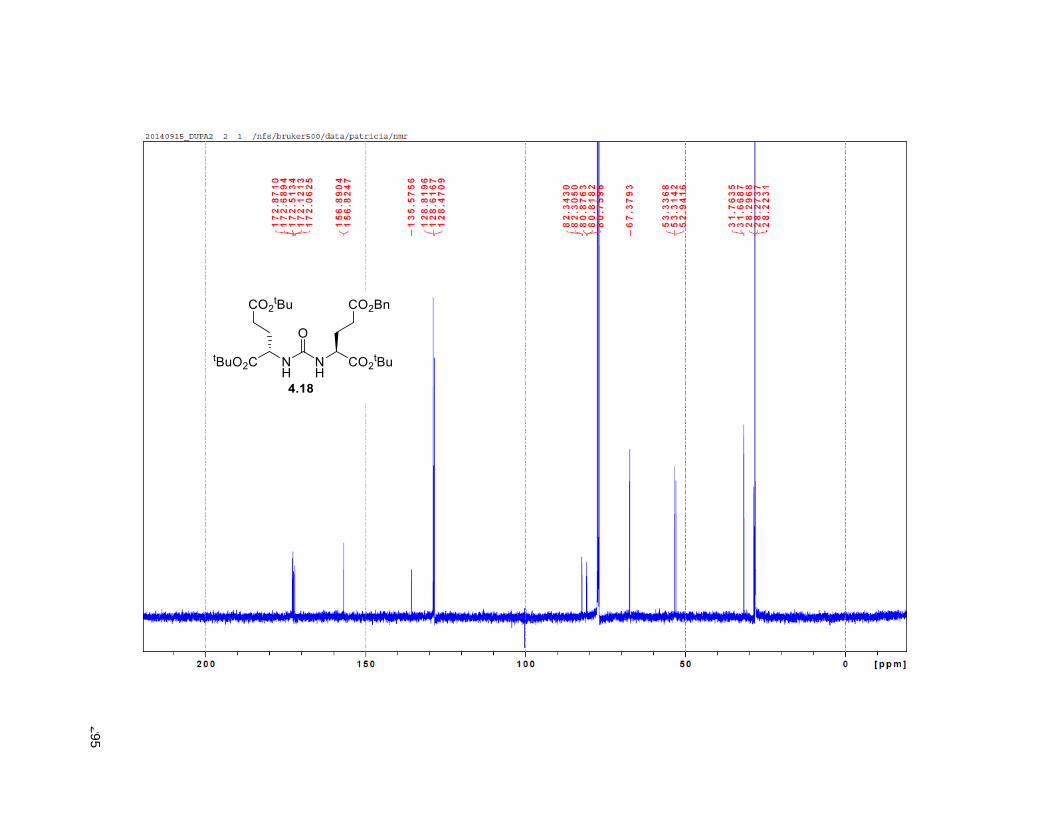
295
295
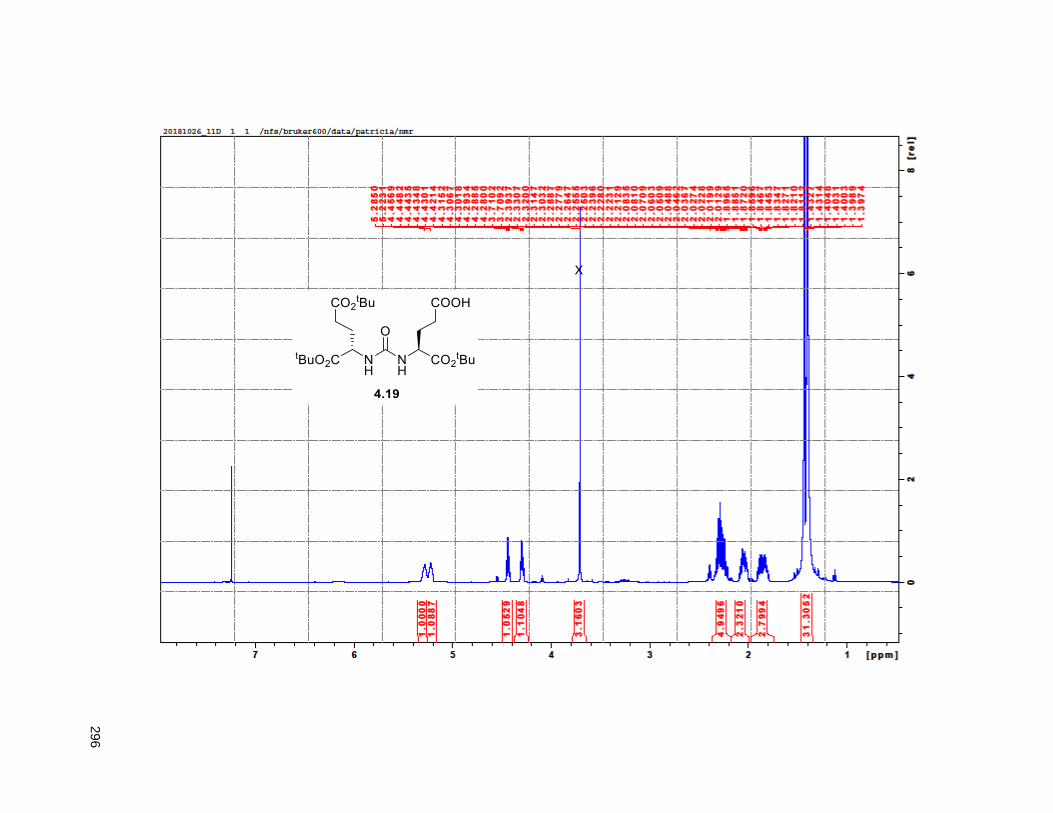
296
296
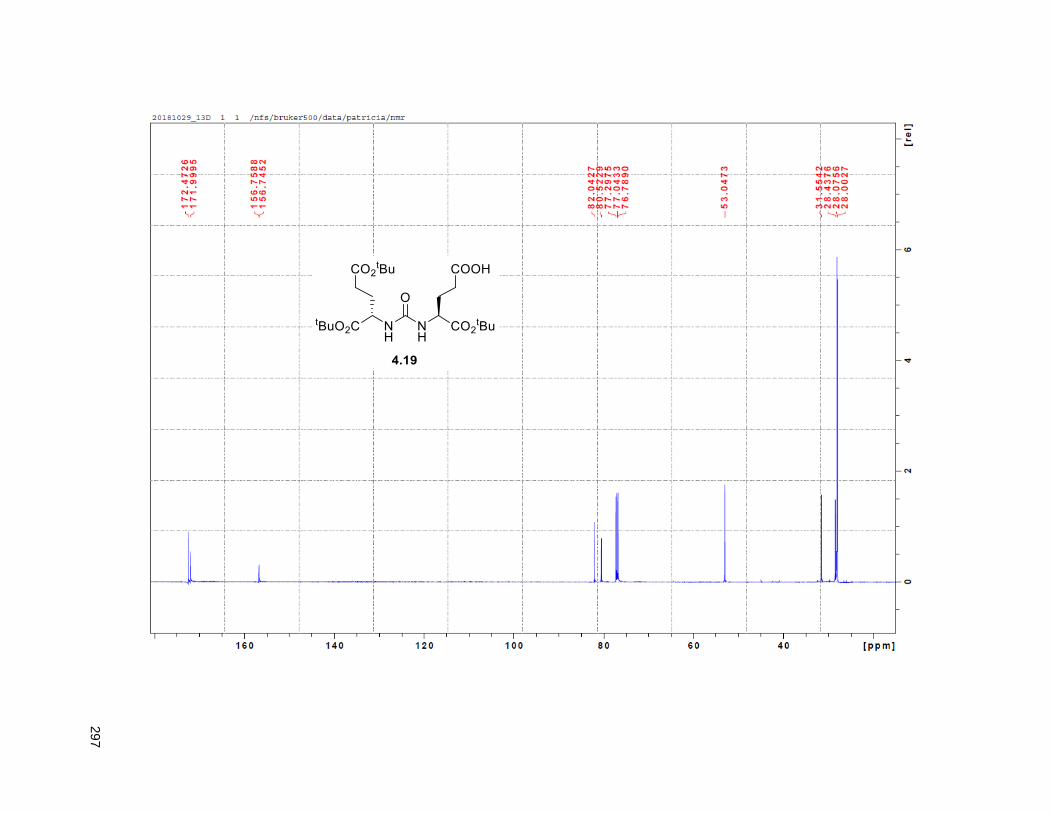
297
297
298
298
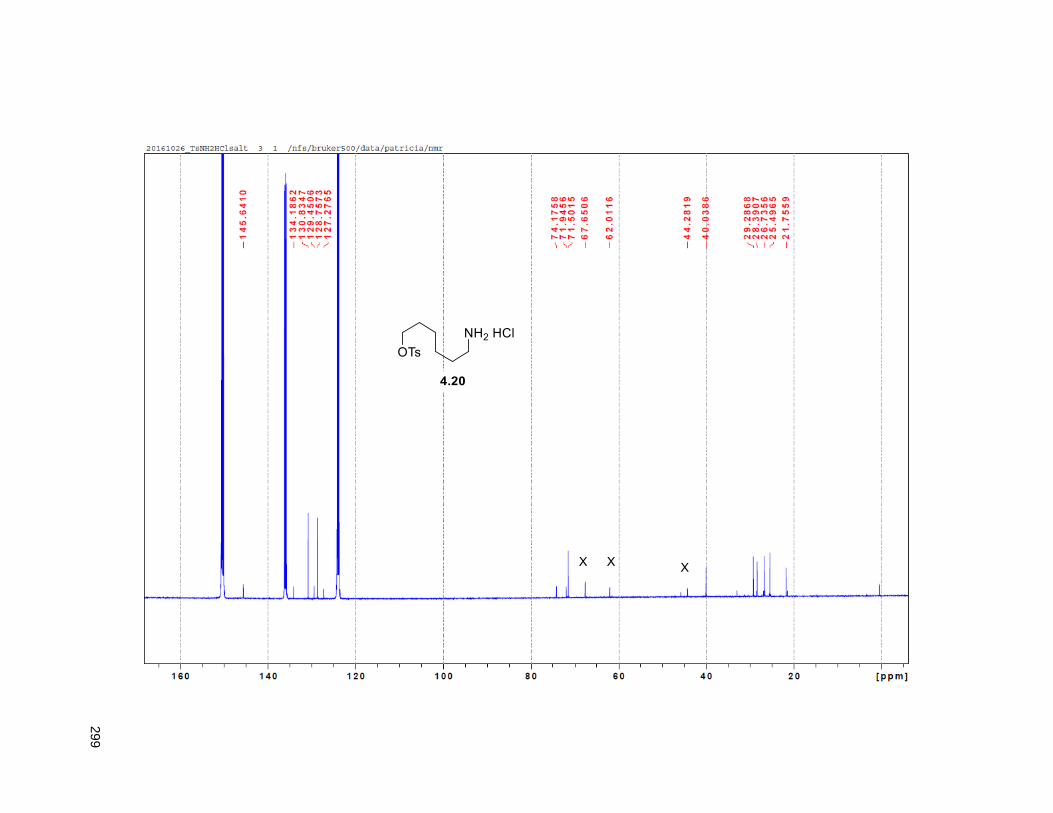
299
299
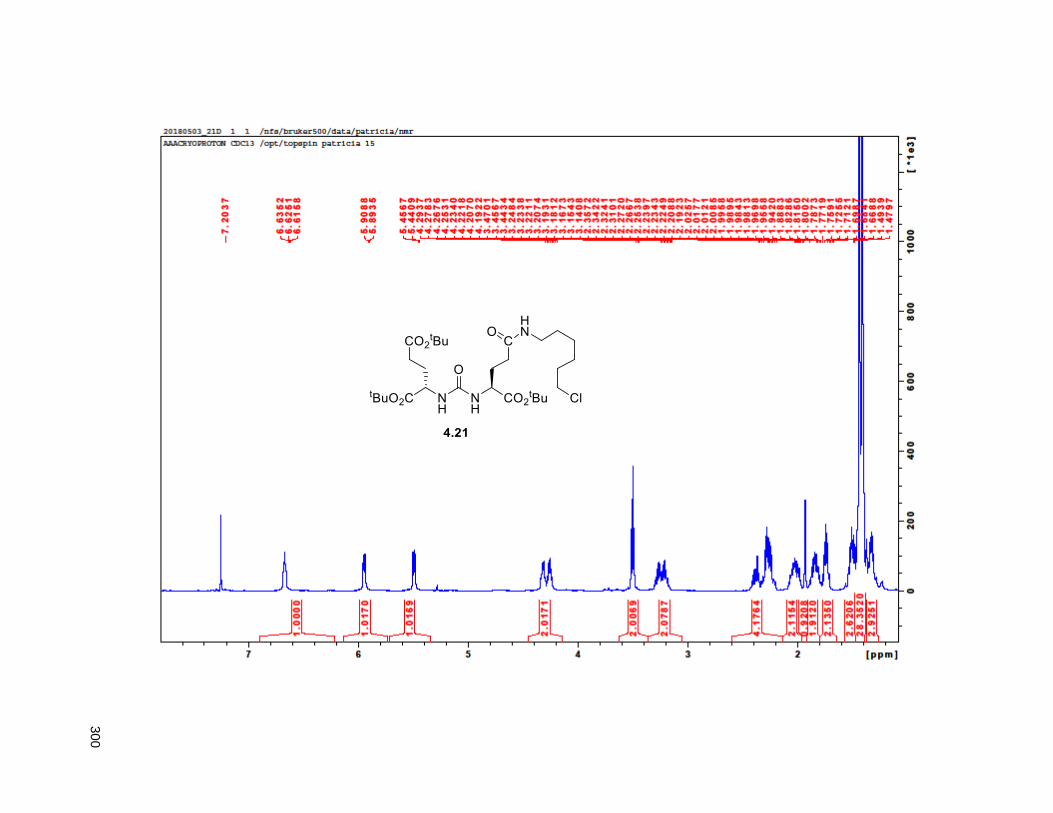
300
300
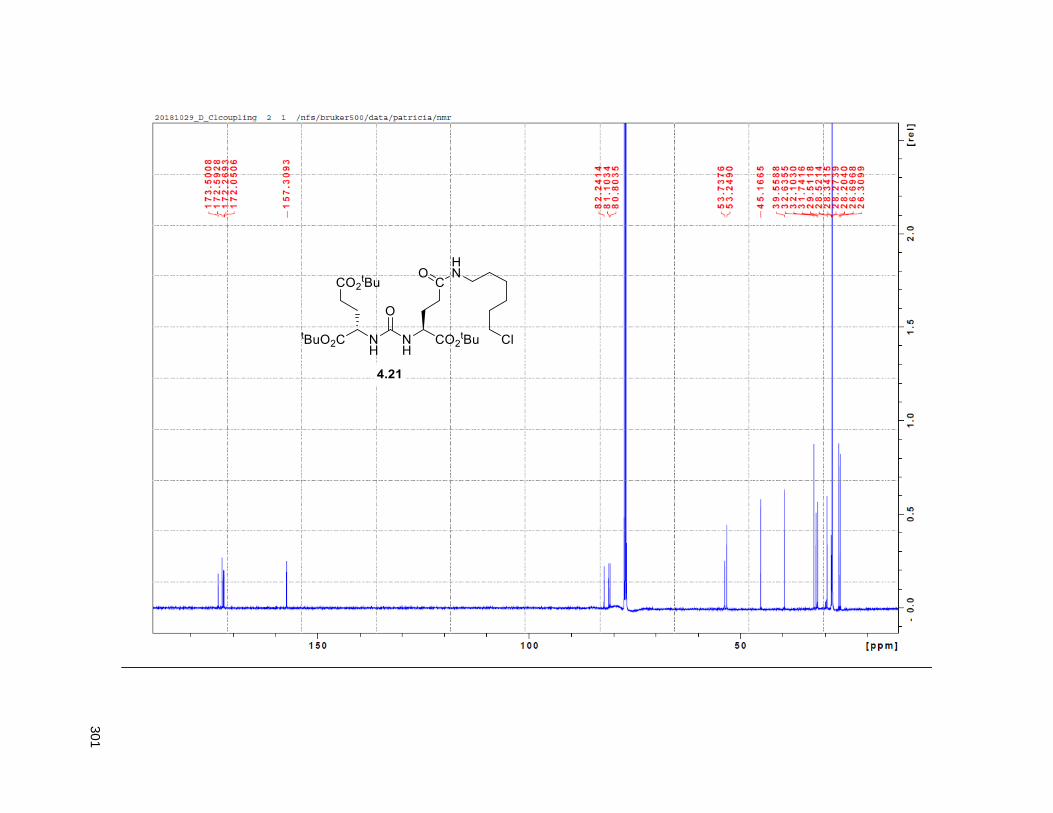
301
301
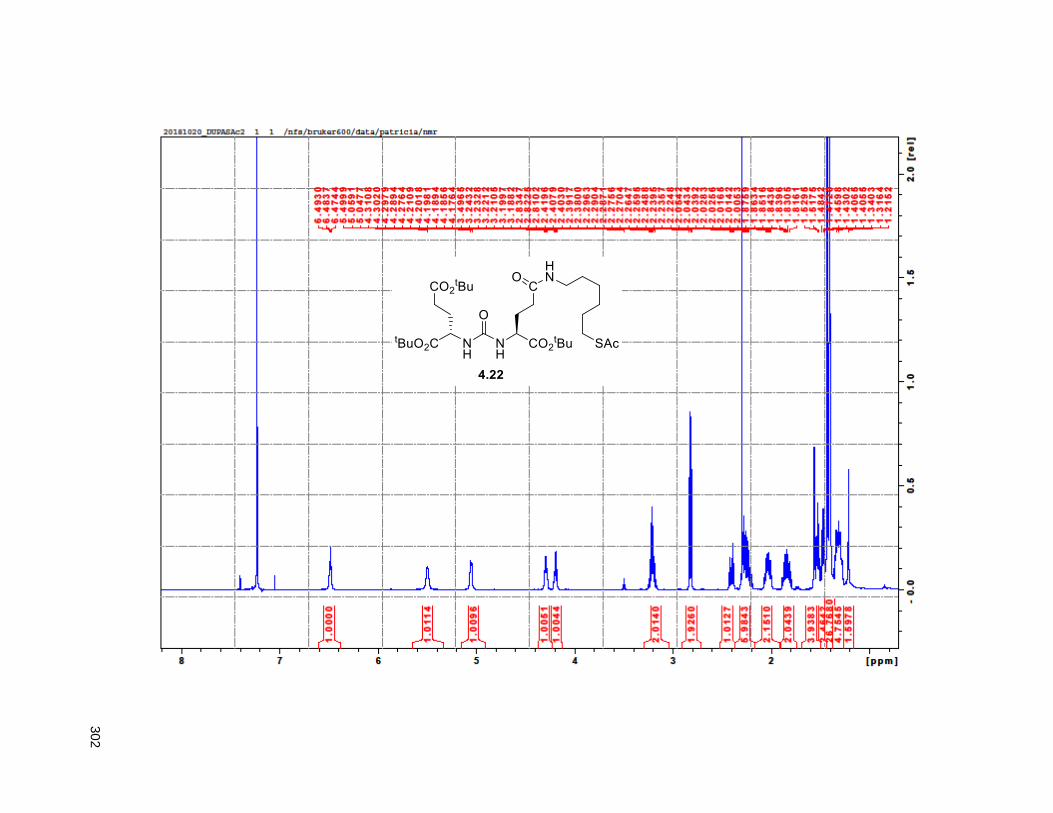
302
302
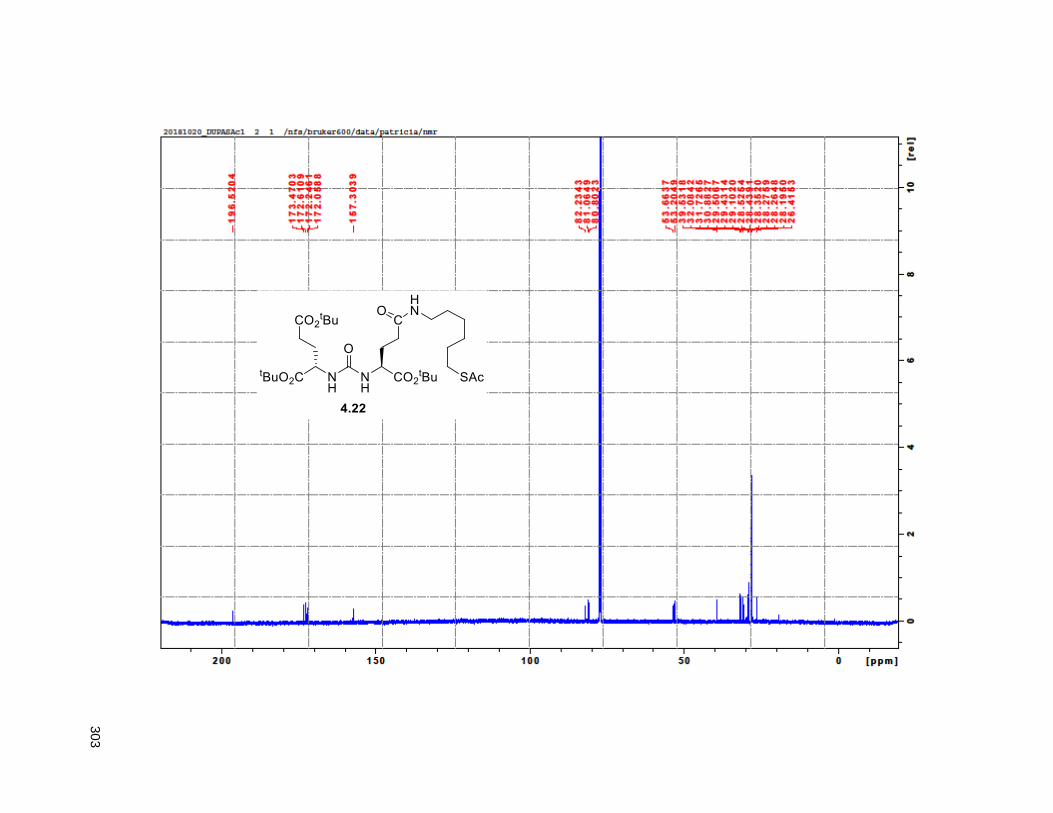
303
303
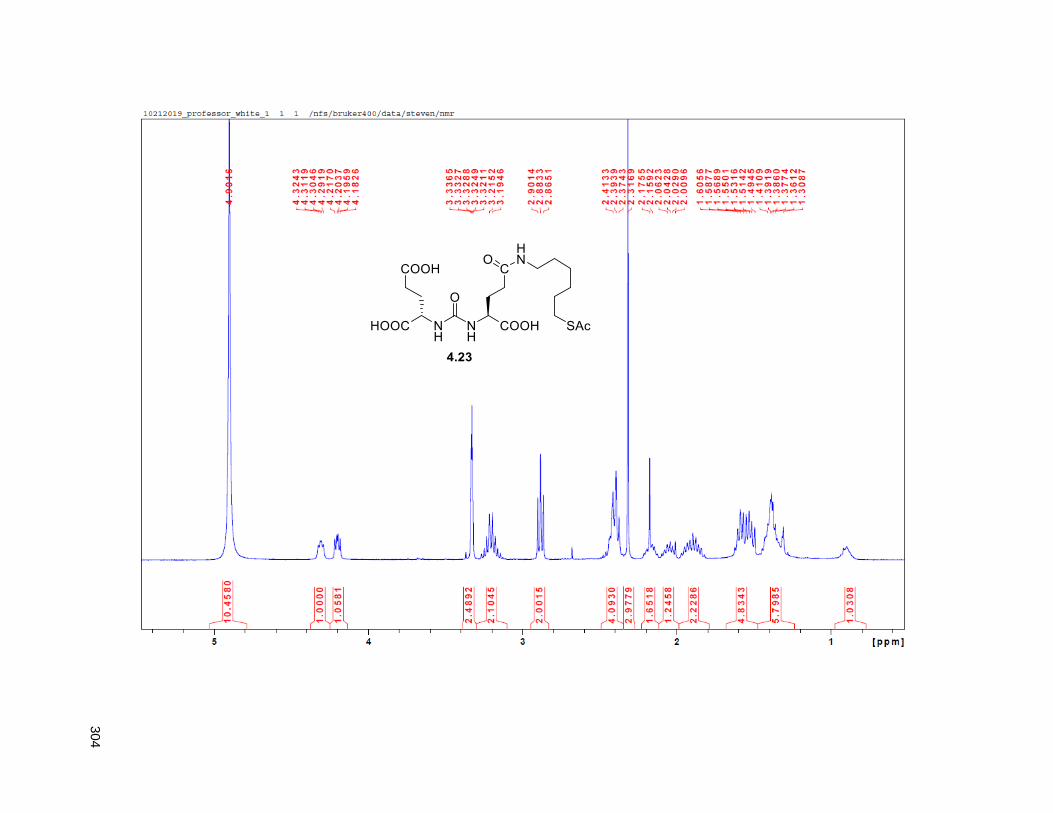
304
304
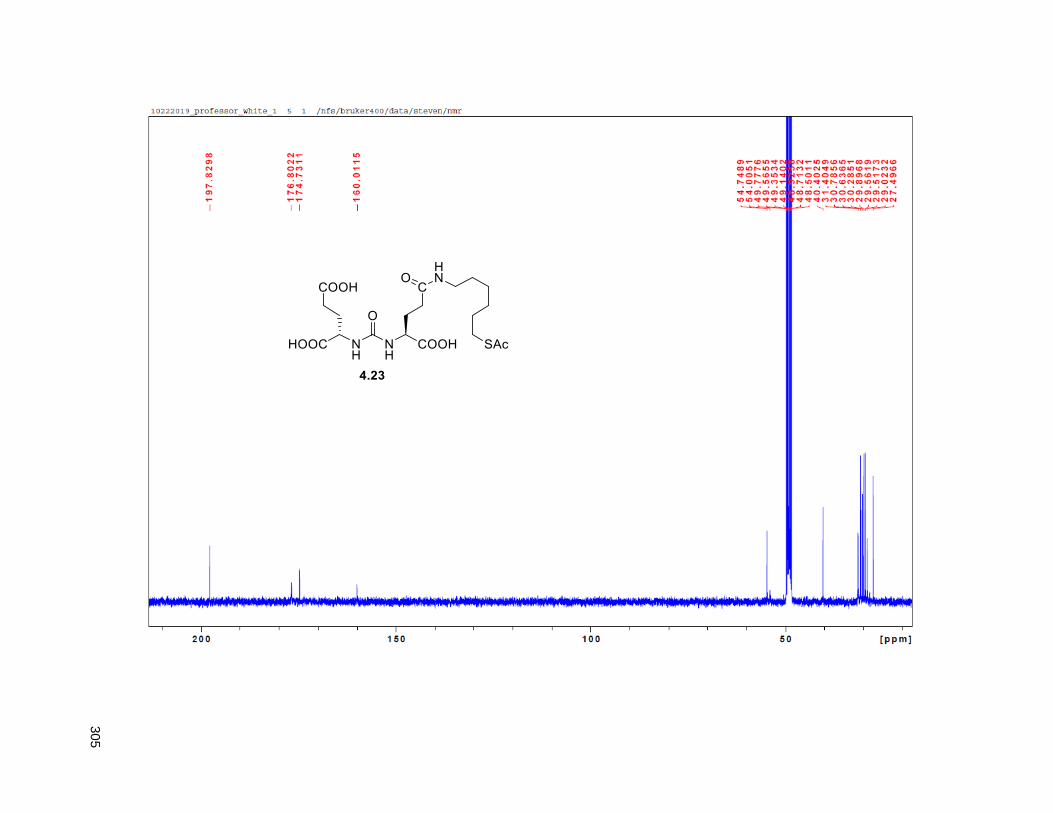
305
305
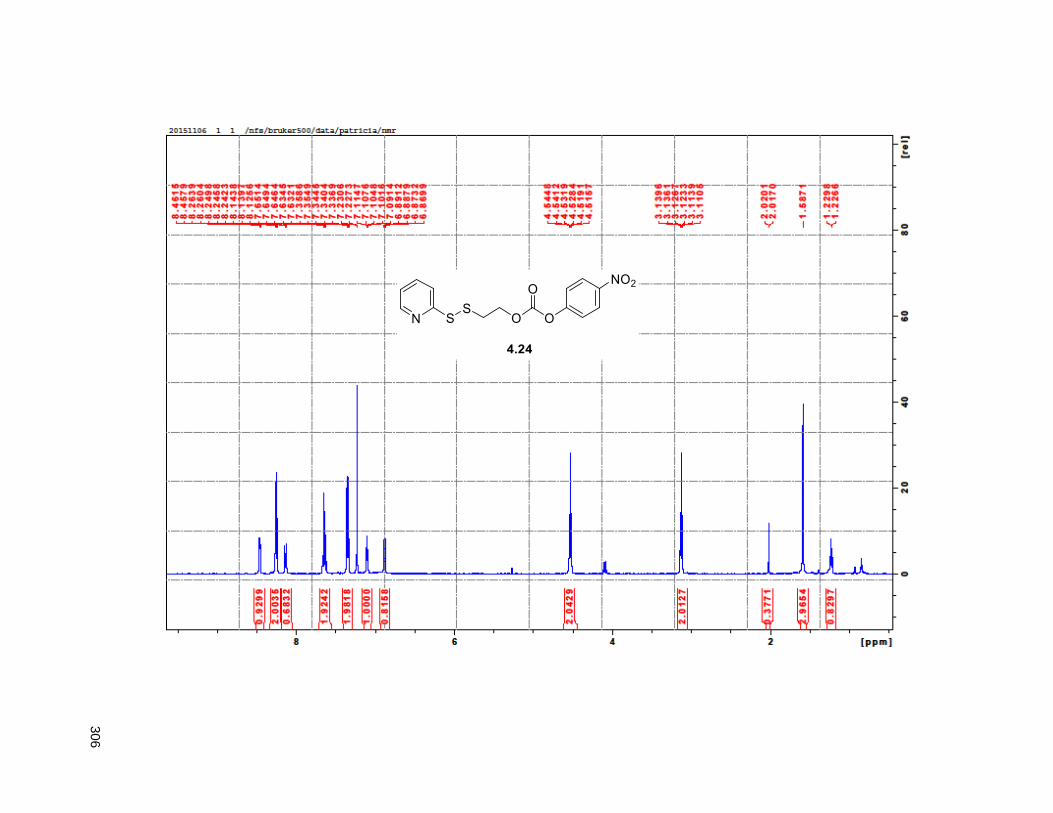
306
306
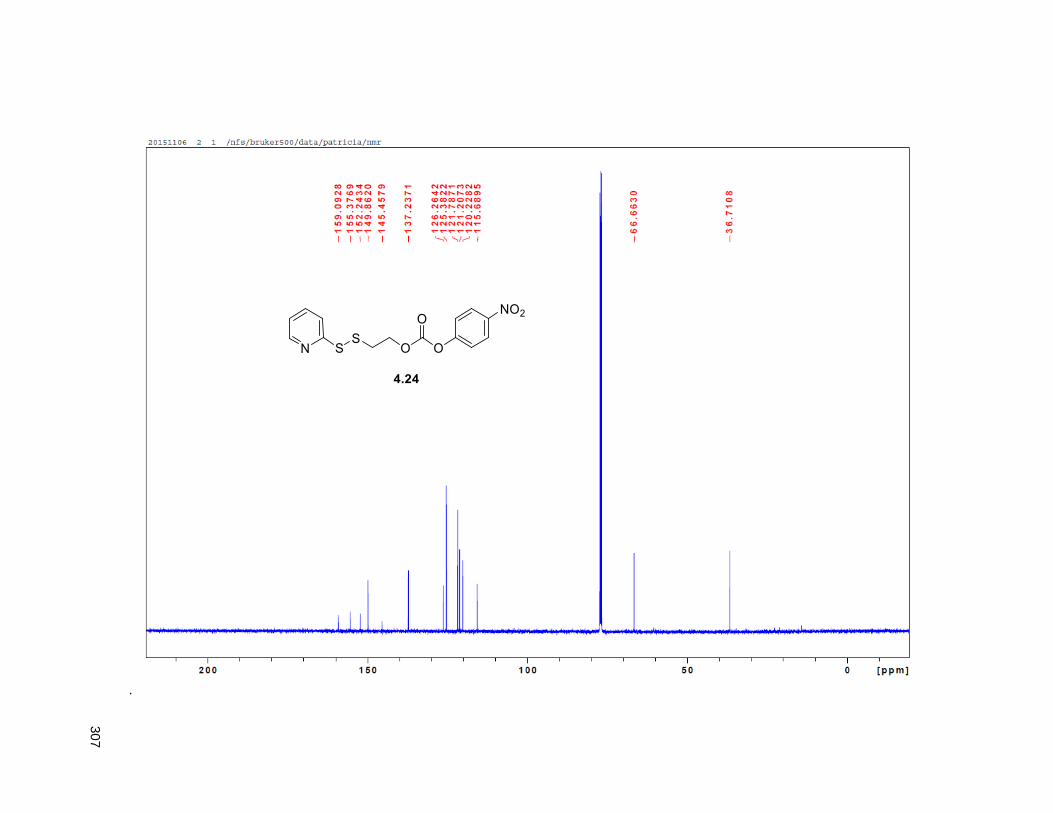
307
307
.
308
308
309
309
310
310
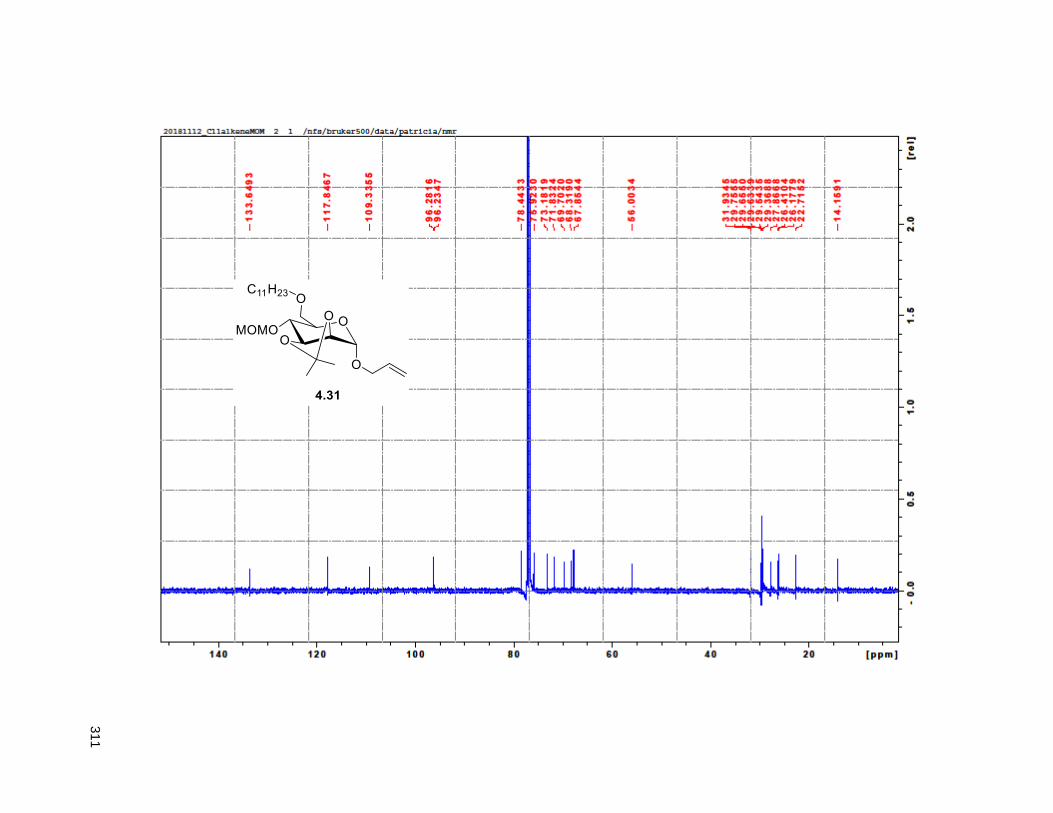
311
311
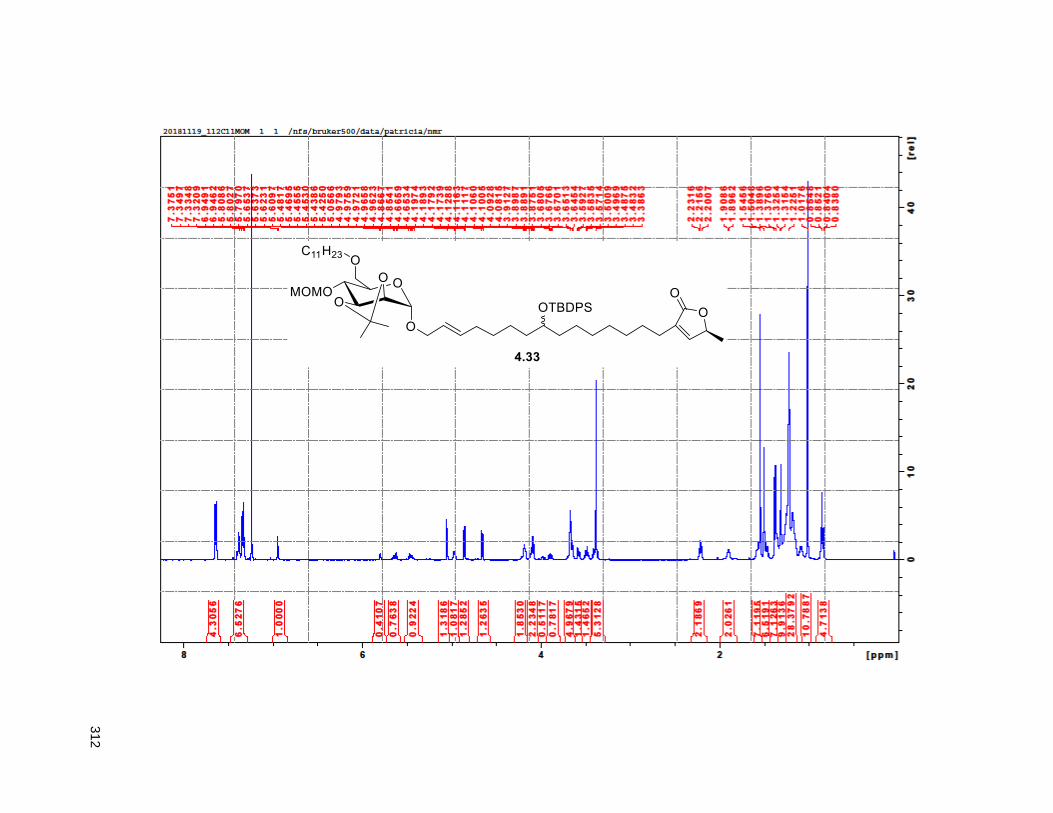
312
312
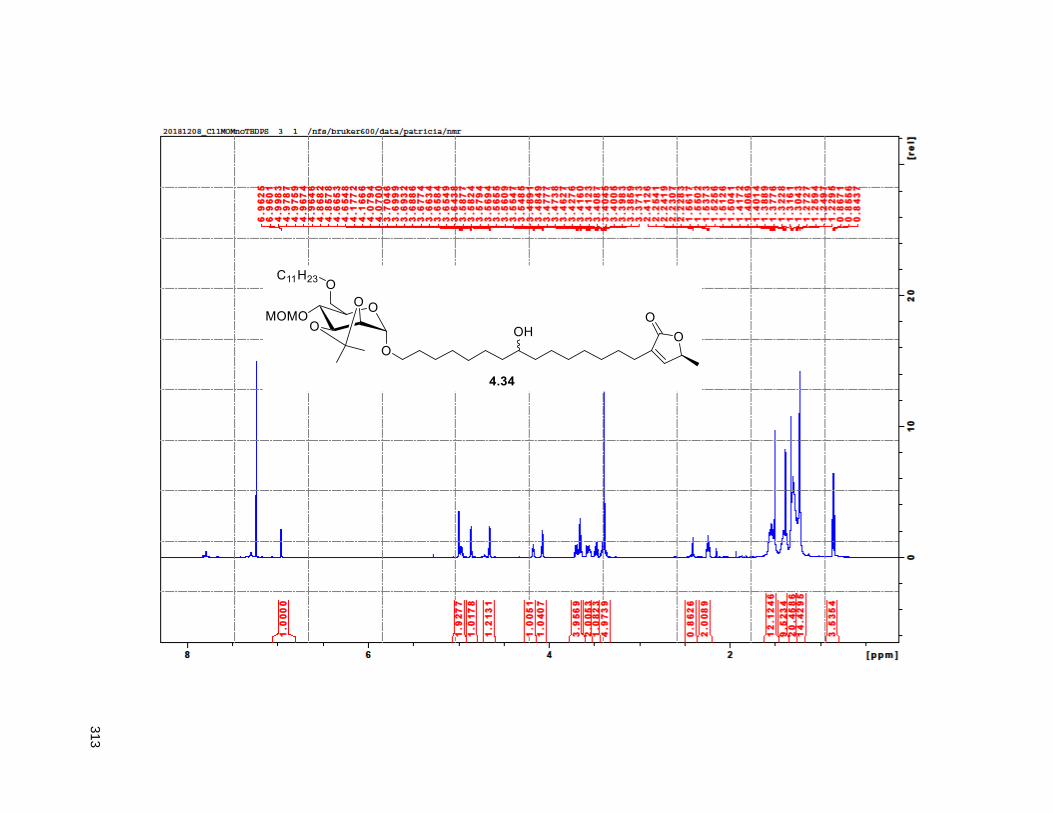
313
313
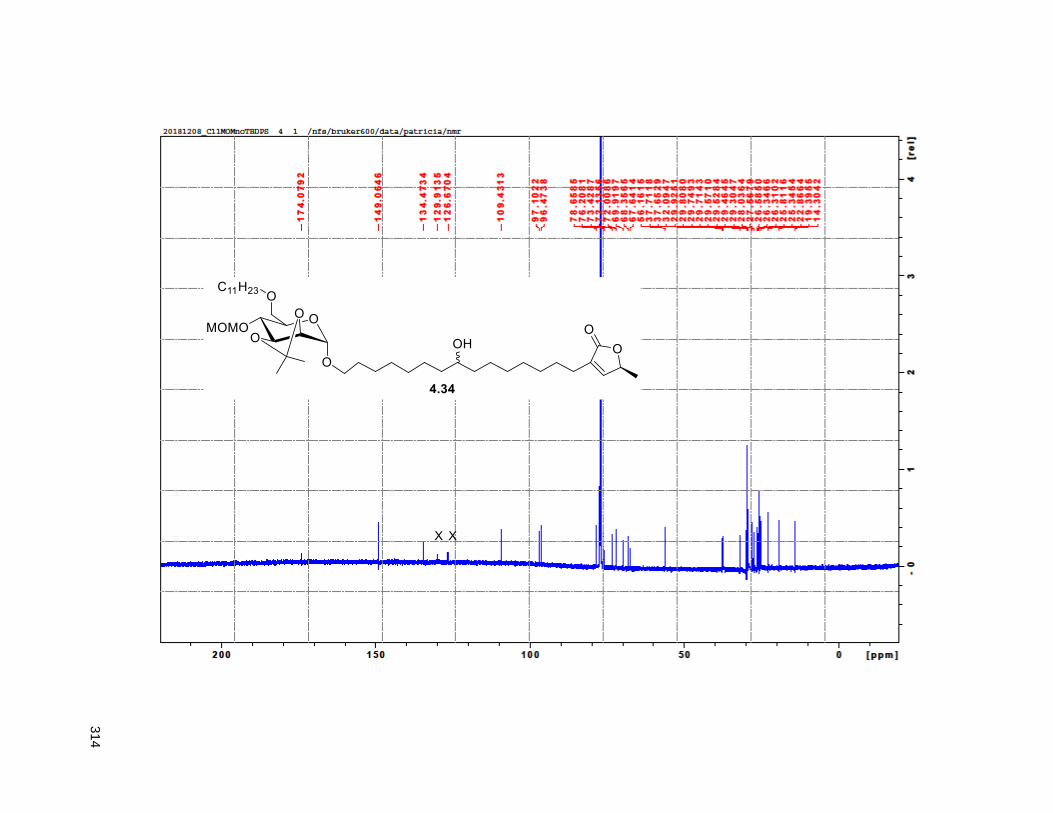
314
314
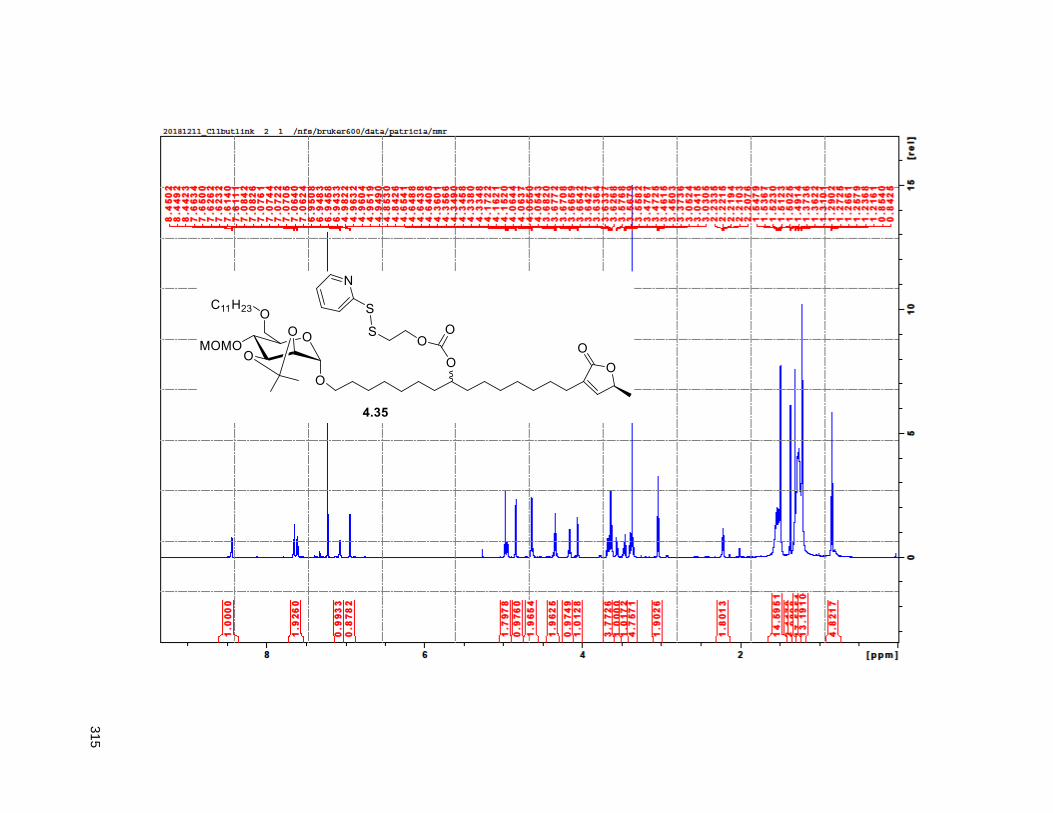
315
315
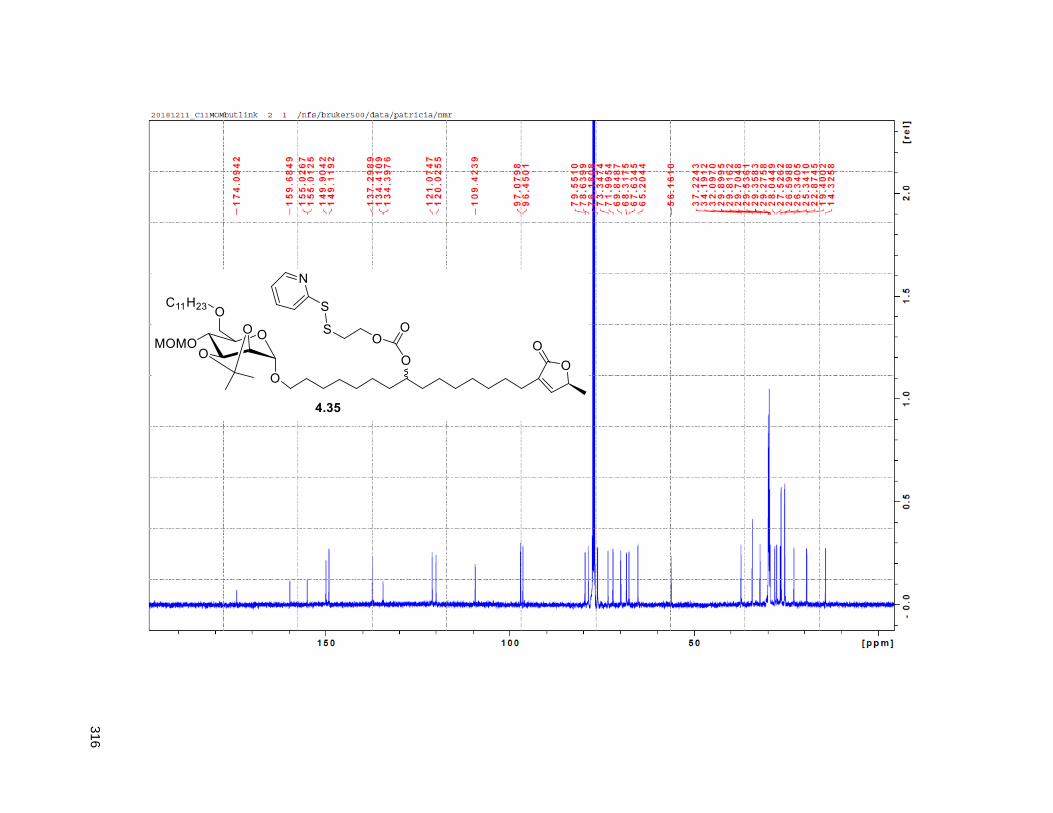
316
316
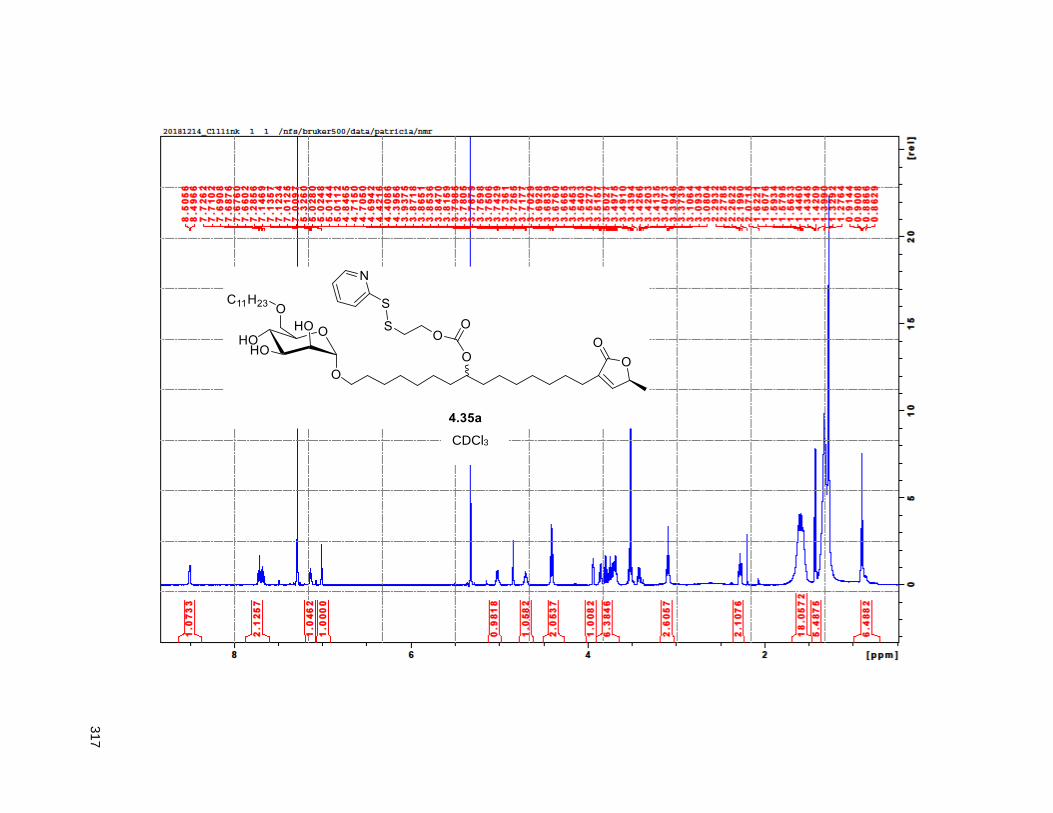
317
317
CDCl3
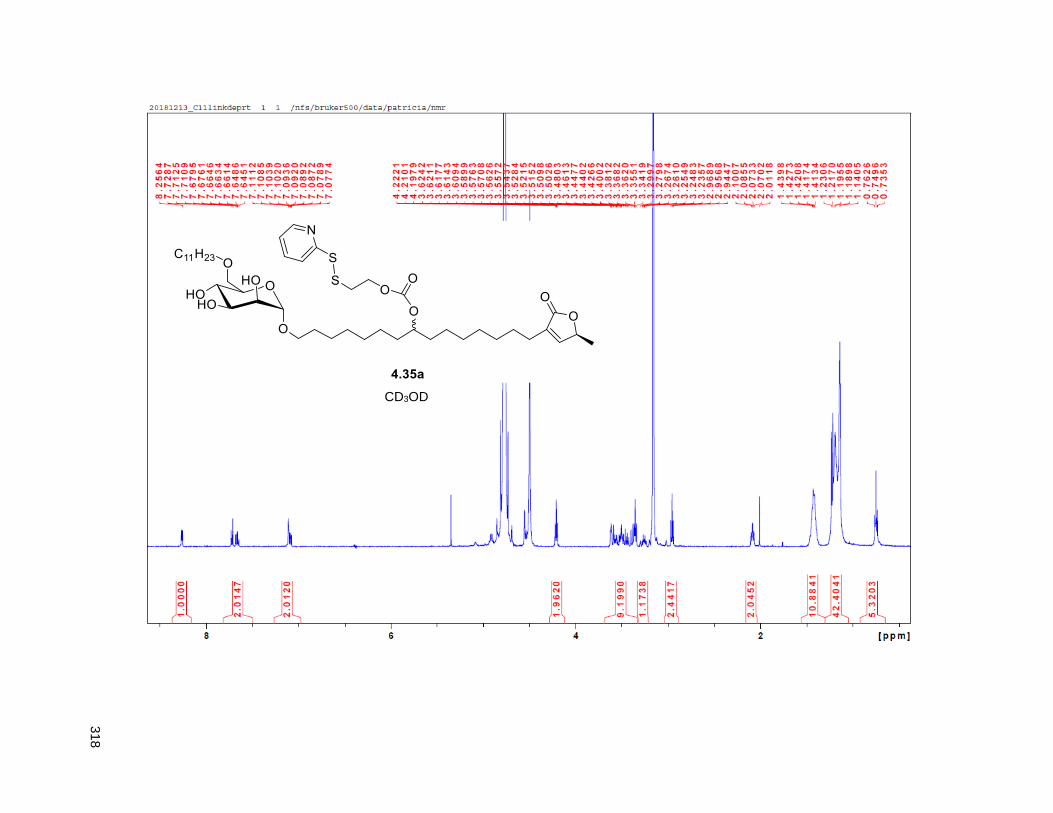
318
318
CD3OD
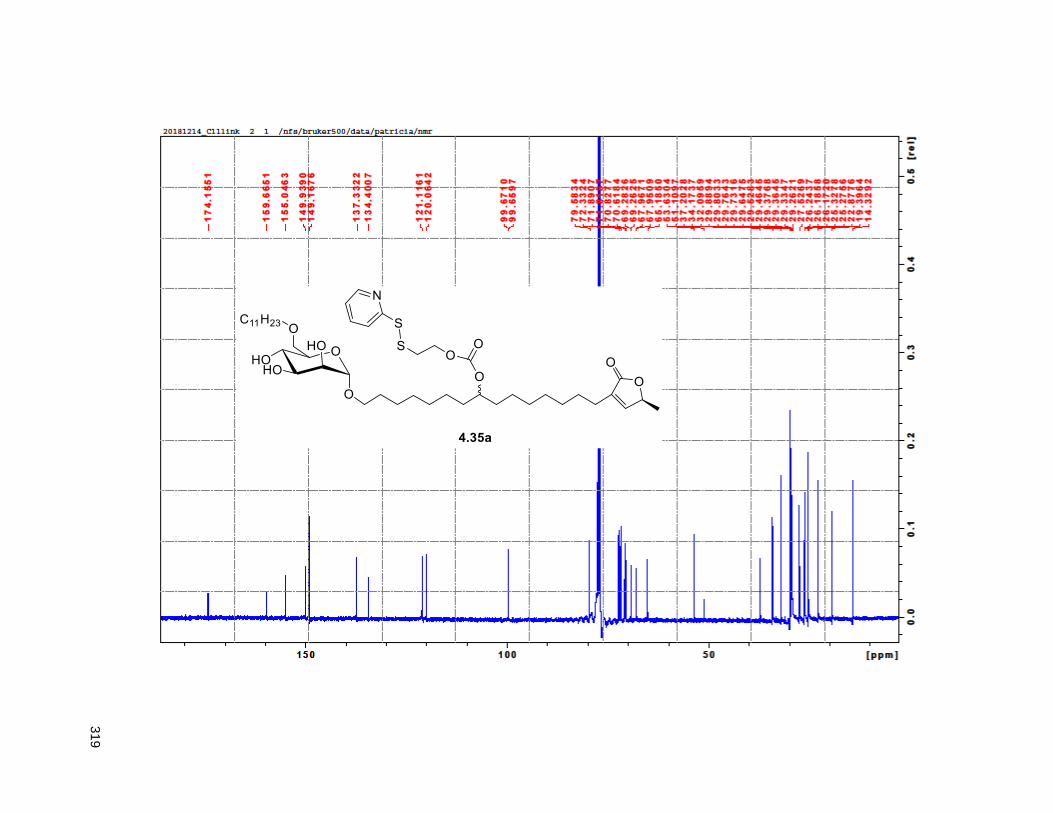
319
319

320
320
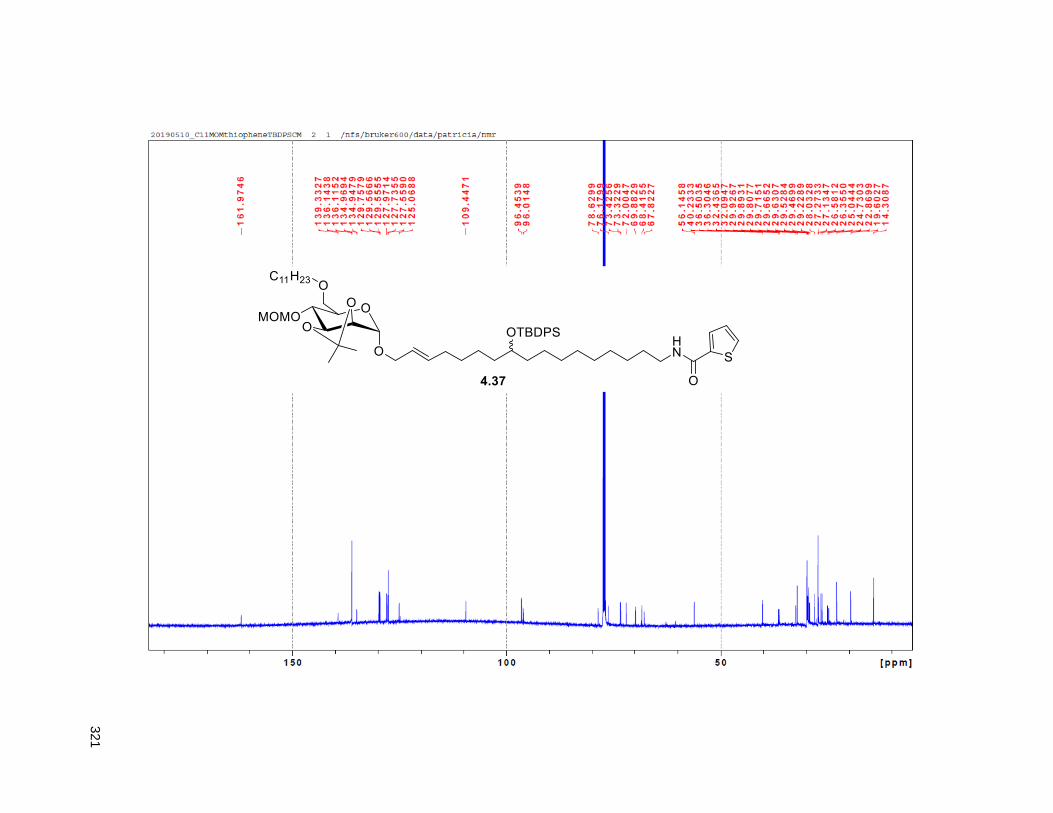
321
321
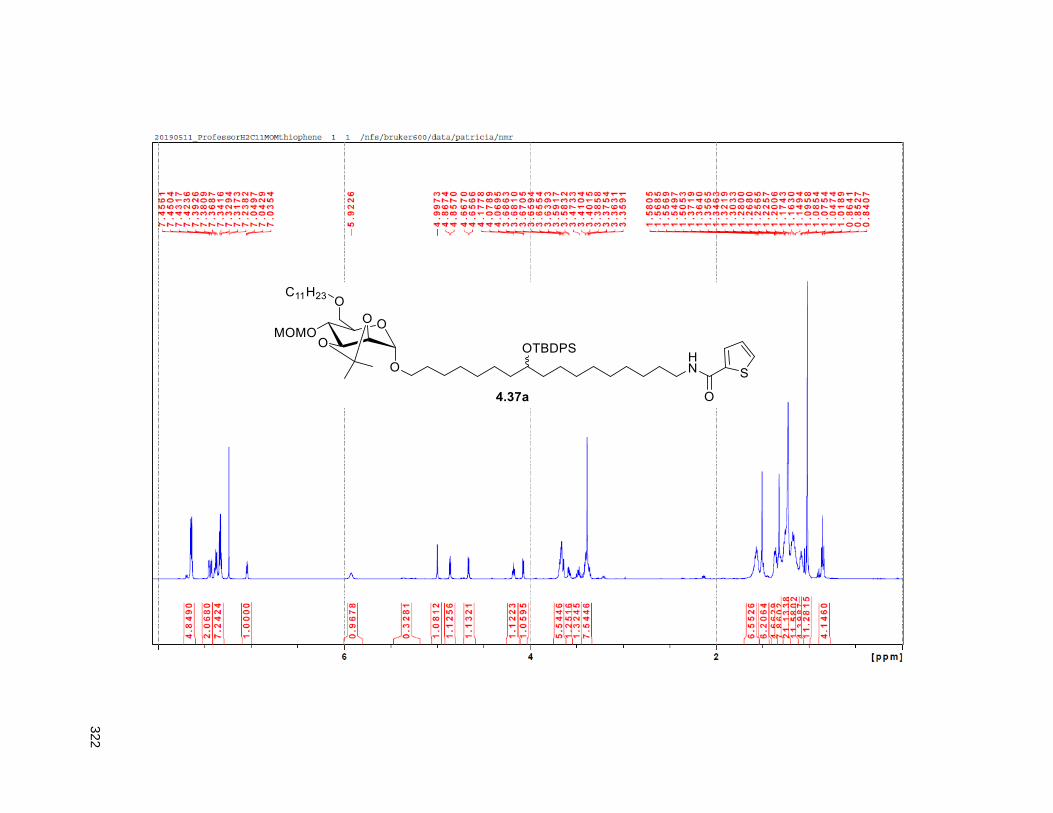
322
322

323
323
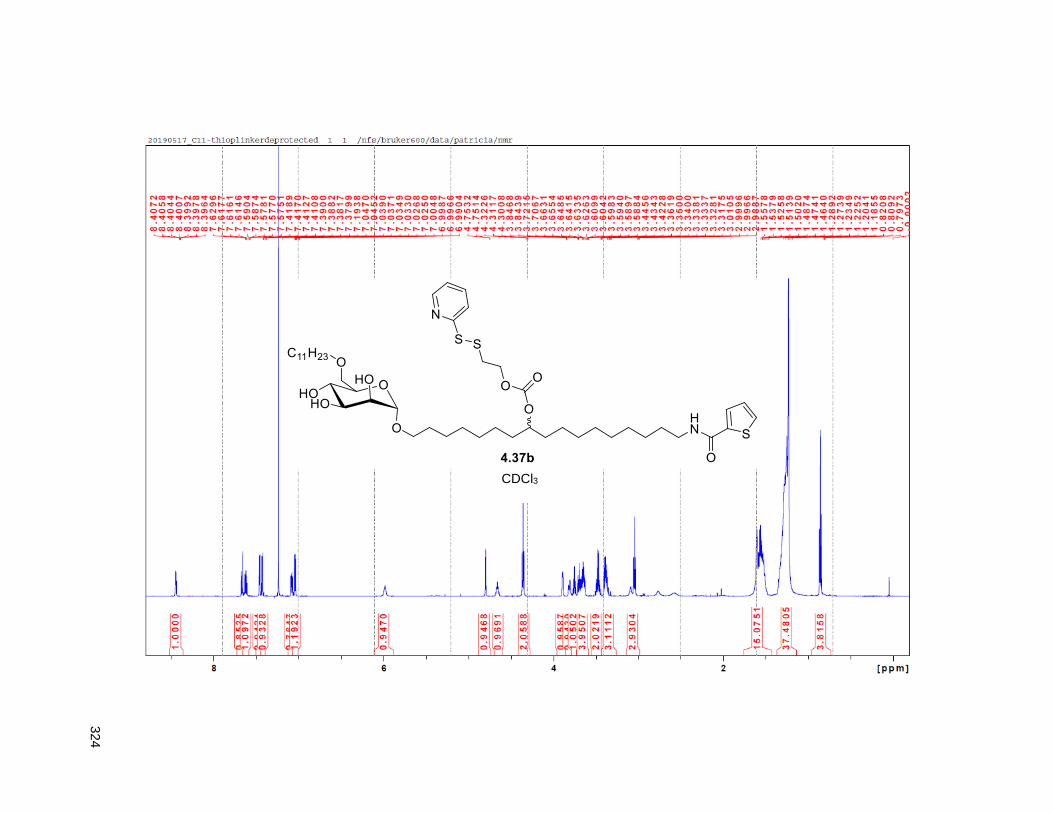
324
324
CDCl3

325
325
CD3OD
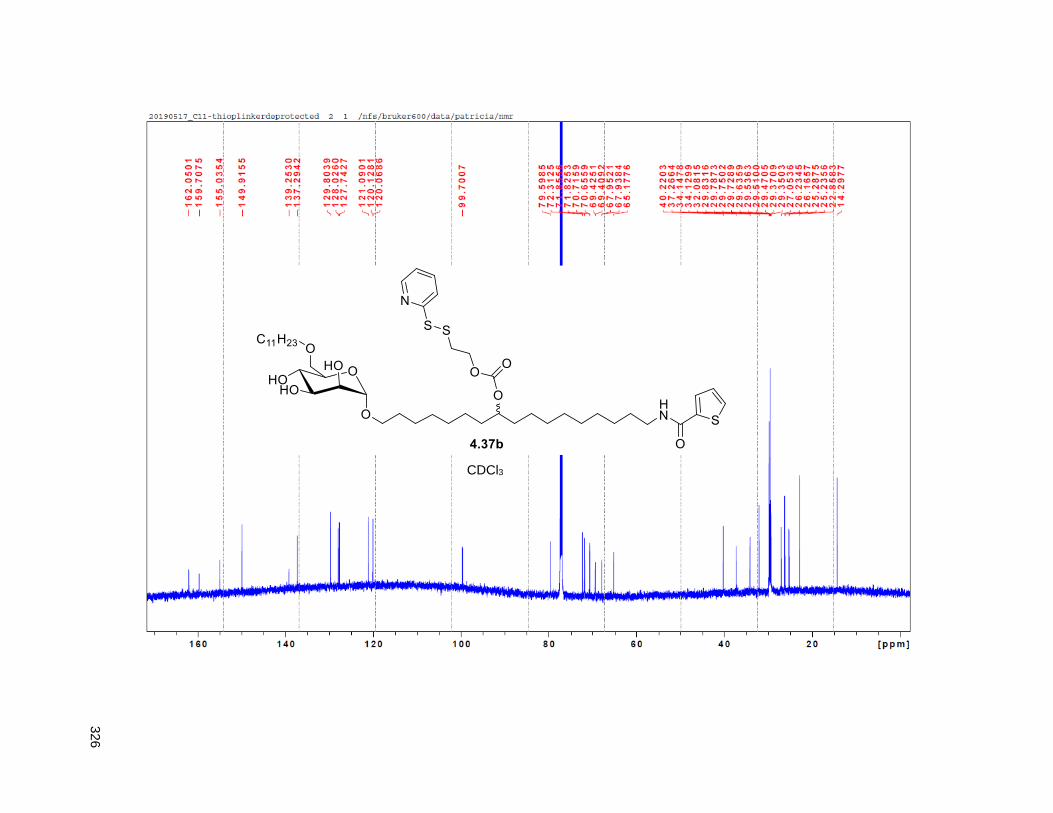
326
326
CDCl3
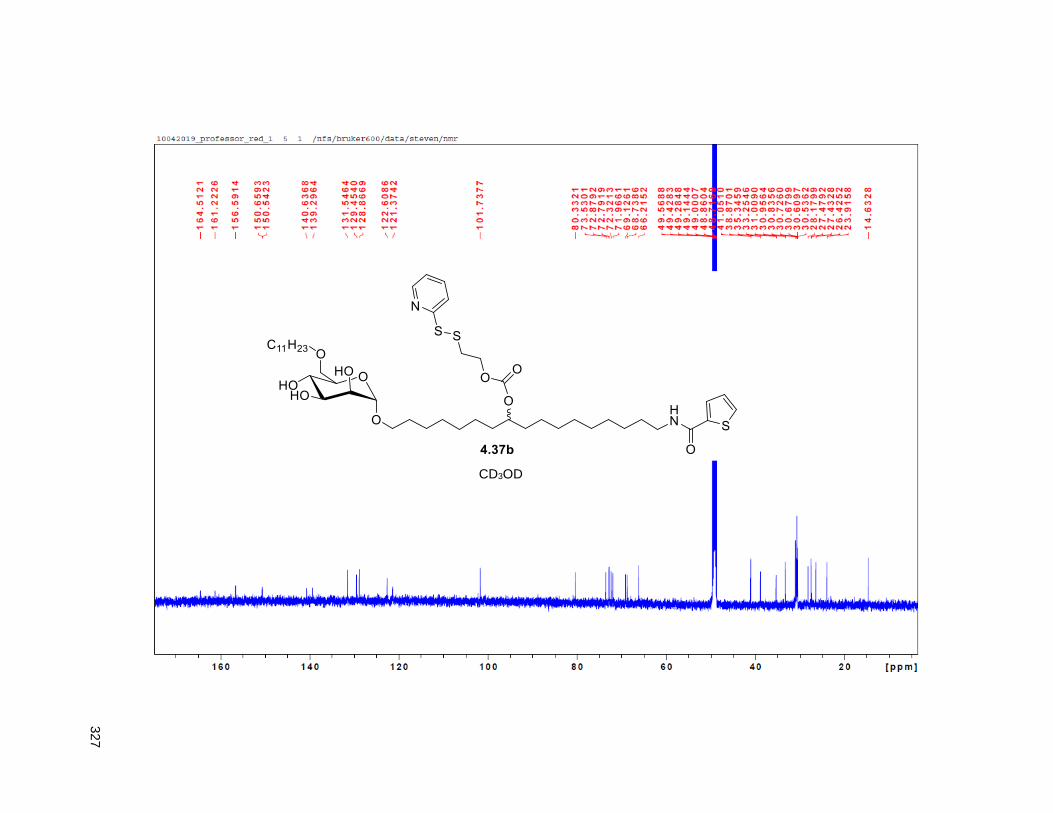
327
327
CD3OD
328
328
COSY of 4.37b
329
329
HSQC of 4.37b
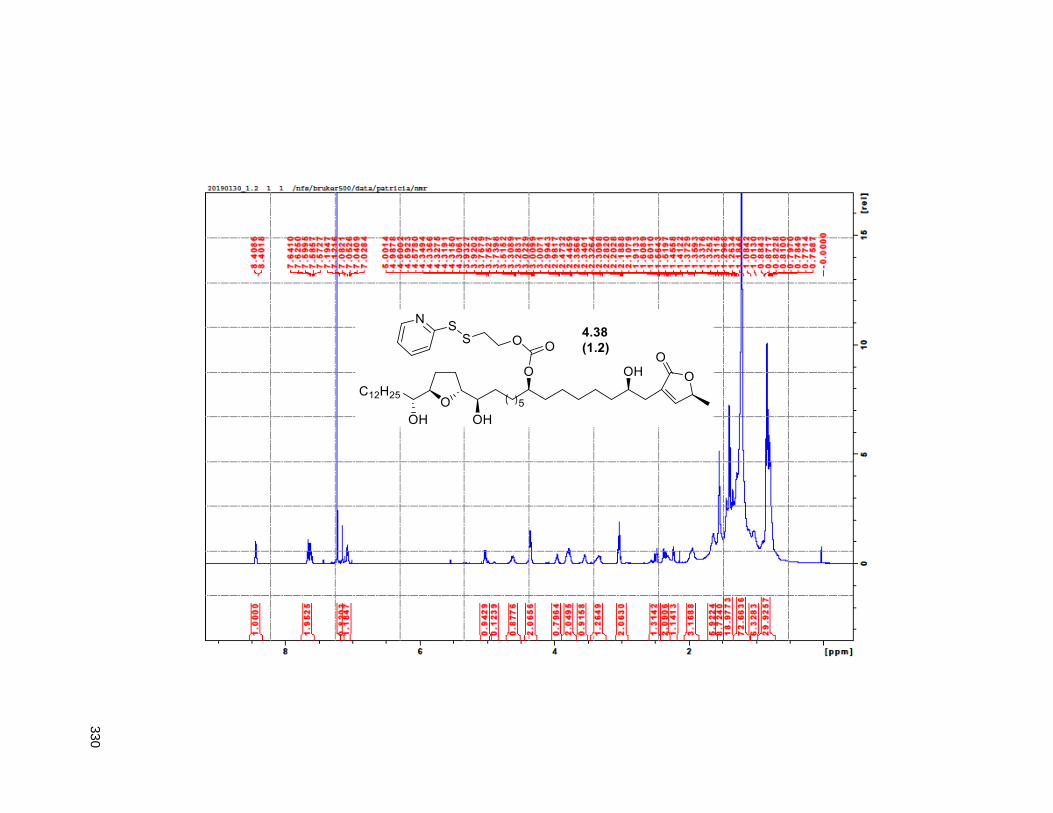
330
330
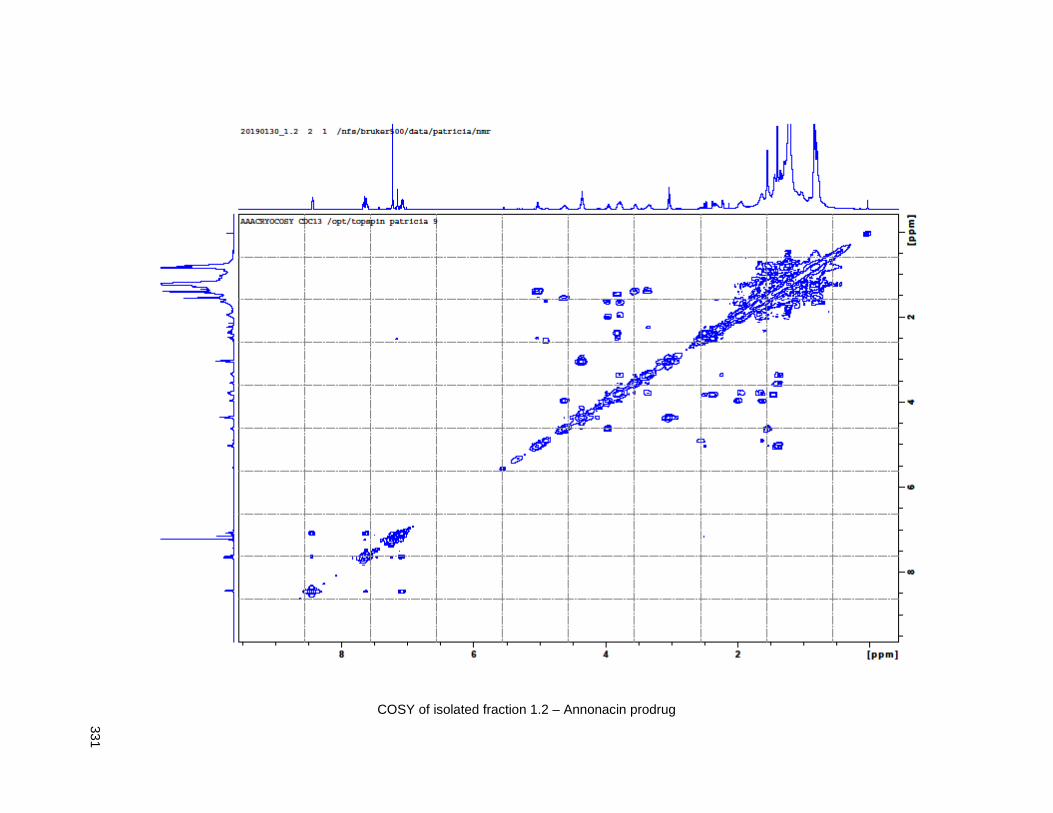
331
331
COSY of isolated fraction 1.2 – Annonacin prodrug

332
332

333
333
COSY of fraction 2.2 – Annonacin prodrug
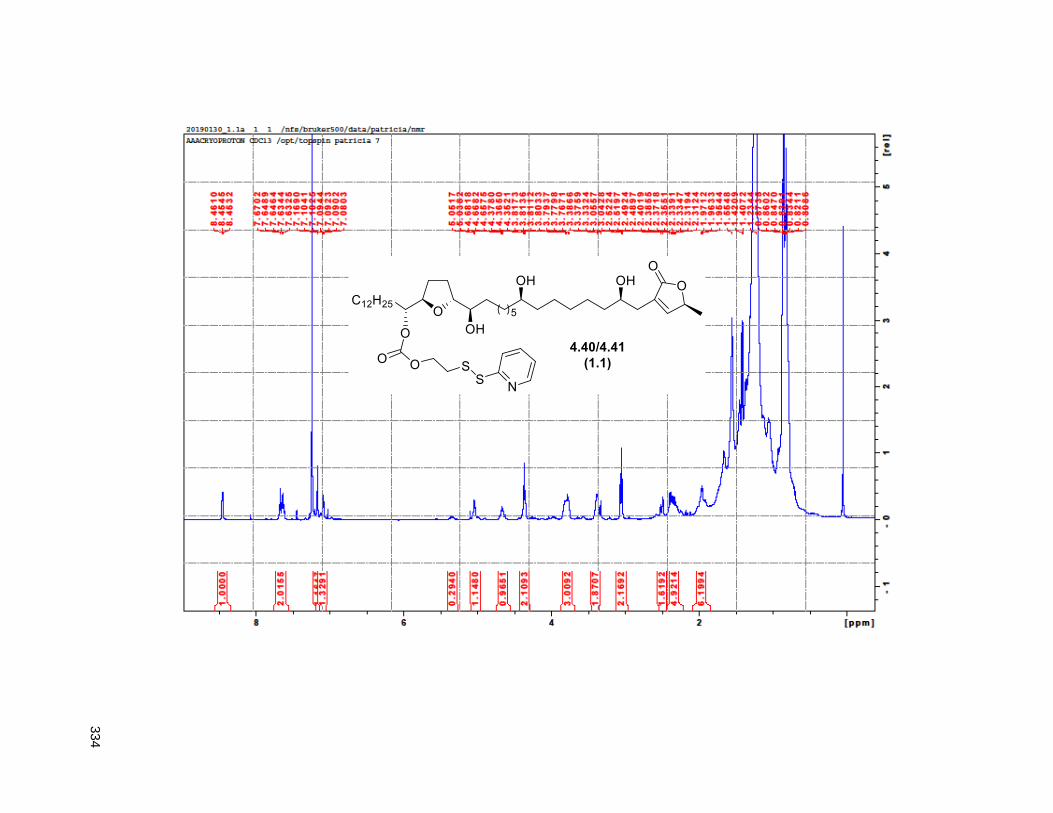
334
334
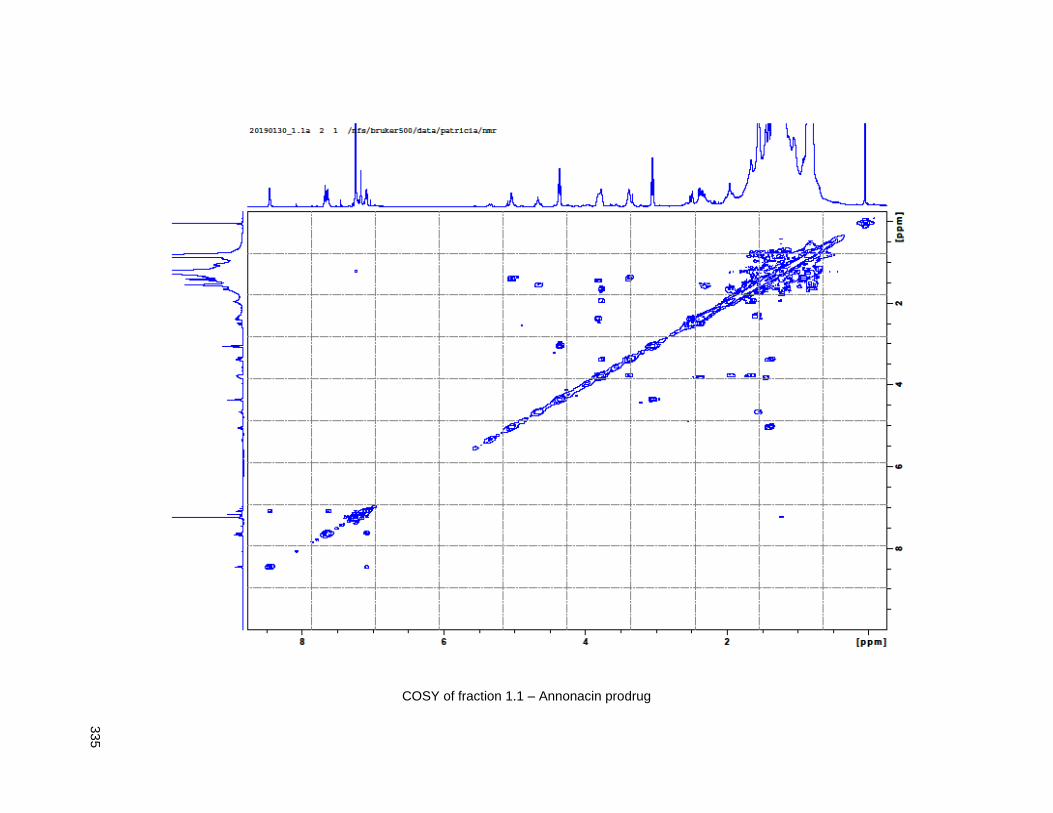
335
335
COSY of fraction 1.1 – Annonacin prodrug
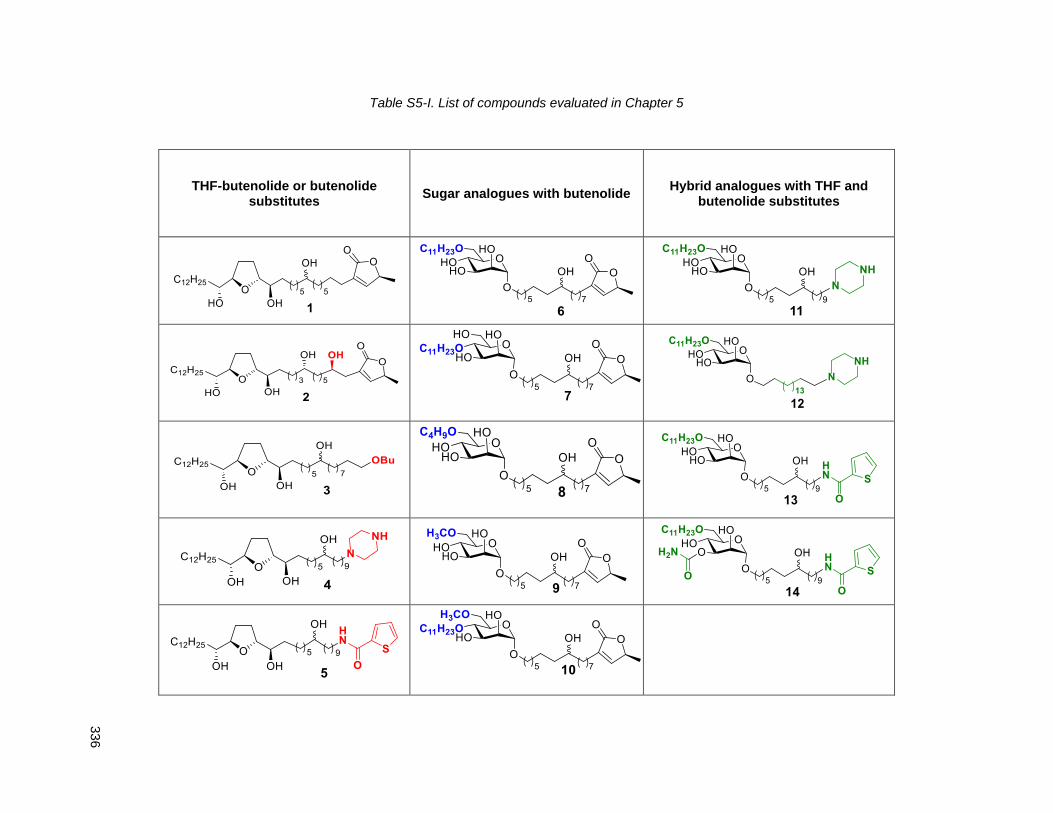
336
336
Table S5-I. List of compounds evaluated in Chapter 5
THF-butenolide or butenolide substitutes
Sugar analogues with butenolide Hybrid analogues with THF and
butenolide substitutes
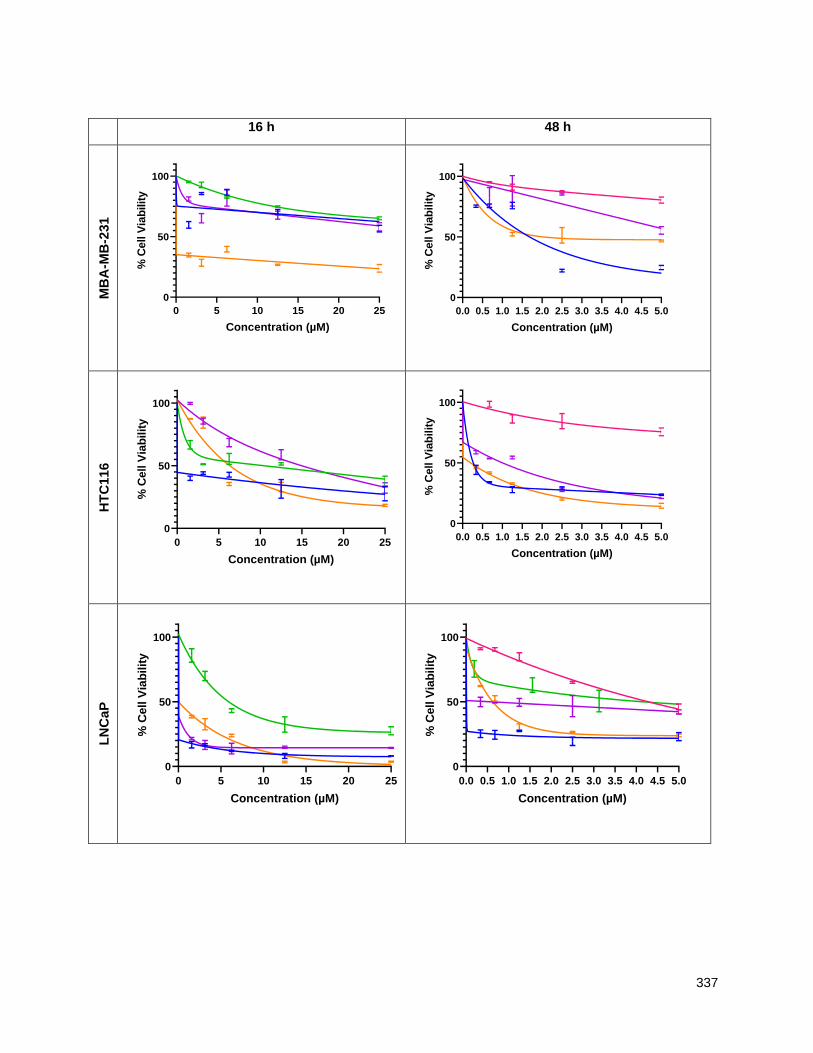
337
16 h 48 h
MB
A-M
B-2
31
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
HT
C116
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
LN
CaP
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
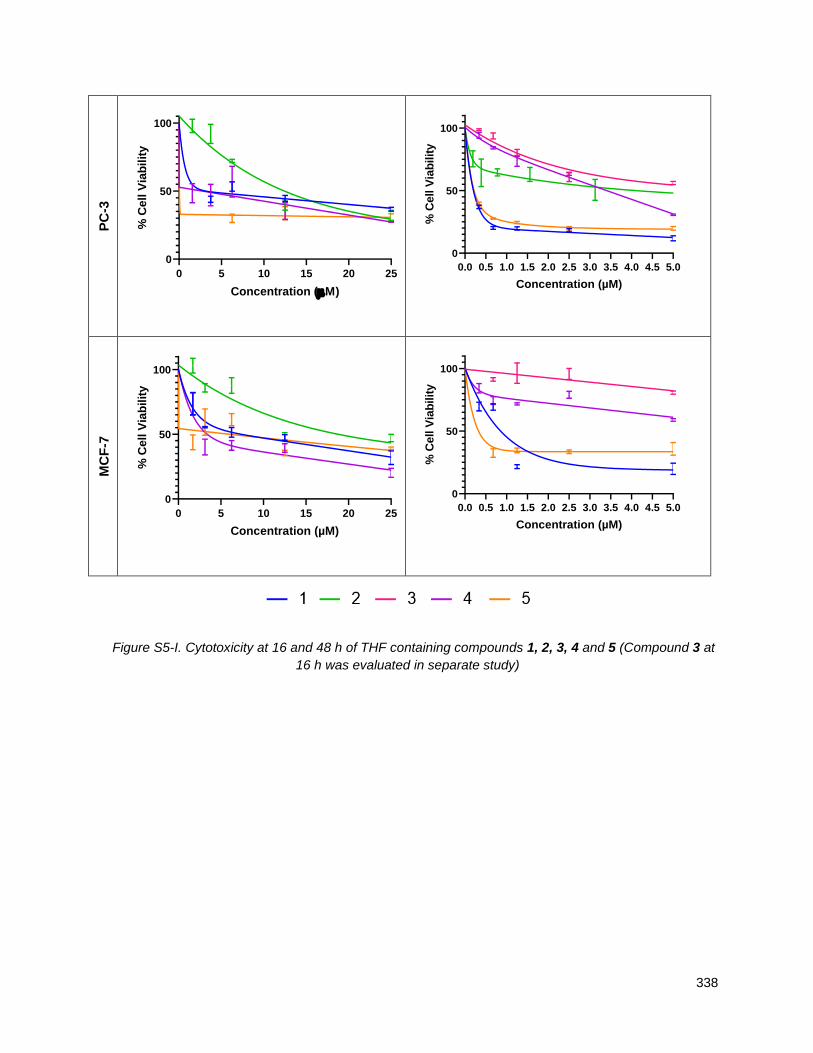
338
Figure S5-I. Cytotoxicity at 16 and 48 h of THF containing compounds 1, 2, 3, 4 and 5 (Compound 3 at
16 h was evaluated in separate study)
PC
-3
0 5 10 15 20 25
0
50
100
Concentration (M)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
MC
F-7
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
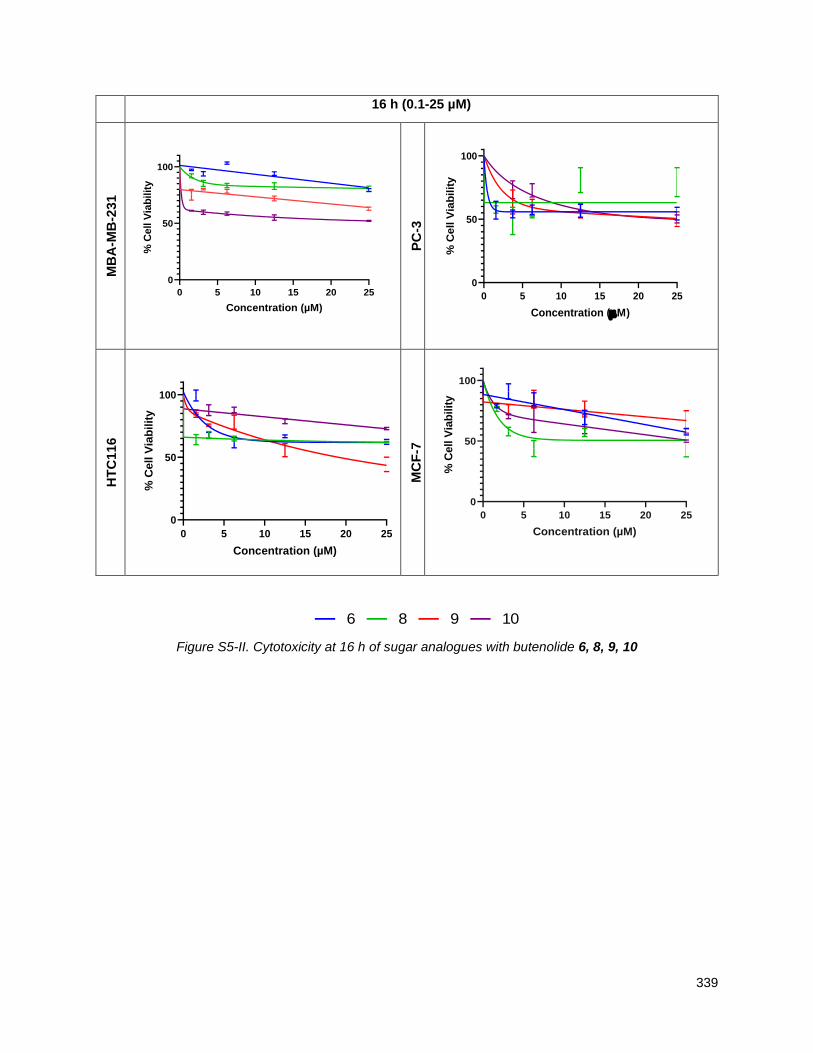
339
16 h (0.1-25 µM) M
BA
-MB
-231
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
PC
-3
0 5 10 15 20 25
0
50
100
Concentration (M)
% C
ell V
iab
ilit
y
HT
C116
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
M
CF
-7
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
1
70
75
80
85
90
95
100
Data 1
6 8 9 10
Figure S5-II. Cytotoxicity at 16 h of sugar analogues with butenolide 6, 8, 9, 10

340
16 h 48 h M
BA
-MB
-231
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
HT
C116
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
LN
CaP
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
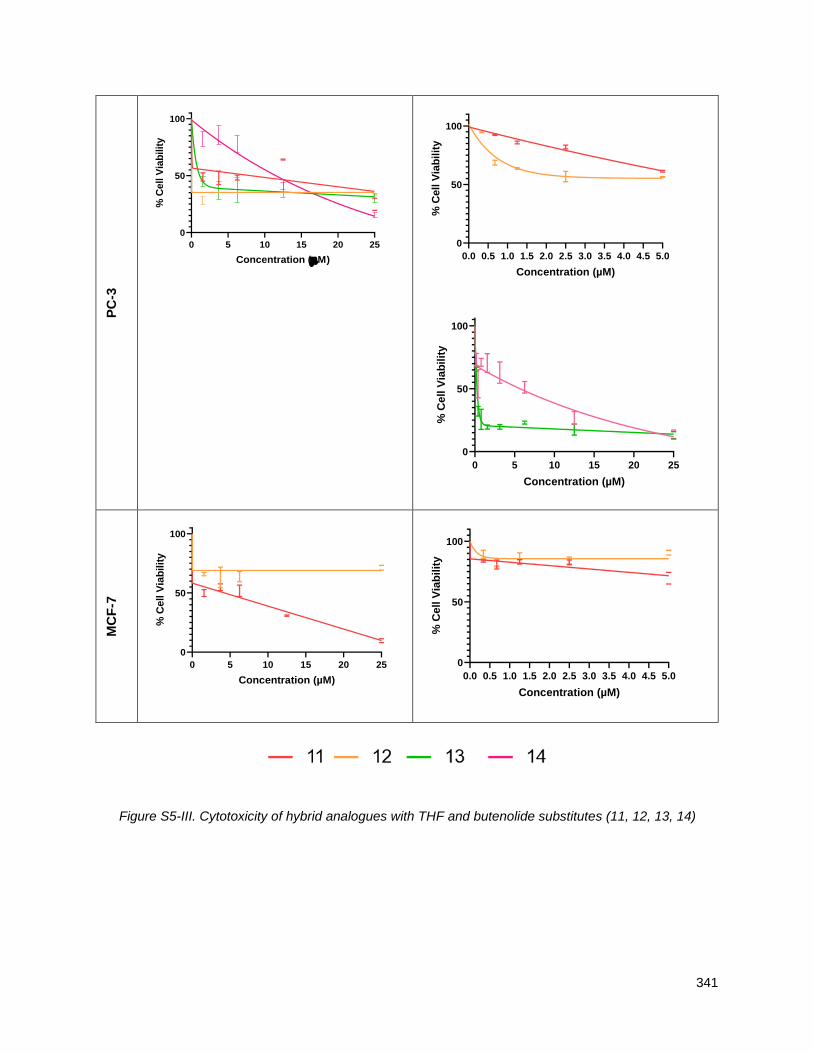
341
PC
-3
0 5 10 15 20 25
0
50
100
Concentration (M)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
MC
F-7
0 5 10 15 20 25
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
50
100
Concentration (µM)
% C
ell V
iab
ilit
y
Figure S5-III. Cytotoxicity of hybrid analogues with THF and butenolide substitutes (11, 12, 13, 14)
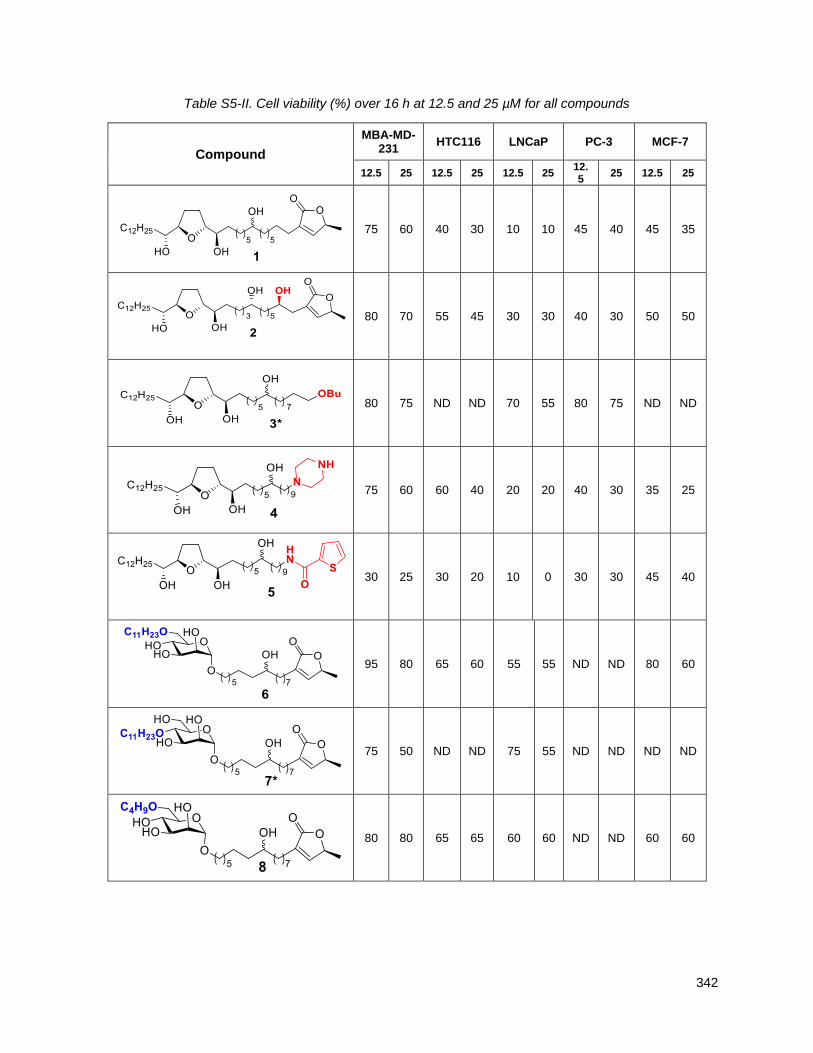
342
Table S5-II. Cell viability (%) over 16 h at 12.5 and 25 µM for all compounds
Compound
MBA-MD-231
HTC116 LNCaP PC-3 MCF-7
12.5 25 12.5 25 12.5 25 12.5
25 12.5 25
75 60 40 30 10 10 45 40 45 35
80 70 55 45 30 30 40 30 50 50
80 75 ND ND 70 55 80 75 ND ND
75 60 60 40 20 20 40 30 35 25
30 25 30 20 10 0 30 30 45 40
95 80 65 60 55 55 ND ND 80 60
75 50 ND ND 75 55 ND ND ND ND
80 80 65 65 60 60 ND ND 60 60
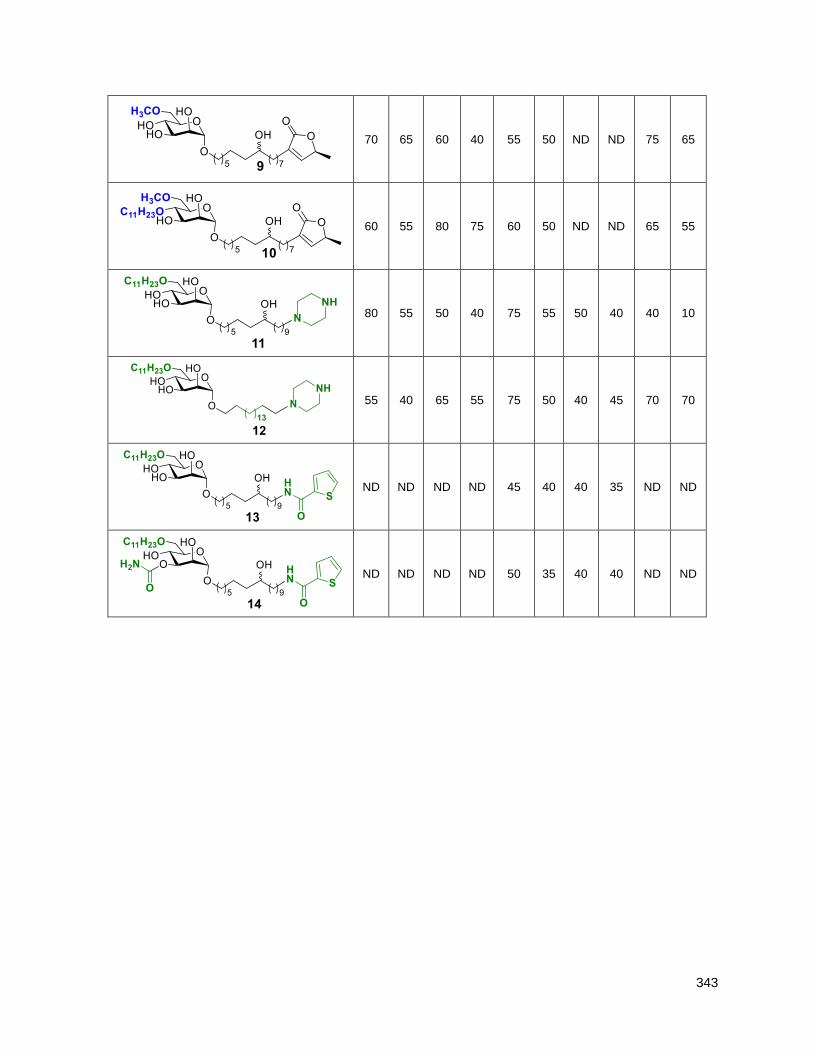
343
70 65 60 40 55 50 ND ND 75 65
60 55 80 75 60 50 ND ND 65 55
80 55 50 40 75 55 50 40 40 10
55 40 65 55 75 50 40 45 70 70
ND ND ND ND 45 40 40 35 ND ND
ND ND ND ND 50 35 40 40 ND ND

344
REFERENCES

345
1. Jiaju, Z., Guirong, X., Xinjian Y., Encyclopedia of Traditional Chinese Medicines, 2011, p.
2. Annona species (Annonaceae): a rich source of potential antitumor agents? Tundis, R.; Xiao, J.; Loizzo, M.R. Ann N Y Acad Sci. 2017, 1398, 30-36.
3. Uvaricin, a new antitumor agent from Uvaria accuminata (Annonaceae). Tempesta, M.S.; Kriek, G.R.; Bates, R.B. 1982, 47, 3151-53.
4. Annonaceous Acetogenins: A Review. Rupprecht, J.K.; Hui, Y.-H.; McLaughlin, J.L. Journal of Natural Products. 1990, 53, 237-78.
5. Recent advances in annonaceous acetogenins. Zeng, L.; Ye, Q.; Oberlies, N.H.; Shi, G.; Gu, Z.-M.; He, K.; McLaughlin, J.L. Natural Product Reports. 1996, 13, 275-306.
6. Paw Paw and Cancer: Annonaceous Acetogenins from Discovery to Commercial Products. McLaughlin, J.L. Journal of Natural Products. 2008, 71, 1311-21.
7. Goniothalamus species: a source of drugs for the treatment of cancers and bacterial infections? Wiart, C. Evidence-based complementary and alternative medicine : eCAM. 2007, 4, 299-311.
8. Historic perspectives on Annonaceous acetogenins from the chemical bench to preclinical trials. Liaw, C.C.; Wu, T.Y.; Chang, F.R.; Wu, Y.C. Planta Med. 2010, 76, 1390-404.
9. Syntheses of Acetogenins of Annonaceae: A New Class of Bioactive Polyketides. Figadere, B. Accounts of Chemical Research. 1995, 28, 359-65.
10. Stereoselective Synthesis of cis-2,5-Disubstituted THFs: Application to Adjacent Bis-THF Cores of Annonaceous Acetogenins. Fujioka, H.; Maehata, R.; Wakamatsu, S.; Nakahara, K.; Hayashi, T.; Oki, T. Organic Letters. 2012, 14, 1054-57.
11. a) Stereoselective Synthesis of Oligo-Tetrahydrofurans. Koert, U. Synthesis. 1995, 1995, 115-32; b) Hoppe, R.; Scharf, H.D., Annonaceous Acetogenins - Synthetic Approaches Towards a Novel Class of Natural Products, 1995, p. 1447-64; c) Recent progress on the total synthesis of acetogenins from Annonaceae. Li, N.; Shi, Z.; Tang, Y.; Chen, J.; Li, X. Beilstein J Org Chem. 2008, 4, 48; d) Acetogenins from Annonaceae: recent progress in isolation, synthesis and mechanisms of action. Bermejo, A.; Figadère, B.; Zafra-Polo, M.-C.; Barrachina, I.; Estornell, E.; Cortes, D. Natural Product Reports. 2005, 22, 269-303. 12. Medicinal chemistry of Annonaceous acetogenins: design, synthesis, and biological evaluation of novel analogues. Kojima, N.; Tanaka, T. Molecules. 2009, 14, 3621-61.
13. Synthesis ofcis-Solamin Using a Permanganate-Mediated Oxidative Cyclization. Cecil, A.R.L.; Brown, R.C.D. Organic Letters. 2002, 4, 3715-18.
14. Oxidative Cyclization of Diols Derived from 1,5‐Dienes: Formation of Enantiopure cis‐Tetrahydrofurans by Using Catalytic Osmium Tetroxide; Formal Synthesis of (+)‐cis‐Solamin. Donohoe, T.J.; Butterworth, S. Angew. Chem. 2005, 117, 4844-46.

346
15. Toward Chemical Libraries of Annonaceous Acetogenins. Total Synthesis of Trilobacin. Sinha, S.C.; Sinha, A.; Yazbak, A.; Keinan, E. J. Org. Chem. 1996, 61, 7640-41.
16. Studies of Palladium-Catalyzed Cross-Coupling Reactions for Preparation of Highly Hindered Biaryls Relevant to the Korupensamine/Michellamine Problem. Hoye, T.R.; Ye, Z. J. Am. Chem. Soc. 1996, 118, 1801-02.
17. 1,2-O-Isopropylidene-5-alkene templates for the synthesis of oligo-tetrahydrofurans. Dabideen, D.; Ruan, Z.; Mootoo, D.R. Tetrahedron. 2002, 58, 2077-84.
18. New Strategy for the Construction of a Monotetrahydrofuran Ring in Annonaceous Acetogenin Based on a Ruthenium Ring-Closing Metathesis: Application to the Synthesis of Solamin. Prestat, G.; Baylon, C.; Heck, M.-P.; Grasa, G.A.; Nolan, S.P.; Mioskowski, C. The Journal of Organic Chemistry. 2004, 69, 5770-73.
19. Tanaka, A.; Makabe, H.; Tanimoto, H.; Oritani, T., Total Synthesis of (8'R)- and (8'S)-Corossoline, Heterocycles, 1996, p.
20. A Convergent Synthesis of(+)-Parviflorin,(+)-Squamocin K, and(+)-5S-Hydroxyparviflorin. Trost, B.M.; Calkins, T.L.; Bochet, C.G. Angew. Chem., Int. Ed. Engl. 1997, 36, 2632-35.
21. Total syntheses of squamocin A and squamocin D. Emde, U.; Koert, U. Tetrahedron Lett. 1999, 40, 5979-82.
22. Formal Synthesis of Uvaricin via Palladium-Mediated Double Cyclization. Burke, S.D.; Jiang, L. Org. Lett. 2001, 3, 1953-55.
23. Stereoselective syntheses of rolliniastatin 1, rollimembrin, and membranacin. Keum, G.; Hwang, C.H.; Kang, S.B.; Kim, Y.; Lee, E. J. Am. Chem. Soc. 2005, 127, 10396-99.
24. Total Synthesis and Preliminary Biological Evaluation of cis-Solamin Isomers. Cecil, A.R.L.; Hu, Y.; Vicent, M.J.; Duncan, R.; Brown, R.C.D. The Journal of Organic Chemistry. 2004, 69, 3368-74.
25. Total Synthesis of the Annonaceous Acetogenin (+)-Asimicin. Development of a New Bidirectional Strategy. Marshall, J.A.; Hinkle, K.W. The Journal of Organic Chemistry. 1997, 62, 5989-95.
26. Total synthesis and structural confirmation of (+)-longicin. Hanessian, S.; Giroux, S.; Buffat, M. Org. Lett. 2005, 7, 3989-92.
27. Synthesis of murisolin, (15R, 16R, 19R, 20S)-murisolin A, and (15R, 16R, 19S, 20S)-16,19-cis-murisolin and their inhibitory action with bovine heart mitochondrial complex I. Hattori, Y.; Kimura, Y.; Moroda, A.; Konno, H.; Abe, M.; Miyoshi, H.; Goto, T.; Makabe, H. Chem.–Asian J. 2006, 1, 894-904.
28. Total synthesis of gigantetrocin A. Wang, Z.M.; Tian, S.K.; Shi, M. Chirality. 2000, 12, 581-89.

347
29. Synthesis of the non-classical acetogenin mucocin: a modular approach based on olefinic coupling reactions. Zhu, L.; Mootoo, D.R. Org. Biomol. Chem. 2005, 3, 2750-54.
30. Total Synthesis of the Nonadjacent Bis-Tetrahydrofuran Annonaceous Acetogenin Squamostatin-D. Marshall, J.A.; Jiang, H. J. Org. Chem. 1998, 63, 7066-71.
31. Total synthesis of jimenezin via an intramolecular allylboration. Bandur, N.G.; Brückner, D.; Hoffmann, R.W.; Koert, U. Org. Lett. 2006, 8, 3829-31.
32. First Total Synthesis of Mosin B. Maezaki, N.; Kojima, N.; Sakamoto, A.; Iwata, C.; Tanaka, T. Org. Lett. 2001, 3, 429-32.
33. Total Synthesis of the Cytotoxic Threo, Trans, Erythro, Cis, Threo Annonaceous Acetogenin Trilobin. Marshall, J.A.; Jiang, H. J. Org. Chem. 1999, 64, 971-75.
34. Total Synthesis of Trilobin. Sinha, A.; Sinha, S.C.; Sinha, S.C.; Keinan, E. J. Org. Chem. 1999, 64, 2381-86.
35. Synthesis and antitumor activity of C-9 epimers of the tetrahydrofuran containing acetogenin 4-deoxyannoreticuin. Wang, F.; Kawamura, A.; Mootoo, D.R. Bioorg Med Chem. 2008, 16, 8413-8.
36. Synthesis of annonacin isolated from Annona densicoma. Oasa, M.; Hattori, Y.; Konno, H.; Makabe, H. Biosci Biotechnol Biochem. 2010, 74, 1274-5.
37. Modular assembly of cytotoxic acetogenin mimetics by click linkage with nitrogen functionalities. Mao, C.; Han, B.; Wang, L.-S.; Wang, S.; Yao, Z.-J. MedChemComm. 2011, 2, 918-22.
38. Critical role of a methyl group on the gamma-lactone ring of annonaceous acetogenins in the potent inhibition of mitochondrial complex I. Kojima, N.; Abe, M.; Suga, Y.; Ohtsuki, K.; Tanaka, T.; Iwasaki, H.; Yamashita, M.; Miyoshi, H. Bioorg Med Chem Lett. 2013, 23, 1217-9.
39. Structure-activity relationships of hybrid annonaceous acetogenins: powerful growth inhibitory effects of their connecting groups between heterocycle and hydrophobic carbon chain bearing THF ring on human cancer cell lines. Kojima, N.; Fushimi, T.; Tatsukawa, T.; Yoshimitsu, T.; Tanaka, T.; Yamori, T.; Dan, S.; Iwasaki, H.; Yamashita, M. Eur J Med Chem. 2013, 63, 833-9.
40. Total synthesis of 14,21-diepi-squamocin-K. Liu, C.-W.; Yeh, T.-C.; Chen, C.-H.; Yu, C.-C.; Chen, C.-S.; Hou, D.-R.; Guh, J.-H. Tetrahedron. 2013, 69, 2971-76.
41. New cytotoxic annonaceous acetogenin mimetics having a nitrogen-heterocyclic terminal and their application to cell imaging. Chen, Y.-J.; Jin, S.; Xi, J.; Yao, Z.-J. Tetrahedron. 2014, 70, 4921-28.
42. Thiophene-3-carboxamide analogue of annonaceous acetogenins as antitumor drug lead. Kojima, N.; Fushimi, T.; Tatsukawa, T.; Tanaka, T.; Okamura, M.; Akatsuka, A.; Yamori, T.; Dan, S.; Iwasaki, H.; Yamashita, M. Eur J Med Chem. 2014, 86, 684-9.

348
43. Total synthesis of muricadienin, the putative key precursor in the solamin biosynthesis. Adrian, J.; Stark, C.B. Org Lett. 2014, 16, 5886-9.
44. Synthesis of dansyl-labeled probe of thiophene analogue of annonaceous acetogenins for visualization of cell distribution and growth inhibitory activity toward human cancer cell lines. Kojima, N.; Suga, Y.; Matsumoto, T.; Tanaka, T.; Akatsuka, A.; Yamori, T.; Dan, S.; Iwasaki, H.; Yamashita, M. Bioorg Med Chem. 2015, 23, 1276-83.
45. Total Synthesis of (+)-Goniodenin. Ueda, T.; Suzuki, A.; Sasaki, M.; Hoshiya, N.; Uenishi, J. J Org Chem. 2016, 81, 12374-81.
46. Convergent synthesis of stereoisomers of THF ring moiety of acetogenin thiophene analogue and their antiproliferative activities against human cancer cell lines. Matsumoto, T.; Kojima, N.; Akatsuka, A.; Yamori, T.; Dan, S.; Iwasaki, H.; Yamashita, M. Tetrahedron. 2017, 73, 2359-66.
47. Total Synthesis of Two Possible Diastereomers of Natural 6-Chlorotetrahydrofuran Acetogenin and Its Stereostructural Elucidation. Takamura, H.; Katsube, T.; Okamoto, K.; Kadota, I. Chemistry. 2017, 23, 17191-94.
48. Convergent Total Synthesis of Asimicin via Decarbonylative Radical Dimerization. Kawamata, T.; Yamaguchi, A.; Nagatomo, M.; Inoue, M. Chemistry. 2018, 24, 18907-12.
49. A General Diastereoselective Strategy for Both cis- and trans-2,6-Disubstituted Tetrahydropyrans: Formal Total Synthesis of (+)-Muconin. Srinivas, B.; Reddy, D.S.; Mallampudi, N.A.; Mohapatra, D.K. Org Lett. 2018, 20, 6910-14.
50. Membrane Conformations and Their Relation to Cytotoxicity of Asimicin and Its Analogues. Shimada, H.; Grutzner, J.B.; Kozlowski, J.F.; McLaughlin, J.L. Biochemistry. 1998, 37, 854-66.
51. Makabe, H.; Miyawaki, A.; Takahashi, R.; Hattori, Y.; Konno, H.; Abe, M.; Miyoshi, H. Tetrahedron Lett. 2004, 45, 973-77.
52. Mechanistic Study of Tetrahydrofuran- acetogenins In Triggering Endoplasmic Reticulum Stress Response-apotoposis in Human Nasopharyngeal Carcinoma. Juang, S.H.; Chiang, C.Y.; Liang, F.P.; Chan, H.H.; Yang, J.S.; Wang, S.H.; Lin, Y.C.; Kuo, P.C.; Shen, M.R.; Thang, T.D.; Nguyet, B.T.; Kuo, S.C.; Wu, T.S. Sci Rep. 2016, 6, 39251.
53. Mitochondrial complex I inhibitors, acetogenins, induce HepG2 cell death through the induction of the complete apoptotic mitochondrial pathway. de Pedro, N.; Cautain, B.; Melguizo, A.; Vicente, F.; Genilloud, O.; Pelaez, F.; Tormo, J.R. J Bioenerg Biomembr. 2013, 45, 153-64.
54. Annonaceous acetogenins mediated up-regulation of Notch2 exerts growth inhibition in human gastric cancer cells in vitro. Li, Y.; Ye, J.; Chen, Z.; Wen, J.; Li, F.; Su, P.; Lin, Y.; Hu, B.; Wu, D.; Ning, L.; Xue, Q.; Gu, H.; Ning, Y. Oncotarget. 2017, 8, 21140-52.
55. The alternative medicine pawpaw and its acetogenin constituents suppress tumor angiogenesis via the HIF-1/VEGF pathway. Coothankandaswamy, V.; Liu, Y.; Mao, S.C.; Morgan, J.B.; Mahdi, F.; Jekabsons, M.B.; Nagle, D.G.; Zhou, Y.D. J Nat Prod. 2010, 73, 956-61.

349
56. A novel thiophene-3-carboxamide analog of annonaceous acetogenin exhibits antitumor activity via inhibition of mitochondrial complex I. Akatsuka, A.; Kojima, N.; Okamura, M.; Dan, S.; Yamori, T. Pharmacol Res Perspect. 2016, 4, e00246.
57. a) Graviola: a novel promising natural-derived drug that inhibits tumorigenicity and metastasis of pancreatic cancer cells in vitro and in vivo through altering cell metabolism. Torres, M.P.; Rachagani, S.; Purohit, V.; Pandey, P.; Joshi, S.; Moore, E.D.; Johansson, S.L.; Singh, P.K.; Ganti, A.K.; Batra, S.K. Cancer Lett. 2012, 323, 29-40; b) Selective growth inhibition of human breast cancer cells by graviola fruit extract in vitro and in vivo involving downregulation of EGFR expression. Dai, Y.; Hogan, S.; Schmelz, E.M.; Ju, Y.H.; Canning, C.; Zhou, K. Nutr Cancer. 2011, 63, 795-801. 58. Synergistic interactions among flavonoids and acetogenins in Graviola (Annona muricata) leaves confer protection against prostate cancer. Yang, C.; Gundala, S.R.; Mukkavilli, R.; Vangala, S.; Reid, M.D.; Aneja, R. Carcinogenesis. 2015, 36, 656-65.
59. Mode of action of bullatacin: A potent antitumor and pesticidal Annonaceous acetogenin. Ahammadsahib, K.I.; Hollingworth, R.M.; McGovren, J.P.; Hui, Y.H.; McLaughlin, J.L. Life Sciences. 1993, 53, 1113-20.
60. Antitumor activity and toxicity relationship of annonaceous acetogenins. Chen, Y.; Chen, J.W.; Zhai, J.H.; Wang, Y.; Wang, S.L.; Li, X. Food Chem Toxicol. 2013, 58, 394-400.
61. Possible relation of atypical parkinsonism in the French West Indies with consumption of tropical plants: a case-control study. Caparros-Lefebvre, D.; Elbaz, A. The Lancet. 1999, 354, 281-86.
62. Analysis of cytotoxic activity at short incubation times reveals profound differences among Annonaceus acetogenins, inhibitors of mitochondrial Complex I. de Pedro, N.; Cautain, B.; Melguizo, A.; Cortes, D.; Vicente, F.; Genilloud, O.; Tormo, J.R.; Pelaez, F. J Bioenerg Biomembr. 2013, 45, 145-52.
63. Bullatacin--in vivo and in vitro experience in an ovarian cancer model. Holschneider, C.H.; Johnson, M.T.; Knox, R.M.; Rezai, A.; Ryan, W.J.; Montz, F.J. Cancer Chemother Pharmacol. 1994, 34, 166-70.
64. Cytotoxic bistetrahydrofuran annonaceous acetogenins from the seeds of Annona squamosa. Chen, Y.; Chen, J.W.; Li, X. J Nat Prod. 2011, 74, 2477-81.
65. Qayed, W.; Aboraia, A.; Abdel-Rahman, H.; Youssef, A., Annonaceous Acetogenins as a new anticancer agent, 2015, p. 24-35.
66. Annonaceous acetogenins from the leaves of Annona montana. Wang, L.Q.; Min, B.S.; Li, Y.; Nakamura, N.; Qin, G.W.; Li, C.J.; Hattori, M. Bioorg Med Chem. 2002, 10, 561-5.
67. Antitumor activity of annonaceous acetogenins in HepS and S180 xenografts bearing mice. Chen, Y.; Chen, J.W.; Xu, S.S.; Wang, Y.; Li, X.; Cai, B.C.; Fan, N.B. Bioorg Med Chem Lett. 2012, 22, 2717-9.
68. Synthesis and tumor cell growth inhibitory activity of biotinylated annonaceous acetogenins. Shi, J.F.; Wu, P.; Jiang, Z.H.; Wei, X.Y. Eur J Med Chem. 2014, 71, 219-28.

350
69. Annona muricata: Is the natural therapy to most disease conditions including cancer growing in our backyard? A systematic review of its research history and future prospects. Gavamukulya, Y.; Wamunyokoli, F.; El-Shemy, H.A. Asian Pacific Journal of Tropical Medicine. 2017, 10, 835-48.
70. Definition of crucial structural factors of acetogenins, potent inhibitors of mitochondrial complex I. Takada, M.; Kuwabara, K.; Nakato, H.; Tanaka, A.; Iwamura, H.; Miyoshi, H. Biochim Biophys Acta. 2000, 1460, 302-10.
71. Natural-Product Hybrids: Design, Synthesis, and Biological Evaluation of Quinone–Annonaceous Acetogenins. Hoppen, S.; Emde, U.; Friedrich, T.; Grubert, L.; Koert, U. Angewandte Chemie International Edition. 2000, 39, 2099-102.
72. Heterocyclic Analogues of Squamocin as Inhibitors of Mitochondrial Complex I. On the Role of the Terminal Lactone of Annonaceous Acetogenins. Duval, R.A.; Lewin, G.; Peris, E.; Chahboune, N.; Garofano, A.; Dröse, S.; Cortes, D.; Brandt, U.; Hocquemiller, R. Biochemistry. 2006, 45, 2721-28.
73. a) Three new anti-proliferative Annonaceous acetogenins with mono-tetrahydrofuran ring from graviola fruit (Annona muricata). Sun, S.; Liu, J.; Kadouh, H.; Sun, X.; Zhou, K. Bioorg Med Chem Lett. 2014, 24, 2773-6; b) Isolation of three new annonaceous acetogenins from Graviola fruit (Annona muricata) and their anti-proliferation on human prostate cancer cell PC-3. Sun, S.; Liu, J.; Zhou, N.; Zhu, W.; Dou, Q.P.; Zhou, K. Bioorg Med Chem Lett. 2016, 26, 4382-5. 74. Catalytic hydrogenation of annonaceous acetogenins. Cortes, D.; Myint, S.H.; Harmange, J.C.; Sahpaz, S.; Figadere, B. Tetrahedron Letters. 1992, 33, 5225-26.
75. 4-deoxyannomontacin and (2,4-cis and trans)-annomontacinone, new bioactive mono-tetrahydrofuran annonaceous acetogenins from Goniothalamus giganteus. Alali, F.; Zeng, L.; Zhang, Y.; Ye, Q.; Hopp, D.C.; Schwedler, J.T.; McLaughlin, J.L. Bioorg Med Chem. 1997, 5, 549-55.
76. The first total synthesis of 4-deoxyannomontacin. Yu, Q.; Wu, Y.; Ding, H.; Wu, Y.-L. Journal of the Chemical Society, Perkin Transactions 1. 1999, 1183-88.
77. Synthesis and antitumor activity of C-9 epimers of the tetrahydrofuran containing acetogenin 4-deoxyannoreticuin. Wang, F.; Kawamura, A.; Mootoo, D.R. Bioorganic & medicinal chemistry. 2008, 16, 8413-18.
78. Synthesis of Nonadjacently Linked Tetrahydrofurans: An Iodoetherification and Olefin Metathesis Approach. Zhu, L.; Mootoo, D.R. Organic Letters. 2003, 5, 3475-78.
79. Synthesis and anti-tumor activity of carbohydrate analogues of the tetrahydrofuran containing acetogenins. Bachan, S.; Tony, K.A.; Kawamura, A.; Montenegro, D.; Joshi, A.; Garg, H.; Mootoo, D.R. Bioorg Med Chem. 2013, 21, 6554-64.
80. Total Synthesis of the Nonadjacently Linked Bis-tetrahydrofuran Acetogenin Bullatanocin (Squamostatin C). Zhu, L.; Mootoo, D.R. The Journal of Organic Chemistry. 2004, 69, 3154-57.
81. Synthesis of solamin type mono-THF acetogenins using cross-metathesis. Konno, H.; Makabe, H.; Hattori, Y.; Nosaka, K.; Akaji, K. Tetrahedron. 2010, 66, 7946-53.

351
82. Wang, Z.-M.; Zhang, X.-L.; Sharpless, K.B.; Sinha, S.; Sinha-Bagchi, A.; Keinan, E., A General Approach to γ-Lactones via Osmium-Catalyzed Asymmetric Dihydroxylation. Synthesis of (-)- and (+)-Muricatacin, 1992, p. 6407–10.
83. Acetal Templates for the Synthesis of trans-2,5-Disubstituted Tetrahydrofurans. Zhang, H.; Mootoo, D.R. The Journal of Organic Chemistry. 1995, 60, 8134-35.
84. Asymmetric Dihydroxylation−Haloetherification Strategy for the Synthesis of Tetrahydrofuran-Containing Acetogenins. Zhang, H.; Seepersaud, M.; Seepersaud, S.; Mootoo, D.R. The Journal of Organic Chemistry. 1998, 63, 2049-52.
85. Synthesis, Characterization, and Evaluation of 10-Undecenoic Acid-Based Epithio Derivatives as Multifunctional Additives. Geethanjali, G.; Padmaja, K.V.; Sammaiah, A.; Prasad, R.B.N. Journal of Agricultural and Food Chemistry. 2014, 62, 11505-11.
86. a) A short history of SHELX. Sheldrick, G. Acta Crystallographica Section A. 2008, 64, 112-22; b) SHELXT - Integrated space-group and crystal-structure determination. Sheldrick, G. Acta Crystallographica Section A. 2015, 71, 3-8; c) Sheldrick, G.M., An Integrated System for Solving, Refining, and Displaying Crystal Structures from Diffraction Data, University of Göttingen, Göttingen, Federal Republic of Germany, 1981, p. 87. Use of intensity quotients and differences in absolute structure refinement. Parsons, S.; Flack, H.D.; Wagner, T. Acta Crystallographica Section B. 2013, 69, 249-59.
88. Enantiopure Simple Analogues of Annonaceous Acetogenins with Remarkable Selective Cytotoxicity towards Tumor Cell Lines. Zeng, B.-B.; Wu, Y.; Yu, Q.; Wu, Y.-L.; Li, Y.; Chen, X.-G. Angewandte Chemie International Edition. 2000, 39, 1934-37.
89. Analogues of cytotoxic squamocin using reliable reactions: new insights into the reactivity and role of the α,β-unsaturated lactone of the annonaceous acetogenins. Duval, R.A.; Poupon, E.; Romero, V.; Peris, E.; Lewin, G.; Cortes, D.; Brandt, U.; Hocquemiller, R. Tetrahedron. 2006, 62, 6248-57.
90. Acetogenins from Annonaceae, inhibitors of mitochondrial complex I. Zafra-Polo, M.C.; Gonzalez, M.C.; Estornell, E.; Sahpaz, S.; Cortes, D. Phytochemistry. 1996, 42, 253-71.
91. Essential structural factors of annonaceous acetogenins as potent inhibitors of mitochondrial complex I. Miyoshi, H.; Ohshima, M.; Shimada, H.; Akagi, T.; Iwamura, H.; McLaughlin, J.L. Biochim Biophys Acta. 1998, 1365, 443-52.
92. Squamotacin: an annonaceous acetogenin with cytotoxic selectivity for the human prostate tumor cell line (PC-3). Hopp, D.C.; Zeng, L.; Gu, Z.; McLaughlin, J.L. J Nat Prod. 1996, 59, 97-9.
93. a) Expression of the glucose transporters GLUT1, GLUT3, GLUT4 and GLUT12 in human cancer cells. Barron, C.; Tsiani, E.; Tsakiridis, T. BMC Proceedings. 2012, 6, ; b) Mitochondrial metabolism inhibitors for cancer therapy. Ramsay, E.E.; Hogg, P.J.; Dilda, P.J. Pharm Res. 2011, 28, 2731-44; c) The carbamoylmannose moiety of bleomycin mediates selective tumor cell targeting. Bhattacharya, C.; Yu, Z.; Rishel, M.J.; Hecht, S.M. Biochemistry. 2014, 53, 3264-6.

352
94. a) Recent applications of olefin metathesis and related reactions in carbohydrate chemistry. Roy, R.; Das, S.K. Chemical Communications. 2000, 519-29; b) Alkenyl O- and C-glycopyranoside homodimerization by olefin metathesis reaction. Dominique, R.; K. Das, S.; Roy, R. Chemical Communications. 1998, 2437-38. 95. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Srinivasarao, M.; Galliford, C.V.; Low, P.S. Nat Rev Drug Discov. 2015, 14, 203-19.
96. Ligand-Targeted Drug Delivery. Srinivasarao, M.; Low, P.S. Chem Rev. 2017, 117, 12133-64.
97. Folate-modified Annonaceous acetogenins nanosuspensions and their improved antitumor efficacy. Hong, J.; Sun, Z.; Li, Y.; Guo, Y.; Liao, Y.; Liu, M.; Wang, X. International journal of nanomedicine. 2017, 12, 5053-67.
98. Anticancer agents interacting with membrane glucose transporters. Granchi, C.; Fortunato, S.; Minutolo, F. Medchemcomm. 2016, 7, 1716-29.
99. Deglycosylated bleomycin has the antitumor activity of bleomycin without pulmonary toxicity. Burgy, O.; Wettstein, G.; Bellaye, P.S.; Decologne, N.; Racoeur, C.; Goirand, F.; Beltramo, G.; Hernandez, J.F.; Kenani, A.; Camus, P.; Bettaieb, A.; Garrido, C.; Bonniaud, P. Sci Transl Med. 2016, 8, 326ra20.
100. Modified bleomycin disaccharides exhibiting improved tumor cell targeting. Madathil, M.M.; Bhattacharya, C.; Yu, Z.; Paul, R.; Rishel, M.J.; Hecht, S.M. Biochemistry. 2014, 53, 6800-10.
101. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Chang, S.S.; O'Keefe, D.S.; Bacich, D.J.; Reuter, V.E.; Heston, W.D.; Gaudin, P.B. Clin Cancer Res. 1999, 5, 2674-81.
102. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Liu, H.; Rajasekaran, A.K.; Moy, P.; Xia, Y.; Kim, S.; Navarro, V.; Rahmati, R.; Bander, N.H. Cancer Res. 1998, 58, 4055-60.
103. a) Prostate-specific membrane antigen targeted imaging and therapy of prostate cancer using a PSMA inhibitor as a homing ligand. Kularatne, S.A.; Wang, K.; Santhapuram, H.K.; Low, P.S. Mol Pharm. 2009, 6, 780-9; b) Synthesis and biological analysis of prostate-specific membrane antigen-targeted anticancer prodrugs. Kularatne, S.A.; Venkatesh, C.; Santhapuram, H.K.; Wang, K.; Vaitilingam, B.; Henne, W.A.; Low, P.S. J Med Chem. 2010, 53, 7767-77; c) The Rise of PSMA Ligands for Diagnosis and Therapy of Prostate Cancer. Afshar-Oromieh, A.; Babich, J.W.; Kratochwil, C.; Giesel, F.L.; Eisenhut, M.; Kopka, K.; Haberkorn, U. J Nucl Med. 2016, 57, 79S-89S; d) Comparison of Prostate-Specific Membrane Antigen-Based 18F-DCFBC PET/CT to Conventional Imaging Modalities for Detection of Hormone-Naive and Castration-Resistant Metastatic Prostate Cancer. Rowe, S.P.; Macura, K.J.; Ciarallo, A.; Mena, E.; Blackford, A.; Nadal, R.; Antonarakis, E.S.; Eisenberger, M.A.; Carducci, M.A.; Ross, A.E.; Kantoff, P.W.; Holt, D.P.; Dannals, R.F.; Mease, R.C.; Pomper, M.G.; Cho, S.Y. J Nucl Med. 2016, 57, 46-53; e) Glu-Ureido-Based Inhibitors of Prostate-Specific Membrane Antigen: Lessons Learned During the Development of a Novel Class of Low-Molecular-Weight Theranostic Radiotracers. Kopka, K.; Benesova, M.; Barinka, C.; Haberkorn, U.; Babich, J. J Nucl Med. 2017, 58, 17S-26S. 104. Design, synthesis, and preclinical evaluation of prostate-specific membrane antigen targeted (99m)Tc-radioimaging agents. Kularatne, S.A.; Zhou, Z.; Yang, J.; Post, C.B.; Low, P.S. Mol Pharm. 2009, 6, 790-800.

353
105. Chemical Control over Immune Recognition: A Class of Antibody-Recruiting Small Molecules That Target Prostate Cancer. Murelli, R.P.; Zhang, A.X.; Michel, J.; Jorgensen, W.L.; Spiegel, D.A. Journal of the American Chemical Society. 2009, 131, 17090-92.
106. Synthesis and Biological Evaluation of EC20: A New Folate-Derived, 99mTc-Based Radiopharmaceutical. Leamon, C.P.; Parker, M.A.; Vlahov, I.R.; Xu, L.-C.; Reddy, J.A.; Vetzel, M.; Douglas, N. Bioconjugate Chemistry. 2002, 13, 1200-10.
107. Design and synthesis of releasable folate-drug conjugates using a novel heterobifunctional disulfide-containing linker. Satyam, A. Bioorganic & medicinal chemistry letters. 2008, 18, 3196-99.
108. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Dubikovskaya, E.A.; Thorne, S.H.; Pillow, T.H.; Contag, C.H.; Wender, P.A. Proceedings of the National Academy of Sciences. 2008, 105, 12128.
109. Annonacin in Asimina triloba fruit: implication for neurotoxicity. Potts, L.F.; Luzzio, F.A.; Smith, S.C.; Hetman, M.; Champy, P.; Litvan, I. Neurotoxicology. 2012, 33, 53-8.
110. DUPA Conjugation of a Cytotoxic Indenoisoquinoline Topoisomerase I Inhibitor for Selective Prostate Cancer Cell Targeting. Roy, J.; Nguyen, T.X.; Kanduluru, A.K.; Venkatesh, C.; Lv, W.; Reddy, P.V.N.; Low, P.S.; Cushman, M. Journal of Medicinal Chemistry. 2015, 58, 3094-103.
111. A novel synthesis of tri-, di-, and mono-9-acridinyl derivatives of tetra-, tri-, and di-amines. Hansen, J.B.; Buchardt, O. Journal of the Chemical Society, Chemical Communications. 1983, 162-64.
112. Systematic construction of a monotetrahydrofuran-ring library in annonaceous acetogenins by asymmetric alkynylation and stereodivergent tetrahydrofuran-ring formation. Kojima, N.; Maezaki, N.; Tominaga, H.; Asai, M.; Yanai, M.; Tanaka, T. Chemistry. 2003, 9, 4980-90.
113. Duval, R.; Poupon, E.; Romero, V.; Peris, E.; Lewin, G.; Cortes, D.; Brandt, U.; Hocquemiller, R., Analogues of cytotoxic squamocin using reliable reactions: new insights into the reactivity and role of the α,β-unsaturated lactone of the annonaceous acetogenins, 2006, p. 6248-57.




















