
Sorption characteristics of CTMA–bentonite complexes as controlled by surfactant packing density
7
Colloids and Surfaces A: Physicochem. Eng. Aspects 294 (2007) 221–227 Sorption characteristics of CTMA–bentonite complexes as controlled by surfactant packing density Runliang Zhu, Lizhong Zhu ∗ , Liheng Xu Department of Environmental Science, Zhejiang University, Hangzhou 310028, China Received 27 June 2006; received in revised form 8 August 2006; accepted 8 August 2006 Available online 14 August 2006 Abstract This work was to examine the relationship between the configuration and sorption characteristics of surfactant–clay complexes. Various amounts of cetyltrimethylammonium bromide (CTMAB) were intercalated into the bentonite matrixes with different layer charges. Packing densities of the adsorbed surfactants and sorption characteristics of the obtained CTMA–bentonite complexes towards phenol and naphthalene were examined. Experimental results indicated that packing density of the adsorbed surfactant was proportional to the surfactant loading amount and layer charge of the bentonite, and sorption capacities of these complexes had a close relationship with the surfactant packing density. That was, with the increase of surfactant packing density, the organic-carbon normalized sorption coefficient (K oc ) first rose till the maximum, and then began to decrease as the packing density further increased. This could be interpreted that increase of surfactant packing density would render the surfactant phases more hydrophobic environment, and the hydrophobic affinity of the surfactant phases towards the solutes thus increased accordingly. However, in the high surfactant packing density region, the densely packed surfactants reduced the available free space for the solutes, resulting in decrease of sorption capacity for these complexes. Hence, with the increase of surfactant packing density, the adsorbed surfactants would form a series of partition phases showing different affinity to the solutes. © 2006 Elsevier B.V. All rights reserved. Keywords: Sorption; Organic clay; Surfactant; Hydrophobic organic compounds; Partition phase; Layer charge 1. Introduction Numerous studies have investigated the sorption characteris- tics of surfactant–clay complexes towards hydrophobic organic compounds (HOCs) because of their wide variety of applications [1–7]. The hydrophobic medium created by the long alkyl chain of the adsorbed surfactant shows high affinity to HOCs, and thus the complexes are considered as potential sorbents in wastew- ater treatment [1–4], appropriate landfill liner [5] and effective barrier to prevent down-gradient pollution of groundwater and aquifer from organic pollutants [6,7]. Partition of HOCs to the organic phase created by the adsorbed surfactants was considered to be the predominant mechanism controlling sorption of HOCs on surfactant–clay complexes [4,8], and the sorption coefficient, K d (the ratio of solutes concentration on the sorbents to that in solution), was ∗ Corresponding author. Tel.: +86 571 88273733; fax: +86 571 88273450. E-mail address: [email protected] (L. Zhu). suggested to be proportional to the amount of the adsorbed surfactant [9,10]. However, an increasing number of inves- tigations have observed that sorption characteristics of these complexes are strongly dependent on their configurations, and the organic-carbon content (f oc ) normalized sorption coefficient (K oc = K d /f oc ) can be quite different accordingly [11–14]. Smith et al. [11] observed that the surfactant–clay complexes prepared from the surfactant with relatively longer alkyl chain had larger K oc for HOCs. Esumi et al. [12] found that silica modified from the surfactant with two alkyl chains had larger K oc than that with one or three alkyl chains in sorption of 2-naphthol. Even for the surfactant–clay complexes synthesized from the same surfac- tant, K oc can be greatly influenced by the surfactant loading amount [3,13,14]. Although the idea that K oc can vary with the configurations of the surfactant–clay complexes has gradually been accepted, the relationship between the configuration and sorption characteris- tics of these complexes is still ambiguous. Sorption properties of the surfactant–clay complexes should be ascribed to their struc- tural difference. In fact, microstructures of the surfactant–clay 0927-7757/$ – see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.colsurfa.2006.08.016
Transcript of Sorption characteristics of CTMA–bentonite complexes as controlled by surfactant packing density

A
oaEooamtop©
K
1
tc[otaba
amcs
0d
Colloids and Surfaces A: Physicochem. Eng. Aspects 294 (2007) 221–227
Sorption characteristics of CTMA–bentonite complexesas controlled by surfactant packing density
Runliang Zhu, Lizhong Zhu ∗, Liheng XuDepartment of Environmental Science, Zhejiang University, Hangzhou 310028, China
Received 27 June 2006; received in revised form 8 August 2006; accepted 8 August 2006Available online 14 August 2006
bstract
This work was to examine the relationship between the configuration and sorption characteristics of surfactant–clay complexes. Various amountsf cetyltrimethylammonium bromide (CTMAB) were intercalated into the bentonite matrixes with different layer charges. Packing densities of thedsorbed surfactants and sorption characteristics of the obtained CTMA–bentonite complexes towards phenol and naphthalene were examined.xperimental results indicated that packing density of the adsorbed surfactant was proportional to the surfactant loading amount and layer chargef the bentonite, and sorption capacities of these complexes had a close relationship with the surfactant packing density. That was, with the increasef surfactant packing density, the organic-carbon normalized sorption coefficient (Koc) first rose till the maximum, and then began to decreases the packing density further increased. This could be interpreted that increase of surfactant packing density would render the surfactant phasesore hydrophobic environment, and the hydrophobic affinity of the surfactant phases towards the solutes thus increased accordingly. However, in
he high surfactant packing density region, the densely packed surfactants reduced the available free space for the solutes, resulting in decreasef sorption capacity for these complexes. Hence, with the increase of surfactant packing density, the adsorbed surfactants would form a series ofartition phases showing different affinity to the solutes. 2006 Elsevier B.V. All rights reserved.
s; Pa
sstct(efKtost
eywords: Sorption; Organic clay; Surfactant; Hydrophobic organic compound
. Introduction
Numerous studies have investigated the sorption characteris-ics of surfactant–clay complexes towards hydrophobic organicompounds (HOCs) because of their wide variety of applications1–7]. The hydrophobic medium created by the long alkyl chainf the adsorbed surfactant shows high affinity to HOCs, and thushe complexes are considered as potential sorbents in wastew-ter treatment [1–4], appropriate landfill liner [5] and effectivearrier to prevent down-gradient pollution of groundwater andquifer from organic pollutants [6,7].
Partition of HOCs to the organic phase created by thedsorbed surfactants was considered to be the predominant
echanism controlling sorption of HOCs on surfactant–clayomplexes [4,8], and the sorption coefficient, Kd (the ratio ofolutes concentration on the sorbents to that in solution), was
∗ Corresponding author. Tel.: +86 571 88273733; fax: +86 571 88273450.E-mail address: [email protected] (L. Zhu).
a
trttt
927-7757/$ – see front matter © 2006 Elsevier B.V. All rights reserved.oi:10.1016/j.colsurfa.2006.08.016
rtition phase; Layer charge
uggested to be proportional to the amount of the adsorbedurfactant [9,10]. However, an increasing number of inves-igations have observed that sorption characteristics of theseomplexes are strongly dependent on their configurations, andhe organic-carbon content (foc) normalized sorption coefficientKoc = Kd/foc) can be quite different accordingly [11–14]. Smitht al. [11] observed that the surfactant–clay complexes preparedrom the surfactant with relatively longer alkyl chain had largeroc for HOCs. Esumi et al. [12] found that silica modified from
he surfactant with two alkyl chains had larger Koc than that withne or three alkyl chains in sorption of 2-naphthol. Even for theurfactant–clay complexes synthesized from the same surfac-ant, Koc can be greatly influenced by the surfactant loadingmount [3,13,14].
Although the idea that Koc can vary with the configurations ofhe surfactant–clay complexes has gradually been accepted, the
elationship between the configuration and sorption characteris-ics of these complexes is still ambiguous. Sorption properties ofhe surfactant–clay complexes should be ascribed to their struc-ural difference. In fact, microstructures of the surfactant–clay
2 ysico
celslssnfsihiwept
ipsspmliPastspaH
2
2
MI[inCv
2
tcpKtb
dibt[[atwudwv16L
loa7nftpCrC
2
Topswg
2
t3ciTiciatc
22 R. Zhu et al. / Colloids and Surfaces A: Ph
omplexes have been extensively studied in many fields. Forxample, with the increase of alkyl chain length or surfactantoading amount, researchers have observed that the adsorbedurfactants will adopt progressively from liquid-like to solid-ike conformation [15,16]. One would ask then, as the adsorbedurfactant adopted different conformation, what difference theorption characteristics of these hybrids would be? Unfortu-ately, researches about the influence of the adsorbed sur-actants microstructure on the sorption characteristics of theurfactant–clay complexes were far from enough. One interest-ng phenomenon should be noted was that, above limited reportsad implied that Koc values had a close relationship to the pack-ng density of the intercalated surfactants [11–13], although thisas not generally stated in these respective reports. This enlight-
ns us that packing density of the adsorbed surfactant is oneroper factor to bridge the microstructure and sorption charac-eristics of the surfactant–clay complexes.
The objective of this work was to further examine thenfluence of surfactant packing density on Koc, and toresent the possible explanations for the observed relation-hip between microstructure and sorption characteristics of theurfactant–clay complexes. Series of CTMA–bentonite com-lexes were synthesized by controlling both cetyltrimethylam-onium bromides (CTMAB) loading amount and bentonite
ayer charge. Layer charge of bentonites was controlled by heat-ng the Li+ saturated bentonite at different temperatures [17].henol and naphthalene were selected as representatives of polarnd nonpolar organic compounds, respectively, to examine theorption characteristics of the obtained complexes. Conforma-ion of the adsorbed surfactant was characterized with FTIRpectra. Affinity of the surfactant micelle and the obtained com-lexes towards the organic solutes was also compared to providedditional information to the interaction of surfactants withOCs.
. Materials and methods
.1. Materials
The natural bentonite was primarily calcium form from Inner-ongolia, China, with montmorillonite purity more than 95%.
ts structural formula is Na0.02K0.02Ca0.39 [Fe0.45Mg1.10Al2.51]Si7.91Al0.09]O20(OH)4·nH2O. The total cation exchange capac-ty (CEC) was 108 meq 100 g−1. CTMAB, LiCl, phenol andaphthalene were of analytical grade, obtained from Shanghaihemical Co., China. The critical micelle concentration (CMC)alue of CTMAB was 0.9 mmol L−1 [18].
.2. Preparation of CTMA–bentonite complexes
The natural Ca2+ bentonite was first Li+ saturated [17], andhe obtained product was referred as Li-108Bent. The reduced-harge bentonites were prepared by heating the Li-108Bent sam-
le at 110, 120 and 135 ◦C for 24 h. According to the Hofmann-lemen effect [17], small Li+ can migrate into the bentonite lat-ice upon heating at proper temperature and cannot be exchangedy other cations. As a result, CEC of the heated Li+ bentonite
a
sw
chem. Eng. Aspects 294 (2007) 221–227
ecreases. Amount of migrated Li+ cations (i.e., loss of CEC)s just proportion to the heating temperature. Thereby, CEC ofentonite can be controlled with this method. CEC values ofhe products were determined with the [Co(NH3)6]3+ method19]. Briefly, 1.0 g bentonite samples were mixed with 20 mLCo(NH3)6]Cl solution with a concentration of 0.025 mol L−1,nd shaken at 20 ◦C for 24 h for cationic exchange. The concen-ration of the [Co(NH3)6]Cl solutions after cationic exchangeere determined at 474 nm on a spectrometer. The CEC val-es of the samples were thus calculated from the concentrationifference, and the obtained value for the original bentoniteas 110 meq 100 g−1, well in agreement with the calculatedalue from the structural formula. For the samples obtained at10, 120 and 135 ◦C, the measured CEC values were 97, 85,5 meq 100 g−1, respectively. The products were referred to asi-97Bent, Li-85Bent and Li-65Bent accordingly.
CTMA–bentonite complexes were synthesized with the fol-owing procedures: a 5.0 g amount of bentonite with vari-us layer charges was dispersed in 200 mL of distilled water,nd then a desired amount of CTMAB varying from 158 to90 mg g−1 bentonite was added to saturate 40–200% of theatural bentonite’s CEC. The suspensions were stirred at 50 ◦Cor 10 h. The products were washed with distilled water for 5imes, and dried at 80 ◦C. The final products were grounded toass 100 mesh sieves. The obtained products were denoted asTMA-xBent (x = CEC of bentonite), e.g., CTMA-97Bent rep-
esented the produces that were obtained from the bentonite withEC of 97 meq 100 g−1.
.3. Characterization
Organic-carbon content was analyzed with a SHIMADZUOC-V CPH organic-carbon analyzer. The natural bentonite hadrganic-carbon content less than 0.04%. FTIR spectra using KBrressed disk technique on a Thermo Nicolet Nexus 670 FTIRpectrophotometer. FTIR spectra in the range of 4000–400 cm−1
ere recorded with a resolution of 4 cm−1, and 64 interfero-rams were collected.
.4. Procedures of sorption
Sorption experiments were carried out with batch equilibra-ion technique. 0.05–0.3 g (to obtain the removal rates within0–90%) of the complexes was combined with 20 mL solutionontaining 5–200 mg L−1 phenol or 0.5–10 mg L−1naphthalenen 25 mL centrifuge tubes, and sealed with Teflon-lined caps.he tubes were shaken at 25 ◦C for 4 h (preliminary experiments
ndicated 2 h were enough to obtain sorption equilibrium). Afterentrifugation at 4000 rpm for 20 min, the solute concentrationn solution was detected with UV spectrophotometer (with thebsorbance wavelength 270 nm for phenol and 219 nm for naph-halene, respectively). The sorbed amounts of the solutes werealculated by the concentration difference between the initial
nd equilibrium concentration.Control experiments (without addition of complexes) demon-trated that loss of phenol during the period of experimentsas negligible, and loss of naphthalene was less than 3%.
Physicochem. Eng. Aspects 294 (2007) 221–227 223
O(foro
2
0cawaaws
3
3b
aCfiftsfsfhs
aTsow[cbrstwfssaw6ν
Ft
tcoes
ipTammtd
R. Zhu et al. / Colloids and Surfaces A:
rganic-carbon analysis of another series control of sampleswithout addition of solutes) demonstrated that the released sur-actant amounts were far below its CMC. Thus, solubilizationf the solutes by the released surfactants was negligible, and theeleased surfactants had no apparent influence on the detectingf the solutes.
.5. Solubility enhancement of organic solutes by CTMAB
CTMAB solutions with various concentrations (ranging from.1 to 20 CMC) were placed in centrifuge tubes with Teflon-linedaps, and the solutes were subsequently added to the tubes inmount more than required to saturate the solution. The samplesere shaken at 25 ◦C for 48 h on a gyratory shaker at 150 rpm,
nd then centrifuged at 4000 rpm for 20 min to completely sep-rate the undissolved portion. The concentrations of the solutesere determined by UV spectrophotometer, and their apparent
olubility was calculated.
. Results and discussion
.1. Packing density of the adsorbed surfactant onentonites
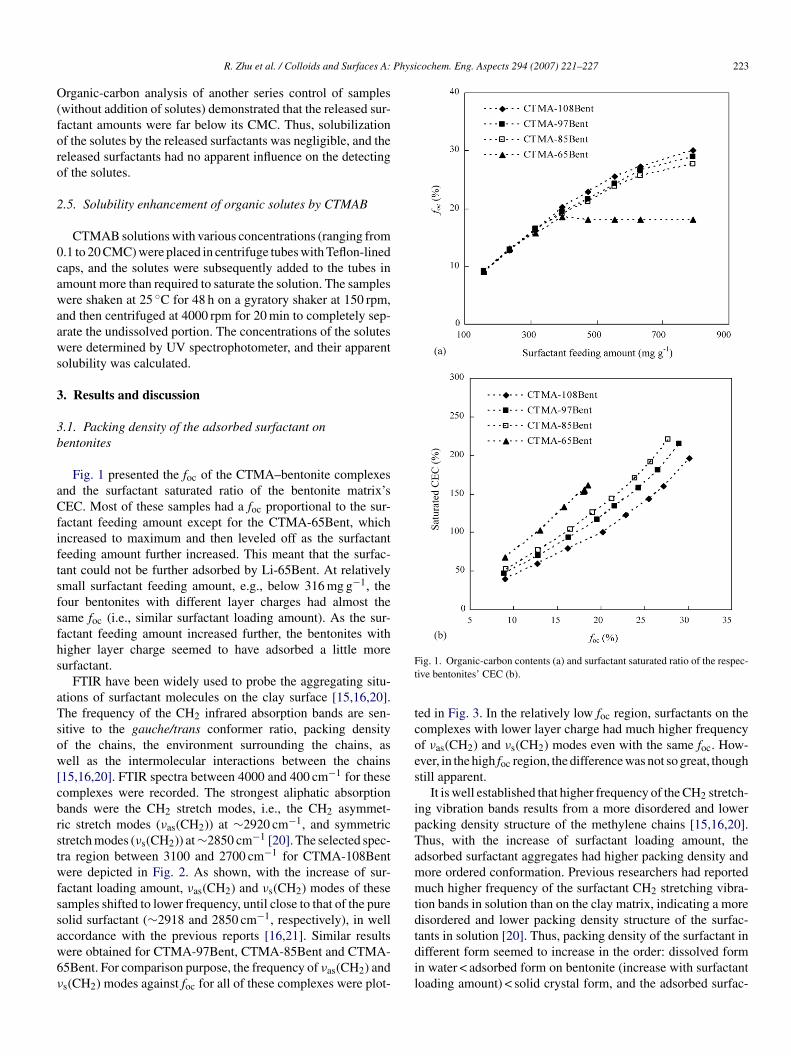
Fig. 1 presented the foc of the CTMA–bentonite complexesnd the surfactant saturated ratio of the bentonite matrix’sEC. Most of these samples had a foc proportional to the sur-
actant feeding amount except for the CTMA-65Bent, whichncreased to maximum and then leveled off as the surfactanteeding amount further increased. This meant that the surfac-ant could not be further adsorbed by Li-65Bent. At relativelymall surfactant feeding amount, e.g., below 316 mg g−1, theour bentonites with different layer charges had almost theame foc (i.e., similar surfactant loading amount). As the sur-actant feeding amount increased further, the bentonites withigher layer charge seemed to have adsorbed a little moreurfactant.
FTIR have been widely used to probe the aggregating situ-tions of surfactant molecules on the clay surface [15,16,20].he frequency of the CH2 infrared absorption bands are sen-itive to the gauche/trans conformer ratio, packing densityf the chains, the environment surrounding the chains, asell as the intermolecular interactions between the chains
15,16,20]. FTIR spectra between 4000 and 400 cm−1 for theseomplexes were recorded. The strongest aliphatic absorptionands were the CH2 stretch modes, i.e., the CH2 asymmet-ic stretch modes (νas(CH2)) at ∼2920 cm−1, and symmetrictretch modes (νs(CH2)) at ∼2850 cm−1 [20]. The selected spec-ra region between 3100 and 2700 cm−1 for CTMA-108Bentere depicted in Fig. 2. As shown, with the increase of sur-
actant loading amount, νas(CH2) and νs(CH2) modes of theseamples shifted to lower frequency, until close to that of the pureolid surfactant (∼2918 and 2850 cm−1, respectively), in well
ccordance with the previous reports [16,21]. Similar resultsere obtained for CTMA-97Bent, CTMA-85Bent and CTMA-5Bent. For comparison purpose, the frequency of νas(CH2) ands(CH2) modes against foc for all of these complexes were plot-tdil
ig. 1. Organic-carbon contents (a) and surfactant saturated ratio of the respec-ive bentonites’ CEC (b).
ed in Fig. 3. In the relatively low foc region, surfactants on theomplexes with lower layer charge had much higher frequencyf νas(CH2) and νs(CH2) modes even with the same foc. How-ver, in the high foc region, the difference was not so great, thoughtill apparent.
It is well established that higher frequency of the CH2 stretch-ng vibration bands results from a more disordered and loweracking density structure of the methylene chains [15,16,20].hus, with the increase of surfactant loading amount, thedsorbed surfactant aggregates had higher packing density andore ordered conformation. Previous researchers had reporteduch higher frequency of the surfactant CH2 stretching vibra-
ion bands in solution than on the clay matrix, indicating a moreisordered and lower packing density structure of the surfac-
ants in solution [20]. Thus, packing density of the surfactant inifferent form seemed to increase in the order: dissolved formn water < adsorbed form on bentonite (increase with surfactantoading amount) < solid crystal form, and the adsorbed surfac-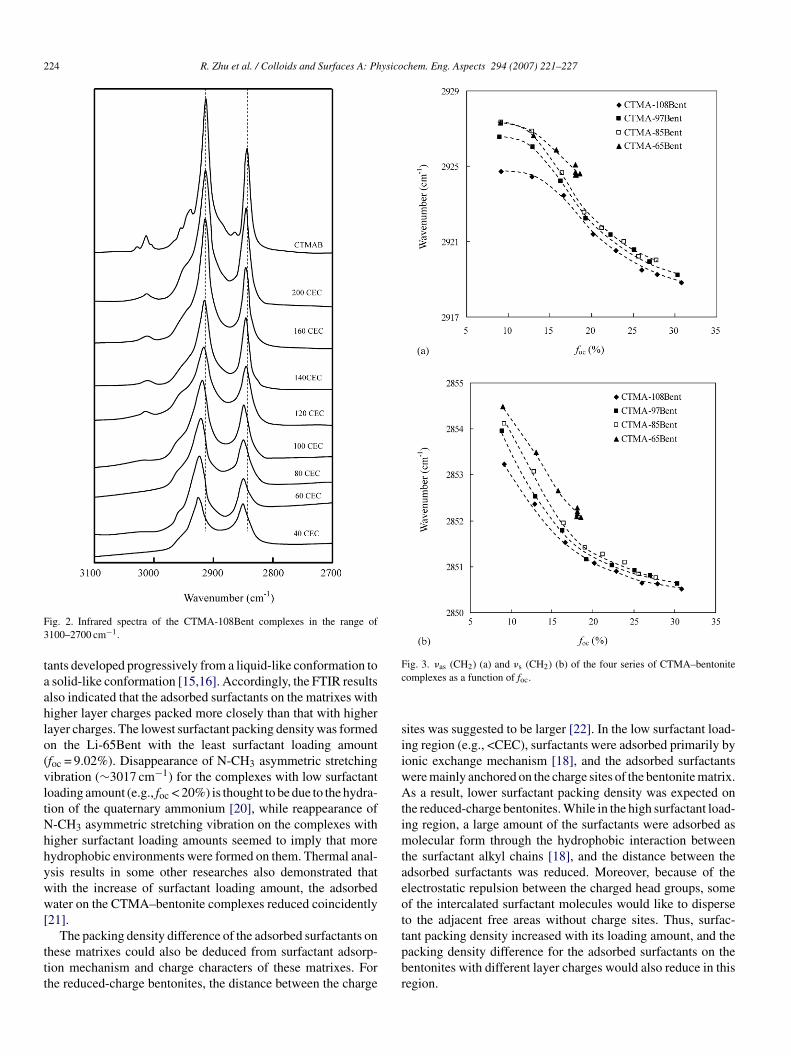
224 R. Zhu et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 294 (2007) 221–227
F3
taahlo(vltNhhyww[
ttt
Fc
siiwAtimtaeot
ig. 2. Infrared spectra of the CTMA-108Bent complexes in the range of100–2700 cm−1.
ants developed progressively from a liquid-like conformation tosolid-like conformation [15,16]. Accordingly, the FTIR resultslso indicated that the adsorbed surfactants on the matrixes withigher layer charges packed more closely than that with higherayer charges. The lowest surfactant packing density was formedn the Li-65Bent with the least surfactant loading amountfoc = 9.02%). Disappearance of N-CH3 asymmetric stretchingibration (∼3017 cm−1) for the complexes with low surfactantoading amount (e.g., foc < 20%) is thought to be due to the hydra-ion of the quaternary ammonium [20], while reappearance of-CH3 asymmetric stretching vibration on the complexes withigher surfactant loading amounts seemed to imply that moreydrophobic environments were formed on them. Thermal anal-sis results in some other researches also demonstrated thatith the increase of surfactant loading amount, the adsorbedater on the CTMA–bentonite complexes reduced coincidently
21].
The packing density difference of the adsorbed surfactants onhese matrixes could also be deduced from surfactant adsorp-ion mechanism and charge characters of these matrixes. Forhe reduced-charge bentonites, the distance between the charge
tpbr
ig. 3. �as (CH2) (a) and �s (CH2) (b) of the four series of CTMA–bentoniteomplexes as a function of foc.
ites was suggested to be larger [22]. In the low surfactant load-ng region (e.g., <CEC), surfactants were adsorbed primarily byonic exchange mechanism [18], and the adsorbed surfactantsere mainly anchored on the charge sites of the bentonite matrix.s a result, lower surfactant packing density was expected on
he reduced-charge bentonites. While in the high surfactant load-ng region, a large amount of the surfactants were adsorbed asolecular form through the hydrophobic interaction between
he surfactant alkyl chains [18], and the distance between thedsorbed surfactants was reduced. Moreover, because of thelectrostatic repulsion between the charged head groups, somef the intercalated surfactant molecules would like to disperseo the adjacent free areas without charge sites. Thus, surfac-
ant packing density increased with its loading amount, and theacking density difference for the adsorbed surfactants on theentonites with different layer charges would also reduce in thisegion.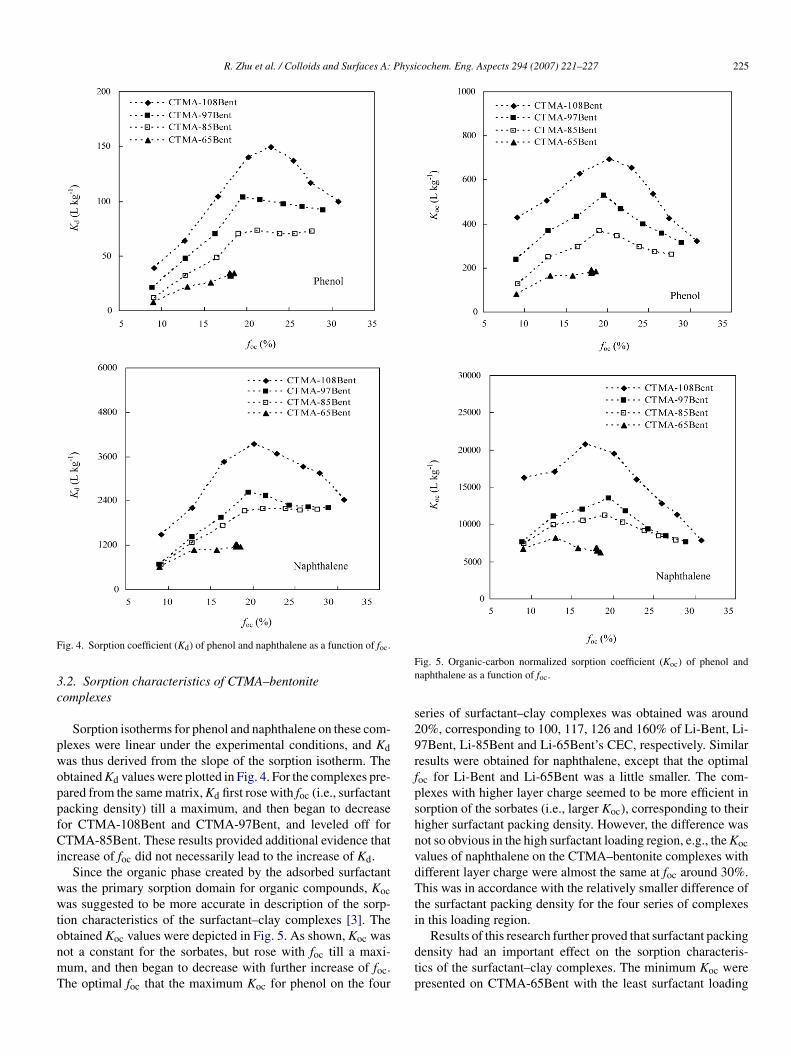
R. Zhu et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 294 (2007) 221–227 225
F
3c
pwoppfCi
wwtonmT
Fn
s29rfpshnvdTti
ig. 4. Sorption coefficient (Kd) of phenol and naphthalene as a function of foc.
.2. Sorption characteristics of CTMA–bentoniteomplexes
Sorption isotherms for phenol and naphthalene on these com-lexes were linear under the experimental conditions, and Kdas thus derived from the slope of the sorption isotherm. Thebtained Kd values were plotted in Fig. 4. For the complexes pre-ared from the same matrix, Kd first rose with foc (i.e., surfactantacking density) till a maximum, and then began to decreaseor CTMA-108Bent and CTMA-97Bent, and leveled off forTMA-85Bent. These results provided additional evidence that
ncrease of foc did not necessarily lead to the increase of Kd.Since the organic phase created by the adsorbed surfactant
as the primary sorption domain for organic compounds, Kocas suggested to be more accurate in description of the sorp-
ion characteristics of the surfactant–clay complexes [3]. The
btained Koc values were depicted in Fig. 5. As shown, Koc wasot a constant for the sorbates, but rose with foc till a maxi-um, and then began to decrease with further increase of foc.he optimal foc that the maximum Koc for phenol on the fourdtp
ig. 5. Organic-carbon normalized sorption coefficient (Koc) of phenol andaphthalene as a function of foc.
eries of surfactant–clay complexes was obtained was around0%, corresponding to 100, 117, 126 and 160% of Li-Bent, Li-7Bent, Li-85Bent and Li-65Bent’s CEC, respectively. Similaresults were obtained for naphthalene, except that the optimaloc for Li-Bent and Li-65Bent was a little smaller. The com-lexes with higher layer charge seemed to be more efficient inorption of the sorbates (i.e., larger Koc), corresponding to theirigher surfactant packing density. However, the difference wasot so obvious in the high surfactant loading region, e.g., the Kocalues of naphthalene on the CTMA–bentonite complexes withifferent layer charge were almost the same at foc around 30%.his was in accordance with the relatively smaller difference of
he surfactant packing density for the four series of complexesn this loading region.
Results of this research further proved that surfactant packingensity had an important effect on the sorption characteris-ics of the surfactant–clay complexes. The minimum Koc wereresented on CTMA-65Bent with the least surfactant loading
2 ysicochem. Eng. Aspects 294 (2007) 221–227
aspa
btattmaseAtWtatpd
mmwtreopoKitop
fsidpfatdrsHnattide
Fc
3C
wTi
wsiaKa(o1ion
c
K
wbaSn
26 R. Zhu et al. / Colloids and Surfaces A: Ph
mount, corresponding to the most low surfactant packing den-ity. While the maximum Koc for phenol and naphthalene wereresented on CTMA-108Bent with medial surfactant loadingmount (i.e., foc = 20.1% and 16.6%, respectively).
Increase of Koc with surfactant packing density also noticedy some previous researchers [3,23]. However, few microstruc-ure evidences were presented for their explanations. As statedbove, in the low surfactant packing density region (low surfac-ant loading amount or low layer charge), the adsorbed surfac-ants were primarily anchored on the charge sites of the clay
atrix. Hence, a portion of the adsorbed surfactants existeds monomers or small isolated patches in the matrix interlayerpace or surface, and were surrounded by a relatively hydrophilicnvironment (e.g., water or hydrated inorganic cations) [15,24].s a result, the hydrophobic affinity of the adsorbed surfac-
ant towards organic sorbates was low, resulting in small Koc.ith the increase of surfactant packing density (higher surfac-
ant loading amount or layer charge), a larger portion of thedsorbed surfactants were combined with each other, and rela-ively bulk organic phases were formed [24]. The bulk organichases created a more hydrophobic environment to accommo-ate the solutes, resulting in larger Koc.
However, as the surfactant packing density increased to auch higher level (high surfactant loading amount region),ost of the interlayer spaces of the bentonite matrix were filledith the adsorbed surfactants. In the confined clay interlayers,
he available free space to accommodate the organic sorbateseduced greatly, and the densely packed surfactants could notxpand as freely as the loosely packed ones. As a result, therganic sorbates could not penetrate into the density organichase easily. Hence, it is reasonable for the observed decreasef Koc in high surfactant packing density region. Decrease ofoc for the highly packed organic phase had also been observed
n some previous reports [13,14,21]. Li and Bowman [13] foundhat above the monolayer coverage, increase of CTMAB leveln the zeolite surface would form a higher density bound organichase, which resulted in the decrease of Koc.
Above analysis implied that there are primarily twoactors that influence the sorption characteristics of theurfactant–clay complexes. The hydrophobic affinity, originat-ng from hydrophobic environment in the organic phase, is theriving force for the sorption of HOCs on surfactant–clay com-lexes. The steric hindrance, rising from the reduced availableree space in the densely packed organic phase, reduces theccessibility of HOCs to surfactant–clay complexes. Magni-ude of both of them directly relates to the surfactant packingensity. Variation of surfactant packing density changes theelative magnitude of these two factors, resulting in variousorption capacities of the surfactant–clay complexes towardsOCs. In fact, increase of surfactant alkyl chain length andumber also results in higher surfactant packing density [25],nd thus Koc increases coincidently [11,12]. However, forhe surfactant with three alkyl chains, the adsorbed surfac-
ants are packed too closely, and the magnitude of hindrances larger than that of the hydrophobic affinity, resulting inecrease of Koc [12]. These obtained results further support ourxplanations.abma
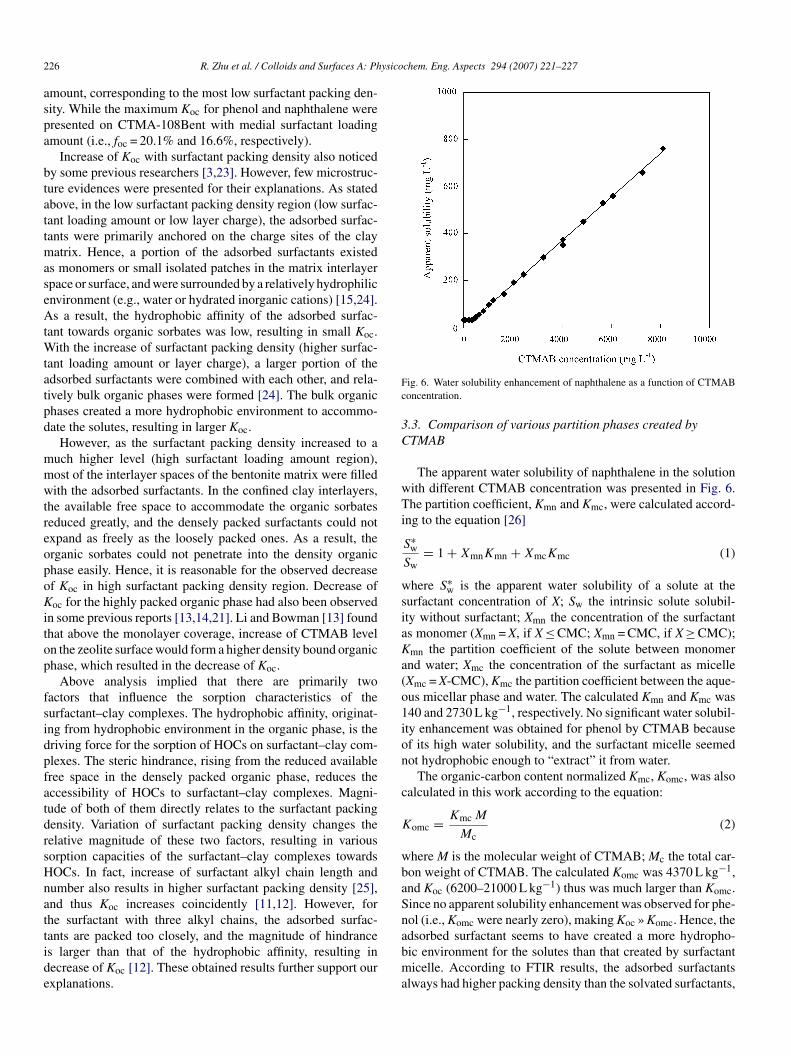
ig. 6. Water solubility enhancement of naphthalene as a function of CTMABoncentration.
.3. Comparison of various partition phases created byTMAB
The apparent water solubility of naphthalene in the solutionith different CTMAB concentration was presented in Fig. 6.he partition coefficient, Kmn and Kmc, were calculated accord-
ng to the equation [26]
S∗w
Sw= 1 + XmnKmn + XmcKmc (1)
here S∗w is the apparent water solubility of a solute at the
urfactant concentration of X; Sw the intrinsic solute solubil-ty without surfactant; Xmn the concentration of the surfactants monomer (Xmn = X, if X ≤ CMC; Xmn = CMC, if X ≥ CMC);mn the partition coefficient of the solute between monomernd water; Xmc the concentration of the surfactant as micelleXmc = X-CMC), Kmc the partition coefficient between the aque-us micellar phase and water. The calculated Kmn and Kmc was40 and 2730 L kg−1, respectively. No significant water solubil-ty enhancement was obtained for phenol by CTMAB becausef its high water solubility, and the surfactant micelle seemedot hydrophobic enough to “extract” it from water.
The organic-carbon content normalized Kmc, Komc, was alsoalculated in this work according to the equation:
omc = Kmc M
Mc(2)
here M is the molecular weight of CTMAB; Mc the total car-on weight of CTMAB. The calculated Komc was 4370 L kg−1,nd Koc (6200–21000 L kg−1) thus was much larger than Komc.ince no apparent solubility enhancement was observed for phe-ol (i.e., Komc were nearly zero), making Koc » Komc. Hence, the
dsorbed surfactant seems to have created a more hydropho-ic environment for the solutes than that created by surfactanticelle. According to FTIR results, the adsorbed surfactantslways had higher packing density than the solvated surfactants,

Physi
ttprafcth
bpf[idhbspotpd
4
mchaistti
oc
A
S
R
[[[[[[[[[[[[[[[
R. Zhu et al. / Colloids and Surfaces A:
hus the obtained larger Koc than Komc further supported the ideahat closely packed surfactants would be a more hydrophobichase for HOCs. Indeed, the role of charged clay matrix can beegarded as to balance the charge of the surfactant’s head group,nd to rearrange the surfactants into a more densely packed con-ormation. For the adsorbed surfactants that adopt a liquid-likeonformation, their sorption characteristics seems to resemblehe micelles, that is, lower hydrophobic affinity and lower stericindrance.
While for the solid-like surfactant phase, their partitionehaviors were more different from the bulk liquid partitionhases, e.g., higher hydrophobic affinity and steric hindrance. Inact, the densely packed solid partition phase, e.g., lipid bilayer27] and plant cuticular materials [28], have been widely stud-ed in many fields. These partition phases are of fundamentallyifferent from the bulk liquid partition phase. The highly packedydrocarbon aggregates cannot expand freely, and the accessi-ility of the sorbates is thus limited [27,28]. Hence, the solid-likeurfactant phase behaves similarly to the densely packed solidartition phase. In conclusion, as the adsorbed surfactants devel-ps progressively from liquid-like to solid-like conformations,he surfactants phase will resemble from bulk liquid partitionhase to densely packed solid partition phase accordingly, ren-ering them various sorption characteristics.
. Conclusion
Surfactant packing density plays an important role in deter-ining the sorption characteristics of the CTMA–bentonite
omplexes. With the increase of surfactant packing density,ydrophobic affinity of the complexes towards HOCs increasesnd accessibility of HOCs to the complexes decreases accord-ngly. As a result, the surfactant phase created by the adsorbed
urfactant will develop progressively from bulk liquid-like parti-ion phase to densely packed solid-like partition phase, renderinghe complexes various sorption characteristics. It is necessar-ly to control the surfactant packing density in the purpose[[[[
cochem. Eng. Aspects 294 (2007) 221–227 227
f designing surfactant–clay complexes with optimal sorptionapacity.
cknowledgement
This work was supported by grants from the National Naturalcience Foundation of China (50378081).
eferences
[1] N. Yıldız, R. Gonulsen, H. Koyuncu, A. Calımlı, Coll. Surf. A 260 (2005)87.
[2] S. Andini, R. Cioffi, F. Montagnaro, F. Pisciotta, L. Santoro, Appl. ClaySci. 31 (2006) 126.
[3] S.L. Bartelt-Hunt, S.E. Burns, J.A. Smith, J. Coll. Interf. Sci. 266 (2003)251.
[4] R.S. Juang, S.H. Lin, K.H. Tsao, J. Coll. Interf. Sci. 254 (2002) 234.[5] I.M.C. Lo, J. Environ. Eng. -ASCE. 127 (2001) 756.[6] J.F. Lee, J.R. Crum, S.A. Boyd, Environ. Sci. Technol. 23 (1989) 1365.[7] J.M. Brixie, S.A. Boyd, J. Environ. Qual. 6 (1994) 1283.[8] S.A. Boyd, S. Sun, J.F. Lee, M.M. Mortland, Clays Clay Miner. 36 (1988).[9] B. Chen, L. Zhu, J. Environ. Sci. 13 (2001) 129.10] S.Y. Lee, S.J. Kim, S.Y. Chung, C.H. Jeong, Chemosphere 55 (2004) 781.11] J.A. Smith, P.R. Jaffe, C.T. Chiou, Environ. Sci. Technol. 24 (1990) 1167.12] K. Esumi, M. Matoba, Y. Yamanaka, Langmuir 12 (1996) 2130.13] H.Z. Li, R.S. Bowman, Environ. Sci. Technol. 32 (1998) 2278.14] L. Zhu, B. Chen, S. Tao, C.T. Chiou, Environ. Sci. Technol. 37 (2003) 4001.15] R.A. Vaia, R.K. Teukolsky, E.P. Giannelis, Chem. Mater. 6 (1994) 1017.16] H. He, F.L. Ray, J. Zhu, Spectrochim. Acta Part A 60 (2004) 2853.17] J. Hrobarikova, J. Madejova, P. Komadel, J. Mater. Chem. 11 (2001) 1452.18] S. Xu, S.A. Boyd, Langmuir 11 (1995) 2508.19] X. Hu, G. Lu, Y. Yang, Chin. J. Anal. Chem. 28 (2000) 1402.20] K.S. Kung, K.F. Hayes, Langmuir 9 (1993) 163.21] B. Chen, L. Zhu, J. Zhu, B. Xing, Environ. Sci. Technol. 39 (2005) 6093.22] J.J. Stevens, S.J. Anderson, S.A. Boyd, Clays Clay Miner. 44 (1996) 88.23] S. Sun, P.R. Jaffe, Environ. Sci. Technol. 30 (1996) 2906.24] S.Y. Lee, S.J. Kim, Clays Clay Miner. 50 (2002) 435.
25] M.A. Osman, M. Ploetze, P. Skrabal, J. Phys. Chem. B. 108 (2004) 2580.26] D.K. Kile, C.T. Chiou, Environ. Sci. Technol. 23 (1989) 832.27] L.R. De Young, K.A. Dill, Biochemistry 27 (1988) 5289.28] B. Chen, E.J. Johnson, B. Xing, L. Zhu, B. Chefetz, Environ. Sci. Technol.39 (2005) 6138.




















