
Steroidal glycosides from the leaves of Ruscus colchicus: Isolation and structural elucidation based...
12
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
Transcript of Steroidal glycosides from the leaves of Ruscus colchicus: Isolation and structural elucidation based...

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Steroidal glycosides from the leaves of Ruscus colchicus: Isolationand structural elucidation based on a preliminary liquidchromatography�electrospray ionization tandem mass spectrometry profiling
Angela Perrone a, Tamara Muzashvili b, Assunta Napolitano a, Alexandre Skhirtladze b,Ether Kemertelidze b, Cosimo Pizza a, Sonia Piacente a,*
a Dipartimento di Scienze Farmaceutiche, Università degli Studi di Salerno, Via Ponte Don Melillo, I-84084 Fisciano, Italyb Institute of Pharmacochemistry, P. Sarajishvili Street 36, 0159 Tbilisi, Georgia
a r t i c l e i n f o
Article history:Received 4 June 2009Received in revised form 19 August 2009Available online 19 September 2009
Keywords:RuscusSteroidal glycosidesLC–MS profile
a b s t r a c t
An HPLC–ESIMSn method, based on high-performance liquid chromatography coupled to electrospraypositive ionisation multistage ion trap mass spectrometry, has been used as an effective tool to rapidlyidentify and guide the isolation of target saponins from the ethanol extract of the leaves of Ruscus colchi-cus Y. Yeo. Twenty-two steroidal glycosides, including seventeen furostanol, four spirostanol and onecholestane glycosides, were online identified. Subsequently, compounds were isolated and their struc-tures were established by the extensive use of 1D- and 2D-NMR experiments. The structures identifiedby MS were fully consistent with those elucidated by NMR data. Sixteen steroidal glycosides, includingthirteen furostanol, two spirostanol and one cholestane glycosides, were identified along with fourknown furostanol and two spirostanol glycosides. The saponin profile shows that the furostanol glyco-sides are the main constituents of R. colchicus extract, unlike the other Ruscus species, for which the spi-rostanol derivatives generally are reported as the major compounds. Moreover, for the first time acholestane glycoside has been isolated from R. colchicus.
� 2009 Elsevier Ltd. All rights reserved.
1. Introduction
Ruscus genus (Liliaceae), native to the Mediterranean, Southernand Western Europe, comprises perennial, rhizomatous and ever-green shrubs. The hydroalcoholic extract of Ruscus aculeatus L. rhi-zomes is traditionally used as vascular preventive and tonic inpharmaceutical preparations for disorders involving the venoussystem, including venous fragility or varicose veins, and clinicaldata also revealed positive effects on circulation (Bouskela et al.,1993a,b). In particular, the extracts are used in the preventionand treatment of venous insufficiency (Bouskela et al., 1993a,b).Flavonoids and steroidal saponins are the major classes of chemicalcompounds isolated from R. aculeatus (Kartnig et al., 1991; Mimakiet al., 1998a,b,c,d, 1999a). Ruscus steroidal saponins, mainly pres-ent in the rhizomes and roots of the plant, are considered to bethe active compounds of the extracts (Bombardelli et al., 1971,1972).
Besides R. aculeatus, also Ruscus colchicus Y. Yeo, Ruscus ponticusWor., Ruscus hypoglossum L. and Ruscus hypophyllum L. have beeninvestigated. Particularly, diosgenin, ruscogenin, neoruscogeninand their glycosides along with furostanol glycosides have been
isolated from the roots and rhizomes of R. ponticus, R. hypophyllum,R. hypoglossum and R. colchicus (Pkheidze et al., 1971; Panova et al.,1974; Korkashvili and Kikoladze, 1991; de Combarieu et al., 2002;Mimaki et al., 1999b, 2008a,b). A series of cholestane glycosideshave been isolated only from R. hypoglossum and R. hypophyllum(Mimaki et al., 1999b, 2008a,b). R. colchicus Y. Yeo is a shrub-likeplant with upright stems and with lanceolate leaves-like cladodes,distributed in humid subtropical areas in Abkhazia, mostly in Adj-arian and Gurian forests. The leaves of R. colchicus are used by thelocal population for feeding of live-stock in order to yield moremilk and to increase its fat content.
We carried out the phytochemical study of the leaves of R. col-chicus Y. Yeo and, to the best of our knowledge, this is the first re-port on the occurrence of steroidal saponins in the leaves of aRuscus species.
Saponins analysis by HPLC–UV is usually difficult since most ofthem lack a strong UV chromophore (Oleszek, 2002). Since theproblematic detection of saponins by UV absorbance makesdifficult their isolation by traditional chromatographic techniques,improved methods are needed to guide their purification. HPLC–ESIMSn has been extensively applied and plays an increasinglyimportant role in the online analysis of natural products (Wolfend-er et al., 2003; Li et al., 2006; Geng et al., 2007; Huang et al., 2007).HPLC–MS methods have been extensively used in the analysis of
0031-9422/$ - see front matter � 2009 Elsevier Ltd. All rights reserved.doi:10.1016/j.phytochem.2009.08.016
* Corresponding author. Tel.: +39 089969763; fax: +39 089969602.E-mail address: [email protected] (S. Piacente).
Phytochemistry 70 (2009) 2078–2088
Contents lists available at ScienceDirect
Phytochemistry
journal homepage: www.elsevier .com/locate /phytochem

Author's personal copy
triterpene saponins (Fuzzati et al., 1997; Cui et al., 2000; Kite et al.,2003; Oleszek and Bialy, 2006), while only few reports occurr ontheir application to the analysis of steroidal saponins (de Comba-rieu et al., 2002, 2003; Wang et al., 2006; Liu et al., 2006).
HPLC–ESIMSn experiments were performed on the ethanol ex-tract of the leaves of R. colchicus. The mass fragmentation pathwaysof different types of steroidal glycosides were studied and the pat-terns of the mass fragmentation summarized. On the basis of theresults of the online screening by HPLC–ESIMSn, 22 compounds,including 16 new compounds, were isolated, and their structureswere unambiguously elucidated by NMR spectroscopic data.
2. Results and discussion
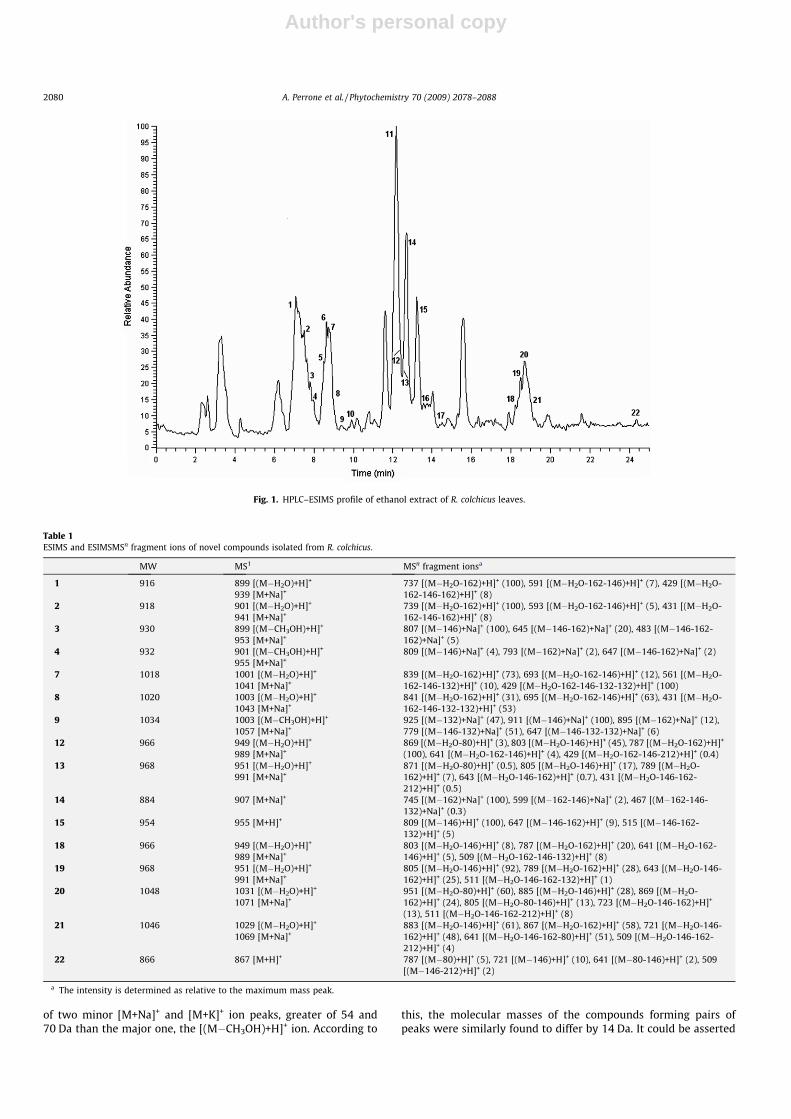
To study the general chromatographic behaviour of saponins,the ethanol extract of R. colchicus leaves was analysed by HPLC–
ESIMSn using positive electrospray ionisation (ESI) with a mobilephase consisting of an aqueous acetonitrile gradient without addi-tion of acid (Fig. 1). Using these conditions, the spirostanol glyco-sides generated [M+H]+ or [M+Na]+ ions, the cholestane glycosideyielded an [M+H]+ ion, while the furostanol glycosides, possessinga labile hydroxy or methoxy group at C-22, produced intense[(M�ROH)+H]+ ions (Table 1) (Kite et al., 2007). Owing to thisbehaviour, the chromatogram showed pairs of peaks having thesame m/z value but different retention times. According to litera-ture data (Kite et al., 2007), in each peak pair, the positive full spec-trum of the earlier eluting peak showed a major [(M�H2O)+H]+ ion,
with two minor ions differing from the first in 40 Da and 56 Da,respectively, and corresponding to [M+Na]+ and [M+K]+ ions. Onthe contrary, the analysis of the full spectrum of the later elutingpeak having the same m/z value allowed us to observe the presence
-D-glcO
1 R1 = H R2 = CH2
OH O
OR1 OR2
O
HOHO OH
OH
-D-glc
OOHO OH
OH
O
OHHO
HO
-L-rha
OHOHO
OH
OOH
OO
-D-xyl
-L-ara
2 R1 = H R2 = -H -CH3
4 R1 = CH3 R2 = -H -CH3
3 R1 = CH3 R2 = CH2
-D-glcHO
7 R1 = H R2 = CH2
O O
OR1 OR2
O
HOHO OH
OH
O
OHHO
HO
-L-rha
8 R1 = H R2 = -H -CH3
9 R1 = CH3 R2 = -H -CH3
OOR3
HOO
-L-ara
-D-glcR1O
12 R1 = H R2 = CH2 R3 = SO3H
O O
OHO
R2
O
HOHO OH
OH
O
OHHO
HO
-L-rha
13 R1 = H R2 = -H -CH3 R3 = SO3H
18 R1 = SO3H R2 = CH2 R3 = H
19 R1 = SO3H R2 = -H -CH3 R3 = H
OOSO3H
HOO
-L-ara
O
O O
O
OHHO
HO
-L-rhaSO3H
O
OOH
HOO
-L-ara
HO
O O
O
OHHO
HO
-L-rha
OO
-D-glc
O
HOHO OH
OH
22
14
OHOHO
O
-D-xyl
-D-glc
O
15
O O
O
HOHO OH
OHO
OHHO
HO
-L-rha
OH
21 R1 = SO3H R2 = CH2 R3 = SO3H
20 R1 = SO3H R2 = -H -CH3 R3 = SO3H
SO3H
A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088 2079
Author's personal copy
of two minor [M+Na]+ and [M+K]+ ion peaks, greater of 54 and70 Da than the major one, the [(M�CH3OH)+H]+ ion. According to
this, the molecular masses of the compounds forming pairs ofpeaks were similarly found to differ by 14 Da. It could be asserted
Table 1ESIMS and ESIMSMSn fragment ions of novel compounds isolated from R. colchicus.
MW MS1 MSn fragment ionsa
1 916 899 [(M�H2O)+H]+
939 [M+Na]+737 [(M�H2O-162)+H]+ (100), 591 [(M�H2O-162-146)+H]+ (7), 429 [(M�H2O-162-146-162)+H]+ (8)
2 918 901 [(M�H2O)+H]+
941 [M+Na]+739 [(M�H2O-162)+H]+ (100), 593 [(M�H2O-162-146)+H]+ (5), 431 [(M�H2O-162-146-162)+H]+ (8)
3 930 899 [(M�CH3OH)+H]+
953 [M+Na]+807 [(M�146)+Na]+ (100), 645 [(M�146-162)+Na]+ (20), 483 [(M�146-162-162)+Na]+ (5)
4 932 901 [(M�CH3OH)+H]+
955 [M+Na]+809 [(M�146)+Na]+ (4), 793 [(M�162)+Na]+ (2), 647 [(M�146-162)+Na]+ (2)
7 1018 1001 [(M�H2O)+H]+
1041 [M+Na]+839 [(M�H2O-162)+H]+ (73), 693 [(M�H2O-162-146)+H]+ (12), 561 [(M�H2O-162-146-132)+H]+ (10), 429 [(M�H2O-162-146-132-132)+H]+ (100)
8 1020 1003 [(M�H2O)+H]+
1043 [M+Na]+841 [(M�H2O-162)+H]+ (31), 695 [(M�H2O-162-146)+H]+ (63), 431 [(M�H2O-162-146-132-132)+H]+ (53)
9 1034 1003 [(M�CH3OH)+H]+
1057 [M+Na]+925 [(M�132)+Na]+ (47), 911 [(M�146)+Na]+ (100), 895 [(M�162)+Na]+ (12),779 [(M�146-132)+Na]+ (51), 647 [(M�146-132-132)+Na]+ (6)
12 966 949 [(M�H2O)+H]+
989 [M+Na]+869 [(M�H2O-80)+H]+ (3), 803 [(M�H2O-146)+H]+ (45), 787 [(M�H2O-162)+H]+
(100), 641 [(M�H2O-162-146)+H]+ (4), 429 [(M�H2O-162-146-212)+H]+ (0.4)13 968 951 [(M�H2O)+H]+
991 [M+Na]+871 [(M�H2O-80)+H]+ (0.5), 805 [(M�H2O-146)+H]+ (17), 789 [(M�H2O-162)+H]+ (7), 643 [(M�H2O-146-162)+H]+ (0.7), 431 [(M�H2O-146-162-212)+H]+ (0.5)
14 884 907 [M+Na]+ 745 [(M�162)+Na]+ (100), 599 [(M�162-146)+Na]+ (2), 467 [(M�162-146-132)+Na]+ (0.3)
15 954 955 [M+H]+ 809 [(M�146)+H]+ (100), 647 [(M�146-162)+H]+ (9), 515 [(M�146-162-132)+H]+ (5)
18 966 949 [(M�H2O)+H]+
989 [M+Na]+803 [(M�H2O-146)+H]+ (8), 787 [(M�H2O-162)+H]+ (20), 641 [(M�H2O-162-146)+H]+ (5), 509 [(M�H2O-162-146-132)+H]+ (8)
19 968 951 [(M�H2O)+H]+
991 [M+Na]+805 [(M�H2O-146)+H]+ (92), 789 [(M�H2O-162)+H]+ (28), 643 [(M�H2O-146-162)+H]+ (25), 511 [(M�H2O-146-162-132)+H]+ (1)
20 1048 1031 [(M�H2O)+H]+
1071 [M+Na]+951 [(M�H2O-80)+H]+ (60), 885 [(M�H2O-146)+H]+ (28), 869 [(M�H2O-162)+H]+ (24), 805 [(M�H2O-80-146)+H]+ (13), 723 [(M�H2O-146-162)+H]+
(13), 511 [(M�H2O-146-162-212)+H]+ (8)21 1046 1029 [(M�H2O)+H]+
1069 [M+Na]+883 [(M�H2O-146)+H]+ (61), 867 [(M�H2O-162)+H]+ (58), 721 [(M�H2O-146-162)+H]+ (48), 641 [(M�H2O-146-162-80)+H]+ (51), 509 [(M�H2O-146-162-212)+H]+ (4)
22 866 867 [M+H]+ 787 [(M�80)+H]+ (5), 721 [(M�146)+H]+ (10), 641 [(M�80-146)+H]+ (2), 509[(M�146-212)+H]+ (2)
a The intensity is determined as relative to the maximum mass peak.
Fig. 1. HPLC–ESIMS profile of ethanol extract of R. colchicus leaves.
2080 A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088

Author's personal copy
that these 22-methyl ethers of furostanol saponins are naturallyoccurring in the leaves of R. colchicus, because no methanol wasused in the extraction procedure neither in the HPLC–ESIMS mo-bile phase (Kite et al., 2007). Moreover, tune conditions used forpositive ESIMS yielded full HPLC–ESIMS spectra of furostanol com-pounds in which ion peaks due to spontaneous in-source collision-induced dissociation (CID) occurred (Fig. 2). These product ionswere due to the subsequent losses of the sugar units starting from[(M�ROH)+H]+ ion until to the aglycon ion peak. Thereby, the anal-ysis of these full HPLC–ESIMS spectra, in agreement with HPLC–ESIMSMS data, allowed a preliminary identification of the detectedcompounds. Beside, further analysis of the chromatogram allowedus to observe the presence of two pairs of peaks, i.e. 12–18, and13–19, each displaying in the full ESIMS spectrum the same[(M�H2O)+H]+ ion, but slightly differing in tandem mass spectra.The analysis of HPLC–ESIMSn experiments for each compound ofa pair showed two different fragmentation pathways, consistentwith two molecules differing for the sulfation site (Table 1). In eachpeak pair, the earlier eluting peak corresponded to the compoundsulfated on the hydroxy group of a sugar unit, while the later elut-ing one to the compound sulfated on the aglycon moiety.
As a result of the online screening by HPLC–ESIMSn, 22 com-pounds were detected (Table 1). On the basis of these results, the22 compounds were isolated to unambiguously elucidate theirstructures by NMR experiments.
Therefore, the ethanol extract was fractionated by silica gel col-umn chromatography. The comparison between the HPLC–ESIMSprofile of the EtOH extract with those of each chromatographicfraction prompted us to screen fractions B–D and F by reversed-phase HPLC, yielding compounds 1–22 (see Section 3).
The sugar composition was determined by acid hydrolysis ofthe crude saponin mixture as D-glucose, D-xylose, L-rhamnoseand L-arabinose and the absolute configurations of the sugar unitswere assigned on the basis of their optical rotation values (Eskan-der et al., 2006).
The positive HRMALDITOFMS spectrum of 1 showed an[M+Na]+ ion peak at m/z 939.4569, supporting the molecular for-
mula C45H72O19Na (calc. for C45H72O19Na, 939.4561). Accordingto the HPLC–ESIMS results, the full positive ESIMS spectrum of 1showed the characteristic pattern of a furostanol saponin with a la-bile hydroxy group at C-22, displaying an intense [(M�H2O)+H]+
ion at m/z 899 together with two minor [M+Na]+ and [M+K]+ ionsat m/z 909 and 955, respectively. The ESIMSMS spectrum of theion at m/z 899 showed a fragmentation pattern in agreement withthe presence of two hexose and one deoxy-hexose moieties, as de-scribed in Table 1. The 1H NMR spectrum of 1 showed signals fortwo tertiary methyl groups at d 0.89 (3H, s) and 1.08 (3H, s), a sec-ondary methyl group at d 1.05 (3H, d, J = 6.6 Hz), exomethyleneprotons at d 5.12 and 4.96 (each 1H, br s), an olefinic proton at d5.60 (1H, br d, J = 5.7 Hz), two methine proton signals at d 3.62(1H, m) and 3.36 (1H, dd, J = 11.9, 3.9 Hz), indicative of secondaryalcoholic functions, two methylene proton signals at d 4.37 and4.15 (each 1H, d, J = 12.1 Hz), ascribable to a primary alcoholicfunction, along with three anomeric protons at d 4.88 (1H, d,J = 1.2 Hz), 4.42 (1H, d, J = 7.5 Hz) and 4.30 (1H, d, J = 7.5 Hz). The13C NMR spectrum displayed for the aglycon signals ascribable toa hemiacetal function at d 113.3, three secondary alcoholic func-tions at d 78.8, 75.9, and 82.3, and one primary alcoholic functionat d 72.5, suggesting the occurrence of a furostanol skeleton (Skhir-tladze et al., 2006). On the basis of the HSQC and HMBC correla-tions, the aglycon moiety of compound 1 was identified asfurosta-5,25(27)-diene-1b,3b,22a,26-tetrol. The configuration ofthe hydroxy group at C-22 was established to be a from the ROESYcorrelations between H-20 (d 2.25) and H-23a (d 1.94) and H-23b(d 1.56). It was evident from the 1H and 13C NMR data that the su-gar chain of 1 consisted of three sugar units. The chemical shifts ofall the individual protons of the three sugar units were ascertainedfrom a combination of 1D-TOCSY and DQF-COSY spectral analysis,and the 13C chemical shifts of their relative attached carbons wereassigned unambiguously from the HSQC spectrum (Table 2). Thesedata showed the presence of two b-glucopyranosyl units (d 4.42and 4.30) and one a-rhamnopyranosyl unit (d 4.88). Glycosidationshifts were observed for C-4glcI (d 79.4), C-3 (d 75.9) and C-26 (d72.5). An unambiguous determination of the sequence and linkage
Fig. 2. Full positive HPLC–ESIMS spectrum of compound 1. Legend: a = [(M�H2O)+H]+; b = [(M�H2O-162)+H]+; c = [(M�H2O-162-146)+H]+; d = [(M�H2O-162-146-162)+H]+.
A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088 2081

Author's personal copy
sites was obtained from the HMBC spectrum, which showed keycorrelation peaks between the proton signal at d 4.42 (H-1glcI)and the carbon resonance at d 75.9 (C-3), d 4.88 (H-1rha) and d79.4 (C-4glcI), and the proton signal at d 4.30 (H-1glcII) and the car-bon resonance at d 72.5 (C-26). On the basis of all these evidences,the structure of the new compound 1 was established as 26-O-b-D-glucopyranosyl-furosta-5,25(27)-diene-1b,3b,22a,26-tetrol3-O-[a-L-rhamnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside].
Compound 2 exhibited an [M+Na]+ ion peak at m/z 941.4722 inthe positive HRMALDITOFMS spectrum, corresponding to themolecular formula C45H74O19Na. Positive full and tandem massspectrometric experiments permitted to identify compound 2 asa furostanol glycoside. In particular, collision-induced dissociationof the [(M�H2O)+H]+ ion at m/z 901 yielded a fragmentation pat-tern similar to that of compound 1 (Table 1), but showing eachproduct ion increased of 2 Da. This result was coherent with afurostanol derivative of 1 in which the exomethylene group wasreduced. In fact, the 1H and 13C NMR data of 2 in comparison tothose of 1 clearly suggested that 2 differed from 1 only by thereplacement of the exomethylene group with a secondary methylgroup at C-27 (dH 0.98, dC 17.0). Thus, the aglycon of 2 was estab-lished as (25R)-furost-5-ene-1b,3b,22a,26-tetrol. The C-25 config-uration was deduced to be R based on the difference of chemicalshifts (dab = da–db) of the geminal protons at H2-26 (dab =0.45 ppm). It has been described that dab is usually >0.57 ppm in25S compounds and <0.48 in 25R compounds (Agrawal, 2004).Therefore, compound 2 was identified as the new (25R)-26-O-b-D-glucopyranosyl-furost-5-ene-1b,3b,22a,26-tetrol 3-O-[a-L-rha-mnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside].
The positive HRMALDITOFMS spectra of compounds 3 and 4showed an [M+Na]+ ion peaks at m/z 953.4728 and 955.4883,respectively. In agreement with the HPLC–ESIMS results, the posi-tive ESIMS spectrum of each compound showed a major[(M�CH3OH)+H]+ ion together with minor [M+Na]+ and [M+K]+
ions, corresponding to the 22-methyl ether furostanol derivativesof 1 and 2, respectively (Table 1). This spectra interpretation was
confirmed by the NMR results. In fact, it was apparent from theNMR data (1H, 13C, 1D-TOCSY, DQF-COSY, HSQC, HMBC, ROESY)of compounds 3 and 4 that these compounds differed from 1 and2, respectively, only by the presence of a methoxy group insteadof a hydroxy group at C-22. Therefore, compound 3 was deducedto be 26-O-b-D-glucopyranosyl-22a-methoxy-furosta-5,25(27)-diene-1b,3b,26-triol 3-O-[a-L-rhamnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside] and compound 4 was established as (25R)-26-O-b-D-glucopyranosyl-22a-methoxy-furost-5-ene-1b,3b,26-triol3-O-[a-L-rhamnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside]. Com-pounds 3 and 4 are 22-O-methyl ethers derivatives of compounds1 and 2, respectively.
The molecular formula of compound 7 was established to beC49H78O22Na by HRMALDITOFMS (m/z 1041.4890 [M+Na]+, calc.for C49H78O22Na, 1041.4883). Positive full mass spectrum of 7 dis-played a major ion at m/z 1001, together with two minor ions atm/z 1041 and 1057, greater of 40 and 56 Da than the major one,respectively. Thereby, compound 7 was identified as a 22-hydrox-yfurostanol molecule. According to HPLC–ESIMSMS, positive tan-dem mass experiment yielded product ions originating fromneutral losses of one hexose, one deoxy-hexose, and two pentoseunits, and the same aglycon ion of compound 1. In fact, the 1Hand 13C NMR chemical shifts of the aglycon moieties of 7 and 1 werealmost superimposable (see Table 2) confirming the same aglyconportion. Additionally for 7, resonances of anomeric protons wereobserved in the 1H NMR spectrum at d 5.34 (1H, d, J = 1.2 Hz),4.44 (1H, d, J = 7.5 Hz), 4.33 (1H, d, J = 3.7 Hz) and 4.30 (1H, d,J = 7.5 Hz). Complete assignments of the 1H and 13C NMR signalsof the sugar portion were accomplished by HSQC, HMBC, DQF-COSYand 1D-TOCSY experiments which led to the identification of one a-rhamnopyranosyl (d 5.34) unit, one b-xylopyranosyl (d 4.44) unit,one a-arabinopyranosyl (d 4.33) unit and one b-glucopyranosyl (d4.30) unit. Once again, direct evidence of the sugar sequence andthe linkage sites was derived from HSQC and HMBC experiments.The glycosidation shifts on C-1 (d 84.6), C-26 (d 72.6), C-2ara (d74.0) and C-3ara (d 85.2) indicated the linkage sites. Key correlation
Table 213C NMR data of the aglycon moieties of compounds 1–4, 7–9, 14–15, and 20–22 (600 MHz, CD3OD).
1 2 3 4 7 8 9 14 15 20 21 22
1 78.8 78.9 78.8 78.8 84.6 84.7 84.6 84.6 84.3 83.8 83.9 84.02 40.4 40.4 40.3 40.2 37.1 37.0 37.2 37.1 34.6 34.4 34.4 34.73 75.9 75.8 76.0 75.7 68.9 69.0 69.1 69.1 76.0 75.9 75.9 75.84 39.7 39.9 39.8 39.7 43.4 43.2 43.4 43.3 40.7 40.6 40.5 40.75 139.0 138.7 139.0 138.8 139.3 139.5 139.6 139.8 138.7 139.0 138.9 138.96 126.0 126.2 126.1 126.0 126.2 126.1 126.0 126.0 127.2 126.5 126.5 126.57 32.6 32.7 32.5 32.6 32.7 32.8 32.6 32.5 32.5 32.5 32.5 32.78 33.6 33.6 33.6 33.7 33.9 33.7 33.8 33.8 34.0 33.4 33.5 34.09 52.0 51.8 52.1 51.7 51.1 51.0 51.2 51.4 51.3 51.0 51.1 51.310 43.7 43.6 43.7 43.8 42.8 42.7 42.7 43.5 43.0 43.2 43.1 43.511 24.3 24.5 24.2 24.6 24.4 24.6 24.8 24.6 24.6 24.0 24.1 24.512 40.7 40.9 40.8 41.0 41.1 41.0 40.9 41.2 41.5 40.9 40.9 41.013 41.0 41.2 41.0 41.3 41.0 41.2 41.2 41.5 42.8 41.0 41.0 41.214 57.8 57.6 57.9 57.7 57.8 57.9 57.8 57.9 56.4 57.8 57.7 57.815 32.8 32.8 32.7 32.8 32.6 32.5 32.5 32.5 37.8 32.6 32.5 32.816 82.3 82.4 82.1 82.3 82.4 82.4 82.3 83.0 83.2 82.3 82.3 82.217 65.2 65.3 65.2 65.3 65.1 65.1 65.2 63.0 59.1 64.8 65.1 63.818 16.6 16.8 16.5 16.7 16.8 16.8 16.8 16.6 13.7 16.7 16.7 16.919 13.3 13.2 13.4 13.2 14.9 15.0 15.0 15.1 14.9 14.7 14.7 15.120 40.8 41.0 40.8 41.0 40.8 41.0 41.1 37.0 36.2 41.0 41.0 42.821 15.8 15.7 15.7 15.8 15.8 15.9 15.8 14.1 11.8 16.0 15.8 14.822 113.3 113.2 113.0 113.2 113.3 113.3 113.0 113.8 74.5 113.3 113.3 110.923 32.0 31.1 32.1 31.1 31.8 31.0 31.0 32.5 33.0 31.1 32.5 33.724 28.4 28.8 28.4 28.7 28.4 28.8 28.7 73.2 37.0 28.8 28.4 29.325 146.5 34.8 146.6 34.7 147.4 34.7 34.7 147.0 29.4 34.8 146.8 145.626 72.5 75.2 72.4 75.3 72.6 75.8 75.6 64.3 23.3 75.2 72.4 65.327 112.0 17.0 112.1 17.1 112.3 17.0 17.1 107.8 23.3 17.0 112.0 108.4OMe – – 47.6 47.6 – – 47.5 – – – – –
2082 A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088

Author's personal copy
peaks were observed in the HMBC spectrum of 7 between the pro-ton signal at d 4.33 (H-1ara) and the carbon resonance at d84.6 (C-1),d 5.34 (H-1rha) and d 74.0 (C-2ara), d 4.44 (H-1xyl) and d 85.2 (C-3ara)and the proton signal at d 4.30 (H-1glc) and the carbon resonance atd 72.6 (C-26). Therefore, compound 7 was established as the new26-O-b-D-glucopyranosyl-furosta-5,25(27)-diene-1b,3b,22a,26-te-trol 1-O-{a-L-rhamnopyranosyl-(1 ? 2)-O-[b-D-xylopyranosyl-(1 ?3)]-a-L-arabinopyranoside}.
HRMALDITOFMS experiments permitted to assign to 21 themolecular formula C44H70O24S2Na (calc. for C44H70O24S2Na,1069.3597). Acid hydrolysis of 21, followed by treatment withBaCl2, again demonstrated the presence of a sulfate residue. Onceagain, positive full ESIMS experiments allowed us to attribute tothis compound a 22-hydroxyfurostanol skeleton. In particular,mass tandem spectrum of 21 showed the presence of the[(M�H2O-146-162-80)+H]+ ion, likewise supporting a sulfategroup in the molecular skeleton of 21 (Table 1). On the other hand,product ion at m/z 509 [(M�H2O-146-162-212)+H]+ was in agree-ment with the presence of a second sulfate group on the molecule.The 1H NMR spectrum of 21 showed signals for two tertiary methylgroups at d 0.89 (3H, s) and 1.13 (3H, s), a secondary methyl groupat d 1.05 (3H, d, J = 6.6 Hz), exomethylene protons at d 5.12 and4.97 (each 1H, br s), an olefinic proton at d 5.65 (1H, br d,J = 5.7 Hz), two methine proton signals at d 4.09 (1H, m) and 3.45(1H, dd, J = 11.9, 3.9 Hz), two methylene proton signals at d 4.37and 4.15 (each 1H, d, J = 12.1 Hz) and signals for four anomeric pro-tons at d 5.34 (1H, d, J = 1.2 Hz), 4.44 (1H, d, J = 7.5 Hz), 4.33 (1H, d,J = 3.7 Hz) and 4.30 (1H, d, J = 7.5 Hz). On the basis of DQF-COSY,HSQC and HMBC spectra for the aglycon moiety and by comparisonof these data with those of the aglycon moiety of 7, it was observedthat the aglycon of compound 21 differed from that of 7 only by thepresence of the sulfate group at C-3, as suggested by the downfieldshifts observed for H-3 (d 4.09) and C-3 (d 75.9) signals, consistentwith the presence of a sulfate group. Additionally, the 1H NMRspectrum showed signals for three anomeric protons at d 5.31(1H, d, J = 1.2 Hz), 4.38 (1H, d, J = 3.7 Hz) and 4.30 (1H, d,J = 7.5 Hz). On the basis of 1D-TOCSY, HSQC, HMBC, DQF-COSY cor-relations these sugar units was identified as a-rhamnopyranose (d5.31), b-glucopyranose (d 4.30) and 4-sulfo-a-arabinopyranose (d4.38), as indicated by the downfield shifts of the H-4ara (d 4.57)and C-4ara (d 77.5) signals (Bassarello et al., 2008). The linkage siteswere determined by the HMBC spectrum, which showed key corre-lation peaks between the proton signal at d 4.38 (H-1ara) and thecarbon resonance at d 84.6 (C-1), d 5.31 (H-1rha) and d 75.1 (C-2ara) and the proton signal at d 4.30 (H-1glc) and the carbon reso-nance at d 72.6 (C-26). Therefore, the new structure of 21 wasestablished as 26-O-b-D-glucopyranosyl-3-sulfo-furosta-5,25(27)-diene-1b,3b,22a,26-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-4-sulfo-a-L-arabinopyranoside].
The HRMALDITOFMS of 22 showed a major ion peak at m/z867.3151 [M+H]+, ascribable to the molecular formula C38H59-O18S2 (calc. for C38H59O18S2, 867.3144). Acid hydrolysis of 22,followed by treatment with BaCl2, gave a white precipitate, thusdemonstrating the presence of a sulfate residue. The analysis of fullpositive ESIMS spectrum allowed us to soon ascertain that 22 wasnot a furostanol compound, lacking the typical [(M�ROH)+H]+ ionpeak, but a spirostanol or cholestane glycoside, indeed showing a[M+H]+ ion peak. In tandem mass spectrum product ions due tosubsequent neutral losses of a sulfate group and sugar units werereported. The 1H NMR spectrum of compound 22 showed signalsfor two tertiary methyl groups at d 0.86 (3H, s) and 1.14 (3H, s),a secondary methyl group at d 0.99 (3H, d, J = 6.6 Hz), exomethyl-ene protons at d 4.80 and 4.77 (each 1H, br s), an olefinic protonat d 5.65 (1H, br d, J = 5.7 Hz), two methine proton signals at d4.09 (1H, m) and 3.44 (1H, dd, J = 11.9, 3.9 Hz), two methylene pro-ton signals at d 4.30 and 3.85 (each 1H, d, J = 12.1 Hz), along with
two anomeric protons at d 5.32 (1H, d, J = 1.2 Hz) and 4.37 (1H, d,J = 3.7 Hz).
The NMR data of the aglycon moiety of 22 were in good agree-ment with those reported for spirosta-5,25(27)-diene-1b,3b-diolor neoruscogenin (Mimaki et al., 1998a), except for the downfieldshifts of for H-3 (d 4.09) and C-3 (d 75.8) signals, consistent withthe presence of a sulfate group. Glycosidation shift was observedfor C-1 (d 84.0). Additionally, for the sugar portion the combinationof 1D-TOCSY, HSQC and DQF-COSY data led to identification of onea-rhamnopyranosyl (d 5.32) unit and one 4-sulfo-a-arabinopyr-anosyl (d 4.37) unit. The linkage sites were deduced by the HMBCspectrum which showed key correlation peaks between the protonsignal at d 4.37 (H-1ara) and the carbon resonance at d 84.0 (C-1) andthe proton signal at d 5.32 (H-1rha) and the carbon resonance at d75.1 (C-2ara). Thus the structure of compound 22 was establishedas 3-sulfo-spirosta-5,25(27)-diene-1b,3b-diol 1-O-[a-L-rhamno-pyranosyl-(1 ? 2)-O-4-sulfo-a-L-arabinopyranoside]. While a neo-ruscogenin derivative with a sulfate group on the sugar portion hasbeen previously isolated from R. aculeatus (Mimaki et al., 1998a),this is the first report of a neoruscogenin derivative characterizedby the presence of two sulfate groups both on the aglycon moietyand the sugar portion.
The analysis of HRMALDITOFMS spectrum of 14 allowed to as-sign it the molecular formula C44H69O18 (calc. for C44H69O18,885.4485). Full and tandem mass experiments were in agreementto identify 14 as a spirostanol glycoside, showing a major[M+Na]+ ion peak and minor product ions owing to neutral lossesof sugar units (Table 1). The 1H NMR spectrum of compound 14showed for the aglycon signals for two tertiary methyl groupsat d 0.93 (3H, s) and 1.13 (3H, s), a secondary methyl group atd 0.96 (3H, d, J = 6.6 Hz), exomethylene protons at d 5.18 and4.98 (each 1H, br s), an olefinic proton at d 5.59 (1H, br d,J = 5.7 Hz), three methine proton signals at d 4.26 (1H, dd,J = 11.7, 5.7 Hz), 3.40 (1H, dd, J = 11.9, 3.9 Hz) and 3.37 (1H, m),two methylene proton signals at d 4.24 and 3.89 (each 1H, d,J = 12.1 Hz). The NMR data (1H, 13C, 1D-TOCSY, DQF-COSY, HSQC,HMBC) of compound 14 in comparison with those of neoruscog-enin (Mimaki et al., 1998a) revealed that this aglycon differedfrom neoruscogenin by the presence of an additional secondaryalcoholic function at C-24 (dH 4.26, dC 73.2). The position of 24-OH was established on the basis of HMBC correlations betweenthe proton signals at d 4.24 (H-26a), d 3.89 (H-26b), d 5.18 (H-27a) and d 4.98 (H-27b) and the carbon resonance at d 73.2 (C-24). The C-24S configuration was deduced from the J values(11.7 and 5.7 Hz) and from ROESY correlations between H-16 (d4.48) and H-26a (d 4.24) and H-24 (d 4.26). Thus, the aglyconof 14 was identified as (24S)-spirosta-5,25(27)-diene-1b,3b,24-triol. This is the first time that a spirosta-5,25(27)-diene deriva-tive characterized by the presence of an hydroxy group at C-24in absence of the hydroxy group at C-23 has been reported. More-over, the 1H NMR spectrum showed signals for three anomericprotons at d 5.33 (1H, d, J = 1.2 Hz), 4.27 (1H, d, J = 7.5 Hz) and4.30 (1H, d, J = 3.7 Hz). From the analysis of HSQC, 1D-TOCSYand DQF-COSY spectra one a-rhamnopyranosyl (d 5.33) unit,one b-glucopyranosyl (d 4.27) and one a-arabinopyranosyl (d4.30) unit were identified. The sequence of sugar chain and link-age sites was obtained from HMBC spectrum which showed keycorrelation peaks between the proton signal at d 4.30 (H-1ara)and the carbon resonance at d 84.6 (C-1), d 5.33 (H-1rha) and d75.2 (C-2ara) and between the proton signal at d 4.27 (H-1glc)and the carbon resonance at d 73.2 (C-24). Therefore, the struc-ture of 14 was deduced to be (24S)-24-O-b-D-glucopyranosyl-spi-rosta-5,25(27)-diene-1b,3b,24-triol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-a-L-arabino- pyranoside].
The molecular formula C44H75O20S was assigned to 15 by theanalysis of its HRMALDITOFMS spectrum (calc. for C44H75O20S,
A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088 2083

Author's personal copy
955,4573). Acid hydrolysis of 15, followed by treatment with BaCl2,again demonstrated the presence of a sulfate residue. Positive fullESIMS spectrum showed a major [M+H]+ ion, suggesting a spiros-tane or cholestane nature for 15. As reported in Table 1, ESIMSMSexperiment yielded product ions due to subsequent neutral lossesof the sugar units. The 1H NMR of 15 spectrum showed signals forfive steroidal methyl group at d 0.95 (3H, s), 1.13 (3H, s), 0.95 (3H,d, J = 6.6 Hz), 0.93 (3H � 2, d, J = 6.6 Hz), an olefinic proton at d 5.64(1H, br d, J = 5.7 Hz), three methine proton signals at d 4.09 (1H, m),3.44 (1H, dd, J = 11.9, 3.9 Hz) and 3.74 (1H, m), along with threeanomeric protons at d 5.38 (1H, d, J = 1.2 Hz), 4.21 (1H, d,J = 7.5 Hz) and 4.37 (1H, d, J = 3.7 Hz). The 1H and 13C NMR dataof the aglycon moiety of compound 15 were almost superimpos-able to those of the (22S)-colest-5-ene-1b,3b,16b,22-tetrol, knownas alliosterol (Takaashi et al., 1995), except for the presence of asulfate group at C-3, as indicated by the downfield of H-3 (d4.09) and C-3 (d 76.0). On the basis of 1D-TOCSY, HSQC, HMBC,DQF-COSY correlations the sugar units were identified as a-rhamnopyranose (d 5.38), b-glucopyranose (d 4.21) and b-xylopy-ranose (d 4.37). Direct connectivity information was obtained fromthe HMBC spectrum, which showed the following key correlationpeaks: d 5.38 (H-1rha) and d 77.5 (C-2xyl), d 4.37 (H-1xyl) and d84.3 (C-1) and d 4.21 (H-1xyl) and d 83.2 (C-16). Therefore, thestructure of 15 was established as the new (22S)-16-O-b-D-glucopyranosyl-3-sulfo-colest-5-ene-1b,3b,16b,22-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-b-D-xylopyranoside]. A series ofcholestane glycosides have been previously isolated from severalspecies belonging to the Liliaceae family and particularly from R.hypoglossum (Mimaki et al., 1999b) and R. hypophyllum (Mimakiet al., 2008a), nevertheless this is the first report of a sulfated cho-lestane derivative.
The structures of compounds 8, 9, 12, 13 and 18–20 were estab-lished by NMR experiments and ESIMS and HRMALDITOFMS spec-tra following the same method of analysis shown for compoundsabove reported (see Section 3 and Table 1).
Furthermore, four known furostanol derivatives namely26-O-b-D-glucopyranosyl-furosta-5,25(27)-diene-1b,3b,22a,26-te-trol 1-O-a-L-rhamnopyranosyl-(1 ? 2)-O-a-L-arabinopyranoside(ruscoponticoside E) (5) (Bombardelli et al., 1972), (25R)-26-O-b-D-glucopyranosyl-furost-5-en-1b,3b,22a,26-tetrol 1-O-a-L-rham-no- pyranosyl-(1 ? 2)-O-a-L-arabinopyranoside (ceparoside B)(Yuan et al., 2008) (6), 26-O-b-D-glucopyranosyl-22a-methoxy-furosta-5,25(27)-diene-1b,3b,26-triol 1-O-{a-L-rhamnopyranosyl-(1 ? 2)-O-[b-D-xylopyranosyl-(1 ? 3)]-a-L-arabinopyranoside}(Mimaki et al., 1996) (10) and (25R)-26-O-b-D-glucopyranosyl-fur-ost-5-en-3b,22a,26-triol 3-O-a-L-rhamnopyranosyl-(1 ? 4)-O -b-D-glucopyranoside (Khodakov et al., 1996) (11) were isolated,along with two known spirostanol derivatives namely (23S,24S)-spirosta-5,25(27)-diene-1b,3b,23,24-tetrol 1-O-a-L-rhamno-pyranosyl-(1 ? 2)-O -a-L-arabinopyranoside (16) (Mimaki et al.,1999c) and (23S, 24S)-24-O -b-D-fucopyranosyl-spirosta-5,25(27)-diene-1b,3b, 23,24-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O -a-L-arabinopyranoside] (namonin C) (17) (Le Tranet al., 2001).
In conclusion, the HPLC–ESIMSn method resulted to be a valu-able and effective tool for the online identification of steroidal gly-cosides in the EtOH extract of the leaves of R. colchicus. It providesstructural informations to rapidly screen this kind of compounds,contributing to reduce labor costs. The subsequent guided isolationfollowed by the NMR analysis led to the definitive structure eluci-dation of these compounds. The new furostanol glycosides isolatedare furosta-5,25(27)-diene-1b,3b,22a,26-tetrol derivatives and thecorresponding (25R)-25,27-dihydro-derivatives. Additionally, forthe first time a cholestane glycoside has been isolated from R. col-chicus. Unlike the other Ruscus species, which show as major com-pounds the spirostanol glycosides, the main constituents of this
extract are the furostanol derivatives, making peculiar the saponinprofile of R. colchicus.
3. Experimental
3.1. General
Optical rotations were measured on a JASCO DIP 1000 polarim-eter. IR measurements were obtained on a Bruker IFS-48 spectrom-eter. NMR experiments were performed on a Bruker DRX-600spectrometer (Bruker BioSpinGmBH, Rheinstetten, Germany)equipped with a Bruker 5 mm TCI CryoProbe at 300 K. All 2D-NMR spectra were acquired in CD3OD (99.95%, Sigma–Aldrich)and standard pulse sequences and phase cycling were used forDQF-COSY, HSQC, HMBC and ROESY spectra. The NMR data wereprocessed using UXNMR software. Exact masses were measuredby a Voyager DE mass spectrometer equipped with a 337 nm laserand delay extraction and operated in positive ion reflector mode.Samples were analyzed by MALDITOF mass spectrometry. A mix-ture of analyte solution and a-cyano-4-hydroxycinnamic acid (Sig-ma) was applied to the metallic sample plate and dried. Masscalibration was performed with the ions from adrenocorticotropichormone (ACTH) fragment 18–39 human at 2465.1989 Da and a-cyano-4-hydroxycinnamic acid at 190.0504 Da as internal stan-dard. Ethanol extract was analyzed by on-line HPLC–ESIMSn usinga ThermoFinnigan Spectra System HPLC coupled with an LCQ Decaion trap mass spectrometer (ThermoFinnigan, San Jose, CA, USA).HPLC separation was conducted on a C18 reversed-phase (RP) col-umn (5 lm, 2.1 mm � 250 mm; X-Terra MS C18; Waters, Milford,MA) at a flow rate of 0.2 ml/min. A gradient elution was performedby using H2O (A) and CH3CN (B) as mobile phases, from 20% B to70% B in 25 min. The column effluent was analyzed by ESIMS in po-sitive ion mode and the mass spectra were acquired and processedusing the software provided by the manufacturer. The capillaryvoltage was set at 23 V, the spray voltage at 5 kV and the tube lensoffset at 50 V. The capillary temperature was 280 �C. Data were ac-quired in MS1 and MSn scanning modes. By using a syringe pump(flow rate 5 ll/min), each pure compound dissolved in H2O/CH3CN(1:1) was infused in the ESI source. Positive ESIMSn analyses wereperformed using the same conditions as those for HPLC–ESIMSn
analysis.HPLC separations were carried out on a Waters 590 system
equipped with a Waters R401 refractive index detector, a WatersXTerra Prep MSC18 column (300 � 7.8 mm i.d.) and a Rheodyneinjector.
3.2. Plant material
The leaves of R. colchicus were collected in August of 2006 in thearea of mountain Mtirala, in Khelvachauri region of Adjara, inGeorgia. Samples of R. colchicus were identified by Dr. Jemal Aneli,Department of Botany, Institute of Pharmacochemistry, Tbilisi,Georgia. A voucher specimen (No. 365), has been deposited at thisDepartment.
3.3. Extraction and isolation
The air-dried, powdered leaves (phylloclade) of R. colchicus(150 g) were extracted with 70% EtOH three times at 60 �C. The col-lected alcohol-aqueous extracts have been dried under vacuum(20 g). Raw extract (5 g) was subjected to silica gel column chro-matography (120 � 3 cm, 100/160 lm, Merck) eluting with iso-cratic system chloroform:methanol:water (26:14:3) yieldingseven combined fractions: A (113.8 mg), B (378.4 mg), C(726.3 mg), D (837.1 mg), E (198.6 mg), F (167.7 mg) and G
2084 A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088

Author's personal copy
(188.5 mg). Fractions were analyzed by HPLC–ESIMS, according toalready described conditions. Fractions B–D and F were chromato-graphed by RP-HPLC (7.8 � 300 mm, LiChroprep RP18, 10 lm,XTerra), using different mixtures of MeOH:H2O in isocratic condi-tions. Fraction B was chromatographed by RP-HPLC using MeOH–H2O (52:48) as mobile phase (flow rate 2.0 ml/min) to yield com-pound 16 (2.1 mg, tR = 12.9 min). Fraction C was chromatographedby RP-HPLC using MeOH–H2O (50:50) as mobile phase (flow rate2.0 ml/min) to yield compounds 14 (2.4 mg, tR = 7.0 min) and 17(1.5 mg, tR = 9.5 min). From fraction F compounds 12 (1.8 mg,tR = 8.0 min), 13 (2.3 mg, tR = 8.7 min) and 15 (2.8 mg,tR = 11.5 min) were obtained by RP-HPLC using MeOH–H2O(46:54) as mobile phase (flow rate 2.0 ml/min). Compounds 18(1.4 mg, tR = 11.6 min), 19 (1.7 mg, tR = 12.3 min), 20 (2.0 mg,tR = 10.8 min), 21 (2.7 mg, tR = 11.5 min) and 22 (1.6 mg,tR = 19.7 min) were obtained from the same fraction F usingMeOH–H2O (60:40) as mobile phase (flow rate 2.0 ml/min). Frac-tion D was chromatographed by RP-HPLC using MeOH–H2O(38:62) as mobile phase (flow rate 2.0 ml/min) to yield compounds1 (1.3 mg, tR = 11.1 min), 2 (1.9 mg, tR = 11.5 min), 3 (1.2 mg,tR = 12.0 min), 4 (2.2 mg, tR = 12.8 min), 5 (1.4 mg, tR = 29.5 min),6 (2.9 mg, tR = 30.4 min), 7 (2.0 mg, tR = 31.0 min), 8 (1.1 mg,tR = 32.0 min), 9 (1.5 mg, tR = 33.0 min), 10 (2.6 mg, tR = 34.5 min)and 11 (1.0 mg, tR = 38.0 min).
3.4. 26-O-b-D-glucopyranosyl-furosta-5,25(27)-diene-1b,3b,22a,26-tetrol 3-O-[a-L-rhamnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside] (1)
Amorphous white solid; C45H72O19; ½a�22D �36.4� (c 0.1 MeOH);
IR mKBrmax cm�1: 3480 (>OH), 2932 (>CH), 1273 and 1055 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.60 (1H, br d,
J = 5.7 Hz, H-6), 5.12 (1H, br s, H-27a), 4.96 (1H, br s, H-27b), 4.40(1H, m, H-16), 4.37 (1H, d, J = 12.1 Hz, H-26a), 4.15 (1H, d,J = 12.1 Hz, H-26b), 3.62 (1H, m, H-3), 3.36 (1H, dd, J = 11.9,3.9 Hz, H-1), 1.08 (3H, s, Me-19), 1.05 (3H, d, J = 6.6 Hz, Me-21),0.89 (3H, s, Me-18); for 13C NMR data of the aglycon moiety, see Ta-ble 2; for 13C and 1H NMR data of the sugar portion, see Table 3;HRMALDITOFMS [M+Na]+ m/z 939.4569 (calc. for C45H72O19Na,939.4561).
3.5. (25R)-26-O-b-D-glucopyranosyl-furost-5-ene-1b,3b,22a,26-tetrol3-O-[a-L-rhamnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside] (2)
Amorphous white solid; C45H74O19; ½a�22D �48.5� (c 0.1 MeOH);
IR mKBrmax cm�1: 3477 (>OH), 2938 (>CH), 1270 and 1060 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.61 (1H, br d,J = 5.7 Hz, H-6), 4.40 (1H, m, H-16), 3.83 (1H, m, H-26a), 3.61 (1H,m, H-3), 3.36 (1H, dd, J = 11.9, 3.9 Hz, H-1), 3.38 (1H, m, H-26b),1.07 (3H, s, Me-19), 1.04 (3H, d, J = 6.6 Hz, Me-21), 0.98 (3H, d,J = 6.6 Hz, Me-27), 0.88 (3H, s, Me-18); for 13C NMR data of theaglycon moiety, see Table 2; for 13C and 1H NMR data of the sugarportion, see Table 3; HRMALDITOFMS [M+Na]+ m/z 941.4722 (calc.for C45H74O19Na, 941.4713).
3.6. 26-O-b-D-glucopyranosyl-22a-methoxy-furosta-5,25(27)-diene-1b,3b,26-triol 3-O-[a-L-rhamnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside] (3)
Amorphous white solid; C45H72O19; ½a�22D �55.7� (c 0.1 MeOH);
IR mKBrmax cm�1: 3482 (>OH), 2935 (>CH), 1270 and 1057 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.59 (1H, br d,J = 5.7 Hz, H-6), 5.12 (1H, br s, H-27a), 4.97 (1H, br s, H-27b), 4.42
Table 313C and 1H NMR data (J in Hz) of the sugar portions of compounds 1, 7, 15, 18 and 21 (600 MHz, CD3OD).
1a 7a 15 18a 21b
b-D-GlcI a-L-Ara b-D-Xyl a-L-Ara a-L-Ara-4-SO3H1 102.0 4.42 d (7.5) 100.8 4.33 d (3.7) 100.6 4.37 d (7.5) 100.9 4.30 d (3.7) 100.3 4.38 d (3.7)2 75.0 3.21 dd (9.0, 7.5) 74.0 3.83 dd (8.5, 3.7) 77.5 3.43 dd (9.2, 7.5) 75.2 3.73 dd (8.5, 3.7) 75.1 3.72 dd (8.5, 3.7)3 76.4 3.48 dd (9.0, 9.0) 85.2 3.75 dd (8.5, 3.0) 79.9 3.47 t (9.2) 75.8 3.66 dd (8.5, 3.0) 74.8 3.80 dd (8.5, 3.0)4 79.4 3.55 dd (9.0, 9.0) 70.1 3.99 m 71.6 3.48 m 70.5 3.77 m 77.5 4.57 m5 76.6 3.36 ddd (9.0, 4.5, 2.0) 66.6 3.88 dd (11.9, 2.0)
3.53 dd (11.9, 3.0)67.1 3.86 dd (11.7, 5.2)
3.18 t (11.7)67.1 3.88 dd (11.9, 2.0)
3.52 dd (11.9, 3.0)65.0 4.24 dd (11.9, 2.0)
3.59 dd (11.9, 3.0)6 61.6 3.83 dd (12.0, 2.0)
3.69 dd (12.0, 4.5)
a-L-Rha a-L-Rha a-L-Rha a-L-Rha a-L-Rha1 102.4 4.88 d (1.2) 102.4 5.34 d (1.2) 101.2 5.38 d (1.2) 101.5 5.33 d (1.2) 100.6 5.31 d (1.2)2 72.0 3.86 dd (1.2, 3.2) 72.0 3.93 dd (1.2, 3.2) 72.3 3.91 dd (1.2, 3.2) 72.1 3.91 dd (1.2, 3.2) 72.0 3.92 dd (1.2, 3.2)3 71.9 3.66 dd (3.2, 9.3) 71.9 3.71 dd (3.2, 9.3) 71.9 3.75 dd (3.2, 9.3) 71.9 3.72 dd (3.2, 9.3) 71.6 3.74 dd (3.2, 9.3)4 73.6 3.43 t (9.3) 73.6 3.42 t (9.3) 74.1 3.41 t (9.3) 73.8 3.43 t (9.3) 73.7 3.41 t (9.3)5 70.3 4.00 m 70.3 4.10 m 69.5 4.12 m 69.4 4.11 m 69.1 4.10 m6 17.6 1.30 d (6.0) 17.6 1.28 d (6.0) 18.2 1.28 d (6.0) 18.1 1.28 d (6.0) 18.2 1.28 d (6.0)
b-D-GlcII b-D-Xyl b-D-Glc b-D-Glc b-D-Glc1 103.0 4.30 d (7.5) 106.1 4.44 d (7.5) 106.6 4.21 d (7.5) 103.1 4.31 d (7.5) 103.3 4.30 d (7.5)2 75.0 3.21 dd (9.0, 7.5) 74.6 3.31 dd (9.2, 7.5) 75.3 3.21 dd (9.0, 7.5) 74.9 3.21 dd (9.0, 7.5) 74.9 3.22 dd (9.0, 7.5)3 77.9 3.37 dd (9.0, 9.0) 77.8 3.34 t (9.2) 78.3 3.37 dd (9.0, 9.0) 77.8 3.37 dd (9.0, 9.0) 78.0 3.37 dd (9.0, 9.0)4 71.3 3.31 dd (9.0, 9.0) 70.8 3.54 m 71.6 3.33 dd (9.0, 9.0) 71.3 3.31 dd (9.0, 9.0) 71.2 3.32 dd (9.0, 9.0)5 77.8 3.29 ddd (9.0, 4.5, 2.0) 66.6 3.88 dd (11.7, 5.2)
3.24 t (11.7)77.8 3.28 ddd (9.0, 4.5, 2.0) 77.8 3.29 ddd (9.0, 4.5, 2.0) 77.7 3.29 ddd (9.0, 4.5, 2.0)
6 62.6 3.90 dd (12.0, 2.0)3.69 dd (12.0, 4.5)
62.6 3.89 dd (12.0, 2.0)3.71 dd (12.0, 4.5)
62.6 3.89 dd (12.0, 2.0)3.70 dd (12.0, 4.5)
62.4 3.89 dd (12.0, 2.0)3.70 dd (12.0, 4.5)
b-D-Glc1 103.0 4.30 d (7.5)2 74.9 3.21 dd (9.0, 7.5)3 77.9 3.38 dd (9.0, 9.0)4 71.2 3.31 dd (9.0, 9.0)5 77.9 3.27 ddd (9.0, 4.5, 2.0)6 62.5 3.90 dd (12.0, 2.0)
3.70 dd (12.0, 4.5)
a The chemical shift values of the sugar portions of 2–4, 8–9, 14 and 19 deviate from the experimental values of 1, 7 and 18, respectively, of ±0.03 ppm.b The chemical shift values of the sugar portions of 12–13, 20 and 22 deviate from the experimental values of 21 of ±0.03 ppm.
A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088 2085

Author's personal copy
(1H, m, H-16), 4.37 (1H, d, J = 12.1 Hz, H-26a), 4.15 (1H, d,J = 12.1 Hz, H-26b), 3.62 (1H, m, H-3), 3.35 (1H, dd, J = 11.9,3.9 Hz, H-1), 3.19 (3H, s, OMe), 1.08 (3H, s, Me-19), 1.04 (3H, d,J = 6.6 Hz, Me-21), 0.88 (3H, s, Me-18); for 13C NMR data of theaglycon moiety, see Table 2; for 13C and 1H NMR data of the sugarportion, see Table 3; HRMALDITOFMS [M+Na]+ m/z 941.4728 (calc.for C45H74O19Na, 941.4718).
3.7. (25R)-26-O-b-D-glucopyranosyl-22a-methoxy-furost-5-ene-1b,3b,26-triol 3-O-[a-L-rhamnopyranosyl-(1 ? 4)-O-b-D-glucopyranoside] (4)
Amorphous white solid; C46H76O19; ½a�22D �65.3� (c 0.1 MeOH);
IR mKBrmax cm�1: 3485 (>OH), 2933 (>CH), 1265 and 1065 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.61 (1H, br d,J = 5.7 Hz, H-6), 4.40 (1H, m, H-16), 3.83 (1H, m, H-26a), 3.61 (1H,m, H-3), 3.36 (1H, dd, J = 11.9, 3.9 Hz, H-1), 3.38 (1H, m, H-26b),3.19 (3H, s, OMe), 1.07 (3H, s, Me-19), 1.04 (3H, d, J = 6.6 Hz, Me-21), 0.98 (3H, d, J = 6.6 Hz, Me-27), 0.88 (3H, s, Me-18); for 13CNMR data of the aglycon moiety, see Table 2; for 13C and 1HNMR data of the sugar portion, see Table 3; HRMALDITOFMS[M+Na]+ m/z 941.4883 (calc. for C45H74O19Na, 941.4874).
3.8. 26-O-b-D-glucopyranosyl-furosta-5,25(27)-diene-1b,3b,22a,26-tetrol 1-O-{a-L-rhamnopyranosyl-(1 ? 2)-O-[b-D-xylopyranosyl-(1 ? 3)]-a-L-arabinopyranoside} (7)
Amorphous white solid; C49H78O22; ½a�22D �40.5� (c 0.1 MeOH);
IR mKBrmax cm�1: 3489 (>OH), 2936 (>CH), 1270 and 1060 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.59 (1H, br d,J = 5.7 Hz, H-6), 5.12 (1H, br s, H-27a), 4.96 (1H, br s, H-27b), 4.40(1H, m, H-16), 4.37 (1H, d, J = 12.1 Hz, H-26a), 4.15 (1H, d,J = 12.1 Hz, H-26b), 3.40 (1H, dd, J = 11.9, 3.9 Hz, H-1), 3.37 (1H,m, H-3), 1.13 (3H, s, Me-19), 1.04 (3H, d, J = 6.6 Hz, Me-21), 0.88(3H, s, Me-18); for 13C NMR data of the aglycon moiety, see Table2; for 13C and 1H NMR data of the sugar portion, see Table 3;HRMALDITOFMS [M+Na]+ m/z 1041.4890 (calc. for C45H74O19Na,1041.4883).
3.9. (25R)-26-O-b-D-glucopyranosyl-furost-5-ene-1b,3b,22a,26-tetrol1-O-{a-L-rhamnopyranosyl-(1 ? 2)-O-[b-D-xylopyranosyl-(1 ? 3)]-a-L-arabinopyranoside} (8)
Amorphous white solid; C49H80O22; ½a�22D �58.3� (c 0.1 MeOH);
IR mKBrmax cm�1: 3477 (>OH), 2930 (>CH), 1265 and 1066 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.60 (1H, br d,J = 5.7 Hz, H-6), 4.40 (1H, m, H-16), 3.83 (1H, m, H-26a), 3.41 (1H,dd, J = 11.9, 3.9 Hz, H-1), 3.40 (1H, m, H-26b), 3.37 (1H, m, H-3),1.13 (3H, s, Me-19), 1.04 (3H, d, J = 6.6 Hz, Me-21), 0.99 (3H, d,J = 6.6 Hz, Me-27), 0.87 (3H, s, Me-18); for 13C NMR data of theaglycon moiety, see Table 2; for 13C and 1H NMR data of the sugarportion, see Table 3; HRMALDITOFMS [M+Na]+ m/z 1043.5048(calc. for C45H74O19Na, 1043.5040).
3.10. (25R)-26-O-b-D-glucopyranosyl-22a-methoxy-furost-5-ene-1b,3b,26-triol 1-O-{a-L-rhamnopyranosyl-(1 ? 2)-O-[b-D-xylopyranosyl-(1 ? 3)]-a-L-arabinopyranoside} (9)
Amorphous white solid; C50H82O22; ½a�22D �47.2� (c 0.1 MeOH);
IR mKBrmax cm�1: 3480 (>OH), 2935 (>CH), 1260 and 1072 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.61 (1H, br d,J = 5.7 Hz, H-6), 4.40 (1H, m, H-16), 3.83 (1H, m, H-26a), 3.40 (1H,dd, J = 11.9, 3.9 Hz, H-1), 3.38 (1H, m, H-26b), 3.37 (1H, m, H-3),3.18 (3H, s, OMe), 1.14 (3H, s, Me-19), 1.04 (3H, d, J = 6.6 Hz, Me-21), 0.98 (3H, d, J = 6.6 Hz, Me-27), 0.88 (3H, s, Me-18); for 13CNMR data of the aglycon moiety, see Table 2; for 13C and 1H
NMR data of the sugar portion, see Table 3; HRMALDITOFMS[M+Na]+ m/z 1057.5202 (calc. for C45H74O19Na, 1057.5196).
3.11. 26-O-b-D-glucopyranosyl-furosta-5,25(27)-diene-1b,3b,22a,26-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-4-sulfo-a-L-arabinopyranoside] (12)
Amorphous white solid; C44H70O21S; ½a�22D �42.8� (c 0.1 MeOH);
IR mKBrmax cm�1: 3484 (>OH), 2940 (>CH), 1273 and 1058 (C–O–C); 1H
NMR (CD3OD, 600 MHz) aglycon moiety d 5.60 (1H, br d, J = 5.7 Hz,H-6), 5.12 (1H, br s, H-27a), 4.98 (1H, br s, H-27b), 4.41 (1H, m, H-16), 4.37 (1H, d, J = 12.1 Hz, H-26a), 4.16 (1H, d, J = 12.1 Hz, H-26b),3.40 (1H, dd, J = 11.9, 3.9 Hz, H-1), 3.37 (1H, m, H-3), 1.13 (3H, s,Me-19), 1.05 (3H, d, J = 6.6 Hz, Me-21), 0.88 (3H, s, Me-18); for13C NMR data of the aglycon moiety, see Table 2; for 13C and 1HNMR data of the sugar portion, see Table 3; HRMALDITOFMS[M+Na]+ m/z 989.4038 (calc. for C45H74O19Na, 989.4029).
3.12. (25R)-26-O-b-D-glucopyranosyl-furost-5-ene-1b,3b,22a,26-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-4-sulfo-a-L-arabinopyranoside] (13)
Amorphous white solid; C44H72O21S; ½a�22D �53.4� (c 0.1 MeOH);
IR mKBrmax cm�1: 3482 (>OH), 2936 (>CH), 1270 and 1068 (C–O–C); 1H
NMR (CD3OD, 600 MHz) aglycon moiety d 5.59 (1H, br d, J = 5.7 Hz,H-6), 4.40 (1H, m, H-16), 3.83 (1H, m, H-26a), 3.40 (1H, dd, J = 11.9,3.9 Hz, H-1), 3.38 (1H, m, H-26b), 3.37 (1H, m, H-3), 1.13 (3H, s, Me-19), 1.05 (3H, d, J = 6.6 Hz, Me-21), 0.98 (3H, d, J = 6.6 Hz, Me-27),0.87 (3H, s, Me-18); for 13C NMR data of the aglycon moiety, see Ta-ble 2; for 13C and 1H NMR data of the sugar portion, see Table 3;HRMALDITOFMS [M+Na]+ m/z 991.4192 (calc. for C45H74O19Na,991.4185).
3.13. (24S)-24-O-b-D-glucopyranosyl-spirosta-5,25(27)-diene-1b,3b,24-triol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-a-L-arabinopyranoside] (14)
Amorphous white solid; C44H68O18; ½a�22D �51.0� (c 0.1 MeOH);
IR mKBrmax cm�1: 3481 (>OH), 2944 (>CH), 1272 and 1065 (C–O–C);
1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.59 (1H, br d,J = 5.7 Hz, H-6), 5.18 (1H, br s, H-27a), 4.98 (1H, br s, H-27b), 4.48(1H, m, H-16), 4.26 (1H, dd, J = 11.7, 5.7 Hz, H-1), 4.24 (1H, d,J = 12.1 Hz, H-26a), 3.89 (1H, d, J = 12.1 Hz, H-26b), 3.40 (1H, dd,J = 11.9, 3.9 Hz, H-1), 3.37 (1H, m, H-3), 1.13 (3H, s, Me-19), 0.96(3H, d, J = 6.6 Hz, Me-21), 0.93 (3H, s, Me-18); for 13C NMR dataof the aglycon moiety, see Table 2; for 13C and 1H NMR data ofthe sugar portion, see Table 3; HRMALDITOFMS [M+Na]+ m/z885.4493 (calc. for C45H74O19Na, 885.4485).
3.14. (22S)-16-O-b-D-glucopyranosyl-3-sulfo-colest-5-ene-1b,3b,16b,22-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-b-D-xylopyranoside] (15)
Amorphous white solid; C44H74O20S; ½a�22D �60.2� (c 0.1 MeOH);
IR mKBrmax cm�1: 3481 (>OH), 2944 (>CH), 1275 and 1064 (C–O–C); 1H
NMR (CD3OD, 600 MHz) aglycon moiety d 5.64 (1H, br d, J = 5.7 Hz,H-6), 4.21 (1H, m, H-16), 4.09 (1H, m, H-3), 3.74 (1H, m), 3.44 (1H,dd, J = 11.9, 3.9 Hz, H-1), 1.13 (3H, s, Me-19), 0.95 (3H, d, J = 6.6 Hz,Me-21), 0.95 (3H, s, Me-18), 0.93 (3H � 2, d, J = 6.6 Hz, Me-26, Me-27); for 13C NMR data of the aglycon moiety, see Table 2; for 13Cand 1H NMR data of the sugar portion, see Table 3; HRMALDI-TOFMS [M+H]+ m/z 955.4582 (calc. for C45H74O19Na, 955.4573).
2086 A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088

Author's personal copy
3.15. 26-O-b-D-glucopyranosyl-3-sulfo-furosta-5,25(27)-diene-1b,3b,22a,26-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-a-L-arabinopyranoside] (18)
Amorphous white solid; C44H70O21S; ½a�22D �44.6� (c 0.1 MeOH);
IR mKBrmax cm�1: 3478 (>OH), 2931 (>CH), 1265 and 1069 (C–O–C); 1H
NMR (CD3OD, 600 MHz) aglycon moiety d 5.65 (1H, br d, J = 5.7 Hz,H-6), 5.12 (1H, br s, H-27a), 4.97 (1H, br s, H-27b), 4.41 (1H, m, H-16), 4.37 (1H, d, J = 12.1 Hz, H-26a), 4.09 (1H, m, H-3), 4.15 (1H, d,J = 12.1 Hz, H-26b), 3.45 (1H, dd, J = 11.9, 3.9 Hz, H-1), 1.13 (3H, s,Me-19), 1.05 (3H, d, J = 6.6 Hz, Me-21), 0.89 (3H, s, Me-18); for13C NMR data of the aglycon moiety, see Table 2; for 13C and 1HNMR data of the sugar portion, see Table 3; HRMALDITOFMS[M+Na]+ m/z 989.4038 (calc. for C45H74O19Na, 989.4029).
3.16. (25R)-26-O-b-D-glucopyranosyl-3-sulfo-furost-5-ene-1b,3b,22a,26-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-a-L-arabinopyranoside] (19)
Amorphous white solid; C44H72O21S; ½a�22D �54.9� (c 0.1 MeOH);
IR mKBrmax cm�1: 3481 (>OH), 2936 (>CH), 1271 and 1068 (C–O–C); 1H
NMR (CD3OD, 600 MHz) aglycon moiety d 5.64 (1H, br d, J = 5.7 Hz,H-6), 4.40 (1H, m, H-16), 4.09 (1H, m, H-3), 3.85 (1H, m, H-26a),3.44 (1H, dd, J = 11.9, 3.9 Hz, H-1), 3.36 (1H, m, H-26b), 1.14 (3H,s, Me-19), 1.05 (3H, d, J = 6.6 Hz, Me-21), 0.98 (3H, d, J = 6.6 Hz,Me-27), 0.89 (3H, s, Me-18); for 13C NMR data of the aglycon moi-ety, see Table 2; for 13C and 1H NMR data of the sugar portion, seeTable 3; HRMALDITOFMS [M+Na]+ m/z 991.4194 (calc. forC45H74O19Na, 991.4185).
3.17. (25R)-26-O-b-D-glucopyranosyl-3-sulfo-furost-5-ene-1b,3b,22a,26-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-4-sulfo-a-L-arabinopyranoside] (20)
Amorphous white solid; C44H72O24S2; ½a�22D �59.0� (c 0.1
MeOH); IR mKBrmax cm�1: 3478 (>OH), 2935 (>CH), 1265 and 1063
(C–O–C); 1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.65 (1H,br d, J = 5.7 Hz, H-6), 4.41 (1H, m, H-16), 4.09 (1H, m, H-3), 3.83(1H, m, H-26a), 3.45 (1H, dd, J = 11.9, 3.9 Hz, H-1), 3.40 (1H, m,H-26b), 1.13 (3H, s, Me-19), 1.05 (3H, d, J = 6.6 Hz, Me-21), 0.98(3H, d, J = 6.6 Hz, Me-27), 0.87 (3H, s, Me-18); for 13C NMR dataof the aglycon moiety, see Table 2; for 13C and 1H NMR data ofthe sugar portion, see Table 3; HRMALDITOFMS [M+Na]+ m/z1071.3763 (calc. for C45H74O19Na, 1071.3754).
3.18. 26-O-b-D-glucopyranosyl-3-sulfo-furosta-5,25(27)-diene-1b,3b,22a,26-tetrol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-4-sulfo-a-L-arabinopyranoside] (21)
Amorphous white solid; C44H70O24S2; ½a�22D �46.1� (c 0.1
MeOH); IR mKBrmax cm�1: 3480 (>OH), 2934 (>CH), 1268 and 1060
(C–O–C); 1H NMR (CD3OD, 600 MHz) aglycon moiety d 5.65 (1H,br d, J = 5.7 Hz, H-6), 5.12 (1H, br s, H-27a), 4.97 (1H, br s, H-27b),4.40 (1H, m, H-16), 4.37 (1H, d, J = 12.1 Hz, H-26a), 4.09 (1H, m,H-3), 4.15 (1H, d, J = 12.1 Hz, H-26b), 3.45 (1H, dd, J = 11.9,3.9 Hz, H-1), 1.13 (3H, s, Me-19), 1.05 (3H, d, J = 6.6 Hz, Me-21),0.89 (3H, s, Me-18); for 13C NMR data of the aglycon moiety, see Ta-ble 2; for 13C and 1H NMR data of the sugar portion, see Table 3;HRMALDITOFMS [M+Na]+ m/z 1069.3605 (calc. for C45H74O19Na,1069.3597).
3.19. 3-Sulfo-spirosta-5,25(27)-diene-1b,3b-diol 1-O-[a-L-rhamnopyranosyl-(1 ? 2)-O-4-sulfo-a-L-arabinopyranoside] (22)
Amorphous white solid; C38H58O18S2; ½a�22D �67.1� (c 0.1 MeOH)
IR mKBrmax cm�1: 3484 (>OH), 2940 (>CH), 1277 and 1067 (C–O–C); 1H
NMR (CD3OD, 600 MHz) aglycon moiety d 5.65 (1H, br d, J = 5.7 Hz,H-6), 4.80 (1H, br s, H-27a), 4.77 (1H, br s, H-27b), 4.48 (1H, m, H-16), 4.30 (1H, d, J = 12.1 Hz, H-26a), 4.09 (1H, m, H-3), 3.85 (1H, d,J = 12.1 Hz, H-26b), 3.44 (1H, dd, J = 11.9, 3.9 Hz, H-1), 1.14 (3H, s,Me-19), 0.99 (3H, d, J = 6.6 Hz, Me-21), 0.86 (3H, s, Me-18); for13C NMR data of the aglycon moiety, see Table 2; for 13C and 1HNMR data of the sugar portion, see Table 3; HRMALDITOFMS[M+H]+ m/z 867.3151 (calc. for C45H74O19Na, 867.3144).
3.20. Acid hydrolysis
The crude saponin mixture (1 g) was heated at 60 �C with 1:10.5 N HCl-dioxane (100 ml) for 2 h, and the mixture was thenevaporated in vacuo. The residue was partitioned with CH2Cl2–H2O, and the H2O layer was neutralized with Amberlite MB-3.The H2O layer was then concentrated and passed through a silicagel column, using CHCl3–MeOH–H2O (7:1:1.2, lower layer) aseluting solvent to afford glucose, xylose, arabinose and rhamnose.The D configuration of xylose and glucose and the L configurationof arabinose and rhamnose were established as by comparison oftheir optical rotation values with those reported in the literature(Eskander et al., 2006). The optical rotations were determinedafter dissolving the sugars in H2O and allowing them tostand for 24 h; D-xylose ½a�22
D +10 (c 0.1), L-arabinose ½a�22D +98.7
(c 0.1), D-glucose ½a�22D +54.5 (c 0.1), L-rhamnose ½a�22
D +11.8(c 0.1).
3.21. Detection of the sulfate group
Aliquot (1–2 mg) of compounds 12–13, 15, and 18–22 wasrefluxed with 10% HCl (4 ml) for 4 h and then extracted withEt2O. An aliquot of the aqueous layer of each was treated with70% BaCl2 to give a white precipitate (BaSO4) (Akai et al., 1985).
References
Agrawal, P.K., 2004. NMR spectral investigations, part 51. Dependence of 1H NMRchemical shifts of geminal protons of glycosyloxy methylene (H2-26) on theorientation of the 27-methyl group of furostane-type steroidal saponins. Magn.Reson. Chem. 42, 990–993.
Akai, E., Takeda, T., Kobayashi, Y., Ogihara, Y., 1985. Sulfated triterpenoid saponinsfrom the leaves of Bupleurum rotundifolium L. Chem. Pharm. Bull. 33, 3715–3723.
Bassarello, C., Muzashvili, T., Skhirtladze, A., Kemertelidze, E., Pizza, C., Piacente, S.,2008. Steroidal glycosides from the underground parts of Helleborus caucasicus.Phytochemistry 69, 1227–1233.
Bombardelli, E., Bonati, A., Gabetta, B., Mustich, G., 1971. Glycosides from rhizomesof Ruscus aculeatus. Fitoterapia 42, 127–136.
Bombardelli, E., Bonati, A., Gabetta, B., Mustich, G., 1972. Glycosides from rhizomesof Ruscus aculeatus. II. Fitoterapia 43, 3–10.
Bouskela, E., Cyrino, F.Z.G.A., Marcelon, G., 1993a. Inhibitory effect of the Ruscusextract and of the flavonoid hesperidin methylchalcone on increasedmicrovascular permeability induced by various agents in the hamster cheekpouch. J. Cardiovasc. Pharmacol. 22, 225–230.
Bouskela, E., Cyrino, F.Z.G.A., Marcelon, G., 1993b. Effects of Ruscus extract on theinternal diameter of arterioles and venules of the hamster cheek pouchmicrocirculation. J. Cardiovasc. Pharmacol. 22, 221–224.
Cui, M., Song, F., Zhou, Y., Liu, Z., Liu, S., 2000. Rapid identification of saponins inplant extracts by electrospray ionization multi-stage tandem massspectrometry and liquid chromatography/tandem mass spectrometry. RapidCommun. Mass Spectrom. 14, 1280–1286.
de Combarieu, E., Falzoni, M., Fuzzati, N., Gattesco, F., Giori, A., Lovati, M., Pace, R.,2002. Identification of Ruscus steroidal saponins by HPLC–MS analysis.Fitoterapia 73, 583–596.
de Combarieu, E., Fuzzati, N., Lovati, M., Mercalli, E., 2003. Furostanol saponins fromTribulus terrestris. Fitoterapia 74, 583–591.
Eskander, J., Lavaud, C., Pouny, I., Soliman, H.S.M., Abdel-Khalik, S.M., Mahmoud, I.I.,2006. Saponins from the seeds of Mimusops laurifolia. Phytochemistry 67, 1793–1799.
Fuzzati, N., Pace, R., Papeo, G., Peterlongo, F., 1997. Identification of soyasaponins byliquid chromatography–thermospray mass spectrometry. J. Chromatogr. A 777,233–238.
Geng, P., Zhang, R., Aisa, H.A., He, J., Qu, K., Zhu, H., Abliz, Z., 2007. Fast profiling ofthe integral metabolism of flavonols in the active fraction of Gossypium
A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088 2087

Author's personal copy
herbaceam L. using liquid chromatography/multi-stage tandem massspectrometry. Rapid Commun. Mass Spectrom. 21, 1877–1888.
Huang, X., Song, F., Liu, Z., Liu, S., 2007. Studies on lignan constituents fromSchisandra chinensis (Turcz.) Baill. fruits using high-performance liquidchromatography/electrospray ionization multiple-stage tandem massspectrometry. J. Mass Spectrom. 42, 1148–1161.
Kartnig, T., Bucar, F., Wagner, H., Seligmann, O., 1991. Flavonoids from the aerialparts of Ruscus aculeatus. Planta Med. 57, 85.
Khodakov, G.V., Akimov, Y.A., Shashkov, A.S., Kintia, P.K., Grishkovets, V.I., 1996.Triterpene and steroid saponins isolated from two Melilotus species. Adv. Exp.Med. Biol. 405, 211–222.
Kite, G.C., Howes, M.J.R., Leon, C.J., Simmonds, M.S.J., 2003. Liquid chromatography/mass spectrometry of malonyl-ginsenosides in the authentication of ginseng.Rapid Commun. Mass Spectrom. 17, 238–244.
Kite, G.C., Porter, E.A., Simmonds, M.S.J., 2007. Chromatographic behaviour ofsteroidal saponins studied by high-performance liquid chromatography–massspectrometry. J. Chromatogr. A 1148, 177–183.
Korkashvili, T.Sh., Kikoladze, V.S., 1991. Carbon-13 NMR analysis of furostanolglycosides from Ruscus ponticus. Khim. Prir. Soedin. 3, 435–436.
Le Tran, Q., Tezuka, Y., Banskota, A.H., Tran, Q.K., Saiki, I., Kadota, S., 2001. Newspirostanol steroids and steroidal saponins from roots and rhizomes ofDracaena angustifolia and their antiproliferative activity. J. Nat. Prod. 64,1127–1132.
Li, B., Abliz, Z., Tang, M., Fu, G., Yu, S., 2006. Rapid structural characterization oftriterpenoid saponins in crude extract from Symplocos chinensis using liquidchromatography combined with electrospray ionization tandem massspectrometry. J. Chromatogr. A 1101, 53–62.
Liu, H., Huang, Y., Wang, Q., Zhang, T., Song, Y., 2006. Detection of saponins inextracts from the rhizomes of Paris species and prepared Chinese medicines byhigh performance liquid chromatography–electrospray ionization massspectrometry. Planta Med. 72, 835–841.
Mimaki, Y., Takaashi, Y., Kuroda, M., Sashida, Y., Nikaido, T., 1996. Steroidal saponinsfrom Nolina recurvata stems and their inhibitory activity on cyclic AMPphosphodiesterase. Phytochemistry 42, 1609–1615.
Mimaki, Y., Kuroda, M., Kameyama, A., Yokosuka, A., Sashida, Y., 1998a. Newsteroidal constituents of the underground parts of Ruscus aculeatus and theircytostatic activity on HL-60 cells. Chem. Pharm. Bull. 46, 298–303.
Mimaki, Y., Kuroda, M., Kameyama, A., Yokosuka, A., Sashida, Y., 1998b. Steroidalsaponins from the underground parts of Ruscus aculeatus and their cytostaticactivity on HL-60 cells. Phytochemistry 48, 485–493.
Mimaki, Y., Kuroda, M., Yokosuka, A., Sashida, Y., 1998c. Two new bisdesmosidicsteroidal saponins from the underground parts of Ruscus aculeatus. Chem.Pharm. Bull. 46, 879–881.
Mimaki, Y., Kuroda, M., Kameyama, A., Yokosuka, A., Sashida, Y., 1998d. AculeosideB, a new bisdesmosidic spirostanol saponin from the underground parts ofRuscus aculeatus. J. Nat. Prod. 61, 1279–1282.
Mimaki, Y., Kuroda, M., Yokosuka, A., Sashida, Y., 1999a. A spirostanol saponin fromthe underground parts of Ruscus aculeatus. Phytochemistry 51, 689–692.
Mimaki, Y., Kuroda, M., Obata, Y., Sashida, Y., 1999b. Steroidal glycosides from therhizomes of Ruscus hypoglossum. Nat. Med. 53, 266–270.
Mimaki, Y., Kuroda, M., Ide, A., Kameyama, A., Yokosuka, A., Sashida, Y., 1999c.Steroidal saponins from the aerial parts of Dracaena draco and their cytostaticactivity on HL-60 cells. Phytochemistry 50, 805–813.
Mimaki, Y., Aoki, T., Jitsuno, M., Kiliç, C.S., Cos�kun, M., 2008a. Steroidal glycosidesfrom the rhizomes of Ruscus hypophyllum. Phytochemistry 69, 729–737.
Mimaki, Y., Aoki, T., Jitsuno, M., Yokosuka, A., Kiliç, C.S., Cos�kun, M., 2008b. Steroidalsaponins from the rhizomes of Ruscus hypophyllum. Nat. Prod. Commun. 3,1671–1678.
Oleszek, W.A., 2002. Chromatographic determination of plant saponins. J.Chromatogr. A 967, 147–162.
Oleszek, W., Bialy, Z., 2006. Chromatographic determination of plant saponins – anupdate (2002–2005). J. Chromatogr. A 1112, 78–91.
Panova, D., Nikolov, S., Akhtardzhiev, K., 1974. Steroid sapogenins and sterols fromRuscus hypoglossum. Planta Med. 26, 90–93.
Pkheidze, T.A., Kereselidze, E.V., Kemertelidze, E.P., 1971. Diosgenin, neoruscogenin,and ruscogenin from Ruscus ponticus, Ruscus hypophyllum, and Allium albidum.Khim. Prir. Soedin. 7, 841–842.
Skhirtladze, A., Plaza, A., Montoro, P., Benidze, M., Kemertelidze, E., Pizza, C.,Piacente, S., 2006. Furostanol saponins from Yucca gloriosa L. Rhizomes.Biochem. Syst. Ecol. 34, 809–814.
Takaashi, Y., Mimaki, Y., Kameyama, A., Kuroda, M., Sashida, Y., Nikaido, T., Koike, K.,Ohmoto, T., 1995. Three new cholestane bisdesmosides from Nolina recurvatastems and their inhibitory activity on cAMP phosphodiesterase and Na+/K+
ATPase. Chem. Pharm. Bull. 43, 1180–1185.Wang, M.K., Liang, C.-.P., Wu, Q.-.L., Simon, J.E., Ho, C.-T., 2006. Instrumental
analysis of popular botanical products in the US market. In: Wang, M., Sang, S.,Hwang, L.S., Ho, C.-.T. (Eds.), Herbs: Challenges in Chemistry and Biology.American Chemical Society, Washington, DC, pp. 39–54.
Wolfender, J.L., Ndjoko, K., Hostettmann, K., 2003. Liquid chromatography withultraviolet absorbance–mass spectrometric detection and with nuclearmagnetic resonance spectroscopy: a powerful combination fort he on-linestructural investigation of plant metabolites. J. Chromatogr. A 1000, 437–455.
Yuan, L., Ji, T.F., Wang, A.G., Yang, J.B., Su, Y.L., 2008. Two new furostanol saponinsfrom the seeds of Allium cepa L. Chin. Chem. Lett. 19, 461–464.
2088 A. Perrone et al. / Phytochemistry 70 (2009) 2078–2088




















