SUMO1 Modification Alters ADAR1 Editing Activity
12
Molecular Biology of the Cell Vol. 16, 5115–5126, November 2005 SUMO-1 Modification Alters ADAR1 Editing Activity Joana M.P. Desterro,* Liam P. Keegan, † Ellis Jaffray, ‡ Ron T. Hay, ‡ Mary A. O’Connell, † and Maria Carmo-Fonseca* *Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal; † MRC Human Genetics Unit, Western General Hospital, Edinburgh EH4 2XU, Scotland, United Kingdom; and ‡ Biomolecular Sciences Building, University of St. Andrews, St. Andrews, Fife KY19 9ST, Scotland, United Kingdom Submitted June 15, 2005; Revised July 25, 2005; Accepted August 16, 2005 Monitoring Editor: Peter Walter We identify ADAR1, an RNA-editing enzyme with transient nucleolar localization, as a novel substrate for sumoylation. We show that ADAR1 colocalizes with SUMO-1 in a subnucleolar region that is distinct from the fibrillar center, the dense fibrillar component, and the granular component. Our results further show that human ADAR1 is modified by SUMO-1 on lysine residue 418. An arginine substitution of K418 abolishes SUMO-1 conjugation and although it does not interfere with ADAR1 proper localization, it stimulates the ability of the enzyme to edit RNA both in vivo and in vitro. Moreover, modification of wild-type recombinant ADAR1 by SUMO-1 reduces the editing activity of the enzyme in vitro. Taken together these data suggest a novel role for sumoylation in regulating RNA-editing activity. INTRODUCTION A defining feature of eukaryotic cells is the generation of protein diversity either posttranscriptionally by alternative splicing and RNA editing or posttranslationally by modifi- cation of amino acids in proteins. One of the most recently discovered posttranslational modification mechanism in eu- karyotes involves the covalent attachment of the small ubiq- uitinlike modifier, SUMO, to target proteins. Modification of proteins by SUMO, or sumoylation, plays crucial regulatory roles in eukaryotes. Proteins known to be modified by SUMO include, among others, RanGAP1, PCNA, IB, p53, c-jun, topoisomerases, promyelocytic leukemia protein (PML), Sp100, and the mitogen-activated protein kinase ki- nase 1 (MEKK1). Many SUMO substrates are transcription factors and cofactors, or proteins implicated in DNA repair and replication (reviewed by Hay, 2001; Melchior et al., 2003; Seeler and Dejean, 2003; Hay, 2005). Although it is well established that SUMO can affect target protein function by altering its subcellular localization, activity, or stability, for many substrates the biological functions of sumoylation re- main unknown. Sumoylation is a reversible and highly dynamic process that involves formation of an isopeptide bond between the C-terminus of SUMO and the -amino group of a lysine residue of the target protein. The most intensely studied human form of SUMO is the SUMO-1 protein, which is 48% identical to yeast Smt3 (Bayer et al., 1998; Mossessova and Lima, 2000). In vertebrates there are at least three additional proteins. SUMO-2 and SUMO-3 are 45% identical to SUMO-1 (Saitoh and Hinchey, 2000), and SUMO-4 shows an 86% amino acid homology to SUMO-2 (Bohren et al., 2004). SUMO is conjugated to protein substrates via an ATP-de- pendent enzymatic pathway that is mechanistically similar to ubiquitination. The reaction requires a SUMO protease that removes four amino acids from the C-terminus of the 101-amino acid SUMO-1 precursor to generate the mature form; an heterodimeric SUMO-activating enzyme, SAE1/2; Ubc9, a SUMO-conjugating enzyme that ligates directly to its protein target; and an E3-like SUMO ligase (reviewed Melchior et al., 2003). Three SUMO E3s have been identified so far: the mammalian protein inhibitors of activated STAT (PIAS; Sachdev et al., 2001), the nucleoporin RanBP2 (Azuma and Dasso, 2002; Pichler et al., 2002), and the polycomb group protein PC2 (Kagey et al., 2003). Recent structural data provide novel insights into the mechanism used by E3s to enhance SUMO conjugation (Duda and Schulman, 2005; Reverter and Lima, 2005; Tatham et al., 2005). Removal of SUMO from proteins is carried out by specific cysteine proteases that have both hydrolase and isopepti- dase activity (Li and Hochstrasser, 1999, 2000). Most en- zymes involved in the SUMO pathway are localized in the nucleus, and it is therefore believed that sumoylation is predominantly a nuclear process (Rodriguez et al., 2001; Zhang et al., 2002; Seeler and Dejean, 2003). Here, we describe that proteins modified by SUMO-1 are present in the nucleolus, that SUMO-1 in the nucleolus colocalizes with the RNA-editing enzyme ADAR1, and that this enzyme represents a novel substrate for sumoylation. ADAR1 (adenosine deaminase that acts on RNA) is a member of the family of enzymes that catalyze the conver- sion of adenosine to inosine in double-stranded RNA (dsRNA; reviewed in Keegan et al., 2001; Bass, 2002; Schaub and Keller, 2002). Because inosine acts as guanosine during translation, A-to-I conversion in coding sequences leads to amino acid changes and often entails changes in protein function. In addition to amino acid changes, A-to-I RNA editing can also occur in 5 and 3 UTR (Morse and Bass, This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05– 06 – 0536) on August 24, 2005. Address correspondence to: Joana M. Pinto Desterro (joanadesterro@ fm.ul.pt). Abbreviations used: SUMO, small ubiquitin modifier; ADAR, aden- osine deaminase that act on RNA; SAE, SUMO-activating enzyme; UBC9, SUMO-conjugating enzyme. © 2005 by The American Society for Cell Biology 5115
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of SUMO1 Modification Alters ADAR1 Editing Activity

Molecular Biology of the CellVol. 16, 5115–5126, November 2005
SUMO-1 Modification Alters ADAR1 Editing ActivityJoana M.P. Desterro,* Liam P. Keegan,† Ellis Jaffray,‡ Ron T. Hay,‡Mary A. O’Connell,† and Maria Carmo-Fonseca*
*Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal;†MRC Human Genetics Unit, Western General Hospital, Edinburgh EH4 2XU, Scotland, United Kingdom;and ‡Biomolecular Sciences Building, University of St. Andrews, St. Andrews, Fife KY19 9ST, Scotland,United Kingdom
Submitted June 15, 2005; Revised July 25, 2005; Accepted August 16, 2005Monitoring Editor: Peter Walter
We identify ADAR1, an RNA-editing enzyme with transient nucleolar localization, as a novel substrate for sumoylation.We show that ADAR1 colocalizes with SUMO-1 in a subnucleolar region that is distinct from the fibrillar center, the densefibrillar component, and the granular component. Our results further show that human ADAR1 is modified by SUMO-1on lysine residue 418. An arginine substitution of K418 abolishes SUMO-1 conjugation and although it does not interferewith ADAR1 proper localization, it stimulates the ability of the enzyme to edit RNA both in vivo and in vitro. Moreover,modification of wild-type recombinant ADAR1 by SUMO-1 reduces the editing activity of the enzyme in vitro. Takentogether these data suggest a novel role for sumoylation in regulating RNA-editing activity.
INTRODUCTION
A defining feature of eukaryotic cells is the generation ofprotein diversity either posttranscriptionally by alternativesplicing and RNA editing or posttranslationally by modifi-cation of amino acids in proteins. One of the most recentlydiscovered posttranslational modification mechanism in eu-karyotes involves the covalent attachment of the small ubiq-uitinlike modifier, SUMO, to target proteins. Modification ofproteins by SUMO, or sumoylation, plays crucial regulatoryroles in eukaryotes. Proteins known to be modified bySUMO include, among others, RanGAP1, PCNA, I�B�, p53,c-jun, topoisomerases, promyelocytic leukemia protein(PML), Sp100, and the mitogen-activated protein kinase ki-nase 1 (MEKK1). Many SUMO substrates are transcriptionfactors and cofactors, or proteins implicated in DNA repairand replication (reviewed by Hay, 2001; Melchior et al., 2003;Seeler and Dejean, 2003; Hay, 2005). Although it is wellestablished that SUMO can affect target protein function byaltering its subcellular localization, activity, or stability, formany substrates the biological functions of sumoylation re-main unknown.
Sumoylation is a reversible and highly dynamic processthat involves formation of an isopeptide bond between theC-terminus of SUMO and the �-amino group of a lysineresidue of the target protein. The most intensely studiedhuman form of SUMO is the SUMO-1 protein, which is 48%identical to yeast Smt3 (Bayer et al., 1998; Mossessova andLima, 2000). In vertebrates there are at least three additional
proteins. SUMO-2 and SUMO-3 are �45% identical toSUMO-1 (Saitoh and Hinchey, 2000), and SUMO-4 shows an86% amino acid homology to SUMO-2 (Bohren et al., 2004).SUMO is conjugated to protein substrates via an ATP-de-pendent enzymatic pathway that is mechanistically similarto ubiquitination. The reaction requires a SUMO proteasethat removes four amino acids from the C-terminus of the101-amino acid SUMO-1 precursor to generate the matureform; an heterodimeric SUMO-activating enzyme, SAE1/2;Ubc9, a SUMO-conjugating enzyme that ligates directly toits protein target; and an E3-like SUMO ligase (reviewedMelchior et al., 2003). Three SUMO E3s have been identifiedso far: the mammalian protein inhibitors of activated STAT(PIAS; Sachdev et al., 2001), the nucleoporin RanBP2 (Azumaand Dasso, 2002; Pichler et al., 2002), and the polycombgroup protein PC2 (Kagey et al., 2003). Recent structural dataprovide novel insights into the mechanism used by E3s toenhance SUMO conjugation (Duda and Schulman, 2005;Reverter and Lima, 2005; Tatham et al., 2005).
Removal of SUMO from proteins is carried out by specificcysteine proteases that have both hydrolase and isopepti-dase activity (Li and Hochstrasser, 1999, 2000). Most en-zymes involved in the SUMO pathway are localized in thenucleus, and it is therefore believed that sumoylation ispredominantly a nuclear process (Rodriguez et al., 2001;Zhang et al., 2002; Seeler and Dejean, 2003).
Here, we describe that proteins modified by SUMO-1 arepresent in the nucleolus, that SUMO-1 in the nucleoluscolocalizes with the RNA-editing enzyme ADAR1, and thatthis enzyme represents a novel substrate for sumoylation.
ADAR1 (adenosine deaminase that acts on RNA) is amember of the family of enzymes that catalyze the conver-sion of adenosine to inosine in double-stranded RNA(dsRNA; reviewed in Keegan et al., 2001; Bass, 2002; Schauband Keller, 2002). Because inosine acts as guanosine duringtranslation, A-to-I conversion in coding sequences leads toamino acid changes and often entails changes in proteinfunction. In addition to amino acid changes, A-to-I RNAediting can also occur in 5� and 3� UTR (Morse and Bass,
This article was published online ahead of print in MBC in Press(http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–06–0536)on August 24, 2005.
Address correspondence to: Joana M. Pinto Desterro ([email protected]).
Abbreviations used: SUMO, small ubiquitin modifier; ADAR, aden-osine deaminase that act on RNA; SAE, SUMO-activating enzyme;UBC9, SUMO-conjugating enzyme.
© 2005 by The American Society for Cell Biology 5115

1999), in introns (Higuchi et al., 1993), and at splicing branchsite (Beghini et al., 2000). Editing can also generate a 3� spliceacceptor (Rueter et al., 1999) and relieve a stop codon (Polsonet al., 1996). In mammals there are three ADAR enzymes,termed ADAR1, ADAR2, and ADAR3. Inactivation of edit-ing enzymes in mice (Higuchi et al., 2000) and in the fruit fly(Palladino et al., 2000b) has resulted in profound neurolog-ical phenotypes. All ADAR proteins have a highly con-served catalytic domain at the C-terminus and one to threedsRNA-binding domains. ADAR1 differs from the othermembers of the family in its extended N-terminus that isenriched in RG residues and contains two tandemly ar-ranged Z-DNA-binding domains (Keegan et al., 2001, 2004).In humans, there are two ADAR1 forms: a 150-kDa protein(comprising amino acids 1–1226) that is induced by inter-feron and localizes predominantly in the cytoplasm, and a110-kDa protein (encompassing residues 296-1226) that isconstitutively expressed and localizes to the nucleus (Patter-son and Samuel, 1995; George and Samuel, 1999b, a).
Several lines of evidence suggest that ADAR activity istightly controlled in the cell. ADARs act as dimers andheterodimer formation between different ADAR forms cancontribute to regulate enzyme activity and substrate speci-ficity (Cho et al., 2003; Gallo et al., 2003). In Drosophila, ADARcan edit its own pre-mRNA (Palladino et al., 2000a), whereasin mammals a self-editing process leads to alternative splic-ing of ADAR2 (Rueter et al., 1999). Furthermore, ADAR1expression in mammals is regulated by interferon (Pattersonand Samuel, 1995).
In this work we demonstrate that ADAR1 is modified bySUMO at lysine residue 418. Substitution of this amino acidresidue by arginine, which cannot be modified by SUMO,affects the editing activity of the enzyme. Our results there-fore suggest a novel role for SUMO in regulating ADAR1editing activity.
MATERIALS AND METHODS
AntibodiesEndogenous ADAR1 was detected with rabbit polyclonal antibodies (anti-body 007 and antibody 668; Desterro et al., 2003). Proteins tagged with ahistidine hexamer were detected with an anti-His monoclonal antibody(mAb; Qiagen, Hilden, Germany), and hemaglutinin (HA)-tagged proteinswere detected with the anti-HA mAb 11 (Babco, Richmond, CA). Greenfluorescence protein (GFP) was detected with a mixture of two mouse mono-clonal antibodies (anti-GFP clones 7.1 and 13.1; Boehringer Mannheim, Indi-anapolis, IN) and endogenous SUMO-1 was detected with a rabbit polyclonalantibody (Santa Cruz Biotechnology, Santa Cruz, CA). PML was detectedwith anti-PML mouse mAb (PGM-3; Santa Cruz Biotechnology). Addition-ally, the following antibodies were used to detect nucleolar proteins: anti-B23/nucleophosmin goat polyclonal antibody C-19 (Santa Cruz Biotechnol-ogy); anti-fibrillarin mAb 71B9 (Reimer et al., 1987); anti-UBF rabbit polyclonalantibody E29 (O’Mahony et al., 1992); and auto-immune human anti-RNApolymerase I serum S18 (kindly provided by Dr U. Scheer).
PlasmidsPlasmids expressing full-length hADAR1 (Desterro et al., 2003), the RC con-struct (Herbert et al., 2002), and GFP-SUMO (Gostissa et al., 1999) have beendescribed previously.
Site-directed MutagenesisThe point mutation in the lysine within the SUMO-1 consensus sequence wasgenerated by oligonucleotide-directed mutagenesis using the QuickChangesite directed mutagenesis kit (Stratagene, La Jolla, CA) and the followingoligonucleotides: 5�-GGAACCTGTCATAAGGTTAGAAAACAGGC-3� and5�-GCCTGTTTTCTAACCTTATGACAGGTTCC-3�. The nucleotides changedin this mutagenesis are indicated in bold.
Mutagenesis was performed on ADAR1 cloned in different plasmids,pEGFP, pFlis, and pPICZA but always with the same set of oligonucleotides.All constructs were confirmed by DNA sequencing.
Cell Culture and TransfectionsHeLa and COS7 cells were maintained in DMEM supplemented with 10%fetal calf serum. To inhibit nuclear export, leptomycin B (LMB; Sigma, St.Louis, MO) was added to a final concentration of 50 nM to the tissue culturemedium before fixation. DNA for transfections assays was purified with aQiagen plasmid Midi-prep kit (Qiagen). HeLa subconfluent cells grown onglass coverslips in 35 � 10-mm tissue culture dishes were transiently trans-fected with 1 mg of purified plasmid DNA and FuGene6 reagent (RocheBiochemicals, Indianapolis, IN) according to the manufacturer’s protocol.Cells were analyzed 24–48 h after transfections.
For Ni2�-NTA-agarose pulldowns COS7 cells were transfected with Lipo-fectamine (Invitrogen, Carlsbad, CA) according to the manufacturer’s proto-col. After transfections, cells were seeded in 75-cm2 flasks and the incubationcontinued for an additional 36 h. His-epitope-tagged proteins were isolated asdescribed (Rodriguez et al., 1999).
ImmunofluorescenceCells on coverslips were briefly rinsed with phosphate-buffered saline (PBS),fixed in 3.7% formaldehyde (freshly prepared from paraformaldehyde), di-luted in PBS for 10 min at room temperature, and washed with PBS. The cellswere then permeabilized with 0.5% Triton X-100 for 15 min or 0.05% SDS for10 min at room temperature and washed with PBS. Immunofluorescence andconfocal microscopy was performed as described (Calado et al., 2000).
In Situ HybridizationGluR-B DNA was obtained by SmaI and XbaI digestion of the GluR-B/pRKplasmid (Higuchi et al., 1993) and RC DNA from EcoRI and XbaI digestion ofthe RC plasmid (Herbert et al., 2002). Both fragments were purified, labeledwith digoxigenin-11-dUTP by nick translation, and used as probes for in situhybridization. Cells were fixed and permeabilized as described previously forimmunofluorescence. Immediately before hybridization, cells were incubatedin hybridization mixture for 5 min at 37°C. Cells were hybridized for 4 h at37°C in 50% formamide, 2� SSC, 10% dextran sulfate, 50 nM sodium phos-phate, pH 7.0, with probes at 2 ng/�l. Posthybridization washes were in 50%formamide, 2� SSC (three times for 5 min at 45°C) and in 2� SSC (three timesfor 5 min at 45°C). The sites of hybridization were visualized with cy3anti-digoxigenin secondary antibody (Molecular Probes, Eugene, OR) dilutedin 4� SSC-Tween, 2% bovine serum albumin, and 0.2% gelatin.
MicroscopySamples were examined on a Zeiss LSM 510 microscope (Thornwood, NY)with a Planapochromat 63�/1.4 objective.
Western Blot AnalysisWestern blot analysis of transfected cells was performed with whole cellextracts that were prepared in SDS sample buffer. Lysates were boiled for 10min before electrophoresis on either 8.5 or 10% polyacrylamide gels andtransferred to a nitrocellulose membrane by electroblotting. Anti-His, anti-ADAR1, and anti-GFP were used as primary antibodies. Horseradish perox-idase-conjugated anti-mouse IgG and anti-rabbit IgG (Bio-Rad Laboratories,Richmond, CA) were used as secondary antibodies. Blots were developedwith the enhanced chemiluminescence detection system (Amersham Bio-sciences, Piscataway, NJ).
Expression and Purification of Recombinant ProteinsSUMO-1, Ubc9, and SAE1/SAE2 were expressed and purified from Esche-richia coli B834 as described previously (Desterro et al., 1997; Tatham et al.,2001). Both ADAR wild-type and K418R mutant were overexpressed in theyeast Pichia pastoris and purified as described (Gallo et al., 2003).
In Vitro Expression of ProteinsIn vitro-coupled transcription/translation of ADAR1 proteins was performedwith 1 �g of plasmid DNA and a wheat germ-coupled transcription/trans-lation system according to the manufacturer’s instructions (Promega, Madi-son, WI). [35S]methionine (Amersham Biosciences) was used in the reactionsto generate radiolabeled protein.
In Vitro SUMO-1 Conjugation AssaySUMO-1 conjugation assays were performed in 10 �l reactions containing anATP regenerating system, 1 �l of [35S]methionine-labeled ADAR1 or 10 ng ofeither WT or K418R purified recombinant ADAR1 and purified recombinantSUMO-1, Ubc9, and SAE proteins as previously described (Tatham et al.,2001). Reaction products were analyzed by SDS-PAGE and either detected byWestern blotting analysis using an anti-ADAR1 antibody or the gel was driedbefore overnight exposure to film.
In Vitro EditingThe nonspecific dsRNA substrate, a shorter form of BScat was prepared by invitro transcription as previously reported and the editing assay was per-
J. M. Pinto Desterro et al.
Molecular Biology of the Cell5116
formed as previously described (O’Connell and Keller, 1994). The assaymixture contained dsRNA containing 200 fmoles of 32P-labeled adenosine,and the reaction was performed at 37°C for 60 min with purified recombinantADAR1-WT and ADAR1-K418R.
Analysis of editing of a transcript encoded by the GluR-B mini-gene B13was performed by primer extension assay with the BHS-RT primer specific forhotspot1 as previously described (Melcher et al., 1995). In the assay 10 fmol ofin vitro-transcribed RNA was incubated with either purified recombinantprotein or previously in vitro SUMO-1 modified protein for 1 h at 37°C. Thereaction mixture was then treated with proteinase K for 30 min., phenol/chloroformed was extracted, and ethanol was precipitated. RadioactiveBHS-RT primer, 10 fmol, was added and annealed at 52°C overnight. Subse-quently an RT reaction was performed in the presence of ddTTP and afterethanol precipitation the extension products were electrophoresed on a 15%denaturing polyacrylamide gel. The gel was dried and quantified on a Phos-phorImager.
RESULTS
Proteins Modified by SUMO-1 Localize to the NucleolusTo date, most known sumoylated proteins are either nuclearproteins or proteins that shuttle to the nucleus. Within thenucleus, proteins modified by SUMO have been localized tothe nucleoplasm, PML nuclear bodies and nuclear pore com-
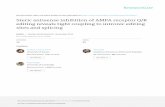
plexes (Melchior et al., 2003; Seeler and Dejean, 2003). Tofurther characterize the subcellular distribution of SUMO-conjugated proteins, a fusion of SUMO-1 to the green fluo-rescence protein (GFP-SUMO-1) was transiently expressedin HeLa cells. GFP-SUMO-1 can replace endogenous SUMO(pmt3) in fission yeast, suggesting that the GFP tag does notinterfere with SUMO-1 function in vivo (Tanaka et al., 1999).Analysis of HeLa cells expressing GFP-SUMO-1 reveals nu-clear staining with accumulation at the nuclear pore com-plexes and in nuclear bodies, as previously described (Fig-ure 1A, arrows and arrowheads). Surprisingly, an additionalnucleolar staining is observed (Figure 1A, dashed lines). Toconfirm that the detection of GFP-SUMO-1 in the nucleoluscorresponds to the presence of modified proteins, HeLa cellswere transfected with GFP-SUMO-1�6. This mutant form ofSUMO-1 lacks the C-terminal Gly-Gly motif and thereforecannot be conjugated to substrates (Johnson et al., 1997).Western blot analysis using antibodies against GFP showsthat GFP-SUMO-1�6 is detected as a single band of theexpected size (Figure 1B, arrow), whereas full-length GFP-SUMO-1 is detected as a high-molecular-weight smear of
Figure 1. GFP-SUMO-1 localizes to the nu-cleolus. (A) HeLa cells expressing GFP fu-sions of both full-length (GFP-SUMO-1) andunconjugatable form of SUMO-1 (GFP-SUMO-1�6). Fluorescence and contrast phaseimages of the same cell are shown side byside. The dashed lines indicate the contour ofthe nucleolus; arrows indicate staining of nu-clear pore complexes; arrowheads point tonuclear bodies. (B) Western blot analysis ofcells expressing both GFP-SUMO-1 and GFP-SUMO-1�6. The blot was probed with anti-bodies against GFP and molecular-weightmarkers (kDa) are shown on the left. (C) Im-munolabeling experiments with anti-PML an-tibody (red staining) of HeLa cells expressingGFP-SUMO-1 confirmed that the nuclear bod-ies enriched in GFP-SUMO-1 correspond toPML bodies but the nucleolar dots do notcolocalize with PML. (D) Subcellular distribu-tion of endogenous SUMO-1. HeLa cells wereimmunolabeled with a rabbit antibodyagainst SUMO-1. The dashed lines indicatethe contour of the nucleolus. (E) HeLa cellsexpressing GFP-SUMO-1 (green staining)were immunolabeled (red staining) with an-tibodies directed against proteins that localizein the fibrillar centers (pol I, UBF), the densefibrillar component (fibrillarin), and the gran-ular component (B23/nucleophosmin). Bar,10 �m.
ADAR1 Is Modified by SUMO-1
Vol. 16, November 2005 5117
conjugated products, indicating that this GFP fusion proteinis effectively conjugated. As shown in Figure 1A, GFP-SUMO-1�6 distributes throughout the cytoplasm and nucle-oplasm, with no concentration at the nuclear periphery,nuclear bodies or the nucleolus. Thus, localization ofSUMO-1 to the nucleolus depends on its ability to bind tosubstrates. This strongly suggests that proteins modified bySUMO-1 are present in the nucleolus. Immunolabeling ex-periments of GFP-SUMO-1-expressing cells with an anti-PML antibody demonstrate that the nucleolar staining doesnot correspond to PML bodies (Figure 1C). Staining of thenucleolus is also detected by immunofluorescence micros-copy using a rabbit polyclonal antibody raised againstSUMO-1 (Figure 1D).
Next, we analyzed in more detail the subnucleolar distri-bution of SUMO-1. In mammalian cells, the nucleolus com-prises three major regions involved in ribosomal biogenesis:the fibrillar centers, the dense fibrillar component, and thegranular component (Carmo-Fonseca et al., 2000). Transcrip-tion of rRNA genes localized at the fibrillar centers producesrRNA precursors (pre-rRNAs) that move away from therDNA template and undergo a series of posttranscriptionalprocessing reactions. The initial processing steps occur whilethe pre-rRNAs reside in the dense fibrillar component,whereas late processing events take place in the granularcomponent. Reflecting the vectorial organization of ribo-
somal synthesis, the fibrillar centers contain proteins re-quired for transcription of the rRNA genes, notably RNApolymerase I (pol I) and the pol I transcription initiationfactor UBF (upstream binding factor); the dense fibrillarcomponent contains proteins involved in early steps of pre-rRNA processing such as fibrillarin, and the granular com-ponent is highly enriched in proteins involved in the assem-bly of preribosomes, an example of which is B23 also callednucleophosmin (Scheer et al., 1993). Double-labeling of HeLacells expressing GFP-SUMO-1 with antibodies specific forRNA polymerase I, UBF, fibrillarin, and nucleophosmin re-veals lack of colocalization (Figure 1E). Thus, the majorfraction of nucleolar proteins modified by SUMO-1 does notassociate with the well-described subnucleolar regions im-plicated in ribosome biogenesis.
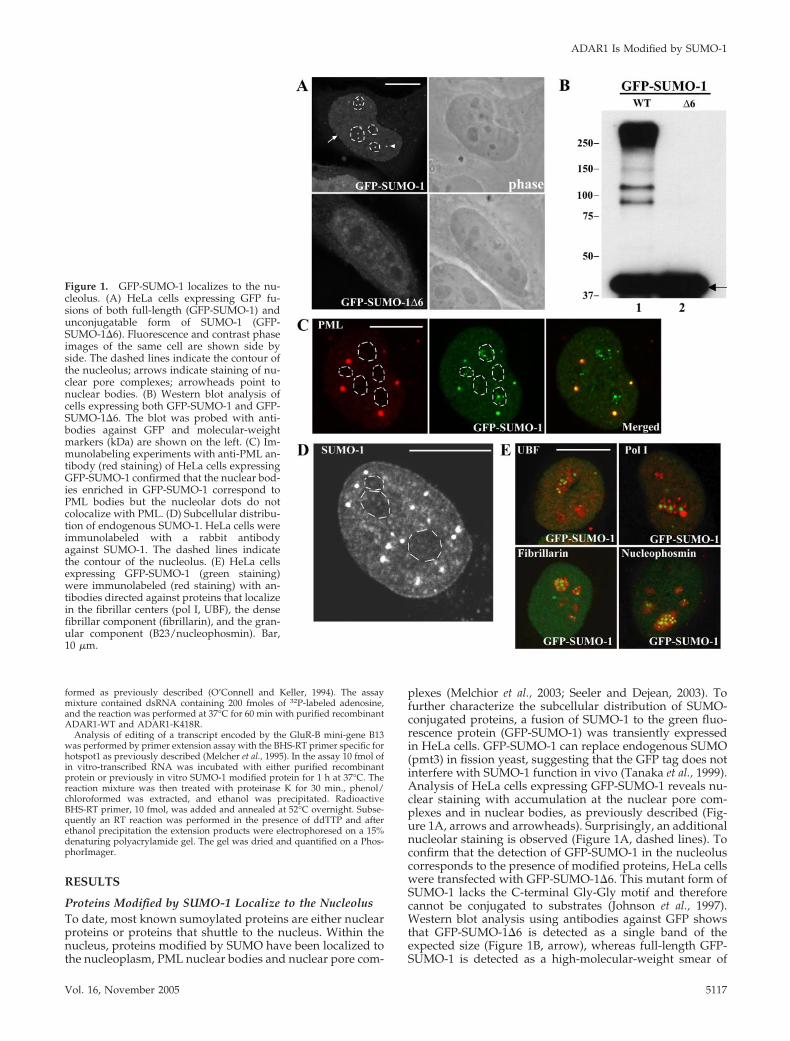
ADAR1 Is Modified by SUMO-1 In Vitro and In VivoWe have recently shown that the RNA-editing enzymesADAR1 and ADAR2 localize transiently to the nucleolus ina region that is distinct from the fibrillar centers, the densefibrillar component, and the granular component (Desterroet al., 2003). We therefore double-labeled HeLa cells express-ing GFP-SUMO-1 with an antibody specific for ADAR1 (Fig-ure 2A). This antibody, which recognizes both forms ofhuman ADAR1, labels the cytoplasm and the nucleoplasm,with additional staining of the nucleolus (Desterro et al.,
Figure 2. Nucleolar ADAR1 colocalizes with GFP-SUMO-1. (A) HeLa cell expressing GFP-SUMO-1 (green staining) and immunolabeledwith an antibody directed against ADAR1 (antibody 007, red staining). This antibody recognizes both the long (150 kDa) and the short (110kDa) forms of endogenous ADAR1. The long form of the protein is predominantly cytoplasmic, whereas the short form is nuclear. In somecell nuclei, ADAR1 is detected in discrete regions within the nucleolus, as previously described (Desterro et al., 2003). Superimposition of redand green images reveals perfect colocalization of SUMO and ADAR1 in the nucleolus. Bar, 10 �m. (B) Schematic representation of thehADAR1 protein (long form, amino acids 1–1226). The most important structural features of the enzyme are indicated: the Z-DNA-bindingdomains (gray); the double-stranded RNA-binding domains (dsRBDs, black); and the deaminase domain (dark gray). Asterisks depict theposition of potential SUMO-1 conjugation sites.
J. M. Pinto Desterro et al.
Molecular Biology of the Cell5118
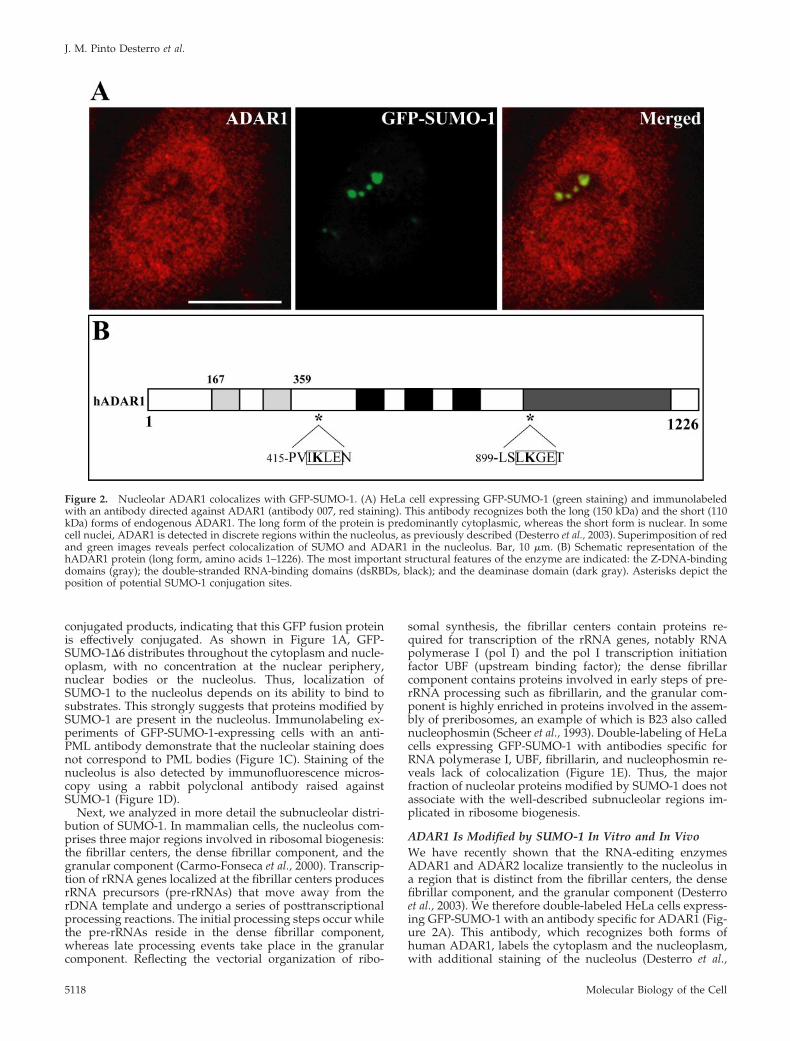
2003). The results show a perfect colocalization at the nucle-olus, raising the possibility that ADAR1 is a target forSUMO-1 conjugation. Most of the proteins modified bySUMO-1 contain the consensus motif �KXE, where � is ahydrophobic large amino acid, K the modified lysine, X anyamino acid, and E a glutamic acid (Rodriguez et al., 2001).Sequence analysis of the long form of ADAR1 (amino acids1–1226) shows two lysines that conform to this consensussequence (Figure 2B). To determine whether any of theselysine residues is a substrate for SUMO-1 modification, 35S-labeled ADAR1 was generated in vitro by a coupled tran-scription/translation reaction and incubated in an ATP-re-generating system with purified recombinant componentsrequired for SUMO modification, SUMO, SUMO-1-activat-ing enzyme (SAE), and UBC9. As a previously describedsubstrate, 35S-labeled Sp100, was used as positive control inthe reaction (Sternsdorf et al., 1997). Analysis of the reactionproducts by SDS-PAGE indicates that a proportion ofADAR1 is converted to a more slowly migrating form that isdependent on the presence of SUMO reaction components(Figure 3A, lanes 2–4). Substitution of SUMO-1 for GST-SUMO-1 alters the mobility of the more slowly migrating
form, which confirms that the mobility shift is due to SUMOmodification (Figure 3A, lane 3). Furthermore, ADAR1 isalso modified by SUMO-2, a SUMO-1-related protein (Fig-ure 3A, lane 4). Analysis of deletion variants of ADAR1reveals that a truncated version of the protein-encompassingamino acid residues 1–442 is modified by SUMO-1 in vitro(Figure 3B, lanes 1 and 2). In contrast, no modification isdetected in a truncated variant that consists of amino acidresidues 442-1226 (Figure 3B, lanes 3 and 4). Thus, it is likelythat ADAR-1 is modified by SUMO-1 on lysine 418. Toconfirm this hypothesis, lysine 418 was mutated to an argi-nine (K418R) and the protein was assayed for SUMO-1modification. ADAR1 containing this single point mutationis no longer modified in vitro by SUMO-1 (Figure 4A, lanes3 and 4). Next we asked whether ADAR1 is modified bySUMO-1 in vivo. To address this question, COS7 cells weretransfected with either ADAR1-WT or ADAR1-K418Rtagged with a C-terminal histidine hexamer, with or withoutSUMO-1 tagged with HA. Cells were lysed directly withguanidine hydrochloride and HIS-tagged proteins were pu-rified by chromatography over Ni2�-NTA agarose. Analysisof eluted proteins by Western blot with an anti-HA antibody
Figure 3. SUMO modification of ADAR1 invitro. (A) In vitro-expressed and 35S-labeledADAR1 was incubated with ATP, recombi-nant SUMO, Ubc9, and SUMO-1-activatingenzyme (SAE). Where indicated, eitherSUMO, SAE, or Ubc9 were omitted (�) fromthe reaction. Reactions were carried out witheither SUMO-1 (1), SUMO-2 (2), or GST-SUMO-1 (1*) as indicated, and Sp100 wasused as positive control. Reaction productswere analyzed by SDS-PAGE and phosphor-imaging. (B) In vitro-expressed and 35S-la-beled C- (1–442) and N-terminal (442–1226)ADAR1 deletions were incubated with ATPand SUMO-1 reaction components (assaymix), and the reaction products were ana-lyzed as in A. The bands corresponding to theSUMO-conjugated forms of ADAR1 are indi-cated (arrowheads). Molecular-weight mark-ers (kDa) are shown on the left.
ADAR1 Is Modified by SUMO-1
Vol. 16, November 2005 5119
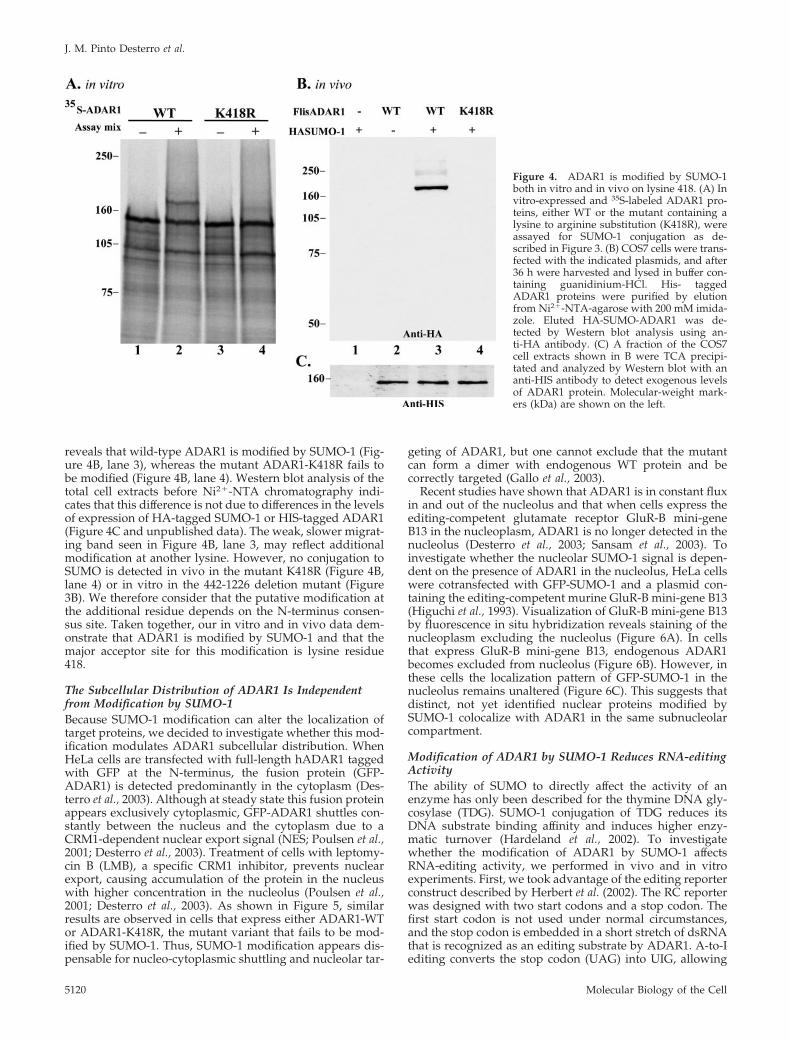
reveals that wild-type ADAR1 is modified by SUMO-1 (Fig-ure 4B, lane 3), whereas the mutant ADAR1-K418R fails tobe modified (Figure 4B, lane 4). Western blot analysis of thetotal cell extracts before Ni2�-NTA chromatography indi-cates that this difference is not due to differences in the levelsof expression of HA-tagged SUMO-1 or HIS-tagged ADAR1(Figure 4C and unpublished data). The weak, slower migrat-ing band seen in Figure 4B, lane 3, may reflect additionalmodification at another lysine. However, no conjugation toSUMO is detected in vivo in the mutant K418R (Figure 4B,lane 4) or in vitro in the 442-1226 deletion mutant (Figure3B). We therefore consider that the putative modification atthe additional residue depends on the N-terminus consen-sus site. Taken together, our in vitro and in vivo data dem-onstrate that ADAR1 is modified by SUMO-1 and that themajor acceptor site for this modification is lysine residue418.
The Subcellular Distribution of ADAR1 Is Independentfrom Modification by SUMO-1Because SUMO-1 modification can alter the localization oftarget proteins, we decided to investigate whether this mod-ification modulates ADAR1 subcellular distribution. WhenHeLa cells are transfected with full-length hADAR1 taggedwith GFP at the N-terminus, the fusion protein (GFP-ADAR1) is detected predominantly in the cytoplasm (Des-terro et al., 2003). Although at steady state this fusion proteinappears exclusively cytoplasmic, GFP-ADAR1 shuttles con-stantly between the nucleus and the cytoplasm due to aCRM1-dependent nuclear export signal (NES; Poulsen et al.,2001; Desterro et al., 2003). Treatment of cells with leptomy-cin B (LMB), a specific CRM1 inhibitor, prevents nuclearexport, causing accumulation of the protein in the nucleuswith higher concentration in the nucleolus (Poulsen et al.,2001; Desterro et al., 2003). As shown in Figure 5, similarresults are observed in cells that express either ADAR1-WTor ADAR1-K418R, the mutant variant that fails to be mod-ified by SUMO-1. Thus, SUMO-1 modification appears dis-pensable for nucleo-cytoplasmic shuttling and nucleolar tar-
geting of ADAR1, but one cannot exclude that the mutantcan form a dimer with endogenous WT protein and becorrectly targeted (Gallo et al., 2003).
Recent studies have shown that ADAR1 is in constant fluxin and out of the nucleolus and that when cells express theediting-competent glutamate receptor GluR-B mini-geneB13 in the nucleoplasm, ADAR1 is no longer detected in thenucleolus (Desterro et al., 2003; Sansam et al., 2003). Toinvestigate whether the nucleolar SUMO-1 signal is depen-dent on the presence of ADAR1 in the nucleolus, HeLa cellswere cotransfected with GFP-SUMO-1 and a plasmid con-taining the editing-competent murine GluR-B mini-gene B13(Higuchi et al., 1993). Visualization of GluR-B mini-gene B13by fluorescence in situ hybridization reveals staining of thenucleoplasm excluding the nucleolus (Figure 6A). In cellsthat express GluR-B mini-gene B13, endogenous ADAR1becomes excluded from nucleolus (Figure 6B). However, inthese cells the localization pattern of GFP-SUMO-1 in thenucleolus remains unaltered (Figure 6C). This suggests thatdistinct, not yet identified nuclear proteins modified bySUMO-1 colocalize with ADAR1 in the same subnucleolarcompartment.
Modification of ADAR1 by SUMO-1 Reduces RNA-editingActivityThe ability of SUMO to directly affect the activity of anenzyme has only been described for the thymine DNA gly-cosylase (TDG). SUMO-1 conjugation of TDG reduces itsDNA substrate binding affinity and induces higher enzy-matic turnover (Hardeland et al., 2002). To investigatewhether the modification of ADAR1 by SUMO-1 affectsRNA-editing activity, we performed in vivo and in vitroexperiments. First, we took advantage of the editing reporterconstruct described by Herbert et al. (2002). The RC reporterwas designed with two start codons and a stop codon. Thefirst start codon is not used under normal circumstances,and the stop codon is embedded in a short stretch of dsRNAthat is recognized as an editing substrate by ADAR1. A-to-Iediting converts the stop codon (UAG) into UIG, allowing
Figure 4. ADAR1 is modified by SUMO-1both in vitro and in vivo on lysine 418. (A) Invitro-expressed and 35S-labeled ADAR1 pro-teins, either WT or the mutant containing alysine to arginine substitution (K418R), wereassayed for SUMO-1 conjugation as de-scribed in Figure 3. (B) COS7 cells were trans-fected with the indicated plasmids, and after36 h were harvested and lysed in buffer con-taining guanidinium-HCl. His- taggedADAR1 proteins were purified by elutionfrom Ni2�-NTA-agarose with 200 mM imida-zole. Eluted HA-SUMO-ADAR1 was de-tected by Western blot analysis using an-ti-HA antibody. (C) A fraction of the COS7cell extracts shown in B were TCA precipi-tated and analyzed by Western blot with ananti-HIS antibody to detect exogenous levelsof ADAR1 protein. Molecular-weight mark-ers (kDa) are shown on the left.
J. M. Pinto Desterro et al.
Molecular Biology of the Cell5120
translation from the first start codon. This results in produc-tion of a fusion protein that contains both HA and GFP tags.If the reporter mRNA is not edited, the stop codon is noteliminated and translation of the messenger starts in thesecond start codon giving rise to a GFP fusion protein thatlacks the N-terminal HA tag (Figure 7A). Therefore, theexpression of an HA-tagged fusion protein is editing-depen-dent. The RC construct was transfected into HeLa cells andthe occurrence of editing was assayed by Western blot anal-ysis with an anti-HA antibody. Cells were cotransfectedwith the RC reporter and either ADAR1-WT or ADAR1-K418R. Both forms of ADAR1 were C-terminally taggedwith a HIS-epitope. No endogenous A-to-I editing of the RCreporter was detected in HeLa cells (Figure 7B, lane 2). Incontrast, editing of the reporter was observed when eitherADAR1-WT or ADAR1-K418R was exogenously expressed.Most important, in cells that express similar levels ofADAR1-WT and ADAR1-K418R (Figure 7, lanes 3 and 4,anti-HIS, arrowhead), the mutant enzyme produces moreHA-tagged fusion protein than the wild-type enzyme (Fig-ure 7, B, anti-HA, and C); thus preventing the attachment ofSUMO to ADAR1 activates RNA editing, suggesting thatsumoylation has an inhibitory effect on ADAR1-editing ac-
tivity. According to this idea, we predicted that increasingthe fraction of wild-type ADAR1 modified by SUMO-1would further reduce editing of the reporter and conse-quently decrease the amount of HA-tagged fusion proteinexpressed in the cell. However, cotransfection of cells with aplasmid encoding SUMO-1 did not alter the expression levelof the HA-fusion protein (unpublished data), most probablybecause overexpression of SUMO-1 is not sufficient to en-hance sumoylation of ADAR1 in vivo. In fact, there is evi-dence that modification of proteins by SUMO is a tightlyregulated process in the cell (Melchior et al., 2003; Marx,2005).
The labile nature of SUMO-1 modification due to the highactivity of SUMO specific proteases and the absence of amechanism to induce SUMO-1 modification in vivo do notfacilitate the investigation of a direct effect of SUMO-1 onADAR1 activity in the cell. We therefore decided to performfurther studies using in vitro systems. Both ADAR1-WT andADAR1-K418R were expressed in the yeast P. pastoris, andthe recombinant proteins containing HIS and FLAG tagswere purified to homogeneity by chromatography over bothNi2�-NTA and FLAG affinity matrices (Ring et al., 2004).Consistent with the in vivo data, the in vitro results indicate
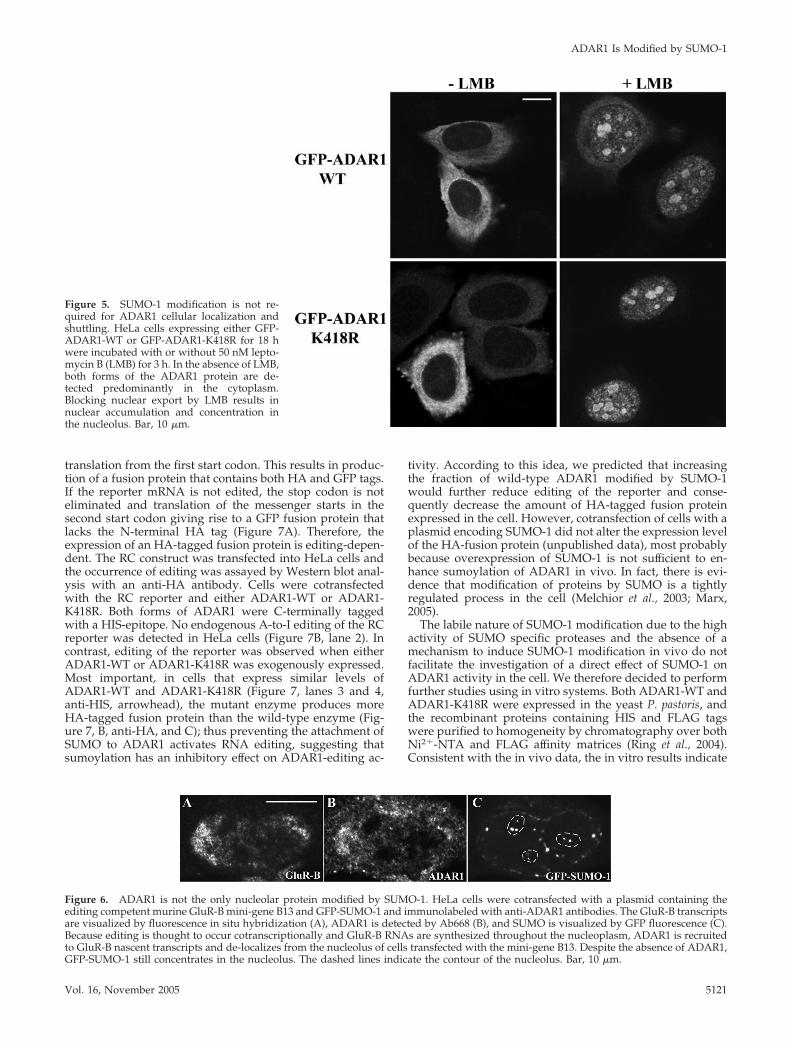
Figure 5. SUMO-1 modification is not re-quired for ADAR1 cellular localization andshuttling. HeLa cells expressing either GFP-ADAR1-WT or GFP-ADAR1-K418R for 18 hwere incubated with or without 50 nM lepto-mycin B (LMB) for 3 h. In the absence of LMB,both forms of the ADAR1 protein are de-tected predominantly in the cytoplasm.Blocking nuclear export by LMB results innuclear accumulation and concentration inthe nucleolus. Bar, 10 �m.
Figure 6. ADAR1 is not the only nucleolar protein modified by SUMO-1. HeLa cells were cotransfected with a plasmid containing theediting competent murine GluR-B mini-gene B13 and GFP-SUMO-1 and immunolabeled with anti-ADAR1 antibodies. The GluR-B transcriptsare visualized by fluorescence in situ hybridization (A), ADAR1 is detected by Ab668 (B), and SUMO is visualized by GFP fluorescence (C).Because editing is thought to occur cotranscriptionally and GluR-B RNAs are synthesized throughout the nucleoplasm, ADAR1 is recruitedto GluR-B nascent transcripts and de-localizes from the nucleolus of cells transfected with the mini-gene B13. Despite the absence of ADAR1,GFP-SUMO-1 still concentrates in the nucleolus. The dashed lines indicate the contour of the nucleolus. Bar, 10 �m.
ADAR1 Is Modified by SUMO-1
Vol. 16, November 2005 5121
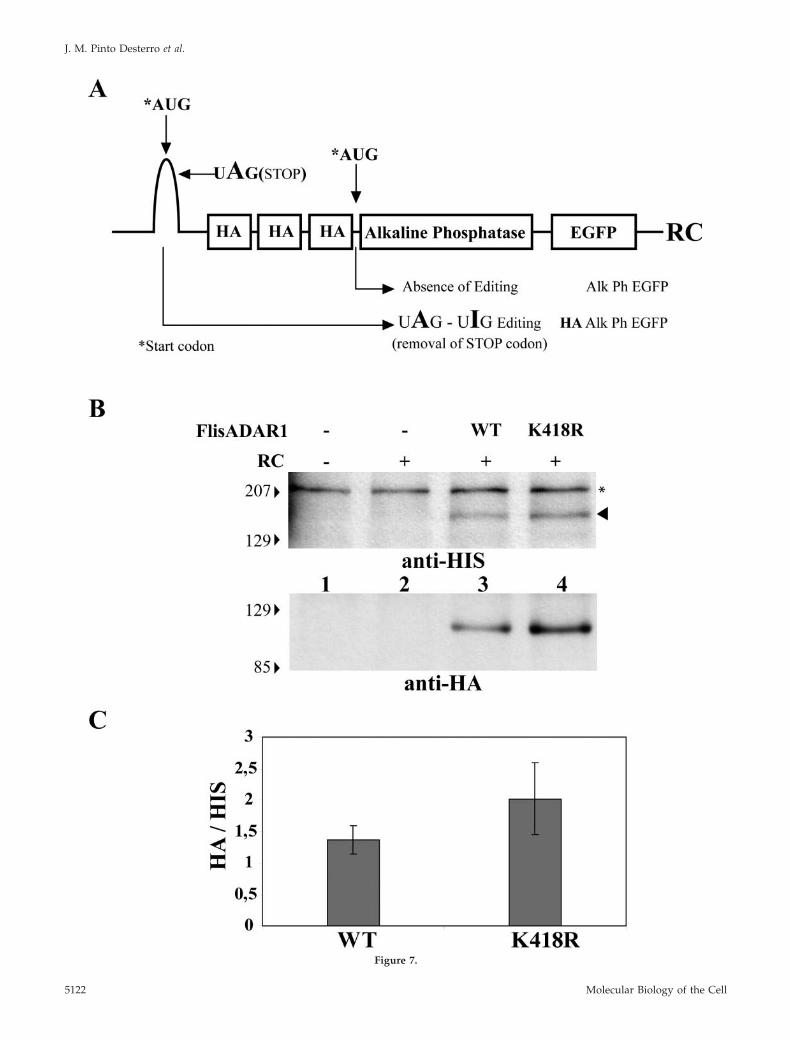
Figure 7.
J. M. Pinto Desterro et al.
Molecular Biology of the Cell5122

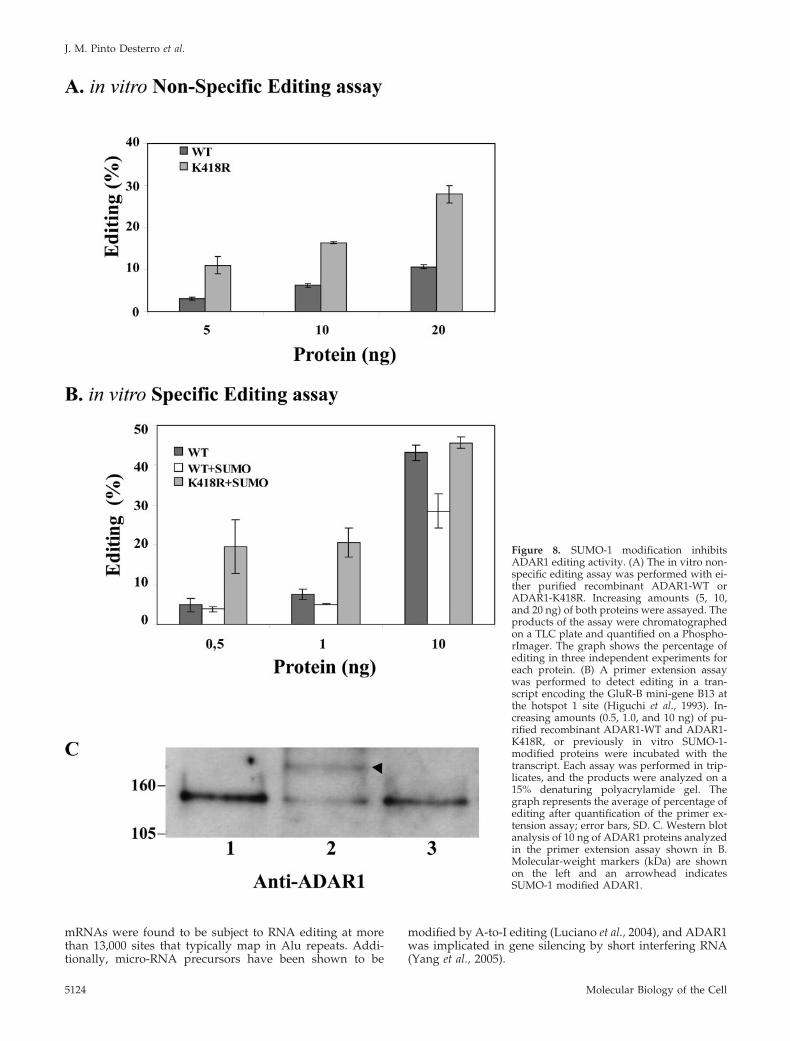
that recombinant ADAR1-WT is consistently less active thanrecombinant ADAR1-K418R in editing a long duplex RNAin a nonspecific assay (Figure 8A) (O’Connell and Keller,1994). Western blot analysis of the recombinant proteinsconfirmed that a fraction of ADAR1-WT is modified bySUMO-1 (unpublished data). This observation prompted usto compare the editing activity of ADAR1-WT and ADAR1-K418R recombinant proteins on a specific substrate, theGluR-B mini-gene B13 (Figure 8B). A primer extension assaywas performed in triplicate on a transcript encoded by theGluR-B mini-gene B13 (Higuchi et al., 1993). As clearlyshown in Figure 8B, following in vitro modification of therecombinant proteins by SUMO-1, the editing activity ofwild-type ADAR1, and not of the K418R mutant, is signifi-cantly reduced. A range of different amounts of recombinantproteins was tested in this assay and reproducible resultswere obtained (Figure 8B). Western blot analysis confirmsthe modification of ADAR1-WT (Figure 8C, lane 2), but notof the mutant ADAR1-K418R (Figure 8C, lane 3). Consistentwith the view that sumoylation is not required for editing,addition of ATP is dispensable for recombinant ADAR1activity in vitro (O’Connell and Keller, 1994), whereas ifSUMO-1 modification was necessary ATP addition wouldbe essential. Taken together these results support a directrole of SUMO-1 modification on reducing the RNA-editingactivity of ADAR1.
DISCUSSION
Posttranslational modification of proteins by SUMO isknown to play a regulatory role in many cellular processes,and the identification of novel SUMO-targeted proteins iscurrently attracting much attention (for recent reviews seeJohnson, 2004; Melchior et al., 2003). In general, only a lim-ited fraction of a certain protein is modified by SUMO in thecell, making it difficult to detect the low abundant pool ofendogenously sumoylated proteins. In this study we showthat GFP-tagged SUMO-1 accumulates in the nucleolus. Incontrast, a GFP-tagged mutant version of SUMO-1 that lacksthe C-terminal amino acids required for covalent attachmentto target proteins fails to localize to the nucleolus (Figure 1,A and B). Similar results were very recently reported byAyaydin and Dasso (2004) who observed that YFP-SUMO-1,but not YFP-SUMO-2 or YFP-SUMO-3, localizes to the nu-cleolus. This strongly suggests that a subset of nucleolarproteins is modified by SUMO. Interestingly, some of theenzymes involved in the sumoylation pathway have beenpreviously localized to the nucleolus, namely the E3SUMO-1 ligase PIAS1 (Valdez et al., 1997), and the SUMO/Smt3-1-specific isopeptidase SMT3IP1 (SENP3; Nishida etal., 2000; Leung et al., 2003). Thus, it is possible that certain
protein targets are reversibly modified by SUMO-1 in thenucleolus.
Noteworthy, SUMO was not detected in recent MS studieson isolated nucleoli from HeLa cells (Scheer et al., 1993;Andersen et al., 2002). However, this is not surprising, takinginto account that many endogenously sumoylated proteinsare present at a level below normal detection limit. More-over, sumoylation is a highly dynamic and reversible reac-tion, making it difficult to preserve SUMO conjugation dur-ing cell fractionation and subcellular purificationprocedures.
The nucleolus is a subnuclear compartment dedicated tothe biogenesis of ribosomes. The nucleolus is where therRNA genes are kept and transcribed and the rRNAs areprocessed and assembled with proteins to form preribo-somes. However, an increasing body of evidence indicatesthat the nucleolus is not exclusively a ribosome factory, butplays additional roles in the cell. According to a currentview, the nucleolus may act as a molecular “safe” or “sink”that regulates protein activity by sequestration (reviewLeung et al., 2003).
DNA topoisomerase I (topo I) is a nuclear protein thatconcentrates in the nucleolus and is modified by SUMO.However, topo I rapidly moves out of the nucleolus, andthis nucleolar delocalization is associated with conjuga-tion of the protein with SUMO (Mo et al., 2002). Thus, todate, no nucleolar proteins modified by SUMO were iden-tified. In the present work we show that GFP-taggedSUMO-1 accumulates in a nucleolar region that is distinctfrom the well-characterized nucleolar domains involvedin ribosomal biogenesis, i.e., the fibrillar center, the densefibrillar component, and the granular component (Figure1E). Rather, GFP-SUMO-1 colocalizes precisely with theRNA-editing enzyme ADAR1 (Figure 2). We further pro-vide in vitro and in vivo evidence that human ADAR1 ismodified by SUMO-1 on lysine residue 418 (Figures 3 and4). Importantly, the nucleolar localization of GFP-SUMO-1 remains unaltered when ADAR1 is no longerdetected in that compartment (Figure 6), arguing thatSUMO-1 modifies additional protein substrates in the nu-cleolus.
Although ADAR1 colocalizes with ADAR2 in the nucleo-lus (Desterro et al., 2003), the ADAR2 protein lacks theamino-terminal region containing the SUMO conjugationsite. Sequence analysis of ADAR2 does not reveal anySUMO-1 consensus motif and ADAR2 is not modified bySUMO-1 in vitro (unpublished data). Because both ADAR1and ADAR2 concentrate in the nucleolus and only ADAR1 ismodified by SUMO, it is unlikely that sumoylation ofADAR1 is required for targeting the enzyme to the nucleo-lus. According to this prediction, a mutant form of ADAR1that is not sumoylated because it contains an arginine sub-stitution of lysine 418 (ADAR1-K418R) localizes to the nu-cleolus similarly to the wild-type protein (Figure 5).
Our work further provides in vivo and in vitro evidencethat modification of ADAR1 by SUMO reduces the RNA-editing activity of the enzyme. ADAR1 can edit RNAs bothin a specific and nonspecific manner, depending on thenature of the substrate. ADAR1 can edit specific transcriptsencoding receptor proteins of CNS and these can result inrecoding events. The best studied specific mammalian sub-strates for ADAR1 are the pre-mRNAs encoding the seroto-nin HT-2c receptor and those encoding ionotropic glutamatereceptor (GluR) subunits. However, more recent studieshave identified widespread A-to-I RNA-editing sites in thehuman transcriptome (Athanasiadis et al., 2004; Kim et al.,2004; Levanon et al., 2004). Approximately 1500 human
Figure 7 (facing page). ADAR K418R has higher ability to edit theRC reporter construct than the WT protein. (A) Schematic represen-tation of the RC-editing reporter construct. (B) HeLa cells weretransfected with the indicated plasmids, and total cell extracts wereprepared 24 h after transfection. Proteins were analyzed by Westernblot with the indicated antibody. The anti-HIS antibody reveals thatthe expression levels of both ADAR1 forms are similar (arrowhead).A nonspecific band that appears with anti-HIS antibody is indicatedwith one asterisk (*). The HA signal indicates that ADAR1 K418Rhas higher editing activity. Molecular-weight markers (kDa) areshown on the left. (C) The relative intensity of the HA bandsnormalized for the intensity of ADAR1 WT and K418R bands (de-tected by anti-HIS antibody) was quantified in three independentexperiments. Error bars, SD.
ADAR1 Is Modified by SUMO-1
Vol. 16, November 2005 5123
mRNAs were found to be subject to RNA editing at morethan 13,000 sites that typically map in Alu repeats. Addi-tionally, micro-RNA precursors have been shown to be
modified by A-to-I editing (Luciano et al., 2004), and ADAR1was implicated in gene silencing by short interfering RNA(Yang et al., 2005).
Figure 8. SUMO-1 modification inhibitsADAR1 editing activity. (A) The in vitro non-specific editing assay was performed with ei-ther purified recombinant ADAR1-WT orADAR1-K418R. Increasing amounts (5, 10,and 20 ng) of both proteins were assayed. Theproducts of the assay were chromatographedon a TLC plate and quantified on a Phospho-rImager. The graph shows the percentage ofediting in three independent experiments foreach protein. (B) A primer extension assaywas performed to detect editing in a tran-script encoding the GluR-B mini-gene B13 atthe hotspot 1 site (Higuchi et al., 1993). In-creasing amounts (0.5, 1.0, and 10 ng) of pu-rified recombinant ADAR1-WT and ADAR1-K418R, or previously in vitro SUMO-1-modified proteins were incubated with thetranscript. Each assay was performed in trip-licates, and the products were analyzed on a15% denaturing polyacrylamide gel. Thegraph represents the average of percentage ofediting after quantification of the primer ex-tension assay; error bars, SD. C. Western blotanalysis of 10 ng of ADAR1 proteins analyzedin the primer extension assay shown in B.Molecular-weight markers (kDa) are shownon the left and an arrowhead indicatesSUMO-1 modified ADAR1.
J. M. Pinto Desterro et al.
Molecular Biology of the Cell5124

Here we show that the mutant form of ADAR1 that is notmodified by SUMO (ADAR1-K418R) is more active than thewild-type enzyme in editing a reporter RNA in vivo, andmodification of the wild-type enzyme by SUMO reducesediting of a GluR-B mini-gene B13 in vitro (Figures 7 and 8).This represents the first indication that sumoylation cancontribute to regulate RNA-editing activity.
ADAR activity is known to be tightly regulated in differ-ent species. In vertebrates, ADARs shuttle between the cy-toplasm, the nucleoplasm, and the nucleolus (Desterro et al.,2003; Sansam et al., 2003), and it is currently thought thatsequestration in the nucleolus contributes to prevent aber-rant editing activity in the nucleoplasm. On the basis of ourobservations that ADAR1 colocalizes with SUMO-1 in thenucleolus and that sumoylation of ADAR1 reduces editingactivity, we propose that the nucleolus represents a “sink”for inactive ADAR1 in the cell. In agreement with this view,it has been recently reported that ADAR2- but not ADAR1-mediated RNA editing occurs in the nucleolus (Vitali et al.,2005). Considering that both ADAR1 and ADAR2 colocalizein the nucleolus, it was unexpected to find that ADAR1 doesnot perform nucleolar RNA editing. This apparent inconsis-tency can be explained by our findings suggesting thatSUMO-1 conjugation renders ADAR1 inactive in the nucle-olus while ADAR2 is not modified by SUMO.
Whether ADAR1 is preferentially sumoylated in the nu-cleolus remains to be established. Another important issueto be addressed concerns the mechanism by which sumoy-lation affects editing activity. Interestingly, ADAR enzymesact as a dimer and dimerization is essential for editingactivity. It remains to be elucidated how the monomers bindto dsRNA and dimerize. Preliminary results for ADAR1have shown that the N-terminal region containing the Z-DNA domain is not required as heterodimers can formbetween the p150 and p110 isoforms of ADAR1 (Cho et al.,2003). However, the minimum region required for thedimerization of Drosophila ADAR is the N-terminus includ-ing and the first dsRNA-binding domain (dsRBD; Gallo etal., 2003). Dimerization affects the enzymatic activity as wellas substrate specificity of ADAR1 and ADAR2 and is essen-tial for editing activity in Drosophila (Gallo et al., 2003).Considering that the SUMO-1 acceptor lysine lies betweenthe Z-DNA and the first dsRBD one could consider SUMOas a stereochemical obstacle for both binding to the dsRNAand subsequent dimerization.
In conclusion, together with the recent finding that SUMOmodifies several heterogeneous nuclear ribonucleoproteins,which are key players in mRNA biogenesis (Li et al., 2004),our results support a novel role for sumoylation in regulat-ing RNA metabolism.
ACKNOWLEDGMENTS
We thank Walter Keller (Biozentrum, Basel, Switzerland) for kindly providinganti-ADAR1 antibodies, Alan Herbert (Boston University School of Medicine)for the RC plasmid and G. Del Sal for the GFP-SUMO plasmid constructs(Laboratorio Nazionale Consorzio Interuniversitario Biotecnologie, Trieste,Italy). This study was supported by grants from “Fundacao para a Ciencia eTecnologia, POCTI/36547/MGI/00” (Portugal), the European Commission“QLG2-CT-2001-01554” and the MRC (United Kingdom).
REFERENCES
Andersen, J. S., Lyon, C. E., Fox, A. H., Leung, A. K., Lam, Y. W., Steen, H.,Mann, M., and Lamond, A. I. (2002). Directed proteomic analysis of thehuman nucleolus. Curr. Biol. 12, 1–11.
Athanasiadis, A., Rich, A., and Maas, S. (2004). Widespread A-to-I RNAediting of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2,e391.
Ayaydin, F., and Dasso, M. (2004). Distinct in vivo dynamics of vertebrateSUMO paralogues. Mol. Biol. Cell 15, 5208–5218.
Azuma, Y., and Dasso, M. (2002). A new clue at the nuclear pore: RanBP2 isan E3 enzyme for SUMO1. Dev. Cell 2, 130–131.
Bass, B. L. (2002). RNA editing by adenosine deaminases that act on RNA.Annu. Rev. Biochem. 71, 817–846.
Bayer, P., Arndt, A., Metzger, S., Mahajan, R., Melchior, F., Jaenicke, R., andBecker, J. (1998). Structure determination of the small ubiquitin-related mod-ifier SUMO-1. J. Mol. Biol. 280, 275–286.
Beghini, A., Ripamonti, C. B., Peterlongo, P., Roversi, G., Cairoli, R., Morra, E.,and Larizza, L. (2000). RNA hyperediting and alternative splicing of hema-topoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum.Mol. Genet. 9, 2297–2304.
Bohren, K. M., Nadkarni, V., Song, J. H., Gabbay, K. H., and Owerbach, D.(2004). A M55V polymorphism in a novel SUMO gene (SUMO-4) differen-tially activates heat shock transcription factors and is associated with suscep-tibility to type I diabetes mellitus. J. Biol. Chem. 279, 27233–27238.
Calado, A., Kutay, U., Kuhn, U., Wahle, E., and Carmo-Fonseca, M. (2000).Deciphering the cellular pathway for transport of poly(A)-binding protein II.RNA 6, 245–256.
Carmo-Fonseca, M., Mendes-Soares, L., and Campos, I. (2000). To be or not tobe in the nucleolus. Nat. Cell Biol. 2, E107–E112.
Cho, D. S., Yang, W., Lee, J. T., Shiekhattar, R., Murray, J. M., and Nishikura,K. (2003). Requirement of dimerization for RNA editing activity of adenosinedeaminases acting on RNA. J. Biol. Chem. 278, 17093–17102.
Desterro, J. M., Keegan, L. P., Lafarga, M., Berciano, M. T., O’Connell, M., andCarmo-Fonseca, M. (2003). Dynamic association of RNA-editing enzymeswith the nucleolus. J. Cell Sci. 116, 1805–1818.
Desterro, J. M., Thomson, J., and Hay, R. T. (1997). Ubch9 conjugates SUMObut not ubiquitin. FEBS Lett. 417, 297–300.
Duda, D. M., and Schulman, B. A. (2005). Tag-team SUMO wrestling. Mol.Cell 18, 612–614.
Gallo, A., Keegan, L. P., Ring, G. M., and O’Connell, M. A. (2003). An ADARthat edits transcripts encoding ion channel subunits functions as a dimer.EMBO J. 22, 3421–3430.
George, C. X., and Samuel, C. E. (1999a). Characterization of the 5�-flankingregion of the human RNA-specific adenosine deaminase ADAR1 gene andidentification of an interferon-inducible ADAR1 promoter. Gene 229, 203–213.
George, C. X., and Samuel, C. E. (1999b). Human RNA-specific adenosinedeaminase ADAR1 transcripts posses alternative exon1 structures that initiatefrom different promoters, one constitutively active and the other interferoninducible. Proc. Natl. Acad. Sci. USA 96, 4621–4626.
Gostissa, M., Hengstermann, A., Fogal, V., Sandy, P., Schwarz, S. E., Scheffner,M., and Del Sal, G. (1999). Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J 18, 6462–6471.
Hardeland, U., Steinacher, R., Jiricny, J., and Schar, P. (2002). Modification ofthe human thymine-DNA glycosylase by ubiquitin-like proteins facilitatesenzymatic turnover. EMBO J 21, 1456–1464.
Hay, R. T. (2001). Protein modification by SUMO. Trends Biochem. Sci. 26,332–333.
Hay, R. T. (2005). SUMO: a history of modification. Mol. Cell 18, 1–12.
Herbert, A., Wagner, S., and Nickerson, J. A. (2002). Induction of proteintranslation by ADAR1 within living cell nuclei is not dependent on RNAediting. Mol. Cell 5, 1235–1246.
Higuchi, M., Maas, S., Single, F. N., Hartner, J., Rozov, A., Burnashev, N.,Feldmeyer, D., Sprengel, R., and Seeburg, P. H. (2000). Point mutation in anAMPA receptor gene rescues lethality in mice deficient in the RNA-editingenzyme ADAR2. Nature 406, 78–81.
Higuchi, M., Single, F. N., Kohler, M., Sommer, B., Sprengel, R., and Seeburg,P. H. (1993). RNA editing of AMPA receptor subunit GluR-B: a base-pairedintron-exon structure determines position and efficiency. Cell 75, 1361–1370.
Johnson, E. S. (2004). Protein modification by SUMO. Annu. Rev. Biochem. 73,355–382.
Johnson, E. S., Schwienhorst, I., Dohmen, R. J., and Blobel, G. (1997). Theubiquitin-like protein Smt3p is activated for conjugation to other proteins byan Aos1p/Uba2p heterodimer. EMBO J. 16, 5509–5519.
Kagey, M. H., Melhuish, T. A., and Wotton, D. (2003). The polycomb proteinPc2 is a SUMO E3. Cell 113, 127–137.
Keegan, L. P., Gallo, A., and O’Connell, M. (2001). The many roles of an RNAeditor. Nat. Rev. Genet. 11, 869–878.
ADAR1 Is Modified by SUMO-1
Vol. 16, November 2005 5125

Keegan, L. P., Leroy, A., Sproul, D., and O’Connell, M. A. (2004). Adenosinedeaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 5,209.
Kim, D. D., Kim, T. T., Walsh, T., Kobayashi, Y., Matise, T. C., Buyske, S., andGabriel, A. (2004). Widespread RNA editing of embedded alu elements in thehuman transcriptome. Genome Res. 14, 1719–1725.
Leung, A. K., Andersen, J. S., Mann, M., and Lamond, A. I. (2003). Bioinfor-matic analysis of the nucleolus. Biochem. J. 376, 553–569.
Levanon, E. Y. et al. (2004). Systematic identification of abundant A-to-Iediting sites in the human transcriptome. Nat. Biotechnol. 22, 1001–1005.
Li, S. J., and Hochstrasser, M. (1999). A new protease required for cell-cycleprogression in yeast. Nature 398, 246–251.
Li, S. J., and Hochstrasser, M. (2000). The yeast ULP2 (SMT4) gene encodes anovel protease specific for the ubiquitin-like Smt3 protein. Mol. Cell. Biol. 20,2367–2377.
Li, T., Evdokimov, E., Shen, R. F., Chao, C. C., Tekle, E., Wang, T., Stadtman,E. R., Yang, D. C., and Chock, P. B. (2004). Sumoylation of heterogeneousnuclear ribonucleoproteins, zinc finger proteins, and nuclear pore complexproteins: a proteomic analysis. Proc. Natl. Acad. Sci. USA 101, 8551–8556.
Luciano, D. J., Mirsky, H., Vendetti, N. J., and Maas, S. (2004). RNA editing ofa miRNA precursor. RNA 10, 1174–1177.
Marx, J. (2005). Cell biology. SUMO wrestles its way to prominence in the cell.Science 307, 836–839.
Melcher, T., Maas, S., Higuchi, M., Keller, W., and Seeburg, P. H. (1995).Editing of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid recep-tor GluR-B pre-mRNA in vitro reveals site-selective adenosine to inosineconversion. J. Biol. Chem. 270, 8566–8570.
Melchior, F., Schergaut, M., and Pichler, A. (2003). SUMO: ligases, isopepti-dases and nuclear pores. Trends Biochem. Sci. 28, 612–618.
Mo, Y.-Y., Yu, Y., Shen, Z., and Beck, W. T. (2002). Nucleolar delocalization ofhuman topoisomerase I in response to topotecan correlates with sumoylationof the protein. J. Biol. Chem. 277, 2958–2964.
Morse, D. P., and Bass, B. L. (1999). Long RNA hairpins that contain inosineare present in Caenorhabditis elegans poly(A)� RNA. Proc. Natl. Acad. Sci.USA 96, 6048–6053.
Mossessova, E., and Lima, C. D. (2000). Ulp1-SUMO crystal structure andgenetic analysis reveal conserved interactions and a regulatory element es-sential for cell growth in yeast. Mol. Cell 5, 865–876.
Nishida, T., Tanaka, H., and Yasuda, H. (2000). A novel mammalian Smt3-specific isopeptidase (SMT3IP1) localized in the nucleolus at interphase. Eur.J. Biochem. 267, 6423–6427.
O’Connell, M. A., and Keller, W. (1994). Purification and properties of double-stranded RNA-specific adenosine deaminase from calf thymus. Proc. Natl.Acad. Sci. USA 91, 10596–10600.
O’Mahony, D. J., Xie, W. Q., Smith, S. D., Singer, H. A., and Rothblum, L. I.(1992). Differential phosphorylation and localization of the transcription fac-tor UBF in vivo in response to serum deprivation. In vitro dephosphorylationof UBF reduces its transactivation properties. J. Biol. Chem. 267, 35–38.
Palladino, M. J., Keegan, L. P., O’Connell, M. A., and Reenan, R. A. (2000a).dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase ishighly developmentally regulated and is itself a target for RNA editing. RNA6, 1004–1018.
Palladino, M. J., Keegan, L. P., O’Connell, M. A., and Reenan, R. A. (2000b).A-to-I pre-mRNA editing in Drosophila is primarily involved in adult ner-vous system function and integrity. Cell 102, 437–449.
Patterson, J. B., and Samuel, C. E. (1995). Expression and regulation byinterferon of a double-stranded-RNA-specific adenosine deaminase from hu-man cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 15,5376–5388.
Pichler, A., Gast, A., Seeler, J. S., Dejean, A., and Melchior, F. (2002). Thenucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108, 109–120.
Polson, A. G., Bass, B. L., and Casey, J. L. (1996). RNA editing of hepatitisdelta virus antigenome by dsRNA-adenosine deaminase. Nature 380, 454–456.
Poulsen, H., Nilsson, J., Damgaard, C. K., Egebjerg, J., and Kjems, J. (2001).CRM1 mediates the export of ADAR1 through a nuclear export signal withinthe Z-DNA binding domain. Mol. Cell. Biol. 21, 7862–7871.
Reimer, G., Raska, I., Tan, E. M., and Scheer, U. (1987). Human autoantibod-ies: probes for nucleolus structure and function. Virchows Arch B Cell Pathol.Incl. Mol. Pathol. 54, 131–143.
Reverter, D., and Lima, C. D. (2005). Insights into E3 ligase activity revealedby a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature 435, 687–692.
Ring, G. M., O’Connell, M. A., and Keegan, L. P. (2004). Purification and assayof recombinant ADAR proteins expressed in the yeast Pichia pastoris or inEscherichia coli. Methods Mol. Biol. 265, 219–238.
Rodriguez, M. S., Dargemont, C., and Hay, R. T. (2001). SUMO-1 conjugationin vivo requires both a consensus modification motif and nuclear targeting.J. Biol. Chem. 276, 12654–12659.
Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., andHay, R. T. (1999). SUMO-1 modification activates the transcriptional responseof p53. EMBO J. 18, 6455–6461.
Rueter, S. M., Dawson, T. R., and Emeson, R. B. (1999). Regulation of alter-native splicing by RNA editing. Nature 399, 75–80.
Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., and Grosschedl, R.(2001). PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1activity by sequestration into nuclear bodies. Genes Dev. 15, 3088–3103.
Saitoh, H., and Hinchey, J. (2000). Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 275,6252–6258.
Sansam, C. L., Wells, K. S., and Emeson, R. B. (2003). Modulation of RNAediting by functional nucleolar sequestration of ADAR2. Proc. Natl. Acad. Sci.USA 100, 14018–14023.
Schaub, M., and Keller, W. (2002). RNA editing by adenosine deaminasesgenerates RNA and protein diversity. Biochemie 84, 791–803.
Scheer, U., Thiry, M., and Goessens, G. (1993). Structure, function and assem-bly of the nucleolus. Trends Cell Biol. 3, 236–241.
Seeler, J. S., and Dejean, A. (2003). Nuclear and unclear functions of SUMO.Nat. Rev. Mol. Cell. Biol. 4, 690–699.
Sternsdorf, T., Jensen, K., and Will, H. (1997). Evidence for covalent modifi-cation of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol. 139, 1621–1634.
Tanaka, K., Nishide, J., Okazaki, K., Kato, H., Niwa, O., Nakagawa, T.,Matsuda, H., Kawamukai, M., and Murakami, Y. (1999). Characterization of afission yeast SUMO-1 homologue, pmt3p, required for multiple nuclearevents, including the control of telomere length and chromosome segregation.Mol. Cell. Biol. 19, 8660–8672.
Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H.,Naismith, J. H., and Hay, R. T. (2001). Polymeric chains of SUMO-2 andSUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9.J. Biol. Chem. 276, 35368–35374.
Tatham, M. H., Kim, S., Jaffray, E., Song, J., Chen, Y., and Hay, R. T. (2005).Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mech-anism for SUMO paralog selection. Nat. Struct. Mol. Biol. 12, 67–74.
Valdez, B. C., Henning, D., Perlaky, L., Busch, R. K., and Busch, H. (1997).Cloning and characterization of Gu/RH-II binding protein. Biochem. Bio-phys. Res. Commun. 234, 335–340.
Vitali, P., Basyuk, E., Le Meur, E., Bertrand, E., Muscatelli, F., Cavaille, J., andHuttenhofer, A. (2005). ADAR2-mediated editing of RNA substrates in thenucleolus is inhibited by C/D small nucleolar RNAs. J. Cell Biol. 169, 745–753.
Yang, W., Wang, Q., Howell, K. L., Lee, J. T., Cho, D. S., Murray, J. M., andNishikura, K. (2005). ADAR1 RNA deaminase limits short interfering RNAefficacy in mammalian cells. J. Biol. Chem. 280, 3946–3953.
Zhang, H., Saitoh, H., and Matunis, M. J. (2002). Enzymes of the SUMOmodification pathway localize to filaments of the nuclear pore complex. Mol.Cell. Biol. 22, 6498–6508.
J. M. Pinto Desterro et al.
Molecular Biology of the Cell5126