
Structure analysis of the novel microporous aluminophosphate IST1 using synchrotron powder...
15
Structure analysis of the novel microporous aluminophosphate IST-1 using synchrotron powder diffraction data and HETCOR MAS NMR Jos e Luis Jord a a , Lynne B. McCusker a, * , Christian Baerlocher a , Cl audia M. Morais b,c , Jo~ ao Rocha b , Christian Fernandez c , Cristina Borges d , Jo~ ao P. Lourenc ßo e , Maria F. Ribeiro d , Zelimir Gabelica f a Laboratorium f€ ur Kristallographie, ETH, CH-8092 Z€ urich, Switzerland b Department of Chemistry, CICECO, University of Aveiro, P-3810-193 Aveiro, Portugal c Laboratoire de Catalyse et Spectrochimie, CNRS UMR 6506, ENSICAEN, F-14050 Caen, France d Instituto Superior T ecnico, Dep. Engenharia Qu ımica, P-1049-001 Lisboa, Portugal e Universidade do Algarve, Faculdade de Ci^ encias e Tecnologia, P-8000 Faro, Portugal f Groupe Securit e et Ecologie Chimiques, ENSCMu, Universit e de Haute Alsace, F-68093 Mulhouse Cedex, France Received 3 June 2003; received in revised form 21 July 2003; accepted 25 July 2003 Abstract A combination of advanced powder diffraction and NMR techniques have allowed the structure of the novel mi- croporous aluminophosphate IST-1 (|(CH 3 NH 2 ) 4 (CH 3 NH þ 3 ) 4 (OH ) 4 | ½Al 12 P 12 O 48 ) to be elucidated. The framework structure was determined in the non-centrosymmetric space group Pca2 1 (a ¼ 9:61523ð1Þ A, b ¼ 8:67024ð1Þ A, c ¼ 16:21957ð2Þ A) from high-resolution synchrotron powder diffraction data using the program FOCUS. Extra framework species were then located on difference electron density maps. A hydroxyl group was found to bridge be- tween two of the framework Al atoms, and one methylamine species, presumably protonated, could be located in the channels where it H-bonds to three framework oxygens. The most unusual feature of the structure is the second methylamine molecule, which bonds directly to a framework Al atom. The structure is entirely consistent with 31 P and 27 Al MAS NMR studies, which showed there to be three P (all 4-coordinate) and three Al (one 4-, one 5- and one 6- coordinate) sites, and with 13 C MAS NMR, which showed there to be two different types of methylamine species in equal amounts. Assignment of the 31 P, 27 Al and 13 C MAS NMR signals could be deduced from the crystallographic data, 31 P- 27 Al HETCOR spectra and ab initio calculations. Ó 2003 Elsevier Inc. All rights reserved. Keywords: Aluminophosphate; FOCUS; IST-1; MAS NMR; Powder diffraction 1. Introduction Since the early work of Flanigen and co-work- ers [1] on AlPO 4 -n molecular sieves, extensive re- search has resulted in the discovery of many novel * Corresponding author. E-mail address: [email protected] (L.B. McCus- ker). 1387-1811/$ - see front matter Ó 2003 Elsevier Inc. All rights reserved. doi:10.1016/S1387-1811(03)00499-2 www.elsevier.com/locate/micromeso Microporous and Mesoporous Materials 65 (2003) 43–57
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Structure analysis of the novel microporous aluminophosphate IST1 using synchrotron powder...

www.elsevier.com/locate/micromeso
Microporous and Mesoporous Materials 65 (2003) 43–57
Structure analysis of the novel microporous aluminophosphateIST-1 using synchrotron powder diffraction data
and HETCOR MAS NMR
Jos�ee Luis Jord�aa a, Lynne B. McCusker a,*, Christian Baerlocher a,Cl�aaudia M. Morais b,c, Jo~aao Rocha b, Christian Fernandez c,
Cristina Borges d, Jo~aao P. Lourenc�o e, Maria F. Ribeiro d, Zelimir Gabelica f
a Laboratorium f€uur Kristallographie, ETH, CH-8092 Z€uurich, Switzerlandb Department of Chemistry, CICECO, University of Aveiro, P-3810-193 Aveiro, Portugal
c Laboratoire de Catalyse et Spectrochimie, CNRS UMR 6506, ENSICAEN, F-14050 Caen, Franced Instituto Superior T�eecnico, Dep. Engenharia Qu�ıımica, P-1049-001 Lisboa, Portugale Universidade do Algarve, Faculdade de Cieencias e Tecnologia, P-8000 Faro, Portugal
f Groupe Securit�ee et Ecologie Chimiques, ENSCMu, Universit�ee de Haute Alsace, F-68093 Mulhouse Cedex, France
Received 3 June 2003; received in revised form 21 July 2003; accepted 25 July 2003
Abstract
A combination of advanced powder diffraction and NMR techniques have allowed the structure of the novel mi-
croporous aluminophosphate IST-1 (|(CH3NH2)4(CH3NHþ3 )4(OH�)4|½½½Al12P12O48���) to be elucidated. The framework
structure was determined in the non-centrosymmetric space group Pca21 (a ¼ 9:61523ð1Þ �AA, b ¼ 8:67024ð1Þ �AA,
c ¼ 16:21957ð2Þ �AA) from high-resolution synchrotron powder diffraction data using the program FOCUS. Extra
framework species were then located on difference electron density maps. A hydroxyl group was found to bridge be-
tween two of the framework Al atoms, and one methylamine species, presumably protonated, could be located in the
channels where it H-bonds to three framework oxygens. The most unusual feature of the structure is the second
methylamine molecule, which bonds directly to a framework Al atom. The structure is entirely consistent with 31P and27Al MAS NMR studies, which showed there to be three P (all 4-coordinate) and three Al (one 4-, one 5- and one 6-
coordinate) sites, and with 13C MAS NMR, which showed there to be two different types of methylamine species in
equal amounts. Assignment of the 31P, 27Al and 13C MAS NMR signals could be deduced from the crystallographic
data,31P-27Al HETCOR spectra and ab initio calculations.
� 2003 Elsevier Inc. All rights reserved.
Keywords: Aluminophosphate; FOCUS; IST-1; MAS NMR; Powder diffraction
* Corresponding author.
E-mail address: [email protected] (L.B. McCus-
ker).
1387-1811/$ - see front matter � 2003 Elsevier Inc. All rights reserve
doi:10.1016/S1387-1811(03)00499-2
1. Introduction
Since the early work of Flanigen and co-work-ers [1] on AlPO4-n molecular sieves, extensive re-
search has resulted in the discovery of many novel
d.

44 J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57
topologies with a diversity of structures and
compositions. Recently, we reported the synthesis
of two new aluminophosphates (IST-1 and IST-2)
prepared in aqueous media using methylamine
(MA) as the main template, either added directly
or generated in situ from methylformamide [2,3].The structure of IST-2 subsequently proved to
have the AEN framework type and a structure
very similar to that of AlPO-53(A) reported by
Kirchner et al. in 2000 [4], but that of IST-1 ap-
peared to be novel.
Short chain amines and in particular methyl-
amine, are rarely used as the sole template in mi-
croporous aluminosilicate or aluminophosphatesyntheses [5], because such small entities, in the
absence of any other structuring agent, tend to
lead to narrow pore structures. In the case of
AlPO-EN3 [6] and its isotypes CFSAPO-1(A) [7],
JDF-2 [8], AlPO4-53(A) [4] and IST-2 [2,3], the
exact role of methylamine, which is added to the
synthesis mixture directly, is unclear. Perhaps it
plays an indirect role by generating hydroxylgroups that can bridge between neighboring Al
atoms and thereby stabilize the topology. Since
IST-1, like these AEN-type materials, does not
collapse upon calcination, but undergoes a struc-
tural transformation above 350 �C to form a dif-
ferent thermally stable microporous material, it
might be expected to have some structural features
in common with IST-2.In order to elucidate the structure of IST-1 and
thereby clarify the role of methylamine in the
synthesis, both high-resolution synchrotron pow-
der diffraction and comprehensive 27Al, and 31P
magic-angle spinning (MAS) NMR experiments
have been performed. Because 27Al is a I ¼ 5=2quadrupolar nucleus, the spectral lines are
broadened by the second-order quadrupolar in-teraction and the �conventional� MAS spectra are,
in general, poorly resolved, so the multiple-quan-
tum (MQ) MAS NMR technique [9] was also
applied.
Recently, it has been shown that the resolution
of the 31P NMR spectra of aluminophosphates can
sometimes be improved by using 27Al decoupling,
thus removing the 27Al–31P scalar coupling [10].HETCOR MAS NMR experiments between
quadrupolar (I) and spin-1/2 (S) nuclei have been
used previously [11,12]. Here we show this to be a
potentially very useful technique for studying the
Al–P connectivity in microporous aluminophos-
phates.
2. Experimental
2.1. Synthesis
A typical synthesis of IST-1 involved heating a
hydrogel having the molar composition 1Al2O3:
1P2O5:1methylamine:1TEAOH:35H2O. The alu-
minum source, pseudoboehmite (PURAL SB,Condea), was added to diluted ortho-phosphoric
acid (Merck, 85% aq. soln.), and the resulting so-
lution was stirred until a homogeneous gel was
obtained. First methylamine (Fluka, 41% aq.
soln.) and then the tetraalkylammonium salt
(TEAOH, Alfa, 35% aq. soln.) was added to the
gel. The mixture was then stirred for 2 h prior to
heating in a Teflon-lined autoclave under staticconditions at 170 �C at autogeneous pressure. The
crystallization time was typically 1 day. The re-
sulting crystalline products were recovered by
centrifugation, washed with distilled water and
dried at 80 �C overnight. The detailed synthesis
operations and variants are described and dis-
cussed elsewhere [3].
The as-synthesized material proved to be pureand highly crystalline, as checked by powder XRD
(Rigaku diffractometer using Ni-filtered CuKaradiation) and SEM (Hitachi F-2400 microscope).
The unit cell composition, derived from bulk ele-
mental analysis for Al and P (ICP, Perkin Elmer
Emission Spectrometer Plasma 400) and N and C
(E.A. 1180 CHNS-O Fisons Instruments) and
from water and methylamine contents determinedindependently by combined TG-DSC (Setaram
TGA 92 microbalance), was |(CH3NH2)7:6-
(H2O)4:4|½½½Al12P12O48���.X-ray powder diffraction and 27Al and 31P MAS
NMR data indicate that IST-1 undergoes an irre-
versible structural reorganization upon removal of
the template at 300 �C to yield a microporous
crystalline phase that is thermally stable up to ca.550 �C. Around 600 �C this phase transforms into
a tridymite-type AlPO4.

J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57 45
2.2. X-ray data collection
High-resolution X-ray powder diffraction data
were collected on the Swiss–Norwegian Beamline
(SNBL) at the European Synchrotron RadiationFacility (ESRF) in Grenoble, France. Data col-
lection parameters are given in Table 1.
The diffraction pattern was indexed on an or-
thorhombic unit cell (a ¼ 9:615 �AA, b ¼ 8:669 �AAand c ¼ 16:220 �AA) using the program N-TREOR
[13]. A careful examination of the diffraction pat-
tern indicated that the reflections 0kl, l ¼ 2nþ 1
and h0l, h ¼ 2nþ 1 were systematically absent, sothe two most probable space groups could be ex-
pected to be Pcam (standard setting Pbcm) or
Pca21.
2.3. NMR experiments
All NMR spectra were recorded on a Bruker
Avance 400 (9.4 T, wide-bore) spectrometer at100.4, 104.2 and 161.9 MHz for 13C, 27Al and 31P,
respectively. The 13C MAS NMR spectrum was
recorded with 40� pulses, a spinning rate of 10 kHz
and 120 s recycle delays. Chemical shifts are quo-
ted in ppm from TMS. The 31P MAS NMR
spectrum was recorded with 40� pulses, a spinning
rate of 10 kHz and 60 s recycle delays. Neither 27Al
nor 1H decoupling improved the resolution of the31P MAS NMR spectrum. Chemical shifts are
quoted in ppm from 85% H3PO4. The single-
quantum 27Al MAS NMR spectrum was measured
using short and powerful radio-frequency pulses
(0.6 ls, equivalent to a 9� pulse angle), spinning
Table 1
X-ray data collection
Synchrotron facility SNBL at ESRF
Wavelength 0.79977 �AA
Diffraction geometry Debye–Scherrer
Analyzer crystal Si 1 1 1
Sample Rotating 1.0 mm capillary
2h range 3–48�Step size 0.004�2hTime per step
3.0–9.9�2h 1.0 s
9.9–17.5�2h 1.7 s
17.5–48.0�2h 9.0 s
rates of 14 kHz and a recycle delay of 1 s.
Chemical shifts are quoted in ppm from
Al(H2O)3þ6 .
The triple-quantum 27Al MAS NMR spectrum
was recorded using the Z-filter three-pulse se-
quence [14]. The lengths of the first and secondhard pulses (radio-frequency magnetic field am-
plitude t1 ¼ 150 kHz) were 3.4 and 1.1 ls, re-
spectively. The length of the third soft pulse (t1 ¼10 kHz) was 11 ls. The MAS rate was tR ¼ 15
kHz. 142 points were acquired in the t1 domain in
increments of ð1=mRÞ ¼ 67 ls. The ppm scale of the
sheared spectrum was referenced to m0 frequency inthe m2 domain and to 1.42 m0 (high-resolutionspectra after shearing) in the m1 domain (reference
Al(H2O)3þ6 ). Numerical simulations of the single
and triple-quantum spectra were carried out with
the inhouse program MASAI (Fernandez).
The 27Al–31P heteronuclear correlation (HET-
COR) spectra were obtained underMAS conditions
using CP for mixing. Efficient cross-polarization
was achieved under a constant radio-frequency fieldof 30 kHz for 31P and by ramping the field between
5 and 10 kHz for 27Al. The experiment was per-
formed using three different values for the offset
frequency on the 27Al nucleus. For each offset value
three contact times values were used. The recycle
delay was 1 s and the MAS rate tR ¼ 10 kHz. 100
points were acquired in the t1 domain in increments
of ð1=mRÞ ¼ 100 ls.Ab initio calculations of 13C NMR chemical
shifts were performed using the Gaussian 98w
package. Molecular structures were optimized at
the HF/6-311G(d,p) standard level, and the mag-
netic properties (from which NMR shifts for C
atoms were obtained) were calculated at the same
level using the GIAO formulation [15] as imple-
mented in G98w.
3. NMR results
3.1. 13C MAS NMR
The 13C MAS NMR spectrum of IST-1, shown
in Fig. 1a, displays two resonances centered at 24.3and 28.0 ppm with a 1:1 intensity ratio.

10152025303540ppm from TMS
-60 -70-40-2060 40 20 0
ppm from Al(H2O)63+
-35-30-25-20-15-10-5 -40
ppm from 85% H3PO4
(a)
(b)
(c)
Fig. 1. (a) 13C, (b) 31P and (c) 27Al MAS NMR spectra of as-
prepared IST-1.
ppm
-5050 0 ppm
- 20
60
40
20
0 S1
S2
S3
-
-
ppm from Al(H2O)63+
Fig. 2. 3Q 27Al MAS NMR spectrum of as-prepared IST-1.
46 J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57
3.2. 31P MAS NMR
The 31P MAS NMR spectrum of IST-1, shown
in Fig. 1b, displays three 31P MAS NMR reso-
nances centered at )16.4, )18.2 and )27.9 ppm in
1:1:1 intensity ratios. 27Al (and 1H) decoupling did
not improve the spectral resolution.
3.3. 27Al MAS NMR
The 27Al MAS NMR spectrum of IST-1, shown
in Fig. 1c, displays three resonances centered at
45.7, ca. 10 and )7.9 ppm in 1:1:1 intensity ratios.
3Q MAS NMR confirms that only three lines are
present (Fig. 2). Simulations of the single- andtriple-quantum spectra (not shown) show that the
isotropic chemical shift of the second-order quad-
rupole powder pattern, S2, centered at 10 ppm isdiso ¼ 21:0 ppm and this resonance was therefore
assigned to five-coordinate Al. The quadrupole
coupling constant and asymmetry parameter are,
respectively, CQ ¼ 4:6 MHz, g ¼ 0:21. The peaks
S3 at 45.7 (diso ¼ 47:3 ppm, CQ ¼ 1:8, g ¼ 0:20)and S1 at )7.9 ppm (diso ¼ �6:3 ppm, CQ ¼ 2:0MHz, g ¼ 0:21) are attributed to four- and six-
coordinate Al, respectively.
3.4. 27Al–31P HETCOR MAS NMR
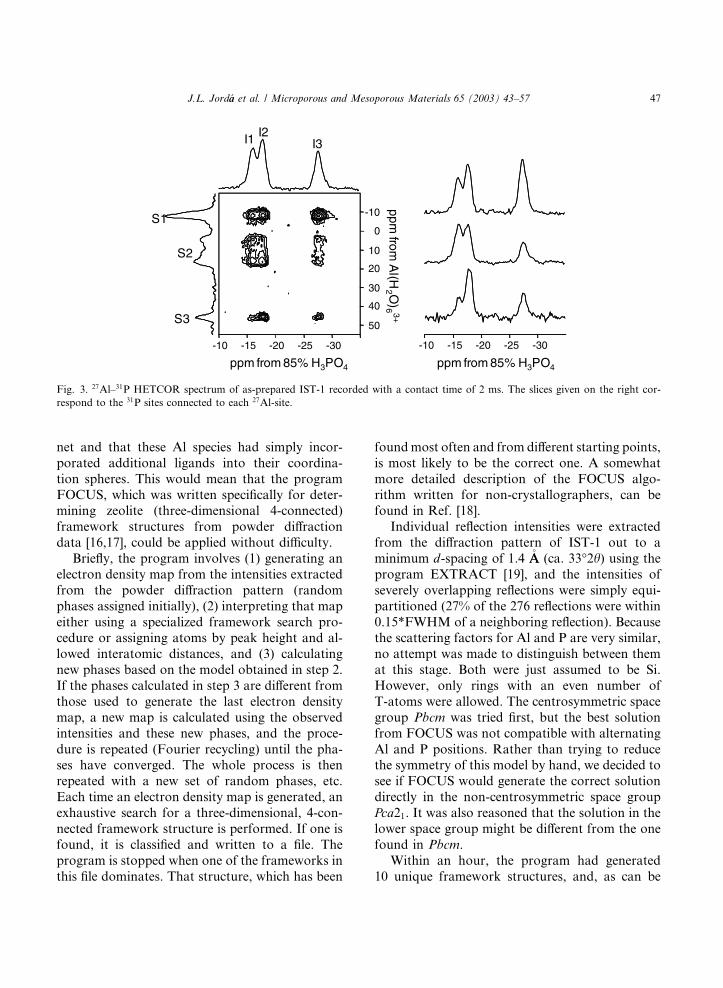
The HETCOR spectrum of IST-1, shown in
Fig. 3, exhibits connectivities between all Al and
all P sites. In order to get a deeper insight into the
Al–P connectivity in this material, a series of
HETCOR spectra at different 27Al–31P cross-polarization contact times and at different 27Al
offsets were recorded. These spectra are identical
with respect to 27Al–31P connectivities. Here only
one of these spectra is shown.
4. Structure determination
Although the NMR data indicated the presence
of 5- and 6-coordinate Al, it was hoped that the
framework could still be regarded as a 4-connected
-10 -15 -20 -25 -30
-10
50
40
30
20
10
0S1
S2
S3
I1 I3I2
ppm from 85% H3PO4
ppm from
Al(H
2 O)6 3+
ppm from 85% H3PO4
-10 -15 -20 -25 -30
Fig. 3. 27Al–31P HETCOR spectrum of as-prepared IST-1 recorded with a contact time of 2 ms. The slices given on the right cor-
respond to the 31P sites connected to each 27Al-site.
J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57 47
net and that these Al species had simply incor-
porated additional ligands into their coordina-
tion spheres. This would mean that the program
FOCUS, which was written specifically for deter-mining zeolite (three-dimensional 4-connected)
framework structures from powder diffraction
data [16,17], could be applied without difficulty.
Briefly, the program involves (1) generating an
electron density map from the intensities extracted
from the powder diffraction pattern (random
phases assigned initially), (2) interpreting that map
either using a specialized framework search pro-cedure or assigning atoms by peak height and al-
lowed interatomic distances, and (3) calculating
new phases based on the model obtained in step 2.
If the phases calculated in step 3 are different from
those used to generate the last electron density
map, a new map is calculated using the observed
intensities and these new phases, and the proce-
dure is repeated (Fourier recycling) until the pha-ses have converged. The whole process is then
repeated with a new set of random phases, etc.
Each time an electron density map is generated, an
exhaustive search for a three-dimensional, 4-con-
nected framework structure is performed. If one is
found, it is classified and written to a file. The
program is stopped when one of the frameworks in
this file dominates. That structure, which has been
foundmost often and from different starting points,
is most likely to be the correct one. A somewhat
more detailed description of the FOCUS algo-
rithm written for non-crystallographers, can befound in Ref. [18].
Individual reflection intensities were extracted
from the diffraction pattern of IST-1 out to a
minimum d-spacing of 1.4 �AA (ca. 33�2h) using the
program EXTRACT [19], and the intensities of
severely overlapping reflections were simply equi-
partitioned (27% of the 276 reflections were within
0.15*FWHM of a neighboring reflection). Becausethe scattering factors for Al and P are very similar,
no attempt was made to distinguish between them
at this stage. Both were just assumed to be Si.
However, only rings with an even number of
T-atoms were allowed. The centrosymmetric space
group Pbcm was tried first, but the best solution
from FOCUS was not compatible with alternating
Al and P positions. Rather than trying to reducethe symmetry of this model by hand, we decided to
see if FOCUS would generate the correct solution
directly in the non-centrosymmetric space group
Pca21. It was also reasoned that the solution in the
lower space group might be different from the one
found in Pbcm.
Within an hour, the program had generated
10 unique framework structures, and, as can be

139
2415
7 6 4 2 1 1 10
20
40
60
80
100
120
140
160
1 2 3 4 5 6 7 8 9 10unique topologies
Fig. 4. The 10 framework topologies generated by FOCUS in the space group Pca21 and their frequency of occurrence.
Table 2
FOCUS input parameters
Framework atoms
Si 24
48 J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57
seen in the histogram shown in Fig. 4, one of thesewas clearly favored over the others. The input
parameters for this FOCUS run are given in
Table 2.
O 48Minimum distances
Si–Si 2.6 �AA
Si–O 1.4 �AAO–O 2.3 �AA
Rings
Minimum ring size 4
Maximum ring size 24
Even loops only
Electron density map interpretation
Maximum number in recycling loop 10
Alternate between largest fragment and
peak height procedures
Reflection data
Fobs
Minimum d-spacing 1.40 �AAscale 0.09
Overlap
Factor (FWHM) 0.15
Treatment Equal mF 2
Reflections
Number input 276
Number used (75% of total intensity) 145
5. Structure completion and Rietveld refinement
The model obtained from the FOCUS runconsisted of six Si atoms. First the Si atoms were
converted to alternating Al and P sites (three of
each), and then the positions of the 12 bridging O-
atoms were calculated using the program KRI-
BER [20]. Finally, the coordinates of all atoms
were optimized using the program DLS-76 [21].
This framework model appeared to be consistent
with the 31P and 27Al NMR data (i.e. each withthree sites of equal occupancy) and to give a rea-
sonable fit to the diffraction pattern, so it was used
as a starting point for structure completion and
Rietveld refinement using the program package
XRS-82 [22]. It was not clear at this point which
set of positions were P-atoms and which Al-atoms.
One of the two possible models was simply as-
sumed.Difference Fourier maps revealed the locations
of two methylamine species. One of these (N(1)–
C(1)) was coordinated (presumably via the N-atom) to a T-atom, so this T-site could be assigned

J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57 49
as Al (Al(1)) and the Al/P ambiguity mentioned
above could be resolved. The other methylamine
species (N(2)–C(2)) was found in the channels,
within hydrogen-bonding distance of three frame-
work oxygens (O(1), O(2) and O(12)). An addi-
tional atom (O(13)) bridging between twoAl-atoms(Al(1) and Al(3)) was also found. This was assumed
to be a hydroxyl group. To maintain charge bal-
ance, the methylamine species within the channels
(N(2)–C(2)) was assumed to be protonated.
This model, with one 6-coordinate, one 5-
coordinate and one 4-coordinate Al-atom (Al(1),
Al(3) and Al(2), respectively) and with three tet-
rahedrally coordinated P-atoms, is in completeagreement with the 27Al and 31P NMR data.
Furthermore, the presence of two types of meth-
ylamine species is consistent with the observation
of two peaks of equal intensity in the 13C NMR
spectrum. The chemical composition of this model
Fig. 5. The coordinations spheres of the framework cations in
as-synthesized IST-1. Note that Al(1) is 6-coordinate, Al(2) 4-
coordinate and Al(3) 5-coordinate, and that all P-atoms are
4-coordinate.
is also consistent with the chemical analysis. The
coordination spheres of the six framework cations
are shown in Fig. 5, and the locations of the
methylamine species in Fig. 6. A simplified draw-
ing of the full framework structure with frame-
work oxygens and methylamine species omitted isgiven in Fig. 7.
Refinement of this structural model converged
with the error indices RF ¼ 0:042 and Rwp ¼ 0:125(Rexp ¼ 0:043). Geometric restraints were used on
the bond distances and angles of the framework
Fig. 6. The location of the extraframework species in as-syn-
thesized IST-1 (a) viewed down the c-axis and (b) viewed down
the b-axis. A hydroxyl group O(13) bridges between Al(1) and
Al(3). One methylamine molecule (N(1)–C(1)) is coordinated
directly to Al(1), while the other is in the channels at hydrogen-
bonding distances from three framework oxygens. This mole-
cule is probably protonated.

Fig. 7. Simplified drawings of the framework structure of IST-1, in which the bridging oxygens have been omitted for clarity, showing
(a) a building unit consisting of four fused 4-rings and two 6-rings, (b) a chain of these building units extended along the a-axis, (c)
chains in (b) related by a 21 axis linked to form a sheet perpendicular to the b-axis, and (d) sheets in (c) linked to form the IST-1
framework structure.
50 J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57
atoms and on the C–N distances of the methyl-amine species (see Table 3). During the course of
the refinement, the weight of these restraints was
reduced to one (i.e. each restraint was given a
weight equivalent to that of a single point on the
diffraction profile). The H-atoms of the methyl-
amine species and of the hydroxyl group weresimulated by increasing the occupancy parameters
of the associated atoms (e.g. the occupancy pa-
rameter for a C of a CH3 group was increased
from 1.0 to 1.5 to approximate the increase from 6
electrons to 9). Neutral scattering factors were

Table 3
Crystallographic data
Chemical composition |(CH3NH2)4(CH3NHþ3 )4(OH�)4|-
½½½Al12P12O48���Unit cell
a 9.61523(1) �AA
b 8.67024(1) �AA
c 16.21957(2) �AASpace group Pca21
Standard peak for peak shape function
(hkl; 2h)002, 5.65�
Peak range (number of FWHM) 20
Number of observations 10 162
Number of contributing reflections 780
Number of geometric restraintsa 82
P–O 1.52(1) �AA 12
Al(1)–O 1.74(3) �AA 4
1.85(5) �AA 1
Al(2)–O 1.74(1) �AA 4
Al(3)–O 1.74(3) �AA 4
1.85(5) �AA 1
C(1)–N(1) 1.469(1) �AA 1
C(2)–N(2) 1.488(1) �AA 1
O–P–O 109.5(1.0)� 18
O–Al(1)–O 90(8)� 8
180(8)� 1
O–Al(2)–O 109.5(1.0)� 6
O–Al(3)–O 90(8)� 6
120(8)� 3
P–O–Al 145(8)� 12
Number of profile parameters 6
Number of structural parameters 71
Rexp 0.043
Rwp 0.125
RF 0.042
a The numbers given in parentheses are the esd�s assumed for each of the restraints. Each restraint was given a weight equivalent to
the reciprocal of its esd.
J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57 51
used for all atoms. A final difference Fourier mapshowed no significant peaks.
Crystallographic details are given in Table 3,
final atomic parameters in Table 4 and selected
interatomic distances and angles in Table 5. The fit
of the profile calculated from the final model to the
experimental data is shown in Fig. 8.
6. Discussion
The framework structure can be viewed in terms
of the [4462] building unit shown in Fig. 7a. The
units are joined through common 4-rings to formchains parallel to the a-axis (Fig. 7b). These chainsare connected in turn to neighboring chains ori-
ented in the opposite direction (related by a 21screw axis) to form sheets parallel to the ac plane
(Fig. 7c). Finally, these sheets are connected to
identical sheets to form a three-dimensional, 4-
connected net (Fig. 7d) with a one-dimensional 10-
ring sinusoidal channel system running parallel tothe a-axis.
The Al atoms in the 4-ring common to neigh-
boring [4462] units are bridged via a hydroxyl
group (O(13)), which is indicated with a dashed

Table 4
Positional, thermal and occupancy parameters for as-synthesized IST-1a
Atom x y z Uiso (·100 �AA2) Occupancyb
Al(1) 0.1950(3) 0.8346(3) 0.3921(2) 0.55(2)c 4.0
Al(2) 0.0171(3) 0.6502(3) 0.1040(2) 0.55(2)c 4.0
Al(3) 0.1780(3) 0.4521(3) 0.3614(2) 0.55(2)c 4.0
P(1) 0.0380(3) 0.3284(3) 0.7909 0.55(2)c 4.0
P(2) 0.1877(3) 0.5844(3) 0.5345(2) 0.55(2)c 4.0
P(3) 0.0259(2) 0.1513(3) 0.4455(2) 0.55(2)c 4.0
O(1) 0.3738(5) 0.8960(6) 0.4306(4) 0.75(4)d 4.0
O(2) 0.1470(6) 0.7320(6) 0.4927(3) 0.75(4)d 4.0
O(3) 0.0188(5) 0.7988(5) 0.3478(4) 0.75(4)d 4.0
O(4) 0.1226(5) 0.0127(6) 0.4409(5) 0.75(4)d 4.0
O(5) 0.2011(5) 0.3396(6) 0.8031(3) 0.75(4)d 4.0
O(6) 0.9740(5) 0.4831(5) 0.8070(4) 0.75(4)d 4.0
O(7) 0.0198(6) 0.2780(5) 0.7014(4) 0.75(4)d 4.0
O(8) )0.0371(5) 0.7890(6) 0.0367(4) 0.75(4)d 4.0
O(9) 0.1931(5) 0.6098(6) 0.0977(4) 0.75(4)d 4.0
O(10) 0.9400(6) 0.4761(6) 0.0856(4) 0.75(4)d 4.0
O(11) 0.0676(5) 0.2760(6) 0.3868(4) 0.75(4)d 4.0
O(12) 0.2282(5) 0.4627(6) 0.4698(3) 0.75(4)d 4.0
O(13) 0.2607(4) 0.6465(5) 0.3459(3) 0.75(4)d 4.5e
C(1) 0.1528(6) 0.9036(8) 0.7281(4) 5.0 6.0e
N(1) 0.2603(7) 0.9621(7) 0.7855(4) 5.0 5.16e
C(2) 0.1442(7) 0.1472(8) 0.1047(4) 5.0 6.0e
N(2) 0.1317(6) 0.1829(7) 0.0146(3) 5.0 5.72e
aNumbers in parentheses are the esd�s in the units of the least significant digit given. Values without an esd were not refined.bOccupancy parameters are given in terms of atoms per unit cell.c Thermal parameters with the same superscript were constrained to be equal.d Thermal parameters with the same superscript were constrained to be equal.e Occupancy increased to approximate the electron density of the associated H atoms.
52 J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57
line in Fig. 7. One of these Al atoms (Al(3)) is 5-
coordinate (four framework oxygens and the hy-
droxyl group), while the other (Al(1)) forms anadditional bond to a methylamine molecule to
become 6-coordinate (see Fig. 5). A second methyl-
amine species is located in the channels parallel to
the c-axis within hydrogen bonding distance of
three framework oxygens (see Fig. 6). It is assumed
that the latter is protonated to balance the charge
of the hydroxyl group.
The presence of hydroxyl groups bridging be-tween Al atoms has been observed in a number of
aluminophosphate framework structures (e.g.
AlPO-21 [23], AlPO-31 [24], AlPO-40 [25], AlPO-
EN3 [6] and its isostructural analogs [4]), and
framework Al atoms have also been observed to
coordinate to water molecules in addition to the
framework oxygens (e.g. VPI-5 [26], AlPO-H2 [27]
and AlPO-H3 [28]). However, the coordination of
an organic molecule to a framework Al during
synthesis is less common.
An |Al6P6O24|-SOD material prepared in thepresence of dimethylformamide (DMF) contains
Al coordinated to the carbonyl oxygen of DMF
and to a water molecule in addition to the four
framework oxygens [29], an |Al16P16O64|-APD
material (APO-CJ3) contains Al coordinated to
the oxygens of two ethanolamine species in addi-
tion to the four framework oxygens [30], and a
layered aluminophosphate material prepared inthe presence of oxalic acid contains Al coordinated
to the two oxygens of an oxalate group in addition
to four oxygens bridging to P atoms [31]. There
appear to be no reports in the literature in which
an amine used during synthesis is coordinated to a
framework Al. However, it has been suggested
from NMR data that an amine coordinates to Al
temporarily during the synthesis of AlPO-21 [32].

Table 5
Selected interatomic distances (�AA) and angles (�) for as-synthesized IST-1
Al(1)–O(1) 1.91(1) P(1)–O(5) 1.58(1)
Al(1)–O(2) 1.92(1) P(1)–O(7) 1.53(1)
Al(1)–O(3) 1.87(1) P(1)–O(6) 1.50(1)
Al(1)–O(4) 1.87(1) P(1)–O(3) 1.54(1)
Al(1)–O(13) 1.90(1)
Al(1)–N(1) 2.10(1)
Al(2)–O(8) 1.71(1) P(2)–O(2) 1.50(1)
Al(2)–O(9) 1.73(1) P(2)–O(12) 1.54(1)
Al(2)–O(10) 1.71(1) P(2)–O(10) 1.57(1)
Al(2)–O(7) 1.73(1) P(2)–O(9) 1.55(1)
Al(3)–O(11) 1.90(1) P(3)–O(4) 1.52(1)
Al(3)–O(12) 1.83(1) P(3)–O(11) 1.50(1)
Al(3)–O(6) 1.80(1) P(3)–O(8) 1.57(1)
Al(3)–O(5) 1.79(1) P(3)–O(1) 1.54(1)
Al(3)–O(13) 1.88(1)
C(1)–N(1) 1.48(1) N(2)–O(2) 2.80(1)
C(2)–N(2) 1.50(1) N(2)–O(12) 2.87(1)
N(2)–O(1) 2.84(1)
O–Al(1)–O,N O–P(1)–O
Min 86(1) Min 104(1)
Max 94(1) Max 113(1)
Min (axial) 173(1)
Max (axial) 177(1)
O–Al(2)–O O–P(2)–O
Min 104(1) Min 106(1)
Max 114(1) Max 112(1)
O–Al(3)–O O–P(3)–O
Min (axial-equatorial) 84(1) Min 105(1)
Max (axial-equatorial) 98(1) Max 112(1)
Min (equatorial-equatorial) 111(1)
Max (equatorial-equatorial) 132(1)
Axial-axial 169(1)
Al(1)–O(1)–P(3) 147(1) Al(2)–O(7)–P(1) 142(1)
Al(1)–O(2)–P(2) 136(1) Al(2)–O(8)–P(3) 144(1
Al(1)–O(3)–P(1) 132(1) Al(2)–O(9)–P(2) 142(1)
Al(1)–O(4)–P(3) 154(1) Al(2)–O(10)–P(2) 137(1)
Al(3)–O(5)–P(1) 138(1) Al(3)–O(11)–P(3) 150(1)
Al(3)–O(6)–P(1) 134(1) Al(3)–O(12)–P(2) 129(1)
Al(1)–O(13)–Al(3) 125(1)
J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57 53
Guest molecules that fit the framework voidspace particularly well are generally referred to as
‘‘templates’’ [33] and they also tend to act as
structure directing agents. In other cases, when the
genesis of the porous structure is governed by
synthesis variables other than the presence of guest
molecules (e.g. hydrophobic clathrasils, zeosils,
silica-rich zeolite frameworks or some AlPO�s), the
stabilization of such (generally neutral) frame-works can be enhanced by (generally neutral) guest
molecules of small size, that only fill the channels
and cavities, where they are weakly linked to the
framework atoms through Van der Waals type
interactions [34]. Neither methylamine species in
IST-1 fills the void structure, but their participa-
tion in the nucleation and growth mechanism of

20 25 30 35 40 45
5 10 15 20 25 30 35 40 45
Fig. 8. The observed (top), calculated (middle) and difference (bottom) profiles for the Rietveld refinement of as-synthesized IST-1. To
show more detail, the scale for the second half of the pattern has been increased by a factor of 5 in the inset.
54 J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57
the AlPO4 framework and their structure-directing
role is readily apparent from the final structure.
Both types of methylamine species serve to stabi-
lize the final topology: one by expanding the co-
ordination sphere of one of the framework Alatoms, and the other through hydrogen bonding
with framework oxygens. A similar behavior was
observed in the case of zeolite Nu-10, where
polyamines act both electrostatically (in the prot-
onated form) and as pore fillers [35].
From the HETCOR spectrum and the crystal-
lographic data given in Table 6, the assignment of
Table 6
Al–O–P connectivities in IST-1 obtained from powder X-ray
diffraction
P(1) P(2) P(3)
Al(1) 1 1 2
Al(2) 1 2 1
Al(3) 2 1 1
27Al and 31P NMR lines is as follows. S1, S2 and
S3 correspond to 6-coordinated, 5-coordinated
and 4-coordinated aluminum, respectively and,
hence, are unambiguously attributed to crystallo-
graphic sites Al(1), Al(3) and Al(2), respectively.The assignment of the 31P NMR resonance can be
based on relative peak intensities of the 27Al–31P
HETCOR spectrum. It should be noticed here that
due to the complicated spin dynamics involved in
spin locking and polarization transfer of the
quadrupolar nuclei, the spectra generally are not
quantitative [36]. In particular, the transfer
strongly depends on the strength of the quadru-polar interaction. However, in the present assign-
ment, only the relative intensities between different
P atoms corresponding to the same Al atom, in
which case the influence of the quadrupolar cou-
pling constant is reduced, are used.
The relative intensities of S1-I cross-peaks are
1:1:1.7, for I1, I2 and I3, respectively, and thus, I3

J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57 55
is assigned to P(3). The remaining peak intensities
for 5-coordinated and (S2) 4-coordinated Al (S3)sites are consistent with the connectivities shown in
Table 6 (2.1:1:1 and 1:2:1.2, respectively). Clearly,
site I1 corresponds to P(1) and I2 to P(2). It should
be noted that these relative intensities did notchange significantly for the different offsets and
contact times used in the HETCOR experiments.
In order to assign the resonances in the 13C
MAS NMR spectrum to the two proposed loca-
tions of the methylamine species, ab initio calcu-
lations of the 13C NMR chemical shifts were
carried out; selected results are summarized in
Table 7. In aqueous solution, the experimentallymeasured chemical shift of methylamine is ca. 27.5
ppm. The calculated chemical shifts for an isolated
Table 7
Molecule
Methylamine+ 3H2O
Protonated methylamine+ 1H2O
Protonated methylamine+ 2H2O
Protonated methylamine+ 3H2O
Octahedral Al complex of methyl
and 1PO4 tetrahedron (capped wi
Protonated methylamine H-bonde
of 2PO4 tetrahedra (capped with
Same as above but protonated m
different position relative to frame
methylamine and for this molecule surrounded by
three water molecules are 26.6 and 25.3 ppm, re-
spectively. In aqueous solution, the experimental
chemical shift measured for protonated methyl-
amine is 26.2 ppm, very close to the value calcu-
lated for this molecule surrounded by three watermolecules (26.6 ppm). In addition, while isolated
protonated methylamine gives a calculated shift of
32.6 ppm, the addition of water molecules signifi-
cantly decreases the shift.
When methylamine is linked through the N-
atom to octahedral aluminum (see Table 7) the
calculated shift is 24.5 ppm, which is very close to
the shift of the IST-1 13C NMR resonance at 24.3ppm. When a protonated methylamine is hydro-
gen bonded to two ‘‘framework’’ oxygens of two
13C calculated shift (ppm)
25.3
29.1
26.1
26.6
amine, water, 2OH�
th OH�)
24.5
d to 2 framework O
waters)
28.6
ethylamine in a
work O
29.4

56 J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57
PO4 tetrahedra (see Table 7), the calculated shift is
28.6 or 29.4 ppm, depending on the orientation of
methylamine. This is close to ca. 28.0 ppm we
observe experimentally for one of the IST-1 reso-
nances, and much less than the value calculated for
isolated methylamine (32.6 ppm). In short, hy-drogen-bonding of methylamine decreases the shift
relative to protonated methylamine, but the exact
value depends somewhat on the orientation of
the molecule with respect to the framework. It is
concluded that the 13C NMR peak at 24.3 ppm
is most likely to be due to methylamine coor-
dinated to framework Al, and that at 28.0 ppm
to protonated methylamine hydrogen-bonded tothe framework. Thus, in these points, the NMR
evidence is fully consistent with the crystal struc-
ture.
7. Conclusions
A combination of advanced powder diffractionand NMR techniques have allowed the structure
of the novel aluminophosphate molecular sieve
IST-1 to be elucidated. It appears to be the first
example of a microporous material in which
methylamine molecules are linked directly through
their N atoms to framework Al atoms, probably in
the early stages of the structure hydrothermal
genesis. Additional methylammonium species areincorporated into the structure via hydrogen bonds
to framework oxygens, and the charge-balancing
hydroxyl group bridges between framework Al
atoms in the same 4-ring. All three extra-frame-
work species seem to stabilize the framework struc-
ture. The IST-1 framework, like that of IST-2 and
other AEN-type aluminophosphates, transforms
to another structure when water and methylamineare released upon heating.
Acknowledgements
Experimental assistance from the staff of the
Swiss–Norwegian Beamlines at the European
Synchrotron Radiation Facility in Grenoble isgratefully acknowledged. This work was sup-
ported in part by the Swiss National Science
Foundation. JLJ thanks the Spanish MEC for
their support in the form of a postdoctoral grant.
The Portuguese authors thank FCT, POCTI and
FEDER for financial support and Dr. P. Claro for
help with 13C NMR shifts calculations.
References
[1] S.T. Wilson, B.M. Lok, C.A. Messina, C.A. Cannan, E.M.
Flanigen, J. Am. Chem. Soc. 104 (1982) 1146–1147.
[2] C. Borges, M.F. Ribeiro, C. Henriques, M.T. Duarte, J.P.
Lourenc�o, Z. Gabelica, Stud. Surf. Sci. Catal. 135 (2001)
194 (CD-ROM ref. 02-P-33).
[3] C. Borges, M.F. Ribeiro, C. Henriques, M.T. Duarte, J.P.
Lourenc�o, J. Rocha, Z. Gabelica, to be submitted.
[4] R.M. Kirchner, R.W. Grosse-Kunstleve, J.J. Pluth, S.T.
Wilson, R.W. Broach, J.V. Smith, Micropor. Mesopor.
Mater. 39 (2000) 319–332.
[5] Z. Gabelica, S. Valange, Micropor. Mesopor. Mater. 30
(1999) 57–67.
[6] J.B. Parise, Stud. Surf. Sci. Catal. 24 (1985) 271–278.
[7] H. He, Y. Long, J. Incl. Phenom. 5 (1987) 591–599.
[8] A.M. Chippindale, A.V. Powell, R.H. Jones, J.M. Thomas,
A.K. Cheetham, Q. Huo, R. Xu, Acta Crystallogr. C 50
(1994) 1537–1540.
[9] L. Frydman, J.S. Harwood, J. Am. Chem. Soc. 117 (1995)
5367–5368.
[10] L. Delevoye, C. Fernandez, C.M. Morais, J.-P. Amoureux,
V. Montouillout, J. Rocha, Solid State Nucl. Magn.
Reson. 23 (2003) 1–13.
[11] R.R. Ernst, G. Bodenhausen, A. Wokaun, Principles of
Nuclear Magnetic Resonance in One and Two Dimensions,
Clarendon Press, Oxford, 1997.
[12] C.A. Fyfe, H. Grodney, K.T. Mueller, K.C. Wong-
Moon, T. Markus, J. Am. Chem. Soc. 114 (1992) 5876–
5878;
C.A. Fyfe, K.T. Mueller, H. Grodney, K.C. Wong-Moon,
J. Phys. Chem. 97 (1993) 13484–13485.
[13] A. Altomare, C. Giacovazzo, A. Guagliardi, A.G.G.
Moliterni, R. Rizzi, P.E. Werner, J. Appl. Crystallogr. 33
(2000) 1180–1186.
[14] J.P. Amoureux, C. Fernandez, S. Steuernagel, J. Magn.
Reson. A 123 (1996) 116–118.
[15] R. Ditchfield, Mol. Phys. 27 (1974) 789–807;
K. Wolinski, J.F. Hinton, P. Pulay, J. Am. Chem. Soc. 112
(1990) 8251–8260;
K. Wolinski, A.J. Sadlej, Mol. Phys. 41 (1980) 1419–1430.
[16] R.W. Grosse-Kunstleve, L.B. McCusker, Ch. Baerlocher,
J. Appl. Crystallogr. 30 (1997) 985–995.
[17] R.W. Grosse-Kunstleve, L.B. McCusker, Ch. Baerlocher,
J. Appl. Crystallogr. 32 (1999) 536–542.
[18] L.B. McCusker, Ch. Baerlocher, R.W. Grosse-Kunstleve,
S. Brenner, T. Wessels, Chimia 55 (2001) 497–504.
[19] Ch. Baerlocher, EXTRACT. A Fortran program for the
extraction of integrated intensities from a powder pattern,

J.L. Jord�aa et al. / Microporous and Mesoporous Materials 65 (2003) 43–57 57
Institut f€uur Kristallographie, ETH, Zurich, Switzerland,
1990.
[20] R. Bialek, KRIBER. Crystallographic computation pro-
gram, Institut f€uur Kristallographie, ETH, Zurich, Switzer-
land, 1991.
[21] Ch. Baerlocher, A. Hepp, W.M. Meier, DLS-76. Distance
least squares refinement program, Institut f€uur Kristallog-
raphie, ETH, Zurich, 1977.
[22] Ch. Baerlocher, A. Hepp, XRS-82. The X-ray Rietveld
system, Institut f€uur Kristallographie, ETH, Zurich, Swit-
zerland, 1982.
[23] J.M. Bennett, J.M. Cohen, G. Artioli, J.J. Pluth, J.V.
Smith, Inorg. Chem. 24 (1985) 188–193.
[24] G. Mali, A. Meden, A. Ristic, N.N. Tusar, V. Kaucic,
J. Phys. Chem. B 106 (2002) 63–69.
[25] V. Ramaswamy, L.B. McCusker, Ch. Baerlocher, Micro-
por. Mesopor. Mater. 31 (1999) 1–8.
[26] L.B. McCusker, Ch. Baerlocher, E. Jahn, M. B€uulow,Zeolites 11 (1991) 308–313.
[27] H.X. Li, M.E. Davis, J. Chem. Soc. Faraday Trans. 89
(1993) 951–965.
[28] J.J. Pluth, J.V. Smith, Acta Crystallogr. C 42 (1986) 1118–
1120.
[29] L. Vidal, J.L. Paillaud, Z. Gabelica, Micropor. Mesopor.
Mater. 24 (1998) 189–197.
[30] K. Wang, J. Yu, G. Zhu, Y. Zou, R. Xu, Micropor.
Mesopor. Mater. 39 (2000) 281–289.
[31] P. Lightfoot, Z.A.D. Lethbridge, R.E. Morris, D.S.
Wragg, A. Wright, �AA. Kvick, G.B.M. Vaughan, J. Solid
State Chem. 143 (1999) 74–76.
[32] Z. Liu, W. Xu, G. Yang, R. Xu, Micropor. Mesopor.
Mater. 22 (1998) 33–41.
[33] B.M. Lok, T.R. Cannan, C.A. Mesina, Zeolites 3 (1983)
282–291.
[34] B. Marler, Zeolites 7 (1987) 393–397.
[35] C. Pellegrino, R. Aiello, Z. Gabelica, ACS Symp. Ser. 398
(1989) 161–175.
[36] A.J. Vega, Solid State Nucl. Magn. Reson. 1 (1992) 17–32;
S.M. De Paul, M. Ernst, M.J. Shore, F. Stebbins, A. Pines,
J. Phys. Chem. B 101 (1997) 3240–3249;
W. Sun, J.T. Stephen, L.D. Porter, Y. Wu, J. Magn.
Reson. A 116 (1995) 181–188.




















