
Multilayer hydrogel coatings to combine hemocompatibility and antimicrobial activity
Upload
independentCategory
view
2download
0
RESEARCH PAPER
Stabilizer specific interaction of gold nanoparticleswith a thermosensitive polymer hydrogel
A. Murugadoss Æ Aslam Khan ÆArun Chattopadhyay
Received: 2 February 2009 / Accepted: 29 May 2009 / Published online: 27 June 2009
� Springer Science+Business Media B.V. 2009
Abstract We report the experimental results on
temperature-dependent studies of interactions between
a novel biocompatible thermosensitive polymer hydro-
gel and different stabilizing agent capped gold nano-
particles (Au NPs) with particle size ranging from 5 to
20 nm. Stabilizing agents such as thioglycolic acid,
tryptophan, and phenylalanine have been used as
capping agents for Au NPs. The poly-N-isopropyl
acrylamide-co-acrylic acid (pNIPAm-AAc) with
3.0 ± 0.7 lm in size was synthesized by radical
polymerization of a selected mixture of N-isopropyl
acrylamide (NIPAm), methylene-bis-acrylamide and
acrylic acid (AAc). The capped Au NPs were mixed
with a solution of pNIPAm-AAc hydrogel. The
temperature-dependent properties of the mixture were
studied by UV–vis spectroscopy, dynamic light
scattering based particle size analysis, and transmis-
sion electron microscopy (TEM). The observations
indicated change in the lower critical solution
temperature (LCST) depending on the nature of the
stabilizer, with hydrophobic ones lowering the value
while hydrophilic stabilizers increasing the same.
Also, the optical absorption due to Au NPs, when
stabilized with hydrophobic groups, reduced signifi-
cantly at above LCST along with significant blue shift
of wavelength maximum.
Keywords Gold � Nanoparticles �Hydrogel � Thermosensitive � Spectroscopy �Drug delivery
Introduction
Stimuli responsive materials such as hydrogels are
important as delivery vehicles for futuristic nanoscale
drugs. The changes in size, shape, and solubility in a
solvent in response to changes in stimuli such as
temperature, pH, ionic, or other chemical and biolog-
ical species make hydrogels ideal for drug delivery and
controlled release of bioactive molecules (Juodkazis
et al. 2000; Chen et al. 2007; Langer and Tirrell 2004).
The advent of nanoscale materials in therapeutics and
medical diagnostics has brought about newer chal-
lenges in conjugating hydrogels with nanomaterials for
targeted delivery (Retama et al. 2007; Robinson et al.
2000; Francois et al. 2007). In this regard, nanoparti-
cles (NPs) could be chemically bonded to the three-
dimensional structure of the gel and thus the hybrid gel
Electronic supplementary material The online version ofthis article (doi:10.1007/s11051-009-9668-0) containssupplementary material, which is available to authorized users.
A. Murugadoss � A. Chattopadhyay (&)
Department of Chemistry, Indian Institute of Technology
Guwahati, Guwahati 781039, India
e-mail: [email protected]
A. Khan � A. Chattopadhyay
Centre for Nanotechnology, Indian Institute
of Technology Guwahati, Guwahati 781039, India
123
J Nanopart Res (2010) 12:1331–1348
DOI 10.1007/s11051-009-9668-0

would act as both the delivery vehicle as well as the
drug itself. On the other hand, chemically or biolog-
ically functionalized NPs could also be encapsulated to
hydrogels that are held by electrostatic or van der
Waals forces and the functionalized NPs could thus be
released in response to a stimulus. Understanding the
functioning of both kinds of hybrids is important for the
development of better drugs and delivery systems.
An important thermosensitive polymer that has
been extensively studied is poly (N-isopropyl acryl-
amide) (pNIPAm), the lower critical solution temper-
ature (LCST) of which can be changed from 32 �C up
to 60 �C by various degrees of copolymerization with
acrylic acid (AAc) (Das et al. 2007; Jones and Lyon
2003; Berndt et al. 2005; Nayak and Lyon 2005). In
addition to tunable LCST and large volume change
accompanying transition, AAc copolymerized in the
backbone of pNIPAm offers a robust architecture
which responds very well to environmental changes in
temperature, pH, ionic strengths, and solvents (Berndt
et al. 2005). These advantages have consequently
contributed to the significant scientific and techno-
logical literature related to pNIPAm-AAc copolymers
as environmentally sensitive delivery system (Heras
Alarcon et al. 2005; Gan and Lyon 2001).
On the other hand, recent developments in the
controlled syntheses of inorganic NPs such as Au
NPs and quantum dots (Qdots) bring additional
potential for their use as drugs such as in hyperther-
mia treatment or in imaging at the cellular level (Jain
and Stroh 2004; Storh et al. 2005). However, these
materials also need appropriate delivery vehicle in
order for their controlled release and targeted deliv-
ery. In this respect, pNIPAm has been proposed as the
stimuli responsive carrier of the NPs (Lu et al. 2006).
There have been some recent reports on the temper-
ature-dependent study of Au functionalized pNIPAm
using the surface plasmon resonance (SPR) of Au
NPs as the probe (Shan and Tenhu 2007; Yusa et al.
2007; Karg et al. 2007). However, these studies were
limited to chemical attachment of the NPs on the
polymer by appropriately modifying the backbone of
the polymer. On the other hand, there are no reports
of temperature-dependent interactions between func-
tionalized Au NPs and pNIPAm polymer or its
derivative. This is important as understanding the
interaction between the functionalized NPs and the
carrier polymer, and its temperature sensitivity may
play pivotal roles in futuristic drug delivery and
chemical sensors (Kim et al. 2006). Also, if a simple
technique such as UV–vis spectroscopy could indi-
cate the interaction between stabilized NPs and the
hydrogel that would broaden the scope of such
studies in future. This is all more important against
the backdrop of substantial literature reports on
interactions of organic molecules and ions with
hydrogels and accompanying volume phase transi-
tions (Zhang et al. 2002; Matsukaa and Ando 1996).
Herein, we report the study of temperature-
dependent interactions of pNIPAm-AAc with Au
NPs, which were functionalized by different stabiliz-
ing agents, to study their effect on the volume phase
transitions of the hydrogel. The principal aim of the
studies has been to understand the stabilizer specific
interactions between the polymer and stabilized Au
NPs and their temperature dependence. The temper-
ature-dependent properties of the polymer alone,
polymer functionalized Au NPs, and tryptophan,
thioglycolic acid, or phenylalanine-capped Au NPs
dispersed in the polymer medium were investigated.
The results indicate that the SPR of the stabilized Au
NPs could act as a sensitive indicator for the change
in the environment of such systems. In addition, the
absorption characteristics of the NPs could them-
selves act as clear indicators of LCST. Essentially,
the results demonstrate that functionalized Au NPs
could be used for probing LCST transition of the
polymer. In addition, knowledge about the details of
immediate environment of the polymer, before and
after the LCST transition could possibly be obtained.
Experimental section
Materials
N-isopropyl acrylamide (NIPAm, 97% purchased
from Sigma-Aldrich Chemical Co.), acrylic acid
(AAc, 99% from Sigma-Aldrich), N, N0-methylene-
bis-acrylamide (MBA, 99% from Sigma-Aldrich),
potassium perdisulphate (KPS, 98% from Merck),
hydrogen tetrachloroaurate (HAuCl4, 17% Au in HCl
from Sigma-Aldrich), L-tryptophan (99% from
Sigma-Aldrich), thioglycolic acid (98% from
Sigma-Aldrich), L-phenylalanine (95% from Merck)
and sodium borohydride (95%, from Merck) were
used as received without further purification. Milli-Q
grade water was used in all the experiments.
1332 J Nanopart Res (2010) 12:1331–1348
123

Methods
Synthesis of PNIPAm-AAc hydrogel
Typically, 50 mL of aqueous NIPAm (987 mg) was
taken in a 100 mL three-necked round-bottom flask.
To this solution 63.0 lL of acrylic acid (0.126% V/V)
and 300.0 lL (4.9% W/V) of methylene-bis-acryl-
amide (molar ratio of NIPAm: AAc = 93:7) were
added under magnetic stirring. These mixtures were
kept in an oil bath at 70 �C and were carefully
degassed by bubbling nitrogen for 30 min. When the
reaction mixtures were turned into homogeneous
solution, 50.0 lL (5% W/V) of potassium perdisul-
phate was introduced to this solution under the same
condition and the reaction was allowed to proceed for
4 h. In the end, the product was centrifuged at
20,000 rpm to remove the unreacted monomers and
excess potassium per disulphate followed by washing
with Milli-Q water at above LCST. Interestingly, we
found that at above LCST the pNIPAm-AAc had
settled as a gel. This gel again was kept at room
temperature which got converted into milky white
solution of hydrogel, and this solution was put into a
dialysis tube (Hi-media dialysis bag 12,000 cut off)
and was subjected to dialysis against distilled water
for 2/3 days. The medium was replaced every 1 h for
the first 6 h. The resultant solution was freeze-dried
for storage.
Synthesis of thioglycolic acid-capped Au NPs
In a typical experiment, 50 mL aqueous solution
containing 15.0 mg of trisodium citrate was kept in
ice-water bath under constant magnetic stirring. To
this solution, 600.0 lL of 17.3 mM HAuCl4 solution
was added. This was followed by addition of
1.2 mL of 20.0 mM freshly prepared sodium boro-
hydride (NaBH4) solution. The resultant solution
was dark red in color, indicating formation of Au
NPs. This solution was kept under the same
condition for 2 h.
In a 5 mL beaker containing 14.0 mM of sodium
hydroxide solution, 120.0 lL of thioglycolic acid was
added. The solution was then slowly poured into
citric acid-capped Au NP solution prepared as above.
The mixture was then stirred for 1 h after which
purple colored precipitation appeared. The precipitate
along with the solution was subjected to centrifuga-
tion (1000 rpm at 4 �C). The pellet was washed with
copious amount of ethanol to remove the excess of
uncapped thioglycolic acid, redispersed in 5 mL
water, and was used as the stock solution for further
characterization studies.
Tryptophan- and phenylalanine-capped Au NPs
One hundred micro liters aqueous solution containing
0.1 mM of HAuCl4 was mixed with 0.01 g of solid
NaBH4. This mixture was kept under constant
magnetic stirring for 2 h at room temperature. The
color of the solution was ruby red, indicating the
formation of Au NPs. This was followed by addition
of 10 mL aqueous solution containing 1.0 mM of
L-tryptophan to 90 mL of the above Au NP solution.
The mixture was stirred for 2 h, after which the
solution color turned red indicating possible forma-
tion of tryptophan-capped Au NPs (Selvakannan
et al. 2004). Finally, the solution was subjected to
ultra-speed centrifugation (70,000 rpm at 15 �C), and
the resulting pellet was washed with copious amount
of water to remove the excess, uncoordinated tryp-
tophan. The pellet was redispersed in 5 mL water; the
resulting solution was stable for weeks. In the
preparation of phenylalanine-capped Au NPs, the
same procedure was followed, taking 10 mL of
9.7 mM of L-phenylalanine solution instead of tryp-
tophan solution, which was added into 90 mL ruby
red Au NP solution. All other conditions were the
same as above.
Synthesis of pNIPAm-AAc stabilized Au NPs
composite
Three hundred micro liters of 17.3 mM HAuCl4solution was added to 50 mL of 0.04% (W/V)
polymer hydrogel solution. To this solution, 600 lL
of 20 mM NaBH4 solution was added. This mixture
was then allowed for 3 h stirring at room tempera-
ture. The solution was then centrifuged at a high
speed (20,000 rpm at 37 �C) and lyophilized to
obtain powdered of composite Au NP-hydrogel
products. Finally, 10 mg of composite Au NP-
hydrogel powder was dissolved in 50 mL water,
and subsequent solution was used for further charac-
terization studies.
J Nanopart Res (2010) 12:1331–1348 1333
123

Preparation of different stabilizing agent capped Au
NP-hydrogel composite
Typically, in a 100 mL beaker containing 50 mL of
0.02% polymer hydrogel solution, 2 mL of a partic-
ular stabilizing agent capped Au NP solution as
prepared above was added. The resulting solution
was stirred for 2 h which was used for further
experiments. Finally, the pH of all the solutions was
measured. The pH of the polymer hydrogel solution
was measured to be 5.60; bare Au NP-hydrogel
composite solution to be 5.40, tryptophan-capped Au
NP composite solution to be 5.22, thioglycolic acid-
capped Au NP composite solution to be 4.69, and that
for phenylalanine-capped Au NP composite hydrogel
solution was measured to be 5.27.
Analytical measurements
UV–vis spectra of the polymer hydrogel, composites
of polymer-Au NPs hydrogel, and different stabiliz-
ing agent capped Au NPs with polymer hydrogel
samples were recorded in the range of 250–800 nm,
using a Perkin-Elmer Lambda-45 spectrophotometer
with temperature controlled sample compartment.
The phase transition for polymer hydrogel, compos-
ites of polymer-Au NP-hydrogel, and different sta-
bilizing agent capped Au NPs with polymer hydrogel
were examined in the temperature range of 20–45 �C
with a heating rate of 0.1 �C/min. FTIR spectra of
the samples were recorded using a Perkin-Elmer
Spectrum one spectrometer. The different stabilizing
agent capped Au NPs were dried as a powder and
mixed with KBr, the resulting mixtures were made as
a pellet for FTIR measurements. The 1H-NMR
spectra of the polymer hydrogel in DMSO-d6
(20 mg/mL) at 20 �C were recorded using a Varian
400 MHz spectrometer, with TMS as the internal
reference. The molar mass and polydispersity of the
polymer hydrogel were determined by gel perme-
ation chromatography (GPC) using Waters GPC
2410 equipment fitted with a refractive index detec-
tor. The HPLC grade of tetrahydrofuran was used as
an eluent, and calibrations were carried out with
polystyrene standard. The dynamic light scattering
(DLS) measurements of the polymer hydrogel,
composites of polymer-Au NPs hydrogel, and differ-
ent stabilizing agent capped Au NPs with polymer
hydrogel samples were performed with an LB-550
(HORIBA) DLS measurement system. The volume
phase transition of the above liquid samples was
examined with increasing temperature at heating rate
of 0.1 �C/min. The average values of the particle
sizes obtained from DLS measurements were calcu-
lated in the following way. When the distribution was
uniform and there was only one particle size
distribution, then the values reported by the instru-
ment were reported as it was. On the other hand,
when two type particle size distributions were
obtained, then higher and lower particle size distri-
butions were individually calculated from the data
obtained from the instrument and then reported.
Transmission electron microscopy (TEM) of the
particles was performed using a JEOL-2100 equip-
ment operating at a maximum acceleration voltage of
200 kV. The TEM samples were prepared by placing
a drop of liquid sample on carbon-coated copper grid
(300 mesh) followed by evaporation of the solvent at
room temperature. The grid was then observed under
the TEM.
Results and discussion
The polymer pNIPAm undergoes a coli-to-globule
transition in aqueous solution at an LCST of ca.
32 �C. It is known that LCST can be altered by co-
polymerization with different monomers containing
hydrophilic or hydrophobic groups. The choice of
AAc monomer for co-polymerization here was due
to its easy solubility in water, bioadhesivenesss, and
biocompatibility (Kim et al. 2007). The synthesis of
pNIPAm-AAc was achieved from the free radical
precipitation polymerization reaction (Khan 2008).
The copolymerization of pNIPAm with AAc has
been confirmed by 1H-NMR using DMSO as the
solvent (refer to supplementary information, SI).
The molecular weight of the polymer and their
polydispersity index (PI) were measured by gel
permeation chromatography. The ratio of weight
averaged and number averaged molecular weight
was close to unity (refer to SI), meaning that
molecular size distribution in the hydrogel was
narrow.
Analytical and other studies were pursued by
dissolving pNIPAm-AAc as well as pNIPAm-AAc
stabilized Au NPs in water at fixed compositions. The
polymer only solution was colorless and transparent
1334 J Nanopart Res (2010) 12:1331–1348
123
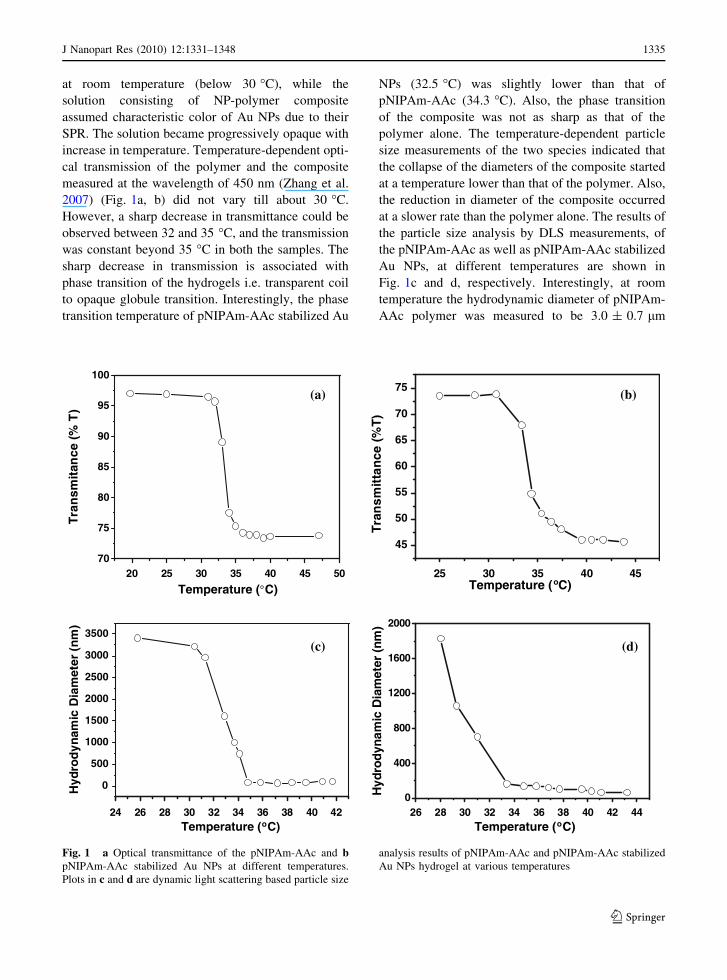
at room temperature (below 30 �C), while the
solution consisting of NP-polymer composite
assumed characteristic color of Au NPs due to their
SPR. The solution became progressively opaque with
increase in temperature. Temperature-dependent opti-
cal transmission of the polymer and the composite
measured at the wavelength of 450 nm (Zhang et al.
2007) (Fig. 1a, b) did not vary till about 30 �C.
However, a sharp decrease in transmittance could be
observed between 32 and 35 �C, and the transmission
was constant beyond 35 �C in both the samples. The
sharp decrease in transmission is associated with
phase transition of the hydrogels i.e. transparent coil
to opaque globule transition. Interestingly, the phase
transition temperature of pNIPAm-AAc stabilized Au
NPs (32.5 �C) was slightly lower than that of
pNIPAm-AAc (34.3 �C). Also, the phase transition
of the composite was not as sharp as that of the
polymer alone. The temperature-dependent particle
size measurements of the two species indicated that
the collapse of the diameters of the composite started
at a temperature lower than that of the polymer. Also,
the reduction in diameter of the composite occurred
at a slower rate than the polymer alone. The results of
the particle size analysis by DLS measurements, of
the pNIPAm-AAc as well as pNIPAm-AAc stabilized
Au NPs, at different temperatures are shown in
Fig. 1c and d, respectively. Interestingly, at room
temperature the hydrodynamic diameter of pNIPAm-
AAc polymer was measured to be 3.0 ± 0.7 lm
20 25 30 35 40 45 5070
75
80
85
90
95
100
)T
%( ecnati
msn ar T
Temperature (°C)25 30 35 40 45
45
50
55
60
65
70
75)
T%( ec
nattims
narT
Temperature (oC)
26 28 30 32 34 36 38 40 42 440
400
800
1200
1600
2000)mn( rete
maiD c i
m anydordy
H
Temperature (oC)24 26 28 30 32 34 36 38 40 42
0
500
1000
1500
2000
2500
3000
3500
)m
n( retemai
D cima
nyd
ordy
H
Temperature (oC)
(b) (a)
(d)(c)
Fig. 1 a Optical transmittance of the pNIPAm-AAc and bpNIPAm-AAc stabilized Au NPs at different temperatures.
Plots in c and d are dynamic light scattering based particle size
analysis results of pNIPAm-AAc and pNIPAm-AAc stabilized
Au NPs hydrogel at various temperatures
J Nanopart Res (2010) 12:1331–1348 1335
123

(at 28 �C), while that of pNIPAm-AAc stabilized Au
NPs was found to be 1.8 ± 0.1 lm (at 28 �C). The
decrease of the hydrodynamic diameter of the
polymer stabilized Au NPs compared to that of
polymer could be ascribed to the reduction of the
electrostatic repulsion among the free –COO- moiety
(Lin and Chiu 2006) in the polymer hydrogel in the
presence of Au NPs. The slow and early change in
volume phase transition of the composite in compar-
ison to the polymer is consistent with the results of
optical transmission studies. The results of the DLS
studies also indicated that Au NPs connected to the
polymer lead to change in collapse behavior of the
polymer and also hasten the volume phase transition
in the composite. It is plausible that the electrostatic
interaction between the carboxylate moieties of the
polymer and Au NPs leads to a structure that favors
coil to globule transition at a temperature lower than
that of the polymer. UV–vis spectra of the pNIPAm-
AAc stabilized Au NPs at different temperatures,
shown in Fig. 2a and b (after normalization of
Fig. 2a), consist of a single peak with absorption
maximum at ca. 530 nm. As is clear from the figure,
kmax did not vary with temperature. At above the
transition temperature, the intensity of the absorption
(after due consideration of the change in background)
decreased, which is possibly due to change in the
immediate environment (and hence refractive index
of the medium) (Morones and Frey 2007) of the Au
NPs upon collapse of the original structure. However,
the lack of shift in the absorption maximum probably
indicates that the interaction between the carboxylate
moieties and Au NPs did not change significantly
upon transition to globule form. It is also interesting
to note that upon increase in temperature and
resulting volume phase transition of the composite
there was no agglomeration of the NPs as is clear
from the UV–vis spectra. This implies that the
interaction between Au NPs and the carboxylate
moieties of the polymer is sufficiently strong so as to
prevent agglomeration due to increase in temperature.
It is important to mention here that the temperature-
dependent optical as well as hydrodynamic behavior
of the composite was completely reversible, indicat-
ing that the composite could be a good model for the
study of the behavior of Au NPs in thermosensitive
hydrogels.
Further, in order to understand the effect of
swelling and de-swelling of the polymer hydrogel
on the structural aspects of Au NPs, TEM measure-
ments of the polymer stabilized Au NPs were carried
out. In this regard, two samples were prepared; one
from a solution that was evaporated at below the phase
transition temperature (at 24.3 �C), while the other
sample was evaporated at above the phase transition
temperature (at 40 �C). Figure 3a–c shows different
views of the TEM images of the polymer stabilized
Au NPs with the sample prepared at below the LCST.
The composite consisted of blobs of particles of about
200 nm in diameters interconnected with each other.
The Au NPs (darker spots) were of 10–20 nm in sizes
and were separated well from each other in the
composite. The particles present in between the blobs
evidenced the presence of polymer in the intervening
region, too. Overall, small and individual NPs
connected to the polymer were present in the
composite. On the other hand, the sample prepared
at above the LCST also consisted of individual NPs
without the presence of any significant agglomeration
(Fig. 3d–f). However, the density of the particles (Au
NPs) appears to be higher in comparison to the sample
prepared at below LCST. This is because at above the
phase transition temperature, the diameters of the
polymer particles and thereby volumes are signifi-
cantly reduced. This increases the apparent density of
the particles. The overall structure of the polymer
350 400 450 500 550 600 650 700 750 350 400 450 500 550 600 650 700 750
0.1
0.2
0.3
0.4 45oC
).u.a( sb
A
Wavelength (nm)
25oC
0.2
0.4
0.6
0.8
1.0
1.2
1.4
).u.a( s
bA
Wavelength (nm)
(a) (b)Fig. 2 a The UV–visible
absorption spectra of
pNIPAm-AAc stabilized Au
NPs at different
temperatures and bnormalized absorption
spectra of (a)
1336 J Nanopart Res (2010) 12:1331–1348
123

seems to be similar, with a slight decrease in the
diameter of the blobs at higher temperature. The
increases in density of the NPs on the blobs of the
composite at higher temperature supports that the
structure indeed collapsed into more compact globule
form at the higher temperature. It may also be
mentioned that at higher temperatures the dispersions
of NPs attached to the polymer might become
inhomogeneous (accompanying volume phase transi-
tion), and hence the particles appear to be close to
each other at certain locations (Fig. 3).
Further probe of the effect of additional agents on
the volume phase transition was pursued using
thioglycolic acid; tryptophan or phenylalanine
Fig. 3 The transmission
electron microscopy (TEM)
images of pNIPAm-AAc
stabilized Au NPs at 25 �C
(a), while b and c are the
expanded views of (a). The
plot in d is TEM picture of
the same at 40 �C, while eand f are the expanded
views of (d)
J Nanopart Res (2010) 12:1331–1348 1337
123

stabilized Au NPs. Thioglycolic acid-capped Au NPs
were generated from citrate stabilized Au NPs using
the legend exchange method. The typical sizes of the
Au NPs generated were 5.0 ± 1.5 nm. The interac-
tion of the –SH group of the thioglycolic acid with
Au NPs resulted in the loss of a peak due to –SH
stretching frequency at 2541 cm-1, which was con-
firmed by FTIR spectroscopy (Lin et al. 2005) (refer
to SI, Figure S2). The acid functionalized Au NPs
were then mixed with pNIPAm-AAc in aqueous
solution. We shall henceforth refer to the composite
of thioglycolic acid-capped Au NPs and pNIPAm-
AAc as pPA-thio-Au for simplicity. The temperature-
dependent UV–vis spectra of pPA-thio-Au are shown
in Fig. 4a. The characteristic SPR band of Au NPs is
present in all the spectra (at all temperatures
recorded). As is clear from the spectra, the spectra
changed little from 25 �C to ca. 34 �C. Also, the
absorption due the NPs did not change in this
temperature range. However, the background scatter-
ing increased significantly at above this temperature
(LCST) indicating phase transition to have occurred.
Interestingly, the absorption due to Au NP only
(obtained after appropriate background correction)
also changed significantly at above the LCST. The
normalized absorption values were obtained as fol-
lows. The absorption values at 350 nm for all
temperatures were first measured. That value was
made to be unity for all temperatures and all other
values were adjusted by dividing them with the value
of absorption at 350 nm for a particular temperature.
The absorption at the maximum wavelength was then
taken as the absorption. Sample normalized absorp-
tion versus wavelength plots is shown in Figure S3
(a) (refer to SI). This method of normalization is
based on the works of Tenhu and coworkers. A plot
of normalized Au NP absorbance at 529 nm (Fig. 4b)
indicated significant decrease in absorbance due to
Au NPs at above the LCST. The value of absorbance
(at kmax) leveled off slowly at above LCST. Also
interesting to note is the blue shift in absorption
maximum (kmax) with as much as 6 nm change with
temperatures (Fig. 4c). For example, the absorption
maximum was 529 nm at 25 �C, while at 36 �C the
absorbance maximum shifted to 525 nm. The blue
shift of absorption maximum could be ascribed to the
changing dielectric constant of the medium surround-
ing the capped Au NPs (Ung et al. 1997). In other
words, when thioglycolic acid-capped Au NPs were
mixed with pNIPAm-AAc in aqueous solution there
can be several types of interaction between the
polymer and thioglycolic acid-capped Au NPs. This
interaction might be either van der Waals or
350 400 450 500 550 600 650 700 750
0.1
0.2
0.3
0.4
0.5
0.646oC
).u.a( s
bA
Wavelength (nm)
25oC
24 26 28 30 32 34 36 38 40 42 44 46
522
523
524
525
526
527
528
529
λxa
m)
mn(
Temperature (oC)
24 26 28 30 32 34 36 38 40 42 44 46
0.60
0.65
0.70
0.75
0.80
0.85
).u.a( s
bA
Temperature (oC)
(b)
(c)
(a)
Fig. 4 a UV–visible spectra of pPA-thio-Au NPs with
increasing temperature. b The plot of normalized maximum
absorbance of the SPR band at different temperatures, while cis the temperature dependence absorption maximum (wave-
length) of SPR band of Au NPs in pPA-thio-Au NPs
1338 J Nanopart Res (2010) 12:1331–1348
123

hydrogen bonding interaction (inter or intra molec-
ular H-bonding). The polymer itself could form H-
bonding among its own –COOH groups and with
water molecules. Similarly, the –COOH groups of the
thioglycolic acid could form H-bonds with water as
well as with the polymer at below LCST. At above
the LCST, the polymer shrinks to a form where water
molecules are expelled from the network, and when
thioglycolic acid-stabilized Au NPs are part of the
polymer structures then the hydrogen bonding would
primarily be between the –COOH groups of the
stabilizer and those (along with amide groups or
–COOH group of AAc moieties) of the polymer.
Thus, a change in the environment might well be
reflected in the observed change in the absorption
characteristic of the Au NPs at above LCST. The shift
in the UV–vis absorption of Au NPs indicated that the
phase transition temperature here was 36 �C which is
different from pure polymer. Interestingly, this
observation implies that the SPR absorption of Au
NPs could act as a good probe for measuring the
phase transition temperature of thermosensitive
polymers.
Further, to understand better the volume phase
transition of the pPA-thio-Au NPs, DLS measure-
ments of the composite hydrogel were carried out at
different temperature. The results are shown in Fig. 5.
At temperatures below LCST, there were two types of
particles sizes: one corresponding to the polymer with
a value of 1.4 ± 0.5 lm, while the other one possibly
was due to thioglycolic acid-stabilized Au NPs with
particles sizes of 116 ± 26 nm when measured at
27 �C. Thus, the average particle sizes mentioned in
the figures are the averages of the values, automati-
cally reported by the software used in measuring the
particle sizes. On other hand at 35.6 �C, the sizes were
reduced down to 116 ± 26 nm. The gradual change in
the particle sizes at above the LCST indicates gradual
de-swelling of the composite in comparison to the
polymer alone. The above results suggest that at
temperatures below LCST, thioglycolic acid-stabi-
lized Au NPs and pNIPAm-AAc hydrogel remain as
individual species in aqueous medium, where inter-
molecular H-bonding is predominant. When the
temperature of the medium is increased, the intramo-
lecular H-bonding in polymer and intermolecular
H-bonding between the polymer and thioglycolic acid-
stabilized Au NPs are favored. At above the LCST,
these H-bondings provide the main force for the
formation of a composite structure consisting of Au
NPs (with the stabilizer) and the polymer. The results
are the formation of particles of uniform sizes and
change in the SPR spectral characteristics of the Au
NPs. Further, the attractive forces leading to composite
formation, at above LCST, between the functionalized
Au NPs and the polymer might be responsible for the
0 1000 2000 3000 4000 5000 60000
5
10
15
)%( yc
neu
qerF
Hydrodynamic diameter (nm)
60 80 100 120 140 160 180 2000
5
10
15
20
)%( yc
neu
qerF
Hydrodynamic diameter (nm)40 60 80 100 120 140 160
0
5
10
15
20
)%( yc
neu
qerF
Hydrodynamic diameter (nm)
28 30 32 34 36 38 40 42
200
400
600
800
1000
1200
1400
)m
n( retemai
D cima
nyd
ordy
H
Temperature (oC)
(b)(a)
(c) (d)
Fig. 5 a Temperature-
dependent hydrodynamic
diameters of the PPA-thio-
Au NPs as measured by
dynamic light scattering
technique. b The histogram
of hydrodynamic diameters
of pPA-thio-Au NPs at
27 �C, c 35.6 �C, and d39 �C obtained by dynamic
light scattering
measurements
J Nanopart Res (2010) 12:1331–1348 1339
123

changes in the SPR peak of the Au NPs. However, the
above investigation of the DLS measurements shows
that there will be a marked attraction between the
thioglycolic acid-capped Au NPs and pNIPAm-AAc
polymer network and this leads to the decreasing of
the hydrodynamic radius of the polymer network gel
toward to the thioglycolic acid-capped Au NPs sizes
with increasing the temperature of the system, which
also leads to the changing the kmax of the SPR toward
to the lower wavelength. In other words, at higher
temperature the water molecules are gradually
removed from polymer hydrogel, and the removal
induces a decrease in total volume of the polymer gel
and brings thioglycolic acid-capped Au NPs very
close to the polymer network leading to the blue shift
of the SPR absorption of the Au NPs in the system. It
must be mentioned here that all of the above behaviors
have been found to be reversible with respect to
change in temperature. Further, TEM analysis of the
pPA-thio-Au NP samples prepared at below the phase
transition temperature (25 �C) and at above the phase
transition temperature (40 �C) was pursued to have a
better understanding of the structural transitions due
to temperature increase. The results are shown in
Fig. 6. At a temperature below the LCST, the Au NPs
could be observed to have been dispersed rather
uniformly all over the polymer (Fig. 6a). Individual
Au NPs could be seen spread over the network of
polymers (Fig. 6c) indicating uniform distribution of
particles in the medium, which upon evaporation
formed such structures. On the other hand, at above
the LCST (at 40 �C), the density of the particles is
much higher and clusters of particles could be seen
deposited over the polymer (Fig. 6d–f). The average
diameter of the Au NPs was found to be 8.0 ± 0.3 nm
in all cases, indicating that there was no agglomera-
tion of particles. The increase in density of the
particles clearly indicates the shrinking of polymer
along with the functionalized Au NPs. This means that
the Au NPs were bound to the polymer at above
LCST, possibly through electrostatic interaction
between the polymer and the stabilizers of Au NPs.
The apparent inconsistency between the particle sizes
as measured from DLS and TEM could originate due
to two factors. First of all, the microviscosity for the
polymer particles and those due to stabilized NPs
would not be identical. They have been assumed to be
the same in the measurement. This would significantly
affect the exact particle size calculation (of the NPs)
from the DLS measurements. In addition, as is clear
from the TEM images there are significant presence of
associated NPs even at lower temperatures. This could
make the apparent particle sizes of the NPs appear
larger than they are.
In addition to thioglycolic acid, we were interested
in studying the effect of amino acids on the volume
phase transition by incorporating of amino acid-
capped Au NPs into pNIPAm-AAc hydrogel solution.
Sastry and coworkers have reported that amino group
can bind more strongly to the surface of Au NPs than
carboxylic acid group (Joshi et al. 2006; Selvakannan
et al. 2003). In addition to their studies, there has been
several report of synthesis of different amino acid-
capped Au NPs, which are useful for biological
applications. The motivation has been that since the
amino acids could have different side chain group—
such as hydrophilic or hydrophobic, polar or non-
polar—interactions of the stabilizer with biological
molecules would also be different. In the present
study, we have taken two different amino acids such
as tryptophan and phenylalanine. Amino acid trypto-
phan contains hydrophilic indole moiety, while
phenylalanine consists of hydrophobic phenyl ring.
Thus, the phase behaviors of the two different amino
acid-stabilized Au NPs with the polymer, at different
temperature, would be potentially interesting. The
capping of Au NPs by amino acids has been confirmed
by FTIR spectroscopy (refer to SI, Figure S4 and S5).
Tryptophan- and phenylalanine-capped Au NPs have
absorption maxima at 543 and 519 nm, respectively.
For subsequent studies, the amino acid-capped Au
NPs were mixed in aqueous solutions containing
pNIPAm-AAc hydrogel. We shall henceforth refer the
resultant composites hybrid hydrogels as pPA-try-Au
NPs (for pPA-AA-tryptophan-capped Au NPs) and
pPA-phe-Au NPs (for pPA-AA-phenylalanine capped
Au NPs). Our observations as detailed below suggest
that the phase transition as well as optical behavior of
the two differently stabilized Au NPs are different.
The temperature-dependent UV–vis spectral changes
and hydrodynamic diameters of pPA-try-Au NPs and
pPA-phe-Au NPs are shown in Figs. 7 and 8, respec-
tively. The as-observed UV–vis spectra are shown in
Figs. 7a and 8a, respectively. The normalized absor-
bances at the maxima are shown in Figs. 7b and 8b,
respectively. It may be mentioned here that to obtain
normalized absorbance from Fig. 7a, the method
similar to the calculations in Fig. 4 was followed
1340 J Nanopart Res (2010) 12:1331–1348
123

(Refer to SI, Figure S3 (b)). On the other hand, the
normalized absorbance values at the maxima from
Fig. 8a were obtained by drawing a baseline between
475 and 555 nm and then measuring the value of
absorbance from the baseline to the maximum
absorption at the kmax (refer to SI, Figure S3 (c)).
For the pPA-try-Au NPs composite, the SPR band of
Au NPs hardly changed with temperature in terms of
intensity (at kmax, Fig. 7c), while there was discern-
ible change in the values of kmax (Fig. 8c). For
example, at below the transition temperature the kmax
of the pPA-try-Au NPs was 543 nm, while at above
the phase transition temperature the kmax was
observed to be 539 nm. The results, however, indicate
Fig. 6 a TEM image of
pPA-thio-Au NPs at 25 �C,
while b and c are the
expanded views of the
picture from (a). The image
in d is the TEM picture of
the same at 40 �C, while eand f are the expanded
views of the picture in (d)
J Nanopart Res (2010) 12:1331–1348 1341
123

behavior similar to the ones of thioglycolic acid as the
stabilizer, although they are less remarkable. Further,
the phase transition temperature as measured from the
UV–vis studies (Fig. 7b and c) occurred at 39 �C,
which is significantly higher than that of the pure
polymer. In addition, the results of measurements of
hydrodynamic diameters (Figs. 7d and 8d, respec-
tively) also support the above results. Essentially,
although the general behavior of the hydrophilic
tryptophan moiety (in the composite) is similar to that
of thioglycolic acid, there are significant differences
as described above, which indicate that the interaction
between the polymer and the stabilized Au NPs could
be substantially different at different temperatures.
The H-bond that could be formed between the indole
group of tryptophan and the carboxylate or amide
moieties of the polymer could possibly be the reason
for the behavior described above. Further evidence in
support of the above conclusion came from the
temperature-dependent UV absorption due to G–G*
transition of the indole moieties of the tryptophan
occurring at 265 nm. It is worth pointing out that the
tryptophan has absorption maxima at 278 nm, which
shifted to 265 nm after the formation of stabilized Au
NPs. This indicates interaction between the Au NPs
and the p-electron cloud of tryptophan (Selvakannan
et al. 2004). Figure 9a shows the temperature-depen-
dent absorption spectrum of a mixture of pPA-try-Au
NPs composites solutions, recorded in the range of
190–420 nm. As is evident from the figure, the band at
265 nm vanished at phase transition temperature
(39 �C) of the pPA-try-Au NPs. It should be men-
tioned here that the band could again be observed
when the temperature reverted back to a temperature
30 32 34 36 38 40 42 44
0
200
400
600
800
1000
1200
1400)m
n( retemai
D cima
n yd
ord y
H
Temperature (oC)30 32 34 36 38 40 42 44 46
539
540
541
542
543
544
λxa
m)
mn(
Temperature (oC)
350 400 450 500 550 600 650 700 750
0.2
0.3
0.4
30oC).u.a( s
bA
Wavelength (nm)
45oC
32 34 36 38 40 42 44
0.90
0.91
0.92
0.93
0.94
0.95
0.96
0.97
0.98
).u.a( s
bA
Temperature (oC)
(b)(a)
(c) (d)
Fig. 7 a Temperature-dependent UV–visible spectra of pPA-
try-Au NPs, while b is the plot of normalized absorbance at
different temperatures obtained from [Figure S3 (b)], and c is
the change of kmax of the surface plasmon band of the pPA-try-
Au NPs from normalized graph of Figure S3 (b). The plot in dis the dynamic light scattering measurement results (hydrody-
namic diameters) of the pPA-try-Au NPs at different
temperatures
1342 J Nanopart Res (2010) 12:1331–1348
123

below LCST. This observation clearly indicates that
there was a significant interaction between the poly-
mer hydrogel and tryptophan-capped Au NPs, which
is reversible with respect to change in temperature. It
might be that encapsulation of the tryptophan moieties
inside the globule of the polymer leads to vanishing of
the absorption.
The results involving pPA-phe-Au NPs were
remarkably different from those mentioned above.
The UV–vis absorption due to SPR of Au NPs
virtually disappeared with increase in temperature
(Fig. 8a, b). Moreover, the changes were more
gradual than in the cases with the other stabilizers.
The changes in the wavelength maxima were also
significant. For example, at room temperature
(25 �C), the kmax was 517 nm, while that at 36 �C
was shifted to 508 nm. Further, the kmax shifted
gradually to still lower wavelengths with increasing
temperatures (Fig. 8c). The phase transition temper-
ature was found to be 36 �C (Fig. 8b, c). It is
important to mention here that the volume phase
transition temperatures of the systems pPA-try-Au
NPs and pPA-phe-Au NPs were found to be 39 and
36.3 �C, respectively, as obtained from temperature-
dependent DLS measurements (Figs. 7d and 8d).
These results are consistent with phase transition
temperatures obtained from temperature-dependent
absorption maxima of the stabilized Au NPs. The
above results suggest that SPR absorption of Au NPs
could act as a probe for understanding the nature of
26 28 30 32 34 36 38 40 42
0.004
0.005
0.006
0.007
0.008
0.009
).u.a( s
bA
Temperature (oC)
26 28 30 32 34 36 38 40 42
506
508
510
512
514
516
λxa
m)
mn(
Temperature (oC)26 28 30 32 34 36 38 40 42
0
200
400
600
800
1000
1200)m
n( retemai
D cima
nyd
ordy
H
Temperature (oC)
300 350 400 450 500 550 600 650 700
0.20
0.24
0.28
0.32
43oC
).u.a( s
bA
Wavelength (nm)
25oC
(a) (b)
(d)(c)
Fig. 8 a Temperature-dependent UV–visible spectra of pPA-
phe-Au NPs. The plot b of normalized absorbance at different
temperatures is based on figure shown in Figure S3 (c) (refer to
SI), while c is the plot of the kmax of the surface plasmon band
of the pPA-phy-Au NPs also calculated from Figure S3 (c).
Here d is the average hydrodynamic diameters of the pPA-phy-
Au NPs at different temperature, measured by dynamic light
scattering method
J Nanopart Res (2010) 12:1331–1348 1343
123

interactions between the stabilizer molecules and
polymer hydrogel. This is especially true in estab-
lishing the LCST of the polymer hydrogel in the
presence of NPs with different stabilizers. Further, a
question may arise about the volume phase transition
temperature of pPA-Try-Au NP composite hydrogel
being higher than that of pPA-phe-Au NP composite
hydrogel. This could be explained in terms of side
chains of the individual amino acids. For example,
tryptophan has more hydrophilic moiety of the indole
ring, which can make the stronger intermolecular H-
bond with polymer hydrogel, as well as with the
surrounding water molecules. When the temperature
is increased gradually, the intermolecular H-bond
between the water and polymer and between the
water and tryptophan may gradually be lost due to the
thermal motion of the water molecules and conse-
quently the volume of the pPA-try-Au NP composite
hydrogel slowly shrinks (Fig. 7d). Stoller et al.
(2004) reported that H-bonding interaction between
the indole-water is more energetically favorable than
that of aromatic ring. They considered that H-
bonding interaction between the heterocyclic ring
and water is much stronger than simple aromatic
benzene ring (Fredericks et al. 1996; Carney et al.
1998). This could be responsible for higher volume
phase transition temperature of the pPA-Try-Au NPs
than that of pPA-phe-Au NPs composite hydrogel.
This was further supported by the results of temper-
ature-dependent UV–vis spectra of NPs in both the
systems. At the phase transition temperature, the SPR
peak of the pPA-phe-Au NPs was more affected than
that of pPA-try-Au NP composite hydrogel. This
could be ascribed to the more hydrophobic nature of
the phenyl ring than indole. Further, the hydrophobic
interaction with the polymer is more dominant in the
phenylalanine capped Au NPs, which leads to
significant shift of the SPR peak in comparison to
tryptophan-capped NPs (Mulvaney 1996; Ung et al.
2001). In addition, phenylalanine contains a highly
hydrophobic side residue that would have different
interaction with the polymer than the highly hydro-
philic thioglycolic acid or tryptophan. Also, the coil
to globule transition of the polymer at above the
transition temperature involves reinforced intramo-
lecular hydrogen bonding and strong van der Waals
interactions (attraction) among its hydrophobic moi-
eties (Zhu et al. 2004). This would lead to localiza-
tion of the stabilized Au NPs into the hydrophobic
pockets of the globule. This is probably the reason for
reduced optical absorption as the NPs that are buried
within the ‘‘particles’’ of the globule. The same may
be the reason for changes in the absorption maxi-
mum. Raula et al. (2003) have shown that both kmax
and absorption of the SPR peak of thermosensitive
hydrogel-coated Au NPs, at above the LCST changes
similar to the above. The reason here has also been
proposed to be due to formation of hydrophobic
pocket after the collapse of the structure into globule
form.
Temperature-dependent particles size histogram
plot of DLS measurements of pPA-try-Au NPs and
pPA-phe-Au NPs indicated specific interactions
between the stabilized Au NPs and the polymer.
The results are shown in Fig. 9 (pPA-try-Au NPs)
and Figure S6 (refer to SI) (pPA-phe-Au NPs). In
these cases, the particles consisted of two distribu-
tions of sizes. The higher particle diameters with
average values of 1.3 ± 0.8 and 1.15 ± 0.9 lm for
pPA-try-Au NPs and pPA-phe-Au NPs, respectively,
in addition to lower particle diameters of values
62 ± 18 and 50 ± 15 nm, respectively for the above.
This means that polymer particles were probably
dispersed separately in the medium from the stabi-
lized Au NPs. However, since the average value of
the polymer particles herein was smaller than those of
polymer alone or polymer stabilized Au NPs, it is
plausible that there were at least some interactions
between the two. Further, at higher temperatures the
particle collapsed into sizes, which are considerably
less than those of all three cases discussed before. For
example, at 39 �C the hydrodynamic diameter of the
pPA-try-Au NPs was 62 ± 18 nm while pPA-phe-Au
NPs showed a value of 52 ± 15 nm at 36 �C. This
means the interactions between phenylalanine-capped
NPs and tryptophan-capped NPs and the polymer led
to globular structures with diameters smaller than that
of the polymer (in its globular form) at above the
LCST. Seida and Nakano (1996) reported the effect
of various organic molecules on the nature of
interaction with the polymer at above and below the
phase transition temperatures. They observed that
organic molecules (phenol and benzoic acid and
aniline) are absorbed much into the polymer at above
the phase transition due to the domination of
hydrophobic interaction. This could be the reason
that when the phenylalanine-capped Au NPs inter-
acted with polymer hydrogel that led to the
1344 J Nanopart Res (2010) 12:1331–1348
123

decreasing of the hydrodynamic diameter of the pPA-
phe-Au NPs than that of pPA-try-Au NPs at above
the phase transition temperature. On the other hand,
strong hydrophobic interactions between phenyl
groups and the hydrophobic backbone of the polymer
may lead to a collapsed structure that is smaller in
diameter than the polymer itself. Temperature-depen-
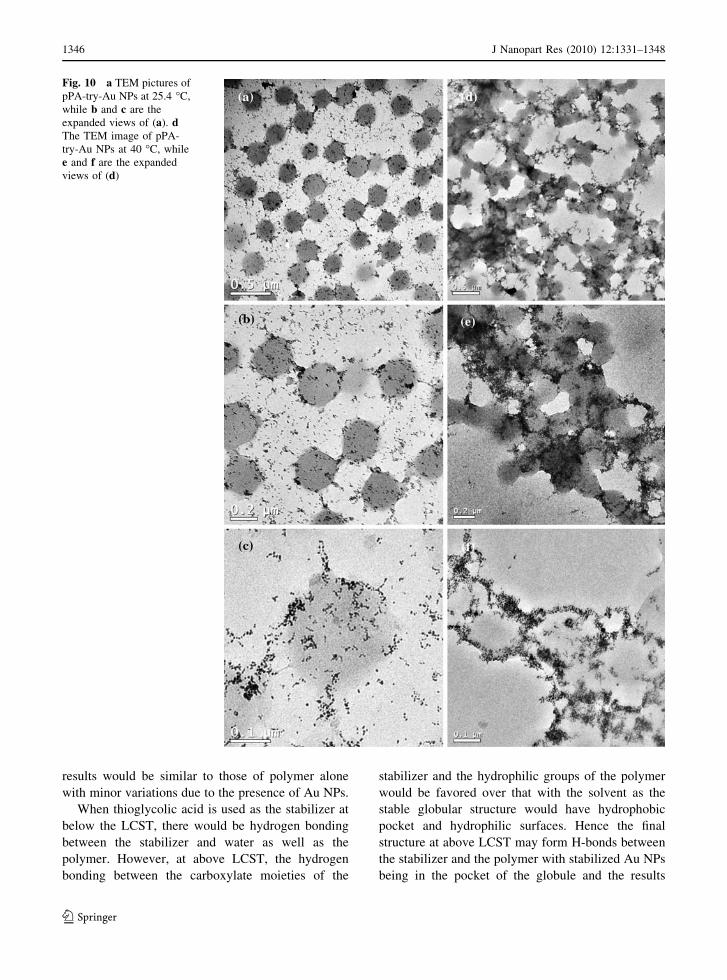
dent TEM measurements pPA-try-Au NPs are shown
in Fig. 10. At below the LCST, the particles were
uniformly distributed all over the solution and hence
the evaporated materials had more or less uniform
distributions of particles (Fig. 8a–c). The NPs had
typical diameters of 8.0 ± 0.6 nm. On the other
hand, when the temperature was increased above
LCST, the particles seem to agglomerate into the
collapsed structure of the polymer. This behavior is
different from the other samples where either poly-
mer or thioglycolic acid-stabilized Au NPs were
used. The reversibility of the structure in solution
(with respect to changes in temperature) indicates
that these particles might not have agglomerated into
lumps of particles only. On the contrary, the inter-
actions between the globular polymer and the stabi-
lizers lead to some sort of collapsed structure with
particles coming very close to each other in compar-
ison to the structures obtained from polymer or
thioglycolic stabilized NPs. Similar results were also
observed in the case of the pPA-phe-Au NPs system
(refer to SI). The TEM pictures are shown in Figure
S7.
Qualitatively, the results mentioned above could
be explained primarily by invoking two factors:
hydrophobic and H-bonding interactions. In the
system involving polymer-capped Au NPs, there is
no extraneous stabilizing agent present and hence no
additional interaction between the species. Thus, the
20 40 60 80 100 120 140 1600
5
10
15
)%( yc
neu
qerF
Hydrodynamic diameter (nm)
0 1000 2000 3000 40000
4
8
12
16
)%( yc
neu
qerF
Hydrodynamic Diameter (nm)
0 500 1000 15000
3
6
9
12
)mn( ycne
uqer
F
Hydrodynamic Diameter (nm)
250 300 350 400
0.4
0.6
0.8
).u.a( s
bA
Wavelength (nm)
(d)(c)
(b)(a)
Fig. 9 a The UV–visible absorption spectra of pPA-try-Au
NPs at different temperatures, measured in the region of 225–
420 nm. b The histogram of hydrodynamic diameters of pPA-
try-Au NPs at 30 �C, c 36 �C, and d 39 �C obtained by
dynamic light scattering measurements
J Nanopart Res (2010) 12:1331–1348 1345
123
results would be similar to those of polymer alone
with minor variations due to the presence of Au NPs.
When thioglycolic acid is used as the stabilizer at
below the LCST, there would be hydrogen bonding
between the stabilizer and water as well as the
polymer. However, at above LCST, the hydrogen
bonding between the carboxylate moieties of the
stabilizer and the hydrophilic groups of the polymer
would be favored over that with the solvent as the
stable globular structure would have hydrophobic
pocket and hydrophilic surfaces. Hence the final
structure at above LCST may form H-bonds between
the stabilizer and the polymer with stabilized Au NPs
being in the pocket of the globule and the results
Fig. 10 a TEM pictures of
pPA-try-Au NPs at 25.4 �C,
while b and c are the
expanded views of (a). dThe TEM image of pPA-
try-Au NPs at 40 �C, while
e and f are the expanded
views of (d)
1346 J Nanopart Res (2010) 12:1331–1348
123

would be different. In the case of phenylalanine
stabilized Au NPs, the final structure in the globule
would possibly involve the stabilized NPs deeply
embedded in the pocket of the globule thereby
reducing even the optical absorption of Au NPs,
which is different from the other cases. Finally, the
general behavior involving the interactions between
stabilized Au NPs and the polymer at below and
above LCST is described schematically in Fig. 11.
Essentially, when Au NPs are stabilized by hydro-
philic moieties then particles are either exposed to the
aqueous medium even after the collapse (at above
LCST) or embedded in the collapsed structure being
bonded to the hydrophilic groups of the polymer. On
the other hand, in the presence of hydrophobic
stabilizers, the particles are embedded deeply inside
the polymer collapsed structure at above LCST.
Conclusion
In the present article, we have been able to report the
use of various functional group stabilized Au NPs on
the volume phase transitions of a thermosensitive
polymer hydrogel. The optical properties of Au NPs
stabilized with different functional groups have been
used as one of the probes to understand the behavior
of volume phase transitions in mixed systems.
Particle size analysis and TEM measurements were
also made to substantiate the conclusions of the
observations. Essentially, when hydrophilic groups
were used in the stabilizer the collapsed structures
consisted of H-bonding between the stabilized Au
NPs and the polymer, with little effect on the
hydrophobic pocket of the globule. On the other
hand, when hydrophobic group was used in the
stabilizer then the observations indicated that the Au
NPs along with the stabilizer were embedded deep
inside the hydrophobic pocket of the globule, dras-
tically changing the optical absorption due to SPR of
Au NPs. That amino acids were used as the stabiliz-
ing agents and their effect on the volume phase
transitions of a biofriendly hydrogel would support
future endeavors involving Au NPs in the drug
delivery systems.
Acknowledgment We thank Central Instrumentation Facility
(IITG) for help in recording TEM. We also thank the
Department of Science and Technology (DST) (No. SR/S5/
NM-01/2005, 2/2/2005-S�F., SR/FT/L-39/2004), Council of
Scientific and Industrial Research (CSIR, 01(2172)/07/EMR-
II), Government of India for financial support. Mr. A.
Murugadoss thanks CSIR for a fellowship.
References
Berndt I, Pedersen JS, Richtering W (2005) Structure of mul-
tiresponsive ‘‘intelligent’’ core–shell microgels. J Am
Chem Soc 127:9372–9373
Carney JR, Hagemeister FC, Zwier TS (1998) The hydrogen-
bonding topologies of indole-(water)n clusters from reso-
nant ion-dip infrared spectroscopy. J Chem Phys
108:3378–3382
Chen H, Gu Y, Hu Y, Qian Z (2007) Characterization of pH-
and temperature-sensitive hydrogel nanoparticles for
controlled drug release. J Pharm Sci Technol 61:303–313
Das M, Sanson N, Fava D, Kumacheva E (2007) Microgels
loaded with gold nanorods: photothermally triggered
volume transitions under physiological conditions. Lang-
muir 23:196–201
Francois NJ, Allo S, Jacobo SE, Daraio ME (2007) Composites
of polymeric gels and magnetic nanoparticles: preparation
and drug release behavior. J Appl Polym Sci 105:647–655
Fig. 11 A schematic representation of the stabilizer specific
interactions of stabilized Au NPs with the thermosensitive
polymer hydrogel at below and above lower critical solution
temperature (LCST). (a) and (b) represent transitions to the
collapsed structure when Au NPs are stabilized by hydrophilic
and hydrophobic groups, respectively
J Nanopart Res (2010) 12:1331–1348 1347
123

Fredericks SY, Jordan KD, Zwie TS (1996) Theoretical char-
acterization of the structures and vibrational spectra of
benzene-(H2O)n (n = 1–3) clusters. J Phys Chem
100:7810–7821
Gan DJ, Lyon LA (2001) Tunable swelling kinetics in core–
shell hydrogel nanoparticles. J Am Chem Soc 123:7511–
7517
Heras Alarcon CDL, Pennadam S, Alexander C (2005) Stimuli
responsive polymers for biomedical applications. Chem
Soc Rev 34:276–285
Jain RK, Stroh M (2004) Zooming in and out with quantum
dots. Nat Biotechnol 22:959–960
Jones CD, Lyon LA (2003) Dependence of shell thickness on
core compression in acrylic acid modified poly(N-iso-
propylacrylamide) core/shell microgels. Langmuir
19:4544–4547
Joshi H, Bhumkar DR, Joshi K, Pokharkar V, Sastry M (2006)
Gold nanoparticles as carriers for efficient transmucosal
insulin delivery. Langmuir 22:300–305
Juodkazis S, Mukai N, Wakaki R, Yamaguchi A, Matsuo S,
Misawa H (2000) Reversible phase transitions in polymer
gels induced by radiation forces. Nature 408:178–181
Karg M, Pastoriza-Santos I, Perez-Juste J, Hellweg T, Liz-
Marzanb LM (2007) Nanorod-coated PNIPAm microgels:
thermoresponsive optical properties. Small 7:1222–1227
Khan A (2008) Preparation and characterization of magnetic
nanoparticles embedded in microgels. Mater Lett 62:898–
902
Kim J, Singh N, Lyon LA (2006) Label-free biosensing with
hydrogel microlenses. Angew Chem Int Ed 45:1446–1449
Kim J, Singh N, Lyon LA (2007) Displacement-induced
switching rates of bioresponsive hydrogel microlenses.
Chem Mater 19:2527–2532
Langer R, Tirrell DA (2004) Designing materials for biology
and medicine. Nature 428:487–492
Lin CL, Chiu WY (2006) Polypyrrole/poly (N-iso-
propylacrylamide-co-acrylic acid) thermosensitive and
electrically conductive composite microgels. J Poly Sci A
44:1648–1659
Lin S, Li M, Dujardin E, Girrard C, Mann S (2005) One-
dimensional plasmon coupling by facile self-assembly of
gold nanoparticles into branched chain networks. Adv
Mater 17:2553–2559
Lu Y, Mei Y, Drechsler M, Ballauff M (2006) Thermosensitive
core-shell particles as carriers for Ag nanoparticles:
modulating the catalytic activity by a phase transition in
networks. Angew Chem Int Ed 45:813–816
Matsukaa S, Ando I (1996) A study of self-diffusion of mol-
ecules in polymer gel by pulsed-gradient spin-echo 1H
NMR. Macromolecules 29:7136–7139
Morones JR, Frey W (2007) Environmentally sensitive silver
nanoparticles of controlled size synthesized with PNIPAm
as a nucleating and capping agent. Langmuir 23:8180–
8186
Mulvaney P (1996) Surface plasmon spectroscopy of nano-
sized metal particles. Langmuir 12:788–800
Nayak S, Lyon LA (2005) Soft nanotechnology with soft
nanoparticles. Angew Chem Int Ed 44:7686–7708
Raula J, Shan J, Nuopponen M, Niskanen A, Jiang H, Kaup-
pinen EI, Tenhu H (2003) Synthesis of gold nanoparticles
grafted with a thermoresponsive polymer by surface-
induced reversible-addition-fragmentation chain-transfer
polymerization. Langmuir 19:3499–3504
Retama JR, Zefeiropoulos NE, Serafinelli C, Reyna RR, Voit
B, Cabarcos EL, Stamm M (2007) Synthesis and charac-
terization of thermosensitive PNIPAM microgels covered
with superparamagnetic c-Fe2O3 nanoparticles. Langmuir
23:10280–10285
Robinson I, Alexander C, Lu LT, Tung LD, Fernig DG, Thanh
NTK (2000) One-step synthesis of monodisperse water-
soluble dual-responsive magnetic nanoparticles. Chem
Commun 460:4602–4604
Seida Y, Nakano YJ (1996) Effect of salt on the property of
adsorption in thermosensitive polymer hydrogel. Chem
Eng Jpn 29:767–769
Selvakannan PR, Mandal S, Phadtare S, Pasricha R, Sastry M
(2003) Capping of gold nanoparticles by the amino acid
lysine renders them water-dispersible. Langmuir 19:3545–
3549
Selvakannan PR, Mandal S, Phadtare S, Gole A, Pasricha R,
Adyanthaya D, Sastry M (2004) Water-dispersible tryp-
tophan-protected gold nanoparticles prepared by the
spontaneous reduction of aqueous chloroaurate ions by the
amino acid. J Colloid Interface Sci 269:97–102
Shan J, Tenhu H (2007) Recent advances in polymer protected
gold nanoparticles: synthesis, properties and applications.
Chem Commun 72:4580–4598
Stoller EJ, Gelpi JL, Velankar S, Golovin A, Orozco M, Luisi
BF (2004) Unconventional interactions between water and
heterocyclic nitrogens in protein structures. Protein
Structure Function and Bioinformatics 57:1–8
Storh JM, Zimmer JP, Drude DG, Levehenko TS, Cohen KS,
Brown EB, Scadden DT, Torchilin VP, Bawendi MG,
Fukumura D (2005) Quantum dots spectrally distinguish
multiple species within the tumor milieu in vivo. Nat Med
11:678–682
Ung T, Giersig M, Dunstan D, Mulvaney P (1997) Spectro-
electrochemistry of colloidal silver. Langmuir 13:1773–
1782
Ung T, Liz-Marzan LM, Mulvaney P (2001) Optical properties
of thin films of Au@SiO2 particles. J Phys Chem B
105:3441–3452
Yusa S, Fukuda K, Yamamoto T, Iwasaki Y, Watanabe A,
Akiyoshi K, Morishima Y (2007) Salt Effect on the heat-
induced association behavior of gold nanoparticles coated
with poly(N-isopropylacrylamide) prepared via reversible
addition-fragmentation chain transfer (RAFT) radical
polymerization. Langmuir 23:12842–12848
Zhang W, Gaberman I, Ciszkowska M (2002) Diffusion and
concentration of molecular probes in thermoresponsive
poly(N-isopropylacrylamide) hydrogels: effect of the
volume phase transition. Anal Chem 74:1343–1348
Zhang J, Chu LY, Li YK, Lee YM (2007) Dual thermo- and
pH-sensitive poly(N-isopropylacrylamide-co-acrylic acid)
hydrogels with rapid response behaviors. Polymer
48:1718–1728
Zhu MQ, Wang LQ, Exarhos GJ, Li ADQ (2004) Thermo-
sensitive gold nanoparticles. J Am Chem Soc 126:2656–
2657
1348 J Nanopart Res (2010) 12:1331–1348
123
Copyright © 2022 FDOKUMEN




















