Structure Re-determination of LASSBio-294 – a cardioactive compound of the N-acylhydrazone class...
19
Structure Re-determination of LASSBio-294 – a cardioactive compound of the N-acylhydrazone class – using X-ray powder diffraction data Fanny N. Costa a , Fabio Furlan Ferreira b , Tiago F. da Silva c,d , Eliezer J. Barreiro c,d , Lídia M. Lima c,d , Delson Braz a , Regina C. Barroso e a Laboratório de Instrumentação Nuclear/COPPE, Universidade Federal do Rio de Janeiro, C.P. 68.509, CEP 21941-972, Rio de Janeiro, RJ, Brazil. b Centro de Ciências Naturais e Humanas (CCNH), Univerisdade Federal do ABC (UFABC), Av. dos Estados, 5001, CEP 09210-580, Santo André, SP, Brazil. c Laboratório de Avaliação e Síntese de Substâncias Bioativas (LASSBio), Faculdade de Farmácia, Universidade Federal do Rio de Janeiro, RJ, Brazil. d Programa de Pós-Graduação em Química, Instituto de Química, Universidade Federal do Rio de Janeiro, Cidade Universitária, Rio de Janeiro, RJ, Brazil e Instituto de Física, Universidade do Estado do Rio de Janeiro (UERJ), CEP 20550-900, Rio de janeiro, RJ, Brazil Many N-acylhydrazone derivatives synthetized in LASSBio ® cannot be prepared as single crystals of sufficient size and/or quality for structure determination to be carried out using single crystal X-ray diffraction techniques. This article highlights the opportunity for determining crystal structures of this class of compounds directly from powder diffraction data. For this task, the crystal structure of LASSBio-294 was re-determined by means of conventional X-ray powder diffraction data and so, compared with the crystal structure already determined for single crystal data. LASSBio-294 is a cardioactive compound of the N-acylhydrazone class, which can become part of the therapeutic interventions designed to decrease exertional fatigue, and, consequently, improve the quality of life of patients suffering from chronic heart failure. Its final crystal structure was refined by means of the Rietveld method (Rietveld, 1967; 1969). This drug crystallizes in a monoclinic (P2 1 /c) space group, with unit cell parameters a = 11.3413(3) Å, b = 12.3573(4) Å, c = 9.0158(3) Å, β = 89.821(2)°, V = 1263.55(7) Å 3 , Z = 4, Z´ = 1 and calc = 1.4419(1) g cm -3 . The goodness-of-fit indicator and R-factors were, respectively: χ 2 = 1.203, R Bragg = 0.696%, R wp = 5.59%, R exp = 4.65% and R p = 4.18%. The molecules in LASSBio-294 are H-bonded along the c-axis involving the atoms N(3)–H(8)···O(4). Key words: N-acylhydrazone, X-ray powder diffractometry, crystal structure determination, Rietveld method. 0885-7156/2013/28(S2)/S2/1/$18.00 ©2013 JCPDS-ICDD S491 S491 Vol. 28, No.S2, September 2013.
Transcript of Structure Re-determination of LASSBio-294 – a cardioactive compound of the N-acylhydrazone class...

Structure Re-determination of LASSBio-294 – a cardioactive compound of the
N-acylhydrazone class – using X-ray powder diffraction data
Fanny N. Costaa, Fabio Furlan Ferreira
b, Tiago F. da Silva
c,d, Eliezer J. Barreiro
c,d, Lídia M.
Limac,d
, Delson Braza, Regina C. Barroso
e
a Laboratório de Instrumentação Nuclear/COPPE, Universidade Federal do Rio de Janeiro, C.P. 68.509, CEP
21941-972, Rio de Janeiro, RJ, Brazil.
b Centro de Ciências Naturais e Humanas (CCNH), Univerisdade Federal do ABC (UFABC), Av. dos Estados,
5001, CEP 09210-580, Santo André, SP, Brazil.
c Laboratório de Avaliação e Síntese de Substâncias Bioativas (LASSBio), Faculdade de Farmácia, Universidade
Federal do Rio de Janeiro, RJ, Brazil.
d Programa de Pós-Graduação em Química, Instituto de Química, Universidade Federal do Rio de Janeiro, Cidade
Universitária, Rio de Janeiro, RJ, Brazil
e Instituto de Física, Universidade do Estado do Rio de Janeiro (UERJ), CEP 20550-900, Rio de janeiro, RJ, Brazil
Many N-acylhydrazone derivatives synthetized in LASSBio® cannot be prepared as
single crystals of sufficient size and/or quality for structure determination to be carried out using
single crystal X-ray diffraction techniques. This article highlights the opportunity for
determining crystal structures of this class of compounds directly from powder diffraction data.
For this task, the crystal structure of LASSBio-294 was re-determined by means of conventional
X-ray powder diffraction data and so, compared with the crystal structure already determined for
single crystal data. LASSBio-294 is a cardioactive compound of the N-acylhydrazone class,
which can become part of the therapeutic interventions designed to decrease exertional fatigue,
and, consequently, improve the quality of life of patients suffering from chronic heart failure. Its
final crystal structure was refined by means of the Rietveld method (Rietveld, 1967; 1969). This
drug crystallizes in a monoclinic (P21/c) space group, with unit cell parameters a = 11.3413(3)
Å, b = 12.3573(4) Å, c = 9.0158(3) Å, β = 89.821(2)°, V = 1263.55(7) Å3, Z = 4, Z´ = 1 and calc
= 1.4419(1) g cm-3
. The goodness-of-fit indicator and R-factors were, respectively: χ2 = 1.203,
RBragg = 0.696%, Rwp = 5.59%, Rexp = 4.65% and Rp = 4.18%. The molecules in LASSBio-294
are H-bonded along the c-axis involving the atoms N(3)–H(8)···O(4).
Key words: N-acylhydrazone, X-ray powder diffractometry, crystal structure determination,
Rietveld method.
0885-7156/2013/28(S2)/S2/1/$18.00 ©2013 JCPDS-ICDD S491S491 Vol. 28, No.S2, September 2013.

I. Introduction
Medicinal Chemistry studies the molecular reasons for the action of drugs in order to
describe the relationship between chemical structure and pharmacological activity, ranking the
different functional contributions (Wermuth, 1996; Nic et al., 2007; Barreiro and Fraga, 2008). It
includes the structural design of new substances that have useful pharmacotherapeutic properties,
able to represent new chemical entities, candidates for prototypes of new drugs to safe use (Lima and
Barreiro, 2005; Viegas-Júnior et al., 2007; Lombardino and Lowe, 2004). This complex task involves
multiple factors responsible for the desired therapeutic response and the resulting of complexity
biological systems, covering a range of concepts from different disciplines and cannot be
performed randomly (Barreiro et al., 2002; Duarte et al., 2007; Barreiro et al., 1997; Evans et al., 1988).
Among the factors involved in the elucidation of the steps to describe the relationship
between chemical structure and pharmacological activity of a prototype of a new drug can be
included the study of atomic-level structure.
The majority of active pharmaceutical ingredients (API) can exist in different solid-state
forms. These solid forms can differ widely in their physicochemical, mechanical, and
biopharmaceutical properties, and thus can influence the quality, safety, and efficacy of the drug
product (Grant, 1999; Haleblian and McCrone, 1969). These properties include particle shape,
size, particle size distribution, surface area, porosity, and surface morphology. A comprehensive
understanding of these properties is essential to understand the behavior of API and drug product
performance (Brittain et al., 1991). For this task a variety of physical techniques
(crystallography, spectroscopy, thermal analysis and microscopy) are useful for characterizing
the solid forms of pharmaceuticals (Yu et al., 1998).
Due to the fact that polycrystalline forms are detected by X-ray diffraction, the powder
diffraction has played a central role in structural physics, chemistry and materials science over
the past twenty-five years. Today, structures of much higher complexity have become accessible
to powder diffractionists (Cheetham, 2006). X-ray powder diffractometry is a powerful
technique for the identification of crystalline solid phases. Every crystalline solid phase has a
unique X-ray powder diffraction (XRPD) pattern, which can form the basis for its identification.
In a powder mixture, each crystalline phase produces its pattern independently of the other
constituents in the mixture. The technique is unique, since it combines absolute specificity with a
high degree of accuracy (Hull, 1919; Shell, 1963).
Fanny N. Costa et al. S492S492 Vol. 28, No.S2, September 2013.

The use of X-rays for determination of three-dimensional atomic arrangements in crystal
structures of organic and inorganic compounds is an important stage in the design of new
substances as well as in the development of the new procedures for the synthesis of the already
known compounds (Chernyshev, 2001). The single crystal X-ray diffraction is the most powerful
approach for the determination of structural information at the atomic level. Nevertheless, it is
important to recall that the requirement for a single crystal sample of appropriate size and quality
imposes a natural limitation on the scope of this technique. Unfortunately, many crystalline
solids can be prepared only as microcrystalline powders, and therefore cannot be studied using
single crystal X-ray diffraction techniques. The most direct approach is to use X-ray powder
diffraction data, although it is important to recognize that the process of carrying out structure
determination from powder diffraction data is substantially more challenging than structure
determination from single crystal diffraction data (Harris and Cheung, 2004). The availability of
reliable procedures for determining crystal structures directly from X-ray powder diffraction data
has the potential to make considerable impact in structural sciences. For this reason, much
research activity in recent years has been devoted to the development and application of new
techniques for carrying out structure determination directly from powder diffraction data, and
has led to significant advances in the scope and power of techniques in this field (Menden, 1998;
Harris et al., 2001; Langford and Louër, 1996; Florence et al., 2005; David et al., 2006a).
The methodology of structure determination from X-ray diffraction data has been
employed as a technique able to define the relative configuration of the compounds, which is
directly related to the biological activity. This technique can be applied to identify polymorphic
phases, to characterize possible intermolecular and intramolecular interactions beyond providing
information about conformational freedom, flatness and also it can be used to distinguish the
correct orientation of the structural groups of the molecule. The physicochemical properties and
the pharmacokinetics characteristics can be understood from this information (Harris, 2002).
A bioactive compound of the N-acylhydrazone (NAH) class (named LASSBio-294)
(Barreiro, 2002; Sudo et al., 2001; Silva et al., 2002; Sudo et al., 2006; Kümmerle et al., 2009)
was synthesized and characterized in LASSBio (Laboratório de Avaliação e Síntese de
Substâncias Bioativas) of the Federal University of Rio de Janeiro (UFRJ). It was shown to
produce a cardioinotropic effect and vasodilatation (Silva et al., 2005). This novel
thienylhydrazone compound (2-thienylidene) 3,4-methylenedioxybenzoylhydrazine, herein
Structure Re-determination of LASSBio-294 – a cardioactive compound S493S493 Vol. 28, No.S2, September 2013.

referred to as LASSBio-294, was initially obtained as part of a program of synthesis of novel
anti-inflammatory leads with an N-acylhydrazone scaffold (Figueiredo et al., 2000).
Subsequently, this compound was identified as a bioisoster of a family of pyridazinone
compounds that increase cyclic adenosine 3’5’ cyclic monophosphate (AMP) levels by inhibiting
selectively the cyclic AMP-specific, low-Km phosphodiesterase 4 (PDE4) (Piaz et al., 1997). A
study was undertaken to investigate the effects of this thienylhydrazone in the contractile
properties of single skeletal muscle fibers of the frog. In addition to demonstrating that
LASSBio-294 has positive inotropic effects in single fibers of phasic muscles of the frog, the
results also showed that the compound reduces fatigue development and accelerates recovery of
maximal tetanic force after fatigue has developed. Some lines of evidence suggest that the effects
of LASSBio-294 on skeletal muscle inotropism and fatigue can be accounted for by
phosphodiesterase inhibition adenosinergic receptors improving the quality of life of patients
suffering from chronic heart failure (LeJemtel et al., 1986) and other physiopathological
conditions in which skeletal muscle dysfunctions are associated with decreased levels of
cytosolic cyclic AMP (Gonzalez-Serratos et al., 2001).
Many N-acylhydrazone (NAH) derivative analogues, synthesized in LASSBio®, planned
by structural modification of the lead compound LASSBio-294 do not create crystals suitable for
single crystal X-ray diffraction. Therefore, the compound LASSBio-294 was elected for our
study as reference crystal structure for the purpose of evaluating the accuracy of the structures
solved using the powder diffraction approach. Since the crystal structure of this compound was
previously determinated (Kümmerle et al., 2009) using single crystal diffraction, the aim of this
work is to demonstrate that through successful re-determination of crystal structure of LASSBio-
294 directly from powder diffraction data we will have the opportunity for determining crystal
structures of other NAH derivatives, unavailable for the study by means of single crystal and
better understanding of pharmacological profiles of these compounds.
In this work, X-ray powder diffraction data and a simulated annealing algorithm
implemented in the DASH software program (David et al., 2006b) were used to re-determine the
crystal structure of LASSBio-294, on the basis of previous procedures (Ferreira et al., 2010;
Ferreira et al., 2011). Thus, a Rietveld refinement of the final crystal structure was conducted
using the Topas Academic v.5 (Coelho et al., 2011) software program providing a satisfactory fit.
Fanny N. Costa et al. S494S494 Vol. 28, No.S2, September 2013.

II. Experimental
A. Materials and Methods
Applying classic and typical strategies of medicinal chemistry to design new molecular
architecture, LASSBio-294 compound was planned as a simplified analogue of pyridazinone
scaffold, known as phosphodiesterase (PDE) inhibitor compounds. Besides that, the inclusion of
the 1,3-benzodioxolyl ring in LASSBio-294 coming from the recognition of attractive natural
biofores in Brazilian abundant natural product, safrole. In addition, the inclusion of the 2-thienyl
ring attached to the imine moiety of NAH, resulted on a novel thienylhydrazone compound (2’-
thienylidene)3,4-methylenedioxybenzoylhydrazine, herein named LASSBio-294 (Barreiro and
Fraga, 2008; Kümmerle et al., 2009).
Briefly, according to the procedure described previously by Lima et al. (2000), LASSBio-294
was synthesized from the starting material safrole (4-allyl-1,2-methylenedioxybenzene). The first
step in the synthesis consisted of a base-catalyzed isomerization of safrole that yielded isosafrole.
By oxidative cleavage, isosafrole was converted into piperonal. Then, by using the Yamada’s
oxidative procedure, piperonal was converted in the methyl 3,4-methylenedioxybenzoate.
Subsequently, the key 3,4-methylenedioxyhydrazine was obtained by means of nucleophilic
substitution of the ester with hydrazine hydrate in ethanol at reflux. Finally, LASSBio-294 was
obtained by condensing the hydrazine derivative with equimolar amounts of thiophene-2-
carboxaldehyde in ethanol by using hydrochloric acid as a catalyst (Gonzalez-Serratos et al., 2001).
The XRPD pattern of LASSBio-294, which was gently hand-grinded in a pestle and
mortar then passed through a 45-µm sieve, was collected on a Panalytical X’Pert Pro
diffractometer operating at 40 kV and 40 mA. Measurements were carried out with Cu Kα
radiation (λ = 1.5418 Å) and diffraction patterns were collected in the 2θ region ranging from 4º
to 50º, with step scans of 0.01º and counting time of 40 s step-1
. Diffraction data were recorded
by a Real Time Multiple Strip (RTMS) X´Celerator detector using a scattered beam graphite
monochromator, a divergence slit of 1º, a Soller slit of 0.04 rad and an antiscattering slit of 3.9 mm.
Structure Re-determination of LASSBio-294 – a cardioactive compound S495S495 Vol. 28, No.S2, September 2013.

III. Results and discussion
A. Indexing and structure determination
The DASH software program (David et al., 2006b) was used for indexing the powder
pattern as well as for the crystal structure determination of LASSBio-294 using conventional X-
ray powder diffraction data. Data from 5.500º to 49.996º (2θ), leading to a real-space resolution
of 1.8242 Å, were used. A Bayesian high-pass filter (David and Sivia, 2001) implemented in
DASH was employed to estimate the background. The first 20 reflections were individually
fitted and used in the indexing procedure by means of the DICVOL91 (Boultif and Louër, 1991)
routine (within DASH), thus yielding the following values for a monoclinic cell: a = 11.3328 Å,
b = 12.3460 Å, c = 9.0151 Å, β = 89.78° and V = 1261.34 Å3. After the unit-cell parameters have
been roughly evaluated, a Pawley fit (Pawley, 1981) was performed and, by using a Bayesian
statistical analysis procedure (Markvardsen et al., 2001), the space group was determined as
being P21/c. The unit-cell volume was calculated by taking into account the individual average
volumes of the atoms from volume increments (Hofmann, 2002). The expected cell volume of
1256.32 Å3 is similar to the value found in the indexing procedure (1261.34 Å
3) and suggested
four molecules per unit cell (Z = 4) and one molecule in the asymmetric unit (Z´ = 1). The
integrated intensities as well as their correlations were extracted by means of a Pawley
refinement, which converged to a χ2 value of 2.25.
The values found via DASH were then used in conjunction with the aforementioned
chemical structure in the process of crystal structure determination by means of a simulated
annealing algorithm (Aarts and Korst, 1991; van Laarhoven and Aarts, 1992). Twenty runs (on a
total of 2.00×107 movements) of the simulated annealing process were globally optimized and
the best result was then considered in the final refinement of the structure. During the simulated
annealing process, ten parameters (three describing the positional coordinates, four, of which
three are independent, describing the molecular orientation and three flexible torsion angles) were
allowed to vary (David et al., 2006b).
Generally, the visual assessment of the goodness of fit between the experimental and
simulated powder pattern and the ratio ‘profile 2/Pawley
2’, which should be, in most cases,
less than 5 (David et al., 2006b), are good indicative of a successful structure solution. Although
in the present case the ratio ‘profile 2/Pawley
2’ (16.19/2.25 7.2) was a little bit greater than
Fanny N. Costa et al. S496S496 Vol. 28, No.S2, September 2013.

5, the best solution found in the simulated annealing runs was considered in the Rietveld
refinement (Rietveld, 1967; 1969) process using the Topas Academic v.5 software program
(Coelho et al., 2011). A reasonable visual fitting as well as statistical values and chemical
structure were obtained. Figure 1 shows the molecular structure of LASSBio-294 with some
H-bonds displayed by cyan lines.
Figure 1. Molecular structure of LASSBio-294 displaying some H-bonds (cyan lines). The symmetry codes are: (i)
.
B. Structure refinement
The background was fitted using a 20-term Chebyschev polynomial. The peak asymmetry
was fitted by the simple axial divergence model of Cheary and Coelho (Cheary and Coelho,
1998a; 1998b). The peak profiles were modeled by the Double-Voigt approach (Balzar, 1993)
with anisotropic peak profiles adjusted using spherical harmonics (Järvinen, 1993) as well as
preferred orientation of the crystals. Bond distances and angles as well as soft planar restraints on
rings were restrained to distances and angles mean values obtained from the MOGUL
Cambridge Structure Databank System (Bruno et al., 2004). Isotropic atomic displacements
(Biso) were constrained to be equal for all non-hydrogen atoms. For hydrogen atoms, Biso values
were constrained to be 1.2 times larger than those of the respective atoms to which they are
connected. Their factional coordinates were refined restraining the distances between them and
the atoms they are connected according to values found in the literature for X-ray diffraction data
that were then modified in Mercury (Macrae et al., 2008) accordingly: C–H = 0.96 Å, N–H =
0.87 Å and O–H = 0.99 Å. The final refined values for the unit-cell parameters after the Rietveld
fit were: a = 11.3413(3) Å, b = 12.3573(4) Å, c = 9.0158(3) Å, β = 89.821(2)°, V = 1263.55(7)
Å3, Z = 4, Z´ = 1 and calc = 1.4419(1) g cm
-3. The goodness-of-fit indicator and R-factors (Toby,
1 12 2
, ,x y z
Structure Re-determination of LASSBio-294 – a cardioactive compound S497S497 Vol. 28, No.S2, September 2013.

2006), RBragg, Rwp, Rexp and Rp were, respectively: χ2 = 1.203, RBragg = 0.696%, Rwp = 5.59%, Rexp
= 4.65% and Rp = 4.18%. Figure 2 displays the final Rietveld refinement.
Figure 2. Final Rietveld refinement of the LASSBio-294 sample. The 2θ region starting at 30º was magnified by a factor of 10 for
better visualization of the Bragg peaks.
C. Structure description, as result from powder data
The crystal structure of LASSBio-294 is comprised by four formula units per unit cell (Z = 4),
accommodating one molecule in the asymmetric unit (Z´ = 1), as represented in Figure 3.
Figure 3. Unit cell representation of LASSBio-294 along the b-axis displaying some hydrogen bonds (cyan lines) between the
atoms N(3)–H(8)···O(4) thus forming a network of molecular aggregates in LASSBio-294 along the c-axis.
Within the unit cell the molecules are held together by short hydrogen bonds between atoms
N(3)–H(8)···O(4) (D–H = 0.871(6) Å, H···A = 2.072(7) Å, D···A = 2.860(7) Å and D–H···A =
Fanny N. Costa et al. S498S498 Vol. 28, No.S2, September 2013.

150.2(6)º; where “D” and “A” are, respectively, hydrogen donor and acceptor, as represented by
cyan lines in Figure 3 (powder data), thus forming a network of molecular aggregates along the c-axis.
The values found by single crystal diffraction were: N(1)–H(10)···O(1) (D–H = 0.876 Å, H···A
= 2.006 Å, D···A = 2.829 Å and D–H···A = 156.1º). The possible complementary π-stacking
interactions between N-acylhydrazone scaffold with thiophenyl ring (C(13) – C(21) = 3.534 Å)
and also N-acylhydrazone scaffold with 1,3-benzodioxoly ring (C(13) – O(17) = 3.386 Å) are
shown in Figure 4.
Figure 4. Unit cell representation of LASSBio-294 displaying some π-stacking interactions (magenta lines) between the atoms
C(13) – C(21) and C(13) – O(17) as well as its distances (Å).
It seems these interactions contribute for the organization of the space arrangement in the unit
cell. The geometry of the crystal structure was validated by means of a Mogul check. All bond
distances and angles are in accordance to the corresponding values found in the CSD database
(Allen, 2002). Also, in order to check the consistency of the obtained results for space group
choice, unit cell parameters, bond distances, angles and torsions, the PLATON software program
was used (Spek, 2003). The ADDSYM routine did not detect any obvious extra crystallographic
symmetry. CCDC ID: 907702 contains the supplementary crystallographic data for this paper.
These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/data_request/cif.
Crystal data as well as details of the structure determination are displayed in Table I.
Structure Re-determination of LASSBio-294 – a cardioactive compound S499S499 Vol. 28, No.S2, September 2013.

Table I. Details from the Rietveld refinement of the crystal structure of LASSBio-294.
Chemical formula C13H10N2O3S
Formula weight (g mol-1) 274.30
Crystal system Monoclinic Space group P21/c (No. 14)
a, b, c (Å) 11.3413(3), 12.3573(4), 9.0158(3)
β (°) 89.821(2) Volume (Å3) 1263.55(7)
Z, Z´ 4, 1
calc (g cm-3) 1.4419(1)
μ (mm-1) 2.344
T (K) 298
Data collection Diffractometer X´Celerator Pro CuKα sealed tube
Monochromator graphite (diffracted beam)
Wavelength (Å) 1.54178
2 range (°) 4 – 50
Step size (°) 0.01
Time per step (s) 40 Refinement
Number of data points 4450
Number of contributing reflections 225
Number of restraints 79 Number of refined parameters 96
Rexp (%) 4.646
Rwp (%) 5.590 RBragg (%) 0.697
χ2 1.203
d-DW 1.507
Structural atomic parameters obtained from the Rietveld refinement are shown in Table II.
Table II. Final coordinates and equivalent isotropic displacement parameters (Uiso = Biso/82) of
all atoms for LASSBio-294 crystal structure.
Atom x/a y/b z/c Biso (Å2)
S(22) 0.5533(4) -0.0214(3) -0.2819(5) 0.0879(2)
O(4) 0.7561(8) 0.3409(5) -0.2424(6) 0.0879(2) O(17) 0.9471(4) 0.5005(4) 0.3390(5) 0.0879(2)
O(19) 0.9261(4) 0.6756(4) 0.2463(5) 0.0879(2)
N(3) 0.7310(7) 0.2444(4) -0.0355(5) 0.0879(2) N(7) 0.6817(7) 0.1600(4) -0.1132(6) 0.0879(2)
C(1) 0.7749(4) 0.3304(3) -0.1122(4) 0.0879(2)
C(2) 0.8144(3) 0.4273(3) -0.0209(4) 0.0879(2) C(5) 0.8021(3) 0.5333(4) -0.0757(4) 0.0879(2)
C(6) 0.8638(2) 0.4104(4) 0.1205(4) 0.0879(2)
C(9) 0.8385(2) 0.6231(4) 0.0094(5) 0.0879(2) C(11) 0.8982(2) 0.4994(3) 0.2008(5) 0.0879(2)
C(13) 0.6730(8) 0.0699(4) -0.0466(6) 0.0879(2)
C(15) 0.8861(3) 0.6020(3) 0.1473(5) 0.0879(2) C(18) 0.6203(6) -0.0247(3) -0.1133(5) 0.0879(2)
C(20) 0.9713(9) 0.6126(4) 0.3686(7) 0.0879(2)
C(21) 0.6217(6) -0.1285(4) -0.0534(6) 0.0879(2) C(25) 0.5687(7) -0.2058(4) -0.1485(6) 0.0879(2)
C(27) 0.5372(10) -0.1601(4) -0.2818(6) 0.0879(2)
H(8) 0.7337(7) 0.2433(4) 0.0610(5) 0.1055(3) H(10) 0.7685(3) 0.5447(4) -0.1721(4) 0.1055(3)
H(12) 0.8719(2) 0.3387(4) 0.1602(4) 0.1055(3) H(14) 0.7020(8) 0.0648(4) 0.0531(6) 0.1055(3)
H(16) 0.8298(2) 0.6957(4) -0.0267(5) 0.1055(3)
H(23) 0.9347(9) 0.6343(4) 0.4601(7) 0.1055(3) H(24) 1.0549(9) 0.6231(4) 0.3753(7) 0.1055(3)
H(26) 0.6552(6) -0.1459(4) 0.0413(6) 0.1055(3)
H(28) 0.5561(7) -0.2803(4) -0.1231(6) 0.1055(3) H(29) 0.5092(10) -0.2008(4) -0.3652(6) 0.1055(3)
Fanny N. Costa et al. S500S500 Vol. 28, No.S2, September 2013.
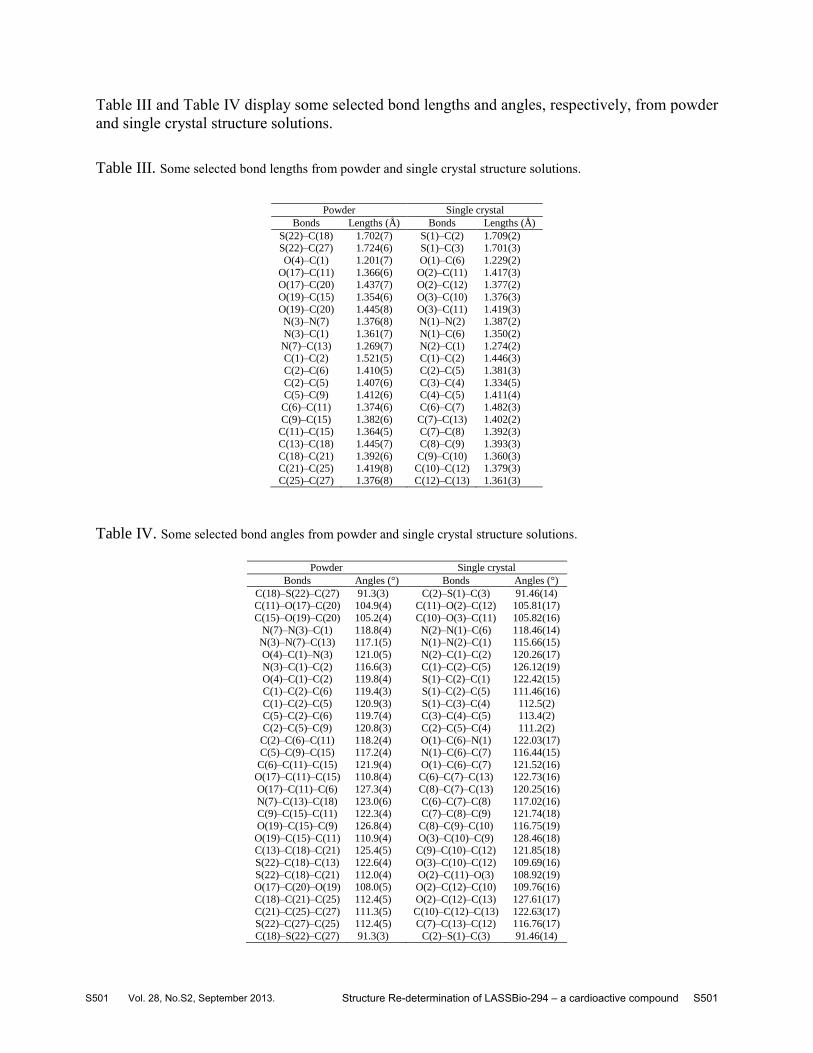
Table III and Table IV display some selected bond lengths and angles, respectively, from powder
and single crystal structure solutions.
Table III. Some selected bond lengths from powder and single crystal structure solutions.
Powder Single crystal
Bonds Lengths (Å) Bonds Lengths (Å)
S(22)–C(18) 1.702(7) S(1)–C(2) 1.709(2) S(22)–C(27) 1.724(6) S(1)–C(3) 1.701(3)
O(4)–C(1) 1.201(7) O(1)–C(6) 1.229(2)
O(17)–C(11) 1.366(6) O(2)–C(11) 1.417(3) O(17)–C(20) 1.437(7) O(2)–C(12) 1.377(2)
O(19)–C(15) 1.354(6) O(3)–C(10) 1.376(3)
O(19)–C(20) 1.445(8) O(3)–C(11) 1.419(3) N(3)–N(7) 1.376(8) N(1)–N(2) 1.387(2)
N(3)–C(1) 1.361(7) N(1)–C(6) 1.350(2)
N(7)–C(13) 1.269(7) N(2)–C(1) 1.274(2) C(1)–C(2) 1.521(5) C(1)–C(2) 1.446(3)
C(2)–C(6) 1.410(5) C(2)–C(5) 1.381(3)
C(2)–C(5) 1.407(6) C(3)–C(4) 1.334(5) C(5)–C(9) 1.412(6) C(4)–C(5) 1.411(4)
C(6)–C(11) 1.374(6) C(6)–C(7) 1.482(3)
C(9)–C(15) 1.382(6) C(7)–C(13) 1.402(2) C(11)–C(15) 1.364(5) C(7)–C(8) 1.392(3)
C(13)–C(18) 1.445(7) C(8)–C(9) 1.393(3)
C(18)–C(21) 1.392(6) C(9)–C(10) 1.360(3) C(21)–C(25) 1.419(8) C(10)–C(12) 1.379(3)
C(25)–C(27) 1.376(8) C(12)–C(13) 1.361(3)
Table IV. Some selected bond angles from powder and single crystal structure solutions.
Powder Single crystal
Bonds Angles (°) Bonds Angles (°)
C(18)–S(22)–C(27) 91.3(3) C(2)–S(1)–C(3) 91.46(14) C(11)–O(17)–C(20) 104.9(4) C(11)–O(2)–C(12) 105.81(17)
C(15)–O(19)–C(20) 105.2(4) C(10)–O(3)–C(11) 105.82(16)
N(7)–N(3)–C(1) 118.8(4) N(2)–N(1)–C(6) 118.46(14) N(3)–N(7)–C(13) 117.1(5) N(1)–N(2)–C(1) 115.66(15)
O(4)–C(1)–N(3) 121.0(5) N(2)–C(1)–C(2) 120.26(17)
N(3)–C(1)–C(2) 116.6(3) C(1)–C(2)–C(5) 126.12(19) O(4)–C(1)–C(2) 119.8(4) S(1)–C(2)–C(1) 122.42(15)
C(1)–C(2)–C(6) 119.4(3) S(1)–C(2)–C(5) 111.46(16)
C(1)–C(2)–C(5) 120.9(3) S(1)–C(3)–C(4) 112.5(2) C(5)–C(2)–C(6) 119.7(4) C(3)–C(4)–C(5) 113.4(2)
C(2)–C(5)–C(9) 120.8(3) C(2)–C(5)–C(4) 111.2(2)
C(2)–C(6)–C(11) 118.2(4) O(1)–C(6)–N(1) 122.03(17) C(5)–C(9)–C(15) 117.2(4) N(1)–C(6)–C(7) 116.44(15)
C(6)–C(11)–C(15) 121.9(4) O(1)–C(6)–C(7) 121.52(16) O(17)–C(11)–C(15) 110.8(4) C(6)–C(7)–C(13) 122.73(16)
O(17)–C(11)–C(6) 127.3(4) C(8)–C(7)–C(13) 120.25(16)
N(7)–C(13)–C(18) 123.0(6) C(6)–C(7)–C(8) 117.02(16) C(9)–C(15)–C(11) 122.3(4) C(7)–C(8)–C(9) 121.74(18)
O(19)–C(15)–C(9) 126.8(4) C(8)–C(9)–C(10) 116.75(19)
O(19)–C(15)–C(11) 110.9(4) O(3)–C(10)–C(9) 128.46(18) C(13)–C(18)–C(21) 125.4(5) C(9)–C(10)–C(12) 121.85(18)
S(22)–C(18)–C(13) 122.6(4) O(3)–C(10)–C(12) 109.69(16)
S(22)–C(18)–C(21) 112.0(4) O(2)–C(11)–O(3) 108.92(19) O(17)–C(20)–O(19) 108.0(5) O(2)–C(12)–C(10) 109.76(16)
C(18)–C(21)–C(25) 112.4(5) O(2)–C(12)–C(13) 127.61(17)
C(21)–C(25)–C(27) 111.3(5) C(10)–C(12)–C(13) 122.63(17) S(22)–C(27)–C(25) 112.4(5) C(7)–C(13)–C(12) 116.76(17)
C(18)–S(22)–C(27) 91.3(3) C(2)–S(1)–C(3) 91.46(14)
Structure Re-determination of LASSBio-294 – a cardioactive compound S501S501 Vol. 28, No.S2, September 2013.

On the basis of an analysis on potential hydrogen interactions performed with PLATON
it was found that atom N(3) is the potential donor of the acceptor atom O(4). The bonds
involving the atoms N(3)–H(8)···O(4) form a network of molecular aggregates in LASSBio-294
along the c-axis, as shown in Figure 3 (in case of single crystal data the network is formed by the
bonds involving atoms N(1)–H(10)···O(1) and N(1)–H(10)···N(2)).
The torsion angle involving the atoms N(3)–C(1)–C(2)–C(5) was –146.4(5)°, being
limited by –120° and –180°. Under this condition, the molecule is arranged in a –anti-clinal (ac)
conformation. The torsion angles involving the atoms C(1)–N(3)–N(7)–C(13), atoms N(7)–
C(13)–C(18)–C(21) and atoms N(7)–N(3)–C(1)–C(2) were, respectively, 160.2(7)°, 171.4(8)°
and 172.7(6)°, all of them being limited by 150° and 180° (for single crystal data the respective
torsion angles were: C(6)–N(1)–N(2)–C(1) = –166.95(17)°, N(2)–C(1)–C(2)–C(5) = –168.3(2)°
and atoms N(2)–N(1)–C(6)–C(7) = –178.25(15)°). In this case, the molecule adopted a +anti-
periplanar (ap) conformation for all torsion angles.
The 5-membered ring system composed by S(22)–C(18)–C(21)–C(25)–C(27) atoms
shows that the highest deviation from the least-squares plane passing through this ring is
0.050(10) Å for C(27) (root mean square error (r.m.s.e.) of contributing atoms is 0.006 Å; the
same analysis performed for single crystal data revealed the highest deviation from the least-
squares plane passing through this ring is 0.006(2) Å for C(5) and r.m.s.e was 0.002 Å). The
other 5-membered ring system, composed by O(17)–O(19)–C(11)–C(15)–C(20) atoms, indicates
that the highest deviation from the least-squares plane passing through this ring is 0.024(10) Å
for C(20) (root mean square error (r.m.s.e.) of contributing atoms is 0.005 Å). Also, the 6-
membered ring system, composed by C(2)–C(5)–C(6)–C(9)–C(11)–C(15) atoms, indicates that
the highest deviation from the least-squares plane passing through this ring is 0.002(3) Å for
C(6) (root mean square error (r.m.s.e.) of contributing atoms is 0.004 Å; the same analysis
performed for single crystal data revealed the highest deviation from the least-squares plane
passing through this ring is 0.007(2) Å for C(7) and r.m.s.e was 0.002 Å) demonstrating that the
rings of the LASSBio-294 molecule are almost planar.
The consistency of the crystal structure geometry was evaluated by a calculation of
intramolecular bond distances and angles using MOGUL, resulting that neither bond distances
nor angles were found as “unusual”. The criteria to confirm the molecular geometry in Mogul
was based on the choice of exact fragments in the CSD database – if the number of exact
Fanny N. Costa et al. S502S502 Vol. 28, No.S2, September 2013.

fragments were less than 15 to bonds, angles and rings, and 40 to torsion angles, the molecular
geometry would be reported as “unusual”.
D. Comparison between crystal structure of LASSBio-294 determined by single
crystal and by powder diffraction
The crystal structure of this compound was previously described in 2009 by Kümmerle et
al. There, X-ray analysis of LASSBio-294 and other three NAH single crystals obtained from an
ethanol saturated solution was carried out to best understand the influence of the methyl group
for the bioprofile of those NAH derivatives. Data pertaining to the X-ray crystallographic
determination of LASSBio-294 by means of single crystal X-ray diffraction was deposited with
CCDC ID: 707596.
Figure 5 shows the solution for LASSBio-294 obtained from powder data (in colors –
present work) overlaid on the single crystal structure (orange) (Kümmerle et al., 2009). This
comparison presents a small difference between these structures but which can be explained by
the slightly variations on bond lengths, bond angles and torsion angles, for example. From a
chemical point of view the structures are quite similar. The relative configuration of E about the
imine double bond was determined in both cases and the preferential flat conformation (amide
hydrogen anti-periplanar to the carbonyl oxygen), in the solid-phase, was also observed. As also
shown in Figure 5, both crystal structures exhibit the orientation of sulfur, of thienyl ring, in the
same direction of carbonyl oxygen.
Figure 5. Overlay of the crystal structure of LASSBio-294 determined directly from powder diffraction data (in colors) and single
crystal diffraction data (orange) (Kümmerle et al., 2009).
Structure Re-determination of LASSBio-294 – a cardioactive compound S503S503 Vol. 28, No.S2, September 2013.

IV. Conclusion
Considering the step of condensation at synthetic route could produce a diasteroisomeric
ratio (E/Z), the X-ray powder diffraction is no doubt a powerful technique providing three-
dimensional structure of the molecule, thus allowing the characterization of the relative
configuration E, of the imine double bond. This information corroborate with results obtained by
single crystal X-ray diffraction (Kümmerle et al., 2009).
The packaging of unit cell was also observed as a result of intermolecular H-bonds involving
peptide bond of the N-acylhydrazone function arranged alternately as well as possible π-stacking
complementary interactions. The high conformational constraint or near planarity of the
compound was also exemplified by the measured dihedral angles. Finally, heteroatoms of the N-
acylhydrazone function and sulfur of thienyl ring orientations in the same direction provides
regions of high electron density permitting interactions with other entities, for example
macromolecules.
The X-ray powder diffraction was successful applied in the crystal structure
determination of LASSBio-294 demonstrating the technique can be applied to solve the
structures of other compounds of NAH class, since many derivatives cannot be prepared as a
crystal suitable for single crystal X-ray diffraction studies. In this context, the three-dimensional
arrangement information accessed by means of this technique contribute for better understanding
the full pharmacodynamic profiles and physicochemical properties of this class of compounds.
Acknowledgments
We would like to thank the Brazilian agencies from financial support and fellowships:
CNPq (# 305186/2012-4; # 477296/2011-4); INCT-INOFAR (CNPq #573.564/2008-6;
FAPERJ E-26/170.020/2008), FAPESP (# 2008/10537-3). We also thank the CBPF (BR) for
experimental support.
Aarts, E. and Korst, J. (1991). Simulated Annealing and Boltzmann Machines: a Stochastic Approach to
Combinatorial Optimization and Neural Computing (John Wiley & Sons, Chichester, UK), 2nd ed., p. 284.
Allen, F. H. (2002). “The Cambridge Structural Database: a quarter of a million crystal structures
and rising,” Acta Crystallogr., Sect. B: Struct. Sci., 58, 380-388.
Fanny N. Costa et al. S504S504 Vol. 28, No.S2, September 2013.

Balzar, D. (1993). “X-Ray-diffraction line broadening - Modeling and applications to high-Tc
Superconductors,” J. Res. Natl. Inst. Stand. Technol. 98, 321-353.
Barreiro, E. J. (2002). “Estratégia de Simplificação Molecular no Planejamento Racional de
Fármacos: A Descoberta de Novo Agente Cardioativo,” Quím. Nova 25, 1172-1180.
Barreiro, E. J., Albuquerque, M. G., Sant´ana, C. M. R., Rodrigues, C. R. and Alencastro, R. B.
(1997). “Modelagem molecular: uma ferramenta para o planejamento racional de
fármacos na Química Medicinal,” Quím. Nova 20, 300-310.
Barreiro, E. J. and Fraga, C. A. M. (2008). Química Medicinal - As Bases Moleculares da Ação
dos Fármacos. (Artmed, Porto Alegre), 2nd ed., p. 243.
Barreiro, E. J Fraga, C. A. M., Miranda, A. I. P. and Rodrigues, C. R.
(2002). “Química medicinal de n-acilidrazonas: novos compostos-protótipos de fármacos
analgésicos, antiinflamatórios e anti-trombóticos,” Quím. Nova 25, 129-148.
Boultif, A. and Louër, D. (1991). “Indexing of powder diffraction patterns for low-symmetry
lattices by the successive dichotomy method,” J. Appl. Crystallogr. 24, 987–993.
Brittain, H. G., Bogdanowich, S. J., Bugay, D. E., DeVincentis, J., Lewen, G., and Newman, A. W.
(1991). “Physical characterization of pharmaceutical solids,” Pharm. Res., 8, 963-973.
Bruno, I. J., Cole, J. C., Kessler, M., Luo, J., Motherwell, W. D. S., Purkis, L. H., Smith, B. R.,
Taylor, R., Cooper, R. I., Harris, S. E. and Orpen, A. G. (2004). “Retrieval of
Crystallographically-Derived Molecular Geometry Information,” J. Chem. Inf. Comput. Sci. 44,
2133-2144.
Cheary, R. W. and Coelho, A. A. (1998a). “Axial divergence in a conventional X-ray powder
diffractometer. I. Theoretical foundations,” J. Appl. Crystallogr. 31, 851-861.
Cheary, R. W. and Coelho, A. A. (1998b). “Axial divergence in a conventional X-ray powder
diffractometer. II. Realization and evaluation in a Fundamental-Parameter profile fitting
procedure,” J. Appl. Crystallogr. 31, 862-868.
Cheetham, A. K. (2006). Structure determination from powder diffraction data: an overview
(International Union of Crystallography / Oxford Science Publications, New York), 1st ed., p. 337.
Chernyshev, V. V. (2001). “Structure determination from powder diffraction,” Russ. Chem.
Bull. Int. Ed. 50, 2273-2292.
Coelho, A. A., Evans, J., Evans, I., Kern, A. and S. Parsons (2011). “The TOPAS symbolic
computation system,” Powder Diffr. 26, s22-s25.
David, W. I. F., Shankland, K., McCusker, L. B. and Baerlocher, C. (2006a). Structure
determination from powder diffraction data. (Oxford University Press, New York), 1st
ed., p. 337 and references therein.
Structure Re-determination of LASSBio-294 – a cardioactive compound S505S505 Vol. 28, No.S2, September 2013.

David, W. I. F., Shankland, K., van de Streek, J., Pidcock, E., Motherwell, W. D. S. and Cole, J. C.
(2006b). “DASH: a program for crystal structure determination from powder diffraction
Data,” J. Appl. Crystallogr. 39, 910-915.
David, W. I. F. and Sivia, D. S. (2001). “Background estimation using a robust Bayesian
Analysis,” J. Appl. Crystallogr., 34, 318-324.
Duarte, C. D., Barreiro, E. J. and Fraga, C. A. M. (2007). “Privileged Structures: A Useful
Concept for the Rational Design of New Lead Drug Candidates,” Mini Rev. Med. Chem. 7, 1108-
1119.
Evans, B. E., Rittle, K. E., Bock, M. G., Dipardo, R. M., Freidinger, R. M., Whitter, W. L., Lundell,
G. F., Veber, D. F., Anderson, P. S., Chang, R. S. L., Lotti, V. J., Cerino, D. J., Chen, T. B.,
Kling, P. J., Kunkel, K. A., Springer, J. P. and Hirshfieldt, J. (1988). “Methods for Drug Discovery:
Development of Potent, Selective, Orally Effective Cholecystokinin Antagonists,” J. Med. Chem.
31, 2235-2246.
Ferreira, F. F., A. C., Antonio, Rosa, P. C. P. and Paiva-Santos, C. O. (2010). “Crystal structure
determination of mebendazole form A using high-resolution synchrotron X-ray powder
diffraction data,” J. Pharm. Sci. 99, 1734-1744.
Ferreira, F. F., Trindade, A. C., Antonio, S. G. and Paiva-Santos, C. O. (2011). “Crystal structure
of propylthiouracil determined using high-resolution synchrotron X-ray powder
diffraction,” CrystEngComm 13, 5474 - 5479.
Figueiredo, J. M., Camara, C. A., Amarante, E. G., Miranda, A. L. P., Santos, F. M., Rodrigues, C. R.,
Fraga, C. A. M. and Barreiro, E. J. (2000). “Design and synthesis of novel potent antinociceptive
agents: N-acetylimidazolyl N-acylhydrazone derivatives,” Bioorg. Med. Chem. 8, 2243-2248.
Florence, A. J., Shankland, N., Shankland, K., David, W. I. F., Pidcock, E., Xu, X., Johnston, A.,
Kennedy, A. R., Cox, P. J., Evans, J. S. O., Steele, G., Cosgrove, S. D. and C. S. Frampton
(2005). “Solving molecular crystal structures from laboratory X-ray powder diffraction data with
DASH: the state of the art and challenges,” J. Appl. Crystallogr, 38, 249-259.
Gonzalez-Serratos, H., Chang, R., Pereira, E. F. R., N. G. Castro, Aracava, Y., Melo, P. A., Lima, P.
C., Fraga, C. A. M., Barreiro, E. J. and E. X. Albuquerque (2001). “A novel
thienylhydrazone, 2-thienylidene3,4-methylenedioxybenzoylhydrazine, increases inotropism and
decreases fatigue of skeletal muscle,” J. Pharmacol. Exp. Ther. 299, 558-566.
Grant, D. J. W. (1999). “Theory and origin of polymorphism”, in Polymorphism in pharmaceutical
solids, edited by H. G. Brittain (New York, Marcel Dekker), Vol. 95, pp. 1-33.
Fanny N. Costa et al. S506S506 Vol. 28, No.S2, September 2013.

Haleblian, J. K. and McCrone, W. J. (1969). “Pharmaceutical applications of polymorphism,” J.
Pharm. Sci. 58, 911-929.
Harris, K. D. M. (2002). “Structure determination of molecular materials from powder
diffraction data,” Curr. Opin. Solid St. Mat. Sci. 6, 125-130.
Harris, K. D. M. and Cheung, E. Y. (2004). “How to determine structures when single crystals
cannot be grown: opportunities for structure determination of molecular materials using powder
diffraction data,” Chem. Soc. Rev. 33, 526-538.
Harris, K. D. M., Tremayne, M. and Kariuki, B. M. (2001). “Contemporary advances in the use of
powder X-ray diffraction for structure determination.” Angew. Chem. Int. Ed., 40, 1626-1651.
Hofmann, D. W. M. (2002). “Fast estimation of crystal densities,” Acta Crystallogr., Sect. B:
Struct. Sci. 58, 489-493.
Hull, A. W. (1919). “A New Method of Chemical Analysis,” J. Am. Chem. Soc. 41, 1168-1175.
Järvinen, M. (1993). “Application of symmetrized harmonics expansion to correction of the
preferred orientation effect,” J. Appl. Crystallogr. 26, 525-531.
Kümmerle, A. E., Raimundo, J. M., Leal, C. M., da Silva, G. S., Balliano, T. L., Pereira, M. A., de
Simone, C. A., Sudo, R. T., Zapata-Sudo, G., Fraga, C. A. M. and E. J. Barreiro
(2009). “Studies towards the identification of putative bioactive conformation of potent
vasodilator arylidene N-acylhydrazone derivatives,” Eur. J. Med. Chem. 44, 4004-4009.
Langford, J. I. Louër, D. (1996). “Powder Diffraction,” Rep. Prog. Phys., 59, 131-234.
LeJemtel, T. H., Gumbardo D,, Chadwick, B., Rutman, H. I. and Sonnenblick, E. H. (1986).
“Milrinone for long-term therapy of severe heart failure: clinical experience with special
reference to maximal exercise tolerance,” Circulation 73, III213–III218.
Lima, L. M. and Barreiro, E. J. (2005). “Bioisosterism: A Useful Strategy for Molecular
Modification and Drug Design,” Curr. Med. Chem. 12, 23- 49.
Lima, P. C., Lima, L. M., Silva, K. C., Léda, P. H., Miranda, A. L., Fraga, C. A. and Barreiro, E. J.
(2000). “Synthesis and analgesic activity of novel N-acylarylhydrazones and isosters, derived
from natural safrole,” Eur. J. Med. Chem. 35, 187-203.
Lombardino, J. G. and Lowe, J. A. (2004). “The role of the medicinal chemist in drug discovery
– then and now,” Nat. Rev. Drug Discov. 3, 853-862.
Macrae, C. F., I. J. Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-
Monge, L., Taylor, R., van De Streek, J. and Wood, P. A. Wood (2008). “Mercury CSD 2.0– new
features for the visualization and investigation of crystal structures,” J. Appl. Crystallogr. 41,
466-470.
Markvardsen, A. J., David, W. I. F., Johnson, J. C. and Shankland, K. (2001). “A probabilistic
Structure Re-determination of LASSBio-294 – a cardioactive compound S507S507 Vol. 28, No.S2, September 2013.

approach to space-group determination from powder diffraction data,” Acta Crystallogr., Sect. A:
Found. Crystallogr. 57, 47-54.
Menden, A. (1998). “Crystal Structure Solution from Powder Diffraction Data – State of the Art
and Perspectives,” Croat. Chem. Acta 71, 615-633.
Nic, M., J. Jirat and B. Kosata (2007). IUPAC Compendium of Chemical Terminology - The
Gold Book. (Blackwell Scientific Publications, Oxford), 2nd ed., p. 1622.
Pawley, G. S. (1981). “Unit-cell refinement from powder diffraction scans,” J Appl.
Crystallogr. 14, 357-361.
Piaz, V. D., Giovannoni, M. P., Castellana, C., Palacios, J. M., Beleta, J., Domenech, T. and Segarra,
V. (1997). “New heterocyclic fused pyridazinones as potent and selective phosphodiesterase
IV inhibitors,” J. Med. Chem. 40, 1417–1421.
Rietveld, H. M. (1967). “Line profiles of neutron powder-diffraction peaks for structure
Refinement,” Acta Crystallogr. 22, 151-152.
Rietveld, H. M. (1969). “A Profile Refinement Method for Nuclear and Magnetic Structures,” J.
Appl. Crystallogr. 2, 65-71.
Shell, J. W. (1963). “X-Ray and Crystallographic Applications in Pharmaceutical Research II,”
J. Pharm. Sci. 52, 24-29.
Silva, A. G., G. Zapata-Sudo, A. E. Kummerle, C. A. M. Fraga, E. J. Barreiro and R. T. Sudo
(2005). “Synthesis and vasodilatory activity of new N-acylhydrazone derivatives,
designed as LASSBio-294 analogues,” Bioorg. Med. Chem. Lett. 13, 3431-3437.
Silva, C. L. M., F. Noël and E. J. Barreiro (2002). “Cyclic GMP-dependent vasodilatory
properties of LASSBio-294 in rat aorta,” Br. J. Pharm. 135, 293-298.
Spek, A. L. (2003). “Single-crystal structure validation with the program PLATON,” J. Appl.
Crystallogr. 36, 7-13.
Sudo, R. T., E. X. Albuquerque, E. J. Barreiro, Y. Aracava, W. M. Cintra, P. A. Melo, F. G.
Noël, G. Z. Sudo, C. L. M. Silva, N. G. Castro, P. D. Fernandes, C. A. M. Fraga and A.
L. P. Miranda (2006). Thienylhydrazon with digitalis-like properties (positive inotropic effects).
Appl. Nr. 10/070,328, US Patent 7,091,238 B1, University of Maryland.
Sudo, R. T., G. Zapata-Sudo and E. J. Barreiro (2001). “The new compound, LASSBio- 294, increases
the contractility of intact and saponin-skinned cardiac muscle from wistar rats,” Br. J. Pharm.
134, 603- 613.
Toby, B. H. (2006). “R factors in Rietveld analysis: How good is good enough?,” Powder Diffr.
21, 67-70.
van Laarhoven, P. J. M. and E. H. L. Aarts (1992). Simulated Annealing: Theory and
Fanny N. Costa et al. S508S508 Vol. 28, No.S2, September 2013.

Applications (Kluwer Academic Publishers, Dordrecht, Holland), 4th ed., p. 198.
Viegas-Júnior, C., A. Danuello, V. S. Bolzani, E. J. Barreiro and C. A. M. Fraga (2007).
“Molecular hybridization: a useful tool in the desing of new drug prototypes,” Curr. Med. Chem.
14, 1829-1852.
Wermuth, C. G. (1996). The Practice of Medicinal Chemistry (Academic Press: New York),
1st ed., p. 968.
Yu, L., S. M. Reutzel and G. A. Stephenson (1998). “Physical characterization of polymorphic
drugs: an integrated characterization strategy,” Pharm. Sci. Tech. Today 1, 118-127.
Structure Re-determination of LASSBio-294 – a cardioactive compound S509S509 Vol. 28, No.S2, September 2013.