
Stratified societies - Medieval world - School of Distance ...
Upload
independentCategory
view
0download
0
METHODOLOGY ARTICLE Open Access
Study of large and highly stratified populationdatasets by combining iterative pruning principalcomponent analysis and structureTulaya Limpiti1, Apichart Intarapanich2, Anunchai Assawamakin3, Philip J Shaw3, Pongsakorn Wangkumhang3,Jittima Piriyapongsa3, Chumpol Ngamphiw3 and Sissades Tongsima3*
Abstract
Background: The ever increasing sizes of population genetic datasets pose great challenges for population structureanalysis. The Tracy-Widom (TW) statistical test is widely used for detecting structure. However, it has not beenadequately investigated whether the TW statistic is susceptible to type I error, especially in large, complex datasets.Non-parametric, Principal Component Analysis (PCA) based methods for resolving structure have been developedwhich rely on the TW test. Although PCA-based methods can resolve structure, they cannot infer ancestry. Model-basedmethods are still needed for ancestry analysis, but they are not suitable for large datasets. We propose a new structureanalysis framework for large datasets. This includes a new heuristic for detecting structure and incorporation of thestructure patterns inferred by a PCA method to complement STRUCTURE analysis.
Results: A new heuristic called EigenDev for detecting population structure is presented. When tested onsimulated data, this heuristic is robust to sample size. In contrast, the TW statistic was found to be susceptible totype I error, especially for large population samples. EigenDev is thus better-suited for analysis of large datasetscontaining many individuals, in which spurious patterns are likely to exist and could be incorrectly interpreted aspopulation stratification. EigenDev was applied to the iterative pruning PCA (ipPCA) method, which resolves theunderlying subpopulations. This subpopulation information was used to supervise STRUCTURE analysis to inferpatterns of ancestry at an unprecedented level of resolution. To validate the new approach, a bovine and a largehuman genetic dataset (3945 individuals) were analyzed. We found new ancestry patterns consistent with thesubpopulations resolved by ipPCA.
Conclusions: The EigenDev heuristic is robust to sampling and is thus superior for detecting structure in largedatasets. The application of EigenDev to the ipPCA algorithm improves the estimation of the number ofsubpopulations and the individual assignment accuracy, especially for very large and complex datasets.Furthermore, we have demonstrated that the structure resolved by this approach complements parametric analysis,allowing a much more comprehensive account of population structure. The new version of the ipPCA softwarewith EigenDev incorporated can be downloaded from http://www4a.biotec.or.th/GI/tools/ippca.
BackgroundAs genotyping platforms incorporate more markers, andthe costs for genotyping keep falling, ever larger andmore complex datasets are being analyzed. The compu-tationally efficient non-parametric methods for analysisof genotypic datasets are thus increasingly being used to
reveal population structure. Resolution of populationstructure reveals evolutionary relationships betweengroups of individuals. Furthermore, population structuremust be accounted for in genome-wide association stu-dies to reduce spurious associations resulting fromancestral differences between cases and controls [1].Principal component analysis (PCA) is a widely used
non-parametric method for population structure analysis,which uses a covariance matrix for eigenanalysis. Theamount and axes of variation among individuals are
* Correspondence: [email protected] Center for Genetic Engineering and Biotechnology, ThailandScience Park, Pathumthani 12120, ThailandFull list of author information is available at the end of the article
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
© 2011 Limpiti et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative CommonsAttribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction inany medium, provided the original work is properly cited.

captured in the eigenvalues and eigenvectors, respectively.Previously, we developed a PCA framework for populationstructure analysis which extended the use of PCA beyondits usual application for visualizing the population struc-ture trend by employing an iterative process to simplifythe pattern of population structure. The iterative methodsused by others, e.g. [2,3] rely on the available ethno-geo-graphical population labels for subjectively grouping indi-viduals, unlike our objective approach.Our framework, which we dubbed iterative pruning
PCA (ipPCA) uses a clustering algorithm to assign indi-viduals into subpopulations without imposing any priorassumptions [4]. ipPCA resolves all subpopulations in apopulation dataset, and thus reports the total number ofprimal subpopulations K in addition to assigning indivi-duals contained within them. The term “population” issynonymous with dataset for ipPCA, which is the entirecollection of individuals available for analysis. The term
“subpopulation” defines a group of individuals assignedby ipPCA in which no further significant substructure ispresent. ipPCA operates by systematically separatingindividuals into two clusters using a clustering algorithmbased on the Euclidean distances between projected datapoints and the cluster centroids. The decision to sepa-rate individuals requires testing of whether significantstructure is present within the dataset (or nested datasetfor subsequent iterations of the algorithm). To test forhomogeneity among groups of individuals, we previouslyproposed using the test statistic as implemented in theEIGENSTRAT/SmartPCA algorithm, which reports theprobability of structure according to Tracy-Widom(TW) distribution [5]. If no significant structure exists,then the individuals under testing belong to a subpopu-lation, thus terminating the iterative clustering process.The ipPCA framework is summarized in Figure 1. Usingdatasets of simulated and real data, we showed how
encode genotypic data elements for PCA
project individuals onto PC space calculated from the
singular vectors of each data matrix
determine structure metric
Encode Data
crossesthreshold
does NOT cross threshold
separate individuals into two clusters Clustering
form two new data matrices according to individual assignments
Check Terminating Criterion
Terminate algorithm
for the data matrix
considered
Figure 1 Outline of the ipPCA framework. The framework consists of three main components. First, the genetic data are encoded, zero-means centered and normalized. Then, individuals are projected onto a space spanned by the principal components of the input data matrix.Next, a structure metric is calculated to decide whether to advance to the clustering step or to terminate the algorithm. When the metric doesnot cross the threshold, a homogenous subpopulation is resolved and subsequently the algorithm terminates. Otherwise, the individuals arebisected. The algorithm iterates until all individuals have been assigned into terminal subpopulations.
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 2 of 11

ipPCA can correctly assign individuals to subpopulationsand infer K. However, the accuracy of ipPCA may beaffected by the stopping criterion. An inappropriate ter-mination criterion leads to under- or over-estimation ofthe number of subpopulations. Moreover, individualassignment errors in early iterations will be com-pounded and carried forward to later iterations.Parametric algorithms for clustering individuals into
subpopulations, e.g., STRUCTURE, frappe, ADMIX-TURE, and BAPS, differ from ipPCA in one crucialaspect, namely the method of assigning individuals intosubpopulation clusters. The aforementioned parametricalgorithms infer ancestral proportions for each indivi-dual separately, and group individuals with similar pat-terns of inferred ancestry. ipPCA and other non-parametric approaches cannot infer ancestry. Thesetechniques attempt to group individuals with similargenetic profiles together. Hence, parametric approachesstill offer important information not seen by non-para-metric analyses. Large and highly structured populationdatasets are however intractable for parametric analysisbecause the number of K ancestral clusters is limited.This is due to the limited number of available samplesused to estimate subpopulation allele frequencies. Inorder to better observe the inherent population struc-ture, a “supervised” structure analysis, with re-sampledindividuals, should be performed. The choice of indivi-duals for such supervised analysis is arbitrary and typi-cally guided by available ethno/geographical labels.Nonetheless, careful selection is needed to ensure thatindividuals being compared have similar ancestries,otherwise the signals of ancestries important for differ-entiating some groups of individuals may be too weak.In this paper, we propose a modification to ipPCA by
introducing a new stopping criterion called EigenDevfor the iterative clustering process which is more robustto spurious patterns in large datasets. The new algo-rithm is termed EigenDev-ipPCA. To distinguishbetween the two algorithms in the ipPCA framework,we refer to the previously proposed algorithm whichuses the TW statistic as the termination criterion asTW-ipPCA in the subsequent sections. Furthermore, wesuggest a new protocol which uses the information fromEigenDev-ipPCA to guide parametric analysis. Usingreal datasets, we demonstrate how this approach canreveal new and structure-informative patterns of ances-try not detectable with unsupervised STRUCTUREanalysis.
MethodsNew ipPCA terminating criterionThe Tracy-Widom (TW) test statistic, which is imple-mented in the EIGENSTRAT/SmartPCA algorithm [5],is used as a stopping criterion for the TW-ipPCA
algorithm. Although this stopping criterion has beenfound to work well for some datasets, we found thatwhen much larger datasets containing roughly >1000individuals were analyzed, the TW-ipPCA resolved farmore subpopulations than were expected. We thereforesuspected that in some cases when sampling is large,the subpopulations resolved may be spurious, i.e., type Ierror. Indeed, as pointed out in [5], the relative samplesizes of the underlying subpopulations affect the TWtest statistic.Besides the type I error we found when using the TW
statistical test for structure, there are other drawbackswhich motivated us to develop an alternative terminat-ing criterion. The first issue is computational difficulty.To obtain the final value of the TW test statistic, toomany unknown parameters need to be estimated. Nobest estimators for these parameters are available, sochoices of estimators affect the result. Instead of usingthe p-values of TW test statistics as thresholds, we pro-pose a new terminating criterion for determiningwhether the data are structured. The new criterion isbased on the eigenvalues of the data matrix and istermed the EigenDev heuristic. The EigenDev heuristicfollows the same assumption as the TW theory, namely,if the first eigenvalue of the data matrix is significantlylarger than the remaining eigenvalues, then substructureexists. However, we extend this observation beyondmerely testing the significance of the first eigenvalue totake into account the remaining variance of the data.This allows us to observe structure in higher dimen-sions. We were inspired to develop EigenDev from theEigenvalue Grads heuristic, which is applied in the sig-nal processing domain [6]. This work showed that if thedata contain only noise and no signal, i.e., non-struc-tured, then there is an excellent linear fit for the eigen-values ranked in descending order. In populationgenetic data, the noise represents the natural geneticvariation within a (sub)population.To test for population structure, the EigenDev statistic
is calculated from the genotypic data. This calculationfirst requires that a data matrix is constructed fromencoded, zero-means and normalized genotypic data, asdescribed in [5]. This matrix contains rows correspond-ing to individuals and columns corresponding to alleles.Thus, biallelic SNP markers are encoded by entries intwo columns, one for each allele, and STRs by the totalnumber of alleles for that marker locus in the dataset.The presence of an allele is encoded as 1 and itsabsence as 0. For missing data, i.e., markers with nogenotypic call, they are encoded as all 0’s.Given the zero-means, normalized genotype data
matrix X (according to [5]) containing m samples withn allele columns per sample, we construct the samplecovariance matrix
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 3 of 11

C =1m
XXT .
The EigenDev value can then be computed from
EigenDev =
√√√√ 1p
p∑i=1
(log(σ̂ 2i ) − log(σ 2
i )) (1)
where
log(σ̂ 2i ) = log(σ 2
1 ) + (i − 1)c (2)
and
c =(log(σ 2
p ) − log(σ 21 )
(p − 1)(3)
where σ 2i , i = 1, ..., p, are the first p eigenvalues of C
ranked in descending order. The quantity in Eq. (1)could be negative in some cases. To militate against thispossibility, the encoded entries are normalized to havezero mean. This step is important to remove the signalfrom the common elements, leaving only the differences(genetic variance) between individuals for eigenanalysis.In all empirical studies on both simulated and real data,we found that 90% of the variance in the data alwaysresults in a positive value and the convexity constraintin question has never been violated. To account for therare cases when negative values are encountered, wehave included a checking step in the algorithm to detectand report negative values. If negative values are found,the parameter p can be adjusted to ensure a positivequantity in the square root. Recall that p < min{m, n} isthe number of eigenvalues used to compute the Eigen-Dev statistic. We also stabilize the variance using logtransformation. If the EigenDev value is large, the groupof individuals being analyzed would comprise more thanone subpopulation and ipPCA progresses to bisect thegroup; otherwise, the EigenDev-ipPCA algorithm termi-nates when the EigenDev value falls below a threshold.
ResultsTestingTo test the EigenDev concept, several datasets were ana-lyzed:
1. A simulated dataset composed of 10,000 indivi-duals from the same population, each containing10,000 SNP markers was used for testing the fit ofTW distribution. It was generated using the GEN-OME tool [7] with the following parameters and thefollowing tree file:
-pop 1 10000 -N tree.txt -C 20 -S 500tree.txt:0 10000
1-11 10000Starting at 10,000 founder population individuals,GENOME generates the first generation with thesame size as the founder. Each individual has 20chromosomes and each chromosome contains500 SNPs.
2. The second dataset was simulated using the sameGENOME parameters as the first dataset but withdifferent tree file:
tree.txt:0 5000 50001-1 2-140 50001-1 1-280 5000 50001-1 2-1100 10000to generate two subpopulations of size 5,000individuals each.
3. The third dataset is the Bovine HapMap Projectcollection of 497 individuals obtained from 19 differ-ent breeds, genotyped for 27203 SNPs. It is publiclyavailable from [8].4. The fourth dataset is publicly available from [9]. Itcontains 3945 individuals comprising 185 differentethno/geographical labels, typed for 1327 markers(consisting of 848 microsatellites, 476 indels, and 3SNPs) from [10].
The ipPCA encoded input matrices from the simu-lated and real complex datasets are also available fordownload from http://www4a.biotec.or.th/GI/tools/ippca.Testing metrics for population structureTo test how TW is affected by sampling, a simulateddataset with no substructure was sampled randomly at20 different sample sizes from 10 to 200 individuals.The corresponding probability-probability (p-p) plotsfor testing the fit of the TW distribution are shown inFigure 2. It is observed that the TW distribution is vio-lated for most of the sample sizes; good fit is observedonly for the sample of 70 individuals. Therefore, thedeviation from TW distribution will give a false detec-tion (type I error), particularly for large sample sizes.On the other hand, the TW test is very sensitive fordetecting structure, since it is based on a non-linearphase change. It is not susceptible to type II error pro-vided sufficient data are available [5]. However, thenon-linearity of the phase change means that an all-or-nothing situation exists where the likelihood of type Icannot be controlled, even across a wide range of p-value thresholds.
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 4 of 11
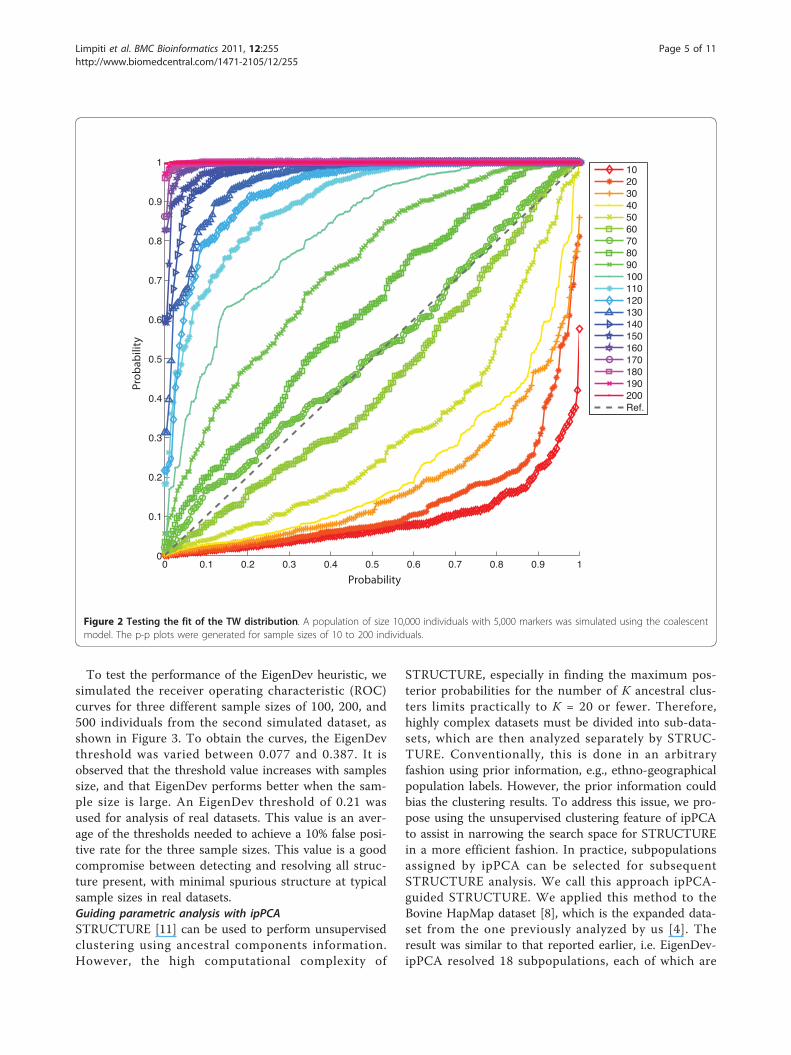
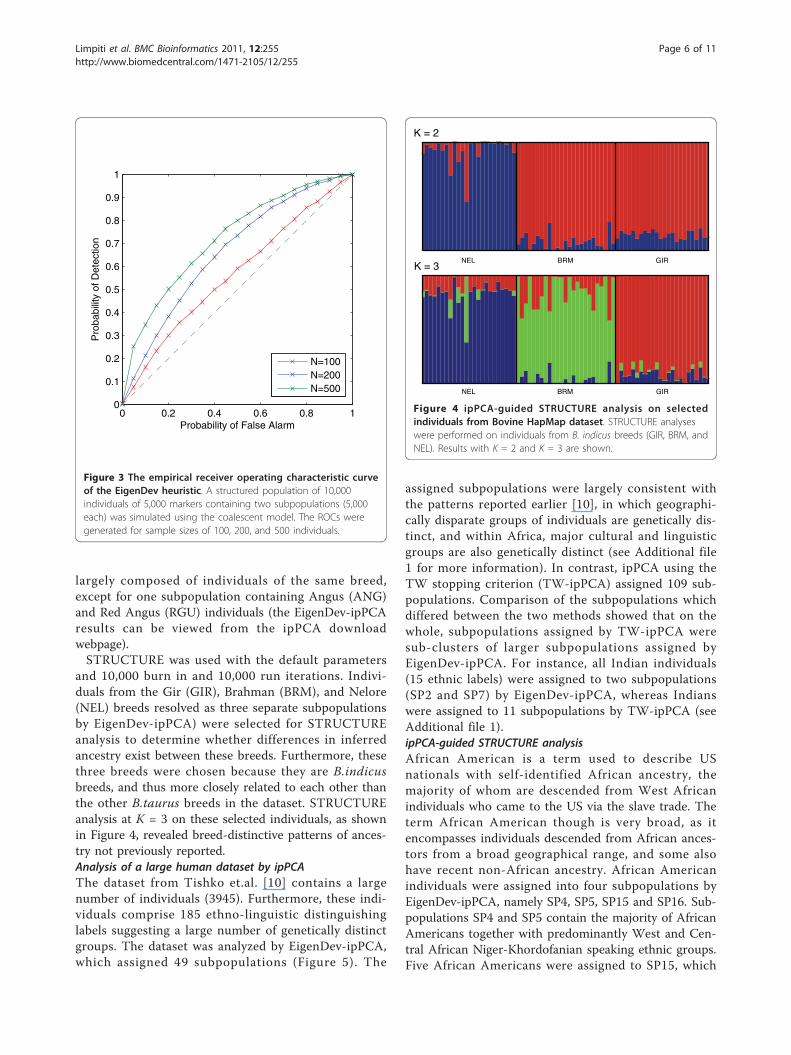
To test the performance of the EigenDev heuristic, wesimulated the receiver operating characteristic (ROC)curves for three different sample sizes of 100, 200, and500 individuals from the second simulated dataset, asshown in Figure 3. To obtain the curves, the EigenDevthreshold was varied between 0.077 and 0.387. It isobserved that the threshold value increases with samplessize, and that EigenDev performs better when the sam-ple size is large. An EigenDev threshold of 0.21 wasused for analysis of real datasets. This value is an aver-age of the thresholds needed to achieve a 10% false posi-tive rate for the three sample sizes. This value is a goodcompromise between detecting and resolving all struc-ture present, with minimal spurious structure at typicalsample sizes in real datasets.Guiding parametric analysis with ipPCASTRUCTURE [11] can be used to perform unsupervisedclustering using ancestral components information.However, the high computational complexity of
STRUCTURE, especially in finding the maximum pos-terior probabilities for the number of K ancestral clus-ters limits practically to K = 20 or fewer. Therefore,highly complex datasets must be divided into sub-data-sets, which are then analyzed separately by STRUC-TURE. Conventionally, this is done in an arbitraryfashion using prior information, e.g., ethno-geographicalpopulation labels. However, the prior information couldbias the clustering results. To address this issue, we pro-pose using the unsupervised clustering feature of ipPCAto assist in narrowing the search space for STRUCTUREin a more efficient fashion. In practice, subpopulationsassigned by ipPCA can be selected for subsequentSTRUCTURE analysis. We call this approach ipPCA-guided STRUCTURE. We applied this method to theBovine HapMap dataset [8], which is the expanded data-set from the one previously analyzed by us [4]. Theresult was similar to that reported earlier, i.e. EigenDev-ipPCA resolved 18 subpopulations, each of which are
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 10
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
102030405060708090100110120130140150160170180190200Ref.
Probability
Prob
ability
Figure 2 Testing the fit of the TW distribution. A population of size 10,000 individuals with 5,000 markers was simulated using the coalescentmodel. The p-p plots were generated for sample sizes of 10 to 200 individuals.
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 5 of 11
largely composed of individuals of the same breed,except for one subpopulation containing Angus (ANG)and Red Angus (RGU) individuals (the EigenDev-ipPCAresults can be viewed from the ipPCA downloadwebpage).STRUCTURE was used with the default parameters
and 10,000 burn in and 10,000 run iterations. Indivi-duals from the Gir (GIR), Brahman (BRM), and Nelore(NEL) breeds resolved as three separate subpopulationsby EigenDev-ipPCA) were selected for STRUCTUREanalysis to determine whether differences in inferredancestry exist between these breeds. Furthermore, thesethree breeds were chosen because they are B.indicusbreeds, and thus more closely related to each other thanthe other B.taurus breeds in the dataset. STRUCTUREanalysis at K = 3 on these selected individuals, as shownin Figure 4, revealed breed-distinctive patterns of ances-try not previously reported.Analysis of a large human dataset by ipPCAThe dataset from Tishko et.al. [10] contains a largenumber of individuals (3945). Furthermore, these indi-viduals comprise 185 ethno-linguistic distinguishinglabels suggesting a large number of genetically distinctgroups. The dataset was analyzed by EigenDev-ipPCA,which assigned 49 subpopulations (Figure 5). The
assigned subpopulations were largely consistent withthe patterns reported earlier [10], in which geographi-cally disparate groups of individuals are genetically dis-tinct, and within Africa, major cultural and linguisticgroups are also genetically distinct (see Additional file1 for more information). In contrast, ipPCA using theTW stopping criterion (TW-ipPCA) assigned 109 sub-populations. Comparison of the subpopulations whichdiffered between the two methods showed that on thewhole, subpopulations assigned by TW-ipPCA weresub-clusters of larger subpopulations assigned byEigenDev-ipPCA. For instance, all Indian individuals(15 ethnic labels) were assigned to two subpopulations(SP2 and SP7) by EigenDev-ipPCA, whereas Indianswere assigned to 11 subpopulations by TW-ipPCA (seeAdditional file 1).ipPCA-guided STRUCTURE analysisAfrican American is a term used to describe USnationals with self-identified African ancestry, themajority of whom are descended from West Africanindividuals who came to the US via the slave trade. Theterm African American though is very broad, as itencompasses individuals descended from African ances-tors from a broad geographical range, and some alsohave recent non-African ancestry. African Americanindividuals were assigned into four subpopulations byEigenDev-ipPCA, namely SP4, SP5, SP15 and SP16. Sub-populations SP4 and SP5 contain the majority of AfricanAmericans together with predominantly West and Cen-tral African Niger-Khordofanian speaking ethnic groups.Five African Americans were assigned to SP15, which
0 0.2 0.4 0.6 0.8 10
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
Probability of False Alarm
Pro
babi
lity
of D
etec
tion
N=100N=200N=500
Figure 3 The empirical receiver operating characteristic curveof the EigenDev heuristic. A structured population of 10,000individuals of 5,000 markers containing two subpopulations (5,000each) was simulated using the coalescent model. The ROCs weregenerated for sample sizes of 100, 200, and 500 individuals.
K = 2
K = 3NEL BRM GIR
NEL BRM GIR
Figure 4 ipPCA-guided STRUCTURE analysis on selectedindividuals from Bovine HapMap dataset. STRUCTURE analyseswere performed on individuals from B. indicus breeds (GIR, BRM, andNEL). Results with K = 2 and K = 3 are shown.
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 6 of 11

SP1
Tugen(1/22)
Borana(3/32)
Hausa (Cameroon)(1/27)
Nuer(18/18)
Shilluk(15/15)
Nyimang(10/17)
Temani(1/21)
SP2
Assamese(25/25)
Bengali(25/27)
Gujarat(49/50)
Hindi(24/28)
Kannada(23/24)
Kashmiri(10/25)
Konkani(40/42)
Malayalam(25/25)
Marathi(25/26)
Marwari(25/25)
Oriya(25/26)
Parsi(1/25)
Punjabi(12/28)
Tamil(26/29)
Telugu(27/27)
SP3
Bedouin(1/46)
Mandinka(22/22)
Yoruba (CEPH)(22/22)
Bantu South(8/8)
Mozabite(2/29)
Bantu (Kenya)(11/11)
SP4
Bassange(20/20)
Yoruba(19/25)
Igbo(23/28)
Igala(17/17)
Gwari(16/22)
Hausa (Nigeria)(16/16)
Fulani (Nigeria)(3/4)
Beja Banuamir(1/23)
Dinka(1/17)
Hadza(2/63)
Sukuma(9/10)
Mbugwe(1/21)
Gogo(4/13)
Podokwo(1/30)
Banen(2/25)
Kotoko(5/17)
Baggara(1/23)
Mandara(4/26)
Giziga(6/24)
Ouldeme(8/26)
Kanuri(3/31)
Bedzan(3/17)
Bakola(5/42)
Iyassa(30/37)
Fang(13/19)
Mabea(12/13)
Yambassa(8/17)
South Tikar(21/21)
North Tikar(7/13)
Ntumu(11/11)
Massa(11/15)
Tupuri(4/22)
Bulu(22/22)
Ashanti(9/15)
Brong(25/26)
Chicago(8/15)
Pittsburgh(18/21)
Baltimore(34/44)
North Carolina(11/18)
Samba’a(6/18)
Yoruba(6/25)
Igbo(5/28)
Gwari(6/22)
Hadza(1/63)
Gogo(1/13)
Ouldeme(1/26)
Kanuri(5/31)
Mvae(1/24)
Baka(2/48)
Bedzan(1/17)
Iyassa(6/37)
Fang(6/19)
Mabea(1/13)
Yambassa(9/17)
North Tikar(6/13)
Tupuri(6/22)
Ashanti(6/15)
Brong(1/26)
Chicago(6/15)
Pittsburgh(1/21)
Baltimore(8/44)
North Carolina(5/18)
SP5
SP6Australian(10/10)
SP7
Bengali(2/27)
Gujarat(1/50)
Hindi(4/28)
Kannada(1/24)
Kashmiri(15/25)
Konkani(2/42)
Marathi(1/26)
Oriya(1/26)
Parsi(24/25)
Punjabi(16/28)
Tamil(3/29)
SP8
Hadza(41/63)
SP9Surui(8/8)
SP10
Kalash(23/23)
SP11
Brahui(25/25)
Balochi(24/24)
Hazara(12/22)
Makrani(25/25)
Sindhi(24/24)
Pathan(24/24)
Burusho(25/25)
Uygur(5/10)
Adygei(2/17)
SP12San(6/6)
SP13
Fiome/Gorowa(19/22)
Gogo(6/13)
Datoga(53/54)
Samba’a(12/18)
Dorobo(10/10)
SP14Hadza(10/63)
SP15
Beja Banuamir(22/23)
Beja Hadandawa(18/19)
Iyassa(1/37)
Chicago(1/15)
Pittsburgh(1/21)
Baltimore(1/44)
North Carolina(2/18)
SP16
Fulani (Nigeria)(1/4)
Dinka(16/17)
Hadza(2/63)
Akie(10/23)
Datoga(1/54)
Sukuma(1/10)
Turu(1/32)
Burunge(1/22)
Gogo(2/13)
Fiome/Gorowa(3/22)
Baggara(1/23)
Pittsburgh(1/21)
Baltimore(1/44)
Maasai Mumonyot(12/12)
Maasai Il’gwesi(21/21)
Samburu(15/18)
Yaaku(19/19)
SP17
Kikuyu(1/22)
Ngambaye(7/30)
Kaba(3/27)
Sara (various)(5/27)
Gbaya(6/15)
Baggara(1/23)
Tupuri(6/22)
Massa(1/15)
Wimbum(2/15)
Batie(3/16)
Baluba(1/6)
Mbum(2/13)
Ngambaye(2/30)
Tupuri(2/22)
Podokwo(3/30)
Fulani Adamawa(3/41)
Fulani Mbororo(3/13)
Wimbum(3/15)
Batie(2/16)
Hausa (Cameroon)(4/27)
Kongo(2/17)
Baluba(1/6)
Mbum(1/13)
Dioula(1/5)
Laka(2/33)
Kanuri(1/31)
Kaba(1/27)
SP18
Samburu(1/18)
Luhya(17/17)
Luo(24/28)
Kikuyu(11/22)
Nandi(1/11)
Sabaot(8/20)
Turkana(12/26)
Ilchamus(2/27)
Bulala(14/15)
Kanembou(5/5)
Tutsi/Hutu(6/8)
Kaba(1/27)
Yakoma(3/6)
Baggara(2/23)
Tupuri(1/22)
Massa(3/15)
Fulani Adamawa(6/41)
Fulani Mbororo(10/13)
Hausa (Cameroon)(2/27)
Sara (various)(1/27)
Podokwo(1/30)
Laka(1/33)
SP19
Samburu(1/18)
Tugen(21/22)
Burji(1/24)
Marakwet(14/14)
Kikuyu(7/22)
Sengwer(21/21)
Okiek(22/22)
Nandi(10/11)
El Molo(5/16)
Sabaot(11/20)
Turkana(13/26)
Pokot(23/23)
Ilchamus(19/27)
Bulala(1/15)SP20
Samburu(1/18)
Burji(23/24)
Wata(6/6)
El Molo(11/16)
Gabra(16/21)
Rendille(27/28)
Turkana(1/26)
Borana(29/32)
Ilchamus(5/27)
Konso(14/14)
Beta Israel(7/17)SP21
Rangi(1/36)
Fulani Adamawa(23/41)
Kanuri(18/31)
Mada(28/28)
Ouldeme(17/26)
Giziga(17/24)
Mandara(21/26)
Baggara(13/23)
Kotoko(12/17)
Zulgo(22/22)
Podokwo(16/30)
SP22
Hadza(2/63)
Turu(1/32)
Mbugwe(3/21)
Maasai (Tanzania)(1/36)
Pare(19/23)
Bedzan(2/17)
Mvae(23/24)
Ngumba(27/27)
Zime(30/30)
Bamoun(31/31)
Banen(23/25)
Bafia(30/30)
Lemande(26/26)
Batanga(20/20)
SP23
SP24
Baka(46/48)
SP25
Bakola(37/42)
Bedzan(11/27)
Hadza(5/63)
Sandawe(2/51)
Iraqw(2/46)
Turu(30/32)
Mbugwe(17/21)
Rangi(35/36)
Burunge(19/22)
Maasai (Tanzania)(33/36)
Akie(10/23)
Pare(3/23)
Mbugu(22/22)
Fulani Adamawa(5/41)
Baggara(5/23)SP26
SP27Karitiana(13/14)
Hazara(1/22)
Cambodian(10/10)
Japanese(29/29)
Han(44/44)
Tujia(10/10)
Yi(10/10)
Miao(10/10)
Daur(1/10)
Mongola(7/10)
Hezhen(3/9)
Xibo(6/9)
Dai(10/10)
Lahu(8/8)
She(10/10)
Naxi(9/9)
Tu(10/10)
SP28
SP29
Hazara(9/22)
Yakut(25/25)
Oroqen(9/9)
Daur(9/10)
Mongola(3/10)
Hezhen(6/9)
Xibo(3/9)
Uygur(5/10)
Melanesian(11/11)
SP30
Papuan(17/17)
SP31
Russian(22/25)
SP32
French(28/28)
Sardinian(28/28)
Orcadian(15/15)
Russian(3/25)
Italian(21/21)
Basque(24/24)
Adygei(15/17)
SP33
SP34Mbuti(13/13)
SP35
Biaka(23/23)
Luo(2/28)
Sabaot(1/20)
Laka(30/33)
Ngambaye(21/30)
Kaba(22/27)
Mbum(10/13)
Yakoma(3/6)
Sara (various)(21/27)
Gbaya(9/15)
Tupuri(3/22)
Podokwo(9/30)
Wimbum(9/15)
Batie(11/16)
Hausa (Cameroon)(20/27)
Kongo(15/17)
Barega(4/4)
Baluba(4/6)
Fulani Adamawa(4/41)
Dioula(4/5)
Kanuri(4/31)
Tutsi/Hutu(2/8)
Giziga(1/24)
Mandara(1/26)
Ewondo(3/3)
Eton(4/4)
SP36
Cape mixed ancestry(2/39)
Venda(3/13)
Xhosa(7/28)
Koma(5/12)
Luo(2/28)
SP37
Cape mixed ancestry(17/39)
Venda(6/13)
!Xun/Kxoe(6/8)
Xhosa(20/28)
Koma(3/12)
Nyimang(2/17)
Kikuyu(1/22)
SP38
Cape mixed ancestry(11/39)
Beta Israel(10/17)
Beja Hadandawa(1/19)
Temani(18/21)SP39
Cape mixed ancestry(1/39)
Dogon(9/9)SP40
Kikuyu(2/22)
Gabra(1/17)
Rendille(1/28)
Ilchamus(1/27)
Wimbum(1/15)
Cape mixed ancestry(8/39)
Venda(4/13)
!Xun/Kxoe(2/8)
Xhosa(1/28)
Koma(4/12)
Temani(2/21)
SP41
Sandawe(49/51)
SP42
Iraqw(44/46)
Burunge(2/22)
Maasai (Tanzania)(2/36)
Akie(3/23)
Pare(1/23)SP43
Columbian(7/7)
Maya(21/21)
Karitiana(1/14)SP44
SP45Pima(14/14)
SP46
Bedouin(21/46)
Druze(5/42)
Bedouin(24/46)
Palestinian(46/46)
SP47
SP48
Druze(37/42)
SP49
Mozabite(27/29)
Figure 5 Population assignments of the Tishkoff et al dataset using the EigenDev-ipPCA method. 49 assigned subpopulations are labeledSP1 to SP49. The height of the bars are proportional to the number of assigned individuals in each subpopulation. The population labels of theassigned individuals are shown to the right of each bar with the number of individuals with the same label in parentheses. To aid visualizationof the individual assignment, the 185 population labels were grouped into 14 color groups reflecting geographical regions. Color gradientswithin the color group denote different population labels. For the complete color scheme, see Figure s3 in the Additional file 1.
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 7 of 11
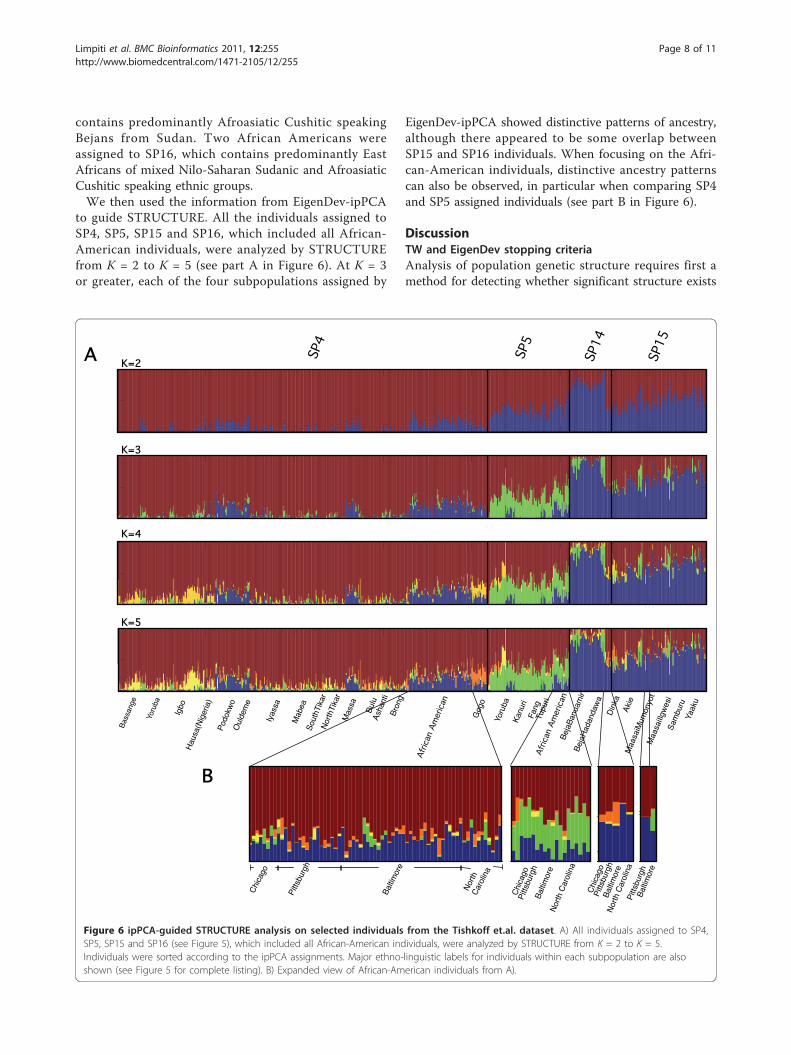
contains predominantly Afroasiatic Cushitic speakingBejans from Sudan. Two African Americans wereassigned to SP16, which contains predominantly EastAfricans of mixed Nilo-Saharan Sudanic and AfroasiaticCushitic speaking ethnic groups.We then used the information from EigenDev-ipPCA
to guide STRUCTURE. All the individuals assigned toSP4, SP5, SP15 and SP16, which included all African-American individuals, were analyzed by STRUCTUREfrom K = 2 to K = 5 (see part A in Figure 6). At K = 3or greater, each of the four subpopulations assigned by
EigenDev-ipPCA showed distinctive patterns of ancestry,although there appeared to be some overlap betweenSP15 and SP16 individuals. When focusing on the Afri-can-American individuals, distinctive ancestry patternscan also be observed, in particular when comparing SP4and SP5 assigned individuals (see part B in Figure 6).
DiscussionTW and EigenDev stopping criteriaAnalysis of population genetic structure requires first amethod for detecting whether significant structure exists
100 200 300 400 500 600
100 200 300 400 500 600
100 200 300 400 500 600
K=2
K=3
K=4
K=5
Chi
cago
Pitt
sbur
gh
Bal
timor
e
Nor
th
Car
olin
a
Bal
timor
eN
orth
Car
olin
a
Pitt
sbur
gh
Chi
cago
Bal
timor
eN
orth
Car
olin
a
Pitt
sbur
gh
Chi
cago
Bal
timor
e
Pitt
sbur
gh
Bas
sang
e
Yoru
ba
Igbo
Hau
sa(N
iger
ia)
Pod
okw
oO
ulde
me
Iyas
sa
Mab
eaS
outh
Tika
rN
orth
Tika
rM
assa
Bul
uA
shan
tiB
rong
Afric
an A
mer
ican
Gog
o
Yoru
baK
anur
iFa
ngTu
puri
Bej
aBan
uam
irB
ejaH
adan
daw
aD
inka
Aki
eM
aasa
iMum
onyo
tM
aasa
iIlgw
esi
Sam
buru
Yaak
u
Afric
an A
mer
ican
A
B
SP4
SP5
SP14
SP15
Figure 6 ipPCA-guided STRUCTURE analysis on selected individuals from the Tishkoff et.al. dataset. A) All individuals assigned to SP4,SP5, SP15 and SP16 (see Figure 5), which included all African-American individuals, were analyzed by STRUCTURE from K = 2 to K = 5.Individuals were sorted according to the ipPCA assignments. Major ethno-linguistic labels for individuals within each subpopulation are alsoshown (see Figure 5 for complete listing). B) Expanded view of African-American individuals from A).
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 8 of 11

in the dataset (or nested dataset for further iterations ofipPCA). The current method to obtain this informationis to test for deviation from the Tracy-Widom distribu-tion of the largest eigenvalue computed from PCA. A p-value lower than 10-12 is considered an acceptablethreshold for significance in rejecting the null hypothesisthat the data belong to a homogenous (sub)population,and thus are structured [5]. The first experiment with asimulated dataset with no structure revealed that signifi-cant deviation from the expected distribution is found,particularly with large sampling (>70 individuals). Weinfer from this result that when the sample size is large,the TW method suffers from type I error because ofthis deviation from the TW distribution. Simply usinglower p-value thresholds may not give better results,since there is a very small range of p-value that is prac-tical [5]. When applied to real datasets, homogenous(sub)populations sampled at high density may be incor-rectly construed as possessing structure. In TW-ipPCA,this would lead to a group of individuals being assignedinto separate subpopulations, when they should actuallybe considered belonging to a single (sub)population.To alleviate the drawbacks of the TW test statistic, we
propose a new termination criterion called EigenDevstatistic that is simpler to compute, has no hidden para-meters and is shown to be more robust to type I error.For simplicity, one could choose a single EigenDev valueto be applied as a universal stopping criterion foripPCA, which needs to be determined empirically. Wedetermined a threshold of 0.21 from data simulation,which was also appropriate for the real datasets analyzedin this paper.
Analyses of Bovine HapMap datasetThe subpopulation assignment by EigenDev-ipPCA sup-ports the accepted notion that cattle breeds have dis-tinctive genetic profiles. The finding that ANG andRGU were assigned together in the same subpopulationsuggests that these breeds are genetically indistinguish-able for the markers available, which was also reportedby other methods [12]. However, the finding that GIR,BRM, and NEL breeds are resolved as separate subpopu-lations by EigenDev-ipPCA is novel, since the earlierunsupervised STRUCTURE analysis in [12] on the entiredataset could not distinguish these breeds. ipPCA-guided STRUCTURE analysis on the Bovine HapMapdataset demonstrated differences in ancestries amongthese breeds, consistent with the assignments by Eigen-Dev-ipPCA. Among these indicine breeds, there is evi-dence (high heterozygosity and unique SNPs) to suggestthat BRM is genetically distinct from others, includingGIR and NEL [12]. These results beg the question, whySTRUCTURE analysis, when done in a EigenDev-ipPCAguided manner, can reveal differences among these
breeds which is not apparent in the unsupervisedSTRUCTURE analysis? The likeliest explanation is thatthe overall number of informative markers is low amongthese indicine breeds in comparison with the others(only 19% of the loci having minor allele frequenciesgreater than 0.3) [12]. In other words, the allele frequen-cies among the indicine breeds are highly correlated incomparison with the taurine breeds. Groups of indivi-duals with highly correlated allele frequencies in com-parison with other groups tend to be merged bySTRUCTURE [11].
Analyses of a large human datasetThe 49 subpopulations assigned by EigenDev-ipPCAeach contain individuals largely sharing the same ethno-linguistic label/affiliation, in accordance with [10,13]. Ofnote, the 426 Indian individuals were assigned to twosubpopulations by EigenDev-ipPCA. This grouping isconsistent with the parametric analysis of these indivi-duals in [13], which showed weak evidence of structure.Hence, the greater degree of stratification resolved byTW-ipPCA compared with EigenDev-ipPCA is likely tobe spurious. The spurious structure resolved by TW-ipPCA is thus attributable to the large sample size (426),which is well above the threshold encountered for type Ierror from the analysis of simulated data.Among the African individuals, subpopulations were
assigned by EigenDev-ipPCA revealing stratification pat-terns not described previously. For instance, Niger-Khordofanian speaking non-Pygmy individuals fromWest and Central Africa could not be distinguishedgenetically in [10], but were assigned to SP3, SP4 andSP5 subpopulations by EigenDev-ipPCA. The assign-ment of the majority African Americans to SP4 and SP5by EigenDev-ipPCA (Figure 5) suggests they have Westand Central African Niger-Khordofanian ancestors, inagreement with [10]. On the other hand, the assignmentof African Americans to different subpopulations byEigenDev-ipPCA is suggestive of significant structureamong these individuals. Supervised STRUCTURE runsperformed in [10] to elucidate African American ances-try could only reveal a subtle clinal pattern of variationamong the African Americans. The EigenDev-ipPCAguided STRUCTURE analysis, however, shows clear dif-ferences in ancestry between SP4 and SP5 AfricanAmericans. The SP15 and SP16 assigned African Ameri-cans also show ancestry distinct from the SP4 and SP5assigned individuals, although given the small numberof individuals assigned to SP15 and SP16, it is not possi-ble to observe significant ancestry differences betweenthese two groups.The EigenDev-ipPCA assignment of some African
Americans to SP15 and SP16 was unexpected. The con-temporary African individuals in these subpopulations
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 9 of 11

are predominantly from Saharan and East Africa. Arecent study of African American ancestry concludedthat some individuals have a major ancestral componentwhich is neither West African Niger-Khordofanian, norEuropean [14]. The possibility that this anomalousancestry is of Saharan or East African may also bereflected in mtDNA haplotypes, since some AfricanAmericans have anomalous haplotypes of unknownAfrican origin [15,16]. The discrepancy between Eigen-Dev-ipPCA guided STRUCTURE and supervisedSTRUCTURE performed in [10] is due to the choice ofindividuals in the analysis. When individuals with inap-propriately diverged allele frequencies from others areused, key ancestral differences will be missed, the sameas was shown in analysis of Bovine data.
ConclusionWe describe EigenDev-ipPCA for analyzing populationstructure. This approach assigns individuals to subpopu-lations and determines the total number of subpopula-tions present. This algorithm incorporates a novelheuristic called EigenDev for detecting substructure,which is applied to the iterative clustering process.EigenDev is robust to population sampling, allowing usto analyze large complex datasets with higher accuracy.The subpopulations assigned by EigenDev-ipPCA revealsoverall genetic relatedness among groups of individuals,which can then be used to guide STRUCTURE. Otherparametric algorithms such as Admixture and frappecould also be used in the same way. Therefore, the com-bination of EigenDev-ipPCA and STRUCTURE arecomplementary and can be used together to perform apowerful population stratification analysis. The softwareboth in Matlab source code (m- file) and executable ver-sions on Windows and Linux (64 bit) are available fordownload at http://www4a.biotec.or.th/GI/tools/ippca.
Additional material
Additional file 1: The detailed analysis and further discussion of theEigenDev-ipPCA results for the Tishkoff et al. dataset.
Acknowledgements and FundingThe authors thank King Mongkut’s Institute of Technology Ladkrabang(KMITL), the National Electronics and Computer Technology Center (NECTEC)and the National Center for Genetic Engineering and Biotechnology(BIOTEC) for financial support. In particular, TL and ST acknowledge thefunding supported by the National Science and Technology DevelopmentAgency (NSTDA). AA was supported by BIOTEC postdoctoral fellowship. JPwas supported by BIOTEC platform technology grant and the ThailandResearch Fund (TRF) new researcher grant (TRG5380028). PJS was funded bythe Bill & Melinda Gates Foundation through the Grand ChallengesExplorations Initiative. ST received the support from BIOTEC platformtechnology and TRF Career Development grant (RSA-54). We thank thereviewers and editors for their constructive comments, which improve the
quality and presentation of the manuscript. Finally, we would like to thankthe authors and the donors who made the real datasets used in this paperavailable.
Author details1Faculty of Engineering, King Mongkut’s Institute of Technology Ladkrabang,Bangkok 10520, Thailand. 2National Electronics and Computer TechnologyCenter, Thailand Science Park, Pathumthani 12120, Thailand. 3National Centerfor Genetic Engineering and Biotechnology, Thailand Science Park,Pathumthani 12120, Thailand.
Authors’ contributionsTL, AI, PJS and ST wrote the manuscript. TL, AI and ST constructed thecomputational improvement scheme of the new algorithm. AA, PJS and JPconceived the ideas to reanalyze the mixed complex datasets. TL, AI, PWand CN conducted all the experiments presented in this work. TL, AA, PJS,JP and ST analyzed the results. AI, PW and CN wrote the EigenDev-ipPCAprogram and made it available in executable formats using a Matlabcompiler. All authors have read and approved the final manuscript.
Competing interestsThe authors declare that they have no competing interests.
Received: 8 October 2010 Accepted: 23 June 2011Published: 23 June 2011
References1. Marchini J, Cardon LR, Phillips MS, Donnelly P: The effects of human
population structure on large genetic association studies. Nat Genet2004, 36(5):512-7.
2. Tian C, Plenge R, Ransom M, Lee A, Villoslada P, Selmi C, Klareskog L,Pulver A, Qi L, Gregersen P, Seldin M: Analysis and application ofEuropean genetic substructure using 300 K SNP information. PLoS Genet2008, 4:e4.
3. Paschou P, Lewis J, Javed A, Drineas P: Ancestry informative markers forfine-scale individual assignment to worldwide populations. J Med Genet2010, 47(12):835-47.
4. Intarapanich A, Shaw PJ, Assawamakin A, Wangkumhang P, Ngamphiw C,Chaichoompu K, Piriyapongsa J, Tongsima S: Iterative pruning PCAimproves resolution of highly structured populations. BMC Bioinformatics2009, 10:382.
5. Patterson N, Price A, Reich D: Population structure and eigenanalysis. PLoSGenet 2006, 2(12):e190.
6. Luo J, Zhang Z: Using Eigenvalue Grads Method to Estimate the Numberof Signal Source. In Proceedings of the 5th International Conference onSignal Processing (WCCC-ICSP 2000). Volume 1. Beijing, China; 2000:223-225.
7. Liang L, Zollner S, Abecasis GR: GENOME: a rapid coalescent-based wholegenome simulator. Bioinformatics 2007, 23(12):1565-7.
8. The BovineHapMap dataset. [http://bfgl.anri.barc.usda.gov/cgi-bin/hapmap/affy2/BulkDownloads].
9. The Tishkoff et. al. dataset. [http://www.sciencemag.org/content/vol0/issue2009/images/data/1172257/DC1/1172257_dataset.zip].
10. Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A,Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, Ibrahim M, Juma AT,Kotze MJ, Lema G, Moore JH, Mortensen H, Nyambo TB, Omar SA, Powell K,Pretorius GS, Smith MW, Thera MA, Wambebe C, Weber JL, Williams SM:The genetic structure and history of Africans and African Americans.Science 2009, 324(5930):1035-44.
11. Pritchard JK, Stephens M, Donnelly P: Inference of Population StructureUsing Multilocus Genotype Data. Genetics 2000, 155:945-59.
12. Consortium TBH: Genome-Wide Survey of SNP Variation Uncovers theGenetic Structure of Cattle Breeds. Science 2009, 324(5926):528-32.
13. Rosenberg N, Mahajan S, Gonzalez-Quevedo C, Blum M, Nino-Rosales L,Ninis V, Das P, Hegde M, Molinari L, Zapata G, Weber J, Belmont J, Patel P:Low levels of genetic divergence across geographically and linguisticallydiverse populations from India. PLoS Genet 2006, 2(12):e215.
14. Bryc K, Auton A, Nelson MR, Oksenberg JR, Hauser SL, Williams S,Froment A, Bodo JM, Wambebe C, Tishkoff SA, Bustamante CD: Genome-wide patterns of population structure and admixture in West Africansand African Americans. Proc Natl Acad Sci USA 2010, 107(2):786-91.
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 10 of 11

15. Salas A, Richards M, Lareu MV, Scozzari R, Coppa A, Torroni A, Macaulay V,Carracedo A: The African diaspora: mitochondrial DNA and the Atlanticslave trade. Am J Hum Genet 2004, 74(3):454-65.
16. Ely B, Wilson JL, Jackson F, Jackson BA: African-American mitochondrialDNAs often match mtDNAs found in multiple African ethnic groups.BMC Biol 2006, 4:34.
doi:10.1186/1471-2105-12-255Cite this article as: Limpiti et al.: Study of large and highly stratifiedpopulation datasets by combining iterative pruning principalcomponent analysis and structure. BMC Bioinformatics 2011 12:255.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit
Limpiti et al. BMC Bioinformatics 2011, 12:255http://www.biomedcentral.com/1471-2105/12/255
Page 11 of 11
Copyright © 2022 FDOKUMEN




















