
Subcellular localization of coagulation factor II receptor-like 1 in neurons governs angiogenesis
12
© 2014 Nature America, Inc. All rights reserved. ARTICLES NATURE MEDICINE ADVANCE ONLINE PUBLICATION F2rl1 is a protease-activated receptor belonging to a family of GPCRs highly expressed throughout the central and the peripheral nervous system 1 . F2rl1, previously known as Par2, is particularly abundant in neurons, where it mediates pain, propagates inflammation and controls cell survival and the release of neurotransmitters 1 . F2rl1 is irreversibly activated by circulating and resident serine proteases that can cleave its N terminus and expose a tethered ligand or by syn- thetic peptides that mimic the tethered ligand 2 . In disease processes, F2rl1 integrates signals from proteases of the coagulation cascade and inflammatory cells to trigger repair by promoting survival and proliferation. In this context, F2rl1 also accelerates the revascu- larization of ischemic tissues 3 and is proangiogenic in the retina 4 . In the developing retina, F2rl1 expression increases proportionally with vessel growth 4 . The anatomic network of blood vessels and neurons evolves devel- opmentally to match neuronal activity with tissue perfusion. This para- digm applies equally to the visual system, where the first appearance of visual-evoked potentials coincides with the development of the human outer vascular plexus (at 25–26 weeks of gestation) 5 . The increased metabolic demand and oxygen consumption of newly functioning neurons contribute to localized regions of hypoxia 6 , particularly at the vascular front of the developing retina, and hypoxia in turn triggers the release of angiogenic factors. Although retinal glial cells participate in the synthesis of hypoxia-driven vascular growth fac- tors under physiologic 7 and pathologic conditions 8 , a selective loss of Vegfa production in astrocytes 9 or Müller cells 10 is relatively dispen- sable during retinal vascular development. Conversely, loss of retinal ganglion cells (RGCs) in transgenic mice (Pou4f2 LacZ-DTA/+ /Six3-Cre) completely abrogates developmental retinal vascular growth while preserving a normal astrocytic network 11 . Hence, neurons have an important role in retinal vascular development by releasing ang- iogenic factors 11 . Analogously, peripheral sensory neurons govern arterial differentiation and vascular branching through vascular endothelial growth factor (Vegfa) secretion 12 . GPCRs, such as F2rl1, classically signal at the plasma membrane or as internalized scaffolds that are subsequently recycled back to the cell surface or sorted for degradation. Many GPCRs have been identified at the cell nucleus, implying nuclear translocation of activated recep- tors 13 . The presence in the nucleoplasm of phospholipids 14 and in the nucleus of membrane-associated signaling factors 13 , β-arrestin 15 , 1 Department of Pediatrics, Centre Hospitalier Universitaire (CHU) Sainte-Justine Research Center, Université de Montréal, Montréal, Québec, Canada. 2 Department of Ophthalmology, Hôpital Maisonneuve-Rosemont Research Center, Université de Montreal, Montreal, Québec, Canada. 3 Department of Pharmacology, Université de Montréal, Montréal, Québec, Canada. 4 Department of Pharmacology and Therapeutics, McGill University, Montréal, Québec, Canada. 5 The Wyss Institute, Harvard University, Boston, Massachusetts, USA. 6 Department of Pharmacology, Sherbrooke University, Sherbrooke, Quebec, Canada. 7 Department of Systems Biology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA. 8 Department of Physiology and Pharmacology, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada. 9 Department of Cardiology, CHU Sainte-Justine Research Center, Université de Montréal, Montréal, Québec, Canada. 10 These authors contributed equally to this work. Correspondence should be addressed to S.C. ([email protected]), G.A. ([email protected]) or J.-S.J. ([email protected]). Received 14 April; accepted 23 July; published online 14 September 2014; doi:10.1038/nm.3669 Subcellular localization of coagulation factor II receptor-like 1 in neurons governs angiogenesis Jean-Sébastien Joyal 1–4,10 , Satra Nim 4,10 , Tang Zhu 1,10 , Nicholas Sitaras 2,3 , José Carlos Rivera 1,2 , Zhuo Shao 4 , Przemyslaw Sapieha 2 , David Hamel 3 , Melanie Sanchez 4 , Karine Zaniolo 1 , Manon St-Louis 4 , Johanne Ouellette 4 , Martin Montoya-Zavala 5 , Alexandra Zabeida 1 , Emilie Picard 1 , Pierre Hardy 1 , Vikrant Bhosle 4 , Daya R Varma 4 , Fernand Gobeil Jr 6 , Christian Beauséjour 3 , Christelle Boileau 3 , William Klein 7 , Morley Hollenberg 8 , Alfredo Ribeiro-da-Silva 4 , Gregor Andelfinger 9 & Sylvain Chemtob 1–4 Neurons have an important role in retinal vascular development. Here we show that the G protein–coupled receptor (GPCR) coagulation factor II receptor-like 1 (F2rl1, previously known as Par2) is abundant in retinal ganglion cells and is associated with new blood vessel formation during retinal development and in ischemic retinopathy. After stimulation, F2rl1 in retinal ganglion cells translocates from the plasma membrane to the cell nucleus using a microtubule-dependent shuttle that requires sorting nexin 11 (Snx11). At the nucleus, F2rl1 facilitates recruitment of the transcription factor Sp1 to trigger Vegfa expression and, in turn, neovascularization. In contrast, classical plasma membrane activation of F2rl1 leads to the expression of distinct genes, including Ang1, that are involved in vessel maturation. Mutant versions of F2rl1 that prevent nuclear relocalization but not plasma membrane activation interfere with Vegfa but not Ang1 expression. Complementary angiogenic factors are therefore regulated by the subcellular localization of a receptor (F2rl1) that governs angiogenesis. These findings may have implications for the selectivity of drug actions based on the subcellular distribution of their targets.
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Subcellular localization of coagulation factor II receptor-like 1 in neurons governs angiogenesis

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
nature medicine advance online publication �
F2rl1 is a protease-activated receptor belonging to a family of GPCRs highly expressed throughout the central and the peripheral nervous system1. F2rl1, previously known as Par2, is particularly abundant in neurons, where it mediates pain, propagates inflammation and controls cell survival and the release of neurotransmitters1. F2rl1 is irreversibly activated by circulating and resident serine proteases that can cleave its N terminus and expose a tethered ligand or by syn-thetic peptides that mimic the tethered ligand2. In disease processes, F2rl1 integrates signals from proteases of the coagulation cascade and inflammatory cells to trigger repair by promoting survival and proliferation. In this context, F2rl1 also accelerates the revascu-larization of ischemic tissues3 and is proangiogenic in the retina4. In the developing retina, F2rl1 expression increases proportionally with vessel growth4.
The anatomic network of blood vessels and neurons evolves devel-opmentally to match neuronal activity with tissue perfusion. This para-digm applies equally to the visual system, where the first appearance of visual-evoked potentials coincides with the development of the human outer vascular plexus (at 25–26 weeks of gestation)5. The increased metabolic demand and oxygen consumption of newly functioning
neurons contribute to localized regions of hypoxia6, particularly at the vascular front of the developing retina, and hypoxia in turn triggers the release of angiogenic factors. Although retinal glial cells participate in the synthesis of hypoxia-driven vascular growth fac-tors under physiologic7 and pathologic conditions8, a selective loss of Vegfa production in astrocytes9 or Müller cells10 is relatively dispen-sable during retinal vascular development. Conversely, loss of retinal ganglion cells (RGCs) in transgenic mice (Pou4f2LacZ-DTA/+/Six3-Cre) completely abrogates developmental retinal vascular growth while preserving a normal astrocytic network11. Hence, neurons have an important role in retinal vascular development by releasing ang-iogenic factors11. Analogously, peripheral sensory neurons govern arterial differentiation and vascular branching through vascular endothelial growth factor (Vegfa) secretion12.
GPCRs, such as F2rl1, classically signal at the plasma membrane or as internalized scaffolds that are subsequently recycled back to the cell surface or sorted for degradation. Many GPCRs have been identified at the cell nucleus, implying nuclear translocation of activated recep-tors13. The presence in the nucleoplasm of phospholipids14 and in the nucleus of membrane-associated signaling factors13, β-arrestin15,
1Department of Pediatrics, Centre Hospitalier Universitaire (CHU) Sainte-Justine Research Center, Université de Montréal, Montréal, Québec, Canada. 2Department of Ophthalmology, Hôpital Maisonneuve-Rosemont Research Center, Université de Montreal, Montreal, Québec, Canada. 3Department of Pharmacology, Université de Montréal, Montréal, Québec, Canada. 4Department of Pharmacology and Therapeutics, McGill University, Montréal, Québec, Canada. 5The Wyss Institute, Harvard University, Boston, Massachusetts, USA. 6Department of Pharmacology, Sherbrooke University, Sherbrooke, Quebec, Canada. 7Department of Systems Biology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA. 8Department of Physiology and Pharmacology, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada. 9Department of Cardiology, CHU Sainte-Justine Research Center, Université de Montréal, Montréal, Québec, Canada. 10These authors contributed equally to this work. Correspondence should be addressed to S.C. ([email protected]), G.A. ([email protected]) or J.-S.J. ([email protected]).
Received 14 April; accepted 23 July; published online 14 September 2014; doi:10.1038/nm.3669
Subcellular localization of coagulation factor II receptor-like 1 in neurons governs angiogenesisJean-Sébastien Joyal1–4,10, Satra Nim4,10, Tang Zhu1,10, Nicholas Sitaras2,3, José Carlos Rivera1,2, Zhuo Shao4, Przemyslaw Sapieha2, David Hamel3, Melanie Sanchez4, Karine Zaniolo1, Manon St-Louis4, Johanne Ouellette4, Martin Montoya-Zavala5, Alexandra Zabeida1, Emilie Picard1, Pierre Hardy1, Vikrant Bhosle4, Daya R Varma4, Fernand Gobeil Jr6, Christian Beauséjour3, Christelle Boileau3, William Klein7, Morley Hollenberg8, Alfredo Ribeiro-da-Silva4, Gregor Andelfinger9 & Sylvain Chemtob1–4
Neurons have an important role in retinal vascular development. Here we show that the G protein–coupled receptor (GPCR) coagulation factor II receptor-like 1 (F2rl1, previously known as Par2) is abundant in retinal ganglion cells and is associated with new blood vessel formation during retinal development and in ischemic retinopathy. After stimulation, F2rl1 in retinal ganglion cells translocates from the plasma membrane to the cell nucleus using a microtubule-dependent shuttle that requires sorting nexin 11 (Snx11). At the nucleus, F2rl1 facilitates recruitment of the transcription factor Sp1 to trigger Vegfa expression and, in turn, neovascularization. In contrast, classical plasma membrane activation of F2rl1 leads to the expression of distinct genes, including Ang1, that are involved in vessel maturation. Mutant versions of F2rl1 that prevent nuclear relocalization but not plasma membrane activation interfere with Vegfa but not Ang1 expression. Complementary angiogenic factors are therefore regulated by the subcellular localization of a receptor (F2rl1) that governs angiogenesis. These findings may have implications for the selectivity of drug actions based on the subcellular distribution of their targets.

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
� advance online publication nature medicine
G protein–receptor kinases16 and regulators of G protein signal-ing17, as well as the existence of nuclear membrane channels and nuclear bodies (with hydrophobic regions18) and electron microscopy evidence of nuclear membrane invaginations19, all point to an intra-nuclear network of membranes and hydrophobic regions that could harbor nuclear-localized GPCRs20. Although translocation of GPCRs to the cell nucleus has been proposed, the mechanism of this translo-cation is unknown21. Moreover, the in vivo physiologic consequences of nuclear-localized GPCR signaling have yet to be established.
In the process of elucidating the contribution of F2rl1 to retinal angiogenesis, we detected its presence at the cell nucleus, leading us to explore how the subcellular distribution of F2rl1 governs its effects on angiogenesis. We show that membrane-activated F2rl1 translocates from the plasma membrane to the cell nucleus using a microtubule-dependent shuttle requiring importin-β1 and Snx11. At the nucleus, activated F2rl1 recruits Sp1 to trigger Vegfa expression and, in turn, neovascularization.
RESULTSF2rl1 is angiogenic in the developing retinaWe first explored whether F2rl1 contributes to retinal vascular develop-ment. Compared to wild-type (WT) mice, F2rl1 knockout (F2rl1−/−) mice showed a modest delay in retinal vas-cular growth and reduced vascular density during early development (Supplementary Fig. 1a–c). Although these results are in part consistent with those reported previously22, our findings diverge from those showing robust effects of F2rl1 stimulation on retinal
angiogenesis (in vivo)4 and neurovascular cell migration (in vitro)23. To eliminate potential confounding compensatory mechanisms in F2rl1−/− mice, we knocked down F2rl1 using intravitreal injections of shRNA-encoded lentivirus (shF2rl1). Knockdown of F2rl1 in neurons of the ganglion cell layer significantly reduced developmental neovas-cularization, whereas treatment with the mouse F2rl1 agonist peptide (F2rl1-AP) SLIGRL triggered retinal neovascularization in WT but not F2rl1−/− animals (Fig. 1a and Supplementary Fig. 1a,d). In a model of ischemic retinopathy, exposure of mice to 75% O2 from postnatal day (P) 7–12 induced a marked vaso-obliteration, which was augmented by shF2rl1 and was diminished by SLIGRL. Physiologic revascularization of the central avascular retina was delayed by shF2rl1, resulting in an increased number of pathologic preretinal neovessels. Conversely, the F2rl1-AP (SLIGRL) improved normal retinal revascularization and in turn decreased the number of pathologic neovessels (Supplementary Fig. 2), confirming a pivotal role for F2rl1 in physiologic retinal angiogenesis.
F2rl1 was localized in RGCs and, to a much lesser extent, in endothe-lial cells, as reported previously24, but was not present in the astroglia of the developing retina (Fig. 1b and Supplementary Fig. 3a–d). We confirmed the predominance of F2rl1 mRNA in RGCs by laser capture microdissection (LCM) of retinal layers (Fig. 1c); F2rl1
a P6
WT
WT
SLIGRL LV.shF2rl1A
WT
F2rl1–/–
P6WT
F2rl1 DAPI MergedVessel
GCL
INL
ONL
Lectin
β3-tubb c
P6
VehPou4f2Z-DTA/cre–/–
Pou4f2Z-DTA/cre–/–Pou4fZ-DTA/Cre-ERTm
Cre-ERTm
SLIGRL
SLIGRL SLIGRL
e
WT
Veh SLIGRL
F2rl1–/–
DAPI DAPI
GCL
β-Gal β-Gal
cre–/–
Pou4f2Z-DTA
LoxP
XCAGG-Cre-ERTM
P0 P1 P4 P6
LoxPLac Z DTAPou4f2
Birth TM SLIGRL FM
Stop
Cre-ERTm
d
IPL
**
Vessel GCL INL ONL
4
3
2
1
0
Pou4f2Z-DTA
****
Vas
cula
r ar
ea a
t P6
chan
ge r
elat
ive
to C
tl (%
)
50
40
30
20
10
+ +–0
cre
20 0**
***
Vas
cula
r ar
ea a
t P6
chan
ge r
elat
ive
to C
tl (%
)15
10
5
0
Veh
icle
SLI
GR
L
LV.s
hScr
am
LV.s
hF2r
l1
–5
–10
–15
–20
***
***
Vas
cula
r de
nsity
at P
6ch
ange
rel
ativ
e to
Ctl
(%)
LV.s
hScr
am
LV.s
h F2r
l1
Veh
icle
SLI
GR
L
0100
–10
–30
–20
80
60
40
20
0
F2r
l1 m
RN
A e
xpre
ssio
n
104 c
opie
s pe
r 10
6 Cyc
lop
hilin
Figure 1 F2rl1 in retinal neurons promotes angiogenesis. (a) Lectin-stained retinal flat mounts of WT and F2rl1−/− mice at P6 after treatment with vehicle or SLIGRL (20 µM) or infection with lentivirus (LV) encoding F2rl1 shRNA (LV.shF2rl1) or scrambled shRNA (LV.shScram). Bar graphs show the changes in vascular area and vascular density. Scale bar, 200 µm. Ctl, control. n = 7–16. (b) F2rl1 retinal expression in WT mice colocalized with β3-tubulin (β3-tub). Scale bar, 20 µm. (c) F2rl1 mRNA expression by LCM of retinal layers. n = 4 mice. INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; NFL, nerve fiber layer. Scale bar, 20 µm. (d) Top, schematic of the experimental strategy. Pou4f2LacZ-DTA/+ (Pou4f2Z-DTA) mice containing a floxed-lacZ-stop–diphtheria A toxin cassette expressed selectively in RGCs were crossed with tamoxifen (Tm)-inducible Cre recombinase mice (CAGG-Cre-ERTm); the mice were injected with Tm at P1 to selectively ablate RGCs during vascular development, treated with SLIGRL at P4 and analyzed by flat mount at P6. Bottom, β-galactosidase (β-Gal) stains of retinal cross-sections. Scale bar, 20 µm. (e) Lectin-stained retinal flat mounts (P6) of Pou4f2LacZ-DTA/+ mice expressing Cre-ERTm or not and treated with vehicle (Veh) or SLIGRL (20 µM). The bar graph shows the changes in vascular area. Scale bar, 200 µm. n = 3–6. All error bars represent the s.e.m. **P < 0.01, ***P < 0.001 compared to WT or vehicle using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test (c) or Student’s t test (a,e).
©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
nature medicine advance online publication �
expression was substantially (3.3-fold) lower in superficial retinal vessels compared to RGCs and was negligible in the inner and outer nuclear layers (Fig. 1c). We then tested the angiogenic contribution of F2rl1 expressed in retinal ganglion cells. In transgenic mice in which RGCs are conditionally ablated during vascular development25 (Pou4f2LacZ-DTA/+/Cre-ERTm mice; Fig. 1d), F2rl1-AP (SLIGRL) failed to promote angiogenesis (Fig. 1e), which is contrary to the results in transgenic mice with intact RGCs (Cre-ERTm/+ or Pou4f2LacZ-DTA/+/cre−/−). There was also a trend towards decreased angiogenesis in RGC-deficient mice (Pou4f2LacZ-DTA/+/Cre-ERTm) compared to controls (Pou4f2LacZ-DTA/+/cre−/−). Ex vivo, when RGCs (but not F2rl1-silenced RGCs) were activated by F2rl1-AP, their conditioned medium induced robust proangiogenic vascular sprout-ing in aortic explants from WT and F2rl1−/− mice (Supplementary Fig. 3e,f). In contrast, direct F2rl1 stimulation with SLIGRL of aortic explants was ineffective. Together these findings point to a key role for F2rl1 on RGCs in stimulating angiogenesis.
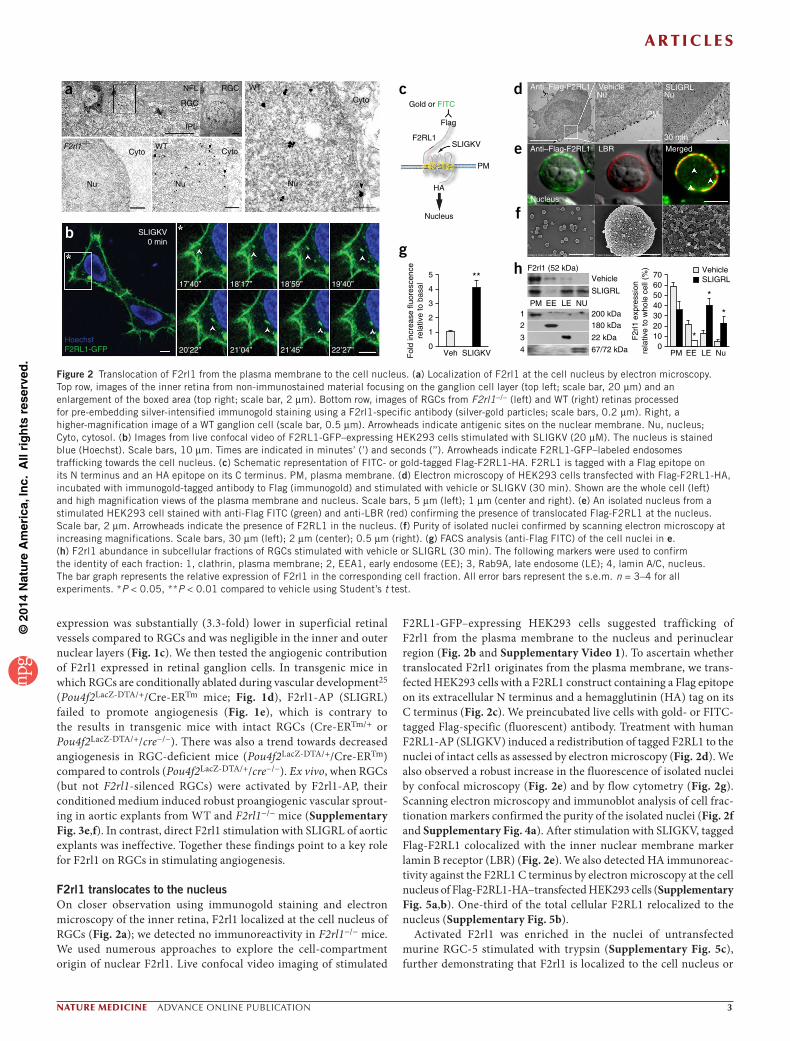
F2rl1 translocates to the nucleusOn closer observation using immunogold staining and electron microscopy of the inner retina, F2rl1 localized at the cell nucleus of RGCs (Fig. 2a); we detected no immunoreactivity in F2rl1−/− mice. We used numerous approaches to explore the cell-compartment origin of nuclear F2rl1. Live confocal video imaging of stimulated
F2RL1-GFP–expressing HEK293 cells suggested trafficking of F2rl1 from the plasma membrane to the nucleus and perinuclear region (Fig. 2b and Supplementary Video 1). To ascertain whether translocated F2rl1 originates from the plasma membrane, we trans-fected HEK293 cells with a F2RL1 construct containing a Flag epitope on its extracellular N terminus and a hemagglutinin (HA) tag on its C terminus (Fig. 2c). We preincubated live cells with gold- or FITC-tagged Flag-specific (fluorescent) antibody. Treatment with human F2RL1-AP (SLIGKV) induced a redistribution of tagged F2RL1 to the nuclei of intact cells as assessed by electron microscopy (Fig. 2d). We also observed a robust increase in the fluorescence of isolated nuclei by confocal microscopy (Fig. 2e) and by flow cytometry (Fig. 2g). Scanning electron microscopy and immunoblot analysis of cell frac-tionation markers confirmed the purity of the isolated nuclei (Fig. 2f and Supplementary Fig. 4a). After stimulation with SLIGKV, tagged Flag-F2RL1 colocalized with the inner nuclear membrane marker lamin B receptor (LBR) (Fig. 2e). We also detected HA immunoreac-tivity against the F2RL1 C terminus by electron microscopy at the cell nucleus of Flag-F2RL1-HA–transfected HEK293 cells (Supplementary Fig. 5a,b). One-third of the total cellular F2RL1 relocalized to the nucleus (Supplementary Fig. 5b).
Activated F2rl1 was enriched in the nuclei of untransfected murine RGC-5 stimulated with trypsin (Supplementary Fig. 5c), further demonstrating that F2rl1 is localized to the cell nucleus or
Gold or FITC
Flag
F2RL1SLIGKV
PM
HA
Nucleus
4
3
2
1
Vehicle
SLIGRL
200 kDa
180 kDa
22 kDa
67/72 kDa
PM
F2rl1 (52 kDa)
EE LE NU
SLIGKV0 min
17’40” 18’17” 18’59” 19’40”
20’22”HoechstF2RL1-GFP 21’04” 21’45” 22’27”
c Anti–Flag-F2RL1
Anti–Flag-F2RL1 LBR Merged
Nucleus
3.0kV 3.1mm ×1.50k SE(U) 3.0kV 3.0mm ×25.0k SE(U) 3.0kV 3.0mm ×70.0k SE(U)2.00µm30.0µm 500µm
Vehicle SLIGRLNu Nu
PM
30 min
PM
d
e
f
gh
b
NFL
RGC
Cyto Cyto
Nu
F2rl1–/–
a
Nu
WT
RGC WT
Nu
Cyto
IPL
**4
5
3
2
1
0Veh SLIGKVF
old
incr
ease
fluo
resc
ence
rela
tive
to b
asal
F2r
l1 e
xpre
ssio
nre
lativ
e to
who
le c
ell (
%)
PM EE LE Nu
706050
VehicleSLIGRL
403020100
*
*
*
Figure 2 Translocation of F2rl1 from the plasma membrane to the cell nucleus. (a) Localization of F2rl1 at the cell nucleus by electron microscopy. Top row, images of the inner retina from non-immunostained material focusing on the ganglion cell layer (top left; scale bar, 20 µm) and an enlargement of the boxed area (top right; scale bar, 2 µm). Bottom row, images of RGCs from F2rl1−/− (left) and WT (right) retinas processed for pre-embedding silver-intensified immunogold staining using a F2rl1-specific antibody (silver-gold particles; scale bars, 0.2 µm). Right, a higher-magnification image of a WT ganglion cell (scale bar, 0.5 µm). Arrowheads indicate antigenic sites on the nuclear membrane. Nu, nucleus; Cyto, cytosol. (b) Images from live confocal video of F2RL1-GFP–expressing HEK293 cells stimulated with SLIGKV (20 µM). The nucleus is stained blue (Hoechst). Scale bars, 10 µm. Times are indicated in minutes’ (’) and seconds (’’). Arrowheads indicate F2RL1-GFP–labeled endosomes trafficking towards the cell nucleus. (c) Schematic representation of FITC- or gold-tagged Flag-F2RL1-HA. F2RL1 is tagged with a Flag epitope on its N terminus and an HA epitope on its C terminus. PM, plasma membrane. (d) Electron microscopy of HEK293 cells transfected with Flag-F2RL1-HA, incubated with immunogold-tagged antibody to Flag (immunogold) and stimulated with vehicle or SLIGKV (30 min). Shown are the whole cell (left) and high magnification views of the plasma membrane and nucleus. Scale bars, 5 µm (left); 1 µm (center and right). (e) An isolated nucleus from a stimulated HEK293 cell stained with anti-Flag FITC (green) and anti-LBR (red) confirming the presence of translocated Flag-F2RL1 at the nucleus. Scale bar, 2 µm. Arrowheads indicate the presence of F2RL1 in the nucleus. (f) Purity of isolated nuclei confirmed by scanning electron microscopy at increasing magnifications. Scale bars, 30 µm (left); 2 µm (center); 0.5 µm (right). (g) FACS analysis (anti-Flag FITC) of the cell nuclei in e. (h) F2rl1 abundance in subcellular fractions of RGCs stimulated with vehicle or SLIGRL (30 min). The following markers were used to confirm the identity of each fraction: 1, clathrin, plasma membrane; 2, EEA1, early endosome (EE); 3, Rab9A, late endosome (LE); 4, lamin A/C, nucleus. The bar graph represents the relative expression of F2rl1 in the corresponding cell fraction. All error bars represent the s.e.m. n = 3–4 for all experiments. *P < 0.05, **P < 0.01 compared to vehicle using Student’s t test.

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
� advance online publication nature medicine
is associated with nuclear membranes. In addition, fractionation of RGC-5 into plasma membrane, nuclei and early and late endosome fractions confirmed a threefold to fourfold enrichment of endog-enous F2rl1 (52 kD) in the nuclear and late endosome fractions after stimulation (Fig. 2h and Supplementary Fig. 4a). Collectively, this evidence suggests that activated F2rl1 translocates from the plasma membrane to the cell nucleus.
Sorting partners shuttle F2rl1 on microtubulesWe next investigated the mechanism for nuclear F2rl1 translocation. The destination of endocytic cargo is influenced by receptor-targeting sequences, association with cargo proteins and cytoskeletal tracks and motors. The sorting of endosomes is location dependent, taking place in segregated compartments, as well as time dependent, involving precisely orchestrated cargo-protein interactions26. In this respect, importins are pivotal nuclear transport proteins of the karyopherin family that recognize and bind nuclear localization targeting sequences.
They have an essential role in protein import across the nucleopore complex, including the nuclear translocation of integral membrane proteins27. Classically, the adaptor protein importin-α binds to the cargo, whereas importin-β1 grants passage to the nucleus through the nucleopore complex; however, importin-β1 is necessary and could be sufficient for nuclear import28. We found that importin-β1 colocalized and coimmunoprecipitated with stimulated F2rl1 in transfected HEK293 cells (Fig. 3a,b and Supplementary Figs. 4b and 6a,b). Silencing of importin-β1 (as well as silencing of importin-α3 or importin-α5) interfered with F2rl1 relocalization to the nucleus (Fig. 3c and Supplementary Fig. 7).
In addition to importins, the role of sorting nexins (Snx) is becoming increasingly recognized in endosomal trafficking. Snx harbor a characteristic Phox domain and have been identified across phyla with conserved endocytic, endosomal-sorting and signaling functions29. SNX1 and SNX2, along with other components of the mammalian retromer, contribute to retrieval of F2R (or PAR1)
+
*
e
+
*
d SLIGKV
SLIGKV
Merged
a Vehicle
SLIGKV
Merged
Merged
Merged
Merged
Importin-β1
Importin-β1
shScram
A
B
C
siImpβ1
*****
****
0 –20 –40 –60
c
Cadaverine
Colchicine
Vinblastine
***
***
0 –20 –40 –60 –80 –100f
***
***
***
0 –20 –40 –60 –80 –100
β-arrestin
C-trunc
NLS-1iL
NLS-3iL
g
Dyn
ein
Tub
ulin
**
**
*
*
Snx11-GFP
α-tubulin
Snx11-GFP
Snx11-GFP
shSNX1
shSNX2
shS
NX
11
DynIK44A
Impβ
1
SN
X11
0 min30 min
b
8
6
4
2
0
300
225
150
75
0Flu
ores
cenc
e (A
U)
1 5 10 15 20Distance (µm)
300F
luor
esce
nce
(AU
)25020015010050
01 3 6 9
Distance (µm)12 15
F2RL1-GFP
Flag-F2RL1
Flag-F2RL1
Decrease in nuclear fluorescencerelative to stimulated F2RL1-GFP (%)
Co-
IP o
f F2R
L1-G
FP
fo
ld c
hang
e re
lativ
e to
0
+
*SLIGKV
Mergedα-tubulin
Figure 3 Transport of F2RL1 by importin-β1 and SNX11. (a) Confocal microscopy of live HEK293 cells expressing GFP-tagged SNX11 co-transfected with Flag-F2RL1 and incubated with Flag-specific antibodies (purple) and then stimulated with vehicle (top) or SLIGKV (bottom) (20 µM). Importin-β1 was labeled using importin-β1–specific antibodies (red). Scale bars, 10 µm. (b) Coimmunoprecipitation (Co-IP) of F2RL1-GFP with importin-β1 (Impβ1), Snx11, dynein and α-tubulin in cells stimulated with SLIGKV at 0 and 30 min. (c) FACS analysis of F2RL1-GFP on isolated nuclei of HEK293 cells previously stimulated with SLIGKV (20 µM); SNX1 and SNX2 were silenced using shRNA; three different shRNAs (A–C) were used to silence SNX11. Importin-β1 was silenced using siRNA (siImpβ1). The bar graphs in c, f and g represent the decrease in nuclear fluorescence relative to stimulated F2RL1-GFP. (d) Colocalization of α-tubulin with F2RL1-GFP and Snx11-GFP in stably transfected SLIGKV-stimulated HEK293 cells. Scale bars, 10 µm. (e) Magnification of the boxed areas in d and line scan (as indicated by the arrows in the micrographs) confirming colocalization of F2RL1-GFP and Snx11-GFP (green) with α-tubulin (red). The line scans display the relative fluorescence of each antibody on the path crossed by the line on the micrograph. Scale bars, 10 µm. AU, arbitrary units. (f) FACS analysis of isolated nuclei of SLIGKV (20 µM)-stimulated HEK293 cells expressing F2RL1-GFP in the presence or absence of DynIK44A, monodansyl cadaverine, colchicine or vinblastine. (g) FACS of isolated nuclei from F2RL1 mutants fused to GFP stably expressed in HEK293 cells stimulated with SLIGKV (20 µM). NLS-3iL, NLS from the third intracellular loop mutated; NLS-1iL, NLS from the first intracellular loop mutated; C-trunc, C-truncated mutant. All error bars represent the s.e.m. n = 3–4 for all experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared to F2RL1-GFP or values without asterisks using one-way ANOVA with Dunnett’s (c) or Tukey’s multiple comparison test (f,g).

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
nature medicine advance online publication �
from the endosome to the trans-Golgi network and prevent its lysosomal degradation30. However, silencing of SNX1 or SNX2 did not affect the relocalization of tagged F2RL1 to the nucleus (Fig. 3c and Supplementary Fig. 6d). Snx11 contains nuclear localization motifs; when expressed in HEK293 cells, Snx11-GFP was found at and around the nucleus, where it colocalized with the late endo-somal marker M6P and importin-β1 (Supplementary Fig. 6c). After F2RL1 stimulation, SNX11 coimmunoprecipitated with F2RL1, along with importin-β1 (Fig. 3a,b and Supplementary Fig. 6a,b). In addition, knockdown of SNX11 abrogated the nuclear transloca-tion of F2RL1-GFP, as detected by FACS analysis of isolated nuclei (Fig. 3c), and Flag-F2RL1 was correspondingly retained largely at or adjacent to the cell membrane, as assessed by confocal imaging (Supplementary Fig. 6d,e).
Sorting nexins and importins have been reported to shuttle on microtubules31. Carrier-protein kinesins move cargo on microtubules towards the plasma membrane and contribute to endosomal sort-ing, whereas dyneins transit towards the juxtanuclear microtubule organizing center and nucleus32,33. Dyneins facilitate the transport of proteins from early to late endosomes and sorting to the endo-cytic recycling compartment34. They also contribute to the nuclear import of regulatory proteins35. We determined whether the micro-tubule–dynein complex contributes to F2RL1 nuclear trafficking. The intracellular distribution of F2RL1 and Snx11 overlapped the microtubule network, as revealed by line-scan analysis (α-tubulin; Fig. 3d,e). F2RL1 coimmunoprecipitated with dynein and α-tubulin after F2RL1-AP stimulation (Fig. 3b and Supplementary Figs. 4b and 6a,b). Concordantly, F2RL1 trafficking to the nucleus was abrogated
b c d100 **
a
80
60
ChI
P r
e-C
hIP
Veg
f pro
mot
erD
NA
cha
nge
inqR
T-P
CR
exp
ress
ion
(%)
40
20
Inpu
t
AV
G
His
tone
H3
Veh
icle
SLI
GR
L
0
ChIP: F2rl1Re-ChIP: Sp1
***
5 VegfaAng1
NS
4
SLI
GR
L-in
duce
d m
RN
Afo
ld e
xpre
ssio
n re
lativ
eto
bas
al 3
2
1
siScr
amsiS
p1
siScr
amsiS
p10
***6
5
4
Sp1
bin
ding
fold
incr
ease
rel
ativ
e to
bas
al
3
2
1
Vehicl
e
SLIGKV
0
12
3
p
Vehicl
e
SLIGKV
Sp1(106 kDa)
pSp1
SLIGRL (min) 0 5
Cell Co-IP
15 30 60
LBR(72 kDa)
Nuclei PAR2
Figure 4 Transcriptional activity of F2rl1 at the nucleus. (a) ChIP-Seq identified a peak to a putative Sp1 consensus binding site within the Vegfa promoter region (Supplementary Fig. 10a,b). Vehicle- or SLIGRL-treated RGC-5 nuclei were analyzed by ChIP with F2rl1 antibodies and then analyzed by re-ChIP with Sp1 antibodies. Recovered DNA fragments were analyzed by qRT-PCR using primers that emcompass the peak discovered by ChIP-Seq. Protein agarose A/G (A/G) alone and antibody to histone H3 were used as controls. (b) Time-dependent coimmunoprecipitation of Sp1 and phosphorylated Sp1 (pSp1) with F2rl1 in RGC-5 nuclei of SLIGRL-stimulated cells (20 µM). LBR served as a nuclear lysate loading control. (c) Electrophoresis and quantification of Sp1 and Sp3 DNA-protein complexes (EMSA) in the nuclei of F2RL1-stimulated cells. 1, Sp1 and Sp3; 2, Sp3; 3, unbound probe; p, labeled probe alone. n = 5 experiments. (d) F2rl1-induced expression of Vegfa and Ang1 in RGC-5 cells silenced for Sp1 (siSp1) or not (siScram) relative to vehicle-treated cells (basal). All error bars represent the s.e.m. n = 3–5 for all experiments. **P < 0.01, ***P < 0.001 relative to vehicle or basal using one-way ANOVA with Tukey’s multiple comparison test (a) or Student’s t test (c,d). NS, not significant.
eNu
F2RL1-E3–GFPLBR
PM
1
2
3
4
*
Colch.
Cadav
.
Vehicl
e
RGC8
6
4
2
0
SLI
GR
L-in
duce
d m
RN
Afo
ld in
crea
se r
elat
ive
to b
asal
**
Colch.
Cadav
.
Vehicl
e
4
3
2
1
0
SLI
GK
V-in
duce
d m
RN
Afo
ld in
crea
se r
elat
ive
to b
asal
b
*****
4 8
Colch.
Cadav
.
Vehicl
e
Colch.
Cadav
.
Vehicl
e
VegfaAng1
HEK
3
2
1
0
6
4
2
0
SLI
GR
L-in
duce
d m
RN
Afo
ld in
crea
se r
elat
ive
to b
asal
c
**
shScr
am
shSnx
11
shScr
am
shSnx
11
VegfaAng1
RGC5
4
3
2
1
0
SLI
GR
L-in
duce
d m
RN
Afo
ld in
crea
se r
elat
ive
to b
asal
d
**
VegfaAng1
Nuclei
0
8
6
4
2
020 45
Prestimulation (min)0 20 45
a3
SLI
GK
V-in
duce
d V
egf m
RN
Afo
ld in
crea
se r
elat
ive
to b
asal
2
1*** ***
0
F2R
L1-G
FP
F2RL1-GFP Mut
β-ar
rest
in
C-t
runc
NLS
-3iL S
LIG
KV
-indu
ced
Ang
1 m
RN
Afo
ld in
crea
se r
elat
ive
to b
asal
3
4
5
2
1
0
F2R
L1-G
FP
F2RL1-GFP Mut
β-ar
rest
in
C-t
runc
NLS
-3iL
mR
NA
exp
ress
ion
fold
incr
ease
rel
ativ
e to
bas
al
fVegfaAng1
5
4
3
2
1
0
F2RL1-NLS
++
+
++
+
++
+
++
+
Vehicle
F2RL1-E3
SLIGKVtat-SLIGKV
***
***
Figure 5 Subcellular location governs gene induction by F2rl1. (a–c) Vegfa and Ang1 mRNA expression in SLIGKV (20 µM)-stimulated F2RL1-GFP–expressing and F2RL1-GFP mutant (Mut)-expressing HEK293 cells (a), F2RL1-GFP–expressing HEK293 (HEK) cells and RGC-5 cells pretreated with vehicle, cadaverine (Cadav.) or colchicine (Colch.) (b) and RGC-5 cells transfected or not with shScram or shSnx11 (c). (d) Vegfa and Ang1 mRNA expression in the nuclei of RGC-5 cells pretreated with SLIGRL for 20 and 45 min before their nuclei were isolated; nuclei were then stimulated (30 min) with vehicle or SLIGRL. (e) Colocalization of GFP-tagged F2RL1-E3 (green) and LBR (red) in HEK293 cells (confocal microscopy). Scale bar, 10 µm. Western blots of the plasma membrane (PM) and nuclear (Nu) fractions in F2RL1-E3–expressing cells. 1, integrin-α5 (PM marker); 2, LBR (nuclear marker); 3, F2RL1-E3 using a GFP-specific antibody; 4, β-actin. (f) Vegfa and Ang1 mRNA responses to vehicle, SLIGKV (20 µM) or tat-SLIGKV (20 µM) in HEK293 cells expressing F2RL1-NLS mutants or F2RL1-E3. All error bars represent the s.e.m. n = 3–7 for all experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared to the corresponding values without asterisks using one-way ANOVA with Tukey’s (a,f) or Dunnett’s (b,d) multiple comparison test or Student’s t test (c).

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
� advance online publication nature medicine
by the microtubule disruptors colchicine and vinblastine36 but was independent of clathrin (dansyl cadaverine)37 and caveolin (dominant-negative dynein, DynIK44A) (Fig. 3f and Supplementary Fig. 6f). F2RL1 therefore travels to the cell nucleus through the microtubule network, and this trafficking involves importin-β1 (as well as importin-α3 and importin-α5) and Snx11.
We next examined which F2RL1 domains are required for nuclear translocation. We made mutations in four regions of the protein: the nuclear localization region (NLS) of either the first (1iL) or third intracellular loop (3iL), the β-arrestin interaction domain or the C terminus (Supplementary Fig. 8a). All F2RL1 constructs preserved functional stimulation-induced calcium transient and MEK-ERK or Akt phosphorylation activity, albeit with different dynamic profiles (Supplementary Fig. 8b,c). We studied mutant F2RL1-GFP–expressing cells stimu-lated with F2rl1-AP using FACS of isolated nuclei (Fig. 3g), sub-cellular fractionation (Supplementary Figs. 4a and 9a) and confocal microscopy (Supplementary Fig. 9b). Both the NLS and C-terminus truncation mutants of F2RL1 failed to translocate to the nucleus after stimulation with F2rl1-AP but instead remained at the plasma membrane (or endosomes) (Supplementary Fig. 9a,b). F2RL1-NLS mutants also failed to coimmunoprecipitate with importin-β1 after stimulation (Supplementary Fig. 9c). These findings suggest a promi-nent role for both the NLS and the C-terminal domains as scaffolds for the assembly of protein complexes that are required for F2RL1 nuclear translocation.
Nuclear F2rl1 recruits Sp1 to bind the Vegfa promoterWe next investigated the potential transcriptional activity of trans-located F2rl1 at the cell nucleus. Plasma membrane receptors clas-sically convey transcriptional effects through second messengers to remote nuclear transcription factors. Because we detected F2rl1 at the nucleus, we explored the possibility that it may form a transcriptional complex, as assessed by chromatin immunoprecipitation (ChIP) of stimulated F2RL1-GFP–expressing HEK293 cells with F2RL1-specific antibodies, followed by short-read sequencing (ChIP-Seq). To focus our analysis on the angiogenic role of F2RL1, we screened for F2RL1 binding to known angiogenic genes and discovered a hit covering a predicted Sp1 binding site of the Vegfa promoter (Supplementary Fig. 10a–c); we also identified binding of F2RL1 to other relevant ang-iogenic gene promoters (Supplementary Table 1). Using stringent fil-tering of Sp1 sites, we found that 227 genes were bound by F2RL1 (data not shown). We then subcloned the Sp1 consensus sequence recovered by the ChIP assay into three different reporter plasmids, which con-firmed its transcriptional activity in response to SLIGKV stimulation;
LV.G
FP
LV.F
2RL1
LV.F
2RL1
-NLS
LV.F
2RL1
-E3
Vas
cula
r ar
eare
llativ
e to
bas
al (
%)
***
***
LV.G
FP
LV.F
2RL1
LV.F
2RL1
-NLS
LV.F
2RL1
-E3
***
*** ***
*
**
***
*
*
Vas
cula
r de
nsity
rela
tive
to b
asal
(%
)m
RN
A e
xpre
ssio
nfo
ld in
crea
se r
elat
ive
to b
asal
a
e f
g
Veh
SLIGRL
Vegfa NeuN MergedWT
GCL
IPL
GCL
IPL
GCL
GCL
IPL
NeuN MergedLV.F2RL1-E3
NFL
GCL
IPL
GCL
IPL
β-III tubulin MergedLV.F2RL1-NLS
3×
2×
NeuN MergedLV.F2RL1
c
F2rl1–/–
GCL
IPL
NFLGCL
IPL
3×
LV.GFPF2rl1–/–
P6
b
LV.F2RL1-NLS
dVegfa NeuN Merged
LV.F2RL1-E3
F2rl1–/–
GCL
IPL
GCL
IPL
LV.F2RL1
LV.F2RL1-E3
LV.F2RL1-NLS
30
70
60
50
40
30
20
10
0
3
2
1
0
0
–10
–20
–30
–40
0
–10
–20
–30
–50
–40
20
10
0
Vessel
5
4
3
2
1
0GCL INL ONLS
LIG
RL-
indu
ced
Veg
f mR
NA
fold
incr
ease
rel
ativ
e to
bas
al
F2rl1–/–WT**
Vegfa Ang1
Figure 6 Nuclear F2rl1 contributes to retinal neovascularization. (a,b) Vegfa expression after stimulation (P6) in retinal ganglion cell layer neurons (NeuN labeled) of WT mice injected intravitreously (P4) with vehicle or SLIGRL (scale bar, 20 µm; a), as confirmed by LCM and quantitative RT-PCR (b). n = 3–4 mice per group. (c) Localization of F2RL1, F2RL1-E3 and F2RL1-NLS mutants (all GFP tagged) in LV-infected retinas; in each case, lower and higher magnification images are shown in the top and bottom rows, respectively. NeuN (red), RGC-layer neuronal marker; β-III tubulin, dendritic and axonal marker. Scale bars, 20 µm. F2RL1-NLS is located at the cell surface of neurons in dendrites and axons (detected in the IPL and NFL), whereas F2RL1-E3 is concentrated at the nucleus. (d) Vegfa expression in retinas infected with LV encoding F2RL1-E3 or F2RL1-NLS. Scale bar, 20 µm. (e) Lectin-stained retinal flat mounts of P6 F2rl1−/− mice intravitreally infected or not with LV encoding GFP, F2RL1, F2RL1-E3 or F2RL1-NLS (all GFP tagged). Scale bar, 200 µm. (f) Bar graphs showing changes in vascular area and vascular density at P6. n = 10–16 mice per group. (g) Vegfa and Ang1 mRNA expression in P6 retinas expressing F2RL1, F2RL1-E3 or F2RL1-NLS compared to GFP-transfected retinas (control). n = 3 mice per group. All error bars represent the s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 compared to control or LV.GFP using one-way ANOVA with Tukey’s multiple comparison test.

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
nature medicine advance online publication �
silencing of Sp1 abolished this effect (Supplementary Figs. 10d and 11a,b). We confirmed the relevance of our ChIP-Seq results to RGCs by showing that stimulated F2rl1 bound to the Vegfa promoter region, as assessed by ChIP-PCR. Using primers encompassing our ChIP-Seq hit (Supplementary Fig. 10b), we tested whether F2rl1 and Sp1 together form a transcriptional complex by performing a ChIP with F2rl1-specific antibodies followed by re-ChIP with Sp1-specific antibodies. We observed a significant enrichment in Sp1-rich DNA fragments from the Vegfa promoter region of stimulated RGCs (Fig. 4a and Supplementary Fig. 10b); in control experiments, ChIP using protein A/G alone or using antibody to histone H3 failed to significantly retrieve the Vegfa promoter DNA fragments.
Consistent with a role in gene induction, native nuclear F2rl1 of RGCs coimmunoprecipitated with phosphorylated Sp1 (active) in a time-dependent manner after F2rl1-AP stimulation (Fig. 4b and Supplementary Fig. 10e). Similarly, F2RL1-GFP, but not the F2RL1-NLS–GFP mutant, was able to recruit phosphorylated Sp1 after stimulation (Supplementary Fig. 10f). Moreover, F2RL1 interacted with Sp1-containing protein complexes that specifically bind Sp1 consensus DNA binding sites, as shown by electrophoresis mobility shift assay (EMSA; Fig. 4c and Supplementary Fig. 12). Silencing of Sp1 expression in RGCs abrogated F2rl1-dependent Vegfa induc-tion (Fig. 4d and Supplementary Fig. 10d) and cell proliferation (Supplementary Fig. 11c). Altogether, these data indicate that F2rl1 forms a transcriptional complex with activated Sp1 at the nucleus that regulates Vegfa expression38.
Expression of distinct genes controlled by F2rl1 at the nucleus and plasma membraneTo examine F2rl1 subcellular signaling specificity, we identified angiogenic genes regulated by F2rl1 that did not show F2rl1 binding in the nuclear ChIP-Seq assay (i.e., Ang1). In RGCs and stably trans-fected HEK293 cells, stimulation of F2rl1 induced the expression of Ang1, in addition to that of Vegfa (Fig. 5a,b). To confirm that nuclear F2rl1 induces Vegfa expression, we pretreated RGCs with SLIGRL to induce F2rl1 nuclear translocation and then isolated their nuclei at 20 and 45 min after stimulation39. Treatment of the isolated nuclei with SLIGRL resulted in time-dependent expression of Vegfa but not Ang1 (Fig. 5d). In contrast, stimulation of F2rl1 in conditions that interfere with its nuclear translocation (cells transfected with the F2RL1 NLS and C-truncation mutants, cells treated with the microtubule disrup-tor colchicine or cells in which Snx11 was silenced) prevented Vegfa expression but preserved that of Ang1 (Fig. 5a–c and Supplementary Fig. 10g). Hence, stimulated F2rl1 that relocates to the cell nucleus induces Vegfa, whereas F2rl1 at or close to the plasma membrane triggers Ang1 expression.
To further ascertain whether nuclear F2rl1 exerts specific func-tions, we explored whether retention of F2rl1 at the nucleus would reproduce these effects. Accordingly, we introduced a nuclear reten-tion signal to the C terminus of F2RL1. The E3 region of adenovirus type 5 targets transmembrane receptors to the nucleus40. F2RL1-GFP fused to E3 (F2RL1-E3) localized at the nucleus to the exclusion of the plasma membrane (Fig. 5e and Supplementary Fig. 13a). To stimu-late nuclear F2RL1-E3, we fused the internalization peptide sequence tat (GRKKRRQRRRPPQ) to SLIGKV (tat-SLIGKV). Tat-SLIGKV triggered calcium transients (Supplementary Fig. 13b) and induced Vegfa but not Ang1 expression in F2RL1-E3–expressing cells (Fig. 5f). Conversely, in cells expressing the F2RL1-NLS mutants that impede nuclear translocation, SLIGKV caused rapid calcium transients and increased Ang1 but not Vegfa expression (Fig. 5f and Supplementary
Fig. 13b). In intact F2RL1-expressing cells, SLIGKV triggered cal-cium transients (Supplementary Fig. 13b), as well as expression of both Ang1 and Vegfa (Fig. 5a,b). These findings corroborate distinct functions for F2rl1 at the plasma membrane and cell nucleus.
Nuclear F2rl1 exhibits an angiogenic phenotype in vivoWe next investigated the differential effects of F2rl1 localized to either the nucleus or plasma membrane of retinal ganglion neurons on reti-nal angiogenesis. During neovascularization, Vegfa causes differentia-tion, proliferation and migration of the vascular endothelium, whereas Ang1 fosters the maturation of newly formed vessels41. Intravitreal injections of SLIGRL resulted in localized production of Vegfa in the ganglion cell layer of WT (but not F2rl1−/−) mice, as assessed by immunofluorescence (Fig. 6a and Supplementary Fig. 14a) and quan-titative RT-PCR of laser capture microdissected ganglion cell layers (Fig. 6b). In contrast, RGC-deficient mice (Pou4f2LacZ-DTA/+/Cre-ERTm) failed to produce Vegfa after F2rl1 stimulation (Supplementary Fig. 14b). To distinguish plasma-membrane from nuclear F2RL1 functions in vivo, we used F2rl1−/− mice infected with F2RL1-encoded lentivirus injected intravitreally, as such a lentivirus has a propensity for targeting retinal ganglion cell layer neurons11,42. As anticipated, the F2RL1-NLS mutant localized at the plasma membrane of ganglion cell layer neurons and was accordingly detected in their axons and dendrites populating the nerve fiber layer and inner plexiform layer, respectively. In contrast, F2RL1-E3 was nearly exclusively localized to the nuclei of ganglion cell layer neurons (Fig. 6c and Supplementary Fig. 14c,d). Intraretinally transfected native F2RL1 increased the expression of both Vegfa and Ang1 and, in turn, promoted develop-mental angiogenesis (Fig. 6e–g). Retinas transfected with the F2RL1-NLS mutant exhibited increased Ang1 but negligable Vegfa expression associated with curtailed retinal neovascularization (Fig. 6d–g); these observations are in line with the actions of Ang1 on vessel quiescence and microangiopathy41. Retinas transfected with F2RL1-E3 showed decreased Ang1 but increased Vegfa expression, resulting in increased developmental angiogenesis (Fig. 6d–g).
DISCUSSIONIn exploring the role of F2rl1 in retinal angiogenesis, we found that unique functions of the receptor are segregated according to its subcellular distribution in neurons. We showed that F2rl1 from the plasma membrane translocates to the nucleus on microtubules together with Snx11 and importins. At the nucleus, F2rl1 forms a transcriptional complex with Sp1 that regulates Vegfa expression, whereas surface F2rl1 triggers the expression of a complementary angiogenic gene, Ang1. Nuclear F2rl1 in retinal ganglion neurons contributes to developmental angiogenesis, providing in vivo evidence for physiologic functions conferred by receptor subcellular localiza-tion (Supplementary Fig. 15).
The coagulation factors FVIIa and FXa are known agonists of F2rl1. These serine proteases bind tissue factor to activate F2rl1 and exert angiogenic effects43. The cytoplasmic domain of tissue factor modu-lates F2rl1 signaling in vessels43. F2rl1 is also widely distributed in neurons1, which are devoid of tissue factor44. However, numerous tissue-specific serine proteases are able to activate F2rl1; examples include members of the kallikrein-related peptidase family (such as KLK6)45, as well as a brain-derived trypsin-like serine protease (P22)46, β-tryptase47 and membrane-type serine proteases 1 and 3 (MT-SP1 and MT-SP3)48. Notably, MT-SP1 and F2rl1 share a similar tissue distribution, have important roles in angiogenesis49 and are stimulated by hypoxia and inflammation50.

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
� advance online publication nature medicine
Early in the course of our studies, we detected F2rl1 at the nuclei of retinal neurons. We used various approaches to show that the activated receptor reaches the nucleus by translocation from the plasma mem-brane: (i) live-confocal imaging of F2RL1-GFP nuclear trafficking; (ii) colocalization by confocal microscopy of FITC-tagged Flag-F2RL1 activated at the plasma membrane with the inner nuclear membrane (marked by LBR), as confirmed by electron microscopy; (iii) nuclear fluorescence of activated cell surface-tagged F2rl1, quantified by FACS and corroborated by cell fractionation; (iv) pharmacological blockade of F2rl1 translocation to the nucleus with microtubule inhibitors; and (v) prevention of the nuclear translocation of F2RL1 by mutagenesis of its NLS or its C terminus. Therefore, unlike GPCRs that localize to the nucleus in an agonist-independent manner51, F2rl1 traffics to the nucleus after activation at the cell surface.
We found that translocation of F2rl1 to the nucleus required micro-tubules. The microtubule motor dynein attaches to cargo proteins and powers long-range retrograde journeys to the nucleus; regulatory proteins and secondary messengers use microtubules to reach the cell nucleus. Numerous receptors, including transferin31 and nerve growth factor receptor-1 (ref. 52), as well as the GPCRs cannabi-noid-1 (ref. 53), somatostatin-2A (ref. 54) and DFrizzled-2 (ref. 21), associate with dynein and microtubules en route to the perinuclear region. Microtubules that are associated with the nuclear envelope allow the transition of proteins from early to late endosomes33.
We also found that the two putative nuclear localization domains determine the intracellular destination of F2RL1. NLSs are recognized by importins to promote nuclear transport, which has been described for soluble as well as integral membrane proteins, including GPCRs13. Proteins are ushered by importin-α and importin-β1 from the endo-plasmic reticulum through the nuclear pore complex to reach the inner nuclear membrane27,28. Native F2RL1, but not the F2RL1-NLS mutants, was able to recruit importin-β1 after activation. Truncation of the C terminus of F2RL1 also abrogated its nuclear translocation. The C terminus harbors a binding site for β-arrestin-1, which can translocate to the nucleus and mediate gene transcription15. However, mutation of the F2RL1 docking site for β-arrestin-1 did not prevent nuclear translocation, suggesting that other proteins assemble on the F2RL1 C terminus to promote its nuclear translocation.
The sorting nexin Snx11 was also needed for the nuclear transloca-tion of F2rl1. Snx11 was abundantly expressed at the cell nucleus and, together with importin-β1 and dynein, forms a protein complex that is necessary for the nuclear translocation of F2rl1. The association of Snx4 with dynein to mediate the transport of transferrin from early endosomes to perinuclear recycling endosomes is a recent example of the key role of Snx in long-range cargo transport31. Snx11 and Snx3 are members of a Snx subfamily that lacks a BAR domain, distinguish-ing them from the prototypical mammalian retromer proteins Snx1 and Snx2. By analogy with Snx3 (ref. 55), we speculate that Snx11 may prevent lysosomal degradation of its endosomal cargo while traveling to the nucleus.
The transcriptional activity of GPCRs in isolated nuclei has been widely reported13,51,56,57. Although traditional signaling machinery has been described at the nucleus13,16,17, nuclear GPCR signaling remains ill defined. We found that F2rl1 could form a nuclear tran-scriptional complex with the transcription factor Sp1, as shown by ChIP, coimmunoprecipitation and DNA binding complex (EMSA) assays. This transcriptional complex in turn bound consensus sites for Sp1 and Sp3 (ref. 38) in the Vegfa promoter region, increasing its expression. All conditions tested that prevent F2rl1 nuclear transloca-tion also abolished Vegfa induction while preserving the transcription
of plasma membrane F2rl1–regulated Ang1. Distinct angiogenic genes are therefore conditionally transcribed depending on the subcellular localization of F2rl1.
Developmental angiogenesis is finely regulated by cooperative angiogenic signals. Both Vegfa and Ang1 have pivotal functions in growth and maturation during vascular development: the former promotes differentiation, proliferation and migration of the vascular endothelium7, whereas the latter recruits pericytes and stabilizes nas-cent vasculature41. Deletion of either gene is embryonically lethal58,59. Conditional suppression of Ang1 between embryonic day (E) 10.5 and E12.5 results in an increased overall number of vessels and dis-organized vascular beds60. We observed a similar phenotype in mouse retinas overexpressing nuclear F2RL1-E3, which produce relatively less Ang1 while maintaining high levels of Vegfa. Conversely, high levels of Ang1 in the presence of lower levels of Vegfa appeared to stunt retinal vascular growth in F2RL1-NLS mutant–expressing mice. Neuronal F2rl1 therefore maintains the equilibrium between retinal vascular growth and maturation. Preventing nuclear translocation of neuronal F2rl1 tilts the balance towards early vascular maturation and results in impaired vascular growth, whereas unopposed nuclear F2rl1 activity triggers excessive Vegfa-mediated neovascularization. Hence, F2rl1 separately governs two major angiogenic cues depending on its subcellular localization, segregating the regulation of comple-mentary growth factors. We provide new evidence for the physiologic role of a nuclear GPCR in vivo that expands our understanding of GPCR signaling governed by subcellular localization. Our findings have implications for the design of more selective drugs based on the subcellular distribution of their targets.
METHODSMethods and any associated references are available in the online version of the paper.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
ACKNOWLEDGMENTSThis study was supported by grants from the Canadian Institutes of Health Research (CIHR; P.S., F.G. Jr., C. Beauséjour, M.H., A.R.-d.-S., G.A. and S.C.), the March of Dimes Birth Defects Foundation (S.C.), the Foundation Fighting Blindness (J.-S.J. and P.S.), Fonds de la Recherche en Santé du Québec (J.-S.J., P.S., G.A. and S.C.), Le Réseau de Recherche en Santé de la Vision (P.S. and S.C.), the National Eye Institute (W.K.) and the Robert Welch Foundation (W.K.). J.-S.J. is supported by the Burroughs Wellcome Fund Career Award for Medical Scientists and the Canadian Child Health Clinician Scientist Program (CIHR). G.A. is a recipient of a CIHR Clinician-Scientist Scholarship. P.S. and S.C. hold Canada Research Chairs. S.C. holds the Leopoldine Wolfe Chair in translational research in age-related macular degeneration. We thank G. Bourque and L. Létourneau (Genome Quebec Innovation Center, McGill University) for advice on ChIP-Seq analysis, C. Brown (Life Sciences Imaging Facility, McGill University) for live imaging advice, D. Barbaz (Department of Pharmacology, Sherbrooke University) for support with scanning EM and M. Chai-An (Department of Biochemistry and Molecular Biology, University of Texas MD Anderson Cancer Center) for breeding Pou4f2LacZ-DTA/+ mice. We thank N. Agarwal (National Eye Institute, US National Institutes of Health) for providing RGC-5 cells and N. Bunnett (Department of Physiology, University of California) for providing F2RL1-GFP and Flag-F2RL1-HA plasmids.
AUTHOR CONTRIBUTIONSJ.-S.J., S.N., T.Z., N.S. and S.C. conceived and designed the experiments. J.-S.J., S.N., T.Z., N.S., J.C.R., Z.S., P.S., D.H., M.S., K.Z., J.O., M.M.-Z., A.Z., E.P., V.B., F.G. Jr., C. Beauséjour, A.R.-d.-S., J.O. and M.S.-L. performed experiments. P.S., P.H., D.R.V., F.G., M.H., A.R.-d.-S. and G.A. provided expert advice. C. Boileau provided F2rl1−/− mice. W.K. provided Pou4f2LacZ-DTA/+ mice. G.A. and C. Beauséjour provided gene expression vectors. All authors analyzed the data. J.-S.J. and S.C. wrote the paper.

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
a r t i c l e s
nature medicine advance online publication �
COMPETING FINANCIAL INTERESTSThe authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
1. Saito, T. & Bunnett, N.W. Protease-activated receptors: regulation of neuronal function. Neuromolecular Med. 7, 79–99 (2005).
2. Adams, M.N. et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol. Ther. 130, 248–282 (2011).
3. Milia, A.F. et al. Protease-activated receptor-2 stimulates angiogenesis and accelerates hemodynamic recovery in a mouse model of hindlimb ischemia. Circ. Res. 91, 346–352 (2002).
4. Zhu, T. et al. Proangiogenic effects of protease-activated receptor 2 are tumor necrosis factor-α and consecutively Tie2 dependent. Arterioscler. Thromb. Vasc. Biol. 26, 744–750 (2006).
5. Hughes, S., Yang, H. & Chan-Ling, T. Vascularization of the human fetal retina: roles of vasculogenesis and angiogenesis. Invest. Ophthalmol. Vis. Sci. 41, 1217–1228 (2000).
6. Cringle, S.J., Yu, P.K., Su, E.N. & Yu, D.Y. Oxygen distribution and consumption in the developing rat retina. Invest. Ophthalmol. Vis. Sci. 47, 4072–4076 (2006).
7. Gerhardt, H. et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161, 1163–1177 (2003).
8. Scott, A. et al. Astrocyte-derived vascular endothelial growth factor stabilizes vessels in the developing retinal vasculature. PLoS ONE 5, e11863 (2010).
9. Weidemann, A. et al. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia 58, 1177–1185 (2010).
10. Bai, Y. et al. Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J. Pathol. 219, 446–454 (2009).
11. Sapieha, P. et al. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 14, 1067–1076 (2008).
12. Mukouyama, Y.S., Shin, D., Britsch, S., Taniguchi, M. & Anderson, D.J. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell 109, 693–705 (2002).
13. Gobeil, F. et al. G-protein–coupled receptors signalling at the cell nucleus: an emerging paradigm. Can. J. Physiol. Pharmacol. 84, 287–297 (2006).
14. Maraldi, N.M. et al. Morphological evidence of function-related localization of phospholipids in the cell nucleus. Adv. Enzyme Regul. 32, 73–90 (1992).
15. Kang, J. et al. A nuclear function of β-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell 123, 833–847 (2005).
16. Zhang, Y. et al. Nuclear effects of G-protein receptor kinase 5 on histone deacetylase 5–regulated gene transcription in heart failure. Circ Heart Fail 4, 659–668 (2011).
17. Panicker, L.M. et al. Nuclear localization of the G protein β5/R7-regulator of G protein signaling protein complex is dependent on R7 binding protein. J. Neurochem. 113, 1101–1112 (2010).
18. Zimber, A., Nguyen, Q.-D. & Gespach, C. Nuclear bodies and compartments: functional roles and cellular signalling in health and disease. Cell. Signal. 16, 1085–1104 (2004).
19. Fricker, M., Hollinshead, M., White, N. & Vaux, D. Interphase nuclei of many mammalian cell types contain deep, dynamic, tubular membrane-bound invaginations of the nuclear envelope. J. Cell Biol. 136, 531–544 (1997).
20. Linde, N. & Stick, R. Intranuclear membranes induced by lipidated proteins are derived from the nuclear envelope. Nucleus 1, 343–353 (2010).
21. Mathew, D. et al. Wingless signaling at synapses is through cleavage and nuclear import of receptor DFrizzled2. Science 310, 1344–1347 (2005).
22. Uusitalo-Jarvinen, H. et al. Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler. Thromb. Vasc. Biol. 27, 1456–1462 (2007).
23. Zhu, T. et al. Cortactin activation by FVIIa/tissue factor and PAR2 promotes endothelial cell migration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R577–R585 (2011).
24. Luo, W., Wang, Y. & Reiser, G. Two types of protease-activated receptors (PAR-1 and PAR-2) mediate calcium signaling in rat retinal ganglion cells RGC-5. Brain Res. 1047, 159–167 (2005).
25. Cho, J.-H., Mu, X., Wang, S.W. & Klein, W.H. Retinal ganglion cell death and optic nerve degeneration by genetic ablation in adult mice. Exp. Eye Res. 88, 542–552 (2009).
26. Maxfield, F.R. & McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5, 121–132 (2004).
27. King, M.C., Lusk, C.P. & Blobel, G. Karyopherin-mediated import of integral inner nuclear membrane proteins. Nature 442, 1003–1007 (2006).
28. Lusk, C.P., Blobel, G. & King, M.C. Highway to the inner nuclear membrane: rules for the road. Nat. Rev. Mol. Cell Biol. 8, 414–420 (2007).
29. Cullen, P.J. Endosomal sorting and signalling: an emerging role for sorting nexins. Nat. Rev. Mol. Cell Biol. 9, 574–582 (2008).
30. Gullapalli, A., Wolfe, B.L., Griffin, C.T., Magnuson, T. & Trejo, J. An essential role for Snx1 in lysosomal sorting of protease-activated receptor-1: evidence for retromer-, Hrs-, and Tsg101-independent functions of sorting nexins. Mol. Biol. Cell 17, 1228–1238 (2006).
31. Traer, C.J. et al. Snx4 coordinates endosomal sorting of TfnR with dynein-mediated transport into the endocytic recycling compartment. Nat. Cell Biol. 9, 1370–1380 (2007).
32. Caviston, J.P. & Holzbaur, E.L. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 16, 530–537 (2006).
33. King, M.C., Drivas, T.G. & Blobel, G. A network of nuclear envelope membrane proteins linking centromeres to microtubules. Cell 134, 427–438 (2008).
34. Aniento, F., Emans, N., Griffiths, G. & Gruenberg, J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 123, 1373–1387 (1993).
35. Roth, D.M., Moseley, G.W., Glover, D., Pouton, C.W. & Jans, D.A. A microtubule-facilitated nuclear import pathway for cancer regulatory proteins. Traffic 8, 673–686 (2007).
36. Thyberg, J. & Moskalewski, S. Microtubules and the organization of the Golgi complex. Exp. Cell Res. 159, 1–16 (1985).
37. Ray, E. & Samanta, A.K. Dansyl cadaverine regulates ligand induced endocytosis of interleukin-8 receptor in human polymorphonuclear neutrophils. FEBS Lett. 378, 235–239 (1996).
38. Pagès, G. & Pouysségur, J. Transcriptional regulation of the vascular endothelial growth factor gene—a concert of activating factors. Cardiovasc. Res. 65, 564–573 (2005).
39. Gobeil, F. et al. Nitric oxide signaling via nuclearized endothelial nitric-oxide synthase modulates expression of the immediate early genes iNOS and mPGES-1. J. Biol. Chem. 281, 16058–16067 (2006).
40. Ghosh, K. & Ghosh, H.P. Role of the membrane anchoring and cytoplasmic domains in intracellular transport and localization of viral glycoproteins. Biochem. Cell Biol. 77, 165–178 (1999).
41. Augustin, H.G., Koh, G.Y., Thurston, G. & Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 10, 165–177 (2009).
42. Joyal, J.-S. et al. Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A. Blood 117, 6024–6035 (2011).
43. Belting, M. et al. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat. Med. 10, 502–509 (2004).
44. Eddleston, M. et al. Astrocytes are the primary source of tissue factor in the murine central nervous system. A role for astrocytes in cerebral hemostasis. J. Clin. Invest. 92, 349–358 (1993).
45. Vandell, A.G. et al. Protease-activated receptor dependent and independent signaling by kallikreins 1 and 6 in CNS neuron and astroglial cell lines. J. Neurochem. 107, 855–870 (2008).
46. Sawada, K., Nishibori, M., Nakaya, N., Wang, Z. & Saeki, K. Purification and characterization of a trypsin-like serine proteinase from rat brain slices that degrades laminin and type IV collagen and stimulates protease-activated receptor-2. J. Neurochem. 74, 1731–1738 (2000).
47. Payne, V. & Kam, P.C. Mast cell tryptase: a review of its physiology and clinical significance. Anaesthesia 59, 695–703 (2004).
48. Takeuchi, T. et al. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem. 275, 26333–26342 (2000).
49. Aimes, R.T. et al. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb. Haemost. 89, 561–572 (2003).
50. Qiu, D., Owen, K., Gray, K., Bass, R. & Ellis, V. Roles and regulation of membrane-associated serine proteases. Biochem. Soc. Trans. 35, 583–587 (2007).
51. Marrache, A.M. et al. Proinflammatory gene induction by platelet-activating factor mediated via its cognate nuclear receptor. J. Immunol. 169, 6474–6481 (2002).
52. Riccio, A., Pierchala, B.A., Ciarallo, C.L. & Ginty, D.D. An NGF-TrkA–mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 277, 1097–1100 (1997).
53. Osborne, K.D., Lee, W., Malarkey, E.B., Irving, A.J. & Parpura, V. Dynamic imaging of cannabinoid receptor 1 vesicular trafficking in cultured astrocytes. ASN Neuro. 1, AN20090040 (2009).
54. Lelouvier, B. et al. Dynamics of somatostatin type 2A receptor cargoes in living hippocampal neurons. J. Neurosci. 28, 4336–4349 (2008).
55. Strochlic, T.I., Schmiedekamp, B.C., Lee, J., Katzmann, D.J. & Burd, C.G. Opposing activities of the Snx3-retromer complex and ESCRT proteins mediate regulated cargo sorting at a common endosome. Mol. Biol. Cell 19, 4694–4706 (2008).
56. Bhattacharya, M. et al. Nuclear localization of prostaglandin E2 receptors. Proc. Natl. Acad. Sci. USA 95, 15792–15797 (1998).
57. Savard, M. et al. Expression of endogenous nuclear bradykinin B2 receptors mediating signaling in immediate early gene activation. J. Cell. Physiol. 216, 234–244 (2008).
58. Carmeliet, P. et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380, 435–439 (1996).
59. Suri, C. et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87, 1171–1180 (1996).
60. Jeansson, M. et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Invest. 121, 2278–2289 (2011).

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
nature medicine doi:10.1038/nm.3669
ONLINE METHODSAnimals. Mice were used in compliance with the Animal Care Committee of CHU Sainte-Justine and the Canadian Council on Animal Care guidelines. Wild-type (WT) C57BL/6 and F2rl1−/− (stock 004993) mice were obtained from Jackson Laboratory and used at P4, P6 and P10. Pou4f2LacZ-DTA/+ mice (provided by W.K.) contain a floxed-lacZ-stop–diphtheria A toxin cassette expressed selectively in RGCs (Pou4f2 or Brn3b promoter) and were crossed with tamoxifen (Tm)-inducible Cre recombinase mice (CAGG-Cre-ERTm; Jackson Laboratory, stock 004682) to generate temporal and geographical conditional RGC ablation. Pou4f2LacZ-DTA/+/Cre-ERTm pups (as well as con-trols (Pou4f2LacZ-DTA/+ or Cre-ERTm)) were injected with tamoxifen (50 µg intraperitoneally) on P1 to selectively ablate RGCs during vascular devel-opment (P3–P4)25. Intravitreal injections were performed with vehicle or SLIGRL (mouse F2rl1-AP; Elim Biopharm) at P4 or lentiviruses expressing GFP, GFP-fused F2RL1, F2RL1m-NLS, F2RL1-E3 or shRNA at P2. Retinas were collected at P6.
Cells. RGC-5 cells (mouse RGCs; kindly provided by N. Agarwal) were terminally differentiated in the presence of staurosporine (1 µM) for 12 h, which effectively confers a post-mitotic neuronal state61. HEK293T and HEK293 cells (American Type Culture Collection) were cultured in DMEM (ATCC) supplemented with 10% FBS, as reported previously39.
shRNAs and siRNAs. shRNA sequences against F2rl1, Snx1, Snx2 and Snx11 and control shRNAs were obtained from Open Biosystems, and siRNA sequences against importin-β1 and Sp1 and the scrambled siRNA were obtained from Ambion (Ambion Silencer TM siRNA Construction kit).
F2RL1 mutant cloning strategies. The cytosolic tail of the adenovirus type 5 E3-11.6k gene (corresponding to amino acids 56–112) was commercially cloned into a pcDNA3 plasmid (Top Gene Technologies) and subcloned into the BamH1 and Age1 sites of the F2RL1–enhanced GFP (eGFP)-N1 vector (kindly provided by N. Bunnett)62 immediately after the cytosolic tail of F2RL1 and preceding eGFP. The Flag-F2RL1-HA11 vector was also kindly provided by N. Bunnett63. C-terminal truncated F2RL1 was PCR cloned by removing the C terminus, starting at amino acid 363. The F2RL1 β-arrestin1 mutant was cloned by the Kunkel method64 using the primer 5′-AACGCTCTCCTTTGCCGAGCCG-TCCGCGCCGTAAAGCAGATGCAAGTATCC-3′, which contains mutations changing serine and threonine to alanines at amino acid positions 363 and 366. F2RL1-NLSm-1iL and F2RL1-NLSm-3iL were PCR cloned by changing lysines to asparagines at amino acid positions 105, 106 and 107 for the first intracellular loop and 280, 281 and 283 for the third intracellular loop. The sense primer used for F2RL1-NLSm-1iL was 5′-TCCGAACTAATAATAATCACCCT-GCTGTGATT-3′, and the sense primer used for F2RL1-NLSm-3iL was 5′-GGATGAAAACTCAGAGAATAATAGGAATAGGGCCATCAAA-3′. The anti-sense primer used for both F2RL1-NLSm-1iL and -3iL was 5′-CGGTGGGCCC-CGATAGGAGGTCTTAA-3′.
Lentivirus production. Third-generation lentiviruses were produced by transiently transfecting lentivector and packaging vectors into HEK293T cells, as described previously65. Briefly, HEK293T (107) cells were seeded and transfected overnight with pV-SVG, pMDL and pREV plasmids and lentivectors in DMEM complete medium (Invitrogen). The supernatant was then replaced with fresh complete DMEM and incubated for an additional 30 h. Secreted viruses were filtered (0.22 µm) and ultracentrifuged at 50,000g (2 h, 10 °C; L8-70M, Beckman). The viruses were then resuspended in PBS, aliquoted (10 µl) and stored at –80 °C. The lentviral titers quantified using the p24 ELISA kit (Clontech) were as follows : LV.GFP, 2.4 ng µl−1; LV.F2RL1-GFP, 1.8 ng µl−1; LV.F2RL1-NLSm, 2.6 ng µl−1; LV.F2RL1-E3, 2.3 ng µl−1; LV.shScram, 3.15 ng µl−1; LV.shF2rl1, 2.2 ng µl−1; and LV.shSnx11, 2.7 ng µl−1.
Intravitreal injections and developmental growth assessment. WT C57BL/6, F2rl1−/− or Pou4f2LacZ-DTA/+/Cre-ERTm pups (or the controls, Pou4f2LacZ-DTA/+ or Cre-ERTm) were injected at P4 with vehicle or SLIGRL (20 µM final intra-vitreal concentration) or at P2 with LV containing F2RL1-GFP, F2RLl1 mutants (fused to GFP), or scrambled shRNA or shRNA targeting F2rl1. Eyes were fixed
in 4% paraformaldehyde for 1 h at room temperature. Retinas were dissected, with careful removal of the hyaloid vessels, and stained overnight at room temperature with fluoresceinated isolectin B4 (I21413, Molecular Probes) in 1 mM CaCl2 in PBS. Lectin-stained retinas were whole mounted onto Superfrost/Plus slides (Fisher Scientific) with the photoreceptor side down and embedded in SlowFade Antifade reagent (Invitrogen). For quantification of retinal vascular area and density, 20 images of each whole-mounted retina were obtained at 10× magnification on a Zeiss AxioObserver.Z1. Images were merged into a single file using the MosiaX option in the AxioVision 4.6.5 software (Zeiss). Retinal vascular area and density (quantified in two to four regions per retina) were measured at P6 (refs. 11,66).
O2-induced retinopathy. This model serves as a proxy for human ocular neovascular diseases, such as retinopathy of prematurity and diabetic retinopathy, that are characterized by a late phase of destructive pathological angiogenesis67,68. WT C57BL/6 mice were exposed to 75% oxygen from P7 to P12. After return to room air, hypoxia-driven neovascularization develops from P14 onwards69,70. Intravitreal injections were performed at P12, and the retinas were collected at P17. Retinas were dissected, fixed for 1 h in paraformaldehyde and stained overnight with fluoresceinated isolectin B4. Vaso-obliterated areas were assessed as the retinal area devoid of vasculature over the total retinal area. Neovascularization was analyzed using the SWIFT_NV method71. All mice weighing less than 5.5 g at P17 were excluded from the study, eliminating runty animals72.
LCM. Eyes were immediately embedded in optimal cutting temperature com-pound (OCT), frozen in liquid nitrogen and cross-sectioned (16 µm) onto a MembraneSlide 1.0 PEN NF (Zeiss). Sections were stained for isolectin (1:50 in 1 mM CaCl2) and dehydrated with 70%, 90% and 100% ethanol washes. Retinal vessels and layers were dissected using LCM with a Zeiss (Observer.Z1) Palm Microbeam LCM microscope system and collected directly into RNA-stabilizing buffer from the RNeasy Micro kit (Qiagen, Chatsworth, CA). RNA was extracted from microdissected tissues using the RNeasy kit (Qiagen), and real-time PCR was performed with the generated cDNA.
Microvascular sprouting from aortic explants. Microvascular sprouting experiments using aortic explants from WT and F2rl1−/− mice were prepared as described previously1. After infection with LV shScram or shF2rl1, RGCs were stimulated with vehicle or SLIGRL (24 h), and their cell medium (RGC condi-tioned medium) was centrifuged and filtered (0.22 µm) to remove cell debris. Aortic rings obtained from WT C57BL/6 or F2rl1−/− aortas were sectioned into 1-mm rings and embedded in Matrigel for 48 h. RGC-conditioned medium or SLIGRL was incubated with the growth factor–reduced Matrigel-embedded aortic rings with nascent vascular sprouts and photographed daily. Vascular sprouting area was assessed using ImageJ.
Snx11-specific antibody production. Polyclonal antibodies to Snx11 were custom generated by Open Biosystems using the epitope CGWAQEERQSTSHLAKGDQ. After immunization, rabbit serum was purified by affinity chromatography.
Immunocytochemistry. Immunocytochemical experiments were performed as described previously11 with antibodies to F2rl1 (Invitrogen, 35-2300, 1:500) and its rodent cleavage-activation site sequence 30-GPNSKGR↓SLIGRLDTP-46 (B5 antibody, from M.H., 1:500)73, GFP (Molecular Probes, A1122, 1:500), importin-β1 (Santa Cruz, C-19, 1:200), NeuN (Millipore, MAB377, 1:100), Thy1.1 (Millipore, MAB1406, 1:200), β-III tubulin (ECM Biosciences, TP1691, 1:500), and Vegfa (Santa Cruz Biotechnology, sc-152, 1:400) and with TRITC-conjugated lectin endothelial marker (Griffonia simplicifolia, Sigma-Aldrich L5364, 1:200) followed by Alexa Fluor–conjugated secondary antibodies (488, 596, 647, Invitrogen; 1:1,000). Negative controls were exposed to secondary anti-bodies. Sections were assessed by epifluorescence and confocal microscopy.
Immunofluorescence and confocal microscopy. F2RL1-GFP–expressing HEK293 cells were maintained at 37 °C, 0.5% CO2 for live imaging with a white laser Leica SP5X multispectral-multiphoton confocal system. Nuclei of live cells were stained with Hoechst (2 µg ml−1) shortly before stimulation with SLIGKV

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
nature medicinedoi:10.1038/nm.3669
(20 µM). Other cells were fixed, permeabilized in the presence of taxol (5 µM) and blocked with the appropriate serum before incubation with antibodies; their nuclei were stained with DAPI. Confocal images of fixed cells were obtained using a Zeiss LSM 510 microscope.
Electron microscopy. Specimens for electron microscopy were prepared as described previously39,74. A pre-embedding immunogold protocol was applied on the retinas of WT C57BL/6 or F2rl1−/− mice fixed by vascular perfusion with a mixture of 4% paraformaldehyde, 15% picric acid (v/v) and 0.1% glutaraldehyde in 0.1 M PB. For ultrastructural immunocytochemistry, tissues and cells were incubated with mouse monoclonal antibodies to F2rl1 (Invitrogen, 35-2300, 1:600) and subsequently with a goat anti-mouse IgG Fab fragment conjugated to 1.4-nm gold particles (1:200, Nanoprobes, 2002); tissue was then processed for silver enhancement using a Nanoprobes HQ Silver #2012 kit. Negative con-trols were performed on tissue sections from F2rl1−/− retinas using conditions identical to those used for WT mice and by omitting the primary antibodies for experiments using cells. Specimens were examined with a transmission electron microscope (Philips CM120) equipped with a Gatan digital camera.
Scanning electron microscopy. The purity and integrity of HEK293 nuclear fractions were confirmed by high-resolution scanning electron microscopy, as reported previously39.
RT-PCR. RT-PCR was performed on HEK293 cells, RGCs and dissected reti-nas. mRNA was extracted with TRIzol (Invitrogen), and synthesized cDNA was amplified using Taq DNA polymerase (Invitrogen, Molecular Probes). Primers were designed (Alpha DNA) for the sequences described in Supplementary Table 2. Results were normalized against cyclophilin A or 18S mRNA levels (QuantumRNA 18S universal Standard, Ambion).
Isolation of subcellular fractions and purified nuclei. Subcellular fractions and nuclei were isolated from RGCs and F2RL1-GFP– or F2RL1-GFP mutant–expressing HEK293 cells, stimulated with vehicle or agonist peptide (20 µM, 30 min) and treated with cycloheximide (50 mM) to arrest protein synthesis 2 h before stimulation. Confluent RGCs and stably transfected HEK293 cells (two 15-cm plates) were rinsed with PBS and harvested in 10 ml ice-cold homog-enizing buffer (HB; 0.25 M sucrose, 25 mM KCl, 5 mM MgCl2 and 20 mM Tris-HCl, pH 7.4). Pelleted cells were resuspended in HB (0.8 ml, 4 °C ) contain-ing protease inhibitor cocktail (Complete, EDTA-free, Roche), homogenized with a 26.5-gauge needle (15 strokes) and centrifuged (700g, 10 min, 4 °C). The pellet contained the nuclear fraction, and the supernatant was used to isolate other subcellular fractions.
To isolate subcellular fractions, a discontinuous OptiPrep density gradient (Sigma-Aldrich, D1556) was prepared containing 2 ml of 2.5%, 3.5 ml of 10%, 3.5 ml of 20% and 3.5 ml of 30% OptiPrep solution. Non-nuclear lysate (0.9 ml) was mixed with 50% OptiPrep (2.1 ml), yielding a 35% OptiPrep solution loaded on the OptiPrep gradient. After ultracentrifugation (130,000g, 4 °C, 22 h), the OptiPrep gradient was separated into 13 fractions of decreasing density.
Similarly, the nuclear fraction (pellet) was resuspended in HB (1 ml, con-taining protease inhibitor cocktail) with OptiPrep (1 ml, 50%) to obtain a 25% OptiPrep solution that was loaded on a new Opti-gradient tube (8 ml of 30% and 5.5 ml of 35% OptiPrep solution). After centrifugation (20,000g, 4 °C, 1 h), the OptiPrep gradient was separated into 13 fractions of decreasing density.
Corresponding fractions were then separated by SDS-PAGE and identified by immunoblot analysis of known plasma membrane (clathrin), early (EEA1) and late endosomes (Rab9A) or nuclear (lamin A/C) markers. Respective fractions were pooled to characterize the subcellular expression of F2rl1 after stimulation.
Flow cytometry. HEK293 cells stably transfected with F2RL1, Flag-F2RL1 and F2RL1 mutants (fused to GFP) were treated with SLIGKV (20 µM) for 30 min before nuclear isolation. HEK293 cells transfected with Flag-F2RL1were first incubated with FITC-tagged antibody to Flag (1:200, Santa Cruz Biotechnology, sc-7787) for 15 min in DMEM (37 °C) before stimulation. The nuclear prepara-tions were processed by flow cytometry using FACSCalibur, and the results were analyzed by CellQuest software (BD Biosciences).
Coimmunoprecipitation experiments and western blot analyses. HEK293 cells expressing GFP-tagged F2RL1, F2RL1-GFP mutants and eGFP-N1 (control; Clontech Laboratories) were lysed and immunoprecipitated (200 µg) using the magnetic GFP-Trap-M kit (Chromotek). Traditional immunoprecipitations were also performed using antibodies (2 µg) to F2rl1, Snx11, importin-β1, dynein (Abcam, ab6304), Sp1 (Cell Signaling, 5931) and IgG (R&D Systems, MAB0041, control) from soluble lysates of F2RL1-GFP– or SNX11-GFP–expressing HEK293 cells treated with SLIGKV (20 µM, 30 min). Immune complexes were immobilized by adding 50 µl of protein A (Sigma), washing three times with lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM eth-ylene glycol tetraacetic acid, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4 and 1 µM ml−1 leupeptin) and incubating with 30 µl of Laemmli SDS-sample buffer (Boston BioProducts, BP-110R). Western blot analyses for F2rl1, Snx1, Snx2, Snx11, importin-β1, α-tubulin, dynein and Sp1 were performed as described39. Total lysates represented 25% of the input (50 µg). Specific proteins were revealed with chemiluminescent reagent (PerkinElmer Life Sciences) and signal was exposed on X-ray films.
Measurement of Ca2+ signals. Cellular Ca2+ signals were measured using Fura-2 AM (Calbiochem) as described previously2. Cells were washed with prewarmed HBSS and calcium buffer (2 ml), incubated with Fura-2 AM (4 µl, 1 h at 37 °C) and resuspended in HBSS and calcium buffer. Cell calcium signals were measured by spectrofluorometry (LS50, PerkinElmer Life Sciences). The calcium concentrations were calculated according to the methods of Grynkiewicz et al.75.
ChIP and short-read sequencing. F2RL1-GFP–expressing HEK293 cells stimu-lated with SLIGKV (20 µM, 30 min) were fixed with formaldehyde (0.75%, 10 min) and quenched with glycine (125 mM) before nuclear isolation. Nuclei were sonicated using a S220x Covaris sonicator (3 min; Covaris), yielding frag-ment sizes of 300–350 bp. After centrifugation (14,000 r.p.m., 5 min), ChIP was performed on the supernatant that was then incubated overnight (4 °C) with F2rl1 antibodies (5 µg) together with protein A/G agarose beads (Sigma, P3296 and P9424) or beads alone; the remaining nuclear sample served as the input control. Isolated protein–DNA complexes were washed and digested with with 6 ng ml−1 RNaseA (Fermentas, EN0531) and 0.01µg ml−1 proteinase K (Fermentas, EO0491) (4 h, 65 °C). Precipitated ChIP DNA was isolated using a kit (GFX PCR DNA and Gel Band purification kit, Qiagen) and bioanalyzed with an Agilent DNA 1000 kit (Agilent Technologies). The ChIP DNA concentrations ranged from 20 to 40 ng µl−1.
Short-read sequencing of ChIP DNA fragments (150 ng) was processed using the Solexa/Illumina Genome Analyzer II platform (McGill University and Genome Quebec Innovation Canter). Short reads were mapped to a human reference genome (Feb. 2009, GRCh37/hg19) using the ELAND module of the Illumina Genome Analyzer IIx. Sequences that could not be mapped unambigu-ously or that mapped to simple or complex repeats were removed from the analy-sis. Results were converted to BED files (using the ChIP-Seq mini 2.0.1 suite76) for processing and visualization in the UCSC Genome Browser (https://genome.ucsc.edu/). MACS 1.4.2 rc2 (ref. 77) settings were used for peak detection, and the output was filtered to remove peaks overlapping repeat and low-complexity regions by more than 50%, as well as peaks distant from potential transcriptional start sites (−10 kb to +500 bp). We used stringent filtering of overlapping Sp1 sites with a motif matrix corresponding to a CCCC[GT]CCCC sequence for Sp1. Key regulators were identified in the functional screen with motif matrices provided by PATCH 1.0, which uses TRANFSAC 6.01 to search for transcription factor patterns78. The raw sequence data is publically available in GEO (acces-sion code GSE58660).
To explore whether F2rl1 and Sp1 form a transcriptional complex in stimu-lated RGC nuclei (SLIGRL, 20 µM, 30 min), F2rl1 ChIP was followed by re-ChIP with Sp1-specific antibodies as described79. Isolated protein–DNA complexes eluted from F2rl1-ChIP were incubated with Sp1 antibodies (2 µg) and A/G agarose beads in re-ChIP buffer (25 µl, 37 °C, 30 min; 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1, and 10 mM dithiothreitol). After washing the agarose beads three times, re-ChIP DNA was eluted (50 µl; 1% SDS and 0.1 M NaHCO3) and quantified (Nanodrop 1000, Thermo Scientific).

©20
14 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
nature medicine doi:10.1038/nm.3669
Primers that emcompass the peak discovered by ChIP-Seq were designed, and 1 ng of re-ChIP DNA was used to confirm enrichment of that peak by qRT-PCR.
EMSA. Crude nuclear protein preparations and EMSA were performed as described previously80. Briefly, EMSA was carried out by incubating 3 × 104 counts per minute of 5′ end–labeled (32P-γATP) double-stranded oligonucleo-tides bearing high-affinity binding sites for the transcription factor Sp1 (5′-GATCATATCTGCGGGGCGGGGCAGACACAG-3′) with SLIGKV-stimulated (20 µM, 30 min) F2RL1-GFP–expressing HEK293 nuclear lysates. Supershift experiments were conducted by adding Sp1- and F2RL1-antibodies (400 ng) to the reaction mixtures.
Luciferase assay. HEK293 F2RL1-GFP–expressing cells were transfected with empty PGL3 vector (Promega), with a PGL3 vector containing a DNA fragment obtained by ChIP (Supplementary Fig. 11a), or with pCDNA3 LacZ (Invitrogen), with or without transfection with siRNA (0.6 µg) targeting Sp1 or a scrambled sequence. Each plate was transfected with Lipofectamine (Invitrogen) with 1 µg of PGL3 and pCDNA3 LacZ plasmids. A dual-luciferase reporter assay system (Promega) was used to monitor luciferase activity using a Sirius single-tube luminometer (Berthold). The pCDNA3 LacZ vector was co-transfected to monitor the transfection efficiency. All luciferase results are reported as relative light units (RLUs), representing the average Photinus pyralis firefly luminosity divided by the average β-galactosidase activity.
Proliferation assay by [3H]thymidine incorporation. F2RL1-GFP–expressing HEK293 cells (4 × 104 cells per well) in 24-well plates were transfected with scrambled or Sp1-targeting siRNA and stimulated with SLIGKV (20 µM) or vehicle for 12 h. [3H]thymidine (1 µCi ml−1) was then incubated with the cells (18 h). Lysed cells were transferred into a scintillation flask and analyzed by a liquid scintillation counter. The data represent the c.p.m. of [3H]thymidine incorporation normalized to the untreated control.
Statistical analyses. Data are presented as the means ± s.e.m. Two-tailed independent Student’s t tests were used when comparing two groups, using Welch’s correction where appropriate to account for significantly different variances. Comparisons between multiple groups were made using one-way analysis of variance followed by post-hoc Dunnett’s or Tukey’s multiple comparisons test among means. Gender did not affect our retinal phenotype and was therefore not considered. Values more than two standard deviations from the mean were considered outliers and were excluded. P < 0.05 was considered as statistically significant.
61. Frassetto, L.J. et al. Kinase-dependent differentiation of a retinal ganglion cell precursor. Invest. Ophthalmol. Vis. Sci. 47, 427–438 (2006).
62. Déry, O., Thoma, M.S., Wong, H., Grady, E.F. & Bunnett, N.W. Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein. β-arrestin–dependent endocytosis of a proteinase receptor. J. Biol. Chem. 274, 18524–18535 (1999).
63. Roosterman, D., Schmidlin, F. & Bunnett, N.W. Rab5a and rab11a mediate agonist-induced trafficking of protease-activated receptor 2. Am. J. Physiol. Cell Physiol. 284, C1319–C1329 (2003).
64. Kunkel, T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. U S A 82, 488–492 (1985).
65. Dull, T. et al. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 (1998).
66. Sirinyan, M. et al. Hyperoxic exposure leads to nitrative stress and ensuing microvascular degeneration and diminished brain mass and function in the immature subject. Stroke 37, 2807–2815 (2006).
67. Stahl, A. et al. The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 51, 2813–2826 (2010).
68. Sapieha, P. et al. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 120, 3022–3032 (2010).
69. Connor, K.M. et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 (2009).
70. Smith, L.E. et al. Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 (1994).
71. Stahl, A. et al. Computer-aided quantification of retinal neovascularization. Angiogenesis 12, 297–301 (2009).
72. Stahl, A. et al. Postnatal weight gain modifies severity and functional outcome of oxygen-induced proliferative retinopathy. Am. J. Pathol. 177, 2715–2723 (2010).
73. Kong, W. et al. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc. Natl. Acad. Sci. USA 94, 8884–8889 (1997).
74. McLeod, A.L., Krause, J.E., Cuello, A.C. & Ribeiro-da-Silva, A. Preferential synaptic relationships between substance P-immunoreactive boutons and neurokinin 1 receptor sites in the rat spinal cord. Proc. Natl. Acad. Sci. USA 95, 15775–15780 (1998).
75. Grynkiewicz, G., Poenie, M. & Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 (1985).
76. Johnson, D.S., Mortazavi, A., Myers, R.M. & Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 (2007).
77. Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
78. Matys, V. et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 31, 374–378 (2003).
79. Truax, A.D. & Greer, S.F. ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol. Biol. 809, 175–188 (2012).
80. Zaniolo, K., Desnoyers, S., Leclerc, S. & Guérin, S.L. Regulation of poly(ADP-ribose) polymerase-1 (PARP-1) gene expression through the post-translational modification of Sp1: a nuclear target protein of PARP-1. BMC Mol. Biol. 8, 96 (2007).




















