
Stereoselective functionalization of α-amino acids - Adelaide ...
205
\(.---l.93 I S tereo selective Functionalization of cr-Amino Acids A Thesis Submitted Towards the Degree of Doctor of Philosophy by Craig Anthony Hutton B.Sc. (Hons.) Department of Chemistry University of Adelaide April 1993 Awarde d lqq 3
-
Upload
khangminh22 -
Category
Documents
-
view
5 -
download
0
Transcript of Stereoselective functionalization of α-amino acids - Adelaide ...

\(.---l.93
I
S tereo selective Functionalizationof cr-Amino Acids
A Thesis
Submitted Towards the
Degree of
Doctor of Philosophy
by
Craig Anthony HuttonB.Sc. (Hons.)
Department of Chemistry
University of Adelaide
April 1993
Awarde d lqq 3

Contents
Acknowledgements
Statement
Publications
Abstract
Introduction
Results and Discussion
Chapter 1 Regiocontrolled Side Chain Bromination of
N-Phthaloyl Protected o-Amino Acid Derivatives
Chapter 2 Stereocontrolled Synthesis of Homochiral
Hydroxy- a-Amino Acid Derivatives
Chapter 3 Towards a Synthesis of Chloramphenicol
Chapter 4 Scope and Limitations of the Elaboration of
B romo- a-Amino Acid Derivatives
Chapter 5 Synthesis of Each Stereoisomer ofp-Deuteriophenylalanine
Chapter 6 Stereochemical Course of the reaction of
(S)- and (R)-Phenylalanine with
(S) -Phenylalanine Ammonia Lyase
Conclusion
Experimental
References
i
ü
iiiiv
105
IT2
115
166
1
27
45
72
78
94
Appendices 104lo+

I
ACKNOWLEDGEMENTS
Many thanks go to Dr. Chris Easton for his guidance over the past few
years. I consider myself fortunate to have had a supervisor who provided
continual optimism, inspiration, enthusiasm and support, as well as sincere
friendship.
I would like to thank the staff and students of my department for providing
an enjoyable working environment. Particular thanks are extended to the
members of my research group and the members of lab 3, Kathy, Jeff and
Steve, for their inspiring discussions. Thanks also to Caroline Ward and
Darren Schliebs for proof reading parts of this thesis.
Special thanks go to Pete Roselt, "Barney" Miller and Eng Wui Tan for
their insights and friendship"
I would also tike to thank my family for their encouragement throughout
the years of my education. Last but not least, thanks go to Kathy for keeping
me sane, especially while I was writing this thesis.

l1
STATEMENT
This work contains no material which has been accepted for the award of
any other degree or diploma in any university or other tertiary institution, and,
to the best of my knowledge and belief, contains no material previously
published or written by another person, except where due reference has been
made in the text.
I give consent to this copy of my thesis, when deposited in the University
Library, being available for loan or photocopying.
Craig Hutton (B.Sc. Hons.) April 14 1993

u1
PUBLICATIONS
Some of the work described in this thesis has been reported in the
following publications:
"Synthesis of Homochiral Hydroxy-a-Amino Acid Derivatives", Easton, C.
J., Hutton, C. 4., Tan, E. W., and Tiekink, E. R. T., Tetrahedron Lett., 1990,
31, 7059.
"Regioselective Functionalization of N-Phthaloyl-Substituted Amino Acid
and Peptide Derivatives", Easton, C. J., Hutton, C. 4., Rositano, G., and Tan,
E. W., J. Org. Chem.,1991,56,5614"
"Synthesis and Molecular Structure of Stable Derivatives of (E)- and
(Z)-Dehydrophenylalanine", Easton, C. J., Hutton, C. 4., Roselt, P. D., and
Tiekink, E. R. T., Aust. J. Chem.,I99I,44,681.
"Crystal Structure of N-Phthaloyl-B-Phenylserine Methyl Ester", Easton,
C. J., Hutton, C.4., and Tiekink, E. R. T.,Z.Krist.,1993,203,310.

lv
ABSTRACT
The cl-position of N-phthaloyl-cr-amino acid derivatives has been shown to
be deactivated towards hydrogen atom abstraction, such that bromination
occurs regiospecifically via the most stable side chain radical.
N-Phthaloyl derivatives of B-bromovaline, y-bromoleucine and
y-bromohomophenylalanine have been converted to the conesponding hydroxy
amino acid derivatives. B-Bromo-phenylalanine and tyrosine derivatives
convert stereoselectively to (25)-threo-þ-hydroxy-phenylalanine and tyrosine
derivatives. It has been found that the stereoslectivity is much greater when the
carboxyl group is protected as an amide rather than as an ester, which is
attributable to the amide substituent providing greater intramolecular
stabilization of intermediate carbocations. Deprotection of B-hydroxy-
phenylalanine derivatives yietds the free amino acid ( 2 S 3 R ) -
B-hydroxyphenylalanine without loss of chiral integrity. A B-hydroxy-
p-nitrophenylalanine derivative which is a potential precursor to
chloramphenicol has been synthesized.
lH N.m.r. spectroscopic correlations have been ascertained which allow the
determination of the relative stereochemistry of B-bromo- and B-hydroxy-
phenylalanine and tyrosine derivatives.
B-Bromophenylalanine derivatives have been converted to 3,4-disubstituted
p-lactams and B-lactones with control of both relative and absolute
stereochemistry. Elaboration of 1-bromo-leucine and homophenylalanine
derivatives has enabled the introduction of functionality at the B- and
ô-positions of leucine derivatives and at the B-position of homophenylalanine
derivatives. B-Bromophenylalanine derivatives have also been converted to
deuteiurn labetleC phenylalanine deri.,,atives. These labelled compounds have
been deprotected to provide each of the stereoisomets of
B-deuteri-qphenylalanine with ca.987o d.e. and 997o 2[rincorporation. The

v
B-deuteriophenylalanines have been used to determine the stereochemical
course of the reactions of (S)- and (R)-phenylalanine with the enzyme
(S)-phenylalanine ammonia lyase. The enzyme catalyzes the stereospecific
removal of the pro-S hydrogen from (S)-phenylalanine, whereas the pro-R
hydrogen is preferentially abstracted from the unnatural substrate,
(R)-phenylalanine.

Introduction 1
INTRODUCTION
cx,-Amino acids (1) occur in nature as the monomeric building blocks of
peptides and proteins, and in many other species.l Although only twenty amino
acids occur commonly in proteins there are currently more than five hundred
known naturally occurring amino acids.2'3 Biosynthesis of many of the non-
proteinogenic amino acids is presumed to proceed viø functionalization of the
side chains of proteinogenic amino acids.4
H¡N
RH(1)
In addition to the fundamental biochemical and physiological significance
of amino acids, both natural and unnatural amino acids have found importance
in the pharmaceutical and agricultural industries,5,6 aS flavours,7 taste
enhancers and sweeteners.S In addition to their industrial uses, major advances
in the understanding of enzyme mechanisms,g protein conformalis¡5lo-l2 ¿n¿
regulatory interactions of peptidesl3 have placed the study of amino acid
chemistry at the forefront of chemical research.l4 As a result of the wide
spectrum of applications of amino acids, their economic impact is quite
significant, with a multi-bitlion dollar indusfry existing for their production
either by extraction from natural sources or by chemical synthesis.
Given their widespread natural occurrence and physiological importance,
intense interest has been focussed on the synthesis and subsequent studies of
cx,-amino acids. Many uncommon amino acids are unavailable from natural
sources in sufficient quantities for thorough structural and biochemical testing,
and it is often only through synthesis of these physiologically important
compoun$s that it becomes possible to confirm their structure and function.
+

Introduction 2
Biochemical tabelling studies,15,16 development of enzyme inhibitorslT-20 and
conformational restriction studiesl0-12 arc but a few of the areas where the
synthesis of rare and unnatural amino acids has provided insight into the
functioning of biological systems.
One approach to the synthesis of chiral unconunon cr-amino acids involves
the elaboration of proteinogenic a-amino acids through manipulation of pre-
existing side-chain functionality. This technique utilizes the important fact that
the proteinogenic cr-amino acids ¿ìre a remarkably cheap and readily available
source of enantiomerically pure starting materials. Examples of this approach
include modification of the hydroxyl moiety of serine,2t-24 and modification of
aspartic and glutamic acids vía eithq Barton decarboxylation procedures25,26
or anionic intermediates.2T Vederas and co-ws¡þsrc22-24 have detailed the
preparation of p-substituted amino acids vía ring opening reactions of
p-lactones derived from serine (Scheme 1). (S)-N-tert-Butoxycarbonylserine
(2a) and (S)-N-benzyloxycarbonylserine (2b) were converted to the
corresponding B-lactones (3a) and (3b) with dimethyl azidodicarboxylate
RNH lht,.
DMADNrnI' RNH
Nu
Nu
co2H
Nu----+PPh3
(2) (3) (4)
(a)R=Boc= Me3CO-CO-
(b) R = Cbz= PhCH2O-CO-
Nu = nucleophile +
(s)
cF3co2H
o HtNrr,". coi+
Scheme 1
o
(ó)
Nu

Introduction 3
(DMAD) and triphenylphosphine. Ring opening with various nucleophiles gave
the corresponding N-protected B-substituted alanine derivatives (4).
Alternatively, the B-lactone (3a) was deprotected by treatment with
ftifluoroacetic acid, with subsequent ring opening leading directly to the free
amino acids (6). This method was found to be useful for the synthesis of mono-
B-substituted amino acid derivatives, but not of p,p-disubstituted amino acid
derivatives.2S
Complementary to the modification of amino acid derivatives via the
manipulation of pre-existing functionality is the direct functionalization of the
side chains of proteinogenic amino acid derivatives. This concept not only
utilizes the inherent chirality of ø-amino acids but also the fact that many, if
not most of the naturally occurring uncommon ø-amino acids are derived from
the proteinogenic o-amino acids viq functionalization of the side chains.
Ohfune and Simamoto29 have developed a novel synthesis of (S)-threo-
B-hydroxy-O-methyltyrosine (10) by direct benzylic oxidation of the
K2S20s
CuSOa
MeO MeO0)
Ac
o
Ac
(8)
i) Ba(OH)2ü) Boc2O
+H3Nza,.. coi
.,rrrOH i)Ptoz/02
BocNHlht,.OH
.,rrtlOH
ü) cF3co2H
(e)MeO(10)
Scheme 2
MeO

Introduction 4
N-tert-butoxycarbonyltyrosinol derivative (7) (Scheme 2). Oxidation of the
tyrosinol derivative (7) with potassium persulfate-copper sulfate gave the cyclic
carbamate (8) with high threo-selectivity. Subsequent hydrolysis and
reprotection of the amino group gave the diol (9), with oxidation and removal
of the tert-butoxycarbonyl (Boc) protecting group giving (S)-threo-p.hydroxy-
O-methyltyrosine (10) in reasonable overall yield. However, this procedure is
rather limited by its lack of generality, with attempts to synthesize the
corresponding (S)-threo-isomer of p-hydroxyphenylalanine (25) yía an
analogous pathway being unsuccess ful.29
Direct functionalization of the side chains of amino acids via fuee radical
halogenation has also been reported, and has gained renewed interest in recent
years. This approach is exemplified by the y-chlorination and subsequent
elaboration of lysine (Scheme 3), as described by Kollonitsch and
co-workers.30-32 Ultraviolet irradiation of a concentrated hydrochloric acid
solution of lysine (11), with the concurrent introduction of chlorine gas, led to
the formation of y-chlorolysine (12). Subsequent elaboration of the chloride
(12) gave the hydroxylysine derivative (13).
+H¡N
cl2 / HCI
hv iÐ H*
co2H+
H¡N+
H¡N
-oHÐ
+ +NH3 NH3
(13)
Scheme 3
The regioselectivity of hydrogen atom abstraction reactions is determined
by several factors, including radical stability, steric effects and polar effects.33
(1 1) (12)

Introduction 5
Radical stability is important when there is extensive carbon-hydrogen bond
breaking, and consequently significant radical character, in the transition state.
Steric factors can influence regioselectivity by hindering the approach of the
hydrogen atom abstractor or by consfraining the conformation of the product
radical. Polar effects refer to activating/deactivating effects brought on by
partial charge development in the transition state of the hydrogen atom
abstraction.33'34 Polar effects were used to explain the regioselectivity
observed in the chlorination of lysine (11) described above" Hydrogen atom
abstraction by the electrophilic chlorine atom creates a partial positive charge
in the transition state on the carbon from which the hydrogen is being
abstracted. The cr- and e-aminium groups and the carboxyl group aÍe
inductively electron withdrawing, and therefore deactivate adjacent positions to
attack by the elecfrophilic chlorine radical" Consequently, reaction occurs at
the position farthest from both the a- and e-positions, that is, at the y-position.
Polar effects have been used to explain the regioselectivity observed in several
radical processes in biological systems" The biosynthesis of
(S)-Ê-hydroxyvaline (37) involves enzymic oxidation at the B-position of a
valine residue in a peptide, and there is strong evidence that the oxidation
involves radical intermediates,3S with the observed regioselectivity arising due
to polar effecß.36
The widespread application of radical reactions to the functionalization of
o-amino acids is limited by the virtual insolubility of amino acids in the
organic solvents generally used in such processes. This problem can be
overcome by suitable protection of the amino acids, however such protection
has been shown to markedly effect the regioselectivity of radical reactions.
Whereas hydrogen atom abstraction from lysine (11) was shown in an acidic
environment to occur at the y-position, due to deactivation by the q,- and
e-substituents, hydrogen atom transfer reactions of N-acyl-cr-amino acid
derivative! generally favour formation of the conesponding a-centred radicals

Introduction 6
(Figure 1).37'38 Shbitization of radicals of this type results from overlap of the
semi-occupied p-orbital of the radical with the n-orbitals of the electron
donating (dative) amido substituent and the electron withdrawing (capto)
carboxy substituent. This type of radical is extremely stable and is referred to
as a captodative radical,39 although the terms merostabilízeda} and push-pull4l
radicals have also been used. Radical functionalization of N-acyl-protected
o-amino acid derivatives is therefore limited due to reaction occurring at the
o-centres rather than on the side-chains. Formation of the cr,-centred radicals
also desfroys the chirality at that position, leading to formation of enantiomeric
mixtures of products.
Ro
R'
Figure 1. cx,-Centred radical formed by hydrogen atom abstraction from anN-acylamino acid.
In contrast, N"lV-diacyl-protected amino acid derivatives undergo hydrogen
atom abstraction at the o-position much less readily. Whereas treatment of
N-benzoylglycylglycine methyl ester (14) with N-bromosuccinimide (NBS)
results in hydrogen atom abstraction and subsequent bromine incorporation at
the N-terminal glycine residtre, to give the bromide (15), reaction of
N-phthaloylglycylglycine methyl ester (16) with NBS results in bromination of
the C-terminal glycine residue to give the bromide (17) (Scheme 4).42
Similarly, reaction of the N-acyl-N'-methylpiperazinedione (18) with one
equivaient of NBS resulted in specific incorporation of bromine at the
cr-position of the N-methylglycine residue to give the bromide (19) (Scheme
5).+r i'
o
NI
H

PhCONH
(14)
PhthN
(16)
PhthN phthalimido
NAc
NBSPhCONH
NBS
Introduction 1
Br
(1s)
Br
CO2Me
Â"o*r^"orr"PhrtrN
Scheme 4
CONH
(r7)
NAcNBS
MeN MeN
(18) Br
(1e)
Scheme 5
It was found that with N-phthaloyl-protected cr,-amino acid derivatives
hydrogen atom abstraction at the cx,-centres is deactivated to such an extent that
reaction occurs on the side chains if suitably stabilized radicals can be formed.
Accordingly, bromination of N-phthaloylvaline methyl ester (20) and
N-phthaloylphenylalanine methyl ester (22) resulted in regiospecific formation
of the p-bromides (21) and (23), respectively (Scheme 6).42
The bromination of N-phthaloylamino acid derivatives was envisaged to be
an effective method for the direct introduction of functionality onto the side
chains of various proteinogenic amino acids. The generality of side-chain
bromination reactions of N-phthaloyl-protected cr-amino acid derivatives and

Introduction 8
the basis of the deactivating effect of the N-phthaloyl-protecting group is
therefore the subject of the investigation described in Chapter 1 of the Results
and Discussion of this thesis.
PhthN PhthN Me
NBS
Br
(20) (2r)
PhthN PhthN
NBSBr
(22) (23)
Scheme 6
The brominated amino acid derivatives described in Chapter 1 of the
Results and Discussion were envisaged to be suitable precursors to a wide
variety of substituted q-amino acids. One such important class of substituted
amino acids is that of the hydroxyamino acids, especially the p-hydroxy-
a-amino acids, which are ubiquitous in nature and play essential physiological
roles. Examples of naturally occurring B-hydroxy-q,-amino acids include the
common amino acids serine, threonine and the 4-hydroxyproline (24).
B-Hydroxyphenylalanine (25) and p-hydroxytyrosine (26) have been implicated
as precursors in the biosynthesis of the hypertensive agents epinephrine (27b)
and norepinephrine (27a) (adrenalin and noradrenalin),44 and of the antibiotic

Introduction 9
chloramphenicol (28¡,+s and as components of peptidases46 and esterases.4
Chloramphenicol (28), isolated from Streptomyces venezuelae in 1947, was one
of the first broad spechum antibiotics to be used medicinally.aT It is also the
only naturally occurring antibiotic which is economically produced on an
industrial scale by synthesis rather than by fermentation.48
+H¡N
+ coi
HO
(24)
RNHCI
.,rrttOHcr
OH
(21)
ozN
R
R=HR=OH
(2s)(26)
H
OH
(a) R=H(b) R=Me (28)
p-Hydroxy-o-amino acids have also found many synthetic applications, for
example in the synthesis of B-lactam antibioliçs.49'5O Millers0 has converted
various hydroxamate derivatives of B-hydroxyamino acids (29) to the
corresponding p-lactams (30), and then elaborated these B-lactams (30) in the
synthesis of a large number of different p-lactam antibiotics, including
penicillins, cephalosporins and monobactams (Scheme 7).

Introduction IO
RCONH -oR3 RCO o
plOH Bl 3
R
(2e)
penicillins monobactams
cephalosporins
Scheme 7
More recently, B-hydroxyamino acids have received attention due to thei¡
common occurence in a family of biologically active compounds known as the
"cyclic peptides". One of the most important groups in this family is the
vancomycin group of antibiotics.5l Vancomycin (31) was isolated in 1956
from Streptomyces orientalis52 and is currently employed in widespread
clinical üse,53 especially in the treatment of severe staphylococcal infections
such as septicaemia. Vancomycin (31) has also been shown to be an effective
antibiotic in many cases of p-lactam resistance.S3 A tricyclic heptapeptide,
vancomycin (31) contains two B-hydroxytyrosine (26) residues (shown bold),
one with the (253R)-stereochemistry, and the other with the (2R3R)-
stereochemistry.
Although vancomycin-resistant bacterial strains are rare, they aÍe
becoming more common,53 and attention has been focussed on finding even
more potent antibiotics. Currently other cyclic peptide antibiotics are most
promising, with lysobactin (32) being found to be four times as potent as
vancomycin (31) and effective over a much wider range of bacterial
infections.54 Lysobactin (32) was isolated in 1988 from a Lysobacter strain,
and contains five p-hydroxyamino acid residues, three of which are residues
of uncommon hydroxyamino acids; namely B-hydroxyphenylalanine (25),
(30)

CIsugar\o CI
Introduction 11
Ho
+NH2Me
o
oo H
N NHNH
CO; o HzN
o
OH
(31)
p-hydroxyleucine (33) and B-hydroxyasparagine QÐ (shown bold). It isinteresting to note that the three uncommon B-hydroxyamino acids present in
lysobac tin (32) all po s ses s the ( 2 S ) + hr e o - stet eochemi s try.
HHzN
oo
o
o
NH
o
OHHN
HN
NH
HN
HN
oHN
NH
NH
HzN
oo
NH
H
(32)
o
HNL NH2

Introduction 12
+ +H3Nr,,...
"?oH "?oH
NHz
(34)
Another clinically important cyclic peptide is cyclosporin A, an
immunosuppressant undecapeptide which plays a very critical role in many
chemotherapeutic drug treatments and bone marrow transplants.5 5
Cyclosporin A contains an unusual B-hydroxy-cr-amino acid residue, MeBmt
(35), which has been shown to be essential to the action of the drug.s6 Various
other cyclic peptide drugs also contain hydroxyamino acid residues.
BouvardinsT contains a B-hydroxytyrosine (26) residue with the (2535)-
stereochemistry, echinocandins C and D58 each contain (253R)-
B-hydroxyhomotyrosine (36) and (254R)-4-hydroxyproline (24) residues, and
phomopsin 459'60 contains a (253R)-B-hydroxyisoleucine (37) residue and a
B-hydroxyphenylalanine (25) residue with the (2S3S)-stereochemistry. The
majority of the hydroxyamino acids found in these cyclic peptides, with the
exceptions of MeBmt (35) and p-hydroxyhomotyrosine (36), are hydroxylated
derivatives of proteinogenic amino acids, and are presumably formed by
functionalization of the corresponding amino acid residues following their
incorporation into the peptides.a
Hydroxyamino acids are also present in toxic peptides,6l-65 ¿¡¡1i1u¡¡1e¡
peptides,tr and are themselves of interest as enzyme inhibitors.6T For example,
B-hydroxyphenylalanine (25) has been shown to inhibit Neisseria gonorrhoeae
bacterial strains68 and the lactose operon in Escherichia coli.69 Although rale,
lS)-y-hydroxyleucine (38) is a naturally occurring amino acid, being found in
the mushroom toxin phalloine,To and (S)-Þ-hydroxyvaline (39) has been found
H
(33)

Introduction 13
in the hydrolysate of a number of peptides, including the antibiotic
Berninamycin A.7l
MeNH ¡¡3N/2r,.. coi
"raoH1¡rN2ø,.
(37)
(3s)
*1tNlrr,,.
OH
(38) (3e)
A multitude of syntheses of rare and unnatural ø-amino acids has been
developed .2,14,72-74 The most useful syntheses are those which address the
stereochemistry of the amino acids, at both the cr-centre and at chiral centres on
the side chains. Preparation of chiral cr,-amino acids has been achieved using
methods such as the resolution of racemates, asymmetric synthesis, and
synthesis from optically active starting materials from the so-called "chiral
pool". Optically active (R)-Þ-hydroxyvaline Ø3) was first prepared by
reaction of benzylamine with p,B-dimethylglycidate (40) to give the alcohol
(41), followed by removal of the benzyl group and reprotection of the amine to
give the alcohol (42). Resolution of the racemic product with
(-)-a-methylbenzylamine followed by removal of the protecting group gave
(R)-p-hydroxyvaline (a3) (Scheme 8).7s Most current syntheses of optically
+
+
(36)
+ +H

Introduction 14
active hydroxyamino acid derivatives, however, utilize asymmetric synthesis or
chiral starting materials to avoid tedious resolution steps.
PhcH2NH o;benzylamine
(40)(41)
i) H} Pdlcii) NaOH, PhCOCI
+ coi PhCONHÐ (-)-PhCH(Me)NH2
iÐ H* lIl2o
(43) (42)
Scheme 8
Many asymmetric syntheses of B-hydroxyamino acids via condensation of
glycine equivalents with aldehydes have been reported.T6-88 One of the most
general and versatile methods for the synthesis of functionalized cr-amino acids
has been developed by Schollkopf and collaborators.S0-8s The general protocol
involves the coupling of two amino acids to give a piperazinedione, with
subsequent bis-lactim ether formation by treatment with trimethyloxonium
tetrafluoroborate. For example, the bis-lactim ether (45) was produced viø
condensation of (S)-O,O' -dimethyl-a-methyldopa (44) and ethyl glycinate,
followed by treatment with trimethyloxonium tetrafluoroborate. Subsequent
ffeatment with n-butyl lithium followed by succesive additions of acetone and
methyl iodide gave the substituted piperazinedione (46) which was converted in
several steps to (R)-B-hydroxyverline (43) with an enantiomeric cxcess (e.e.) of
907o (Scheme 9).82

Introduction 15
+H3N coi
\ llll¡'i) H2NCH2CO2EI
ü) Me3OBFa
(45)OMe
(M)Ð BuLi
ü) acetone
üi) MeI
\ llltr,'N OMe
(43)hydrolysis
(46)
Scheme 9
Other practical glycine equivalents for the asymmetric synthesis of
B-hydroxy-cr-amino acids have been developed by Seebach and co-workers86'87
and by Evans and Weber.88 Evans and Weber's procedure involves the
formation of a chiral oxazolidinone thiocyanate (47) from (S)-phenylalaninol.
Condensation of the tin enolate of the thiocyanate (47) with an aldehyde gives
the thionourethane (48), with cleavage of the chiral auxiliary giving the
oxazolidinone (49). Hydrolysis of the oxazolidinone (49) then gives the
corresponding B-hydroxy-o-amino acid (50) (Scheme 10). The unusual amino
acid present in cyclosporin A, MeBmt (35), was produced in 947o e.e., and
(253R)-þ-hydroxyleucine (33) was produced ín 987o e.e. vía this route.88
AIN
Ar
N

Introduction 16
ooN
oo
N
R
i) Sn(OTf)2
-__-->
NCS ü) RCHO
KOH / H2O
i) Mg(OMe)2/Ilv4rIOHiÐ H2o2
R
N
(47)
(s0)
(48)
o
H+
RN
(4e)
Scheme 10
However, these procedures are obviously limited to the synthesis of
B-substituted a-amino acid derivatives. Also, although the above examples give
products of high enantiomeric excess there is currently a strong demand,
particularly in the pharmaceutical industry, for compounds which are
enantiomerically pure" This arises from the fact that in most cases only one
enantiomer of an administered drug is responsible for the desired therapeutic
effects, while it is often found that the other enantiomer exhibits detrimental
side effects. Probably the most striking example is that of the drug
thalidomide, in which it was found that the required activity resided in the
(R)-enantiomer (51), whereas the (S)-enantiomer was responsible for the
disasterous teratogenic effects.39'90 Similarly, treatment of Wilson's disease
and bitiary crrhosis with (R/-penicillamine (52) is extremeiy effective,9t Oor 'n
contrast the (S)-antipode of penicillamine causes optic atrophy, which can lead
to blindnoss.92

o +
Introduction 17
SH
(50)
Ho
N
o
(s2)(51)
Various enantiospecific syntheses of functionalized amino acids have
therefore been developed by modification of chirally pure starting materials
from the so-called "chi¡al pool". Most of the compounds comprising the chiral
pool are isolated from biological sources; examples include the proteinogenic
c¡-amino acids, carbohydrates, terpenes and tartaric acid. Tanner and co-
workers93 have reported the synthesis of B-substituted aspaftic acid derivatives
from (+)- and (-)-tartaric acid. Rao and co-workers94 have utilized the
inherent chirality in D-glucose (53) to develop a synthesis of MeBmt (35).
Although the procedure is very long, requiring over 17 steps from D-glucose
(53), it represents a general method for the stereospecific synthesis of
p-hydroxy-c¿-amino acids (50) (Scheme 1l).
R
+++
-+
OH
(s3)
OH
(54)
Scheme 11
The amenability of halogenated amino acid derivatives to further
elaboration has been well documented. Examples include the elaboration of
iodoalanine derivatives to give 4-hydroxy-ø-amino acids2l and the synthesis of

Introduction 18
yhydroxylysine (13) as described u5ons.2e-31 The side-chain halogenation of
proteinogenic amino acid derivatives and subsequent manipulation of the
introduced functionality is therefore a powerful approach to the synthesis of
rare and unnatural amino acids. The scope of elaboration of the side-chain
brominated amino acid derivatives, prepared as described in Chapter 1, is the
subject of the studies described in Chapters 2, 3, 4 and 5 of the Results and
Discussion of this thesis. The importance of hydroxyamino acids both as
physiologically active compounds and in the stereocontrolled syntheses of other
biologically active molecules has been detailed above. The conversion of the
bromoamino acid derivatives described in Chapter I to the corresponding
hydroxyamino acid derivatives was therefore investigated. Mild, non-basic
conditions were considered to be required for these reactions to reduce the
possibility of racemization at the o-cenffe, and to avoid undesirable reactions of
the phthalimido group due to its base-sensitivity. The above requirements
preclude the use of sfrong nucleophiles such as hydroxide to effect substitution
of the bromide. However, silver salts have been shown to induce substitution of
alkyl halides under very mild conditions.9s-97 Silver ion induced reactions of
alkyl halides have been shown to occur with graded S¡1-S¡.,r2 character, with the
varying degree of character dependent on the solvent, silver salt and bromide
structure in each reaction.95'97 The conversion of bromoamino acid derivatives
to the corresponding hydroxyamino acid derivatives, with control of both
relative and absolute stereochemistry, was therefore investigated using silver
nitrate induced hydrolysis reactions, with particular attention focussed on
maximizing the stereoselectivity of the reactions. Methods for the deprotection
of the hydroxyamino acid derivatives to give the corresponding free amino
acids, via procedures which avoid racemization, were also studied. The results
of the above studies are discussed in Chapter 2 of this thesis.
Various syntheses of the natural product chloramphenicol (28) have been
reported.4S Many are based on the condensation of glycine equivalents with

Introduction 19
p-nitrobenzaldehyde, with subsequent resolution steps required. The synthetic
methods developed in Chapters 1 and 2 of the Results and Discussion of this
thesis indicated a potential new method for the short, stereoconfrolled synthesis
of chloramphenicol (28), starting from (R)-phenylalanine (55) or itsderivatives. The results of this investigation are described in Chapter 3 of the
Results and Discussion of this thesis.
coi
(ss)
The brominated amino acid derivatives described in Chapter 1 of this thesis
are also suitable for elaboration to a wide variety of functionalized amino acid
derivatives. The scope and limitations of such further elaborations of the
bromoamino acids were therefore investigated. Stereocontrolled cyclization
reactions of the bromoamino acid derivatives to give lactone and lactam
products suitable for elaboration to known antibiotics were studied, as well as
various methods for the synthesis of unstable Ê,T- and y,õ-dehydroamino acids,
which have been found to be potent enzyme inhibitors9S'99 and precursors to
other functionalized amino acid derivatives.2 The results of these investigations
are described in Chapter 4 of the Results and Discussion of this thesis.
The free radical side chain bromination of N-phthaloyl-a-amino acid
derivatives is limited to incorporation of the bromine atom only at the position
on the side chain which gives the most stable radical. Transfer of functionality
from one position on the side chain to other, non-activated positions was
therefore investigated. The aim of this work was to further demonstrate the
applications of side-chain bromination and subsequent elaboration of
+

Introduction 20
N-phthaloyl-cr,-amino acid derivatives to the stereocontrolled synthesis of
functionalized amino acids. The results of this work are also described in
Chapter 4 of this thesis.
The bromoamino acid derivatives described in Chapter 1 of the Results and
Discussion are also suitable for conversion to unnatural amino acid analogues,
such as isotopically-labelled amino acids. The utility of labelled amino acids in
the study of enzyme reactions includes the use of 13C-labelled substrates to
determine the stereochemistry of enzyme-catalyzed decarboxylation reactions,16
and the use of deuterium-labelled tyrosine in the determination of the
stereochemical course of tyramine oxidation by the enzyme semicarbazide-
sensitive amine o*i¿¿ss.l00 The mechanism of the enzyme phenylalanine
ammonia lyase (PAL) has also been determined with the aid of labelling
experimen1s.15,101-105 Numerous studies of PAL have been conducted,l06-110
particularly by Havir and Hanson.lOT-l10 PAL is a plant enzyme which
catalyses the elimination of ammonia and a proton from (S)-phenylalanine (56)
to give trqns-cinnamic acid (57) (Scheme 12)" It has been postulated that this
enzyme represents a switch in plants for re-routing (S)-phenylalanine (56) from
protein synthesis to secondary plant metabolites such as phenylpropanoids,
alkaloids and flavanoid compounds. 1@
+HrNrr,,,. coi
PAL NH3+
(56)
Scheme 12
(st)

Introduction 2l
The enzyme consists of four identical subunits, which each possess an
N-terminal serine residue. The enzyme tetramer contains two active sites,
indicating that two subunits combine to form each of the active sites. Labelling
studies have indicated that the N-terminal serine residues (58) are post-
ribosomally modified, with two undergoing a,B-elimination to give
dehydroalanine residues (60) and the other two most probably being oxidised to
give serine-aldehyde residues (59).101-l0s A dehydroalanine residue (60) then
reacts with a serine-aldehyde residue (59) to give a conjugated electrophilic
prosthetic group (61) in each of the active sites (Scheme 13). Upon binding of
(S)-phenylalanine (56) in an active site, the amino group of (S)-phenylalanine
(56) reacts with and becomes covalently bound to the electrophilic cenre (61).
Formation of the enamine (62) then establishes an elecfton sink for facile C-N
bond cleavage of the substrate, such that removal of a B-proton and subsequent
elimination occurs to give trans-cinnamic acid (57) and the amino-enzyme
complex (63). The enzyme then releases ammonia, and in doing so reforms the
activated electrophilic centre (61) in the active site (Scheme 14).
Enz
-H2 ¡f2Nh,,. -EnzEnz
(se) -Hzo
OH -Hzo
(s8) HzN -Enz
(61)
(60)
-Enz
Enz= enzyme
Scheme 13

(56)
(s7)
Introduction 22
Enzo
Enz
o
Enz
Enz
Enz
(61)
Enz
Ph
Ph
Ph
/a-HLB,
o(
N
HN=
NH¡
B lHEnz
Enz
(
HN
=
Enz
Enz
( HN
HzN
o
Enz
HN
HN
o)
Enz
(
OHEnz
( HN
Hcoz
H
(62)BI
Scheme 14
Enz
(63)
HN

Introduction 23
Battersby and co-workersl5 have reported a study of the stereochemical
course of the elimination catalyzed by PAL using the deuterium-labelled
phenylalanines (65a) and (65b). The synthesis of the labelled phenylalanine
derivatives (65a) and (65b) was performed by hydrogenation of the deuteriated
dehydrophenylalanine derivative (64), followed by removal of the protecting
group, to give a racemic mixture of the p-deuteriated phenylalanines (65a) and
(65c). The N-chloroacetyl derivatives of the racemic mixture of (65a) and
(65c) were resolved by treament with hog kidney-acylase-I to give (253R)-
3-deuteriophenylalanine (65a), and the N-chloroacetyl derivative of (2R35)-
3-deuteriophenylalanine (65c), which was subsequently hydrolysed to give
(2R35)-3-deuteriophenylalanine (65c). Racemization at the cr-centre of the
deuteriated phenylalanine (65c) by treatment with acetic anhydride lacetic acid
gave a mixture of the deuteriated phenylalanines (65b) and (65c) (Scheme
15¡.ts The deuteriated phenylalanines (65a), (65b) and (65c) produced viø this
procedure were isolated with enantiomeric excesses of ca. 867o and deuterium
incorporation of 9OVo"
DD
D N-benzoyl-glycine
Ph N
Ph
NaOH
cot
NHCOPh
(64)
i) H2lPd
ii) hydrolysis
DD
=
D
= Ac2O / cozPh
NH¡-1-NH.+)
(65a)
NH"^¡ '^ -J1
Ph
(6sb)
AcOH
Scheme 15
(65c)
+

Introduction 24
Kirby and Michaellll reported a very similar procedure at about the same
time, and Ife and Haslaml12 also reported a synthesis of (2S3R)- and (2535)'
3-deuteriophenylatanine (65a) and (65b), from (S)-c-deuteriobenzyl alcohol
(66) (Scheme 16). These and related synthesesl13,l14 are limited by their lack
of enantioselectivity, and require enzyme-catalyzed resolution steps to obtain
homochiral products.
NHAcD
ErO2C
i) HBrÆI2OiÐ crcH2cocl
-+
Phüi) Acylase I
AcNH
(66)
D
I
PCI 3
NHAc i) HBrÆI2OiÐ clcH2coclEtO2C
Ph C1Ph
üi) Acylase IAcNH
Ts = SO2C6Ha-p-Me
Scheme 16
Battersby and co-workersl5 used the deuterium labelled phenylalanines
(65) to determine that PAL catalyses the elimination of the pro-S hydrogen of
(S)-phenylalanine (56) (Figure 2), together with ammonia, to give trans-
cinnamic acid (57). Reaction of (253R)-3-deuteriophenylalanine (65a, 907o 2II)
with the enzyme gave deuteriated trans-cinnamic acid (67) with 88+47o
deuterium incorporation, indicating a highty stereoselective elimination of the
3-pro-S hydrogen from (S)-phenylalanine (56) (Scheme 17). Treatment of the
(2SiS)-isomer (65b, 907o 2H) with the enzyme gave cinnamic acid (57) with
8+27o do¡terium incorporation, supporting the above conclusion. Hanson and
D
?
Ph
(65a)
(65b)
D
=
D

Introduction 25
¡¡uu¡l10 showed that (R)-phenylalanine (55) is also metabolized by PAL, but at
a, rate one five thousandth that of lS)-phenylalanine (56). However,
(R)-phenylalanine (55) does act as a competitive inhibitor of the enzyme,
indicating that it binds effectively to the active site. Hanson and Havirl l0
suggested that when (R)-phenylalanine (55) is bound to the active site the
groups on the enzyme are poorly situated to play an effective chemical role,
however no stereochemical studies of the reaction of PAL with
(R)-phenylalanine (55) have been reported.
+g3Nzø,,.
PAL
D D
(65a) (6t¡
+
co2H
g3N/ø,,.
(6sb)
PAL
Scheme 17
+
HR
(56)
H
(s7)
¡¡3NZr,.. cot
Fig ure. 2. D e s ig natio n of theo3,í#;i lr.;"f il
of (S) - phen y I al ani ne ( 5 6 )

Introduction 26
The above syntheses of the deuteriated phenylalanines (65) are limited
because they are not stereospecific and/or require enzymic resolution steps to
obtain enantiomerically pure products. The conversion of
B-bromophenylalanine derivatives described in Chapter 1 of this thesis to all
four stereoisomers of B-deuteriated phenylalanine (65) was therefore studied.
The results of this investigation are described in Chapter 5 of the Results and
Discussion of this thesis.
The use of the deuteriated phenylalanines (65) in a study of the reactions of
both lS)- and (R)-phenylalanine, (56) and (55), with the enzyme PAL is
described in Chapter 6 of the Results and Discussion. Particular attention was
focussed on determining the stereochemical course of the reaction of the
B-deuteriated (R)-phenylalanines (65c) and (65d) with PAL, with the interest
being to determine how the enzyme metabolizes a compound with the opposite
stereochemistry to that of its normal subsffate.

Chapter I
RESULTS AND DISCUSSION: CHAPTER 1
Regiocontrolled Side Chain Bromination of N-Phthaloyl-Protected
s-Amino Acid Derivatives
The starting materials required for the study of side chain bromination of
N-phthaloylamino acid derivatives were synthesized as described below. The
N-phthaloyl-protected (S)-valine, (S)-leucine, (S)-phenylalanine and
(RS)-homophenylalanine derivatives (68)-(70) and (76) were synthesized
according to the procedure of Sheehan, Chapman and Roth,ll5 r¡¡þiçþ involved
heating equimolar mixtures of finely groung phthalic anhydride and the
corresponding amino acids at 145-150o for 30 minutes (Schemes 18 and 19).
Temperature control was important as it has been reported that racemization
can occur at temperatures above 180o.115 Although this procedure is suitable
for the preparation of many N-phthaloyl-a-amino acids, it is not applicable for
protection of amino acids with hydroxyl groups on the side chains.l16
Phthaloylation of (S)-tyrosine was therefore performed by addition of
(S)-tyrosine to a. boiling solution of phthalic anhydride in
N,N-dimethylformamide (DMF), with boiling continued for 10 minutes, giving
(S)-N-phthaloyltyrosine (71) tn 787o yield.
+¡I3Nzzr,.. phthatic
anhydride
CO; tnrhN¿¿,,,.MeOH
HCI
(68) R = CHMez
(69) R = CHzCHMez
(70) R: CHzPh
(71) R = CHzCoHq-p-OH
(72) R = CHMez
(73) R = CHzCHMez
(74) R = CHzPh
(75) R = CHzCoH¡-P-OH
..........-..-.-'-RRR
27
Scheme l8

+H¡N coi PhthN
Chapter I 28
PhthN CO2Me
phthalicanhydride
MeOH
HCI
(77)
Scheme 19
Optical rotations of the N-phthaloylamino acids (68)-(71) were consistent
with literature values,115,117 indicating that racemization at the q-centres had
not occurred. The N-phthaloylamino acids (68)-(71) and (76) were then
esterified by treatment with methanolic hydrogen chloride solution, which was
prepared by the slow addition of thionyl chloride to cooled, dry methanol
under anhydrous conditions (Schemes 18 and 19), to give the esters (72)-(75)
and (77) in good yields.
(S)-N-Phthaloylphenylalanine (70) was also protected as the corresponding
tert-butylamide (78). Initially synthesis of the amide (78) was attempted by
treatment of (S)-N-phthaloylphenytalanine (70) with thionyl chloride and
pyridine in refluxing carbon tetrachloride (CCl¿) to give the corresponding
acid chloride, followed by addition of tert-butylamine. However, this
procedure was shown to cause racemization at the o-centre. Analysis of the
product by high performance liquid chromatography (HPLC) using a column
with (S)-phenylglycine as the chiral stationary phase showed the presence of
two compounds, with retention times of 2l and 22 minutes. Subsequently the
amide (78) was synthesized under milder conditions in order to prevent
racemization. Accordingly, a solution of (S)-N-phthaloylphenylalanine (70) in
dichloromethane at 0o was treated with triethylamine then ethylchloroformate,
(76)

Chapter I 29
followed by the addition of tert-butylamine, giving the product (78) in 937o
yield (Scheme 20). Analysis of this material by HPLC using conditions
identical to those previously described showed the presence of only the
(S)-enantiomer (78), which eluted with a retention time of 2l minutes. The
(S)-enantiomer (78) was therefore shown to have a shorter retention time on
the column than the corresponding (R)-enantiomer, which is consistent with
previous results using columns with (S)-phenylglycine as the chiral stationary
phase.lta
nhthN¿r,,,. NHCMe3
Ð NEt,ü) EIOCOCI
iii) t-BuNH2
(70) (78)
Scheme 20
Attempted amidation of the tyrosine derivative (71), under conditions
identical to those described for the preparation of the phenylalaninamide (78),
proved difficult due to its limited solubility in the reaction solvent,
dichloromethane. The phenolic group was therefore acetylated by treatment of
the tyrosine derivative (71) in refluxing acetic anhydride to give (S)-O-acetyl-
N-phthaloyltyrosine (79) in 797o yield" This compound had much increased
solubility in dichloromethane, and the amidation reaction therefore proceeded
smoothly via treatment of the tyrosine derivative (79) with triethylamine,
ethylchloroformate and tert-butylamine, to give the fully protected tyrosine
derivative (80) in 937o yield (Scheme 2l).

PhthN co2Hhh,.
(71)
Ac2O
Chapter I 30
PhthN co2H
(7e)
Ð NEr3ü) ETOCOCIiii) r-BuNH2
PhthN CONHCMe3
(80)
Scheme 21
Reactions of the (S)-valine and (S)-phenylalanine ester derivatives (72) and
(74) with NBS in CCI¿ to give the corresponding B-bromoamino acid
derivatives (81) and (82) (Scheme 22) were conducted as reported by Easton,
Tan and Hay.az The B-bromovaline derivative (81) was isolated as a white
solid in 837o yieLd. The B-bromophenylalanine derivative (82) was obtained as
a 1:1 ratio of the diastereomers (82a) and (82b), as determined by lH n.m.r.
spectroscopy, which showed peaks at õ 3.55 and 3.82 colresponding to their
methyl ester resonances.
The reaction to give the bromide (81) presumably proceeds viø abstraction
of the hydrogen atom from the p-position of the valine derivative (12) by a
bromine atom to give the tertiary radical (83) and hydrogen bromide.
Hydrogen bromide then reacts with NBS to give succinimide and molecular
bromine. Reaction of the radical (83) with bromine then gives the
B-bromoVåline derivative (81) and a bromine atom, which continues the radical

Chapter I 3I
.¡uin.119 Similarly, reaction of the phenylalanine derivative (74) would
proceed vía the benzylic radical (84) to give the p-bromide (82) as a mixture of
diastereomers. The lack of asymmetric induction in the formation of the
bromide (82) can be attributed to the exfremely low activation energy of such
halogen hansfer reactions.34
PhthN ththN¿¿r,,. CO2Me
NBS
Br
Q2) (8 1)
,hthN¿¿r,.. CO2Me tnrhN¿r,,,. CO2Me
NBS pl2
(74) (82)
(a) Rl=Br, R2=H(b) Rl=H, R2=Br
Scheme 22
PhthNnnr¡Nzrr,,. CO2Me
(83)(84)
The bromide diastereomers (82a) and (82b) were separated by tiactional
crystallization from hexane/dichloromethane, giving diastereomer (82a) in 427o
i"

Chapter I 32
yield (m.p. 142-143") and diastereomer (82b) in 407o yield (m.p. l2I-122").
The lH n.m.r. spectrum of diastereomer (82a) showed doublets at õ 5.52 and
6.02 due to the cr- and p-protons, respectively, as well as a singlet at ô 3.55 due
to the ester moiety. Diastereomer (82b) showed doublets at ô 5.59 and 5.91
due to the o- and p-protons, respectively, and a singlet at ô 3.82 due to the ester
moiety. The assignment of peaks due to the cr- and B-protons was based on
comparison of the lH n.m.r. spectral data of the bromides (82a) and (82b) with
that of the non-brominated compound (74). Substitution of a methylene proton
with bromine is reported to cause a downfield shift of the adjacent proton of
2.3 ppm.rz0 Hence, as the signals corresponding to the p-protons of the
phenylalanine derivative (74) occur at ca. õ 3.6, the peaks corresponding to the
P-protons of the bromides (82a) and (82b) would be expected to occur at ca.
õ 5.9. This is in close agreement with the observed values of õ 5.91 and 6.02.
The signals due to the a-protons of the bromides (82a) and (82b) therefore
occur at ô 5.52 and 5.59, respectively, being shifted downfield by ca.0.3-0.4
ppm from that of the non-brominated compound (74). These assignments were
confirmed using heterocorrelation n.m.r. spectroscopy" The 13C n.m.r.
spectrum of the bromide (82b) was obtained and the peaks were assigned to
their corresponding carbon atoms.120 The signal corresponding to the
a-carbon occurs at õ 57.0, and the signal corresponding to the B-carbon occurs
at õ 47.6. In the heterocorrelation n.m.r. spectrum, a strong coupling is
observed between the cr,-carbon and the doublet at ô 5.59, indicating that the
resonance observed at ô 5.59 in the lH n.m.r. spectrum corresponds to the
a-proton. Similarly, there is a strong coupling between the p-carbon and the
doublet at ô 5.91, indicating that the resonance observed at õ 5.91 in the
lH n.m.r. spectrum corresponds to the B-proton.
It was of interest to determine whether bromination also occurs with thc
amide-protected phenylalanine derivative (78). Treatment of the
phenylal4ninamide (78) with NBS under similar conditions as for bromination

Chapter I 33
of the ester (74), but using a3:1 mixture of CCI+ and dichloromethane to
achieve complete dissolution of the amide (78), did indeed give the
p-bromophenylalanine derivative (85) (Scheme 23), as a 1:1 ratio of the
diastereomers (85a) and (85b). To verify that no racemization at the cr-centre
had occurred during the bromination reaction, the diastereomeric mixture of
the bromides (85a) and (85b) was reduced to the phenylalaninamide derivative
(78), by treatment with tributyltin hydride (Scheme 23), and the product was
analysed by TIPLC as described above. Only the (S)-enantiomer (78) was
eluted, with a retention time of 21 minutes, indicating that both bromination
and reduction occurred withoutracemization at the g-centre.
ththN¿¿r,,. CONHCMe3 PhthN
NBS
p1Bu3SnH 2
(78) (8s)
(a) Rl=Br, R2=H(b) Rl=H, R2=Br
Scheme 23
The bromoamide diastereomers (85a) and (85b) were separated by
fractional recrystallization from hexane/iso-propanol, giving diastereomer
(85a) ín 4IVo yield (m.p. 188-191') and diastereomer (85b) in 43Vo yield (m.p.
208-209")" Both of the bromides (85a) and (85b) were identified using mass
spectrometry, each giving molecular ions at mlz 428 and 430 in equal
abundance, and using lH n.m.r. spectroscopy. Diastereomer (85a) showed
doublet resonances at õ 5.22 and 6.17, due to the c¿- and B-protons,
respectively. Diastereomer (85b) showed doublet resonances at õ 5.32 and
6.05, also due to the cr- and p-protons, respectively.i'

Chapter I 34
The relative stereochemistry of the B-bromophenylalanine ester and amide
derivatives (82a), (82b), (85a) and (85b) was determined by converting them to
the corresponding dehydrophenylalanine derivatives (86)-(89). Potassium
fluoride was chosen as the appropriate base for these ¡e¿ç¡iq¡s.121 Treatment
of the bromoamide (85a) with potassium fluoride and 18-crown-6 in
acetonitrile produced the (Z)-dehydrophenylalanine derivative (86) in 84Vo
yield, with none of the (E)-dehydrophenylalanine derivative (87) present.
Treatment of the other bromide diastereomer (85b) under the same conditions
gave the (E)-dehydrophenylalanine derivative (87) in 57Vo yield, and a small
amount of the (Z)-isomer (86) (97o) (Scheme 24). The stereochemistry of each
of the dehydrophenylalanine derivatives (86) and (87) was determined by X-ray
crystallographic analysis (Appendices 3 and 4). lH N.m.r. spectroscopy
showed the signal corresponding to the vinylic proton of the
(E)-dehydrophenylalanine derivative (87) at ô 7.05, whereas that of the
(Z)-dehydrophenylalanine derivative (86) occurred downfield at õ 7.59.
Consistent with an earlier repoÍ,rz2 this observation was the first in the present
tnrhN¿¿r,,. CONHCMe3 PhthN
.,?8, K+F-
18-crown-6
(85a) (86)
tn,hN¿rr,,. CONHCMe3 PhthN CONHCMe3
K+F-
18-crown-6
(8sb)
Br
Scheme 24
(87)
+ (86)

Chapter I 35
work of a trend in which the signals corresponding to the vinylic protons of
various (Z)-dehydrophenylalanine derivatives consistently occurred downfield
of the signals attributed to the vinylic protons of the corresponding
(E)-isomers.
The elimination of hydrogen bromide from the bromides (85a) and (85b)
is stereoselective and is therefore most probably proceeding via an anti-
elimination mechanism as the major pathway. On this basis the bromide (85a)
which gave only the (Z)-dehydrophenylalanine derivative (86) on elimination
of hydrogen bromide must have the (253R)-configuration, and the bromide
(85b) which gave the (E)-dehydrophenylalanine derivative (87) as the major
product must have the ( 2 S 3 S )- stereochemistry.
Treatment of the bromoester (82a) with potassium fluoride and
18-crown-6 in acetonitrile under similar conditions to those described above
produced the (Z)-dehydrophenylalanine derivative (88) in 88Vo yield, with none
of the (E)-dehydrophenylalanine derivative (89) present. The stereochemistry
of the (Z)-isomer (88) was confirmed by X-ray crystallographic analysis
(Appendix 5). Treatment of the bromide diastereomer (82b) under the same
conditions gave a 1:2 mixture of the (E)-dehydrophenylalanine derivative (89)
and the (Z)-dehydrophenylalanine derivative (88), in 88Vo overall yield
(Scheme 25). The isomers (88) and (89) were inseparable by chromatography,
but were partially separated by fractional crystallization from hexane/ethyl
acetate to give a pure sample of the (Z)-dehydrophenylalanine derivative (88).
The (E)-isomer (89) was always contaminated with residual amounts of the
(Z)-isomer (88), and consequently a crystal structure of the (E)-isomer (89)
could not be obtained. The stereochemistry of the (E)-isomer (89) was
confirmed by comparison of its lH n.m.r. spectrum with that of the
corresponding (Z)-isomer (88). The signal due to the vinylic proton of the
(E,)-isomer (89) occurred at õ 7.23, whereas the vinylic proton signal of the

Chapter I 36
(Z)-isomer (88) occurred downfield at ô 8.12, consistent with the trend
described above.
PhthN CO2Me
K+F-
18-crown-6
(82a) (88)
tnthN¿rr,,. PhthN
K+F- + (88)Br 18-crown-6
(82b) (8e)
Scheme 25
To determine whether the (Z)-dehydrophenylalanine derivatives (86) and
(88), formed during reactions of the corresponding erythro-bromides (85b)
and (82b) with potassium fluoride, were produced viø isomerization of the
corresponding (E)-isomers (87) and (89), the (E)-isomers (87) and (89) were
re-subjected to the original reaction conditions. No conversion of the
(E)-dehydrophenylalanine derivatives (87) and (89) to the corresponding
(Z)-isomers (86) and (88) was detected in the reaction mixtures by 1¡¡ n.m.r.
spectroscopy. Formation of the (Z)-isomers (86) and (88) is therefore
attributed to removal of the acidic cr-hydrogens from the bromides (85b) and
(82b), respectively, by fluoride ion, to give the conesponding stabilized
c-anions, followed by rotation about the respective Co-CÞ bonds, with
subsequent elimination of bromide ion. Formation of the (Z)-isomers (86) and

Chapter I 37
(88) is not unexpected, as the (E)-isomers of dehydrophenylalanine derivatives
are generally much less stable than the corresponding (Z)-isomers.l23,l24 A
number of isomerizations of (E)-dehydrophenylalanine derivatives to the
corresponding (Z)-dehydrophenylalanine derivatives have been reported; for
example, the (E)-dehydrophenylalanine derivative (90) isomeized to the the
correspondtng (Z)-isomer on heating in chloroform and, more rapidly, on
standing in neat trifluoroacaliç ¿gld.12s
CbzNH
(e0)
Evidently, elimination of hydrogen bromide from the erythro-bromides
(85b) and (82b) does not proceed exclusively via an E2 mechanism, but also vlø
an El.6 mechanism to some extent. The decreased selectivity in the elimination
of hydrogen bromide from the ester (82b) compared to the amide (85b) can be
attributed to the greater acidity of the a-hydrogen of the ester (82b), such that
the E1ç5 mechanism occurs to a greater extent. However, the major product
still arises from anti-elimination, and therefore the relative stereochemisfry of
the bromoesters (82a) and (82b) and the bromoamides (85a) and (85b) can still
be inferred. Thus, the bromide (82a) which gave the
(Z)-dehydrophenylalanine derivative (88) on elimination of hydrogen bromide
must have the (253R)-configuration, and the bromide (82b) which gave the
(E)-dehydrophenylalanine derivative (89) must have the (253 S )-
stereochemistry.
Reactions of the (S)-leucine and (RS)-homophenylalanine derivatives (73)
and (77) ¡vere also studied, with the aim of investigating the generality of the

Chapter I 38
side-chain bromination procedure. Treatment of the leucine derivative (73)
with one equivalent of NBS in refluxing CCla, with irradiation by a 250W
mercury lamp for 2 h, gave the y-bromoleucine derivative (91) (Scheme 26) in
8I7o yield after chromatography. Identification of the y-bromoleucine
derivative (91) was achieved using lH n.m.r. spectroscopy, which showed
singlet resonances at ô 1.76 and 1.84 corresponding to the diastereotopic
y-methyl groups, and by mass $pectrometry which gave molecular ions at mlz
353 and 355 ofequal abundance.
PhthN PhthN
NBSBr
(73) (e1)
PhthN PhthN
NBSBr
(77) (e2)
Scheme 26
Treatment of the homophenylalanine derivative (77) under identical
conditions gave the corresponding 7-bromohomophenylalanine derivative (92)
(Scheme 26) as an approximately l:1.5 ratio of diastereomers. The bromide
(92) was identified using mass spectrometry, which showed molecula¡ ions at
mlz 401 and 403 in equal abundance, and using lH n.m.r. spectroscopy. Two
resonances were observed at near identical chemical shifts at õ 4.91, due to the

Chapter I 39
y-protons of the diastereomers of the bromide (92), with each resonance
existing as a triplet due to the presence of two hydrogens on the adjacent
p-carbon. A riplet at ô 5.10 and a doublet of doublets at ô 5.21 were also
observed, attributable to the cr,-protons of the diastereomers of the bromide
(92). Assignment of the peaks due to the a- and y-protons was made by
comparison of the lH n.m.r. spectral data of the bromide (92) with that of the
non-brominated compound (77), as for the bromophenylalanine derivative (82)
described above. As the signal due to the y-protons of the homophenylalanine
derivative (77) is observed at ca. õ 2.6, the signal due to the y-protons of the
diastereomers of the bromide (92) would be expected to occur at cq. 6 4.9,120
indicating that the peaks observed at ô 4.91 are due to the y-protons of the
diastereomers of the bromide (92). The signals corresponding to the q,-protons
of the diastereomers of the bromide (92) therefore occur at õ 5.10 and 5.21,
being shifted downfield by ca" 0.2-0.3 ppm from that of the non-brominated
compound (77).
Attempted separation of the diastereomers of the bromohomophenylalanine
derivative (92) by fractional recrystallization and through chromatography on
silica was unsuccessful, with mixtures always being obtained.
Having developed procedures for the regioselective bromination of the
above N-phthaloylamino acid derivatives (72)-(74), (77) and (78), similar
bromination of the tyrosine derivatives (75) and (80) was investigated. The
challenge in radical bromination of tyrosine derivatives is to avoid oxidation of
the phenolic group, which is a facile process.126 Initially, attempted
bromination of fS)-N-phthaloyltyrosine methyl ester (75) was unsuccessful,
with no identifiable products being formed. Presumably the phenolic group
was oxidized, giving a complex mixture of products. In order to overcome this
oxidation the nhenol was nrotected as the corresnondins acetate. Treatment of'--r --__ f _ --_ f __ - ----t - - o' "'
(S)-N-phthaloyltyrosine methyl ester (75) with acetic anhydride, triethylamine
and a catalytic amount of N/V-dimethylaminopyridine (DMAP) gave the acetate

Chapter I 40
(93) in 87Vo yield. Subsequent treatment of the protected tyrosine derivative
(93) with NBS under standard conditions for 2 h gave a near quantitative yield
of the B-bromotyrosine derivative (94) (Scheme 21) as a 1:1 ratio of
diastereomers.
tnrhN¿r¿,..
NBSp1
p2
(e4)
(a) Rl =Br, R2=H(b) Rl=H, R2=Br
Scheme 27
The bromide (94) was identified using mass spectrometry, which showed
molecular ions at mlz 445 and 447 in equal abundance. The bromide (94)
slowly decomposed on standing in air, or in solution, and was therefore
characterized using high resolution mass spectrometry, which gave an ion at
mlz 445.018 corresponding to the molecular ion with a calculated mass of
445.016, rather than by elemental analysis. The bromide (94) was also
characterized as the corresponding B-hydroxytyrosine derivative (I22a) as
described in Chapter 2 of the Results and Discussion of this thesis. Due to their
instability the diastereomers of the bromide (94) were not able to be separated
by fractional recystallization, but enrichment of diastereomer (94a) was
achieved by chromatography on silica, which enabled the assignment of the 1H
n.m.r. spectra of each of the diastereomers (94a) and (94b). Diastereomer
(94a) showed doublet resonances at õ 5.46 and 6.03, due to the s- ancl
B-protons, respectively. Diastereomer (94b) showed doublet resonances at
ô 5.56 and" 5.93, also due to the cr- and B-protons, respectively.
(e3)

Chnpter I 4I
Treatment of the tyrosinamide derivative (80) with one equivalent of NBS
under similar conditions to those described above for bromination of the
tyrosine derivative (93) gave a near quantitative yield of the
p-bromotyrosinamide derivative (95) (Scheme 28), again as a 1:1 ratio of
diastereomers. The bromide (95) was identified using mass spectrometry,
which showed molecular ions at mlz 486 and 488 in equal abundance. In a
similar manner to that for the bromoester (94), the bromoamide (95) was
characterized using high resolution mass spectrometry, giving an ion at mlz
486.080 corresponding to the molecular ion with a calculated mass of 486.079,
and also by conversion to the B-hydroxytyrosinamide derivative (124) as
described in Chapter 2 of the Results and Discussion of this thesis. Enrichment
of diastereomer (95a) was acheived by chromatography on silica, which
enabled the assignment of the lH n.m.r. spectra of each of the diastereomers
(95a) and (95b). Diastereomer (95a) showed doublet resonances at ô 5.17 and
6.22, due to the a- and B-protons, respectively" Diastereomer (95b) showed
doublet resonances at ô 5.30 and 6.08, also due to the cr- and B-protons,
respectively.
PhthNhtr,. CONHCMe3 tnthNr¿r,,. CMe3
NBSp1
p2
AcO(80) (e5)
(a) Rl=Br, R2=H(b) Rl=H, R2=Br
Scheme 28
Although the diastereomers of the tyrosyl bromides (94) and (95) were not
separated and their relative configurations determined independently,I

Chapter I 42
comparison of their lH n.m.r. spectral data with that of the diastereomers of
the phenylalanyl bromides (82) and (85) enabled the ascription of the lH n.m.r.
spectra of the threo-bromides (94a) and (95a) and of the corresponding
erythro-bromides (94b) and (95b) (Appendix l). It can be seen from the data
in Appendix I that the lH n.m.r. spectra of the bromotyrosine ester
diastereomers (94a) and (94b) correlate very closely with those of the
bromophenylalanine ester diastereomers (82a) and (82b). The tyrosyl bromide
(94a) which exhibits its methyl ester resonance at ô 3.56 is therefore assumed to
be the threo-isomer, and similarly the bromide (94b) with resonance at ô 3.81
is assumed to be the erythro-isomer. Similar comparison of the lH n.m.r.
spectra of the diastereomers of the bromoamide derivatives (95) and (85)
enabled stereochemical assignment of the spectra of the tyrosyl bromide
diastereomers (95a) and (95b)"
Indeed a general trend was seen in the lH n.m.r. spectra for atl the
B-bromo-phenylalanine and tyrosine derivatives (82), (85), (94) and (95), and
those described in latter chapters of this thesis. The signal corresponding to the
carboxylate protecting group, be it ester or amide, occurs at lower chemical
shift for the threo-diastereomer than for the corresponding erythro-
diastereomer. Also, the threo-bromides exhibit the B-proton signal at higher
chemical shift, the cr-proton signal at lower chemical shift, and a larger
coupling constant between the c¡- and p-protons, than the corresponding values
for the erythro-isomers. These effects may be explained by studying, as an
example, the most favoured conformations of the threo- and erythro-bromides
(82a) and (82b). The most favoured of the conformations of the bromides
(82a) and (82b) is presumably staggened with the a- and B-protons anti to each
other (Figure 3). This minimizes the number of gauche interactions between
the bulky groups. It can be seen that when the threo-isomer (82a) adopts this
conformation, the phenyl group is situated close to the ester group, and the
shielding effect of the phenyl group may explain the lower chemical shift of the

Chapter I 43
ester group of the threo-isomer (82a) compared to that of the erythro-tsomer
(82b). Also, in the case of the erythro-bromide (82b), æ,æ-stacking may occur
between the aromatic rings of the phthalimido and phenyl groups. This would
cause rotation of the Co-CÞ bond such that the dihedral angle between the a-
and B-protons was less than 180o, thereby explaining the lower coupling
constant observed between the a- and B-protons for the erythro-diastereomer
(S2b) than for the threo-diastereomer (82a) (Figure 3).120
Ph Br
PhthN PhthN
HH
Ph
H
threo-isomer
(82a)
erythro-ísomer
(82b)
Figure 3. Newman projection of the preferred conformation of the
B-bromophenylalanine derivatives (82a) and (82b).
In summary, it has been shown that the protection of various cr-amino
acids as their N-phthaloyl derivatives enables the regioselective radical
bromination of the side chain of these protected cr-amino acids, rather than
functionalization at the o-centre (which occurs with amido- or carbamate-
protected cr,-amino acids). It is apparent, therefore, that the presence of the
phthaloyl protecting group strongly impedes the formation of the
corresponding cx,-centred radical. The destabilization of the cr-centred radical is
attributed to several steric and electronic factors. To obtain maximum
captodative stabilization of the radical (97), the molecule must lie in a planar
orientation such thai maximum orbital overlap between ihe imide, single
electron, and ester orbitals occurs. In this orientation it is clearly seen that
severe steric interactions exist between the phthalimido and ester carbonyl

Chapter I 44
H oI
N NOMe Me
I I Ro R
(e6)
groups, compared to the negligible steric interactions that exist in the benzoyl-
protected amino acid radical (96). The phthalimido group would therefore
twist out of the plane of the sp2-hybridized a-carbon, and would not participate
in stabilization of the radical. Also, the nitrogen electron pair of the imide (97)
is substantially more delocalized than that of the amide (96). The nitrogen
electron pair of the imide (97) is therefore less available for dative radical
stabilization. Thus, abstraction of the cr-hydrogen does not normally occur in
N-phthaloyl-protected ø-amino acids, instead abstraction occurs from the most
reactive position on the side chain if the resultant radical is appropriately
stabilized" Therefore the side chain bromination of various amino acid
derivatives can be achieved, and with retention of the inherent chirality at the
cr-position.
oX
(e7)

Chapter 2 45
RESULTS AND DISCUSSION: CHAPTER 2
Stereocontrolled Synthesis of Homochiral Hydroxy-cr-Amino Acid
Derivatives
The brominated amino acid derivatives (81), (82), (85), (91), (92), (94)
and (95) described in Chapter 1 of the Results and Discussion of this thesis were
thought to be suitable precursors for the synthesis of hydroxy amino acids, the
utility of which has been described in the Introduction. Hence, the synthesis of
the p-hydroxyvaline derivative (100) from the p-bromovaline derivative (81)
was initially attempted. As this transformation involves a nucleophilic
substitution ata tertiary centre, it was presumed that a method which proceeds
via an S¡1 mechanism was required. Silver ion is a strong halophile and has
been shown to promote the substitution of tertiary alkyl halides,95-97 hence
silver nitrate was chosen as a suitable reagent to encourage substitution of the
bromide (81). Many studies of the mechanism of silver ion induced
substitutions of alkyl halides have shown that these reactions occur with graded
S¡1-S¡2 character,9s-97 but that substitution of tertiary alkyl halides can
normally be assumed to proceed via carbocation-type intermediates.95
Kornblum and Hardies9T have reported that substitution of
(S)-a-phenylethylchloride (98) occurs with varying amounts of inversion and
retention of configuration, depending on the solvent and silver salt used in the
reaction. Rearrangement of benzylic halides upon treatment with silver nitrate
provides evidence for carbocation intermediates in these reactions,95 whereas
reaction of (S)-cr-phenylethylchloride (98) with silver nitrite to give
(R)-o-phenylnitroethane (99) with complete inversion of configuration in
acetonitrile (Sc.he.me. 29) indicate.s the importance, of the. rearwarcl displacement
process, or S¡2 nature, of these reactions.95 The low reactivity of neopentyl
halides with silver nitrate compared to the reaction of n-butyl halides also

Chapter 2 46
provides evidence for the S¡2 nature of silver nitrate induced substitutions of
alkyl ¡u1i¿s5.127
Ag+ Nor-
(ee)
.rrrtlCl
(e8)
Scheme 29
Treatment of the bromide (81) with 1.5 equivalents of silver nitrate, in a
1:1 mixture of acetone and water, gave the p-hydroxyvaline derivative (100) in
437o yiel{ as well as the p,y-dehydrovaline derivative (102) (347o) and a small
amount (87o) of the cr,p-dehydrovaline derivative (101) (Scheme 30). The
p-hydroxyvaline derivative (100) was identified using lH n.m.r. spectroscopy,
which showed singlet resonances at ô 1.31 and 1.53, corresponding to the two
B-methyl groups, a singlet at ô 4.91 attributable to the G-proton, and a broad
singlet at õ 4.4I due to the hydroxyl proton.
PhthN CO2Me
+
,^r^*r+CO2Me
Br
AgNO3
acetone / water
tnrhN¿¿¿,.. CO2Me
OH
(100)
(101)
(102)
OnrhN¿¿r,..
+(8 1)
Scheme 30

Chapter 2 41
Identification of the cr,p-dehydrovaline derivative (101) was also achieved
using lH n.m.r. spectroscopy, which showed the absence of any o-proton
resonances, with singlets at õ 1.88 and 2.43 conesponding to the vinylic methyl
groups. The p,1-dehydrovaline derivative (102) was identified by comparison
with the 1i¡"¡¿¡u¡s.128
Using lH n.m.r. spectroscopy with a chiral shift reagent, it was shown that
formation of the alcohol (100) occurred without racemization at the cr-position.
An authentic racemic sample of p-hydroxy-N-phthaloylvaline methyl ester was
prepared via an identical procedure as for the chiral compound (100), but
starting with (RS)-valine" The lH n.m.r. spectrum of a solution of the racemic
material and the chiral shift reagent Eu(hfc)3,rzg in deuteriochloroform,
resolved the enantiomers, showing four singlet resonances corresponding to the
two p-methyl groups of each of the two enantiomers" Two methyl ester
resonances were observed, as well as two hydroxyl proton peaks and two
o-proton peaks. The lH n.m.r. spectrum of a solution of Eu(hfc)3 and the
hydroxyvaline derivative (100), synthesized from (S)-valine, showed
resonances corresponding to only one enantiomer, indicating that the
hydroxyvaline derivative (100) was homochiral at the cr,-position" The
hydroxyl and a-proton peaks of the (S)-enantiomer (100) were observed
upfield of the conesponding peaks of the (R)-enantiomer, and the peaks
corresponding to the p-methyl groups of the (S)-isomer (100) resonated with a
smaller chemical shift difference than the corresponding peaks for the
(R)-isomer.
Presumably silver ion abstracts bromide from the valine derivative (81), to
give the carbocation (103), which is then either quenched by water to give the
alcohol (100), or an cr,- or y-proton is abstracted, leading to formation of the
cr,p-dehydrovaline derivative (101) and p,y-dehydrovaline derivative (102),
respectively (Scheme 31). The greater yield of the p,1-dehydrovaline
derivative (I02) compared to the o,p-dehydrovaline derivative (101) can be
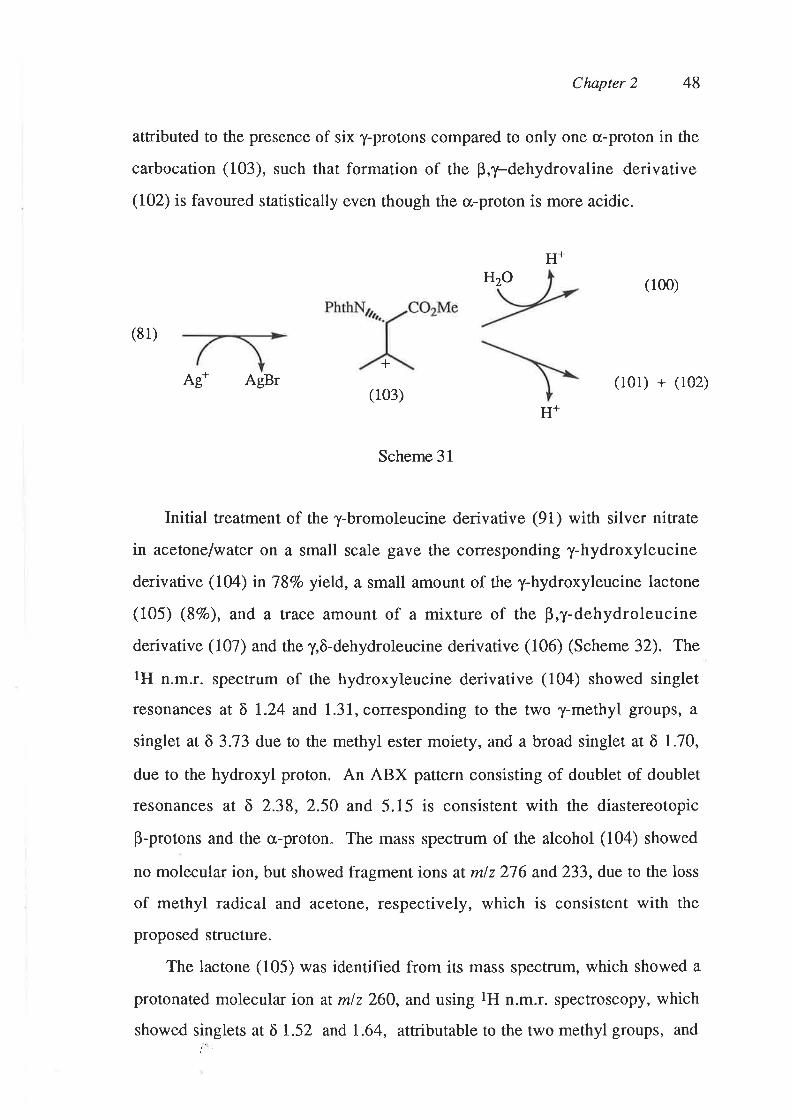
Chapter 2 48
atfibuted to the presence of six y-protons compared to only one o-proton in the
carbocation (103), such that formation of the B,y-dehydrovaline derivative
(102) is favoured statistically even though the cr-proton is more acidic"
(100)
(81)
+Ag* AgBr (101) + (102)
(103)
Scheme 31
Initial treatment of the y-bromoleucine derivative (91) with silver nitrate
in acetonelwater on a small scale gave the corresponding y-hydroxyleucine
derivative (104) in 787o yield, a small amount of the y-hydroxyleucine lactone
(105) (87o), and a trace amount of a mixture of the p,y-dehydroleucine
derivative (107) and the y,ô-dehydroleucine derivative (106) (Scheme 32). The
lH n.m.r. spectrum of the hydroxyleucine derivative (104) showed singlet
resonances at õ I.24 and 1.31, corresponding to the two T-methyl groupS, â
singlet at õ 3.73 due to the methyl ester moiety, and a broad singlet at õ 1.70,
due to the hydroxyl proton. An ABX pattern consisting of doublet of doublet
resonances at ô 2.38, 2.50 and 5.15 is consistent with the diastereotopic
p-protons and the cr,-proton" The mass specffum of the alcohol (104) showed
no molecular ion, but showed fragment ions at mlz 276 and 233, due to the loss
of methyl radical and acetone, respectively, which is consistent with the
proposed structure.
The lactone (105) was identified from its mass spectrum, which showed a
protonated molecular ion at mlz 260, and using lH n.m.r. spectroscopy, which
showed singlets at ô I .52 and I.64, attributable to the two methyl groups, and
HIHzo
H+

Chnpter 2 49
nhthN¿¿¿.,. CO2Me PhthNrr,,.
(106)
+
Ph
BrAgN03
acetone lwater
(104) (10s)
PhthN CO2Me PhthN CO2Me
+(e1)
+
(107)
Scheme 32
an ABX pattern consisting of doublets of doublets at E 2.46,2.60 and 5.25,
consistent with the diastereotopic p-protons and the G-proton. The melting
point of the lactone (105) was identical to that reported in the literature.l3O
The p,y-dehydroleucine derivative (107) and the 1,õ-dehydroleucine derivative
(106) were inseparable by chromatography on silica or on silver nitrate
impregnated silica, a method described for the separation of isomeric
alkenes.13r They were identified using mass spectrometry, which showed a
peak at mlz 273 corresponding to the molecular ions of each of the
dehydroleucine derivatives (106) and (107), and using lH n.m.r" spectroscopy,
with the p,y-dehydroleucine derivative (107) exhibiting a doublet resonance at
õ 5.66, conesponding to the cr-proton, coupled to only one p-proton. The
signal due to the vinylic p-proton was observed at õ 5.82 as a doublet of
multiplets, indicating that the B-proton was coupled to the ü-proton and also vi¿
long-range coupling to the vinylic methyl groups. The y,ô-dehydroleucine
cferivative (106) exhibited singlets at ô 4.65 and 4.69, coresponding to the
terminal vinylic protons, with an ABX system comprising of doublets of
doublets at õ 5.10,3.10 and 2.86 corresponding to the cr-proton andÌ

Chapter 2 50
diastereotopic p-protons. Singlets were observed at õ 1.J6, L.77 and 1J8
which were attributed to the y-methyl group of the y,õ-dehydroleucine
derivative (106) and the y-methyl groups of the p,y-dehydroleucine derivative
(107).
By analogy with reaction of the bromovaline derivative (81),
hydroxylation of the bromoleucine derivative (91) is presumed to proceed viø
the tertiary carbocation (108), with subsequent attack by water leading to
formation of the hydroxyleucine derivative (104), or abstraction of either a õ-
or p-proton leading to formation of the dehydroleucine derivatives (106) or
(107), respectively. Formation of the lactone (105) presumably occurs via
cyclizatíon of the alcohol (104) with loss of methanol. Indeed, the alcohol
(104) slowly converted to the lactone (105) on standing in deuteriochloroform
for extended periods, and further reactions of the alcohol (104) described in
Chapter 4 of the Results and Discussion of this thesis also gave small amounts of
the lactone (105) as a byproduct, providing further evidence for the conversion
of the alcohol (104) to the lactone (105)" The lactone (105) was shown to be
homochiral by comparison with the literature,l30 ¿nd it was concluded that the
alcohol (104) must therefore also be homochiral.
The conversion of the bromide (91) to the alcohol (104) was shown to be
susceptible to a concentration effect. 'When the reaction was performed with
the bromide (91) concentration greater than 0.1M, the lactone (105) was
produced in greater than 607o yield, with only a trace of the alcohol (104).
However, it was found that if the reaction was performed under high dilution
conditions (<0.02M of the bromide (91)), the hydroxyleucine derivative (104)
was consistently isolated in greater thanTO%o yield.
It can be seen that in the reaction of the leucyl bromide (91) with silver
nitrate, the yield of the hydroxyleucine derivative (104) is much greater than
that of the elimination products (106) and (107). In contrast, reaction of the
valyl bromide (81) with silver nitrate occurs to give the substitution product

Chapter 2 5l
(100) and the elimination products (101) and (102) with approximately equal
propensity. This result can be attributed to the leucine y-carbocation (108)
being less hindered than the valine p-carbocation (103), due to the fact that the
y-position is further from the bulky phthalimido and ester moieties. That is, in
the synthesis of the hydroxyvaline derivative (100), steric hindrance at the
p-position of the valine carbocation (103) disfavours substitution, such that
elimination of a y-proton to give the p,y-dehydrovaline derivative (IOZ)
competes effectively with substitution. In contrast, as there is less steric
hindrance at the 1-position of the leucine carbocation (108), substitution to give
the hydroxyleucine derivative (104) is the major reaction pathway.
CO2Me
(108)
+
The method described above therefore represents an expeditious route for
the synthesis of homochiral p-hydroxyvaline and yhydroxyleucine derivatives,
with the chirality at the cr,-centre predetermined by that of the corresponding
chiral, commercially available amino acids.
Synthesis of the y-hydroxyhomophenylalanine derivative (109) was also
performed. Treatment of a 1.5:1 mixture of the diastereomers of the
bromohomophenylalanine derivative (92) with silver nitrate in acetone lwater
gave the y-hydroxyhomophenylalanine derivative (109) (Scheme 33) in 8O7o
yield after chromatography, as a 1.5:1 ratio of diastereomers. A mixture of the
lactones (110) and (111) was also obtained, which was separated by fractional
crystallization to give the cis-lactone (110) in37o yield and the trans-lactone
(111) in27o yield. The y-hydroxyhomophenylalanine derivative (109) was
'ir

Chapter 2 52
identified from its mass spectrum, which showed a molecular ion at mlz 339,
and using lH n.m.r. spectroscopy. The major diastereomer (109a) exhibited a
singlet at õ 3.70 corresponding to the methyl ester moiety, and a broad doublet
of doublets at ô 4.92 conesponding to the y-proton, coupled to the two
p-protons and broadened by coupling to the hydroxyl group. A doublet of
doublets at ô 5.08 was attributable to the G-proton, coupled to the adjacent
p-protons" The minor diastereomer (109b) exhibited a singlet at õ 3.71, a
broad doublet of doublets at ô 4.57, and a doublet of doublets at ô 5.25,
attributable to the methyl ester moiety, the 1-proton and the o-proton,
respectively.
PhthN
+
PhthN PhthN CO2Me
Br AgNo3
o
(110)
acetone/watero
PhthN
+(e2) (10e)
(111)
Scheme 33
The lactones (110) and (111) were identified from their mass spectra, with
the trans-lactone (111) giving a peak at mlz 307, corresponding to the

Chnpter 2 53
molecular ion, and the cis-lactone (110) giving a peak af mlz 308,
corresponding to the protonated molecular ion. The relative stereochemistry of
the lactones (110) and (111) was inferred on the basis of their lH n.m.r.
spectra. Altman et a1.132 have reported that in the lH n.m.r. spectra of related
2,4-disubstituted butyrolactones the chemical shift difference between the
diastereotopic geminal protons is greater for the cis-isomer than for the
corresponding trans-isomer. The lH n.m.r. spectrum of the lactone (111)
showed an ABMX splitting system, with resonances at õ 2.85 (J - 10.6, 12.0
and 12.5 Hz) and õ 2.93 (J - 6.2, 9.2 and 12.5 Hz) conesponding to the
geminal protons. The lactone (110) also showed an ABND( splitting system,
with resonances atõ2.65 (J =3.3,9.8 and 13.l IJz) and ô 3.12 (J = 8.7,9.4 and
13.1 Hz) again corresponding to the geminal protons. On this basis the lactones
(111) and (110) were assigned the trans- and cis-stereochemistry, respectively.
As the diastereomers of the alcohol (109), produced from a 1.5:1 mixture
of the diastereomers of the bromide (92),werc also formed in a 1.5:1 ratio, it
seems likely that substitution of the bromide (92) proceeds viø an S¡2
mechanism. However, as separation of the diastereomers of the bromide (92)
was unsuccessful, the mechanism of this reaction could not be determined
unambiguously. It may be that substitution of the bromide (92) occurs with
substantiat S¡1 character, and fortuitously provides the diastereomers of the
alcohol (109) in the same ratio as that of the sta¡ting bromide (92).
Presumably formation of the lactones (110) and (111) occurs vía
cyclization of the diastereomers of the alcohol (109), with elimination of
methanol, as for production of the leucine y-lactone (105).
It was perceived that the synthesis of p-hydroxyphenylalanine derivatives
could be performed via an analogous procedure to that used in the formation of
the hydroxyvaline, hydroxyleucine and hydroxyhomophenylalanine derivatives
(100), (104) and (109). However, as substitution at the p-carbon of
phenylal4nine introduces a chiral centre adjacent to the G-centre, some

Chapter 2 54
diastereoselectivity was anticipated if the substitution reactions were to proceed
via an S¡1 mechanism. As silver nitrate induced substitutions of alkyl halides
have been reported to proceed with graded SNl-SN2 character, substitution
reactions of the phenylalanine bromide (82) were initially performed on the
separated diastereomers (82a) and (82b) in order to minimize the
stereochemical complexity of the reactions.
Treatment of the threo-bromide (82a) with silver nitrate in acetone lwater
gave a 2:1 mixture of the threo- and erythro-þ-hydroxyphenylalanine
derivatives (112a) and (112b) (Scheme 34). The diastereomeric ratio was
determined by integration of the methyl ester signals of the alcohols (112a) and
(112b) in the 1H n.m.r. spectrum of the crude reaction mixture.
Recrystallization of the crude product gave the threo-þ-hydroxyphenylalanine
derivative (lI2a) in 617o yield, the stereochemistry of which was determined
unambiguously using X-ray crystallographic analysis (Appendix 6).
Purification of the erythro-isomer (112b) in the mother liquor from small
amounts of the threo-isomer (112a) and other impurities was achieved by
HPLC using a Crg reverse-phase column. Repeated elutions of the mother
liquor and collection of the appropriate components provided a small amount of
the pure erythro-isomer (112b) for charactenzation purposes.
PhthN¿¿r,,. CO2Me PhthN CO2Me PhrhNhr,,.
AgN03
Bracetone/water
H
+
(It2a) (1 12b)(82a)
Scheme 34

Chapter 2 55
The lH n.m.r. spectrum of the threo-þ-hydroxyphenylalanine derivative
(llza) showed a doublet resonance at õ 5.51, corresponding to the o-proton, a
doublet of doublets at õ 5.71, due to the B-proton, and a doublet at ô 5.13, due
to the hydroxyl proton. The cr- and p-protons were coupled by 4.6 Hz, and the
hydroxyl and p-protons were coupled by IO.4 Hz" Exchange with D2O
removed the peak corresponding to the hydroxyl proton, and simplified the
p-proton resonance to a doublet, being coupled only to the cr-proton. FAB
mass spectrometry of the alcohol (Il2a) showed a protonated molecular ion at
mlz 326. The lH n.m.r. spectrum of the erythro-isomer (112b) showed a
doublet at õ 4.95 corresponding to the cr-proton, a doublet of doublets at õ 5.46
corresponding to the p-proton, and a doublet at õ 4.27 due to the hydroxyl
proton. The a- and p-protons were coupled by 8.4 Hz, and the hydroxyl and
B-protons were coupled by 2.3 Hz.
The lH n.m.r. spectra of the threo- and erythro-isomers (ll2a) and (112b)
follow a general trend seen for all the p-hydroxyphenylalanine and
p-hydroxytyrosine derivatives described in this thesis, as seen in Appendix 2.
The chemical shifts of the o-, Þ-, and hydroxyl protons of the threo-isomets
are all significantly higher than those of the corresponding erythro-isomers.
Also, the threo-isomers each exhibit a smaller coupling constant between their
c¿- and p-protons, and a larger coupling constant between their p- and hydroxyl
protons, than is observed for the corresponding erythro-isomers.
PhthN o PhthN oHp
Ph
threo-ísomer
(rr2a)
Ph oOMe
HÞ
erythro-iwmer
(1 12b)
H
Figure 4. Hydrogen bonded conformations of the alcohols (112a) and (112b).

Chapter 2 56
The p-hydroxyphenylalanine derivatives (112a) and (112b) may adopt
stable hydrogen-bonded chair conformations as shown in Figure 4. The
presence of these conformations may explain the observed trends in the lH
n.m.r. spectra described above. The dihedral angle between the cr- and
p-protons of the threo-isomer (ll2a) in this conformation would be ca.60". In
contrast, the dihedral angle between the cr- and p-protons of the erythro-isomer
(112b) would be ca. 180", hence the coupling constant between the cr- and
p-protons of the threo-isomer (ll2a) would be smaller than that observed for
the corresponding erythro-isomer (112b). Similarly, the dihedral angle
between the p- and hydroxyl protons in the threo-isomer (ll2a) would be ca.
180o, whereas in the erythro-isomer (112b) it would be ca" 60", thereby
explaining the greater coupling constant between the p- and hydroxyl protons
of the threo-isomer (Il2a) than that of the erythro-isomer (112b)"
The major product of the hydroxylation of the threo-bromide (82a) is
produced with retention of chirality. This is consistent with abstraction of
bromide by silver ion to give the carbocation (113)" The carbocation (113)
may adopt a preferred conformation as shown in Figure 5. In this
conformation, steric interactions betwen the phenyl group and the phthalimido
Hzo
HPhthN
H Ph
OMe
(1 13)
Figure 5. The carbocation (113) in its preferred conformation.
and ester groups are minimized. Molecular modelling studies using MM2
calculations in the modelling program PCMODEL (Serena Software,

Chapter 2 57
Bloomington, Indiana, 1990) provide supporting evidence for this preferred
conformation of the carbocation (113). The ester moiety then blocks one face
of the carbocation (113), such that water attacks preferentially from the
opposite face, giving rise to the threo-isomer (ll2a) as the major product.
Treatment of the erythro-bromide (82b) under the same conditions as
described for the hydroxylation of the threo-bromide (82a) gave only the
threo-hydroxyphenylalanine derivative (ll2a), ín 93Vo yield (Scheme 35).
None of the erythro-isomer (112b) was observed.
"nrhN¿r¿,..Me PhthNfht,. CO2Me
AgNO3
Br acetone lwater
(82b) (rr2a)
Scheme 35
If hydroxylation of each of the bromides (82a) and (82b) proceeded vía the
carbocation (113) then identical ratios of the alcohols (llza) and (112b) would
be observed. Substitution of the bromides (82a) and (82b) must therefore
proceed via different mechanisms. The preferred conformation of the threo-
bromide (82a) is shown in Scheme 36" From this conformation, abstraction of
bromide by silver ion occurs easily, as the phenyl and p-hydrogen substituents
are already in the required orientation to give the most stable conformation of
the carbocation (113). There also exists the potential for stabilization of the
developing carbocation (113) by the ester carbonyl group, as the ester moiety is
situated in the plane of the developing unoccupied orbital. Also, SN2
substitution of the bromide (82a) is disfavoured as the ester moiety blocks
attack of water from behind the carbon-bromine bond. Hence, formation of the
"?ott
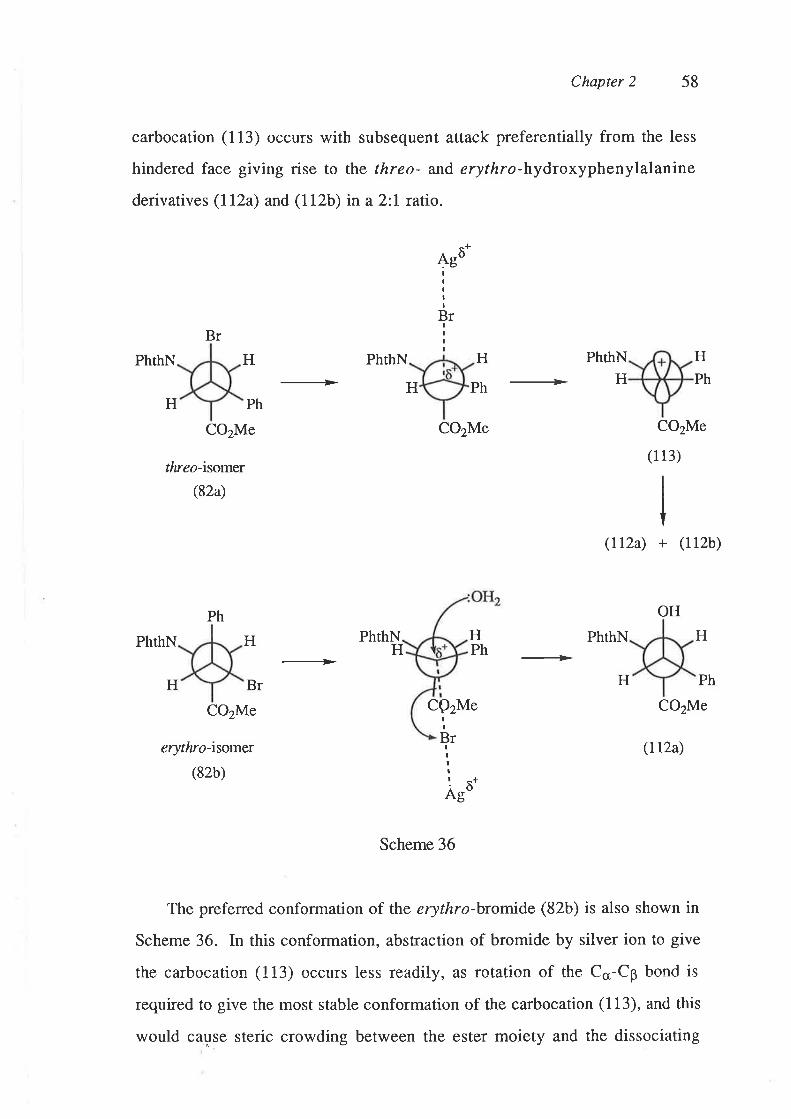
Chapter 2 58
carbocation (113) occurs with subsequent attack preferentially from the less
hindered face giving rise to the threo- and erythro-hydroxyphenylalanine
derivatives (112a) and (112b) in a2:I ratio.
PhthN PhthN PhthN
# H Ph-_+
CO2Me CO2Me CO2Me
threo-isomer
(82a)
OH
PhthN PhthN HPh
PhthN HH +.#
Br H Ph
CO2Me CO2Me
Br
^+
4goIIIIt
BrIIIt
HPhH
HH
PhH
(
2b
3)
(11
11
I+I2a)(1 )
Ph
H
H
erythro-isomer
(82b)
(IrZa)
ô*
The preferred conformation of the erythro-bromide (82b) is also shown in
Scheme 36. In this conformation, abstraction of bromide by silver ion to give
the carbocation (113) occurs less readily, as rotation of the Co-CÊ bond is
required to give the most stable conformation of the carbocation (113), and this
would ca-use steric crowding between the ester moiety and the dissociating
Cp2Me
Scheme 36
II
BrIItII
Ag

Chapter 2 59
bromine atom. In contrast, attack of water from behind the breaking carbon-
bromine bond is not hindered. It is therefore presumed that reaction of the
erythro-bromide (82b) occurs via an S¡2-type mechanism, where the silver ion
initially coordinates to the bromine, and assists in breaking the carbon-bromine
bond, as water attacks from the other side to give the t hr e o -
p-hydroxyphenylalanine derivative (112a) stereospecifically. These results are
consistent with many observations concerning substitutions of alkyl halides in
the presence of silver s¿l¡5.95-97 It is therefore not surprising that in the
reaction of the threo-bromide (82a) with silver nitrate, where S¡2 attack is
sterically disfavoured, substitution appears to proceed via the carbocation
intermediate (113), whereas in the reaction of the erythro-bromide (82b),
where S¡2 attack by water can occur, the product is formed with complete
inversion of configuration.
As hydroxylation of each of the bromides (82a) and (82b) gives rise to the
threo-hydroxyphenylalanine derivative (ll2a) as the major product, it was
decided to carry out this and further hydroxylation reactions using I :1
diastereomeric mixtures of the appropriate bromides, thereby avoiding their
separation. Treatment of a 1:1 mixture of the diastereomers of the
phenylalanyl bromide (82) under the standard conditions described above gave
the threo-p-hydroxyphenylalanine derivative (lI2a) and erythro-
B-hydroxyphenylalanine derivative (1 12b) in the expected 5:1 ratio.
In order to check the hypothesis for the observed diastereoselectivity in the
hydroxylation of the bromides (82a) and (82b) a more bulky ester group was
used, anticipating that it would increase the stereoselectivity of the reaction.
Hence the synthesis of the phenylalanine tert-butyl ester derivative (114) was
performed. Treatment of a solution of (S)-N-phthaloylphenylalanine (70) in
dichloromethane wíth tert-butanol, dicyclohexylcarbodiimide (DCC) and a
catalytic amount of 4-(N,N-dimethylamino)-pyridine (DURP¡t33 gave the
desired ester (114) in 757o yield. Bromination of the tert-butyl ester (114) was

Chapter 2 60
performed under identical conditions as described for that of the methyl ester
(74),and gave a 1:1 mixture of the bromide diastereomers (115a) and (115b)
(Scheme 37).
PhthN nhthN¿¿r,,. cMqr-BuOH NBSDCCDMAP
....'....+p1
(114)
Scheme 37
Separation of the bromides (115a) and (115b) was achieved by fractional
recrystallization from hexane/dichloromethane, giving diastereomer (115a) in
4l7o yield and diastereomer (115b) in 39Vo yield. Determination of the
configuration of the bromides (115a) and (115b) was accomplished by
comparison of their lH n.m.r. spectral data with that of the bromoesters (82a)
and (82b) and the bromoamides (85a) and (85b), as described in Chapter 1
(Appendix 1)" The lH n.m.r. specftum of the bromide (115a) showed a singlet
at ô 1.16, corresponding to the tert-butyl group, and doublets at ô 5.47 and
6.06, conesponding to the o- and p-protons, respectively, with a coupling
constant between those protons of 11 .4 Hz. The lH n.m.r. specÍum of the
bromide (115b) showed a singlet at õ 1.48, corresponding to the tert-bufyl
group, and doublets at õ 5.49 and 5.85, corresponding to the cr- and B-protons,
respectively, with a coupling constant between those protons of 10.4 Hz. The
bromide (115a) whose tert-butyl group signal occurred at ô 1.16 was therefore
assumed to be the threo-isomer, and the bromide (115b) whose tert-butyl group
signal occured at õ 1.48 was assumed to be the erythro-isomer, with the lH
n.m.r. speptral data following the trend described in Chapter 1.
(70) (1 1s)
(a) Rl =Br, R2=H(b) Rl=H, R2=Br

Chapter 2 61
PhthN CMe3 PhthN nnrhN¿r,,,.
AgN03
-+pl acetone/water
+
(1 1s)
(a) Rl=Br, R2=H(b) Rl =H, R2=Br
(1 16a) (1 16b)
Scheme 38
Hydroxylation of a 1:1 mixture of the bromide diastereomers (115a) and
(115b) was performed under standard conditions as previously described to
give amixture of the alcohols (116a) and (116b) (Scheme 38). Integration of
thetert-butyl ester signals of the alcohols (116a) and (116b) in the lH n.m.r.
spectrum of the crude reaction mixture indicated that the alcohols (116a) and
(116b) were produced in an 8:1 ratio. The threo-isomer (116a) was isolated in
SlVo yietd after recrystallization and was identified using lH n.m.r.
spectroscopy. A singlet was observed at ô 1.52 due to the tert-butyl group"
Doublet resonances at ô 5.25 and 5.44 were attributed to the hydroxyl and
q-protons, respectively. A doublet of doublets at ô 5.67 was attributed to the
p-proton, coupled to the ct,-proton by 4.6 Hz" and to the hydroxyl proton by
10.6 Hz. The chemical shifts and coupling constants of the c-, p- and hydroxyl
protons correlated closely to those of the threo-þ-hydroxyphenylalanine methyl
ester derivative (ll2a) (Appendix 2). Signals corresponding to Íhe erythro-
isomer (116b) were observed in the lH n.m.r. spectrum of the crude product.
Doublet resonances were observed at õ 4.95 and 5.49, corresponding to the cr-
and p-protons, respectively. The hydroxyl proton was observed as a broad
singlet at õ 4.60, and the tert-butyl ester signal was observed at õ 1.28.
It can be seen that there is an approximately two-fold increase in the
diastereoselectivity observed in the substitution of the tert-butyl ester bromide

Chapter 2 62
(115) compared to that observed in the substitution of the methyl ester bromide
(82). Presumably, by analogy with the reaction of the erythro-bromide methyl
ester (82b), the reaction of the erythro-bromide (115b) occurs with complete
inversion of stereochemistry. The reaction of the threo-bromide (115a) must
therefore occur to give an approximately 4:l ratío of the threo- and erythro-
alcohols (116a) and (116b), such that an 8:1 ratio of the alcohols (116a) and
(116b) is produced from a 1:1 mixture of the bromides (115a) and (115b)" The
increased selectivity observed for reaction of the terl-butyl ester derivative
(115) can be attributed to the larger tert-butyl group more effectively blocking
one face of the intermediate carbocation (117), than the methyl group in the
methyl ester,,carbocation (113), and provides further evidence for the
hypothesis described above explaining the preferential formation of the threo-
p-hydroxy-phenylalanine deriv atív e (l l2a).
H
(1 17)
In order to further examine the above hypothesis, hydroxylation of the
bromophenylalaninamide (85) was also performed (Scheme 39), under the
standard conditions. The threo-p-hydroxyphenylalaninamide (1 18a) was
isolated in excellent yield (937o) after recystallization, and was identified using
FAB mass spectrometry, which showed a protonated molecular ion at mlz 367,
and rH n.m.r. spectroscopy (see Appendix 2) as described below. No peaks
atftibutable to the erythro-isomer (118b) could be identified in the 300MHz
lH n.m.r. spectrum of the crude reaction mixture, indicating that the
hydroxylation reaction had proceeded to give the threo-isomer (118a) in

Scheme 39
> 95Vo diastereomeric excess. To calculate the diastereomeric excess more
accurately, the lH n.m.r. spectrum of the erythro-þ-hydroxyphenylalanine
derivative (118b) was required" The synthesis of the erythro-isomer (118b)
was proposed yi¿ racemizatton at the p-centre of the threo-isomer (118a). This
was achieved by oxidation of the alcohol (118a) to the ketone (119), with
subsequent reduction of the ketone (119) to give a mixture of the threo- and
erythro-atcohols (118a) and (118b) (Scheme 40).
ththN¿¿,,..
CrO" / H+(118a)
-'+
PhrhN CONHCMe3
plB2
(85)
(a) Rl=Br, R2=H(b) Rl=H, R2:Br
AgN03
acetone/water
Chapter 2 63
CONHCMe3
(1 18a)
+ (118a)
(1 18b)
CMe3
NaBHa
(1 1e)
Scheme 40
Treatment of the alcohol (118a) with Jones reagentl34 gave the ketone
(119) which was identified using mass spectroscopy, which showed a peak at
mlz 364 corresponding to the molecular ion, and using lH n.m.r" Spectroscopy.
A singlet was observed at õ 6.04, which was attributed to the cr-proton, with no
other siglals observed between õ 2-l " The crude ketone was reduced by

Chapter 2 64
treatment with a mixture of sodium borohydride in ethanol, with the reaction
being quenched by the addition of dilute hydrochloric acid after 10 minutes.
Care was taken to ensure that the reaction time was kept to a minimum, or the
yield of the alcohols (118a) and (118b) was greatly reduced, presumably due to
reduction of the phthalimido group.l3s The product was isolated in 847o yield
as an approximately 1.2:l ratio of the threo- and erythro-diastereomers (118a)
and (118b). It is possible that reduction of the ketone (119) with sodium
borohydride may be accompanied by racemization at the o-centre, due to the
acidic nature of the cr -hydrogen of the ketone (1 19), but as the
oxidation/reduction of the threo-a\cohol (118a) was only performed to obtain
the lH n.m.r" spectrum of the erythro-alcohol (118b) the chirality at the
cl,-centre is of no consequence. The lH n.m.r. spectrum of the threo-isomer
(118a) showed a singlet at ô 1.30, attributable to the tert-butyl group, doublets
at ô 4.41 and 5.11 due to the hydroxyl and a-protons, respectively, and a
doublet of doublets at õ 5.63 due to the p-proton. The lH n.m.r. spectrum of
the erythro-isomer (118b) exhibited a singlet at ô 1.15, due to the tert-butyl
group, characteristic doublets at õ 4.25 and 4.60, due to the hydroxyl and
G-protons, respectively, and a doublet of doublets at ô 5.39, due to the
B-proton. The stereochemistry of the alcohols (118a) and (118b) was
determined by correlation of their 1H n.m.r. spectra with other
p-hydroxyamino acid derivatives, as shown in Appendix 2, which indicated that
the product formed upon hydroxylation of the bromoamide (85) was the threo-
isomer (118a).
The lH n.m.r. spectrum of the crude product from reaction of the
bromoamide (85) with silver nitrate in acetone lwater is shown in Figure 6. No
peak corresponding to the tert-butyl group of the erythro-\somer (118b) can be
seen. Indeed, one of the two 13C satellites of the peak due to rhe tert-butyl
group of the threo-isomer (118a) can be seen at õ 1.08 and, as the natural
abundance of the 13C isotope is ca. l.l 7o, the area of this peak constitutes about

Chapter 2 65
0.5Vo of the area of the signal due to the tert-butyl group of the threo-isomer
(118a). If it is assumed that a peak being of one tenth the area of the l3C
satellite can be visualized, then it can be determined that the amount of the
erythro-isomer (118b) present in the crude mixture is less than 0.057o of that
of the threo-isomer (118a). Therefore the threo-alcohol (118a) is produced in
greater Íhan 99.9Vo diastereomeric excess (d.e.).
tert-butyl signal of (118a)
\
13C satellite
chemical shift oftert-búyl signal of (118b)
13C satellite
1.5 1.)PPM
Figure 6. lH n.m.r. of the crude product from substitution of the bromide (85)"
Within the limits of detection the hydroxylation of the bromoamide (85)
occurs diastereospecificalty, giving only the threo-þ-hydroxyphenylalanine
derivative (118a). Much greater selectivity is seen in the hydroxylation of the
bromoamide (85) than the bromoester (82), with the increase in selectivity
being at least 100-fold. This pronounced increase in selectivity may be due to
stabilization of the intermediate carbocation (120) by the amide carbonyl

Chapter 2 66
group. The carbonyl oxygen of an amido group is about 106 times more basic
than the carbonyl oxygen of an esls¡.136 Stabilization of the amide carbocation
(I2O) as shown in Figure 7 may therefore lock the carbocation (120) into a
specific conformation, such that water can only attack from one side to give the
thr eo -þ-hydroxyphenylalanine derivative ( I 1 8a) s tereospecifically.
PhthN
H
H
\ ô*NHCMe3
(120)
Figure 7. The carbocation (120) in the preferred conformation, withstabilization by the amido substituent.
It can be seen from the above examples that bromination of N-phthaloyl-
protected phenylalanine derivatives at the p-position and subsequent treatment
of the bromides with silver nitrate in acetone lwater provides a fast and efficient
method for the production of threo-þ-hydroxyphenylalanine derivatives with
control of stereochemistry at both the a- and p-centres.
To determine the generality of the method described above for the
synthesis of the p-hydroxyphenylalanine derivatives (ll2), (116) and (118),
hydroxylation reactions were also carried out with the p-bromotyrosine
derivatives (9a) and (95). Treatment of a 1:1 mixture of the diastereomers of
the bromotyrosine ester (94) under the standard hydroxylation conditions
proceeded to give the p-hydroxytyrosine derivative (121) (Scheme 4l) in 867o
yield, as a 6:1 ratio of diastereomers, consistent with the 5:1 ratio observed in
the hydroxylation of the phenylalanine derivative (82). The diastereomeric
ratio was determined using lH n.m.r. spectroscopy, as for the phenylalanine
derivatives (112a) and (1I2b), with the major alcohol (lzla) exhibiting peaks at
I

PhrhN
Blp2
(e4)
(a) Rl=Br, R2=H(b) Rl =H, R2=Br
AgN03
acetone lwater
Chapter 2 67
plB2
(r2r)
(a) Rl=OH, R2=H(b) Rl=H, R2=OH
Ph
MeOH /p-TsOH
Ph CO2Me PhthN CO2Me
+
(122a) (r22b)
Scheme 41
E 2.L9 and 3.78 due to the acetyl and methoxycarbonyl groups, repectively, and
the minor alcohol (121b) giving peaks at õ 2.17 and 3.14 due to the acetyl and
methoxycarbonyl groups, respectively. The FAB mass spectrum of a mixture
of the hydroxytyrosine derivatives (121a) and (121b) showed an ion at mlz 384,
corresponding to the protonated molecular ions. However, the hydroxytyrosine
derivatives (121a) and (121b) were unstable and deacetylated slowly on
standing in air or in solution. Hencg, the crude product was deacetylated by
treatment with a catalytic amount of p-toluenesulfonic acid in methanol
(Scheme 41) to give a 6:1 ratio of the hydroxytyrosine derivatives (122a) and
(122b)" The threo-isomer (L22a) was isolated in 727o yield after
recrystallization, and was identified using FAB mass spectrometry, which
showed a protonated molecular ion at mlz 342, and using lH n.m.r.
spectroscopy. Characteristic doublet resonances were observed at ô 5.02 and
HO

Chapter 2 68
5.44, due to the p-hydroxyl and a-protons, respectively, and a doublet of
doublets was observed at õ 5.64, due to the p-proton. The o- and p-protons
were coupled by 4.8 Hz, and the hydroxyl and p-protons were coupled by 10.3
tlz. Purification of the erythro-isomer (122b) was performed by HPLC using a
C1g reverse-phase column, as for the phenylalanine derivative (112b). The
lH n.m.r. spectrum of the erythro-isomer (122b) showed characteristic doublet
resonances at E 4.27 and 4.97, due to the p-hydroxyl and G-protons,
respectively, and a doublet of doublets at ô 5.49, due to the p-proton. The a-
and p-protons were coupled by 8.6 Hz, and the hydroxyl and p-protons were
coupled by 2.1Hz. The lH n.m.r. spectra of the p-hydroxytyrosine derivatives
(122a) and (122b) correlate very closely with those of the
p-hydroxyphenylalanine derivatives (1 l2a) and (112b), and were used to assign
the stereochemistry of the p-hydroxytyrosine derivatives (122a) and (I22b)
(Appendix 2).
p1 AgNO3
acetone/water
PhCMe3
",?oH
(es)
(a) Rl=Br, R2=H(b) Rl =H, R2=Br
(123)
MeOH /p-TsOH
CONHCMe3
",?oH
Scheme 42
(r24)

Chapter 2 69
Hydroxylation of the bromotyrosinamide (95) was also performed under
the standard conditions, giving the alcohol (123) (Scheme 42). Deacetylation of
the alcohol (123) gave the threo-þ-hydroxytyrosinamide (124) in 937o yield
after recrystallization. The hydroxytyrosine derivative (124) was identified
using FAB mass spectrometry, which showed a protonated molecular ion at mlz
383, and from its lH n.m.r. spectrum, which showed characteristic doublets at
õ4.12 and 5.11 due to the hydroxyl and cr-protons, respectively, and a doublet
of doublets at õ 5.62 due to the p-proton. The coupling constant between the p-
and hydroxyl protons was 7 .5 Hz, and the coupling constant between the cr- and
p-protons was 6.9 Hz. A singlet at õ 1 .27 was attributed to the tert-butyl
group. No peaks near ô 1"15 were observed in the 300MHz lH n.m.r.
spectrum of the crude product, which is where the peak due to the tert-butyl
group of the erythro-ísomer would be expected by comparison with the spectral
data of the phenylalanine derivative (118b). This indicates that the
hydroxylation reaction had proceeded in greater than 957o d.e., and it is
probable that the hydroxylation reaction occurs with greater than 99.97o
stereospecificity, as in the case of the formation of the hydroxyphenylalanine
derivative (118a).
The above procedure is therefore also applicable to the synthesis of threo-
B-hydroxytyrosine derivatives with known chirality at both the a- and
p-positions. As for the syntheses of the phenylalanine derivatives (112a),
(116a) and (118a), the diastereoselectivity observed in the reactions to give the
tyrosine derivatives (121a) and (L23) can be attributed to preferential attack
from one face of each of the corresponding intermediate carbocations. The
greater selectivity seen in the reaction to give the tyrosinamide (123) compared
to that observed in the synthesis of the tyrosine methyl ester (121a) is consistent
with the stereoselectivity observed in the reactions to give the corresponding
phenylalanine derivatives (ll2a) and (118a), and provides supporting evidence
for the effect of the amido substituent to increase the diastereoselectivity"

Chapter 2 lO
The above procedures for the synthesis of hydroxyamino acid derivatives
are of limited utility unless these derivatives can be converted to the
corresponding free hydroxy-a-amino acids without racemization at the chiral
centres. Hydrolysis of the hydroxyphenylalanine ester derivative (112a) and
the hydroxyphenylalanine amide derivative (1 18a) to (253R)-p-hydroxy-
phenylalanine (25a) was therefore investigated. Initially a mild two-step
procedure for the hydrolysis of the ester (lI2a) was examined. Removal of
phthaloyl protecting groups by treatment with hydrazine is an established
procedu¡s.137 Treatment of the hydroxy ester (llza) with a 2M solution of
hydrazine hydrate in ethanol to remove the phthaloyl group, followed by mild
acid hydrolysis of the ester in 1N HCI solution, gave the hydrochloride salt of
the amino acid (25a). Treatment of a solution of the hydrochloride salt in
ethanol with excess aniline resulted in crystallization of the free amino acid
(25a) in 8O7o yield (Scheme 43). A more vigorous but one-step procedure was
also performed, by treatment of the ester (ll2a) in a refluxing 2:1 mixture of
6N hydrochloric acid and glacial acetic acid. This gave the hydrochloride salt
of the amino acid (25a), which was treated with aniline in ethanol to give the
free amino acid (25a) in 937o yield (Scheme 43)" Identification of the free
amino acid (25a) was achieved by comparison of its lH n.m.r. spectrum with
that of a commercially available racemic sample, and by mass spectrometry,
which showed a protonated molecular ion at mlz 182. An optical rotation value
of -49.7" for the free amino acid (25a) obtained via the one-step hydrolysis was
determined in 6N HCI solution at 16o, which is consistent with literature
values,l38 indicating that the hydrolysis had occurred without racemization.
Hydrolysis of the amide (118a) was performed under conditions identical
to those described for the one-step hydroysis of the ester (112a), to give the
free amino acid (25a) in 78Vo yield (Scheme 44). The optical rotation value of
the free amino acid (25a) obtained from hydrolysis of the amide (118a) again
indicated that no racemízation had occurred.

Chapter 2 7l
PhthNr. either Ð NH2NH2iÐ lN HCriiÐ PhNHÆroH
CO;
or Ð 6N HCI /AcOHii) PtrNH2ÆtOH
(rr2a) (25a)
Scheme 43
Ð 6N HCI / AcOH(25a)
ü) PhNHr/EtOH
(1 18a)
Scheme 44
Hence, the side chain bromination of N-phthaloyl amino acid derivatives
followed by treatment of the bromides with silver nitrate and water provides an
efficient route to the synthesis of hydroxyamino acid derivatives, with control
of both relative and absolute stereochemistry, with subsequent deprotection
steps providing the corresponding homochiral hydroxy-substituted free amino
acids.
+
"%oll

Chapter 3 12
RESULTS AND DISCUSSION: CHAPTER 3
Towards a Synthesis of Chloramphenicol
The development of the methods for the synthesis of p-hydroxyamino acid
derivatives described in Chapters 1 and 2 of the Results and Discussion of this
thesis prompted an investigation of the synthesis of the alcohol (129) as a
potential precursor to the antibiotic chloramphenicol (28). It was envisaged
that the alcohol (I29) could be prepared from (R)-p-nitrophenylalanine (125) as
outlined in Scheme 45, using a route analogous to that described for the
synthesis of the hydroxyphenylalanine derivative (ll2a). This work was
conducted concurrently with that described in Chapter 2 of the Results and
Discussion of this thesis, and consequently the potential benefits of utilizing
amide rather than ester derivatives of phenylalanine in the synthesis of
p-hydroxyphenylalanine derivatives were not exploited. Although outside the
scope of the present work, the alcohol (129) can be expected to be a suitable
precursor for the synthesis of chloramphenicol (28). Indeed, Chenevert and
ThiboutotaT have elaborated a racemic sample of the amino alcohol (130) to
chloramphenicol (28).
Protection of the commercially available amino acid (125) was
accomplished via the same method as described for the protection of
(S)-phenylalanine. Treatment of a mixture of phthalic anhydride and
(R)-p-nitrophenylalanine (IZ5) at 140' for 30 minutes gave the N-phthaloyl-
protected amino acid (L26) in 817o yieId. The reaction time and temperature
were both kept to a minimum, as prolonged heating caused discolouration of
the mixture and reduced the yield of the product (126). Esterification was
perforrned by treatment of the protected amino acid (126) with a methanolic
solution of hydrogen chloride, giving the ester (121) in 860/o yield (Scheme 45).

Chapter 3 13
+H
PhrhN
(a) Rl =O) Rl=
PhthN
PhrhN
ozN
ozN
ozN
ozN
(r2s) (126)
p2
PhrhN
CI
HN
(t27)
(28)
(128)
Br, R2=HH, R2=Br
(r29)
--
ozN
ozN
Scheme 45
R
CI
ozN
HzN
(130)
CO2Me

Chapter 3 14
Bromination of the protected amino acid (127) was achieved by treatment
with a refluxing solution of NBS in CCI¿ for 4 hours, while the mixture was
irradiated with a 250W mercury lamp, and gave a 1:1 ratio of the bromide
diastereomers (128a) and (128b) in quantitative yield. The reaction time
required to effect bromination of the p-nitrophenylalanine derivative (I27) was
twice that required for bromination of the corresponding phenylalanine
derivative (74). This is presumably due to the strong electron withdrawing
effect of the p-nitro substituent, such that hydrogen atom abstraction from the
benzylic position by the electrophilic bromine radical, with consequent
development of a partial positive charge at that position, is disfavoured.
The bromides (128a) and (128b) were separated by fractional
recrystallization, giving diastereomer (128a) in 44Vo yield, and diastereomer
(128b) in 407o yield. Identification of the bromides (128a) and (128b) was
achieved using mass spectrometry, which showed molecular ions at rnlz 432 and
434 of equal abundance in each case. lH N.m.r. spectroscopy showed
characteristic doublets for diastereomer (128a) at õ 5.51 and 6.O2, due to the cr-
and p-protons, respectively, with a coupling constant between these protons of
II.2 llz" A singlet was observed at ô 3.59, due to the methyl ester moiety.
Diastereomer (128b) showed doublets at ô 5.59 and 5.9J, due to the cr- and
p-protons, respectively, with a coupling constant between these protons of 10.3
Hz. The signal due to the methyl ester group was observed at ô 3.83. By
analogy with the corresponding values obtained for bromides described in
Chapter 1 of the Results and Discussion of this thesis (Appendix 1) this data
indicates that the bromide (128a) has the threo-stereochemistry, and the
bromide (128b) has the erythro-stereochemistry. The stereochemical
assignments were confirmed by X-ray crystallographic analysis of the erythro-
bromide. (128b) (Appe.ndix 7).
Initially hydroxylation of a mixture of the bromides (128a) and (128b) was
attempte{ using the same conditions as for the preparation of the

Chaprer 3 75
p-hydroxyphenylalanine derivatives (llZa) and (112b). However, after stirring
a 1:1 mixture of the bromides (128a) and (128b) and silver nitratè in
acetone/water solvent for 16 hours at room temperature, no reaction had
occurred, and the bromides (128a) and (128b) were recovered. The lack of
reactivity of the bromides (128a) and (128b) under these conditions is
attributable to the electron withdrawing effect of the p-nitro group such that
formation of the benzylic carbocation (131) is disfavoured.
PhthN Me
+ H
ozN(131)
It was thought that more vigorous conditions were required to achieve
substitution of the bromides (128a) and (128b). Accordingly, the threo-
bromide (128a) was treated with silver nitrate in acetone/water at 50" for 2
days. However, under these conditions the (Z)-dehydrophenylalanine
derivative (132) was isolated in high yield, with none of the alcohol (I29)
formed (Scheme 46). Treatment of the erythro-bromide (128b) under
conditions as described above for the reaction of the threo-bromide (128a) gave
the threo-p-hydroxy-p-nitrophenylalanine derivative (L29) in 657o yield, with
I57o of the (Z)-dehydrophenylalanine derivative (132) and lOTo of the
(E)-dehydrophenylalanine derivative (1 33) (Scheme 47).
PhthN Me PhthN
AgNO3
acetone/water
500, 2d
Me
ozN
(128a)
Br
Scheme 46
ozN
(r32)

Chapter 3 76
PhthN PhthN CO2Me+ (r29)
AgNO3,a
Br acetone/water
500,2d+ (132)
ozN
(128b) (133)
Scheme 47
The alcohol (129) was purified by column chromatography on silica and
was identified using lH n.m.r. spectroscopy. Characteristic doublets were
observed at ô 5.34 and 5.53, due to the hydroxyl and G-protons, respectively.
A doublet of doublets was observed at ô 5.79, corresponding to the p-proton,
which was coupled by 10.0 Hz to the hydroxyl proton and by 4.4 Hz to the
o-proton. The lH n.m.r. spectrum of the alcohol (129) is very similar to that
of the threo-þ-hyd¡oxyphenylalanine derivative (llZa), and indicates that the
B-hydroxy-p-nitrophenylalanine derivative (I29) has the threo-stereochemistry
(Appendix 2). The dehydrophenylalanine derivatives (132) and (133) were
identified using mass spectrometry, with each giving a molecular ion at mlz
352, and from their lH n.m.r. spectra. The (Z)-isomer (132) showed a singlet
at ô 8.13 corresponding to the vinylic proton. The singlet signal due to the
vinylic proton of the (E)-isomer (133) was observed at õ 7.28, upfield of that
of the (Z)-isomer (132), consistent with the trend described in Chapter I of the
Results and Discussion of this thesis.
Presumably S¡1 substitution of the bromides (128a) and (128b) does not
occur as formation of the intermediate carbocation (131) is disfavoured due to
the electron withdrawing effect of the p-nitro group. The conformations of the
bromides (128a) and (128b) in which S¡2 substitution would be expected to
occur most readily are shown in Figure 8, such that attack from behind the
carbon-bromine bond is not hindered. These conformations, however, are also
the conformations required for E2 anti-elimination of hydrogen bromide from
Noz

Chapter 3 77
the p-nitrophenylalanine derivatives (128a) and (128b). Thus, in the reaction
of the threo-bromide (128a), elimination is the predominant pathway and the
(Z)-dehydro-p-nitrophenylalanine derivative (I32) is formed. In the reaction
of the erythro-bromide (128b), E2 elimination of hydrogen bromide is less
favourable as this produces the less stable (E)-dehydro-p-nitrophenylalanine
derivative (133).r23,r24 Substitution to give the alcohol (129) therefore
competes effectively with elimination of hydrogen bromide. The substitution
reaction of the erythro-bromide (128b) to give the alcohol (129) is analogous to
the formation of the threo-p-hydroxyphenylalanine derivative (112a) from the
ery thro -bromide (82b).
p-O2N-Ph H H Ph-p-N02
PhthN PhthN CO2Me
HH
BrBr
threo-isomer
(128a)
erythro-ísomet
(128b)
Figure 8. Conformations of the bromides (128a) and (128b) most susceptible toboth S¡¡2 andE2 pathwâys
Formation of the alcohol (129) was not always reproducible, the yield
ranging between I0 - 65Vo, with the dehydrophenylalanine derivatives (132)
and (133) making up the remainder of the products. Various conditions were
employed in an attempt to maximize the yield of the alcohol (129), by adjusting
the temperature, solvent ratio and amount of silver nitrate added, but optimum
conditions were not found. However, further work in this area would be
expected to provide a system which gives the alcohol (129) in a reasonable and
consistent yield.

Chapter 4 18
RESULTS AND DISCUSSION: CHAPTER 4
Scope and Limitations of the Elaboration of Bromo-cr-Amino Acid
Derivatives
Having developed procedures for the synthesis of p-hydroxyamino acids
with control of relative and absolute stereochemistry, the scope and limitations
of further elaboration of side chain brominated N-phthaloyl-cr,-amino acid
derivatives were investigated. It was conceived that the bromoamides (85a) and
(85b) could be converted to 3,4-disubstituted p-lactams. Formation of
B-lactams via cyclization of p-bromopropionamides has been reported,l39 as
has cyclization of other p-substituted amides, such as those derived from
p-hydroxy-cr-amino u.1¿5.140 Cyclízation via N-C4 bond formation was
expected to provide a method for the stereocontrolled synthesis of
3,4-disubstituted p-lactams, with the stereochemistry of the products
predetermined by that of the precursor cr,p-disubstituted amides.
Treatment of the bromide (85a) with a solution of potassium amide in
ammonia, prepared by the addition of potassium and a catalytic amount of
ferric nitrate to liquid ammonia,l4l gave the trans-p-lactam (134) in good
yield, with a small amount of the (Z)-dehydrophenylalanine derivative (86)
(Scheme 48). The mass spectrum of the p-lactam (134) showed a molecular ion
at mlz 348. The lH n.m.r. spectrum showed doublets at ô 4.96 and 5.01, due to
the C4 and C3 protons, respectively. The coupling constant between these
protons was 2.3 Hz, consistent with the 3,4-disubstituted p-lactam (134)
pos ses sing the t rans - stereochemistry. t+z
Presumably the bromide (85a) reacts initially by loss of the amide proton.
The product anion can then undergo an intramolecular S¡¡2 cyclization to form
the trans-þ-lactam (134) yia substitution at the benzylic position. Alternatively,
intermole ular proton transfer affords the corresponding cr-anion which reacts

Chapter 4 79
by elimination of bromide to form the (Z)-dehydrophenylalanine derivative
(86). The intermolecular nature of the proton transfer to give the a-anion was
determined by performing the above reaction with varying concentrations of
the bromide (85a). It was found that as the concentration of the bromide (85a)
decreased, the ratio of the p-lactam (134) to the (Z)-dehydrophenylalanine
derivative (86) increased. The rate of the proton transfer reaction is therefore
dependent on the concentration of the bromide (85a), indicating a bimolecular
reaction, whereas cyclization to give the p-lactam (134) is unimolecular with
the rate being independent of the concentration of the bromide (85a).
CMe3PhrhN
K+ NH2-(86)+
Br
(85a) (134)
Scheme 48
Treatment of the bromide (85b) under conditions identical to those used in
the preparation of the p-lactam (I34) from the bromide (85a) did not give the
cis-p-lactam (136), but instead gave the B-aminophenylalanine derivative (135)
in 93Vo yield (Scheme 49). The amine (135) was identified from its FAB mass
spectrum, which showed a protonated molecular ion at mlz 366, and from its
lH n.m.r. spectrum, which showed doublet resonances at õ 4.63 and 5.77, due
to the cr- and p-protons, respectively. The amine (135) is presumed to have the
threo-stereochemistry, assuming the reaction proceeds vi¿ an SN2 mechanism
based on analogy with the reactions of the erythro-bromides (82b) and (85b) to
give the correspondíng ihreo-p-hydroxyphenylalanine derivatives (i 12a) and
(118a), as described in Chapter 2 of the Results and Discussion of this thesis.

Chapter 4 80
ththN¿¿,... CMe3 CONHCMe3
either K+NH2-
or NH4OHrfrf'?l.tn
2
(8sb) (13s)
Scheme 49
The difference in the above reactions of the bromides (85a) and (85b) may
be explained by examining the preferred conformations of the bromides (85a)
and (85b) (Figure 9a)" It can be seen that the preferred conformation of the
threo-bromide (85a) has the amido group in the required orientation for
displacement of bromide, and hence ring closure to form the trans-p-lactam
(134) occurs readily. The erythro-bromide (85b), however, must undergo
rotation of the Co-CÞ bond from its preferred conformation for the molecule
to be aligned for cyclization. This results in severe steric repulsion between the
PhthN CONHCMe3 PhthN CONHCMe3
threo-isomer erythro-isomer
(85a) (8sb)
Figure 9a. Preferred conformations of the bromides (85a) and (85b).
H
PhrhN NHCMe3
Figure 9b. Conformation of the bromide (85b) required to form the
cis-p-lactam (136).
HHBrPhPhBr
H
Ph
H
Br

Chapter 4 81
(136)
phthalimido, phenyl and tert-búylamido groups (Figure 9b), such that
formation of the cis-p-lactam (136) is disfavoured. Substitution of the erythro-
bromide (S5b) at the benzylic position to give the threo-p-aminophenylalanine
derivative (135) is therefore the most favourable process.
The amine (135) was also produced by treatment of a solution of the
erythro-bromide (85b) in tetrahydrofuran (TFß) with concentrated aqueous
ammonia. Treatment of the threo-bromide (85a) under identical conditions
gave only the (Z)-dehydrophenylalanine derivative (86). The difference
between these reactions is presumably a result of the relative stabilities of the
(Z)- and (E)-dehydrophenylalanine derivatives (86) and (87). anti-Elimination
of hydrogen bromide from the threo-bromide (85a) gives the
(Z)-dehydrophenylalanine derivative (86), whereas anti-elimination from the
erythro-bromide (85b) is less favourable as the product would be the less stable
(E)-isomer (87). Substitution of Íhe erythro-bromide (85b) at the benzylic
position to give the threo-p-aminophenylalanine derivative (135) therefore
occurs preferentially.
Having produced the trans-p-lactam (134) from the bromoamide (85a) it
was of interest to ascertain whether synthesis of the corresponding p-lactone
(139a) could be achieved. p-Lactones have recently received much interest due
to their antibiotic propertiesr43-r4s and their potential as precursors to a wide
variety of substituted amino acids.22-24 Various syntheses of p-lactones from
p-bromoacids have been reported, with most involving treatment of the
bromoacids with mild bases or silver salts.146 It was envisaged that the
synthesis.of the phenylalanine B-lactones (139a) and (139b) could be achieved

Chnpter 4 82
viø intramolecular substitution of the p-bromo acids (l3la) and (137b). This
procedure is analogous to the reactions of the bromides (82a) and (82b) to give
the alcohols (ll2a) and (1lzb), except that it involves intramolecular
substitution by the carboxylic acid group rather than intermolecular substitution
by water. Bromination of (S)-N-phthaloylphenylalanine (70) without
protection of the carboxylic acid group was therefore investigated.
Accordingly, (S)-N-phthaloylphenylalanine (70) was treated with NBS
under the standard conditions as described for the preparation of the bromides
(82a) and (82b) from the ester (74), and gave the p-bromophenylalanine
derivative (131) in987o yield, as a 1:1 ratio of diastereomers (Scheme 50).
Attempts to separate the diastereomers (137a) and (137b) by fractional
crystallization from methanol/water resulted in decomposition of the bromide
(137a) to give p-phthalimidostyrene (138) (Scheme 51), which crystallized as
large yellow prisms, with the other bromide (137b) remaining in solution. The
lH n.m.r. spectrum of the bromide (137b) showed doublets at õ 5.64 and 5.86
attributable to the cr- and p-protons, respectively, while the corresponding
resonances of the bromide (137a) were observed at ô 5.55 and 5.97"
Correlation of the lH n.m.r. spectral data of the bromoacids (131a) and (137b)
with that of the corresponding bromoesters (82a) and (82b) (Appendix 1)
indicated that the bromoacid (137b) had the erythro-stereochemistry and that
the bromide (137a) had the threo-stereochemistry.
PhthN PhrhN lth,,
NBSB1
p2
(r3t)(a) Rl=Br, R2=H(b) Rl =H, R2=Br
(70)
Scheme 50

Chnpter 4 83
Presumably only the threo-bromide (I37a) undergoes a decarboxylative
anti-elimination, to give the trans-alkene (138) (Scheme 51), because the
prefened conformation of fhe threo-bromoacid (137a) has the carboxylic acid
group anti to the bromine (Figure 10), in the required alignment for
elimination to occur. Decarboxylative anti-elimination from the erythro-
bromide (137b) is less favourable as rotation from the preferred conformer is
required (Figure 10). The facile conversion of the bromide (137a) to the
alkene (138) is presumably facilitated by the formation of an extended
conjugated rc-system in the alkene (138). Due to the lability of the bromide
(137 a) further reactions were carried out with mixtures of the diastereomers
(r37a) and (137b).
Ph
PhthN PhthN co2H
HHBrPhBr
HH
threo-isomer
(137a)
erythro-isomer
(137b)
Figure 10. Preferred conformations of the bromoacids (137a) and (137b).
co2H PhthN
.rrtt
Br
CO2 + HBr
(r37a) (138)
Scheme 51
Treatment of a mixture of the bromides (131a) and (137b) with silver
carbonate. in acetone gave a mixture of products which was analyzed using

Chapter 4 84
lH n.m.r. spectroscopy and shown to consist of the cis-p-lactone (139b), the
trans-þ-Iactone (139a) and a small amount of the trans-alkene (138). The cis-
p-lactone (139b) exhibited doublets at ô 6.33 and 6.70, due to the o- and
p-protons, with a coupling constant of 9.2 H:z. The trans-p-lactone (139a)
exhibited doublet signals at õ 5.67 and 5.92, due to the a- and p-protons, with a
coupling constant between these protons of 4.5 H:z. Determination of the
stereochemisry of the lactones (139a) and (139b) was based on the values of
the coupling constants between the a- and p-pro¡sns.147,148 Periodic analysis of
the mixture using lH n.m.r. spectroscopy showed that the trans-p-lactone
(139a) was slowly decomposing with a concomitant increase in the amount of
the alkene (138).
PhthN
Ag2CO3
Br
(r37a)(139a)
PhthN
Ag2C03
Br
(137b) (13eb)
Scheme 52
Presumably the threo-bromide (I3la) cyclizes to give the trans-lactone
(139a) and the erythro-bromide (137b) cyclizes to give the cis-lactone (139b)
(Scheme 52). The trans-lactone (139a) then undergoes a facile retro-[2+2]-
cycloaddition to give the alkene (138) and carbon dioxide (Scheme 53). In
Ph
o
o

Chapter 4 85
contrast, steric repulsion between the phthalimido and phenyl groups in the cis-
lactone (139b) presumably causes the four-membered ring to twist out of
planarity, such that there is not sufficient orbital overlap to enable
cycloreversion of that compound.
Attempted separation of the mixture of the products (138), (139a) and
(139b) by chromatography on silica resulted in total decomposition of the
rrans-þ-lactone (139a), while the cis-p-lactone (139b) was isolateÀin16%o yield
and the alkene (138) was isolated in 67Vo yield. The low yield of the cis-
p-lactone (139b) suggests that this compound is slowly decomposing on silica,
but the absence of the cis-alkene (141), expected from a concerted process,
indicates that the decomposition is probably occurring via a stepwise acid-
catalyzed process, as shown in Scheme 53. Cleavage of the O-C4 bond gives
the benzylic cation (140) which then undergoes decarboxylation to give the
trans-alkene (138).
Ph,hNr,,..
- coz(138)o
-+
(139a)
oH+
#PhrhNrr,,.. - coz
-H+(138).#
o+ H
(140)(13eb)
Scheme 53

Chapter 4 86
PhthN
(141)
The potential for utilizing the y-functionality in the amino acid derivatives
(91) and (92) to introduce functionality at the p-position of homophenylalanine
derivatives and the p- and y-positions of leucine derivatives was also studied.
Radical bromination of alkyl bromides has been shown to occur
regioselectively, giving the corresponding vicinal dibromides in high yields.las
Incorporation of the second bromine atom occurs at the most substituted vicinal
position, in accordance with the factors governing radical stability. The
selctivity of these reactions has been attributed to a neighbouring group effect,
where formation of the radical on the vicinal carbon is stabilized by the
adjacent bromine substituent.
Bromination of the y-bromohomophenylalanine derivative (92) was
initially attempted, as vicinal bromination can only occur at the p-position. The
bromide (92) was initially treated with two equivalents of NBS and a catalytic
amount of benzoyl peroxide in refluxing CCla, with irradiation by a 250W
mercury lamp for 4 hours. However, no products were observed and the
starting material (92) was recovered. The reaction was then repeated at reflux
in chlorobenzene, such that the temperature of the reaction was increased.
After 4 hours, lH n.m.r. spectroscopy indicated that only the starting material
(92) was present. Increasing the reaction time to as long as 3 days gave no
reaction, with the starting material (92) being isolated in good yield. The lack
of reactivity of the bromide (92) is attributable to severe hindrance at the
B-position due to the presence of the bulky o-substituents.
The lack of reactivity of the homophenylalanine bromide (92) at the
p-position suggested that this procedure may be applicable to the selective

Chapter 4 87
functionalization of the leucine derivative (91) at a primary ô-position.
Accordingly, the bromide (92) was treated under the same conditions as
previously described for reaction of the bromohomophenylalanine derivative
(92) (Scheme 54). Analysis of the reaction mixture using mass spectrometry
indicated the presence of the dibromide (I42), with peaks at mlz 431, 433 and
435 in a l:2:l ratio, corresponding to the molecular ion, as well as peaks at mlz
372,374 and376 in a 1;2:I ratio, corresponding to the loss of CO2Me from the
molecular ion. The dibromide (142) was purified through repeated
chromatography on silica, and its structure was determined using lH n.m.r.
spectroscopy. Resonances characteristic of the methyl ester groups of the
diastereomers of the dibromide (142) were observed at E 3.15 and 3.76.
Overlapping resonances were observed between ô 5.16-5.30, corresponding to
the a-protons of the diastereomers of the dibromide (142)" Resonances
between õ2.43-3.02 are consistent with the p-protons, with singlet resonances
at ô 1.93 and 1.86 attributable to the õ-methyl groups. A multiplet between
õ 3.68-3.85 is attributable to the ô-methylene protons Integration of the
singlets attributable to the õ-methyl groups indicated that the diastereomers of
the bromide (142) were produced in a ca. l:1 ratio.
PhthN
NBSBr Br
(e1) (r42)
Scheme 54
The selective bromination of the bromoleucine derivative (91) at a primary
ô-position in preference to reaction at the secondary B-position is consistent
PhthN¿rr,,.
Br

Chapter 4 88
with the explanation of the lack of reactivity of the bromohomophenylalanine
derivative (92) described above.
Alternative methods for the introduction of functionality at the Ê- and
ô-positions of the bromoamino acid derivatives (91) and (92) were also studied.
Conversion of the bromides (91) and (92) to the corresponding dehydroamino
acid derivatives (106), (107) and (146) was envisaged as one such method. The
p,ydehydroamino acid derivatives (107) and (146) are vinylglycine analogues,
themselves receiving attention as synthetic targets due to their propensity for
enzyme inhibition.9,98, I 50- 1 52
Elimination reactions were initially performed on the bromo-
homophenylalanine derivative (92), as less side reactions were expected with
this compound than with the bromoleucine derivative (91), where the
possibility of both p,y- and y,ô-elimination exists. Accordingly, the
bromohomophenylalanine derivative (92) was treated with one equivalent of
1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in THF" The hindered base DBU
has been reported to promote regioselective dehydrohalogenation of various
alkyl halides, such that the most substituted alkene is the major product.l53
However, none of the p,y-dehydrohomophenylalanine derivative (146) was
produced, instead the methanophenylalanine derivative (143) was isolated, in
72Vo yield (Scheme 55)" The cyclic product (143) was identified using
PhthNPhthN
DBU......................................-
Br
(e2)
Scheme 55
(143)

Chapter 4 89
lH n.m.r. spectroscopy, which showed an ABX splitting pattern consisting of
doublets of doublets at ô 2.2J, 2.43 and 3.38, corresponding to the
diastereotopic methylene protons and the benzylic proton, respectively. Only
one diastereomer of the cyclic product (143) was isolated, and X-ray
crystallographic analysis of this material (Appendix 8) indicated that the phenyl
and phthalimido groups were cis to each other" Presumably the base, DBU,
abstracts the a-proton from the bromide (92)" Cyclization of the resultant
cr-anion to give the cyclopropyl amino acid derivative (143) then occurs
diastereoselectively, although the basis for this selectivity is not obvious.
The reaction of the bromide (92) with DBU was also performed in the
presence of silver nitrate, anticipating that formation of the benzylic
carbocation through abstraction of bromide by silver ion may increase the
acidity of the p-protons" However, the cyclopropyl amino acid derivative (143)
was again isolated in good yield, with a tÍace of the 1-hydroxy-
homophenylalanine derivative (109) also being produced, presumably due to a
small amount of water present in the reaction mixture.
As elimination of hydrogen bromide from the bromophenylalanine
derivative (92) proceeded via a,y-elimination to give the cyclic product (143)
rather than via p,1-elimination, a different route to the formation of the
dehydroamino acid derivatives (106), (107) and (146) was formulated.
Elimination of acetates and xanthates under pyrolytic conditions has been
reported to proceed cleanly and in good yield.ls¿ Consequently the acetates
(lM) and (145) were synthesized from the corresponding hydroxyamino acid
derivatives (10a) and (109), the syntheses of which are described in Chapter 2
of the Results and Discussion of this thesis. Treatment of a solution of the
hydroxyleucine derivative (104) in dichloromethane with acetyl chloride,
pyridine and a catalytic amount of DMAP gave the acetate (lM) in 7O7o yield,
and the leucine lactone (105) in 167o yield (Scheme 56). The acetate (144) was
identified using FAB mass spectrometry, which showed a protonated molecular

Chapter 4 90
ion at mlz334, and from its lH n.m.r. spectrum, which was similar to that of
the alcohot (104) but showed an additional singlet resonance corresponding to
the acetyl group" The lactone (105) is presumably formed by cyclization of the
alcohol (104)"
PhthNhtq.
AcCI + (10s)pyr /DMAP
Scheme 56
Pyrolysis of the acetate (L44) was performed under reduced pressure (0.08
mm), with the acetate distilled (ca.250") through a pyrolysis tube packed with
quartz fragments and heated to ca.450". Purification of the crude product by
chromatography on silica gave a mixture of the y,õ-dehydroleucine derivative
(106) and the p,y-dehydroleucine derivative (107) in a 1.6:1ratio, in847o yield
(Scheme 57).
tnrhN¿¿,,..
45tr
(104)
(t44)
Me
(I44)
+---+
(106) (107)
Scheme 57
Although the more substituted p,y-dehydroleucine derivative (107) is
thermodynamically more stable, formation of the y,ö-dehydroleucine derivative
(106) is favoured statistically as there are six õ-protons compared to two

Chapter 4 9l
p-protons in the acetate (144). These factors are consistent with the
dehydroamino acid derivatives (106) and (107) being produced in the observed
1.6:1 ratio.
Synthesis of the homophenylalanine acetate (145) from the alcohol (109)
(Scheme 58) was carried out under conditions identical to those described for
acetylation of the hydroxyleucine derivative (104). The acetate (145) was
produced in74%o yield, with small amounts of the cis-p-lactone (1lO) (1Vo) and
the trans-p-lactone (111) (zEo) also isolated. The diastereomers of the aceta;te
(145) were sepÍuated by fractional crystallization and identified using lH n.m.r.
spectroscopy, with one diastereomer exhibiting a singlet resonance at õ 1.94
due to its acetyl group, and the other diastereomer giving rise to a singlet at
ô 2.09 for that substituent.
PhthN PhthN
AcCl Ac + (110)
+ (111)
pyrlDMAP
(10e) (r4s)
Scheme 58
Pyrolysis of the acetoxyhomophenylalanine derivative (145) was initially
performed as described for that of the acetoxyleucine derivative (144).
However, the reaction did not proceed to completion, with approximately 507o
of the acetate (145) recovered. Consequently, pyrolysis of the acetate (145)
was performed at the higher temperature of 520' (Scheme 59). lH N.m.r"
specfioscopy of the crude reaction mixture indicated the presence of the trans-
p,y-dehydrohomophenylalanine derivative (146). A doublet signal was

Chnpter 4 92
observed at ô 5.58, attributable to the o-proton. Another doublet was observed
at õ 6.68, attributable to the vinylic y-proton, with a coupling constant of 15.9
Hz characteristic of a trans-substituted alkene. A doublet of doublets was
observed at õ 6.74 corresponding to the vinylic p-proton, coupled to the
G-proton by 7.1 Hz and to the yproton by 15.9 }Iz. Further interpretation of
the lH n.m.r. spectrum of the crude reaction mixture indicated the formation of
the cr,p-dehydrohomophenylalanine derivative (147). A doublet was observed
atõ3.52, attributable to the T-protons of the alkene (147) coupled to the vinylic
p-proton. The signal due to the vinylic P-proton presumably occurs between õ
7.19-7.43, beneath the complex multiplet observed due to the phenyl groups of
the two alkenes (146) and (147). The a,p-dehydrohomophenylalanine
derivative (147) was presumed to have the (Z)-configuration, based on the
general relative stability of (E)- and (Z)-dehydroamino acid derivatives, as
described in Chapter 1 of the Results and Discussion of this thesis. Integration
of the peaks due to the methyl ester groups of the alkenes (146) and (147)
indicated that they were produced in a ca. l:1 ratio. Attempted separation of
these compounds by chromatography on silica and silver nitrate-impregnated
silica proved unsuccessful.
PhthN PhthN PhthN CO2Me
s?tr +
(146) (147)(14s)
Scheme 59

Chapter 4 93
Presumably pyrolysis of the acetate (145) proceeds initially to give the
trans-þ,y-dehydrohomophenylalanine derivative (146)" This alkene (146) then
undergoes partial thermal isomerization to give the more stable
cr,p-dehydrohomophenylalanine derivativ e Oa7 ) "
In conclusion, it can be seen that regiospecific bromination of the side
chains of N-phthaloyl-o-amino acid derivatives and subsequent elaboration of
this functionality via various methods has the potential to provide
stereocontrolled syntheses of a myriad of functionalized cr-amino acid
derivatives. For example, the erythro-bromoamide (85b) has been converted to
the trans-3,4-disubstituted p-lactam (I34) with control of relative and absolute
configuration. Synthesis of the corresponding cÍs-B-lactam (136) from the
threo-bromoamide (85a) was not possible, but the bromide (85a) was converted
to the corresponding threo-p-aminophenylalanine derivative (135). The
bromoacids (137a) and (137b) have been converted to the corresponding
disubstituted p-lactones (139a) and (139b), although isolation of the trans-
p-lactone (139a) was not possible due to its lability. Bromination of the
bromoleucine derivative (91) occurred regiospecifically to give the
y,ô-dibromoleucine derivative (I42). Elaboration of the bromoleucine and
bromohomophenylalanine derivatives (91) and (92) provided the dehydroamino
acid derivatives (106), (107) and (146), suitable for the synthesis of naturally
occurring side-chain functionalized leucine and homophenylalanine derivatives.

Chapter 5 94
RESULTS AND DISCUSSION: CHAPTER 5
Synthesis of Each Stereoisomer of B-Deuteriophenylalanine
It was anticipated that the side chain brominated amino acid derivatives
described in Chapter 1 of the Results and Discussion of this thesis could be
converted to deuterium labelled amino acids, which have found important use
in the study of enzyme catalyzed reactions. To examine this potential, the
synthesis of deuteriated phenylalanine derivatives was studied, with interest
focussed on the development of stereoselective routes to all four stereoisomers
of B-deuteriophenylalanine (65a)-(65d).
Initial attempts to synthesize deuteriated phenylalanine derivatives involved
reduction of the bromides (85a) and (85b) with tri-n-butyltin deuteride" As
reduction of each of the bromides (85a) and (85b) would proceed via the sÍìme
intermediate benzylic radical (150), separation of the diastereomers (85a) and
(85b) was not required. It was anticipated that the intermediate radical (150)
would adopt a preferred conformation (Figure 11) similar to that of the
carbocation (120), with subsequent delivery of the deuterium atom from the
stannane preferentially from one face of the sp2-hybridized radical leading to
the stereoselective formation of the deuterium labelled phenylalanine
derivatives (148a) and (148b). Although radical reactions were once thought to
be impractical for use in asymmetric synthesis, much work in this area has been
reported in recent years, with a variety of radical reactions with very high
stereoselectivity being developed. 155- 1s7
Consequently, a mixture of the bromides (85a) and (85b) was freated with
one equivalent of tri-n-butyltin deuteride in refluxing benzene for 4 hours.
The reaction proceeded in high yield and with 957o 2Et-incorporation, but with
only low stereoselectivity, producing the deuterides (148a) and (148b) in a2:l
ratio. The reaction was then carried out at 25", in an attempt to increase the

Chapter 5 95
selectivity of the reduction, however this required the addition of a catalytic
amount of AIBN and a reaction time of 3 days to achieve a high yield of the
deuterides (1a8a) and (148b). The stereoselectivity of the reaction increased
marginally, giving the deuterides (148a) and (148b) in a 3:1 ratio (Scheme 60).
Reduction of a 1:1 mixture of the bromoesters (82a) and (82b) under
NHCMe3 PhthN CONHCMe3
B1Bu3SnD pl
2 p2
(8s)
(a) Rl=Br" R2=H(b) Rl =H, R2=Br
(148)
(a) R1 =D, R2=H(b) Rl=H, R2=D
Scheme 60
nhthN¿rr,..
pl Bu3SnD pl2 R2
(82)
(u) Rl =Br, R2=H(b) Rl=H, R2=Br
(l4e)
(a) Rl=D, R2=H(b) Rt=H, R2=D
Scheme 61
identical conditions as described for the reduction of the amides (85a) and (85b)
at25" gave the corresponding deuterides Qa9$ and (149b) in 6l%o yield, in a
2:I ratío (Scheme 61).
The lH n.m.r. spectrum of a mixture of the deuterides (148a) and (148b)
showed doublet resonances at õ 3.54 and 3.46, coresponding to the respective

Chapter 5 96
B-protons, and integration of these signals was used to determine the
diastereomeric ratio of the deuterides (148a) and (148b). The lH n.m.r.
spectrum of the material obtained from reduction of the bromoesters (82a) and
(82b) showed doublets at ô 3.59 and 3.53 for the respective p-protons, with
integration of these peaks used to determine the diastereomeric ratio of the
deuterides (I49a) and (1a9b).
The greater selectivity observed in the reduction of the amide (85) than the
ester (82) is consistent with the greater selectivity observed in the
hydroxylation reactions of the bromoamide (85) than the corresponding
bromoester (82), as described in Chapter 2 of the Results and Discussion of this
thesis. It is presumed therefore, that the amide substituent more effectively
blocks one face of the intermediate radical (150) (Figure l l) than does the ester
moiety in the radical (84), such that the stannane delivers the deuterium atom
with greater selectivity in the reduction of the bromoamide (85) than the
bromoester (82). On this basis, it was assumed that the reduction of the
bromides (82) and (85) occurs selectively to give the threo-deuterides Qa9a)
and (148a) as the major products, analogous to the preferential formation of the
threo-alcohols (112a) and (118a) from the bromides (82a) and (85a)" These
assignments were confirmed by experiments described below"
PhrhN
H
CONHCMe3
(1s0)
Figure 11. The radicat (150) in the preferred conformation for deuteriumatom abstraction from tributyltin deuteride.
It was thought that a less reactive deuterium atom donor would result in
higher stereoselectivity in the reductions of the bromides (82) and (85),
H
Ph

Chaprer 5 91
Germanium hydrides have been reported to be more selective reducing agents
than the cofresponding tin hydrides, as the germanium-hydrogen bond is
stronger than the tin-hydrogen bond, thereby increasing the activation energy
required for reaction to occur.158 Reductions of benzylic bromides with
ftiphenylgermanium hydride have been reported to proceed effectively,l59 ¿¡d
consequently reductions of the bromoamide (85) and bromoester (82) were
attempted. Triphenylgermanium deuteride was synthesized by reduction of
triphenylgermanium chloride with lithium aluminium deuteride (997o ztt¡.
However, upon treatment of the bromoamide (85) and bromoester (82) with
triphenylgermanium deuteride, no reduction was observed, even when high
temperatures and activation with ultraviolet light were employed, and the
starting materials (85) and (82) were recovered. The absence of reactivity
under these conditions may be attributed to considerable steric hindrance
inherent in these systems.
As the radical reductions of the bromides (85) and (82) with tri-n-butyltin
deuteride did not proceed stereospecifically, other methods of reduction were
investigated. Sodium borohydride (NaBIIa) effectively reduces benzylic
bromides,l60 þor¡¡syer NaBII¿ has also been used to deprotect phthalimi¿e5.135
The bromide (82b) was therefore treated with NaBH4 in order to determine
whether reduction of the benzylic bromide or the phthalimide group would
occur preferentially. The alcohols (151) and (152) were isolated, indicating
that preferential reduction of the phthatimido group had occurred (Scheme 62).
Evidence for the alcohols (151) and (152) was obtained from their lH n.m.r.
spectra. The secondary alcohol (151) showed doublets at õ 4.28 and 5.75,
colresponding to the cr- and p-protons, respectively. Singlets at õ 4.70 and
5.77 were attributable to the hydroxyl proton and the isoindole ring proton.
The primary alcohol (152) showed doublets at ô 4.70 and 5.92, corresponding
to the cr- and p-protons, respectively. The diastereotopic benzylic protons
exhibited,-4n AB sptitting pattern, with doublets observed at ô 4.69 and 4.86.

NaBHa(82b)
OH
Br
Chnpter 5 98
CO2Me
Br
N
+
(151) (rs2)
Scheme 62
Sodium cyanoborohydride (NaCNBH3) is a less reactive reducing agent
than NaBH4, and its zinc chloride complex is reported to reduce benzylic
halides without affecting amides e¡ i¡¡ide5.161 Accordingly, the bromide (82b)
was treated with the sodium cyanoborohydndelzinc chloride complex, however
no reaction occurred and the starting material (82b) was recovered. The
absence of reactivity may again be attributed to the inherent steric hindrance in
this system.
Efforts were then directed to the catalytic reduction of the bromides (82a)
and (82b) over a palladium catalyst. Initialty, hydrogenolysis of a mixture of
(82a) and (82b) was performed using a 5Vo patladium on carbon catalyst, with
ethyt acetate aS the solvent, under an atmosphere of hydrogen, to examine
whether reduction of the bromides (82a) and (82b) was effected under these
conditions. This reaction proceeded to give the reduced product (74) in good
yield (Scheme 63).
PhthN
pl Hz, Pdlc.'.............................._*
R
CO2Me
2
(82)
(a) Rl=Br, R2=H(b) Rl=H, R2=Br
Scheme 63
(7 4)

Chapter 5 99
Having established benzylic reduction, deuteriolysis of a mixture of the
bromides (82a) and (82b) was then investigated, conducting the reaction under
an atmosphere of deuterium. The reduced product was isolated with only 58Vo
deuterium incorporation, indicating that some scrambling had occurred
between the deuterium gas and ethyl acetate. It was anticipated that a
deuteriated solvent system was required to prevent hydrogen-deuterium
scrambling. Accordingly, treatment of a mixture of the bromides (82a) and
(82b) in a mixture of TFIF and D2O at room temperature, using palladium on
carbon catalyst under an atmosphere of deuterium, proceeded to give a mixture
of the B-deuteriated phenylalanine derivatives (149a) and (149b) with >957o
2H1-incorporation.
Atthough little has been reported on the stereochemical course of the
catalytic reduction of benzylic halides, Berti and co-wo¡¡e¡5162,163 have shown
that hydrogenolysis of 2-phenyl-2-halocyclohexanols occurs with retention of
configuration. The deuteriolysis reactions were therefore repeated on the
separated bromide diastereomers (82a) and (82b) in an attempt to produce the
corresponding deuteriated phenylalanine derivatives (149a) and (149b)
stereoselectively. Reduction of the bromide (82a) under the conditions
described above for reduction of a mixture of the bromides (82a) and (82b)
was indeed stereoselective, producing the deuterides (149a) and (149b) in a
13:1 ratio. Reduction of the bromide (82b) was also stereoselective, giving a
1:13 ratio of the deuterides (149a) and (149b) (Scheme 64). The
stereoselectivity of these reactions was determined as described above for that
of the deuterides (1a9a) and (149b) obtained by reduction of the bromides
(82a) and (82b) with ni-n-butyttin deuteride. The deuteriated phenylalanine
derivatives (149a) and (149b) were produced with ca.99%o 2[t-incorporation,
as determined using rnass spectrometry.
The temperature of the deuteriolysis reactions was then decreased in order
to increas-g the stereoselectivity. Deuteriolysis of the bromide (82b) at 5o

PhthN
Dz, Pd/C
D2, Pdlc
6N HCI...q
AcOHD
Chapter 5 100
+*¡rNrr,.. CO;
,rrtD
(65a)
+t¡rNrr,,.
(65b)
PhthN/r,,.
...q
(82a)
(82b)
Br
Br
(t49a)
PhthNrr,.. PhthN/r,,. coi6N HCI
AcOHD D
(14eb)
Scheme 64
proceeded in24 hours to give the deuterides (149b) and (I49a) in a27:1 ratio.
Below 0",2Hr-methanol was employed as the solvent, as D2O crystallized from
the TIIF/D2O mixture. When the reaction of the bromide (82b) was pedormed
at -20" in 2Hr-methanol, deuteriolysis proceeded with almost complete
stereospecificity (Table 1). The (2S3S)- and (253R)-deuterides (149b) and
Qa9a) were produced in a 100:1 ratio, indicating a diastereomeric excess of
987o, as determined by lH n.m.r. spectroscopy" Analogous reactions of the
threo-bromide (82a) produced the (2S3RJ- and (253Í)-deuterides (149a) and
(149b) in the corresponding ratios as for reduction of the erythro-bromide
(82b). However, precise determination of the diastereomeric excess of the
(253R)-deuteride (149a), produced from the reduction of the threo-bromide
(82a) at -20", was not possible using lH n.m.r. spectroscopy, as resonances
corresponding to a 13C satellite of the methyl ester group of (I49a) and the
methyl ester group of the bromide (82a) present in trace amounts overlapped
with the signal corresponding to the p-proton of the minor (2539)-deuteride
(149b). However, it could be determined that the deuteride (149a) was,nt ,

"101
'!t.ìì.
Chnpter 5
't :.,. .
produced in >95Vo diastereomeric excess. Further experiments described below
detailing the deprotection of the deuteride (149a), and experiments described in
Chapter 6 of the Results and Discussion of this thesis, enabled a more accurate
determination of the stereochemical purity of the deuteride (149a). When the
deuteriolysis was attempted at temperatures below -30o, no reaction was
observed after a period of 7 days.
1 3 1
27 I
1 00 1
no reaction
<r2h
24h
3d
7d
25"
50
-20"
-300
THF/DzO
TFIF/DzO
MeOD
MeOD
diastereo-selectivitytimetemperaturesolvent
Table 1. Diastereoselectivity of the deuteriolysis ofthe bromides (82a) and (82b).
The deuteriophenylalanine derivatives (149a) and (149b) are of limited
utility unless they can be converted to the corresponding free amino acids (65a)
and (65b), without loss of stereochemical integrity. Accordingly, deprotection
of the deuterides Qa9a) and (149b) was investigated. The deuterides (I49a)
and (149b) were hydrolyzed in a 2:1 mixture of 6N HCI and acetic acid, with
subsequent treatment with aniline in ethanol giving the corresponding free
amino acids (65a) and (65b) (Scheme 64). The lH n.m.r. specfrum of the free
amino acid (65a) showed doublet signals at õ 3.92 and 3.21, conesponding to
the a- and p-protons respectively. The coupling constant between these protons
was 4.9 Hz, which is consistent with the coupling constant of 4.5 Hz reported by
Ife and Haslaml12 for the (253R)-deuteride (65a). The lH n.m.r. spectrum of
the free amino acid (65b) showed doublet signals at 83.92 and 3.03,

Chapter 5 lO2
corresponding to the cr- and p-protons respectively, with a coupling constant
between these protons of 7.9 Hz, consistent with the value of 8.5 Hz reported by
Ife and Haslaml12 for the (2S3S)-deuteride (65b). Assignments of the
stereochemistry of the free amino acids (65a) and (65b) were based on the
coupling constants between the a- and p-pro1e¡s.164 Presumably the most
stable conformers of the deuterides (65a) and (65b) have a staggered
orientation, with the phenyl and carboxyl groups lying anti to each other, such
that steric interactions are reduced (Figure 12). It can be seen that in this
conformation (253R)-deuteriophenylalanine (65a) would have a smaller
coupling constant between the cr,- and B-protons than would (2535)-
deuteriophenylalanine (65 b).
D
+H¡N
DH
(65a) (65b)
Figure 12. Preferred conformations of the deuterides (65a) and (65b).
The conversion of the bromides (82a) and (82b) to (253R)- and (2535)-
deuteriophenylalanine (65a) and (65b), respectively, confirms that the
deuteriolysis reactions of the bromides (82a) and (82b) proceed preferentially
with retention of configuration. The diastereomeric excess of each of the free
amino acids (65a) and (65b) was determined to be >98Vo using lH n.m.r.
spectroscopy, indicating that no racemization had occurred during deprotection.
Mass specffometric analysis of the amino acids (65a) and (65b) indicated that
each contained ca. 997o2H1at the B-position.
The (2R3S)- and (2R3R)-þ-bromophenylalanine derivatives (82c) and
(82d) were synthesized via identtcal procedures to those used for the (253R)-
HH
Ph
+H¡N
HPh

Chapter 5 103
and (2S3S)-B-bromides (82a) and (82b), but using (R)-phenylalanine (55) as the
starting material (Scheme 65). Thus, all four stereoisomers of B-bromo-N-
phthaloylphenylalanine methyl ester (82a)-(82d) have been synthesized with
known relative and absolute configuration at both cr- and B-centres.
+PhttrN
i) phthalicanhydride
B2ü) MeOH /
HCI
(s5)
(82c)
D2, Pd/C
Scheme 65
(149c)
..rat
(14ed)
NBS...*
6N HCI
AcOH
6N HCI
AcOH
(82)
(c) Rl = Br, R2
(d) Rl = H, R2
D
(65c)
+H
(65d)
=fl=Br
PhthN+
H¡N
Br
CO2Me
D2,Pdlc
coi
"4DBr D
(82d)
Scheme 66
Deuterioiysis of the bromides (82c) and (82d) was carried ouL aL -20" in
2Hr-methanol, as described for the reactions of the bromides (82a) and (82b),
and gave,.the corresponding deuteriated phenylalanine derivatives (149c) and

Chapter 5 LM
(149d). Subsequent deprotection of these compounds was achieved as described
for the deuterides (I49a) and (149b), to give the deuterium labelled free amino
acids (65c) and (65d) (Scheme 66). The deuterides (65c) and (65d) were
produced in 98Vo d.e., and with cø. 99Vo 2H1-incorporation, as for the
production of the deuterides (65a) and (65b).
Hence a procedure has been developed for the synthesis of all four
stereoisomers of p-deuteriophenylalanine (65a)-(65d), homochiral at the
q,-cenffe, with 987o diastereomeric excess and 99Vo 2H1-incorporation.

Chapter 6 105
RESULTS AND DISCUSSION: CHAPTER 6
Stereochemical Course of the Reactions of (S)- and
(R)-Phenylalanine with (S)-Phenylalanine Ammonia Lyase
The highly stereoselective synthesis of all four stereoisomers of
p-deuteriophenylalanine (65a)-(65d), as described in Chapter 5 of the Results
and Discussion of this thesis, enabled an investigation of the stereochemical
course of the reaction of both (S)- and (R)-phenylalanine, (56) and (55), with
the enzyme (S)-phenylalanine ammonia lyase (PAL).
In accord with Battersby's studieslS Eeatment of (253R)-3-deuterio-
phenylalanine (65a) with PAL in sodium borate buffer at pH 8.7 gave
deuteriated trans-cinnamic acid (67) (Scheme 67). The deuteriated cinnamic
acid (67) was identified using lH n.m.r. spectroscopy, with a broad singlet
observed at ô 6.46 corresponding to the cr-vinylic proton, consistent with
literature values.l5 A fiace of non-deuteriated trans-cinnamic acid (57) was
observed, with integration of the relevant peaks in the lH n.m.r. spectrum
indicating that the product contained 987o deterium at the p-position. The
presence of ZVo non-deuteriated cinnamic acid (57) is consistent with the
production of the deuteriophenylalanine derivative (65a) in 987o diastereomeric
excess and with 997o 2E1-incorporation, as described in Chapter 5 of the
Results and Discussion of this thesis. This result infers that specific removal of
the 3-pro-S hydrogen from (S)-phenylalanine (56) occurs during reaction with
the enzyme, with l7o of the non-deuteriated cinnamic acid (57) arising from
unlabelled (S)-phenylalanine (56), and the remainder arising from the lVo
(253 S)-deuteriophenylalanine (65b) in the sample.
Similar treatment of (2S3S)-3-deuteriophenylalanine (65b) with PAL gave
trans-cinnamic acid (57) (Scheme 67), providing further evidence for the above
conclusion. The product (57) was identified by comparison of its lH n.m.r.

Chnpter 6 106
spectrum with that of an authentic sample, giving doublets atõ 6.41 and 7.81,
corresponding to the cr- and B-vinylic protons, respectively, with a coupling
constant of 16.0 FIz being characteristic of a trans-sttbstituted u1¡e¡s.120 No
signals attributable to deuteriated trans-cinnamic acid (67) were observed.
Approximately I7o deuteriated cinnamic acid (67) is expected from this
reaction, due to l%o of the contaminant (253R)-deuteriophenylalanine (65a).
However, the signal in the lH n.m.r" spectrum due to the ø-proton of
deuteriated trans-cinnamic acid (67) occurs as a broad singlet atõ 6.46, and is
presumably not visible under the doublet signal due to the a-proton of cinnamic
acid (57).
t¡rNzrr,,.
PAL
+
D
(65a) (67)
+H¡N¿¿,,.
PAL
H
(6sb) (s7)
Scheme 67
These results are in agreement with the conclusion of Battersby and
co-workers15 that the elimination reaction catalyzed by PAL proceeds viø
abstraction of the 3-pro-S hydrogen of lS)-phenylalanine (56) with anti-
elimination of the amino group leading to formation of trans-cinnamic acid

Chapter 6 lO7
(57). Battersby and co-workersl5 used deuteriated phenylalanines which
contained 907o z}lr at the p-position, and which were of less than 93c/o
stereochemical purity, such that the deuteriated trans-cinnamic acid (67)
obtained from reaction of the (253R)-deuteride (65a) contained only 88+4Vo
2IJt" Similarly, the trans-cinnamic acid (57) obtained from reaction of the
(2S3S)-deuteride (65b) with PAL contained 8+2Vo z}It. The results described
above confirm that the minor impurities in Battersby's studies are in fact due to
impurities in the substrates, and are not due to a lack of enzyme
stereospecificity.
Atthough (R)-phenylalanine (55) is a subsffate of PAL,I10 nq mechanistic
or stereochemical studies have been reported with this compound.
(R)-Phenylalanine (55) binds effectively to the active site, but is metabolized at
only one five thousandth the rate of (S)-phenylalanine (50).tto We therefore
wished to confirm the product from the reaction of (R)-phenylalanine (55) with
PAL, and then investigate the stereochemical course of the reaction, with the
interest being to examine how the enzyme manages to metabolize a substrate
with the opposite chiratity to its normal substrate. Initially, (R,)-phenylalanine
(55) was treated with PAL and trans-cinnamic acid (57) was isolated in
reasonable yield. Treatment of (2R35)-deuteriophenylalanine (65c) with PAL
gave deuteriated cinnamic acid (67) with 927o 2Hr-incorporation, indicating
that the enzyme preferentially abstracts the 3-pro-R hydrogen from
(R)-phenylalanine (55) (Scheme 68). Treatment of (2R3R,)-deuterio-
phenylalanine (65d) with the enzyme confirmed this result, giving non-
deuteriated cinnamic acid (57) as the major product (Scheme 68).
These results can be explained by examining the conformation of both
(S)-phenylalanine (56) and (R)-phenylalanine (55) in the active site.
trans-Cinnamic acid (57) and benzoic acid have been shown to inhibit the
enzyme,lOT'IO8 indicating that the active site contains a hydrophobic pocket for
binding oi{ Fe aromatic ring, and positively charged residues, possibly arginine,

Chapter 6 108
to accommodate binding of the carboxyl group. As described in the
Introduction, the active site also contains a prosthetic group for covalent
attachment of the amino group. For anti-elimination to occur, a B-hydrogen
must be antiperiplanar to the nitrogen atom. The phenyl and carboxyl groups
must also be qnti to each other, such that they aÍe trans to each other in the
product cinnamic acid (57). Therefore, when (S)-phenylalanine (56) binds in
the active site, it must adopt a conformation in which the amino, carboxyl and
phenyl groups and the 3-pro-S hydrogen all lie in a plane (Figure 13). A basic
residue such as histidine is presumably situated in an appropriate position to
abstract the 3-pro-S hydrogen with anti-elimination leading to formation of
tans -cinnamic acid (57).
PAL
D 927ozH1
(65c) (67)
co2H
PAL
H 277o 2H1
(6sd) (s7)
Scheme 68
H
+H
+coiH¡N
+
(5s)
coi
pro-R

Chapter 6 109
H H
Ph- B-Enz
..r¡lll NH
oEnz-HN
HR o- HR
Figure 13" (S)-Phenylalanine (56) in the active site of the enzyme PAL.
llntr. Enz-HN
Ph Hn_/- B-EnzH H
Figure 14. (R)-Phenylalanine (55) in the active site of the enzyme PAL.
(R)-Phenylalanine (55) can also be situated in the active site with the
phenyl, amino and carboxyl groups bound in the same way as that for
(S)-phenylalanine (56), even though the chirality at the c-centre is reversed
(Figure 14). In this conformation, the phenyl and carboxyl groups are in the
required anti onentation, and the amino group and pro-R hydrogen are also
antiperiplanar. The pro-R hydrogen of (R)-phenylalanine (55) occupies a
similar position in the active site to that which the pro-S hydrogen of
(S)-phenylalanine (56) occupies. Thus, abstraction of the pro-R hydrogen from
(R)-phenylalanine (55) occurs preferentially, wîth anti-elimination again giving
trans -cinnamic acid (57).
However, it can be seen that the reaction of the enzyme with
(R)-phenylalanine (55) is not completely stereospecific, as is the case with
(S)-phenylalanine (56). Reaction of the (2R35)-deuteride (65c) occurs wíth ca.
87o remov"SL of the B-deuterium atom to give the cinnamic acid (67) with only
Ba*
NH

Chapter 6 110
927o 2[rincorporation. Similarly, reaction of the enzyme with the (2R3R)-
deuteride (65d) occurs with 277o removal of the B-hydrogen atom to give the
cinnamic acid (57) with2TVo 2H1-incorporation. The difference between these
ratios is attributable to two deuterium isotope effects, and indicates that
although the pro-R hydrogen is preferentially abstracted from
(R)-phenylalanine (55), presumably ca. l57o of the reaction occurs vic
abstraction of the pro-S hydrogen. Reaction of the (2R35)-deuteride (65c) with
PAL occurs with only 8Vo (rather than l57o) removal of the p-deuterium due
to a deuterium isotope effect of ca. 2, and similarly reaction of the (2R3R)-
deuteride (65d) occurs with 27Eo (rather than l57o) removal of the B-hydrogen
atom, again due to a deuterium isotope effect of ca.2. As no cis-cinnamic acid
is produced from this reaction, concerted removal of the pro-S hydrogen and
elimination of ammonia must proceed via a syn-elimination. Thus,
(R)-phenylalanine (55) must be distorted in the active site and situated in such a
way that the basic residue is sufficiently close to the pro-S hydrogen, or another
basic residue in the active site may be close enough to the pro-S hydrogen to
abstract it, with elimination then leading to the production of trans-cinnamic
acid (57) (Figure 16). It is interesting to note that if a second basic residue was
to exist in proximity to the pro-S hydrogen of (R)-phenylalanine (55) in the
active site (Figure 16), it would not interfere with binding of the natural
subsffate, (S)-phenylalanine (5O Gf Figure 13).
B
ll tt t t,.
H
Ho-
Figurp"16. Abstraction of the pro-S hydrogen of (R)-phenylalanine (55).

chapter 6 111
In conclusion, it has been demonstrated that the elimination of a p-proton
and ammonia from (S)-phenylalanine (56) catalyzedby the enzyme PAL occurs
with stereospecific removal of the pro-S hydrogen. In contrast, the elimination
of a p-proton and ammonia from (R)-phenylalanine (55) catalyzed by the
enzyme PAL occurs with stereoselective removal of the pro-R hydrogen. The
pro-R hydrogen of (R,)-phenylalanine (55) occupies the same position in the
active site as does the pro-S hydrogen of (S)-phenylalanine, and is therefore
preferentially abstracted. However, the unnatural substrate lR)-phenylalanine
(55) presumably is distorted such that the elimination catalyzed by PAL is not
completely stereospecific.

Conclusion II2
CONCLUSION
The work described in this thesis has shown that N-phthaloyl-protected
cr-amino acid derivatives are deactivated towards hydrogen abstraction at the a-
position. This effect has been attributed to steric and electronic factors.
Bromination of various N-phthaloyl amino acid derivatives has been shown to
occur regiospecifically via the most stable side chain radicals"
The versatility of brominated amino acid derivatives has been exploited in
the synthesis of a variety of homochiral hydroxy-G-amino acids and their
derivatives" Stereoselective syntheses of (S)-threo-þ-hydroxy-phenylalanine
and tyrosine derivatives have been developed" Incidentally, the (S)-threo-
isomers of p-hydroxy-a-amino acids are the predominant stereoisomers which
occur in nature. Deprotection procedures have been developed to provide the
free amino acid (253R)-þ-hydroxyphenylalanine, a component of various
naturally occurring peptide antibiotics. The methods are suitable for the
preparation of unnatural and rare amino acid derivatives. A p-hydroxy-
p-nitrophenylalanine methyl ester derivative, suitable for elaboration to the
antibiotic chloramphenicol, has also been prepared. In future work,
exploitation of the potential benefits of using p-bromoamino acid derivatives
with the carboxyl group protected as an amide rather than an ester, discovered
during the course of the present study, should provide an even more efficient
route to the synthesis of chloramphenicol.
Procedures have been formulated for the stereocontrolled synthesis of
trans-3, -disubstituted p-lactams and threo-þ-aminophenylalanine derivatives
with control of relative and absolute stereochemistry. A suitable method for
the introduction of functionality at the ô-position of leucine derivatives, the
least activated of the positions on the side chain, has been developed. Methods
have also been developed for the introduction of functionality at the p-position
of leucine and homophenylalanine derivatives.

Conclusion I 13
A stereoselective route which provides each of the stereoisomers of
p-deuteriophenylalanine with very high diastereoselectivity and deuterium
incorporation has been developed. The stereoisomers of p-deuterio-
phenylalanine have been used to determine the stereochemical course of the
reaction of both lS)- and (R)-phenylalanine with the enzyme (S)-phenylalanine
ammonia lyase (PAL)" It has been determined that PAL catalyzes the
stereospecific removal of the pro-S hydrogen from the natural substrate
(S)-phenylalanine, whereas reaction with (R,)-phenylalanine occurs with
stereoselective removal of the pro-R hydrogen" The contrasting reactions of
the natural substrate and its antipode with PAL were attributed to both
compounds occupying the active site of the enzyme with the amino, carboxyl
and phenyl groups bound at specific sites. The p ro-R hydrogen of
(R)-phenylalanine then occupies the same spacial position in the enzyme active
site as does the pro-S hydrogen of (S)-phenylalanine when it is bound.
The above syntheses are all enantiospecific, except where indicated in the
text, with the chirality determined by that of the starting amino acids"
Although homochirality of each individual compound has not been determined
independently, it is implied by inference with representative examples. The
chiral integrity of a bromophenylalanine derivative has been established
through analysis of a reduced derivative using HPLC with a chiral stationary
phase. An hydroxyvaline derivative has been shown to be homochiral using
lH n.m.r. spectroscopy with a chiral shift reagent, which also implies the
homochirality of the precursor bromide. Comparison of a leucine lactone with
an authentic sample confirms the chiral integrity of the precursor hydroxy- and
bromo-leucine derivatives. Optical rotation of a p-hydroxyphenylalanine free
amino acid and comparison with an authentic sample has established the
homochirality of the corresponding hydroxy- and bromo-phenylalanine
derivatives. X-Ray crystallographic analysis of a hydroxyphenylalanine
derivative and a bromo-p-nitrophenylalanine derivative has also established the

Conclusion ll4
chirality of these compounds and of their precursors. Reactions of
deuteriophenylalanine stereoisomers with PAL confirm the homochirality of
these compounds and also of the corresponding precursors.
In summary, ûew procedures have been developed for the synthesis of
functionalized cr-amino acid derivatives with control of both relative and
absolute stereochemistry, and the use of such compounds in the study of
biochemical systems has been illustrated.

Experimental 115
EXPERIMENTAL
General
Melting points were determined on a Kofler hot-stage under a Reichert
microscope and are unconected.
Elemental analyses were carried out by Canadian Microanalytical Service
Ltd., New Westminster, Canada, and by Chemical and Micro Analytical
Services Pty. Ltd., Victoria, Australia.
Analytical thin layer chromatography was performed using Merck
Kieselgel 60 Fzs+ silica on aluminium backing plates. Preparative
chromatography was performed using positive pressure flash
chromatography165 or dry column flash chromatographyl66 using Merck
Kieselgel 60 (230-400 mesh ASTM).
High performance liquid chromatography (HPLC) was carried out using a
V/aters 60004 solvent pump, a Waters U6K injector and a Waters model 441
absorbance detector operating at 254 nm, in conjunction with an ICI DP-700
data station. Analyses were performed using a Waters Radial Pak normal phase
10 pm silica column (100 x 8 mm), eluting with a gradient of hexanelethyl
acetate, or where specified, a Waters Nova Pak C13-reverse phase 10 pm silica
column (100 x 8 mm) eluting with a gradient of methanol/water, or a Regis
Pirkle Covalent (S)-Phenylglycine column (250 x 4.6 mm), eluting with a
gradient of hexane/iso-propanol.
Infrared spectra were recorded on an Hitachi 270-30 spectrophotometer or
a Jasco A-I02 spectrophotometer using the 1603 cm-l band of polystyrene as a
reference. Spectra were recorded as solutions in dichloromethane unless
otherwise specified.
Nuclear magnetic resonance spectra were recorded on either a Bruker
CXP-300 " or Bruker ACP-300 spectrometer. Proton nuclear magnetic

Experimental 116
resonance (lH n.m.r.) specna were recorded at 300 MHz, and carbon nuclear
magnetic resonance (13C n.m.r.) spectra were recorded at75.5 MHz. Spectra
were either recorded in deuteriochloroform using tetramethylsilane (TMS) as
an internal standard or in deuterium oxide (DzO) using sodium 12,2,3,3-2Ha)3-
(trimethylsilyl)propionate (TSP) as an internal standard. Chemical shifts are
quoted as õ in parts per million downfield from the internal standard.
Multiplicities are abbreviated to; s, singlet; d, doublet; t, triplet; q, quartet; m,
multiplet; br, broad.
Electron impact mass spectra were recorded with an AEI MS-30 double
focussing mass spectrometer operating at 70 eV or on a Vacuum Generators
ZAB 2ÉIF mass specfrometer. Fast atom bombardment (FAB) mass spectra
were recorded on a Vacuum Generators ZAB 2FIF mass spectrometer.
All solvents were distilled before use. Anhydrous diethyl ether and
tetrahydrofuran (THF) were obtained by distillation from sodium
benzophenone ketyl. Drying and purification of other solvents and reagents
was performed using standard laboratory procedures.l6T'168 Organic solutions
were dried by the addition of anhydrous magnesium sulfate, unless otherwise
specified.

Experimental II7
(S)-N-Phthaloylvaline (68)
A mixture of (S)-valine (10.0 g, 85 mmol) and finely ground phthalic
anhydride (12.6 g, 85 mmol) was heated in an oil bath at 145-150o for 30 min.
The mixture was allowed to cool, then the solid was dissolved in hot methanol
(40 rnl). The solution was filtered and then water (60 ml) was added. The title
compound (68) crystallized on cooling and was isolated as fine colourless
crystals (18.8 g,89Vo), m.p. 116-117" (tit.t6q 118-119'), [a]¡22 -6go (c, 1 in
acetone) (lit.ttz [ct]o23 -69.4" (c, 1 in ethanol)). lH n.m.r. (300 MHz, CDCI3) ô
0.93, d, J 6.7 Hz,3}J, Me; 1 .l'7, d, J 6.7 Hz,3H., tr..fe'; 2.71, m, H3; 4.65, d, J
8.3 Hz, H2:7.14-1.90, m, 4H, Phth; 10.68, br s, CO2H.
(S)-N-Phthaloylleucine (69)
A mixture of (S)-leucine (10.0 g, 26.2 mmol) and finely ground phthalic
anhydride (11.3 9,26.3 mmol) was heated in an oil bath at 145-150" as
described for the synthesis of (S)-N-phthaloylvaline (68) from (S)-valine. The
títle compound (69) was isolated as a white solid (19.07 g,967o), m.p. l2I-I22"(lit.tzo 120-121"), [a]o23 -25" (c, 1 in acetone) (li1.t6e ¡s¿lrzz -24" (c, I inethanol)). lH n.m.r. (300 MHz, CDCI3) õ 0.94, d, J 6.6 Hz,3H, Me; 0.97, d, J
6.6 Hz,3H, Me'; 1.53, m, H4: I.96, m, H3; 2.39, m, H3'; 5.02, dd, J 4.2H2,
lI .5 Hz, II2; 7 .16, m, 2H, Phth; 7 .89, m, 2H, Phth; 9.64, br s, CO2H.
(S)-N-Phthaloylphenylalanine (70)
A mixture of (S)-phenylalanine (10.0 g, 60.5 mmol) and finely ground
phthalic anhydride (9.0 g, 60.8 mmol) was heated in an oil bath at 145-150" as
described for the synthesis of (S)-N-phthaloylvaline (68) from (S)-valine. The
title compound (70) was isolated as fine colourless crystals (17.08 g,967o),

Experimental 118
m.p. 187' (lit.trt 187'), [cr]o2r -212" (c, 1 in acetone) (lit.tts [a]o -212o (c, 1.9
in ethanol)). tH n.m.r. (300 MHz, CDCI3) õ 3.59, d, J 8.5 }{z,2H,CHz;5.23,t,
J 8.5 Hz, II2;7.16, s,5H, Ph;7.62-7.77,m,4H, Phth; 10.68, br s, CO2H.
(S)-N-Phthaloyltyrosine (71)
(S)-Tyrosine (10.0 g, 55.2 mmol) and phthalic anhydride (8.2 g, 55.3
mmol) were dissolved in boiling DMF (50 nìl) and the solution was boiled for a
further l0 min. The mixture was then filtered, and the fìlrate was poured into
water (500 mt) with vigorous stirring. The precipitate was collected by
filtration and was recrystallized from methanol/water to give the title
compound (71) as a white solid (13.32 g,787o), m.p. 163-165'(lit.t6e 173-
I74"), [cr]o23 -234" (c, 1 in acetone) (lit.16e [a]o22 -232' (c, 1 in ethanol)). lH
n.m.r. (300 MHz, CDCI¡) ô 3.58, d, J 8.4 Hz,CHz;5.21,t,J 8.4H2,ÍI2;6.91,
d, J 8.6 Hz,2H, ArH; 7.I8, d, J 8.6 Hz,2H, ArH; 7.67-7.8I, m,4H, Phth; 9.05,
br s, COzH.
(S)-N-Phthaloylvaline Methyl Ester (72)
(S,)-N-Phthaloylvaline (68) (6.0 g, 24.3 mmol) was treated with dry
methanol (100 rrìl) which had been pre-treated with thionyl chloride (1 ml, 14
mmol). The solution was stirred under anhydrous conditions for 16 h, then the
solvent was removed under reduced pressure. The residue was taken up in
dichloromethane, and the organic solution was washed with I07o NazCO¡ and
water, then was dried. Removal of the solvent under reduced pressure and
recystallizatíon of the residue from hexane/ethyl acetate gave the title
compound (72) as a viscous oil (5.52 g,87Vo), [a]p23 -68" (c, 1 in acetone). lH
n.m.r. (300 MHz, CDCI3) õ 0.92, d, J 6.8 IJz,3}J, Me; 1.16, d, J 6.8 Hz,3H,

Experimental 119
Me'; 2.77 , m, H3; 3.72, s, 3H, OMe; 4.58, d, J 8.3 Hz, H2; J .78, m, 2H, Phth;
7.88, m, 2H, Phth.
(S)-N-Phthaloylleucine Methyl Ester (73)
(s)-N-Phthaloylleucine (69) (19.07 g, 73.O mmol) was dissolved in dry
methanol (150 rnl) which had been pre-treated with thionyl chloride (2.8 9,24
mmol). The solution was stirred under anhydrous conditions for 16 h, then the
solvent was removed under reduced pressure. The residue was taken up in
dichloromethane (150 ml), and the solution was washed with l07o NazCO¡ (100
ml) and water (100 mt). The organic phase was dried, then was concenfated
under reduced pressure" Distillation (170o, 0.05 mm, block) gave the title
compound (73) as a clear viscous oit (18.90 g, 94 7o), [o]p23 -23" (c, 1.0 in
acetone). 1H n.m.r. (300 MHz, CDCI3) ô 0.93, d, J 6.8 Ilz,3H, Me; 0.96, d, J
6.6 H2, 3H, Me'; I.49, m, H4; 1.97, ddd, J 4.4 }l;2, I0.3 }lz, I4.3 Hz, }l3; 2.34,
ddd, J 4.1 Hz, II.6 Hz, 14.3 Hz,H3';3.73, s, 3H, OMe; 4.96, dd, J 4.4 Hz, lI.6
Hz, ÍI2; I .7 5, m, 2}l, Phth; 7 .87, m, 2H, Phth.
(S)-N-Phthaloylphenylalanine Methyl Ester (74)
(S)-N-Phthatoylphenylalanine (70) (5.0 g,16.9 mmol) was dissolved in dry
methanol (100 rnl) which had been pre-treated with thionyl chloride (1 ml, 14
mmol), as described for the preparation of (S)-N-phthaloylvaline methyl ester
(72) from (S)-N-phthaloylvaline (68). The title compound (74) was isolated as
colourless crystals (4.48 g,867o), m.p. 123-125' (lit.l72 Il3-114"), [a]o2: -
212" (c, 1.0 in acetone). lH n.m.r. (300 MHz, CDCI3) õ 3.58, dd, J 10.9 Hz,
14.3 Hz,lf3;3.63, dd, J 5.6 Hz,14.3 Hz,H3';3.80, s, 3H, OMe; 5.20, dd, J 5.6

Experímental l2O
Hz, 10.9 }lz, H2;7.I8, s, 5H, Ph;1.70, m,2H, Phth; 1.79, m,2fJ, Phth.
lH n.m.r. spectral data consistent with literature uu1u"r.173
(S)-N-Phthaloyltyrosine Methyl Ester (75)
lS)-N-Phthaloyltyrosine (71) (8.0 g,25.7 mmol) was dissolved in dry
methanol (100 rnl) which had been pre-treated with thionyl chloride (1 ml), and
the solution was stirred under anhydrous conditions for 16 h. The solvent was
removed under reduced pressure, then the residue was taken up in
dichloromethane. The solution was washed with IOTo Na2CO3 and water, then
was dried. Removal of the solvent under reduced pressure and recrystallization
of the residue from hexane/dichloromethane gave a ffanslucent gel, which upon
drying in air and over PzOs gave the title compound (75) as a white powder
(6.73 g,8l7o), m.p. 101-104' (Iit.130 98-99"). lH n.m.r. (300 .I..f.}Jz, CDCI¡) õ
3.44, dd, J 11.0 Hz, l4.4Hz, H3; 3.50, dd, J 5.7 Hz,I4.4 Hz, H3'; 3.77, s, 3H,
OMe; 5.10, dd, J 5.7 Hz,II.0Hz,I12;6.62, d, J 8.6 }lz,2H, ArH; 6.98, d, J 8.6
Hz, zIJ, ArH; 7 .66-7 .78, m, 4H, Phth"
N-Phthaloylhomophenylalanine (7 6)
A mixtu¡e of homophenylalanine (4.7 g,26.2 mmol) and finely ground
phthalic anhydride (3.9 9,26.3 mmol) was heated in an oil bath at 145-150" as
described for the synthesis of (S,)-N-phthaloylvaline (68) from (S)-valine. The
title compound (76) was isolated as a white solid (7.29 g,907o),m.p. 144-145"
(lit.tz+ 142.5-144"). lH n.m.r. (300 MHz, CDCI3) õ 2.56-2.77 , m, 4H, CIlzCftz;
4.93, m, II2; 7 .A9-7 .29, m, 5II, Ph; 7 .7 5, m, 2II, Phth; 7 .86, m, 2H, Phth; 9.43,
br s, CO2H.

Experimental I2l
N-Phthaloylhomophenylalanine Methyl Ester (77)
N-Phthaloylhomophenylalanine (76) (7.0 9,22.7 mmol) was dissolved in
dry methanol (100 ml) which had been pre-ffeated with thionyl chloride (1 ml,
14 mmol). The solution was stirred under anhydrous conditions for 16 h, then
the solvent was removed under reduced pressure. The residue was taken up in
dichloromethane, and the solution was washed with l07o NazCO3, then with
water, and was dried. Removal of the solvent under reduced pressure and
purification by chromatography on silica, followed by distillation (17 5" ,
0.03mm, block), gave the title compound (77) as a clear oil (6.73 g, 927o),
(Found: C, 70.5; H,5.3; N,4.4. CrqHrzNO4 requires C,70.6: H, 5.3; N,4.37o).
lH n.m.r. (300 MHz, CDCI3) õ2.52-2.71, ffi, 4}l,CHzCHz;3J72, s,3H, OMe;
4.88, m, lH, ÍÐ;7.08-7.22,m,5H, Ph; 7.74,m,2H, Phth; J.84,m,2H, Phth"
v max 1794, 17 44, 1720, I 388, 1098, 708 cm-l. Mass spectrum; ml z 324 (M+H+,
27o),323 (M*', I),307 (-CH4, 2),262 (7), 219 (100), 187 (85), 132 (24), 109
(30), 104 (21),9t (21),76 (t2).
(R S) -N- f e r t -B uty l-¡rr ø- p h t h a I o y I p h e n y I a I an i n a m i d e
Thionyl chloride (12.1g, 0.10 mol) and pyridine (0.1 ml) were added to a
suspension of (S)-N-phthaloylphenylalanine (70) (10.0 9,34 mmol) in CCI¿
(100 ml). The mixture was refluxed for 3 h, allowed to cool, then the solvent
and excess thionyt chtoride were removed under reduced pressure. The residue
was suspended in CCl¿ (100 ml) and tert-butylamine (6.2 9,85 mmol) was
added slowly. The mixture was refluxed for I h, then the solvent and excess
tert-butylamine were removed under reduced pressure. The residue was taken
up in ethyl acetate, then was washed with I07o NazCO¡ and water. The solution
was dried, then the solvent was removed under reduced pressure. The residue

Experimental I22
was recrystallized from hexane/ethyl acetate to give the title compound as white
crystals (10.3 g,87Vo), m.p.194-195". Spectral data identical to that described
below for the (S)-enantiomer (78).
Elution of this compound through a Regis Pirkle HPLC column showed the
presence of two compounds, with retention times of 2I and 22 min, eluting
with a gradient of hexane and iso-propanol (5-407o iso-propanol). The
material obtained viø this method was therefore assumed to be racemic.
(S) -N -terf-Butyl-Nø-phthaloylphenylalaninamide (78)
To a solution of (S)-N-phthaloylphenylalanine (70) (8.0 9,27.1 mmol) and
triethylamine (2.74 9,2J.1 mmol) in dichloromethane (100 ml) was added
ethyl chloroformate (2.94 9,27.1 mmol). The mixture was stirred for 10 min,
then was cooled to 0". tert-Búylamine (1.98 9,27.1 mmol) was added, and the
mixture was stirred at 0o for 15 min. The mixture was allowed to warm to
room temperature, then was stirred for a further 30 min. The mixture was
filtered, the filtrate was washed with water, then was dried. The solvent was
removed under reduced pressure and the residue was recrystallized from
hexane/ethyl acetate to give the títle compound (78) as a white solid (8.8 g,
937o), m.p. 186-188" (Found: C, 72.0; H, 6.3; N, 8.I. CztHzzNzO¡ requires C,
72.0: H,6.3; N, 8.07o)" lH n.m.r. (300 MHz, CDCI3) ô 1.30, s,9H, CMe¡; 3.48,
dd, J 10.0 IJz, l4.l Ha H3; 3.56, dd, J 6.7 Hz, l4.l Hz,H3'; 4.99, dd, J 6.7 Hz,
10.0 Hz, H2:5.62, br s, NH; 7.18, s,5H, Ph;7.68-1.80, m,4H, Phth. Vmax
3300, 1775, 1720,1660, 1510, 1380, 720 cm-r" Mass spectrum; mlz 350 (M*',
lTVo),279 (l),251 (50), 250 (54),232 (26),203 (10), 188 (7)' 173 (19),167
(29), 160 (20), r49 (100), 104 (41), 9r (16),77 (33).
Elution of this compound through a Regis Pirkle HPLC column showed the
presence of only the (S)-enantiomer (78). This enantiomer (78) had a retention

Experimental I23
time of 21 min, eluting with a gradient of hexane and iso-propanol (5-407o iso-
propanol).
(S) -O -
^cetyl-N-phthaloyltyrosine (79)
A solution of (S)-N-phthaloyltyrosine (71) (3.0 g, 9.6 mmol) in acetic
anhydride (20 ml) was refluxed for 4 h. The solution was allowed to cool, then
was poured into water (200 ml) and stirred vigorously for 10 min. The
mixture was cooled on ice and the solid was isolated by filtration, washed with
water, and dried over P2O5. Recrystallization of the solid gave the title
compound (79) as a white powder (2.68 9,797o), m.p. 186-187" (Found M+',
mlz 353.091. CleHlsNOo requires mlz 353.090¡. t1¡ n.m.r. (300 MHz, CDCI¡)
õ2.23, s, 3H, OAc; 3.58, d, J 8.3 }lz,2H, CHz;5.22, t, J 8.3 H:z,IÐ;6.92, d, J
8.4H2,2H, ArH;7.18, d, J 8.4 IIz,2H, ArH; 7.67-7.80, m,4H, Phth; 9.09, br
s, COzH. v*a* 3480, 3100-2600 br, 17J5, 1760, 1720,1510, 1390,1195, 1020,
9I5,720 cm-l. Mass specfium; mlz 353 (M*', l7o),311 (-CHzCO, 5), 288
(r4),273 (4),227 (5),2r3 (5), 187 (6), r73 (13), 164 (100), r47 (PhrhNH,
24), r30 (10), 121 (11), 107 (64), r04 (13), 91 (t0),71 (17),76 (17), 43 (30).
(S) -O -A c etyl- N - terf-b utyl -¡¡r ø-p hthaloyltyrosinamide (8 0)
To a solution of (S)-O-acetyl-N-phthaloyltyrosine (79) (2.0 9,5ó7 rnrnol)
and triethylamine (574 mg,5.67 mmol) in dichloromethane (20 ml) was added
ethyl chloroformate (615 mg, 5.67 mmol). The mixture was stirred for 10
min, then was cooled to 0". tert-Butylamine (415 mg, 5.67 mmol) was added,
and the mixture was stirred at 0" for 15 min. The mixture was allowed to warm
to room temperature, then was stirred for a further 30 min. The mixture was
filtered, the filtrate was washed with water, then was dried. The solvent was

Experimental 124
removed under reduced pressure and the residue was recrystallized from
hexane/ethyl acetate to give the title compound (80) as a white solid (2.15 g,
937o), m.p.212-213' (Found: C, 67.2; H, 5.8; N, 6.7. C23H2aN2O5 requires C,
67.6; H,5.9; N,6.97o). lH n.m.r. (300 MHz, CDCI3) ô 1.31, s,9H, CMe¡; 2.24,
s, 3H, OAc; 3.50, dd, J 10.0 Hz,l4.2Hz,H3;3.56, dd, J 6.8 IJ'z,I4.2Hz,H3';
4.98, dd, J 6.8 Hz, 10.0 Hz, H2:5.87, br s, NH; 6.92, d, J 8.5 Hz,2}I, ArH;
7.I9, d, J 8.5 }Iz,2H, ArH; 7.69-7.8I, m,4H, Phth" vnlu* 3430,1762, 1718,
1688, 1510, 1392, 1198, 960,ll9 cm-l. Mass spectrum; mlz 408 (M+',98o),
308 (4), 266 (59),248 (24), 2t9 (56), r92 (29), r13 (20), 163 (23), 162 (22),
r47 (31), 130 (25), t20 (27), t07 (68), 104 (5r),76 (53),43 (100).
(S)-3-Bromo-N-phthaloylvaline Methyl Ester (81)
To a solution of (S)-N-phthaloylvaline methyl ester (72) (1.0 g, 3.83
mmol) in CCI¿ (30 rnl) was added NBS (0.68 g, 3.83 mmol), and the mixture
was refluxed for 2 h while irradiated with a 250W mercury latnp. The mixture
was allowed to cool, then was filtered and the filtrate was washed with water
(30 mt). The organic phase was dried and then concenfrated under reduced
pressure. Recrystallization of the residue from hexane/dichloromethane gave
the títle compound (81) as a white solid (0.87 g,83Vo), m.p. 124-126". rH
n.m.r. (300 MHz, CDCI3) ô 1.99, s, 3H, Mle;2.15, s, 3H, Me'; 3.7I, s, 3H,
OMe; 5.I7, s, H2; 7.81, m,2H, Phth; 7.92,m,2H, Phth. lH n.m.r. spectral
data consistent with literature values.42

Experimental I25
(253R)-3-Bromo-N-phthaloylphenylalanine Methyl Ester (82a) and
(2535)-3-Bromo-N-phthaloylphenylalanine Methyl Ester (82b)
To a solution of (S)-N-phthaloylphenylalanine methyl ester (74) (10.0 g,
32.3 mmol) in CCla (150 ml) was added NBS (5.76 g,32.4 mmol), and the
mixture was refluxed for 2 h while irradiated with a 250W mercury lamp" The
mixture was allowed to cool, then was filtered and the filtrate was washed with
water (100 ml). The organic phase was dried, then was concentrated under
reduced pressure to give the title compounds (82a) and (82b) (12.55 g, lffi%o)
as a 1:l mixture. Fractional recrystallization from hexane/dichloromethane
(ca.2:1,200 ml) gave the threo-diastereomer (82a) (5.27 g,427o), m.p. 142-
143'(Found: C, 55.7;H,3.7; N, 3.6. ClsHlaBrNOa requires C, 55.8; H,3.7;
N, 3.67o). lH n.m.r. (300 MIIZ, CDCI3) ô 3.55, s, 3H, OMe; 5.52, d, J 1l.2Hz,
lI2;6.02, d, J 1l.2Hz,}{3;7.34-7.61, m, 5H, Ph; 7.79-7.97, m, 4H, Phth. Vmax
1778, 1755, 1709,708 cm-I. Mass spectrum; mlz 389 and 387 (M*', 67o),330
(-COzMe, 8), 328 (-CO2Me, 8), 308 (-Br, 65), 307 (-HBr, 18), 276 (89),249
(92),248 (100), 242 (32),24A Q2\218 (59), 190 (46), t62 (39), r3O (22),
105 (46), 103 (28), 77 (Il). Further recrystallization gave the erythro-
diastereomer (82b) (4.98 9,407o), m.p. l2l-122" (Found: C, 56.2; H, 3.7; N,
3.7. CrsHlaBrNOa requires C, 55.8; H,3.7: N, 3.67o). lH n.m.r. (300 MHz,
CDCI¡) ô 3.82, s, 3H, OMe; 5.59, d, J 10.5 Hz, H2,5.91, d, J 10.5 Hz, H3;
7 .lI-7 .37 , m, 5H, Ph; 7 .63-7 .72, m,4H, Phth. 13C n.m.r. (75.5 N.4Hz, CDCI¡)
õ 47.6, 53.1, 51.0, 123.6, 128.1, 128.6, 128.9, 130.9, 134.3, 137.r, 166.3,
167.2. v^u* 1774, 1758, 1718, 727 cm-r. Mass spectrum; mlz 389 and 387
(M*', ZVo),330 (-COzMe, 5), 328 (-CO2Me, 5), 308 (-8r,32),276 (59),249
(79),248 (85),242 (24),240 (24),2t8 (56), 190 (62), 169 (tt), 167 (11), l6l
(26), 130 (33), 104 (82), r02 (64),76 (100).

Experimental 126
(2 S 3 R) -3- B romo -N - te rt-buty¡ -¡¿ø- phthaloylp henylalaninamide (S5a)
and(2 S 3 S ) -3- B romo - N - te rt-buty¡ -¡y a-p hthaloylpheny lalaninami de (S5b )
To a solution of ( s ) -N -tert-butyl-Nø-phthaloylphenylalaninamide (78) (3.5
g, 10.0 mmol) in CCl¿/dichloromethane (3:1, 60 ml) was added NBS (1 .87 g,
10.5 mmol), and the mixture was refluxed for 2 h while irradiated with a
250W mercury lamp. The mixture was allowed to cool, then was filtered and
the filtrate was washed with water (60 rnl). The organic phase was dried, then
was concenfrated under reduced pressure to give the title compounds (85a) and
(85b) (4.17 g, 977o) as a 1:l mixture" Fractional recrystallization from
hexane/iso-propanol gave the erythro-diastereomer (85b) (1.84 g,43Vo), m.p.
208-209" (Found: C, 58,6; H, 5.0; N, 6.8. CztHztBrNzO¡ requires C, 58.8; H,
4.9; N, 6.57o). lH n.m.r. (300 MHz, CDC13) ô 1.40, s, 9H, CMe¡; 5.32, d, J
Il.4 IJz, I{2; 6.05, d, J 1I.4 Hz, H3; 6.41, br s, NH; 7.11-7.38, m, 5H, Ph;
7.62-7 .72, m, 4H, Phth" v'nu* 3350, 1700, 1530, 1450, 1365, 710 cm-l. Mass
spectrum; mlz 430 and 428 (M*', 57o), 415 (-Me, 0.2), 413 (-Me, 0.2),375
(0.6), 313 (0.6),330 (2),328 (2),249 (rW), 232 (6),220 (3),204 (5), 165 (2),
130 (3), 104 (8), I02 (7), 16 (6). Further recrystallization gave the threo-
diastereomer (85a) (I.75 g,4l%o), m.p. 188-191" (Found: C,58.8; H,4.9; N,
6.6. CztH21BrN2O3 requires C, 58.8; H, 4.9; N, 6.57o). lH n.m.r. (300 MHz,
CDCI¡) õ 1.02, s, 9H, CMq; 5.22, d, J 11.8 Hz, H2;5.76, br s, NH; 6.17, d,I11.8 Hz, H3;1.34-7.62, m,5H, Ph; l.7l-7.96, m, 4H, Phth. vn'u^ 3375 , 1J75,
I'705, 1530, 1380, 120 cm-r. Mass spectrum; mlz 430 and 428 (M*', IVo),4I5
(-Me, 0.l), 413 (-Me, 0.1), 375 (0.2), 373 (0.2), 330 (1), 328 (1), 249 (IO0),
232 (r0),220 (3),204 (4),165 (2),130 (3), 104 (r2),102 (10), 76 (13).

Experimental 127
Treatment of QS3R)- and (2535)-3-Bromo- N-tert-butyl-¡¡rø-phthaloylphenylalaninamide (85a) and (85b) with tri-n-butyltinhydride
A 1:1 mixture of the bromides (85a) and (85b) (1:1, 100 m9,0.23 mmol)
was dissolved in benzene (5 ml) under an atmosphere of nitrogen.
Tri-z-butyltin hydride (67 mg,0.23 mmol) was added, and the solution was
refluxed for 4 h. The solvent was removed under reduced pressure, then
hexane (10 ml) was added to dissolve the tri-n-butyltin bromide. The
suspension was stirred for 5 min, then the reduced compound (78) was isolated
by filtration as a white solid (71 mg,887o). Spectral characteristics of this
material were identical to those described above.
Elution of this compound through a Regis Pirkle HPLC column showed the
presence of only the (S)-enantiomer (78). This enantiomer (78) had a retention
time of 2I min, eluting with a gradient of hexane and iso-propanol (5-407o iso-
propanol).
(Z) -N - te rf-B utyl-Na-phthaloyl-ø,p-d ehydrop henylalaninamid e (86)
A solution of 18-crown-6 in acetonitrile (1.15M,25m1) was treated with
anhydrous potassium fluoride (0.11 9,2.3 mmol), and the mixture was stirred
vigorously at reflux for 30 min. The threo-bromide (85a) (0.50 g, 1.2 mmol)
was then added and the mixture was stirred at reflux for a further 30 min, after
which the mixture was allowed to cool and was then concentrated under
reduced pressure. The residue was taken up in ethyl acetate, and the suspension
was washed with water, dried, then concentrated under reduced pressure.
Recrystallization of the residue from hexane/ethyl acetate afforded the title
compound (86) as coiouriess crystals (0.36 9,847o), m.p. 2i5' (Found: C,72.6;
H, 5.8; N, 8.0. C21H2oN2O3 requires C,72.4; H, 5.8; N, 8.07o). lH n.m.r. (300
}d}Jz, CDCI3) ô 1.40, s, 9H, CMe3; 5.9J, br s, NH; 7.25, m,5H, Ph; 7.59, s,

Experimental I28
vinylic-H; 7.75, m,2H, Phth; 7.87, m,2H, Phth. v*u* 3350, 1780, 1720, 1660,
1630, 1525 cm-l. Mass spectrum; mlz 348 (M*', I07o),334 (6),321 (4),294
(67),277 (36),264(1),248 (100), 165 (8), 162 (8), 145 (5), 104 (88),76(32).
(B) -N -terf-Butyl-Nø'phthaloyl-cr,B-dehydrophenylalaninamide (87)
Treatment of the erythro-bromide (85b) with the 18-crown-6 complex of
potassium fluoride in acetonitrile, as described for the synthesis of (Z)-N-tert-
butyl-Nø-phthaloyl-a,B-dehydrophenylalaninamide (86) from the thre o-
bromide (85a), afforded a 5:l mixture of (E)- and (Z)-N-tert-búyl-Nd-
phthaloyl-o,p-dehydrophenylalaninamide (87) and (86). The isomers (86) and
(87) were separated by fractional recrystallization from a dilute solution of
hexane/ethyl acetate to give the (Z)-isomer (86) (9Vo), identical to the sample
described above, and the (E)-isomer (87) (517o), m.p.185-186o (Found: C,
12.5; H, 5.8; N, 8.0. C21H26N2O3 requires C, 72.4; H, 5.8; N, 8.07o). lH
n.m.r. (300 MHz, CDCI3) õ 1.22, s, 9H, CMe¡; 5.50, br s, NH; 7.05, s, vinylic-
H;7.40, m,5H, Ph; 7.85, m,4H, Phth. v^u*3420, lJ15, 1660, 1510 cm-l.
Mass spectrum; mlz348 (M*').
(Z)-N-Phthaloyl-ø,B-dehydrophenylalanine Methyl Ester (88)
Treatment of the threo-bromide (82a) with the 18-crown-6 complex of
potassium fluoride in acetonitrile, as described for the synthesis of (Z)-N-tert-
butyl-Nø-phthaloyl-o,B-dehydrophenylalaninamide (86) from the threo-
bromide (85a), afforded the title compound (88) as colourless crystals (887o),
m.p. 136-137o (Found: C, 70.6;H,4.2; N, 4.4. CrsHr¡NO4 requires C,70.4iH,
4.3; N, 4.67o). lH n.m.r. (300 MHz, CDCIg) õ 3.82, s,3H, OMe; 7.26-7.42,m,
5H, Ph; 7.18, m,2H, Phth; 7.90, m,2P,., Phth; 8.12, s, vinylic-H. vru* 1780,

Experimental 129
1120, 1640,1600 cm-l. Mass spectrum; mlz 307 (M*', 1007o),279 (-CO),248
(-COzMe) ,247 .
(E')-N-Phthaloyl-a,p-dehydrophenylalanine Methyl Ester (89)
Treatment of the erythro-bromide (82b) with the 18-crown-6 complex of
potassium fluoride in acetonitrile, as described for the synthesis of (Z)-N-tert-
butyl-Nø-phthaloyl-cr,B-dehydrophenylalaninamide (86) from the thre o-
bromide (85a), afforded a 2:l mixture of (Z)- and (E)-N-phthaloyl-o,0-
dehydrophenylalanine methyl ester (88) and (89). Recrystallization of the
mixture from hexane/ethyl acetate gave the (Z)-isomer (88) (34Eo). The
(E)-isomer (89) could not be obtained pure, but 5:1 mixture of the (E)- and
(Z)-isomers (89) and (88) was isolated as a clear oil. The (E)-isomer (89) had
1H n.m.r. (300 MHz, CDCI3) õ3.74, s,3H, OMe; 7.23, s, vinylic-H;7.29-7.49,
m, 5H, Ph; 7.80, m,2}J, Phth; 7.93, m,2}{, Phth" vru^ 1780, 1724, 1645,
1600, 1560 cm-l. Mass spectrum; mlz 307 (M*', LjOVo), 279 (-CO),248
(-COzMe) ,247 .
(S)'4-Bromo-N-phthaloylleucine Methyl Ester (91)
To a solution of (S)-N-phthaloylleucine methyl ester (73) (10.3 g, 37.5
mmol) in CCI¿ (150 mt) was added NBS (6.67 9,37.5 mmol), and the mixture
was refluxed for 2 h white inadiated with a 250W mercury lamp" The mixture
was allowed to cool, then was filtered and the filtrate was washed with water
(100 ml). The organic phase was dried, then was concentrated under reduced
pressure. The residue was purified by chromatography on silica to give the
bromide (91) as a viscous oil which crystallized on storage at -20o overnight.

Experimental 130
Recrystallization from ethanol/water gave the title compound (91) as colourless
crystals (10.8 E, 8l7o), m.p. 63-64" (Found: C, 50.9; H, 4.6; N, 4.0.
C15H16BrNOa requires C, 50.8; H,4.6;N,4.07o).lH n.m.r. (300 MHz, CDCI3)
õ 1.76, s,3H, Me; 1.84, s,3H, Me'; 2.85, m,2}l,CH2;3.74, s,3H, OMe; 5.25,
dd, J 4.4 FI2,8.0 Hz, }l2;7.75-7.91, m, 4H, Phth. vmax 1748, 1719, 1269,
1230, Il34,7Il cm-r. Mass spectrum; mlz 355 and 353 (M*', 0.3Vo) 354 (-H,
0.6),352 (-H, 0.6), 296 (-COzMe, 1), 294 (-COzMe, 1), 274 (-8r,2),215 (64),
214 (60),200 (74), t74 (35), 160 (100), l3O (26), 104 (38), 76 (49).
4-Bromo-N-phthaloylhomophenylalanine Methyl Ester (92)
To a solution of N-phthaloylhomophenylalanine methyl ester (77) (7.5 g,
23.2 mmol) in CCI¿ (150 ml) was added NBS (4.15 g, 23.3 mmol), and the
mixture was refluxed for 2 h while inadiated with a 250W mercury lamp" The
mixture was allowed to cool, then was filtered and the filrate was washed with
water (100 ml). The organic phase was dried and then concenffated under
reduced pressure. The residue was purified by chromatography on silica to
give an approximately 1 .2:l ratio of the diastereomers of the title compound
(92) as a viscous oil which crystallized on standing (7.88 g, 847o), m.p. 112-
118" (Found: C,56.8; H,4.0; N,3.5. C19H16BrNOa requires C, 56.1; H,4.0;
N,3.57o). lH n.m.r. (300 MHz, CDCI3) ô (one diastereomer) 3.12-3.27,m,2H,
CHz;3.75, s,3H, OMe; 4.91,t,J 7.6H2,}J4;5.21, dd, J 5.8 Hz, 9.4H2,H.2;
7.05-7.35, m, 5H, Ph; 7.73, m, 2H, Phth; '7.79, m, 2H, Phth; (other
diastereomer) ô 2.99-3.31, m, 2H, CHz; 3.73, s, 3H, OMe; 4.91, t, J 7 .6 Hz, H4;
5.10, t, J 7.5 H:z, H:2; 7.05-7.38, m, 5H, Ph; 7.76, m, 2H, Phth; 7.88, m,2H,
Phth. v,nax1774, 1744, 1716, 1264, 1216,122,704 cm-l. Mass spectrum: mlz
403 and 401 (M+', 0.57o), 402 (-H, 1), 400 (-H, l), 369 (2), 367 (2),342 (5),

Experimental l3I
340 (5), 322 (-Br, l00),262 (76),219 (68), 187 (31), 175 (15), 148 (24), 132
(33), 130 (42), tO5 (37), tO4 (57),91 (29).
(S)-O-Acetyl-N-phthaloyltyrosine Methyl Ester (93)
To a solution of (S)-N-phthaloyltyrosine methyl ester (75) (5.0 g, 15.4
mmol) in dichloromethane (20 ml) was added triethylamine (2.33 g, 23.0
mmol), acetic anhydride (2.35 g, 23.0 mmol) and DMAP (100 mg, 0.82
mmol). The solution was stirred at ambient temperature for t h, then water
(20 ml) was added, and the mixture was stirred vigorously for a further 10
min. The organic phase was separated, washed with water, dried, and then the
solvent was removed under reduced pressure. The solid residue was
recrystallized from hexane/ether to give the title compound (93) as colourless
needles (4.91 g,877o) m.p. 86-87o (Found: C, 65.3; H,4.5: N, 3.8. C29H17NO6
requires C, 65.4; H, 4.7; N, 3.87o). lH n.m.r. (300 MHz, CDCI3) õ 2.22, s, 3H,
OAc; 3.55, dd, J 10.5 Hz, 14.4 Hz,H3;3.59, dd, J 6.0 Hz, 14.4 Hz,H'3';3.78, s,
3H, OMe; 5.16, dd, J 6.0 Hz, 10.5 Hz,IÐ;6.92, d, J 8.6 Hz, 2H, ArH; 7.18, d, J
8.6 Hz, 2H, ArH; 7.68-7.80, m, 4H, Phth. v^u* 1775,1760, 1720, 1610, 1510,
1390, 1190, 915,7I5 cm-l. Mass specffum; mlz367 (M*',2Vo),325 (9),308
(-OAc, 3),266 (8), 248 (3),220 (11), 178 (100), 147 (16), 122 (71), I2l (M),
r07 (39),105 (34).
(253R)-O-Acetyl-3-bromo-N-phthaloyltyrosine Methyl Ester Qaa)and(2535)-O-Acetyl-3-bromo-N-phthaloyltyrosine Methyl Ester (94b)
To a solution of (S)-O-acetyl-N-phthaloyltyrosine methyl ester (93) (1.0 g,
2.7 mmol) in CCI+ (50 ml) was added NBS (520 mg, 2.9 mmol), and the

Experimental I32
mixture was refluxed for 2 h while irradiated with a 250W mercury lamp. The
mixture was allowed to cool, then was filtered and the filtrate was washed with
water (50 mt). The organic phase was dried, then was concentrated under
reduced pressure to give thetitle compounds (9aa) and (94b) as a 1:1 mixture
(1.18 g, 977o) (Found: y7+',mlz 445.018. CzoH168rNO6 requhes mlz
445.016). lH n.m.r. (300 MHz, CDCI3) threo-isomer (94a) õ 2.30, s, 3H, OAc;
3.56, s, 3H, OMe; 5.46, d, J II.2 Hz, fl2;6.03, d, J lI.2 Hz, H3:- J .I4, d, J 8.6
Hz, 2H, ArH; 7.6I, d, J 8.6 H:z, 2H, ArH; 7.78-7.96, m, 4H, Phth; erythro-
isomer (94b) õ2.I9, s,3H, OAc; 3.81, s,3H, OMe; 5.56, d, J 10.4 Hz,II2;
5.93, d, J 10.4 Hz,}I3;6.93, d,I 8.6H2,2H, ArH; 7.37, d,J 8.6H2,2H, ArH;
7.64-7.73, m, 4H, Phth. Mass spectrum; mlz 447 and 445 (M*', 0.37o),405
(0.3), 403 (0.3), 388 (-CO2Me, 0.6), 386 (-CO2Me, 0.6),366 (-Br, 8), 334
(r7),324 (5), 306 (5),292 (16),264 (100), 218 (r2), r87 (7), t63 (9), 147
(10), 138 (r4),tzl (35), 104 (22),86 (38),76 (19).
( 2 S 3 R) - O- A ce ty l - 3 - b r o mo - N - t e r t -buty¡ - ¡y ø- p ht hal o y I ty r o s in am i d e
(95a) and
(2 S 3 S ) - O- A ce ty l - 3 - b r o mo -N - t e rt -butyl -¡y ø- p h tha I o y lty r o s in a m id e
(esb)
To a solution of ( S )-O -acetyl-Nø-phthaloyltyrosinamide (80) (500m g, 0.82
mmol) in CCl¿/dichloromethane (4:I,20 ml) was added NBS (150 mg, 0.84
mmol), and the mixture was refluxed for 2 h while inadiated with a 250W
mercury lamp. The mixture was allowed to cool, then was filtered and the
filtrate was washed with water (20 ml). The organic phase was dried, then was
concentrated under reduced pressure to give the title compounds (95a) and
lq5h\ âs e l'l mixtrrre (57R ms 97q"\ lFound: M+'. mlz 486.080.\-_-, _ \-- _ ---ò,, -,,-/ \- __-__-'.
C1;.}J23BrNzOs requires m12486.079). t1¡ n.m.r. (300 MHz, CDCI3) threo-
isomer (9.5a) ô 1.05, s,9H, CMe3; 2.31, s,3H, OAc; 5.17, d, J ll.8Hz,H2;

Experimental 133
5.87, br s, NH; 6.22, d, J 11.8 flz,}{3; J .I5, d, J 8.6 }Jz,2}I, ArH; J.62, d, J 8.6
Hz,2IJ, ArH;7.77-7.94, m,4H, Phth; erythro-isomer (95b) ô 1.38, s,9H,
CMe3; 2.20, s, 3 H, OAc; 5.30, d, J 11.5 Hz,fI3i 6.08, d, J I1.5 Hz,I12;6.41,
br s, NH; 6.95, d, J 8.6 Hz,2H, ArH; 7.39, d, J 8.6 Hz,2H, ArH; 7.62-7.73,m,
4H, Phth. Vmax 1780, 1726, 1675, 1608, 1394, 1160, 720 cm-r. Mass
spectrum; mlz 488 and 486 (M*', 27o), 446 (l), 444 (1), 403 (14), 388 (1), 386
(1), 361 (10), 308 (3t),265 (100), 248 (5), r2t (11), 118 (10), 104 (r4),76
(e).
Treatment of (S)-3-Bromo-N-phthaloylvaline Methyl Ester (81)
with silver nitrate in acetone/water.
To a solution of the bromide (8I) (1.2 g,3.52 mmol) in acetone (50 ml)
was added a solution of silver nitrate (0.90 g, 5.3 mmol) in water (50 rnl). The
resultant mixture was stirred in the dark for 24 h, then the yellow precipiøte
was removed by filtration. The filtrate was concentrated under reduced
pressure, and the residue was extracted with dichloromethane (2 x 40ml). The
combined extracts were dried, and the solvent was removed under reduced
pressure. The products were separated by chromatography on silica and
recrystallized from hexane/dichloromethane to give the p-hydroxyvaline
derivative (100) as colourless crystals (0.42 g, 437o), m.p" 79-80" (Found: C,
60.6; H, 5.5; N, 5.1. ClaHrsNOs requires C, 60.6; H, 5.5; N, 5.17o). lH n.m.r.
(300 MHz, CDCI3) õ 1.31, s,3H, Me; 1.53, s,3H, N.4e'i3.77, s,3H, OMe; 4.41,
br s, OH; 4.9I, s, H2; 7.78-7.93, m,4H, Phth.v*u* 3544,1767,1725, 1275,
lI7 cm-r. Mass spectrum; mlz 262 (-Me, l}Vo),246 (5), 230 (28),219 (100),
188 (74), 187 (98), 160 (14)" The o,B-dehydrovaline derivative (101) was
isolated as colourless crystals (0.08 g,87o), m.p. 8l-82" (Found: C,64.7i H,
5.1; N, 5.4. Cr+Hr:NO¿ requires C, 64.8; H, 5.1; N, 5.4Vo). lH n.m.r. (300
MHz, CDCI¡) ô 1.88, s, 3H, M.e:2.43, s, 3H, Me'; 3.68, s, 3H, OMe; 7.75,m,

Experimental 134
4H, Phth. vn.,u* 1728 br, l22J,l2O cm-t. Mass spectrum; mlz 259 (M+',IVo),
227 (-MeOH, 55), 132 (87), 104 (100), 76 (43)" The p,y-dehydrovaline
derivative (I02) was isolated as a clear oil (0.30 g, 34Vo) (Found: y4+', mlz
259.086. C1aH13NO4 requires mlz 259.085). lH n.m.r (300 MHz, CDCI3) ô
t.92, s,3H, y-Me; 3.79, s,3H, OMe; 5.11, s, lH; 5.14, s, lH; 5.38, s, lH; 7.73-
7.91, m, 4H, Phth. v*u* (neat) 1780, 1748, 1728, 1470, I44O, 1386, 1293,
1113, 915,717 cm-l. Mass spectrum; mlz259 (M*', 87o),227 (-MeOH,20),
200 (-COzMe, 100).
Treatment of (S)-4-Bromo-N-phthaloylleucine Methyl Ester (91)
with silver nitrate in acetone/water.
To a solution of the bromide (91) (18.9 9,53.4 mmol) in acetone (1.5 L)
was added a solution of silver nitrate (13.5 g,79.9 mmol) in water (1.0 L)"
The resultant mixture was stirred in the dark for 24 h, then the yellow
precipitate was removed by filtration" The filtrate was concentrated under
reduced pressure, and the residue was extracted with dichloromethane (2 x
400rnl). The combined extracts were dried, and the solvent was removed under
reduced pressure to give a pale yellow oil. Fractional crystallization from
ether gave the lactone (105) as a white solid (1.16 g,\Vo), m.p. 132-133'
(Ht.l¡o 131'). lH n.m.r. (300 MHz, CDCI3) ô 1.52, s, 3H, Me; 1.64, s,3H, Me';
2.46, dd, J 9.7 }{z, 12.3 }Iz, }I4; 2.60, dd, J lI.5 Hz, 12.3 Hz, H4'; 5.25, dd, J
9.7 IJz, II.5 Hz,H3;7.75-7.88, m, 4H, Phth. Mass spectrum; mlz 260 (M+H+,
l7o),215 (-COz, 82),200 (100), 160 (72), 130 (22), 104 (34), 76 (51).
Concentration of the filtrate and purification by chromatography on silica gave
the y-hydroxyleucine derivative (104) as a clear, viscous oil which was
crystallized from hexane/clichloromethane to afford eolourless crystals (12.1 g,
787o), m.p.7l-72", ¡u1orc -25.9" (c, 0.5 in EIOH) (Found C, 61.8; H, 5.9; N,
4.8. CrsHlzNOs requires C, 61.8; H, 5.9; N, 4.8¡. t1¡ n.m.r. (300 MHz, CDCI¡)

Experimental 135
õ I.24, s, 3H, Me; 1.31, s, 3H, Me'; l.70,br s, OH; 2.38, dd, J 8.8 Hz,I5.l Hz,
H3;2.50, dd, J 4.0 Hz, 15JH:z,H3';3.J3, s,3H, OMe; 5.15, dd, J 4.0 Hz, 8.8
Hz, IJ2; 7.74, m,2H, Phth; 7.86, m,2H, Phth. v*u* 3514, 1770, 1743, 1707,
1272, 1230, 1161, 720 cm-r. Mass spectrum; mlz 276 (M+'-Me, 77o),233
(-Me2CO, l7),215 (29),2W (41), 174 (100). The y,õ- and B,y-dehydroleucine
derivatives (106) and (107) were isolated as an inseparable mixture (291 mg,
27o¡. r11n.m.r (300 MHz, CDCI3) 7,õ-dehydroleucine derivative (106) õ 1.78,
s,3H, Me; 2.86, brdd, J 4.0}J.2,I4.3H2, H3; 3.10, dd,J l2.3Hz,14.3Hz,I{3'i
3.76, s, 3H, OMe; 4.65, br s, vinylic-H; 4.69, br s, vinylic-H; 5.10, dd, J 4.0
Hz,l2.3 }{z,I{2;7.71-7.89, m, 4H, Phth: p,1-dehydroleucine derivative (107) õ
1.76, s, 3H, Me; I.77, s,3H, Me'; 3.76, s,3H, OMe; 5.66, d, J 9.2Hz,IÐ;5.82"
br d, J 9.2H2,H3;7.71-7.89, m,4H, Phth. vmax 1777,1746, I7I8, 1392,
1188, 1104 cm-l" Mass spectrum; mlz273 (M*',27o),241 (6),2I4 (-CO2Me,
100), 196 (16), 190 (8), 160 (10), 130 (19), 126 (13), r04 (24),16 (28).
Treatment of (S)-4-Bromo-N-phthaloylhomophenylalanine MethylEster (92) with silver nitrate in acetone/water.
To a solution of the bromide (92) (9.0 g, 22.4 mmol) in acetone (600 mt)
was added a solution of silver nitrate (5.67 9,33.6 mmol) in water (a00 ml).
The resultant mixture was stirred in the dark for 24 h, then the yellow
precipitate was removed by filtration. The filtrate was concentrated under
reduced pressure, and the residue was extracted with dichloromethane (2 x
250m1). The combined exfracts were dried, and the solvent was removed under
reduced pressure to give a pale yellow oil. Separation of the products by
chromatography on silica gave the y-hydroxyhomophenylalanine derivative
(109) as a 1.5:1 ratio of diastereomers, as a clear, viscous oil (6.06 g,8O7o)
(Found ¡11+', mlz 339.1I2. CeHlzNOs requires mlz 339.lII). lH n.m.r. (300
MHz, CDCI¡) ô (major isomer) 2.54-2.76,m,2H, CHz; 3.70, s,3H, OMe; 4.92,

Experimental 136
br dd, J 5.2 }lz, 6.5 Hz,}{4;5.08, dd, J 5.3 f{2,8.8 IIz, H2:7.II-7.32, m, 5H,
Ph; 7.67-7.79, m,4H, Phth; (minor isomer) õ2.54-2.16, D, 2H, CH2;3.71, s,
3H, OMe;4.57, br dd, J 4.2 Hz, 8.8 }lz,}I4;5.25, dd, J 4.7 Hz, l0.4Hz,H2;
7.ll-7.32, m,5H, Ph;7.7I-7.85, m,4H, Phth. vn'u* 3600,3480, 17'74, 1752,
1719, 1614, 1426, 1280, 1184, 1118, 918,719 cm-l. Mass spectrum: mlz 339
(M*', 37o),338 (4),322 (-OH, 65), 308 (-OMe, l5),262 (16),219 (100), 187
(36), 174 (54), 160 (20), 148 (r3)" r32 (19), 1 16 (26), 105 (36), 77 (r7).
Further recystallization gave the cis-lactone (110) (198 mg, 3Vo), m.p. 140-
143" (Found M+H+, mlz 308.092. C13H1¿NO+ requires mlz 308.092¡. r¡¡ n.m.r.
(300 MHz, CDCI3) õ2.65, ddd, J 3.3H2,9.8H2, 13.I}{z,H4;3.l2,ddd,J 8.7
Hz,9.4Hz,13.L}Iz, H4'; 5.1'l,dd,J 9.4Hu9.8Hz,, H5;5.93, dd, J 3.3 Hz,8.J
Hz, IJ3; 7.33-7.54, m, 5H, Ph; 7.74-7.90, m, 4H, Phth. v'nu* 3005, 1790, 1780,
1730, 1720, 1395 cm-l. Mass spectrum; mlz 308 (M+H+), 245,239, 148, 116
(I007o). Further recrstallization gave the trans-lactone (1ll) (129 mg,2Vo),
m.p. 143-144'(Found C, 70.2;H,4.2; N, 4.6. CrsHr:NO4 requires C,70.4;H,
4.3; N,4.6¡. tg n.m.r. (300 MHz, CDCI3) ô 2.85, ddd, J l0.6Hz,l2.OIJz,l2.5
Hz,IJ4;2.93, ddd,J 6.2H2,9.2H2, I2.5Hz,}l4';5.31, dd, J 9.2H2, I2.0Hz,
H5; 5.50, dd, 6.2 Hz, 10.6 Hz, H3; 1.33-7.56, m, 5H, Ph; 7.77-7.93, m, 4H,
Phth. vr* 3000,1795,1780, 1730,1390, 1095 cm-I. Mass spectrum; mlz307
(M*.), 245, I48,116 $mEo).
(253R) -3-Hydroxy-N-phthaloylphenylalanine Methyl Ester (ll2a)and(2535)-3-Hydroxy-N-phthaloylphenylalanine Methyl Ester (112b)
To a solution of a 1:1 mixture of the bromides (82a) and (82b) (I.09,2.6
mmol) in acetone (40 nìl) was added a solution of silver nitrate (660 mg, 3.9
mmol) in water (40 ml), and the resultant mixture was stirred in the dark for
24 h. Tþe precipitate was removed by filtration and the filtrate was

Experimental 137
concentrated under reduced pressure. The residue was extracted with
dichloromethane, the organic phase was dried, and then the solvent was
removed under reduced pressure to give a crude mixture of the (253R)- and
(2535)-3-hydroxyphenylalanine derivatives (112a) and (lI2b) in a 5:1 ratio
(769 mg, 92Eo)" Recrystallization from hexane/dichloromethane gave the
(253R)-þ-hydroxyphenylalanine derivative (l l2a) as colourless crystals (633
mg,75Vo), m.p. 185-186", [o]o16 -67.0" (c, 0.6 in EtOH) (Found: C, 66.7i H,
4.7; N, 4.4. ClsHrsNOs requires C, 66.5; H, 4.7; N, 4.3Vo). lH n.m.r. (300
MHz, CDCI¡) ô 3.86, s,3H, OMe; 5.13, d, J 10.4 Hz, OH; 5.51, d, J 4.6H2,ÍÐ;
5.71, dd, J 4.6 IJ.z, 10.4 }lz, Il3: I .14-l .34, m, 5H, Ph; 7 .69-1.80, m, 4H, Phth.
Vmax 3580, 3420 br, 1780, 1745" 1715, 1390, 1215, 1020, 120 cm-r. Mass
spectrum (FAB); mlz 326 (M+H+, 77Vo),308 (-HzO, 100), 248 (54),219 (19),
t60 (27), 149 (23), r3t (21),105 (48), 104 (33), 9t (42)"
Elution of the mother liquor through a reverse-phase HPLC column gave
the (253R)- and (2535)-hydroxyphenylalanine derivatives (112a) and (112b), at
retention times of 10.5 and 11 min, respectively. The (2S3S)-isomer (112b)
was collected from repeated injections (ca" 5 mg) m.p" 110-111'. lH n.m.r"
(300 MHz, CDCI3) E 3.72, s, 3H, OMe; 4.2J , d, I 2.3 Hz, OH; 4.95, d, I 8.4 Hz,
II2; 5.46, dd, J 2.3 Hz, 8.4 }{z, H3; 7 .ll-l .21 , m, 5H, Ph; 7 .60-7 .70, m, 4H,
Phth.
(S)-N-Phthaloylphenylalanine tert-Butyl Ester (114)
To a solution of (S)-N-phthaloylphenylalanine (70) (l .0 g,3.2 mmol) in
dichloromethane (25 ml) was added DCC (730 mg, 3.5 mmol), /¿rl-butanol
(260 mg,3.5 mmol) and DMAP (39 mg, 0.32 mmol). The solution was stirred
at room temperature for 16 h, then the dicyclohexylurea was removed by
filtration. filfrate was washed with 57o acetic acid solution (2 x 25 ml),

Experimental 138
and water (2 x 25 ml). The organic phase was dried, then the solvent was
removed under reduced pressure to give the crude tert-butyl ester (114) as a
viscous oil. The crude product was purified by chromatography on silica,
yielding the ester (114) as a clear oil which solidified on standing.
Recrystallization from hexane/ether gave the title compound (114) as large
colourless crystals (0.85 g, 75Vo), m.p. 95-96" (Found ¡4+', mlz 351.148.
CzrHzrNO4 requires mlz 351.147)" lH n.m.r. (300 MHz, CDCI3) ô 1.45, s, 9H,
CMe¡; 3.51, dd, J 10.7 }lz, l4.3}Jz,H3i 3.55, dd, J 6.0 I{z,l4.3Hz,H3;5.07,
dd, J 6.0 }lz, 10.7 Hz,IÐ;7.I0-7.2I, m, 5H, Ph;7.66-7.79, m,4H, Phth. Vmax
1780, I745, 1720,1390, 710 cm-1. Mass specffum; mlz 352 (M+H+, 27o),35I
(I), 296 (29), 295 (53), 250 (-COzCMq,73),232 (28), 148 (100), I3O (12),
104 (14), 91 (8), 77 (9),57 (25).
(253R)-3-Bromo-N-phthaloylphenylalanine tert-Butyl Ester (115a)
and(2535)-3-Bromo-N-phthaloylphenylalanine tert-Butyl Ester (115b)
To a solution of (S)-N-phthaloylphenylalanine tert-butyl ester (114) (7W
mg, 2.0 mmol) in CCI¿ (50 ml) was added NBS (355 mg,2.0 mmol), and the
mixture was refluxed for 2 h while irradiated with a 250W mercury lamp. The
mixture was allowed to cool, then was filtered and the filtrate was washed with
water (50 ml). The organic phase was dried, then was concentrated under
reduced pressure to give the title compounds (115a) and (115b) (858 mg,
l00%o) as a 1:1 mixture, as a pale yellow oil which partially crystallized on
standing overnight. Fractional recystallization from hexane/dichloromethane
gave the threo-díastereomer (115a) (348 mg, 4l7o), m.p. 152-153"" lH n.m.r.
(300 MHz, CDCI3) ô l 16, s, 9H, CMe:; 5.4J, d, J 11 .4 H2,H2;6.06, d. J Il.4
Hz,H3;7.34-7.59, m, 5H, Ph;7.77-7.96, m,4H, Phth. v*u^ 1780,1J38, 1120,
1420, 1390, 1050, 840, 110 cm-l. Mass spectrum; mlz 431 and 429 1N'{+',

Experimental 139
.02Vo),375 (9),373 (9),330 (-COzCMe3, lI),328 (-COzCMe3, 11),294 (4),
216 (2),249 (93),248 (94),232 (r5),220 (lO),204 (44), 165 (7), 130 (8), 104
(38), 102 (32),76 (36), 57 (100). Further recrystallization gave the erythro-
diastereomer (115b) (337 mg,397o), D.P. 118-119". lH n.m.r. (300 MHz,
CDCI¡) õ 1.48, s, 9H, CMe3; 5.49, d, J 10.4 Hz,}f2;5.85, d, J 10.4 }l.z,fI3i
7.10-7.36, m, 5H, Ph;7.62-7.72, m,4H, Phth. vru^ 1780, l'748, 1726, 1390,
715 cm-l. Mass spectrum; mlz43l and 429 (M+',.017o),315 (7),373 (1),330
(-COzCMe3, 10), 328 (-COzCMe3, l0),294 (6),276 (3),249 (75),248 (100)'
232 (tt),220 (8), 204 (28), 165 (5), 130 (6), 104 (27), r02 (25),76 (26), 51
(66).
(253R) -3-Hydroxy-N-phthatoylphenylalanine tert-Butyl Ester
(116a) and (253S) -3 - Hydroxy -N-phthaloylphenylalanine tert-Butyl
Ester (116b)
To a solution of a 1:1 mixture of the bromides (115a) and (115b) (430 mg,
1.0 mmol) in acetone(12 ml) was added a solution of silver nitrate (255 mg,
1.5 mmol) in water (8 mt), and the resultant mixture was stined in the dark
for 24 h" The precipitate was removed by filtration and the filtrate was
concentrated under reduced pressure. The residue was extracted with
dichloromethane, the organic phase was dried, and then the solvent was
removed under reduced pressure to give a crude mixture of the (253R)- and
(2535)-hydroxyphenylalanine derivatives (116a) and (116b) in an 8:1 ratio.
Recrystallization from hexane/dichloromethane gave the threo-B-
hydroxyphenylalanine derivative (116a) as colourless crystals (297 mg,8I7o),
m.p. 125-127" (Found: C,68.7; H,5.9; N, 3.8. CzrHztNOs requires C, 68.7; H,
5.8; N, 3.87o). lH n.m.r. (300 MHz, CDC13) ô 1.52, s, 9H, CMe¡; 5.25, d, J
lO.6 ldz, OIgr; 5.44, d, J 4.6 Hrz, H2;5.67, dd, J 4.6 Hz, 10.6 Hz, H3; I .15-7 .36,

Experimental l4O
m, 5H, Ph;7.67-7.79, m,4H, Phth. v'nu* 3440, 1785, 1738, 1712, 1604, 1552,
1392, 1194, 1108, 844, 120 cm-1. Mass spectrum (FAB); mlz 368 (M+H+,
87o),312 (44),294 (-MqCOH, 55),216 (23),266 (-COzCMe3, 10), 250 (100),
232 (tt),205 (13), 160 (19), 105 (2t), 9r (20),87 (25), 51 (93). The lH
n.m.r. spectrum of the crude product showed peaks attributable to the erythro-
isomer (116b); õ 1.28, s, 9H, CMe¡; 4.60, br s, OH; 4.95, d, J 8.0 Hz,IÐ;5.49,
d, J 8.0 IJz,H3;7.I5-7.36, m,5H, Ph;7.67-7.79,m,4H, Phth.
(2 S 3 R) -3- Hy droxy - N -te rt-butyl-N a-phthaloylp heny lalaninamid e
(118a)
To a solution of a 1:l mixture of the bromides (85a) and (85b) (500 mg,
1.17 mmol) in acetone (20 ml) was added a solution of silver nitrate (300 mg,
1.76 mmol) in water (20 ml). The mixture was stirred in the dark for 24 h,
then was filtered through celite and the filnate was concentrated under reduced
pressure. To the residue was added dichloromethane (50 ml) and water (30
ml), the organic phase was then dried, and the solvent was removed under
reduced pressure. The crude product was then recystallized from
hexane/dichloromethane to give the title compound (118a) as colourless crystals
(398 mg,937o), m.p. 195-197" (Found: C,68.8; H,6.0; N,7.7. CztHzzNzO¿
requires C, 68.8; H, 6.1 ; N,7 .77o). lH n.m.r. (300 lvftlz, CDCI3) õ 1.30, s, 9H,
CMer; 4.41,d,J 8.2 Hz, OH; 5.11, d, J 6.2H2,H2;5.63,dd,16.2H2,8.2H2,
H3; 5.95, br s, NH; 7.20-7.38, m, 5H, Ph;7.68-1.81, m, 4H, Phth. Vmax 3352,
3275, 1778, llÙ4, 1644,717 cm-r. Mass specrum (FAB); mlz 361 (M+H+,
68Vo),307 (3), 289 (2),266 (4),260 (-PhCHO, 18),250 (100)' 232 (1), 187
(10), 160 (17), 154 (24), 136 (18), 107 (8), 105 (6),11 (6). In the lH n.m.r.
spectrum of the crude product, the l3C sateiiites of the tert-butyl signal of the
threo-alcohot (118a) were observed at õ 1.08 and 1.51 (J 126.7 Hz), but no

Experimental l4l
peak atüibutable to the tert-butyl group of the erythro-alcohol (118b) (ô 1.15)
was observed.
N - te rt-Butyl- 3 - oxo -¡¡r ø-phthaloylp henylalaninamide ( 1 19)
Jones reagen¡l34 was added dropwise to a solution of the threo-alcohol
(118a) (500 mg, 1.37 mmol) in acetone (20 ml) with vigorous stirring until the
red/brown colour persisited (c¿. 0.5 ml). The reaction was stirred for a
further 15 min, then the solvent was removed under reduced pressure, and the
residue was partitioned between dichloromethane and water. The organic layer
was washed with lOTo Na2CO3, then with water, and was dried. The solvent
was removed under reduced pressure and the residue was eluted through a
short silica column with ethyl acetate. The appropriate fractions were
combined and the solvent was removed under reduced pressure.
Recrystallization of the residue from hexane/ethyl acetate gave the title
compound (1 19) as a white solid (332 mg, 677o)" lH n.m.r. (300 MHz, CDCI:)
õ 1.34, s, 9H, CMe3; 6.04, s, H2; 7.21-1.56, m,5H, Ph;7.73-7.90, m,4H,
Phth. Mass spectrum; mlz 364 (M*', 27o),308 (l),291 (2),265 40),221 (6),
147 (PhthNH,72), 105 (PhCO, 43), 104 (87), 103 (58), 76 (100)"
(2 S 3 S) -3 -Hy droxy - N -te rt-butyl -Nø- p hthaloy lp h eny lalanin amid e
(1 18b) and (2S3R) -3-Hydroxy -N-f ert-butyl-¡¡rø-phthaloylphenylalaninamide (118a)
To a solution of the ketone (119) (100 mg,0.27 mmol) in ethanol (5 ml)
was added NaBtIa (10 mg, 0.26 mmol). The mixture was stirred at room
temperature for 10 min, then was quenched by the addition of dilute HCI (ca. I
rrìl). The solvent was removed under reduced pressure, and the residue was

Experimental 142
partitioned between dichloromethane and water. The organic layer was washed
with I07o Na2CO3, then with water, then was dried and the solvent was
removed under reduced pressure" Purification of the mixture by
chromatography on silica gave a, mixture of the threo- and erythro-alcohols
(118a) and (118b) in a 1.2:l ratio (84 mg, 847o). lH n.m.r. (300 MIIZ, CDCI3)
erythro-alcohol (118b) ô 1.15, s,9H, CMe¡; 4.25,d,J 4.9 Hz, OH; 4.60,d,J
8.3 Hz,lÐ; 5.39, dd, J 4.9 H2,8.3 Hz,H3:.1.25-7.54, m, 5H, Ph; 7.68-7.84, m,
4H, Phth. lH n.m.r. specfral data of the threo-isomer (118a) identical to that
described above.
(253R)-O-Acetyl-3-hydroxy-N-phthaloyltyrosine Methyl Ester(lzla) and (2S3S) -O - A cetyl -3 - hy droxy -N- p hthaloyltyrosine MethylEster (121b)
To a solution of a 1:1 mixture of the bromides (9aa) and (94b) (1.18 g,
2.65 mmol) in acetone (40 ml) was added a solution of silver nitrate (680 mg,
4.0 mmol) in water (40 ml), and the resultant mixture was stirred in the dark
for 24 h" The mixture was then filtered, and the filtrate was concentrated
under reduced pressure. The residue was extracted with dichloromethane, the
organic phase was washed with water, dried, and then the solvent was removed
under reduced pressure to give a crude mixture of the hydroxytyresine
derivatives (121a) and (12lb) in a 6:l ratio (870 mg, 867o) (Found: M+'-HzO,
mlz 365.090. CzoHrsNOo requires m12365.090). lH n.m.r. (300 MHz, CDCI3)
threo-ísomer (121a) õ 2.19, s, 3H, OAc; 3.J8, s, 3H, OMe; 4.98, br s, OH;
5.48, d, J 5.0 Hz,IÐ;5.67, d, J 5.0 Hz, H3; 7.00, d, J 8.6 Hz,2H, ArH; 7.38, d,
J 8.6 IJz,2H, ArH; 7.64-7.17, m,4H, Phth; erythro-isomer (12lb) ô 2.17, s,
3H, OAc; 3.74,s, 3H, OMe; 4.98, br s, OH; 5.02,d, J 8.1 IIz,ÍI2; 5.54, d, J
8.1 Hz, H3;6.94, d, J 8.7 Hz,2H, ArH; J.34, d, J 8.7 Hz,2H, ArH; 7.64-1.77,
m,4H, Phth. v.¿^ 3580,3410, 1775, l'752, ll12, 1552, 1392, 1184, 1118,

Experimental I43
852,710 cm-l. Mass spectrum (FAB); mlz 384 (M+H+, l37o),366 (-HzO,79),
334 (100), 324 (-OAc, 7), 306 (35), 292 (38), 264 (58), 219 (34), 187 (23),
160 (r5), t54 (37), 136 (33), r07 (t7), 105 (13), t04 (12),89 (17), 77 (t9).
The crude mixture of the alcohols (l2la) and (121b) was found to decompose
slowly on standing and was therefore de-acetylated without purification.
(253R)-3-Hydroxy-N-phthaloyltyrosine Methyl Ester (L22a) and
(2535)-3-Hydroxy-N-phthaloyltyrosine Methyl Ester (122b)
A crude mixture of the acetylated hydroxytyrosine derivatives (121a) and
(121b) (870 mg, 2.27 mmol) was dissolved in MeO[VH2O (20:1,20 ml) and
p-toluenesulfonic acid monohydrate (100 mg 0.5 mmol) was added. The
solution was stirred at room temperature for 6 h, then the solvent was removed
under reduced pressure. The residue was partitioned between ethyl acetate and
water, the organic phase was washed twice with l07o Na2CO3, dried, and then
the solvent was removed under reduced pressure to give the hydroxytyrosine
derivatives (I22a) and (122b) in a 6:1 ratio (741 mg 967o)" Recrystallization of
the crude product from hexane/ethyl acetate gave the (253R)-p-hydroxy-
tyrosine derivative (I22a) as a white solid (556 mg, 727o), m.p. 200-201",
[cr]o16 -70.7" (c, 0.4 in EtOH) (Found: C,63.3;H,4.4; N, 4.0. CrsHrsNOo
requires C,63.3;H,4.4; N, 4.17o). L11n.m.r. (300 MHz, CDCI3) ô 3.85, s, 3H,
OMe; 4.76, s, Ar-OH; 5.02, d, J 10.3 Hz,3-OH;5.44, d, J 4.8 Hz,}l2;5.64,dd,
J 4.8 Hz, lO.3 Hz,Il3; 6.11, d, J 8.6 }Jz,2}l, ArH; 7.I9, d, J 8.6 Hz,2H, ArH;
7.70-7.81, m, 4H, Phth. v*a* 3582,3405, 1775, ll50, 1714,1516, 1392, lll2,
850, 715 cm-l. Mass spectrum (FAB); mlz 342 (M+H+, 27o),324 (-H20, 10),
307 (r2),292 (ll),289 (1),264 (5),232 (r4),231 (17), 219 (9), 154 (100),
r37 (63), 136 (77),107 (30), 105 (12), 89 (29),77 (3t).

Experimental I44
Elution of the mother liquor through a reverse-phase HPLC column gave
the (253R)- and (2535)-hydroxytyrosine derivatives (122a) and (122b), at
retention times of 10.5 and 10 min, respectively. The (2,S3S)-isomer (122b)
was collected from repeated injections (ca. lmg). lH n.m.r. (300 MHz, CDCI3)
õ 3.78, s, 3H, OMe; 4.27, d, I 2.1 Hz, 3-OH; 4.84, br s, Ar-OH; 4.97, d, J 8.6
Hz, H2; 5.49, dd, J 2.1 Hz, 8.6 }{z, H3; 6.65, d, J 8.5 Hz, 2H, ArH; 7 .2O, d, I8.5 Hz, 2H, ArH; 7.67-7.77, m, 4H, Phth.
(2 S 3 R) -O-Acetyl-3 -hydroxy -N -te rt-butyl-Nø-phthaloyltyrosinamid e
(r23)
To a solution of a 1:1 mixture of the bromides (95a) and (95b) (200 mg,
0.41 mmol) in acetone (10 ml) was added a solution of silver nitrate (105 mg,
0.62 mmol) in water (10m1). The resultant mixture was stirred in the dark for
24 h, then the filtered and the filtrate was concenhated under reduced pressure.
The residue extracted with dichloromethane, the organic phase was dried, then
the solvent was removed under reduced pressure. Recrystallization from
hexane/dichloromethane afforded the title compound (I23) as colourless
crystals (153 mg, 887o), m.p. 202-204" (Found: M+'-HzO, mlz 406.154"
CztHzzNzO5 requires m\2,406.153). lH n.m.r. (300 MHz, CDCI3) õ 1.31, s,
9H, CMe3;2.26, s, 3H, OAc; 4.43, d, J 8.1 Hz, OH; 5.08, d, J 6.3 Hz,}l2:5.63,
dd, J 6.3 Hz, 8. I }Iz, H3; 5.95, br s, NH; 7 .03, d, J 8.6 Hz, 2}{, ArH; J .40, d, J
8.6H2,2H, ArH; 7.69-1.82, m,4H, Phth. v.u* 3600,3440,3390, 1775,l7l0,
1685, 1515, 1390, 1220, 860, 720 cm-I. Mass spectrum (FAB); mlz 425
(M+H+, 437o),308 (67), 266 (100),248 (4), 187 (16), 160 (26), 154 (23), 136
(37), t07 (18), 105 (10), 89 (r7),77 (22).

Experimental 145
(2 S 3 R) -3- Hy d rox y - N - te rt-b utyl -N ø- p hthaloy ltyrosinami d e (124)
The hydroxytyrosine derivative (I23) (90 mg, 0.21 mmol) was dissolved
in MeOHÆI2O (10:1, 10 ml). p-Toluenesulfonic acid monohydrate (10 mg, 0.05
mmol) was added, and the solution was stirred at 50o for 16 h. The solvent was
removed under reduced pressure, and the residue was partitioned between ethyl
acetate and water. The organic phase was dried, then the solvent was removed
under reduced pressure. Recrystallization of the crude product from
hexane/chloroform gave the title compound (124) as a white solid (73 mg,
937o), m.p. 214-215' (Found: C, 65.7: H,5.8; N, 7 .2. CztHzzNzOs requires C,
66.0; H, 5.8; N, 7.3Vo).1H n.m.r. (300 MHz, CDCI3) õ 1.21, s, 9H, CMe3; 4.72,
d, J 7 .5 Hz, 3-OH; 5.1 1, d, J 6.9 Hz, H2; 5.62, dd, J 6.9 Hz, '7 .5 Hz, H3; 6.4I,
br s, NH; 6.79, d, J 8.5 Hz,2IJ, ArH; 7.24,d, J 8.5 Hz,2H, ArH;7.J1-7.82,m,
4H, Phth; 7.97, s, Ar-OH. vma* 3590,3546, 3410, 1772, 1712, 1685, 1366,
855, 717 cm-l. Mass spectrum (FAB); mlz 383 (M+H+, I87o),307 (3),289
(-CoH+OH, 4),267 (48), 266 (100), 260 (16), 187 (15), 160 (15), 154 (26), 136
(17), t07 (8), 105 (8), 91 (10), 77 (9).
(25 3 R) -3-Hydroxyphenylalanine (25a)
Method 1
(2 S 3 R ) -3 -Hydroxy-N-phthaloylphenylalanine methyl ester ( I l2a) (250 mg,
0.77 mmol) was dissolved in a2:I mixture of 6N HCI and acetic acid (10 ml).
The solution was refluxed for 5 h, then was cooled and evaporated to dryness
under reduced pressure. The residue was taken up in water (10 ml) and the
suspension was filtered to remove phthalic acid. The filnate was evaporated to
dryness under reduced pressure, then the residue was dissolved in ethanol (7
ml) and a solution of aniline (0.7 ml) in dichloromethane (10 ml) was added.
The solutien was stored at 0o overnight, and the title compound (25a) was

Experimental 146
isolated as a white powder (129 mg,93Vo), m.p. 192-195", [o]o16 -49.7+O.5"
(c, 0.4 in 6N HCI) (lit.t¡s [a]p2o -50.2+2" (c,2 ín 6N HCI)). lH n.m.r. (300
MHz, DzO) ô 3.95, d, J 4.4 Hz, H2; 5.29, d, J 4.4 }lz, }{3; J.47, s, 5H, Ph.
Spectral data consistent with literature uu1uss.175
Method 2
To a solution of (253R)-3-hydroxy-N-phthaloylphenylalanine methyl ester
(ll2a) (250 mg, 0.77 mmol) in ethanol (5 ml) was added a solution of
hydrazine hydrate in ethanol (2M,0.77 mI). The mixture was refluxed for 2 h,
then was cooled and lÙVo aqueous HCI (1 rnl) was added" The mixture was
refluxed for a further t h, then was cooled and filtered. The white solid was
washed with ethanol, then the filtrate and washings were combined and
evaporated to dryness under reduced pressure. The residue was dissolved in
ethanol (7 ml) and a solution of aniline (0.7 ml) in dichloromethane (10 ml)
was added. The solution was stored at 0o overnight, and the title compound
(25a) was isolated as a white powder (111 mg, 807o). Spectral data identical to
that described above.
Method 3
The hydroxyphenylalaninamide derivative (118a) (500 mg, 1.37 mmol)
was hydrolyzedin a2:1 mixture of 6N HCI and acetic acid, as described for the
hydrolysis of the alcohol (ll2a), and gave the title compound (25a) as a fine
white powder (194 mg, 787o). Spectral data identical to that described above.

Experímental I47
(R)-N-Phthaloyl-p-nitrophenylalanin e (126)
A mixture of (R)-p-nitrophenylalanine monohydrate (2.5 g, 11.0 mmol)
and finely ground phthalic anhydride (1.63 g, 11.0 mmol) was heated in an oil
bath at 140o for 30 min. The resultant yellow gum was dissolved in hot
methanol (30 ml), filtered, and then water (20 ml) was added. The product was
isolated by filtration and dried over P2O5 to give the title compound (126) as a
pale yellow crystalline solid (3.01 g,8I7o), m.p. 206-207" (Iit.t76 204.7o). tH
n.m.r. (300 MHz, CDCI3) ô 3.69, dd, J 10.9 Hz, 14.4 Hz, H3; 3.75, dd, J 5.7
H4 I4.4 Hz, H3'; 5.26, dd, I 5.7 IJz, 10.9 Hz,IÐ; 7 "48, d, J 8.7 Hz,2H, ArH;
7.80, s,4H, Phth; 8.07, d, J 8.7 IJz,2H, ArH"
(R)-N-Phthaloyl-p-nitrophenylalanine Methyl Ester (L27)
(R)-N-Phthaloyl-p-nitrophenylalanine (126) (2.5 g, 7.35 mmol) was
dissolved in dry methanol (50 ml) which had been pre-treated with thionyl
chloride (400 mg, 3.36 mmol). The solution was stirred under anhydrous
conditions for 16 h, then the solvent was removed under reduced pressure. The
residue was taken up in dichloromethane, and was washed with l07o Na2CO3
and water, then was dried" Removal of the solvent under reduced pressure and
recystallization of the product from hexane/dichloromethane gave the title
compound (127) as a white solid (2.24 g,867o), h.p. l2l-122" (Found M+',
mlz 354.087. CraHr¿NzOo requires mlz 354.085¡. tg n.m.r. (300 MHz, CDCI¡)
ô 3.71, dd, J lO.9 Hz, I4.3 }lz, H.3; 3.77, dd, J 5.5 Ilz, 14.3 Hz, H3'; 3.81, s,
3H, OMe; 5.31, dd, J 5.5Il.2, 10.9H2,}I2;7.45, d, J 8.6 Hz,2}l, ArH; 7.12-
7.82, m,4H, Phth; 8.06, d, J 8.6 Hz,2H, ArH. v^u* 1775,I750, 1715, 1600,
1520, 1390,1345, t240, 860,120 cm-1. Mass spectrum; mlz 354 (M*', I27o),
295 (-COzMe,37),278(14),218 (36),207 (100), 190 (37), ll6 (25), 130 (33),
104 (n),16 (2r).

Experimental 148
(2R35)-3-Bromo-N-phthaloyl-p-nitrophenylalanine Methyl Ester(128a) and
(2R3 R) -3-Bromo-N-phthaloyl-p-nitrophenylalanine Methyl Ester(128b)
To a solution of (R)-N-phthaloyl-p-nitrophenylalanine methyl ester (I27)
(2.20 9,6.24 mmol) in CCI¿ (40 mt) was added NBS (1.20 9,6.74 mmol), and
the mixture was refluxed for 4 h while irradiated with a 250W mercury lamp.
The mixture was allowed to cool, then was filtered and the filtrate was washed
with water (40 ml). The organic phase was dried, then was concentrated under
reduced pressure to give the title compounds (128a) and (128b) (2.69 g, 1007o)
as a 1:1 mixture. Fractional recrystallization from hexane/dichloromethane
gave the threo-diastereomer (128a) (1.11 g, 447o), m.p. 198-201" (Found: C,
49.8; H, 3.0; N, 6.5. ClgHl3BrNzOo requires C, 49.9; H, 3.0; N, 6.57o). lH
n.m.r. (300 MHz, CDCI¡) ô 3.59, s, 3H, OMe; 5.51, d, J 1l.2Hz,ÍÐ; 6.02, d, J
II.2Hz,}I3;1.78, d, J 8.8 Hz,2H, ArH; 7.82-7.99, m,4H, Phth; 8.27, d, J 8.8
Hz, 2H, ArH. v^a* I77 5, 1750, 1720, 1600, 1525, 1340, 1215, 1 100, 820, 7 15
cm-l. Mass spectrum; mlz 434 and 432 (M*', 27o),375 (-CO2Me, 6), 373
(-CO2Me, 6), 353 (-Br, 4), 352 (9),321 (6),294 (29),293 (11),287 (IO),285
(r0),247 (7),2t9 (16), 218 (100), 190 (30), 130 (18), 104 (40),76 (37).
Further recrystallization gave the erythro-diastereomer (128b) (1.07 g, 407o),
m.p. 195-197' (Found: C, 49.8; H, 3.0; N, 6.6. ClsHl3BrNzOe requires C,
49.9; H, 3.0; N, 6.57o). 1H n.m.r. (300 MHz, CDCI3) ô 3.83, s, 3H, OMe; 5.59,
d, J 10.3 I{z,H2; 5.9J, d, J 10.3 }lz,}l3;7.56, d, J 8.7 H¿2H, ArH; 7 .68-1.76,
m, 4H, Phth; 8.07, d, J 8.7 }Jz,2IJ, ArH. v^u* 1775, L755, 1720, 1605, 1525,
1390, 1350, 855,720 cm-l. Mass spectrum; mlz 434 and 432 (M*', l7o),375
(-COzMe, 3), 373 (-CO2Me, 3), 353 (-Br, 6), 352 (3), 321 (7), 294 (20),293
(r2),287 (3), 285 (3),247 (5),219 (15), 218 (100), t90 (29),130 (16), 104
(28),76 (26).

Experimental 149
Treatmen t of (2 R3S) -3 - B rom o -N- p hthaloy l-p-nitrop henylalanine
Methyl Ester (128a) with silver nitrate in acetone/water.
To a solution of the threo-bromide (128a) (50 mg, 0.115 mmol) in acetone
(3 mt) was added a solution of silver nitrate (25 mg 0.15 mmol) in water (2
rnl). The resultant mixture was stirred at 50o in the dark for 16 h. The mixture
was then filtered and the fitrate was concentrated under reduced pressure. The
residue extracted with dichloromethane, the organic phase was dried, then the
solvent was removed under reduced pressure. Recrystallization of the residue
from hexane/dichloromethane gave the (Z)-p-nitro-dehydrophenylalanine
derivative (I32) as large prisms (34 mg, 84Vo), m.p. 133-134' (Found; I\z[+',
mlz 352.068. CrsHrzNzOo requires mlz 352.070¡. t1¡ n.m.r. (300 MHz, CDCI3)
ô3.87, s,3H, OMe; 7.55, d, J 8.8 }{z,2H, ArH; 7.83,m,2}l, Phth; 7-92,m,2}{,
Phth; 8.13, s, vinylic-H; 8.16, d, J 8.8 Hz, ArH. vru* 1780,1720,1600, 1530,
1345 cm-l. Mass spectrum; mlz352 (M*', 90Vo),324 (63),293 (-COzMe, 41),
292 (46),247 (24),218 (15), 190 (18), 166 (2r),104 (100),76 (73).
Treatment of (2R3R)-3-Bromo-N-phthaloyl-p-nitrophenylalanineMethyl Ester (12Sb) with silver nitrate in acetone/water.
To a solution of the erythro-bromide (128b) (50 mg, 0.115 mmol) in
acetone (3 ml) was added a solution of silver nitrate (25 mg,0.15 mmol) in
water (2 mt). The resultant mixture was stirred at 50" in the dark for 16 h.
The mixture was then filtered and the filtrate was concentrated under reduced
pressure. The residue extracted with dichloromethane, the organic phase was
dried, then the solvent was removed under reduced pressure. Separation of the
products by chromatography on silica gave the B-hydroxy-p-nirophenylalanine
derivative (129) which was recrystallized from hexane/dichloromethane and
obtained,:t .otou.less needles (27 mg, 637o), m.p. 183-185". lH n.m'r- (300

Experimental 150
MHz, CDCI¡) ô 3.89, s, 3H, OMe; 5.34, d, J 10.0 Hz, OH; 5.53, d, J 4.4 Hz,
IÐ; 5.79, dd, J 4.4 Hz, 10.0 Hz, }I3;7 .54, d, J 8.9 Hz,2H, ArH; 7 .73-7.82, m,
4H, Phth; 8.13, d, J 8.9 Hz,2H ArH. v,nu* 3604,3421, 1779, I752, 1714,
1614, 1526, 1392, 1352, 1182 cm-I. Mass spectrum (FAB); mlz 371 (M+H+,
97o), 353 (-HzO, 3),321 (3),301 (11), 289 (9),219 (3), 154 (100), 137 (66),
136 (79), 107 (28),89 (33),77 (31). A mixture of the (Z)- and
(E)-dehydrophenylalanine derivatives (132) and (133) was obtained as a viscous
oil (3:2, 257o)" lH n.m.r. (300 MHz, CDCI3) (E)-isomer (133) õ 3.72, s, 3H,
OMe; 7.28, s, vinylic-H; 7.60, d, J 8.7 }Jz, 2}J, ArH; 7 .80-1.98, m, 4H, Phth;
8.26, d,I 8.7 }{z,2H, ArH. Spectral data for the (Z)-isomer (132) identical to
that described above.
(3 S4S)-l-tert-Butyl-4-phenyl-3-phthalimidoazetidin-2-one (134) and(Z) -N - te rf- B utyl -N ø-p hthaloyl - cr, B -d ehy drop henylalaninamid e (8 6)
Potassium amide solution was prepared by adding potassium (19 mg, 0.48
mmol) and ferric nitrate (1 mg) to liquid ammonia (200m1). The solution was
cooled to -78", then a solution of the bromide (85a) (200 mg, 0.47 mmol) in
dichloromethane (10m1) was added dropwise. The mixture was stirred at -78"
for 3 h, then the ammonia was allowed to evaporate overnight. The residue
was partitioned between dichloromethane and water, the organic layer was
separated, then washed with water. The solution was dried, and the solvent was
removed under reduced pressure. The mixture was chromatographed on silica
to give the (Z)-dehydrophenylalanine derivative (86) (22 mg,277o), identical to
the sample described above, and the trans-þ-Iactam (134) (51 mg, 637o), m.p.
243-245" (Found C,1L8; H, 5.8; N, 8.1. CzrHzoNzOs requires C,72.4;, H, 5.8;
N, 8"07o). lH n.m.r. (300 MHz, CDCI3) ô 1.36, s, 9H, CMe3; 4.96, d. J 2.3 Hz,
H4; 5.01, d, J 2.3 lFrz,H3;7.39, s,5H, Ph; 7.72-7.87, m, 4H, Phth. v^u^ 2920,
1745, 1120, 1465, 1390, 710 cm-l. Mass spectrum; mlz 348 (M*', 27o),333

Experimental 151
(-Me, 1), 305 (2),291 (4),276 (3),249 (100), 232 (5),220 (9),204 (14), 165
(5), t32 (5), 104 (28), tjz (34),76 (27).
(2 S 3 R) -3- Am ino -N - t e rt -butyl -¡¿ø-phthaloylphenylalaninamid e ( 135)
Method 1
Potassium amide solution was prepared by adding potassium (11 mg, 0.28
mmol) and ferric nitrate (1 mg) to liquid ammonia (30 ml). The solution was
cooled to -78o, then a solution of the bromide (85b) (100 mg, 0.23 mmol) in
dichloromethane (5ml) was added dropwise. The mixture was stirred for 2 h,
then the ammonia was allowed to evaporate overnight. The residue was
partitioned between dichloromethane and water, then the organic layer was
washed with water, and dried. The solvent was removed under reduced
pressure, and the residue was recrystallized from hexane/dichloromethane to
give the title compound (135) as a white solid (78 mg, 937o), n.P. 150-152"
(Found C,68.9; H,6.5; N, 11.4. CzrHz¡N¡O3 requires C,69.0; H,6.3; N,
lI.57o). lH n.m.r. (300 MHz, CDCI3) õI.44, s, 9H, CMe3; 4.63, d, J 6.8 Hz,
IÐ;5.7i, br s, lH, NH; 5.77, d, J 6.8 Hz,IF.13;6.03, br s, lH, NH'; 6.73, br s,
lH, CONH;7.26-7.41, h,5H, Ph; 7.46-8.02,m,4}l, Phth. v,nu* 3360,3310,
3170,2900, 1660, 1640, 1520, 1450, 750 cm-l. Mass spectrum (FAB); mlz
366 (M+H+,1007o),249 (29),130 (53), 120 (30), 105 (15), 103 (17), 57 (49).
Method 2
The bromide (85b) (100 mg, 0.23 mmol) was dissolved in TÉIF (10 ml)
and then concentrated aqueous ammonia solution (0.5m1) was added. The
solution was stirred for 18 h, then the THF was removed under reduced
pressure. The residue was partitioned between dichloromethane and water, and
the aqueous phase was re-extracted with dichloromethane. The combined

Experimental I52
extracts were dried, and the solvent was removed under reduced pressure
Purification of the product by chromatography on silica gave the title
compound (135) (55 mg, 66Vo), identical to the sample produced above.
Treatment of (2 S 3R) -3 - B ro mo - N - te rú-b utyl-N a-p hthal oyl -
phenylalaninamide (85a) with aqueous ammonia in THF.
Treatment of a solution of the bromide (85a) in TIIF with aqueous
ammonia, as described above for the treatment of the bromide (85a), gave the
(Z)-dehydrophenylalaninamide derivative (86) (887o), with spectral data
identical to that described above"
(253 R) -3-Bromo-N-phthaloylphenylalanine (L37 a) and
(253 R) -3-Bromo-N-phthaloylphenylalanine (137b)
lS)-N-Phthaloylphenylalanine (70) (5.0 g, I7 mmol) was dissolved in a
mixture of CCI¿ and dichloromethane (10:1, 150rnl). NBS (3.15 g, 18 mmol)
was added, and the mixture was refluxed for 2 h while irradiated with a 250W
mercury lamp. The mixture was cooled, filtered, and the solid was washed
with CCI¿. The filtrate was then washed with water, dried, then the solvent was
removed under reduced pressure to give a pale yellow gum. Dissolution of the
gum in CCla and removal of the solvent under reduced pressure at room
temperature gave the title compounds (137a) and (137b) as a 1:1 mixture, as a
pale yellow solid (6.2 g,987o) (Found: C, 52.8; H,3.2; N, 3.5. C17H12BrNOa
requires C,54.6:H,3.2;N,3.7Vo). lH n.m.r. (300 MHz, CDCI3) threo-isomer
(131a) ô5.55, d, J 1l.2Hz,fI2;5.97, d, J 1I.2Hz,H3;6.64, br s, CO2H;7.10-
7.55, m,5H, Ph;7.76-7.95, m,4H, Phth; erythro-isomer (137b) ô 5.64, d, J
I0.4IJz, H2; 5.86, d, J 10.4 Hz,Il3:7.I0-7.31, m,5H, Ph;7.61-7.80, m,4H,t"

Experimental 153
Phth; 8.03, br s, CO2H. vn'u* 3450 br, 1798, 1718, 1608, 1168, 1104, 924,868
cm-l. Mass spectrum; mlz 375 and 373 (M*'), 249 (l'ffiVo),232,220,204.
Treatment of a mixture of (253R)-3-Bromo-N-phthaloyl-phenylalanine (137a) and (253R)-3-Bromo-N-phthaloyl-phenylalanine (137b) with silver carbonate.
To a solution of a 1:1 mixture of the bromides (I37a) and (137b) (6.2 g,
16.6mmol) in acetone (100 ml) was added silver carbonate (900 mg, 8.5
mmol)" The mixture was stirred for 3 h, then was filtered through celite and
the solvent was removed under reduced pressure. Analysis of the crude
mixture using lH n.m.r. specFoscopy indicated the presence of the cis-B-lactone
(139b), the trans-B-lactone (139a) (õ 5.67, d, I 4.5 Hz, H3:. 5.92, d, J 4.5}{2,
H4), and trans-2-phthalimidostyrene (138) in a 2:l;l ratio. Attempted
separations of the compounds by fractional recrystallization or chromatography
on silica resulted in the decomposition of the trans-B-lactone (139a) to give
2-phthalimidostyrene (138). Consequently, separation of the products by
chromatography on silica gave trans-2-phthatimidostyrene (138) (2.769, 67Vo),
m.p. 185-187". lH n.m.r. (300 MHz, CDCI3) õ 7.36, d, I 15.2 Hz, lH;7.36-
1.38, m, 3H, ArH; 7.46-7.49, m, 2H, ArH; 7.65, d, J I5.2 }lz, l}I; 7.74-1.93"
m, 4H, Phth. Further elution gave the cis-B-lactone (139b) (170 mg, 167o),
m.p. 123-125o.lH n.m.r. (300 MHz, CDC13) õ 6.33, d,J 9.2Hz,H4i 6.70, d,J
9.2 Hz, H3; 7.25, s, 5H, Ph;1.72-7.87, m, 4H, arom. Vmax 1840, 1180, 1760
cm-l. Mass spectrum; mlz 24911vf+'-CO2, 1007o),232 (9),220 (20),2O4 (28),
165 (11), 133 (7), 130 (4), I n Q),104 (39), 102 (51),76 (76)"

Experimental 154
(25)-4,5-Dibromo-N-phthaloylleucine Methyl Ester (I42)
The y-bromoleucine derivative (91) (100 mg, 0.28 mmol) was dissolved in
chlorobenzene (2 ml). NBS (100 mg, 0.56 mmol) and a catalytic amount of
benzoyl peroxide (5 mg) were added, and the mixture was refluxed while
irradiated with a 250W mercury lamp for 4 h. Inadiation was stopped and the
mixture was refluxed for a further 12 h. The mixture was cooled and the
solvent was removed under reduced pressure. Purification of the residue by
chromatography on silica gave the title compound (I42) as a pale yellow oil (64
mg,537o). lH n.m.r. (300 MHz, CDCI¡) ô 1.86, s, 1.5H, Y-Me; 1.93, s, 1.5H,
y-Me'; 2.46, dd, J 9.7 Hz, J I2.3 Hz, 0.5H, H.3;2.59, dd, J Il.6 }{z, I2.3 Hz,
0.5H, H3';2.87-3.02, m, lH, CHCIIz; 3.68-3.85, m,2H, CHzBr; 3.75, s, 1.5H,
OMe; 3.76, s, 1.5H, OMe; 5.16-5.30, m, lH, H2;7.27-7.58, m,5H, Ph;7.74-
7.94, m,4H, Phth. Mass spectrum; mlz 435,433 and 431 (M+', 1,2 and lVo),
376 (-COzMe, 28), 374 (-COzMe, 56), 372 (-COzMe, 28), 354 (-Br, 16),352 (-
Br, 16), 294 (38),292 (38),272 (Il),270 (ll),257 (13),242 (12),215 (45),
2r4 (75),200 (52),186 (40), 160 (47),130 (41), 105 (33), 104 (100), 76 (50).
(Z)-N-Phthaloyl-o,B-methanophenylalanine Methyl Ester (143)
To a solution of the y-bromohomophenylalanine derivative (92) (100 mg,
0.25 mmol) in TIIF (2 ml) was added 1,8-diazabicyclo[5.4.0]undec-7-ene
(DBU) (38 mg, 0.25 mmol). The mixture was heated at reflux for t h, then
was cooled and dituted with dichloromethane (20 ml). The solution was washed
with water, dried, then the solvent was removed under reduced pressure. The
residue was chromatographed on silica, eluting with hexanell,2-dichloroethane,
tc give a viscous oil which was crystallized from hexane./dichloromethane to
give the title compound (143) as colourless crystals (58 mg,727o), m.p. 131-
132' (Fouqd: C, 10.9;H,4.9; N, 4.4. CrsHrsNO4 requires C,71.0; H, 4.7; N,

Experimental 155
4.47o). lH n.m.r. (300 MHz, CDCI3) õ2.27, dd, J 6.6 Il2,9.9 Hz,Il,4;2.43, dd,
J 6.6 H2,8.5 Hz,H4';3.38, dd,J 8.5 Hz,9.9}{z,H'3;3.12, s,3H, OMe; 7.07-
7.17, m, 5H, Ph;1.60-7.85, m, 4H, Phth. vmax 1182, L724, 1440, 1404, 1272,
732 cm-t. Mass specfrum; mlz32l (M*').
Acetylation of (S)-a-Hydroxy-N-phthaloylleucine Methyl Ester
(104).
To a solution of y-hydroxyleucine derivative (104) (11.5 g,39.5 mmol) in
dichloromethane (40 ml) was added pyridine (3.13 g,39.5 mmol) and DMAP
(0.48 g,3.9 mmol). Acetyt chloride (20 rnl) was then added, and the mixture
was stirred under nitrogen for 16 h. The solvent was removed under reduced
pressure, and the residue was taken up in ether. The solution was then washed
successivety with dilute HCl, water and 10% NazCO¡" The organic phase was
dried, and the solvent was removed under reduced pressure to give a clear oil.
The products were separated by dry column flash chromatography on silica, to
give the lactone (105) (1.62 g, 167o), identical to the sample described above,
and (S)-4-acetoxy-N-phthatoylleucine methyl ester (144). Further purification
of the acetate (144) by distittation using a Kugelrohr apparatus (200", 0.04mm)
yielded a clear oil which partially crystallized on storage at -20o overnight.
Repeated trituration with hexane/ether and vacuum filtration gave the acetate
(I44) as a white solid (9.24 g,707o), m.p.85-87" (Found: C,61.3; H,5.9; N,
4.2. CnHrqNOe requires C, 61.3; H, 5.7; N, 4.2Vo). lH n.m.r. (300 MHz,
CDCI¡) ô 1.48, s, 3H; 1.57, s, 3H; 1.59, s, 3H;2.52, dd, J 2.8 þ¡z,15.4Hz,f{3
2.82, dd, J 10.1 Hz, 15.4Hz,H3':3.'74, s,3H, OMe; 5.11, dd, J 2.8}{2, IO-l
Hz,rr2i l.l5-7.92, m, 4H, Phth" v^u* l7'14,1738,1718, 1390, 1145, 1020,110
cm-l. Mass spectrum (FAB); mlz 334 (M+H+, 37o),274 (-OAc, 90), 260 (10)'
214 (59),185 (47), r27 (34),93 (100).

Experimental 156
Pyrolysis of (S)-4-Acetoxy-N-phthaloylleucine Methyl Ester (144).
(S)-4-Acetoxy-N-phthaloylleucine methyl ester (144) (5.0g, 15.0 mmol)
was distilled (ca.250",0.5 mm) through a quartz pyrolysis tube packed with
quartz fragments, which had been preheated to 450o. The distillate was
obtained as a viscous yellow-green oil, and was purified by dry cloumn flash
chromatography. The y,ô- and B,y-dehydroleucine derivatives (106) and (107)
were obtained as a mixture in a 1.6:1 ratio, as a pale yellow oil which
crystallized on standing overnight (3.42 g,84Vo). Spectral characteristics of the
y,ô- and B,y-dehydroleucine derivatives (106) and (107) were identical to those
described above.
Acetylation of 4-Hydroxy-N-phthaloylhomophenylalanine MethylEster (145).
To a solution of the y-hydroxyhomophenylalanine derivative (109) (5.0 g,
14.7 mmol) in dichloromethane (20 ml) was added pyridine (1.16 g, I4.7
mmol) and DMAP (0.18 g, I.47 mmol). Freshly distilled acetyl chloride (10
ml) was then added, and the mixture was stirred under nitrogen for 6 h. The
solvent was removed under reduced pressure, and the residue was taken up in
ether, then washed successively with dilute HCl, water and L07o NazCO¡. The
organic phase was dried, then the solvent was removed under reduced pressure
to give a clear oil which crystallized on standing overnight. Fractional
recrystallization from hexane/ethyl acetate gave the cis-B-lactone (110) (304
mg, 77o), and the trans-þ-Iactone (77 mg, 27o), identical to the samples
described above. Further recrystallization gave one diastereomer of (S)-4-
acetoxy-N-phthatoylhomophenylalanine methyl ester (145) (2.21 9,397o), m.p.
137-140".IH n.m.r. (300 MHz, CDCI3) ô 1.94, s, 3H, OAc; 2.83,m,2H,CH2;
3.72, s,3H, OMe; 4.90, dd, J 6.2 H2,8.1 Hz, H4; 5.85, t, J 6.6 Hz, H2;7.18-

Experimental 157
7.45, m, 5H, Ph;7.73-7.90, m,4H, Phth. vru* 1780,1745,1715,1395,1025
cm-1. Mass spectrum; mlz38I (M*', O.lVo),338 (-Ac, ll),322 (-CO2Me, 1),
308 (3), 262 (r3),219 (100), t87 (86),114 (43), r32 (28), 130 (27),115 (81),
107 (44), 105 (52), 77 (33),76 (33), 43 (92). Further recrystallizatíon gave
the other diastereomer of the acetate (145) (1.51 g,277o), m.p. I25-I27"
(Found: C,66.1; H, 5.1; N, 3.6. CzrHrqNO6 requires C,66.L; H, 5.0; N,3.77o)"
rH n.m.r. (300 MHz, CDCI3) õ 2.09, s, 3H, OAc; 2.80, ddd, J 4.9 IIz,,9.7 }Iz,
l4|7 IJz,H.3;2.86, ddd, J 4.9 Hz, ll.0 Hz, 14.7 Hz,}{3';3.74, s, 3H, OMe;
5.I4, dd, J 4.9 Hz, 1 1.0 Hz, H4: 5.62, dd, J 4.9 Hz, 9.7 HaIÐ; 7 .18-'7 .30, m,
5H, Ph; 7.74-7.87, m, 4H, Phth. v*u* 1778, 1746, 1722, 1600, 1555, 1390,
1066, 772 cm-r. Mass spectrum; mlz 381(M+', 0.57o),338 (-Ac, l4), 308 (3),
262 (r5),2t9 (100), 187 (84), t74 (28),115 (36), 105 (67), 77 (16),16 (16),
43 (s0).
Pyrolysis of 4-Acetoxy-N-phthaloylhomophenylalanine Methyl Ester(14s).
A 1.5:1 mixture of the diastereomers of 4-acetoxy-N-phthaloyl-
homophenylalanine methyl ester (145) (1.0 g,2.62 mmol) was distilled (250",
0.3 mm) and passed through a pyrolysis tube (520') as for pyrolysis of the
acetoxyleucine derivative (I44). Purification of the crude residue by flash
chromatography on silica gave a 1:1 mixture of the B,1-dehydro-
homophenylalanine derivative ( 146) and the a,B-dehydrohomophenylalanine
derivative (141) (560 mg, 667o), as a clear oil. lH n.m.r. (300 MHz, CDCI3)
p,y-dehydrohomophenylalanine derivative (146) õ 3.80, s, 3H, OMe; 5.58, d, J
7.1Hz, IJr2; 6.68, d, J 15.9 }lz,H4; 6.74, dd, J 7.1 Hz, 15.9 Hz,H3;7.19-7.43,
m, 5H, Ph;7.73-1.95, m, 4I{, Phth; a,B-dchydrohomophenylalanine derivative
(I41) õ 3.52, d,J 7.4IJr2,2H, CHzPh; 3.77, s,3H, OMe; 7.19-7.43, m,6H, H3
and Ph; 7:73-7.95, m,4H, Phth. vnlu^ 1780,1745, l'715, 1605, 1560, 1390,

Experímental 158
850,720 cm-I. Mass spectrum; mlz32l (M*', 57o),3O5 (5),289 (-MeOH,78),
262 (-CO2Me, 54),245 (20),230 (19), 147 (12), 132 (29), 115 (100), 104
(71), 9l (rt),76 (44).
Treatment of ( 2 S 35) -3 - B ro mo -N - te rf-bu tyl -N ø-p hthaloyl -
phenylalaninamide (85b) with NaBHa
To a solution of the bromide (85b) (100 mg, 0.23 mmol) in 8O7o aqueous
ethanol (10 rnt) was added NaBtIa (40 mg, 1.06 mmol), and the mixture was
stirred at room temperature for 12 h. The reaction was quenched by the
addition of dilute HCl, then the mixture was concentrated under reduced
pressure. To the concentrate was added water (40 ml), then the mixture was
extracted with dichloromethane (2x40 mt). The combined organic phases were
dried and the solvent was removed under reduced pressure" Separation of the
products by chromatography on silica gave the secondary alcohol (151) (36 mg,
367o)" lH n.m.r. (300 MHz, CDCI3) õ 1.40, s, 9H, CMe3; 4.28,d,I7.II{z,lI2;
4.70, s, OH; 5.75, d, J 7.1 IIz, }I3:- 5.74, s, ring-H; 6.51, br s, NH; 7.22-7.37,
m, 5H, Ph; 7.63-7.71, m, 3H, ArH; 7.86-7.88, m, lH, ArH. Further elution
gave the primary alcohol (152) (21 mg, 2l7o). lH n.m.r. (300 MHz, CDCI3) ô
1.41, s, 9H, CMe3; 4.69, d, J 12.5 IIz, CHOH; 4.70, d, J 7 .2 Hz, H2; 4.86, d, J
12.5 }Jz, CII'OH; 5.28, d, J 7 .2 IIz, H3; 6.34, br s, NH; 7 .23-7 .54, m, 8H, ArH;
8.04-8.07, m, lH, fuH.

Experimental 159
Treatment of a mixture of (253R)- and (2535)-3-Bromo'N-tert-butyl-Nø-phthaloytphenylalaninamide (85a) and (85b) withtri-z-butyltin deuteride.
To a solution of a 1:1 mixture of the bromides (85a) and (85b) (200 mg,
0.47 mmol) in benzene (10 ml) was added tri-n-butyltin deuteride (275 mg,
0.93 mmol) and AIBN (40 mg, 0.24 mmol). The solution was stined at room
temperature for 3 d, then the solvent was removed under reduced pressure.
The residue was taken up in hexane, and the suspension was stirred for 5 min.
The white sotid was isolated by filtration and was washed repeatedly with
hexane to remove traces of stannanes. The solid was recrystallized from
hexane/ethyl acetate to give the deuteriophenylalaninamidd derivative (148) as a
3:1 ratio of diastereomers (102 mg, 637o), m.p. 186-187". lH n.m.r. (300
MIIz, CDCI¡) ô 1.30, s,9H, CMe3; 3.46,d,110.2H2,0.25H,H3;3.54,d,J 6.4
Hz, 0.75H, H3'; 4.99, d, J 10.2 Hz, 0.25 H, H2: 4.99, d, J 6.4 Hz, O.l5H, H2;
l.l3-7.21, m, 5H, Ph;7.67-7.80, m, 4H, ArH.
Treatment of a mixture of (2S3R)- and (2535)-3-Bromo'N-phthaloylphenylalanine Methyl Ester (82a) and (82b) withtri-z-butyltin deuteride.
Treatment of a 1:1 mixture of the bromides (82a) and (82b) with
tri-r-butyltin deuteride and AIBN, as described above for treatment of a
mixture of the bromoamides (85a) and (85b), gave the deuteriophenylalanine
derivative (149) as a2:1 ratio of diastereomers (98 mg, 617o), m.p. I22-I24".
lH n.m.r. (300 MHz, CDCI3) õ 3.53, d, J 11.7 }J2,0.35H, }l3;3-59, d, J 4-8H2,
0.65H, F-l3';3.77, s,3H, OMe; 5.16, d, J 11.7 H2,0.35H, }{2:5.16, d, J 4-8H2,
0.65H, H2;7.11-7.19, m,5H, Ph;7.65-7.78, m,4H, ArH.

Experimental 160
(253R)-3-Deuterio-N-phthaloylphenylalanine Methyl Ester Ga9a)
A mixture of the threo-bromide (82a) (1.0 g, mmol) and 5Vo Pd/C (100
mg) in 2Hr-methanol (99.57o 2H,20 ml) was stirred at -20" under an
atmosphere of deuterium gas for 3 d in a refrigerated bath. The palladium
catalyst was removed by filftation, and the solvent was removed under reduced
pressure. The residue was taken up in dichloromethane and washed with l07o
NazCO¡, then dried, and the solvent was removed under reduced pressure.
Recrystallization of the residue from hexane/ethyl acetate gave the title
compound (149a) (987o d.e.) as colourless prisms (728 mg,9I7o), m.p. 124-
126"" lH n.m.r. (300 MHz, CDCI3) õ 3.59, d, J 4.8 Hz,}J3;3.77, s,3H, OMe;
5.16, d, J 4.8 llz,}l2;7.I1-7.19, m,5H, Ph; 7.65-7.78, m,4H, Phth. Mass
spectrum; mlz 310 (M+', 997o 2}J).
(2535)-3-deuterio-N-phthaloylphenylalanine Methyl Ester (149b)
Treatment of a solution of the erythro-bromide (82b) (1.0 g, mmol) in
2Hr-methanol (99.5Vo D,20 ml) wíth 57o PdlC under an atmosphere of
deuterium gas, as described for the preparation of the deuteriophenylalanine
derivative (I49a) from fhe threo-bromide (82a), gave the title compound
(149b) as colourless prisms (717 mg,90Vo), m.p. 125-121". tH n.m'r. (300
MHz, CDCI3) ô 3.53, d, J 1I.J }Jz,H3;3.17, s, 3H, OMe; 5.16, d, J 11.7 Hz,
II2; 7.Il-7.19, m, 5H, Ph; 1.65-7.78, m, 4H, Phth. Mass spectrum; mlz 310
(M*.,997o2Hù.
(R) -N-Phthaloylp heny lalanine
Treatment of (R)-phenylalanine (55) with phthalic anhydride, as described
above for the preparation of (S)-N-phthaloylphenylalanine (70) from

Experimental 161
(S)-phenylalanine (56), gave the title compound as colourless crystals (87Vo),
m.p. 185-186' (lit.lls 180-180.5). lH n.m.r. spectrum identical to that of the
(S)-enantiomer (70).
(R)-N-Phthaloylphenylalanine Methyl Ester
Treatment of (R)-N-phthaloylphenylalanine with a methanolic solution of
hydrogen chloride, as described for the preparation of ( S ) - N -
phthaloylphenylalanine methyl ester (74) from (^S)-N-phthaloylphenylalanine
(70), gave the títle compound as large colourless prisms (937o), m.p. 123-126".
lH n.m.r. spectrum identical to that of the (S)-isomer (74).
(2R35)-3-Bromo-N-phthaloylphenylalanine Methyl Ester (82c) and(2R3R)-3-Bromo-N-phthaloylphenylalanine Methyl Ester (82d)
Treatment of (R)-N-phthaloylphenylalanine methyl ester with NBS, as
descibed for the preparation of the bromides (82a) and (82b) from
(S)-N-phthaloylphenylalanine methyl ester (74), gave the title compounds (82c)
and (82d) as colourless crystals" The threo-bromide (82c) was isolated in MVo
yield, m.p. 139-142", and had spectral data identical to that of the (253R)-
isomer (82a). The erythro-bromide (82d) was isolated in 43%oyield, m.p. 120-
I21", and had spectral data identical to that of the (2535,)-isomer (82b).
(2R35)-3-Deuterio-N-phthaloylphenylalanine Methyl Ester (1a9c)
Treatment of a solution of the threo-bromide (82c) (500 mg, mmol) in
2Hr-methanol (99.5Vo 2H, l0 mt) with 57o Pd/C under an atmosphere of

Experimental 162
deuterium gas, as described for the preparation of the deuteriophenylalanine
derivative (I49a) from the threo-bromide (82a), gave the title compound
(149c) as colouless crystals (372 mg, 93Vo), m.p. 125-126" " lH n.m.r.
spectrum identical to that of the (253R)-enantiomer (149a)" Mass spectrum;
mlz310 (M+',99Vo2H).
(2R3R)-3-Deuterio-N'phthaloylphenylalanine Methyl Ester (149d)
Treatment of a solution of the erythro-bromide (82d) (500 mg, mmol) in
2Hr-methanol (99.5Vo 2}I, 10 ml) with 5Vo Pd/C under an atmosphere of
deuterium gas, as described for the preparation of the deuteriophenylalanine
derivative Qaga) from the threo-bromide (82a), gave the title compound
(149d) as colouless crystals (358 mg, 907o), m.p. 124-126". lH n.m.r.
spectrum identicat to that of the (2SiS)-enantiomer (149b). Mass spectrum;
mlz310 (M+',997o2Hù.
(253 R) -3-Deuteriophenylalanine (65a)
A solution of the threo-deuteriophenylalanine derivative (I49a) (500 mg,
1.6 mmol) in 6N HCl/acetic acid (2:I,30 ml) was refluxed for 6 h. The
mixture was allowed to cool, then was evaporated to dryness under reduced
pressure. The residue was taken up in water (30 ml) and the phthalic acid was
removed by filtration. The filtrate was evaporated to dryness, then the residue
was dissolved in dry ethanot (15 ml). Aniline (1.5 ml) was added and the
solution was left to crystallize. The precipitate was isolated by filtration and
was washed with acetone to give the title compound (65a) as a white powder
(223 mg,837o),m.p.272-276".IH n.m.r. (300 MHz, DzO) õ 3.21 ,d,14.9H2,
H3; 3.92, d, J 4.9 Hz, IJZ: 7.24-7.38, m, 5H, Ph. Mass spectrum; mlz 167

Experimental 163
(M+H+, 99Vo 2}t1, l77o), I2l (42),93 (11), 92 (PhCHO+, 38), 74 (100),28
(r2).
(2535)-3-Deuteriophenylalanine (65b)
Treatment of the erythro-deuteriophenylalanine derivative (149b) (500
mg, 1.6 mmol) with 6N HCl/acetic acid, as described for the preparation of
(253R)-deteriophenylalanine (65a) from the deuteride (149a), gave the title
compound (65b) as a white powder (228 mg, 857o), m.p. 270-275". lH n.m.r.
(300 MHz, DzO) õ 3.03, d, J 7 .9 Hz, }J3; 3.92, d, J 7 .9 Hz, H2; 7 .24-7 .38, m,
5H, Ph. Mass spectrum; mlz 167 (M+H+, 997o 2H1, 4O7o), 121 85), 93 (23),92
(PhcHo+, 100), 74 (95),28 (20).
(2R35)-3-Deuteriophenylalanine (65c)
Treatment of the threo-deuteriophenylalanine derivative Oa9c) (100 mg,
0.32 mmol) with 6N HCl/acetic acid, as described for the preparation of
(253R)-deteriophenylalanine (65a) from the deuteride (149a), gave the title
compound (65c) as a white powder (44 mg, 827o), m.p. 268-212". Spectral
data identical to that of the (253R)-enantiomer (65a).
(2R3R)-3-Deuteriophenylalanine (65d)
Treatment of the erythro-deuteriophenylalanine derivative (149d) (250
mg, 0.81 mmol) with 6N HCl/acetic acid, as described for the preparation of
(253R)-deteriophenylalanine (65a) from the deuteride (149a), gave the title

Experimentnl I&
compound (65d) as a white powder (109 mg, SlVo), m.p. 270-273". Spectral
data identical to that of the (2S3S)-enantiomer (65b).
Treatment of (253R)-3-Deuteriophenylalanine (65a) with PAL
Sodium borate buffer (0.04M, pH 8.7) was prepared by dissolving sodium
tetraborate decahydrate (26.7 g, 0.07 mol) in water (1.75 L). The pH was
reduced to 8.7 by the addition of conc. IICI (ca. 7 ml). To a solution of
(253R)-3-deuteriophenylalanine (65a) (33 mg, 0.20 mmol) in sodium borate
buffer (25 ml) was added PAL (Sigma, from Rhodotorula glutinis, grade 1,
solution in 607o glycerol, 3 mM Tris-HCl, pH 7.5; 0.2 ml, 0.5 units). The
solution was stirred at 30o in a constant temperature water bath for 20 h. The
solution was acidified to pH 1 by the addition of conc. HCl, then was extracted
with dichloromethane (2x25 ml). The organic phase was dried and the solvent
was removed under reduced pressure Recrystallization of the residue gave
trans-3-deuteriocinnamic acid (67) (15.9 mg,547o) m.p. 135-137". lH n.m.r.
(300 MI{z, CDCI3) ô 6.46, br s, 0.98H,H.2; 6.47, d, J 16.0}J2,0.02H, H2;7.41-
7.44, m, 3H, ArH; 7.55-7.58, m,2Il, ArH; 7.81, d, J 16.0 }{2,0.02H, H3. lH
n.m.r. spectroscopic analysis indicated that the product (67) contained 98Vo
2H1-incorporation.
Treatment of (2535)-3-Deuteriophenylalanine (65b) with PAL
Treatment of (2S3S)-3-deuteriophenylalanine (65b) (33 mg, 0.20 mmol) in
sodium borate buffer (25 ml) with PAL (0.2 ml, 0.5 units), as described above
for the treatment of (253R)-3-deuteriophenylalanine (65a) with PAL, gave
trans-cinnamic acid (57) (17.8 mg, 607o) m.p. 134-136' (lit.t7e I32o). lH

Experímental 165
n.m.r. (300 MHz, CDCI3) õ 6.47, d, J 16.0 }{z,II2; 7.41-1.44, m, 3H, ArH;
7.55-7.58, m,2}J., ArH; 7.81, d, J 16.0 Hz,H3.
Treatment of QR3S)-3-Deuteriophenylalanine (65c) with PAL
Treatment of (2R35)-3-deuteriophenylalanine (65c) (33 mg, 0.20 mmol) in
sodium borate buffer (25 ml) with PAL (0.2 ml, 0.5 units), as described above
for the treatment of (253R)-3-deuteriophenylalanine (65a) with PAL, but
stirring the solution for 8 days, gave trans-3-deuteriocinnamic acid (67) (20.6
mg,707o) m.p. 134-I37". lH n.m.r. (300 MHz, CDCI3) ô 6.46, br s, 0.92H,
IÐ; 6.47 , d, J 16.0 Hz, 0.08H , I12; 7 .41-7 .44, m, 3H, ArH; 7 .55-7 .58, m, 2}J,
ArH; 7.81, d, J 16.0 Hz, 0.08H, H3. lH n.m.r. spectroscopic analysis indicated
that the product (67) contained 927o 2H1-incorporation.
Treatment of (2R3R)-3-deuteriophenylalanine (65d) with PAL
Treatment of (2R3R)-3-deuteriophenylalanine (65d) (33 mg, 0.20 mmot)
in sodium borate buffer (25 ml) with PAL (0.2 ml, 0.5 units), as described
above for the treatment of (2R35)-3-deuteriop[enylalanine (65c) with PAL,
gave trons-cinnamic acid (57) (17.4 mg, 597o) m.p. 135-136'. lH n.m.r. (300
lvffIz, CDCI¡) õ 6.46, br s, 0.27H,IÐ; 6.47, d, J 16.0 Hz,0.73H,If2;7.4I-7 .M,
m,3H, ArH; 7.55-7.58,m,2H, ArH; 7.81, d, J 16.0 }Iz,0.l3H. H3. lH n.m.r.
spectroscopic analysis indicated that the product (57) contained 217o 2I{r
incorporation.

1
2
3
4
References 166
REFERENCES
Greenstein, J. P., and Winitz,M., Chemistry of the Amino Acids, John
Wiley and Sons Inc., New York, 196I, vols. 1-3.
(a) Wagner, I., and Musso, H., Angew. Chem., Int. Ed. EngI., 1983,22,
816; (b) Ohfune, Y., Acc. Chem. Res., 1992,25, 360, and references
cited therein.
Williams, R. M., Sinclair, P. J., Zha\ D., and Chen, D., J. Am. Chem
9oc.,1988, II0, 1547.
(a) Hammond, S. J., Williamson, M. P., Williams, D. H., Boeck, L. D.,
and Marconi, G. G., .f. Chem. Soc., Chem. Commun., 1982,344; (b)
Hammond, S.J., Williams, D.H., and Nielsen, R. V., J. Chem. Soc.,
Chem. Commun", 1983, 116; (c) Lin, T.-S., and Kolattukudy, P. E.,
Eur. J. Biochem., 1980, 106,341.
5. Meerakami, Y., Hisaeda, Y., and Ohno, T., Chem. Lett., 1987,1351.
6. Suzuki, F., and Baker, H. 4., J" BioI. Chem., 1966,244,878.
Kleeman, 4., Leuchtenberger, W., Hoppo, B., and Tanner, H., "Amino
Acids" in Ullmann's Encyclopedia of Industríal Chemistry, VCH
Verlagsgesellschaft, Weinheim, 1985, vol. A-2, pp. 57-97 .
Mazur, R. H., Schlatter, J. M., and Goldkamp, A. H., J. Am. Chem. Soc.,
1969,9L,2684.
7
8

9
Reþrences 167
(a) Rando, R. R., Acc. Chem. Res., 1915,8, 28; (b) Abeles, R. H., Pure
Appl. Chem.,1980, 53, 149.
10" Poisel, H., and Schmidt, U., Angew. Chem., Int. Ed" EngI., 1976, 15,
294.
11. Shimohigashi, Y., Stammer, C. H., and Costa, T., "Synthetic Enkephalin
Analogues: Designing New Drugs of Pharmacological Interest" in
Synthetic Peptides in Biotechnology, A.R. Liss, Inc., 1988, pp.203-223.
12. Mapelli, C., Kimura, H., and Stammer, C. H., Int. J. Peptíde Protein
Res., 1986, 28,347.
13. Hruby, V. J., Life 9ci.,1982,31, 189.
14. Williams, R. M., Synthesis of Optícally Active a-Amino Acíds,
Pergammon Press, Oxford, UK, 1989"
15 Wightman, R. H., Staunton, J., Battersby, A. R., and Hanson, K. R.,.f.
Chem. Soc., Perkin Trans. I , 1972,2355.
16. Thomas, N. R., and Gani, D., Tetrahedron, 199I, 47 , 497 .
I7. Kollonitsch, J., Perkins, L. M., Patchett, A. 4., Doldouras, G. 4.,
Marburg, S., Duggatr, D.E., Maycock, A. L., and Aster, S. D., Nature,
1978,274,906.
18. Versteeg, D. H. G., Palkouts, M., Van der Gugten, J., Wijnen, J. L. M.,
Smeets, G. W. M., and DeJong, W., Prog. Brain Res., 1971, 47, lll.

References 168
19. Walsh, C., and Wang, 8., Biochemistry, 1978,17, 1313.
20. Kollonitsch, J., and Barash, L., J. Am. Chem. Soc., I976,98, 5591.
2I. Jackson, R. F. W., Wood, 4., and Wythes, M. J., Synlett, 1990,735.
22. Arnold, L. D., Kalantar, T. H., and Vederas, J. C., J" Am. Chem. Soc.,
1985, L07,7105.
23 Arnold, L. D., Drover, J. C. G., and Vederas, J. C., J. Am. Chem. Soc.,
1987, 109" 4649"
24 Arnold, L. D., Muy, R. G., and Vederas, J. C., J. Am. Chem. Soc., 1988,
1^1.0, 2231.
25 (a) Barton, D. H. R., Herve, Y., Potier, P., and Thierry,J.Tetrahedron,
1988, 43,4297; (b) Barton, D. H. R., Herve, Y., Potier, P., and
Thierry, J., Tetrahedron, 1988, 44, 5419.
26 (a) Ohfunê, Y., and Nishio, H., Tetrahedron Lett., 1984,25, 4I3; (b)
Fushiya, S., Nakatsuyama, S., Sato, Y.and Nozoe, 5., Heterocycles,1981,
15, 819.
27 (a) Baldwin, J. E., Moloney, M. G., and North, M., Tetrahedron, 1989,
45,6309; (b) Sardina, F. J., Paz, M. M., Fernandez-Megia, E., de Boer,
R. F., and Alvarez, M. P., Tetrahedron Lett., 1992, 33, 4637 .
Pu, Y., Martin, F. M., and Vederas, J. C.,J. Org. Chem., 1991,56,
1280.
28

References 169
29. Shimamoto, K., and Ohfune, Y., Tetrahedron Lett., 1988, 29, 5171.
30. Kollonitsch, J., Rosegay, 4., and Doldouras, G. 4., J. Am. Chem. Soc.,
1964,86,1857.
31. Kollonitsch, J., Scott, A. N., and Doldouras, G. A., J. Am. Chem. Soc.,
1966,88,3624.
32. Fujita, Y., Kollonitsch, J., and Witkopp, 8., J. Am. Chem. Soc., 1965,
97, 2030.
33. Russell, G. 4., in Free Radícals, Kochi, J. K. (Ed.), Wiley, New York,
1973, vol. l, pp.275-331.
34 Poutsma, M. L., in Free Radícals, Kochi, J.K. (Ed.), Wiley, New York,
1973, vol.2, pp. 159-229.
35 (a) Ito, Y., Ohashi, Y., Kawabe, S., Abe, H., and Okuda, T., J. Antibiot.,
1972, 25,360; (b) Konishi, M., Ohkuma, H., Sakai, F., Tsuno, T.,
Koshiyamâ, H., Naito, T., and Kawaguchi, H., J. Am. Chem. Soc., 1981,
I03, I24l; (c) Arnold, E., and Clardy, 1., J. Am. Chem. Soc., 1981,
r03, t243.
36. Bowman, N. J., Hay, M. P., Love, S. G., and Easton, C.J.,J. Chem
Soc., Perkin Trans. l, 1988,259.
37 . Neta, P., ancl Fessenclen, R. W'., .l . Phys. Chem., 1971,75,738.

References I7O
38. Tanaguchi, H., Hatano, H., Hasegawa, H., and Maruyama, '1., J. Phys
Chem., 1970,74,3063.
39 (a) Viehe, H. G., Merenyi, R., Stella, L., and Janousek, 2., Angew
Chem., Int. Ed. Engl., 1979,18, 917; (b) Viehe, H. G., Janousek, 2.,
and Merenyi, R., Acc. Chem. Res.,1985, 18, 148.
q (a) Baldock, R. W., Hudson, P., Katritsky, A. R., and Soti, F.,
Heterocycles,1973,L,67; (b) Baldock, R. W., Hudson, P., Katritsky, A.
R., and Soti, F., J. Chem. Soc., Perkin Trans. I, I9'74, 1422.
4I. Balaban, A. T., Rev. Roum. Chím., I9ll,16,725.
42. Easton, C. J., Tan, E. W., and Hay, M. P., J" Chem" Soc., Chem"
Commun.,1989, 385.
Badran, T. !V., Ph.D. Thesis, University of Adelaide, 1991, pp. 58-63.
Rosenmund, K. W., and Dornsaft, H., Chem. 9er.,1919,52, 1734.
(a) Hahn, F. E., in Antibiolics, Gottlieb, D., and Shaw, P. D. (Eds.),
Springer-Verlag, Heidelberg, 1967, vol. 1, p. 308; (b) Pestka, S., in
Antibiotics, Corcoran, J. W., and Hahn, F. E. (Eds.), Springer-Verlag,
Heidelberg,I975, vol.3, p.310: (c) Pongs, O., in Antibiotics, Hahn, F.
E. (Ed.), Springer-Verlag, Heidelberg, 1979, vol.5, part 1, p.26.
46. Krant, J., Ann. Rev. Ùiochem.,1977,46,331.
43
44
45
47 Che¡evert, R., and Thiboutot,5., Synth¿sis, 1989, 444.

48
51.
References I7l
(a) Ehrhart, G., Siedel, W., and Nahm, H., Chem. Ber., 1957,90, 2088;
(b) Johnson, F., in The Total Synthesis of Natural Products, ApSimon, J.
(Ed.), Wiley, NewYork, 1973, vol. l, p. 457; (c) Suzuki, M., 'f .
Antibiot. (Japan), Ser. 8,1961, 14,323.
49 Sahu, D. P., Mashava, P., Manhas, M. S., and Bose, A. K., J. Org
Chem.,1983, 48,1142.
50. Miller, M. J., Acc. Chem. Res., 1986, 19,49"
Williams, D. H., Acc. Chem. Res., 1984, 17,364, and references cited
therein.
52 McCormick, M. H., Stark, W. M., Pittenger, G. F., Pittenger, R, C., and
McGuire G. M., Antibiot. Annu.,1955-56, 606.
53. (a) Cafferkey, M. T., Hone, R., and Keane, C. T., .,f. Antimícrob.
Chemother., 1982,9,69; (b) Watanakunakorn, C., Rev. Inf. Dis., 1981,
3, 5210; (c) Horowitz, H. W., Handwerger, S., van Korn, K. G., and
Wormser, G. P., Lancet, 1981, vol.Il(857I), 1329.
54 (a) O'sullivan, J., McCullough, J. 8., Tymiak, A. 4., Kirsch, D. R.,
Trejo, W. H., and Principe, P. A.,.1. Antibiot., 1988, 41, 1740; (b)
Bonner, D. P., O'Sullivan, J., Tanaka, S. K., Clark, J. M., and Whitney,
R. R., J. Antibiol., 1988, 4L, 1745; (c) Tymiak, A. 4., McCormick, T.
J., and Unger, S. E., J. Org. Chem.,1989, 54, 1149.
55. Cyclosporir¿ A, White, D. J. G. (Ed.), Biomedical, Amsterdam, 1982

57
References 172
56. Wenger, R. M., Angew. Chem., Int. Ed. Engl., 1985, 24,77.
Jolad, S. D., Hoffmann, J.J., Torrance, S. J., Wiedhopf, R. M., Cole, J
R., Arora, S. K., Bates, R. 8., Gargiulo, R. L., and Kriek, G. R., J. Am
Chem. Soc.,1977, 99, 8040.
Traber, R., Keller-Juslen, C., Loosli, H.-R., Kuhn, M., and von
'Wartburg, A., Helv. Chim. Acta, 1979, 62, 1252.
58
59. Culvenor, C. C. J., Cockruñ, P. 4., Edgar, J. 4., Frahn, J. L., Gorst-
Allman, C. P., Jones, A. J., Marasas, W. F. O., Murray, K. E., Smith, L.
W., Steyn, P. S., Vleggaar, R., and'Wessels, P. L., J. Chem. Soc., Chem.
Commun.,1983, 1259"
60. Mackay, M. F., Van Donkelaar, 4., and Culvenor, C. C. J., J" Chem
Soc., Chem. Commun., 1986,1219.
Wiedland, T ., N at urwi s s e ns c haft e n, 197 2, 59, 225
V/iedland, T., and Hofer, A., Liebigs Ann. Chem., 1958, 619,35.
Pfeander, P., and Wiedland, T., Liebigs Ann. Chem., 1966,700, 126.
Wiedland,T., Liebigs Ann. Chem.,1981, 1445.
Buku, 4., Faulstich, H., Wiedland, T., and Dabrowski, J., Proc. Natl
Acad" Sci. [/SA, 1980, 71,2370.
61.
62"
63.
64.
65.

67
68
69
References 173
66. Morita, H., Kondo, K., Hitotsuyanagi, Y., Takeyâ, K., Itokawa, H.,
Tomioka, N., Itai, 4., and Iitaka, Y., Tetrahedron, 1991, 47,2757.
DiBello, I. C., Dorling, P., Fellows, L., and Winchester, 8., FEBS Lett.,
1984, L76,61.
Hendry, A. T., and Dillon, J. R., Can. J. Microbiol.,1984,30, 1319.
Janecek, J., Jiresova, M., Technikova, 2., and Spizek, J., Folia
M icrobiol., 197 l, 16, 367 .
70. Wiedland, T., Mannes, K., and Schopf, 4., Liebigs Ann. Chem., 1958,
617, r52.
7I. Liesch, J. M., Milington, D.S., Pandey, R. C., and Reinhart, K. L.,J"
Am. Chem. Soc., 1976,98,8237.
72 "cr-Amino Acid Synthesis", Tefiahedron Symposia in Print, O'Donnell,
M. J., (Ed.), Tetrqhedron, 1988,44,5253.
Williams, R. M., and Hendrix, J. 4., Chem. Rev., 1992,92,889.
Coppola, G. M., and Schuster, H. F., Asymmetric Synthesís:
Constructíon of Chiral Molecules Using Amíno Acids, John Wiley and
Sons, New York, 1987.
13
74
75. Oh-Hashi, J., and Harada, K., Bull. Chem. Soc. Jpn, t966,39,2281.

l6
77
References 174
Guanti, G., Banfi, L., Narisano, E., and Scolastico, C., Tetrahedron,
1988,44,3671.
Belokon, Y. N., Bulychev, A. G., Vitt, S. V., Struchkov, Y. T.,
Batsanov, A. S., Timofeeva, T. V., Tsyryapkitr, V. 4., Ryzhov, M. G.,
Lysova, L.4., Bakhmutov, V. I., and Belikov, V. M., J. Am. Chem.
Soc., 1985, 107, 4252.
Casella, L., Jommi, G., Montanari, S., and Sisti, M., Tetrahedron Lett.,
1988,29,2067.
78
79 Ito, Y., Sawamura, S., and Hayashi, T., J. Am. Chem. \oc.,1986, 108,
6405.
80 Schollkopf, U., Groth, U., and Deng, C., Angew" Chem., Int. Ed. Engl.,
1981,20,798.
81. Schollkopf, U., Hartwig, W., Pospischil, K.-H., and Kehne, H., Synthesis,
1981,966.
Schollkopf, U., Nozulak, J., and Groth, U., Synthesis, 1982, 868.
Schotlkopf, U., and Groth,U., Angew. Chem., Int. Ed. Engl., 198I,20,
977.
Schollkopf, IJ., and Groth,U., Synthesis, 1983 , 673.
Schollkopf, U., Tiller, T., and Bardenhagen, J., Tetrahedron, 1988,44,
s29,3.
82.
83.
84.
85.

References I75
86. Seebach, D., Imwinkelried, R., and Weber, T., Modern Synthetic
Methods, Springer-Verlag, Heidelberg, 1986, vol. 4, 182.
87. Seebach, D., Juaristi, 8., Miller, D. D., Schickli, C., and'Weber, T.,
Helv. Chim. Acta, 1987 ,70,237 .
(a) Evans, D. 4., and Weber, A. E., J. Am. Chem. Soc., 1986, L08,
6757; (b) Evans, D.4., and Weber, A. E., J. Am. Chem. Soc., 1987,
109, 7151.
88
89 Mellin, G. W., and Katzenstein, M., New Engl. J. Med., 1962,267,
1 184.
90. von Blaschke, G., Kraft, H. P., Finkentscher, K., and Kohler, F.,
Arzneim.-F orsch.l Drug Res., 1979, 29, 1640.
9I. Remington's Pharmqceutical Sciences, l6th ed., Osol, ,A.,. (Ed.), Mack
Publishing, Easton, PA, 1980, 1170.
92 Csaky, T.2., Cutting's Handbook of Pharmacology, 6th ed., Appelton-
Century-Crofts, New York, 1979, 16l "
93 Tanner, D., Birgersson, C., and Dhaliwal, H. K., Tetrahedron Lett.,
1990,31, 1903.
Rao, A. V. R., Yadav, J. S., Chandresekhar, S., and Rao, C. S
Tetrahedron Lett., 1989, 30,6769"
94

References 176
95 Kornblum, N., Smiley, R. A., Blackwood, R. K., and Iffland, D. C., J.
Am. Chem" Soc., 1955, 77, 6269"
96. Pocker, Y., and.Kevill, D. N., J. Am. Chem. Soc., 1965,87, 4760.
97. Korblum, N., and Hardies, D. E., J. Am. Chem" Soc., 1966,88, 1707.
98.'Walsh, C.,Tetahedron, 1982,38, 871.
99. Walsh, C., Ann. Rey. Bíochem.,1984,53, 493.
100. Scaman, C. H., and Palcic, M. M., Biochemístry,1992,31,6829.
101. Hanson, K. R., and Havir, E. 4., Arch. Biochem. Biophys., 1970,I4L,
1.
102. Havir, E. A., Reid, P. D., and Marsh, H. V. Jr., Plant Physiol., I971,48,
130.
103. Havir, E. 4., and Hanson, K. R., Biochemistry, I975,14, 1620.
104. Hodgins, D. S., J. Bíol. Chem., 197I,266,2977.
105. Parkhurst, J. R., and Hodgins, D. 5., Arch. Biochem. Bíophys., 1972,
I52, 597.
106. Walsh, C., Enzymatic Reactíon Mechanisms, W. H. Freeman & Co., San
Francisco , 1979, pp. 563-567.

References ll7
lO1. Hanson, K. R., and Havir, E. 4., "The Enzymic Elimination of
Ammonia" inThe Enzymes,3rd ed., Boyer, P. D. (Ed.), Academic Press,
New York,1972, vol. 7, pp. 75-166.
108. Hanson, K. R., and Havi¡, 8.,A.., "Phenylalanine Ammonia Lyase" inThe
Biochemistry of Plar?ts, Stumpf, P. K., and Conn, E. E. (Eds.), Academic
Press, New York, 1981, vol. 7,pp.577-625.
109. Havir, E. 4., and Hanson, K. R., Biochemistry,1968, 7, 1896.
110. Havir, E. 4., and Hanson, K. R., Biochemístry, 1968,7 , 1904"
111. Kirby, G. W., and Michael, J., J. Chem. Soc., PerkinTrans. 1,1973,
115.
lI2. Ife, R., and Haslan, E., J. Chem. Soc. (C), 197I,2818.
113. Bartl, K., Cavalar, C., Krebs, T., Ripp, E., Retey, J., Hull, 'W. E.,
Gunther, H., and Simon, H., Eur. J" Bíochem., 1977,72,247 .
ll4. Nagai, U., and Kobayashi, J., Tetrahedron Lett., 1976,33,2873
115. Sheehan, J" C., Chapman, D. W., and Roth, R. W., J. Am. Chem. Soc.,
1952,74,3822.
116. Sheehan, J. C., Goodman, M., and Hess, G. P., J. Am. Chem.\oc.,1956,
78, 1367.

Reþrences 178
Il7. Fling, M., Minard, F. N., and Fox, S. W., .f. Am. Chem. Soc., 1947, 69,
2466.
118. (a) Pirkle, W.H., and Welch, C.J., J. Org. Chem., 1984,49, 138, and
references cited therein; (b) Pirkle, W. H., Finn, J. M., Schreiner, J. L.,
and Hamper, J. C., f . Am. Chem. Soc., 1981, L03,3964; (c) Pirkle, W.
H., and House, D. W., J. Org. Chem.,1979,44, 1957.
119. Adam, J., Gosselain, P. 4., and Goldfinger,P.,Nature,1953,l7L,704.
I20. Williams, D. H., and Fleming, I., Spectroscopic Methods in Organic
Chemistry, 3rd ed., McGraw Hill Book Company, Maidenhead, U.K.,
1980.
I2I. Liotta, C. L., and Hanis, H. P., J. Am. Chem. Soc., 1974,96,2250.
122" Srinivasan, 4., Richards K. D., and Olsen, R. K., Tetrahedron Lett.,
1976,33,891.
123" Poisel, H., and Schmidt, U., Chem. Ber., 1915,108,2547 "
I24. Srinivasan, 4., Stephenson, R. W., and Olsen, R. K., J. Org. Chem.,
1977,42,2256.
125. Nitz, T. J., Holt, E. M., Rubin,8., and Stammer, C.H., J. Org. Chem.,
1981,46,2667.

References I79
126. (a) Waters, Vy'. A, Mechanism of Oxidatíon of Organic Compounds,
Methuen and Co. Ltd., London, 1964; (b) Walling, C., Free Radicals in
Solution, John Wiley and Sons, New York,1957.
I27. Hammond, G. S., Hawthorne, M. F., 'Waters, J, H., and Graybill, B. M.,
J. Am. Chem. 5oc., 1960, 82, 704.
128. Griesbeck, A. G., and Mauder, H., Angew. Chem., Int. Ed. EngI., 1992,
31,73.
129. Goering, H. L., Eikenberry, J. N., Koermer, G. S., and Lattimer, C. J.,
J. Am. Chem. Soc., 19J4,96, 1493.
130. Clarke, S., Hider, R. C., and John, D. I., J. Chem. Soc., PerkinTrans. l,1973,230.
131. Jain, T. C., and McCloskey, J.9., Tetrahedron,I975,3l,22ll
132. Altman, J., Gilboa, H., and Ben-Ishai, D., Tetrahedron, 1971,33,3173
133. Hassner, A., and Alexanian, Y ., Tetrahedron Lett., 1978, M75.
I34. Bowden, K., Heilbron, I. M., Jones, E. R., and Weedon, B. C., J. Chem.
Soc., 1946,39.
135. Osby, J. O., Martin, M. G., and Ganem,B.,Tetrahedron Lett.,1984,25,
2093

References 180
136. March, J., Advanced Organic Chemistry, 3,d ed.,Iohn Wiley and Sons,
New York, 1985, pp.220-222.
137. Sheehan, J. C., and Frank, V.S., J. Am. Chem.5oc.,1949, 71, 1856.
138. Vogler, K., Helv. Chim. Acta,1950, 33,2I1I.
I39. Takahata, H., Ohnishi, Y., Takehara, H., Tsuritani, K., and Yamazaki,
T., Chem. Pharm. Bull. Jpn., 1981, 29, 1063
140. Miller, M. J., and Mattingly, P. G., Tetrahedron, 1983, 39,2563.
l4l. Easton, C. J., J. Chem. Soc., PerkinTrans.l, 1985, 153.
I42. Barrow, K. D., and Spotswood, T. M., Tetrahedron Lett., 1965,3325"
143. Wells, J. S., Trejo, W. H., Principe, P. 4., and Sykes, R. 8., J. Antibiot.,
1984,37,802.
144. Lowe, C., Pu, Y., and Vederas, J. C., J. Org. Chem.,1992,57, 10.
I45. Herbert, R. 8., and Knaggs, A. R., J" Chem. Soc., PerkinTrans. 1,1992,
103 and 109.
146. Zaugg, H. E., Org. React., 1954,8, 305.
147. Danheiser, R. L,., and Nowick, J. S., J Org. Che.m.., 1991, 56, 1176.

References 181
148. Cardani, S., DeTomâ, C., Gennari, C., and Scolastico, C., Tetrahedron,
1992,48, 5551.
I49. Thaler, W., ./. Am. Chem. Soc., 1963,85,2607
150. Rando, R. R., Nature (London), 1974,250, 586
151. Castelhano,4.L., Plivra, D.H., Taylor, G. J., Hsieh, K. C., and Krantz,
A., J. Am. Chem. Soc., 1984, 106,2734.
I52. Scannell, J. P., Preuss, J. L., Demney, T. C., Sello, L. H., Williams, T.,
and Stempel, 4., J. Antibiot., 1972,25, I22.
153. V/olkoff, P., J. Org. Chem., 1982,47, 1944.
154. DePuy, C. H., and King, R. W., Chem. Rev., 1960,431.
155. Kopping, B., Chatgilialoglu, C.,Znhnder, M., and Giese,8", J. Org.
Chem., 1992,57,3994; (b) Giese, 8., Damm, W., Wetterich, F., and
Zeitz, H.-G., Tetrahedron Lett., 1992,33, 1863; (c) Giese, 8., Damm,
W., Roth, M., and 7nhnder,M., Synlett, 1992,441.
156. Mok, P. L. H., and Roberts, 8., J. Chem. Soc., Chem. Commun., 1991,
151.
I57. Guindon, Y., Yoakifl, C., Lemieux, R., Boisvert, L., Delorme, D., and
Lavallee, J.-F., Tetrahedron Lett., 1990, 31,2845.

Reþrences I82
158. Lusztyk, J., Maillard, B., Lindsay, D. 4., and Ingold, K. U., J. Am
Chem. \oc.,1983, 105, 3578.
159. Beckwith, A. L. J., and Pigou, P. E., Aust. J. Chem.,1986,39, 1151.
160. Bell, H. M., and Brown, H. C., J. Am. Chem. Soc., 1966,88, 1473.
161.
162.
Kim, S., Kim, Y. J., and Ahn, K. H., Tetrahedron Lett., 1983, 24,3369"
Berti, G., Bottari, F., Macchia, 8., and Macchia, F., Tetrahedron, 1966,
22, lgg.
163. Berti, G., Macchia, 8., Macchia, F., and Monti, L., J. Org. Chem., 1968,
33,4M5.
164. Cavanaugh, J. R., J. Am. Chem. Soc., 1968,90, 4533.
165. Still, \V. C., Kahn, M., and Miffa, A., J. Org. Chem.,1978,43,2923
166. Harwood, L. M., Aldrichimica Acta., 1985, 18,25
161. Perrin, D. D., Armarego, W. L. F., and Perrin, D. R., Purification of
Laboratory Chemicals,2nd ed., Pergammon, Oxford, 1980.
168. Vogel, A. I., A Textbook of Practical Organic Chemistry including
Quantitative Organic Analysis, 3,d ed., Longman, London, 1977.
t69. Hoogwater, D. 4., Reinhoundt, D. N., Lie, T. S., Gunneweg, J. J., and
Beyerman, H. C., Recl. Trav. Chim. Pays-Bas., 19J3,92,819.

References 183
170. Wunsch, E., Drees, F., and Jentsch, J., Chem. Ber., 1965, 98, 803
I7l. Hoffmann, E., and Schiff-Shenhav, H., J.Org. Chem., 1962,27, 4686.
172. Banks, G. R., Cohen, D., Pattenden, G. E., and Thomas, J. A. G
J. Chem Soc. (C), 1967, 126"
173. (a) Schwyzer, R., and Ludescher, U., Biochemistry, 1968,7,2514; (b)
Greenlee, W. J., and Thorsett, E. D., J. Org. Chem., 1981, 46, 5351"
174. Horton, W. J., and Thompson, G., J. Am. Chem.Ïoc.,1954,76, 1909.
I75. Marchand, J., Pais, M., and Jarreau, F.-X., Bull. Soc. Chím. Fr., 1971,
3742.
176. Schwyzer, R., and Caviezel, M., Helv. Chím. Acta.,I97l,54, 1395.
177. Ville, 4., Comte,P.,Zwingelstein, G., and Favre-Bonvin, J., Bull. Soc
Chim. Fr.,1958, 1352.

Appendices 184
Appendix 1
lH n.m.r. spectral data of the p-bromophenylalanine derivatives (82), (85),
(115), (128) and (137), and the p-bromotyrosine derivatives (9a) and (95).
tnrhN¿¿,,,. R an,hNrrr,..
.,.tf
Bf Br
R' R'
(82a)
(9aa)
(1 15a)
(128a)
(85a)
(95a)
(r37a)
(82b)
(e4b)
(1 15b)
(128b)
(85b)
(e5b)
(137b)
R=OMe, R'=HR = OMe, R' = OAc
R = OCMe¡, R' = H
R = OMe, R' = NOz
R = NHCMo3, R' = H
R = NHCMe3, R' = OAc
R= OH, R'= H
R=OMe, R'=HR = OMe, R' = OAc
R = OCMe¡, R' = H
R = OMe, R' = NOz
R = NHCMe3, R' = H
R = NHCMe3, R' = OAc
R=OH, R'=H
8.031.381.401.483.833.813.82R
erythro-
ßomers to.411.5LL.4to.410.310.410.5Jnß
5.866.086.055.8.55.975.935.91p
5.645.30s.325.495.595.565.59ct
(r37b)(e5b)(85b)(1 15b)(128b)(94b)(82b)
6.641.05r.o21.163.593.563.55R
threo-
rsomers rt.211.811.8tt.4tr.2rt.2tt.2Jaß
5.976.226.t76.066.O26.036.02p
5.555.175.225.475.515.465.52c[
037a\(95a)(85a)(1 15a)(t28a)(o4a\(82a)

Appendices 185
Appendix 2
lH n.m.r. spectral data of thep-hydroxyphenylalanine derivatives (1 LZ),QI6), (1 I 8) and (129)
and the p-hydroxytyrosine derivatives (121), (I22), (I23) and (I24).
R R
R' R'
(rr2a)(t22a)(r2La)(tze)(1 16a)(1 18a)(r24)(r23)
OMe,OMe,OMe,OMe,OCMe3,NHCMe3,
_H=OH= OAc= NOz_H_H=OH= OAc
(rrzb(r22b(tzrb
R'= HR'= OHR' = OAc
Me,Me,Me,
) R=O) R=O) R=O
RRRRR
R_R_R_R_R_ft=R_R-
(116b) R=OCMe¡, R'=H(118b) R=NHCMe3,R'=H
NHCMe3, RNHCMe3, R
1 15t.283.743.783.72R
erythroisomers
4.254.604.984.274.27OH
4.92.t2.3Ja.on
s.395.495.545.495.46p
8.38.08.28.68.4Ja,ß
4.614.955.024.974.95c[
(1 18b)(116b)(121b)(r22b)(112b)
1 3 1t.271.30t.523.893.783.853.86R
tlveo-isomers
4.434.724.41.5.255.344.985.O25.13OH
8.17.58.210.610.010.310.4Json
5.63s.625.635.675.795.675.645.7 |p
6.36.96.24.64.45.04.84.6Ja.ß
5.085.1 15.115.445.535.485.445.51c[
( r23)(124\(1 I 8a)(1 16a)(129\(I2la\(122a\(ll2a\

Appendices 186
Appendix 3
Crystal sEucture of
(Z )-N - t e rt- butyl-Nø-phthaloyl-o, B-dehydrophenylalaninamide ( 86)
o(21) c(32)
N(2) c(31)
o(28)
c(2) c(3)
N(3)c(1)
c(4)c(4a)
o(1)
C(4c) c(4b)

Appendices 187
Bond distances (,Å,) for
(Z ) -N - t e rt-butyl-Nø-phthaloyl-a, p-dehydrophenylalaninamide ( 86)
c (1)c (28)c (21)c (1)H (3N)c (3)H (3)c (48)H(41)H (43)H (4s)H (47)H(4e)c (23)c (241c (2s)c(26tc (271c (28)c (36)a(32',,H (33)H (34)H (3s)
o (1)o (28)N (2)N (3)N (3)c(2,c (3)c (4)c (4À)c (4À)c (48)c(4c)c (4c)c(22',,c(23)c (241c(2s)c(261c (21\c (31)c(32)c (33)c(34)c (3s)
1 .236 (3 )
1 - 211 (3)1.4ls (3)1.337 (3)o.824 (26'.,1 .336 (3 )
o -962 (2411.s01(4)0-9?00-9700-9?00-9700.9701 -386 (3)1.385 (4)1.383 (4)1.390(4)1.383 (3)1 .4 86 (3)r.390 (3)o -921 (2610:933 (32)0.960 (31)I .019 (29)
c (21)c (21c (28)c(4)c(21c (31)c(44)c(4c)H(421H(44)H(45)H (48)c(221c (211H (23)11(241H (2s)H(261c(321c (33)c(34)c(3s)c(36)H (36)
1.20s (3)1- 431 (3)1. 404 (3)1.480 (3)r. s02 (3)1.{62(3)1.s21(4)r.s11(4)0. 9700. 9?00.9?00.9701.48s (3)r.38s (3)0. 930 (30)0.930 (29)0. 935 (36)1.011 (27)1.401 (3)r-3?5(4)r.364 (4)1.390(4)1.379(4)o.941 (211
(2oN
N
N
cccccccccccccccccccc
2
2
3
1
3
4
4)4À)
( 4B)(48)( 4c)(2rl(221(231(241(2st(261(31)(32 )
(33)(34 )
(3s )
(36)
Bond angles (deg.) for
(Z )-N - te rt-butyl-Nø-phthaloyl-o, B-dehydrophenylalaninamide ( 86)
c (2rlc (281H (3NlN (3)c(2)c(3)c (31)H(3)c (¡¡B)c (4clc (4clH 142lH (43)H ({3)H ({slH (r6lH({6)H(48)H ({9}H({e}c(221c (23)c(2'tlH (231c (2slH (24)H (2s)c l21lH(26)c (28)N (21c (2-r)c (36)c (33)H (32)H (33)c (3st
, H (34)H (3s)
; c (3s)H (36)
- N(2)- N(2)- N(31- c(1)- c(1)- c(2)- c(3)- c(31- c({)- c(4)- c(4)- c({À)- c(4À)- c({À)- c(48)- c(48)- c(48)- c(4c)- c(4cl- c(4cl- c(21)- c(221- c(221- c(231- c(241- cl24l- c(2s)- c(261- c(26)- c(2?)- c (2B)- c (28)- c (311- c(32¡- c(321- c(33)- c(34)- c(34)- c (3s)- c(36)- c(36)
- c(2t- c(21'- c(tl- o(1)- N(3)- N(2)- c(2)- c(3r.1- N(3)- N(3)- c(48)- c(4)- c(4)- H (42)- c(4)- c({)- H ({s)- c(4)- c(4)- H (48)- o(2r)- c(211- c(23)- cl22l- c(23)- c (2s)- c(24l.- c(2s)- c(2?)- c(221- o (28)- N(2)- c(3)- c(3rl- c (331
- c (32)- c(33)- c (15)- c(34)- c(31)- c (35)
109109109109r09129L29l2lr2lt22L22L20rl7r22108L24t05119t2Ltl9tl7tt912312012tlt 9
L25 - 6 (21r1r.6(2)tt?.6(18)L24 -2 (21tr5.?(2)r22 -5 (21t29 -8 (21r1{-2(14)r10.3(2)109-5 (2)lll.9(3)r09.3 (2)109.6 (2)109. s (-)r09.3(2)
l2(2(2
(2t(-)(2t(2t(-)
2lr8)2Ll2trsl
)))
8)
c (28)c({)H (3N)c(2)c(t)c (3)H (3)c (4À)c (48)c (4cl¡{(41)H (42)H (431H(44)H (4s)H(46)H (4?)H ({8)H (49)N(2tc (221c (2't,c(241H (23)H (24)c(26)H (2slH (26)c (26)c (28)c (2?)c (32)c (36)H (32)c (34)H (33)H (3{)c (36)H (3slH (r6)
- N(2)- N(31- N(3)- c(11- c(2)- c(21- c(3)- c(4)- c(4)- c(4)- c (4Àl- c(4Al- c(4À)- c(48'- c(4Bl- c (4Bl- c(4c)- c(4c)- c(4cl- c(21t- c (211
- c(221- c (231
- c (23t- c(24'- c (2s)- c (2s)- c (26)- c(211- c(211- c (28)- c(31)- c(3r)- c(32)- c (33)- c (33)- c(34)- c (3s,- c (3s)- c(36)
- c(21- c(r)- c({}- o(11- N(21- c(1t- c(21- N(3)- c(4À)- c (4À)- c(4t- H (4r)- H({r}- c(41- H ({4}- H (¡r4)- c(41- H (47'- H(¡tl)- o(2r,- N(2)- c(2r)- c(221- c(24',,- c (23)- c (24'- c (26)- c (25)- cl22l- c(26)- o (28)- c(3t- c{32)- c(31)
c(12)- c(r4)- c (33)
c (]{)- c (36)
c(31)
t22t2611st201l-tr20ll6106108).0 9
1.0 9
109I09109I09t09109109109125105108tl6L2l115l2Ilr8t20121130r29\231ì?119t20l2ltr7Ì20tl9t19
3 t2te (2tI (18)1(2tI (2)3 t2t0(r4)1(2'ts (3)I (3)5 l2ts (-)s (-)6 l2',ts (-)s (-)5 (2)s (-)s (-)o (2't4l2lI (2)6 l2t6(r8tB(T8)o(2t2(2rl8(r5)5 l2tI (2)4 12tt (2)6 t2t5(r5)6 (3)8(r9)2(ì8)0(3)4 (r6)r (ì7)
3 t2t6 (2)9l2l2 (210 (3)2 (1s)3 (19)? (3)1(18)5(rs)I (2tB(11)

Appendices 188
Appendix 4
Crystal structure of
( E )-N -t ert-B utyl-Nø-phthaloyl-cr, p-dehydrophenylalaninamide (87)
C(4c)
c(4b)
C(aa) c(4)N(3)
o(1)c(31) c(32)
c(1)
c(2) c(3)
o(28)
N(2)
o(21)

Appendices 189
Bond distances (,Ä.) for
( E ) -N -t e rt-B utyl-Nø-phthaloyl-cr, p-dehydrophenylalaninamide (87)
c (1)c (28)c (21)c (1)H (3N)c (3)H (3)c (48)H (41)H (43)H (4s)H (47)H (4e)c (23)c (241c (2s)c (26)c (211c (28)c (36)H (32)H (33)H (34)H (3s!
o (1)o(28)N (2)N (3)N (3)c(21c (3)c(4)c(4À)c (4À)c(48)c(4c)c (4c)c(221c(23)c(241c (2s)c(261c(2'tlc(31)c (32)c(33)c(34)c(3s)
1.216 (4)1.197 (4)1 .407 (s)r.340 (s)0.8s4 (4e)1.331(s)0 - BB6 (33)1 . s20 (6)1-046 (71)1.040 (64)1-10s(63)I .008 (43)o - e26 (s9)1.376(s)1.399 (6)I .312 (6',t
1.3?s (s)1-392 (A',)
1.486 (s)1.381(s)1 .007 (s0)o.es3(47)0. e83 (s2)0.92s (4s)
c(2L',1c (21c (28)c(4)c(21c (31)c (4À)c (4c)H(421H(44)H(46)H (48)c (221c(211H (23)H(241H (25)H (261c (32)c (33)c (34)c (3s)
o (21)N(2)N (2)N (3)c (1)c (3)c(4)c (4)c (4A)c (48)c (48)c (4c)c (21)c(22',tc (231c (241c (2s)c(26)c (31)c(32)c (33)c(34)c (3s)c (36)
1-205(4)r - 428 (411- 406 (4)1- 4 67 (s)1 - s0s (s)1 - 4?5 (5)1 - s21 (6)1 - s1s (s)0.80s(68)0. 909 (s3 )
1.002 (so)0- 966 (4s)r.49s(s)1-3?1(s)0.941(36)1.000 (43)1.045 (3s)0 - 933 (36)1.388 (s)1-380(6)r - 360 (7)1.3se (8)1.394 (6)0. 887 (39 )
c (36)H (36)
ond angles (deg.) fo
( E )-N + e n-B utyl Nø-phthaloyl-a, p-dehydrophenylalaninamide ( 87)
c (211c (28)H (3N)N (31c(2)c (31
c (31)H (3)c (4Blc (¡lclc(4c)H (421H (431H(431H (4s)H ({6}H(46)H (48)H (4slH(49)c (221c (23)c(211H (23)c (251H(24)H (2s)c t21lH (261c (28)N (2)
c (36)c (33)H (32)H (33)c (35)H (34)
l" H (3s)c (3s)H (36)
- N(2)- N(2)- N(3)- c(1)- c (t)- c(21- c(3)- c(3)- c(4'- c({)- c(41- c (4À)- c(4À)- c(4Àt- c(48)- c({B}- c ({B}- c({cl- c(4cl- c (¡rc)- c(211- c(221- c(221- c(231- c(2{}- c (2{}- c (2sl- c(26)- c (26)- cl21l- c (281
- c (28)- c(3rl- c (32)- c (32)- c (33)- c (34 )
- c(34)- c(351- c(36)- c (36)
- c(2t- c (2rl- c(r)- o(11- N(3)- N(2)- c(2)- c (31)- N(3)- N(3)- c (48)- c(4)- c(4)- H (42)- c(4)- c(4)- H (4s)- c(4)- c(4)- H ({8}- o (21)- c (21)- c(23)- c(221- c (23)- c (2s)- c(24)- c (2s)- c(211- c(22t- o (28)- N(2)- c(3)- c (3r)- c (33)- c (32)- c (33)- c (3s)- c(34)- c(3rt- c(3s¡
r2s-9(3)ll2. r (3)rr6.3(30)124.9 (3)lts-3 (31
rr9.5(3)121.3(4)rr4.2(r?)106.2 (4tr09-8(31109-0(4)111.1(39)104 - 4 (35)Llt-3(52)lo5-3(32)1O8.6 (26)r01.4(38)107.0(23)1tr-6(38)106.4 (421129-3 (3)129.9(3)122-0(3)r19.6(21)r22 .3 (41121.r(24)120- 0 (20)lr'7.¡t(41lI?.o(22)109 - 6 (3)r24.3(31104.9 (l)122.5(4)Ì20-4 (s)r19.1(28)rr9 8(241120-6(s)125.8(28t130-4(321tI9_7(5)114.8(25)
c (28)c(4tH (3N)c (21c(11c(3)H (3)c (4À.)c(4alc(4clH(4r)H (421H ({3)H({4)H (4s)H(461H (47)H ({8}H (491N (2)c l22lc(2llc (241H (23)R (2{)c (26)H (2slH (26)c (26)c (2Blc (271c (32)c (36'H (32)c(34)H (33)H(34)c (36)H (3slH (361
- N(2)- N(lt- N(3t- c(1)
c(21- c(2)- c(3)- c(4t- c(4)- c(4)- c(4À)- c (4À)- c(4Àl- c (48)- c(48)- c(,[B)- c(4cl- c (4c)- c(4c)- c(2Ì)- c(2r)- c(221- c(231- c(23)- cl24'l- c (2s)- c(2s)- cl26l- cl21l- cl2ll- c (28)
c(31)c(3t)
- c(32)- c(33)- c (31)- c(14)- c(3s)- c (351
- c(36)
- ct2lc(I)
- c(4)o(I)N(2)
- c(r)- c(2)- N(3t- c(4À)- c(4À)
c(4)- H (41)- H(4r)- c(4)- H(44)- H(44)- c(4)- n(47)- H(41)- o(2t)- N(2)- c(21)- cl22't- c(24)- c(231- c(24)- c(26)- c(25)- cl22l- c(25)- o(28)
c(l)- c(12)- c (31)- c(32)
c(34tc(ll)
- c(34)c{16)c(lU
l2l-7(3tì26.r(4trls.B(3r)rl9 8(3)D.1.5(3)126.8(3)rr8.5(r-r)r09.-r(3)rrr.3(s)1r0-r(511r2.6(36)rr0.9(58t106.3(49)r04.0(32)1t4.2({7)t22-3(42t1r0.8(21)IO0.6 (34 )
119.0({01125.4(3)ro5-3 (3t108- r (3)1r6.1(41t24.3(2r)rl6-?(25)r20-8(4)118-9(2O)t25 .5 l22l121-3(3)I29-I(4)r30.9(3)1r8.6(3)118 9(4)120.5(28)120.0(5)tzo 2 l24lÌ1r.6(28t120 r (s))o9 2 (33)t25 . 6 125',t

Appendices 190
Appendix 5
Crystal sfructures of
(Z ) -N -phthaloyl- a, p-dehydrophenylalanine methyl ester (8 8)
I'aOla
028a Ol'aC75a 7a ClaC)-8a
N2aC22'
C24aC23a ah O2la C3a
C31a
C32a C36a
C33a C35a
C34a
bolb
a5b cz6bo28b orb
c28b 1b
Q4b N2bczb
cz3b cz2bc21b 021b
c3b
c31b
c32b c36b
c34b
c35b

Appendices 191
Bond distances (,Ä.) for
(Z)-N -phthaloyl-o, B-dehydrophenylalanine methyl ester (88)
atom
o (1a)
o (1' a)
o(1'a)
o(1'b)
o(1,b)
o (Ib)
O (21a)
o (21b)
o (2Bb)
o (2Ba)
N (2a)
N (2a)
N (2a)
N (2b)
N (2b)
N (2b)
c (1a)
C(1'a)
c(1,a)
c(1,a)
c(rb)
c (r, b)
c (t' b)
c ( r'b)
c (2a)
c (2b)
disLance
1.1?6 (6)
1.315 (5)
1.449 (?)
1.320 (5)
1.441(?)
1.191 (5)
1.187 (s)
1.188 (s)
1.188 (s)
1.201 (st
1.412 (5)
1.407 (6)
1.3?9 (6)
1.41? (5)
1.404 (s)
1.465(6)
1.07(4)
1.07 (6)
0.Bs(s)
r.47216',,
0.90(s)
1.05 (s)
0.92(6)
1.311(6)
1.318 (6)
di stance
1.443 (6)
0.99(4)
1.443 (6)
0.98 (3)
r.472('t',,
r - 412 (6\
1.371 (6)
1.3?4 (6)
r.3't2 (6',,
1.3s6 (6)
1.380 (B)
0.9s (s)
1.3?0(?)
0-91(4)
1.360 (7)
1.02 (41
1.349(8)
0.98 (4)
1.376(?)
r.13 (5)
1.375 (7)
0.95(4)
1.363 (6)
0.9s(4)
r.3?0 (6)
0.99 (4)
distance
1.466 (6)
1.46s(6)
r.379 (?)
1.377 (6)
1.3?s(6)
1.383 (6)
1.3?3 (B)
0.94 (s)
1.36s (7)
0.92 (3)
1.3s6 (7)
0.93(4)
1.352 (B)
1.06 (5)
1.341(?)
r-.01 (4)
1.3s6 (7)
0.94 (4)
1.361(?)
1.02 (s)
1.3?5 (?)
0.99(4)
r.0B(4)
0.9? (3)
atom
C (1a)
c (1a)
C (1'a)
c (1b)
c(1,b)
c(rb)
c (21a)
c (21b)
c(2Bb)
C (28a)
c l2a')
c (21a)
C (2Ba)
c (2b)
c (2lb)
c (28b)
c (2al
H (1' a)
H (1', b)
H(1'c)
c (2b)
H (1'd)
H (1'e)
H(1'f)
c(3a)
c (3b)
atom
c (3b)
c (3b)
c (3a)
C (3a)
c (21a)
c (2lb)
c(22b')
c(22b1
C (22a)
C (22a!
c (23a)
c (23a)
c (23b)
c (23b)
c (24b)
c (24b1
c (24a)
c (24a!
c (25a)
c (25a)
c (25b)
c (25b)
c (26b)
c (26b)
C (26a)
C (26a)
atom
c (3Ib)
H (3b)
C (3la)
H (3a)
C (22aI
c(22bt
c (23b)
c(2'tb')
C (23a)
C (2'l a)
c (24a!
H (23a)
c (24b',
H (23b)
c (25b)
H (24b)
C (25a )
H (24a',
C (26a)
H (25a)
c (26c'l-
H (25b)
c (z'tbl
H (26b)
C (27 al
H (26a)
atom
C (2'l al
c (27b)
C (31a)
C(31a)
c ( 3lb)
c ( 31b)
C (32a)
c (32a)
c (32b)
c (32b)
c (33b)
c (33b)
C (33a)
c (33a)
C (34a)
C (34a)
c ( 34b)
c ( 34b)
c (3sb)
c (3sb)
c (35a)
C (35a)
C (36a)
c(36b)
atom
c (2Ba)
c (2Bb)
c (32a)
c (36a)
c (32b)
c (36b)
c (33a)
H (32a)
c (33b)
H (32b)
c (34b)
H (33b)
C (34a)
H (33a)
c (35a)
H (34a)
c (35b)
H ( 34b)
c (36b)
H (35b)
C (36a)
H (35a)
H (36a)
H (36b)
1 5943

Appendices 192
Bond angles (deg.) for
(Z ) - N -phthalo yl-cr, p-dehydro phenylalanine methy I es ter ( 8 8 )
c(r.t otl'¿r ctl'ål
ctlbt o(l'br c(r'bt
c{2åt Nl2å) ct2låt
ct2àl Nl2¿r c{20àl
C(21.1 N(2.¡ ct28ål
c{2b} Nt2bt ct2tbl
ct2b) N(2b) c(28Þl
c(2rb) N(2br c(28b1
o(r.l c(l.l o(¡'.1
o{l.l C(ròl C{2.1
O(l'ål C{1.¡ Cl2¡l
olr'àl c(r'.t fl(r'àl
O(l'.1 C(1'¡r [(l'Þl
Oll'.1 Cl¡'.1 H(l'c)
trlr'àl C{l'.t BII'Þl
r(l'¡, c(l'¡t HtI'c'
f,(l,bt c(1,ât H(1,c)
o(r,bt c(rbt o{tbl
o(r'bt c(rb¡ c(2b)
o(lb) c(lbl c(2b)
o(1.b, c(r,bt H{1,d)
o(r'bt c(r'bt B(r'e,
o(r'b) c(r'b) x(r'rl
É(l'dl CII'bl H(¡'et
Í(l'dt c(l'bt fl(l'ft
H(Ì'êl C(1'bl H(l'f)
Ct2a.l C(21¡) H(21.1
c{22br c(2lbt c(2{bl
c{22bt c(2lbt r{21Þl
c(?rþt c(2lbr fl(23b)
ct2lbt c(2abt cl25bl
ct2lbt c(2rbt fl(2{bl
c t25bì c (2abt H (2{Þl
c(21.! C(2aÀl C(25å)
ct2làt c(2ràt ü{2.Àl
c(25à' C(2aâl H(2a¡l
c(2.!l ct25¡) c(26à)
Ct2a.l C(25.1 H(25¡)
c{26åì c(25àl ¡(25¡l
c(24bt c(25b, cl26bl
c{2{Þt ct25bt fl(25b)
ct25bt c(25b1 f(25b)
c(25b) c(26bt c{2?b)
c(25b, c(26bt fl(26b1
c(2?bl c(26bt H(26b1
C(25.1 ct26àl Ct2?å)
c(25¿) c(26ål 8(26à)
C(21ôì c(26Àl ff{26à)
C{22Àl C(21¿l Cl26ål
¡ræ ¡tú .tm
N(2al C{2.1 C(l¿l
N(2¡l c(2¡) c(l.l
cll¿l c(2.) c(lál
N{2bt c(2bt c(lbt
N(2b) C(2b) Ctfbl
c (rb, c (2bt c (3bl
ct2b) c(lb) c(3rÞ)
c(2bt c(3bt x(3bt
c(3lb) c{3b} f(fbl
C(2¡, c(là, c(3r.)
C(2.1 C(3.1 ff(3àt
c(fl¡) c(3.1 a(3.1
o121., C(21¡l N(2¡)
o(2r¡l c(21¡t c(22r)
N(2¡) C(21¡l Cl22àl
o(21b, c(21b) N(2b)
o(2lb) c(2rb) c(22b1
N(2bt c(2rb, ct22b)
c(2rbt c(22b) c(23bt
c(2lbt c(22b) c(27b)
c(23b) c(22bt c(2?Þl
c(21.t c(22à) c(23à)
c(21à) c(22àl c(21.1
c(23ål cl22¡) c(27öl
C(22.t C(23¡) C(2iât
c(22.1 cl23¡) f,(23¡l
c(22bt c(2lbt c(2lbì
c(26bt c(2rbt c(2rbl
o(28¡, c(28àl N(2âì
o(28âl c(28¡l c(21àl
N(2àl c(28äl C(21àl
o(28bt ct28bl N(2bl
o(28b) c(28Þ' C(2?br
N(2bl c(28bt c{2?bl
C(3.1 c(31.) c(32!)
c(3!l c(31â) cl36àl
c(32.1 c{3ràl cl36àl
c(3bt c(3rbt c(32b'
c(3b, ct3rbl c(36b1
c(32bt c(lrbt c(36b1
c{31.1 c(32àl C(33.1
c(31.1 c(32å) H(32.1
c(3!.1 c{32!} f,(32.1
c(3rbt c(32b1 c(33b)
c(3lbt c(32bt fl(32b)
c(33b, c(l2b) fl{32b,
c(l2b) c(33b1 c(3tbl
c(32bt c(3lbl ¡(33b1
c(lrb) c(33b) l(llb)
c(32.1 c(33a1 c{34.)
c(32ò) c(33å) ff133¿l
c (34à) c (33å) I (314,
c22 C21 C2S
tt5.a(5)
ll5_5({l
r2a.2{5t
r23.6 (5t
rrr 3(a)
120-9(.t
t22 -6 tll
1rÌ.0{ll
r2r.l t6t
r2l 3 (6)
r12-5(51
r0? (31
r03 {. I
r0{ {a)
Il2 (a)
r09 {5t
rl9 (6,
lzr.0 (5)
r11.9(5t
l2{.1(51
r03(r,
r0l (lt
rr? (5,
rrr (5t
12t (6)
97 (5t
129 (l)
t¡?.å{5t
r2l (ì
l¡8 tll
r20.9(61
r2r (2)
ll9 (2)
r22.6 (6)
rr9 (lt
r¡8 (31
r20.5 (6)
12? (31
rr2(ll
r21.7 (51
tr9 (3)
1r9 (31
lrl-a (5)
r22 (3)
r20 (3t
1r1.0(6t
r22 (3t
r21 (3t
r22 5 t5'
¡08.0(5)
r29.5(5t
¡2r -2 (5)
r20-9 (6t
It2.5 f5)
r2J.6t5t
r23.9t5t
tt2. I (al
r2{ - l (51
t23.2 tsl
r32.5(5)
11t (2)
rr4 (2)
13r.9(5)
rrr (2,
lla (2)
12a-?(51
130.r(6)
¡0a .5 (5,
12a - ? (5)
r29.9 (s)
r0s.a (a¡
r30.0(st
r08.9(5'
r20.9 (51
129.? (6'
109.5t5t
120. I (5'
116-6(61
lla (!t
t08 t (r)
r30.6 t5r
¡2{. t (51
L29 2t6l
r06.5(st
¡23- 3 (5r
r30. ? {s,
r06-o(4t
125.I (6'
rr7.2(5t
117.5 (51
L25.2 tsl
!r6.9(s)
rr6- E (5ì
l2t.lf5t
lr9 (3t
tt9 (3)
l2t.r(5t
r20 (2,
rr9 (2t
t20.4 (6)
l1l (3t
r22 13)
l¡9 a (6t
116 t3)
r2l(3)
C(ll.l
c(ls.t
c (l3bt
c (33b1
c (35b1
c{3abl
c (lrbl
c (36b)
c(3{¡l
c ( 31.1
c (36.t
c (3r.)
c ( 31¡,
c (35rt
c (3rbl
c(3lbl
c (35b)
c ( la.l
cflaàl
c(labl
c(3abt
ct3{b)
c ( l5bt
c(35b1
c (l5bt
c(35àt
cll5ål
c (35.)
c(36àl
c (l6rl
c (16à I
c{16b,
c (36b)
c (36b1
{lla¿l
H(la¿l
c(l5bl
ff ( t{bt
n ( labt
c(l6bl
H (lsbt
I (l5bt
c(16Àl
H(15àt
f ( 15.1
c(15¿l
fl(36àl
H (36¿l
cll5bl
H (36bt
H (36b)
tl9(2t
120 t2,
tl9 6(6r
ltltlt
l2l l3r
r20_2 {6t
t20 f l¡
rl9(lr
r20 2 t6¡
r2l o)
rt6(3t
t20-r(61
t20 12l
tl9(2t
l2r - 5 t6t
tl7 (21
l2t {2t
ct26.t c(2?àt c(28a,
ct¿2bt c{21Þr c(25b1
c(33ål c{34a1 C(15ål

Appendices 193
Appendix 6
Crystal structure of
( 2 S 3 R ) -3 -hydroxy-N-phthaloylphenylalanine meth yl ester (l l2a)
q32)
c(31)
o(21)c(21) q3)
c(?21 o(3)
q2)o(1)
c42n q28)c(l)o(28)
c(1)o(1)

Appendices 194
Bond distances 1Å¡ for
( 2 S 3 R ) -3 -hydroxy-N-ph thaloy lphenylal anine methyl ester (l l2a)
c (1)c (1')c (3)c (29')c(2tlc(2tH(1'B)H(21H (3)c(221c (271c (24tc (2s)c(26tc (27')c (32)H (32)H (33)H(34)H (3s)H (36)
ooooN
cccccccccccccccc
1)1')3)28t2',)
1)1')2t3)2tl22)
1.192(8)1.443 (8)1.419 (9)1.203 (9)1.411 (8)1.537 (10)0.970 (-)1.014(64)0.931 (s2)1.494 (10)1.378 (10)1.38s (10)1.380 (12)1.36?(11)1.388 (9)1.375 (10)0.970 (-)0 - 970 (-)0.970 (-)0.970 (-)0.9?0 (-)
c (1)H (3)c(zL',c (2')c (28)H (1'A)H(1'C)c (3)c (31)c (23',)H(23)H(24',)H(2slH(26',)c(28tc (36)c (33)c (34)c (3s)c (36)
(1')(3)(2L)(2')(2t(1')(1')(2',)
(3)(22',)qz3t(24t
oooNNccccccccccccccc
1.320 (8)0.949(6)1.207(8)1.448 (9)1.39s (9)0.970 (-)0.9?0 (-)1. s46 (9)1 - 500 (10)1.378 (10)0.9?0 (-)0.970 (-)0.970 (-)0.9?0 (-)1.496 (10)1.401 (10)1.406 (11)1.386 (14)1. 354 (14 )
1.363(11)
23t24',)2st26t31)32t33)34)3s)36)
2sl26273132333435
)
)
)
)
)
)
)
Bond angles (deg.) for
( 2 S 3 R ) -3 -hydroxy-N-phthaloylphenylalanine methyl es ter (l l2a)
c(1'lc(2r)c (28)c(2)H (r 'À)H(I'B)H (1,C)c (1)H (2tc (3)c(2)H (3)c (311N (2)c (221c l2', tH (23)c (24'c (251H (25)c (26)c(2?)c 126lc (28)c (211c(32)c(36)c (33)H (33)c(34'c (35)H (35)c (36)H (36)
o(1,)N(2)N(2)c (1)c (1,tc (1,)c(1,)c (2,c(21c(21c (3)c (3)c(3)c (2r)c(21)c(221c (23)c(23)c(24)c (25)c(2s)c (26tc 127',c(271c (28)c(31)c(31)c(32)c(33)c(33)c(34)c (35¡c (3s)c (36)
-c-c-c-o-o-H-H-N-c-c-o-c-c-o-N-c-c-H-c-c-H
-c-c-o-c
1
2I
1r6L2LllrL25109109109110118113lr2106111r23105r08T2L121L2L118119117t2I130LZ9120119120L20120119118118r.2 0
6 (6)2 t6t3 (6)r (7)4 (4)s (-)s (-)5 (5)9 (36)0 (6)? (6)4 (31)4 (6)9(7)9 (6)1(6)6 (s)I (s)3 (8)9 (s)1(s)0 (8)o (?)5(?)3 (7)0 (7)4 (?)2 (8)4 (6)4(-tt4 (10)6(7)7 (6)4 (5)
c (3)c(28)o(1')c(2)H (1'¡ B)H(r,c)H(1,C)H az','c(3)c (3)H (3)c(31)c(31)c (221c (23)c(211c l24tH(24tc (25)c(25)H (26tc(211c(28tN (2)c(211c (36tH(32)c (331c(341H(34)c (35)c (36)c (3s)H(36)
o(3)N (2)c(1)c(1)c (1,)c(1,1c(1,1c(2tc (2)c (2'tc (3)c (3)c (3)c (21)c(22tc(22tc(23)c (241c (24tc (25tc (26tc (261ct21tc (28)c (28tc (31tc(32)c(32tc (33)c(34)c(34)c (3s)c (36)c(36)
- H(3)- c(2)- o(1)- o (1')- o(1')- o (1,)- H(1'Bì- N(2)- N (2)- H(2)- o(3)- o(3)- H(3)- o(21)- c (2r)- c(23)- c(22t- c(23)- H (24 )
- cl24,- c (25)- Hl26t- cl22l- o(28)- N(2)- c(3)- c(3r)- H(32t- c(32)- cf ?1ì- H (34)- c(34)- c(3r)- c (35)
92.4 (5)r2?.0(6)r25. 5 (7 )
r09.3(6)r09.s(s)109-5(4)109.5(-)99.6 (36t
rr,4-5(6199.s(33)
r06.9 (32)114.4(6)104.2 (31)r30.2 (?)129.8(7tr22 .L ('t Irt6.6(8)1r9.4 (5'119.3(5)t22 .0 ('t Ir2r.4 (51
121.6 (5)r08.5(6)L24 .-t l-t I106.0 (61r20.4 (?)119-8(5)r20.0(6)rr9.3(9)r20.3 (7)r20.3(?)L22 -'t (L0lr19.0(9)t20.6(6)
r,)(1'À)(t'À)(2t(1)(1)(3)(2t(21(2Ll(21(21)122',(23 )(23t(24t(251(2s)(22t(26t(28 )
(31- c(32)- c (31)- c(32)- H rllì- c(33)- c(34)- H(3s)- c (31)

Appendices 195
Appendix 7
Crystal structure of
(2 S 3 S ) -3 -bromo-N-ph thaloyl-p - ninophenylalanine methy I es ter ( I 28 b)
o1B13
cl'C1
o2r01' c46
c4L
czl N2t
c22 e8 c4z c44c43o28 o44'a3
c27 N44o.44
e6
es
C3
c2
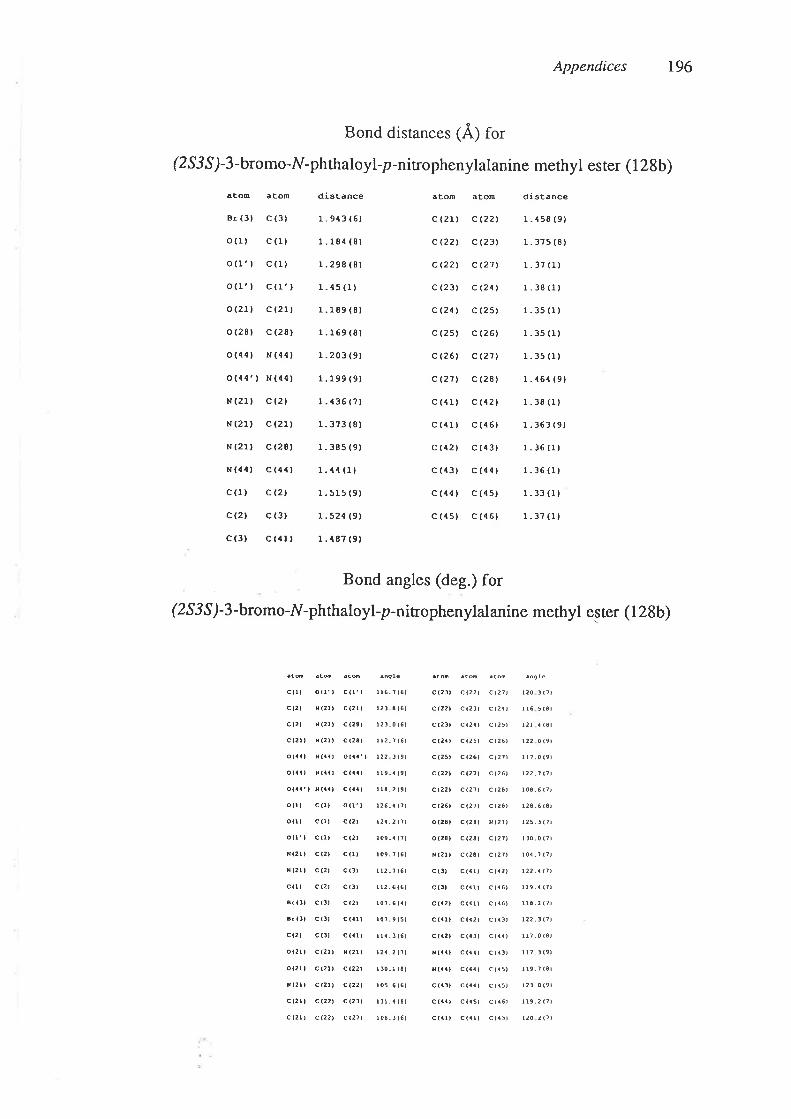
Appendices 196
Bond distances 1Å¡ for
( 2 s 3 s ) -3 -bro mo-N-phthaloyt-p -nirophenylalanine merhyl es ter ( I 28 b)
Bond angles (deg.) for
(2 S 3 S ) -3 -bro mo-N-phthatoyt-p -nitrophenylalanine merhyl es ter ( 1 28b)
at om
Br(31
o(r)
o(1')
o(1't
o(2r)
o(28t
o(4¡tl
o(4{, )
N (21)
N (21)
N (21)
N(¡l/tl
c(1)
c (21
c (3!
a Com
c (3)
c (rl
c (r)
c(1')
c (21)
c (281
N (44)
N(441
c (2)
c (2r,)
c (28)
c(4¿¡)
cl2',
c (3)
c({1)
distance
r _ 943 (6t
1,184(8)
1.298(8)
1.45(1)
1.189(81
1.169 (8t
1.203 (9t
1. r.99 (9)
1.436(?)
r.3?3(8)
r.385 (9)
1. ¡t4 (]. )
1.515 (9)
1. s24 (9)
1. ¡¡87 (91
a Lom
c (21t
c (221
c (22t
c (23)
c (24',
c (25)
c (26)
c l21l
c(41)
c (41)
c (421
c (43)
c(44)
c (45)
a Èom
c l22t
c (23)
c(211
c 124't
c (25)
c t26t
c tz't I
c(28)
c (421
c (46)
c(43)
c(44)
c (4s)
c (46)
distance
r-458(91
r..3?5(8)
r.37(l)
r.38(1)
r-35(r)
r.35(1)
1.35 (r )
1.464 (9)
r.38(rl
1.363 (9)
r.36(r)
r.36(11
r-33(rt
1.37(r)
c(U
c (2t
c (2t
c(2rt
o(aal
o({al
o(at't
o(U
o(u
ou'tN(2rt
N (2r'
c(lt
Br(31
B!(ll
c(2t
o(2u
ot21,
N(2tt
c(2lt
c(2r,
olt't
N(2tt
N(2tt
x(21)
N(aal
N(til
N(ail
c(r)
c(1t
c(1)
c (21
c 12l
c(2t
ct3t
c (lt
cot
c(2U
c(2ll
c(2rl
cl22l
c(22)
c(r'I
ct2rl
c(20t
c(2El
otta'I
c(aal
c(aat
o(¡')
c(2t
c(2t
ctU
c(lt
c(lt
c(2t
c(.u
c{(rl
f(2rl
ct22l
c a22l
c(23'
ct21l
¡t6.?(61
r2l-r(51
r21.0(61
rr2-t{51
r2z I {9t
tl9-a {9,
ltl-2{9,
126-a (71
¡2a.2(?l
109-a t?)
r09.7 (61
It2-? (6)
rr2-5(6)
r0?. 6 (a )
¡0r-9(5t
rra,I (6t
l2a-2t?l
rl0-1 (8t
r05- 6 (6t
rlr,a(8t
r06.1(6t
c(2lt
ct22l
ct23t
c(2at
c t25l
ct22l
ct22l
c (26)
o(28t
o(2St
N(2¡)
c(3t
c(!t
c(a2r
c(.rt
c (42)
x({ll
x({Í
c(a3t
c(aat
c(alt
c(22t
c(2lt
c(2al
ct25t
c (261
c{2?l
c(271
c(2ll
c(28t
c(281
c (28,
c(at)
c(a¡l
c(a¡t
c({21
c({lt
c(att
c(aal
c(aal
c(.5t
c(a6l
c{¿?t
c(2al
c(251
c(261
cl21l
c(261
c{2El
c(20t
N(2lt
c(2lt
c(2rl
c({2,
c(a5t
c(a6,
c(r3t
c(ial
ctall
c{{51
c(a5l
c(a6t
cta5l
120 I tìt
rr6 Stst
I2l a (€t
r22 0 (91
llì.0t9t
122.7 t1l
106.6 (71
r20-6 f 8t
125.I trt
tl0.0t?r
104.1{rl
122-t (?¡
t¡9.a (lt
t1,.2 f ?t
122. I {rt
tr?.0ttt
ll?-lt9l
rr9_rl8r
r2l_ 0 (9t
119.2 (ll
r20 2 (l)

Appendices l9l
Appendix I
Crystal structure of
(Z ) - N -phthaloyl-cr, p-meth anophenylalanine meth yl es ter ( 1 43 )
c(13)c(14)
c(t5)
c(16)
N(10)
c(3)
c(l)
C(12) o(u)
c(1
c(l8)CQI)
c(17)
o(30)
o(18) c(2)
o(31) c(22)c(30)
c(31)

Appendices 198
Bond distances (Å) for
( Z ) - N -phth aloyl-o, p- methanophenylalanine methyl ester ( I 43 )
c (11)c (30)c (30)c (11)c(21c (30)c (3)H (31)c (12)c (17)c (14)c (1s)c (16)c (1?)c (23)tl(221H (23)H (24l-H (2s)H (26)H (312)
o(11)o (30)o (31)N (10)c (1)c (1)c (21c (3)c (11)c (r2',c (13)c(14)c (ls)c (16)c (22'c(221c(23)c (241c (2s)c (26)c (31)
1.200(B)1.347 (10)1.186(9)1.39s(9)1.s40 (11)1.476 (10)r - 411 (rz',,r .o12 ('l9l1.491 (9)1.393 (10)1.394 (12)1.373 (13)L.362 (Lzl1.3?9 (10)1.39s(-)1 .203 (70)0.965 (72)0.879 (7 9)1.133 (72)1.111 (73)1.067 (82)
c (18)c (31)c (1)c(18)c (3)H(21c(2LlH (32)c(13)H (13)H(14)H (1s)H (16)c (18)c (21)c (241c (2s)c (26',,
c (21)H (311)H (313)
o (18)o (30)N (10)N (10)c (1)c (2')c (2'tc(3)c (12)c (13)c (14)c (1s)c (16)c (17)c(221c (23)c (241c (2s)c (26'c (31)c (31)
1.194 (B)1.460(14)1.431 (9)1.410 (9)1.506(11)0.936(79)1.483 (10)0. 942 (86)1. 382 (11 )
0.9s9(?6)0-865(7?)o. 89? (82 )
1.002 (75)1.503 (11 )
1.39s (-)1.3es (-)1.395 (-)1.39s (-)1.3es (-)0.?41 (91)0. 863 (82 )
Bond angles (deg.) for
( Z ) -N -phthaloy l-cr, p-meth anophenylalanine methyl es ter ( I 43 )
c (3r)c (r6.tc (2tc(3tc (3olH(2)c (3)c (21tc (21H (311H (321
N (r0lc (r2tc (r?)H (r3)c (1llc (rslH (rstc(16tc (r7)c(r5lc (r8)c (r7lc (2rtA t22lH (23'c (25'H (2{)H (2s)c (21)H (26)c(26)o(31)c(t)H (312)H (313)rì (rr3)
o (30,N (r0tc(11c(llc(r)c(21c(2)c(21c (3tc (31c (3¡c(rltc(rr)c(r2lc(13)c(131c(1{)c(151c (rslc (r6lc(r7lc(r7tc(r8)cl22lc(221c (231c(2{)c(2{}c (251c(261c (25)c(2r)c (30tc (30)c(31'c(31'c(3r)
- c (30)- c(r)- N (r0l- c(21- c(2t- c(rt- H(2t- H(2)- c(11- cl2l- c(2)- o(rr)- N (r0l- c(lr)- c (r2t- H (r3l- c(r3l- c(lrl- rcsl- c(r5l- c(121- c(161- o(l8t- c (23)- c(2rl- c1221- c(231 .
- c(2sl- c(24)- c(2st- c (2r)- c(2)- o(30)- o(3r)- o(301- o(30t- H (3121
t15.0(8t125.2(6!rr8. o (l)58. 0 (st
rrs.l(71105. I (53trr3.{ (51t116.4 (s2)
62 .2 16lr11.6 (a3,11{.1 (s2)r25.3 (?)ro6- 9 ('r)107. 9 (l)rrs.7({91r21 .1 l19lr20.9 (91ro9.o(5?l127.8(5?lrt6.9 (9)12r. s (81130. { (8t129.i(81120. o (-l1rr.4 (331112.8({2)r20.0 (-)118.2 (¡¡6)r2o_2(r6tr2o.o(-l1r8-7(36)t21.7 ({}12{-o(9)125.1(9)l0't - 4 (¡t5)r00-3(55)129-6(86)
c (rrlc (r8tc(3)c (3olc (3olc (31c(2r)c(2rtH (3rlH (321H (321c (r2lc (r3lc (r7lc(r{lH(r{lc (rs¡c(r6lH (r6tc(r7lc(101N (lotc (171H (221c(2{}H (231H(2rtc(261H (2slH(26'cl22lc(26)c(r)H (31r)H(3t2tH (3rl)
(r0l(ro'(1)(r)(Ðt2t121t2t(3'(3t(3t(tl)(12t(L2t( r3l(1t¡(rlt(1s)( 161(16'(1?l(r8t(t8t(22t(23 I(23t(21 I(2s I(2st126t(2rl(21)(301(31t
- c(rt- c(rrl- N (10)- N (ro)- c(31- c(rt- c(1t- c(3t- c(11- c(rl- n(3r'- o(1rl- c(rrl- c(r3l- c(l2)- c(r3t- H(r.l- c(rrl- c (rsl- H(r6t- c(r2l- o(r8l- N (r0l- c(231- c122,- c(2{)- c(23)- c(2{l- c(261- c(2s)- ct2t- c(221- o (301
- o (lot- H(3rrt- H(3rrl
L22 -2 16lrrr.2(6trl'r.3(71tr7.5(71r17.s(7159.8(61
122.{ (5}125. ? (8)tr5.3({{}lIl.9 (52t12r.r (66tr27. 8 (81
131. r (8)12r.o(7)1r6.9(9)rr2.5 (621126.6(621t22.1 l9l126. r ({8}116. 4 ( ¡l8lr07.9 (ll12{. s (8tr05.8 (?lr28.3(3{tr2o.0 (-tr25.0({21121. ? (l6lr 20. 0 (-lrr9.8(36)r21.2(36)rr8.2 ({}i20.0 (-)rt0.9(81r0l. 9 ( rtt02-9(45)r0-r-{(65}
-ll-l¡-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c-c- c(3r)- c(31)




















