
Congenital brain abnormalities: Pictorial essay - JournalAgent
Upload
khangminh22Category
view
1download
0
Structural Brain Abnormalities in Temporomandibular Disorders
by
Massieh Moayedi
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Institute of Medical Science University of Toronto
© Copyright by Massieh Moayedi 2012

ii
Structural Brain Abnormalities in Temporomandibular Disorder
Massieh Moayedi
Doctor of Philosophy
Institute of Medical Science University of Toronto
2012
Abstract
Temporomandibular disorders (TMD) are a family of prevalent chronic pain disorders affecting
masticatory muscles and/or the temporomandibular joint. There is no unequivocally recognized
peripheral aetiology for idiopathic TMD. The central nervous system (CNS) may initiate and/or
maintain the pain in idiopathic TMD due to sustained or long-term nociceptive input that induces
maladaptive brain plasticity, and/or to inherent personality-related factors that may reduce the
brain's capacity to modulate nociceptive activity. The main aim of this thesis is to determine
whether there are structural neural abnormalities in patients with TMD, and whether these
abnormalities are related to TMD pain characteristics, or to neuroticism. The specific aims are to
delineate in TMD: (1) gray matter (GM) brain abnormalities and the contribution of pain and
neuroticism to abnormalities; (2) the contribution of abnormal brain GM aging in focal cortical
regions associated with nociceptive processes; and (3) abnormalities in brain white matter and
trigeminal nerve and the contribution of pain. In groups of 17 female patients with TMD and 17
age- and sex- matched controls, magnetic resonance imaging revealed that patients with TMD
had: (1) thicker cortex in the somatosensory, ventrolateral prefrontal and frontal polar cortices

iii
than controls, (2) cortical thickness in motor and cognitive areas that was negatively related to
pain intensity, orbitofrontal cortical thickness that was negatively correlated to pain
unpleasantness, and thalamic GM volume correlated to TMD duration, (3) an abnormal
relationship between neuroticism and orbitofrontal cortical thickness, (4) abnormal GM aging in
nociceptive, modulatory and motor areas, (5) widespread abnormalities in white matter tracts in
the brain related to sensory, motor and cognitive functions, (6) reduced trigeminal nerve integrity
related to pain duration, and (7) abnormal connectivity in cognitive and modulatory brain
regions. In sum, this thesis demonstrates for the first time abnormalities in both peripheral nerve
and CNS in patients with TMD.

Acknowledgments This thesis is dedicated to my father, who instilled in me a deep appreciation for learning, an
insatiable curiosity to understand the world, the drive to do so, and the humbleness to realize that
I may not understand everything. I owe my successes to his unwavering love and support.
I am grateful to Dr. Karen Davis for her mentorship. As a supervisor, Karen has a unique
understanding of her students’ needs and addressing them. Her training is unparalleled: the
emphasis and careful study of the body of literature is truly humbling. She has a deep
understanding of the scientific method, and her meticulous methodology maintains a level of
rigour and a high standard of quality in the work produced in her lab. Karen’s greatest skill is
transferring this deep appreciation to her students. Her patience and flexibility allow students to
find themselves a niche within the field, and to develop an expertise. Her rigour ensures that the
questions are addressed appropriately, and that studies are well-conducted. The greatest thing I
will take away from Karen’s lab is a strong sense of integrity. This is the greatest gift a young
scientist can be given – Thank you, Karen.
I am also very grateful to my committee members, Drs. Adrian Crawley, Barry Sessle and
Howard Tenenbaum, for their guidance and direction. They have helped me tremendously in the
development of the questions posed in this thesis, through helping me navigate the literature, and
understand complex concepts. I would like to especially thank Adrian for all of the time he spent
with me outside the lab, and the friendship we have developed. I would also like to thank Dr.
Mary Pat McAndrews – an unofficial committee member who has provided me with much
feedback, support and was always ready to lend an ear.
I also thank Dr. Bruce Freeman and Dr. Michael Goldberg for patient recruitment at the Mount
Sinai Dental Clinic, and Mr. Geoff Pope, Mr. Keith Ta and Mr. Eugen Hlasny for scanning our
subjects and helping us troubleshoot.
I would like to express my sincerest gratitude to my lab mates. I have had the opportunity to
work with several brilliant and hardworking colleagues. Udi Blankstein and Jerry Chen and I
joined the Davis lab in 2007, and we shared many experiences together. Working alongside them
created a very positive working environment, filled with ideas, and laughter. Keri Taylor and
Javeria Hashmi were PhD candidates when we joined the lab. They provided us with much

v
guidance, leadership and support. They also helped us stay on track and helped us develop a
deeper understanding of science. I am very grateful for time and the many conversations we had
together.
I would especially like to thank Irit Weissman-Fogel who was a post-doctoral fellow in our lab.
Without her hard work, dedication, meticulousness and patience, this thesis would have never
been completed. Her contributions to our data collection, study design and analysis were
enormous. Irit was also a unique role model – I will never forget her discipline at work, and her
amazing ability to maintain such a work-life balance. Her dedication to her work and her family
is nothing short of admirable.
During my training, the lab had a turnover, and the Davis lab was full of new faces and
colleagues. Drs. Nathalie Erpelding and Tim Salomons have been phenomenal colleagues and
are great friends. Both have helped me academically, and have become some of my closest
friends. Qi Wu, Danielle DeSouza, Aaron Kucyi, Gang Wang, and Ruma Goswami are great
additions to the lab, full of ideas. It’s been a real pleasure working alongside such brilliant minds
for the past five years, and I will take away many pleasant memories.
I would like to acknowledge the funding agencies that have made my research and this thesis
possible: The Ontario government for awarding me an Ontario Graduate Scholarship for the first
year of my graduate studies; the Canadian Institutes of Health Research (CIHR), for awarding
me a Banting and Best Canada Graduate Scholarship – Doctoral Research Award; the CIHR
Strategic Training Initiatives in Health Research programs: Pain: Molecules to Community and
Cell Signaling in Mucosal Inflammation and Pain for their support. This project was funded by
operating grants from the Canadian Institutes of Health Research and funds from Karen Davis’
Canada Research Chair.
I would like to thank my friends for their distractions, their support and their love. Your
encouragement got me through this!
I would also like to acknowledge Massey College – a truly unique place at the heart of the
University of Toronto. Heartfelt thanks to John Fraser and Elizabeth McCallum for making
Massey so special.

vi
Lastly, and most importantly, I thank my family. Albaloo, you are the most important person in
my life. You are the best role model. Thank you so much for everything. Chebli – what can I
say? No one is quite like you. You’re brilliant and I envy your creativity. Siamak – thank you for
being a groundstone, and a source of unwavering support, and always being the voice of reason. I
would like to thank my mom for all of her support, from close and afar. Mercedes and Debbie –
thank you both for your support. Lizbeth – merci pour ton support, ton amitié, et tes aiguilles. I
love you all!

Human beings are members of a whole, In creation of one essence and soul. If one member is afflicted with pain, Other members uneasy will remain. If you've no sympathy for human pain, The name of human you cannot retain!
-Sa’adi, Golestan, 1258

viii
It is a shame that we possess such insufficient knowledge concerning the character of pain – those symptoms which represent the essential part of all bodily suffering of man.
-A. Goldscheider, Ueber den Schmerz, 1894

ix
Table of Contents
Acknowledgments .......................................................................................................................... iv!
Table of Contents ........................................................................................................................... ix!
List of Tables ............................................................................................................................. xviii!
List of Figures .............................................................................................................................. xix!
List of Appendices ....................................................................................................................... xxi!
List of Abbreviations .................................................................................................................. xxii!
Chapter 1 Introduction and General Aims ...................................................................................... 1!
Chapter 2 Literature Review ........................................................................................................... 4!
2.1.! What is pain? ......................................................................................................................... 4!
2.1.1.! Historical perspectives and theories of pain ........................................................... 5!
2.1.1.1.! Specificity Theory ................................................................................... 5!
2.1.1.2.! Intensity theory of Pain ......................................................................... 11!
2.1.1.3.! Pattern Theory ....................................................................................... 12!
2.1.1.4.! Gate Control Theory of Pain ................................................................. 12!
2.1.2.! Multidimensional aspects of pain ......................................................................... 15!
2.1.3.! Chronification of pain ........................................................................................... 16!
2.1.3.1.! Sensitization, Hyperalgesia and Allodynia ........................................... 16!
2.1.3.2.! The role of glia in sensitization ............................................................. 18!
2.1.4.! Functional pain syndromes ................................................................................... 18!
2.1.4.1.! Central abnormalities in functional pain syndromes ............................. 19!
2.1.5.! Pain-cognition interactions ................................................................................... 19!
2.1.6.! Salience and pain .................................................................................................. 22!
2.1.7.! Pain-motor interactions ......................................................................................... 23!
2.1.8.! Neuroticism and pain ............................................................................................ 24!

x
2.2.! Temporomandibular disorders ............................................................................................ 25!
2.2.1.! Historical perspectives .......................................................................................... 25!
2.2.2.! Prevalence and social costs ................................................................................... 25!
2.2.3.! Clinical aspects ..................................................................................................... 26!
2.2.3.1.! Sensory abnormalities ........................................................................... 27!
2.2.3.2.! Motor abnormalities .............................................................................. 34!
2.2.3.3.! Cognitive abnormalities ........................................................................ 34!
2.3.! Anatomy and physiology of orofacial pain and sensation .................................................. 35!
2.3.1.! Peripheral receptors and sensory afferents ........................................................... 35!
2.3.2.! Orofacial nervous system: the trigeminal nerve ................................................... 37!
2.3.3.! White matter pathways ......................................................................................... 40!
2.3.4.! Nociceptive pathways and mechanisms ................................................................ 43!
2.3.4.1.! Peripheral nociceptive mechanisms ...................................................... 43!
2.3.4.2.! Spinal nociceptive pathways and mechanisms ...................................... 44!
2.3.4.3.! Trigeminal nociceptive pathways and mechanisms .............................. 47!
2.3.4.4.! Trigeminal motor pathways ................................................................... 49!
2.3.4.5.! Supraspinal nociceptive and pain regions ............................................. 52!
Thalamus ...................................................................................................... 54!
Primary somatosensory cortex .................................................................... 55!
The parasylvian cortex: S2 and insula ......................................................... 57!
The cingulate cortex ..................................................................................... 59!
Prefrontal cortex .......................................................................................... 62!
Motor regions .............................................................................................. 63!
2.3.5.! Descending modulation ........................................................................................ 66!

xi
2.3.5.1.! Descending modulation pathways ......................................................... 68!
2.3.5.2.! Diffuse noxious inhibitory controls ....................................................... 69!
2.4.! Structural brain imaging ..................................................................................................... 71!
2.4.1.! MRI ....................................................................................................................... 71!
2.4.1.1.! What is magnetic resonance? ................................................................ 71!
2.4.1.2.! MR signals ............................................................................................. 74!
Magnetization time (T1) ............................................................................... 74!
Signal decay time (T2) ................................................................................. 75!
Spatial Encoding .......................................................................................... 75!
Contrast ....................................................................................................... 76!
2.4.2.! Gray matter imaging ............................................................................................. 77!
2.4.2.1.! Voxel-based morphometry .................................................................... 77!
2.4.2.2.! Cortical thickness .................................................................................. 78!
2.4.3.! White matter imaging ........................................................................................... 79!
2.4.3.1.! Diffusion tensor imaging ....................................................................... 80!
2.4.3.2.! Between Group Comparisons of White Matter: Tract-based spatial statistics 85!
2.4.3.3.! Tractography ......................................................................................... 87!
Streamline (deterministic) tractography ...................................................... 88!
Probabilistic tractography ........................................................................... 89!
2.5.! Brain imaging of pain ......................................................................................................... 91!
2.5.1.! Functional neuroimaging of pain .......................................................................... 91!
2.5.1.1.! Imaging acute pain ................................................................................ 92!
S1 ................................................................................................................. 93!

xii
S2/Parietal operculum ................................................................................. 94!
Insula ........................................................................................................... 95!
Cingulate cortex ........................................................................................... 96!
Prefrontal cortex .......................................................................................... 97!
Cortical and subcortical motor regions ....................................................... 98!
2.5.1.2.! Imaging salience and pain ..................................................................... 98!
2.5.1.3.! Chronic pain .......................................................................................... 99!
2.5.2.! Structural imaging of pain .................................................................................. 101!
2.5.2.1.! Acute Pain ........................................................................................... 101!
2.5.2.2.! Chronic Pain ........................................................................................ 102!
Gray matter abnormalities ......................................................................... 102!
White matter abnormalities ........................................................................ 105!
2.6.! Neuroplasticity .................................................................................................................. 111!
2.6.1.! Use-dependent neuroplasticity ............................................................................ 112!
2.6.2.! Neuroplasticity and pain ..................................................................................... 113!
2.6.3.! Age-related neuroplasticity ................................................................................. 115!
2.6.4.! Microstructural basis of neuroplasticity .............................................................. 116!
Chapter 3 Rationale, Specific Aims and Hypotheses ................................................................. 118!
3.1.! Study I. Contribution of chronic pain and neuroticism to abnormal forebrain gray matter in TMD patients ............................................................................................................... 118!
3.2.! Study II. Age-related gray matter abnormalities in patients with TMD ........................... 120!
3.3.! Study III. White matter brain abnormalities in temporomandibular disorder ................... 121!
Chapter 4 General Methods ........................................................................................................ 123!
4.1.! Overview of Project .......................................................................................................... 123!

xiii
4.2.! Subject Recruitment .......................................................................................................... 123!
4.3.! Questionnaires ................................................................................................................... 124!
4.3.1.! Edinburgh Handedness Inventory ....................................................................... 124!
4.3.2.! NEO-Five Factor Inventory-Short Form ............................................................ 125!
4.3.3.! Pain Catastrophizing Scale ................................................................................. 125!
4.3.4.! Patient Interview ................................................................................................. 125!
4.4.! MR Imaging ...................................................................................................................... 126!
4.4.1.! Study design ........................................................................................................ 126!
4.4.2.! Imaging parameters ............................................................................................. 126!
4.4.3.! Gray Matter Analysis .......................................................................................... 127!
4.4.3.1.! Cortical Thickness Analysis ................................................................ 127!
4.4.3.2.! Voxel-Based Morphometry ................................................................. 130!
4.4.4.! White matter analysis .......................................................................................... 130!
4.4.4.1.! Tract-based spatial statistics ................................................................ 131!
4.4.4.2.! TBSS with other DTI metrics .............................................................. 131!
4.4.4.3.! Probabilistic tractography .................................................................... 132!
Chapter 5 STUDY I: Contributions of chronic pain and neuroticism to abnormal forebrain gray matter in TMD ............................................................................................................... 133!
5.1.! Introduction ....................................................................................................................... 133!
5.2.! Methods ............................................................................................................................. 135!
5.2.1.! Subjects ............................................................................................................... 135!
5.2.2.! Questionnaires ..................................................................................................... 135!
5.2.3.! Imaging parameters ............................................................................................. 136!
5.2.4.! Structural brain imaging analysis ........................................................................ 136!
5.2.4.1.! Cortical thickness analysis .................................................................. 136!
5.2.4.2.! Subcortical analysis with VBM ........................................................... 137!

xiv
5.2.5.! Statistical analyses .............................................................................................. 137!
5.2.5.1.! Demographics ...................................................................................... 137!
5.2.5.2.! Gray matter group differences ............................................................. 138!
CTA ............................................................................................................ 138!
VBM ........................................................................................................... 139!
5.2.5.3.! Clinical correlates ................................................................................ 139!
5.2.5.4.! Neuroticism effects .............................................................................. 139!
5.3.! Results ............................................................................................................................... 140!
5.3.1.! Patient demographics .......................................................................................... 140!
5.3.2.! Group differences: S1 and frontal thickening in TMD ....................................... 140!
5.3.3.! Effect of chronic pain intensity and unpleasantness ........................................... 141!
5.3.4.! Effect of TMD chronicity ................................................................................... 141!
5.3.5.! Effect of neuroticism ........................................................................................... 141!
5.4.! Discussion ......................................................................................................................... 154!
5.4.1.! S1 and thalamic gray matter increases in TMD .................................................. 154!
5.4.2.! Cortical thickening in cognitive and modulatory regions in TMD ..................... 154!
5.4.3.! Relationship between chronic pain intensity and cortical thickness in M1 and aMCC .................................................................................................................. 155!
5.4.4.! Interaction between neuroticism and group in prefrontal gray matter ................ 157!
5.4.5.! Study limitations ................................................................................................. 157!
5.5.! Conclusions ....................................................................................................................... 158!
Chapter 6 STUDY II: Age-related gray matter abnormalities in temporomandibular disorder . 159!
6.1.! Introduction ....................................................................................................................... 159!
6.2.! Methods ............................................................................................................................. 160!
6.2.1.! Subjects ............................................................................................................... 160!
6.2.2.! Imaging & Analysis ............................................................................................ 161!

xv
6.2.3.! Global age effects ............................................................................................... 161!
6.2.4.! Age-by-group effects in gray matter ................................................................... 161!
CTA ...................................................................................................................... 161!
VBM ..................................................................................................................... 162!
6.2.5.! Contribution of TMD duration to age-related gray matter abnormalities ........... 162!
6.3.! Results ............................................................................................................................... 163!
6.3.1.! Patient characteristics .......................................................................................... 163!
6.3.2.! Global age effects ............................................................................................... 163!
6.3.3.! Focal age effects ................................................................................................. 163!
6.3.4.! The contribution of TMD duration to gray matter age effects ............................ 164!
6.4.! Discussion ......................................................................................................................... 174!
6.4.1.! Whole brain gray matter atrophy ........................................................................ 174!
6.4.2.! Use-dependent plasticity ..................................................................................... 174!
6.4.3.! Cellular basis of changes in gray matter ............................................................. 176!
6.4.4.! Study limitations ................................................................................................. 177!
6.5.! Conclusion ........................................................................................................................ 177!
Chapter 7 STUDY III: White matter brain and trigeminal abnormalities in TMD .................... 178!
7.1.! Introduction ....................................................................................................................... 178!
7.2.! Methods ............................................................................................................................. 179!
7.2.1.! Subjects ............................................................................................................... 179!
7.2.2.! Questionnaires ..................................................................................................... 180!
7.2.3.! Imaging parameters ............................................................................................. 180!
7.2.4.! DTI pre-processing ............................................................................................. 181!
7.2.4.1.! Other DTI metrics ............................................................................... 181!
7.2.5.! Assessment of CNV ............................................................................................ 181!
7.2.6.! Tract-based spatial statistics ............................................................................... 182!

xvi
7.2.7.! Probabilistic tractography ................................................................................... 183!
7.2.8.! Statistical analyses .............................................................................................. 183!
7.2.8.1.! Whole brain white matter .................................................................... 183!
7.2.8.2.! Mask analysis ...................................................................................... 184!
7.2.8.3.! Quantitative Tractography: connection probability ............................ 185!
7.3.! Results ............................................................................................................................... 185!
7.3.1.! Patient characteristics .......................................................................................... 185!
7.3.2.! Trigeminal nerve FA ........................................................................................... 186!
7.3.3.! Patients have lower white matter FA .................................................................. 186!
7.3.4.! Probabilistic tractography ................................................................................... 187!
7.3.5.! White matter FA related to TMD pain characteristics ........................................ 188!
7.4.! Discussion ......................................................................................................................... 201!
7.4.1.! Trigeminal nerve abnormalities .......................................................................... 201!
7.4.2.! Abnormal sensorimotor tracts in TMD ............................................................... 202!
7.4.3.! Cognitive interference of pain and decreased modulation in TMD .................... 202!
7.4.4.! Abnormal connectivity in TMD .......................................................................... 203!
7.4.5.! Intense and prolonged TMD may drive plasticity in white matter ..................... 203!
7.4.6.! Cellular basis of changes in FA .......................................................................... 204!
7.4.7.! Study limitations ................................................................................................. 205!
7.5.! Conclusion ........................................................................................................................ 205!
Chapter 8 General Discussion ..................................................................................................... 206!
8.1.! Pain-driven abnormalities along nociceptive pathways .................................................... 207!
8.2.! Abnormalities in motor regions ........................................................................................ 209!
8.3.! Abnormalities in cognitive-modulatory brain regions ...................................................... 211!
8.4.! Neuroticism and TMD ...................................................................................................... 214!
8.5.! Basis of structural brain abnormalities ............................................................................. 216!

xvii
8.6.! Study limitations ............................................................................................................... 217!
8.7.! Future directions ............................................................................................................... 218!
8.8.! Conclusions ....................................................................................................................... 220!
References ................................................................................................................................... 221!
Appendices .................................................................................................................................. 305!
Copyright Acknowledgements .................................................................................................... 319!

xviii
List of Tables
Table 2-1: Summary of quantitative sensory testing studies in TMD .......................................... 30
Table 2-2: Number of quantitative sensory testing studies testing sensory thresholds and tolerance in TMD .......................................................................................................................... 33!
Table 2-3: Summary of regions with gray matter abnormalities in chronic pain ....................... 107
Table 5-1: Patient demographics ................................................................................................ 143!
Table 5-2: Group differences in cortical thickness ..................................................................... 145!
Table 5-3: Cortical thickness negatively correlates with TMD pain intensity or unpleasantness ............................................................................................................................. 146!
Table 6-1 Distribution of subject ages ........................................................................................ 166
Table 6-2: Age-related group differences in cortical thickness and subcortical gray matter
volume ......................................................................................................................................... 167!
Table 6-3: Contributions of TMD duration to the age-gray matter relationships ....................... 168
Table 7-1: Group differences in whole brain skeletonised white matter. ................................... 189!
Table 7-2: White matter regions in patients with TMD with significantly lower fractional anisotropy compared to controls ................................................................................................. 190!
Table 7-3: Group differences in mean diffusivity (MD), radial diffusivity (RD) and axial diffusivity (λ1) in clusters with significant group differences in FA .......................................... 191!

xix
List of Figures
Figure 2-1: Descartes’ line drawing of the pain system. ................................................................ 7
Figure 2-2: Schematic of the Gate Control Theory of Pain mechanisms ..................................... 14
Figure 2-3: Schematic of diffusion tensor imaging, and summary of eigenvectors and metrics .. 42
Figure 2-4: Diffusion tensor imaging reconstruction of Meynert’s classification of white matter tracts in the brain ........................................................................................................................... 84
Figure 2-5: White matter skeleton from tract-based spatial statistics ........................................... 87
Figure 4-1: Template deformation process for skull-stripping in FreeSurfer ............................ 128
Figure 4-2: Cutting planes overlaid on sections through white matter-labeled volume ............. 128
Figure 4-3 White surface overlaid on original T1 scan .............................................................. 129
Figure 4-4 Pial surface overlaid on original T1 scan .................................................................. 129
Figure 4-5 Original (left), gray/white boundary (middle), and pial surface (right) reconstructions of a left hemisphere ..................................................................................................................... 129
Figure 5-1: Masks used to restrict analyses ................................................................................ 147!
Figure 5-2: Cortical thickening in TMD ..................................................................................... 149!
Figure 5-3: TMD pain intensity and unpleasantness are negatively correlated to modulatory regions ......................................................................................................................................... 150 !
Figure 5-4: TMD duration is related to plasticity in the thalamus .............................................. 152!
Figure 5-5: Patients show an abnormal positive relationship between OFC thickness and neuroticism .................................................................................................................................. 153!
Figure 6-1: Total gray matter volume and correlations with age and duration ........................... 169!
Figure 6-2: Age-by-group interactions in cortical thickness ...................................................... 171!
Figure 6-3: Group differences in age effects within subcortical gray matter volume ................ 172!
Figure 6-4: Summary of age-related gray matter abnormalities ................................................. 173!
Figure 7-1: Schematic of diffusion tensor imaging, and summary of eigenvectors and metrics 182!
Figure 7-2: Colour orientation maps of the trigeminal nerves .................................................... 192!
Figure 7-3: Trigeminal nerve fractional anisotropy abnormalities in TMD ............................... 193!

xx
Figure 7-4: Group differences in mean fractional anisotropy between patients with TMD and controls ........................................................................................................................................ 195!
Figure 7-5: Group differences in fractional anisotropy between patients with TMD and controls ........................................................................................................................................ 196!
Figure 7-6: Clusters with significant group differences in fractional anisotropy ....................... 197!
Figure 7-7: Abnormal white matter connectivity in TMD .......................................................... 198!
Figure 7-8: Regions with group differences in fractional anisotropy are also correlated with TMD characteristics .................................................................................................................... 200
Figure 8-1 Proposed model of neural basis of sensory abnormalities in TMD .......................... 209
Figure 8-2 Proposed model of neural basis of motor abnormalities in TMD ............................. 211
Figure 8-3 Proposed model of the neural basis for cognitive abnormalities in TMD ................ 213
Figure 8-4 Proposed model for the neural basis of modulatory system abnormalities in TMD . 216

xxi
List of Appendices
Appendix I: Recruitment Letter .................................................................................................. 293
Appendix II: Consent forms ........................................................................................................ 294!
Appendix III: Edinburgh Handedness Inventory ........................................................................ 299
Appendix IV: Pain Catastrophizing scale ................................................................................... 300
Appendix IV: TMD pain assessment .......................................................................................... 301
Appendix V: McGill Pain Questionnaire .................................................................................... 304!
Appendix VI: McGill Pain Questionnaire results for patients with TMD .................................. 305!
Appendix VIII: Making Sense of Gray Matter Abnormalities in Chronic Orofacial Pain ......... 306!

List of Abbreviations
AAOP American Association of Orofacial Pain ACC Anterior cingulate cortex ADC Apparent diffusion coefficient aMCC Anterior mid-cingulate cortex AMH Aδ-mechano-heat sensitive afferent Amyg Amygdala ASSET Array Spatial Sensitivity Encoding Technique B0 Magnetic field BA Brodmann’s Area BCE Before Common Era BET Brain Extraction Tool BG Basal ganglia Cb Cerebellum CBP Chronic back pain cCMA Caudal cingulate motor area CH Cluster headache CL Centrolateral nucleus of the thalamus CM Centromedian nucleus of the thalamus CMA Cingulate motor area CMH C-mechano-heat sensitive afferent CNS Central nervous system CNV Cranial nerve five (trigeminal nerve) CNVII Facial nerve CNIX Glossopharyngeal nerve CNX Vagus nerve CPP Chronic pelvic pain CPTH Chronic posttraumatic headache CRPS Complex regional pain syndrome CSF Cerebrospinal fluid CTA Cortical thickness analysis CTTH Chronic tension-type headache DBM Deformation-based morphometry DCML Dorsal-column medial-leminiscal pathway dlPFC Dorsolateral prefrontal cortex DLPT Dorsolateral pontomesencephalic tegmentum dmPFC Dorsomedial prefrontal cortex DNIC Diffuse noxious inhibitory controls

xxiii
dODF Diffusion orientation density functions dPCC Dorsal posterior cingulate cortex DTI Diffusion tensor imaging DWI Diffusion-weighted imaging EC/ExC External/Extreme capsules EEG Electroencephalography FA Fractional anisotropy FDR False discovery rate FDT FSL Diffusion Toolbox fMRI Functional magnetic resonance imaging FMS Fibromyalgia fODF Fibre orientation density functions FSL FMRIB's Software Library FSPGR Fast spoiled gradient echo FWE Family-wise error FWHM Full-width half-maximum GLM General linear model GMV Gray matter volume GP Globus pallidus HC Hippocampus Hip OA Hip osteoarthritis HPC Heat-pinch-cold HT Hypothalamus HTM High-threshold mechanoreceptors HypH Hypnic headache IASP International Association for the Study of Pain IBS Irritable bowel syndrome IC Internal capsule ICAL Anterior limb of the internal capsule ICBM International Consortium for Brain Mapping iTL Inferior temporal lobe LC Locus coeruleus LEP Laser-evoked potentials LTM Low-threshold mechanoreceptors M1 Primary motor cortex MCC Mid-cingulate cortex MCS Motor cortex stimulation

xxiv
MD Mean diffusivity MD Medial dorsal thalamus MDvc Ventral caudal portion of the medial dorsal nucleus of the thalamus MEG Magnetoencephalography MeT Mesencephalic nucleus of the trigeminal nerve MNI Montreal Neurological Institute MOH Medication-overuse headache mPFC Medial prefrontal cortex MR Magnetic resonance MRI Magnetic resonance imaging MSN Main sensory nucleus NC Caudate nucleus NEO-FFI NEO-Five Factor Inventory NRM Nucleus raphe magnus NS Nociceptive-specific ODF Orientation density functions OFC Orbitofrontal cortex PAG Periaqueductal gray PCS Pain catastrophizing scale PDF Probability density function PET Positron-emission tomography Pf Parafascicular nucleus of the thalamus PFC Prefrontal cortex pgACC Pregenual anterior cingulate cortex PHG Parahippocampal gyrus PIFP Persistent idiopathic facial pain plIC Posterior limb of the internal capsule PMC Premotor cortex pMCC Posterior mid-cingulate cortex PMv Ventral premotor cortex PNS Peripheral nervous system PO Posterior nucleus of the thalamus PPC Posterior parietal cortex pTL Posterior temporal lobe Put Putamen PVD Provoked vestibulodynia QST Quantitative sensory testing RA Relative anisotropy

xxv
rCBF Regional cerebral blood flow rCMA Rostral cingulate motor area RCZ Rostral caudal zone RD Radial diffusivity rf Radiofrequency RF Reticular formation of the brainstem RFT Random field theory Rheum Arth Rheumatoid arthritis ROI Region of interest RVM Rostral ventromedial medulla S Signal S1 Primary somatosensory cortex S2 Secondary somatosensory cortex SC Subcoeruleus sgACC Subgenual cingulate cortex SM Submedian nucleus of the thalamus SMA Supplementary motor area SN Substantia nigra SPECT Single-photon emission computed tomography SPM Statistical parametric mapping STG Superior temporal gyrus STN Spinal trigeminal nucleus STT Spinothalamic tract SVC Small-volume correction T1 Spin-lattice relaxation time or Magnetization time T2 Spin-spin relaxation time or Signal decay time TBSS Tract-based spatial statistics TE Echo time Thal Thalamus TI Inversion time TIV Total intracranial volume TL Temporal lobe TMD Temporomandibular disorder TMD-RDC Temporomandibular disorder research diagnostic criteria TMJ Temporomandibular joint TMS Transcranial magnetic stimulation TN Trigeminal neuralgia TNP Trigeminal neuropathic pain TR Repetition time TTT Trigeminothalamic tract uODF Uncertainty orientation density functions

xxvi
V1 Ophthalmic branch of the trigeminal nerve V1 Primary visual cortex V2 Maxillary branch of the trigeminal nerve V3 Mandibular branch of the trigeminal nerve VBM Voxel-based morphometry VBSNC Trigeminal brainstem nuclear complex Vc Subnucleus caudalis Vi Subnucleus interpolaris VL Ventrolateral nucleus of the thalamus vlPFC Ventrolateral prefrontral cortex vmPFC Ventromedial prefrontal cortex vmPO Posterior region of the ventromedial nucleus of the thalamus Vo Subnucleus oralis vPCC Ventral posterior cingulate cortex VPI Ventroposterioinferior nucleus of the thalamus VPL Ventroposteriolateral nucleus of the thalamus VPM Ventroposteriomedial nucleus of the thalamus vSTR Ventral striatum WDR Wide-dynamic range γ Gyromagnetic ratio λ1 Longitudinal diffusivity ρ Proton density τ Diffusion time ω0 Precession rate

1
Chapter 1 Introduction and General Aims
Temporomandibular disorders (TMD) comprise clinical problems involving the structures of and
around the temporomandibular joint (TMJ), the masticatory musculature, or both [1,418]. TMD
represent the most common orofacial chronic pain disorder [285], and are primarily characterized
by spontaneous pain, or pain associated with jaw function, which can affect mandibular range of
motion and produce TMJ sounds during jaw function. Pain from TMD can arise from the
muscles of mastication, the TMJ, or both, and, in general, when combined (i.e., muscular and
TMJ pain) the chief complaint tends to be muscular in nature [402].
There have not yet been any comprehensive epidemiological studies of TMD in the Canadian
population, and so this thesis will refer to data from the United States. TMD are estimated to
affect between 3-20 percent of the United States’ adult population [269,539,540,562]. Of these,
only a small proportion of persons suffering from TMD pain seek treatment, and are seen at
tertiary and quaternary medical facilities, such as pain clinics [269,350]. It has been estimated
that TMD cost $4 billion annually due to lost-wages and medical treatment. TMD 1.5-9 times
more prevalent in women [113,201,284,285,312,721] and a nationwide epidemiological study in
the United States reported that 84% of persons with TMD were women [284].
In some cases, there is no clear aetiological evidence for TMD pain, and the pain is considered
idiopathic [283,286,287,653]. In this group of patients, it has been suggested that abnormal
function in the central nervous system (CNS) may initiate or maintain TMD pain [757]. One line
of evidence for abnormal CNS function is that TMD symptomatology, including persistent pain,
allodynia, and hyperalgesia occurs not only in the orofacial region, but also in other body sites
[305,314,390,565,756,883]. Patients with TMD also show greater temporal summation of pain to
repetitive noxious heat stimuli [563], and dysfunctional diffuse noxious inhibitory controls
(DNIC) [113,491,756]. These centrally-mediated processes provide further evidence for CNS
dysfunction in TMD and provide evidence for the involvement of ascending nociceptive
pathways and/or descending pain-modulatory pathways [515]. Additionally, patients with TMD
can exhibit cognitive [370,379,380] and motor dysfunction [837] possibly related to
abnormalities in brain regions associated with these functions [799,800,839,928].

2
There are two main routes by which the CNS may contribute to the development and/or
maintenance of chronic pain conditions such as TMD. One possibility is that long-term
nociceptive input into the brain induces maladaptive brain plasticity, which may play a role in
maintaining pain [12,213,576,950]. This maladaptive plasticity suggests that patients with TMD
may be unable to habituate to increased nociceptive activity, which may be related to a reduced
capacity of the brain to dampen pain by descending (top-down) controls [95].
The second route by which the CNS may contribute to the development and/or maintenance of
chronic pain relates to inherent personality-related factors that reduce the brain’s capacity to
modulate nociceptive input. This poor control of pain might lead to the development of
vulnerability towards the development of chronic pain. For example, there is evidence that
neuroticism may be associated with pain-related suffering [408], pain sensitivity [177,373,907],
nerve injury outcomes as well as neuropathic pain [848], inhibition of negative thoughts [178],
and the development of TMD [315].
Structural magnetic resonance imaging (MRI) and diffusion-weighted imaging (DWI) can be
used to investigate the structure of CNS gray and white matter, respectively. Indeed, many
structural MRI studies have demonstrated differences in gray matter in populations of patients
with chronic pain associated with pain-related characteristics (e.g., intensity, unpleasantness, or
duration) (see [98,206,303,356,575,764,772]). Furthermore, structural MRI studies have reported
accelerated loss of gray matter in the brains of patients with chronic pain [27,502]. These age-
related changes in chronic pain may be related to the cumulative effect of pain over time (i.e.,
pain duration) or other pre-existing factors that may alter the normal pattern of aging in TMD. In
addition to abnormalities in central gray matter found in the presence of chronic pain, previous
studies examining measures of white matter integrity in other clinical conditions with sensory
abnormalities and/or chronic pain reported abnormalities in white matter tracts related to sensory,
modulatory and cognitive functions [156,356,558,847]. Furthermore, these studies reported
white matter abnormalities correlated with clinical findings in the patients. Therefore, studying
the correlation between TMD characteristics and measures of white matter integrity can provide
insight into whether chronic pain drives changes in white matter microstructure.
With regard to the second route by which the CNS may contribute to the development and/or
maintenance of chronic pain, pre-existing abnormalities may be a factor for the development of

3
TMD. These structural abnormalities in the CNS could be related to personality traits that are
related to chronic pain, such as neuroticism [315]. However, not all patients with chronic pain
have high neuroticism scores, and not all persons with neuroticism have chronic pain [179].
Therefore, neuroticism alone is not sufficient to develop chronic pain. Rather, there may exist an
abnormal relationship between neuroticism and brain structure, which may impair function in
pain-modulatory brain regions, such as the orbitofrontal cortex (OFC) [954] or the medial
prefrontal cortex (mPFC) [245,386] and this could facilitate or maintain chronic pain. However,
the precise mechanism by which neuroticism can contribute to TMD development remains to be
elucidated.
Given the poor understanding of the mechanisms underlying TMD, it is challenging to develop
rational treatment approaches for this chronic pain condition. Moreover, it is becoming clearer
that TMD may be mediated by central mechanisms. However, while there are some data
demonstrating neuropsychological and cognitive, as well as psychosocial contributors to TMD,
there are little data demonstrating whether there are underlying neuroanatomical changes that
could support the notion that TMD is a centrally-mediated pain condition.
Therefore, the main aims of this thesis are:
1. To examine gray matter abnormalities in patients with idiopathic TMD and to
determine the contribution of pain-related characteristics and neuroticism;
2. To determine whether the cumulative effect of idiopathic TMD interacts with the
normal aging process in focal cortical regions associated with nociceptive processes;
3. To evaluate white matter brain and trigeminal nerve (cranial nerve five: CNV)
abnormalities in patients with idiopathic TMD and to determine the contribution of
pain-related characteristics.

4
Chapter 2 Literature Review
2.1. What is pain?
The current definition of pain, established by International Association for the Study of Pain
(IASP) in 1986, defines pain as “an unpleasant sensory and emotional experience associated with
actual or potential tissue damage, or described in terms of tissue damage, or both.” It is important
to recognize that pain and nociception are not synonymous. Nociception, based on the 2011
IASP taxonomy [110], is “the neural process of encoding noxious stimuli” and is initiated by the
activation of peripheral receptors that preferentially respond to stimuli in the noxious range,
referred to as nociceptors. It includes the neural activity in the peripheral nervous systems (PNS)
and CNS, elicited by a noxious stimulus. In contrast to nociception, pain requires a conscious
perception.
Pain is a complex, multidimensional experience involving sensory, affective, cognitive,
attentional, motivational, and motor components [593]. The sensorial dimension of pain relates
to the discriminability of pain qualities, location, intensity and duration of pain. Pain is inherently
salient. Based on the context, our psychological and affective state, we initiate a response to
noxious stimulus. Therefore, different people respond to pain differently.
Pain can be categorized based on its temporal profile: short-term pain is known as acute pain,
and lasts from seconds to days. Acute pain plays an essential role in signaling actual or potential
tissue damage [442]. However, chronic pain persists more than three to six months, and is not
necessarily related with tissue damage [791]. The aetiology of chronic pain is not as evident as it
is in acute pain. Often, chronic pain persists beyond the time necessary for the tissue to heal
normally following injury and, in these cases, it is believed that nociceptive input and/or the CNS
maintain pain [576]. The IASP has devised a classification scheme for chronic pain [600], based
on five axes: (1) the region of the pain, (2) the system affected, (3) temporal characteristics of the
pain, (4) time since onset, and (5) the aetiology of the pain.

5
2.1.1. Historical perspectives and theories of pain
A number of theories have been postulated to describe mechanisms underlying pain perception.
These theories date back several centuries, and even millennia [483,729]. This thesis will
primarily focus on theories that were postulated since the 17th century.
2.1.1.1. Specificity Theory
Specificity theory refers to the presence of dedicated pathways for each somatosensory modality.
The fundamental tenet of the specificity theory is that each modality has a specific receptor and
associated sensory fibre (primary afferent) that is sensitive to one specific stimulus [273]. For
instance, the model proposes that non-noxious mechanical stimuli are encoded by low-threshold
mechanorecpetors (LTM), which are associated with dedicated primary afferents that project to
mechanoreceptive second-order neurones in the spinal cord or brainstem (depending on the
source of the input). These second-order neurones project to higher mechanoreceptive areas in
the brain. Similarly, noxious stimuli would activate a nociceptor, which would project to higher
pain centres through a pain fibre. These ideas have been emerging over several millennia, but
were experimentally tested, and formally postulated as a theory in the nineteenth century by
physiologists in Western Europe. This thesis will briefly review some of the key ideas that have
led to the development of specificity theory.
René Descartes was one of the first western philosophers to describe a detailed pain pathway in
humans. Descartes’ manuscript Treatise of Man (originally written in French) was illustrated,
edited and published posthumously, first in Latin in 1662 [241], and then in French in 1664
[242]. In Treatise of Man, based on the French edition by Louis La Forge (who was also one of
the illustrators), Descartes describes pain as a perception that exists in the brain and makes the
distinction between the neural phenomenon of sensory transduction (today known as
nociception) and the perceptual experience of pain. What is essential to the development of
Descartes’ theory is his description of nerves, which he perceived as hollow tubules that convey
both sensory and motor information. This understanding of neural function was by no means
novel. In the 3rd century BCE, Herophilus demonstrated the existence of sensory and motor
nerves, and Erasistratus demonstrated that the brain influenced motor activity [729]. Half a
millennium later, Galen demonstrated that sectioning the spinal cord caused sensory and motor
deficits [651]. Within the spirit of scientific enquiry that resurfaced in the renaissance,

6
anatomical studies by Vesalius published in 1543 reiterated and confirmed Galen’s findings
[651]. In relation to this, Galen had postulated that three conditions be met for perception: (1) an
organ must to be able to receive the stimulus, (2) a connection from the organ to the brain, and
(3) a processing centre that converts the sensation to a conscious perception [729]. Descartes
contributed to Galen’s model by postulating that a gate existed between the brain and the tubular
structures (the connections), which was opened by a sensory cue [242]. A sensory cue would
“tug” on the tube, which would then open a gate between the tube and the brain. The opening of
this gate would then allow “Animal Spirits” (an extension of the Greek pneuma1) to flow through
these tubes and within the muscles to move them. Although this sensory system was not specific
to pain, La Forge’s drawing (based on Descartes’ concept) of a foot near a flame is one of the
most ubiquitous figures in neuroscience (see Figure 2-1). This example describes the pathway for
promptly moving one’s foot away from a hot flame. In the figure (and its description in the text)
the heat of the flame near the foot activates a fibril (or fibre) within the nerve tubule that
traverses up the leg, to the spinal cord and finally to the brain. Descartes compared this fibre to a
cord attached to a bell – by pulling on the other end of cord, the bell will ring. The proverbial
bells in this case are the pores that line the ventricles in the brain. Once these pores open in
response to the sensory input, the Animal Spirits were thought to flow through the tubule and
elicit a motor response. This motor response included turning the head and the eyes to see the
flame, and raising the hands and folding the body away from the flame for protection. Descartes
conceived that there are many of these fibrils and that their movements elicit the sensations. For
example, the perception of pain would be felt in the brain when there is a significant “tug” on the
fibre, which caused it to sever. In contrast, a tug of the same magnitude that doesn’t cause the
fibre to break would evoke a tickling (or tingling – Descartes uses the French word
chatouillement) perception. Although Descartes’ figure of the boy and the flame suggests that
there is a dedicated pain pathway, a closer read of the text indicates that Descartes believed that
the pattern and rate of firing (intensity of tugging) of a fibre provided the adequate information to
the brain about the stimulus intensity and quality.
1In ancient Greek medicine, the concept of pneuma represents air, breath, or motion. It was also called the breath of
life and the spirit of man. [87] Bernard C, Magendie F. Leçon d’ouverture du Cours de Médecine du Collège de France. Paris: Baillière, 1856.

7
Figure 2-1: Line drawing of the pain system by Florentius Schuyl (left) and Louis La Forge (right), based on Descartes description in Treatise of Man: The fire (A) is close to the foot (B). Particles from the fire move and press the skin, and tug on the fibril (C), which opens the pore (d, e) where the fibril terminates. Opening the pore is akin to tugging on a rope attached to a bell, thus ringing the bell. The open pore allows the “animal spirits” to flow from the cavity (F) into the fibril and, part of them activate the muscles to move the foot away from the fire, and part of them activate the muscles to turn the eyes and the head toward the fire to look at it, and part of them are used to bring forth the hands and fold the body to protect it. (Image on the left is reproduced from and the image on the right and text is reproduced from [242], out of copyright; translated by Massieh Moayedi).

8
The concept of a dedicated pain pathway (also known as specificity theory) was developed by
Charles Bell in his landmark essay Idea of a new anatomy of the brain; submitted for the
observation of his friends, first published as a conference proceeding in 1811, and later
reproduced in a journal [79]. In this essay, Bell provided an alternative perspective about the
organization of the nervous system. First he suggested that the brain is not “common sensorium”
as suggested by Descartes, which was the accepted model of the brain at the time. Instead, he
provided anatomical evidence that the brain was a heterogeneous structure, a theory first
postulated by Willis in the seventeenth century [729]. He then suggested that nerves were
bundles of heterogeneous neurones that have specialized functions, and that their bundling was
only for ease of distribution. Thus, Bell spoke of different sensory neurones for different types of
stimuli, motor neurones, and so-called vital neurones that are wired to the mind, rather than the
brain. He did, however, maintain that perception of stimulus (such as vision and nociception) is
different than the perceptual experience (e.g., colour and pain, respectively). Importantly for
specificity theory, Bell states:
…that while each organ of sense is provided with a capacity of receiving certain changes; to be played upon it, as it were, yet each is utterly incapable of receiving the impressions destined for another organ of sensation. It is also very remarkable that an impression made on two different nerves of sense, though with the same instrument, will produce two distinct sensations; and the ideas resulting will only have relation to the organ affected [79].
This is the fundamental tenet of specificity theory, which postulates that there is a dedicated fibre
that leads up a dedicated pain pathway to the sensory modality’s region of the brain. This model,
therefore, suggests that a pathway specific to pain exists.
François Magendie was a French physician considered by some as the father of experimental
physiology [87,773,827]. Magendie made substantial contributions to neurophysiology,
including reiterating Bell’s findings regarding the existence of both motor and sensory nerves,
and that these have separate paths to and from the spinal cord (the ventral and dorsal roots,
respectively) [827]. This differentiation of spinal nerves is known as the Bell-Magendie Law,
which is a fundamental aspect of the organization of the nervous system.

9
Concurrently in Germany, Johannes Müller published a Manual of Physiology, which echoed
Charles Bonnet’s manual published a century earlier [729]. Müller’s manual, published in 1840,
sought to summarize and synthesize findings in physiology. The purpose of this synthesis was to
understand how different stimuli were so clearly sensed and how the brain could distinguish
them from one another. He, like Bonnet, concluded that specific receptors have must have
specific energy of stimulation, and that there were infinite numbers and types of fibres, each to a
specific sensory stimulus; e.g., there is a specific fibre for the smell of bananas, and another for
the scent of an apple, and yet another for the scent of an orange. Furthermore, because of a sense
organ’s specific energy, the sensory neurone will only encode a single perceptual quality. For
example, if a warm fibre is artificially stimulated by electric current, the brain will perceive
warmth. In line with these findings, Erasmus Darwin (Charles Darwin’s grandfather) provided
the first evidence for a set of specific nerves for the perception of heat [204].
The discovery of specific cutaneous touch receptors such as Pacinian corpuscles [148],
Meissner’s corpuscles [149], Merkel’s discs [394], and Ruffini’s end-organs [393] in the latter
half of the nineteenth century provided further evidence that specific sensory qualia were
encoded by dedicated nerve fibres (see Section 2.3.1). However, there remained a debate about
the nature of pain as part of the five senses. An end-organ specific to pain stimuli (nociceptor)
had not yet been discovered. In contrast to the idea of a dedicated pain pathway, it was argued
that pain was different than the other senses in that it is inherently unpleasant [106,199]. These
ideas persisted from Plato and Aristotle’s writings of pain as an emotion [767]. This inherently
makes pain the antithesis of pleasure, and, because pleasure is a characteristic of the mind (i.e.,
an emotion), it was inferred that pain was also a characteristic of the mind, and not a percept of
the body.
Further evidence for the specificity theory came from Schiff’s and Woroschiloff’s findings of a
pain pathway in the spinal cord in a series of experiments between 1854 and 1859. These
findings built upon Charles-Édouard Brown-Séquard’s observations that sensory fibres decussate
in the spinal cord [18,199]. Schiff and Woroschiloff established the presence of two pathways
through observations of the effect of incisions at different levels of the spinal cord: the
anterolateral pathway for pain and temperature, and the posterior bundles for tactile sensibility
[199,729]. However, Schiff and Woroschiloff noted that the tactile pathway did not decussate at
the level of the spinal cord. These findings were supported by a case study reported by William

10
Richard Gowers, a physician in London. In this report, Gowers reported that a patient with a
bullet wound to the gray matter of the spinal cord lost the sense of pain and temperature, but not
touch [729]. He concluded that there were specific pathways for pain and temperature, separate
from that of touch. However, those who held onto the Aristotelian dogma argued strenuously
against specificity theory. They insisted that pain is a quality of all senses, a percept of the mind.
Only when Blix and Goldscheider published their findings of sensory spots on the skin
independently did specificity theory gain momentum, and did pain become a recognized sense
[199]. Sensory spots were defined as tiny areas of the skin that elicit a specific sensation when
touched. These sensory spots were specific to warmth, cold, pressure or pain. However, both
Blix and Goldscheider moved away from specificity theory some years later, and moved towards
the intensity theory of pain, a concurrent theory (see section 2.1.1.2).
Between 1894 and 1896, Max von Frey’s carried out experiments that advanced the specificity
theory. Von Frey indicated that there were four somatosensory modalities: cold, heat, pain and
touch and that all of the other skin senses were derivatives of these four modalities. To test this
idea, he developed his now well known von Frey hairs (termed an aesthesiometer) that consisted
of a hair, usually from a human, but sometimes he used a horsehair or a hog bristle, attached to a
wooden stick [682]. By measuring the hair’s diameter, length and the maximal weight precisely
it could support (maximal tension) without breaking off of the stick, it was possible to measure
the force applied to a very specific spot. Today, von Frey hairs are made of fine nylon filaments
of varying thicknesses (and hence stiffness) to deliver different forces and pressures upon
bending. Using these hairs, he could carefully determine the pressure required to elicit a
sensation at each of the skin spots identified by Blix and Goldscheider. Further, his experimental
setup allowed him to determine which spots responded to innocuous pressure, and which ones
responded noxious pressure. Von Frey demonstrated that there were distinct spots for innocuous
pressure and for noxious pressure. He presented a model of the skin that comprised of a “mosaic
of distinct tactile, cold, warm, and pain spots distributed across the skin with distinctive regional
variation” [683]. Von Frey related the distribution of the pressure points to the distribution of
Meissner’s corpuscles (see section 2.3.1), whereas pain points were related to the distribution of
free nerve endings in the skin (see section 2.3.3.1). Despite these remarkable findings, the
specificity theory made a number of assumptions about the anatomical basis of somesthesis. For
instance, when von Frey postulated the theory, pain receptors had yet to be identified, nor were

11
the peripheral pathways and brain centers specific to pain sensation established, as well as other
factors (for a review, see [199,729]).
Charles Scott Sherrington addressed some of the assumptions of the specificity theory in his
proposed framework of nociception. He applied a Darwinian (i.e., evolutionary) approach to
describe the specificity of neurones, which included the four basic modalities recognized by von
Frey. He stated that “the main function of the receptor is […] to lower the excitability threshold
of the arc for one kind of stimulus and heighten it for all others” [796,797]. This approach
resolved the divide between the intensity theory (see below) and specificity theory [729]. He also
coined the term “nocicipient” [795] to describe the specificity of the cutaneous end-organ for
pain, later termed nociceptor [796]. Sherrington developed a framework that advanced the
specificity theory of pain even further. However, the nociceptor had yet to be identified
definitively.
The discovery of myelinated primary afferent fibres that respond only to mechanical noxious
stimuli occurred much later – in 1967 [124]. Soon thereafter, Bessou and Perl [89] discovered
nociceptive unmyelinated afferent fibres: polymodal nociceptors and high-threshold
mechanoreceptors (HTM). These findings revolutionized the field of pain research, and helped
advance and develop a number of theories of pain. Since Sherrington’s endorsement of the
specificity theory of pain, this became the dominant theory at the time. However, its popularity
waned with the postulation of the Gate Control Theory of pain (see Section 2.1.1.4) postulated
by Melzack and Wall [595].
2.1.1.2. Intensity theory of Pain
The intensive (or summation) theory of pain (now referred to as the intensity theory) has been
postulated at several different times throughout history. First postulated in the 4th century BCE
by Plato in his œuvre Timaeus [698], the theory defines pain not as a unique sensory experience,
but rather as an emotion that occurs when a stimulus is stronger than usual. Centuries later,
Erasmus Darwin reiterated this concept in Zoonomia [204]. One hundred years after Darwin,
Wilhelm Erb also suggested that pain occurred in any sensory system when sufficient intensity
was reached, rather than being a stimulus modality in its own right [199]. Arthur Goldscheider
further advanced the intensity theory, based on an experiment performed by Bernhard Naunyn in
1859 (cited in: [199]). These experiments showed that repeated tactile stimulation (below the

12
threshold for tactile perception) produced pain in patients with syphilis who had degenerating
dorsal columns. When this stimulus was presented to patients 60-600 times per second, they
rapidly developed what they described as unbearable pain. Naunyn reproduced these results in a
series of experiments with different types of stimuli, including electrical stimuli. It was
concluded that there must be some form of summation that occurs, in order for the subthreshold
stimuli to become unbearably painful. Goldscheider’s suggested a neurophysiological model to
describe this summation effect: repeated subthreshold stimulation or suprathreshold hyper-
intensive stimulation could cause pain. Goldscheider suggested that the increased sensory input
would converge and summate in the gray matter of the spinal cord. This theory competed with
the specificity theory of pain, which was championed by von Frey. However, the intensity theory
lost support with Sherrington’s evolutionary framework for specificity theory; and postulated the
existence of sensory receptors that are specialized to respond to noxious stimuli, for which he
coined the term “nociceptor”.
2.1.1.3. Pattern Theory
In an attempt to overhaul theories of somaesthesis (including pain), J. P. Nafe postulated a
“quantitative theory of feeling” [636]. This theory ignored findings of specialized nerve endings
and many of the observations supporting the specificity and/or intensive theory of pain. The
pattern theory stated that any somaesthetic sensation occurred by a specific and particular pattern
of neural firing, and that the spatial and temporal profile of firing of the peripheral nerves
encoded the stimulus type and intensity. Lele, Sinclair and Weddell [531] championed this
theory, and added that cutaneous sensory nerve fibres, with the exception of those innervating
hair cells, are the same. To support this claim, they cited work that had shown that distorting a
nerve fibre would cause action potentials to discharge in any nerve fibre, whether encapsulated
or not. Furthermore, intense stimulation of any of these nerve fibres would cause the percept of
pain [807,924].
2.1.1.4. Gate Control Theory of Pain
In 1965, Ronald Melzack and Patrick (Pat) David Wall proposed a theory that would
revolutionize pain research: the Gate Control theory of pain. The Gate Control theory recognized
the experimental evidence that supported specificity theory and pattern theory, and provided a
model that could explain these seemingly opposed findings. In a landmark paper, Melzack and

13
Wall [595] carefully discussed the shortcomings of the specificity and pattern theories – the two
dominant theories of the era. The Gate Control theory attempted to bridge the gap between
intensity and specificity theory with a framework based on the aspects of each theory that had
been corroborated by physiological data. Specifically, Melzack and Wall accepted that there are
nociceptors (pain fibres) and touch fibres, and proposed that that these fibres synapse in two
different locations within dorsal horn of the spinal cord: the substantia gelatinosa and the
“transmission” cells. The model (see Figure 2-2) proposed that signals produced in primary
afferents from stimulation of the skin were transmitted to three regions within the spinal cord: (1)
the substantia gelatinosa, (2) the dorsal column, and (3) a group of cells that they called
“transmission” cells. They proposed that the gate in the spinal cord is the substantia gelatinosa in
the dorsal horn, which modulates the transmission of sensory information from the primary
afferent neurones to “transmission cells” in the spinal cord. This gating mechanism is controlled
by the activity in the large and small fibers. Large fiber activity inhibits (or closes) the gate,
whereas small fiber activity facilitates (or opens) the gate. Activity from descending fibres from
supraspinal regions to the dorsal horn could also modulate this gate. When nociceptive
information reaches a threshold that exceeds the inhibition elicited, it “opens the gate” and
activates pathways that lead to the experience of pain and its related behaviours. Therefore, the
Gate control theory of pain provided a neural basis for the findings that supported and in fact
helped to reconcile the apparent differences between the pattern and specificity theories of pain.

14
Figure 2-2: Schematic of the Gate Control Theory of Pain mechanisms. L – large diameter fibres; S – small diameter fibres. The fibres project to the substantia gelatinosa (SG) and first central transmission (T) cells. The inhibitory effect exerted by SG on the afferent fibre terminals is increased by activity in L fibres and decreased by activity in S fibres. The central control trigger is represented by a line running from the large-fibre systerm to the central control mechanisms; these mechanisms, in turn, project back to the gate control system. The T cells project to the entry cells of the action system. +, Excitation; –, inhibition. (Figure and legend are reproduced with permission from: [595]).

15
2.1.2. Multidimensional aspects of pain
Melzack and Casey [593] described pain as being multidimensional and complex, with sensory-
discriminative, affective-motivational and a cognitive-evaluative components. Additionally,
recent work has shown that pain can also affect and interact with motor systems [23]. The
concept of pain as a multidimensional experience has been described in ancient texts, dating as
far back as the Syriac Empire (circa 200 BCE). In the Book of Medicines [122], it is suggested
that pain is the product of bile and phlegm mingled with cold and heat. These simple
combinations occur in the brain, and, according to Syriac medicine, pain is a product of the brain
(a concept which has passed the test of time, and that we still hold true today). Different types of
pains would thus arise from differential combinations of these substances affecting the type of
pain. It is noteworthy that the concepts of bile and phlegm, and even those of cold and hot were
understood in a different paradigm of philosophical thought – these are not the simple
compounds we know today, but are rather used as a classification of the world. For instance,
certain foods make the body “cool”, whereas others make the body “warm.” These concepts are
not unique to the Syrians, since they follow a long tradition of ancient medicine passed down
from the Egyptians (who were the first to record medical texts, e.g., The Papyrus Ebers [120]), to
the Greeks (e.g., most-famously, Hippocrates and Galen), to the Babylonians and to the
Assyrians.
The contemporary definition used by the IASP is based on the divisional (multidimensional)
definition proposed by Melzack and Casey, in 1968 [593]. These dimensions include the
sensory-discriminative (intensity, location, quality, and duration), the affective-motivational
(unpleasantness and the subsequent flight response) and the cognitive-evaluative (appraisal,
cultural values, context and cognitive state) dimensions of pain. These three dimensions are not
independent, but rather, interact with one another. They are, however, partially dissociable: the
cognitive state of a person can modulate one or both of these dimensions of pain perception. In
general, the more intense a noxious stimulus is, the more unpleasant it will be [279]. However,
there are exceptions to this rule: hypnosis has been shown to modulate pain unpleasantness
without affecting intensity – that is, the person felt the pain, but was not as bothered by the
sensation [500,590,714-716,937]. This is an example of how the cognitive state can modulate the
percept of the affective-motivational component of pain and can be referred to as cognitive

16
modulation. Cognitive modulation of pain is reflected in the effects of placebo, nocebo
[171,172,295,908], cognitive behavioural therapy [752,753,793] and other treatments for chronic
pain.
Theories about chronic pain have continued to evolve as knowledge accumulates concerning the
structure and function of pathways underlying pain perception and pain modulation. Recent
advances in neuroimaging and cellular and molecular medicine have vastly expanded our
understanding of pain, and some of these key findings relevant to this thesis will be discussed
further when anatomical pathways and brain regions implicated in pain are described (see
Sections 2.3).
2.1.3. Chronification of pain
An important issue that is not fully understood is the way in which acute pain becomes chronic.
For example, how does pain change from being a beneficial signal of a potential threat that
engages protective mechanisms, to become a state where pain itself becomes a threat,
dysfunction, despair, and much suffering.
2.1.3.1. Sensitization, Hyperalgesia and Allodynia
When the skin is injured and pain is evoked, the tissue can in some situations become tender and
inflamed. The tenderness can be associated with two processes: allodynia and hyperalgesia.
Allodynia is defined as a “pain due to a stimulus that does not normally provoke pain” [110]. The
definition further specifies that allodynia is not related to any specific mechanism; rather it
specifies an unexpected behavioural response (pain) to a normally innocuous stimulus.
Furthermore, to demonstrate allodynic responses, it is preferred that responses be tested in
unaffected body areas to show normal responses, where possible. Because the normative
response to the stimulus is not painful, and the allodynic response is painful, “allodynia involves
a change in the quality of the sensation” [110]. Hyperalgesia is defined as a “increased pain from
a stimulus that normally provokes pain” [110]. There are two types of hyperalgesia: primary (at
the injury site) and secondary (outside the site of injury). These types of hyperalgesia can be
described with the following example. When glabrous skin (non-hairy skin, like the palm of the
hand) is burned, the burn spot (primary zone) becomes more sensitive to heat and mechanical
stimuli – a stimulus that was mildly painful before the burn is subsequently perceived as being

17
much more painful [609]. In normal cases, it is believed that allodynia and hyperalgesia provide
warning signs, and are likely related to nocifensive behaviours – that is, behaviours that are
meant to protect the damaged limb, prevent further damage, and ultimately promote healing.
The perceptual states of hyperalgesia and allodynia are related to the physiological process
known as sensitization, which is defined as “increased responsiveness of nociceptive neurones to
their normal input, and/or recruitment of a response to normally subthreshold inputs” [110].
There are different mechanisms of sensitization, depending on the type of tissue and/or injury
(chemical, mechanical or heat) [610]. This sensitization process can result from threshold related
changes in nociceptors (i.e., lower firing threshold to sensory stimuli and/or an increased firing
rate to suprathreshold stimuli, spontaneous firing and/or an expansion of the receptive field).
Evidence suggests that primary hyperalgesia occurs as a result of nociceptors becoming
sensitized [609,853]. Similarly, evidence supports that allodynia in the primary zone is also
related to sensitization. Peripheral sensitization occurs as a result of the prolonged activation of
peripheral nociceptors, or exposure to the algesic chemicals, which leads to the release of a
number of chemical messengers that reduce nociceptive neurones’ threshold (for a
comprehensive review, see: [515,951]). This process of sensitization causes amplification of the
response to a noxious stimulus as well as to non-noxious thermal and mechanical stimuli. In the
example of the burn described above, the region around the burn (secondary zone) is more
sensitive to mechanical stimuli, but not to heat stimuli [718]. In fact, the secondary zone is less
sensitive to heat stimuli than normal skin. Peripheral sensitization does not fully explain
secondary hyperalgesia and there is now much evidence that secondary hyperalgesia is largely
mediated by central mechanisms [512], although there may also be some peripheral contribution.
Specifically, neurones in the CNS show enhanced responses to mechanical and thermal stimuli in
the secondary zone [806], i.e. a central sensitization. The IASP defines central sensitization as an
“increased responsiveness of nociceptive neurones in the central nervous system to their normal
or subthreshold afferent input.” These abnormal neuronal states are thought to be, in part, the
neural basis for hyperalgesia and allodynia. This state of hyperalgesia is likely related to
dysfunctional endogenous pain-modulatory mechanisms (see section 2.3.6).

18
2.1.3.2. The role of glia in sensitization
There are many more glial cells in the nervous system than there are neurones. Classically, it was
believed that glia only served as support or structural cells. However, recent evidence has
suggested that these cells play a much more significant role in both the PNS and CNS
processing. In the context of pain, recent evidence suggests that glia are important to the
pathogenesis of pain [159]. Specifically, satellite glial cells in dorsal root ganglia and trigeminal
ganglia are implicated in nociceptive conduction and transmission, and microglia and astroglia in
the CNS are implicated in sensitization and chronification of pain [159]. However, microglia and
astroglia have no effect on normal nociceptive processing, they are only implicated in
pathological states [614,921]. Specific mechanistic details of the role of glia in sensitization are
outside of the scope of this thesis (for a comprehensive review, see: [159]). Microglia are also
activated after inflammation, and are implicated in CNS inflammatory processes [159].
Taken together, it is possible that prolonged nociceptive input may induce central sensitization, a
process that is mediated by neurones and glia. Over time, this sensitization can lead to changes in
the function and structure of the nervous system (i.e., plasticity – see section 2.6). Normally,
there is a delicate balance between nociceptive and antinociceptive signaling in the central
nervous system. For instance, persistent nociceptive input is accompanied by an increase in
activity in pain-modulatory pathways. This functional change is considered a form of beneficial
plasticity, or change in the nervous system (see Section 2.6). However, in some cases plasticity
can be harmful, or maladaptive. For instance, it is believed that in persons who develop chronic
pain, the supraspinal modulatory pathways become dysfunctional or ineffective. This
maladaptive plasticity can contribute to the development and/or maintenance of chronic pain,
even in the absence of a noxious stimulus [576] (see Section 2.6.2). Furthermore, pain sensitivity
and a reduction in the ability to modulate pain may be related to pre-existing abnormalities in the
CNS.
2.1.4. Functional pain syndromes
Pain without a known physiological or “organic” cause is diagnosed as functional pain disorders.
Functional pain syndromes include the most prevalent of all pain conditions, which can be
divided into two categories: somatic or visceral pain syndromes. Somatic pain syndromes include
TMD, vulvodynia, low-back pain and fibromyalgia (FMS). Visceral pain syndromes include

19
irritable bowel syndrome (IBS), chronic cardiac pain syndrome, and internal cystitis (or painful
bladder syndrome). Functional pain syndromes are prevalent in 15-20% of adults worldwide
[562]. Relevant to this thesis is the prevalence of TMD, which is the most prevalent of these
functional pain syndromes (discussed in more detail in Section 2.2.3).
2.1.4.1. Central abnormalities in functional pain syndromes
Functional pain disorders are more prevalent in women than in men [581,582]. Furthermore,
some functional pain disorders are more likely to have increased co-morbidity in women than in
men [582]. Functional pain syndromes, such as TMD, are diagnosed as such because of a lack of
a clear physiological cause for the pain. This means that there are no consistent findings for the
source of the pain, and there are no clear peripheral abnormalities. Functional pain syndromes
do, however, share some common pathophysiological features, including changes to
neurobiological, physiological and anatomical aspects of the CNS. A number of behavioural
studies have also shown that patients with functional pains are hypersensitive to experimental
pain [581]. These patients also have dysfunctional pain-modulatory systems (e.g., [491,693]),
and, in some cases, have cognitive deficits (e.g., [365,412,455,489,666]). There is also a greater
degree of comorbidity with mood and anxiety disorders in patients suffering from functional pain
disorders [637]. With the lack of peripheral aetiological factors, these behavioural abnormalities
have led researchers to suspect that central mechanisms contribute to and mediate functional pain
syndromes. Findings specific to idiopathic TMD are described in further detail in section 2.2.3,
and support the hypothesis that central sensitization may be the underlying pathophysiological
processes underlying functional pain syndromes. These hypotheses have been corroborated by a
number of functional and structural neuroimaging studies, and are described in further detail in
Section 2.5.2.
2.1.5. Pain-cognition interactions
There is a two-way interaction between cognitive processes and pain in that cognitive processes
can interfere with and modulate pain, and pain can interfere with and modulate cognitive
processes (e.g., [123,193,290,529,776,778,781,893,935]). The interaction is best illustrated by a
common example: that of a football player who injures his ankle, but doesn’t feel the pain until
after the game. This suggests that pain can be attenuated when a person is distracted or engaged
in a cognitively-demanding task [851]. Conversely, pain can hamper cognitive performance (e.g.

20
reduced task accuracy or slower task reaction times) [713]. The cognitive interference of pain
may be due to the possibility that pain has a cognitive load, and that there could be limited
cognitive resources in the brain. Therefore, the brain must prioritize between competing tasks,
such as pain and a cognitive task. More recent work has demonstrated that cognition-pain
interactions are the product of complex interactions between two modes of attention selections:
bottom-up and top-down [529]. The bottom-up attention selection is related to the salience of
pain in that pain can be attention demanding because it is behaviourally relevant. Interestingly,
the salience of pain is partly relevant to its novelty [530,632,886]. This process of habituation is
likely related to top-down attention selection processes. These processes allow the redirection of
attention to other behaviourally relevant stimuli, and thus reduce the attention-grabbing (or
salience) effect of pain [529,892].
Several seminal studies in the 1990s demonstrated the interaction of pain and cognitive
performance in healthy subjects, showing that their task performance decreased (i.e., longer
reaction times) when concurrently presented with a noxious stimulus [192-194]. These studies
indicate that pain can interrupt or detract from the ability to perform a cognitive task, and thus
suggests that it is difficult to disengage from the painful stimulus. Later studies showed that
difficult tasks, which required presumably more cognitive resources, could briefly reduce
subjective pain ratings. Therefore, pain can affect cognitive performance, and cognitive
performance can modulate pain ratings (for a comprehensive review see [778]). These processes
have been summarized in the neurocognitive model of attention to pain [529]. This model
describes a “bottom-up capture of attention by pain”, whereby pain grabs one’s attention to
signal a relevant and threatening stimulus. However, this bottom-up process can be modulated by
top-down factors, such as cognitive load, emotional state and motivation. Inherent to this form of
modulation is the context of the noxious stimulus. If a subject is in an emotional state he/she may
report heightened pain, as he/she is more focused on stimuli with a negative valence.
Alternatively, a subject may report a heat stimulus as less painful if the stimulus is perceived as
beneficial, such as a heating pad, or a hot tub in a therapeutic setting.
In the context of chronic pain, a number of studies have shown that chronic pain negatively
impacts cognitive abilities, including concentration, learning, memory, and attentional focus
[26,250,275,365,369,455,489,666,928]. Eccleston and colleagues [289] showed that chronic pain
patients could briefly be disengaged from pain during attentional switches. They further showed

21
that pain has a cognitive load, and that only when there is heavy use of cognitive resources, do
pain ratings decrease. Taken together, these findings suggest that the brain has a limited
cognitive resource, where a more salient task can overcome pain – however, only for a brief
period of time. Over time, patients report getting tired, and not being able to focus on the task,
and reducing the subject’s ability to block out pain from consciousness. In a later study,
Eccleston and colleagues [292] showed that the cognitive interference effect of chronic pain was
related to levels of negative affect and somatic awareness. In fact, it has been shown that
negative affect can enhance perceived pain [936].
A study by Harman and Ruyak [409] reported that people with persistent low-level pain have
cognitive deficits, indicative of the interference of pain. There is evidence of functional and
structural plasticity that occurs in chronic pain (see Section 2.5.1 and 2.5.2, respectively and
2.6.2): over time, pain can change the connectivity of brain regions, which can lead to cognitive
impairment. Another possible mechanism is the aforementioned limited resource model – the
cognitive load of pain reduces resources to perform a task. These mechanisms are not mutually
exclusive, as prolonged nociceptive input can alter the connectivity of cognitive regions, and
further reduce resources available for cognitive processing.
In line with the limited resource model, is the concept of cognitive branching – the ability to put
a pending task “on hold” to execute a more salient (or rewarding) task [497]. Therefore, the
cognitive branching model provides a functional basis for the limited resource model, and
provides a model that explains reductions in pain ratings when engaged in a cognitive task, or
reduced cognitive performance whilst receiving prolonged nociceptive input. Specifically, if the
cognitive branching system is limited, as is suggested by Koechlin and Hyafil [497], then the
system can be overloaded, and affect task performance. This explains findings that tasks that
require little cognitive engagement do not modulate pain ratings, and that low noxious stimulus
intensities do not have a significant effect on task performance [776]. Conversely, as cognitive
processes increase in difficulty, pain ratings can be modulated; or if nociceptive input increases,
cognitive performance can be negatively affected. For example, Weissman-Fogel and colleagues
[928] found no group differences in task performance between controls and patients with TMD
in a neutral Stroop task (no interference). However, when presented with a more difficult
interference counting Stroop task, patients showed sluggish reaction times – their cognitive
branching system was either overwhelmed, or prioritized pain over the Stroop task. Finally, this

22
effect was modulated by an emotional counting Stroop task. Subjects were asked to report the
number of TMD-pain related words visually presented. Specifically, these words were: ache,
tight, annoying, pain, stress, hurt, clench, anxious, throb, unpleasant, pressure, and tiring. The
patient group reported that the TMD pain words affected their pain intensity and unpleasantness
during the Stroop task, whereas controls did not. The increase of the patients’ pain rating affected
the TMD group’s reaction times for the Stroop task.
In sum, pain and cognition have a complex interaction that can be modulated by affective state,
context and motivation. Pain is inherently salient, but its behavioural relevance can be up- or
downregulated based on these factors. Furthermore, chronic pain can alter brain structure,
connectivity and function, which may further impact cognitive abilities.
2.1.6. Salience and pain
Pain plays an essential role in survival – it signals a potential or actual injury. Because of the
behavioural relevance of pain, noxious stimuli are inherently salient. Furthermore, studies have
shown that consciousness is required to perceive pain. Specifically, these studies have
investigated the neural correlates of nociception in subjects in a persistent vegetative state using
neuroimaging techniques (these techniques are discussed in detail in Section 2.5). We must
clearly indicate that these studies were not investigating pain (recall that the definition of pain
requires that a person report pain – it is a subjective perceptual experience), and so, these studies
were comparing neural correlates of normally noxious stimuli in persistent vegetative state
patients to pain perception in the control group. One study found that the regions along the
ascending nociceptive pathways, involved in the sensory-discriminative dimension (midbrain,
thalamus and primary somatosensory cortex (S1)), showed activation, but the other regions that
showed activation in the control group (including the secondary somatosensory cortex (S2),
insula, cingulate, prefrontal cortex (PFC) were not activated [516]. Conversely, Kassubek and
colleagues [479] found that the patients did have activation in brain regions involved in
nociception and pain modulation. However, it is worth noting that recent studies have shown that
some, but not all, patients in a vegetative state show brain activity in response to stimuli in the
environment [517,659]. Therefore, it is possible that in a subset of these patients, where
consciousness may persist, pain perception and modulation occurs. However, in patients that

23
have lost consciousness, nociception occurs [768], but there is no evidence that they feel pain, as
regions underlying different dimensions of pain do not show activation.
2.1.7. Pain-motor interactions
Pain-motor interactions are a hallmark of pain-related behaviours, the most obvious example
being nocifensive behaviours. Descartes portrayed nocifensive behaviour in the Treatise of Man
[242] in his famous depiction of the boy burned by the fire, and Charles Darwin describes
nocifensive behaviours in his text Expression of Emotion in Man and Animal [203]. There is no
doubt that the sensory and motor system interact, a concept supported by the many sensorimotor
reflexes observed in animals and humans. However, the interactions of pain and motor functions
are not as clear. Furthermore, the effects of prolonged (or chronic) pain on motor output are
equally ambiguous. A number of experimental pain paradigms have attempted to shed light onto
these topics. For instance, it has been shown that noxious input inhibits the motor cortex
excitability [3,157,301,302,638,856]. Similarly, motor cortex stimulation has been shown to
modulate pain [103,117,118,301,331,342,344,345,535,548,657,689]. Furthermore, a study by
Boudreau and colleagues [109] reported that motor cortex plasticity is inhibited by noxious
stimulation of the intraoral cavity. Therefore, it seems that motor learning is modulated, in this
case inhibited, by a tonic noxious stimulus.
A number of theories have been postulated to explain the relationship between pain and motor
output. The two dominant theories are the vicious cycle theory, first postulated by Travell [866],
and the pain adaptation model, postulated by Lund and colleagues [556]. The vicious cycle
theory suggests that the presence of a reciprocal relationship between pain and dysfunction: pain
causes muscular dysfunction, which increases pain, creating a vicious cycle. This theory states
that, over time, the muscle hyperactivity contributes to and/or maintains pain. Specifically,
tonically hyperactive muscles become ischaemic, which in turn leads to an increase in algesic
chemicals within the musculature. This has been supported by findings of increased motor
activity in experimental models of muscle pain [431]. However, some studies of orofacial pain
have failed to demonstrate that pain leads to muscular hyperactivity [634,675]. Therefore, Lund
and colleagues [556] postulated the pain adaptation model, which suggests that there is both
muscle hyper- and hypo-activity. Specifically, the model states that there is a decrease in activity
of muscles that are painful or that hurt with movement, and that antagonist muscles have

24
increased activity. However, more recent experimental findings have not supported the concept
of uniform inhibition of a painful muscle. Instead, they have shown variable patterns of change
in activity both spatially and temporally (for a review, see: [431]). To address these
inconsistencies, Hodges and colleagues propose a “new theory of motor adaptation to pain”
[431], which maintains the basic premise that pain modifies muscular activity as a nocifensive
mechanism. This theory proposes that the body adapts to muscular pain is several ways. When
there is pain or threat of pain, there is a redistribution of activity within a muscle, and between
muscles, so that force is maintained. This redistribution occurs upon the recruitment of new
motor units, and changes the mechanical behaviour of the muscle including increased stiffness
and altered load distribution. In the short term, this reduces movement of the muscle and
decreases the load on the muscle, which is beneficial and promotes healing. In the long term,
however, these changes can lead to increased load, decreased movement and decreased
variability, which can increase the risk of injury.
2.1.8. Neuroticism and pain
Psychological factors have been associated with sensitivity to pain, and the development and
maintenance of chronic pain. Inherent personality-related factors reduce the brain’s capacity to
effectively modulate nociceptive input and pain perception, likely affecting the cognitive
modulation system (discussed in Section 2.1.2 and Section 2.3.6). Of particular interest to this
thesis is the personality dimension of neuroticism [178]. Neuroticism has been defined as a stable
personality trait associated with heightened sensitivity and/or processing of negative affective
stimuli [178,179,907]. Therefore, it is not surprising that patients with high neuroticism scores
have a lower pain threshold and pain tolerance, and are more distressed by pain in experimental
settings and chronic pain [408]. Specifically, neuroticism has been associated with pain-related
suffering [35,408], pain sensitivity [177,373,907], nerve injury outcomes and the development of
neuropathic pain [848], and TMD [315]. However, not all chronic pain patients have high
neuroticism scores, and not all persons with neuroticism have chronic pain [179]. Therefore,
neuroticism alone is not sufficient to develop chronic pain; however it can contribute to the
severity of the pain reported by chronic pain patients.

25
2.2. Temporomandibular disorders
2.2.1. Historical perspectives
Reports of TMD-like symptoms date back 1500 years when Hippocrates described a method for
reducing dislocated mandibles [423]. These treatments often led to TMD-like disorders,
including arthritis and infection of the TMJ. The first documented surgeries for disc
displacements in the TMJ occurred in the nineteenth century, and shortly thereafter, articular disc
removal was used to manage TMD symptoms [587]. In the late 1800s and early 1900s, dentists
primarily attributed TMD symptoms to problems with the muscles of mastication and their
contribution to malocclusion. By 1934, malocclusion was formally considered to be the cause of
TMD symptomology [181]. Costen’s range of symptoms, that far exceeds the recognized
pathophysiological understanding of TMD today, led to the development of numerous treatments
and devices to treat TMD and other symptoms believed to be related to malocclusion (for a
review of these, see [587]). However, this range of symptoms was criticized for being too broad
[587]. More recent, randomized control research studies have refuted the use of occlusal devices
in the treatment of TMD, suggesting that the positive effects of these sorts of treatments are
related to the patients’ expectation of the treatment [174,882]. Therefore, it has been suggested
that less invasive and less expensive methods of treatment may be as efficacious in pain
management as occlusal splints [872]. Despite an exceptional historical presence and extensive
research, little is known of TMD pathophysiology, and few treatments are effective. Today,
TMD continues to be a prevalent pain syndrome that poses a significant financial burden on
healthcare and society.
2.2.2. Prevalence and social costs
TMD are estimated to affect between 3-20 percent of the United States’ adult population
[269,539,540,562]. Of these, only a small proportion of persons suffering from TMD pain seek
treatment and end up at tertiary and quaternary medical facilities such as pain clinics [350].
TMD are 1.5-9 times more prevalent in women [113,201,284,285,312,721]; a study found that
84% of persons with TMD were women in a nationwide epidemiological study in the United
States [284]. It has been estimated that 5.3 million people in the United States seek medical
attention for a TMD-related problem per year [349]. Furthermore, TMD physical symptoms
(e.g., pain) and psychological symptoms (e.g., depression) have been reported as more severe in

26
women than in men. The predominance of TMD symptoms in women has been attributed to
differences in sex hormones, such as oestrogen, as well as the use of oral contraceptives. Older
(55-74 years old) and younger (< 20 years old) subjects report fewer TMD-like symptoms than
subjects in reproductive years (20-55 years old). The annual incidence of TMD is 2-4%, with
0.1% going on to develop chronic pain. There are different disease outcomes for different
patients, with 31% of cases persisting over 5 years, 33% whose pain resolves, and 36% who have
recurring TMD. The direct healthcare costs for treatment of TMD in the United States have been
estimated at $2 billion. However, TMD is often co-morbid with other disorders, such as FMS,
IBS, anxiety, and depression, and may have greater economic impact based on indirect costs
(such as missing work, unemployment, disability, limitations, etc.). The estimated total of both
direct and indirect costs in the United States exceeds $4 billion dollars per annum [350]. Taken
together, then, it is clear that TMD are a prevalent disorder with a significant economic burden
on society.
2.2.3. Clinical aspects
TMD comprise of a cluster of clinical problems involving the structures of, and around, the TMJ,
the masticatory musculature, or both [1,418]. TMD are primarily characterized by spontaneous
pain and/or pain associated with jaw function, which can affect mandibular range of motion and
produce TMJ sounds (e.g., crepitus, clicking) during jaw function. TMD pain can also spread to
the muscles of the neck. The most common diagnostic criteria (and classification scheme) for
TMD is the TMD-Research Diagnostic Criteria (RDC) established by Dworkin and LeResche in
1992 [285]. The American Association of Orofacial Pain (AAOP) has adopted this classification
scheme. Based on the TMD-RDC, there are two primary types of painful TMD: myogenous
(pain generated by the masticatory muscles) and arthrogenous (pain generated by the TMJ)
[285]. Myogenous TMD, often called masticatory muscle pain, is diagnosed on the basis of
patient history and clinical examination. Arthrogenous TMD conditions are usually caused by
displacement or derangement of the TMJ disc, but also by inflammation within or around the
joint. Hence, the pain in arthrogenic TMD can be attributed to inflammatory processes that occur
as a result of intra-articular events or anatomical factors including disk displacement – although
extra-articular events can also cause TMJ inflammation [646]. Additionally, TMD can be
classified as idiopathic or as post-traumatic. Idiopathic TMD have no clear aetiology for pain
onset and/or maintenance, although, as discussed above, there are a large number of factors that

27
have been considered in the past to play an important aetiological role. However, over time, and
with increasing research, these factors have either been put into question or even eliminated. In
fact, this is why it has been suggested that TMD is generally an idiopathic condition. In contrast,
post-traumatic TMD are caused by traumatic events in the craniofacial region. These traumatic
events can be as serious as head injury, hyperextension-flexion injury (e.g., following a motor
vehicle accident), or as minor as a dental treatment (e.g., a long dental procedure) [81].
2.2.3.1. Sensory abnormalities
Sensory function in TMD has been assessed with a variety of psychophysical measures and
quantitative sensory testing (QST) approaches to determine detection and pain thresholds to
experimental stimuli across thermal, mechanical, electrical and chemical modalities. The details
of these studies are provided in Table 2-1, and a summary of the findings is provided in Table 2-
2. In general, these studies demonstrate that patients with TMD can have sensory abnormalities
such as hyperalgesia, reduced pain tolerance and reduced DNIC.
QST studies have demonstrated that patients with TMD have hyperalgesic responses2 to acute
experimental pain in the trigeminal region as well as in extra-trigeminal regions, as measured by
pain thresholds. Specifically, studies investigating pressure pain thresholds in TMD have
reported that patients with TMD have reduced pain thresholds to noxious pressure stimuli in the
trigeminal region, and outside the trigeminal region [42,139,305,565,841]. Interestingly, one of
these studies did not find significant group differences in pressure pain thresholds between
controls and patients with TMD, but did so when they exposed study participants to a
psychosocial stressor [139]. Most studies investigating heat thresholds in the trigeminal region
and outside the trigeminal region reported that patients with TMD have lower heat pain
thresholds than controls [313,491,563-565,841]. However, two studies failed to find differences
in heat pain thresholds in the arm between the TMD group and controls [113,313]. Also, five
studies have investigated pain thresholds to experimental ischaemic pain in the arm of patients
with TMD versus controls. Three of these studies reported reduced pain thresholds in patients
with TMD versus controls [313,563,564], whereas two studies [113,180] did not identify any
differences between patients with TMD and controls. Patients with TMD also have lower
2 Whether decreased pain thresholds are related to hyperalgesia or allodynia, or both remains a contentious issue.

28
pinprick pain thresholds in the orofacial region [42]. Four studies have investigated electrical
pain thresholds in the orofacial region, and one study examined these thresholds outside the
orofacial region [42,211,391,792]. Of these, only one identified lower pain thresholds to electric
stimuli in the orofacial region [42]. Together, these studies demonstrate that, in most cases,
patients with TMD have reduced sensory thresholds to noxious stimuli across different
modalities. Furthermore, patients with TMD show prolonged pain and increased peak pain in
response to hypertonic saline injected into muscles in the trigeminal and extra-trigeminal regions
[841]. These hyperalgesic responses may be related to either peripheral sensitization or central
sensitization, or both (see Section 2.1.2.1).
In addition to pain thresholds, some studies have investigated pain tolerance in TMD. It is
believed that pain tolerance reflects a more integral view of the pain system, and its various
dimensions. Because pain tolerance reflects “how much” pain a subject can withstand, this
measure not only captures the sensory-discriminative dimension of pain, but also cognitive-
evaluative and affective-motivational dimensions. Pain tolerance data are variable so it is not
clear what mechanistic factors underlie this attribute seen in patients with pain. Specifically, two
studies [42,841] have reported reduced pressure pain tolerance in trigeminal and extra-trigeminal
regions of TMD patients compared to controls. Four studies have investigated ischemic pain
tolerance, and two studies [563,564] reported lower pain tolerance in patients with TMD
compared to controls, and two studies failed to find any group differences [90,313,563,564]. Five
studies investigated heat pain tolerance in TMD, and four of these identified lower pain tolerance
in patients with TMD compared to controls, compared to controls, whereas one of these studies
failed to identify any group differences. Three studies tested pain tolerance to experimental
electrical stimulation in trigeminal and extra-trigeminal regions, and found no differences
between patients with TMD and controls [211,391,792]. In sum, there are variable findings with
regard to pain tolerance in patients with TMD.
Taken together, there seems to be variability in the findings of quantitative sensory testing
studies. This may be due to the heterogeneity of TMD, which was the motivation for the
development of the TMD-RDC. Therefore, it is difficult to compare studies conducted prior to
the establishment of the RDC, or that do not use the RDC to select patients to studies that have
abided by the RDC.

29
Temporal summation is the production of a gradual and progressive increase in pain from the
repeated painful stimuli. A typical protocol to test for temporal summation involved delivering
painful stimuli at 3s inter-stimulus interval [711] in a short timeframe [31]. It is believed that
temporal summation is related to “wind-up” – a process of increased nociceptive firing of dorsal
horn neurones to repeated noxious stimulation, related to central sensitization [915]. Studies have
demonstrated that the degree of temporal summation is related to central hyperexcitability: if the
subjects have central sensitization, they have increased temporal summation. Two studies have
reported augmented temporal summation in patients with TMD, compared to pain-free controls
[42,565]. One study by Sarlani and colleagues reported that women with TMD had greater
temporal summation than men with TMD, and healthy controls [755]. Therefore, these studies
support the hypothesis that patients with TMD may have central sensitization.
In support of central sensitization in TMD, King and colleagues [491] reported that patients with
TMD had dysfunctional endogenous pain modulation systems. The authors used an experimental
method that modulates pain via descending inhibition: a counter-stimulation method. Under
normal circumstances, when a conditioning noxious stimulus is administered to a remote body
part, it activates the descending modulation system. This activation of the modulation pathway
attenuates pain responses to a second focal stimulus, a process called DNIC [519,521]. In King et
al’s [491] study, patients with TMD did not report less pain to a second focal noxious stimulus,
but rather reported higher pain ratings than when the second stimulus was administered without
the conditioning stimulus. Similarly, Maixner and colleagues [563] reported that patients with
TMD could not effectively engage anti-nociceptive mechanisms when given a remote noxious
stimulus, which would be expected to dampen their spontaneous TMD pain. In this regard, it has
been shown that in concert with findings showing that patients with TMD have increased pain
sensitivity outside the orofacial region, they are also unable to modulate pain perception when a
second noxious stimulus is presented.
In sum, there is evidence that patients with TMD have both peripheral and/or central
sensitization, as well as dysfunctional anti-nociceptive systems.

30
Table 2-1: Summary of quantitative sensory testing studies in TMD
Stimulus TMD subtype*
Stimulus laterality Body site n patients
(# females) n controls (# females)
Pain threshold
Pain tolerance
Temporal summation Other Ref
Pressure myofascial Bilateral V1a, V3
b and wrist 20 (20) 20 (20) !
! ! !![305]
Ischaemia combined Leftc
arm 36 (N/A) 80 (N/A)
! ! ! ![313]
Heat forearm "! ! ! !
Heatd myofascial Left palm 14 (14) 28 (28) ! ! ! !!!DNIC!
[491]
Heat Combined
Left forearm
23 (23) 24 (24)
! ! ! !
[565] Left palm ! ! " !Bilateral face ! ! ! !
Pressure Bilateral face and forearm ! ! ! !
Heat Combined Left forearm 64 (64) 23 (23)
! ! ! !![564]
Ischaemia ! ! ! !!Heat combined Left forearm 52 (44) 23 (19) ! ! ! !
[563] Ischaemia ! ! ! !Heat
myofascial Bilateral
masseter and anterior tibialis
22 (16) 21 (15)
! ! ! !!
[841] Pressure ! ! ! !!
Hypertonic Saline
! !
"!peak!pain!"!pain!duration!
Heat combined Left forearm 20 (20) 42 (20) "! "! ! ! [113] Ischaemia Right arm "! "! ! !

31
Stimulus TMD subtype*
Stimulus laterality Body site n patients
(# females) n controls (# females)
Pain threshold
Pain tolerance
Temporal summation Other Ref
Ischaemia combined Non-dominant arm
28 (10) 20 (20) "! "! ! !!
[180]
Pressure myofascial Bilateral
masseter and temporalis muscles and finger
35 (33) 35 (33)
!e ! ! !
[139]
Pressure
arthralgia
Bilateral
TMJ, non-dominant finger
43 (19) 20 (17)
! ! ! !!
[42] Tactilef TMJ and
face ! ! ! !!
Electric
Ipsilateral to pain in TMD, random in controls
TMJ
! ! " !!
Electric myofascial N/A incisor tooth pulp 12 (11) 12 (10) "! "! ! !
[792]
Electric combined Bilateral masseter 30 (19) 31 (18) "! "! ! ! [391]
Electric myofascial Bilateral masseter and forearm
12 (10) 12 (10) "! "! ! !!
[211]
a - Frontalis muscle, which receives afferent input from the ophthalmic branch of the trigeminal nerve (V1) b - Masseter muscle, which receives afferent input from the ophthalmic branch of the trigeminal nerve (V1) c - No information was provided about pain laterality or handedness.

32
d - The study investigated thermal heat pain thresholds with and without a cold water immersion conditioning stimulus e - Group differences only found when a psychosocial stressor was presented f - Tactile stimuli were two von Frey filaments; 5.16g and 84.96g for tactile and pin-prick stimuli, respectively * "combined" refers to patients with both myofascial and articular (joint) pain or a mixed population of myofascial TMD or joint TMD.
Abbreviations: DNIC – diffuse noxious inhibitory controls; TMJ – temporomandibular joint; V1 – ophthalmic branch of the trigeminal
nerve; V3 – mandibular branch of the trigeminal nerve; - no difference; ! – decrease

33
Table 2-2: Number of quantitative sensory testing studies testing sensory thresholds and tolerance in TMD
Data are based on studies presented in Table 2-1. ! indicates a decrease in TMD compared to controls. Blank cells indicate that the measure
was not tested or reported.
Stimulus (n studies)
Trigeminal Region Extra-trigeminal Regions
Threshold Tolerance Threshold Tolerance
!! no change ! !! no
change not
tested ! !! no change ! !! no
change not
tested Electric (4) 1 3 0 3 1 0 1 0 1 3 Ischaemia (5) 5 3 2 2 2 1 Pressure (5) 5 2 3 5 0 2 3 Thermal heat (7) 2 1 1 5 2 4 1 2 Pin-prick (1) 1 0 1 1

34
2.2.3.2. Motor abnormalities
Some studies have shown that patients with TMD have motor abnormalities in the orofacial
region, although there is much discordance across studies. For instance, three studies reported
that patients with TMD have hyperactivity in their muscles of mastication, whereas other studies
did not find any differences when comparing patients to controls (for a review, see [197,832]).
Furthermore, some studies have shown that there are individual differences in muscle reactivity
to pain [634,675]. One study that tested the excitability of central motor controls of masticatory
pathways in patients with TMD, and found that central motor pathways were not hyperexcitable
[195]. These variable findings between the interaction of TMD-related pain and orofacial motor
function may be related to a unique experience of pain, reflective of the heterogeneity of TMD.
In addition, some patients with TMD have limited unassisted jaw opening (with or without pain)
[539], whether they were symptomatic or asymptomatic [286]. Therefore, in the context of TMD,
it seems that the revised pain adaptation model (see section 2.1.6) best explains the observed
variability in motor abnormalities. This model accounts for individual differences in changes in
motor output to maintain homeostasis – the muscle, or region maintains contraction force, and
promotes nocifensive behaviour. Based on these findings, Svensson and Graven-Nielsen [839]
proposed that TMD pain interferes with sensorimotor integration and inhibits motor output to the
craniofacial region. They also suggested that differential patterns of excitation of masticatory
muscles occur in a phase-dependent manner to dampen jaw movements. Specifically, there can
be increased excitation of antagonistic muscles during contraction of the agonistic (painful)
muscle. Additionally, there is increased activity in the antagonistic phases of muscle movement
and decreased excitation during agonist muscle contraction, presumably to reduce agonistic
muscle activity, and thus pain [374]. In contrast to this proposed model, Cairns, Sessle and Hu
[133] reported that the injection of noxious chemicals into the TMJ increased both agonist and
antagonist muscle activity.
2.2.3.3. Cognitive abnormalities
There is extensive literature about the cognitive dimension of pain, and interactions between
cognition and pain in general (see section 2.1.4). A few studies have examined cognitive
performance in TMD and reported various cognitive deficits. For instance, Goldberg and
colleagues [370] showed that post-traumatic TMD patients performed poorly on simple and

35
complex reaction time tasks, as well as neuropsychological tests testing interference effects,
compared to patients with idiopathic TMD. In two follow-up studies, Grossi et al. [379,380]
determined that a patient’s performance on neuropsychological test, including he simple and
multiple-choice reaction-time tests, California Verbal Learning Tests, the Brown-Peterson
Consonant Trigram Auditory Memory Test, Sleep Assessment Questionnaire, and Beck
Depression Inventory, as well as fatigue and energy level assessments (100-mm visual analog
scale), predicted their treatment outcome. Weissman-Fogel and colleagues [928] used a cognitive
interference task, the counting Stroop task, and found that patients with idiopathic (non-
traumatic) TMD had sluggish reaction times compared to controls. Together, these studies
suggest that patients with TMD may have cognitive deficits. However, although there is evidence
that patients with TMD have a variety of cognitive deficits, it is not clear whether these deficits
existed prior to the onset of TMD pain or are caused by TMD pain.
2.3. Anatomy and physiology of orofacial pain and sensation
In order to understand concepts related to the possibly neurological and pathophysiological
aspects of orofacial pain and TMD in particular, it is important to describe the ascending sensory
and nociceptive pathways from sensory transduction in peripheral receptors, to transmission to
the brainstem, and supraspinal levels. The emphasis will be on the trigeminal system but some
differences with the spinal (i.e., in the trunk, neck and limbs) system are also noted.
Sensory information, such as touch, temperature, proprioception, kinaesthesia, itch and pain are
processed by the somatosensory system. In general, peripheral receptors transduce stimulus
energy and encode them as neural signals. These neural signals are propagated from the
periphery to the CNS via primary afferent neurones. The cell bodies of these afferents are
generally located in the periphery – with the notable exception of the mesencephalic nucleus of
the CNV (see below). These peripheral neurones project to central neurones. There are two main
routes for orofacial sensory information to travel from the brainstem to the brain: the trigeminal
lemniscus, and the trigeminothalamic tract (TTT) for craniofacial information.
2.3.1. Peripheral receptors and sensory afferents
The nervous system monitors the state of the internal and external milieus. There are many types
of specialized peripheral receptors, each with specific properties to encode different stimuli

36
[428]. Receptors in muscles, deep tissues, joints and tendons provide sensory information about
movement and limb position [573]. These include Golgi tendon organs and muscle spindles.
Other aspects of the somatosensation, such as touch, flutter, vibration and skin indentation are
encoded by encapsulated nerve endings that are either fast adapting (Meissner corpuscles for
flutter, Pacinian corpuscles for vibration) or slowly adapting (Merkel discs for touch, Ruffini
endings for stretch – both are related to skin indentation) [573]. Sensations of pain and
temperature in the skin and muscles are encoded by free-nerve endings [941]. There are also
receptors specific to hair follicles, which are mechanically-sensitive (mechanoreceptors) [346].
These nerve fibres are inside the hair follicle and wrap around the sheath. These rapidly adapting
fibres encode movements of the hair. Information regarding position of a limb is provided by
proprioceptors. In relation to this investigation, the TMJ is innervated by proprioceptors that
provide information about jaw position and the force being applied by masticatory muscles
[843].
Each receptor is the peripheral ending of a primary afferent fibre. The peripheral somatosensory
primary afferents can be classified based on their conduction velocity, a classification system
developed by Erlanger and Gasser [297]. The free nerve endings have unmyelinated small-
diameter C-fibres with slow conduction velocities (~< 2 m/s) [612], and Aδ fibres, which have a
conduction velocity of ~2-30 m/s [612]. The Merkel discs and encapsulated endings are
generally associated with large-diameter myelinated fibres (Aβ) with conduction velocities of
~30-70 m/s [449]. These axons transmit information pertaining to touch and proprioception from
the periphery to the CNS. Muscle spindles and Golgi spindle afferents that carry proprioceptive
and movement information are large myelinated fibres (Aα), which have a conduction velocity
of 70-120 m/s. Receptors that respond preferentially to noxious stimuli are called nociceptors
[796] and lack a receptive structure or corpuscle, but rather are free nerve endings [826]. Primary
afferents with free nerve endings respond to mechanical, thermal and/or chemical stimuli [941].
In cutaneous tissues, LTM and thermoreceptors (receptors that detect changes in temperature)
also have free nerve endings [941]. Similarly, LTM in skeletal muscles have free nerve endings
[598]. Free nerve endings have small-myelinated Aδ fibre or unmyelinated C-fibre axons fibres.
For instance, a subpopulation of unmyelinated fibres (C) detects stimuli that warm the skin [462]
and a subpopulation of small-myelinated fibres (Aδ) detects stimuli that cool the skin [202].

37
There are also some reports that a small number of Aß afferents may have nociceptive functions
[255].
Nociceptors can respond to a single modality (mechanical, thermal, chemical) or to multiple
modalities (e.g., mechano-heat, polymodal). Mechanical nociceptors respond to suprathreshold
mechanical stimuli [681], and thermal nociceptors respond to noxious temperatures [202].
Polymodal nociceptors respond to various modalities of noxious stimuli, including mechanical,
thermal and chemical stimuli [612]. Furthermore, nociceptors can be classified based on the type
of afferent fibre to which they are associated, Aδ or C fibres. There are also C fibre mechano-
heat sensitive afferents (CMH) and Aδ-mechano-heat sensitive afferents (AMH). It is believed
that these different types of nociceptors encode the different qualia of pain, such as burning,
aching, pricking, prickle [612]. The CMH and AMH are considered polymodal nociceptors, and
some respond to noxious chemical stimuli [224].
There are other classification schemes for primary afferents, including nociceptors, but they are
outside the scope of this thesis.
2.3.2. Orofacial nervous system: the trigeminal nerve
The CNV is a mixed nerve with both sensory and motor components. However, the sensory
segment is much larger than the motor portion [884]. The CNV has three main branches: the
ophthalmic branch (V1), the maxillary branch (V2) and the mandibular branch (V3).
V1 and V2 are entirely sensory nerves, while V3 is comprised of both sensory and motor fibres
[843]. The V1 innervates the scalp, forehead, upper eyelid, the cornea and the eye’s conjunctive
tissue, the nose, the nasal mucosa, the frontal sinuses and the dura and blood vessels of the
meninges. The V2 innervates the lower eyelid, the cheek, the nostrils and nasal mucosa, the upper
lip, the upper teeth and gingiva, the palate, the roof of the pharynx, the maxillary, ethmoid and
sphenoid sinuses and some of the meninges. The third branch of CNV, V3, innervates the lower
lip, the lower teeth and gingiva, the chin and parts of the jaw, parts of the external ear, and some
of the meninges. Somatosensory information from the floor of the oral cavity is also encoded by
V3. CNV is not the only nerve that encodes signals from the orofacial region. In relation to this,
somatosensory information from certain parts of the oral cavity, ear and the meninges is encoded
by the facial nerve (cranial nerve VII; CNVII), the glossopharyngeal nerve (cranial nerve IX;

38
CNIX) and the vagus nerve (cranial nerve X; CNX). Sensory fibres from CNVII, CNIX and
CNX enter the brainstem and synapse onto the same nuclei as CNV (see below).
The sensory aspect of the nerve receives input from receptors that encode touch, nociceptive,
proprioceptive and temperature stimuli from the face, facial and masticatory muscles and the oral
cavity [784]. Aδ and C fibres in the orofacial region, especially in the TMJ and muscles of
mastication are very similar to those present in the spinal nociceptive system [784]. There are
some notable tissues that are innervated by the trigeminal nerve, such as the tooth pulp, the
cornea and the dura regions that are only (or predominantly) innervated by nociceptors, but do
not have other somatosensory receptors. For instance, Davis and Dostrovsky [216] have
demonstrated that stimulation of the middle meningeal artery and the sagittal sinus activates
central nociceptive neurones in the trigeminal system.
The sensory fibres of the three branches of the CNV converge at the trigeminal ganglion, where
the cell bodies of afferent fibres are located. The trigeminal ganglion is similar to the dorsal root
ganglion in the spinal somatosensory system in terms of markers associated with nociception,
and nociception-related receptors [9,33,82,487,569]. From the ganglion, a single sensory root
emerges and enters the CNS at the level of the pons. Primary afferent fibres terminate in the
principal sensory nucleus (or main sensory nucleus; MSN) and the spinal trigeminal nucleus
(STN). There is another specialized trigeminal nucleus in the brainstem: the mesencephalic
nucleus of the CNV (MeT) (see below). Together, the MSN, the STN and the MeT form the
trigeminal brainstem sensory nuclear complex (VBSNC). Unlike the dorsal horn of the spinal
cord, the VBSNC is comprised of several nuclei with unique cytoarchitectonic and
organizational features [82].
The MSN is similar to the dorsal column (gracile and cuneate) nuclei where large-diameter
myelinated primary afferents that encode touch and position information from the body synapse
onto second-order fibres. Like these nuclei, the MSN maintains a somatotopic map of the regions
from which it receives touch and position information, namely the orofacial region [411]. The
second-order fibres from the MSN bifurcate, and a larger branch of these second-order fibres
forms the trigeminal lemniscus, which decussates and runs parallel to the medial lemniscus. The
medial lemniscus, which carries touch and position from the body, terminates at the

39
ventroposterior lateral (VPL) nucleus of the thalamus. The trigeminal lemniscus terminates in the
ventroposterior medial (VPM) nucleus of the thalamus.
The STN is a long nucleus that is located between the pons and upper cervical spinal cord
(around C2) and is comprised of three subnuclei, organized rostro-caudally: oralis, interpolaris
and caudalis (Vo, Vi and Vc, respectively) [656]. Vc is cytoarchitectonically similar to the spinal
dorsal horn [656], and has been termed the medullary dorsal horn [366]. A-β fibres bifurcate and
terminate in the MSN and along the STN, and Aδ and unmyelinated C fibres terminate
predominantly in the STN at the level of Vc and to the upper cervical spinal cord dorsal horn.
Interestingly, some Aδ primary afferents carrying pain and temperature information synapse onto
second-order fibres in Vo and Vi, but this does not occur with C-fibres [947]. There are several
somatotopic maps along the VBSNC. Specifically, somatotopy is maintained in the dorsoventral
plane of each nucleus: the mandibular region is encoded in the dorsal segment, then the
maxillary region, and, ventrally, the ophthalmic region. In Vc, the somatotopic organization has
been shifted, and has an “onion-skin” arrangement, where oral regions are represented rostrally,
and lateral regions of the face are represented more caudally. There is a second somatotopic
representation along the mediolateral axis of Vc where the head is inverted [453,801]. Second-
order fibres from the STN form the TTT and, as implied by the name, project to the thalamus,
while maintaining a somatotopic organization. These pathways are described in greater detail in
Section 2.3.3.1.
A unique feature of the trigeminal system is that the cell bodies of primary afferents that encode
proprioceptive information from the masticatory muscles and periodontal ligaments are located
within the central nervous system in the MeT [167,176,549]: this is the only site where soma of
afferent neurones occur within the CNS. The MeT fibres bifurcate in the brainstem. One branch
of these fibres forms a reflex loop: it synapses on to the trigeminal motor nucleus, which has
efferent fibres that projects back to the periphery. These loops form the basis of orofacial reflex
arcs (such as the jaw jerk reflex) [399-401,411,557]. The other branch of these fibres projects to
other brainstem targets via the tract of Probst [138]. For instance, some of these axons project to
the MSN where they synapse onto second-order neurones [918].

40
2.3.3. White matter pathways
This section will provide a brief overview of the major white matter tracts in the brain, especially
those related to brain regions implicated to nociception and pain processing and modulation will
be described, particularly in terms of the functional neuroanatomy of these tracts (although there
has actually been little work on the functional anatomy of white matter in humans).
White matter tracts are classically divided into three groupings: (1) projection fibres, that connect
the cortex with subcortical, brainstem and other regions; (2) commissural fibres, that connect
cortical regions from one hemisphere to the other hemisphere; and (3) association fibres that
connect cortical regions within a hemisphere [147] (See Figure 2-3).
Today, it is recognized that there are several types of white matter tracts in the brain, including
long association fibre pathways, striatal pathways, commissural fibres, and projection fibres
[237,760] (see Figure 2-3). There are also short “U” association fibres that connect neighbouring
gyri [237,311].
The corona radiata contain ascending and descending projection fibres that include motor tracts
that emerge from the primary motor cortex (M1), supplementary motor area (SMA) and
premotor cortex (PMC) and project to the basal ganglia, the brainstem motor nuclei
(corticobulbar tracts) and the spinal cord (corticospinal tract; CST) [147,644]. There are also
corticobulbar tracts that emerge from various PFC regions that are involved in various
homeostatic functions. Of particular interest are tracts from the PFC that project to the
periaqueductal gray (PAG), which is implicated in pain modulation. The corona radiata also
includes ascending tracts that project from the thalamus to the cortex, forming the
thalamocortical tracts (TCT) [644]. Furthermore, there are corticothalamic tracts that run parallel
to the TCT.
Somatosensory (including nociceptive) input from the orofacial region is mainly transported to
the CNS via the CNV. The primary afferents project to the VBSNC. Second-order neurones form
the TTT and the trigemino-bulbar tracts, which project to the thalamus and the various brainstem
nuclei (see above). Third-order neurones from the various thalamic nuclei project to cortical
regions, forming the TCT. For instance, tracts from VPM project to S1 and S2. These tracts
ascend to these cortical targets through the corona radiata, via the posterior limb of the internal

41
capsule (plIC) [237]. The MD thalamus projects to the cingulate cortex and the insular cortex via
the anterior corona radiata (ACR), which course through the anterior limb of the internal capsule
(ICAL) [237]. Similarly, there are corticothalamic tracts, which run parallel to the TCT.
Association tracts are intrahemispherical and project between distant cortical regions. For
example, the cingulum connects the entire cingulate cortex, and is connected to every lobe of the
brain: the medial frontal, insular, parietal, occipital, temporal lobes [147,644]. The superior
longitudinal fasciculus is comprised of three major tracts (I, II, III). It runs antero-posteriorly,
and connects frontal regions along the sylvian fissure, and the insula to the parietal and temporal
lobes [147], and form most of the external and extreme capsules (EC/ExC). Another association
fibre is the uncinate fasciculus, which connects the OFC to the temporal lobe, including the
hippocampus and amygdala [146].
Animal studies have demonstrated that the trigemino-bulbar (as well as the spinobulbar) tracts
project to supraspinal regions via the hypothalamus [258], thereby bypassing the canonical
thalamocortical projections. Therefore, it is likely that these fibres provide input to the circuit of
Papèz [664], the limbic circuit of the brain. This circuit includes the hippocampus, the
mammillary bodies, the anterior thalamic nucleus, and the cingulate gyrus [644]. These gray
matter regions are connected through several white matter tracts: the fornix, the
mammillothalamic tract, the thalamocingulate tract, and the cingulum. The hippocampus projects
to the hypothalamus via collaterals of the fornix [644]. The mammillary bodies receive input
from braintstem nuclei, including the parabrachial nuclei, and further project to the anterior
nucleus of the thalamus via the mammillothalamic tract. The thalamus projects to the cingulate
gyrus via the thalamocingulate tract. The cingulum bundle courses the entire length of the
cingulate gyrus, and projects posteriorly to the hippocampus [644,664]. Anteriorly, there are
reciprocal projections with the PFC [644]. Therefore, the circuit of Papèz may be implicated in
processing the affective-motivational dimension of pain. Furthermore, key parts of the circuit of
Papèz, especially the cingulate cortex, are connected to other brain regions implicated in pain
[21,23].
Commissural tracts comprise of interhemispherical tracts. The corpus callosum contains fibres
that project from one hemisphere to the other, including the frontal, parietal, occipital and
temporal lobes, and so is thought to transmit information between the hemispheres. Little is

42
known about the role of the corpus callosum in the context of pain, but one study has
demonstrated abnormalities in the anterior body of the corpus callosum of patients with CRPS
[356]. The bilateral amygdalae and the ventromedial temporo-occipital cortices are connected by
the anterior commissure.
Figure 2-3: Diffusion tensor imaging reconstruction of Meynert’s classification of white matter
tracts in the brain. The tracts are superimposed on medial and lateral images of the brain.
Reproduced with permission from: [147].

43
2.3.4. Nociceptive pathways and mechanisms
The ascending nociceptive pathways can broadly be divided into three components: primary
sensory afferents that transmit nociceptive information from the periphery to the CNS; second-
order neurones that transmit nociceptive information to the thalamus; and third-order neurones
that transmit nociceptive information from the thalamus to cortical brain regions. There are also
collateral projections from trigeminal brainstem nuclei to other brainstem nuclei, including the
reticular formation (RF) and other brainstem nuclei. Here, the peripheral nociceptive
mechanisms and both the spinal nociceptive pathways and the trigeminal nociceptive pathways
will be described.
2.3.4.1. Peripheral nociceptive mechanisms
Free nerve endings that respond preferentially to noxious stimuli are called nociceptors
[89,124,796]. The transductive properties of nociceptors are based on the receptor molecules
and/or ion channels on the free nerve endings.
Some nociceptors are specialized and have specific response properties (unimodal), whereas
other types of nociceptors (polymodal) respond to several noxious stimuli. Unimodal nociceptors
are the basis of different pain qualia, such as burning, stinging, and aching. Polymodal
nociceptors, for example, can either be C or Aδ (and some Aß) afferents that respond to intense
mechanical and heat stimuli (CMH, AMH), and can also respond to chemical stimuli [224,944].
Furthermore, AMHs can be classified as type I and type II [870]. Type I AMH cells (Aß or Aδ
fibres) have low mechanical thresholds and a high heat threshold. Type II AMH cells (Aδ fibres)
have high mechanical thresholds and moderate heat thresholds. Another class of nociceptive
afferents is C and Aδ fibres that do not respond to mechanical stimulation, or have very high
mechanical thresholds, and are thus called mechanically insensitive afferents or silent
nociceptors [611]. There are several types of C fibre nociceptors: CMH cells, mechanically-
insensitive C fibres nociceptors and polymodal nociceptors.
Another feature of the different fibres is the quality of pain they encode. Type I AMH fibres are
present in both hairy and glabrous skin, and respond to sustained high-intensity heat stimuli (i.e.,
they are slowly adapting to the noxious thermal stimulus). Type II AMH fibres are rapidly
adapting fibres and are also present in hairy and glabrous skin [444,863]. CMH cells respond to

44
noxious heat stimuli near the pain threshold, and the neural response increases with the stimulus
temperature. Because of the different conduction velocities of the fibres, the percepts from the
different fibres are felt at different times: myelinated Aδ neural responses reach the CNS before
neural responses from unmyelinated C fibres. For instance, when a noxious heat stimulus is
presented to glabrous skin, Aδ (type II AMH) fibres give rise to first-pain, which is a sensation
of pricking, sharp and aching pain. This sensation is followed by second pain, which is a
sensation of dull or burning pain elicited by C fibres [136,543,710,711,867,870]. It is also
noteworthy that stimulation of glabrous skin does not elicit a dual percept of pain; the absence of
type II AMH fibres eliminated the possibility of a first-pain sensation [612].
Muscles and joints also have nociceptors. It is believed that the primary cause of muscular pain is
ischaemia (reduced blood flow and oxygenation), which activates nociceptors [572].
Furthermore, the gradual accumulation of metabolites formed by muscular exercise can also
sensitize nociceptors and induce allodynia [375,376,432]. The percept of muscular pain is
usually mediated by Aδ and C fibres, which are HTM [81], which can be sensitized by algesic
chemicals [941]. There are also mechanically insensitive nociceptors that respond to algesic
chemicals (chemonociceptors) [480]. The TMJ is also innervated by nociceptors. Specifically,
the TMJ and its periarticular connective tissues are innervated by nociceptors. Furthermore,
evidence suggests that the articular disk contains low-threshold mechanoreceptors [34].
Another distinction in pain qualities is based on the spread of the pain. Specifically, cutaneous
pain is often localized, whereas muscle pain is can be diffuse [482,805,946]. This is caused by
the size of the receptive field of a single nociceptor and less dense innervation of muscles. The
receptive field of cutaneous nociceptors is in the mm2 range [89,483], whereas that of muscle
nociceptors is in the cm2 range [504,599]. Further, muscular and cutaneous pains have several
different characteristics. For instance, deep muscular pain is reported as more unpleasant [838]
and it has a different quality than cutaneous pain. These differences can be attributed to different
neurophysiological phenomena in the peripheral innervation of muscles and skin, as well as
differential central pathways encoding nociceptive information from these tissues.
2.3.4.2. Spinal nociceptive pathways and mechanisms
Primary afferents encoding innocuous and nociceptive information from the skin, the viscera,
muscles and joints enter the central nervous system at the dorsal horn of the spinal cord. These

45
tracts join Lissauer’s tract, posterolateral to the spinal cord, and send collaterals into the dorsal
horn at various levels of the spinal cord [941]. Thermoreceptive and nociceptive primary
afferents synapse onto second order neurones in the dorsal horn of the spinal cord. Rexed
[727,728] identified several layers in the spinal cord based on cytoarchitectonic features. He
identified six layers in the dorsal horn, three layers in the intermediate region and ventral horn
and one layer surrounding the central canal. The dorsal horn layers can further be classified as
the superficial layers (laminae I, or the marginal zone, and II, or the substantia gelatinosa) and
the deep layers (laminae III-V, or the nucleus proprius, and lamina VI) [410,720,941]. Cutaneous
Aδ fibres terminate in laminae I, II, and V, and cutaneous C-fibres synapse onto laminae I and II.
Deep tissue, including muscles and joints, and visceral afferents synapse onto various layers.
There are two paths for HTM afferents: one set terminates exclusively onto lamina I, and the
other set terminates onto laminae I, IV, and V [857]. Unmyelinated nociceptive afferents from
the deep tissues project to lamina I and deeper laminae, but not lamina II. There are two types of
nociresponsive second-order neurones: nociceptive-specific (NS) neurones and wide-dynamic
range (WDR) neurones, which predominate in laminae I/II and V/VI. A small proportion of
nociceptors also synapses onto second-order cells in the other laminae (III, IV and VI). Fibres
that encode tactile information other than pain and temperature bifurcate upon their entry to the
spinal dorsal horn, and form an ascending branch which forms the dorsal column and a
descending branch that synapses to fibres in laminae IV and V [857].
There are two types of second-order neurones that receive input from primary nociceptive
afferent fibres: NS and WDR cells. NS neurones respond exclusively to noxious stimuli [160].
WDR neurones respond to innocuous stimuli but have greater responses noxious stimuli. The NS
cells include the polymodal heat-pinch-cold (HPC) cells [397]. Second-order neurones form the
STT. The second-order fibres originating in the marginal zone are mostly modality-specific, and
include thermoreceptive (which respond to cooling), HPC, and NS cells [20,397,964,967]. These
fibres form 50% of the fibres in STT [185]. The substantia gelatinosa (lamina II) receives
nociceptive input and is mostly comprised of interneurones which project to laminae I and V and
to adjacent spinal segments, and thus these cells do not contribute to the STT. STT neurones
originating from deeper laminae (the border of IV/V, the lateral aspect of VI, and a smaller
proportion from VII and VIII) [24] of the spinal cord are mostly WDR cells [943]. There are two
STT pathways: the lateral and the medial STT [545]. The lateral STT is a monosynaptic tract that

46
encodes sensory-discriminative aspects of pain, which project directly to the ventrolateral
thalamus in a somatotopically organized fashion. The lateral STT receives input mostly from the
superficial layers of the dorsal horn [545]. The medial STT is comprised of monosynaptic and
polysynaptic tracts that encode affective-motivational aspects of pain, and receives its input from
deeper laminae of the dorsal horn. The medial STT projects to the medial thalamus as well as
several brainstem nuclei (see below). The receptive fields of the medial STT neurones are
typically large, unspecific and not related to stimulus intensity [363], and there is no somatotopic
organization [102]. Unlike the neurones in lamina I, lamina V cells do not maintain somatotopic
organization [913,941,942,944]. Furthermore, lamina I cells have small receptive fields
compared to lamina V cells, which have more extensive receptive fields. Therefore, it is likely
that lamina I cells are more involved in the stimulus localization, whereas lamina V cells, which
have a graded response to innocuous and noxious stimuli, are likely more involved in intensity
coding.
As mentioned above, lamina II neurones receive input from primary nociceptive afferents, but
rather than ascend along the STT, these cells play a largely modulatory role in the spinal cord.
Specifically, lamina II cells comprise mostly of interneurones that modulate second-order
neurones originating in lamina I and V. These cells may, in fact, represent the gate discussed in
the Gate Control theory of pain (see Section 2.1.1.4). In addition to the STT, there are other
spinal nociceptive pathways, but these are outside the scope of this thesis.
About 85% of the STT decussates and 15% of the fibres remain ipsilateral to the input [258,545].
Both pathways ascend the spinal cord to project to several thalamic nuclei. For instance, there are
STT projections to ventroposterior region of the thalamus, including VPL and the inferior
ventroposterior thalamus (VPI) [258]. STT fibres originating from lamina I also project to the
ventromedial aspect of the posterior (vmPO) nucleus and the ventrocaudal aspect of the medial
dorsal (MDvc) nucleus of the thalamus [184]. Lamina V and deep laminae neurones also project
to the centromedian (CM) and centrolateral (CL) nucleus of the thalamus [258]. Canonically,
these formed the lateral and medial pain systems. The lateral system included projections to VPI
and VPL and vmPO and their cortical projections. The medial pain system includes MDvc and
CL thalamus [869].

47
As mentioned above, the medial STT has collaterals that project to several brainstem nuclei,
which form the spinobulbar tract [184]. There are four main regions where this tract projects to:
(1) the catecholamine cell groups (A1-A7), the parabrachial nuclei, the PAG and the brainstem
RF. The role of these regions is pain is discussed in further detail in Section 2.3.6. There are
other spinal nociceptive pathways, including the spinohypothalamic pathway, the postsynaptic
dorsal column system, and the spinocervicothalamic systems [258], but specific details about
these pathways are outside the scope of this thesis.
2.3.4.3. Trigeminal nociceptive pathways and mechanisms
The trigeminal nociceptive system includes primary afferents from peripheral nociceptors.
Nociceptors are specific receptors in the skin and muscles and joints that are free nerve endings.
Different nociceptors encode different types of noxious stimuli, such as mechanical, thermal
(heat and cold) and chemical stimuli, and some encode more than one of these stimuli
(polymodal nociceptors) (see Section 2.3.3.1).
Small myelinated Aδ and unmyelinated C primary nociceptive afferent fibres have their cell
bodies in the trigeminal ganglion, and project to the VBSNC [784]. Nociceptive afferents
synapse all along the VBSNC, including the three subnuclei of the STN and MSN [214,216,784].
Specifically, these project mostly to Vc, although small-myelinated primary nociceptive afferents
(Aδ) fibres have been shown to synapse (in lesser density) along the entire VBSNC, including Vo
and Vi, and MSN [453]. The laminar organization of the Vc is similar to that of the spinal dorsal
horn. Cutaneous nociceptors from the orofacial region project to the outermost lamina (lamina I).
Dense connections within and between the STN nuclei, termed deep fibre bundles of the VBSNC
[367,452,639], link different nuclei within the complex that have a common somatotopic
representation [82]. Although the role of these connections has yet to be established, evidence
suggests that the connections from Vc have a facilitatory effect on the other nuclei. Evidence for
the facilitatory role of these interneurones comes from studies reporting that inhibiting or
lesioning Vc reduced or abolished the excitability of rostral portions of the STN to noxious
stimuli in the trigeminal region, and the rostral nuclei showed increased responses to innocuous
stimuli in the periphery [82,83,452]. There is also evidence of ascending tracts in the deep fibre
bundles [82,215,450,452,501] that have a modulatory effect of rostral regions of the STN onto
Vc [425].

48
Similar to the spinal dorsal horn, primary afferents have specific targets within the Vc (i.e., Vc)
[784]. For instance, LTM project to the deeper layers of Vc (laminae III – VI) [453]. Small-
diameter nociceptive afferents synapse onto laminae I, III, V and VI of Vc. Many of the
aforementioned local circuit neurones, which modulate rostral regions of the STN emerge from
lamina II [784]. This lamina, akin to the spinal substantia gelatinosa modulates other neurones in
the Vc. Second-order neurones in the STN can be categorized like those in the spinal dorsal horn:
NS neurones, which receive input from nociceptive Aδ- and C-fibres, and WDR neurones, which
receive convergent input from neurones which encode both innocuous and nociceptive
somatosensory information [784]. Both cell types show graded responses to noxious stimulus
intensity, or as more of the receptive field is stimulated [784]. NS and WDR neurones
predominate in laminae I/II and V/VI [783-785]. NS cells have ipsilateral, localized receptive
fields in the skin, whereas WDR neurones have larger, more-complex receptive fields, with
tactile and, often, pinch-sensitive areas [784]. There are also NS and WDR in more rostral
portions of VBSNC, including Vo, Vi and nuclei outside the VBSNC, such as interstitial and
paratrigeminal islands [784]. However, as these neurones show a graded response to stimuli in
the oral cavity and perioral regions, they are outside of the scope of this thesis.
Additionally, most NS and WDR neurones in Vc receive converging input from both cutaneous
afferents and from afferents supplying other structures in the craniofacial region, including dura,
cerebrovasculature, the cornea, the tooth pulp and, relevant to this thesis, the muscles of
mastication and TMJ [17,141,214,586,597,645,708,787,788]. Furthermore, some of these
neurones also receive afferent input from other cranial nerves and upper cervical cord neurones
[784]. Together, these converging afferent inputs provide a neural substrate for the poor
localization, spread and referral of pain in painful conditions of the TMJ and deep craniofacial
tissues [784].
As in the STT, the TTT projects to the thalamus, as implied by its name. Sensory fibres from the
VBSNC synapse onto third-order neurones in the VPM, the posterior nucleus (PO), the
parafascicular nuclei (Pf), and the submedial nucleus (SM) of the thalamus [257] (for further
discussion, see 2.3.5). Other tracts emerging from second-order neurones in the Vc project to
brainstem and brain regions involved in nociception and pain modulation (see Section 2.3.5 and
2.3.6). These include projections to the brainstem RF, the PAG, the hypothalamus, brainstem

49
motor nuclei and brainstem autonomic nuclei [784]. The brainstem targets likely form the neural
basis for sensorimotor and autonomic reflex loops.
2.3.4.4. Trigeminal motor pathways
Abnormalities in motor output in the trigeminal system have been associated with parafunctional
habits, including bruxism (grinding) and clenching of the jaw [628]. These habits have been
associated with pain in the orofacial region, and with TMD [840]. Furthermore, patients with
TMD show abnormal motor function in the orofacial region (see Section 2.1.6), and so an
understanding of the interaction of sensory and motor pathways is essential to this thesis.
The sensory and motor systems interact and show connections throughout the CNS [361]. These
interactions allow for rhythmic motor activity (such as walking and chewing), reflexes (such as
withdrawal from a noxious stimulus) and smooth execution of a voluntary task. These
interactions are the neural basis of feedback loops. Feed-forward loops allow for the use of
sensory information and cues in the external environment to prepare for motor output. These
loops are used for rapid actions, such as catching a ball [361]. The sensory visual information
about the trajectory of a ball provides necessary information for the motor system to properly
anticipate where the ball can be caught. Conversely, feedback loops allow for sensory
information to modulate or correct motor output to attain a desired output. The sensory signal is
compared to a reference, and if these do not match, error signals are produced to correct the
proper trajectory. These loops are used for slow movements, e.g., such as reaching for an object
[361].
The motor aspect of the CNV supplies the muscles of mastication, including the masseter
muscles, the temporalis muscles, the anterior digastrics, the medial mylohyoids, the medial
pterygoids, and the lateral pterygoids [912]. The motor fibres originate in the trigeminal motor
nucleus in the brainstem, and receive input from supraspinal motor centres and sensory
information from afferents in the brainstem [555]. The trigeminal motor nucleus is located in the
lateral pons, medial to the MSN. Motor efferents emerge from the trigeminal motor nucleus and
leave the pons at the ventral surface, forming the motor root of the CNV. The motor root is much
smaller than and medial to the sensory root [256]. Other cranial nerves also innervate the
masticatory muscles. For instance, CNVII innervates the posterior digastric muscle (as well as

50
other facial muscles), CNX innervates the palatoglossus part of the tongue, and the rest of the
tongue is innervated by CNXII [644].
There are three levels of parallel and hierarchical motor control: the cortex, the brainstem and the
spinal cord (or in the case of the trigeminal system, the sensory and motor nuclei of the CNV)
[361]. At the most basic level, the trigeminal sensory and motor nuclei mediate simple reflexes,
such as the jaw-jerk reflex. This monosynaptic reflex occurs when there is pressure on the
mandibular teeth. Proprioceptive information of the mandible is relayed to the trigeminal MeT
nucleus in the midbrain via primary afferents in the third branch of the sensory root of the CNV
[176,644]. These neurones’ cell bodies are located in the MeT nucleus, and some collaterals
synapse directly onto the trigeminal motor nucleus. The trigeminal motor nucleus projects
efferent fibres to the masseter muscle, the internal pterygoid, and the temporalis muscle, which
contract and depress the mandible.
The brainstem motor nuclei are also responsible for repetitive (or rhythmic) motor behaviour,
such as mastication (or chewing) [644]. In the case of mastication, it is believed that a central
pattern generator maintains the rhythmic behaviour. The controlled and patterned contraction of
muscles to produce rhythmic mandibular motion can be broken down into three different stages
[555]: the preparatory, reduction and pre-swallowing stages. The first step is breaking food into
chewable pieces. In some cases, such as a steak, this is done before the food reaches the mouth.
In other cases, such as biting into an apple or a sandwich, this step occurs at the mouth. In this
stage, the digastric muscles (jaw-opening muscles) are very active, and the masseter and the
temporalis muscle show little activity, but are involved as they are responsible for fast-closing.
The chewing (or reduction) cycle is characterized by three phases: opening and fast-closing and
slow-closing. The slow-closing phase occurs when teeth come in contact with food, and muscles
move the jaw laterally to grind food between the molars. The final stage of chewing – pre-
swallowing – consists of fast-closing, slow-closing, and three sequential opening phases. These
coordinated movements rely heavily on sensory feedback – location of food in the oral cavity,
state of the food, position of the jaw, lips, and tongue, etcetera. These sensory cues modulate the
rhythmicity of masticatory muscles while chewing. Taken together, there is evidence that a
central pattern generator at the level of the trigeminal motor and MeT nuclei exists, and
coordinates muscular activity during chewing [555,644]. The central pattern generator in the
brainstem can be modulated by corticobulbar tract (motor efferent tracts from motor cortical

51
regions) [555]. Further modulation can come from RF of the brainstem, which receives
convergent input from the cortex, and the VBSNC. It is important to distinguish that reflexes and
pattern generators do not require input from upper motor neurones (corticobulbar tracts), but
cannot function without this input [555].
The next level of motor control comes from the brainstem – namely, the RF [361]. As described
above, the RF modulates the central pattern generator based on sensory cues and efferents from
cortical regions and other brain regions such as the amygdala and the hypothalamus. Other
brainstem motor regions that project to the trigeminal motor nucleus include the red nucleus
[368], and the vestibular nucleus [256]. These findings suggest that motor output to the orofacial
muscles can be modulated by a number of different sources. The cortical regions represent the
highest level of motor control: voluntary motor movement [361,644]. These movements are
initiated in the brain. Specifically, M1 (Brodmann’s Area 4 (BA4)) is where voluntary motor
output is initiated. M1 receives dense input from the S1 (BA 1, 2, 3a, and 3b) [674,706,747,748],
and two regions involved in motor planning: the SMA, and the PMC (both regions are
designated as BA6) [644]. It has been suggested that somatosensory input into M1 may be
related to motor learning. In line with these findings, it has been shown that acute nociceptive
input inhibits motor learning, and that patients with TMD have abnormal motor output (see
Section 2.2.1.2).
M1, SMA and PMC have motor efferents that project to the trigeminal motor nucleus either
directly, via corticobulbar tracts, or indirectly, via the RF [644]. In primates, M1 is
somatotopically organized: the map is organized ventrolateral-dorsomedially, with the toes along
the medial wall, and the hand region in the middle of gyrus, and the orofacial region is
represented on the ventrolateral surface of the gyrus [644]. The corticobulbar tracts originate at
the lateral aspect of the M1 (or SMA, or PMC), descend along the genu of the internal capsule
(IC), and course through the cerebral peduncles in the brainstem [644]. There is both ipsilateral
and contralateral contribution (contralateral being dominant) to the corticobulbar tracts, which
decussate at the level of the cerebral peduncles in the midbrain, and synapse onto lower motor
neurones in the trigeminal motor nucleus. Therefore, the sensory and motor systems are highly
interconnected, and sensory information modulates motor output at several levels within the CNS
[556,644].

52
2.3.4.5. Supraspinal nociceptive and pain regions
Peripheral and brainstem nociceptive pathways project to the thalamus and other brain regions
implicated in pain. Herein, the subcortical and cortical brain targets of second-order neurones of
the TTT and its collaterals that bypass the thalamus will be described. Additionally, this thesis
will describe experimental findings from animal studies and electrophysiological studies
implicating these regions in nociception and the putative role of these regions in the experience
of pain. Neuroimaging studies describing neural correlates of acute and/or chronic pain will be
discussed in Section 2.5.
It is important to recognize that it has not always been clear that the cerebral cortex is involved in
pain perception. It has been proposed that the brain is where perceptions occur (c.f., sensations) –
even Aristotle recognized that perception was different from sensation. Sensation means to
encode stimuli, and perception is the interpretation of this sensation. Aristotle posited that
sensations were a property of the physical being, whereas he relegated percepts to the soul (or the
mind) [810]. Nonetheless, until the mid-twentieth century, there was little evidence for the cortex
to be involved in pain perception. For instance, Hitzig [426] stated that pain is a subcortical
phenomenon. Head and Holmes [417] found little evidence that the cortex is involved in pain – a
patient with extensive cortical damage still felt pain, and pain unpleasantness. In fact, they
suggested that, in the case of central pain – a condition where a stroke affects the thalamus – pain
arises because the thalamus is released from cortical control [514]. Cushing could not produce a
painful response in patients with electrical stimulation of the cortex [196], and so thought that
pain was a percept of the thalamus. Penfield and Boldrey echoed these findings in their paper in
1934 [677], stating that electrical stimulation of the brain did not elicit painful responses.
However, in a later study, they found that stimulating the postcentral gyrus (S1) did elicit painful
responses, but that these responses were rare – 11 of 800 stimulations produced a “painful”
experience, none of which were intense enough for the patient to object [679]. Additionally, they
suggested that the removal of cortex does not relieve pain [678]. From these findings, they
concluded that pain has little cortical representation. In support of this hypothesis, Adrian [6]
could not identify any impulses reaching the cerebral cortex from noxious or thermal stimulation
in the periphery in several animals.

53
Conversely, to treat a patient with thalamic syndrome (or central pain), procaine was injected
into the somatosensory cortex of a patient, who reported pain relief for a period of 2 months
[845]. Further, Russel and Horsley [746] excised a portion of S1 (in the area of the arm), and the
subject reported a loss of sensation and analgesia in the affected limb, however not all sensory
qualities were abolished. Similarly, a number of studies reported that patients who suffered
cortical lesions, especially in the region of S1, had altered pain sensations – in the absence of
thalamic damage (reviewed by [571]). Electrical stimulation of the cortex provided variable
results, as some groups found that stimulating S1 caused the patient to experience pain, whereas
several other studies S1 stimulation did not elicit pain. In one of these cases, stimulation of S1
reproduced the patients’ pain, and resection of S1 alleviated their pain [542,571]. Furthermore,
Dusser de Barenne and McCulloch showed that the function of VPL and S1 were interdependent,
and when deeper layers of S1 or VPL were ablated, the spontaneous electrical activity in the
other was diminished [281].
Although other theories have postulated that the percept of pain occurs in the brain, none had
incorporated a specific role for it in their model. However, no study has shown that pain is solely
due to a single area in the brain. As Marshall [571] suggested, S1 (and likely other cortical
regions) are involved in pain, but may not be the “endpoint” of pain – that S1 receives and
processes nociceptive input (likely sensory-discriminative information); however, this is not the
region where the nociceptive input is perceived as pain. Rather, this information is processed and
“sent” to other brain regions where the percept of pain occurs. To address this point, Melzack
suggested that a network of brain regions working together is required for the percept of pain
[592-594]. This pain “neuromatrix”, as he called it, is supported by the multidimensional
experience of pain. This theory required the concerted activation of several brain areas to
produce the experience of pain, each set underlying a dimension of pain. Therefore, it was
proposed that to have a conscious experience of pain, many brain regions, each related to a
dimension of pain (cognitive, modulatory, sensory-discriminative, affective, motor) are required.
This theory provides an explanation for the finding that stimulating a single brain region does not
consistently produce pain. However, as will be described, to some extent in this section, and in
further detail in Section 2.5, few (if any) studies have been able to show that these regions of the
brain are, effectively, functionally connected – a fundamental tenet of a “pain matrix”. In fact,

54
the concept of the so-called “pain matrix” idea has now fallen out of vogue, and other theories
about cortical brain regions have been postulated, and will be discussed herein.
Thalamus
The STT (and the TTT) project to the thalamus. Interestingly, the TTT ascends bilaterally, albeit
the contralateral thalamus receives a greater density of projections [784]. These tracts project to
the ventroposterior nuclear group (ventrobasal complex in subprimates) in a somatotopic fashion.
In fact, the lateral STT projects to the lateral portion (called the VPL) [351] and the TTT projects
to the medial portion (called the VPM) [340]. These projections maintain the somatotopic
organization of the VBSNC. The ventral STT projects to other thalamic nuclei, including MD, Pf
and CL [869]. Gaze and Gordon [351] identified NS neurones in the thalamus of cats and
monkeys for the first time. Similarly, Perl and Whitlock [684] identified NS neurones in the
ventrobasal thalamus of cats and monkeys. These neurones showed responses only to noxious
stimuli, and had a specific (c.f. large or diffuse) receptive field in the contralateral body.
Similarly, Applebaum and colleagues [29] confirmed these findings with antidromic mapping of
VPL neurones in the upper lumbar region of the spinal cord. There are also WDR neurones in the
thalamus of humans and animals [142,527]. Electrophysiological recordings and stimulation
studies in humans undergoing neurosurgery have confirmed that the human thalamus receives
nociceptive input, and these inputs are topographically organized in the ventroposterior nucleus
[223,396,534]. The somatotopic organization of STT and the ventroposterior thalamus suggests
that these nuclei may be related to the sensory-discriminative aspect of nociception, i.e., the
location of the stimulus, and the graded response to nociceptive stimulus suggests that this region
is implicated in magnitude encoding, i.e., the intensity of a stimulus. These concepts were
supported by findings that stimulating VPL results in localized pain in humans [415].
Additionally, some STT neurones projecting to VPL have collateral projections that synapse onto
CL [30,362,589].
Furthermore, Perl and Whitlock [684] demonstrated that the STT also projects to CM and
intralaminar nuclei, which only receive nociceptive input and have bilateral receptive fields, and
the posterior nuclear group of the thalamus, which has diffuse receptive fields. These findings
were confirmed by several tracing studies [25,102,129,485,488,603,701]. Additionally,
connections from the STT to the VPI were also found [25,85,701].

55
The STT and TTT also have topographical input to MDvc: the STT projects to the anterior
portion, and the TTT projects more caudally [186,341]. There is also evidence of a pain- and
temperature-specific nucleus – the VMpo [188]. Neurones that responded to cold perception, in
the vicinity and had some of the same properties attributed to VMpo were located in the human
thalamus [223]. Blomqvist et al. [101] found a site in the human thalamus with STT projections
in the vicinity of VMpo.
Projections of the TTT are similar to those of the STT. As mentioned above, these ascending
tracts project to different nuclei within the ventroposterior thalamus: the VPM and VPL,
respectively. The VPM receives input from all levels of the VBSNC, however, only dorsal MSN
neurones project to the ipsilateral VPM, whereas ventral MSN and STN project to the
contralateral VPM [126,127,140,574,618,719,811,812,831,861,948,957]. Furthermore, there are
also projections to the PO and MDvc [127,341], and projections to CL and CM have also been
identified [948].
Finally, the ventrolateral nucleus of the thalamus (VL) receives input from deep dorsal horn, via
the STT [85]. These projections overlap with cerebellar projections to VL. Third-order neurones
project from VL to the motor cortex, and so it has been suggested that these inputs are related to
sensorimotor integration, or the motor dimension of pain [794].
In summary, the thalamus receives thermal and nociceptive information from the STT and TTT.
The tracts project to several nuclei within the thalamus, which have various cortical targets.
These various projections suggest that the different targets contribute to various dimensions of
pain. These concepts are discussed in further detail herein.
Primary somatosensory cortex
In primates, S1 is located along the postcentral gyrus, and is comprised of four cytoarchitectonic
rostrocaudally organized regions: BA 3a, 3b, 1, and 2. These regions have functional differences:
areas 3b and 1 receive cutaneous tactile input, and areas 3a and 2 respond to proprioceptive
input. Furthermore, each of the four regions in the somatosensory cortex has a complete body
(homuncular) map: the foot is located medially, and the face is located on the ventrolateral bank
[196,652,679].

56
S1 receives input from the somatosensory thalamus. Specifically, in primates, there is evidence
of projections from the VP thalamus to each of the cytoarchitectonic regions of S1 [644]. As
information is processed from BA 3b to BA 1, the size of the neuronal receptive field increases,
and the neurones have more complex response properties, which is an indication of integration
and modulation of sensory input within S1 [644]. Similarly, BA 3a projects to M1, BA 2 and S2.
The receptive fields in BA 2 and S2 are larger and the response properties are more complex than
in BA 3a. Furthermore, there is evidence of projections from S1 to other cortical regions,
including reciprocal connections between 3b, 1, and 2 and the posterior parietal cortex (PPC, BA
5 and 7), and contralateral somatosensory cortex. Area 3a projects to M1. There are also
projection to distant cortical regions, including the insula, the cingulate cortex and the parietal
lobule and operculum [644]. There is also evidence for descending projections from S1 to
subcortical regions [644].
As discussed above, the role of the S1 in pain processing is contentious. Some of the earliest
documented evidence comes from a study by Dusser de Barenne in 1916 where he reported that
cats showed an allodynic response (which he called hyperalgesic) to noxious stimuli in the
periphery by applying strychnine, an alkaloid that blocks inhibitory signaling [825], to S1 cortex
[280]. In humans, electrical stimulation of S1 only sometimes elicited a feeling of pain in the
patient (e.g., [679]). Similarly, in some but not all cases, phantom limb pain was resolved after
S1 ablation [293,383,542,833] (reviewed in: [296,571,845]). Furthermore, resection of S1 was
ineffective in relieving pain in other pain disorders. Conversely, patients with Rolandic seizures
(epilepsy in the region of S1 and M1) reported pain [961], and lesions to parts of S1 caused a
disorder similar to thalamic syndrome [941].
Studies in non-human primates support the aforementioned findings in humans undergoing
neurosurgery – that S1 has a role in nociception. For instance, Peele showed that ablating
specific BA in S1 or PPC in monkeys led to a loss of nociception for nine months, and a
permanent loss of the ability to locate a noxious stimulus [676]. Furthermore,
electrophysiological studies in S1 in non-human primates have demonstrated that nociceptive
regions of the thalamus project to BA 1 and 2 [485,631]. There is also evidence of somatotopic
organization [270,484]. Kenshalo and Isensee [486] identified neurones in areas 3b and 1 that
responded maximally to pinching in the receptive field, and a proportion of these showed a

57
graded response to noxious heat stimuli in the receptive field. Furthermore, there is evidence that
area 3a neurones contribute to allodynic and hyperalgesic phenomena [929].
Specific to the trigeminal system – and of particular interest to this thesis – Biedenbach and
colleagues electrically stimulated the tooth pulp in non-human primates, and reported graded
responses in the face area of S1, mainly in area 3a [91]. Similarly, Chudler and colleagues found
cells that in the somatotopic region of the face in BA 1 and 2, which also showed a graded
response to nociceptive input [161]. Interestingly, Biedenbach and colleagues suggested that S1
cortex is likely essential for localizing noxious stimuli on the body, but also observed responses
in neurones outside S1, and so suggested that S1 contributes to pain perception, but this role is
not exclusive to S1.
The parasylvian cortex: S2 and insula
The parasylvian cortex includes several regions: S2, insular cortex, and the ventrolateral
prefrontal cortex (vlPFC, which will be discussed later). S2 is comprised of two adjacent
cytoarchitectonic regions: BA 40 and 43, which are in the ventrolateral bank of the postcentral
gyrus, and within the parietal operculum [644]. The insula is a very large, heterogeneous region.
It is comprised of two long sulci in the posterior portion, and three short sulci in the anterior
portion [39,40]. The insula can be divided into three subregions: the anterior, middle and
posterior insula, based on functional and anatomical features.
S2 receives dense connections from S1, and some input from the VP thalamus. S2 also has a
somatotopic organization, but much less detailed than that of S1 [735,737,930,941,952]. S2
maintains this somatotopic organization in its projections to M1 [859]. Evidence from non-
human primate studies has shown that, in addition to performing integrative aspects of
somatosensory processing [294,644], neurones within S2 respond to noxious stimulation
[65,736,930], and that these neurones show somatotopic organization and graded response to
nociceptive laser-evoked potentials [65]. Electrophysiological studies in humans have shown that
S2 responds to noxious stimulation [336,338,583,734,871,890,953]. Frot and colleagues have
demonstrated that S2 shows graded responses to increasing intensity of non-noxious and noxious
somatosensory stimuli [335]. Taken together, it would seem that S2 is implicated in processing
the sensory-discriminative dimension of pain. However, the receptive fields in S2 are larger and
more complex than those of S1, and so it is possible that S2 plays a more integrative role and

58
outputs this information to motivational and motor regions to respond to a noxious stimulus
accordingly.
The insula is implicated in a variety of functions. Evidence for this comes from anatomical
studies that have revealed that the insula receives input from primary sensory, limbic, motor, and
other brain areas, and projects to a number of brain regions [40]. The insula is considered to be a
visceral sensory and visceromotor area [40]. Further evidence for the role of the insula in several
systems comes from neuroimaging studies – the insula is one of the most commonly activated
brain regions in various experimental paradigms [956]. Craig has suggested that the insula is the
homeostatic/interoceptive centre of the brain [187 10572,190]. It has also been suggested that the
insula is a node in a general salience network [263,265,616]. In terms of pain perception, the
insula is considered as a part of the medial pain system [869]– albeit it has also been suggested
that it may be part of the lateral pain system (i.e., its role in sensory-discriminative dimensions of
pain are under debate). The dorsal posterior subregion of the insula receives direct input from
VMpo, a nucleus in the thalamus that exclusively receives thermal and nociceptive input [188].
Evidence for nociceptive input to the insula comes from studies of electrical stimulation and
recordings of the insular cortex [7,337,583,658,688]. Further evidence comes from lesion
studies, where patients suffered sensory loss after a lesion of the insula (Obrador, 1957, cited in:
[343,377,532]) and surrounding areas [92]. Another lesion study found that the patient showed
hypalgesic responses to noxious stimuli after an ischaemic stroke affected the parasylvian cortex
(insula, parietal operculum and S1) [233]. Greenspan and Winfield [378] reported that a patient
with a tumour that was compressing the posterior insula and the parietal operculum had
increased pain thresholds to heat, cold, and mechanical noxious stimuli. Conversely, Starr and
colleagues [829] found that two patients with insular lesions showed increased heat pain intensity
ratings. Greenspan and colleagues [377] showed that patients with lesions to the parietal
operculum, where the insula was spared, had decreased discrimination of noxious stimuli,
whereas in patients where the insula was damaged, there was decreased motivation to escape the
noxious stimuli (it was less aversive).
Because of the diverse input into the insula, a number theories about its role in pain perception
have been postulated. For instance, it has been postulated that the insula is a multidimensional
integration site for pain [116]. This suggests that the many dimensions of pain are integrated
and/or interoceptive region [183]. Alternatively, Coghill [169] suggested that because of the

59
behavioural relevance of pain, and its essential role is survival, there are several “backup” pain
intensity coders, including S1 and the insula. In line with these findings, it has recently been
suggested that the insula is a central magnitude estimator for the brain [48], and that there is a
also pain-specific magnitude estimator in the insula. These estimators have differential
connections and are hubs of cortical and subcortical networks, respectively. An alternative
perspective is that the insula may be a salience detector in the brain (for a more comprehensive
discussion of these ideas, see [616], and Section 2.5.1.1)
The cingulate cortex
The cingulate cortex surrounds the corpus callosum, anterior to the genu, dorsal to the body and
posterior to the splenium. In 1937, Papèz [664] included the cingulate cortex as part of the limbic
circuit he identified, the brain’s emotional circuit. Although this remains true today, many other
functions have been ascribed to the cingulate cortex, including cognitive, motor, limbic
nociceptive, pain-modulatory, visuospatial and memory [899]. However, there are several
subregions within the cingulate that are specialized for these functions, although, like many other
associative brain regions, there is much integration and overlap of function between adjacent
subregions [899].
There are many classification systems used to differentiate subregions within the cingulate
cortex, which add to the complexity of comparing studies that use different species or different
nomenclatures. In this thesis the nomenclature developed by Vogt and colleagues [905] will be
used, whenever possible. It is noteworthy that the region previously referred to as the anterior
cingulate cortex (ACC) has now been divided into two regions: the mid-cingulate cortex (MCC)
and the ACC. This nomenclature was developed based on a multimodal approach, including
eletrophysiological properties of cells, cytoarchitectonic studies, lesion studies and neuroimaging
studies [900,905]. The subdivisions of the cingulate include the ACC, MCC and the posterior
cingulate cortex, and the following description of these regions comes from: [899]. The ACC can
be further subdivided into the subgenual ACC (sgACC; BA 25) and the perigenual ACC
(pgACC; BA 32, 24 a, b, c). The MCC can be further subdivided into the anterior MCC (aMCC;
BA 33’, 32' and 24 a', b', c'), which includes the rostral cingulate motor area (rCMA) and the
posterior MCC (pMCC; BA 32' and 24 a', b', c'), which includes the caudal cingulate motor area
(cCMA). The PCC is also divided into dorsal (dPCC; BA 31, 23) and ventral (vPCC) areas

60
[906]. There is additionally the rostral caudal zone (RCZ; BA 29, 30), which is ventral and
anterior to the dPCC [900]. This thesis will focus on the ACC and MCC, whereas the PCC and
RCZ are outside the scope of this thesis.
It is worth prefacing the specific descriptions of the roles of the ACC and the MCC with the
observation that these regions are among the most often activated in brain imaging studies,
across various paradigms [956]. Therefore, descriptions of specific findings may be related to
broader functions, such as salience, rather than specific functions.
Tracing studies in non-human primates have elucidated thalamocingulate connections. The ACC
receives the ventral anterior, CL, Pf, and other midline and intralaminar nuclei of the thalamus
[798]. There are some, albeit a small proportion, projections from the MD nucleus. Specifically,
the anterior thalamus projects to the BA 32 region of pgACC and sgACC. MD thalamus projects
to both subregions of the ACC (all three BAs). The ACC has been implicated in modulating
emotional, mood state and visceromotor functions in humans. In monkeys, the ACC projects to
subcortical and brainstem structures involved in these functions, including the hippocampus and
bed nucleus of the stria terminalis, hypothalamus and the PAG and parabrachial nucleus. There
are also extensive projections from area 24 to the MD thalamus [803]. The sgACC is
interconnected to several frontal brain regions, including the pgACC, the OFC, vlPFC, and
mPFC. The pgACC receives input from the sgACC, MCC, PCC, the insula, the frontal pole, the
OFC, mPFC, the frontal operculum, and the dorsolateral prefrontal cortex (dlPFC).
The MCC includes the CMA, which has direct projections to motor neurones in the ventral horn
of the spinal cord [623,691]. This subregion is involved in the planning and execution of
volitional movement. The MCC receives input from VL, VPL, CL, CM-Pf, and MD [798].
Corticothalamic projections from the MCC synapse terminate onto ventroanterior nucleus (VA)
and MD. Tracing studies in non-human primates have also elucidated anatomical connections
between the MCC and other cortical regions, including the insula, the temporal, parietal, frontal
and limbic association areas. The MCC connects to the sgACC, pgACC, the OFC, pre-
supplementary motor areas, mPFC, PCC, PMC, frontal operculum, the vlPFC and the dlPFC
[686]. Additionally, the CMA receives input from M1 and more ventral regions of the MCC
[416].

61
Of relevance to this thesis are the intrinsic connections (via the cingulum bundle) within the
cingulate cortex, which modulate and coordinate attention, cognitive processes and response
behaviour [622,901]. Additionally, these intrinsic connections are modulated by input from and
output to the dlPFC – a region involved in selecting information about the sensory environment
(attention, self-monitoring, personality, working memory) and decision-making. Therefore, the
connection between the dlPFC and the cingulate cortex is of particular interest in the context of
pain processing (heightened awareness of pain) and pain modulation. Another important
connection is between the OFC and the cingulate [150,686]. The OFC is involved in autonomic
and emotional processes, and goal-directed behaviour [742]. It is believed that the OFC and
dlPFC modulate sgACC output (potentially via the MCC’s intrinsic connections) to subcortical
and brainstem nuclei that are involved in pain modulation (see Section 2.3.6).
Lesion and electrophysiological studies in humans have implicated the MCC in pain processing.
Specifically, patients who had large regions of their cingulate cortex lesioned to relieve
intractable pain (a procedure called “cingulotomy” [50]) showed attenuated emotional responses
to pain – they still perceived pain, but it was no longer distressing [327,326]. These findings
suggest that the cingulate cortex is implicated in the affective dimension of pain processing.
Later studies reported equivocal post-cingulotomy outcomes for pain relief
[50,51,414,696,938,939]: from 23% [439] to 75% [327]. This variability is likely due to the
location and size of the lesion within the MCC [733]. In addition to changes in the affective
dimension of pain post-cingulotomy, Davis and colleagues [217] report that a patient with a
relatively small MCC lesion showed hyperpathic intensity and unpleasantness responses to
noxious thermal stimuli, suggesting that the cingulate may also have a role in the sensory-
discriminative (or intensity coding) dimension of pain. Furthermore, Dierssen and colleagues
elicited a painful response in the contralateral body and face of a patient with phantom limb pain
when they stimulated the MCC (case V.G.D., point no. 8 in [254]).
In line with these findings, Hutchison and colleagues [440] identified neurones in the human
MCC that respond to painful cutaneous stimuli, and Lenz and colleagues [533] reported laser-
evoked potentials in the MCC as measured by microelectrode recordings to noxious laser stimuli
in the periphery. Iwata and colleagues [451] identified neurones in the CMA that are
nociresponsive, which are involved in attention to pain, and nocifensive behaviour. Similarly, in
a reward and pain-avoidance cued task, Koyama and colleagues [499] found that cells in the

62
MCC (likely the CMA) were implicated in stimulus prediction and anticipation, and response
selection. Taken together, these findings suggest that the cingulate cortex is implicated in the
affective-motivational dimension of pain: as pain becomes more bothersome or aversive, there is
greater motivation to escape it, and these processes are encoded within the MCC.
However, it is essential to note that the MCC is involved in a number of complex tasks, and
therefore, may serve a more general role in the brain: salience detection and response selection.
Neurones in the MCC are activated during tasks that have a cognitive and/or emotional load and
that are attention demanding, such as mental arithmetic, word generation, and Stroop tasks
[218,229]. Furthermore, cingulotomy increased errors in motor output in a monetary reward task
[940]. Therefore, the MCC may, in fact, be a generalized system for orienting the self to a more
salient stimulus, and choosing an appropriate response, whether it is to avoid a noxious stimulus,
or to increase rewarding behaviour.
Prefrontal cortex
The PFC is a large, heterogeneous region of the brain comprised of a number of subregions
involved in various higher cognitive and limbic functions. Broadly defined, it is believed that the
PFC is central to the cognitive-evaluative dimension of pain. Specifically, various regions of the
PFC have been implicated in cognitive modulation of pain; however, most of these data come
from neuroimaging studies, and so will be discussed in Section 2.5.1 and 2.5.2. Non-imaging
evidence to support a role for the PFC in pain perception and/or modulation is paltry. One line of
evidence comes from an extreme surgical procedure: frontal lobotomies, which were used to
relieve intractable pain, amongst other conditions [154,288,330,880,923]. However, the extent
and specificity of these surgeries is unclear, and regions such as the cingulate cortex – which is
known to be involved in pain perception and modulation – could have been destroyed.
Furthermore, a tracing study that injected murine VBSNC neurones with a neuroanatomical
tracer, horseradish peroxidase (a retrograde tracer), did not identify any cortical targets in the
PFC [570]. Additionally, a later study injected a viral tracer (an anterograde, trans-synaptic
neural tracer) into the murine tooth pulp, and failed to identify any infected cells in the PFC.
Additionally, Dum and colleagues [277] injected the STT with a viral tracer, and also failed to
identify any infected cells in the PFC. However, tracing studies identified corticofugal tracts
from the PFC to brainstem regions involved in pain modulation in the rats [70,406,536], cats

63
[823] and monkeys [405,520], and stimulation of the PFC in rats modulates nociceptive input
[403,404]. Further, anaesthetization of the PFC of rats reduced pain modulation, as measured by
the jump reflex threshold [175]. In a recent histological study of a rat model of spared-nerve
injury, Metz and colleagues [608] reported that there are morphological and functional changes
in pyramidal neurones in the mPFC. Specifically, they found increased dendritic branching,
increased spine density in the nerve injury group, compared to the sham group. Additionally,
these structural changes were associated to functional changes in synaptic currents, which
correlated with tactile thresholds in the injured limb. More recently, Ji and Neugebauer [460]
have attempted to elucidate the neural underpinnings of observed abnormal mPFC activity in
neuroimaging studies of pain [47,356] (see Section 2.5.1). They found that cells in the mPFC
were effectively inhibited in a murine chronic pain model, and may be related to cognitive
deficits in chronic pain [461]. Additionally, there is evidence that transcranial stimulation of the
dlPFC modulates pain in experimental pain paradigms [310] and in chronic pain
[114,649,754,802]. Taken together, it seems that the PFC does not play a role in pain perception,
but rather may be involved in cognitive appraisal and evaluation of pain, and its subsequent
modulation.
Motor regions
Pain and motor interactions have been described in Section 2.1.6, and the anatomical pathways
pertinent to this thesis are described in 2.3.2. This section will describe tracing and
electrophysiological studies in animals and humans that have implicated suprabulbar motor
regions in pain. In general, nociceptive input into motor regions is believed to serve two
purposes: (1) orient the body toward the threat (noxious stimulus) and (2) initiate nocifensive
behaviour (e.g., avoid the stimulus). Several lines of evidence have implicated suprabulbar motor
regions in pain. The first line of evidence comes from a study that investigated dorsal column
stimulation in monkeys [53]. This study found that the stimulation of the dorsal column
modulated nociceptive input into the Pf thalamic nucleus, which indicated that modulation of
nociceptive input occured at the level of the thalamus, rather than in (or in addition to) the spinal
cord.
Further evidence for supraspinal motor-pain interaction comes from unexpected findings that M1
stimulation (motor cortex stimulation; MCS) reduced spontaneous hyperactive firing in a model

64
of central pain in the cat [874]. Interestingly, stimulation of S1 did not have an effect on
hyperactivity in the thalamus. Furthermore, studies have demonstrated that MCS in cats
suppresses nociceptive activity in the spinal and medullary dorsal horns [182,789]. MCS has
become a prevalent therapy for intractable neuropathic pain; however, the mechanism of action
remains unclear. It is believed that MCS recruits distant antinociceptive brain regions through
corticocortical connections [643]. Similarly, there is ample evidence that transcranial magnetic
stimulation (TMS) – a less invasive method to stimulate the cortex – of the M1 is effective tool
to modulate neuropathic pain (e.g., [8,331,463,643,821]). Additionally, there is likely activation
of CST that can modulate nociceptive input (as postulated in the Gate Control theory of pain, see
Section 2.1.1.4).
Several studies have investigated the interaction of pain and motor learning in the brain. For
instance, one study found that a novel tongue-protrusion task is associated with acute and
reversible neuroplastic changes in the face region of M1, as measured by TMS [41,842].
However, noxious stimulation of the oral cavity disrupts learning the motor task, and related
neuroplasticity in M1 [109].
Another line of evidence for motor-pain interactions in the brain comes from viral tracing studies
of the STT and that have identified infected neurones in subcortical and cortical motor regions.
For instance, Dum and colleagues [277] found that, in addition to S1, S2, and the posterior
insula, M1 (BA 4), the ventral premotor cortex (PMv; BA 6) and the CMA (BA 32) receive input
from thalamic targets of the STT.
The basal ganglia are a group of subcortical nuclei that have classically been associated to motor
behaviour [13,239,640,945]. However, anatomical, functional and behavioural studies have
elucidated several roles of the basal ganglia. The basal ganglia are organized in a number of
parallel and segregated anatomical circuits forming functional loops [665] and the role of these
loops is integrated to mediating goal-directed behaviours [388]. In the context of nociception and
pain, there are a number of findings suggesting that the basal ganglia have a complex, and yet
undetermined role. For instance, Dum and colleagues [277] reproduced (but did not present)
findings that subcortical structures, including the basal ganglia receive direct input from the
anterolateral tract of the spinal cord. Previous studies demonstrated that nocireponsive neurones
project directly to the globus pallidus and the putamen [642], and that neurones in the globus

65
pallidus respond to nociceptive input [88,164]. Furthermore, the caudate nucleus of the rat and
the cat has neurones that respond to noxious mechanical stimuli in the periphery and that are
somatotopically organized [164,544,732,769]. Also, there is evidence that striatum is densely
populated with opiate receptors in rats [38] and humans [97]. In line with these findings,
stimulation of the caudate nucleus has been shown to produce analgesic effects in the monkey
[550].
Further support for the role of the basal ganglia in pain comes from evidence that patients with
movement disorders related to the dysfunction of the basal ganglia, such as Parkinson’s disorder,
often report pain [162,385,398,624,635,707,759,965]. Furthermore, depletion of dopamine in the
substantia nigra (the region affected in Parkinson’s disorder) in mice leads to hyperalgesic
responses to noxious stimuli in the periphery [163]. Stimulation of the striatum also modulates
orofacial pain: stimulating dopaminergic neurones has been shown to modulate the nociceptive
jaw-opening reflex in rat [55,78].
In a recent study of patients with lesions in the putamen, Starr and colleagues [828] found that
patients showed lower cold pain detection thresholds (i.e., less sensitive), lower pain intensity
ratings to suprathreshold heat pain and decreased pain intensity ratings for prolonged noxious
heat stimuli on the affected side (contralateral to the lesion), compared to the affected side and to
controls. However, the patients’ tactile thresholds remained unchanged. Therefore, it seems that
damage to the putamen causes decreased pain sensitivity, but not tactile thresholds, suggesting a
role in the putamen in the sensory-discriminative dimension of pain.
The nucleus accumbens, integral to the reward circuit in the mammalian brain [388], is another
region within the basal ganglia that has recently been implicated in pain processing. The concept
of the reward pathway encoding aversive stimuli is reminiscent of the age-old dichotomy of
pleasure and pain, and can be dated back to Platonic and Aristotelian ideas [199]. It has been
reported that, in rats, the nucleus accumbens is pronociceptive, and that inhibiting the nucleus’
function with opioids induces antinociception [352].
It is tempting to conclude that the basal ganglia are involved in determining a behavioural
response to pain - the motivational and nocifensive dimensions of pain. However, the substantial
input of nociceptive information from different regions of the brain and the extensive
connectivity of the basal ganglia suggest that these subcortical nuclei may have a more

66
prominent role in pain perception. For instance, it has been suggested that the basal ganglia may
be the site of convergence of the different dimensions of pain (i.e., pain integration) [828];
however, more research is required to substantiate this theory. It is noteworthy that sensory
convergence has been demonstrated in several nuclei within the basal ganglia [162,165].
Finally, there is evidence that changes to sensory input to the brain, including prolonged
nociceptive input, can modulate M1 excitability and output. Specifically, when a peripheral
nerve is transected in mice, neuroplasticity occurs in M1 [2,4,328,329]. However, it is possible
that these changes in M1 reflect changes in S1 cortex, which has dense projections to M1 [41].
Furthermore, evidence in humans suggests that acute pain in the periphery can decrease M1
excitability [3,157,301,302,744] and acute intraoral pain can prevent learning a novel tongue-
protrusion task [109].
In sum, motor regions in the brain receive nociceptive input, have nociresponsive neurones and,
in some cases, are modulated by nociceptive input. The most obvious role for motor regions in
the context of pain is to initiate a motor response to pain (nocifensive behaviour), however,
evidence suggests that they may have a more prominent role in pain integration, and pain
modulation.
2.3.5. Descending modulation
The perception of pain is modulated by several psychological factors, including attention, arousal
and expectation, as well as the context in which the stimulus is presented [309]. This can be
illustrated by two examples: athletes injured during a sporting event and a soldier wounded in
combat. In both cases, the injured person often reports less pain than persons sustaining the same
injury in another context [153]. This variability in the perception of pain has been associated to
endogenous pain-modulatory systems, which are the neural basis by which attention, arousal,
expectation and cognitive processes alter the perception of pain. It has been suggested that these
networks modulate the transmission of nociceptive information to suppress or enhance
nocifensive responses to increase the chance of survival [309]. In the example of a soldier in
combat, suppressing pain reduces pain-related distraction, and facilitates escape from combat.
Conversely, the pain-modulatory systems can enhance sensitivity to pain, and enhance
nocifensive behaviour, which promote healing [309].

67
It has long been recognized that pain-modulatory mechanisms exist. In 1911, Head and Holmes
[417] proposed that the thalamus was the site of pain perception, and corticothalamic
connections modulated the nociceptive processing in the thalamus. Further, they suggested that
such inhibitory mechanisms were present at lower levels in the sensory pathway. Evidence
suggests that both sensory input and motor output can be modulated by supraspinal regions. For
instance, in 1915, Sherrington and Sowton demonstrated that, in a decerebrated animal, spinal
transection reduced stretch reflexes and enhanced flexor reflexes. In 1954, Hagbarth and Kerr
[389] demonstrated that supraspinal mechanisms do influence afferent (sensory) input from the
periphery to the spinal cord in the cat. Specifically, stimulation of M1, S1, S2, ACC, RF and
ventral anterior part of the cerebellar vermis depressed the dorsal root reflex, suggesting
modulatory effects of brain regions onto the spinal cord. It was also shown that cortical
stimulation could inhibit cutaneous sensory input to the VBSNC [e.g., 421]. In 1964, Kuypers
demonstrated that there are several tracts emerging from the brainstem that terminate onto the
dorsal horn [507]. However, the Gate Control theory of pain [595] (See Section 2.1.1.4) was the
first to propose that there were supraspinal regions that modulated nociceptive input from the
periphery at the level of the spinal cord.
Animal studies have demonstrated that there are, indeed, specific nuclei in the brainstem that
have a modulatory effect on dorsal horn nociresponsive neurones (for a comprehensive review,
see: [420,521,580,726]. The first study to demonstrate this was by Reynolds [730], in 1969. In
this study, electrical stimulation of the mesencephalic central gray matter in rats provided an
analgesic effect. Specifically, during an invasive surgical procedure, a laparotomy, the rats did
not show aversive behaviour, without paralysis. The potent analgesic effect of stimulating the
brainstem, specifically the periaqueductal gray (PAG), has been confirmed in the rat, cat, non-
human primate and in humans [580] [437]. This stimulation-produced analgesia is selective, in
that animals remain alert and respond normally to environmental stimuli, with the exception of
nociceptive stimuli – these responses are abolished [309]. Furthermore, if a noxious stimulus is
presented outside the region of analgesia, the animal responds with typical pain-related responses
[580]. Studies have demonstrated that stimulation-produced analgesia of the PAG is opiate-
based. Specifically, studies have found that injecting morphine, and other opiates induces
modulation [252,307,309,613]. Furthermore, naloxone, an opioid antagonist, abolishes the
analgesic effect when injected into the PAG [5,873,959] or the third ventricle [958]. The

68
analgesic effect of the stimulation of the PAG is mediated through a descending pathway from
the brainstem (via the rostral ventromedial medualla (RVM)) to the dorsal horn, and has been
demonstrated to inhibit nociceptive reflexes (e.g., the tail flick in rats), and nociceptive neurones
in the spinal and medullary dorsal horns [260,354,786,789].
It is noteworthy that descending pathways not only inhibit somatosensory input and pain
transmission, but can also play a facilitatory role at the spinal level [353,613,725]. It is believed
that this facilitatory role may be related to the chronification of pain [724].
2.3.5.1. Descending modulation pathways
There are several descending pathways that can modulate nociceptive input and pain
transmission. In general, these pathways are modulated by brain regions through corticobulbar
tracts and other tracts originating from subcortical nuclei that terminate in the brainstem
[95,309,420]. The best characterized of these pathways involves a circuit connecting the PAG to
nuclei within the RVM, including the nucleus raphe magnus (NRM), and the spinal dorsal horn,
via the dorsolateral funiculus [58]. Another modulatory region is the dorsolateral
pontomesencephalic tegmentum (DLPT), which connects to the cuneiform nucleus in the
midbrain, and the locus coeruleus/subcoeruleus (LC/SC), and other noradrenergic nuclei.
The PAG receives input from the ascending fibres from spinal lamina I, via the
spinomesencephalic tract [43,441,481,596] and the VBSNC [77,567,931], from the cerebral
cortex, the hypothalamus and other brainstem nuclei, such as the RVM, including the NRM, and
the nucleus cuneiformis [77]. There are direct projections from the PFC (with the exception of
the medial OFC), ACC and the insula to the PAG [406,405,413]. There are also some
corticobulbar connections to the PAG from M1 and the PMC (BA 4 and 6, respectively)
[405,619]. Furthermore, the amygdala has dense projections to the PAG. The amygdala, in turn,
is modulated by several cortical and subcortical regions. Furthermore, the PAG receives dense
input from the hypothalamus, and stimulation and opioid injection to the hypothalamus have
analgesic effects [433,566,731]. There are also cortical projections to the PAG via the anterior
pretectum [158,960]. In addition to its descending projections, and the afferent input from brain
regions to the PAG, the PAG has ascending efferents to MD thalamus and the OFC [135,168].

69
The RVM is another brainstem region essential for descending pain modulation. The RVM is a
group of nuclei that are the primary relay site for projections from the PAG to the spinal (or
medullary) dorsal horn [420]. The most important nucleus in the RVM in the context of pain
modulation is the NRM. For instance, inhibiting NRM neurones abolishes the analgesic effect of
PAG stimulation [10,71,355], whereas stimulation of the NRM produces analgesic effects by
inhibiting nociceptive neurones in the spinal dorsal horn [253,308,469]. The RVM projects to the
spinal dorsal horn via the DLF [58]. Therefore, the RVM is essential for the analgesic effect of
the PAG to occur. Therefore, the PAG is an important antinociceptive region, which receives
convergent input from ascending tracts, cortical regions and other brainstem regions. The PAG
projects to the RVM, which directly modulates nociceptive neurones in the STN and the spinal
dorsal horn.
Recent studies have also found that descending projections from the PAG to the spinal dorsal
horn and Vc, via the RVM, can also be facilitatory, i.e., these projections have a pronociceptive
effect, rather than an antinociceptive effect [654,835]. It has been suggested that these
mechanisms may contribute to the chronification of pain [704].
In addition to the PAG-RVM descending pathway, a parallel pathway exists in the brainstem: the
DLPT pathway [272,309,354]. The components of the DLPT include the nucleus cuneiformis,
which is adjacent to the PAG and is similar in function to the PAG in terms of pain modulation,
and pontine noradrenergic nuclear groups, including the LC/SC. It is believed that this pathway
modulates deeper laminae in dorsal horn, as it has moderate projections to these laminae,
compared to few projections to the superficial dorsal horn [166]. The LC/SC projects directly to
the dorsal horn via the ventrolateral funiculus, and electrical stimulation of these nuclei has an
inhibitory effect on nociceptive cells [726]. This pathway is separate from that of the PAG-RVM
pathway because inhibition of the RVM does not disrupt the analgesic effects of the LC/SC
[468].
2.3.5.2. Diffuse noxious inhibitory controls
It has long been known that a stimulus perceived as painful can diminish or mask a pain arising
from elsewhere in the body. For instance, Hippocrates stated in his Aphorisms: “of two pains
occurring together, not in the same part of the body, the stronger weakens the other” [424]. This
effect has been described as counter-irritation analgesia, and only occurs when “distant” body

70
parts are affected, i.e., extrasegmental body parts. In 1979, Le Bars and colleagues [520]
proposed the DNIC hypothesis. This hypothesis states that nociceptive signals can be inhibited
by nociceptive input from another body part, that projects to a different segment of the spinal
cord. To test this hypothesis, the sural nerve RIII reflex was recorded in subjects who were
presented with a painful “conditioning” stimulus (a hot water bath) [521]. Normally, when the
sural nerve is electrically stimulated, a painful pinprick sensation is felt in the dermatome of the
nerve and a knee flexor muscle nociceptive (RIII) reflex is evoked. It has been proposed that
DNIC is a spinal cord/STN mediated process [251]. However, when a noxious stimulus was
applied to the hand, the reflex and the perceived pain intensity of the sural nerve stimulation was
decreased. DNIC has also been demonstrated in animal models. For example, in the
anaesthetized rat, the C fibre reflex which is strongly inhibited by heterotopic noxious
stimulation [521]. It is believed that DNIC is a spinally-mediated process [521], and WDR
neurones, mainly in lamina V of the spinal cord, appear to be key for the effect of DNIC [519].
However, DNIC does not occur in animal models where the spinal cord is sectioned, or in
tetraplegic human subjects [739]. This suggests that supraspinal regions contribute to DNIC.
Specifically, there is evidence that the dorsal reticular nucleus in the caudal medulla contribute to
DNIC [519,896]. This nucleus receives afferent nociceptive input from the spinal cord, as well as
convergent input from PAG, RVM, RF, the hypothalamus, the amygdala, M1, S1, OFC, and the
insula [309]. The dorsal reticular nucleus projects to the thalamus, suggesting a role in both
nociceptive-signal transmission as well as antinociception [621]. Similarly to the descending
modulation pathways, it has been shown that DNIC may also have a facilitatory role for
nociceptive-signal transmission, but this is outside the scope of this thesis. For a review of these
concepts, see [546,895]. DNIC has also been demonstrated in the orofacial region [917].
Furthermore, a remote noxious stimulus can affect jaw muscle activity [131,132,894].
In sum, there are both negative and positive feedback loops that are consistently modulating
nociceptive-signal transmission to the brain. These loops are comprised of a network of
brainstem nuclei that are modulated by cortical regions. This delicate balance of inhibition and
facilitation can be very useful, based on the context of the nociceptive stimulus. Cortical regions
can upregulate the inhibitory effects in order to block out excess or unwanted stimuli.
Conversely, in a dangerous or threatening environment, when a subject is hypervigilant, the
facilitatory mechanisms can be upregulated as a protective mechanism.

71
2.4. Structural brain imaging
With the advent of functional neuroimaging, there has been a large increase of studies
investigating the function of the brain in health and disease. These studies have vastly improved
our understanding of brain function, and its relationship to perception, cognition and behaviour.
However, brain function is intimately related to brain structure, and so, studying neural structure
can provide a fundamental framework for understanding brain function. Aristotle, often
considered the father of comparative anatomy [145], described the basic tenet of physiology as
the structure-function relationship [100]. For Aristotle, function is what is of importance,
structure is secondary, and thus form and function are not separate disciplines, but are
complementary [100]. In the context of the brain, functional MRI (fMRI) has provided a wealth
of functional information, but the study of brain structure supporting brain function is only just
burgeoning [145]. Scientists have used the structure-function relationship to understand normal
function and abnormalities in disease. In a recent editorial, Catani [145] criticizes this approach
as reverse logic. He argues that form begets function, or, in evolutionary terms, the form comes
from the necessity for a function. Therefore, modification of form causes dysfunction. Only by
studying “form” (or anatomy) can we understand function, and dysfunction.
MRI technologies can be used to examine both gray and white matter structure in humans. These
techniques will be discussed below.
2.4.1. MRI
2.4.1.1. What is magnetic resonance?
A brief overview of the concepts related to MRI signal production, analysis and applications to
structural brain imaging will now be presented. For a comprehensive review of the basic
concepts of magnetic resonance (MR) and MRI, see [119,438] – the information reviewed below
was derived from these sources.
The concept of MR is based on the interaction of an atomic nucleus that possesses a spin and an
applied magnetic field (B). Nuclear spin angular momentum is an intrinsic property of an atom.
Most atoms have a nuclear spin, meaning that they are spinning at a constant velocity around an
axis. Nuclear spin angular momentum is a vector quantity, and it is conserved for different
elements. The human body is primarily comprised of water and fat, which contain hydrogen

72
atoms and so the 1H nucleus is an ideal choice for MRI. The 1H nucleus is a single proton
(termed the protium, and will be referred to as a proton herein), and has a spin of ½, and is the
most abundant hydrogen isotope (c.f. deuterium) for hydrogen. Also, its response to an applied
magnetic field is one of the most robust observed in nature.
A proton has a magnetic moment, which is parallel to the axis of rotation. The orientation of the
spin and the changes induced to it by a magnetic field provide the basis for the MR signal.
However, in MR, it is the population, ensemble or collection of spins (e.g., in a tissue) that is
key. The net magnetic moment of a population of spins (i.e., the vector sum of the proton
moments) is zero, and so no net magnetization is observed in a tissue. However, when the tissue
is placed into a magnetic field (B0), the protons begin to precess about the magnetic field. The
proton is slightly tilted away from the axis of B0, but the axis of rotation is parallel to B0. This
precession is caused by the interaction of B0 and the spinning proton. The rate of precession (ω0)
is proportional to the strength of the magnetic field and can be summarized by the Larmor
equation:
ω0 = γB0
where ω0 has units of Rad/s, B0 has units in Tesla (T; 1T is roughly five orders of magnitude
greater than the magnetic field of the Earth, and three orders of magnitude greater than that of a
refrigerator magnet), and γ is the gyromagnetic ratio, which is a constant for each nucleus. In the
case of protons, γ = 267.52 x 106 Rad/s/T (≈ 42.577 MHz/T).
Another feature of spins is that they have two possible states when they are in a magnetic field:
“spin-up” and “spin-down”. These are the two quantum-allowed states, although they can make
transitions between these two states, there are no intermediate states. These two states have
different energies associated with them (potential energy). The spin-up configuration is
associated with a greater potential energy than the spin-down configuration. Transitions between
the spin states require a specific energy, produced by molecular motion and collision. The energy
required for the transition to occur must be quantized – they must have the correct amplitude to
induce the “flip”. In an ideal system and at rest, there are an equal number of spin-up and spin-
down positions, and the net magnetization is zero. However, when the ensemble is placed in an
external magnetic field, the spins return to their lower energy level configuration due to
interactions with surroundings. The spin-down configuration is of lower energy, and is parallel to

73
B0, whereas the spin-up configuration is of higher energy, and is anti-parallel to B0. This effect is
called the Zeeman effect. Therefore, a small excess of the spins will try to flip to the lower
energy state. This process requires time because collisions are required to produce the quanta of
energy for the flip. Over time, the ensemble will reach an equilibrium state with the applied
magnetic field, and the transition probability of the spins becomes balanced because a small
excess of the spins have flipped to the lower energy state. This number of protons at each energy
level is governed by the Boltzmann distribution:
Nup/Ndown= e-∆E/kT
where Nup is the number of protons in the spin-up configuration, Ndown is the number of protons
in the spin-down configuration, and k is the Boltzmann constant, which is 1.381 x 10-23 J/K.
When a vector sum of spins in the transverse plane (perpendicular to B0) is performed, the net
magnetization is zero because of the random distribution of the position of the perpendicular
component of the protons’ precession. However, along the longitudinal axis (parallel to B0), there
is a net magnetization parallel to the field. This means that the tissue is polarized and magnetized
when placed in a field B0, and the level of net magnetization is M, which is parallel to B0. This
induced magnetization, M, is the basis of the MR signal.
A rotating frame is defined as a Cartesian plane rotating about an axis, which remains stationary.
If we assume that B0 is parallel to the z-axis, and that the xy-plane is rotating at the Larmor
frequency with the proton, then we observe what looks like a stationary spin. If we sum these
vectors, the resultant vector resembles a vertical magnetization. A time-varying magnetic field
creates an electric field. The created electrical signal is what we measure as the MR signal.
However, in the summated vector of M is not time-varying – recall that the transverse vectors
cancel each other, and the longitudinal vectors are stationary. To induce a time-varying magnetic
field, a new magnetic field (B1) is introduced at an axis perpendicular to B0 and M. B1 rotates at
the Larmor rate. M will rotate about the axis of B1 (and therefore perpendicular to B1,) at a given
frequency (ω1 = γB1). We can control the angle of rotation by setting the duration and amplitude
of B1. This rotation of the spins produces a time-varying magnetic field. For instance, if we rotate
the spins 90˚, then the spins are now precessing about B0 in the transverse plane. Given that a
rotating magnetic field induces a voltage in a loop of wire, if a loop of wire is placed around the
ensemble of the rotating spins, and attached to an amplifier, signal within the frequency range of

74
radio signals (3 kHz to 300 GHz) can be detected. The recorded signal is the summated
magnetization of the difference of the up and down spins of the entire population.
2.4.1.2. MR signals
The second magnetic field that is introduced to rotate M is a short pulse of radiofrequency (rf),
also called an excitation pulse. The excitation pulse is comprised of several frequencies within a
narrow range, or bandwidth. During the pulse, the protons in the sample absorb the energy of a
given frequency (ω0), and after a given time, depending on the T1, reemit this energy at the same
frequency. When the pulse is turned off, the protons begin to realign themselves to their stable
state, and they reemit the energy they absorbed in order to realign. As more protons undergo
relaxation and reemit their absorbed energy, the transverse magnetization of the protons creating
the signal is lost, and the signal decays.
Relaxation is a fundamental feature of MR, and relaxation time is measured for an ensemble,
rather than individual protons. For example, relaxation times for cerebrospinal fluid (CSF), gray
matter and white matter within the brain are different, and we can measure the relaxation time for
the entire tissue, rather than individual water, protein or fat cells. There are two relaxation times
that we can measure: T1 and T2.
Magnetization time (T1)
After the spins have been rotated, symbolized by M, the protons have a natural tendency to
return to their stable state – relaxation. The relaxation time “T1” represents the time required for
M to return to 63% of its original value (M0) before the rf pulse, in the z-axis (along B0). This
relaxation follows an exponential recovery function:
M(t) = M0(1-e-TR/T1)
where TR is the time between the rf pulse (repetition time), and T1 is a constant and is specific to
the tissue. T1 represents the time it takes for tissues to relax after magnetization. This relaxation
is named a “spin lattice” relaxation. The motion of the surrounding spins provides a fluctuating
magnetic field that enables spins to be flipped back to their stable state.
The mobility of atoms varies based on the size of the molecule it is part of, and its interactions
with the immediate environment. The consequence of this is that different tissues magnetize at

75
different rates. For instance, smaller molecules, such as water (the main component of CSF),
have a very long T1 (up to several seconds), whereas larger molecules, such as fat (which is the
main component of myelin, and thus white matter), have a very short T1 (less than a second).
Grey matter has an intermediate T1, between that of CSF and white matter. These differences in
T1 provide a useful contrast mechanism in MRI.
Signal decay time (T2)
The relaxation time “T2” refers to the time required for the transverse component of M to return
to 37% of its initial value. When M is rotated into the xy-plane, the spins of the protons are in
phase. However, over time, the signal decays because the spins begin to dephase. This loss of
coherence occurs because of molecular interactions with the protons, and because of the transfer
of energy from one spin to another. The signal decay can be summarized by the equation:
! ! = !"#(!)
!!!!!!!!!!!!!!!!= !!!!!"/!!
Where “TE” is time from excitation (or echo time), and is an adjustable parameter that represents
the time after the rf pulse during which the signal data are collected. T2 is also tissue specific,
and is slower in small molecules (the signal from CSF, which is mostly water, decays over
several seconds), and faster in larger molecules (e.g., gray matter and white matter, where the
signal decays in less than a hundred milliseconds). Again, differences in T2 allow for a contrast
useful to MRI. Further, by increasing TE, the contrast between tissues can be increased but less
signal is actually received as it decays.
Spatial Encoding
The creation of an image requires a process that encodes signals in tissues in 3D space. The
Larmor relation states that the signal’s frequency is related to the magnetic field affecting the
protons. For instance, if we maintain a homogenous magnetic field, B0, in the bore of an MRI,
there would be a single frequency present in the MR signal. However, if we vary the field with a
gradient coil, then frequency encodes location. To do so, we need to have different frequencies
representing different points in space. Therefore, we introduce a gradient pulse that distorts the
magnetic field, and produces a gradient in the magnetic field. The effect is that at one end of the

76
gradient there is slightly less magnetic field strength (B < B0), in the middle there is still a net
magnetic field strength, which is equal to B0 (B = B0), and at the other end there is a slightly
higher magnetic field strength (B > B0). In a simplified model, this will affect the frequency of
the nuclear spins – they are precessing at a slower rate at the lower end, and precessing faster at
the higher end, compared to the middle, where B = B0. This is done in three orthogonal planes to
localize signals in all three dimensions. The relationship between frequency and B, allows us to
encode spatial position as a function of precession frequency.
Contrast
Given that the relaxation times, T1 and T2, are specific to tissues, and that there are parameters
that are specific to each relaxation time’s contrast (TR and TE, respectively), we can vary these
parameters to adjust the contrast in MR images. The signal we obtain be summarized by:
where ρ is proton density, k is an instrument specific calibration factor.
We can experimentally adjust TR, which affects the T1 contrast. Alternatively, we can adjust TE,
which affects the T2 contrast. If we set low values for the TR and the TE, then the contrast of our
image will be T1-weighted. However, if we have a long TR and TE, the image will be T2-
weighted. Water molecules that interact with small molecules have relatively low signal in a T1-
weighted image, and relatively high signal in a T2-weighted image. Conversely, water molecules
that interact with larger molecules have relatively high signal in a T1-weighted scan, and a
relatively low signal in a T2-weighted scan. Therefore, in a T1-weighted scan white matter is
bright, and CSF is dark. The opposite occurs in a T2-weighted scan: white matter is dark and
CSF is bright.
In summary, rf pulses convert nuclear magnetization to signal. Rf pulses add energy by
displacing longitudinal equilibrium. The contribution of intrinsic tissue properties, T1 and T2,
can be manipulated by adjusting the timing of TR and TE, respectively.

77
2.4.2. Gray matter imaging
Structural studies of the human brain used to be limited to post-mortem investigation and
sometimes biopsies (e.g., [844]). However, for the past few decades, it has been possible to use
MRI to examine brain structure in living humans non-invasively. Structural MRI can be used to
measure many structural features including the thickness of the cortical mantle, the gyrification
index, and other measures of interest, at a very high spatial resolution: the millimeter scale. As
newer technologies emerge, even greater spatial resolution can be achieved, but these are outside
the scope of this thesis (for a review, see: [282]).
Structural MRI analysis of gray matter is commonly performed on T1-weighted images, as these
acquisitions can be optimized to increase the contrast between gray matter and white matter. In a
T1-weighted image, the CSF is dark (almost black, due to the long T1 relaxation time), the white
matter is bright (due to the myelin sheath surrounding axons) and the intensity of gray matter is
between that of CSF and white matter. Many automated and semi-automated techniques have
been developed to characterize gray matter differences within a population and between
populations. These measures include the shape of gray matter structures, gray matter density (the
relative amount of gray matter within a voxel) and cortical thickness. Brain imaging software
packages typically include toolboxes that use algorithms to segment the three tissue classes in the
brain (gray matter, white matter and CSF), and perform statistical analyses. Currently, the two
most common methods are voxel-based morphometry (VBM) and cortical thickness analysis
(CTA). Specific information about the methods and the relative advantages and limitations of
each are discussed below.
2.4.2.1. Voxel-based morphometry
One of the first automated toolboxes to analyze gray matter was Christian Gaser’s VBM toolbox,
implemented in the Statistical Parametric Mapping (SPM). A VBM analysis of brains from a
group of subjects includes the following stages: 1) segment high-resolution brain images into
gray matter, white matter and CSF; 2) spatially normalize the brains to a common standard
stereotactic space; 3) apply a spatial filter to “smooth” the gray matter segments (this effectively
blurs the gray matter to improve alignment) [36,37]; and 4) perform voxelwise parametric or
non-parametric statistics. Using either statistical method, the results are corrected for multiple
comparisons. More recently, Jacobian modulation has been added to the processing pipeline

78
prior to the smoothing step so that findings can be considered as volumes, rather than relative
gray matter density [36,371]. To do so, for each brain the software calculates the deformation for
normalization to the standard space (stereotactic) template – a Jacobian determinant, and then
modulates each voxel in standard space to account for the expansion that occurs. VBM is a
widely used tool in neuroimaging, and its popularity can be attributed to its relative ease of use,
and the little processing power required. However, this technique has been criticized because it is
sensitive to misalignment, especially in the cortex [104,210]. Therefore, results in cortical
regions should be interpreted with caution. Nonetheless, the alignment of subcortical gray matter
regions is robust, and thus more reliable.
2.4.2.2. Cortical thickness
It has been suggested that cortical thickness (rather than gray matter volume) may provide a
better measure to examine changes or variability of the cortical mantle and so a technique called
CTA was developed. CTA measures the thickness of the cortex in millimeters, which is a well-
defined physical measure. However, CTA is limited to the cortex, as implied by its name, and
thus we must use it in conjunction with VBM to get a full picture of gray matter in the brain.
Nonetheless, CTA is purportedly superior to VBM because of improved alignment of cortical
gray matter, especially gyral folding patterns. This method inflates the cortical gray matter sheet
into a sphere, and aligns the gyri and sulci based on landmarks. Several software packages offer
a CTA toolbox, and the most common one is FreeSurfer. The analysis pipeline relies on some of
the basic principles of VBM, but with a few differences. First, high-resolution T1-weighted brain
images are aligned to a different standard stereotactic space than the one used in VBM, which
uses the Montreal Neurological Institute’s standard template of 152 brains (MNI152). FreeSurfer
uses the Talairach and Tournoux [844] brain as a template for normalization. Then, the brains
undergo intensity normalization to remove coil-related inhomogeneities. The image is then skull-
stripped, and the subcortical structures are identified and labeled, which aids in the segmentation
of white matter from gray matter. The pial surface is then defined, and the hemispheres are
separated. The software then measures the distance at every point between the edge of the white
matter surface and the edge of the pial surface, which represents the thickness of the cortex. This
cortical sheet is then parcellated and the individual gyri and sulci are identified and labeled using
an atlas that is based on manual parcellations [243,318,321]. Parametric univariate statistics can
be used to perform group-level analyses on cortical thickness. To do so, the brains must first be

79
aligned to one another. This is done by inflating the cortical surface into a sphere, and iterative
alignment algorithms, which use landmarks and the individual parcellation to align specific sulci
and gyri [319,320]. These steps allow for better alignment of the cortex, and thus overcome the
limitations of VBM.
2.4.3. White matter imaging
DWI with MRI can be used to non-invasively evaluate human white matter. The study of white
matter paths is called brain hodology, and comes from the Greek hodos, which means path. Brain
hodology in humans has mainly been on post-mortem studies of white matter, which date back to
antiquity. Notably, in the seventeenth century, Steno stated:
To say that the white matter is but a uniform substance like wax in which there is no hidden contrivance, would be too low an opinion of nature’s finest masterpiece. We are assured that wherever in the body there are fibres, they everywhere adopt a certain arrangement among themselves, created more or less according to the function for which they are intended [translated by 760,830]
Neuroscientists in the nineteenth century then attempted to elucidate structure-function
relationship [145,306]. However, these scientists recognized that local structure-function
relationships could not fully explain dysfunction in the brain; they understood that distant brain
regions interacted to produce behaviour. This is exemplified by Wernicke’s statement that
sensory and motor regions must work together through connections (i.e., white matter tracts) in
the brain [145]. Déjérine was a French neurologist, medical historian, and neuroanatomist, whose
investigation of white matter in the brain is the most comprehensive of the time – and some
would argue until today [607]. He agreed with Wernicke, proposing that the key to
understanding brain function relied on understanding the connections of association fibres within
the brain [145,237]. More recently, the concept of studying the anatomic basis of behaviour, and
functional relevance in anatomic circuits has reemerged [147,760], notably by Geschwind
[359,360] and Kuypers (e.g., [387,663]). Geschwind proposed that “brain function is the result of
effective communication between structures geographically distributed around the nervous
system”[761]. Recently, Mesulam [607] suggested that the function of a neurone is defined by its
connections, and that understanding the connection pattern of the cortex is the only way to
understand how the brain functions. Therefore, in addition to functional and gray matter studies
of the brain, elucidating the connectional anatomy, and the integrity of these tracts in the brain

80
will provide essential insight into the connections (or disconnections) of patients with chronic
pain. The advents of DWI and diffusion tensor imaging (DTI) have provided a method to
essentially perform in vivo dissections of the white matter in humans [60], and to elucidate
abnormal white matter structure related to clinical correlates in disorders, such as chronic pain.
Furthermore, of particular relevance to this thesis, is that DTI (see below) can also be used to
examine peripheral nerves [429], such as the CNV in trigeminal neuralgia [525] (discussed
below and in Chapter 7).
2.4.3.1. Diffusion tensor imaging
DTI is a mathematical model that is fit to DWI data. DWI is a brain imaging technique that can
be used to visualize white matter. Specifically, DWI is an MR modality that is sensitive to the
diffusion of water molecules [63]. The following description of DWI and DTI are based on
information from: [52,60,62,63,67,522,524,626,660]. Diffusion is the process of molecules
moving, and is the result of Brownian motion. All molecules, when suspended in a liquid or gas,
show random translation movements (i.e., Brownian motion), resulting from thermal energy of
the molecules. The diffusion of water molecules can be summarized by Einstein’s equation:
X2= 2DTd
where X2 is the average mean-squared diffusion distance (in m2) along one direction, Td is the
diffusion time (in s), and D (in m2/s) is the diffusion coefficient. DWI’s ability to discern
microstructural features of a tissue is based on the notion that water molecules sample the
microscopic environment at a very high resolution (much higher than the average MRI
resolution), roughly 10 µm per 50 ms [524]. This sampling includes collisions, interactions and
crossing of water molecules with cell membranes, fibres and other macrostructural features, such
as macromolecules. A DWI provides a summary of the bulk motion of a diffusion molecule
within a voxel, which can reveal structural and geometric organization of a tissue [60,523]. The
molecule of choice in human DWI is the water molecule, because 90% of the protons in the
human body are located in water molecules. Diffusion occurs in all three dimensions of space,
and when unhindered, will diffuse equally in all directions (the expected vector-sum of
displacement is zero), a process known as isotropic diffusion. When diffusion is hindered, e.g.,
by a tissue, we observe anisotropic diffusion. Anisotropic diffusion of water has been observed
in muscle, spinal cord and cerebral white matter [524]. Einstein’s equation does not apply to

81
anisotropic diffusion, and we have to use a symmetric tensor, , to describe diffusion in this
scenario:
It is noteworthy that in the scenario of anisotropic diffusion, we assume that diffusion is
Gaussian in all directions.
Anisotropic diffusion in brain white matter has generated much interest in understanding the
connectional anatomy of the brain. Specifically, water molecules within or between white matter
tracts have anisotropic diffusion, as the bulk diffusion is parallel to the main axis of the fibre
tract. This anisotropy comes from the geometrical structure of the axon – it is tubular in shape,
and many axons form a fibre tract. Specifically the diffusion of water is restricted by cellular
barriers, such as myelin, cell membranes, and macromolecules (such as microtubules and
neurofilaments), and the molecules tend to diffuse along the primary axis of the tract, whereas
diffusion perpendicular to this axis is impeded. MRI acquisition parameters can be adjusted to
acquire images that provide information about the diffusion of water, and thus, the connectivity,
integrity and orientation of white matter in the brain (see below). This thesis will briefly review
the basic principles of DWI signal generation and image acquisition (for a comprehensive
review, see: [522,524,626]).
MRI can be used to acquire several different types of image contrasts (T1, T2; see section
2.4.1.2). As described above, the MR signal equation can be summarized as:
S = kρM0 (1-e-TR/T1) e-TE/T2
where S is the acquired signal, TR is the repetition time, TE is the echo time, ρ is proton density
and T1 and T2 represent relaxation times specific to tissues. This equation can be modified to
include a diffusion term, bD:
S = kρM0 (1-e-TR/T1) e-TE/T2 e-bD
€
D
€
D =
Dxx Dxy Dxz
Dxy Dyy Dyz
Dxz Dyz Dzz
"
#
$ $ $
%
&
' ' '

82
where the b, or b-factor, summarizes all of the gradient effects, and D is the diffusion tensor. To
measure the diffusion of water molecules, we must acquire two different b-values.
Therefore, for Sb we obtain:
!! = !"!!(1− !!!"!!)!!
!"!!!!!"
and for Sb0, we obtain:
We can then solve the equation for D:
where Sb is a signal acquired from the scanner with a given value for b and Sb0 is the signal
acquired with a b-factor equal to zero. The signal acquired from b = 0 is not sensitive to
diffusion, and is referred to as a non-diffusion-weighted image. All of the other terms in the
signal equation are kept constant, and so cancel out.
The signal (S) that we acquire is generated. To do so, we apply a phase encoding gradient pulse
along a particular direction, using different combinations of the x, y, z gradients. After the rf
pulse is applied (for a given duration), the protons which are precessing about the z-axis begin to
dephase. We then apply a second re-focusing pulse, which is the same as the first pulse, but in
the opposite gradient direction. This paired-pulse sequence sensitizes the signal to diffusion.
Specifically, the refocusing pulse can only rephase the protons if they have not moved. If the
water molecules diffuse (i.e., change location between the first and second rf pulse), then this
motion will disrupt the phase-gradient. This imperfect rephasing is measured as signal loss. As
previously mentioned, by using two different b-values we can acquire two different signals,
which allows us to solve for D. The b-value is related to γ, the strength of the gradient pulse
(Gi), the length of time we apply the gradient (δ), and the time between the application of
gradients ∆). These relationships can be summarized by the following equation:
D =− ln Sb Sb0( )
b− b0

83
which can be simplified to:
This equation demonstrates that the simplest way to modify the b-value is by manipulating the
time between the two gradient pulses.
In a DTI experiment, a symmetric b-factor tensor (b-matrices) is calculated for each DWI
acquired with a different b-value (b ≠ 0 and b = 0). Usually the b ≠ 0 is acquired at a b-value
equal to 1000 s/mm2. We then solve for D and the result is all 6 components of tensor model,
which can be summarized in an image of the apparent diffusion coefficient (ADC). Therefore,
the signal ratio equation becomes:
The intensity of a voxel in the ADC image represents the direction of the bulk diffusion of water
molecules with regard to the direction of the applied gradient. In the ADC image, the brighter the
voxel intensity, the more diffusion has occurred. However, complex features in microstructure,
such as crossing fibres, can produce erroneous measures of diffusion. Thus, to properly
determine the principal diffusion direction within a voxel, an enormous number of directions
should be sampled. However, this is not a feasible approach, as MRI scanning is costly, the
hardware is limited, and the subject cannot undergo such long scan durations. Therefore, to
overcome these limitations, a tensor model is used to calculate the bulk direction of water
diffusion within a voxel. In a tensor model, diffusion only needs to be acquired along six
directions; however, the more directions acquired, the better the signal-to-noise ratio of the
image, which will improve the precision of the DTI estimation, and hence the angular resolution.
Some imaging studies acquire up to 120 directions, although 60 directions has now become
standard practice. The tensor (D) is a positive, definite tensor, and describes a three-dimensional
ellipsoid in displacement space, called the diffusion-ellipsoid. The three diagonal terms of the
tensor model (TX, Dyy and Dzz) represent the three axes of the ellipsoid. These relative
€
b = γ 2 G t'( )dt '0
t∫[ ]
2
dt0
te∫
b = γ 2Gi2δ 2 (Δ−δ 3)
€
ln SbSb0
"
# $
%
& ' = −b(ADC)

84
diffusivities of water along these axes can be expressed by three eigenvalues (λ1, λ2, λ3), and
their orientation is represented by three corresponding eigenvectors (ν1, ν2, ν3) (see Figure 2-4).
For a comprehensive review of the tensor model, see: [60,61]. These values allow us to calculate
several useful measures (or contrasts), such as mean diffusivity (MD), fractional anisotropy (FA),
and radial diffusivity (RD) [62,494,524,626] (for the biological basis of these, see 2.6.4), and
their respective equations are presented in Figure 2.4.
Figure 2-4: Schematic of diffusion tensor imaging, and summary of eigenvectors and metrics.
Lambda (λ) represents each of the eigenvalues that make up the tensor model.
In DTI analysis, FA is the most commonly used measure, as it is scaled between 0 and 1, where
0 represents complete isotropic diffusion, and 1 represents complete anisotropic diffusion.
Assuming that these measures represent white matter integrity along a tract (see below), then we
can evaluate white matter integrity within a population, or compare between populations (see
next section). By combining FA as a threshold and the primary eigenvector, ν1, then we can
determine the orientation of white matter tracts, at each voxel.
Despite the great advances that DTI has afforded us, there are a number of inherent limitations
and challenges that must be overcome in data acquisition and analysis. For instance, as DTI

85
measures the relative displacement of water molecules, it is very sensitive to head motion and
physiological motion. To minimize head motion, the subject’s head is padded, and to minimize
physiological motion, some investigators measure respiration rate and cardiac rhythm during the
scan, and control for these during analysis. Furthermore, to correct for relatively small amounts
of motion during a scan, some scanners collect several non-diffusion-weighted images at several
points during the acquisition, interspersed between the diffusion-encoding runs (usually at a ratio
of 5:1 for diffusion-encoding directions: non-diffusion-weighted images). These non-diffusion
encoding directions are aligned to one another, and the transformation matrix is applied to the
DWIs between these images. Other motion correction algorithms also exist (for a review of
these, and other denoising methods, see: [694]). Another source of noise is the production of
eddy-currents artifacts, which occur due to residual gradient effects and the rapid switching of
gradient pulses. Most motion-correction toolboxes also correct for these artifacts.
There are some notable limitations with the diffusion tensor model. For instance, some voxels
may appear to have low FA values despite their location in white matter. We would expect
voxels in gray matter and CSF to have low FA, as there are few barriers and little geometric
organization that restrict the diffusion of water. Regions of complex white matter organization,
such as crossing fibres or fibre dispersion, cause these relative hypointensities of FA in white
matter [14]. Another limitation of DTI is that it cannot resolve the direction of the fibre, but only
its orientation in three-dimensional space [14]. Therefore, one cannot make any directional
interpretations about whether a tract is connecting to or coming from a gray matter region.
2.4.3.2. Between Group Comparisons of White Matter: Tract-based spatial statistics
One of the major challenges in applying DTI to study clinical populations has been the
development of an approach to compare white matter metrics between subjects, given the
intersubject variability, but now, several approaches have been developed. For instance, some
groups use an average value (or summary measure) of FA, MD and RD across the whole brain
[112,152]. Yet, summary measures of white matter integrity are not very informative, as there is
no information about the location of the white matter abnormality. This method is useful when
investigating a diffuse disease, such as multiple sclerosis [722]. Other groups have used a VBM-
style approach (see Section 2.4.1.2) to delineate regions of white matter abnormality between
subject cohorts, or to identify regions where white matter correlates with a behavioural measure

86
within a group of subjects [36,37]. However, VBM-style analyses have been heavily criticized
for their lack of reproducibility (due to the parameters that can be selected) and its lack of
accuracy (due to misalignment) [151,210,813]. In the former case, one example is smoothing.
Two studies have shown that results vary largely based on the selection of the size of the
smoothing kernel [466,667]. Another limitation of VBM-style white matter analysis arises when
aligning white matter tracts for comparison between subjects. To obtain a reliable measure, it is
essential to compare the same tracts between subjects, and to compare the same part of the tract
(as FA is not the same at the edge of a tract, compared to the centre of the tract), the so-called
correspondence issue [813,815]. These issues have been encountered in several studies (e.g.,
[80,804,822]. Another white matter analysis approach has been to confine the analysis to specific
white matter regions-of-interest (ROI). ROI analyses of white matter can be beneficial, in that
they are very sensitive to changes in measures of white matter integrity, so long as the ROI is
specific. Furthermore, this approach is not very computationally intensive. However, delineation
of an ROI requires expert knowledge of the anatomy, in order to identify appropriate landmarks
to draw reliable and reproducible ROI [151].
To address some of the issues of these white matter analysis methods, Smith and colleagues
developed tract-based spatial statistics (TBSS), a toolbox implemented in the Oxford Centre for
Functional MRI of the Brain’s (FMRIB) software library (FSL) [813,815,818]. TBSS is a fully-
automated program that has a similar pipeline to that of VBM, but it has been optimized for
white matter analysis. TBSS uses FA to align subjects to a common template using a non-linear
algorithm. A standard FA template (FMRIB58_FA) with 58 subjects in the MNI152 standard
space was developed for this purpose. Alternatively, FSL allows the creation of a study-specific
template for populations where a standard-space template is not suitable, such as a pediatric
population, or in the case of severe pathology. TBSS then calculates a group mean FA map, and
“thins” this map to create a skeletonized mean FA image (see Figure 2-5). This skeletonized FA
map represents the centre of the white matter tracts common to all subjects. The highest FA
value of each subject’s tract, deemed to be the centre of the tract, is projected onto the skeleton at
every voxel, in order to overcome any residual misalignments. Voxelwise univariate statistical
tests can then be performed on the skeletonized FA map. Furthermore, once alignment is
achieved, other indices of white matter integrity, or shape can be projected onto the skeleton for
analysis. Therefore, TBSS effectively addresses the limitations of the other methods described.

87
However, restricting the analysis to the peak FA value may reduce sensitivity to changes outside
the centre of the tract.
Figure 2-5: White matter skeleton from tract-based spatial statistics. The skeleton is shown in
green, and is overlaid onto the MNI152 standard brain in FSL.
2.4.3.3. Tractography
Another method to evaluate white matter in the brain with DTI is tractography. In general, this
method allows us to perform in vivo “dissections” of white matter tracts (i.e., visualize the tract),
and to delineate the anatomical connectivity of the brain. The concept of tractography is based on
the signal information collected in a DWI scan and the tensor model utilized in DTI. Briefly,
when we apply the tensor model to a DWI data set, we obtain eigenvalues and eigenvectors for
each voxel in the image, which represent the orientation and magnitude of water diffusion within
the voxel. When diffusion is restricted, by a biological tissue for instance, then the diffusion is
anisotropic. We can use the orientation information within each voxel to reconstruct white matter
pathways in the brain, called tractography. The fundamental assumption of white matter
tractography is that the primary eigenvector (υ1) is parallel to the primary axis of the white
matter tract. It is, however, noteworthy that this assumption is often violated in regions of
complex white matter (e.g., crossing fibres and fibre dispersions). More complex algorithms
have been developed to address these issues, and to increase the sensitivity and reliability of DTI.
Two of the most commonly used methods, deterministic and probabilistic tractography, are
described in further detail below. Although there are other, more complex methods being
developed, these are outside the scope of this thesis.

88
Streamline (deterministic) tractography
Deterministic tractography algorithms use the orientation information within voxels to track the
path of white matter tracts [14]. The resultant image is a tractogram, which is based on the
primary (or major) eigenvector within each voxel. The tractogram is initiated from a seed
location (a single voxel, or a group of voxels of interest). Because the algorithms use a threshold
for FA to distinguish between gray and white matter, the seed is usually located within the white
matter [14]. If the seed were in the gray matter, or in a voxel with FA below the threshold, then
tracts could not be generated. Several restraints can be placed on the algorithm to remove
spurious connections, and to ensure that the tracts are consistent with the predicted pattern of
connectivity [14].
There are several deterministic tractography algorithms that can produce tractograms. Most of
these methods utilize a streamline approach. This approach calculates the stepwise progression of
the tract (or the path), beginning from a seed. The progression of the path is based on the
direction of the primary eigenvector. The termination of a tract is based on an arbitrarily selected
parameter [465]. For instance, an FA value is selected where the primary eigenvalue is well
defined, e.g., FA ≥ 0.15. Another parameter used to terminate a tract is the curvature angle,
which is also arbitrarily chosen. The specific aspects of deterministic algorithms are outside the
scope of this thesis, but for a detailed review of these methods see: [14].
There are both advantages and disadvantages to deterministic tractography. The most significant
advantage is that fewer diffusion-encoding directions are required to produce tractograms,
compared to other algorithms, such as probabilistic tractography, or diffusion spectrum imaging
(see below). Furthermore, the analysis is not as computationally intensive as some of the other
methods (including probabilistic tractography). However, deterministic tractography can only be
used as a qualitative means of visualizing tracts in the brain, although the images can serve as
masks to perform quantitative analysis, e.g., evaluate measures of white matter microstructure,
such as FA. The images produced with deterministic tractography are visually appealing, but this
can instill a false sense that the visualized tracts are accurate. However, there are many potential
sources of errors in deterministic tractography. As previously discussed, DTI is very sensitive to
motion and other sources of noise. Even small perturbations in the image can lead to tracking
spurious connections. The largest source of error in tractography is tract dispersion, or the

89
variance of trajectories, which increases from the seed because there are an increasing number of
potential paths [14]. Another source of error comes from tract deviation, or errors in tract
position. These are related to the step size, especially in regions where the curvature of the tract
is high. In general, a larger step size will increase the chance of error [518]. Another source of
error in deterministic tractography is that of crossing fibres, as discussed above. Crossing fibres
can lead to false-negatives in the tractograms [145]. For instance, most deterministic algorithms
are not able to resolve tracts within the CST emerging from the lateral motor cortex and
projecting to the pyramids in the brainstem [14]. This is the result of the crossing fibres in the
centrum semiovale, which contains association fibres, callosal fibres and short U-shaped fibres,
which project in different directions. As a result of the multiple fibre directions in voxels within
this region, the primary eigenvalue is diminished, and the tractography algorithms fail to track
the rest of the tract. Therefore, experiments that seek to resolve tracts using deterministic
algorithms should have strong hypotheses and predictions based on known anatomical pathways,
from animal studies and human post-mortem studies.
Probabilistic tractography
Probabilistic (or stochastic) tractography is a method that attempts to address some of the
limitations of deterministic tractography. Specifically, probabilistic tractography acknowledges
that there are inherent errors that occur in tract-tracing algorithms, and accounts for this
variability. Therefore, the inherent variability in the data is included in probability estimates of a
tract [668]. To do so, algorithms use probability density functions (PDF), which considers the
expected distribution of the possible fibre orientations, at the level of the voxel [671]. The PDF
are used instead of discrete measures of fibre orientation. An advantage of probabilistic
tractography, compared to deterministic tractography, is that the PDF allow tractography to
continue in a region where deterministic tractography would normally stop. Another advantage
of probabilistic tractography is that we can quantitatively compare the connectivity of tracts
based on the probability of connections.
The PDF is comprised of three orientation density functions (ODFs): the diffusion ODF (dODF),
the fibre ODF (fODF) and the uncertainty ODF (uODF) [72]. The dODF and the fODF are
biophysical properties of the tissue that is being measured. The fODF describes the proportion of
the fibres in each direction. For instance, as in the example above, we have two orthogonal fibres

90
in a voxel, the resultant fODF will appear as a cross, in the direction of the fibres [862]. The
fODF is useful in that it contains a proportion, which can be used to estimate the connectivity of
a tract connecting A to B, providing a quantitative method that is both useful and biophysically
meaningful [72]. However, DWI measures the diffusion of water within each voxel in the brain,
and not the proportion of fibres, and so the measure we obtain is dODF, a measure of the
orientation of diffusion within a voxel [879]. If water molecules diffused exactly along the axis
of a tract, then the dODF would be exactly equal to the fODF. However, in reality, water
molecules diffuse primarily along the axis of the tract, but there is also diffusion in other
directions. Therefore, the dODF is necessarily broader than the fODF. Whereas the dODF and
fODF are properties of the tissue which can be measured, the uODF is a measure of the error that
can be predicted, or as Behrens puts it: “it [uODF] is a function that describes our belief” [72].
Specifically, the uODF represents the uncertainty that is included in the data because of the noise
inherent to it.
These variables have very important implications in tractography. It can no longer be assumed
that the primary eigenvector is the path of the tract, as there are an infinite number of paths
within each voxel, each associated to a probability of being the correct orientation. To calculate
the probability of connection of two regions, every possible path and its probability must be
considered, and then the probabilities associated with each path connecting the two regions is
summed [75]. Rather than attempt to solve this equation, more reasonable approaches have been
developed that allow us to sample the probability distributions [75,392,465,669-671]. Briefly,
these algorithms sample many paths from a seed at sub-voxel to sub-voxel steps (or for lower
resolution, or larger step size, at a voxel to voxel steps). Each step considers the distribution of
possibilities, resulting in many possible paths, each with a probability. These results are
summarized within the voxel as a proportion of the number of samples that pass through the
voxel [465]. Whereas deterministic algorithms rely on arbitrary FA values or curvature angles to
stop tractography, probabilistic algorithms do not rely on these parameters for termination. In
contrast to deterministic tractography, this allows the algorithm to propagate even when a seed is
in gray matter. The criterion for termination of a tract is usually the angular deviation between
successive steps (c.f. FA values in deterministic tractography), which prevents a tract from
looping back onto itself, or if a streamline enters the same voxel more than once [75,465]. The

91
specific differences between probabilistic tractography algorithms are outside the scope of this
thesis. For a comprehensive review, see: [72,671].
The results of probabilistic tractography should be interpreted with care. These results have a
very specific meaning: a calculated probability is the probability that a connection from a given
seed to a target through the diffusion data exists within the context of the model for the diffusion
signal and the model of connections. These tracts do not provide anatomical evidence for the
existence of a path within the brain. However, if a hypothesis is based on known anatomical
pathways from post-mortem studies in humans (and homologues from tracing studies in
monkeys), then we can infer that the tract we have identified is “real”. Otherwise, we must
interpret the presence or absence of a tract with caution.
Crossing fibres are still a consideration in probabilistic tractography; in fact, it is a shortcoming
of the tensor model. Some groups have developed alternate, non-parametric models to replace
the tensor model, that can better resolve crossing fibres, such as diffusion spectrum imaging
[878,925], q-ball imaging [877,879], spherical deconvolution [862], persistent angular structure
MRI [456], and diffusion orientation transform [661]. However, these are outside the scope of
this thesis (for a review, see: [15]). Some groups have tried to resolve crossing fibres within the
tensor model. The most common approach is to model more than one tensor per voxel: the multi-
tensor model [73]. The number of “fibres” that can be modeled per voxel depends on the signal-
to-noise ratio, and the number of diffusion-encoding directions.
In sum, there are several advantages and limitations to tractography. Nonetheless, when care is
taken to acquire, check and de-noise the data, and seeds are selected carefully, based on known
landmarks, and results correspond to known tracts within the brain, then tractography can be a
very useful and informative tool. Additionally, tractography can be qualitative (visualizing tracts)
and quantitative (calculating the probability of a connection). The latter allows group
comparisons of connectional anatomy.
2.5. Brain imaging of pain
2.5.1. Functional neuroimaging of pain
Functional neuroimaging has confirmed and advanced our understanding of brain regions
implicated in nociception and pain modulation – especially the role of the PFC. With the

92
exception of the PFC, the pain-related brain regions identified with functional neuroimaging are
similar to the brain regions previously identified in animal models and human
electrophysiological studies. These regions, however, are implicated in sensory, motor, attention,
salience, cognitive processes, decision-making, and other functions. Therefore, further research
is required to understand the role of these regions in pain perception and modulation, and further
to determine how pain is integrated.
Functional brain imaging encompasses a number of modalities, including positron emission
topography (PET), single-photon emission computed tomography (SPECT), fMRI, and
magnetoencephalography (MEG). Also, electroencephalogram (EEG) is sometimes considered
as a brain imaging technique when it is coupled with visualization of the brain areas activated.
Each of these modalities has its benefits and shortcomings because of spatial and temporal
resolution, fundamental principles (e.g., indirect hemodynamic measures, direct electrical
measures, etc.), invasiveness, availability, cost, and other scientific and practical issues. Because
this thesis focuses on structural abnormalities in the brains of patients with TMD, findings from
functional neuroimaging will only be briefly described.
2.5.1.1. Imaging acute pain
The first two brain imaging studies to investigate brain regions involved in nociception and pain
perception and modulation in healthy subjects were performed using PET [464,846]. One study
reported that heat pain, compared to an innocuous warm stimulus, evoked an increase in regional
cerebral blood flow (rCBF) in the contralateral thalamus, lentiform nucleus and ACC (MCC,
with the current nomenclature) [464]. The other study reported increased rCBF in S1, S2 and the
MCC [846]. The first fMRI study to investigate the neural correlates of pain perception found
that painful electrical stimulation increased the blood-oxygenation-level-dependent (BOLD)
signal in S1 and MCC [231]. Many studies have since investigated pain-related brain activation,
and some have specifically attempted to identify the neural correlates of the different dimensions
of pain. Several reviews and meta-analyses have summarized these findings
[23,95,143,212,274,304,690,709,714,869]. In general, brain imaging studies of pain have
reported the activation of S1, S2, MCC, insula, prefrontal and motor regions. Relevant to this
thesis, two studies have examined BOLD correlates of pain in the orofacial region. One study by
Ettlin and colleagues [300] reported that electrical stimulation of the tooth in healthy subjects

93
was related to BOLD activity in the cerebellum, superior temporal gyrus, anterior insula, MCC,
vlPFC and dlPFC. Furthermore, they reported that BOLD activity in the posterior parietal cortex,
anterior insula, MCC, cerebellum, and caudate nucleus were correlated to pain intensity ratings.
Another study of the neural correlates of acute orofacial pain reported that acute electrical
stimulation of the tooth activated nuclei believed to be related to trigeminal nociceptive
processing (within the VBSNC) and brainstem nuclei implicated in pain modulation, such as the
RVM [926]. Furthermore, they found significant clusters of activation in S1, S2, anterior insula,
MCC, PMC, M1, SMA and dlPFC. Therefore, it seems that acute pain in the orofacial region
activates the same set of brain regions as acute experimental pain outside the orofacial region.
The putative roles of these regions in pain-related processing as determined by neuroimaging
studies are described below.
S1
It is well known from classic brain stimulation studies in humans and animal electrophysiology
studies that S1 is somatotopically organized (see Section 2.3.5). This S1 homunculus has been
largely confirmed in human brain imaging studies (for a review, see: [23]), generally showing an
inverted organization with the face represented ventrolaterally, the upper limb more dorsolateral,
and the lower limb represented within the medial wall. Of relevance to this thesis, several studies
have demonstrated that the dermatomes of the three branches of the CNV are somatotopically
represented on the ventrolateral surface of S1: V1 is more superior and V3 is more inferior,
although there is some overlap [205,446,630]. Of these studies, one has explicitly tested the
somatotopy of the branches of the CNV with a noxious stimulus, compared to an innocuous
stimulus. Specifically, DaSilva and colleagues [205] found that the representation of noxious and
innocuous stimuli in the receptive fields of V1 and V2 were the same, whereas noxious stimuli in
the receptive field of V3 seemed to have a broader representation than innocuous stimuli in the
same receptive field. This broader cortical representation of V3 overlapped with the
representation of V1 and V2.
Similar to the electrophysiological data presented in Section 2.3.5, neuroimaging data has not
provided a clear role for S1 in pain processing [130]. It is unclear whether S1 is implicated in
pain perception. It has been proposed that the activation of S1 is due to the tactile component of
noxious stimuli, e.g., when a heat probe is used to stimulate the arm, the probe places some

94
pressure on the arm, and fibres that encode innocuous warm temperatures or touch may also be
firing. Furthermore, imaging studies have found vastly different findings in S1 activation in pain
paradigms. Specifically, Talbot et al. [846] reported significant contralateral S1 activation to the
stimulation of the arm, whereas Jones et al. [464] did not find S1 activation. In a later study,
Apkarian et al. [28] found that S1 actually showed deactivation in response to noxious thermal
stimuli. However, in a meta-analysis, Apkarian and colleagues [23] reported that 69% of PET,
76% of fMRI, 10% of EEG and 70% of MEG studies reported an S1 activation in acute pain
paradigms. More recently, MEG [699,855], EEG [885] and diffuse optical tomography [68]
studies have reported S1 responses evoked by painful stimuli. Also, a meta-analysis carried out
by Duerden and Albanese [274] identified S1 as a region that is activated across 140
neuroimaging studies of acute experimental heat and cold pain. Several imaging studies in
humans have found that S1 (in addition to other cortical areas) shows graded activation in
response to the increased intensity of experimental pain stimuli [169,240,629,705]. Also,
Rainville and colleagues [715] reported that when subjects’ perceived pain intensity and
unpleasantness were manipulated under hypnosis, S1 and possibly S2 responses correlated with
perceived pain intensity, whereas the ACC response correlated with the perceived pain
unpleasantness. In sum, neuroimaging studies have provided inconsistent findings with regards
to the role of S1 in nociception and pain processing. However, more recent and sensitive
methods have clearly demonstrated that S1 may, indeed, be implicated in pain perception.
Therefore, neuroimaging studies have replicated many of the previous findings from
electrophysiological and brain stimulation of the role of S1 in nociception and pain perception.
Specifically, neuroimaging studies have confirmed the somatotopic organization of S1, and have
shown that the haemodynamic response in S1 is graded to noxious stimulus intensity. Together,
these findings suggest that S1 is implicated in the sensory-discriminative dimension of pain,
encoding the location and intensity of a noxious stimulus.
S2/Parietal operculum
Although many imaging studies report that S2 is activated during painful stimuli, the findings
have not clarified the role of S2 in pain processing. Apkarian et al. [23] reported that 68% of
PET, 81% of fMRI, 60% of EEG and 95% of MEG studies reported S2 activation in studies
investigating the neural correlates of acute pain in healthy controls. Also, Duerden and
Albanese’s meta-analysis [274] identified S2 as one of the regions that is most commonly

95
activated in neuroimaging studies of experimental pain. However, it is not clear how S2
activations reflect various aspects of the pain experience. It is also unclear whether S2 receives
nociceptive information from the thalamus via a relay through S1 (i.e., serial processing)
[16,407], or directly in parallel to information being sent to S1 [699,702]. Furthermore, evidence
from EEG studies of laser-evoked potentials (LEP) have demonstrated that the early
electrophysiological nociceptive response (N2) is caused by activity in S2/posterior insular
cortex (e.g., [335]), which suggests that there is parallel processing in S1 and S2, although recent
findings have opposed a parallel processing model with regard to the sensory-discriminative
dimension of pain [885].
One study investigating painful LEP with subdural electrodes has demonstrated somatotopic
organization along the anterior-posterior axis, with the foot anterior to the face, within S2 [897].
This organization is different from that of tactile input, and it has been suggested that there may
be a pain somatotopic map independent of the tactile somatotopic map [23].
Insula
The insula also has several somatotopic maps for different noxious stimuli, and within different
subregions [64]. In this somatotopic map, the face is anterior to the foot. The insula was activated
in many neuroimaging studies [187], that investigated many paradigms, including gustation,
emotion, olfaction, empathy, interoception, motor output, attention, language, memory, speech,
and pain [506]. Apkarian et al. [23] reported that 88% of PET, 100% of fMRI, 30% of EEG and
20% of MEG studies reported insular activation in imaging studies of acute pain in healthy
volunteers. Duerden and Albanese’s [274] meta-analysis of 140 experimental pain neuroimaging
studies also identified the insula as one of the most likely activated regions. In a meta-analysis of
the function of the insula, Kurth and colleagues [506] investigated 79 studies that showed insular
activation in imaging studies of acute pain stimulation paradigms in healthy controls, and found
that all of the subregions of the insula were activated. It is, however, noteworthy that this same
study identified that different subregions of the insula showed responses to paradigms that tested
the neural correlates of processes related to pain, such as somatosensation, motor output,
attention, interoception, and emotion, and that these activations generally overlapped with that of
pain.

96
A number of groups have postulated a role for the insula in terms of pain perception. For
instance, evidence that the insula is involved in intensity encoding comes from a study by
Coghill and colleagues [169] who reported that the insula responds in a graded fashion to
increasing intensity of noxious stimuli. Brooks and Tracey [116] suggested that the insula is a
multidimensional integration site for pain. An alternative hypothesis about the role of the insula
proposed by Craig [183] suggested that the mid-posterior insula was a multimodal homeostatic
or interoceptive area. The former hypothesis maintained pain as a separate modality, whereas
Craig’s hypothesis established pain as a subset of homeostatic function. Baliki and colleagues
[48] have posited another hypothesis suggesting that, in addition to integrating the dimensions of
pain perception, the insula is a central, multimodal magnitude estimator and a nociceptive-
specific magnitude estimator. However, the insula has been implicated in a multimodal sensory
stimulus salience detection network, where salience stimuli are encoded and processed to
appropriately orient attention [263-265,445,530,632,633].
In sum, because the insula receives multimodal input, it is an ideal site for integrating
information from the various dimensions of pain – it is the site where nociception becomes pain.
However, it is activated in many other paradigms, and across different modalities [265,956].
Therefore, the insula may best be considered as a region that encodes behaviourally-salient
stimuli, including pain.
Cingulate cortex
The cingulate cortex is a large, heterogeneous brain region that can be subdivided into several
subregions (see Section 2.3.5) [169]. Apkarian et al. [23] reported that 94% of PET, 81% of
fMRI, 100% of EEG and 25% of MEG studies reported cingulate activation in imaging studies
of acute pain in healthy volunteers. Furthermore, Coghill and colleagues demonstrated that the
cingulate, specifically the MCC, showed graded responses to increasing intensity of noxious
stimuli [169]. The different subregions of the cingulate cortex have been implicated in different
dimensions of pain, including the affective, cognitive, modulatory and motor dimensions
[128,860,902,903]. The aMCC/pgACC is a complex, multimodal region that has been implicated
in a number of functions [69]. For instance, this region has been identified as a node in the
salience network, and has been implicated in multimodal salience detection
[213,229,262,263,774,849,927], including detection of noxious stimuli

97
[220,230,231,259,265,440,508,528,632,933]. The cingulate has also been implicated in the
cognitive and affective processing of pain [218,230,529,716,776,777,934,936], and the MCC,
which includes the CMA, is involved in action selection and modulation of motor output in
response to aversive stimuli [790,898,904]. The sgACC has been implicated in pain modulation
[95]. Furthermore, the pgACC and sgACC have been implicated in emotional processing and
depression [299,579,790] (See section 2.3.6).
Prefrontal cortex
The PFC is a large, heterogeneous region, in the frontal lobe anterior to the motor cortex. There
is no evidence suggesting that the PFC receives nociceptive information directly from the
thalamus (see Section 2.3.5). The PFC does, however, receive input from a number of
nociresponsive brain regions, such as the cingulate cortex, the insula, and the somatosensory
cortex [644]. Apkarian et al. [23] reported that 39% of PET, 70% of fMRI studies reported PFC
activation in imaging studies of acute pain in healthy volunteers. Interestingly, no EEG or MEG
studies reported PFC activation. The PFC has been implicated in the cognitive-motivational
dimension of pain. Evidence for this role comes from a number of studies that have investigated
the interaction of pain and cognition of the brain. For instance, the OFC is activated when
subjects are distracted from pain, which suggests that this region is involved in pain modulation
[888]. Furthermore, some studies have found that the dlPFC is implicated in pain modulation
[552], although other studies have not found this. Conversely, neuroimaging studies of persistent
pain, and experimental models of hyperalgesia and allodynia have shown that increased
sensitization is related to increased activity in the dlPFC [56,443,551,775]. Furthermore, one
study has demonstrated that regulation strategies can modulate pain via the vlPFC, which is
involved in top-down cognitive modulation of pain [932]. However, another regulation study did
not find that the vlPFC was implicated in pain modulation [753]. Alternatively, it has been
suggested that the vlPFC is implicated in pain anticipation, rather than modulation [752,908].
The medial PFC has been implicated in the integration of cognitive and motivational dimensions
of pain [935]. Therefore, although the PFC does not receive nociceptive input from the thalamus,
neuroimaging has revealed that the PFC plays an extensive role in the cognitive dimension of
pain, and, through it connections to antinociceptive brainstem regions (see Section 2.3.6), pain
modulation.

98
Cortical and subcortical motor regions
Motor regions are activated in acute pain paradigms in healthy controls, but less reliably than the
aforementioned brain regions [23]. As previously discussed (Section 2.3.5), in the context of
pain, it is believed that motor regions serve two purposes: to orient the body toward the source of
pain and to initiate nocifensive behaviour (e.g., avoid the stimulus). Similarly, another study
reported that hypertonic saline injection into the masseter muscle, an experimental acute pain
model, was related to deactivation in the face region of M1, as measured by fMRI [638]. It is,
however, noteworthy that some studies have not observed activation in M1 during noxious
stimulation [395,744].
The basal ganglia are a set of subcortical nuclei that have been associated with motor function.
However, it is noteworthy that the basal ganglia do receive input from many cortical and
subcortical regions, and form several functional loops [644]. It has been proposed that this
extensive wiring to the brain suggests that the basal ganglia may be implicated in more than just
motor functions [388]. For instance, the basal ganglia are often activated in neuroimaging studies
of experimental pain [107]. It has been suggested that the cortico-thalamo-basal ganglia-cortical
loops may provide a unique anatomical substrate for the integration of the various dimensions of
pain [107]. However, more work is required to further investigate this possibility.
Other motor regions are also commonly activated in brain imaging studies of experimental pain
in healthy volunteers, including the PMC and SMA [23,274]. The PMC has been implicated in
cognitive modulation of motor output, and the SMA has been implicated in motor planning, and
the temporal organization of movements [692]. Therefore, their activation during experimental
pain is likely related to the cognitive and motivational dimensions of pain, with regard to
nocifensive behaviours.
2.5.1.2. Imaging salience and pain
Salience and pain are closely related. Pain is inherently salient and therefore can engage brain
regions that serve as “salience-detectors”. Specifically, salient stimuli have been shown to
activate a right-lateralized brain network, including regions in the frontal, parietal, insular and
cingulate cortex [364,604-606,647]. In an fMRI study, Downar et al. [262-264] identified a
multimodal salience network in humans. In the context of pain, a network of regions, comprised

99
of the MCC, temporoparietal junction, PFC, and the putamen encode stimulus salience.
Similarly, an fMRI study investigating the neural basis of perceptual decision-making in the
context of pain as a perceived threat and increased salience stimulus, reported that the insula
integrated the perceived threat to form a decision about the stimulus [933]. It is thus conceivable
that these regions form a system that appraises, encodes and modulates a salient (and potentially
threatening) stimulus based on the context in which it is presented – or its relative salience.
Another study demonstrated that brain regions that are commonly activated by noxious stimuli
are also activated by equally salient, non-noxious stimuli [530]. Therefore, results from
experimental pain paradigms attempting to investigate the roles of brain regions in pain must be
interpreted with caution, as the aforementioned studies have demonstrated that they are non-
specific to pain, and may be implicated in encoding any behaviourally-relevant stimulus.
2.5.1.3. Chronic pain
Many studies have used functional neuroimaging to investigate the central abnormalities in
chronic pain patients. One of the differences between BOLD-fMRI and PET is that BOLD-fMRI
requires a stimulus, as it relies on differences in activation, whereas PET can assess baseline
function. In terms of imaging chronic pain disorders, fMRI cannot capture brain activity related
to spontaneous pain, or other clinical features. PET can capture these features, but is invasive as
it requires a radioactive tracer. Therefore, some groups use percept-related fMRI, where subjects
are continuously rating a quality of a painful stimulus throughout an fMRI scan [213,226,509]. In
a meta-analysis of all functional neuroimaging studies of pain up to 2005, Apkarian and
colleagues [23] compared noxious stimulus-evoked brain activation in healthy, pain-free subjects
and chronic pain patients. They reported that, in general, chronic pain patients showed less
noxious stimulus-activation of S1, S2, insula, and the ACC/MCC compared to pain-free subjects.
They reported no difference between the groups for the thalamus (although studies have reported
thalamic findings), and that the PFC showed significantly greater activation in chronic pain
patients, compared to normal subjects. However, not all chronic pain disorders have the same
symptomatology. Therefore, it is more likely that different types of chronic pains have different
“brain signatures” or “fingerprints”– the pattern of abnormally functioning brain regions is
specific to the symptom profile of the pain disorders. A challenge for neuroimaging of pain is to
disentangle which abnormally-functioning brain region is specific to a pain disorder, and which
is common to all pain disorders.

100
Another method of analyzing functional brain imaging data is to identify regions that may be
functionally connected [333]. Conceptually, this method is based on the idea that brain regions
do not process information in isolation, but rather process information in different schemes,
either hierarchical, or in parallel. Therefore, these regions are functionally connected during a
task, and in task-free paradigms (or at rest) [213]. Recent evidence has shown that some of these
functionally connected brain regions may be disrupted in chronic pain (e.g., [49,560]).
Few studies have investigated functional brain abnormalities in TMD. The first study to do so,
investigated neural responses to tactile-flutter stimuli on the index finger [641] and found that the
patients reported the stimuli as more intense, but not painful, than the control subjects. Further,
compared to the control subjects, the tactile stimuli evoked less activation in S1, S2 and insula,
and greater activation in the thalamus, MCC, the amygdala, and another region of S1 in the
patients with TMD. This study reported that patients with TMD have both increased and
decreased activation across several regions, including S1, S2 and the insula to a vibratory
stimulus applied outside the orofacial region, compared to controls. Patients with TMD show
increased activation in the MCC, in the contralateral insula and thalamus. Therefore, patients
with TMD show abnormal processing of an innocuous tactile stimulus outside the orofacial
region. It would have been more informative to have also applied a vibratory stimulus to the
orofacial region, to test whether tactile thresholds were different in this region, and the neural
correlates of these differences. However, a vibratory stimulus on the face in an MRI magnet
would likely be a source of noise and therefore difficult to test.
Our laboratory has conducted an fMRI study to investigate brain responses to cognitive and
emotional attention-demanding tasks in patients with TMD [928]. In this study, a group of
patients with idiopathic TMD and a group of age- and sex-matched pain-free controls underwent
fMRI scanning while performing three counting Stroop tasks (neutral, conflict numbers,
emotional). We found that behaviourally, the patients with TMD had sluggish reaction times,
compared to controls, in all three Stroop tasks. The fMRI data indicated that patients had greater
deactivation in the mPFC, OFC, medial temporal gyrus and dlPFC during the neutral Stroop task,
compared to controls. Furthermore, patients showed increased activation in the dlPFC, vlPFC,
insula, ACC, SMA and amygdala during the high conflict Stroop task, compared to controls.
During the emotional Stroop task, patients showed greater activation than controls in the sgACC,
MCC, SMA, PCC, insula, vlPFC, and the inferior parietal lobule. Finally, patients showed less

101
functional connectivity than controls between the dlPFC and the aMCC, and the pgACC and the
amygdala during the high conflict task. During the emotional Stroop task, patients showed
increased connectivity between the dlPFC and the MCC. In sum, these findings suggest that
patients with idiopathic TMD have cognitive deficits related to poorer task performance,
compared to controls. These deficits may be related to abnormal brain function in
cognitive/affective and attention-related brain regions.
Resting state networks reflect the intrinsic connectivity of brain regions when subjects are not
actively engaged in a task. It has been posited that these networks reflect the anatomical
connectivity, as well as the “history” of prior co-activation during task [235,236]. In the context
of TMD, a recent study investigated the connectivity of the insular and cingulate cortices in
myofascial patients with TMD during rest, and during experimental pressure pain in the orofacial
region [448]. These studies reported that during resting state, in patients with TMD compared to
controls, the left anterior insula was more functionally connected to the right pgACC and the left
parahippocampal gyrus, and the right anterior insula was more connected to the right thalamus.
During noxious stimulation, both groups showed greater functional connectivity between the left
anterior insula and the left S2, cerebellum, the left posterior insula showed greater connectivity
to the right dlPFC. Finally, the study reported a group-by-pressure interaction in the functional
connectivity between the left anterior insula and the left pgACC, bilateral frontal polar cortex,
and the right anterior insula with the right pgACC. Therefore, these findings suggest that in TMD
there is increased functional connectivity between sensory and pain-modulatory regions at rest,
and increased connectivity between cognitive and modulatory regions during experimental
pressure pain.
In sum, few studies have used functional neuroimaging in TMD. These studies have found
abnormal function in sensory, motor, cognitive/modulatory and salience-related brain regions.
2.5.2. Structural imaging of pain
2.5.2.1. Acute Pain
To date, only two studies have investigated structural gray matter correlates of acute pain.
Teutsch and colleagues [852] demonstrated that eight days of noxious heat pain stimulation
induced increased gray matter volume in MCC and S1. Furthermore, as subjects habituated to the

102
noxious stimulus over eight days , these gray matter increases resolved (i.e., the gray matter
volume returned to baseline). In a study by Erpelding et al. [298], cortical thickness in S1
correlated with individual subjects’ heat and cold pain sensitivity, and cortical thickness of the
MCC correlated with heat pain sensitivity. To date, the relationship between white matter
microstructure and acute pain thresholds or tolerance has not been studied in healthy subjects.
2.5.2.2. Chronic Pain
Gray matter abnormalities
There are several approaches to investigating gray matter structure in chronic pain patients. For
instance, studies can either investigate the whole brain, or focal areas (ROIs) that have been
hypothesized to show abnormal gray matter structure, or investigate a specific ROI. Furthermore,
the question posed can also vary: (1) do the patients have abnormal structure, compared to a
control group?; and (2) Are there structural neural correlates of behavioural measures, such as
pain characteristics and personality traits? Furthermore, as discussed in Section 2.4.2, there are
various MRI-based analysis methods to measure gray matter. It is noteworthy that studies that
only used CTA to evaluate differences in gray matter structure did not study subcortical
structures.
A summary of all studies examining gray matter structure in chronic pain populations is provided
in Table 2.3. It includes chronic back pain [27,121,764,782], FMS
[125,502,558,712,738,766,949], IBS [98,225,779], migraine [207,490,740,762,889], trigeminal
neuropathic pain [206,382], trigeminal neuralgia [382], TMD [358,963], phantom-limb pain
[268], provoked vestibulodynia [772], complex regional pain syndrome (CRPS) [356], cluster
headache [578], chronic tension-type headache [765], medication-overuse headache [765],
hypnic headache [434], chronic pelvic pain [303], chronic posttraumatic headache [650],
persistent idiopathic facial pain (previously atypical facial pain) [763], hip osteorarthritis [741],
rheumatoid arthritis [919] and pain disorder [887] (a diagnosis based on the DSM-IV, defined as
persistent and chronic pain at one or more sites that cannot be fully explained by a physiological
process or physical disorder). Overall, the most common brain regions showing gray matter
abnormalities were the PFC, insula, the ACC and the MCC. Other regions that were abnormal in
chronic pain included the thalamus, basal ganglia, S1, S2, and the brainstem. Additionally, some
studies demonstrated abnormalities in the temporal lobe and the PCC. In every region, with the

103
notable exception of the basal ganglia and the brainstem, most studies showed a decrease in gray
matter in chronic pain.
Of particular interest to this thesis, three studies used VBM to investigate gray matter
abnormalities in TMD. One study by Gustin and colleagues [382] reported no gray matter
abnormalities in the brain of patients with TMD. Gustin and colleagues did not provide the
diagnostic criteria used to select patients with TMD. Another study, by Younger and colleagues
[963], tested for gray matter abnormalities in myofascial patients with TMD and reported four
main findings: 1) compared to controls, patients with TMD had increased gray matter in the
thalamus, the basal ganglia, insula, the vlPFC, and the brainstem; 2) a negative correlation
between jaw pain and gray matter volume in the dorsal posterior cingulate, the MCC,
SMA/preSMA, and the superior temporal gyrus; 3) a negative correlation between experimental
pressure pain thresholds (as measured by pressure algometry) and brainstem gray matter volume;
and 4) a positive correlation between TMD duration and gray matter volume in the posterior
cingulate, the hippocampus, and the brainstem. Another study by Gerstner and colleagues [358]
investigated myofascial patients with TMD and found that patients with TMD had decreased
gray matter in the vlPFC, MCC, PCC, SMA, insula and the regions within temporal lobe. These
findings seem to conflict with the Younger et al. [962] study, but could be attributed to
methodological differences between the studies. For instance, the age range and the range of
durations of TMD in the Younger patient sample were larger than those of the Gerstner patient
sample. These factors could contribute to changes in gray matter, and they cannot be adequately
controlled with the statistical methods used to evaluate group difference in structural gray matter
analyses. For a complete discussion of these points see: [615].
Interestingly, recent evidence suggests that some gray matter abnormalities in some chronic pain
disorders are caused by the pain, rather than pre-existing abnormalities. For instance, Rodriguez-
Raecke and colleagues [741] showed gray matter abnormalities in several brain region of patients
with primary hip osteoarthritis. These patients underwent surgery, and for the most part, the pain
was resolved. The post-operative scan of a subgroup of these patients revealed that some, but not
all, of the gray matter abnormalities had resolved (i.e., there were no differences between
controls and patients). Four other studies have since demonstrated similar effects – partial
reversal of gray matter changes in brain once patients’ pain has been resolved, in the same [384]
and other chronic pain disorders, including chronic low back pain [782]. However, it remains

104
unclear whether these changes in gray matter are due to the resolution of pain, or are secondary
to the resolution of pain. For instance, it is possible that a person with chronic pain in the hip will
be more sedentary, compared to when they are no longer in pain. Furthermore, pain has been
associated with changes in mood, including increased incidence of depression and anxiety, which
could resolve once the pain is gone. Furthermore, it has recently been shown that medications
can alter the structure of the brain [916,962]. Therefore, when patients are no longer taking pain-
killers, the changes in the brain may resolve.
Furthermore, it is noteworthy that not all of the observed gray matter abnormalities were
abolished with the cessation of pain. This may be related to lasting effects of medications, such
as opiates, which have been shown to produce long-term changes in the pgACC, MCC,
amygdala, the vlPFC, hypothalamus, brainstem, and the hippocampus of patients with chronic
low-back pain [962]. These changes did not show reversal after the patients had ceased managing
their pain with morphine for an average period of 4.7 months. Therefore, there may still be pre-
existing abnormalities that contribute to the development of chronic pain.
Additionally, it is noteworthy that functional pain syndromes, such as IBS, FMS and TMD, are
defined as disorders that disturb normal function without any obvious structural or biochemical
abnormality [246]. Therefore, it is possible that mechanisms other than increased nociceptive
drive from the periphery may be driving gray matter abnormalities in the brain. An alternative
possibility is that gray matter abnormalities pre-date the onset of chronic pain. In this scenario,
gray matter abnormalities are pre-existing vulnerabilities that may contribute to the development
of chronic pain [213]. Evidence for this proposition comes from studies that have identified
regions of gray matter abnormalities related to stable personality traits, such as neuroticism or
pain catastrophizing [98,772], which are related to heightened pain sensitivity (see Section
2.1.7).
In sum, there are both pre-existing and pain-driven gray matter abnormalities in the brain of
chronic pain patients. In general, there appears to be increases in sensory/nociceptive brain
regions, indicative of increased nociceptive drive; decreases in pain modulation brain regions,
indicative of potentially dysfunctional pain-modulatory systems.

105
White matter abnormalities
There have only been eight studies investigating white matter abnormalities in the brain of
chronic pain patients. Of these, two have used VBM to evaluate white matter volume from T1-
weighted images, and four have used DTI to delineate abnormalities in white matter
microstructure, or gray matter microstructure. The two studies that have used DTI to investigate
gray matter [334,558] and the white matter VBM studies [358,553] are omitted from this thesis
because of flaws in the experimental design and methodologies. Of the four remaining studies
that used the approach of DTI, one did not find any group differences between patients with
chronic pelvic pain and controls [303]. The other three that did identify abnormalities are
described below.
The first study to test for abnormalities in white matter microstructure associated with chronic
pain, by DaSilva and colleagues [208], reported decreased FA along tracts between the brainstem
and the thalamus and the thalamus and S1 cortex of patients with migraine. The authors
concluded that there are abnormalities along the ascending nociceptive pathways in migraine
patients.
Geha and colleagues [356] reported white matter abnormalities in patients with CRPS that
consisted of lower FA in the cingulum bundle and the adjacent corpus callosum. This was the
first study to investigate the abnormalities in white matter tracts by elucidating the connectivity
of the region. They reported that abnormal white matter region had fewer connections per unit of
distance in CRPS patients compared to controls.
A study by Chen et al. [156], from our laboratory, investigated white matter abnormalities in
patients with IBS, and reported that patients had increased FA in the fornix and the
external/extreme capsule, adjacent to the insula. It was also found that FA in the anterior insula
and the VPL thalamus were significantly correlated to pain severity. Further, FA in the left insula
correlated with pain unpleasantness and duration. Finally, in patients with IBS, FA in the
cingulum bundle was negatively correlated to the PCS, and a positive correlation between FA in
the MD thalamus and neuroticism. These findings suggest that IBS patients have abnormal
connections in white matter tracts related to nociception and/or cognitive/limbic function.

106
In addition to the aforementioned studies that have investigated white matter abnormalities in the
brains of chronic pain patients, two DTI studies have investigated abnormalities along the CNV
in patients with trigeminal neuralgia. One study by Fujiwara et al. [339] did not find any
significant abnormalities in the FA, MD, or the cross-sectional area of the patients’ CNV. This
study reported that no significant group differences in the ratio of FA of the affected to the
unaffected nerve. However, the study reported significantly more variance in the FA values of
patients’ CNV, compared to controls, and this value was positively correlated to the
affected:unaffected ratio of the cross-sectional area of the CNV. The other study, by Leal et al.
[525], reported that the affected CNV had significantly increased MD, and decreased FA, nerve
volume, and cross-sectional area. Furthermore, the decrease in FA was positively correlated to
the decrease in volume and cross-sectional area of the CNV. Decreased FA and increased MD
indicate that there is increased diffusion, or less organization. Therefore, it is possible that the
CNV has a larger diameter, is inflamed, or damaged. However, the interpretation is limited as
other measures of white matter integrity (RD and λ1) were not provided. These studies indicate
that there are structural abnormalities along the CNV in patients with trigeminal neuralgia, and
suggest that DTI can be used to investigate the CNV for abnormalities in TMD.

107
Table 2-3. Summary of regions with gray matter abnormalities in chronic pain
Chronic pain disorder Method n patients
(# females)n controls
(# females) Thal BG S1 S2 IC ACC MCC M1 PFC Bstem ThresholdMultiple
comparisons correction
Contributing factors Ref
VBM 26 (16) 26 (16) ! ! dl p<0.05 Permutation D [27]VBM 18 (9) 18 (9) " " Put ! ! dl ! pTL,
TL p<0.001 Nonea D, P [764]VBM 8 (3) 8 (4) ! PPC p<0.001 None [121]VBM 18 (10) 16 (8) ! ! !
! vl, dl, OFC " MTL p<0.05 Cluster [782]
VBM 10 (10) 10 (10) ! ! ! m ! PCC, PHG p<0.05 RFT; Cluster t
>3 A, D [502]
VBM 20 (19) 22 (20) ! " " OFC ! STG, " Cb
p<0.05, 0.001 ROI; Unc P [766]
VBM 30 (30) 30 (30) ! HC p<0.05 Permutation, Bonferroni [558]
VBM 30 (30) 20 (20) !! PHG, PCC p<0.0005 None [949]
VBM 14 (14) 14 (14) ! ! vl ! Amyg p<0.001 None De, An, M [125]VBM 5(5) 5(5) ! SMA p<0.05 Permutation [712]VBM 14 (14) 11 (11) ! ! ! PCC p<0.02 FDR [738]CTA VBM 9 (6) 11 (7) ! ! ! ! p<0.001 None [225]CTA VBM 11 (11) 16 (16) ! " HT p<0.05 FDR,
Permutation PCS, D [98]
CTA VBM 56 (56) 49 (49) ! ! vStr, Put " " "
" OFC, dl; ! vl, FP, m
" HC p<0.05, p<0.01
Cluster, None A, An, De [779]
VBM 16 (15) 15 (13) ! ! dl, vl " PAG ! TL p<0.001 None A, D [740]VBM 35 (32) 31 (31) ! ! ! p<0.05 SVC [762]
27 (21) 27 (20) ! ! vl ! TL p<0.05, 0.001
FWE, SVC
11 (9)/16 (12)b ! ! ! ! vl, dl ! Amyg p<0.05 SVC P12 (9) [w/ aura] " p<0.05 ROI12 (7) [w/o aura] " " p<0.05 ROI
VBM 20 (17) 33 (29) ! ! !! dm, dl,
OFC
! PPC, PMC, V1
p<0.05, 0.001
SVC, None D, P [490]
[889]
[207]
Other?
CBP
FMS
IBS
Migraine
CTA 12 (9)
VBM
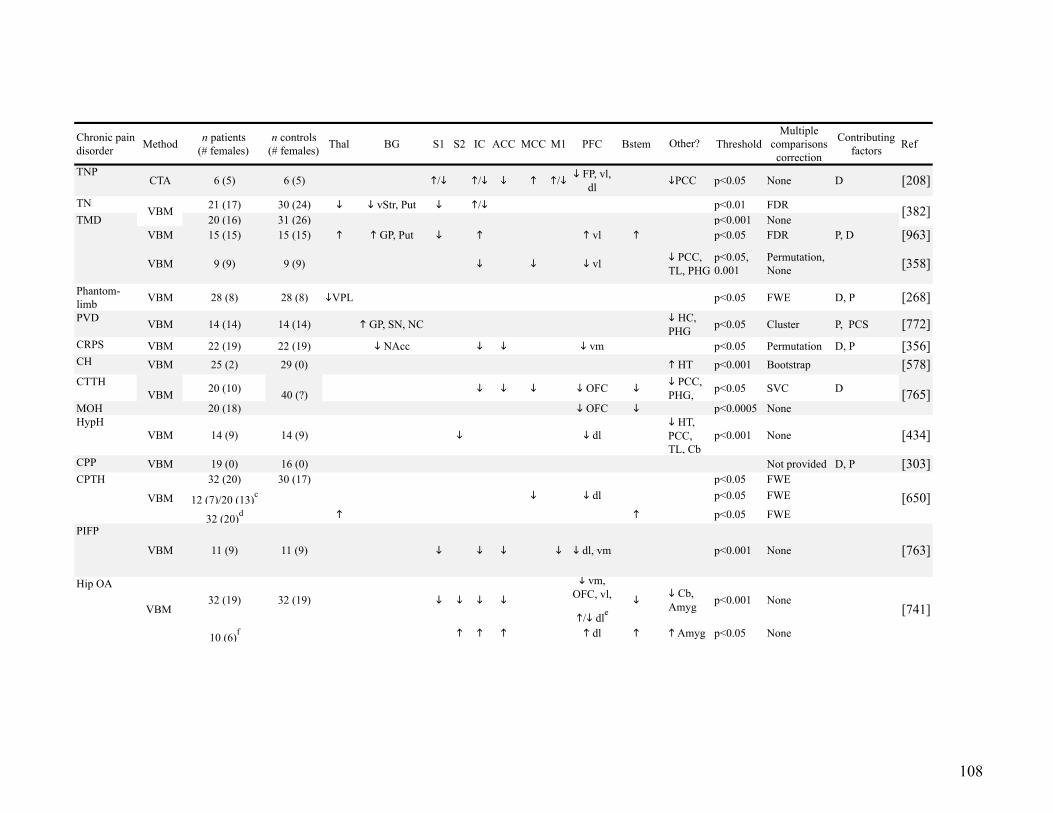
108
Chronic pain disorder Method n patients
(# females)n controls
(# females) Thal BG S1 S2 IC ACC MCC M1 PFC Bstem ThresholdMultiple
comparisons correction
Contributing factors Ref
TNPCTA 6 (5) 6 (5) !/" !/" " ! !/" " FP, vl,
dl "PCC p<0.05 None D [208]TN 21 (17) 30 (24) " " vStr, Put " !/" p<0.01 FDR
20 (16) 31 (26) p<0.001 NoneVBM 15 (15) 15 (15) ! ! GP, Put " ! ! vl ! p<0.05 FDR P, D [963]
VBM 9 (9) 9 (9) " " " vl " PCC, TL, PHG
p<0.05, 0.001
Permutation, None [358]
Phantom-limb VBM 28 (8) 28 (8) "VPL p<0.05 FWE D, P [268]PVD VBM 14 (14) 14 (14) ! GP, SN, NC " HC,
PHG p<0.05 Cluster P, PCS [772]CRPS VBM 22 (19) 22 (19) " NAcc " " " vm p<0.05 Permutation D, P [356]CH VBM 25 (2) 29 (0) ! HT p<0.001 Bootstrap [578]CTTH 20 (10) " " " " OFC "
" PCC, PHG, p<0.05 SVC D
MOH 20 (18) " OFC " p<0.0005 NoneHypH
VBM 14 (9) 14 (9) " " dl" HT, PCC, TL, Cb
p<0.001 None [434]
CPP VBM 19 (0) 16 (0) Not provided D, P [303]32 (20) 30 (17) p<0.05 FWE
12 (7)/20 (13)c " " dl p<0.05 FWE
32 (20)d ! ! p<0.05 FWEPIFP
VBM 11 (9) 11 (9) " " " " " dl, vm p<0.001 None [763]
32 (19) 32 (19) " " " "
" vm, OFC, vl,
!/" dle"
" Cb, Amyg p<0.001 None
10 (6)f ! ! ! ! dl ! ! Amyg p<0.05 None
[765]
[382]
[741]
[650]
VBM
Other?
VBM 40 (?)
Hip OA
VBM
CPTH
VBM
TMD

109
a. the p-value was reduced for some findings, but the lower threshold is not provided b. Between patient groups analysis: Chronic vs. Episodic c. Comparing temporary post-traumatic headache to chronic post-traumatic headache (temporary > chronic) d. Longitudinal study comparing time 1 scans with subjects after the pain has resolved e. Decrease GM on the right, increase GM on the left f. Longitudinal study comparing time 1 scans with post-operative scans, when pain is resolved g. Pain disorder is a diagnosis based on the DSM-IV, defined as persistent and chronic pain at one or more sites that cannot be fully explained by a physiological process or physical disorder."
Abbreviations: Amyg — amygdala; ACC — anterior cingulate cortex; BG — basal ganglia; Cb — cerebellum; CH — cluster
headache; CPP — chronic pelvic pain; CPTH — chronic posttraumatic headache; CRPS — complex regional pain syndrome; CTA —
cortical thickness analysis; DBM — deformation-based morphometry; dlPFC — dorsolateral prefrontal cortex; dmPFC —
dorsomedial prefrontal cortex; FDR — false discovery rate; FWE — family-wise error; GMV — gray matter volume; GP — globus
pallidus; HC — hippocampus; Hip OA — hip osteoarthritis; HT — hypothalamus; HypH — hypnic headache; IBS — irritable bowel
syndrome; IC — insular cortex; iTL- inferior temporal lobe; M1 — primary motor cortex; MCC — midcingulate cortex; MOH —
medication-overuse headache; mPFC — medial prefrontal cortex; MTL — medial temporal lobe; NC — caudate nucleus; OFC —
orbitofrontal cortex; PCC — posterior cingulate cortex; PFC — prefrontal cortex; PHG — parahippocampal gyrus; PIFP — persistent
idiopathic facial pain; PMC — premotor cortex; PPC — posterior parietal cortex; pTL — posterior temporal lobe; Put — putamen;
Chronic pain disorder Method n patients
(# females)n controls
(# females) Thal BG S1 S2 IC ACC MCC M1 PFC Bstem ThresholdMultiple
comparisons correction
Contributing factors Ref
Rheum Arth VBM CTA DBM
31 (22) 25 (17) ! Put, NAcc, NC p<0.05 FDR D [919]
Pain
disorderg VBM 14 (14) 25 (25) " "" vm, OFC
" iTL, PHG p<0.05 FDR D [887]
Other?

110
PVD — provoked vestibulodynia; RFT — random field theory; Rheum Arth — rheumatoid arthritis; ROI — region of interest; S1 —
primary somatosensory cortex; S2 — secondary somatosensory cortex; SMA — supplementary motor area; SN — substantia nigra;
STG — superior temporal gyrus; SVC — small-volume correction; Thal — thalamus; TMD — temporomandibular disorder; TN —
trigeminal neuralgia; TNP — trigeminal neuropathic pain; TL — temporal lobe; V1 — primary visual cortex; vlPFC — ventrolateral
prefrontal cortex; vmPFC — ventromedial prefrontal cortex; vSTR — ventral striatum

111
2.6. Neuroplasticity
It is clear that brain structure is defined by genetics and experience [577]. In fact, it has been
suggested that neuroplasticity is “ the normal ongoing state of the nervous system throughout the
life span” [672], in order to better adapt to the environment [969]. This concept of brain
neuroplasticity was recognized early in the twentieth century. For instance, Cajal described the
concept:
The labor of a pianist [...] is inaccessible for the un-educated man as the acquisition of new skill requires many years of mental and physical practice. In order to fully understand this complex phenomenon it becomes necessary to admit, in addition to the reinforcement of pre-established organic pathways, the formation of new pathways through ramification and progressive growth of the dendritic arborization and the nervous terminals. [134] (cited and translated in [672])
The term plasticity was first used by Ernesto Lugaro in 1906 [554] to describe changes in the
nervous system during maturation, compensatory changes after lesions, and changes related to
learning and memory. He suggested the specific chemicals, today known as neurotrophins, that
are responsible for the development and organization of the nervous system, continue to
reorganize the neuronal connections in the brain. This property of the nervous system, which he
called plastic, occurred through chemical signaling. Lugaro recognized two processes specific to
neurones: conduction of an impulse, and transduction of a neural signal between neurones [86].
In line with Hebbian doctrine [419], Lugaro postulated that transduction would lead to
modifications of the neural circuit: increased firing would enhance a connection by modifying
the synaptic distance [86]. Despite Lugaro’s postulation that the brain remains plastic throughout
life, scientific dogma maintained that only the developing nervous system could undergo
changes. The concept of plasticity was not established until pioneering studies were published in
the second half of the twentieth century. These studies used animal models to demonstrate that
plasticity and “learning” occur at several levels of the nervous established systems, from the
periphery, and the spinal cord, to the brain (see [477]). Today, it is established that plasticity
occurs in the mature nervous system. It is also now known that plasticity can occur rapidly at
several levels of the nervous system. These neuroplastic events can either be beneficial, as in
learning a novel task, or maladaptive, as is the case in chronic pain (discussed below).

112
One of the first studies [914] to report plasticity in the somatosensory system found that after
destruction of the dorsal column neurones in the VPL region of the thalamus that normally
responded to tactile stimulation in the foot changed their receptive fields. The representation of
the arm in VPL expanded to the region that previously represented the foot. Later studies have
confirmed that plasticity occurs throughout the adult nervous system, including in the
somatosensory system (for a complete review, see: [347,475]).
2.6.1. Use-dependent neuroplasticity
One type of beneficial plasticity occurs during learning. This concept was first described
anecdotally by Hebb [419]: rats that were exposed to enriched environments performed better on
tasks requiring complex behaviour, compared to rats raised in cages. In humans, a study by
Pascual-Leone and colleagues [673] reported that learning a novel motor task is associated with
motor cortex plasticity, as measured by TMS. Interestingly, these changes returned to baseline
once the task had been learned, and became part of implicit knowledge. An fMRI study [478]
investigating motor learning (the learning of sequential finger movements) found increased brain
activation in M1 in subjects that have learned a new task, but, in contrast to Pascual-Leone et
al.’s study, these changes persisted for several months. Another example of beneficial plasticity
is a study by Nudo and colleagues [648], which showed that after an experimentally-induced
ischaemic stroke in the hand region of M1 of non-human primates, retraining of the affected
hand preserved the cortical tissue adjacent to the lesion. This preserved tissue underwent
plasticity and contributed to the functional recovery of the hand. These studies demonstrate the
concept of use-dependent plasticity. Motor learning, or retraining, can change the structure and
function of the brain – in these cases, M1.
Longitudinal studies of use-dependent plasticity have found that behavioural changes are related
to both functional and structural gray and white matter changes in the brain
[111,266,267,271,771]. Animal studies in non-human primates have demonstrated that the
nervous system undergoes functional reorganization (changes in cortical representation fields) in
response to massive changes in the environment, such as sensory loss due to deafferentation or
brain lesions [454,458,703]. Furthermore, studies in non-human primates have demonstrated
massive white matter plasticity in response to learning a novel task [422] and brain lesions [200].

113
Other studies have shown that experience-dependent (or use-dependent) plasticity is related to
cellular changes, specifically in dendritic spine density [436,493,865].
Plasticity in the nervous system can include changes to a receptive field (e.g., with increased use
of a limb, its representation in the brain can be expanded), changes in neuronal responses or
firing thresholds, and structural changes in neuronal connections.
However, not all use-dependent neuroplasticity is beneficial [576]. For instance, Rosenkranz and
colleagues [745] compared healthy musicians to a group of patients with musician’s dystonia,
and a group of controls. The authors reported that musicians showed functional plasticity in the
motor cortex. Patients with dystonia, however, had even more changes, which disrupted normal
motor output. This study demonstrates that neuroplasticity can actually interfere with normal
function. Similar mechanisms may occur in patients that develop chronic pain: increased or
prolonged nociceptive input can induce “excess” plasticity, which can itself become the
aetiology for pain (see below).
2.6.2. Neuroplasticity and pain
Normally, pain is a behaviourally-relevant signal that can promote survival by signaling injury or
potential injury. However, in some cases, pain persists long after its usefulness, even after the
injury has healed. This occurs due to a process called the chronification of pain (see Section
2.1.2). In cases of chronic pain, pain is no longer a useful protective signal which is promoting
survival, but rather the pain becomes a disease in itself. This chronification occurs due to
functional and structural changes at several levels of the nervous system, or maladaptive
plasticity.
One level of plasticity is in the periphery at the nociceptor. When injury occurs, pain experience
is heightened by increasing the sensitivity of nociceptors to thermal and mechanical stimuli (i.e.,
peripheral sensitization) [470]. This occurs, in part, because of chemical mediators that are
released at the site of injury, which modify the properties of the nociceptors such as their
threshold. There are also changes in the neurotransmitters produced by these neurones. The
dorsal horn of the spinal cord and the VBSNC also undergo plasticity [159,784,950,951]. For
instance, in phantom limb pain, where the peripheral end of the nerve undergoes changes that
cause pain, blocking conduction along the peripheral nerve at the stump does not eliminate

114
ongoing pain in all patients [96,323]. Furthermore, when a nerve is severed, there is a cascade of
events that occur in the CNS. For instance, microglia are activated, and chemical mediators
(including inflammatory molecules) are released [568]. Interestingly, when these chemical
mediators are inhibited in the spinal cord and VBSNC [159], patients with neuropathic pain
report that their pain is resolved [568]. These factors can initiate or maintain pain after peripheral
nerve injury by sensitizing or disinhibiting second-order neurones. Importantly, immune cells are
not implicated in acute pain conditions, but rather in persistent and chronic pain states (see
[159,568]). Further neuroplastic events occur in suprabulbar regions, in the thalamus and the
cortex. For instance, after peripheral nerve injury (including amputation) in non-human primates,
spinal, brainstem, thalamic and cortical neurones can undergo large-scale reorganization
[324,325,467,472-474,703]. Further evidence for pain-related neuroplasticity comes from a study
that showed that thalamic stimulation in phantom limb patients could elicit their phantom limb
pain [219]. This suggests that neuronal representation of the phantom limb remains in the
thalamus, even after the loss of the limb.
Neuroimaging studies have contributed to our understanding of brain plasticity in response to
repeated noxious stimulation. For instance, healthy subjects who received 20 minutes of noxious
stimulation over eight days habituated behaviourally (i.e., they reported the same stimulus as less
painful) [93,94,852] and showed decreased pain-evoked activation in the thalamus, S2, insula
and the putamen, and increased activation in the sgACC [94]. Structural imaging in these
subjects also revealed increased gray matter volume in the PMC, MCC, S1, inferior parietal
lobule, and the medial temporal gyrus after the eight days of stimulation, compared to baseline
[852]. Although not explicitly tested, it is inferred that these structural and functional changes
observed were related to the observed behavioural changes. Although the functional and
structural changes were located in different regions, the findings nonetheless provide evidence
that repeated noxious stimulation induces functional and structural brain plasticity. Further
support for the concept that increased nociceptive input alters the structure of the brain comes
from studies that have demonstrated that gray matter abnormalities are reversible upon the
successful treatment of the chronic pain disorder [384,741,782]. However, not all gray matter
abnormalities in chronic pain conditions are reversible. It is possible that chronic pain patients
may not have effective pain-modulatory systems. In TMD, evidence suggests that some pain-
modulatory systems, such as DNIC [113,491,756], are dysfunctional. Also, most studies

115
investigating the structure of the brain in chronic pain have reported decreased gray matter in the
pain-modulatory regions [575]. Additionally, it is possible that, to some extent, hypersensitivity
to nociceptive stimuli is related to changes that have occurred in brain structure. In line with this
concept, studies have demonstrated that some patients with TMD show allodynic and
hyperalgesic responses to experimental stimuli, consistent with the concept of central
sensitization (See section 2.2.3). It is possible that the combination of increased barrage of
nociceptive processing, an increase in the activity of pain-facilitatory pathways and decreased
activity in pain-inhibitory pathways induce central sensitization. This central sensitization can
lead to further functional and structural changes that can maintain painful states.
Based on the concept of that pain may drive CNS plasticity, structural abnormalities in chronic
pain patients would correlate with pain characteristics pain, such as duration, intensity and
unpleasantness. For instance, if we expect increased nociceptive activity, then we would expect a
positive correlation between measures of brain structure (volume or thickness in gray matter; FA
in white matter) in nociceptive brain regions and pain duration, intensity and unpleasantness.
Conversely, we would expect a negative correlation between structural measures and pain
characteristics in antinociceptive regions. Alternatively, if there are pre-existing abnormalities in
the brain that predispose subjects to the development of pain, then we would not expect
correlations between structural measures in pain-related brain regions and pain characteristics.
However, in the latter scenario, we can expect that brain structure would be related to personality
factors that predispose persons to developing chronic pain, such as neuroticism.
2.6.3. Age-related neuroplasticity
It has long been recognized that, over time, the brain undergoes atrophy. These age-related
changes have been observed in post-mortem studies [115,191,232,261,427] as well as in brain
imaging studies [322,749-751,911,968]. These studies have found that, in general, the brain
underwent atrophy with age, although some regions were spared, and others showed an increase
with age. For instance, Ziegler and colleagues [968] showed that the MCC and sgACC were
thicker in older adults than in younger adults. Some disease states have been shown to modulate
this aging process. For instance, Alzheimer’s disease has been shown to increase the rate of gray
matter atrophy. Furthermore, two studies have shown that patients with chronic pain, specifically
FMS [502] and chronic back pain [27], also had accelerated gray matter atrophy. It is not known

116
whether this form of plasticity is related to duration of pain, or other factors that may contribute
to accelerated gray matter atrophy.
2.6.4. Microstructural basis of neuroplasticity
MRI-detectable changes occur in the brain as part of the natural aging process, with increased
use (experience, learning and memory), in response to peripheral nerve injury, and amputation.
However, the neural basis of these changes is not well understood. Pakkenberg and Gundersen
[662] reported that whole brain age-related atrophy in humans is due to neuronal loss and
reduced cell packing density; whereas Peters et al. [685] reported that age-related atrophy in
macaques is related to a decrease in the number of neuropils, not neurones. There are also other
proposed hypotheses to explain mechanisms of gray matter change. For instance, rather than
neural loss, there may be glial death [575]. Recent evidence suggests that gray matter loss may
be related to the density of small dendritic spines [278,608], and the remodeling of neuronal
processes [537]. Alternatively, reversible gray matter changes in chronic pain may be caused by
neuroinflammation [238,381,920], and induce MRI-detectable increases in gray matter. This
mechanism could explain both abnormal age-related increases and maintenance of gray matter
volume/thickness. For the observed gray matter losses, however, we cannot rule out cell death as
a factor in age-related gray matter loss – healthy populations lose neurones as they age, and
persons with neurodegenerative diseases suffer increased rates of atrophy related to cell death.
Changes in measures of white matter integrity, as indicated by FA, MD, RD and λ1, each reflect
a different aspect of white matter microstructure. The most commonly computed and assessed
measure of white matter integrity is FA, because it is scaled between 0 and 1, where 0 represents
complete isotropic diffusion, and 1 represents complete anisotropic diffusion, and studies have
demonstrated that a threshold value of 0.2 for FA value can suitably delineate white matter from
gray matter [820]; however, this needs to be checked for specific populations. There could be
macrostructural factors that contribute to reduced FA, such as increased branching, crossing
fibres or larger tracts (more axons) and/or microstructural changes such as cell swelling
(œdema), changes to protein filaments (neurofilament phosphorylation), disruptions to the cell
membranes, and, to a certain extent, decreased myelin [66,67]. However, as FA is a summary
measure, it is not specific to any microstructural features of white matter tracts [695].
Histological studies have demonstrated that RD and λ1 are related to alterations in specific

117
structures [67]. FA can be increased by any decrease in RD, and increases in λ1, and thus RD and
λ1 provide greater insight into the cellular properties underlying the observed changes in FA, and
presumably, white matter tracts. Specifically, RD is related to membrane integrity, and, to some
extent, myelination; whereas λ1 is related to factors that may disrupt the axon or neurofilament
phosphorylation [67].
Recent studies have suggested that chronic pain is associated with neuroinflammation in the
brain [238,381,920], which can lead to changes in both gray matter and white matter that can be
detected by MRI. Therefore, changes in the measures of white matter microstructure may, in
fact, be related to neuroinflammation rather than increases or decreases in the number or size of
cells and/or axons present in the fields being studied. In this scenario, we would expect that
changes in FA are associated to changes in MD and RD, which are markers of inflammation
and/or œdema [63,66].

118
Chapter 3 Rationale, Specific Aims and Hypotheses
The general aim of this thesis is to determine whether patients with TMD have structural
abnormalities in the brain or CNV, compared to healthy, pain-free controls. To this end, three
MRI-based studies have been conducted to assess gray and white matter abnormalities. The
rationale, specific aims and hypotheses for each of these studies are provided below.
3.1. Study I. Contribution of chronic pain and neuroticism to abnormal forebrain gray matter in TMD patients
TMD can be idiopathic in that there may not be any clear peripheral etiological factors
identifiable [283,286,653] (see Chapter 2). In this scenario, it is thought that the CNS may
initiate and/or maintain the pain [757]. Previous structural MRI studies of chronic pain
populations have found both increases and decreases in brain gray matter (See Section 2.5.2).
There are two main routes by which the CNS may contribute to the development and/or
maintenance of chronic pains such as TMD. One possibility is that persistent or long-term
nociceptive input into the brain induces maladaptive brain plasticity that can play a role in
maintaining pain [12,950]. This maladaptive plasticity could hamper a patient’s ability to
habituate to sustained nociceptive activity, possibly related to a reduced capacity of the brain to
dampen pain by descending (top-down) controls [95]. Indeed, many structural MRI studies have
found that chronic pain-related characteristics (intensity, unpleasantness, or duration) can in part
account for gray matter abnormalities in chronic pain populations (See Section 2.5.2, and Table
2-3). Alternatively, inherent personality-related factors that reduce the brain's capacity to
modulate nociceptive activity may contribute to gray matter abnormalities, and represents a
vulnerability to develop chronic pain. For example, there is evidence that neuroticism may be
associated with pain-related suffering (See Section 2.1.7), nerve injury outcomes and neuropathic
pain [848] and inhibition of negative thoughts [178]. However, not all chronic pain patients have
high neuroticism scores, and not all persons with neuroticism have chronic pain [179]. Therefore,
neuroticism alone is not sufficient to develop chronic pain. Rather, the normal relationship
between neuroticism and brain structure and function may be disrupted within regions involved
in pain modulation, and this could facilitate or maintain chronic pain.

119
Therefore, the main aim of Study I is to examine gray matter abnormalities in patients with
idiopathic TMD and to determine the contribution of pain-related characteristics and
neuroticism.
Specific aims:
1. To determine whether patients with TMD have increased gray matter in regions of the
brain implicated in pain perception, compared to healthy, pain-free controls.
2. To determine whether patients with TMD have reduced gray matter in regions of the
brain implicated in pain modulation and motor function, compared to healthy, pain-free
controls.
3. To determine the relative contribution of TMD pain characteristics (duration, intensity
and unpleasantness) to gray matter abnormalities in the brains of patients with TMD.
4. To determine the relative contribution of neuroticism to gray matter abnormalities in the
brain of patients with TMD.
Specific hypotheses:
Compared to healthy, pain-free controls, patients with TMD will have:
1. increased gray matter in brain areas associated with pain perception;
2. decreased gray matter in areas associated with pain modulation and motor function;
3. a positive correlation between gray matter in pain perception regions and pain intensity,
unpleasantness and TMD duration; but a negative correlation between gray matter in
regions implicated in pain inhibition and pain intensity, unpleasantness and TMD
duration;
4. a positive correlation between gray matter in regions implicated in the affective
dimension of pain and neuroticism, but a negative correlation between gray matter in
regions implicated in pain inhibition and neuroticism.

120
3.2. Study II. Age-related gray matter abnormalities in patients with TMD
The brain undergoes structural changes due to normal conditions such as aging. For example,
normal aging is characterized by cortical gray matter atrophy [84,99,372,585,627,824], although
gains have also been reported in some brain areas [322,749]. Brain plasticity also occurs with
dysfunction, injury, in specific diseases, and with chronic pain (see Section 2.5.2). The
cumulative effect of chronic pain has been shown to interact with normal aging processes. For
example, accelerated age-related whole brain gray matter atrophy has been reported in FMS
[502] and chronic back pain [27]. Most aging studies in chronic pain have assessed global gray
matter but little is known about the interaction between chronic pain and age in specific brain
areas.
Although the functional significance of gray matter changes is not understood, MRI and
histological studies indicate that changes can be stimulus-dependent. For instance, studies have
demonstrated gray matter changes in response to learning [267], training [266], and repetitive
noxious stimulation [852]. These data suggest that abnormal gray matter progression in chronic
pain may be related to pain duration. While it is plausible that age-related changes specific to
chronic pain are the product of cumulative pain exposure, this hypothesis has not been tested
empirically in TMD.
Therefore, the main aim of study II is to determine whether the cumulative effect of idiopathic
TMD interacts with the normal aging process in focal cortical regions associated with
nociceptive processes.
Specific aims:
1) To determine whether patients with TMD exhibit greater rates of whole brain gray matter
atrophy, compared to healthy, pain-free controls.
2) To determine whether TMD is associated with abnormal gray matter aging in focal
cortical regions associated with pain processes.
3) The degree to which the cumulative effects of pain (i.e. TMD duration) contributes to the
observed age effects.

121
Specific hypotheses:
1) Patients with TMD will have increased rates of whole brain gray matter atrophy,
compared to healthy, pain-free controls.
2) Age-related gray matter changes will be increased in brain regions implicated in pain
perception of patients with TMD,
3) Abnormal gray matter changes in patients with TMD will be driven by TMD duration,
rather than age.
3.3. Study III. White matter brain abnormalities in temporomandibular disorder
Abnormal CNS function is thought to initiate or maintain TMD pain [757] based on TMD
symptomology such as persistent pain, allodynia, and hyperalgesia (sometimes extending to
regions distant from the face) [305,314,390,565,756,883], enhanced temporal summation of pain
to repetitive noxious heat stimuli [563], and dysfunctional DNIC [113,491,756]. Abnormalities
of these centrally-mediated processes suggest that ascending nociceptive pathways and/or
descending pain-modulatory pathways [515] are affected in TMD. Additionally, patients with
TMD can exhibit cognitive [370,379,380] and motor dysfunction [837] possibly related to
abnormalities in brain regions associated with these functions [799,800,839,928].
Studies (including this thesis) indicate that patients with TMD have gray matter abnormalities in
sensory, motor and cognitive/limbic regions (see Section 2.5.2 and Chapter 5). Our finding of a
correlation of thalamic gray matter with TMD duration [617] supports the concept of long-term
peripherally-induced central plasticity. Therefore, although clinical observations have not clearly
identified gross peripheral abnormalities in TMD, the CNV may indeed undergo changes due to
abnormal persistent activity resulting in microstructural abnormalities.
Previous studies examining white matter in other clinical conditions with sensory abnormalities
and/or chronic pain [156,356,558,847] found white matter abnormalities in brain areas involved
in sensory, modulatory and cognitive functions, and some of these white matter abnormalities
correlated with clinical findings. Therefore, studying the correlation between TMD

122
characteristics and measures of white matter integrity can provide insight into whether chronic
pain drives changes in white matter microstructure.
Therefore, the main aim study III is to evaluate white matter brain and CNV abnormalities in
patients with idiopathic TMD and to determine the contribution of pain-related characteristics.
Specific aims:
1) To determine whether there are white matter abnormalities along the CNV of patients
with TMD compared to pain-free controls.
2) To determine whether there are abnormalities in white matter tracts in the brain
associated with sensory, cognitive or motor functions, compared to healthy controls.
3) To determine the connectivity of main regions of white matter disruption.
4) To determine whether there is a link between white matter microstructure and clinical
characteristics of TMD.
Specific hypotheses:
Patients with TMD will have:
1) Abnormal white matter along the CNV;
2) Abnormal white matter in sensory, cognitive and motor tracts;
3) Abnormal connectivity of white matter regions with abnormal microstructure;
4) Abnormalities in white matter will be correlated to pain characteristics (TMD duration,
pain intensity and pain unpleasantness).

123
Chapter 4 General Methods
This project was conducted in a cohort of female patients with TMD and an age-matched group
of control subjects (see below).
4.1. Overview of Project
All study participants underwent two experimental sessions:
• Questionnaires and task training session – This session consisted of handedness and
personality questionnaires. All subjects were also trained in an fMRI task that is not part of
this thesis. Furthermore, patients underwent an interview to characterize their pain, and
completed pain questionnaires.
• Brain imaging session – This session included acquisition of structural MRI sequences to
assess gray and white matter structure. This session also included acquisition of Stroop task
fMRI [928] and resting state fMRI that are not part of this thesis.
4.2. Subject Recruitment
Patients were recruited by dentists working in the Facial Pain Program at the Mount Sinai
Hospital Dental Clinic. Patients were diagnosed with non-traumatic TMD based on the Mount
Sinai Hospital Dental Clinics’ TMD diagnostic criteria (that are based on the TMD-RDC).
Specifically, inclusion criteria included: 1) Self-report of TMD pain in the masticatory muscles
of greater or equal to 4/10 (with ‘0’ being equivalent to no pain at all and ‘10’ representing
maximal pain) for at least 3 months or pain that is aggravated by mandibular function; and 2)
moderate pain to palpation and/or persisting pain after examination in at least 3 masticatory
muscle sites and/or moderate pain to palpation of the TMJ region and/or limitation in mandibular
movement (opening less than 40 mm). Exclusion criteria included: 1) serious metabolic,
rheumatoid or vascular disorders and other diseases; 2) other craniofacial pain disorders,
previously diagnosed psychiatric disorders (e.g., depression, schizophrenia and attention-deficit
hyperactivity disorder) or self-reported history of an abnormal neurological examination; 3) any
contraindication to MRI scanning (e.g., claustrophobia, pregnancy and metal); and 4) use of
psychotropic drugs. All participants were female because of the increased prevalence of TMD in
females. If patients were eligible to participate in the study, the study was outlined to them, and

124
any of their questions were answered. If there were interested in participating in the study, they
were provided with a letter outlining the general goals and methods of the study, and inviting the
patients to participate (see Appendix I: Recruitment letter). If patients chose to participate,
contact information was collected, so that they had a week to reconsider whether to participate in
the study. Nineteen patients with TMD were initially included in the study, of which two were
later excluded because they did not meet inclusion criteria.
The final study cohort included 17 patients with TMD (age ± SD: 33.1 ± 11.9 years; range: 18 –
59 years). Additionally, a group of 17 age-and sex-matched healthy controls (age ± SD: 32.2 ±
10.1 years; range: 20 – 50 years) was recruited from the University and Hospital environments.
To recruit control subjects, the study was advertised by postings within the hospital. Control
subjects would contact us by telephone or email, and undergo a basic screening over the
telephone. The exclusion criteria for the healthy control cohort were the same as those mentioned
for the patient cohort with the addition of a history of past chronic pain.
All study participants provided informed written consent to procedures approved by the
University Health Network and Mount Sinai Hospital Research Ethics Boards (see Appendix II:
Consent Forms).
One additional control subject was recruited and consented to REB approved procedures to
replace one subject subsequently removed from the subcortical analysis (see below) because of
poor segmentation in the pre-processing pipeline.
4.3. Questionnaires
4.3.1. Edinburgh Handedness Inventory
Handedness was determined using the Edinburgh handedness inventory [655] (see Appendix III).
This questionnaire consists of ten questions about which hand is used to perform everyday
manual activities. Subjects report whether they use their right or left hand, and whether they
could use the opposite hand if they so desired. To tabulate, a laterality quotient is calculated with
the following equation:

125
where X(i,R) is the number of right-handed responses for each item, and X(i,L) is the number of
left-handed responses for each item. If a subject reports that the other hand cannot be used for a
given task, then their response is weighted as 2 responses. The range of possible scores is
between -100 and +100. If subjects scores between < -40 he or she is categorized as left-handed;
if the subject scores between -40 and +40, then he or she is considered ambidextrous; and if the
subject scores > +40, he or she is deemed right-handed.
4.3.2. NEO-Five Factor Inventory-Short Form
All subjects completed the NEO (Neuroticism Extraversion Openness) Five Factor Inventory
(NEO-FFI; Psychological Assessment Resources, Lutz, Florida, USA) [178]. Participants were
asked to indicate the degree to which they agree with a statement on a five-point Likert scale
(strongly disagree, disagree, neutral, agree and strongly agree), each of which is coded to a
number (0–4). A total of 15 of 60 questions probe for aspects of neuroticism in this
questionnaire. Neuroticism was the only personality dimension that was assessed.
4.3.3. Pain Catastrophizing Scale
The Pain Catastrophizing Scale [836] (see Appendix IV) was also collected for all participants of
this study, but they were not included in the analysis within this thesis.
4.3.4. Patient Interview
Patients were interviewed to characterize their TMD-related pain (Appendix V). Patients were
also asked to provide a numerical pain score for their current pain intensity and pain
unpleasantness verbally and were asked to report their average pain intensity and unpleasantness
over the last month before scanning. They were specifically asked the following questions:
“Please rate your current/ average pain/unpleasantness rating over the last month on a scale of 0
to 10 (0 = no pain, 10 = worst pain imaginable)”. The duration of TMD was also documented for
each patient.
H =100∗ i=1
10
ΣX(i,R)−i=1
10
ΣX(i,L)
i=1
10
ΣX(i,R)+i=1
10
ΣX(i,L)

126
Furthermore, patients completed the McGill Pain Questionnaire [591] (see Appendix VI). The
McGill Pain questionnaire was used to assess the different dimensions of pain, and thus to
specify the qualia of pain. The questionnaire comprised 102 words classified into three classes
and sixteen subclasses. Different pain disorders have been related to a specific set of words from
the questionnaire. For the current thesis, the McGill Pain Questionnaire was only collected as a
qualitative tool, and was not further assessed. Results are presented in Appendix VII.
4.4. MR Imaging
4.4.1. Study design
All subjects participated in a 1-hour imaging session that included: 1) a low-resolution
anatomical scan for co-registration of fMRI scans, 2) three “task” fMRI scans (not included in
this thesis), 3) a high-resolution T1-weighted whole brain anatomical scan, 4) a resting state
fMRI scan (not included in this thesis), and 5) two runs of DWI covering the whole head. After
brain scanning, subjects were asked to complete a questionnaire related to the task fMRI scans.
Of note, the task fMRI required subjects to wear special goggles to view images, and to use a
response button box. These devices can increase the inhomogeneity during structural MRI
acquisition and so the goggles and the response button-box were removed from the MRI prior to
structural imaging.
4.4.2. Imaging parameters
Brain imaging data were acquired using a 3.0 T GE Signa HDx MRI system (General Electric,
Milwaukee, WI) fitted with an eight-channel phased-array head coil. Subjects were placed supine
on the MRI table and each subject's head was padded to reduce movement. Each subject was
given a “panic button” which they could use if they felt uncomfortable and wanted to stop the
scan.
For gray matter analysis, a whole brain three dimensional (3D) high-resolution anatomical scan
was acquired with a T1-weighted 3D IR-FSPGR sequence: 128 axial slices, 0.94 × 0.94 × 1.5
mm3 voxels, 256 × 256 matrix size, field of view = 24 × 24 cm, one signal average, flip
angle=20°, TE =5 ms, TR =12 ms, inversion time=300 ms.

127
For white matter analysis, two DWI scans were performed (TR = 14,500 ms, field of view: 24
cm x 24 cm, 128 x 128 matrix, 1.875mm x 1.875 mm in-plane resolution, 3 mm thick axial
slices, with Array Spatial Sensitivity Encoding Technique (ASSET) with a factor of 2, maximum
gradient strength = 40 mT/m, maximum slew rate = 150 T/m/s) acquired along 23 non-collinear,
isotropic directions (b = 1,000 s/mm2). Additionally, 2 non-diffusion-weighted scans (b = 0
s/mm2; b0) were acquired at the beginning of each run. The scans covered from the top of the
head to the second cervical spinal process (C2).
4.4.3. Gray Matter Analysis
Two analysis methods were used to assess gray matter: CTA for cortical analysis [316,538,559],
and VBM for subcortical analysis [36] . We used masks of specific brain regions (i.e., ROI
analysis) to test our specific a priori hypotheses; this focus also reduced the statistical burden
associated with the need to adjust the α level in multiple comparisons to regions of no interest
(masks specific to the different studies are described in Chapters 5 and 6).
4.4.3.1. Cortical Thickness Analysis
FreeSurfer software v 4.5 (http://surfer.nmr.mgh.harvard.edu) was used to assess cortical
thickness [198,316,317,319,320]. There are two automated pipelines embedded into the
FreeSurfer analysis pipeline: the cortical (surface) stream and the subcortical (volumetric)
stream. The subcortical stream is out of the scope of this thesis, and so it will not be discussed
further. The cortical stream is described in [198]. The pipeline is as follows: (1) T1-weighted
scans were normalized to Talairach stereotaxic space [844]. To do so, transformation parameters
are computed “by using gradient descent at various scales to maximize the correlation between
an individual volume and an average volume composed of a large number of previously aligned
brains”[198]. The resultant transformation matrix (from individual space to Talairach space) is
stored for later steps. (2) Anatomical T1-weighted images have MRI-susceptibility artifacts and
RF-field inhomogeneities which cause varying intensities and contrast across the volume. These
need to be corrected for proper segmentation. To do so, white matter is used to correct
inhomogeneities and artifacts, because it is more spatially extensive than gray matter, and thus
has minimal partial voluming issues, and allows for low spatial frequency inhomogeneities to be
estimated. Inhomogeneities are corrected by adjusting for the variability in peak white matter
signal between different slices. This process is called “estimating a B1 bias field” [198]. (3) The

128
skull is removed from the brain image. To do so, the inner surface of the skull is modeled using a
deformation-based tessellated ellipsoid template [198] (see Figure 4-1).
Figure 4-1: Template deformation process for skull-stripping in FreeSurfer. Reproduced with
permission from [198].
(4) Intensity values are used to classify white matter voxels as “white matter” or “not white
matter”. (5) Planes based on the Talairach location of the corpus callosum and the pons are
applied to separate the two hemispheres, remove the cerebellum and the brainstem (see Figure 4-
2).
Figure 4-2: Cutting planes overlaid on sections through white matter-labeled volume.
Reproduced with permission from [198].
(6) The surface that is the border of white matter and gray matter (i.e., the outer surface of the
white matter) is modeled as an initial surface for each hemisphere (see Figure 4.5). The modeled
surface (called “white surface”; see Figure 4-3 and 4-5) undergoes smoothing based on local
MRI intensity values to better reflect the gray/white border. (7) The white matter surface is then
inflated to the gray/CSF surface (the “pial surface”; see Figure 4-4 and 4-5). Cortical thickness is
defined as the distance between the white surface and the pial surface.
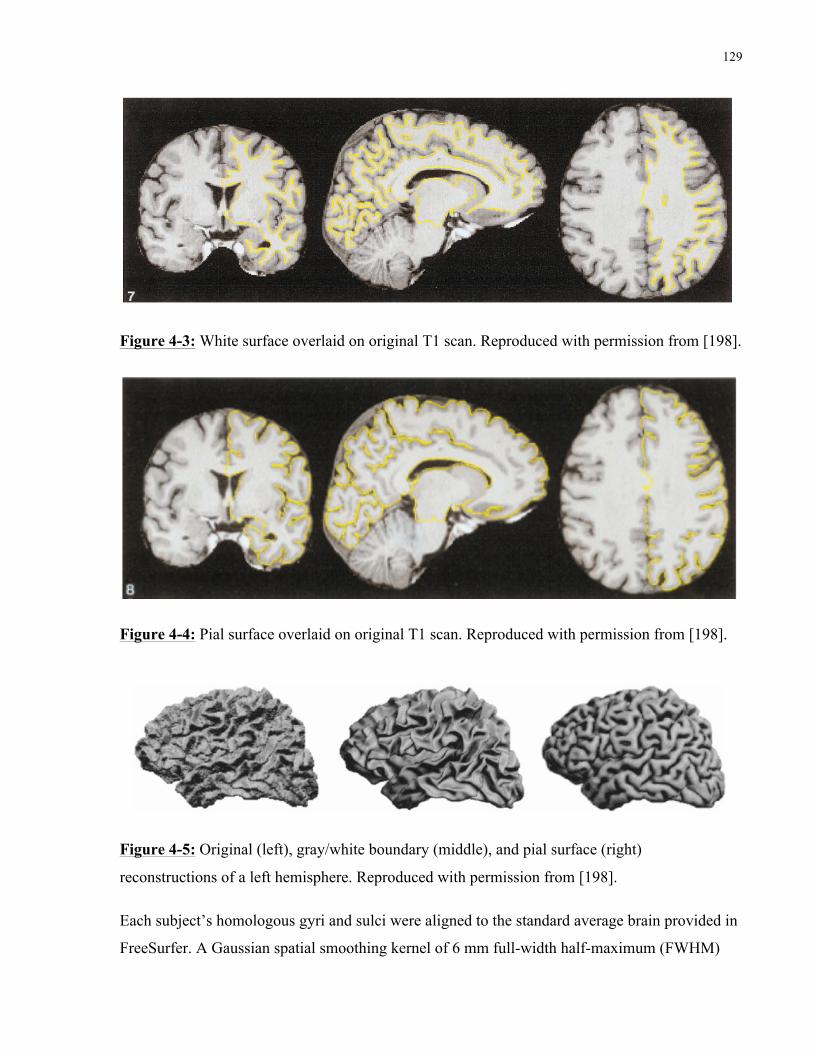
129
Figure 4-3: White surface overlaid on original T1 scan. Reproduced with permission from [198].
Figure 4-4: Pial surface overlaid on original T1 scan. Reproduced with permission from [198].
Figure 4-5: Original (left), gray/white boundary (middle), and pial surface (right)
reconstructions of a left hemisphere. Reproduced with permission from [198].
Each subject’s homologous gyri and sulci were aligned to the standard average brain provided in
FreeSurfer. A Gaussian spatial smoothing kernel of 6 mm full-width half-maximum (FWHM)

130
was applied to compensate for topographical heterogeneity prior to statistical analysis. For this
thesis, the final sampling distance between adjacent points on the cortex (or two vertices) was
0.71 mm.
4.4.3.2. Voxel-Based Morphometry
A VBM analysis was used to measure the gray matter volume of subcortical structures.
Preprocessing of T1-weighted images and statistical analyses were performed in the SPM v5
software package (http://www.fil.ion.ucl.ac.uk/spm/software/spm5/) running under Matlab (The
Mathworks, Inc, Natick, MA). We used the VBM 5.1 toolbox (http://dbm.neuro.uni-
jena.de/vbm) implemented in SPM 5 to perform VBM. The methods are described in [36]. The
pipeline included the following steps: DICOM file format images were converted to the NIfTI-2
file format using the DICOM import tool in SPM. Next, images were normalized to a standard
template (International Consortium for Brain Mapping (ICBM) template), using a 12-parameter
affine transformation. Next, the images were intensity normalized, and segmented based on
tissue types using a Markov random field model and prior probability maps. The segmented gray
matter posterior probability maps, underwent Jacobian modulation. The images were then spatial
smoothed with a 10 mm FWHM Gaussian kernel. An absolute threshold mask of 0.10 (i.e., the
posterior probability of being gray matter must exceed 0.1) was set to restrict the results to gray
matter. All coordinates were converted from Montreal Neurological Institute Stereotaxic Space
(MNI; also the ICBM Standard Template) to Talairach space [844] using Yale BioImage Suite
non-linear MNI to Talairach Coordinate Converter [510]
(http://www.bioimagesuite.org/Mni2Tal/index.html) for results in Chapter 5, and the Lancaster
transform [513] implemented in GingerALE v.2.0.4 (http://www.brainmap.org/ale) in Chapter 6.
4.4.4. White matter analysis
Diffusion-weighted images were converted from DICOM image file format to the NIfTI-1 file
format (for FSL) using the dcm2nii application, as part of Chris Rorden’s MRIcon software
package (http://www.mccauslandcenter.sc.edu/mricro/mricron/dcm2nii.html). The NIfTI-1
diffusion-weighted images were imported into FSL v.4.1.8 (http://www.fmrib.ox.ac.uk/fsl) for
quality control [816]. Pre-processing included eddy current and motion artifact correction using
the FSL diffusion toolbox (FDT) [459]. This was achieved by aligning each 3D volume (each

131
DWI direction forms a 3D image; the image with all of the directions is called a 4D image) to the
b0 (non-diffusion-weighted image) collected at the beginning of each scan. In this thesis, two b0
images were acquired for each run, and the 4D dataset was aligned to the second b0 image. Next,
each subject’s two DWI runs were averaged to a single volume to achieve a greater signal-to-
noise ratio. Then, individual brain masks were created using the Brain Extraction Tool (BET)
[814] to cover the entire brain and exclude the skull, the eyes and other non-brain structures. The
parameters were adjusted for each individual subject to obtain maximal brain coverage, while
excluding as much of the non-brain tissue. These images were then processed through two
different pipelines for (1) voxelwise analysis using TBSS and (2) probabilistic tractography.
4.4.4.1. Tract-based spatial statistics
TBSS v.1.2 [815,817] was used to compare FA between the TMD and healthy control cohorts.
FA maps were created using DTIFIT tool in FDT. For TBSS, FA maps underwent non-linear
registration to a 1 x 1 x 1 mm FA map in standard space (FMRIB58_FA, available in FSL), a
mean image derived from all the subjects was created and thinned to represent the centre of
major white matter tracts common to all subjects, forming a white matter skeleton. Each
subject’s peak FA value perpendicular to the skeleton was then projected onto the skeleton. One
skeleton was created for analysis of group differences that included all study participants, and a
second skeleton based on the patients’ FA maps was created for correlation analyses of TMD
pain characteristics (see below). The mean skeleton images were set at a threshold of FA > 0.2 to
include FA values that are related to white matter [815]. Other DTI metrics were also evaluated
to characterize FA findings (see Figure 7-1). Additionally, a separate patient group analysis was
performed to assess which areas of the skeleton correlate with TMD characteristics (pain
intensity, unpleasantness, duration).
4.4.4.2. TBSS with other DTI metrics
To gain more insight into FA findings, other DTI metrics were assessed in the clusters with
significant group difference. λ1 is thought to reflect diffusion along a tract, and reductions in this
value suggest disruptions along the tract diffusivity. RD is believed to reflect changes in neural
membrane permeability and, to some extent, myelination [66,67]. RD is calculated by averaging
the two radial vectors of the tensor model (λ2 and λ3). Finally, MD was measured because it is
associated to œdema and inflammation. Figure 7-1 provides a schematic of these DTI metrics.

132
DTI metrics (FA, MD, λ1, λ 2 and λ3) are calculated using DTIFIT in the FDT toolbox. The TBSS
skeleton was created based on FA, and peak FA values are projected onto the skeleton. For
statistical analysis of the other DTI metrics, MD, RD and λ1 values from the same voxels are
projected onto the skeleton. To test for group differences, we performed a multivariate analysis
of variance (MANOVA) with each of the DTI metrics as dependent variables, and group as an
independent variable.
4.4.4.3. Probabilistic tractography
We sought to better characterize the corpus callosum cluster identified using cluster-mass
correction (see section 7.3.3) by elucidating its connectivity. Therefore, we performed multi-fibre
probabilistic tractography (http://www.fmrib.ox.ac.uk/fsl/fdt/index.html) [73,74] to determine
pathways passing through this cluster, and to elucidate its connectivity. To do so, the
preprocessed diffusion-weighted images were downsampled in order to create isotropic voxels (3
x 3 x 3 mm). The images were then processed in FDT. Probability density functions (PDF) on up
to 2 principal fibre directions were estimated at each voxel in the brain. Multi-fibre tractography
was used and 5000 samples were drawn from each seed voxel along the PDFs. Significant
clusters from the TBSS group difference analysis were binarised and used as seeds for
tractography. Together, each seed voxel’s streamlines provide an estimate of its connectivity.
When a streamline reaches a voxel with more than one fibre direction, the streamline follows the
direction closest to the direction at which it arrives at the voxel. The pathways generated by the
algorithm represent the number of samples that have passed through a voxel. The pathways
generated in each subject were thresholded at 10 samples (of the 5000 generated from each seed
voxel; i.e., 0.2%) to eliminate spurious connections. These thresholds provided consistent tracts
between subjects. For visualization, each of the subject’s tracts were binarised and overlaid on a
standard brain to produce a probabilistic map of the pathways for controls and patients. The
values in these maps represent the number of subjects who share a common pathway.

133
Chapter 5 STUDY I: Contributions of chronic pain and neuroticism to
abnormal forebrain gray matter in TMD
This study was published in Neuroimage:
Moayedi M, Weissman-Fogel I, Crawley AP, Goldberg MB, Freeman BV, Tenenbaum HC,
Davis KD. Contribution of chronic pain and neuroticism to abnormal forebrain gray matter in
patients with temporomandibular disorder. Neuroimage 2011; 55(1): 277-86
5.1. Introduction
TMD represent the most common orofacial chronic pain disorder, and are more prevalent in
women than in men [721]. TMD can be idiopathic in that there may not be any clear peripheral
etiological factors identifiable [283,286,287,653]. Thus, central mechanisms have been proposed
to account for TMD pain [757].
Although a clear pattern of change has yet to be determined, previous structural MRI studies of
chronic pain populations have found both increases and decreases in gray matter. For instance,
some studies of headache and chronic facial pain populations have found that patients with
chronic pain had gray matter increases in regions likely associated with pain perception
[207,575,650,963]. Additionally, most studies of chronic pain patients have found reduced gray
matter in cortical regions likely associated with antinociception and limbic function
[98,356,575]. Interestingly, some studies have also reported gray matter loss in cortical and
subcortical motor areas [22,575,763]. However, the increases are not limited to regions thought
by some to be associated with pain perception, and the decreases are not limited to pain-
modulatory regions. Although the role of motor regions in pain is not fully established, there is
evidence suggesting these areas play a role in pain modulation [3,117,189,342,345,547]. In
support of this concept are the motor abnormalities that can accompany chronic pain
[471,492,839,928], possibly related to nocifensive behaviour [634].
There are two main routes by which the central mechanisms may contribute to the development
and/or maintenance of chronic pains such as TMD. One possibility is that long-term nociceptive
input into the brain induces maladaptive brain plasticity, which may play a role in maintaining
pain [12,950]. For example, a recent study demonstrated that experimental pain that increased

134
gray matter in nociceptive regions [852], induced pain habituation over time that was
accompanied by decreased activity within nociceptive areas and increased activity within the
antinociceptive system [94]. Chronic pain patients, however, may not be able to adapt in this way
to nociceptive activity. For example, neuroimaging studies of chronic pain have shown
hyperactivity in nociceptive regions, and hypoactivity in antinociceptive regions [23,541].
Chronic pain patients’ inability to habituate to increased nociceptive activity may be related to a
reduced capacity of the brain to dampen pain by descending (top-down) controls [95]. Indeed,
many structural MRI studies have found gray matter differences in chronic pain populations
associated with pain-related characteristics (intensity, unpleasantness or duration)
[27,98,575,741,963].
The second route by which the CNS may contribute to the development and/or maintenance of
chronic pain relates to inherent personality-related factors that reduce the brain’s capacity to
modulate nociceptive input. This poor pain control represents a vulnerability to develop chronic
pain. For example, there is evidence that neuroticism may be associated with pain-related
suffering [408], pain sensitivity [177,373,907], nerve injury outcomes and neuropathic pain
[848] and inhibition of negative thoughts [178]. However, not all chronic pain patients have high
neuroticism scores, and not all persons with neuroticism have chronic pain [179]. Therefore,
neuroticism alone is not sufficient to develop chronic pain. Rather, the normal relationship
between neuroticism and brain structure and function may be disrupted within regions involved
in pain modulation, such as the OFC [954] or the mPFC [245,386] and this could facilitate or
maintain chronic pain.
Thus, in the current study we examined gray matter abnormalities in patients with idiopathic
TMD and focused our investigation on the contribution of pain-related characteristics and
neuroticism. Towards this goal, we measured gray matter in patients who had suffered from
TMD over a range of pain intensities, unpleasantness and for varying durations, and neuroticism
scores. Based on the aforementioned behavioural and neuroimaging studies, we specifically
tested the hypotheses that patients with TMD will have: 1) increased gray matter in areas
associated with pain perception; 2) reduced gray matter in areas associated with pain modulation
and motor function; 3) gray matter positively correlates with pain intensity, unpleasantness and
TMD duration within areas associated with pain perception areas and negatively correlates with
gray matter in areas associated with antinociception; 4) negative correlation between neuroticism

135
and gray matter in regions implicated in pain modulation, and positive correlation in regions
implicated in the affective dimension of pain, because of the interaction between affective
processing and pain modulation in TMD [881].
5.2. Methods
A detailed description of methods is provided in Chapter 4.
5.2.1. Subjects
A group of 17 females with idiopathic TMD (mean age ± SD: 33.1 ± 11.9 years) and 17 healthy
females (mean age ± SD: 32.2 ± 10.1 years) provided informed written consent to procedures
approved by the University Health Network and Mount Sinai Hospital Research Ethics Boards.
All subjects were right-handed. Patients with TMD were screened with the Mount Sinai Hospital
Dental Clinic TMD diagnostic criteria, based on the TMD-RDC [285]. Inclusion criteria
included: 1) TMD pain masticatory muscle region greater or equal to 4/10 for at least 3 months
or pain that is aggravated by mandibular function; 2) moderate pain to palpation and/or persisting
pain after examination in at least 3 muscle sites and/or moderate pain to palpation of the TMJ
region and/or limitation in the mandibular movement (opening less than 40mm). Patients were
asked to remain analgesic-free for 24 hours prior to scanning, as functional data was also being
collected during the scanning session. For both patients and control subjects, exclusion criteria
included: 1) serious metabolic, rheumatoid or vascular disorders and other diseases; 2) other
craniofacial pain disorders, previously diagnosed psychiatric disorders (e.g., depression,
schizophrenia, ADHD) or self-reported history of an abnormal neurological examination; 3) any
contraindication to MRI scanning (e.g., claustrophobia, metal); 4) use of psychotropic drugs. In
addition, healthy controls were not eligible for the study if they had a history of chronic pain.
5.2.2. Questionnaires
Each participant completed the NEO-FFI [178]. The NEO-FFI is a self-report questionnaire
comprising of 60 statements. Participants were asked to indicate the degree to which they agree
with a statement on a five-point scale (strongly disagree, disagree, neutral, agree, strongly agree),
each of which is coded to a number (0-4). A total of 15 of 60 questions probe for aspects of
neuroticism in this questionnaire.

136
Patients were also asked to rate their current pain and unpleasantness and their average pain
intensity and unpleasantness over the last month before scanning with the following instructions:
“Please rate you current/average pain/unpleasantness rating over the last month on a scale of 0 to
10 (0 = no pain, 10 = worst pain imaginable)”. The duration of the patients’ TMD was also
recorded for each patient.
5.2.3. Imaging parameters
Brain imaging data were acquired using a 3T GE MRI system fitted with an eight-channel
phased array head coil. Subjects were placed supine on the MRI table and each subject’s head
was padded to reduce movement. A whole brain three dimensional (3D) high-resolution
anatomical scan was acquired with a T1-weighted 3D IR-FSPGR sequence: 128 axial slices, 0.94
x 0.94 x 1.5 mm3 voxels, 256 x 256 matrix size, field of view = 24 x 24 cm, one signal average,
flip angle = 45°, TE = 5ms, TR = 12ms, TI = 300ms.
5.2.4. Structural brain imaging analysis
Because we were interested in both cortical and subcortical structures, we used two analysis
approaches to measure gray matter from high-resolution MRI images. To evaluate differences in
cortical thickness (measured in mm) we used CTA [316,538,559], and VBM [36] was used to
measure subcortical gray matter density. We used masks of specific brain regions (i.e., ROI
analysis) to test our specific a priori hypotheses; this focus also reduced multiple comparisons to
regions of no interest (see below).
5.2.4.1. Cortical thickness analysis
CTA was carried out using the Freesurfer software (http://surfer.nmr.mgh.harvard.edu); methods
are described in full detail elsewhere (see:[198,316,317,319,320]. Briefly, pre-processing
included intensity normalization, skull stripping, separation of the hemispheres, and gray matter
segmentation. The white matter/gray matter border (i.e., white surface) and gray matter/CSF
border (i.e., pial surface) were identified and modeled as surfaces. The software then calculated
the distance between the two borders at every point on the cortex, for each hemisphere. The gray
matter surface was then warped such that homologous gyri and sulci were aligned across all
subjects. Each individual subject’s cortex underwent automatic anatomical parcellation, and each
sulcus and gyrus was labeled during pre-processing [321]. To restrict our search to regions

137
hypothesized to be involved in TMD chronic pain, we used the cortical parcellations map
implemented into FreeSurfer (aparcs2005) on a standard subject (fsaverage). Two masks were
created: (1) a sensorimotor/pain mask which included S1, S2, M1, and MCC (see Figure 1 a);
and (2) a cognitive/modulatory mask of the frontal lobe that included the OFC, PFC, insula and
the ACC/MCC (see Figure 1 b). The MCC has been implicated in both nociception and pain
modulation [23] and so this region was included in both masks. To compensate for topographical
heterogeneity amongst the different subjects, a 6 mm FWHM Gaussian spatial smoothing kernel
was applied to the data prior to statistical analysis. A vertex represents a point on a two-
dimensional sheet, and, in this study, the distance between two vertices is 0.71 mm.
5.2.4.2. Subcortical analysis with VBM
Image processing and statistical analyses were performed in the SPM5 software package
(http://www.fil.ion.ucl.ac.uk/spm/software/spm5/) running under Matlab (Mathworks). The
VBM analysis used Gaser’s VBM 5.1 toolbox (http://dbm.neuro.uni-jena.de/vbm) within SPM 5;
detailed methodology is described in elsewhere [36]. Briefly, the preprocessing included setting
the origin of the image at the anterior commissure of each subject, spatial normalization to the
ICBM-152 template, gray matter segmentation, Jacobian modulation to adjust for the effects of
spatial normalization, and spatial smoothing with a 10 mm FWHM Gaussian kernel. Total
intracranial volume (TIV) was calculated for each subject from the volume estimates output
during segmentation. An absolute threshold mask of 0.10 was applied to restrict the results to
gray matter only. We constructed a single mask for our subcortical VBM analysis (See figure 1
c). We used prefabricated anatomical masks in WFU Pickatlas
(http://www.nitrc.org/projects/wfu_pickatlas). The subcortical mask was used to investigate
regions that cannot be investigated using CTA, and comprised the basal ganglia, amygdala,
brainstem and thalamus.
5.2.5. Statistical analyses
5.2.5.1. Demographics
Group differences between characteristics of the patients and controls were evaluated using a
multivariate analysis of variance in SPSS 18.0 (http://www.spss.com/). Age and neuroticism
scores were compared between the groups. The significance threshold was set at p < 0.05.

138
5.2.5.2. Gray matter group differences
CTA
To restrict our search to regions hypothesized to be involved in TMD chronic pain, we used the
cortical parcellations map implemented into FreeSurfer (aparcs2005) on a standard subject
(fsaverage). Two masks were created: (1) a sensorimotor/pain mask which included S1, S2, M1,
and the MCC (see Figure 5-1 a); and (2) a cognitive/modulatory mask of the frontal lobe that
included the OFC, PFC, insula and the ACC and MCC (see Figure 5-1 b). The MCC has been
implicated in both nociception and pain modulation [23] and so this region was included in both
masks.
To test our first hypothesis that patients have thicker cortex in perception areas, we ran a general
linear model (GLM) testing for group differences in cortical thickness at every vertex within the
sensorimotor/pain mask (see Figure 5-1a). Age was included as a regressor of no interest. To test
our second hypothesis that patients have thinner cortex in modulatory/cognitive areas, we ran a
second GLM testing for group differences at every vertex within the cognitive/modulatory mask
(see Figure 5-1b). Percent change was calculated using the following formula: ((Mean of
patients’ thickness - mean of controls’ thickness)/mean of controls’ thickness) × 100.
All data were thresholded at an experiment-wide Bonferroni corrected p < 0.05. To do so, we
calculated the corresponding cluster-wide p-value to reach significance and so the experiment-
wide correction was based on conducting 6 tests: (1) thicker cortex in pain perception regions,
(2) thinner cortex in cognitive/modulatory regions, (3) the contribution of pain intensity, (4) the
contribution of pain unpleasantness, (5) the contribution of TMD duration to cortical
abnormalities, and (6) group by neuroticism interactions in cortex. Thus the correction for the
threshold was 0.05/6 = 0.008. Therefore, to achieve a Bonferroni-corrected threshold of p < 0.05,
CTA data were thresholded at image-wide threshold of p < 0.008. Our two masks (see Figure 5-
1) were of different sizes; thus, we used Monte Carlo permutation to calculate an image-wide
threshold for each. For the sensorimotor/pain mask, this was derived from an uncorrected
voxelwise p < 0.01 and 190 contiguous vertices (379 voxels), and for the cognitive/modulatory
mask, this was derived from an uncorrected voxelwise p < 0.01 and 220 contiguous vertices (436
voxels). Monte Carlo simulations were run with 1000 iterations using AlphaSim
(http://afni.nimh.nih.gov/afni/doc/manual/AlphaSim), as we have used previously [847].

139
VBM
We constructed a single mask for our subcortical VBM analysis (See figure 5-1c). We used
prefabricated anatomical masks in WFU Pickatlas (http://www.nitrc.org/projects/wfu_pickatlas).
The subcortical mask was used to investigate regions that cannot be investigated using CTA, and
comprised the basal ganglia, amygdala, brainstem and thalamus. To test both our first and second
hypotheses, we used student’s t-test to test for group differences in each voxel within the
subcortical mask (see Figure 5-1c). The general linear model included age and TIV as covariates
of no interest. All data were thresholded at a voxelwise corrected p < 0.05 using false discovery
rate (FDR), as implemented in SPM 5.
5.2.5.3. Clinical correlates
To assess pain-related plasticity, we correlated pain intensity, pain unpleasantness and TMD
duration to gray matter. For all the correlations, age was included as a regressor of no interest.
However, for the correlation of gray matter to TMD duration we could not include age as a
covariate, as these two explanatory variables (age and TMD duration) are correlated (r = 0.536, p
= 0.027). For the CTA analysis, pain intensity, pain unpleasantness and TMD duration
correlations were performed within the sensorimotor/pain mask and the cognitive/modulatory
mask. For the VBM analysis, we ran correlations in each voxel within our subcortical mask, as
previously described. TIV was included as a covariate of no interest in every model.
5.2.5.4. Neuroticism effects
To determine whether neuroticism may be affected by chronic pain in TMD, we performed a
Pearson correlation between TMD duration and neuroticism scores in the patient group.
To investigate whether neuroticism has an abnormal relationship to gray matter, and thus
contribute to TMD onset, we performed an interaction analysis. Specifically, in CTA, we
performed a group by neuroticism interaction using the FreeSurfer software on a vertex-wise
basis within the cognitive/modulatory mask. In the VBM analysis, to test this hypothesis, we
performed a group by neuroticism interaction in each voxel within our subcortical mask.

140
5.3. Results
5.3.1. Patient demographics
The characteristics of individual patients are shown in Table 5-1. Patients and controls did not
differ in age (patients: mean age ± SD: 33.1 ± 11.9 years; controls: 32.2 ± 10.1 years; p = 0.94)
or in neuroticism scores (patients: mean age ± SD: 19.35 ± 6.84; controls: 18.35 ± 7.95; p =
0.70). Of importance for this study is that there was variability in the durations of TMD, intensity
and unpleasantness of pain that facilitated assessment of these factors in correlation analyses.
The duration of TMD ranged from 0.75 – 30 years (mean ± SD: 9.8 ± 8.2 years), while pain
intensity ranged from 2 – 7 (mean ± SD: 4.3 ± 1.8), and unpleasantness ranged from 1 – 8 (mean
± SD: 5.4 ± 2.1). Interestingly, TMD pain intensity and pain unpleasantness scores were not
significantly correlated (r = 0.40, p = 0.12), unlike the tight correlation of these dimensions in
healthy subjects in acute pain paradigms [717]. Patients reported that their TMD pain was
confined to the TMJ (n = 6), the muscles of mastication (n = 3), or both the joint and muscles (n
= 8). Most patients reported bilateral pain (n = 13), but some reported unilateral pain (three right-
sided, one left-sided). The duration of TMD significantly correlated with the patients’ age (r =
0.536, p = 0.027).
Patient neuroticism scores showed no significant relationship to TMD duration (r = 0.33, p =
0.21).
5.3.2. Group differences: S1 and frontal thickening in TMD
There were prominent group differences (p < 0.05, corrected) in gray matter thickness in the
patients with TMD compared to controls (see Table 5-2, Figure 5-2). In line with our first
hypothesis, the ventrolateral S1 was 22% thicker in the TMD group compared to controls. This
region of thickening was located in a region consistent with the somatosensory representation of
the face [205,630]. However, contrary to our second hypothesis, we did not identify statistically
significant cortical thinning in any pain modulation or motor region. Unexpectedly though, we
identified regions in the left frontal pole and vlPFC that were thicker in the patient group
compared to controls by 17% and 15% respectively (see Table 5-2 and Figure 5-2). The VBM
analysis did not reveal any significant subcortical differences between patients and controls.

141
5.3.3. Effect of chronic pain intensity and unpleasantness
We also examined the relation between pain intensity and pain unpleasantness in gray matter
within our cortical masks. Cortical thinning that correlated with the degree of TMD pain
intensity or unpleasantness was found within three brain regions (see Figure 5-3 and Table 5-3).
A significant negative correlation was found between pain intensity and gray matter thickness in
the aMCC (BA32; r = -0.83), and in the ventrolateral aspect of M1 in a region consistent with the
representation of the face [952] (r = -0.83). Our test of the impact of pain unpleasantness on
cortical thickness within our two masks revealed a significant effect only in left OFC. This
region showed a negative correlation between gray matter thickness and unpleasantness (BA 47;
r = -0.74). Interestingly, in these three brain areas, the patients reporting intense pain had gray
matter that was thinner than the healthy controls, but the individuals with only mild pain had
gray matter either slightly thicker than the controls or within the normal range. Of note is that we
did not identify any significant gray matter subcortical correlations with pain intensity or
unpleasantness.
5.3.4. Effect of TMD chronicity
The subcortical VBM analysis revealed that gray matter in the sensory thalamus was positively
correlated to the length of time that patients had been suffering with TMD (r = 0.91; see Table 5-
4 and Figure 5-4 for details). This region of gray matter increase included the left PO, the left
VL, and bilateral VPM of the thalamus. Interestingly, the patients who had TMD for only a few
years showed gray matter within the normal range (see Figure 5-4), but patients who had TMD
for 7 or more years showed a stronger relationship between duration and thalamic gray matter.
We did not find any significant correlations between TMD duration and gray matter thickness in
the a priori hypothesized cortical ROI.
5.3.5. Effect of neuroticism
Because of the expectation that neuroticism manifests differently in patients than healthy
controls, we tested whether there was neuroticism by group interaction in the
cognitive/modulation mask. The controls showed a negative correlation (r = -0.32) between gray
matter thickness in the left OFC (BA11; Talairach coordinates: -18, 22, -18) and neuroticism
scores, whereas patients show a positive relationship (r = 0.61) in this region. The area of the
interaction in this ROI was 517 mm2 (peak vertex t-score = -2.87; p < 0.05, experiment-wide

142
Bonferroni corrected; see Figure 5-5). We did not identify any significant neuroticism by group
interactions in the subcortical VBM analysis.

143
Table 5-1: Patient demographics.
# Age
TMD
Dur
(yrs)
Pain
Intensity
Pain
Unpl
Pain
Sites
TMD
Laterality
Other
pains Meds
Oral
contraceptive
1 22 2 4 5 J Bilateral Knee pain NSAID Yes
2 20 3 3 3 M, J Bilateral A*, cyc* No
3 24 7 4 8 J Right Headache N, P Yes
4 38 20 7 6 J Bilateral NSAID No
5 42 0.75 2 3 M Bilateral F* No
6 33 4 5 6 M, J Bilateral Shoulder,
Neck
A*, F* No
7 28 17 3 5 M, J Left No
8 34 14 2
8
J Bilateral
A*, F*,
Hy* No
9 50 10 7 6 M Bilateral A, Ch, Di No
10 59 13 7 7 M, J Bilateral Whiplash
(neck and
shoulder),
Gastric
Distress
No
11 18 3 6 8 M, J Bilateral Yes
12 34 15 2 3 M Right Neck,
Sciatica
No
13 52 30 4 5 M, J Bilateral A No
14 31 2 6 5 M, J Right Ovarian
cyst
N No
15 33 17 5 8 M, J Bilateral A, F,
NSAID
No
16 22 1 4 1 J Bilateral Yes
17 23 8 2 5 J Bilateral Yes
Abbreviations: M= masticatory muscles; J=TMJ; NSAID: Non-steroidal anti-inflammatory, over
the counter, as needed; A: Arthrotec; cyc: cyclobenzapine; Ch: Champix; Di: Dixarit
(Clonidine); F: Flexoril; Hy: Hydromorphone; Meds: Medications; N: Naproxen; P: Prevacid;
TMD Dur: TMD duration; Unpl: unpleasantness; yrs: years. The asterisk (*) denotes that
subjects discontinued the use of the drug prior to our study. A and N are also NSAIDs, but were

144
patients were prescribed to take these medications daily- rather than when needed. Gastric
distress was described as “burning in the stomach” by the patient.

145
Table 5-2: Group differences in cortical thickness.
Peak vertex Talairach coordinates are reported. All results are significant at p < 0.05, corrected
for multiple comparisons. Abbreviations: R: right; L: left; BA: Brodmann’s Area; FP: frontal
polar cortex; S1: primary somatosensory cortex; vlPFC: ventrolateral prefrontal cortex.
Contrast Region BA
TAL of Peak Area
(mm2)
Peak
T-score X Y Z
Patients > Controls R S1 2 50 -18 37 1132 5.18
L FP 10 -30 45 6 569 4.87
L vlPFC 9/10 -38 35 4 531 3.41

146
Table 5-3: Cortical thickness negatively correlates with TMD pain intensity or unpleasantness.
Peak vertex Talairach coordinates (TAL) are reported. All results are significant at p < 0.05,
corrected for multiple comparisons. Abbreviations: L: left; aMCC: anterior mid-cingulate cortex;
M1: primary motor cortex; OFC: orbitofrontal cortex.
TMD attribute Region BA
TAL of Peak Area
(mm2)
Peak
T-score X Y Z
Pain Intensity L aMCC 32 -4 18 19 718 - 3.94
L M1 4 -57 -3 13 674 - 4.15
Unpleasantness L OFC 11/47 -27 16 -19 472 - 4.22

147
L R
R L
L R
R L
b)
a)
R L c)

148
Figure 5-1: Masks used to restrict analyses. Three masks were used to restrict the analyses to
regions of interest: a) the sensorimotor/pain mask included cortical sensorimotor regions
(primary and secondary somatosensory cortices (S1, S2), the primary motor cortex (M1), and the
mid-cingulate cortex (MCC), and b) the cognitive/modulatory mask which included the insula,
the prefrontal cortex (PFC), the orbitofrontal cortex (OFC), and the cingulate cortex
(ACC/MCC), c) a subcortical mask used for voxel-based morphometry, included the basal
ganglia, thalamus, amygdala and the brainstem. CTA results are overlaid onto the fsaverage
standard brain in FreeSurfer.

149
Figure 5-2: Cortical thickening in TMD. The CTA GLM group analyses within the cortical
masks identified three cortical regions (shown in blue on the brain images) that showed
significant group differences at a corrected p < 0.05. The top panel shows that the TMD patient
group had thicker right primary somatosensory cortex (S1) (peak T-score = 5.18) compared to
controls, with the effect of age regressed out. The bottom panel shows that the TMD patient
group has thicker ventrolateral (peak T-score = 3.41) and frontal polar cortices (peak T-score =
4.87). All labeled regions are significant at a corrected p < 0.05. The graphs indicate the mean
cortical thickness ± SE (age not regressed out). CTA results are overlaid onto the fsaverage
standard brain in FreeSurfer.
R
L
vlPFC
FP
FP
vlPFC
S1 S1

150
Figure 5-3: TMD pain intensity and unpleasantness are negatively correlated to modulatory
regions. The CTA regressions within the cortical masks in the TMD group identified three
cortical regions (shown in blue on the brain images) that were significantly correlated with
individual pain scores at a corrected p < 0.05. The top panel shows that pain intensity has a
negative correlation to anterior mid-cingulate cortex (aMCC) (r = - 0.83, peak T-score = -3.94).
L
L
M1
aMCC
L
OFC

151
The middle panel shows that pain intensity is negatively correlated to cortical thickness in the
left primary motor cortex (M1) (r = -0.83, peak T-score = -4.15). The bottom panel shows that
pain unpleasantness negatively correlated to cortical thickness in the left orbitofrontal cortex
(OFC; r = -0.75, peak T-score = -4.22), with the effect of age regressed out). All labeled regions
are significant at a corrected p < 0.05. The graphs indicate the mean cortical thickness ± SE for
each subject versus pain intensity (age not regressed out). CTA results are overlaid onto the
fsaverage standard brain in FreeSurfer.

152
Figure 5-4: TMD duration is related to plasticity in the thalamus. The TMD patient group
showed a positive correlation between TMD duration (measured in years) and gray matter in a
cluster in the sensory nuclei in the left thalamus (posterior (PO), ventral lateral (VL), ventral
posterior medial (VPM) (2312 voxels, peak T-score = 7.84, r = 0.91). The finding is significant
at a false-discovery rate corrected p < 0.013. The graph indicates each TMD patient’s gray matter
volume versus her individual TMD duration (in years). The dashed lines represent the mean gray
matter volume of controls ± SE. VBM results are displayed on the single subject standard brain
in SPM.
L

153
Figure 5-5: Patients with TMD showed a positive relationship between OFC thickness and
neuroticism, which is considered to be abnormal. Neuroticism was correlated with changes in the
left orbitofrontal cortex (OFC; peak T-score = 3.97), with the effect of age regressed out. For all
correlations, mean cortical thickness ± SE (age not regressed out) versus the raw neuroticism
score was graphed. CTA results were overlaid onto the fsaverage standard brain in FreeSurfer.
L
OFC

154
5.4. Discussion
This structural imaging study identified striking abnormalities in patients with chronic idiopathic
TMD pain and highlights the contribution of both TMD-related and neuroticism-related factors.
Our key findings were that, compared to controls, patients with TMD had 1) cortical thickening
of the S1, frontal pole and vlPFC, 2) pain intensity-dependent cortical thinning in the aMCC and
M1 and pain unpleasantness-dependent cortical thinning in the OFC, 3) TMD duration-
dependent gray matter increase in the sensory thalamus, and 4) a positive correlation between
cortical thickness in the OFC and neuroticism, in contrast to a normally negative correlation.
5.4.1. S1 and thalamic gray matter increases in TMD
Our finding of S1 thickening in TMD is consistent with other gray matter studies of chronic pain
in the trigeminal system [207]. However, trigeminal chronic pains are heterogeneous, and so may
produce different patterns of gray matter changes specific to the particular symptomology or
pathology. Nonetheless, the convergence of increased S1 thickness suggests that there may be
prolonged or repeated barrage of nociceptive input to the cortex from the thalamus. Evidence for
this hypothesis comes from a study by Teutsch and colleagues [852] that showed persistent
noxious stimulation in healthy subjects can induce increased gray matter in S1. Since we also
found that in patients with TMD there was a positive correlation between thalamic gray matter
and TMD duration, it is possible that sustained trigeminothalamic nociceptive activity over time
leads to increased gray matter in sensory thalamus. The patients included in our study have a
larger range of TMD durations (0.75-30 years) compared to the study by Younger et al. [962] (0-
11 years). Thalamic changes were more apparent in patients with longer reports of TMD
duration, which may explain why our findings are different than those reported by Younger and
colleagues [963]. Increased firing to the thalamus may lead to increased thalamocortical activity
to S1 and plasticity [950]. The cellular basis of cortical thickening (plasticity) is not yet
established but is thought to include increases in synaptic boutons, dendritic branching, glial
cells, and/or neurones [575,608].
5.4.2. Cortical thickening in cognitive and modulatory regions in TMD
We found that patients with TMD had cortical thickening in the frontal pole. These findings
diverge from those of Younger et al. [963] study. A factor that may have contributed to the

155
different findings is that their study examined patients with myofascial TMD, whereas our TMD
group consisted of a somewhat more clinically representative group of mixed muscular and/or
joint TMD.
Activation of the frontal pole has been reported during spontaneous pain in patients with chronic
pain [47]. The frontal pole has also been implicated in a number of complex executive cognitive
functions such as learning behavioural routines [457,496,834], cognitive branching (the ability to
put a pending task on hold to execute an ongoing one) [497], behavioural flexibility/adaptability
[105] and post-hoc monitoring or evaluating decisions based on feedback [875]. Therefore, the
frontal pole may process the cognitive dimension of pain, which suggests that pain has a
cognitive load. We propose that the cognitive load of pain may require constant engagement of
the frontal polar cortex for cognitive branching. That is, chronic pain may need to be put “on
hold” in order for a patient to engage in another competing cognitive load. However, if the
cognitive branching system is limited, a chronic pain load may impact the ability to properly
complete tasks [529]. In support of this, we have recently shown that patients with TMD have
sluggish reaction times to low and high conflict cognitive interference tasks, but not to a simple
sensorimotor task [928]. Further evidence for cognitive branching of pain comes from studies
showing that an increase in cognitive load modulates acute pain perception in healthy controls
[935]. Therefore, one possibility for the thickening of the frontal polar cortex is that chronic
TMD pain bears a cognitive load that the brain needs to ‘put on hold’ in order to address more
immediate environmental demands.
The other frontal area we found to be thicker in TMD than controls was the vlPFC. This region
has been implicated in pain anticipation [752], pain modulation [909], and behavioural inhibition
[32]. There is evidence that patients with TMD are hypervigilant [435,808], which is related to
increased pain anticipation. Therefore, vlPFC thickening may be related to patients’
hypervigilance and increased anticipation to pain.
5.4.3. Relationship between chronic pain intensity and cortical thickness in M1 and aMCC
We found that gray matter in the orofacial region of M1 showed a negative correlation to pain
intensity. Several studies have reported abnormal stimulus-evoked fMRI activity in motor
regions of chronic pain patients, although the implication of these abnormalities is not often

156
discussed (see: [23]). One interpretation of our finding is that there is an adaptive mechanism
that responds to sustained, intense nociceptive activity by dampening motor output to prevent
further damage to the affected region. Evidence for this concept comes from studies of both
acute and chronic orofacial pains . For instance, TMD pain inhibits craniofacial motor function
[839], and dampens motor neurone output [556]. Similarly, acute pain stimuli applied to the
orofacial region can decrease M1 excitability [3,109]. Interestingly, Kirveskari et al. [492]
reported that patients with CRPS had weaker M1 reactivity with increased spontaneous pain
ratings, that is to say that there is a negative correlation between pain intensity and M1
activation. Another possibility is that patients with TMD make fewer jaw movements to avoid
eliciting pain. This reduced jaw activity may lead to atrophy of the orofacial region of the motor
cortex. Conversely, there is evidence to suggest that M1 may play a role in central processing of
pain [11,244,476,785] and pain modulation [189]. For instance, motor cortex stimulation has
been shown to have some, albeit limited, analgesic effect in chronic pain [342,689]. Therefore,
another interpretation of our finding is that M1 may be, in part, involved in descending
modulation of pain.
The other gray matter region that we identified that was negatively correlated to pain intensity is
the aMCC. This finding had also been identified in a previous study of gray matter in TMD
[963]. Experimental acute pain in healthy subjects is associated with activity in the MCC (caudal
BA 24) [690] and NS neurones have been identified in caudal BA 24 [440]. The top-down
descending antinociceptive system is thought to arise from the aMCC (possibly triggered by
activity in cingulate nociceptive neurones) [95]. The aMCC also shows increased rCBF during
analgesic thalamic stimulation [228]. Furthermore, this region shows opioid-related pain
modulation [342].
Therefore, both the M1 and the aMCC have been implicated in descending pain modulation
[689]. We found that patients with thicker cortex in both the aMCC and M1 report lower pain
intensity, whereas patients with thinner cortex in these regions report higher pain intensity.
Therefore, thicker cortex in these regions may provide a greater capacity to modulate TMD pain,
whereas thinner cortex may be related to an impairment of this descending modulation system.

157
5.4.4. Interaction between neuroticism and group in prefrontal gray matter
We investigated two factors that have not been previously examined in imaging studies of TMD:
neuroticism and TMD pain unpleasantness. Neuroticism is described as a personality trait
associated with heightened sensitivity and/or processing of negative affective stimuli
[178,179,907]. We found that in patients with TMD there is a positive correlation between
neuroticism and cortical thickness in the left ventromedial PFC (vmPFC; part of the OFC); an
area that normally shows a negative correlation between gray matter and neuroticism [954].
Neuroticism is considered to be a stable trait across the lifespan [178]. Therefore, our
observation highlights the contribution of neuroticism to abnormal gray matter in the OFC of
patients with TMD and, perhaps, to the development of TMD. An alternative interpretation is
that chronic pain may have modified the relationship between neuroticism and OFC thickness.
However, the lack of correlation of TMD duration and neuroticism scores in our patients with
TMD does not support this hypothesis.
We also found that unpleasantness is negatively correlated to gray matter in an adjacent region of
the OFC. The OFC has been implicated in cognitive reappraisal and emotional regulation [723]
related to interoception and somatoviscero stimuli, for directing our behaviour appropriately
[57,935]. The OFC is also thought to play a role in mental flexibility and adaptability (for a
critical review, see: [770]. Therefore, our finding suggests that patients with TMD may have
abnormal emotional regulation and reappraisal, which may lead to pain behaviours and
exaggerated affective responses to pain.
5.4.5. Study limitations
A few limitations in study design and results should be considered in interpretation of our data.
First, it is not known whether medications used by some of the patients have an effect on gray
matter. Second, some of our patients had co-morbid chronic pains (see Table 5-1), and these
pains (mostly related to TMD) may have contributed to our findings. Third, we cannot
disassociate an age effect from a pure TMD duration effect because of the strong correlation
between age and TMD duration (see Chapter 6). Furthermore, duration (and intensity) may not
fully describe the effect of pain on gray matter structure. Due to the nature of TMD pain, other
factors that we did not collect, such as the number of days of pain in a month may have more

158
explanatory variance. Our patients did not self-report any previous diagnosis of depression or
anxiety, although these exist in some patients with TMD [283,808,850]. Thus we cannot exclude
the contribution of these factors in white matter abnormalities in TMD in general. Finally, we
cannot totally rule out a contribution of ventricular volume to the VBM analysis of subcortical
structures. Despite these limitations, our study does provide considerable evidence in line with
previous findings in the literature about gray matter abnormalities in chronic pain. Further study
limitations are discussed in Section 8.6.
5.5. Conclusions
This study provides evidence for gray matter abnormalities in both the ascending pain and
descending antinociceptive systems as well as motor and cognitive areas in TMD. Further, we
have shown that the personality trait neuroticism and TMD intensity and duration can affect gray
matter, suggesting the presence of both personality-based and chronic pain-related abnormalities.
This structural imaging study identified striking abnormalities in patients with chronic idiopathic
TMD pain and highlights the contribution of both TMD-related and neuroticism-related factors.
Our key findings were that, compared to controls, patients with TMD had 1) cortical thickening
of the S1, frontal pole and vlPFC, 2) pain intensity-dependent cortical thinning in the aMCC and
M1 and pain unpleasantness-dependent cortical thinning in the OFC, 3) TMD duration-
dependent gray matter increase in the sensory thalamus, and 4) a positive correlation between
cortical thickness in the OFC and neuroticism, in contrast to a normally negative correlation.

159
Chapter 6 STUDY II: Age-related gray matter abnormalities in
temporomandibular disorder
This study has been published in Brain Research:
Moayedi M, Weissman-Fogel I, Salomons TV, Crawley AP, Goldberg MB, Freeman BV,
Tenenbaum HC, Davis KD. Abnormal gray matter aging in chronic pain patients. Brain
Research 2012; 1456: 82–93.
6.1. Introduction
There is no doubt that the brain undergoes structural changes as part of a normal aging process.
For example, normal aging is characterized by cortical gray matter atrophy
[84,99,372,585,627,824], although gains have also been reported in some brain areas [322,749].
Brain plasticity also occurs with dysfunction, injury, or specific disease. Chronic pain in
particular is associated with brain plasticity, including reduced gray matter in regions associated
with pain modulation and limbic function, such as the ACC, insula, dlPFC
[98,225,575,577,617,779,780,963], and gray matter increases in regions associated with pain
perception, such as S1 [209,577,617]. Additionally, some studies of chronic pain populations
have identified gray matter decreases in the basal ganglia [741,764-766,772,963] and pain-
related gray matter decreases in other motor regions, such as M1 (see Chapter 5). We have
previously reported that thalamic gray matter volume in patients with TMD is correlated to the
duration of their pain [617] (see Chapter 5). However, it is unclear whether these changes are
caused by the cumulative effect of TMD pain over time, or whether age contributes to the
observed abnormalities.
Chronic diseases such as pain may interact with normal aging processes. For example,
accelerated age-related whole brain gray matter atrophy has been reported in FMS [502] and
chronic back pain [27]. However, most aging studies in chronic pain have assessed global gray
matter and little is known about the interaction between chronic pain and age in gray matter
volume/thickness of specific brain areas.
Although the functional significance of gray matter changes is not understood, MRI and
histological studies indicate that changes can be stimulus-dependent, as has been demonstrated in

160
studies of learning [267], training [266], and repetitive noxious stimulation [852]. These data
suggest that the aforementioned findings of abnormal gray matter progression in chronic pain
may be related to pain duration. While it is plausible that age-related changes specific to chronic
pain are the product of cumulative pain exposure, this hypothesis has not been tested empirically.
Therefore, the aims of this study were to determine 1) whether chronic pain in TMD is associated
with abnormal gray matter aging in focal cortical regions associated with nociceptive processes,
and 2) the degree to which the cumulative effects of pain contributes to age effects.
Prolonged nociceptive activity may disrupt or even reverse normal gray matter atrophy in
nociceptive and motor regions, and increase rates of atrophy in pain-modulatory regions.
Therefore, we hypothesized that normal age-related gray matter changes would be enhanced in
brain regions implicated in pain perception (e.g., the thalamus, S1, posterior insula and MCC,
and suppressed in motor regions (e.g., the M1, PMC, SMA and the basal ganglia) and in regions
implicated in pain modulation (e.g., ACC and the anterior insula) of patients with chronic pain.
6.2. Methods
A detailed description of methods is provided in Chapter 4.
6.2.1. Subjects
Seventeen patients with non-traumatic TMD and 17 pain-free healthy subjects with no prior
history of chronic pain were recruited and provided informed written consent to procedures
approved by the local Research Ethics Boards. All subjects were right-handed. Structural MRI
data in this cohort unrelated to age have been presented [617] (See Chapter 5). Patients with
TMD were screened with the Mount Sinai Hospital Dental Clinic TMD diagnostic criteria, based
on the TMD-RDC [285]. Patient demographics and specific screening criteria have been
previously described elsewhere [617,928] (see Chapter 5 and Table 5-1). All patients reported
the number of years of TMD symptoms, herein described as TMD duration. Exclusion criteria
(for all study participants) included: a history of serious diseases (metabolic, rheumatoid,
vascular), concurrent craniofacial pain disorders, or any contraindication to MRI scanning (e.g.,
claustrophobia, metal).

161
6.2.2. Imaging & Analysis
Each study participant was placed in a 3T GE MRI (Signa HDx) system fitted with an eight-
channel phased array head coil, lying supine with foam padding around their head to reduce
movement. The 3D brain scan was acquired with a T1-weighted scan (IR-prep 3D-FSPGR with
128 axial slices, 0.94 x 0.94 x 1.5 mm3 voxels, 256 x 256 matrix size, field of view = 24 x 24
cm, one signal average, flip angle = 20°, TI = 300 ms, TE = 5 ms, TR = 12 ms).
6.2.3. Global age effects
To assess group differences in global age effects, we estimated whole brain gray matter volume
from tissue segmentation performed in the SPM5 software package
(http://www.fil.ion.ucl.ac.uk/spm/software/spm5/) running under Matlab v.7.0.4 (Mathworks).
We used a general linear model to test for age-by-group interactions for whole brain gray matter.
All statistical analyses were performed in SPSS v.19.0 (www.spss.com).
6.2.4. Age-by-group effects in gray matter
CTA
FreeSurfer software (http://surfer.nmr.mgh.harvard.edu) was used to assess cortical thickness.
Detailed methods are described elsewhere [198,316,317,319,320]. Briefly, T1-weighted scans
underwent intensity normalization, skull stripping and segmentation of each hemisphere based
on tissue type (gray matter, white matter, CSF). Within each hemisphere, the boundary between
each tissue type was modeled as a surface so that the distance between each surface could be
calculated at each point on the cortex. Each subject’s homologous gyri and sulci were aligned to
the standard average brain provided in FreeSurfer. A Gaussian spatial smoothing kernel of 6 mm
FWHM was applied to compensate for topographical heterogeneity prior to statistical analysis.
For this study, the distance between two points on the cortex (or two vertices) was 0.71 mm.
To restrict our search to regions hypothesized to be involved in TMD chronic pain, a single mask
using FreeSurfer’s cortical parcellations was constructed [321] that included the OFC, PFC,
insula, M1, SMA, cingulate cortex, postcentral gyrus and sulcus (including S1 and S2) and the
posterior parietal cortex [23]. Age-by-group interactions in cortical thickness were tested at each
vertex on the cortex within the cortical mask. To correct CTA results for multiple comparisons,
we used a Monte Carlo simulation with 1000 permutations with the AlphaSim software package

162
(http://afni.nimh.nih.gov/afni/doc/manual/AlphaSim), as done previously [98,847]. Thus, we
calculated a mask-wise corrected threshold of p < 0.05 from an uncorrected voxelwise p < 0.01
and a cluster threshold of 178 contiguous vertices (349 voxels).
VBM
VBM analysis was used to measure subcortical structures. One additional control subject was
recruited and consented to REB approved procedures to replace one subject subsequently
removed from the subcortical analysis because of poor segmentation in the pre-processing
pipeline.
Preprocessing of T1-weighted images and statistical analyses were performed in the SPM5
software package (http://www.fil.ion.ucl.ac.uk/spm/software/spm5/) running under Matlab
(Mathworks). We used the VBM 5.1 toolbox (http://dbm.neuro.uni-jena.de/vbm) implemented in
SPM 5 to perform VBM (for details see: [36]). Briefly, images were normalized to a standard
template (ICBM-152), tissue types were then segmented using a Markov random field model,
and underwent Jacobian modulation, followed by spatial smoothing of gray matter with a 10 mm
FWHM Gaussian kernel. We set an absolute threshold mask of 0.10 to restrict the results to gray
matter. All coordinates were converted from MNI space to Talairach space [844] using the
Lancaster transform [513] implemented in GingerALE v.2.0.4 (http://www.brainmap.org/ale).
A gray matter mask of subcortical regions was constructed using the WFU Pickatlas toolbox
(http://www.nitrc.org/projects/wfu_pickatlas). The mask was constructed using the “Sub-lobar”
label mask and the restricted by the “Gray Matter” label mask. The final mask included the
bilateral basal ganglia, amygdala and thalamus. Age-by-group interactions in gray matter volume
were tested for each voxel within the subcortical mask. All significant results are reported at a
voxelwise false discovery rate (FDR) [357] corrected p < 0.05, as implemented in SPM 5.
6.2.5. Contribution of TMD duration to age-related gray matter abnormalities
We examined the degree to which any of the observed age effects are attributed to cumulative
duration of chronic pain. To do this, we performed a forward model multiple linear regression,
which measures the degree to which one independent variable correlates to the dependent
variable. Additional independent variables are added to the equation and the degree to which the

163
independent variable predicts the dependent variable. Age and TMD duration were entered as
explanatory (independent) variables and each of the significant findings as dependent variables.
6.3. Results
6.3.1. Patient characteristics
The mean age of subjects in the patient group (mean ± SD: 33 ± 12 years) was not significantly
different than the control group (CTA analysis cohort: 33 ± 9.8 years, p = 0.94; VBM analysis
cohort: 32 ± 10.1 years, p = 0.81). The range of the controls ages was 20 to 50 years old, and the
age range for patients was 18-59 years old. The distribution of controls’ and patients’ ages is
presented in Table 6-1. Patient characteristic details are found in Table 5-1 [617,928]. Patients
reported having TMD for durations of 0.75- 30 years (mean ± SD: 9.8 ± 8.3 years). Of
importance for this study is that there was variability in the durations of TMD pain that
facilitated assessment of this factor, compared to age.
6.3.2. Global age effects
Both the control and TMD groups showed age-related whole brain gray matter atrophy, and there
were no significant group differences in whole brain gray matter volume (p = 0.88, see Figure 6-
1a). Specifically, there was a significant negative correlation between whole brain gray matter
and age for the controls (r = -0.55, p = 0.024, slope (m) = -3.46 cm3 /year) and for the patients ( r
= -0.72; p = 0.001, m = -5.11 cm3/year). However, there was an accelerated overall whole brain
aging effect in the patients with a significant group interaction of the slopes of the gray
matter/age curves (i.e., rate of change of gray matter with age) (p = 0.0002; see Figure 6-1b).
There was a significant correlation between patients’ duration of TMD and their age (r = 0.54, p
= 0.026; see Figure 6-1c), however TMD duration was not significantly correlated to gray matter
volume in the patient group (r = -0.37, p = 0.14, see Figure 6-1d).
6.3.3. Focal age effects
A significant age-by-group interaction (p < 0.05, corrected for multiple comparisons) was
localized to two focal regions within the right cortex. In one region, on the border of aMCC and
pgACC (BA32), patients had cortical thinning with age (r = -0.54, m = -0.022 mm/year),
whereas controls had age-related cortical thickening (r = 0.76, m = 0.033 mm/year). In another

164
region, the PMC, controls had age-related cortical thinning (r = -0.87, m = -0.035 mm/year),
whereas patients did not have normal atrophy, but rather had a very modest age-related cortical
thickening (r = 0.11, m = 0.002 mm/year) (see Table 6-2 and Figure 6-2 for details).
Three subcortical clusters were identified that had a significant age-by-group interaction (p <
0.05, FDR corrected; see Figure 6-3): the left PO thalamus and the right and left dorsal striatum.
The cluster in the right dorsal striatum extends to VPL, VPM and VL thalamus. Interestingly,
Figure 6-3 illustrates the strong positive correlation between gray matter volume with age in the
TMD group (r = 0.65) in contrast to the weak correlation in the control group in the thalamus (r =
-0.27). Figure 6-3 also illustrates the group differences in the dorsal striatum where the control
group showed a clear age-related gray matter loss (left: r = -0.78; right: r = -0.80), in contrast to
the age effect observed in the TMD group (left: r = 0.42, right: r = 0.33) (see Table 6-2, Figure 6-
3).
6.3.4. The contribution of TMD duration to gray matter age effects
A schematic of the focal aging effects in control and TMD groups is shown in Figure 6-4a (also
see discussion). The contribution of age and duration to the observed age-by-group interactions
was examined because of the observed correlation between age and duration. We performed a
multiple regression analysis with age and duration as independent variables to parse out the
relative contributions of these two factors. To describe our results, we used the standard
annotation for partial correlations, e.g., age • duration, such that age is being correlated to the
dependent variable while regressing out the variance related to duration. The outcomes of these
analyses are shown schematically in Figure 6-4b. In the right thalamus, the partial correlation
coefficient between age and gray matter volume was no longer significant when controlling for
duration (rage = 0.65, p = 0.005; rage • duration = 0.45, p = 0.082), whereas duration remained
significant when age was included in the model (rduration = 0.70, p = 0.002; rduration • age = 0.56 p =
0.026). In the aMCC/pgACC, the age-by-group interaction was driven by the shared variance
between age and duration. When the age was regressed out of the correlation between duration
and cortical thickness, the relationship remained insignificant (rduration = -0.45, p = 0.073 to rduration
• age= -0.22, p = 0.409). Further, when duration was regressed out of the correlation between age
and cortical thickness in the aMCC/pgACC, the relationship was no longer significant (rage = -
0.54, p = 0.027 and rage • duration = -0.39, p = 0.133) (see Table 6-3).

165
In some cases, duration did not contribute to the observed age-by-group interaction. For instance,
in the bilateral dorsal striatum, the partial correlations between age and gray matter in these
regions did not show much change when duration was regressed out. Similarly, when age was
regressed out of the correlation between duration and gray matter, the partial correlations were
not significant (see Table 6-3). In the case of the PMC, we found a suppressive effect [170], i.e.,
when duration was included in the regression model, age became a better predictor of the
interaction as the correlation became more significant. That is, the partial correlation coefficient
of age and thickness in the PMC, controlling for duration, was larger than the zero-order
correlation (zero-order correlation: 0.11, partial correlation: 0.38). Similarly, when correlating
thickness to TMD duration and controlling for the effect of age, the correlation coefficient for the
PMC decreased from -0.36 to -0.50 (see Table 6-3).

166
Table 6-1: Distribution of subject ages
Age range
18-20 21-25 26-30 31-35 36-40 41-45 46-50 51-55 56-60 TOTAL
#Con 1 5 2 2 3 2 2 0 0 17
#Pat 2 4 1 5 1 1 1 1 1 17
Abbreviations: #Con – Number of control subjects; #Pat – Number of patients with TMD

167
Table 6-2: Age-related group differences in cortical thickness and subcortical gray matter
volume. Shown are the statistically significant (p<0.05) group differences in correlation
coefficients of gray matter against age. Peak vertex/voxel Talairach coordinates (TAL) are
reported.
Interaction Region # Voxels Correlation
(age vs. thickness)
Peak TAL Peak
t-score
rpatients rcontrols X Y Z
Cortical
(CTA)
C > P R aMCC/pgACC 451 -0.54 0.76 14 30 20 4.42
C < P R PMC 423 0.11 -0.87 8 12 54 -5.18
Subcortical
(VBM)
C < P L dorsal Striatum 1537 0.42 -0.78 -18 10 10 -4.99
R dorsal Striatum 4415 0.33 -0.80 11 -3 18 -4.65
L Thalamus 264 0.65 -0.27 -21 -23 14 -3.36
Abbreviations: aMCC – anterior mid-cingulate cortex; pgACC – perigenual anterior cingulate cortex; CTA – Cortical thickness analysis; PMC – premotor cortex; VBM – voxel-based morphometry.

168
Table 6-3: Contributions of TMD duration to the age-gray matter relationships. Zero (0) order
correlations and partial correlations are presented for age and TMD duration versus gray matter
thickness/volume. The p-values in the age and duration columns represent the significance of the
correlations.
Significant group
differences
Age Duration
r p r age • duration p r p r duration • age p
R PMC 0.11 0.677 0.38 0.145 -0.36 0.159 -0.50 0.051
R aMCC/ pgACC -0.54 0.027 -0.39 0.133 -0.45 0.073 -0.22 0.409
L Dorsal Striatum 0.42 0.096 0.42 0.109 0.13 0.639 -0.12 0.650
R Dorsal Striatum 0.33 0.204 0.33 0.216 0.09 0.742 -0.10 0.706
R Thalamus 0.65 0.005 0.45 0.082 0.70 0.002 0.56 0.026
Abbreviations: aMCC – anterior mid-cingulate cortex; pgACC – perigenual anterior cingulate cortex; PMC – premotor cortex; S1 – primary somatosensory cortex

169
Figure 6-1: Total gray matter volume and correlations with age and duration: (a) Total gray
matter volume was not significantly different in the TMD group versus control group (p = 0.88).
(b) Significant gray matter volume decreases with age for both controls (3.46cm3/year) and TMD

170
group (5.11cm3/year) with a significant age-by-group interaction between the groups’ slopes (p =
0.0002). (c) TMD duration significantly correlated with patients’ age (r = 0.54, p = 0.026), (d)
but not to total gray matter volume (r = -0.37, p = 0.139). n.s. = not statistically significant
(p>0.05); asterisks (*) = p < 0.05; m = slope (rate of gray matter change in cm3/year).
R

171
Figure 6-2: Age-by-group interactions in cortical thickness. Patients with TMD had age-related
thinning (0.022 mm/year) in the anterior mid-cingulate cortex/perigenual anterior cingulate
cortex (aMCC/pACC), whereas controls show age-related thickening (0.033 mm/year) in this
region. In the premotor cortex (PMC), only the controls had age-related thinning (0.035
mm/year).

172
Figure 6-3: Group differences in age effects within subcortical gray matter volume. Age-by-
group interactions in the thalamus and dorsal striatum (p < 0.05, FDR). The graphs indicate gray
matter volume (corrected for total intracranial volume; TIV) versus age for each subject by
group. Colour bar shows t-statistic values.

173
Figure 6-4: Summary of age-related gray matter abnormalities (a) Schematic diagram of gray
matter regions with normal aging in healthy controls and abnormal aging in TMD. (b) Schematic
diagram summarizing the relative contribution of age and TMD duration to the regions of gray
matter with abnormal aging. The line thickness of each arrow depicts the relative contribution of
age and duration and the plus (+) and minus (-) signs depicts the direction of the relationship.
Abbreviations: aMCC/pACC – anterior mid-cingulate cortex/perigenual anterior cingulate
cortex; PMC – premotor cortex

174
6.4. Discussion
This study is the first to show that chronic pain associated with TMD in females is associated
with abnormal gray matter aging in focal cortical regions associated with pain and motor
processes. We found that patients with TMD have accelerated whole brain gray matter atrophy,
compared to pain-free controls. We also identified three types of aberrant patterns of gray matter
aging in five focal brain regions (Figure 6-4): 1) in the thalamus, patients with TMD gray matter
volume increased with age, whereas gray matter in controls was relatively sustained; 2) in the
aMCC/pgACC, patients with TMD had a progressive loss of cortical thickness with age, whereas
the controls had age-related cortical thickening; 3) in the dorsal striatum and PMC, there was
little change with age in the patients with TMD, whereas the controls had age-related atrophy.
TMD duration added to the age effects in the thalamus and the aMCC/pgACC, suppressed the
age effect in the PMC, but did not contribute to age effects in the dorsal striatum. Abnormal gray
matter aging in TMD may thus be due to the progressive impact of TMD-related factors as well
as inherent factors in individuals with this chronic pain.
6.4.1. Whole brain gray matter atrophy
Our finding of accelerated gray matter atrophy with age in TMD is consistent with previous
studies of gray matter in FMS [502] and chronic back pain [27]. These previous studies
attributed increased rate of gray matter loss to excitotoxicity and inflammatory molecules.
Specifically, they suggest that, because chronic pain is inherently harmful to the body and is
associated with negative affect and increased stress, the inflammatory response is upregulated
centrally, inducing cell death. These processes have been implicated in chronic age-related
diseases (Mattson, 2003), and therefore, it is plausible that they are implicated in age-related gray
matter loss in chronic pain states.
6.4.2. Use-dependent plasticity
We have demonstrated that TMD duration added to the age effects in the thalamus and cingulate
cortex. This form of plasticity is in line with the concept of use-dependent plasticity which
comprises structural and functional changes in the brain in response to increased or decreased
neuronal input [348,758]. For instance, studies in healthy human subjects have found that
training [266] and learning [267] can increase gray matter in the brain. Conversely, limb

175
amputation, and other forms of sensory loss can induce reorganization of the cortical map that
represents the affected limb [601,602] and gray matter loss [268,847]. Of particular interest are
studies that report reversible gray matter changes in nociceptive and antinociceptive regions of
the brain in response to repeated noxious stimulation in healthy subjects [93,852]. Furthermore,
patients with chronic pain show age-independent gray matter changes in both nociceptive and
pain-modulatory regions [98,356,558,575,617,741,772,963], some of which can be reversible
[384,741]. Our finding of progressive thinning in the cingulate cortex and increasing thalamic
gray matter are consistent with the concept of use-dependent plasticity. That is to say that
increased input, activity or use will increase gray matter, and decreased use or activity is
associated with decreased brain gray matter. Therefore, the observed age-by-group interaction in
the thalamus and aMCC/pgACC may, in part, be driven by prolonged nociceptive activity, which
could suppress the normal pattern of aging. These findings are consistent with our previous
report that gray matter in the thalamus is positively correlated with TMD duration, and may be
due to increased nociceptive activity, as previously discussed (see [617]).
In the aMCC/pgACC, we found that the shared variance of both age and TMD duration
contributes to progressive atrophy in the patients. This region receives orofacial nociceptive
input from thalamic nuclei that are part of the STT and TTT [189,276]. The aMCC/pgACC is a
complex, multimodal region that has been implicated in a number of functions [69]. For instance,
this region has been identified as a node in the salience network,!and has been implicated in
aspects of salience [213,229,262,263,774,849,927],!and pain
[220,230,231,259,265,440,508,528,632,933]. The cingulate cortex has also been implicated in
the cognitive and affective processing of pain [218,230,529,716,776,777,934,936], and the MCC
is involved in action selection and modulation of motor output in response to aversive stimuli
[790,898,904]. Therefore, it is possible that the age-related thinning in the MCC is related to
abnormalities in TMD with regard to cognitive and attentional processes related to prolonged
TMD pain [928].
The suppressive relationship identified in the PMC is of particular interest. The PMC is a region
that is often activated in neuroimaging studies of experimental pain [304,511]. In the current
study, age and TMD duration uniquely and non-redundantly predicted variance in gray matter.
When we included duration in the model, both variables (age and duration) better predicted the
progression of cortical thickness over time. Furthermore, age and duration had differential effects

176
on the PMC. Specifically, patients had sustained cortical thickness with age, whereas TMD
duration was related to cortical thinning in the PMC. The PMC receives nociceptive input from
the ventral caudal portion of the MD thalamus [276]. Therefore, we would expect that a barrage
of nociceptive input from the thalamus over an extended period of time could induce gray matter
plasticity in the PMC, as we have observed in the thalamus, rather than the observed
normalization. These paradoxical findings warrant further study to better understand the
relationship of the PMC and aging in the context of chronic pain. It is therefore apparent that
there is abnormal gray matter aging in TMD, independent of how long patients have had TMD.
The findings reported here are in contrast to previous findings that in some chronic pain
conditions central gray matter changes are related to pathology where there exists a peripheral
aetiology, such as osteoarthritis. It has been reported that once the peripheral cause of the pain
has been resolved, gray matter changes resolve [384,741]. There is evidence suggesting that both
central and peripheral mechanisms contribute to pain in TMD [561,757]. Therefore the source of
age-related variance may derive from multiple mechanisms in addition to (dis)use-dependent
plasticity. Interestingly, recent studies have reported that development of some brain regions is
tightly regulated by genes, rather than the environment [680,854]. There is also evidence
suggesting that there is a genetic predisposition to functional chronic pain syndromes [247-
249,809]. It is therefore feasible that there is a genetic contribution to the observed abnormal
aging effects. These findings provide a genetic source of plasticity [137,858] that may co-exist
with other forms of plasticity.
6.4.3. Cellular basis of changes in gray matter
The cellular and molecular basis of MRI-detectable gray matter changes remains to be explained.
However, several hypothetical mechanisms have been postulated, such as neuronal and/or glial
death [575], but recent evidence suggests that, to some extent, gray matter losses are likely
related to density of small dendritic spines [278,608], and remodeling of neuronal processes
[537]. Alternatively, reversible gray matter changes in chronic pain may be caused by
neuroinflammation [238,381,920], and induce MRI-detectable increases in gray matter. This
mechanism could explain both abnormal age-related increases and maintenance of gray matter
volume/thickness. For the observed gray matter losses, however, we cannot rule out that cell
death does not occurring in age-related gray matter losses – healthy populations lose neurones as

177
they age, and persons with neurodegenerative diseases suffer increased rates of atrophy related to
cell death.
6.4.4. Study limitations
One limitiation of this study is that it is not possible to control for the impact of long-term use of
medications in some of the patients on gray and white matter plasticity. Although the effect of
such medications is not well understood, recent evidence suggests that medications commonly
used to manage pain, such as anti-inflammatory medications and opiates, can impact brain
structure [916,962]. Considering that thirteen of seventeen of the patients included in this study
were taking NSAIDs, it is possible that the observed age-related findings (especially those of
gray matter maintenance) may be related to the effect of these drugs. Only one subject was
taking an opiate, hydromorphone, and so this is of less concern to this study.
Our patients did not self-report any previous diagnosis of depression or anxiety, although these
exist in some patients with TMD [283,808,850]. Thus we cannot exclude the contribution of
these factors in white matter abnormalities in TMD in general.
Another limitation of these findings is that it is a cross-sectional examination of structural
abnormalities in TMD. Therefore, the interpretations of “age-driven” and “TMD duration-
driven” abnormalities are limited to parsing out the relative variance that is accounted by each
factor. Therefore, the results warrant further study, and should be interpreted with caution.
6.5. Conclusion
In sum, our findings provide novel evidence that chronic TMD pain patients have abnormal age-
related gray matter changes in cognitive, motor and nociceptive brain regions. Our study
highlights the importance of understanding the effects of age and TMD duration in structural
studies of chronic pain, as progressive changes in gray matter may require differential
therapeutic approaches.

178
Chapter 7 STUDY III: White matter brain and trigeminal abnormalities in
TMD
The findings presented in this chapter are in press in Pain:
Moayedi M, Weissman-Fogel I, Salomons TV, Crawley AP, Goldberg MB, Freeman BV,
Tenenbaum HC, Davis KD. White matter brain and trigeminal nerve abnormalities in
temporomandibular disorder, Pain 2012; (in press).
7.1. Introduction
TMD comprise clinical problems involving the structures of and around the TMJ, the
masticatory musculature, or both [1,418]. TMD represent the most common orofacial chronic
pain disorder, prevalent in ~3-20% of the United States’ adult population [269,539,540,562].,
mostly in women [285,312,539,721]. In idiopathic TMD there is no clear aetiology
[283,286,287,653]. Central mechanisms are thought to initiate or maintain TMD pain [757]
based on TMD symptomology such as persistent pain, allodynia, and hyperalgesia, sometimes
extending to regions distant from the face [305,314,390,565,756,883], enhanced temporal
summation of pain to repetitive noxious heat stimuli [563], and dysfunctional DNIC
[113,491,756]. Abnormalities of these centrally-mediated processes suggest that ascending
nociceptive pathways and/or descending pain-modulatory pathways [515] are affected.
Additionally, patients with TMD can exhibit cognitive [370,379,380] and motor dysfunction
[837] possibly related to abnormalities in brain regions associated with these functions
[799,800,839,928].
Structural brain imaging provides an opportunity to delineate anatomical substrates of CNS
abnormalities in TMD. We reported that patients with TMD have increased cortical thickness in
the orofacial region of the primary somatosensory cortex (S1), the ventrolateral prefrontal cortex
(vlPFC) and the frontal pole [617]. Similarly, Younger and colleagues [963] reported increased
gray matter volume in motor, limbic and sensory regions, including trigeminal brainstem nuclei,
in myofascial TMD. These studies indicate that TMD patients have gray matter abnormalities in
sensory, motor and cognitive/limbic regions. Our finding of a correlation of thalamic gray matter
with TMD duration [617] supports the concept of long-term nociceptive-induced central

179
plasticity, possibly due to increased peripheral nociceptive activity, or dysfunctional
antinociceptive systems. Therefore, the former possibility suggests that although clinical
observations have not clearly identified gross peripheral abnormalities in TMD, the CNV may
indeed undergo changes due to abnormal persistent activity resulting in microstructural
abnormalities.
Previous studies examining white matter in other clinical conditions with sensory abnormalities
and/or chronic pain [156,356,558,847] found white matter abnormalities in brain areas involved
in sensory, modulatory and cognitive functions, and some of these white matter abnormalities
correlated with clinical findings. Therefore, studying the correlation between TMD
characteristics and measures of white matter integrity can provide insight into whether chronic
pain drives changes in white matter microstructure.
The main aim of this study was to evaluate the white matter abnormalities in TMD. Specifically,
we determined whether there are abnormalities along the CNVs or in white matter tracts in the
brain associated with sensory, cognitive or motor functions, compared to healthy controls.
Towards this goal, we used DTI, a brain imaging technique sensitive to the inherent diffusion of
water molecules that allows us to assess white matter microstructure. FA is a DTI metric that
reflects white matter tract integrity or structure [52,59,66,626]. We tested for abnormalities in FA
along the CNV of patients with TMD. Further, we used two approaches to assess white matter in
the CNS: First, we quantified brain FA abnormalities in TMD with TBSS [815], and second we
used probabilistic tractography [74] to determine the connectivity of main regions of white
matter disruption. Finally, we determined whether there is a link between white matter
microstructure and clinical characteristics of TMD.
7.2. Methods
A detailed description of methods is provided in Chapter 4.
7.2.1. Subjects
The study consisted of a cohort of 17 right-handed females with idiopathic TMD (mean age ±
SD: 33.1 ± 11.9 years) and 17 healthy right-handed females (mean age ± SD: 32.8 ± 9.8 years).
Informed written consent was obtained from all study participants for procedures approved by
the University Health Network and Mount Sinai Hospital Research Ethics Boards. Patients were

180
screened with the Mount Sinai Hospital Dental Clinic TMD diagnostic criteria, based on the
TMD-RDC [285]. Inclusion criteria included: 1) pain in the muscles of mastication rated
verbally as at least 4/10 for at least 3 months at the time of evaluation, or pain that is aggravated
by mandibular function; 2) moderate (on a scale of no (0), low (1), moderate (2), or severe (3))
pain to palpation and/or pain persisting post-examination in at least 3 muscle sites and/or
moderate pain to palpation of the TMJ and/or limited mandibular movement (opening < 40mm).
Patients were analgesic-free for 24 hours prior to scanning. Exclusion criteria for all subjects
included: 1) left-handedness; 2) self-report of metabolic, rheumatoid or vascular diseases or
disorders, or any other serious diseases; 3) self-report of commonly co-morbid functional chronic
pain disorders (IBS and FMS); 4) self-report of psychiatric disorders (e.g., depression,
schizophrenia, attention deficit hyperactivity disorder); and 5) self-report history of an abnormal
neurological examination; 6) contraindication to MRI scanning; 7) self-report of substance
abuse. Additionally, healthy controls were excluded if they had a history of chronic pain and
patients were excluded if they had a pain disorder other than TMD.
7.2.2. Questionnaires
Patients provided verbal ratings of their current pain and unpleasantness, as well as their average
pain intensity and unpleasantness ratings over the last month before scanning on a scale of 0 to
10 (0 = no pain, 10 = worst pain imaginable). Specifically, patients provided a numerical pain
score for pain intensity and pain unpleasantness by answering the following questions: 1) “Please
rate the intensity of your average pain over the last month, 0 being no pain and 10 being the
worst pain imaginable?” and 2) “Please rate the unpleasantness of your average pain over the last
month, 0 being not unpleasant and 10 being the most unpleasant pain imaginable?”. The patients
also reported the duration of their TMD symptoms.
7.2.3. Imaging parameters
All subjects underwent scanning in a 3-Tesla GE MRI system fitted with an eight-channel phased
array head coil. Each subject’s head was padded to reduce movement. The DTI acquisition
consisted of two runs of diffusion-weighted scans (TR = 14,500 ms, field of view: 24 cm x 24
cm, 128 x 128 matrix, 1.875mm x 1.875 mm in-plane resolution, 3 mm thick axial slices, with
ASSET with a factor of 2, maximum gradient strength = 40 mT/m, maximum slew rate = 150
T/m/s) acquired along 23 non-collinear, isotropic directions (b = 1,000 s/mm2). Additionally, 2

181
non-diffusion weighted scans (b = 0 s/mm2; b0) were acquired at the beginning of each run. The
scans covered from the top of the head to the second cervical spinal process (C2).
7.2.4. DTI pre-processing
The diffusion-weighted images were imported into FSL 4.1.8 (http://www.fmrib.ox.ac.uk/fsl) for
quality control [816]. Pre-processing included eddy current and motion artifact correction using
FDT [459]. Each subject’s two DTI runs were averaged to a single volume to achieve a greater
signal-to-noise ratio. Then, individual brain masks were created using BET [814]. These images
were then processed through two different pipelines for (1) voxelwise analysis and (2)
tractography.
The preprocessed images were fit with a diffusion tensor model using DTIFIT in FDT v. 2.0. We
then calculated voxelwise values of FA, and other DTI metrics (see below)
7.2.4.1. Other DTI metrics
To gain more insight into FA findings, we also assessed other DTI metrics in the clusters with
significant group difference. λ1 is thought to reflect diffusion along a tract, and reductions in this
value suggest disruptions along the tract diffusivity. RD is believed to reflect changes in
membrane permeability and, to some extent, myelination [66,67]. RD is calculated by averaging
the two radial vectors of the tensor model (λ2 and λ3). Finally, MD was measured because it is
associated to oedema and inflammation. Figure 7-1 provides a schematic of these DTI metrics.
DTI metrics (FA, MD, λ1, λ 2 and λ3) are calculated using DTIFIT in the FDT toolbox. The TBSS
skeleton is created based on FA, and peak FA values are projected onto the skeleton. For
statistical analysis of the other DTI metrics, MD, RD and λ1 values from the same voxels are
projected onto the skeleton. To test for group differences, we performed a multivariate analysis
of variance (MANOVA) with each of the DTI metrics as dependent variables, and group as an
independent variable.
7.2.5. Assessment of CNV
To assess the CNV, we manually drew a ROI (3.75 mm x 3.75 mm x 3 mm) in the axial plane on
each subject’s CNV root in the cisternal space in native space (see Figure 7-1). The nerves were
identified on color orientation maps of the primary eigenvector (λ1) image overlaid onto the FA

182
map. FA, MD, RD, λ1 values (see Figure 7-1) were calculated for each of the DTI volumes and
extracted from the ROI. Independent samples t-tests were used to assess group differences in FA
for each CNV. Furthermore, a paired t-test was utilized to compare left and right CNV FA values
within the patient group. Lastly, FA values derived from the each of patients’ nerve were
correlated with TMD characteristics (pain intensity, unpleasantness and duration). Statistical
significance was set at p < 0.05. All statistical tests were performed in SPSS v. 19.0
(http://www.spss.com/).
Figure 7-1: Schematic of diffusion tensor imaging, and summary of eigenvectors and metrics.
Lambda (λ) represents each of the eigenvectors that make up the tensor model. The equations to
calculate MD, fractional anisotropy (FA) and radial diffusivity (RD) from these eigenvectors are
also provided.
7.2.6. Tract-based spatial statistics
TBSS v.1.2 [815,817] was used to compare FA between the TMD and healthy control cohorts.
Briefly, FA maps underwent non-linear registration to a 1 x 1 x 1 mm FA map in standard space
(FMRIB58_FA, available in FSL), a mean image derived from all the subjects was created and
thinned to represent the centre of major white matter tracts common to all subjects, forming a
white matter skeleton. Each subject’s peak FA value perpendicular to the thinned track was then
projected onto the skeleton. One skeleton was created for analysis of group differences that

183
included all study participants, and a second patient only skeleton was created for correlation
analyses of TMD pain characteristics (see below). The mean skeleton images were set at a
threshold of 0.2 to include FA values that are related to white matter [815]. Other DTI metrics
were also evaluated to characterize FA findings (see Figure 7-1, and below). Additionally, we
performed a separate patient group analysis to assess which areas of the skeleton correlate with
TMD characteristics (pain intensity, unpleasantness and duration).
7.2.7. Probabilistic tractography
Probabilistic tractography was used to assess the connectivity of findings from the TBSS analysis
(http://www.fmrib.ox.ac.uk.fsl/fdt/index.html)[74,76]. First, we downsampled the preprocessed
diffusion-weighted images to create isotropic voxels (3 x 3 x 3 mm). The images were then
processed in FDT. Probability density functions (PDF) on up to two principal fibre directions
were estimated at each voxel in the brain. We then used multi-fibre tractography and drew 5000
samples from each seed voxel along the PDFs. The seed voxels were binarised images of the
significant clusters. Together, each seed voxel’s streamlines provide an estimate of its
connectivity. When a streamline reaches a voxel with more than one fibre direction, the
streamline follows the direction closest to the direction at which it arrives at the voxel. The
pathways generated by the algorithm represent the number of samples that have passed through a
voxel. To eliminate spurious connections, each computed pathway in each subject was
thresholded at 10 samples (of the 5000 generated from each seed voxel) that passed through the
voxels. These thresholded tracts were consistent between subjects. To visualize the findings,
each of the subject’s tracts were binarised and overlaid on a standard brain to produce a
probabilistic map of the pathways for controls and patients. The values in these maps reflect the
number of subjects who share a pathway.
7.2.8. Statistical analyses
7.2.8.1. Whole brain white matter
We assessed group differences in global (whole brain) FA and the white matter skeleton FA. To
do so, we extracted mean FA values across every voxel in the brain and every voxel in the
skeleton for each subject and performed a two-tailed t-test.

184
7.2.8.2. Mask analysis
A mask was created to restrict the analysis to a priori white matter ROI, including the white
matter regions containing the pathways subserving nociceptive, antinociceptive, motor and
cognitive functions. Thus, the mask included the brainstem white matter (including the white
matter that contains the TTT and the CST, TCT (including tracts to S1 and the anterior corona
radiata), the IC containing the CST, the cingulum, the rostrum, genu and the body of the corpus
callosum, the uncinate fasciculus, and the EC/ExC. The mask also included the thalamus, as
white matter courses through this region. The ROI were identified with the Johns Hopkins
University white matter Tractography Atlas, ICBM-DTI-81 white matter Atlas [625,910], the
Harvard-Oxford Cortical Atlas and the Harvard-Oxford Subcortical Atlas, available within FSL
(http://www.cma.mgh.harvard.edu/fsl_atlas.html). The final mask corresponded to voxels within
the FA skeleton.
A between group voxel-wise t-test within the skeleton mask was performed to identify regions
with significant group differences in FA, with age included as a covariate of no-interest. We used
two complementary statistical thresholding methods with 5000 permutations testing in FSL’s
randomise toolbox: (1) threshold-free cluster enhancement (TFCE; correct cluster p < 0.01)
[819], which is sensitive to spatially extensive areas of significant difference; and (2) cluster-
mass correction (p < 0.05), which requires clusters to meet a specific height threshold and,
therefore, is only sensitive to t-values above the threshold, which we set at t > 2.3.
An additional set of post-hoc of analyses was run to characterize regions with significant group
differences in FA. First, we determined whether the findings were related to specific
characteristics of TMD. To do so, we extracted each subject’s mean FA values from significant
group difference clusters. The patients’ FA values from each of these clusters then were
correlated with the TMD pain intensity, unpleasantness and duration in SPSS v. 19.0
(http://www.spss.com/). Statistical significance was set at p < 0.05. Second, we extracted the
mean values of mean diffusivity MD, RD and λ1 for each of the clusters and assessed whether
there were significant group differences between patients with TMD and controls using a
multivariate analysis of variance (MANOVA) with each of the DTI metrics as dependent
variables and group as an independent variable. Statistical significance was set at p < 0.05.

185
All significant TBSS results are thickened with the “tbss_fill” tool implemented in FSL for ease
of visualization. This tool uses a 3mm Gaussian smoothing kernel to thicken significant results to
the white matter tract (as defined by FA > 0.2).
7.2.8.3. Quantitative Tractography: connection probability
We identified that patients and controls had differential connectivity between the corpus
callosum seed and the dlPFC, and between the corpus callosum seed and the frontal pole.
However, these findings were only qualitative, and we therefore sought to quantify these group
differences. To do so, we performed a second tractographic analysis with specified targets based
on the observed differences. Specifically, we thresholded the tractography maps for each group
at 5 of 17 subjects (~30%) (see Figure 7-7A). Subsequently, we binarised the resultant image for
each group, and subtracted them one from another to determine the regions of difference between
the groups. We extracted the tracts for each hemisphere and restricted them to gray matter of the
target region. For example, the tract projecting to the frontal pole was restricted to the frontal
polar cortex. This was done by multiplying the binarised subtraction image to a binarised mask
of the frontal pole (from the Harvard-Oxford Cortical Structural Atlas;
http://www.cma.mgh.harvard.edu/fsl_atlas.html). Similarly, the tracts projecting to the dlPFC
were restricted to the middle frontal gyrus. We then performed tractography between the corpus
callosum seed and these 4 targets (the left and right frontal pole, and the left and right dlPFC).
Next, we extracted the number of samples that project to each target in each subject, and the
number of voxels in the seed that project to each of the targets. Group differences were tested
using two-tailed, independent samples t-tests. Significance was set at p < 0.05
7.3. Results
7.3.1. Patient characteristics
The ages of the patient and control groups were not significantly different (patients: mean age ±
SD: 33.1 ± 11.9 years; controls: 32.8 ± 9.8 years; p = 0.94). Details of the individual patient
demographics have been tabulated in our previous study of this cohort (see Table 5-1; and
[617]). Of importance for this study is that there was variability in the durations of TMD,
intensity and unpleasantness of pain that facilitated assessment of these factors in correlation
analyses. The duration of TMD ranged from 0.75 – 30 years (mean ± SD: 9.8 ± 8.2 years), while
pain intensity ranged from 2 – 7 (mean ± SD: 4.3 ± 1.8), and unpleasantness ranged from 1 – 8

186
(mean ± SD: 5.4 ± 2.1). Patients reported that their TMD pain was confined to the TMJ (n = 6),
the muscles of mastication (n = 3), or both the joint and muscles (n = 8). Most patients reported
bilateral pain (n = 13), but some reported unilateral pain (three right-sided, one left-sided). The
duration of TMD significantly correlated with the patients’ age (r = 0.536, p = 0.027).
7.3.2. Trigeminal nerve FA
We tested the CNV roots, in the cisternal space (see Figure 7-2), for group differences in FA,
MD, RD and λ1. We found that patients with TMD had significantly lower FA in both CNVs
(right and left: p < 0.001) (see Figure 7-3A). FA in the right CNV was also negatively correlated
with TMD duration (r = -0.53, p = 0.028) (See Figure 7-3B). The left CNV was not significantly
correlated with TMD characteristics. MD and RD were significantly higher in both CNVs (right:
p < 0.01; left: p < 0.05), compared to controls (See Figure 7-3C), but there were no significant
group differences in λ1.
7.3.3. Patients have lower white matter FA
To investigate global differences in white matter microstructure, we tested for group differences
in FA across the whole brain and within the white matter skeleton. Compared to controls, the
TMD patient group showed a 3.8% reduction in whole brain FA (mean ± SD: controls = 0.264 ±
0.012; patients = 0.254 ± 0.008; p = 0.003) and 3.6% reduction in whole brain skeletonised FA
(controls = 0.449 ± 0.018; patients = 0.433 ± 0.001; p = 0.006) (see Figures 7-4 and 7-5). We
also evaluated MD, RD and λ1 across the white matter skeleton and found that there was a
significant group difference (Pillai’s trace = 0.416; F(3,30) = 7.117; p< 0.001), and Bonferroni
post-hoc tests revealed that all three DTI metrics were significantly higher in the patient group
compared to the control group (all post-hoc tests: p < 0.05; see Table 7-1 for details).
The mask-wide TBSS analysis (p < 0.01; TFCE corrected) identified white matter clusters that
had 6.6-16.8% lower FA in the patient cohort compared to controls (see Table 7-2 and Figure 7-
5A). Two of these clusters were localized to the right IC, and two were in the right
external/extreme capsule adjacent to the insula. Other significant clusters were at the junction of
the right internal and external/extreme capsules, adjacent to the vlPFC, and in the white matter
adjacent to right S1 and M1. The largest cluster included the bilateral anterior body of the corpus
callosum, and extended to the left cingulum, the bilateral anterior corona radiata, the bilateral

187
ICs, the left external/extreme capsules, the fornix, the left and right thalamus and the brainstem.
Although the thalamus is mostly a gray matter structure, white matter does course through it and
we identified a cluster within the right thalamus with significantly lower FA in patients with
TMD, compared to controls. Individual clusters are shown in Figure 7-6.
Because one of our findings encompassed a large, diffuse region, we performed a secondary
analysis to test for more focal, highly significant group differences in FA. To do so, we used a
cluster-mass correction that requires significant clusters to reach a specified threshold, in this
case t(32)= 2.30. This analysis revealed a single focal cluster in the corpus callosum (peak voxel
MNI coordinates: -4, 26, 9, t(32) = 2.31, cluster size 353 voxels) (see Figure 7-5B).
We also determined whether the white matter tracts with lower FA in patients with TMD also
showed changes in MD, RD and λ1, indicative changes in axonal diffusion, permeability and
myelination. We found that there was a significant between-group difference in these
microstructural variable (Pillai’s trace = 0.958; F(7,26) = 6.182; p = 0.009). Bonferroni post-hoc
tests revealed that all of the clusters with lower FA showed significantly increased MD and RD
(p < 0.05), but there were no significant differences in λ1 (see Table 7-3).
7.3.4. Probabilistic tractography
Because the corpus callosum is a large structure that contains interhemispheric fibres we used
tractography to elucidate the connectivity of our finding of lower FA in the corpus callosum. We
evaluated the relationship between FA and connectivity by determining the connectivity of a
region with lower FA in patients. To do so, we used multi-fibre probabilistic tractography to
determine pathways passing through the corpus callosum finding from the cluster-mass
correction analysis (see Figure 7-5B and 7-7A). We first performed a qualitative analysis to
determine the connectivity of the corpus callosum finding within each group. As shown in Figure
7-7A, there were denser connections between the corpus callosum and the bilateral dlPFC in
controls, compared to patients. Conversely, connections between the corpus callosum and the
bilateral frontal pole were quite dense in patients, but sparse in controls. Quantification of these
connection probabilities verified that significantly fewer voxels from the corpus callosum
connect with the left and right frontal poles in controls, compared to patients with TMD (see
Figure 7-7B and C). We also found that patients with TMD have significantly higher number of
samples (i.e., connection probability) between the corpus callosum and the left frontal pole (p <

188
0.05). There were no significant group differences in the connection probabilities of the corpus
callosum and the right frontal pole. Additionally, we found that patients had significantly lower
connection probability between the corpus callosum and the right dlPFC (p < 0.05), but not the
left dlPFC (p > 0.05) (see Figure 7-7C).
7.3.5. White matter FA related to TMD pain characteristics
We tested whether each of the clusters with significant group differences in FA was correlated
with TMD characteristics (TMD pain intensity, unpleasantness and duration). We found that
three of these clusters showed significant FA-TMD correlations. Specifically, the FA within the
cluster at the junction of the internal and external/extreme capsules (adjacent to the vlPFC) was
significantly negatively correlated with TMD pain intensity (r = -0.49, p = 0.046) (see Figure 7-
8A). We also identified a significant negative correlation between FA in the thalamic cluster and
TMD pain intensity (r = -0.59, p = 0.013) (see Figure 7-8B). Finally, we identified a significant
negative correlation between a cluster in the right IC and both pain intensity (r = -0.72, p =
0.001) and pain unpleasantness (r = -0.63, p = 0.007) (See Figure 7-8C).

189
Table 7-1: Group differences in whole brain skeletonised white matter. Group means ± standard
deviations (SD) are presented for each group. Bonferroni post-hoc p-values are calculated based
on independent-samples t-tests.
Controls Patients
p-value
Mean
(x10-4 mm2/s) SD
Mean
(x10-4 mm2/s) SD
MD 7.3 0.16 7.6 0.12 < 0.05
RD 5.4 0.19 5.6 0.10 <0.05
λ1 11.2 0.22 11.4 0.23 <0.05
Abbreviations: MD – mean diffusivity; RD – radial diffusivity; λ1 – axial diffusivity

190
Table 7-2: White matter regions in patients with TMD with significantly lower fractional
anisotropy compared to controls. Mean and standard deviations of FA values, T-scores, percent
decrease and MNI coordinates are reported for the peak voxel.
Regions
FA
# Voxels T
% decrease
MNI
Controls TMD
x y z Mean St Dev Mean St Dev
Thalamus 0.371 0.0326 0.32 0.0002 305 4.08 13.8 17 -15 12
WM adjacent to S1/M1 0.428 0.0476 0.356 0.0012 129 3.51 16.8 32 -22 38
ICAL 0.564 0.0613 0.526 0.0046 3 3.06 -11.1 14 12 -2
EC/ExC 0.373 0.0335 0.339 0.0059 4 2.90 9.1 33 6 3
EC/ExC 0.399 0.0283 0.366 0.0073 3 2.82 8.3 32 9 1
Internal capsule 0.599 0.0454 0.558 0.0132 581 2.60 6.8 11 1 3
ICAL and EC/ExC 0.513 0.0501 0.456 0.0332 55 2.27 6.6 26 24 11
Diffuse1 0.853 0.0521 0.786 0.0018 10283 3.46 7.8 -5 26 10
1Diffuse cluster includes white matter in the corpus callosum, corticospinal tracts, internal
capsule, external/extreme capsules, fornix, cingulum, anterior corona radiata, cerebellar
peduncle, and pontine tracts.
All clusters are significant at p < 0.05, voxel-wise, mask-wide, FWE corrected after threshold-
free cluster enhancement. Voxels are 1mm3.
Abbreviations: IC – internal capsule; WM – white matter; S1 – primary somatosensory cortex;
M1 – primary motor cortex; ICAL – anterior limb of the internal capsule; EC/ExC –
external/extreme capsules.

191
Table 7-3: Group differences in mean diffusivity (MD), radial diffusivity (RD) and axial
diffusivity (λ1) in clusters with significant group differences in FA. Mean DTI metric values and
p-values are shown for two-tailed t-tests for MD, RD and λ1 values in each cluster from Table 7-2
and the cluster-mass corrected cluster. n.s. indicates that the t-test did not reach significance (p ≥
0.05).
MD (x104 mm2/s) RD (x104 mm2/s) λ1 (x103 mm2/s)
Controls TMD p Controls TMD p Controls TMD p
ICAL 8.0 8.5 <0.001 5.3 6.0 <0.001 1.3 1.3 n.s.
EC/ExC 7.5 7.9 <0.005 6.0 6.5 <0.001 1.0 1.1 n.s.
EC/ExC 7.6 8.0 <0.001 6.2 6.7 <0.001 1.0 1.1 n.s.
ICAL/EC/ExC 7.2 7.6 <0.01 5.0 5.4 <0.001 1.2 1.2 n.s.
S1/M1 6.7 6.9 <0.05 4.8 5.1 <0.001 1.1 1.0 n.s.
Thalamus 7.5 7.8 <0.001 6.0 6.4 <0.001 1.0 1.0 n.s.
IC 7.0 7.2 <0.005 4.1 4.5 <0.001 1.3 1.3 n.s.
Diffuse1 7.6 7.9 <0.001 4.8 5.3 <0.001 1.3 1.3 n.s.
Corpus callosum2 8.2 8.8 <0.05 3.8 4.6 <0.005 1.7 1.7 n.s.
1see Table 7-2 for description of the diffuse cluster.
2Corpus callosum cluster is from the cluster-mass correction.
Abbreviations: IC – internal capsule; ICAL- anterior limb of the internal capsule; EC/ExC –
external/extreme capsules; S1 – primary somatosensory cortex; M1 – primary motor cortex

192
Figure 7-2: Colour orientation maps of the trigeminal nerves. Axial images of three controls and
three patients with the primary eigenvector of the tensor model (λ1) within each voxel. The
eigenvector is colour-coded (green - anterior-posterior, red - left-right, blue - in the inferior-
superior plane) and modulated to the FA map. The location of the trigeminal nerves is indicated
by the yellow arrowheads.

193
Figure 7-3: Trigeminal nerve fractional anisotropy abnormalities in TMD. (A) The trigeminal
nerve roots (within the blue boxes) at the pontine level are shown on an axial slice from a high-
resolution T1- weighted MRI scan. The magnified view of the right nerve is from a diffusion-
weighted scan. The direction of the primary vector of the tensor model (λ1) within each voxel is
color-coded (green - anterior-posterior, red - left-right, blue - in the inferior-superior plane) and

194
the primary vector shown within each voxel. (B) patients with TMD have lower fractional
anisotropy in bilateral trigeminal nerves, compared to controls. (C) Fractional anisotropy is
negatively correlated with TMD duration. (D) Group differences in trigeminal nerve mean
diffusivity (MD), radial diffusivity (RD) and axial diffusivity (λ1). * = p < 0.05; ** = p < 0.01
*** = p < 0.005. CNV = trigeminal nerve.

195
Figure 7-4: Group differences in mean fractional anisotropy (FA) between patients with TMD
and controls. Patients had significantly lower whole brain FA (left panel) and skeleton FA (right
panel). Asterisks (*) indicate p < 0.05.

196
Figure 7-5: Group differences in fractional anisotropy between patients with TMD and controls.
(A) Blue regions indicate areas showing significant reduction in fractional anisotropy in TMD
compared to controls using threshold-free cluster enhancement (TFCE, corrected p < 0.01) and
overlaid on the white matter skeleton (shown in green) (significant clusters have been thickened
for enhanced visualization). Widespread abnormalities are observed in bilateral internal (IC) and
external/extreme capsules (EC/ExC), corpus callosum (CC), cingulum (Cing), thalamic and
brainstem white matter. (B) 3D rendering of the region of reduced FA in the corpus callosum of
patients compared to controls using cluster-mass thresholding (t > 2.3, corrected p < 0.05), the
cluster-mass corrected result in the corpus callosum (CC). A = anterior.

197
Figure 7-6: Clusters with significant group differences in fractional anisotropy. Blue clusters
(filled for visualization purposes) indicate regions of decreased FA in patients compared to
controls. Significant clusters (TFCE, p < 0.01) are displayed on separate brains in the (A)
anterior limb of the internal capsule (ICAL), (B and C) external/capsules (EC/ExC), (D) junction
of the ICAL and the EC/ExC, (E) white matter adjacent to the primary somatosensory and
primary motor cortices (S1/M1), (F) white matter in the thalamus, (G) internal capsule (IC) and
(H) in a diffuse cluster that includes brainstem white matter (cerebellar peduncles, pontine tracts,
corticospinal tracts), IC, EC/ExC, corpus callosum (CC), cingulum (Cing), the fornix and white
matter within the orbitofrontal cortex (OFC).

198
Figure 7-7: Abnormal white matter connectivity in TMD. Qualitative analysis of probabilistic
tractography of the cluster-mass corrected (t > 2.3, p < 0.05) cluster in the left corpus callosum
revealed that (A) this abnormal white matter region has different connections (yellow arrowhead)

199
in TMD and controls. The right panel shows a subtraction map that reveals that patients have
sparser connections between the corpus callosum seed and the frontal pole (blue box), and denser
connections to the dorsolateral prefrontal cortex (dlPFC; green box). The colour bar indicates the
number of subjects (between 5 and 17) contributing to the cluster at each voxel (controls are in
blue-light blue, and patients are in red-yellow). Quantitative tractography (B and C) revealed that
more voxels from the seed region (in green) in the corpus callosum project to the frontal pole in
the patients, compared to controls. The colour bar in (B) represents the proportion of subjects
with projections to the frontal pole in each voxel (controls are in blue-light blue, and patients are
in red-yellow). (C) Also, controls have a higher connection probability between the corpus
callosum and the right dlPFC, whereas patients have a higher probability of connection between
the corpus callosum and the left frontal pole. Graphs show mean number of samples (± SE) that
reach the target in each group (top panel), and the mean number of voxels (± SE) in the seed
mask that have samples that project to the target masks. * = p < 0.05.

200
Figure 7-8: Regions with group differences in fractional anisotropy are also correlated with
TMD characteristics: (A) the junction between the right internal and external/extreme capsules
(ICAL/EC/ExC) adjacent to the ventrolateral prefrontal cortex (vlPFC), (B) thalamic WM (white
matter) and the (C) right internal capsule are negatively correlated with TMD pain intensity. The
right internal capsule is also negatively correlated with TMD unpleasantness. Controls’ mean ±
SE FA for each region is shown in green. * = p < 0.05; ** = p < 0.01

201
7.4. Discussion
This study is the first report to show that both peripheral nerve and CNS white matter
abnormalities contribute to TMD pain. In support of a peripheral contribution, we found that
patients with TMD had lower FA in both CNVs and FA in the right CNV was negatively
correlated with TMD duration. In support of a central component to TMD, we found that patients
with TMD had (1) widespread abnormalities in the microstructure of white matter tracts related
to sensory, motor, cognitive and pain functions, including a focal area of the corpus callosum; (2)
stronger connectivity between the corpus callosum and the frontal pole, but sparser connectivity
with the dlPFC, compared to controls; (3) FA abnormalities that correlated with TMD
characteristics, and (4) reduced brain FA that were associated with greater MD and RD, markers
of inflammation and œdema.
7.4.1. Trigeminal nerve abnormalities
This is the first study to assess the CNV white matter integrity in TMD. We found that patients
with TMD had significantly lower FA and higher MD and RD in both CNVs. We also found that
the FA in the right CNV was negatively correlated with TMD duration. Previously, TMD was
considered idiopathic because there were no observable or major peripheral abnormalities in the
TM joint or muscles [757]. However, the DTI-based imaging technology used here clearly shows
microstructural abnormalities in the CNV. In line with peripheral abnormalities in TMD, one
study has demonstrated that the jaw-jerk reflex is abnormal on the painful side in TMD [332],
and another study found reduced cortical LEPs and reflexive masticatory muscular activity when
the CNV was stimulated in TMD, compared to controls [743]. Therefore, increased nociceptive
firing either from the periphery or from aberrant firing patterns (see below) over time could
affect the microstructure of the CNV and could also contribute to central abnormalities along the
ascending nociceptive system. This concept is supported by findings of increased pain sensitivity
within and outside the trigeminal region [313,491,563-565,841]. Alternatively, there is evidence
of abnormalities in orofacial motor function in TMD [46]. Recent evidence suggests that
nociceptive input can modulate motor output (see Section 2.2.3.2). Therefore, it is possible that
the microstructural abnormalities in the trigeminal nerve may be related to altered motor output
from the CNS.

202
7.4.2. Abnormal sensorimotor tracts in TMD
In addition to the peripheral nerve findings, we also found that patients have lower FA in the
brainstem, white matter coursing through the thalamus, the IC, and tracts adjacent to S1/M1.
Therefore, decreases in FA and increases in MD and RD along the ascending nociceptive
pathways could be induced or maintained by aberrant peripheral input from the CNV, or reduced
antinociceptive activity in the CNS. In support of this concept, are our previous study showing
that in patients with TMD there is a positive correlation between gray matter volume in the
thalamus and TMD duration, and thicker cortex in the orofacial region of S1 (See Chapter 5)
[617], suggesting that increased activity may lead to structural brain changes over time.
Given that DTI cannot distinguish between ascending and descending tracts [63], it is possible
that the abnormal white matter tracts contain descending projection fibres (the corticofugal
tracts) and corticothalamic tracts. The corticofugal tracts comprise the CST, corticobulbar,
corticoreticular, corticopontine tracts [625]. These tracts largely contain motor efferents and so
abnormalities in these tracts may underlie motor abnormalities in TMD, such as the masticatory
muscle hyperactivity, and abnormal jaw motor function under cortical control [837,839].
Furthermore, patients with TMD have been shown to have increased cortical activity in motor
regions and sluggish reaction times during a cognitive task [928].
7.4.3. Cognitive interference of pain and decreased modulation in TMD
We also identified lower FA in the cingulum bundle (adjacent to the mid-cingulate cortex), white
matter tracts coursing to the OFC and sgACC, tracts adjacent to the vlPFC, the anterior corona
radiata (in the ICAL, which projects to the prefrontal cortex) and the EC/ExC adjacent to the right
mid-insula. The so-called medial pain system is comprised of ascending nociceptive fibres that
project to the medial thalamus and further to the cingulate cortex, the insula and the PFC
[234,505,868]. It is believed that these regions contribute to the cognitive-affective dimension of
pain. The insula, mid-cingulate cortex and vlPFC have been implicated in pain perception,
negative emotion, and cognitive function
[23,94,108,116,183,221,220,222,227,304,447,752,790,898]. The OFC and the sgACC are
thought to contribute to the descending pain modulation [95]. Patients with TMD have
widespread pain [305,563,565,883], abnormal DNIC [113,491,756], and have been shown to

203
have sluggish reaction times to a cognitive task [928]. Therefore, the observed microstructural
abnormalities in both the ascending medial pain system and descending modulatory system
provide a plausible neural substrate for abnormal cognitive and anti-nociceptive function in
TMD.
7.4.4. Abnormal connectivity in TMD
Another main finding of this study is that we identified a significant decrease in FA in the corpus
callosum (identified using cluster-mass correction). This is a region that connects
interhemispheric prefrontal regions [966], and this portion of the corpus callosum is also
connected to the cingulate cortex. Similarly, a study examining white matter abnormalities in
complex regional pain syndrome reported decreased FA in a similar region of the corpus
callosum of patients, compared to controls [356].
In our study, probabilistic tractography revealed that patients had differential connectivity
between the corpus callosum and two regions that have been implicated in executive control and
pain perception and/or modulation: the frontal polar cortex and the dlPFC
[19,47,105,144,173,403,404,457,496,497,503,552,620,834,875,876,908,922]. In line with these
findings, we have previously identified structural and functional abnormalities in these regions
[617,928]. Further, studies have shown that pain interferes with cognitive processes [529]. Given
the possibility that cognitive resources are limited, these competing demands can interfere with
one another, and impede cognitive performance. Evidence for the cognitive interference of pain
comes from studies that have shown that acute pain can modulate performance in a cognitive
task [291,529,687,697]. Further evidence comes from studies that have reported that chronic pain
patients have slower reaction times in cognitive tasks [370,379,526,850,928]. Within the context
of these findings, our results may represent that FA differences in the corpus callosum are related
to differential connectivity in the brain, and potentially related to interaction between executive
cognitive function and pain in TMD.
7.4.5. Intense and prolonged TMD may drive plasticity in white matter
We have previously reported that both pre-existing and chronic pain-driven factors contribute to
gray and white matter brain abnormalities in patients with IBS (see: [98,156]. In the current
study, the intensity and unpleasantness of TMD pain also correlated with regions with lower FA

204
in patients. There are two possible explanations of the finding of reduced FA correlated with pain
intensity. Firstly, it is conceivable that abnormalities in white matter existed prior to the onset of
TMD and, therefore, could influence the degree of TMD pain. Secondly, more intense,
unpleasant or longer TMD pain may induce white matter plasticity, reflected as changes in FA.
Evidence that pain induces or is associated with structural brain plasticity comes from recent
studies that have reported that gray matter abnormalities in chronic pain are reversible, once the
source of the pain has been resolved [384,741,782], which suggests that pain can potentially
induce gray matter plasiticity. In this study, decreased FA in the right CNV was related to the
duration of TMD, which provides support for the latter possibility – that is, pain-induced
plasticity. Similarly, the relationship between regions with abnormal FA in the brain (i.e., tracts
adjacent to the vlPFC, within the IC and coursing through the thalamus) and TMD pain intensity
suggest that TMD pain may be driving the decrease in FA, and the observed group difference. As
mentioned above, these findings suggest that increased peripheral firing or abnormal firing in the
CNV of patients with TMD or decreased dysfunctional central antinociceptive systems may alter
its microstructure over time. This represents a particularly novel finding given the emphasis of a
central ætiology for TMD [757].
7.4.6. Cellular basis of changes in FA
The diffuse pattern of white matter abnormality is likely due to the multidimensional nature of
chronic pain that impacts several anatomical pathways. However, our diffuse white matter
findings need to be interpreted with caution due to the global difference in FA we identified
between patients and controls.
The factors that contribute to reduced FA may be macrostructural, such as increased branching,
more crossing fibres or larger tracts (more axons) and/or microstructural changes such as cell
swelling (œdema), changes to protein filaments (neurofilament phosphorylation), disruptions to
the cell membranes, and, to a certain extent, decreased myelin [66,67]. Recent studies have
suggested that chronic pain may induce neuroinflammation in the brain [238,381,920], which can
lead to changes in both gray matter and white matter that can be detected by MRI. Therefore, the
observed changes in white matter may, in fact, be related to neuroinflammation rather than
increases or decreases in the number or size of cells and/or axons present in the fields being
studied. Our study has examined other measures of white matter microstructure (MD, RD and

205
λ1) to better characterize group differences in FA. The neuroinflammation model is supported by
our finding that regions with lower FA in TMD have higher MD and RD, which are markers of
inflammation and/or œdema [63,66].
7.4.7. Study limitations
The patients in this study patients did not self-report any previous diagnosis of depression or
anxiety, although these exist in some patients with TMD [283,808,850]. Thus, the contribution of
these factors in white matter abnormalities in TMD in general cannot be excluded. Another
limitation of the current study is that microstructural abnormalities in white matter tracts cannot
definitively be attributed to functional abnormalities, as this study only investigates structural
abnormalities. For example, the CNV is a mixed cranial nerve, with motor and sensory fibres. It
was not possible to disentangle whether the structural abnormalities are related to sensory
abnormalities, motor abnormalities, or both. The resolution of DTI analysis based on voxels that
are much larger than the width of a single axon. The current methods allow generalized
inferences to be made only about the local microstructure within a voxel, but cannot differentiate
which tracts are affected.
7.5. Conclusion
This is the first study to identify abnormalities in the CNVs of patients with TMD, suggesting a
peripheral contribution to TMD. We have also demonstrated widespread white matter brain
abnormalities in patients with TMD in sensory, motor and cognitive-affective regions, consistent
with a central component to TMD. Finally, we have identified that patients with TMD have
abnormal connectivity of cognitive-affective regions of the brain. In sum, we provide structural
findings consistent with observed behavioural and functional finding in TMD, supporting a
model of central abnormalities. while also present novel evidence of a potential peripheral
contribution to TMD.

206
Chapter 8 General Discussion
This is the first comprehensive study of both gray and white matter structural abnormalities in
idiopathic TMD, and the first to investigate focal age-related gray matter abnormalities in
chronic pain. The main findings of this thesis are that patients with TMD show:
1) Abnormal gray matter in brain areas implicated in somatosensory, motor, cognitive and
modulatory functions, including a) increased cortical thickness in the S1 and frontal
cortex, b) chronic pain intensity or unpleasantness-dependent cortical thinning in the
cingulate, motor, and orbitofrontal cortices, c) pain chronicity-dependent thalamic gray
matter increases, d) abnormal impact of neuroticism on orbitofrontal gray matter;
2) Abnormal gray matter aging in focal brain regions of pain and motor systems, some of
which is impacted by chronic pain chronicity;
3) Widespread brain white matter abnormalities, including a) in tracts containing
connections for sensory, motor, cognitive and pain functions, b) prominently stronger
corpus callosum-frontal pole connectivity and weaker dlPFC connectivity, c) chronic
pain/unpleasantness-dependent abnormalities, d) abnormally high MD and RD levels,
markers of brain neuroinflammation; and
4) Trigeminal nerve abnormalities, exacerbated by TMD chronicity.
Together, these findings suggest that idiopathic TMD are complex, and are accompanied by both
central and peripheral abnormalities, some of which may be pre-existing abnormalities that could
predispose persons to developing TMD.
Several factors can contribute to the observed structural abnormalities in the brains of patients
with TMD. The first possibility is that these changes are “all-or-none”, where having TMD is
accompanied with these brain abnormalities. Another possibility is that the observed changes
occur over time, the prolonged effect of pain progressively affects brain structure. Another
possibility is also related to duration, but different in that the magnitude of pain (intensity or
unpleasantness) can affect the magnitude of change in the brain: higher pain intensity drives
changes in the structure of the brain. Together, these possibilities may be referred to as pain-

207
driven abnormalities. Finally, in contrast to pain-driven abnormalities, there could be pre-
existing brain abnormalities, possibly related to stable personality traits that can predispose
persons to developing TMD. The relative contribution of these factors are discussed below.
8.1. Pain-driven abnormalities along nociceptive pathways
In this thesis, structural abnormalities were identified at several locations along the ascending
nociceptive pathway. This pathway begins with the CNV in the periphery. Nociceptive
information from the TMJ and the muscles of mastication is carried to the CNS via the CNV.
Nociceptive C- and Aδ-fibres project to second-order NS and WDR neurones in the VBSNC
[784]. These cells ascend to various brainstem nuclei, and the thalamus via the TTT, where they
project to third-order neurones, which form thalamocortical tracts [784]. This thesis has reported
decreased FA, and increased MD, and RD along the CNV of patients with idiopathic TMD.
These data suggest that there is increased permeability in the membrane or decreased myelin
insulation [67] (see below). Further, these abnormalities are negatively correlated to TMD
duration in the right CNV– patients with relatively shorter TMD durations have relatively normal
FA values, and these values deviate from normal values as TMD duration increases. These data
suggest that chronicity of TMD pain contributes to changes in the microstructure of the nerve.
Therefore, there may be increased nociceptive input from the periphery, or
decreased/dysfunctional central antinociceptive systems, or progressive abnormalities along the
nerve root, which may cause aberrant firing patterns. In line with the revised pain adaptation
model [431], changes in the recruitment of motor units, over time, can themselves become a
source of pain. Therefore, it is possible that increased nociceptive activity over time alters facial
motor activity. Prolonged alterations in motor activity from the orofacial region can itself
become pathological and a source of nociceptive input. This prolonged input, in turn, may alter
CNV microstructure and structures along the ascending nociceptive pathways.
In support of this hypothesis is the finding that gray matter volume in the thalamus positively
correlated to TMD duration. Similar to our finding in the CNV, with increased TMD chronicity,
patients showed increased gray matter in the thalamus. The age-by-group study confirmed the
finding that thalamic gray matter changes are driven by duration. It is therefore likely that
ascending nociceptive pathways undergo progressive change with long-term TMD. Similarly,
several studies reported pain-driven gray matter structural abnormalities in chronic pain

208
[27,98,206,268,303,356,490,502,740,764-766,772,888,889,919,963] (see Table 2-3). Support for
the concept of chronic pain-driven gray matter changes comes from a study by Teutsch and
colleagues [852], who reported increased S1 and MCC gray matter volume after eight
consecutive days of 20-minute paradigm of acute nociceptive stimulation to the volar forearm.
Subjects had thicker S1 and MCC. Additionally, this thesis reports abnormalities along white
matter tracts connecting the various levels of the ascending trigeminal nociceptive system – the
TTT, white matter coursing through the thalamus, TCT coursing through the somatomotor region
of the IC and adjacent to the somatosensory and motor cortices. Furthermore, the white matter
tracts within the thalamus and the motor region of the IC are related to pain intensity and/or
unpleasantness. For a summary schematic of abnormalities along the ascending sensory (and
nociceptive) pathway, see Figure 8-1.
In sum, this thesis has demonstrated structural abnormalities at several points along the
ascending nociceptive pathway – including key gray matter regions implicated in nociception,
and the tracts that connect them.

209
Figure 8-1: Proposed model of neural basis of sensory abnormalities in TMD. Altered
nocicieptive activity in the periphery may be related to decreased FA in CNV, and TTT in the
brainstem. This altered activity also leads to progressive increases in thalamic GM volume,
reduced FA in the TCT and increased cortical thickness in the orofacial region of S1. Brain
outline modified from the PHT500Y laboratory manual, University of Toronto.
8.2. Abnormalities in motor regions
The pain and motor systems interact at several levels within the nervous system (see Section
2.1.6.). This thesis reports pain-related structural abnormalities in gray matter and white matter
associated to motor function in patients with TMD. Specifically, the thickness of the orofacial
region of M1 was negatively correlated to pain intensity in patients with TMD. Similarly,
patients with TMD had lower FA in white matter tracts associated to motor regions, and FA in
these regions was negatively correlated to pain intensity. Furthermore, our lab has previously
reported that the same group of patients with TMD had increased cortical activity in motor
regions and sluggish reaction times during a cognitive task [928]. Finally, the age-by-group
analysis revealed abnormal aging of the PMC and the dorsal striatum in TMD, compared to
healthy controls.

210
There are three possible mechanisms that may explain the observed abnormalities in motor
regions and tracts (see Figure 8-2): (1) nociceptive input dampens motor output to prevent
further damage; (2) motor cortex modulates pain intensity; or (3) reduced movement to avoid
pain may affect motor cortex thickness. Future work should specifically address the pain-motor
interaction in TMD to determine which of these mechanisms is occurring in TMD.
Previous studies have reported that nociceptive input inhibited motor cortex excitability
[3,157,301,302,638,856], and that motor cortex stimulation modulated the perceived intensity of
pain in chronic pain [103,117,118,301,331,342,344,345,535,548,657,689]. Furthermore, a study
by Boudreau and colleagues [109] reported that task training-related motor cortex plasticity was
inhibited by noxious stimulation of the intraoral cavity. Additionally, several studies have
reported abnormal stimulus-evoked fMRI activity in motor regions of chronic pain patients,
although the implication of these abnormalities is not often discussed (see: [23]). For instance,
Kirveskari et al. [492] reported that patients with CRPS had weaker M1 reactivity with increased
spontaneous pain ratings, and a negative correlation between pain intensity and M1 activation.
Therefore, studies in experimental pain paradigms and in chronic pain patients have
demonstrated that nociceptive input modulates M1 function. Furthermore, evidence suggests that
M1 activity can modulate pain perception.
Alternatively, the observed relationship between pain intensity and M1 thickness may be related
to the possibility that patients with TMD make fewer jaw movements to avoid eliciting pain.
Based on the concepts of use-dependent plasticity (or in this case disuse-dependent plasticity),
this reduction in jaw activity may lead to atrophy of the orofacial region of the motor cortex
[576].

211
Figure 8-2: Proposed model for the neural basis of motor abnormalities in TMD. Altered
nociceptive input inhibits motor cortex output in an intensity dependent manner, and reduces FA
in corticocortical tracts connecting S1 and M1. Decreased motor output causes disuse-related
plasticity along corticofugal tracts (including corticobulbar tracts) and the motor portion of the
CNV.
8.3. Abnormalities in cognitive-modulatory brain regions
This thesis has demonstrated structural abnormalities in brain areas that have been implicated in
cognitive/modulatory functions, providing a structural framework that could underlie the
negative impact of chronic pain on cognitive abilities, including concentration, learning,
memory, and attentional focus [26,250,275,365,369,455,489,666,928]. Specifically, we have
reported thicker cortex in the vlPFC. Furthermore, patients with TMD had reduced measures of
white matter integrity in white matter adjacent to the vlPFC. The vlPFC has been implicated in
various aspects of pain, including anticipation [752] and modulation [909], as well as
behavioural inhibition [32]. Furthermore, studies have reported hypervigilance in patients with
TMD [435,808], which is related to increased pain anticipation. Therefore, the observed
structural abnormalities in the vlPFC may be related to patients’ hypervigilance and increased
anticipation to pain (see Figure 8-3).

212
Another key finding of this thesis was that the left frontal polar cortex was thicker in patients
with TMD, than in controls. Additionally, probabilistic tractography revealed that the frontal
polar cortices were more connected interhemispherically – the right and left frontal poles were
more connected in TMD than in pain free controls. The frontal pole can be activated during
spontaneous pain in patients with chronic pain [47]. Additionally, the frontal pole has also been
implicated in a number of complex executive cognitive functions such as learning behavioural
routines [457,496,834], cognitive branching (the ability to put a pending task on hold to execute
an ongoing one) [497], behavioural flexibility/adaptability [105] and post-hoc monitoring or
evaluating decisions based on feedback [875]. The concept of cognitive branching would suggest
that there is a limited resource of working memory, and that tasks that are more behaviourally-
relevant (or salient) are prioritized [44,45,155,495,497,498]. Specifically, the frontal polar cortex
monitors the relevant salience (or value) of a given stimulus and select behavioural outcomes
accordingly. Given that pain is an inherently salient stimulus [265,530,933], it is conceivable that
pain engages the frontal pole. Further, a study by Harman and Ruyak [409] reported that people
with persistent low-level pain related to their work habits had cognitive deficits, indicative of an
interference effect of pain. Therefore, the frontal pole may process the cognitive dimension of
pain, which suggests that pain has a cognitive load. In the short term, as in cases of acute pain,
the frontal pole may direct behaviour to protect the subject. However, in chronic pain conditions,
this region may be continuously engaged. Therefore, when a person with chronic pain is
distracted, or performing a demanding cognitive task, the frontal pole may be “putting the pain
on hold”, allocating resources to the other, competing cognitive demands (see Figure 8-3).
Accordingly, Eccleston and colleagues [289] showed that chronic pain patients could briefly be
disengaged from pain during attentional switches. Further support for this hypothesis comes from
studies that demonstrate that distraction can reduce pain perception
[123,430,584,588,687,864,888,891,935,955]. These studies have demonstrated that distraction-
based modulation of pain requires the recruitment of antinociceptive brain regions. However,
these systems may not be effective in patients with TMD in two ways: (1) there is evidence of
dysfunctional antinociceptive systems (see Section 2.2.3.1) [305,491], and (2) the cognitive
branching model suggests that cognitive resources are limited – there are only so many tasks that
can be juggled [497]. Therefore, the cognitive load of chronic pain could impact the a patient’s
ability to properly complete tasks [529]. In support of this idea, we have recently shown that
patients with TMD have sluggish reaction times to low and high conflict cognitive interference

213
tasks, but not to a simple sensorimotor task [928]. Therefore, in the context of use-dependent
plasticity [576], one possible explanation for the observed thickening of the frontal pole and the
increased white matter connectivity between the bilateral frontal poles is that chronic TMD pain
bears a cognitive load that the brain needs to ‘put on hold’ in order to address more immediate
environmental demands.
Figure 8-3: Proposed model of the neural basis for cognitive abnormalities in TMD. Altered
nociceptive activity alters the structure and connectivity of cognitive brain regions, including the
vlPFC and the frontal pole.
Additionally, this thesis found that patients with TMD have abnormal gray matter aging in the
aMCC/pgACC. Furthermore, these patients have lower FA in the cingulum bundle (adjacent to
the MCC). This region receives orofacial nociceptive input from thalamic nuclei that are part of
the STT and TTT systems [189,276]. The aMCC/pgACC is a complex, multimodal region that
has been implicated in a number of functions [69]. For instance, this region has been identified as

214
a node in the salience network,!and has been implicated in aspects of salience
[213,229,262,263,774,849,927],!and pain [230,231,259,265,440,508,528,632,933]. Furthermore,
the cingulate cortex has also been implicated in the cognitive and affective processing of pain
[218,230,529,716,776,777,934,936], and the MCC is involved in action selection and modulation
of motor output in response to aversive stimuli [790,898,904]. Therefore, it is possible that these
structural abnormalities in the cingulate cortex are related to cognitive and attentional
abnormalities in TMD.
Patients with TMD also had widespread white matter abnormalities in white matter tracts related
to pain-modulatory brain regions. Specifically, patients had lower FA and increased MD and RD
in white matter tracts coursing to the OFC and sgACC, the anterior corona radiata (in the ICAL,
which projects to the prefrontal cortex) and the external/extreme capsule adjacent to the right
mid-insula. The insula, mid-cingulate cortex have been implicated in pain perception, negative
emotion, and cognitive function [23,94,108,116,183,222,227,304,447,752,790,898]. The OFC
and sgACC are thought to contribute to descending pain modulation [95]. Furthermore, patients
with TMD had abnormal connectivity to the dlPFC, a region implicated in pain modulation
[552], selecting information about the sensory environment (attention, self-monitoring,
personality, working memory) and decision-making [54,700] (see Figure 8-4). Patients with
TMD have widespread acute pain hypersensitivity [305,563,565,883], abnormal DNIC
[113,491,756], while having sluggish reaction times to a cognitive task [928].
In sum, this thesis provides a potential structural neural basis for cognitive deficits and
dysfunctional pain-modulatory circuits in patients with idiopathic TMD.
8.4. Neuroticism and TMD
It is possible that some factors predispose persons to develop TMD. A recent prospective study
by Fillingim and colleagues [315] reported that certain psychological traits could, to some extent,
predict TMD onset, including neuroticism. In the current thesis, we report that in patients with
TMD there is a positive correlation between neuroticism and cortical thickness in the left vmPFC
(part of the OFC); an area that normally shows a negative correlation between gray matter and
neuroticism [954]. Neuroticism has been defined as a stable personality trait associated with
heightened sensitivity and/or processing of negative affective stimuli [178,179,907]. Therefore, it
is not surprising that patients with high neuroticism scores have lower pain threshold and pain

215
tolerance, and are more distressed by pain in experimental settings and chronic pain [408].
Neuroticism has been associated with pain-related suffering [35,408], pain sensitivity
[177,373,907], nerve injury outcomes and the development of neuropathic pain [848], and TMD
[315]. However, the mechanism by which inherent personality-related factors reduce the brain’s
capacity to effectively modulate nociceptive input and pain perception, remains unclear.
Furthermore, not all chronic pain patients have high neuroticism scores, and not all persons with
neuroticism have chronic pain [179]. Therefore, neuroticism alone is not sufficient to develop
chronic pain, however it can contribute to the severity of pain in chronic pain patients. Therefore,
our observation highlights the contribution of neuroticism to abnormal gray matter in the OFC of
patients with TMD and, perhaps, to the development of TMD (see Figure 8-4). Similarly, in IBS
patients, neuroticism correlated to FA in the left MD thalamus, but not in controls [156]. An
alternative interpretation is that chronic pain may have modified the relationship between
neuroticism and OFC thickness. However, the lack of correlation of TMD duration and
neuroticism scores in our patients with TMD does not support this hypothesis.
We also found that unpleasantness is negatively correlated to gray matter in an adjacent region of
the OFC. The OFC has been implicated in cognitive reappraisal and emotional regulation [723]
related to interoception and somatovisceral stimuli, for directing our behaviour appropriately
[57,935]. The OFC is also thought to play a role in mental flexibility and adaptability (for a
critical review, see: [770]. Therefore, our finding suggests that patients with TMD may have
abnormal emotional regulation and reappraisal, which may lead to pain behaviours and
exaggerated affective responses to pain.

216
Figure 8-4: Proposed model for the neural basis of modulatory system abnormalities in TMD.
The relationship of neuroticism and cortical thickness in the OFC alters the structure and
antinociceptive function in pain modulatory brain regions, including the sgACC. The dlPFC is
also has reduced white matter connectivity in patients with idiopathic TMD, which may be
another substrate for dysfunctional pain modulation networks in TMD.
8.5. Basis of structural brain abnormalities
The cellular and molecular bases of MRI-detectable gray and white changes in chronic pain are
not known but there is now evidence to support several hypotheses. Mechanisms of gray matter
change include neuronal and/or glial death [575], but recent evidence suggests that, to some
extent, gray matter losses are likely related to density of small dendritic spines [278,608], and the
remodeling of neuronal processes [537]. Alternatively, reversible gray matter changes in chronic
pain may be caused by neuroinflammation [238,381,920], and induce MRI-detectable increases
in gray matter. This mechanism could explain abnormal thicknening (or maintenance) of gray
matter volume/thickness. For the observed gray matter losses, however, we cannot rule out that
cell death is not occurring – healthy populations lose neurones as they age, and persons with
neurodegenerative diseases suffer increased rates of atrophy related to cell death.

217
The factors that contribute to reduced FA may be macrostructural, such as increased branching,
more crossing fibres or larger tracts (more axons) and/or microstructural changes such as cell
swelling (œdema), changes to protein filaments (neurofilament phosphorylation), disruptions to
the cell membranes, and, to a certain extent, decreased myelin [66,67]. Furthermore, the
neuroinflammation hypothesis for gray matter changes can also apply to white matter changes.
Therefore, the observed changes in white matter may, in fact, be related to neuroinflammation
rather than increases or decreases in the number or size of cells and/or axons present in the fields
being studied. This thesis has examined other measures of white matter microstructure (MD, RD
and λ1) to better characterize group differences in FA. The neuroinflammation model is
supported by the finding that regions with lower FA in TMD have higher MD and RD, which are
markers of inflammation and/or œdema [63,66].
8.6. Study limitations
A few limitations in study design and results should be considered in interpretation of the data
reported in this thesis. Some of the limitations specific to each study have been discussed within
the relevant chapters.
First, the patients that were recruited for this project were deemed to have TMD based on
longstanding criteria used by the TMD clinicians at the Mount Sinai Hospital Pain Clinic and
represent a spectrum of idiopathic TMD seen clinically. However, this categorization does not
strictly adhere to the standard TMD-RDC, as described by Dworkin and LeResche [285].
Second, it is not known whether there is an impact of long-term use of medications in some of
the patients on gray and white matter plasticity. Recent evidence suggests that medications
commonly used to manage pain, such as anti-inflammatory medications and opiates, can impact
brain structure [916,962]. Only one subject was taking an opiate, hydromorphone, and so this is
of less concern to this study, but considering that thirteen of seventeen of the patients included in
this study were taking NSAIDs, it is possible that the observed age-related findings – especially
those of gray matter maintenance, may be related to the effect of these drugs..
Furthermore, it is not possible to totally rule out a contribution of ventricular volume to the VBM
analysis of subcortical structures. Additionally, some of the patients had co-morbid chronic pains
(see Table 5-1), and these pains (mostly related to TMD) may have contributed to our findings.

218
Another limitation is that duration (and intensity) may not fully describe the effect of pain on
gray and white matter structure. Due to the nature of TMD pain, other factors that were not
collected, such as the number of days of pain in a month may have more explanatory variance.
Patients also did not self-report any previous diagnosis of depression or anxiety, although these
exist in some patients with TMD [283,808,850]. Thus the contribution of these factors to gray
and white matter abnormalities in TMD cannot be ruled out.
Most of the patient sample had bilateral pain, but our findings tended to be unilateral. These
inconsistencies can simply be due to statistical thresholds. However, future studies should limit
patient recruitment to unilateral pain to determine the laterality of CNS abnormalities.
Another limitation of this thesis is that it is a cross-sectional examination of structural
abnormalities in TMD. Therefore, the interpretations of “pain-driven” and “pre-existing”
abnormalities are limited, and should be taken with caution. Furthermore, quantitative sensory
testing was not performed on the patient group recruited for this study, and so we cannot directly
relate our findings of structural abnormalities to these behavioural abnormalities.
Despite these limitations, this thesis does provide considerable evidence in line with previous
findings in the literature about gray and white matter abnormalities in chronic pain.
8.7. Future directions
This thesis has clearly demonstrated structural gray and white matter abnormalities in the brain
of patients with TMD. Future studies could build on these findings to provide more insight into
the potential underlying mechanisms. For example, behavioural measure of pain sensitivity in
these patients (obtained through quantitative sensory testing) would be useful to link the
structural abnormalities with perceptual abnormalities. Another hypothesis put forward by this
thesis is that patients with TMD (and potentially all chronic pain patients) have deficient
cognitive abilities due to the cognitive load of pain overloading the cognitive branching system.
Therefore, future studies could conduct cognitive tests that have been used to establish the
presence of a cognitive branching system in the brain to assess whether patients with TMD do
have deficient branching.
It was also demonstrated that pain intensity is related to abnormal structure in the motor cortex.
However, whether this abnormality is related to function has not been determined. Therefore, in

219
the future, motor function could be tested in the patients with TMD in the orofacial region and
elsewhere in the body. Future studies could also investigate the functional brain abnormalities
related to abnormal motor behaviour. In line with this experiment, we have clearly demonstrated
abnormalities along the CNV of patients with TMD. Nerve conduction studies could also be used
to test whether the sensory or motor aspect of the nerve is affected, and how these structural
abnormalities affect nerve function.
Furthermore, other imaging modalities can be used to assess the molecular basis of structural
abnormalities in the brain. For instance, magnetic resonance spectroscopy can be used to assess
whether there are differences in markers of specific neural structure and viability, and relate
these to structural abnormalities.
Animal models of TMD should be used to investigate the relationship between MRI-detectable
structural changes in the brain, and pain-related behaviours. These structural abnormalities could
be further characterized with genetic, cellular, electrophysiological and molecular methods.
Functional brain imaging with arterial-spin labeling, an imaging modality that allows baseline
imaging, like PET but not invasive, could be used to assess spontaneous pain in patients with
TMD. This would provide insight into which brain regions are implicated in pain processing, and
abnormally activated in TMD pain.
Prospective studies of TMD could also provide insight about the risk factors and development of
TMD. Patients with TMD could be followed over time to assess whether they respond to
treatment, or not. The factors that contribute and/or predict whether patients are treatment-
responders and non-responders could be assessed. Neuroimaging at several time points could
elucidate how the structure and function of the brain are affected by the cumulative effect of
pain, and how these abnoramltities may vary between the groups.
Fewer patients with idiopathic TMD respond to treatment, compared to patients with post-
traumatic TMD. Therefore, another study could compare whether the same functional and
structural abnormalities occur in patients with idiopathic and post-traumatic TMD. The study
could provide insight about peripheral pain-driven abnormalities (in post-traumatic TMD) and
central mechanisms that contribute to TMD.

220
8.8. Conclusions
In sum, this thesis provides novel evidence for both peripheral nerve and CNS abnormalities that
contribute to TMD pain. In support of a peripheral contribution, it was found that patients with
TMD had lower FA in both CNVs and FA in the right CNV was negatively correlated with TMD
duration. In support of CNS abnormalities, structural abnormalities in gray and white matter
were found along the ascending nociceptive, descending pain-modulatory, motor and cognitive
pathways. Furthermore, it was demonstrated that some, but not all of the observed structural
abnormalities are pain-driven. Other brain abnormalities do not show a relationship to pain
characteristics, but rather to factors independent of pain, such as age and neuroticism, a stable
personality trait. The latter findings suggest the presence of pre-existing abnormalities that may
be a predisposing risk factor to the development of TMD.

221
References
[1] National Institutes of Health Technology Assessment Conference on Management of
Temporomandibular Disorders. Bethesda, Maryland, April 29-May 1, 1996. Proceedings.
Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;83:49-183.
[2] Adachi K, Lee JC, Hu JW, Yao D, Sessle BJ. Motor cortex neuroplasticity associated with
lingual nerve injury in rats. Somatosens Mot Res 2007;24:97-109.
[3] Adachi K, Murray GM, Lee J-C, Sessle BJ. Noxious lingual stimulation influences the
excitability of the face primary motor cerebral cortex (face MI) in the rat. J Neurophysiol
2008;100:1234-1244.
[4] Adachi K, Murray GM, Lee JC, Sessle BJ. Noxious lingual stimulation influences the
excitability of the face primary motor cerebral cortex (face MI) in the rat. J Neurophysiol
2008;100:1234-1244.
[5] Adams JE. Naloxone reversal of analgesia produced by brain stimulation in the human. Pain
1976;2:161-166.
[6] Adrian ED. Afferent discharges to the cerebral cortex from peripheral sense organs. J Physiol
1941;100:159-191.
[7] Afif A, Hoffmann D, Minotti L, Benabid AL, Kahane P. Middle short gyrus of the insula
implicated in pain processing. Pain 2008;138:546-555.
[8] Ahdab R, Ayache SS, Brugieres P, Goujon C, Lefaucheur JP. Comparison of "standard" and
"navigated" procedures of TMS coil positioning over motor, premotor and prefrontal
targets in patients with chronic pain and depression. Neurophysiol Clin 2010;40:27-36.
[9] Aigner M, Robert Lukas J, Denk M, Ziya-Ghazvini F, Kaider A, Mayr R. Somatotopic
organization of primary afferent perikarya of the guinea-pig extraocular muscles in the
trigeminal ganglion: a post-mortem DiI-tracing study. Exp Eye Res 2000;70:411-418.
[10] Aimone LD, Gebhart GF. Stimulation-produced spinal inhibition from the midbrain in the
rat is mediated by an excitatory amino acid neurotransmitter in the medial medulla. J
Neurosci 1986;6:1803-1813.

222
[11] Al-Chaer ED, Feng Y, Willis WD. Visceral pain. A disturbance in the sensorimotor
continuum? Pain Forum 1998;7:117-125.
[12] Albanese MC, Duerden EG, Rainville P, Duncan GH. Memory Traces of Pain in the Human
Cortex. J Neurosci 2007;27:4612-4621.
[13] Aldridge JW, Anderson RJ, Murphy JT. The role of the basal ganglia in controlling a
movement initiated by a visually presented cue. Brain Res 1980;192:3-16.
[14] Alexander AL. Deterministic white matter tractography. In: DK Jones, editor. Diffusion
MRI: Theory, Methods and Applications. New York: Oxford University Press, 2011. pp.
383-395.
[15] Alexander DC, Seunarine KK. Mathematics of crossing fibres. In: DK Jones, editor.
Diffusion MRI: Theory, Methods and Applications. New York: Oxford University Press,
2011. pp. 451-464.
[16] Allison T, McCarthy G, Wood CC, Darcey TM, Spencer DD, Williamson PD. Human
cortical potentials evoked by stimulation of the median nerve. I. Cytoarchitectonic areas
generating short-latency activity. J Neurophysiol 1989;62:694-710.
[17] Amano N, Hu JW, Sessle BJ. Responses of neurons in feline trigeminal subnucleus caudalis
(medullary dorsal horn) to cutaneous, intraoral, and muscle afferent stimuli. J
Neurophysiol 1986;55:227-243.
[18] Aminoff MJ. Brown-Séquard and His Work on the Spinal Cord. Spine 1996;21:133-140.
[19] Andersen E. Periaqueductal gray and cerebral cortex modulate responses of medial thalamic
neurons to noxious stimulation. Brain Res 1986;375:30-36.
[20] Andrew D, Krout KE, Craig AD. Differentiation of lamina I spinomedullary and
spinothalamic neurons in the cat. J Comp Neurol 2003;458:257-271.
[21] Apkarian AV. Thalamic anatomy and physiology of pain perception: connectivity, somato-
visceral convergence and spatio-temporal dynamics of nociceptive information coding.
In: JM Besson, G Guilbaud, H Ollat, editors. Forebrain areas involved in pain processing.
Paris: John Libbey Eurotext, 1995. pp. 93-118.
[22] Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol
2009;87:81-97.

223
[23] Apkarian AV, Bushnell MC, Treede RD, Zubieta J. Human brain mechanisms of pain
perception and regulation in health and disease. Eur J Pain 2005;9:463-484.
[24] Apkarian AV, Hodge CJ. Primate spinothalamic pathways: III. Thalamic terminations of the
dorsolateral and ventral spinothalamic pathways. J Comp Neurol 1989;288:493-511.
[25] Apkarian AV, Shi T. Squirrel monkey lateral thalamus. I. Somatic nociresponsive neurons
and their relation to spinothalamic terminals. J Neurosci 1994;14:6779-6795.
[26] Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN,
Chialvo DR. Chronic pain patients are impaired on an emotional decision-making task.
Pain 2004;108:129-136.
[27] Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic
back pain is associated with decreased prefrontal and thalamic gray matter density. J
Neurosci 2004;24:10410-10415.
[28] Apkarian AV, Stea RA, Manglos SH, Szeverenyi NM, King RB, Thomas FD. Persistent
pain inhibits contralateral somatosensory cortical activity in humans. Neurosci Lett
1992;140:141-147.
[29] Applebaum AE, Beall JE, Foreman RD, Willis WD. Organization and receptive fields of
primate spinothalamic tract neurons. J Neurophysiol 1975;38:572-586.
[30] Applebaum AE, Leonard RB, Kenshalo DR, Jr., Martin RF, Willis WD. Nuclei in which
functionally identified spinothalamic tract neurons terminate. J Comp Neurol
1979;188:575-586.
[31] Arendt-Nielsen L, Brennum J, Sindrup S, Bak P. Electrophysiological and psychophysical
quantification of temporal summation in the human nociceptive system. EurJApplPhysiol
1994;68:266-273.
[32] Aron AR, Fletcher PC, Bullmore ET, Sahakian BJ, Robbins TW. Stop-signal inhibition
disrupted by damage to right inferior frontal gyrus in humans. Nat Neurosci 2003;6:115-
117.
[33] Arvidsson J. Somatotopic organization of vibrissae afferents in the trigeminal sensory nuclei
of the rat studied by transganglionic transport of HRP. J Comp Neurol 1982;211:84-92.

224
[34] Asaki S, Sekikawa M, Kim YT. Sensory innervation of temporomandibular joint disk. J
Orthop Surg (Hong Kong) 2006;14:3-8.
[35] Asghari A, Nicholas MK. Personality and pain-related beliefs/coping strategies: a
prospective study. Clin J Pain 2006;22:10-18.
[36] Ashburner J, Friston KJ. Voxel-based morphometry--the methods. Neuroimage
2000;11:805-821.
[37] Ashburner J, Friston KJ. Voxel-based morphometry. In: KJ Friston, editor. Statistical
parametric mapping the analysis of funtional brain images. Amsterdam ; Boston:
Elsevier/Academic Press, 2007. pp. 92-98.
[38] Atweh SF, Kuhar MJ. Autoradiographic localization of opiate receptors in rat brain. III. The
telencephalon. Brain Res 1977;134:393-405.
[39] Augustine JR. The insular lobe in primates including humans. Neurological Res 1985;7:2-
10.
[40] Augustine JR. Circuitry and functional aspects of the insular lobe in primates including
humans. Brain Res Rev 1996;22:229-244.
[41] Avivi-Arber L, Martin R, Lee JC, Sessle BJ. Face sensorimotor cortex and its
neuroplasticity related to orofacial sensorimotor functions. Arch Oral Biol 2011.
[42] Ayesh EE, Jensen TS, Svensson P. Somatosensory function following painful repetitive
electrical stimulation of the human temporomandibular joint and skin. Exp Brain Res
2007;179:415-425.
[43] Azkue JJ, Mateos JM, Elezgarai I, Benitez R, Lazaro E, Streit P, Grandes P. Glutamate-like
immunoreactivity in ascending spinofugal afferents to the rat periaqueductal grey. Brain
Res 1998;790:74-81.
[44] Badre D, D'Esposito M. Is the rostro-caudal axis of the frontal lobe hierarchical? Nat Rev
Neurosci 2009;10:659-669.
[45] Badre D, Hoffman J, Cooney JW, D'Esposito M. Hierarchical cognitive control deficits
following damage to the human frontal lobe. Nat Neurosci 2009;12:515-522.

225
[46] Bakke M, Hansdottir R. Mandibular function in patients with temporomandibular joint pain:
a 3-year follow-up. Oral surgery, oral medicine, oral pathology, oral radiology, and
endodontics 2008;106:227-234.
[47] Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN, Parrish TB, Apkarian AV.
Chronic pain and the emotional brain: specific brain activity associated with spontaneous
fluctuations of intensity of chronic back pain. J Neurosci 2006;26:12165-12173.
[48] Baliki MN, Geha PY, Apkarian AV. Parsing pain perception between nociceptive
representation and magnitude estimation. J Neurophysiol 2009;101:875-887.
[49] Baliki MN, Geha PY, Apkarian AV, Chialvo DR. Beyond feeling: chronic pain hurts the
brain, disrupting the default-mode network dynamics. J Neurosci 2008;28:1398-1403.
[50] Ballantine HT, Jr., Cassidy WL, Flanagan NB, Marino R, Jr. Stereotaxic anterior
cingulotomy for neuropsychiatric illness and intractable pain. J Neurosurg 1967;26:488-
495.
[51] Ballantine HT, Jr., Giriunas I. Treatment of intractable psychiatric illness and chronic pain
by stereotactic cingulotomy. In: HH Schmidek, WH Sweet, editors. Operative
neurosurgical techniques, Vol. Second Edtion. Toronto: Grune and Stratton, 1988. pp.
1069-1075.
[52] Bammer R. Basic principles of diffusion-weighted imaging. Eur J Radiol 2003;45:169-184.
[53] Bantli H, Bloedel JR, Thienprasi P. Supraspinal interactions resulting from experimental
dorsal column stimulation. J Neurosurg 1975;42:296-300.
[54] Barbas H. Anatomic basis of cognitive-emotional interactions in the primate prefrontal
cortex. Neurosci Biobehav Rev 1995;19:499-510.
[55] Barcelo AC, Filippini B, Pazo JH. The Striatum and Pain Modulation. Cell Mol Neurobiol
2011.
[56] Baron R, Baron Y, Disbrow E, Roberts TP. Brain processing of capsaicin-induced
secondary hyperalgesia: a functional MRI study. Neurology 1999;53:548-557.
[57] Barrett LF, Mesquita B, Ochsner KN, Gross JJ. The experience of emotion. Ann Rev
Psychol 2007;58:373-403.

226
[58] Basbaum AI, Fields HL. Endogenous pain control systems: brainstem spinal pathways and
endorphin circuitry. Annu Rev Neurosci 1984;7:309-338.
[59] Basser PJ, Jones DK. Diffusion-tensor MRI: theory, experimental design and data analysis -
a technical review. NMR Biomed 2002;15:456-467.
[60] Basser PJ, Mattiello J, LeBihan D. Estimation of the effective self-diffusion tensor from the
NMR spin echo. J Magn Reson B 1994;103:247-254.
[61] Basser PJ, Mattiello J, LeBihan D. MR diffusion tensor spectroscopy and imaging. Biophys
J 1994;66:259-267.
[62] Basser PJ, Ozarslan E. Anisotropic diffusion: from the apparent diffusion coefficient to the
apparent diffusion tensor. In: DK Jones, editor. Diffusion MRI : theory, methods, and
application. New York: Oxford University Press, 2011. pp. 79-91.
[63] Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by
quantitative-diffusion-tensor MRI. J Magn Reson B 1996;111:209-219.
[64] Baumgartner U, Iannetti GD, Zambreanu L, Stoeter P, Treede RD, Tracey I. Multiple
somatotopic representations of heat and mechanical pain in the operculo-insular cortex: a
high-resolution fMRI study. J Neurophysiol 2010;104:2863-2872.
[65] Baumgartner U, Tiede W, Treede RD, Craig AD. Laser-evoked potentials are graded and
somatotopically organized anteroposteriorly in the operculoinsular cortex of anesthetized
monkeys. J Neurophysiol 2006;96:2802-2808.
[66] Beaulieu C. The basis of anisotropic water diffusion in the nervous system - a technical
review. NMR Biomed 2002;15:435-455.
[67] Beaulieu C. The biological basis of diffusion. In: H Johansen-Berg, TEJ Behrens, editors.
Diffusion MRI. London: Elsevier, 2009. pp. 105-126.
[68] Becerra L, Harris W, Grant M, George E, Boas D, Borsook D. Diffuse optical tomography
activation in the somatosensory cortex: specific activation by painful vs. non-painful
thermal stimuli. PLoS One 2009;4:e8016.
[69] Beckmann M, Johansen-Berg H, Rushworth MF. Connectivity-based parcellation of human
cingulate cortex and its relation to functional specialization. J Neurosci 2009;29:1175-
1190.

227
[70] Beckstead RM. An autoradiographic examination of corticocortical and subcortical
projections of the mediodorsal-projection (prefrontal) cortex in the rat. J Comp Neurol
1979;184:43-62.
[71] Behbehani MM, Fields HL. Evidence that an excitatory connection between the
periaqueductal gray and nucleus raphe magnus mediates stimulation produced analgesia.
Brain Res 1979;170:85-93.
[72] Behrens TE, Jbabdi S. MR diffusion tractography. In: H Johansen-Berg, TE Behrens,
editors. Diffusion MRI. London: Academic Press, 2009. pp. 334-351.
[73] Behrens TE, Johansen-Berg H, Jbabdi S, Rushworth MFS, Woolrich M. Probabilistic
diffusion tractography with multiple fibre orientations. What can we gain? Neuroimage
2007;23:144-155.
[74] Behrens TE, Johansen-Berg H, Woolrich MW, Smith SM, Wheeler-Kingshott CA, Boulby
PA, Barker GJ, Sillery EL, Sheehan K, Ciccarelli O, Thompson AJ, Brady JM, Matthews
PM. Non-invasive mapping of connections between human thalamus and cortex using
diffusion imaging. Nat Neurosci 2003;6:750-757.
[75] Behrens TE, Woolrich MW, Jenkinson M, Johansen-Berg H, Nunes RG, Clare S, Matthews
PM, Brady JM, Smith SM. Characterization and propagation of uncertainty in diffusion-
weighted MR imaging. Magn Reson Med 2003;50:1077-1088.
[76] Behrens TEJ, Woolrich MW, Walton ME, Rushworth MFS. Learning the value of
information in an uncertain world. Nat Neurosci 2007;10:1214-1221.
[77] Beitz AJ. The organization of afferent projections to the midbrain periaqueductal gray of the
rat. Neuroscience 1982;7:133-159.
[78] Belforte JE, Pazo JH. Striatal inhibition of nociceptive responses evoked in trigeminal
sensory neurons by tooth pulp stimulation. J Neurophysiol 2005;93:1730-1741.
[79] Bell C, Shaw A. Reprint of the "Idea of a New Anatomy of the Brain," with Letters, &c. J
Anat Physiol 1868;3:147-182.
[80] Bengtsson SL, Nagy Z, Skare S, Forsman L, Forssberg H, Ullen F. Extensive piano
practicing has regionally specific effects on white matter development. Nat Neurosci
2005;8:1148-1150.

228
[81] Benoliel R, Sharav Y. Masticatory myofacial pain, and tension-type and chronic daily
headache. In: Y Sharav, R Benoliel, editors. Orofacial pain & Headache. New York:
Elsevier, 2008. pp. 109-148.
[82] Bereiter DA, Hargreaves KM, Hu JW. Trigeminal mechanisms of nociception: peripheral
and brainstem organization. In: A Basbaum, MC Bushnell, editors. Science of Pain. San
Diego: Elsevier, 2009. pp. 434-460.
[83] Bereiter DA, Hirata H, Hu JW. Trigeminal subnucleus caudalis: beyond homologies with
the spinal dorsal horn. Pain 2000;88:221-224.
[84] Bergfield KL, Hanson KD, Chen K, Teipel SJ, Hampel H, Rapoport SI, Moeller JR,
Alexander GE. Age-related networks of regional covariance in MRI gray matter:
Reproducible multivariate patterns in healthy aging. Neuroimage 2009;49:1750-1759.
[85] Berkley KJ. Spatial relationships between the terminations of somatic sensory and motor
pathways in the rostral brainstem of cats and monkeys. I. Ascending somatic sensory
inputs to lateral diencephalon. J Comp Neurol 1980;193:283-317.
[86] Berlucchi G. The origin of the term plasticity in the neurosciences: Ernesto Lugaro and
chemical synaptic transmission. J Hist Neurosci 2002;11:305-309.
[87] Bernard C, Magendie F. Leçon d’ouverture du Cours de Médecine du Collège de France.
Paris: Baillière, 1856.
[88] Bernard JF, Huang GF, Besson JM. Nucleus centralis of the amygdala and the globus
pallidus ventralis: Electrophysiological evidence for an involvement in pain processes. J
Neurophysiol 1992;68:551-569.
[89] Bessou P, Perl ER. Response of cutaneous sensory units with unmyelinated fibers to
noxious stimuli. J Neurophysiol 1969;32:1025-1043.
[90] Bhalang K, Sigurdsson A, Slade GD, Maixner W. Associations among four modalities of
experimental pain in women. J Pain 2005;6:604-611.
[91] Biedenbach MA, Van Hassel HJ, Brown AC. Tooth pulp-driven neurons in somatosensory
cortex of primates: role in pain mechanisms including a review of the literature. Pain
1979;7:31-50.

229
[92] Biemond A. The conduction of pain above the level of the thalamus opticus. AMA Arch
Neurol Psychiatry 1956;75:231-244.
[93] Bingel U, Herken W, Teutsch S, May A. Habituation to painful stimulation involves the
antinociceptive system--a 1-year follow-up of 10 participants. Pain 2008;140:393-394.
[94] Bingel U, Schoell E, Herken W, Buchel C, May A. Habituation to painful stimulation
involves the antinociceptive system. Pain 2007;131:21-30.
[95] Bingel U, Tracey I. Imaging CNS modulation of pain in humans. Physiology 2008;23:371-
380.
[96] Birbaumer N, Lutzenberger W, Montoya P, Larbig W, Unertl K, Tîpfner S, Grodd W, Taub
E, Flor H. Effects of regional anesthesia on phantom limb pain are mirrored in changes in
cortical reorganization. J Neurosci 1997;17:5503-5508.
[97] Blackburn TP, Cross AJ, Hille C, Slater P. Autoradiographic localization of delta opiate
receptors in rat and human brain. Neuroscience 1988;27:497-506.
[98] Blankstein U, Chen J, Diamant NE, Davis KD. Altered brain structure in irritable bowel
syndrome: potential contributions of pre-existing and disease-driven factors.
Gastroenterology 2010;138:1783-1789.
[99] Blinkov SM, Glezer II. The human brain in figures and tables: a quantitative handbook.
New York: Plenum Press, 1968.
[100] Blits KC. Aristotle: form, function, and comparative anatomy. Anat Rec 1999;257:58-63.
[101] Blomqvist A, Zhang ET, Craig AD. Cytoarchitectonic and immunohistochemical
characterization of a specific pain and temperature relay, the posterior portion of the
ventral medial nucleus, in the human thalamus. Brain 2000;123 Pt 3:601-619.
[102] Boivie J. An anatomical reinvestigation of the termination of the spinothalamic tract in the
monkey. J Comp Neurol 1979;186:343-369.
[103] Bonicalzi V, Canavero S. Motor cortex stimulation for central and neuropathic pain (Letter
regarding Topical Review by Brown and Barbaro). Pain 2004;108:199-200; author reply
200.

230
[104] Bookstein FL. "Voxel-based morphometry" should not be used with imperfectly registered
images. Neuroimage 2001;14:1454-1462.
[105] Boorman ED, Behrens TE, Woolrich MW, Rushworth MF. How green is the grass on the
other side? Frontopolar cortex and the evidence in favor of alternative courses of action.
Neuron 2009;62:733-743.
[106] Boring EG. Sensation and perception in the history of experimental psychology. New
York, London,: D. Appleton-Century Company, 1942.
[107] Borsook D, Upadhyay J, Chudler EH, Becerra L. A key role of the basal ganglia in pain
and analgesia--insights gained through human functional imaging. Mol Pain 2010;6:27.
[108] Botvinick MM, Cohen JD, Carter CS. Conflict monitoring and anterior cingulate cortex: an
update. Trends Cogn Sci 2004;8:539-546.
[109] Boudreau S, Romaniello A, Wang K, Svensson P, Sessle BJ, Arendt-Nielsen L. The effects
of intra-oral pain on motor cortex neuroplasticity associated with short-term novel
tongue-protrusion training in humans. Pain 2007;132:169-178.
[110] Boyd D, Butler M, Carr DB, Cohen MJ, Devor M, Dworkin R, Greenspan JD, Jensen TS,
King S, Koltzenburg M, Loeser JD, Merskey H, Okifuji A, Paice J, Serra J, Treede RD,
Woda A. IASP Pain Terminology. An update on the IASP Taskforce on Taxonomy. Part
III: Pain Terms, A Current List with Definitions and Notes on Usage. In: JD Loeser,
editor. IASP Taxonomy, Vol. 2011. Seattle: IASP Press, 2011.
[111] Boyke J, Driemeyer J, Gaser C, Buchel C, May A. Training-induced brain structure
changes in the elderly. J Neurosci 2008;28:7031-7035.
[112] Bozzali M, Franceschi M, Falini A, Pontesilli S, Cercignani M, Magnani G, Scotti G,
Comi G, Filippi M. Quantification of tissue damage in AD using diffusion tensor and
magnetization transfer MRI. Neurology 2001;57:1135-1137.
[113] Bragdon EE, Light KC, Costello NL, Sigurdsson A, Bunting S, Bhalang K, Maixner W.
Group differences in pain modulation: pain-free women compared to pain-free men and
to women with TMD. Pain 2002;96:227-237.

231
[114] Brighina F, Piazza A, Vitello G, Aloisio A, Palermo A, Daniele O, Fierro B. rTMS of the
prefrontal cortex in the treatment of chronic migraine: a pilot study. J Neurol Sci
2004;227:67-71.
[115] Brody H. Organization of the cerebral cortex. III. A study of aging in the human cerebral
cortex. J Comp Neurol 1955;102:511-516.
[116] Brooks JC, Tracey I. The insula: a multidimensional integration site for pain. Pain
2007;128:1-2.
[117] Brown JA, Barbaro NM. Motor cortex stimulation for central and neuropathic pain: current
status. Pain 2003;104:431-435.
[118] Brown JA, Pilitsis JG. Motor cortex stimulation for central and neuropathic facial pain: a
prospective study of 10 patients and observations of enhanced sensory and motor
function during stimulation. Neurosurgery 2005;56:290-297.
[119] Brown MA, Semelka RC. MRI : basic principles and applications. Hoboken, N.J.: Wiley-
Blackwell, 2010.
[120] Bryan CP. The Papyrus Ebers. Translated from the German version by Cyril P. Bryan ...
With an introduction by Professor G. Elliot Smith. [With plates.]. London: Geoffrey Bles,
1930.
[121] Buckalew N, Haut MW, Morrow L, Weiner D. Chronic pain is associated with brain
volume loss in older adults: preliminary evidence. Pain Med 2008;9:240-248.
[122] Budge EATW. The book of medicines : ancient Syrian anatomy, pathology and theraputics
[sic]. London: Kegan Paul, 2002.
[123] Buhle J, Wager TD. Performance-dependent inhibition of pain by an executive working
memory task. Pain 2010;149:19-26.
[124] Burgess PR, Perl ER. Myelinated afferent fibres responding specifically to noxious
stimulation of the skin. J Physiol 1967;190:541-562.
[125] Burgmer M, Gaubitz M, Konrad C, Wrenger M, Hilgart S, Heuft G, Pfleiderer B.
Decreased gray matter volumes in the cingulo-frontal cortex and the amygdala in patients
with fibromyalgia. Psychosom Med 2009;71:566-573.

232
[126] Burton H, Craig AD, Jr. Distribution of trigeminothalamic projection cells in cat and
monkey. Brain Res 1979;161:515-521.
[127] Burton H, Craig AD, Poulos DA, Molt JT. Efferent projections from temperature sensitive
recording loci within the marginal zone of the nucleus caudalis of the spinal trigeminal
complex in the cat. J Comp Neurol 1979;183:753-777.
[128] Bush G, Luu P, Posner MI. Cognitive and emotional influences in anterior cingulate
cortex. Trends Cogn Sci 2000;4:215-222.
[129] Bushnell MC, Duncan GH. Sensory and affective aspects of pain perception: Is medial
thalamus restricted to emotional issues. Exp Brain Res 1989;78:415-418.
[130] Bushnell MC, Duncan GH, Hofbauer RK, Ha B, Chen JI, Carrier B. Pain perception: is
there a role for primary somatosensory cortex? Proc Natl Acad Sci U S A 1999;96:7705-
7709.
[131] Cadden SW, Van der Glas HW, Lobbezoo F, Van der Bilt A. Effects of remote noxious
stimulation on exteroceptive reflexes in human jaw-closing muscles. Brain Research
1996;726:189-197.
[132] Cadden SW, Van Der Glas HW, Van Der Bilt A. Modulation of jaw reflexes by remote
noxious stimulation and mental state: possible association with psychological
measurements of mental stress and occupation. J Oral Rehabil 1999;26:952-961.
[133] Cairns BE, Sessle BJ, Hu JW. Evidence that excitatory amino acid receptors within the
temporomandibular joint region are involved in the reflex activation of the jaw muscles. J
Neurosci 1998;18:8056-8064.
[134] Cajal SRy. Textura des sistema nerviosa del hombre y de Los Vertebrados. S.l.: Nicolas
Moya, 1904.
[135] Cameron AA, Khan IA, Westlund KN, Cliffer KD, Willis WD. The Efferent projections of
the periaqueductal gray in the rat: A phaselous vulgaris -leucoagglutinin study. I.
Ascending projections. J Comp Neurol 1995;351:568-584.
[136] Campbell JN, LaMotte RH. Latency to detection of first pain. Brain Research
1983;266:203-208.

233
[137] Cannon TD, van Erp TG, Bearden CE, Loewy R, Thompson P, Toga AW, Huttunen MO,
Keshavan MS, Seidman LJ, Tsuang MT. Early and late neurodevelopmental influences in
the prodrome to schizophrenia: contributions of genes, environment, and their
interactions. Schizophr Bull 2003;29:653-669.
[138] Capra NF, Dessem D. Central connections of trigeminal primary afferent neurons:
topographical and functional considerations. Crit Rev Oral Biol Med 1992;4:1-52.
[139] Carlson CR, Reid KI, Curran SL, Studts J, Okeson JP, Falace D, Nitz A, Bertrand PM.
Psychological and physiological parameters of masticatory muscle pain. Pain
1998;76:297-307.
[140] Carpenter MB, Hanna GR. Fiber projections from the spinal trigeminal nucleus in the cat. J
Comp Neurol 1961;117:117-131.
[141] Carstens E, Kuenzler N, Handwerker HO. Activation of neurons in rat trigeminal
subnucleus caudalis by different irritant chemicals applied to oral or ocular mucosa. J
Neurophysiol 1998;80:465-492.
[142] Casey KL. Unit analysis of nociceptive mechanisms in the thalamus of awake squirrel
monkey. J Neurophysiol 1966;29.
[143] Casey KL. Concepts of pain mechanisms: the contribution of functional imaging of the
human brain. Prog Brain Res 2000;129:277-287.
[144] Casey KL. Central pain: distributed effects of focal lesions. Pain 2004;108:205-206.
[145] Catani M. From hodology to function. Brain 2007;130:602-605.
[146] Catani M. The functional anatomy of white matter: from postmortem dissections to in vivo
virtual tractography. In: DK Jones, editor. Diffusion MRI: Theory, Methods and
Applications. Oxford: Oxford University Press, 2011. pp. 5-18.
[147] Catani M, ffytche DH. The rises and falls of disconnection syndromes. Brain
2005;128:2224-2239.
[148] Cauna N, Mannan G. The structure of human digital pacinian corpuscles (corpus cula
lamellosa) and its functional significance. J Anat 1958;92:1-20.

234
[149] Cauna N, Ross LL. The fine structure of Meissner's touch corpuscles of human fingers. J
Biophys Biochem Cytol 1960;8:467-482.
[150] Cavada C, Compa§y T, Tejedor J, Cruz-Rizzolo RJ, Reinoso-Su†rez F. The anatomical
connections of the macaque monkey orbitofrontal cortex. A review. Cereb Cortex
2000;10:220-242.
[151] Cercignani M. Strategies for patient-control comparison of diffusion MR data. In: DK
Jones, editor. Diffusion MRI : theory, methods, and application. New York: Oxford
University Press, 2011. pp. 485-499.
[152] Cercignani M, Iannucci G, Rocca MA, Comi G, Horsfield MA, Filippi M. Pathologic
damage in MS assessed by diffusion-weighted and magnetization transfer MRI.
Neurology 2000;54:1139-1144.
[153] Cervero F, Laird JMA. Mechanisms of touch-evoked pain (allodynia): A new model. Pain
1996;68:13-23.
[154] Chapman WP, Rose AS, Solomon HC. Measurements of heat stimulus producing motor
withdrawal reaction in patients following frontal lobotomy. Association For Research In
Nervous and Mental Disease 1948;XXVII:754-768.
[155] Charron S, Koechlin E. Divided representation of concurrent goals in the human frontal
lobes. Science 2010;328:360-363.
[156] Chen J, Blankstein U, Diamant N, Davis KD. White matter abnormalities in irritable bowel
syndrome and relation to individual factors. Brain Res 2011;1392:121-131.
[157] Cheong JY, Yoon TS, Lee SJ. Evaluations of inhibitory effect on the motor cortex by
cutaneous pain via application of capsaicin. Electromyogr Clin Neurophysiol
2003;43:203-210.
[158] Chiang CY, Chen IC, Dostrovsky JO, Sessle BJ. Anterior pretectal nucleus-induced
modulatory effects on trigeminal brainstem somatosensory neurons. Neurosci Lett
1992;134:233-237.
[159] Chiang CY, Dostrovsky JO, Iwata K, Sessle BJ. Role of glia in orofacial pain.
Neuroscientist 2011;17:303-320.

235
[160] Christensen BN, Perl ER. Spinal neurons specifically excited by noxious or thermal
stimuli: Marginal zone of the dorsal horn. Journal of Neurophysiology 1970;33:293-307.
[161] Chudler EH, Anton F, Dubner R, Kenshalo DR, Jr. Responses of nociceptive S1 neurons in
monkeys and pain sensation in humans elicited by noxious thermal stimulation: Effect of
interstimulus interval. J Neurophysiol 1990;63:559-569.
[162] Chudler EH, Dong WK. The role of the basal ganglia in nociception and pain. Pain
1995;60:3-38.
[163] Chudler EH, Lu Y. Nociceptive behavioral responses to chemical, thermal and mechanical
stimulation after unilateral, intrastriatal administration of 6-hydroxydopamine. Brain Res
2008;1213:41-47.
[164] Chudler EH, Sugiyama K, Dong WK. Nociceptive responses in the neostriatum and globus
pallidus of the anesthetized rat. J Neurophysiol 1993;69:1890-1903.
[165] Chudler EH, Sugiyama K, Dong WK. Multisensory convergence and integration in the
neostriatum and globus pallidus of the rat. Brain Res 1995;674:33-45.
[166] Clark FM, Proudfit HK. Projections of neurons in the ventromedial medulla to pontine
catecholamine cell groups involved in the modulation of nociception. Brain Res
1991;540:105-115.
[167] Cody FW, Lee RW, Taylor A. A functional analysis of the components of the
mesencephalic nucleus of the fifth nerve in the cat. J Physiol 1972;226:249-261.
[168] Coffield JA, Bowen KK, Miletic V. Retrograde tracing of projections between the nucleus
submedius, the ventrolateral orbital cortex, and the midbrain in the rat. J Comp Neurol
1992;321:488-499.
[169] Coghill RC, Sang CN, Maisog JM, Iadarola MJ. Pain Intensity Processing Within the
Human Brain: A Bilateral, Distributed Mechanism. J Neurophysiol 1999;82:1934-1943.
[170] Cohen J, Cohen P, West SG, Aiken LS. Applied Multiple Regression/Correlation Analysis
for the Behavioural Sciences. Mawhaw: Lawrence Erlbaum Associates, Inc, 2003.
[171] Colloca L, Benedetti F. Placebos and painkillers: is mind as real as matter? Nat Rev
Neurosci 2005;6:545-552.

236
[172] Colloca L, Benedetti F, Porro CA. Experimental designs and brain mapping approaches for
studying the placebo analgesic effect. Eur J Appl Physiol 2008;102:371-380.
[173] Condes-Lara M, Omana Zapata I, Leon-Olea M, Sanchez-Alvarez M. Dorsal raphe and
nociceptive stimulations evoke convergent responses on the thalamic centralis lateralis
and medial prefrontal cortex neurons. Brain Res 1989;499:145-152.
[174] Conti PC, dos Santos CN, Kogawa EW, de Castro Ferreira Conti A, de Araujo Cdos R.
The treatment of painful temporomandibular joint clicking with oral splints: a
randomized clinical trial. J Am Dent Assoc 2006;137:1108-1114.
[175] Cooper SJ. Anaesthetisation of prefrontal cortex and response to noxious stimulation.
Nature 1975;254:439-440.
[176] Corbin KB, Harrison F. Function of Mesencephalic Root in Fifth Cranial Nerve. J
Neurophysiol 1940;3.
[177] Costa PT. Influence of the Normal Personality Dimension of Neuroticism on Chest Pain
Symptoms and Coronary Artery Disease. Am J Cardiol 1987;60:J26-33.
[178] Costa PT, McCrae RR. Revised NEO personality (NEO-PI-R) and NEO five factor
inventory (NEO-FFI) professional manual. Book Revised NEO personality (NEO-PI-R)
and NEO five factor inventory (NEO-FFI) professional manual. City: Psychology
Assessment Resources, 1992.
[179] Costa PT, McCrae RR, Zonderman AB, Barbano HE, Lebowitz B, Larson DM. Cross-
Sectional Studies of Personality in a National Sample: 2. Stability in Neuroticism,
Extraversion, and Openness. Psych Aging 1986;1:144-149.
[180] Costello NL, Bragdon EE, Light KC, Sigurdsson A, Bunting S, Grewen K, Maixner W.
Temporomandibular disorder and optimism: relationships to ischemic pain sensitivity and
interleukin-6. Pain 2002;100:99-110.
[181] Costen JB. A syndrome of ear and sinus symptoms dependent upon disturbed function of
the temporomandibular joint. Ann Otol Rhinol Laryngol 1934;43:1-15.
[182] Coulter JD, Maunz RA, Willis WD. Effects of stimulation of sensorimotor cortex on
primate spinothalamic neurons. Brain Res 1974;65:351-356.

237
[183] Craig AD. How do you feel? Interoception: the sense of the physiological condition of the
body. Nat Rev Neurosci 2002;3:655-666.
[184] Craig AD. Pain mechanisms: labeled lines versus convergence in central processing. Ann
Rev Neurosci 2003;26:1-30.
[185] Craig AD. Distribution of trigeminothalamic and spinothalamic lamina I terminations in
the macaque monkey. J Comp Neurol 2004;477:119-148.
[186] Craig AD. Human feelings: why are some more aware than others? Trends Cogn Sci
2004;8:239-241.
[187] Craig AD. Once an island, now the focus of attention. Brain Struct Funct 2010;124:395-
396.
[188] Craig AD, Bushnell MC, Zhang ET, Blomqvist A. A thalamic nucleus specific for pain and
temperature sensation. Nature 1994;372:770-773.
[189] Craig AD, Dostrovsky JO. Processing of nociceptive information at supraspinal levels. In:
TL Yaksh, et al., editors. Anesthesia: Biologic foundations. Philadelphia: Lippincott-
Raven Publishers, 1997. pp. 625-642.
[190] Craig ADB. How do you feel--now? The anterior insula and human awareness. Nat Rev
Neurosci 2009;10:59-70.
[191] Creasey H, Rapoport SI. The aging human brain. Ann Neurol 1985;17:2-10.
[192] Crombez G, Baeyens F, Eelen P. Sensory and temporal information about impending pain:
the influence of predictability on pain. Behav Res Ther 1994;32:611-622.
[193] Crombez G, Eccleston C, Baeyens F, Eelen P. The disruptive nature of pain: an
experimental investigation. Behav Res Ther 1996;34:911-918.
[194] Crombez G, Eccleston C, Baeyens F, Eelen P. Habituation and the interference of pain
with task performance. Pain 1997;70:149-154.
[195] Cruccu G, Frisardi G, Pauletti G, Romaniello A, Manfredi M. Excitability of the central
masticatory pathways in patients with painful temporomandibular disorders. Pain
1997;73:447-454.

238
[196] Cushing H. A note upon the faradic stimulation of the postcentral gyrus in conscious
patients. Brain 1909;32:44-53.
[197] Dahlstrom L. Electromyographic studies of craniomandibular disorders: a review of the
literature. J Oral Rehabil 1989;16:1-20.
[198] Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and
surface reconstruction. Neuroimage 1999;9:179-194.
[199] Dallenbach KM. Pain: history and present status. Am J Psychol 1939;52:331-347.
[200] Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, Stowe AM, Nudo RJ.
Extensive cortical rewiring after brain injury. J Neurosci 2005;25:10167-10179.
[201] Dao TT, LeResche L. Gender differences in pain. J Orofac Pain 2001;14:169-195.
[202] Darian-Smith I, Johnson KO, Dykes R. "Cold" fiber population innervating palmar and
digital skin of the monkey: responses to cooling pulses. J Neurophysiol 1973;36:325-346.
[203] Darwin C. The Expressions of the Emotions in Man and Animals. London: J. Murray,
1901.
[204] Darwin E, Darwin RW. Zoonomia; or, The laws of organic life. London,: J. Johnson, 1794.
[205] DaSilva AF, Becerra L, Makris N, Strassman AM, Gonzalez RG, Geatrakis N, Borsook D.
Somatotopic activation in the human trigeminal pain pathway. J Neurosci 2002;22:8183-
8192.
[206] DaSilva AF, Becerra L, Pendse G, Chizh B, Tully S, Borsook D. Colocalized structural
and functional changes in the cortex of patients with trigeminal neuropathic pain. PLoS
One 2008;3:e3396.
[207] DaSilva AF, Granziera C, Snyder J, Hadjikhani N. Thickening in the somatosensory cortex
of patients with migraine. Neurology 2007;69:1990-1995.
[208] DaSilva AF, Granziera C, Tuch DS, Snyder J, Vincent M, Hadjikhani N. Interictal
alterations of the trigeminal somatosensory pathway and periaqueductal gray matter in
migraine. Neuroreport 2007;18:301-305.
[209] DaSilva AFM, Granziera C, Snyder J, Hadjikhani N. Thickening in the somatosensory
cortex of patients with migraine. Neurology 2007;69:1990-1995.

239
[210] Davatzikos C. Why voxel-based morphometric analysis should be used with great caution
when characterizing group differences. Neuroimage 2004;23:17-20.
[211] Davidson RM, Gale EN. Cutaneous sensory thresholds from skin overlying masseter and
forearm in MPD patients and controls. J Dent Res 1983;62:555-558.
[212] Davis KD. Studies of pain using fMRI. In: MC Bushnell, KL Casey, editors. Pain Imaging,
Vol. 18. Seattle: IASP Press, 2000. pp. 195-210.
[213] Davis KD. Neuroimaging of pain: what does it tell us? Curr Opin Support Palliat Care
2011;5:116-121.
[214] Davis KD, Dostrovsky JO. Activation of trigeminal brain-stem nociceptive neurons by
dural artery stimulation. Pain 1986;25:395-401.
[215] Davis KD, Dostrovsky JO. Effect of trigeminal subnucleus caudalis cold block on the
cerebrovascular-evoked responses of rostral trigeminal complex neurons. Neurosci Lett
1988;94:303-308.
[216] Davis KD, Dostrovsky JO. Responses of feline trigeminal spinal tract nucleus neurons to
stimulation of the middle meningeal artery and sagittal sinus. J Neurophysiol
1988;59:648-666.
[217] Davis KD, Hutchison WD, Lozano AM, Dostrovsky JO. Altered pain and temperature
perception following cingulotomy and capsulotomy in a patient with schizoaffective
disorder. Pain 1994;59:189-199.
[218] Davis KD, Hutchison WD, Lozano AM, Tasker RR, Dostrovsky JO. Human anterior
cingulate cortex neurons modulated by attention- demanding tasks. J Neurophysiol
2000;83:3575-3577.
[219] Davis KD, Kiss ZHT, Luo L, Tasker RR, Lozano AM, Dostrovsky JO. Phantom sensations
generated by thalamic microstimulation. Nature 1998;391:385-387.
[220] Davis KD, Kwan CL, Crawley AP, Mikulis DJ. fMRI of the anterior cingulate cortex
during painful, thermal and motor tasks in individual subjects. Neuroimage 1998;7:S426.
[221] Davis KD, Kwan CL, Crawley AP, Mikulis DJ. Functional MRI study of thalamic and
cortical activations evoked by cutaneous heat, cold and tactile stimuli. J Neurophysiol
1998;80:1533-1546.

240
[222] Davis KD, Kwan CL, Crawley AP, Mikulis DJ. fMRI of cortical and thalamic activations
correlated to the magnitude of pain. In: M Devor, MC Rowbotham, Z Wiesenfeld-Hallin,
editors. Progress in pain research and management, Proceedings of the 9th World
Congress on Pain, Vol. 9. Seattle: IASP Press, 2000. pp. 497-505.
[223] Davis KD, Lozano AM, Manduch M, Tasker RR, Kiss ZHT, Dostrovsky JO. Thalamic
relay site for cold perception in humans. J Neurophysiol 1999;81:1970-1973.
[224] Davis KD, Meyer RA, Campbell JN. Chemosensitivity and sensitization of nociceptive
afferents that innervate the hairy skin of monkey. J Neurophysiol 1993;69:1071-1081.
[225] Davis KD, Pope G, Chen J, Kwan CL, Crawley AP, Diamant NE. Cortical thinning in IBS:
implications for homeostatic, attention, and pain processing. Neurology 2008;70:153-
154.
[226] Davis KD, Pope GE, Crawley AP, Mikulis DJ. Neural correlates of prickle sensation: a
percept-related fMRI study. Nat Neurosci 2002;5:1121-1122.
[227] Davis KD, Pope GE, Crawley AP, Mikulis DJ. Perceptual illusion of "paradoxical heat"
engages the insular cortex. J Neurophysiol 2004;92:1248-1251.
[228] Davis KD, Taub E, Duffner F, Lozano AM, Tasker RR, Houle S, Dostrovsky JO.
Activation of the anterior cingulate cortex by thalamic stimulation in patients with
chronic pain: a positron emission tomography study. J Neurosurg 2000;92:64-69.
[229] Davis KD, Taylor KS, Hutchison WD, Dostrovsky JO, McAndrews MP, Richter EO,
Lozano AM. Human anterior cingulate cortex neurons encode cognitive and emotional
demands. J Neurosci 2005;25:8402-8406.
[230] Davis KD, Taylor SJ, Crawley AP, Wood ML, Mikulis DJ. Functional MRI of pain- and
attention-related activations in the human cingulate cortex. J Neurophysiol 1997;77:3370-
3380.
[231] Davis KD, Wood ML, Crawley AP, Mikulis DJ. fMRI of human somatosensory and
cingulate cortex during painful electrical nerve stimulation. Neuroreport 1995;7.
[232] Davis PJM, Wright EA. A new method for measuring cranial cavity volume and its
application to assessment of cerebral atrophy at autopsy. Neuropathol Appl Neurobiol
1977;3:341-358.

241
[233] Davison C, Schick W. Spontaneous pain and other subjective sensory disturbances. Arch
Neurol Psychiatry 1935;34:1204–1237.
[234] de Leeuw R, Davis CE, Albuquerque R, Carlson CR, Andersen AH. Brain activity during
stimulation of the trigeminal nerve with noxious heat. Oral Surg Oral Med Oral Pathol
Oral Radiol Endod 2006;102:750-757.
[235] Deco G, Corbetta M. The dynamical balance of the brain at rest. Neuroscientist
2011;17:107-123.
[236] Deco G, Jirsa VK, McIntosh AR. Emerging concepts for the dynamical organization of
resting-state activity in the brain. Nat Rev Neurosci 2011;12:43-56.
[237] Dejerine J. Anatomie des centres nerveux. Paris,: Reuff, 1895.
[238] DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune Activation and Neuroinflammation in
Chronic Pain and Opioid Tolerance/Hyperalgesia. Neuroscientist 2004;10:40-52.
[239] DeLong MR. Activity of pallidal neurons during movement. J Neurophysiol 1971:414-
427.
[240] Derbyshire SWG, Jones AKP, Gyulai F, Clark S, Townsend D, Firestone LL. Pain
processing during three levels of noxious stimulation produces differential patterns of
central activity. Pain 1997;73:431-445.
[241] Descartes R. De Homine - Figuris et latinitate donatus Florentio Schuyl. Leiden: Leffer &
Moyardus, 1662.
[242] Descartes R, Clerselier C, La Forge Ld, Schuyl F. L'homme ... et un traitté De la formation
du foetus du mesme autheur. Paris: Charles Angot, 1664.
[243] Destrieux C, Fischl B, Dale A, Halgren E. Automatic parcellation of human cortical gyri
and sulci using standard anatomical nomenclature. Neuroimage 2010;53:1-15.
[244] Dettmers C, Adler T, Rzanny R, Van Schayck R, Gaser C, Weiss T, Miltner WH, Bruckner
L, Weiller C. Increased excitability in the primary motor cortex and supplementary motor
area in patients with phantom limb pain after upper limb amputation. Neurosci Lett
2001;307:109-112.

242
[245] DeYoung CG, Hirsh JB, Shane MS, Papademetris X, Rajeevan N, Gray JR. Testing
predictions from personality neuroscience: brain structure and the big five. Psychol Sci
2010;21:820-828.
[246] Diamant NE. Overview of functional gut disorders: a challenge. Scandinavian journal of
gastroenterology Supplement 1995;213:1-6.
[247] Diatchenko L, Anderson AD, Slade GD, Fillingim RB, Shabalina SA, Higgins TJ, Sama S,
Belfer I, Goldman D, Max MB, Weir BS, Maixner W. Three major haplotypes of the
beta2 adrenergic receptor define psychological profile, blood pressure, and the risk for
development of a common musculoskeletal pain disorder. Am J Med Genet B
Neuropsychiatr Genet 2006;141B:449-462.
[248] Diatchenko L, Nackley AG, Slade GD, Fillingim RB, Maixner W. Idiopathic pain
disorders--pathways of vulnerability. Pain 2006;123:226-230.
[249] Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D,
Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W. Genetic basis for
individual variations in pain perception and the development of a chronic pain condition.
Hum Mol Genet 2005;14:135-143.
[250] Dick B, Eccleston C, Crombez G. Attentional functioning in fibromyalgia, rheumatoid
arthritis, and musculoskeletal pain patients. Arthritis Rheum 2002;47:639-644.
[251] Dickenson AH, Le Bars D. Diffuse noxious inhibitory controls (DNIC) involve
trigeminothalamic and spinothalamic neurones in the rat. Exp Brain Res 1983;49:174-
180.
[252] Dickenson AH, Le Bars D. Morphine microinjections into periaqueductal grey matter of
the rat: effects on dorsal horn neuronal responses to C-fibre activity and diffuse noxious
inhibitory controls. Life Sci 1983;33 Suppl 1:549-552.
[253] Dickenson AH, Oliveras JL, Besson JM. Role of the nucleus raphe magnus in opiate
analgesia as studied by the microinjection technique in the rat. Brain Res 1979;170:95-
111.
[254] Dierssen G, Odoriz B, Hernando C. Sensory and motor responses to stimulation of the
posterior cingulate cortex in man. J Neurosurg 1969;31:435-440.

243
[255] Djouhri L, Lawson SN. Abeta-fiber nociceptive primary afferent neurons: a review of
incidence and properties in relation to other afferent A-fiber neurons in mammals. Brain
Res Brain Res Rev 2004;46:131-145.
[256] Dodd J, Kelly J. Trigeminal system. In: ER Kandel, A Schwartz, TM Jessell, editors.
Principles of Neural Science. New York: McGraw-Hill, 2000. pp. 701-710.
[257] Dostrovsky J, Craig AD. The thalamus and nociceptive processing. In: A Basbaum, MC
Bushnell, editors. Science of Pain. San Diego: Elsevier, 2009. pp. 635-654.
[258] Dostrovsky JO, Craig AD. Ascending projection systems. In: A McMahon, M
Koltzenburg, editors. Wall and Melzack's Textbook of Pain. Edinburgh, UK: Churchill
Livingstone, 2006. pp. 187-204.
[259] Dostrovsky JO, Hutchison WD, Davis KD, Lozano A. Potential role of orbital and
cingulate cortices in nociception. In: JM Besson, G Guilbaud, H Ollat, editors. Forebrain
areas involved in pain processing. Paris: John Libbey Eurotext, 1995. pp. 171-181.
[260] Dostrovsky JO, Shah Y, Gray BG. Descending inhibitory influences from periaqueductal
gray, nucleus raphe magnus, and adjacent reticular formation. II. Effects on medullary
dorsal horn nociceptive and nonnociceptive neurons. J Neurophysiol 1983;49:948-960.
[261] Double KL, Halliday GM, Kril JJ, Harasty JA, Cullen K, Brooks WS, Creasey H, Broe
GA. Topography of brain atrophy during normal aging and Alzheimer's disease.
Neurobiol Aging 1996;17:513-521.
[262] Downar J, Crawley AP, Mikulis DJ, Davis KD. A multimodal cortical network for the
detection of changes in the sensory environment. Nat Neurosci 2000;3:277-283.
[263] Downar J, Crawley AP, Mikulis DJ, Davis KD. A cortical network for the detection of
novel events across multiple sensory modalities. Neuroimage 2001;13:S310.
[264] Downar J, Crawley AP, Mikulis DJ, Davis KD. A cortical network sensitive to stimulus
salience in a neutral behavioral context across multiple sensory modalities. J
Neurophysiol 2002;87:615-620.
[265] Downar J, Mikulis DJ, Davis KD. Neural correlates of the prolonged salience of painful
stimulation. Neuroimage 2003;20:1540-1551.

244
[266] Draganski B, Gaser C, Busch V, Schuierer G, Bogdahn U, May A. Neuroplasticity:
changes in grey matter induced by training. Nature 2004;427:311-312.
[267] Draganski B, Gaser C, Kempermann G, Kuhn HG, Winkler J, Buchel C, May A. Temporal
and spatial dynamics of brain structure changes during extensive learning. J Neurosci
2006;26:6314-6317.
[268] Draganski B, Moser T, Lummel N, Ganssbauer S, Bogdahn U, Haas F, May A. Decrease
of thalamic gray matter following limb amputation. Neuroimage 2006;31:951-957.
[269] Drangsholt MT, LeResche L. Temporomandibular disorder pain. In: IK Crombie, PR
Croft, SJ Linton, L LeResche, M Von Korff, editors. Epidemiology of pain. Seattle: IASP
Press, 1999. pp. 202-233.
[270] Dreyer DA, Metz CB, Schneider RJ, Whitsel BL. Differential contributions of spinal
pathways to body representation in postcentral gyrus of Macaca mulatta. J Neurophysiol
1974;37:119- 145.
[271] Driemeyer J, Boyke J, Gaser C, Buchel C, May A. Changes in gray matter induced by
learning--revisited. PLoS One 2008;3:e2669.
[272] Dubner R, Bennett GJ. Spinal and trigeminal mechanisms of nociception. Annu Rev
Neurosci 1983;6:381-418.
[273] Dubner R, Sessle BJ, Storey AT. The neural basis of oral and facial function. New York:
Plenum Press, 1978.
[274] Duerden EG, Albanese MC. Localization of pain-related brain activation: A meta-analysis
of neuroimaging data. Hum Brain Mapp 2011.
[275] Dufton BD. Cognitive failure and chronic pain. Int J Psychiatry Med 1989;19:291-297.
[276] Dum R, Levinthal D, Strick P. The Spinothalamic System Targets Motor and Sensory
Areas in the Cerebral Cortex of Monkeys. J Neurosci 2009;29:14223-14235.
[277] Dum RP, Levinthal DJ, Strick PL. The Spinothalamic System Targets Motor and Sensory
Areas in the Cerebral Cortex of Monkeys. Journal of Neuroscience 2009;29:14223-
14235.

245
[278] Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WGM, Lou W, Rapp PR, Morrison JH.
Selective changes in thin spine density and morphology in monkey prefrontal cortex
correlated with age-related cognitive impairment. J Neurosci 2010;30:7507-7515.
[279] Duncan GH, Bushnell MC, Lavigne GJ. Comparison of verbal and visual analogue scales
for measuring the intensity and unpleasantness of experimental pain. Pain 1989;37:295-
303.
[280] Dusser de Barenne JG. Experimental researches on sensory localisations in the cerebral
cortex. Q J Exp Physiol Cogn Med Sci 1916;9:355-390.
[281] Dusser de Barenne JG, McCulloch WS. Functional interdependence of sensory cortex and
thalamus. J Neurophysiol 1941;4:304-310.
[282] Duyn JH. The Future of Ultra-High Field MRI and fMRI for study of the human brain.
Neuroimage 2011;in press.
[283] Dworkin SF. Perspectives on the interaction of biological, psychological and social factors
in TMD. J Am Dent Assoc 1994;125:856-863.
[284] Dworkin SF, Huggins KH, LeResche L, Von Korff M, Howard J, Truelove E, Sommers E.
Epidemiology of signs and symptoms in temporomandibular disorders: clinical signs in
cases and controls. J Am Dent Assoc 1990;120:273-281.
[285] Dworkin SF, Leresche L. Research diagnostic criteria for temporomandibular disorders:
review, criteria, examinations and specifications, critique. J Craniomandib Disord
1992;6:301-355.
[286] Dworkin SF, Massoth DL. Temporomandibular disorders and chronic pain: disease or
illness? J Prosthet Dent 1994;72:29-38.
[287] Dworkin SF, Turner JA, Wilson L, Massoth D, Whitney C, Huggins KH, Burgess J,
Sommers E, Truelove E. Brief group cognitive-behavioral intervention for
temporomandibular disorders. Pain 1994;59:175-187.
[288] Dynes JB, Poppen JL. Lobotomy for intractable pain. J Am Med Assoc 1949;140:15-19.
[289] Eccleston C. Chronic pain and attention: a cognitive approach. Br J Clin Psychol 1994;33 (
Pt 4):535-547.

246
[290] Eccleston C. The attentional control of pain: Methodological and theoretical concerns. Pain
1995;63:3-10.
[291] Eccleston C, Crombez G. Pain demands attention: a cognitive-affective model of the
interruptive function of pain. Psychol Bull 1999;125:356-366.
[292] Eccleston C, Crombez G, Aldrich S, Stannard C. Attention and somatic awareness in
chronic pain. Pain 1997;72:209-215.
[293] Echols DH, Colclough JA. Abolition of painful phantom foot by resection of the sensory
cortex. J Am Med Assoc 1947;134:1476-1477.
[294] Eickhoff SB, Jbabdi S, Caspers S, Laird AR, Fox PT, Zilles K, Behrens TE. Anatomical
and functional connectivity of cytoarchitectonic areas within the human parietal
operculum. J Neurosci 2010;30:6409-6421.
[295] Eippert F, Bingel U, Schoell ED, Yacubian J, Klinger R, Lorenz J, Buchel C. Activation of
the opioidergic descending pain control system underlies placebo analgesia. Neuron
2009;63:533-543.
[296] Erasmus JFP. Studies on pain - a review of theories and observations. S Afr Med J
1948;22:683-691.
[297] Erlanger J, Gasser HS. The action potential in fibres of slow con- duction in spinal roots
and somatic nerves. A J Physiol 1930;92:43-82.
[298] Erpelding N, Moayedi M, Davis KD. Cotical correlates of temperature and pain sensitivity.
Book Cotical correlates of temperature and pain sensitivity. City, 2011.
[299] Etkin A, Egner T, Kalisch R. Emotional processing in anterior cingulate and medial
prefrontal cortex. Trends Cogn Sci 2011;15:85-93.
[300] Ettlin DA, Brugger M, Keller T, Luechinger R, Jancke L, Palla S, Barlow A, Gallo LM,
Lutz K. Interindividual differences in the perception of dental stimulation and related
brain activity. Eur J Oral Sci 2009;117:27-33.
[301] Farina S, Tinazzi M, Le Pera D, Valeriani M. Pain-related modulation of the human motor
cortex. Neurol Res 2003;25:130-142.

247
[302] Farina S, Valeriani M, Rosso T, Aglioti S, Tamburin S, Fiaschi A, Tinazzi M. Transient
inhibition of the human motor cortex by capsaicin-induced pain. A study with
transcranial magnetic stimulation. Neurosci Lett 2001;314:97-101.
[303] Farmer MA, Chanda ML, Parks EL, Baliki MN, Apkarian AV, Schaeffer AJ. Brain
functional and anatomical changes in chronic prostatitis/chronic pelvic pain syndrome. J
Urol 2011;186:117-124.
[304] Farrell MJ, Laird AR, Egan GF. Brain activity associated with painfully hot stimuli applied
to the upper limb: a meta-analysis. Hum Brain Mapp 2005;25:129-139.
[305] Fernandez-de-las-Penas C, Galan-del-Rio F, Fernandez-Carnero J, Pesquera J, rendt-
Nielsen L, Svensson P. Bilateral widespread mechanical pain sensitivity in women with
myofascial temporomandibular disorder: evidence of impairment in central nociceptive
processing. J Pain 2009;10:1170-1178.
[306] ffytche DH, Catani M. Beyond localization: from hodology to function. Phil Trans R Soc
B 2005;360:767-779.
[307] Fields HL. Understanding how opioids contribute to reward and analgesia. Reg Anesth
Pain Med 2007;32:242-246.
[308] Fields HL, Anderson SD. Evidence that raphe-spinal neurons mediate opiate and midbrain
stimulation-produced analgesias. Pain 1978;5:333-349.
[309] Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain
modulation. In: S McMahon, M Koltzenburg, editors. Wall and Melzack's Textbook of
Pain. Edinburgh, UK: Churchill Livingstone, 2006. pp. 125-142.
[310] Fierro B, De Tommaso M, Giglia F, Giglia G, Palermo A, Brighina F. Repetitive
transcranial magnetic stimulation (rTMS) of the dorsolateral prefrontal cortex (DLPFC)
during capsaicin-induced pain: modulatory effects on motor cortex excitability. Exp
Brain Res 2010;203:31-38.
[311] Filley CM. Neurobiology of white matter disorders. In: DK Jones, editor. Diffusion MRI:
Theory, Methods and Applications. Oxford: Oxford University Press, 2011. pp. 19-30.
[312] Fillingim RB, Maixner W. Sex-related factors in temporomandibular disorders. In: RB
Fillingim, editor. Sex, Gender, and Pain, Vol. 17. Seattle: IASP Press, 2000. pp. 309-325.

248
[313] Fillingim RB, Maixner W, Kincaid S, Sigurdsson A, Harris MB. Pain sensitivity in
patients with temporomandibular disorders: relationship to clinical and psychosocial
factors. Clin J Pain 1996;12:260-269.
[314] Fillingim RB, Maixner W, Kincaid S, Silva S. Sex differences in temporal summation but
not sensory-discriminative processing of thermal pain. Pain 1998;75:121-127.
[315] Fillingim RB, Ohrbach R, Greenspan JD, Knott C, Dubner R, Bair E, Baraian C, Slade
GD, Maixner W. Potential Psychosocial Risk Factors for Chronic TMD: Descriptive Data
and Empirically Identified Domains from the OPPERA Case-Control Study. J Pain
2011;12:T46-60.
[316] Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic
resonance images. Proc Natl Acad Sci U S A 2000;97:11050-11055.
[317] Fischl B, Liu A, Dale AM. Automated manifold surgery: constructing geometrically
accurate and topologically correct models of the human cerebral cortex. IEEE Trans Med
Imaging 2001;20:70-80.
[318] Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, Van der Kouwe A,
Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole
brain segmentation: Automated labeling of neuroanatomical structures in the human
brain. Neuron 2002;33:341-355.
[319] Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: Inflation, flattening,
and a surface-based coordinate system. Neuroimage 1999;9:195-207.
[320] Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a
coordinate system for the cortical surface. Hum Brain Mapp 1999;8:272-284.
[321] Fischl B, van der KA, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ,
Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM. Automatically
parcellating the human cerebral cortex. Cereb Cortex 2004;14:11-22.
[322] Fjell AM, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH,
Greve DN, Fischl B, Dale AM, Walhovd KB. High consistency of regional cortical
thinning in aging across multiple samples. Cereb Cortex 2009;19:2001-2012.

249
[323] Flor H, Nikolajsen L, Staehelin Jensen T. Phantom limb pain: a case of maladaptive CNS
plasticity? Nat Rev Neurosci 2006;7:873-881.
[324] Florence SL, Kaas JH. Large-scale reorganization at multiple levels of the somatosensory
pathway follows therapeutic amputation of the hand in monkeys. J Neurosci
1995;15:8083-8095.
[325] Florence SL, Taub HB, Kaas JH. Large-scale sprouting of cortical connections after
peripheral injury in adult macaque monkeys. Science 1998;282:1117-1121.
[326] Foltz EL, White LE. The role of rostral cingulumotomy in "pain" relief. Int J Neurol
1968;6:353-373.
[327] Foltz EL, White LE, Jr. Pain "relief" by frontal cingulumotomy. J Neurosurg 1962;19:89-
100.
[328] Franchi G. Persistence of vibrissal motor representation following vibrissal pad
deafferentation in adult rats. Exp Brain Res 2001;137:180-189.
[329] Franchi G, Veronesi C. Short-term reorganization of input-deprived motor vibrissae
representation following motor disconnection in adult rats. J Physiol 2006;574:457-476.
[330] Freeman W, Watts JW. Pain of organic disease relieved by prefrontal lobotomy. Lancet
1946;1:953-955.
[331] Fregni F, Freedman S, Pascual-Leone A. Recent advances in the treatment of chronic pain
with non-invasive brain stimulation techniques. Lancet Neurol 2007;6:188-191.
[332] Frisardi G, Chessa G, Sau G, Frisardi F. Trigeminal electrophysiology: a 2 x 2 matrix
model for differential diagnosis between temporomandibular disorders and orofacial pain.
BMC musculoskeletal disorders 2010;11:141.
[333] Friston KJ. Functional and effective connectivity in neuroimaging: a synthesis. Hum Brain
Mapp 1994;2:56-78.
[334] Frokjaer JB, Olesen SS, Gram M, Yavarian Y, Bouwense SA, Wilder-Smith OH, Drewes
AM. Altered brain microstructure assessed by diffusion tensor imaging in patients with
chronic pancreatitis. Gut 2011;60:1554-1562.

250
[335] Frot M, Magnin M, Mauguiere F, Garcia-Larrea L. Human SII and posterior insula
differently encode thermal laser stimuli. Cereb Cortex 2007;17:610-620.
[336] Frot M, Mauguiere F. [Operculo-insular responses to nociceptive skin stimulation in
humans. A review of the literature]. Neurophysiol Clin 1999;29:401-410.
[337] Frot M, Mauguiere F. Dual representation of pain in the operculo-insular cortex in humans.
Brain 2003;126:438-450.
[338] Frot M, Rambaud L, Guenot M, Mauguiere F. Intracortical recordings of early pain-related
CO2-laser evoked potentials in the human second somatosensory (SII) area. Clin
Neurophysiol 1999;110:133-145.
[339] Fujiwara S, Sasaki M, Wada T, Kudo K, Hirooka R, Ishigaki D, Nishikawa Y, Ono A,
Yamaguchi M, Ogasawara K. High-resolution diffusion tensor imaging for the detection
of diffusion abnormalities in the trigeminal nerves of patients with trigeminal neuralgia
caused by neurovascular compression. J Neuroimaging 2011;21:e102-108.
[340] Fukushima T, Kerr FW. Organization of trigeminothalamic tracts and other thalamic
afferent systems of the brainstem in the rat: presence of gelatinosa neurons with thalamic
connections. J Comp Neurol 1979;183:169-184.
[341] Ganchrow D. Intratrigeminal and thalamic projections of nucleus caudalis in the squirrel
monkey (Saimiri sciureus): A degeneration and autoradiographic study. J Comp Neurol
1978;178:281-311.
[342] Garcia-Larrea L, Maarrawi J, Peyron R. Thalamocingulate Mechanisms of Precentral
Cortex Stimulation for Central Pain. In: B Vogt, editor. Cingulate Neurobiology and
Disease. Oxford: Oxford University Press, 2009. pp. 438-449.
[343] Garcia-Larrea L, Perchet C, Creac'h C, Convers P, Peyron R, Laurent B, Mauguiere F,
Magnin M. Operculo-insular pain (parasylvian pain): a distinct central pain syndrome.
Brain 2010;133:2528-2539.
[344] Garcia-Larrea L, Peyron R, Mertens P, Grégoire MC, Lavenne F, Bonnefoi F, Mauguiäre
F, Laurent B, Sindou M. Positron emission tomography during motor cortex stimulation
for pain control. Stereotact Funct Neurosurg 1997;68:141-148.

251
[345] Garcia-Larrea L, Peyron R, Mertens P, Gregoire MC, Lavenne F, Le Bars D, Convers P,
Mauguiere F, Sindou M, Laurent B. Electrical stimulation of motor cortex for pain
control: a combined PET-scan and electrophysiological study. Pain 1999;83:259-273.
[346] Gardner EP, Martin JH, Jessell TM. The bodily senses. In: ER Kandel, JH Schwartz, TM
Jessell, editors. Principles of Neural Science. New York: McGraw-Hill, 2000. pp. 432-
450.
[347] Garraghty PE, Kaas JH. Functional reorganization in adult monkey thalamus after
peripheral nerve injury. Neuroreport 1991;2:747-750.
[348] Garraghty PE, Muja N. Possible use-dependent changes in adult primate somatosensory
cortex. Brain Res 1995;686:119-121.
[349] Gatchel RJ, Garafalo JP, Ellis E, Holt C. Major psychological disorders in acute and
chronic TMD: an initial examination. J Am Dent Assoc 1996;127:1356-1374.
[350] Gatchel RJ, Stowell AW, Wildenstein L, Riggs R, Ellis E, 3rd. Efficacy of an early
intervention for patients with acute temporomandibular disorder-related pain: a one-year
outcome study. J Am Dent Assoc 2006;137:339-347.
[351] Gaze RM, Gordon G. The representation of cutaneous sense in the thalamus of the cat and
monkey. Q J Exp Physiol Cogn Med Sci 1954;39:279-304.
[352] Gear RW, Levine JD. Nucleus accumbens facilitates nociception. Exp Neurol
2011;229:502-506.
[353] Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev 2004;27:729-737.
[354] Gebhart GF, Jones SL. Effects of morphine given in the brain stem on the activity of dorsal
horn nociceptive neurons. Prog Brain Res 1988;77:229-243.
[355] Gebhart GF, Sandkuhler J, Thalhammer JG, Zimmermann M. Quantitative comparison of
inhibition in spinal cord of nociceptive information by stimulation in periaqueductal gray
or nucleus raphe magnus of the cat. J Neurophysiol 1983;50:1433-1445.
[356] Geha PY, Baliki MN, Harden RN, Bauer WR, Parrish TB, Apkarian AV. The brain in
chronic CRPS pain: abnormal gray-white matter interactions in emotional and autonomic
regions. Neuron 2008;60:570-581.

252
[357] Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional
neuroimaging using the false discovery rate. Neuroimage 2002;15:870-878.
[358] Gerstner G, Ichesco E, Quintero A, Schmidt-Wilcke T. Changes in regional gray matter
and white matter volume in patients with myofascial-type temporomandibular disorders:
a voxel-based morphometry study. J Orofac Pain 2011;25:99-106.
[359] Geschwind N. Disconnexion syndromes in animals and man. I. Brain 1965;88:237-294.
[360] Geschwind N. Disconnexion syndromes in animals and man. II. Brain 1965;88:585-644.
[361] Ghez C. Motor systems in the brain. In: ER Kandel, A Schwartz, TM Jessell, editors.
Principles of Neural Science. New York: McGraw-Hill, 2000. pp. 533-537.
[362] Giesler GJ, Jr. , Spiel HR, Willis WD. Organization of spinothalamic tract axons within the
rat spinal cord. J Comp Neurol 1981;195:243-252.
[363] Giesler GJ, Jr., Katter JT, Dado RJ. Direct spinal pathways to the limbic system for
nociceptive information. Trends Neurosci 1994;17:244-250.
[364] Gitelman DR, Alpert NM, Kosslyn S, Daffner K, Scinto L, Thompson W, Mesulam MM.
Functional imaging of human right hemispheric activation for exploratory movements.
Ann Neurol 1996;39:174-179.
[365] Glass JM, Park DC. Cognitive dysfunction in fibromyalgia. Curr Rheumatol Rep
2001;3:123-127.
[366] Gobel S, Hockfield S, Ruda MA. An anatomical analysis of the similarities between
medullary and spinal dorsal horns. In: Y Kawamura, R Dubner, editors. Oral-Facial
Sensory and Motor Functions. Tokyo: Quintessence, 1981. pp. 211-223.
[367] Gobel S, Purvis MB. Anatomical studies of the organization of the spinal V nucleus: the
deep bundles and the spinal V tract. Brain Res 1972;48:27-44.
[368] Godefroy JN, Thiesson D, Pollin B, Rokyta R, Azerad J. Reciprocal connections between
the red nucleus and the trigmeinal nuclei: a retrograde and anterograde tracing study.
Physiol Res 1998;47:489-500.

253
[369] Goldberg M, Mock D, Ichise M, Proulx G, Gordon A, Shandling M, Tsai S, Tenenbaum
HC. Neuropsychological Deficit in and Clinical Features of Post-Traumatic TMD. J
Orofac Pain 1996;10.
[370] Goldberg MB, Mock D, Ichise M, Proulx G, Gordon A, Shandling M, Tsai S, Tenenbaum
HC. Neuropsychologic deficits and clinical features of posttraumatic temporomandibular
disorders. J Orofac Pain 1996;10:126-140.
[371] Goldszal AF, Davatzikos C, Pham DL, Yan MX, Bryan RN, Resnick SM. An image-
processing system for qualitative and quantitative volumetric analysis of brain images. J
Comput Assist Tomogr 1998;22:827-837.
[372] Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS. A voxel-
based morphometric study of ageing in 465 normal adult human brains. Neuroimage
2001;14:21-36.
[373] Goubert L, Crombez G, Van Damme S. The role of neuroticism, pain catastrophizing and
pain-related fear in vigilance to pain: a structural equations approach. Pain 2004;107:234-
241.
[374] Graven-Nielsen T, Arendt-Nielsen L. Human models and cliical manifestations of
musculoskeletal pain and pain-motor interactions. In: T Graven-Nielsen, L Arendt-
Nielsen, S Mense, editors. Fundamentals of Muskuloskeletal Pain. Seattle: IASP Press,
2008. pp. 155-188.
[375] Graven-Nielsen T, Jansson Y, Segerdahl M, Kristensen JD, Mense S, Arendt-Nielsen L,
Sollevi A. Experimental pain by ischaemic contractions compared with pain by
intramuscular infusions of adenosine and hypertonic saline. Eur J Pain 2003;7:93-102.
[376] Graven-Nielsen T, Mense S. The peripheral apparatus of muscle pain: evidence from
animal and human studies. Clin J Pain 2001;17:2-10.
[377] Greenspan JD, Lee RR, Lenz FA. Pain sensitivity alterations as a function of lesion
location in the parasylvian cortex. Pain 1999;81:273-282.
[378] Greenspan JD, Winfield JA. Reversible pain and tactile deficits associated with a cerebral
tumor compressing the posterior insula and parietal operculum. Pain 1992;50:29-39.

254
[379] Grossi ML, Goldberg MB, Locker D, Tenenbaum HC. Reduced neuropsychologic
measures as predictors of treatment outcome in patients with temporomandibular
disorders. J Orofac Pain 2001;15:329-339.
[380] Grossi ML, Goldberg MB, Locker D, Tenenbaum HC. Irritable bowel syndrome patients
versus responding and nonresponding temporomandibular disorder patients: a
neuropsychologic profile comparative study. Int J Prosthodont 2008;21:201-209.
[381] Guo LH, Schluesener HJ. The innate immunity of the central nervous system in chronic
pain: the role of Toll-like receptors. Cell Mol Life Sci 2007;64:1128-1136.
[382] Gustin SM, Peck CC, Wilcox SL, Nash PG, Murray GM, Henderson LA. Different Pain,
Different Brain: Thalamic Anatomy in Neuropathic and Non-Neuropathic Chronic Pain
Syndromes. J Neurosci 2011;31:5956-5964.
[383] Gutierrez-Mahoney CGd. The treatment of painful phantom limb by removal of post-
central cortex. J Neurosurg 1944;1:156-162.
[384] Gwilym SE, Filippini N, Douaud G, Carr AJ, Tracey I. Thalamic atrophy associated with
painful osteoarthritis of the hip is reversible after arthroplasty: a longitudinal voxel-based
morphometric study. Arthritis Rheum 2010;62:2930-2940.
[385] Ha AD, Jankovic J. Pain in Parkinson's disease. Mov Disord 2011.
[386] Haas BW, Constable RT, Canli T. Stop the sadness: Neuroticism is associated with
sustained medial prefrontal cortex response to emotional facial expressions. Neuroimage
2008;42:385-392.
[387] Haaxma R, Kuypers HG. Intrahemispheric cortical connexions and visual guidance of
hand and finger movements in the rhusus monkey. Brain 1975;98:239-260.
[388] Haber SN, Knutson B. The Reward Circuit: Linking Primate Anatomy and Human
Imaging. Neuropsychopharmacology 2009;35:4-26.
[389] Hagbarth KE, Kerr DI. Central influences on spinal afferent conduction. J Neurophysiol
1954;17:295-307.
[390] Hagberg C, Hagberg M, Kopp S. Musculoskeletal symptoms and psychosocial factors
among patients with craniomandibular disorders. Acta Odontol Scand 1994;52:170-177.

255
[391] Hagberg C, Hellsing G, Hagberg M. Perception of cutaneous electrical stimulation in
patients with craniomandibular disorders. J Craniomandib Disord 1990;4:120-125.
[392] Hagmann P, Thiran JP, Jonasson L, Vandergheynst P, Clarke S, Maeder P, Meuli R. DTI
mapping of human brain connectivity: statistical fibre tracking and virtual dissection.
Neuroimage 2003;19:545-554.
[393] Halata Z. The ultrastructure of the sensory nerve endings in the articular capsule of the
knee joint of the domestic cat (Ruffini corpuscles and Pacinian corpuscles). J Anat
1977;124:717-729.
[394] Halata Z, Grim M, Bauman KI. Friedrich Sigmund Merkel and his "Merkel cell",
morphology, development, and physiology: review and new results. Anat Rec A Discov
Mol Cell Evol Biol 2003;271:225-239.
[395] Halkjaer L, Melsen B, McMillan AS, Svensson P. Influence of sensory deprivation and
perturbation of trigeminal afferent fibers on corticomotor control of human tongue
musculature. Exp Brain Res 2006;170:199-205.
[396] Halliday AM, Logue V. Painful sensations evoked by electrical stimulation in the
thalamus. In: GG Somjen, editor. Neurophysiology studied in man. Amsterdam: Excerpta
Medica, 1972. pp. 221-230.
[397] Han ZS, Zhang ET, Craig AD. Nociceptive and thermoreceptive lamina I neurons are
anatomically distinct. Nat Neurosci 1998;1:218-225.
[398] Hanagasi HA, Akat S, Gurvit H, Yazici J, Emre M. Pain is common in Parkinson's disease.
Clin Neurol Neurosurg 2011;113:11-13.
[399] Hannam AG, Matthews B. Reflex jaw opening in response to stimulation of periodontal
mechanoreceptors in the cat. Arch Oral Biol 1969;14:415-419.
[400] Hannam AG, Matthews B, Yemm R. Changes in the activity of the masseter muscle
following tooth contact in man. Arch Oral Biol 1969;14:1401-1406.
[401] Hannam AG, Matthews B, Yemm R. The response of the masseter muscle following tooth
contact in man. J Physiol 1969;203:25P-26P.

256
[402] Hapak L, Gordon A, Locker D, Shandling M, Mock D, Tenenbaum HC. Differentiation
between musculoligamentous, dentoalveolar, and neurologically based craniofacial pain
with a diagnostic questionnaire. J Orofac Pain 1994;8:357-368.
[403] Hardy SG. Analgesia elicited by prefrontal stimulation. Brain Res 1985;339:281-284.
[404] Hardy SG, Haigler HJ. Prefrontal influences upon the midbrain: a possible route for pain
modulation. Brain Res 1985;339:285-293.
[405] Hardy SG, Leichnetz GR. Cortical projections to the periaqueductal gray in the monkey: a
retrograde and orthograde horseradish peroxidase study. Neurosci Lett 1981;22:97-101.
[406] Hardy SG, Leichnetz GR. Frontal cortical projections to the periaqueductal gray in the rat:
a retrograde and orthograde horseradish peroxidase study. Neurosci Lett 1981;23:13-17.
[407] Hari R, Karhu J, Hamalainen M, Knuutila J, Salonen O, Sams M, Vilkman V. Functional
organization of the human first and second somatosensory cortices: A neuromagnetic
study. Eur J Neurosci 1993;5:724-734.
[408] Harkins SW, Price DD, Braith J. Effects of ex- traversion and neuroticism on experimental
pain, clinical pain, and illness behavior. Pain 1989;36:209-218.
[409] Harman K, Ruyak P. Working through the pain: a controlled study of the impact of
persistent pain on performing a computer task. Clin J Pain 2005;21:216-222.
[410] Harmann PA, Carlton SM, Willis WD. Collaterals of the spinothalamic tract cells to the
periaqueductal gray: A fluorescent double-labeling study in the rat. Brain Res
1988;441:87-97.
[411] Harrison F, Corbin KB. The central pathway for the jaw-jerk. Am J Physiol 1941;135:439-
445.
[412] Hart RP, Wade JB, Martelli MF. Cognitive impairment in patients with chronic pain: the
significance of stress. Curr Pain Headache Rep 2003;7:116-126.
[413] Harting JK, Updyke BV, Van Lieshout DP. Corticotectal projections in the cat:
anterograde transport studies of twenty-five cortical areas. J Comp Neurol 1992;324:379-
414.

257
[414] Hassenbusch SJ, Pillay PK, Barnett GH. Radiofrequency cingulotomy for intractable
cancer pain using stereotaxis guided by magnetic resonance imaging. Neurosurgery
1990;27:220-223.
[415] Hassler R. Dichotomy of facial pain conduction in the diencephalon. In: R Hassler, AE
Walker, editors. Trigeminal neuralgia. Philadelphia: Saunders, 1970. pp. 123-138.
[416] Hatanaka N, Tokuno H, Hamada I, Inase M, Ito Y, Imanishi M, Hasegawa N, Akazawa T,
Nambu A, Takada M. Thalamocortical and intracortical connections of monkey cingulate
motor areas. Journal of Comparative Neurology 2003;462:121-138.
[417] Head H, Holmes G. Sensory disturbances from cerebral lesions. Brain 1911;34:102-254.
[418] Health NIo. Management of temporomandibular disorders. National Institutes of Health
Technology Assessment Conference Statement. J Am Dent Assoc 1996;127:1595-1606.
[419] Hebb DO. Spontaneous neurosis in chimpanzees; theoretical relations with clinical and
experimental phenomena. Psychosom Med 1947;9:3-19.
[420] Heinricher MM. The brainstem and nociceptive modulation. In: AI Basbaum, MC
Bushnell, editors. Science of Pain. San Diego: Academic Press, 2009. pp. 593-626.
[421] Hernandez-Peon R, Hagbarth KE. Interaction between afferent and cortically induced
reticular responses. J Neurophysiol 1955;18:44-55.
[422] Hihara S, Notoya T, Tanaka M, Ichinose S, Ojima H, Obayashi S, Fujii N, Iriki A.
Extension of corticocortical afferents into the anterior bank of the intraparietal sulcus by
tool-use training in adult monkeys. Neuropsychologia 2006;44:2636-2646.
[423] Hippocrates. Instruments of reduction. Book Instruments of reduction. City: D.C.
Stevenson, Web Atomics, 400 B.C.E.
[424] Hippocrates. The genuine works of Hippocrates. Book The genuine works of Hippocrates.
City: The Sydenham Society, 1849.
[425] Hirata H, Okamoto K, Bereiter DA. GABA(A) receptor activation modulates corneal unit
activity in rostral and caudal portions of trigeminal subnucleus caudalis. J Neurophysiol
2003;90:2837-2849.

258
[426] Hitzig E. Hughlings Jackson and the Cortical Motor Centres in the Ligh of Physiological
Research. Brain 1900;23:545-581.
[427] Ho KC, Roessmann U, Straumfjord JV, Monroe G. Analysis of brain weight. II. Adult
brain weight in relation to body height, weight, and surface area. Arch Pathol Lab Med
1980;104:640-645.
[428] Hoagland H. Specific afferent impulses and cutaneous sensitivity. J Gen Psychol
1932;6:276-295.
[429] Hodaie M, Quan J, Chen DQ. In vivo visualization of cranial nerve pathways in humans
using diffusion-based tractography. Neurosurgery 2010;66:788-796.
[430] Hodes RL, Howland EW, Lightfoot N, Cleeland CS. The effects of distraction on
responses to cold pressor pain. Pain 1990;41:109-114.
[431] Hodges PW, Tucker K. Moving differently in pain: a new theory to explain the adaptation
to pain. Pain 2011;152:S90-98.
[432] Hoheisel U, Reinohl J, Unger T, Mense S. Acidic pH and capsaicin activate
mechanosensitive group IV muscle receptors in the rat. Pain 2004;110:149-157.
[433] Holden JE, Van Poppel AY, Thomas S. Antinociception from lateral hypothalamic
stimulation may be mediated by NK(1) receptors in the A7 catecholamine cell group in
rat. Brain Res 2002;953:195-204.
[434] Holle D, Naegel S, Krebs S, Gaul C, Gizewski E, Diener HC, Katsarava Z, Obermann M.
Hypothalamic gray matter volume loss in hypnic headache. Ann Neurol 2011;69:533-
539.
[435] Hollins M, Harper D, Gallagher S, Owings EW, Lim PF, Miller V, Siddiqi MQ, Maixner
W. Perceived Intensity and Unpleasantness of Cutaneous and Auditory Stimuli: An
Evaluation of the Generalized Hypervigilance Hypothesis. Pain 2009;141:215-221.
[436] Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and
cell-type-specific spine growth in the neocortex. Nature 2006;441:979-983.
[437] Hosobuchi Y. Combined electrical stimulation of the periaqueductal gray matter and
sensory thalamus. Applied Neurophysiology 1983;46:112-115.

259
[438] Huettel SA, McCarthy G, Song AW. Functional magnetic resonance imaging. Sunderland,
Mass.: Sinauer Associates, 2009.
[439] Hurt RW, Ballantine HT. Stereotactic anterior cingulate lesions for persistent pain: a report
of 68 cases. Clin Neurosurg 1973;21:334-351.
[440] Hutchison WD, Davis KD, Lozano AM, Tasker RR, Dostrovsky JO. Pain-related neurons
in the human cingulate cortex. Nat Neurosci 1999;2:403-405.
[441] Hylden JL, Hayashi H, Bennett GJ. Lamina I spinomesencephalic neurons in the cat
ascend via the dorsolateral funiculi. Somatosens Res 1986;4:31-41.
[442] Iadarola JM, Caudle RM. Good pain, bad pain. Science 1997;278:239-240.
[443] Iadarola MJ, Berman KF, Zeffiro TA, Byas-Smith MG, Gracely RH, Max MB, Bennett
GJ. Neural activation during acute capsaicin-evoked pain and allodynia assessed with
PET. Brain 1998;121:931-947.
[444] Iannetti GD, Leandri M, Truini A, Zambreanu L, Cruccu G, Tracey I. Adelta nociceptor
response to laser stimuli: selective effect of stimulus duration on skin temperature, brain
potentials and pain perception. Clin Neurophysiol 2004;115:2629-2637.
[445] Iannetti GD, Mouraux A. From the neuromatrix to the pain matrix (and back). Exp Brain
Res 2010;205:1-12.
[446] Iannetti GD, Porro CA, Pantano P, Romanelli PL, Galeotti F, Cruccu G. Representation of
different trigeminal divisions within the primary and secondary human somatosensory
cortex. Neuroimage 2003;19:906-912.
[447] Iannetti GD, Zambreanu L, Cruccu G, Tracey I. Operculoinsular cortex encodes pain
intensity at the earliest stages of cortical processing as indicated by amplitude of laser-
evoked potentials in humans. Neuroscience 2005;131:199-208.
[448] Ichesco E, Quintero A, Clauw DJ, Peltier S, Sundgren PM, Gerstner GE, Schmidt-Wilcke
T. Altered Functional Connectivity Between the Insula and the Cingulate Cortex in
Patients With Temporomandibular Disorder: A Pilot Study. Headache 2011;doi:
10.1111/j.1526-4610.2011.01998.x. [Epub ahead of print].
[449] Iggo A, Muir AR. The structure and function of a slowly adapting touch corpuscle in hairy
skin. J Physiol 1969;200:763-796.

260
[450] Ikeda M, Tanami T, Matsushita M. Ascending and descending internuclear connections of
the trigeminal sensory nuclei in the cat. A study with the retrograde and anterograde
horseradish peroxidase technique. Neuroscience 1984;12:1243-1260.
[451] Iwata K, Kamo H, Ogawa A, Tsuboi Y, Noma N, Mitsuhashi Y, Taira M, Koshikawa N,
Kitagawa J. Anterior cingulate cortical neuronal activity during perception of noxious
thermal stimuli in monkeys. J Neurophysiol 2005;94:1980-1991.
[452] Jacquin MF, Chiaia NL, Haring JH, Rhoades RW. Intersubnuclear connections within the
rat trigeminal brainstem complex. Somatosens Mot Res 1990;7:399-420.
[453] Jacquin MF, Renehan WE, Mooney RD, Rhoades RW. Structure-function relationships in
rat medullary and cervical dorsal horns. I. Trigeminal primary afferents. J Neurophysiol
1986;55:1153-1186.
[454] Jain N, Florence SL, Qi HX, Kaas JH. Growth of new brainstem connections in adult
monkeys with massive sensory loss. Proc Natl Acad Sci U S A 2000;97:5546-5550.
[455] Jamison RN, Sbrocco T, Parris WC. The influence of problems with concentration and
memory on emotional distress and daily activities in chronic pain patients. Int J
Psychiatry Med 1988;18:183-191.
[456] Jansons KM, Alexander DC. Persistent Angular Structure: new insights from diffusion
MRI data. Dummy version. Inf Process Med Imaging 2003;18:672-683.
[457] Jenkins IH, Brooks DJ, Nixon PD, Frackowiak RSJ, Passingham RE. Motor sequence
learning: A study with positron emission tomography. J Neurosci 1994;14:3775-3790.
[458] Jenkins WM, Merzenich MM. Reorganization of neocortical representations after brain
injury: a neurophysiological model of the bases of recovery from stroke. Prog Brain Res
1987;71:249-266.
[459] Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and
accurate linear registration and motion correction of brain images. Neuroimage
2002;17:825-841.
[460] Ji G, Neugebauer V. Pain-related deactivation of medial prefrontal cortical neurons
involves mGluR1 and GABAA receptors. J Neurophysiol 2011.

261
[461] Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, Neugebauer V. Cognitive impairment
in pain through amygdala-driven prefrontal cortical deactivation. J Neurosci
2010;30:5451-5464.
[462] Johnson KO, Darian-Smith I, LaMotte C, Johnson B, Oldfield S. Coding of incremental
changes in skin temperature by a population of warm fibers in the monkey: correlation
with intensity discrimination in man. J Neurophysiol 1979;42:1332-1353.
[463] Johnson S, Summers J, Pridmore S. Changes to somatosensory detection and pain
thresholds following high frequency repetitive TMS of the motor cortex in individuals
suffering from chronic pain. Pain 2006;123:187-192.
[464] Jones AKP, Brown WD, Friston KJ, Qi LY, Frackowiak RSJ. Cortical and subcortical
localization of response to pain in man using positron emission tomography. Proc R Soc
Lond B: Biol Sci 1991;244:39-44.
[465] Jones DK. Studying connections in the living human brain with diffusion MRI. Cortex
2008;44:936-952.
[466] Jones DK, Symms MR, Cercignani M, Howard RJ. The effect of filter size on VBM
analyses of DT-MRI data. Neuroimage 2005;26:546-554.
[467] Jones EG, Pons TP. Thalamic and brainstem contributions to large-scale plasticity of
primate somatosensory cortex. Science 1998;282:1121-1125.
[468] Jones SL. Descending noradrenergic influences on pain. Prog Brain Res 1991;88:381-394.
[469] Jones SL, Sedivec MJ, Light AR. Effects of iontophoresed opioids on physiologically
characterized laminae I and II dorsal horn neurons in the cat spinal cord. Brain Res
1990;532:160-174.
[470] Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature 2001;413:203-210.
[471] Juottonen K, Gockel M, SilÇn T, Hurri H, Hari R, Forss N. Altered central sensorimotor
processing in patients with complex regional pain syndrome. Pain 2002;98:315-323.
[472] Kaas JH. Sensory loss and cortical reorganization in mature primates. Prog Brain Res
2002;138:167-176.

262
[473] Kaas JH, Florence SL. Mechanisms of reorganization in sensory systems of primates after
peripheral nerve injury. Adv Neurol 1997;73:147-158.
[474] Kaas JH, Florence SL, Jain N. Subcortical contributions to massive cortical
reorganizations. Neuron 1999;22:657-660.
[475] Kaas JH, Qi HX, Burish MJ, Gharbawie OA, Onifer SM, Massey JM. Cortical and
subcortical plasticity in the brains of humans, primates, and rats after damage to sensory
afferents in the dorsal columns of the spinal cord. Exp Neurol 2008;209:407-416.
[476] Kanda M, Mima T, Oga T, Matsuhashi M, Toma K, Hara H, Satow T, Nagamine T,
Rothwell JC, Shibasaki H. Transcranial magnetic stimulation (TMS) of the sensorimotor
cortex and medial frontal cortex modifies human pain perception. Clin Neurophysiol
2003;114:860-866.
[477] Kandel ER, Spencer WA. Cellular neurophysiological approaches in the study of learning.
Physiol Rev 1968;48:65-134.
[478] Karni A, Meyer G, Jezzard P, Adams MM, Turner R, Ungerleider LG. Functional MRI
evidence for adult motor cortex plasticity during motor skill learning. Nature
1995;377:155-158.
[479] Kassubek J, Juengling FD, Els T, Spreer J, Herpers M, Krause T, Moser E, Lucking CH.
Activation of a residual cortical network during painful stimulation in long-term
postanoxic vegetative state: a 15O-H2O PET study. Journal of the Neurological Sciences
2003;212:85-91.
[480] Kaufman MP, Rybicki KJ, Waldrop TG. Effect of ischemia on responses of group III and
IV afferents. J Appl Physiol 1984;57:644-650.
[481] Keay KA, Feil K, Gordon BD, Herbert H, Bandler R. Spinal afferents to functionally
distinct periaqueductal gray columns in the rat: an anterograde and retrograde tracing
study. J Comp Neurol 1997;385:207-229.
[482] Kellgren JH. Referred Pains from Muscle. Br Med J 1938;1:325-327.
[483] Kenins P. The functional anatomy of the receptive fields of rabbit C polymodal
nociceptors. J Neurophysiol 1988;59:1098-1115.

263
[484] Kenshalo DR, Iwata K, Sholas M, Thomas DA. Response properties and organization of
nociceptive neurons in area 1 of monkey primary somatosensory cortex. J Neurophysiol
2000;84:719-729.
[485] Kenshalo DR, Jr., Giesler GJ, Jr., Leonard RB, Willis WD. Responses of neurons in
primate ventral posterior lateral nucleus to noxious stimuli. J Neurophysiol
1980;43:1594-1614.
[486] Kenshalo DR, Jr., Isensee O. Responses of primate SI cortical neurons to noxious stimuli. J
Neurophysiol 1983;50:1479-1496.
[487] Kerr FW. The Divisional Organization of Afferent Fibres of the Trigeminal Nerve. Brain
1963;86:721-732.
[488] Kerr FWL, Lippman HH. The primate spinothalamic tract as demonstrated by anterolateral
cordotomy and commissural myelotomy. Adv Neurol 1974;4:147-156.
[489] Kewman DG, Vaishampayan N, Zald D, Han B. Cognitive impairment in musculoskeletal
pain patients. Int J Psychiatry Med 1991;21:253-262.
[490] Kim JH, Suh SI, Seol HY, Oh K, Seo WK, Yu SW, Park KW, Koh SB. Regional grey
matter changes in patients with migraine: a voxel-based morphometry study. Cephalalgia
2008;28:598-604.
[491] King CD, Wong F, Currie T, Mauderli AP, Fillingim RB, Riley JL, III. Deficiency in
endogenous modulation of prolonged heat pain in patients with Irritable Bowel Syndrome
and Temporomandibular Disorder. Pain 2009;143:172-178.
[492] Kirveskari E, Vartiainen NV, Gockel M, Forss N. Motor cortex dysfunction in complex
regional pain syndrome. Clin Neurophysiol 2010;121:1085-1092.
[493] Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes
synapse formation in the adult neocortex in vivo. Nat Neurosci 2006;9:1117-1124.
[494] Koay CG, Nevo U, Chang LC, Pierpaoli C, Basser PJ. The elliptical cone of uncertainty
and its normalized measures in diffusion tensor imaging. IEEE transactions on medical
imaging 2008;27:834-846.
[495] Koechlin E. Frontal pole function: what is specifically human? Trends Cogn Sci
2011;15:241; author reply 243.

264
[496] Koechlin E, Corrado G, Pietrini P, Grafman J. Dissociating the role of the medial and
lateral anterior prefrontal cortex in human planning. Proc Natl Acad Sci U S A
2000;97:7651-7656.
[497] Koechlin E, Hyafil A. Anterior Prefrontal Function and the Limits of Human Decision-
Making. Science 2007;318:594-600.
[498] Koechlin E, Ody C, Kouneiher F. The architecture of cognitive control in the human
prefrontal cortex. Science 2003;302:1181-1185.
[499] Koyama T, Kato K, Tanaka YZ, Mikami A. Anterior cingulate activity during pain-
avoidance and reward tasks in monkeys. Neurosci Res 2001;39:421-430.
[500] Kropotov JD, Crawford HJ, Polyakov YI. Somatosensory event-related potential changes
to painful stimuli during hypnotic analgesia: Anterior cingulate cortex and anterior
temporal cortex intracranial recordings. Int J Psychophysiol 1997;27:1-8.
[501] Kruger L, Saporta S, Feldman SG. Axonal transport studies of the sensory trigeminal
complex. In: DJ Anderson, B Matthews, editors. Pain in the Trigeminal Region. New
York: Elsevier, 1977. pp. 191-201.
[502] Kuchinad A, Schweinhardt P, Seminowicz DA, Wood PB, Chizh BA, Bushnell MC.
Accelerated brain gray matter loss in fibromyalgia patients: premature aging of the brain?
J Neurosci 2007;27:4004-4007.
[503] Kulkarni B, Bentley DE, Elliott R, Youell P, Watson A, Derbyshire SW, Frackowiak RS,
Friston KJ, Jones AK. Attention to pain localization and unpleasantness discriminates the
functions of the medial and lateral pain systems. Eur J Neurosci 2005;21:3133-3142.
[504] Kumazawa T, Perl ER. Primate cutaneous receptors with unmyelinated (C) fibres and their
projection to the substantia gelatinosa. J Physiol 1977;73:287-304.
[505] Kupers RC, Svensson P, Jensen TS. Central representation of muscle pain and mechanical
hyperesthesia in the orofacial region: a positron emission tomography study. Pain
2004;108:284-293.
[506] Kurth F, Zilles K, Fox PT, Laird AR, Eickhoff S. A link between the systems: functional
differentiation and integration within the human insula revealed by meta-analysis. Brain
Struct Funct 2010;214:519-534.

265
[507] Kuypers HG. The Descending Pathways to the Spinal Cord, Their Anatomy and Function.
Prog Brain Res 1964;11:178-202.
[508] Kwan CL, Crawley AP, Mikulis DJ, Davis KD. An fMRI study of the anterior cingulate
cortex and surrounding medial wall activations evoked by noxious cutaneous heat and
cold stimuli. Pain 2000;85:359-374.
[509] Kwan CL, Diamant NE, Pope G, Mikula K, Mikulis DJ, Davis KD. Abnormal forebrain
activity in functional bowel disorder patients with chronic pain. Neurology
2005;65:1268-1277.
[510] Lacadie CM, Fulbright RK, Rajeevan N, Constable RT, Papademetris X. More accurate
Talairach coordinates for neuroimaging using non-linear registration. Neuroimage
2008;42:717-725.
[511] Lamm C, Decety J, Singer T. Meta-analytic evidence for common and distinct neural
networks associated with directly experienced pain and empathy for pain. Neuroimage
2011;54:2492-2502.
[512] LaMotte RH, Shain CN, Simone DA, Tsai EFP. Neurogenic hyperalgesia: Psychophysical
studies of underlying mechanisms. J Neurophysiol 1991;66:190-211.
[513] Lancaster JL, Tordesillas-Gutierrez D, Martinez M, Salinas F, Evans A, Zilles K,
Mazziotta JC, Fox PT. Bias between MNI and Talairach coordinates analyzed using the
ICBM-152 brain template. Hum Brain Mapp 2007;28:1194-1205.
[514] Langworthy OR, Fox HM. Thalamic Syndrome: Syndrom of the posterior cerebral artery;
a review. Arch Intern Med 1937;60:203-224.
[515] Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by
central neural plasticity. J Pain 2009;10:895-927.
[516] Laureys S, Faymonville ME, Peigneux P, Damas P, Lambermont B, Del Fiore G,
Degueldre C, Aerts J, Luxen A, Franck G, Lamy M, Moonen G, Maquet P. Cortical
processing of noxious somatosensory stimuli in the persistent vegetative state.
Neuroimage 2002;17:732-741.
[517] Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related
disorders. Lancet Neurol 2004;3:537-546.

266
[518] Lazar M, Alexander AL. An error analysis of white matter tractography methods: synthetic
diffusion tensor field simulations. Neuroimage 2003;20:1140-1153.
[519] Le Bars D. The whole body receptive field of dorsal horn multireceptive neurones. Brain
Res Brain Res Rev 2002;40:29-44.
[520] Le Bars D, Dickenson AH, Besson JM. Diffuse noxious inhibitory controls (DNIC). I.
Effects on dorsal horn convergent neurones in the rat. Pain 1979;6:283-304.
[521] Le Bars D, Willer JC. Diffuse noxious inhibitory controls (DNIC). In: AI Basbaum, MC
Bushnell, editors. Science of Pain. San Diego: Academic Press, 2009. pp. 763-773.
[522] Le Bihan D. Magnetic resonance diffusion imaging: introduction and concepts. In: DK
Jones, editor. Diffusion MRI. New York: Oxford University Press, 2011. pp. 57-78.
[523] Le Bihan D, Breton E, Lallemand D, Grenier P, Cabanis E, Laval-Jeantet M. MR imaging
of intravoxel incoherent motions: application to diffusion and perfusion in neurologic
disorders. Radiology 1986;161:401-407.
[524] Le Bihan D, Mangin JF, Poupon C, Clark CA, Pappata S, Molko N, Chabriat H. Diffusion
tensor imaging: concepts and applications. J Magn Reson Imaging 2001;13:534-546.
[525] Leal PR, Roch JA, Hermier M, Souza MA, Cristino-Filho G, Sindou M. Structural
abnormalities of the trigeminal root revealed by diffusion tensor imaging in patients with
trigeminal neuralgia caused by neurovascular compression: a prospective, double-blind,
controlled study. Pain 2011;152:2357-2364.
[526] Lee DM, Pendleton N, Tajar A, O'Neill TW, O'Connor DB, Bartfai G, Boonen S,
Casanueva FF, Finn JD, Forti G, Giwercman A, Han TS, Huhtaniemi IT, Kula K, Lean
ME, Punab M, Silman AJ, Vanderschueren D, Moseley CM, Wu FC, McBeth J. Chronic
widespread pain is associated with slower cognitive processing speed in middle-aged and
older European men. Pain 2010;151:30-36.
[527] Lee J, Dougherty PM, Antezana D, Lenz FA. Responses of neurons in the region of human
thalamic principal somatic sensory nucleus to mechanical and thermal stimuli graded into
the painful range. J Comp Neurol 1999;410:541-555.
[528] Lee MC, Mouraux A, Iannetti GD. Characterizing the cortical activity through which pain
emerges from nociception. J Neurosci 2009;29:7909-7916.

267
[529] Legrain V, Damme SV, Eccleston C, Davis KD, Seminowicz DA, Crombez G. A
neurocognitive model of attention to pain: behavioral and neuroimaging evidence. Pain
2009;144:230-232.
[530] Legrain V, Iannetti GD, Plaghki L, Mouraux A. The pain matrix reloaded: a salience
detection system for the body. Prog Neurobiol 2011;93:111-124.
[531] Lele PP, Sinclair DC, Weddell G. The reaction time to touch. J Physiol 1954;123:187-203.
[532] Lenz FA, Rios M, Chau D, Krauss GL, Zirh TA, Lesser RP. Painful stimuli evoke
potentials recorded from the parasylvian cortex in humans. J Neurophysiol 1998;80:2077-
2088.
[533] Lenz FA, Rios M, Zirh A, Chau D, Krauss G, Lesser RP. Painful stimuli evoke potentials
recorded over the human anterior cingulate gyrus. J Neurophysiol 1998;79:2231-2234.
[534] Lenz FA, Seike M, Lin YC, Baker FH, Rowland LH, Gracely RH, Richardson RT.
Neurons in the area of human thalamic nucleus ventralis caudalis respond to painful heat
stimuli. Brain Res 1993;623:235-240.
[535] Leo RJ, Latif T. Repetitive transcranial magnetic stimulation (rTMS) in experimentally
induced and chronic neuropathic pain: a review. J Pain 2007;8:453-459.
[536] Leonard CM. The prefrontal cortex of the rat. I. Cortical projection of the mediodorsal
nucleus. II. Efferent connections. Brain Res 1969;12:321-343.
[537] Lerch JP. Maze training in mice induces MRI-detectable brain shape changes specific to
the type of learning. Neuroimage 2011;54:2086-2095.
[538] Lerch JP, Evans AC. Cortical thickness analysis examined through power analysis and a
population simulation. Neuroimage 2005;24:163-173.
[539] Leresche L. Epidemiology of temporomandibular disorders: implications for the
investigation of etiologic factors. Crit Rev Oral Biol Med 1997;8:291-305.
[540] LeResche L, Dworkin SF, Sommers EE, Truelove EL. An epidemiologic evaluation of two
diagnostic classification schemes for temporomandibular disorders. J Prosthet Dent
1991;65:131-137.

268
[541] Lev R, Granovsky Y, Yarnitsky D. Orbitofrontal disinhibition of pain in migraine with
aura: An interictal EEG-mapping study. Cephalalgia 2010;30:910-918.
[542] Lewin W, Phillips CG. Observations on partial removal of the post-central gyrus for pain. J
Neurol Neurosurg Psychiatry 1952;15:143-147.
[543] Lewis T, Pochin E. The double pain response of the human skin to a single stimulus. Clin
Sci 1937;3:67-76.
[544] Lidsky TI, Labuszewski T, Avitable MJ, Robinson JH. The effects of stimulation of
trigeminal sensory afferents upon caudate units in cats. Brain Res Bull 1979;4:9-14.
[545] Lima D. Ascending pathways: anatomy and physiology. In: A Basbaum, MC Bushnell,
editors. Science of Pain. Slovenia: Academic Press, 2009. pp. 477-526.
[546] Lima D, Almeida A. The medullary dorsal reticular nucleus as a pronociceptive centre of
the pain control system. Prog Neurobiol 2002;66:81-108.
[547] Lima MC, Fregni F. Motor cortex stimulation for chronic pain. Neurology 2008;70:2329-
2338.
[548] Lima MC, Fregni F. Motor cortex stimulation for chronic pain: systematic review and
meta-analysis of the literature. Neurology 2008;70:2329-2337.
[549] Linden RW. Properties of intraoral mechanoreceptors represented in the mesencephalic
nucleus of the fifth nerve in the cat. J Physiol 1978;279:395-408.
[550] Lineberry CG, Vierck CJ. Attenuation of pain reactivity by caudate nucleus stimulation in
monkeys. Brain Res 1975;98:119-134.
[551] Lorenz J, Cross DJ, Minoshima S, Morrow TJ, Paulson PE, Casey KL. A unique
representation of heat allodynia in the human brain. Neuron 2002;35:383-393.
[552] Lorenz J, Minoshima S, Casey KL. Keeping pain out of mind: the role of the dorsolateral
prefrontal cortex in pain modulation. Brain 2003;126:1079-1091.
[553] Luerding R, Weigand T, Bogdahn U, Schmidt-Wilcke T. Working memory performance is
correlated with local brain morphology in the medial frontal and anterior cingulate cortex
in fibromyalgia patients: structural correlates of pain-cognition interaction. Brain
2008;131:3222-3231.

269
[554] Lugaro E. Modern problems in psychiatry. Translated by David Orr, and R. G. Rows,
Manchester: The University Press, 1909.
[555] Lund JP. Mastication and its control by the brain stem. Crit Rev Oral Biol Med 1991;2:33-
64.
[556] Lund JP, Donga R, Widmer CG, Stohler CS. The pain-adaptation model: a discussion of
the relationship between chronic musculoskeletal pain and motor activity. Can J Physiol
Pharmacol 1991;69:683-694.
[557] Luo P, Wong R, Dessem D. Ultrastructural basis for synaptic transmission between jaw-
muscle spindle afferents and trigeminothalamic neurons in the rostral trigeminal sensory
nuclei of the rat. J Comp Neurol 1995;363:109-128.
[558] Lutz J, Jager L, de Quervain D, Krauseneck T, Padberg F, Wichnalek M, Beyer A, Stahl R,
Zirngibl B, Morhard D, Reiser M, Schelling G. White and gray matter abnormalities in
the brain of patients with fibromyalgia: a diffusion-tensor and volumetric imaging study.
Arthritis Rheum 2008;58:3960-3969.
[559] MacDonald D, Kabani N, Avis D, Evans AC. Automated 3-D extraction of inner and outer
surfaces of cerebral cortex from MRI. Neuroimage 2000;12:340-356.
[560] Mainero C, Boshyan J, Hadjikhani N. Altered functional magnetic resonance imaging
resting-state connectivity in periaqueductal gray networks in migraine. Ann Neurol
2011;70:838-845.
[561] Maixner W. Biopsychosocial and Genetic Risk Factors for Temporomandibular Joint
Disorders and Related Conditions. In: T Graven-Nielsen, L Arendt-Nielsen, S Mense,
editors. Fundamentals of Muskuloskeletal Pain. Seattle: IASP Press, 2008.
[562] Maixner W. Temporomandibular Joint Disorders. In: EA Mayer, MC Bushnell, editors.
Functional Pain Syndromes. Seattle: IASP Press, 2009.
[563] Maixner W, Fillingim R, Booker D, Sigurdsson A. Sensitivity of patients with painful
temporomandibular disorders to experimentally evoked pain. Pain 1995;63:341-351.
[564] Maixner W, Fillingim R, Kincaid S, Sigurdsson A, Harris MB. Relationship between pain
sensitivity and resting arterial blood pressure in patients with painful temporomandibular
disorders. Psychosom Med 1997;59:503-511.

270
[565] Maixner W, Fillingim R, Sigurdsson A, Kincaid S, Silva S. Sensitivity of patients with
painful temporomandibular disorders to experimentally evoked pain: evidence for altered
temporal summation of pain. Pain 1998;76:71-81.
[566] Manning BH, Morgan MJ, Franklin KB. Morphine analgesia in the formalin test: evidence
for forebrain and midbrain sites of action. Neuroscience 1994;63:289-294.
[567] Mantyh PW. The ascending input to the midbrain periaqueductal gray of the primate. J
Comp Neurol 1982;211:50-64.
[568] Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat
Rev Neurosci 2005;6:521-532.
[569] Marfurt CF. The somatotopic organization of the cat trigeminal ganglion as determined by
the horseradish peroxidase technique. Anat Rec 1981;201:105-118.
[570] Marfurt CF, Turner DF. Sensory nerve endings in the rat oro-facial region labeled by the
anterograde and transganglionic transport of horseradish peroxidase: a new method for
tracing peripheral nerve fibers. Brain Res 1983;261:1-12.
[571] Marshall J. Sensory disturbances in cortical wounds with special reference to pain. J
Neurol Neurosurg Psychiatry 1951;14:187-204.
[572] Martens MA, Moeyersoons JP. Acute and recurrent effort-related compartment syndrome
in sports. Sports Med 1990;9:62-68.
[573] Martin JH, Jessell TM. Modality coding in the somatic sensory system. In: ER Kandel, JH
Schwartz, TM Jessell, editors. Principles of Neural Science. Norwalk, Connecticut:
Appleton & Lange, 1991. pp. 341-352.
[574] Matsushita M, Ikeda M, Okado N. The cells of origin of the trigeminothalamic,
trigeminospinal and trigeminocerebellar projections in the cat. Neuroscience
1982;7:1439-1454.
[575] May A. Chronic pain may change the structure of the brain. Pain 2008;137:7-15.
[576] May A. Experience-dependent structural plasticity in the adult human brain. Trends Cogn
Sci 2011;15:475-482.

271
[577] May A. Structural brain imaging: a window into chronic pain. Neuroscientist 2011;17:209-
220.
[578] May A, Ashburner J, Buchel C, McGonigle DJ, Friston KJ, Frackowiak RS, Goadsby PJ.
Correlation between structural and functional changes in brain in an idiopathic headache
syndrome. Nat Med 1999;5:836-838.
[579] Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, Silva JA,
McGinnis S, Glass TG, Martin CC, Fox PT. Cingulate function in depression: A potential
predictor of treatment response. Neuroreport 1997;8:1057-1061.
[580] Mayer DJ, Price DD. Central nervous system mechanisms of analgesia. Pain 1976;2:379-
404.
[581] Mayer EA, Bushnell MC. Synthesis. In: EA Mayer, MC Bushnell, editors. Functional Pain
Syndromes. Seattle: IASP Press, 2009.
[582] Mayer EA, Labus JS, Berkley KJ. Sex differences in pain. In: JB Becker, KJ Berkley, N
Geary, E Hampson, JP Herman, EA Young, editors. Sex Differences in the Brain from
Genes to Behavior. Oxford: Oxford University Press, 2008. pp. 371-396.
[583] Mazzola L, Isnard J, Mauguiere F. Somatosensory and pain responses to stimulation of the
second somatosensory area (SII) in humans. A comparison with SI and insular responses.
Cereb Cortex 2006;16:960-968.
[584] McCaul KD, Haugtvedt C. Attention, distraction, and cold-pressor pain. Journal of
Personality and Social Psychology 1982;43:154-162.
[585] McGinnis SM, Brickhouse M, Pascual B, Dickerson BC. Age-Related Changes in the
Thickness of Cortical Zones in Humans. Brain Topogr 2011.
[586] McHaffie JG, Larson MA, Stein BE. Response properties of nociceptive and low-threshold
neurons in rat trigeminal pars caudalis. Journal of Comparative Neurology 1994;347:409-
425.
[587] McNeill C. History and evolution of TMD concepts. Oral Surg Oral Med Oral Pathol Oral
Radiol Endod 1997;83:51-60.
[588] McRae K, Hughes B, Chopra S, Gabrieli JD, Gross JJ, Ochsner KN. The neural bases of
distraction and reappraisal. J Cogn Neurosci 2010;22:248-262.

272
[589] Mehler WR. Some neurological species differences - a posteriori. Ann N Y Acad Sci
1969;167:424-468.
[590] Meier W, Klucken M, Soyka D, Bromm B. Hypnotic hypo- and hyperalgesia: Divergent
effects on pain ratings and pain-related cerebral potentials. Pain 1993;53:175-181.
[591] Melzack R. The McGill pain questionnaire: Major properties and scoring methods. Pain
1975;1:277-299.
[592] Melzack R. Pain - an overview. Acta Anaesthesiol Scand 1999;43:880-884.
[593] Melzack R, Casey KL. Sensory, motivational, and central control determinants of pain: a
new conceptual model. In: D Kenshalo, editor. The Skin Senses. Springfield: C.C.
Thomas, 1968. pp. 423-439.
[594] Melzack R, Loeser JD. Phantom body pain in paraplegics: evidence for a central "pattern
generating mechanism" for pain. Pain 1978;4:195-210.
[595] Melzack R, Wall PD. Pain mechanisms: a new theory. Science 1965;150:971-979.
[596] Menetrey D, Chaouch A, Binder D, Besson JM. The origin of the spinomesencephalic tract
in the rat: an anatomical study using the retrograde transport of horseradish peroxidase. J
Comp Neurol 1982;206:193-207.
[597] Meng ID, Hu JW, Benetti AP, Bereiter DA. Encoding of corneal input in two distinct
regions of the spinal trigeminal nucleus in the rat: cutaneous receptive field properties,
responses to thermal and chemical stimulation, modulation by diffuse noxious inhibitory
controls, and projections to the parabrachial area. J Neurophysiol 1997;77:43-56.
[598] Mense S, Hoheisel U. Morphology and functional types of muscle nociceptors. In: T
Graven-Nielsen, L Arendt-Nielsen, S Mense, editors. Fundamentals of Muskuloskeletal
Pain. Seattle: IASP Press, 2008. pp. 3-18.
[599] Mense S, Meyer H. Different types of slowly conducting afferent units in cat skeletal
muscle and tendon. J Physiol 1985;363:403-417.
[600] Merskey H. Classification of chronic pain. Descriptions of chronic pain syndromes and
definitions of pain terms. Seattle: IASP Press, 1994.

273
[601] Merzenich MM, Kaas JH, Wall J, Nelson RJ, Sur M, Fellman D. Topographic
reorganization of somatosensory cortical areas 3B and 1 in adult monkeys following
restricted deafferentation. Neuroscience 1983;8:33-55.
[602] Merzenich MM, Nelson RJ, Stryker MP, Cynader MS, Schoppmann A, Zook JM.
Somatosensory cortical map changes following digit amputation in adult monkeys. J
Comp Neurol 1984;224:591-605.
[603] Mesulam MM. Tracing neural connections of human brain with selective silver
impregnation. Observations on geniculocalcarine, spinothalamic, and entorhinal
pathways. Arch Neurol 1979;36:814-818.
[604] Mesulam MM. The functional anatomy and hemispheric specialization for directed
attention. The role of the parietal lobe and its connectivity. Trends Neurosci 1983;6:384-
387.
[605] Mesulam MM. Large-scale neurocognitive networks and distributed processing for
attention, language, and memory. Ann Neurol 1990;28:597-613.
[606] Mesulam MM. Spatial attention and neglect: parietal, frontal and cingulate contributions to
the mental representation and attentional targeting of salient extrapersonal events. Phil
Trans R Soc B 1999;354:1325-1346.
[607] Mesulam MM. Foreword. In: JD Schmahmann, DN Pandya, editors. Fiber pathways of the
brain. New York: Oxford University Press, 2006. pp. ix-x.
[608] Metz AE, Yau HJ, Centeno MV, Apkarian AV, Martina M. Morphological and functional
reorganization of rat medial prefrontal cortex in neuropathic pain. Proc Natl Acad Sci U S
A 2009;106:2423-2428.
[609] Meyer RA, Campbell JA. Myelinated nociceptive afferents account for the hyperalgesia
that follows a burn to the hand. Science 1981;213:1527-1529.
[610] Meyer RA, Campbell JN, Raja SN. Peripheral neural mechanisms of nociception. In: PD
Wall, R Melzack, editors. Textbook of pain, Vol. 3. New York: Churchill Livingstone,
1994. pp. 13-44.
[611] Meyer RA, Davis KD, Cohen RH, Treede RD, Campbell JN. Mechanically insensitive
afferents (MIAs) in cutaneous nerves of monkey. Brain Res 1991;561:252-261.

274
[612] Meyer RA, Ringkamp M, Campbell CM, Raja SN. Peripheral mechanisms of cutaneous
nociception. In: S McMahon, M Koltzenburg, editors. Wall and Melzack's Textbook of
Pain. Edinburgh, UK: Churchill Livingstone, 2006. pp. 3-35.
[613] Millan MJ. Descending control of pain. Progress in Neurobiology 2002;66:355-474.
[614] Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat
Rev Neurosci 2009;10:23-36.
[615] Moayedi M, Desouza D, Erpelding N. Making sense of gray matter abnormalities in
chronic orofacial pain--synthesizing divergent findings. J Neurosci 2011;31:12396-
12397.
[616] Moayedi M, Weissman-Fogel I. Is the insula the "how much" intensity coder? J
Neurophysiol 2009;102:1345-1347.
[617] Moayedi M, Weissman-Fogel I, Crawley AP, Goldberg MB, Freeman BV, Tenenbaum
HC, Davis KD. Contribution of chronic pain and neuroticism to abnormal forebrain gray
matter in patients with temporomandibular disorder. Neuroimage 2011;55:277-286.
[618] Mogami H, Kuroda K, Hayakawa T, Akagi K. Ascending paths from the spinal trigeminal
nucleus and its adjacent structures. In: R Dubner, editor. Orofacial Sensory and Motor
Mechanisms. New York: Appleton-Century-Croft, 1971. pp. 205-228.
[619] Monakow KH, Akert K, Kunzle H. Projections of precentral and premotor cortex to the red
nucleus and other midbrain areas in Macaca fascicularis. Exp Brain Res 1979;34:91-105.
[620] Monconduit L, Bourgeais L, Bernard JF, Le Bars D, Villanueva L. Ventromedial thalamic
neurons convey nociceptive signals from the whole body surface to the dorsolateral
neocortex. J Neurosci 1999;19:9063-9072.
[621] Monconduit L, Desbois C, Villanueva L. The integrative role of the rat medullary
subnucleus reticularis dorsalis in nociception. Eur J Neurosci 2002;16:937-944.
[622] Morecraft RJ, Tanji J. Cingulofrontal interactions and the cingulate motor areas. In: BA
Vogt, editor. Cingulate neurobiology and disease. New York: Oxford University Press,
2009. pp. 114-144.

275
[623] Morecraft RJ, Van Hoesen GW. Cingulate input to the primary and supplementary motor
cortices in the rhesus monkey: evidence for somatotopy in areas 24c and 23c. J Comp
Neurol 1992;322:471-489.
[624] Moreno CB, Hernandez-Beltran N, Munevar D, Gutierrez-Alvarez AM. Central
neuropathic pain in Parkinson's disease. Neurologia 2011.
[625] Mori S, Wakana S, van Zijl PC, Nagae-Poetscher LM. MRI Atlas of Human White Matter.
Amsterdam: Elsevier Science, 2005.
[626] Mori S, Zhang J. Principles of diffusion tensor imaging and its applications to basic
neuroscience research. Neuron 2006;51:527-539.
[627] Morrison JH, Hof PR. Life and death of neurons in the aging cerebral cortex. Int Rev
Neurobiol 2007;81:17.
[628] Moss RA, Villarosa GA, Cooley JE, Lombardo TW. Masticatory muscle activity as a
function of parafunctional, active and passive oral behavioural patterns. J Oral Rehabil
1987;14:361-370.
[629] Moulton EA, Keaser ML, Gullapalli RP, Greenspan JD. Regional intensive and temporal
patterns of functional MRI activation distinguishing noxious and innocuous contact heat.
J Neurophysiol 2005;93:2183-2193.
[630] Moulton EA, Pendse G, Morris S, Aiello-Lammens M, Becerra L, Borsook D. Segmentally
arranged somatotopy within the face representation of human primary somatosensory
cortex. Hum Brain Mapp 2009;30:757-765.
[631] Mountcastle VB, Powell TP. Neural mechanisms subserving cutaneous sensibility, with
special reference to the role of afferent inhibition in sensory perception and
discrimination. Bull Johns Hopkins Hosp 1959;105:201-232.
[632] Mouraux A, Diukova A, Lee MC, Wise RG, Iannetti GD. A multisensory investigation of
the functional significance of the "pain matrix". Neuroimage 2011;54:2237-2249.
[633] Mouraux A, Iannetti GD. Nociceptive laser-evoked brain potentials do not reflect
nociceptive-specific neural activity. J Neurophysiol 2009;101:3258-3269.
[634] Murray GM, Peck CC. Orofacial pain and jaw muscle activity: A new model. J Orofac
Pain 2007;21:263-278.

276
[635] Mylius V, Brebbermann J, Dohmann H, Engau I, Oertel WH, Moller JC. Pain sensitivity
and clinical progression in Parkinson's disease. Mov Disord 2011;26:2220-2225.
[636] Nafe JP. A quatitative theory of feeling. J Gen Psychol 1929;2:199-211.
[637] Naliboff BD, Rhudy JL. Anxiety and functional pain disorders. In: EA Mayer, MC
Bushnell, editors. Functional Pain Syndromes: Presentation and Pathophysiology. Seattle:
IASP Press, 2009. pp. 185-213.
[638] Nash PG, Macefield VG, Klineberg IJ, Gustin SM, Murray GM, Henderson LA. Changes
in human primary motor cortex activity during acute cutaneous and muscle orofacial
pain. J Orofac Pain 2010;24:379-390.
[639] Nasution ID, Shigenaga Y. Ascending and descending internuclear projections within the
trigeminal sensory nuclear complex. Brain Res 1987;425:234-247.
[640] Nauta WJ, Mehler WR. Projections of the lentiform nucleus in the monkey. Brain Res
1966;1:3-42.
[641] Nebel MB, Folger S, Tommerdahl M, Hollins M, McGlone F, Essick GK.
Temporomandibular disorder modifies cortical responses to tactile stimulation. J Pain
2010;11:1083-1094.
[642] Newman HM, Stevens RT, Apkarian AV. Direct spinal projections to limbic and striatal
areas: anterograde transport studies from the upper cervical spinal cord and the cervical
enlargement in squirrel monkey and rat. J Comp Neurol 1996;365:640-658.
[643] Nguyen JP, Nizard J, Keravel Y, Lefaucheur JP. Invasive brain stimulation for the
treatment of neuropathic pain. Nat Rev Neurol 2011.
[644] Nieuwenhuys R, Voogd J, van Huijzen C. The Human Central Nervous System. Berlin
Heidelberg: Springer, 2008.
[645] Nishida Y, Yokota T. Corneal representation within the trigeminal subnucleus caudalis and
adjacent bulbar lateral reticular formation of the cat. The Japanese journal of physiology
1991;41:551-565.
[646] Nitzan D, Benoliel R, Heir G, Dolwick F. Pain and dysfunction of the temporomandibular
joing. In: Y Sharav, R Benoliel, editors. Orofacial pain & headache. New York: Elsevier,
2008. pp. 149-192.

277
[647] Nobre AC, Sebestyen GN, Gitelman DR, Mesulam MM, Frackowiak RSJ, Frith CD.
Functional localization of the system for visuospatial attention using positron emission
tomography. Brain 1997;120:515-533.
[648] Nudo RJ, Wise BM, SiFuentes F, Milliken GW. Neural substrates for the effects of
rehabilitative training on motor recovery after ischemic infarct. Science 1996;272:1791-
1794.
[649] O'Reardon JP, Fontecha JF, Cristancho MA, Newman S. Unexpected reduction in migraine
and psychogenic headaches following rTMS treatment for major depression: a report of
two cases. CNS Spectr 2007;12:921-925.
[650] Obermann M, Nebel K, Schumann C, Holle D, Gizewski ER, Maschke M, Goadsby PJ,
Diener HC, Katsavara Z. Gray matter changes related to chronic posttraumatic headache.
Neurology 2009;73:978-993.
[651] Ochs S. A history of nerve functions : from animal spirits to molecular mechanisms.
Cambridge, UK ; New York: Cambridge University Press, 2004.
[652] Ogino Y, Nemoto H, Goto F. Somatotopy in human primary somatosensory cortex in pain
system. Anesthesiology 2005;103:821-827.
[653] Ohrbach R, Dworkin SF. Five-year outcomes in TMD: relationship of changes in pain to
changes in physical and psychological variables. Pain 1998;74:315-326.
[654] Okada-Ogawa A, Porreca F, Meng ID. Sustained morphine-induced sensitization and loss
of diffuse noxious inhibitory controls in dura-sensitive meduallry dorsal horn neurons. J
Neurosci 2009;29:15828-15835.
[655] Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory.
Neuropsychologia 1971;9:97-113.
[656] Olszewski J. On the anatomical and functional organization of the spinal trigeminal
nucleus. J Comp Neurol 1950;92:401-413.
[657] Osenbach RK. Motor cortex stimulation for intractable pain. Neurosurg Focus 2006;21:E7.
[658] Ostrowsky K, Magnin M, Ryvlin P, Isnard J, Guenot M, Mauguiere F. Representation of
pain and somatic sensation in the human insula: a study of responses to direct electrical
cortical stimulation. Cereb Cortex 2002;12:376-385.

278
[659] Owen AM, Schiff ND, Laureys S. A new era of coma and consciousness science. Prog
Brain Res 2009;177:399-411.
[660] Ozarslan E, Nevo U, Basser PJ. Anisotropy induced by macroscopic boundaries: surface-
normal mapping using diffusion-weighted imaging. Biophys J 2008;94:2809-2818.
[661] Ozarslan E, Shepherd TM, Vemuri BC, Blackband SJ, Mareci TH. Resolution of complex
tissue microarchitecture using the diffusion orientation transform (DOT). Neuroimage
2006;31:1086-1103.
[662] Pakkenberg B, Gundersen HJ. Neocortical neuron number in humans: effect of sex and
age. J Comp Neurol 1997;384:312-320.
[663] Pandya DN, Kuypers HG. Cortico-cortical connections in the rhesus monkey. Brain Res
1969;13:13-36.
[664] Papèz JW. A proposed circuit for emotion. Arch Neurol Psychiatry 1937;38:725-746.
[665] Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico- basal
ganglia-thalamo-cortical loop. Brain Res Rev 1995;20:91-127.
[666] Park DC, Glass JM, Minear M, Crofford LJ. Cognitive function in fibromyalgia patients.
Arthritis Rheum 2001;44:2125-2133.
[667] Park HJ, Westin CF, Kubicki M, Maier SE, Niznikiewicz M, Baer A, Frumin M, Kikinis
R, Jolesz FA, McCarley RW, Shenton ME. White matter hemisphere asymmetries in
healthy subjects and in schizophrenia: a diffusion tensor MRI study. Neuroimage
2004;23:213-223.
[668] Parker GJ. Analysis of MR diffusion weighted images. Br J Radiol 2004;77 Spec No
2:S176-185.
[669] Parker GJ, Alexander DC. Probabilistic anatomical connectivity derived from the
microscopic persistent angular structure of cerebral tissue. Phil Trans R Soc B
2005;360:893-902.
[670] Parker GJ, Haroon HA, Wheeler-Kingshott CA. A framework for a streamline-based
probabilistic index of connectivity (PICo) using a structural interpretation of MRI
diffusion measurements. J Magn Reson Imaging 2003;18:242-254.

279
[671] Parker GJM. Probabilistic fibre tracking. In: DK Jones, editor. Diffusion MRI: Theory,
Methods and Applications. New York: Oxford University Press, 2011. pp. 396-409.
[672] Pascual-Leone A, Amedi A, Fregni F, Merabet LB. The plastic human brain cortex. Ann
Rev Neurosci 2005;28:377-401.
[673] Pascual-Leone A, Grafman J, Hallett M. Modulation of cortical motor output maps during
development of implicit and explicit knowledge. Science 1994;263:1287-1289.
[674] Pavlides C, Miyashita E, Asanuma H. Projection from the sensory to the motor cortex is
important in learning motor skills in the monkey. J Neurophysiol 1993;70:733-741.
[675] Peck CC, Murray GM, Gerzina TM. How does pain affect jaw muscle activity? The
Integrated Pain Adaptation Model. Aust Dent J 2008;53:201-207.
[676] Peele TL. Acute and chronic parietal lobe ablations in monkeys. J Neurophysiol
1944;7:269-286.
[677] Penfield W. The Influence of the Diencephalon and Hypophysis Upon General Autonomic
Function. Can Med Assoc J 1934;30:589-598.
[678] Penfield W. Some observations on the cerebral cortex of man. Proc R Soc B
1947;134:329-347.
[679] Penfield W, Boldrey E. Somatic motor and sensory representation in the cerebral cortex of
man as studied by electrical stimulation. Brain 1937;60:389-443.
[680] Peper JS, Brouwer RM, Boomsma DI, Kahn RS, Hulshoff Pol HE. Genetic influences on
human brain structure: a review of brain imaging studies in twins. Hum Brain Mapp
2007;28:464-473.
[681] Perl ER. Myelinated afferent fibres innervating the primate skin and their response to
noxious stimuli. J Physiol 1968;197:593-615.
[682] Perl ER. Pain and the discovery of nociceptors. In: C Belmonte, F Cervero, editors.
Neurobiology of nociceptors. New York: Oxford University Press, 1996. pp. 5-36.
[683] Perl ER, Kruger L. Nociception and Pain: Evolution of concepts and observations. In: L
Kruger, editor. Pain and Touch. San Diego, CA: Academic Press, 1996. pp. 180-212.

280
[684] Perl ER, Whitlock DG. Somatic stimuli exciting spinothalamic projections to thalamic
neurons in cat and monkey. Exp Neurol 1961;3:256-296.
[685] Peters A, Morrison JH, Rosene DL, Hyman BT. Feature article: are neurons lost from the
primate cerebral cortex during normal aging? Cereb Cortex 1998;8:295-300.
[686] Petrides M, Pandya DN. Dorsolateral prefrontal cortex: comparative cytoarchitectonic
analysis in the human and the macaque brain and corticocortical connection patterns.
European Journal of Neuroscience 1999;11:1011-1036.
[687] Petrovic P, Ingvar M. Imaging cognitive modulation of pain processing. Pain 2002;95:1-5.
[688] Peyron R, Frot M, Schneider F, Garcia-Larrea L, Mertens P, Barral FG, Sindou M, Laurent
B, Mauguiere F. Role of operculoinsular cortices in human pain processing: converging
evidence from PET, fMRI, dipole modeling, and intracerebral recordings of evoked
potentials. Neuroimage 2002;17:1336-1346.
[689] Peyron R, Garcia-Larrea L, Deiber MP, Cinotti L, Convers P, Sindou M, Mauguiäre F,
Laurent B. Electrical stimulation of precentral cortical area in the treatment of central
pain: Electrophysiological and PET study. Pain 1995;62:275-286.
[690] Peyron R, Laurent B, Garcia-Larrea L. Functional imaging of brain responses to pain. A
review and meta- analysis. Neurophysiol Clin 2000;30:263-288.
[691] Picard N, Strick PL. Motor areas of the medial wall: A review of their location and
functional activation. Cereb Cortex 1996;6:342-353.
[692] Picard N, Strick PL. Imaging the premotor areas. Curr Opin Neurobiol 2001;11:663-672.
[693] Piche M, Bouin M, Arsenault M, Poitras P, Rainville P. Decreased pain inhibition in
irritable bowel syndrome depends on altered descending modulation and higher-order
brain processes. Neuroscience 2011;195C:166-175.
[694] Pierpaoli C. Artifacts in diffusion MRI. In: DK Jones, editor. Diffusion MRI : theory,
methods, and application. New York: Oxford University Press, 2011. pp. 303-318.
[695] Pierpaoli C, Jezzard P, Basser PJ, Barnett A, Di Chiro G. Diffusion tensor MR imaging of
the human brain. Radiology 1996;201:637-648.

281
[696] Pillay PK, Hassenbusch SJ. Bilateral MRI-guided stereotactic cingulotomy for intractable
pain. Stereotact Funct Neurosurg 1992;59:33-38.
[697] Pincus T, Morley S. Cognitive-processing bias in chronic pain: a review and integration.
Psychol Bull 2001;127:599-617.
[698] Plato. Timaeus. Book Timaeus. City: Project Gutenberg, 1998.
[699] Ploner M, Gross J, Timmermann L, Schnitzler A. Pain processing is faster than tactile
processing in the human brain. J Neurosci 2006;26:10879-10882.
[700] Pochon JB, Levy R, Poline JB, Crozier S, LehÇricy S, Pillon B, Deweer B, Le Bihan D,
Dubois B. The role of dorsolateral prefrontal cortex in the preparation of forthcoming
actions: an fMRI study. Cereb Cortex 2001;11:260-266.
[701] Poggio GF, Mountcastle VB. The Functional Properties of Ventrobasal Thalamic
Neuronsstudied in Unanesthetized Monkeys. J Neurophysiol 1963;26:775-806.
[702] Pons TP, Garraghty PE, Mishkin M. Serial and parallel processing of tactual information in
somatosensory cortex of rhesus monkeys. J Neurophysiol 1992;68:518-527.
[703] Pons TP, Garraghty PE, Ommaya AK, Kaas JH, Taub E, Mishkin M. Massive cortical
reorganization after sensory deafferentation in adult macaques. Science 1991;252:1857-
1860.
[704] Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation.
Trends in Neurosciences 2002;25:319-325.
[705] Porro CA, Cettolo V, Francescato MP, Baraldi P. Temporal and intensity coding of pain in
human cortex. J Neurophysiol 1998;80:3312-3320.
[706] Porter LL, Sakamoto K. Organization and synaptic relationships of the projection from the
primary sensory to the primary motor cortex in the cat. J Comp Neurol 1988;271:387-
396.
[707] Potvin S, Grignon S, Marchand S. Human evidence of a supra-spinal modulating role of
dopamine on pain perception. Synapse 2009;63:390-402.

282
[708] Pozo MA, Cervero F. Neurons in the rat spinal trigeminal complex driven by corneal
nociceptors: receptive-field properties and effects of noxious stimulation of the cornea. J
Neurophysiol 1993;70:2370-2378.
[709] Price DD. Psychological and neural mechanisms of the affective dimension of pain.
Science 2000;288:1769-1772.
[710] Price DD, Dubner R. Mechanisms of first and second pain in the peripheral and central
nervous systems. J Invest Dermatol 1977;69:167-171.
[711] Price DD, Hu JW, Dubner R, Gracely RH. Peripheral suppression of first pain and central
summation of second pain evoked by noxious heat pulses. Pain 1977;3:57-68.
[712] Puri BK, Agour M, Gunatilake KD, Fernando KA, Gurusinghe AI, Treasaden IH.
Reduction in left supplementary motor area grey matter in adult female fibromyalgia
sufferers with marked fatigue and without affective disorder: a pilot controlled 3-T
magnetic resonance imaging voxel-based morphometry study. The Journal of
international medical research 2010;38:1468-1472.
[713] Quevedo AS, Coghill RC. Attentional modulation of spatial integration of pain: evidence
for dynamic spatial tuning. J Neurosci 2007;27:11635-11640.
[714] Rainville P. Brain mechanisms of pain affect and pain modulation. Curr Opin Neurobiol
2002;12:195-204.
[715] Rainville P, Carrier B, Hofbauer RK, Bushnell MC, Duncan GH. Dissociation of sensory
and affective dimensions of pain using hypnotic modulation. Pain 1999;82:159-171.
[716] Rainville P, Duncan GH, Price DD, Carrier B, Bushnell MC. Pain affect encoded in human
anterior cingulate but not somatosensory cortex. Science 1997;277:968-971.
[717] Rainville P, Feine JS, Bushnell MC, Duncan GH. A psychophysical comparison of sensory
and affective responses to four modalities of experimental pain. Somatosens Mot Res
1992;9:265-277.
[718] Raja SN, Campbell JN, Meyer RA. Evidence for different mechanisms of primary and
secondary hyperalgesia following heat injury to the glabrous skin. Brain 1984;107:1179-
1188.

283
[719] Ralston HJ, III. The synaptic organization in the dorsal horn of the spinal cord and the
ventrobasal thalamus of the cat. In: R Dubner, editor. Orofacial Sensory and Motor
Mechanisms. New York: Appleton-Century-Croft, 1971. pp. 229-250.
[720] Ralston HJ, III, Ralston DD. The distribution of dorsal root axons in laminae I, II and III of
the macaque spinal cord: a quantitative electron microscope study. J Comp Neurol
1979;184:643-684.
[721] Ramírez LM, Sandoval GP, Ballesteros LE. Temporomandibular disorders: referred
cranio-cervico-facial clinic. Medicina oral, patología oral y cirugía bucal 2005;10 Suppl
1:E18-26.
[722] Rashid W, Hadjiprocopis A, Griffin CM, Chard DT, Davies GR, Barker GJ, Tofts PS,
Thompson AJ, Miller DH. Diffusion tensor imaging of early relapsing-remitting multiple
sclerosis with histogram analysis using automated segmentation and brain volume
correction. Mult Scler 2004;10:9-15.
[723] Ray RD, Ochsner KN, Cooper JC, Robertson ER, Gabrieli JD, Gross JJ. Individual
differences in trait rumination and the neural systems supporting cognitive reappraisal.
Cogn Affect Behav Neurosci 2005;5:156-168.
[724] Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain 2002;100:1-
6.
[725] Ren K, Dubner R. Pain facilitation and activity-dependent plasticity in pain modulatory
circuitry: role of BDNF-TrkB signaling and NMDA receptors. Mol Neurobiol
2007;35:224-235.
[726] Ren K, Dubner R. Descending control mechanisms. In: AI Basbaum, MC Bushnell,
editors. Science of Pain. San Diego: Academic Press, 2009. pp. 724-762.
[727] Rexed B. The cytoarchitectonic organization of the spinal cord in the cat. J Comp Neurol
1952;96:414-495.
[728] Rexed B. A cytoarchitectonic atlas of the spinal cord in the cat. J Comp Neurol
1954;100:297-379.
[729] Rey R. The history of pain. Cambridge, Mass.: Harvard University Press, 1995.

284
[730] Reynolds DV. Surgery in the rat during electrical analgesia induced by focal brain
stimulation. Science 1969;164:444-445.
[731] Rhodes DL, Liebeskind JC. Analgesia from rostral brain stem stimulation in the rat. Brain
Res 1978;143:521-532.
[732] Richards CD, Taylor DC. Electrophysiological evidence for a somatotopic sensory
projection to the striatum of the rat. Neurosci Lett 1982;30:235-240.
[733] Richter EO, Davis KD, Hamani C, Hutchison WD, Dostrovsky JO, Lozano AM.
Cingulotomy for psychiatric disease: microelectrode guidance, a callosal reference
system for documenting lesion location, and clinical results. Neurosurgery 2004;54:622-
628.
[734] Rios M, Treede R, Lee J, Lenz FA. Direct Evidence of Nociceptive Input to Human
Anterior Cingulate Gyrus and Parasylvian Cortex. Curr Rev Pain 1999;3:256-264.
[735] Robinson CJ, Burton H. Organization of somatosensory receptive fields in cortical areas
7b, retroinsula, postauditory and grnaular insula of M. fascicularis J Comp Neurol
1980;192:69-92.
[736] Robinson CJ, Burton H. Somatic submodality distribution within the second
somatosensory (SII), 7b, retroinsular, postauditory, and granular insular cortical areas of
M. fascicularis. J Comp Neurol 1980;192:93-108.
[737] Robinson CJ, Burton H. Somatotographic organization in the second somatosensory area
of M. fascicularis. J Comp Neurol 1980;192:43-67.
[738] Robinson ME, Craggs JG, Price DD, Perlstein WM, Staud R. Gray matter volumes of
pain-related brain areas are decreased in fibromyalgia syndrome. J Pain 2011;12:436-443.
[739] Roby-Brami A, Bussel B, Willer JC, Le Bars D. An electrophysiological investigation into
the pain-relieving effects of heterotopic nociceptive stimuli. Probable involvement of a
supraspinal loop. Brain 1987;110 ( Pt 6):1497-1508.
[740] Rocca MA, Ceccarelli A, Falini A, Colombo B, Tortorella P, Bernasconi L, Comi G, Scotti
G, Filippi M. Brain gray matter changes in migraine patients with T2-visible lesions: a 3-
T MRI study. Stroke 2006;37:1765-1770.

285
[741] Rodriguez-Raecke R, Niemeier A, Ihle K, Ruether W, May A. Brain gray matter decrease
in chronic pain is the consequence and not the cause of pain. J Neurosci 2009;29:13746-
13750.
[742] Rolls ET, Grabenhorst F. The orbitofrontal cortex and beyond: from affect to decision-
making. Prog Neurobiol 2008;86:216-244.
[743] Romaniello A, Cruccu G, Frisardi G, Arendt-Nielsen L, Svensson P. Assessment of
nociceptive trigeminal pathways by laser-evoked potentials and laser silent periods in
patients with painful temporomandibular disorders. Pain 2003;103:31-39.
[744] Romaniello A, Cruccu G, McMillan AS, Arendt-Nielsen L, Svensson P. Effect of
experimental pain from trigeminal muscle and skin on motor cortex excitability in
humans. Brain Res 2000;882:120-127.
[745] Rosenkranz K, Williamon A, Butler K, Cordivari C, Lees AJ, Rothwell JC.
Pathophysiological differences between musician's dystonia and writer's cramp. Brain
2005;128:918-931.
[746] Russel CK, Horsley V. Note on apparent re-representation in the cerebral cortex of the type
of sensory representation as it exists in the spinal cord. Brain 1907;29:137-151.
[747] Sakamoto T, Arissian K, Asanuma H. Functional role of the sensory cortex in learning
motor skills in cats. Brain Res 1989;503:258-264.
[748] Sakamoto T, Porter LL, Asanuma H. Long-lasting potentiation of synaptic potentials in the
motor cortex produced by stimulation of the sensory cortex in the cat: a basis of motor
learning. Brain Res 1987;413:360-364.
[749] Salat DH, Buckner RL, Snyder AZ, Greve DN, Desikan RS, Busa E, Morris JC, Dale AM,
Fischl B. Thinning of the cerebral cortex in aging. Cereb Cortex 2004;14:721-730.
[750] Salat DH, Lee SY, van der Kouwe AJ, Greve DN, Fischl B, Rosas HD. Age-associated
alterations in cortical gray and white matter signal intensity and gray to white matter
contrast. Neuroimage 2009;48:21-28.
[751] Salat DH, Tuch DS, Greve DN, van der Kouwe AJ, Hevelone ND, Zaleta AK, Rosen BR,
Fischl B, Corkin S, Rosas HD, Dale AM. Age-related alterations in white matter

286
microstructure measured by diffusion tensor imaging. Neurobiol Aging 2005;26:1215-
1227.
[752] Salomons TV, Johnstone T, Backonja M, Shackman AJ, Davidson RJ. Individual
differences in the effects of perceived controllability on pain perception: critical role of
the prefrontal cortex. J Cogn Neurosci 2007;19:993-1004.
[753] Salomons TV, Johnstone T, Backonja MM, Davidson RJ. Perceived controllability
modulates the neural response to pain. J Neurosci 2004;24:7199-7203.
[754] Sampson SM, Rome JD, Rummans TA. Slow-frequency rTMS reduces fibromyalgia pain.
Pain Med 2006;7:115-118.
[755] Sarlani E, Garrett PH, Grace EG, Greenspan JD. Temporal summation of pain
characterizes women but not men with termporomandibular disorders. J Orofac Pain
2007;21:309-317.
[756] Sarlani E, Greenspan JD. Evidence for generalized hyperalgesia in temporomandibular
disorders patients. Pain 2003;102:221-226.
[757] Sarlani E, Greenspan JD. Why look in the brain for answers to temporomandibular
disorder pain? Cells Tissues Organs 2005;180:69-75.
[758] Schallert T, Kozlowski DA, Humm JL, Cocke RR. Use-dependent structural events in
recovery of function. Adv Neurol 1997;73:229-238.
[759] Scherder E, Wolters E, Polman C, Sergeant J, Swaab D. Pain in Parkinson's disease and
multiple sclerosis: Its relation to the medial and lateral pain systems. Neurosci Biobehav
Rev 2005;29:1047-1056.
[760] Schmahmann JD, Pandya DN. Fiber pathways of the brain. New York: Oxford University
Press, 2006.
[761] Schmahmann JD, Pandya DN. Preface. In: JD Schmahmann, DN Pandya, editors. Fiber
pathways of the brain. New York: Oxford University Press, 2006. pp. xi-xii.
[762] Schmidt-Wilcke T, Ganssbauer S, Neuner T, Bogdahn U, May A. Subtle grey matter
changes between migraine patients and healthy controls. Cephalalgia : an international
journal of headache 2008;28:1-4.

287
[763] Schmidt-Wilcke T, Hierlmeier S, E. L. Altered regional brain morphology in patients with
chronic facial pain. Headache 2010;50:1278-1212-1285.
[764] Schmidt-Wilcke T, Leinisch E, Ganssbauer S, Draganski B, Bogdahn U, Altmeppen J,
May A. Affective components and intensity of pain correlate with structural differences
in gray matter in chronic back pain patients. Pain 2006;125:89-97.
[765] Schmidt-Wilcke T, Leinisch E, Straube A, Kampfe N, Draganski B, Diener HC, Bogdahn
U, May A. Gray matter decrease in patients with chronic tension type headache.
Neurology 2005;65:1483-1486.
[766] Schmidt-Wilcke T, Luerding R, Weigand T, Jurgens T, Schuierer G, Leinisch E, Bogdahn
U. Striatal grey matter increase in patients suffering from fibromyalgia--a voxel-based
morphometry study. Pain 2007;132 Suppl 1:S109-116.
[767] Schmitter AM. 17th and 18th Century Theories of Emotions. In: EN Zalta editor. Book
17th and 18th Century Theories of Emotions. City, 2010.
[768] Schnakers C, Chatelle C, Vanhaudenhuyse A, Majerus S, Ledoux D, Boly M, Bruno MA,
Boveroux P, Demertzi A, Moonen G, Laureys S. The Nociception Coma Scale: a new
tool to assess nociception in disorders of consciousness. Pain 2010;148:215-219.
[769] Schneider JS, Lidsky TI. Processing of somatosensory information in striatum of behaving
cats. J Neurophysiol 1981;45:841-851.
[770] Schoenbaum G, Roesch MR, Stalnaker TA, Takahashi YK. A new perspective on the role
of the orbitofrontal cortex in adaptive behaviour. Nat Rev Neurosci 2009;10:885-892.
[771] Scholz J, Klein MC, Behrens TE, Johansen-Berg H. Training induces changes in white-
matter architecture. Nat Neurosci 2009;12:1370-1371.
[772] Schweinhardt P, Kuchinad A, Pukall CF, Bushnell MC. Increased gray matter density in
young women with chronic vulvar pain. Pain 2008;140:411-419.
[773] Sechzer JA. The ethical dilemma of some classical animal experiments. Ann N Y Acad Sci
1983;406:5-12.
[774] Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H, Reiss AL, Greicius
MD. Dissociable intrinsic connectivity networks for salience processing and executive
control. J Neurosci 2007;27:2349-2356.

288
[775] Seifert F, Maihofner C. Representation of cold allodynia in the human brain--a functional
MRI study. Neuroimage 2007;35:1168-1180.
[776] Seminowicz DA, Davis KD. Interactions of pain intensity and cognitive load: the brain
stays on task. Cereb Cortex 2007;17:1412-1422.
[777] Seminowicz DA, Davis KD. Pain enhances functional connectivity of a brain network
evoked by performance of a cognitive task. J Neurophysiol 2007;97:3651-3659.
[778] Seminowicz DA, Davis KD. A re-examination of pain-cognition interactions: implications
for neuroimaging. Pain 2007;130:8-13.
[779] Seminowicz DA, Labus JS, Bueller JA, Tillisch K, Naliboff BD, Bushnell MC, Mayer EA.
Regional gray matter density changes in Brains of Patients with Irritable Bowel
Syndrome. Gastroenterology 2010;139:48-57.
[780] Seminowicz DA, Laferriere AL, Millecamps M, Yu JSC, Coderre TJ, Bushnell MC. MRI
structural brain changes associated with sensory and emotional function in a rat model of
long-term neuropathic pain. Neuroimage 2009;47:1007-1014.
[781] Seminowicz DA, Mikulis DJ, Davis KD. Cognitive modulation of pain-related brain
responses depends on behavioral strategy. Pain 2004;112:48-58.
[782] Seminowicz DA, Wideman TH, Naso L, Hatami-Khoroushahi Z, Fallatah S, Ware MA,
Jarzem P, Bushnell MC, Shir Y, Ouellet JA, Stone LS. Effective treatment of chronic low
back pain in humans reverses abnormal brain anatomy and function. J Neurosci
2011;31:7540-7550.
[783] Sessle BJ. Recent developments in pain research: central mechanisms of orofacial pain and
its control. J Endod 1986;12:435-444.
[784] Sessle BJ. Acute and chronic craniofacial pain: brainstem mechanisms of nociceptive
transmission and neuroplasticity, and their clinical correlates. Crit Rev Oral Biol Med
2000;11:57-91.
[785] Sessle BJ. Mechanisms of oral somatosensory and motor functions and their clinical
correlates. J Oral Rehabil 2006;33:243-261.

289
[786] Sessle BJ, Dubner R, Greenwood LF, Lucier GE. Descending influences of periaqueductal
gray matter and somatosensory cerebral cortex on neurones in trigeminal brain stem
nuclei. Can J Physiol Pharmacol 1976;54:66-69.
[787] Sessle BJ, Hu JW. Mechanisms of pain arising from articular tissues. Can J Physiol
Pharmacol 1991;69:617-626.
[788] Sessle BJ, Hu JW, Amano N, Zhong G. Convergence of cutaneous, tooth pulp, visceral,
neck and muscle afferents onto nociceptive and non-nociceptive neurones in trigeminal
subnucleus caudalis (medullary dorsal horn) and its implications for referred pain. Pain
1986;27:219-235.
[789] Sessle BJ, Hu JW, Dubner R, Lucier GE. Functional properties of neurons in cat trigeminal
subnucleus caudalis (medullary dorsal horn). II. Modulation of responses to noxious and
nonnoxious stimuli by periaqueductal gray, nucleus raphe magnus, cerebral cortex, and
afferent influences, and effect of naloxone. J Neurophysiol 1981;45:193-207.
[790] Shackman AJ, Salomons TV, Slagter HA, Fox AS, Winter JJ, Davidson RJ. The
integration of negative affect, pain and cognitive control in the cingulate cortex. Nat Rev
Neurosci 2011;12:154-167.
[791] Sharav Y, Benoliel R. The diagnostic process. In: Y Sharav, R Benoliel, editors. Orofacial
pain & headache. New York: Elsevier, 2008. pp. 1-17.
[792] Sharav Y, McGrath PA, Dubner R. Masseter inhibitory periods and sensations evoked by
electrical tooth pulp stimulation in patients with oral-facial pain and mandibular
dysfunction. Arch Oral Biol 1982;27:305-310.
[793] Sharp TJ. Chronic pain: a reformulation of the cognitive-behavioural model. Behav Res
Ther 2001;39:787-800.
[794] Sherman SM, Guillery RW. Exploring the thalamus and its role in cortical function.
Cambridge, MA: MIT Press, 2001.
[795] Sherrington CS. Qualitative difference of spinal reflex corresponding with qualitative
difference of cutaneous stimulus. J Physiol 1903;30:39-46.
[796] Sherrington CS. Observations on the scratch-reflex in the spinal dog. J Physiol 1906;34:1-
50.

290
[797] Sherrington CS. Man on his nature. Cambridge, England: Cambridge University press,
1955.
[798] Shibata H, Yukie M. Thalamocingulate connections in the monkey. In: BA Vogt, editor.
Cingulate Neurobiology and Disease. New York: Oxford University Press, 2009. pp. 96-
111.
[799] Shibukawa Y, Ishikawa T, Kato Y, Shintani M, Zhang ZK, Jiang T, Tazaki M, Shimono
M, Kumai T, Suzuki T, Kato M, Nakamura Y. Cortical dysfunction in patients with
temporomandibular disorders. J Oral Biosci 2009;51:65-71.
[800] Shibukawa Y, Ishikawa T, Kato Y, Zhang Z-K, Jiang T, Shintani M, Shimono M, Kumai
T, Suzuki T, Kato M, Nakamura Y. Cerebral cortical dysfunction in patients with
temporomandibular disorders in association with jaw movement observation. Pain
2007;128:180-188.
[801] Shigenaga Y, Chen IC, Suemune S, Nishimori T, Nasution ID, Yoshida A, Sato H,
Okamoto T, Sera M, Hosoi M. Oral and facial representation within the medullary and
upper cervical dorsal horns in the cat. J Comp Neurol 1986;243:388-408.
[802] Short EB, Borckardt JJ, Anderson BS, Frohman H, Beam W, Reeves ST, George MS. Ten
sessions of adjunctive left prefrontal rTMS significantly reduces fibromyalgia pain: A
randomized, controlled pilot study. Pain 2011;152:2477-2484.
[803] Siegel A, Troiano R, Royce A. Differential projections of the anterior and posterior
cingulate gyrus to the thalamus in the cat. Exp Neurol 1973;38:192-201.
[804] Simon TJ, Ding L, Bish JP, McDonald-McGinn DM, Zackai EH, Gee J. Volumetric,
connective, and morphologic changes in the brains of children with chromosome 22q11.2
deletion syndrome: an integrative study. Neuroimage 2005;25:169-180.
[805] Simone DA, Marchettini P, Caputi G, Ochoa JL. Identification of muscle afferents
subserving sensation of deep pain in humans. J Neurophysiol 1994;72:883-889.
[806] Simone DA, Sorkin LS, Oh U, Chung JM, Owens C, LaMotte RH, Willis WD. Neurogenic
hyperalgesia: Central neural correlates in responses of spinothalamic tract neurons. J
Neurophysiol 1991;66:228-246.

291
[807] Sinclair DC. Cutaneous sensation and the doctrine of specific energy. Brain 1955;78:584-
614.
[808] Slade GD, Diatchenko L, Bhalang K, Sigurdsson A, Fillingim RB, Belfer I, Max MB,
Goldman D, Maixner W. Influence of psychological factors on risk of
temporomandibular disorders. J Dent Res 2007;86:1120-1125.
[809] Slade GD, Diatchenko L, Ohrbach R, Maixner W. Orthodontic Treatment, Genetic Factors
and Risk of Temporomandibular Disorder. Semin Orthod 2008;14:146-156.
[810] Slakey TJ. Aristotle on sense perception. The Phil Rev 1961;70:470-484.
[811] Smith RL. The ascending fiber projections from the principal sensory trigeminal nucleus in
the rat. J Comp Neurol 1973;148:423-445.
[812] Smith RL. Axonal projections and connections of the principal sensory trigeminal nucleus
in the monkey. J Comp Neurol 1975;163:347-375.
[813] Smith S, Kindlmann G. Cross-subject Comparison of local Diffusion MRI Parameters. In:
H Johansen-Berg, TE Behrens, editors. Diffusion MRI. London: Academic Press, 2009.
pp. 148-174.
[814] Smith SM. Fast robust automated brain extraction. Hum Brain Mapp 2002;17:144-158.
[815] Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, Watkins
KE, Ciccarelli O, Cader MZ, Matthews PM, Behrens TE. Tract-based spatial statistics:
voxelwise analysis of multi-subject diffusion data. Neuroimage 2006;31:1487-1505.
[816] Smith SM, Jenkinson M, Woolrich M, Beckmann CF, Behrens TE, Johansen-Berg H,
Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy R, Saunders J, Vickers J,
Zhang Y, De Stefano N, Brady JM, Matthews PM. Advances in functional and structural
MR image analysis and implementation as FSL. Neuroimage 2004;23:208-220.
[817] Smith SM, Johansen-Berg H, Jenkinson M, Rueckert D, Nichols TE, Miller KL, Robson
MD, Jones DK, Klein JC, Bartsch AJ, Behrens TE. Acquisition and voxelwise analysis of
multi-subject diffusion data with tract-based spatial statistics. NatProtoc 2007;2:499-503.
[818] Smith SM, Johansen-Berg H, Jenkinson M, Rueckert D, Nichols TE, Miller KL, Robson
MD, Jones DK, Klein JC, Bartsch AJ, Behrens TEJ. Acquisition and voxelwise analysis

292
of multi-subject diffusion data with tract-based spatial statistics. Nat Protoc 2007;2:499-
503.
[819] Smith SM, Nichols TE. Threshold-free cluster enhancement: addressing problems of
smoothing, threshold dependence and localisation in cluster inference. Neuroimage
2009;44:83-98.
[820] Snook L, Plewes C, Beaulieu C. Voxel based versus region of interest analysis in diffusion
tensor imaging of neurodevelopment. Neuroimage 2007;34:243-252.
[821] Soler MD, Kumru H, Pelayo R, Vidal J, Tormos JM, Fregni F, Navarro X, Pascual-Leone
A. Effectiveness of transcranial direct current stimulation and visual illusion on
neuropathic pain in spinal cord injury. Brain 2010;133:2565-2577.
[822] Sommer M, Koch MA, Paulus W, Weiller C, Buchel C. Disconnection of speech-relevant
brain areas in persistent developmental stuttering. Lancet 2002;360:380-383.
[823] Sotnichencko TS. Convergence of the descending pathways of motor, visual and limbic
cortex in the cat di- and mesencephalon. Brain Res 1976;116:401-415.
[824] Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW.
Mapping cortical change across the human life span. Nat Neurosci 2003;6:309-315.
[825] Squire LR. David R. Curtis. In: LR Squire editor. Book David R. Curtis, Vol. 5. City:
Academic Press, 2006. pp. 170-225.
[826] Stacey MJ. Free nerve endings in skeletal muscle of the cat. J Anat 1969;105:231-254.
[827] Stahnisch FW. Francois Magendie (1783-1855). J Neurol 2009;256:1950-1952.
[828] Starr CJ, Sawaki L, Wittenberg GF, Burdette JH, Oshiro Y, Quevedo AS, McHaffie JG,
Coghill RC. The contribution of the putamen to sensory aspects of pain: insights from
structural connectivity and brain lesions. Brain 2011;134:1987-2004.
[829] Starr PA, Vitek JL, DeLong M, Bakay RAE. Magnetic resonance imaging-based
stereotactic localization of the globus pallidus and subthalamic nucleus. Neurosurgery
1999;44:303-313.
[830] Steno N, Andrault R. Discours sur l'anatomie du cerveau (1669). Paris: Classiques Garnier,
2009.

293
[831] Stewart WA, King RB. Fiber Projections from the Nucleus Caudalis of the Spinal
Trigeminal Nucleus. J Comp Neurol 1963;121:271-286.
[832] Stohler CS. Craniofacial pain and motor function: pathogenesis, clinical correlates, and
implications. Crit Rev Oral Biol Med 1999;10:504-518.
[833] Stone TT. Phantom limb pain and central pain; relief by ablation of a portion of posterior
central cerebral convolution. Arch Neurol Psychiatry 1950;63:739-748.
[834] Strange BA, Henson RN, Friston KJ, Dolan RJ. Anterior prefrontal cortex mediates rule
learning in humans. Cereb Cortex 2001;11:1040-1046.
[835] Sugiyo S, Takemura M, Dubner R, Ren K. Trigeminal transition zone/rostral ventromedial
medulla connections and facilitation of orofacial hyperalgesia after masseter
inflammation in rats. J Comp Neurol 2005;493:510-523.
[836] Sullivan MJ, Bishop SR, Pivik J. The pain catastrophizing scale: development and
validation. Psychol Assessment 1995;7:524-532.
[837] Svensson P. What can human experimental pain models teach us about clinical TMD?
Arch Oral Biol 2007;52:391-394.
[838] Svensson P, Beydoun A, Morrow TJ, Casey KL. Human intramuscular and cutaneous pain:
Psychophysical comparisons. Exp Brain Res 1997;114:390-392.
[839] Svensson P, Graven-Nielsen T. Craniofacial muscle pain: review of mechanisms and
clinical manifestations. J Orofac Pain 2001;15:117-145.
[840] Svensson P, Jadidi F, Arima T, Baad-Hansen L, Sessle BJ. Relationships between
craniofacial pain and bruxism. J Oral Rehabil 2008;35:524-547.
[841] Svensson P, List T, Hector G. Analysis of stimulus-evoked pain in patients with
myofascial temporomandibular pain disorders. Pain 2001;92:399-409.
[842] Svensson P, Romaniello A, Wang K, Arendt-Nielsen L, Sessle BJ. One hour of tongue-
task training is associated with plasticity in corticomotor control of the human tongue
musculature. Exp Brain Res 2006;173:165-173.
[843] Tal M, Devor M. Anatomy and neurophysiology of orofacial pain In: Y Sharav, R
Benoliel, editors. Orofacial pain & headache. New York: Elsevier, 2008. pp. 19-44.

294
[844] Talairach J, Tournoux P. Co-planar stereotaxic atlas of the human brain. New York:
Thieme Medical Publishers Inc., 1988.
[845] Talairach J, Tournoux P, Bancaud J. Traitement chirurgical central de la douleur, du
thalamus (non compris) au cortex parietal. Acta Neurochirurgica 1959;7: 48-143.
[846] Talbot JD, Marrett S, Evans AC, Meyer E, Bushnell MC, Duncan GH. Multiple
representation of pain in human cerebral cortex. Science 1991;251:1355-1358.
[847] Taylor KS, Anastakis DJ, Davis KD. Cutting your nerve changes your brain. Brain
2009;132:3122-3133.
[848] Taylor KS, Anastakis DJ, Davis KD. Chronic Pain and Sensorimotor Deficits Following
Peripheral Nerve Injury. Pain 2010;15:582-591.
[849] Taylor KS, Seminowicz DA, Davis KD. Two systems of resting state connectivity between
the insula and cingulate cortex. Hum Brain Mapp 2009;30:2731-2745.
[850] Tenenbaum HC, Mock D, Gordon AS, Goldberg MB, Grossi ML, Locker D, Davis KD.
Sensory and affective components of orofacial pain: is it all in your brain? Crit Rev Oral
Biol Med 2001;12:455-468.
[851] Terkelsen AJ, Andersen OK, Molgaard H, Hansen J, Jensen TS. Mental stress inhibits pain
perception and heart rate variability but not a nociceptive withdrawal reflex. Acta Physiol
Scand 2004;180: 405-414.
[852] Teutsch S, Herken W, Bingel U, Schoell E, May A. Changes in brain gray matter due to
repetitive painful stimulation. Neuroimage 2008;42:845-849.
[853] Thalhammer JG, LaMotte RH. Spatial properties of nociceptor sensitization following heat
injury of the skin. Brain Res 1982;231:257-265.
[854] Thompson PM, Cannon TD, Narr KL, van Erp T, Poutanen VP, Huttunen M, Lonnqvist J,
Standertskjold-Nordenstam CG, Kaprio J, Khaledy M, Dail R, Zoumalan CI, Toga AW.
Genetic influences on brain structure. Nat Neurosci 2001;4:1253-1258.
[855] Timmermann L, Ploner M, Haucke K, Schmitz F, Baltissen R, Schnitzler A. Differential
coding of pain intensity in the human primary and secondary somatosensory cortex. J
Neurophysiol 2001;86:1499-1503.

295
[856] Tinazzi M, Zarattini S, Valeriani M, Romito S, Farina S, Moretto G, Smania N, Fiaschi A,
Abbruzzese G. Long-lasting modulation of human motor cortex following prolonged
transcutaneous electrical nerve stimulation (TENS) of forearm muscles: evidence of
reciprocal inhibition and facilitation. Exp Brain Res 2005;161:457-464.
[857] Todd AJ, Koerber HR. Neuroanatomical substrates of spinal nociception. In: SB
McMahon, M Koltzenburg, editors. Wall and Melzack's Textbook of Pain. Edingburgh,
UK: Churchill Livingstone, 2006. pp. 73–90.
[858] Toga AW, Thompson PM, Sowell ER. Mapping brain maturation. Trends Neurosci
2006;29:148-159.
[859] Tokuno H, Tanji J. Input organization of distal and proximal forelimb areas in the monkey
primary motor cortex: a retrograde double labeling study. J Comp Neurol 1993;333:199-
209.
[860] Torta DM, Cauda F. Different functions in the cingulate cortex, a meta-analytic
connectivity modeling study. Neuroimage 2011;56:2157-2172.
[861] Torvik A. The ascending fibers from the main trigeminal sensory nucleus; an experimental
study in the cat. Am J Anat 1957;100:1-15.
[862] Tournier JD, Calamante F, Gadian DG, Connelly A. Direct estimation of the fiber
orientation density function from diffusion-weighted MRI data using spherical
deconvolution. Neuroimage 2004;23:1176-1185.
[863] Towell AD, Purves AM, Boyd SG. CO 2 laser activation of nociceptive and non-
nociceptive thermal afferents from hairy and glabrous skin. Pain 1996;66:79-86.
[864] Tracey I, Ploghaus A, Gati JS, Clare S, Smith S, Menon RS, Matthews PM. Imaging
attentional modulation of pain in the periaqueductal gray in humans. J Neurosci
2002;22:2748-2752.
[865] Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-
term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature
2002;420:788-794.
[866] Travell J. Pain and disability of the shoulder and arm. J Am Med Assoc 1942;120:417-422.
[867] Treede RD. Peripheral acute pain mechanisms. AnnMed 1995;27:213-216.

296
[868] Treede RD, Apkarian AV, Bromm B, Greenspan JD, Lenz FA. Cortical representation of
pain: functional characterization of nociceptive areas near the lateral sulcus. Pain
2000;87:113-119.
[869] Treede RD, Kenshalo DR, Gracely RH, Jones AKP. The cortical representation of pain.
Pain 1999;79:105-111.
[870] Treede RD, Meyer RA, Raja SN, Campbell JN. Evidence for two different heat
transduction mechanisms in nociceptive primary afferents innervating monkey skin. J
Physiol 1995;483:747-758.
[871] Treede RD, Vogel H, Rios M, Krauss G, Lesser RP, Lenz FA. Pain-related evoked
potentials from parasylvian cortex in humans. Electroencephalogr Clin Neurophysiol
Suppl 1999;49:250-254.
[872] Truelove E, Huggins KH, Mancl L, Dworkin SF. The efficacy of traditional, low-cost and
non-splint therapies for temporomandibular disorders. J Am Dent Assoc 2006;137:1099-
1107.
[873] Tsou K, Jang CS. Studies on the Site of Analgesic Action of Morphine by Intracerebral
Micro-Injection. Sci Sin 1964;13:1099-1109.
[874] Tsubokawa T, Katayama Y, Yamamoto T, Hirayama T, Koyama S. Chronic motor cortex
stimulation for the treatment of central pain. Acta Neurochir Suppl (Wien) 1991;52:137-
139.
[875] Tsujimoto S, Genovesio A, Wise SP. Evaluating self-generated decision in the frontal
polar cortex of monkeys. Nat Neurosci 2010;13:120-126.
[876] Tsujimoto T, Ogawa M, Tsukada H, Kakiuchi T, Sasaki K. Decline of the monkey's limbic
and prefrontal activity during task repetition. Neurosci Lett 2000;283:69-72.
[877] Tuch DS. Q-ball imaging. Magn Reson Med 2004;52:1358-1372.
[878] Tuch DS, Reese TG, Wiegell MR, Makris N, Belliveau JW, Wedeen VJ. High angular
resolution diffusion imaging reveals intravoxel white matter fiber heterogeneity. Magn
Reson Med 2002;48:577-582.
[879] Tuch DS, Reese TG, Wiegell MR, Wedeen VJ. Diffusion MRI of complex neural
architecture. Neuron 2003;40:885-895.

297
[880] Tucker WI, Dynes JB. Indications for lobotomy. Lahey Clin Bull 1949;6:90-96.
[881] Turner JA, Dworkin SF, Mancl L, Huggins KH, Truelove EL. The roles of beliefs,
catastrophizing, and coping in the functioning of patients with temporomandibular
disorders. Pain 2001;92:41-51.
[882] Turp JC, Komine F, Hugger A. Efficacy of stabilization splints for the management of
patients with masticatory muscle pain: a qualitative systematic review. Clin Oral Investig
2004;8:179-195.
[883] Turp JC, Kowalski CJ, O'Leary N, Stohler CS. Pain maps from facial pain patients indicate
a broad pain geography. J Dent Res 1998;77:1465-1471.
[884] Usunoff KG, Marani E, Schoen JH. The trigeminal system in man. Adv Anat Embryol Cell
Biol 1997;136:I-X, 1-126.
[885] Valentini E, Hu L, Chakrabarti B, Hu Y, Aglioti SM, Iannetti GD. The primary
somatosensory cortex largely contributes to the early part of the cortical response elicited
by nociceptive stimuli. Neuroimage 2011.
[886] Valentini E, Torta DM, Mouraux A, Iannetti GD. Dishabituation of Laser-evoked EEG
Responses: Dissecting the Effect of Certain and Uncertain Changes in Stimulus Modality.
J Cogn Neurosci 2011.
[887] Valet M, Gundel H, Sprenger T, Sorg C, Muhlau M, Zimmer C, Henningsen P, Tolle TR.
Patients with pain disorder show gray-matter loss in pain-processing structures: a voxel-
based morphometric study. Psychosom Med 2009;71:49-56.
[888] Valet M, Sprenger T, Boecker H, Willoch F, Rummeny E, Conrad B, Erhard P, Tolle TR.
Distraction modulates connectivity of the cingulo-frontal cortex and the midbrain during
pain - an fMR1 analysis. Pain 2004;109:399-408.
[889] Valfre W, Rainero I, Bergui M, Pinessi L. Voxel-based morphometry reveals gray matter
abnormalities in migraine. Headache 2008;48:109-117.
[890] van Buren JM. Sensory responses from stimulation of the inferior Rolandic and Sylvian
regions in man. Journal of Neurosurgery 1983;59:119-130.
[891] Van Damme S, Crombez G, Eccleston C. Disengagement from pain: the role of
catastrophic thinking about pain. Pain 2004;107:70-76.

298
[892] Van Damme S, Crombez G, Eccleston C. Coping with pain: a motivational perspective.
Pain 2008;139:1-4.
[893] Van Damme S, Legrain V, Vogt J, Crombez G. Keeping pain in mind: a motivational
account of attention to pain. Neuroscience and biobehavioral reviews 2010;34:204-213.
[894] Van der Glas HW, Cadden SW, Van der Bilt A. Mechanisms underlying the effects of
remote noxious stimulation and mental activities on exteroceptive jaw reflexes in man.
Pain 2000;84:193-202.
[895] van Wijk G, Veldhuijzen DS. Perspective on diffuse noxious inhibitory controls as a model
of endogenous pain modulation in clinical pain syndromes. J Pain 2010;11:408-419.
[896] Villanueva L, Bouhassira D, Le Bars D. The medullary subnucleus reticularis dorsalis
(SRD) as a key link in both the transmission and modulation of pain signals. Pain
1996;67:231-240.
[897] Vogel H, Port JD, Lenz FA, Solaiyappan M, Krauss G, Treede RD. Dipole source analysis
of laser-evoked subdural potentials recorded from parasylvian cortex in humans. J
Neurophysiol 2003;89:3051-3060.
[898] Vogt BA. Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev
Neurosci 2005;6:533-544.
[899] Vogt BA. Regions and subregions of the cingulate cortex. In: BA Vogt, editor. Cingulate
neurobiology and disease. New York: Oxford University Press, 2009. pp. 5-30.
[900] Vogt BA. Regions and subregions of the cingulate cortex. In: BA Vogt, editor. Cingulate
Neurobiology and Disease. New York: Oxford University Press, 2009. pp. 1-30.
[901] Vogt BA, Pandya DN. Cingulate cortex of the rhesus monkey: II. Cortical afferents. J
Comp Neurol 1987;262:271-289.
[902] Vogt BA, Sikes RW. The medial pain system, cingulate cortex, and parallel processing of
nociceptive information. Prog Brain Res 2000;122:223-235.
[903] Vogt BA, Sikes RW. Cingulate nociceptive circuitry and roles in pain processing: the
cingulate premotor pain model. In: BA Vogt, editor. Cingulate neurobiology and disease.
New York: Oxford University Press, 2009. pp. 312-338.

299
[904] Vogt BA, Sikes RW, Vogt LT. Anterior cingulate cortex and the medial pain system. In:
BA Vogt, M Gabriel, editors. Neurobiology of cingulate cortex and limbic thalamus: A
comprehensive handbook. Boston: Birkhauser, 1993. pp. 313-344.
[905] Vogt BA, Vogt L, Farber NB, Bush G. Architecture and neurocytology of monkey
cingulate gyrus. J Comp Neurol 2005;485:218-239.
[906] Vogt BA, Vogt L, Laureys S. Cytology and functionally correlated circuits of human
posterior cingulate areas. Neuroimage 2006;29:452-466.
[907] Wade JB, Dougherty LM, Hart RP, Rafii A, Price DD. A canonical correlation analysis of
the influence of neuroticism and extraversion on chronic pain, suffering, and pain
behavior. Pain 1992;51:67-73.
[908] Wager TD, Rilling JK, Smith EE, Sokolik A, Casey KL, Davidson RJ, Kosslyn SM, Rose
RM, Cohen JD. Placebo-induced changes in FMRI in the anticipation and experience of
pain. Science 2004;303:1162-1167.
[909] Wager TD, Scott DJ, Zubieta JK. Placebo effects on human mu-opioid activity during
pain. Proc Natl Acad Sci U S A 2007;104:11056-11061.
[910] Wakana S, Caprihan A, Panzenboeck MM, Fallon JH, Perry M, Gollub RL, Hua K, Zhang
J, Jiang H, Dubey P, Blitz A, van ZP, Mori S. Reproducibility of quantitative
tractography methods applied to cerebral white matter. Neuroimage 2007;36:630-644.
[911] Walhovd KB, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH,
Greve DN, Fischl B, Dale AM, Fjell AM. Consistent neuroanatomical age-related volume
differences across multiple samples. Neurobiol Aging 2009.
[912] Walker HK. Cranial Nerve V: The Trigeminal Nerve. In: HK Walker, WD Hall, JW Hurst,
editors. Clinical Methods: The History, Physical, and Laboratory Examinations. Boston,
1990.
[913] Wall PD. The laminar organization of dorsal horn and effects of descending impulses. J
Physiol 1967;188:403-423.
[914] Wall PD, Egger MD. Formation of new connexions in adult rat brains after partial
deafferentation. Nature 1971;232:542-545.

300
[915] Wall PD, Woolf CJ. Muscle but not cutaneous C-afferent input produces prolonged
increases in the excitability of the flexion reflex in the rat. J Physiol 1984;356:443-458.
[916] Walther K, Bendlin BB, Glisky EL, Trouard TP, Lisse JR, Posever JO, Ryan L. Anti-
inflammatory drugs reduce age-related decreases in brain volume in cognitively normal
older adults. Neurobiol Aging 2011;32:497-505.
[917] Wang K, Svensson P, Sessle BJ, Cairns BE, Arendt-Nielsen L. Painful conditioning
stimuli of the craniofacial region evokes diffuse noxious inhibitory controls in men and
women. J Orofac Pain 2010;24:255-261.
[918] Wang N, May PJ. Peripheral muscle targets and central projections of the mesencephalic
trigeminal nucleus in macaque monkeys. Anat Rec 2008;291:974-987.
[919] Wartolowska K, Hough MG, Jenkinson M, Andersson J, Wordsworth BP, Tracey I.
Structural brain changes in rheumatoid arthritis. Arthritis Rheum 2011.
[920] Watkins LR, Maier SF, Goehler LE. Immune activation: the role of pro-inflammatory
cytokines in inflammation, illness responses and pathological pain states. Pain
1995;63:289-302.
[921] Watkins LR, Milligan ED, Maier SF. Spinal cord glia: new players in pain. Pain
2001;93:201-205.
[922] Watson A, El-Deredy W, Iannetti GD, Lloyd D, Tracey I, Vogt BA, Nadeau V, Jones AK.
Placebo conditioning and placebo analgesia modulate a common brain network during
pain anticipation and perception. Pain 2009;145:24-30.
[923] Watts JW, Freeman W. Psychosurgery for the relief of unbearable pain. J Int Coll Surg
1946;9:679-683.
[924] Weddell G. Somesthesis and the chemical senses. Annu Rev Psychol 1955;6:119-136.
[925] Wedeen RP, Udasin I, Fiedler N, D'Haese P, De Broe M, Gelpi E, Jones KW, Gochfeld M.
Urinary biomarkers as indicators of renal disease. Ren Fail 1999;21:241-249.
[926] Weigelt A, Terekhin P, Kemppainen P, Dorfler A, Forster C. The representation of
experimental tooth pain from upper and lower jaws in the human trigeminal pathway.
Pain 2010;149:529-538.

301
[927] Weissman-Fogel I, Moayedi M, Taylor KS, Pope G, Davis KD. Cognitive and default-
mode resting state networks: do male and female brains "rest" differently? Hum Brain
Mapp 2010;31:1713-1726.
[928] Weissman-Fogel I, Moayedi M, Tenenbaum HC, Goldberg MB, Freeman BV, Davis KD.
Abnormal cortical activity in patients with temporomandibular disorder evoked by
cognitive and emotional tasks. Pain 2011;152:384-396.
[929] Whitsel BL, Favorov OV, Li Y, Quibrera M, Tommerdahl M. Area 3a neuron response to
skin nociceptor afferent drive. Cereb Cortex 2009;19:349-366.
[930] Whitsel BL, Petrucelli LM, Werner G. Symmetry and connectivity in the map of the body
surface in somatosensory area II of primates. J Neurophysiol 1969;32:170-183.
[931] Wiberg M, Westman J, Blomqvist A. The projection to the mesencephalon from the
sensory trigeminal nuclei. An anatomical study in the cat. Brain Res 1986;399:51-68.
[932] Wiech K, Kalisch R, Weiskopf N, Pleger B, Stephan KE, Dolan RJ. Anterolateral
prefrontal cortex mediates the analgesic effect of expected and perceived control over
pain. J Neurosci 2006;26:11501-11509.
[933] Wiech K, Lin CS, Brodersen KH, Bingel U, Ploner M, Tracey I. Anterior insula integrates
information about salience into perceptual decisions about pain. J Neurosci
2010;30:16324-16331.
[934] Wiech K, Ploner M, Tracey I. Neurocognitive aspects of pain perception. Trends Cogn Sci
2008;12:8.
[935] Wiech K, Seymour B, Kalisch R, Stephan KE, Koltzenburg M, Driver J, Dolan RJ.
Modulation of pain processing in hyperalgesia by cognitive demand. Neuroimage
2005;27:59-69.
[936] Wiech K, Tracey I. The influence of negative emotions on pain: Behavioural effects and
neural mechanisms. Neuroimage 2009;47:987-994.
[937] Wik G, Fischer H, Bragee B, Finer B, Fredrikson M. Functional anatomy of hypnotic
analgesia: a PET study of patients with fibromyalgia. Eur J Pain 1999;3:7-12.
[938] Wilkinson HA. Cingulotomy. J Neurosurg 2009;110:607.

302
[939] Wilkinson HA, Davidson KM, Davidson RI. Bilateral anterior cingulotomy for chronic
noncancer pain. Neurosurgery 1999;45:1129-1134.
[940] Williams ZM, Bush G, Rauch SL, Cosgrove GR, Eskandar EN. Human anterior cingulate
neurons and the integration of monetary reward with motor responses. Nat Neurosci
2004;7:1370-1375.
[941] Willis WD, Jr. The Pain System. The neural basis of nociceptive transmission in the
mammalian nervous system. In: PL Gildenberg, editor. Pain and Headache, Vol. 1. Basel:
S.Karger, 1985. pp. 1-346.
[942] Willis WD, Jr., Coggeshall RE. Sensory mechanisms of the spinal cord, Vol. 2. New York:
Plenum, 1991.
[943] Willis WD, Trevino DL, Coulter JD, Maunz RA. Responses of primate spinothalamic tract
neurons to natural stimulation of the hindlimb. J Neurophysiol 1974;37:358-372.
[944] Willis WD, Westlund KN. Neuroanatomy of the pain system and of the pathways that
modulate pain. J Clin Neurophysiol 1997;14:2-31.
[945] Wilson SAK. Croonian Lectures: On some disorders of motility and of muscle tone with
special reference to the corpus striatum. Lancet 1925;206:268-276.
[946] Witting N, Svensson P, Gottrup H, Arendt-Nielsen L, Jensen TS. Intramuscular and
intradermal injection of capsaicin: a comparison of local and referred pain. Pain
2000;84:407-412.
[947] Woda A. Pain in the trigeminal system: from orofacial nociception to neural network
modeling. J Dent Res 2003;82:764-768.
[948] Woda A, Azerad J, Guilbaud G, Besson JM. [A microphysiologic study of thalamic
projection of the cat's dental pulp (author's transl)]. Brain Res 1975;89:193-213.
[949] Wood PB, Glabus MF, Simpson R, Patterson JC, 2nd. Changes in gray matter density in
fibromyalgia: correlation with dopamine metabolism. J Pain 2009;10:609-618.
[950] Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science
2000;288:1765-1769.

303
[951] Woolf CJ, Salter MW. Plasticity and pain: role of the dorsal horn. In: KL McMahon, M
Koltzenburg, editors. Textbook of Pain Philadelphia: Elsevier, 2006. pp. 91-106.
[952] Woolsey CN. Organization of somatic sensory and motor areas of the cerebral cortex. In:
HF Harlow, CN Woolsey, editors. Biological and biochemical basis of behavior.
Madison: University of Wisconsin Press, 1958.
[953] Woolsey CN, Erickson TC, Gilson WE. Localization in somatic sensory and motor areas
of human cerebral cortex as determined by direct recording of evoked potentials and
electrical stimulation. J Neurosurg 1979;51:476-506.
[954] Wright CI, Williams D, Feczko E, Barrett LF, Dickerson BC, Schwartz CE, Wedig MM.
Neuroanatomical correlates of extraversion and neuroticism. Cereb Cortex 2006;16:1809-
1819.
[955] Yamasaki H, Kakigi R, Watanabe S, Naka D. Effects of distraction on pain perception:
magneto- and electro-encephalographic studies. Brain Res Cogn Brain Res 1999;8:73-76.
[956] Yarkoni T, Poldrack RA, Nichols TE, Van Essen DC, Wager TD. Large-scale automated
synthesis of human functional neuroimaging data. Nature methods 2011;8:665-670.
[957] Yasui Y, Itoh K, Mizuno N, Nomura S, Takada M, Konishi A, Kudo M. The posteromedial
ventral nucleus of the thalamus (VPM) of the cat: direct ascending projections to the
cytoarchitectonic subdivisions. J Comp Neurol 1983;220:219-228.
[958] Yeung JC, Rudy TA. Sites of antinociceptive action of systemically injected morphine:
involvement of supraspinal loci as revealed by intracerebroventricular injection of
naloxone. J Pharmacol Exp Ther 1980;215:626-632.
[959] Yeung JC, Yaksh TL, Rudy TA. Effect on the nociceptive threshold and EEG activity in
the rat of morphine injected into the medial thalamus and the periaqueductal gray.
Neuropharmacology 1978;17:525-532.
[960] Yoshida A, Sessle BJ, Dostrovsky JO, Chiang CY. Trigeminal and dorsal column nuclei
projections to the anterior pretectal nucleus in the rat. Brain Res 1992;590:81-94.
[961] Young GB, Blume WT. Painful epileptic seizures. Brain 1983;106 (Pt 3):537-554.
[962] Younger JW, Chu LF, D'Arcy NT, Trott KE, Jastrzab LE, Mackey SC. Prescription opioid
analgesics rapidly change the human brain. Pain 2011;152:1803-1810.

304
[963] Younger JW, Shen YF, Goddard G, Mackey SC. Chronic myofascial temporomandibular
pain is associated with neural abnormalities in the trigeminal and limbic systems. Pain
2010;149:222-228.
[964] Yu XH, Zhang ET, Craig AD, Shigemoto R, Ribeiro-da-Silva A, De Koninck Y. NK-1
receptor immunoreactivity in distinct morphological types of lamina I neurons of the
primate spinal cord. J Neurosci 1999;19:3545-3555.
[965] Zambito Marsala S, Tinazzi M, Vitaliani R, Recchia S, Fabris F, Marchini C, Fiaschi A,
Moretto G, Giometto B, Macerollo A, Defazio G. Spontaneous pain, pain threshold, and
pain tolerance in Parkinson's disease. J Neurol 2011;258:627-633.
[966] Zarei M, Johansen-Berg H, Smith S, Ciccarelli O, Thompson AJ, Matthews PM.
Functional anatomy of interhemispheric cortical connections in the human brain. J Anat
2006;209:311-320.
[967] Zhang ET, Craig AD. Morphology and distribution of spinothalamic lamina I neurons in
the monkey. J Neurosci 1997;17:3274-3284.
[968] Ziegler DA, Piguet O, Salat DH, Prince K, Connally E, Corkin S. Cognition in healthy
aging is related to regional white matter integrity, but not cortical thickness. Neurobiol
Aging 2010;31:1912-1926.
[969] Zilles K. Neuronal plasticity as an adaptive property of the central nervous system. Ann
Anat 1992;174:383-391.

305
Appendices

306
Appendix I: Recruitment Letter

307
Appendix II: Consent forms

308
Consent Form
Title: Pain, Temperature and Tactile Perceptions in Normal Subjects and in
Patients with Neurological Disorders
Investigators: K.D. Davis, Tel.: 416-603-5662
Co-Investigators: H.C. Tenenbaum
M. Goldberg
Introduction:
You are being asked to take part in a research study. Please read this explanation about the study and its
risks and benefits before you decide if you would like to take part. You should take as much time as you
need to make your decision. You should ask the study doctor or study staff to explain anything that you
do not understand and make sure that all of your questions have been answered before signing this
consent form. Before you make your decision, feel free to talk about this study with anyone you wish.
Participation in this study is voluntary.
Purpose:
We are studying the brain mechanisms of touch, temperature, pain sensation and visual and auditory
perception. Therefore, we will assess your perception of various stimuli presented and also obtain images
of your brain with the magnetic resonance imaging (MRI) scanner during the stimulation.
Procedures:
There are two parts to the project, which may take place on separate days or a single day, and will require
approximately 2 hours of your time in total. In the first part, we will ask about your health, your pain and
also conduct some very basic personality and cognitive tests to measure your brain’s ability to process
information. This will take approximately 30-40 minutes. The second part includes some additional

309
questionnaires and a MRI (magnetic resonance imaging) scan (at the Toronto Western Hospital) to take
pictures of your brain while lying still and also while viewing words that may be related to your pain. The
MRI will take approximately 1-1.5 hours.
As part of these procedures you will be trained to rate the intensity of a stimulus and to describe its
quality. In some sessions the ratings will consist of a verbal or written indication of the stimulus. In other
sessions you may be trained to use a computer to rate the stimulus. You may also be asked to watch visual
images or listen to sounds. You may have your skin marked with water soluble ink in studies in which we
deliver mechanical, thermal and/or electrical stimuli to a small area of your skin. Following each stimulus
you will be asked for a rating. For those studies, mechanical (pressure) stimuli will consist of one or more
of several types such as slender nylon filaments, cotton swabs, tuning forks and pins. The thermal stimuli
will be delivered with a probe that is precisely controlled and may be warm, cold or painful. You may be
asked to place you hand into a cold water bath. The electrical stimuli will be delivered by electrodes
placed on your skin. The visual images will be presented on a screen and the auditory stimuli will be
heard through headphones.
To image your brain, you will be placed into the narrow tube of the MRI machine. The MRI makes loud
banging noises when it is taking pictures. At any time, if you are experiencing distress, you can freely
communicate with the MR technician through a microphone in the MRI. You are free to terminate the
procedure if you feel extreme claustrophobia.
Risks:
There are no major risks associated with the experimental protocols other than the discomfort of the
painful stimuli. At any time during these sessions, you are free to withdraw from any stimulus and are
free to terminate a session.
Benefits:
You will not receive any direct benefit from being in this study. Information learned from this study may
help other people with chronic pain in the future.
Voluntary Participation:
Your participation in this study is voluntary. You may decide not to be in this study, or to be in the study
now and then change your mind later. You may leave the study at any time without affecting your current
or future medical care.

310
Confidentiality:
If you agree to join this study, the study doctor and his/her study team will look at your personal health
information and collect only the information they need for the study. Personal health information is any
information that could be used to identify you and includes your:
• name, • address, • date of birth, • new or existing medical records, that includes types, dates and results of medical tests or
procedures.
The information that is collected for the study will be kept in a locked and secure area by the study doctor
for 7 years. Only the study team or the people or groups listed below will be allowed to look at your
records. Your participation in this study also may be recorded in your medical record at this hospital.
Representatives of the Mount Sinai Hospital Research Ethics Board may look at the study records and at
your personal health information to check that the information collected for the study is correct and to
make sure the study followed proper laws and guidelines.
All information collected during this study, including your personal health information, will be kept
confidential and will not be shared with anyone outside the study unless required by law. You will not be
named in any reports, publications, or presentations that may come from this study.
If you decide to leave the study, the information about you that was collected before you left the study
will still be used. No new information will be collected without your permission.
In Case You Are Harmed in the Study:
If you become ill, injured or harmed as a result of taking part in this study, you will receive care.
The reasonable costs of such care will be covered for any injury, illness or harm that is directly a
result of being in this study. In no way does signing this consent form waive your legal rights nor
does it relieve the investigators, sponsors or involved institutions from their legal and
professional responsibilities. You do not give up any of your legal rights by signing this consent
form.
Expenses Associated with Participating in the Study:

311
You will receive remuneration for your time at approx. $30-40/hour or $75/study.
Questions:
If you have any questions regarding these procedures please contact Dr. Karen D. Davis at 416-603-5662.
For any questions regarding your rights as a research subject please contact Dr. Heslegrave, (Chair, UHN
and Mount Sinai Hospital Research Ethics Boards) at 416-340-4557 or 416-586-4875. This person is not
involved with the research project in any way and contacting him will not affect your participation in the
study.
Consent:
This study has been explained to me and any questions I had have been answered.
I know that I may leave the study at any time. I agree to take part in this study.
Print Study Participant’s Name Signature Date
(You will be given a signed copy of this consent form)
My signature means that I have explained the study to the participant named above. I have answered all
questions.
Print Name of Person Obtaining Consent Signature Date
312
Appendix III: Edinburgh Handedness Inventory
Edinburgh Handedness Inventory Page 1 of 1
Handedness Questionnaire
Instructions
Please indicate your preferences in the use of hands in the following activities. If you are really indifferent, select “Either”. Where the preference is so strong that you would never try to use the other hand select “No”.
When: Which hand do you prefer? Do you ever use the other hand?
Writing: L R Either Yes No Drawing: L R Either Yes No
Throwing: L R Either Yes No Using scissors: L R Either Yes No
Using a toothbrush: L R Either Yes No Using a knife (without fork): L R Either Yes No
Using a spoon: L R Either Yes No Using a broom (upper hand): L R Either Yes No
Striking a match: L R Either Yes No Opening a box (lid): L R Either Yes No
Thank you for your responses.
Adapted from: Mark S. Cohen
Based on: Oldfield, R.C. 1971

313
Appendix IV: Pain Catastrophizing Scale
< Copyright � 1995 Michael JL Sullivan
PCS Client No.: __________ Age: _____ Sex: M(__) F(__) Date: _______________
Everyone experiences painful situations at some point in their lives. Such experiences may include headaches, tooth pain, joint or muscle pain. People are often exposed to situations that may cause pain such as illness, injury, dental procedures or surgery. We are interested in the types of thoughts and feelings that you have when you are in pain. Listed below are thirteen statements describing different thoughts and feelings that may be associated with pain. Using the following scale, please indicate the degree to which you have these thoughts and feelings when you are experiencing pain. 0 – not at all 1 – to a slight degree 2 – to a moderate degree 3 – to a great degree 4 – all the time When I’m in pain …
1� I worry all the time about whether the pain will end.
2� I feel I can’t go on.
3� It’s terrible and I think it’s never going to get any better.
4� It’s awful and I feel that it overwhelms me.
5� I feel I can’t stand it anymore.
6� I become afraid that the pain will get worse.
7� I keep thinking of other painful events.
8� I anxiously want the pain to go away.
9� I can’t seem to keep it out of my mind.
10� I keep thinking about how much it hurts.
11� I keep thinking about how badly I want the pain to stop.
12� There’s nothing I can do to reduce the intensity of the pain.
13� I wonder whether something serious may happen.
…Total

314
Appendix V: TMD pain assessment
Date: ……./……./…….
Patient Name:
Patient No.:
Date of Birth: ……./……./…….
What is your highest level of Education:
Phase of the menstrual cycle
What day did your last period start: ……./……./…….
Oral contraceptive Y N
Site and Radiation:
ES HS UNI M
1 2 3 4
Preauricular
Intra-auricular
Temporal
Massetric

315
1. How long have you had TMD? _________________________
2. Describe how your pain feels? _________________________
3. McGill Pain Questionnaire
4. Frequency: Constant-………….
Occasional- ………………… per week
………………… per day
Pain History:
Worse in a.m.? Y N
Worse in p.m.? Y N
Responsive to analgesics? Y N
Current medication/treatment
Other Pain Conditions?
Aggravating factors?
Relieving factors?
Y N _______________________________________
Y N arthritis, fibromyalgia, IBS _____________________
Y N eating, chewing, yawning, talking,____________________
Y N rest, warmth, cold, chewing,__________________________

316
Other symptoms?
Ear symptoms?
Sleep affected?
Clenching/Bruxism
Do you have pain in regions in other parts of your body?
Current Pain Rating: NPS 0 (no pain)-10 (most intense pain imaginable)
Intensity- _________
NPS (0 (not unpleasant)-10 (most unpleasant pain imaginable)
Unpleasantness- ________
History of symptoms: Since onset, pain has improved, stayed the same, worsened?
Has the nature of pain changed since onset?
Y N clicking, locking, limited mouth opening, _________________
Y N pop ________________________________________________
Y N
Y N Prevention, disturbance, wake early a.m.______________________
Y N Specify____________________________________________

317
Appendix VI: McGill Pain Questionnaire

318
Appendix VII: McGill Pain Questionnaire results for Patients with TMD
Table A-1: McGill Pain Questionnaire result for patients with TMD
Abbreviations for “Evaluative” words selected: A – annoying; T – troublesome; M – miserable; I – intense; U - unbearable
Sub
Number of Words Pain
Intensity Sensory
(Q.1-10)
Affective
(Q.11-15)
Evaluative
(Q.16)
Miscellaneous
(Q17-20)
Total
1 10 1 1 I 3 15 1
2 7 0 2 AT 1 10 2
3 5 0 0 1 6 2
4 7 2 3 ATM 1 13 3
5 7 1 0 2 10 2
6 10 1 3 ATI 4 18 2
7 9 1 1 A 4 15 1
8 13 1 3 TIU 4 21 2
9 12 2 4 AMIU 5 23 2
10 10 5 4 ATMI 5 24 2
11 10 0 0 1 11 1
12 7 2 2 AT 4 15 3
13 11 0 0 4 15 2
14 5 4 2 AM 1 12 2
15 11 2 1 T 2 16 2
16 11 2 1 A 5 19 2
17 10 0 3 ATI 4 17 1

319
Copyright Acknowledgements

320
We acknowledge the following copyright permissions:
The American Association for the Advancement of Science for permission to reprint the figure
“Wall and Melzack’s Gate Control Theory of Pain mechanism” (Figure 2-2) from [595].
Oxford University Press for permission to reprint the figure “Diffusion tensor imaging
reconstruction of Meynert’s classification of white matter tracts in the brain” (Figure 2-4) from
[147].
Elsevier Limited for permission to reprint 5 figures (Figures 4-1, 4-2, 4-3, 4-4 and 4-5) from
[198].
Elsevier Limited for permission to reprint the published articles: [617]:
Moayedi M, Weissman-Fogel I, Crawley AP, Goldberg MB, Freeman BV, Tenenbaum HC,
Davis KD. Contribution of chronic pain and neuroticism to abnormal forebrain gray matter in
patients with temporomandibular disorder. Neuroimage 2011; 55(1): 277-86
and
Moayedi M, Weissman-Fogel I, Salomons TV, Crawley AP, Goldberg MB, Freeman BV,
Tenenbaum HC, Davis KD. Abnormal gray matter aging in chronic pain patients. Brain
Research 2012; 1456: 82–93.
Copyright © 2022 FDOKUMEN




















