
Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo...
33
For Peer Review Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo D178N mutation in the PRNP gene Journal: American Journal of Medical Genetics Part B: Neuropsychiatric Genetics Manuscript ID: NPG-09-0272.R1 Wiley - Manuscript type: Research Article Date Submitted by the Author: Complete List of Authors: Alzualde, Ainhoa; BioDonostia Institute Moreno, Fermin; Hospital Donostia - Osakidetza, Neurology; CIBERNED Martinez-Lage, Pablo; Fundacion ACE Ferrer, Isidre; IDIBELL-Hospital Universitari de Bellvitge, Anatomical Pathology; CIBERNED Gorostidi, Ana; BioDonostia Institute, Neurosciences; CIBERNED Otaegui, David; BioDonostia Institute, Neurosciences Blazquez, Lorea; BioDonostia Institute, Neurosciences; CIBERNED Atares, Begoña; Hospital Txagorritxu - Osakidetza, Anatomical Pathology Cardoso, Sergio; University of the Basque Country, DNA Bank Martinez de Pancorbo, Marian; University of the Basque Country, DNA Bank Juste, Ramon; NEIKER-Tecnalia, Animal Health Rodriguez-Martinez, Ana Belen; NEIKER-Tecnalia, Animal Health Indakoetxea, Begoña; Hospital Donostia, Neurology; CIBERNED Lopez de Munain, Adolfo; Hospital Donostia - Osakidetza, Neurology; CIBERNED Keywords: prion protein, de novo mutation, sporadic CJD, genetic counseling John Wiley & Sons, Inc. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
Transcript of Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo...

For Peer Review
Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo D178N
mutation in the PRNP gene
Journal: American Journal of Medical Genetics Part B: Neuropsychiatric
Genetics
Manuscript ID: NPG-09-0272.R1
Wiley - Manuscript type: Research Article
Date Submitted by the Author:
Complete List of Authors: Alzualde, Ainhoa; BioDonostia Institute Moreno, Fermin; Hospital Donostia - Osakidetza, Neurology; CIBERNED Martinez-Lage, Pablo; Fundacion ACE Ferrer, Isidre; IDIBELL-Hospital Universitari de Bellvitge, Anatomical Pathology; CIBERNED Gorostidi, Ana; BioDonostia Institute, Neurosciences; CIBERNED Otaegui, David; BioDonostia Institute, Neurosciences Blazquez, Lorea; BioDonostia Institute, Neurosciences; CIBERNED Atares, Begoña; Hospital Txagorritxu - Osakidetza, Anatomical Pathology Cardoso, Sergio; University of the Basque Country, DNA Bank Martinez de Pancorbo, Marian; University of the Basque Country, DNA Bank Juste, Ramon; NEIKER-Tecnalia, Animal Health Rodriguez-Martinez, Ana Belen; NEIKER-Tecnalia, Animal Health Indakoetxea, Begoña; Hospital Donostia, Neurology; CIBERNED Lopez de Munain, Adolfo; Hospital Donostia - Osakidetza, Neurology; CIBERNED
Keywords: prion protein, de novo mutation, sporadic CJD, genetic counseling
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics

For Peer Review
1
Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease
carrying a de novo D178N mutation in the PRNP gene.
Running head title: PRNP somatic mosaicism in an apparently sCJD
A. Alzualde1,7*
, F. Moreno 2,7*
, P. Martinez-Lage3, I. Ferrer
8,7, A. Gorostidi
1,7, D.
Otaegui 1
, L. Blázquez 1,7
, B. Atares, 4
S. Cardoso,5
M. Martínez de Pancorbo, 5
R. Juste
6, A.B. Rodríguez-Martínez
6, B. Indakoetxea
2,7, A. Lopez de Munain
1,2,7.
* Both authors contributed equally
Corresponding author:
Ainhoa Alzualde PhD
Ilundain Fundazioa- Instituto Biodonostia
Unidad Experimental, Edificio materno-infantil,-3 azul, Hospital Donostia,
Pº Dr. Beguiristain s/n, 20014, San Sebastián, Spain
e-mail: [email protected]
Telephone number: +34 943007061; Fax number: +34 943007061
1 Unidad Neurociencias, Instituto de Investigación Biodonostia, San Sebastián, Spain
2 Servicio de Neurología, Hospital Donostia, Osakidetza, San Sebastián, Spain
3Fundación ACE, Barcelona, Spain
4 Servicio de Anatomía Patológica, Hospital de Txagorritxu, Osakidetza, Vitoria, Spain
5 Departamento de Zoología y Dinámica Celular, Facultad de Farmacia, Banco de ADN,
Universidad del País Vasco- Euskal Herriko Unibertsitatea, Vitoria-Gasteiz, Spain
6Instituto Vasco de Investigación y Desarrollo Agrario, NEIKER, Derio, Spain
7CIBERNED (Centro de Investigación Biomédica en Red de Enfermedades
Neurodegenerativas), Instituto de Salud Carlos III, Spain
Page 1 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
2
8Institut de Neuropatologia, Servei Anatomia Patològica, IDIBELL-Hospital
Universitari de Bellvitge, Universitat de Barcelona, Hospitalet de LLobregat, Spain
Page 2 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
3
ABSTRACT
Transmissible spongiform encephalopathies (TSEs) are a group of rare fatal
neurodegenerative disorders. Creutzfeldt-Jakob disease (CJD) represents the most
common form of TSE and can be classified into sporadic, genetic, iatrogenic and variant
forms. Genetic cases are related to prion protein gene mutations but they only account
for 10-20% of cases. Here we report an apparently sporadic CJD case with negative
family history carrying a mutation at codon 178 of prion protein gene. This mutation is
a de novo mutation as the parents of the case do not show it. Furthermore the presence
of three different alleles (wild type 129M-178D and 129V-178D and mutated 129V-
178N), confirmed by different methods, indicates that this de novo mutation is a post-
zygotic mutation that produces somatic mosaicism. The proportion of mutated cells in
peripheral blood cells and in brain tissue was similar and was estimated at
approximately 97%, suggesting that the mutation occurred at an early stage of
embryogenesis. Neuropathological examination disclosed spongiform change mainly
involving the caudate and putamen, and the cerebral cortex, together with proteinase K-
resistant PrP globular deposits in the cerebrum and cerebellum. PrP typing was
characterized by a lower band of 21 kDa. This is the first case of mosaicism described
in prion diseases and illustrates a potential etiology for apparently sporadic
neurodegenerative diseases. In light of this case, genetic counseling for inherited and
sporadic forms of transmissible encephalopathies should take into account this
possibility for genetic screening procedures.
Keywords: sporadic CJD; prion protein; de novo mutation; genetic counseling
Page 3 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
4
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases,
comprise a group of rare fatal neurodegenerative disorders characterized by the
accumulation of a pathological isoform of the cellular prion protein (PrPC), called PrP
Sc
(PrP scrapie), generated by post-translational modification of PrPC (Prusiner ,1991).
Scrapie and bovine spongiform encephalopathy are the most frequent prion diseases in
sheep and cattle, respectively (Dearmond and Prusiner ,1995). Creutzfeldt-Jakob disease
(CJD) is the most common human TSE and may occur as sporadic (sCJD), genetic
(gCJD) and iatrogenic (iCJD) diseases (Budka et al., 2003; Ironside et al., 2008;
Ironside and Head ,2008; Ricketts and Pergami ,2003). Another form, the new variant
of CJD (vCJD), is caused by the transmission of the bovine spongiform encephalopathy
to humans (Ironside et al., 2003). Sporadic CJD accounts for 80 to 90% of all cases,
with an estimated incidence of 1-1.5 per million and year (Budka et al., 2003).
Genetic CJD is associated with various mutations within the human prion protein gene
(PRNP) (Dearmond and Prusiner ,1995; Parchi and Gambetti ,1995). One of the most
frequent mutations is located at codon 178 (Kovacs et al., 2005) and consists of an A →
G transition that results in an amino acid change from aspartic acid to asparagine. The
D178N point mutation is linked to two distinct phenotypes: Fatal Familial Insomnia
(FFI) which is associated with methionine at codon 129, and genetic Creutzfeldt-Jakob
disease D178N (gCJD D178N) when associated with a valine at codon 129 (Goldfarb et
al., 1992).
According to The European Creutzfeldt-Jakob disease Surveillance System a
collaborative surveillance project (Kovacs et al., 2005), 455 (10.2%) of all 4,441 TSE
cases registered in Europe between 1993 and 2002 were genetic. The D178N mutation
was present in 82 cases (18.02% of all genetic cases) including 16 with gCJD (D178N-
Page 4 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
5
129V) and 66 with FFI (D178N-129M). Interestingly, up to 47% of all genetic cases
showed no family history (Kovacs et al., 2005). In China, Zheng and colleagues found
PRNP mutations in 5 of 185 patients with sporadic neurodegenerative dementias (Zheng
et al., 2008). These sporadic-genetic cases could be explained partially by mutations
that appear de novo (Dagvadorj et al., 2002). Since de novo mutations can affect germ
lines and somatic cells, mosaicism may explain sCJD cases that carry a mutation in
PRNP. Somatic mosaicism has not yet been described in prion diseases.
Here we report the first description of a genetic CJD patient with a de novo mutation in
the 178 codon of PRNP showing a somatic mosaicism caused by a post-zygotic
mutation who presented a clinical phenotype of sCJD.
Page 5 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
6
MATERIAL AND METHODS
DNA was extracted from whole peripheral blood cells (PBC) by standard procedures
for genomic studies. DNA was also obtained from nine brain regions (frontal, temporal,
occipital and parietal cortices, cerebellum, caudate nucleus, brainstem, thalamus and
striatum) using QIAmp DNA mini kit (QIAGEN) following the recommendations of the
supplier.
PRNP amplification and sequencing
The PRNP open reading frame (ORF) was amplified by PCR in two different amplicons
using 5’ORF-PRNP Fw and Rev and 3’ORF-PRNP Fw and Rev primers (Table 1) and
Phusion® Hot Start High-Fidelity DNA Polymerase (Fynnzymes, Finland). PCR
products were sequenced in both directions using M13(-21) universal primers on an
ABI 3130 system with Big Dye v3.1 following the manufacturer’s protocol (Applied
Biosystems, USA).
Determination of the linkage phase of codons 178D/N-129M/V
A fragment of 300 bp was amplified using primers PORF-L and PORF-H (Table 1)
followed by double restriction enzyme digestion, using enzymes Tth111I and
HpyCH4IV (New England Biolabs, USA), as described elsewhere (Rodriguez-Martinez
et al., 2008).
To further characterise the genomic region surrounding the PRNP gene, four
microsatellite loci (MsP1, MsP2, MsP4 and MsP5) (Rodriguez-Martinez et al., 2005)
and five single nucleotide polymorphisms (rs2756271, rs6037932, rs13045348,
rs6116474 and rs6116475) (Rodriguez-Martinez et al., 2008) were analysed in the
patient and the parents.
Page 6 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
7
PCR product cloning and sequencing
A fragment of PRNP which includes the sequence for codon 129 and codon 178 was
amplified by PCR using primers that we designed (PRNP Fw and PRNP Rev) (Table 1).
The PCR product was A tailed, purified and ligated to a pGEM-T vector (pGEM-T and
pGEM-T Easy Vector Systems, Promega, USA). These vectors were transformed into
One Shot TOP 10 Competent E. coli (Invitrogen, USA) and cells were plated and
incubated in agar (1% ampicilin) at 37ºC overnight. The colonies were picked and
incubated in liquid media (1% ampicillin) 4-5 hours. A PCR was performed with
PGEM-T Fw and Rev primers (Table 1) and those that had the vector inserted were
sequenced on an ABI 3130 system.
Assessment of biological paternity
Paternity was verified by the analysis of 15 microsatellite loci (AmpFlSTR®
Identifiler™ PCR Amplification Kit, Applied Biosystems); 10 of them were further
confirmed using the AmpFlSTR® SGM Plus™ PCR Amplification Kit (Applied
Biosystems). The index of paternity was calculated from the probability of paternity
obtained by Familias v1.6 software (Egeland et al., 2000).
Allelic dosage
Two different methods were used to estimate the proportion between wild-type 129V-
178D and mutant 129V-178N alleles:
A. Quantitative PCR SNP detection. A fragment of PRNP was amplified as mentioned
in the paragraph PCR product cloning and sequencing. Amplification product was
digested by BsaAI restriction enzyme which cuts the amplified product corresponding
Page 7 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
8
to 129V, thus allowing discrimination between 129V and 129M allele fractions.
Digestion product was run and the DNA fragment of 311bp which corresponds to 129V
allele was purified from the agarose gel using QIAquick Gel Extraction Kit (Qiagen).
129V allele fractions were analyzed using a 7900HT (Applied Biosystems) and a
Custom TaqMan SNP genotyping assay (Applied Biosystems) to detect the D178N
mutation. An approach based on Ct numbers was used to calculate the percentage of
wild type (178D) and mutated alleles (178N) as follows (Bai and Wong ,2004):
proportion of mutant = 1/(1+1/2∆Ct
) where ∆Ct = Ctwild-type
- Ctmutant
). qPCR analyses
were performed in 9 independent PCRs for each tissue (brain and PBC) and in triplicate
for each PCR. The father’s sample was also processed in triplicate. The approach was
validated with experimental samples containing 80%, 60%, 40% and 20% of mutant
allele generated by mixing known amounts of cloned plasmid DNA. Likewise, controls
containing 100% of mutated and 100% of wild-type alleles were included.
B. AFLP analysis on ABI 3130 system. A labeled 129V allele fraction was obtained as
indicated above but using a tag PRNP Rev primer marked with HEX fluorochrome
instead of PRNP Rev primer. The labeled 129V allele population was digested for 3
hours with Tth111I restriction enzyme, which cuts the fragments corresponding to
178D. This digestion was analyzed on an ABI 3130 system, and the generated data with
GeneMapper software (Applied Biosystems).
Neuropathological study
At autopsy, most of the brain was fixed in buffered 10% formalin for a month
at room temperature. Then, selected tissue blocks were immersed in formic
acid for 1 h, post-fixed in formalin and embedded in paraffin. The sections
were stained with haematoxylin and eosin, periodic acid Shiff (PAS) and
Page 8 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
9
Klüver-Barrera, and processed for immunohistochemistry following the
EnVision + system peroxidase procedure (Dako, Dakopats, Barcelona, Spain)
for glial fibrillary acidic protein, CD68 for microglia, hyper-phosphorylated
tau epitopes (antibody AT8), phosphorylated neurofilament epitopes, β-
amyloid 1-40 and β-amyloid 1-42, α-synuclein, ubiquitin, TDP-43, αB-
crystallin, and prion protein (antibody 3F4) both without and with pre-
treatment with proteinase K.
Western blot analysis
Fresh frozen samples of the frontal, occipital and parietal cortices, cerebellum, and
striatum obtained at autopsy and stored at -80ºC until use were processed for gel
electrophoresis and western blotting (Hill et al., 2006) using the 3F4 monoclonal
antibody for specific staining of PrP. Samples were pre-incubated with proteinase K to
reveal PrPSc
.
Page 9 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
10
RESULTS
Case report
The patient was a 46-year-old lawyer, right-handed, and with no family history of
dementia. He presented with an 18-month history of anxiety, emotional lability,
depressed mood, irritability and insomnia. He responded well to treatment with
venlafaxine and gabapentin but one year later he started to show forgetfulness, difficulty
in recalling recent events, and occasional spatial disorientation. At the first visit, he
showed signs of left corticospinal failure, and he had difficulties performing the tandem
gait test. Formal testing showed reduced verbal fluency, mild anomia, moderate
memory deficits, severe deficit in visual-perspective and visual-constructive abilities,
and mild dysexecutive syndrome. Standard brain Magnetic Resonance Imaging (MRI)
and single photon emission computerized tomography were reported as normal. 18
FDG-
PET (18-Fluoro Deoxy Glucose-Positron Emission Tomography) scan showed cortical
and left thalamic hypometabolism (Supplementary Figure 1). CerebroSpinal fluid was
normal with negative 14-3-3 protein. A new brain MRI including diffusion-weighted
sequences showed cortical hyper-intensities involving frontal, cingulate, temporal,
parietal and occipital regions (Supplementary Figure 2). Two and a half years after the
onset, he developed generalized bradykinesia, postural-kinetic ataxia and tremor of the
left hand, focal myoclonus, and corticospinal signs with generalized brisk tendon
reflexes and left extensor plantar response. Gait was slightly impaired, and accompanied
by instability. Spontaneous speech was non-fluent with some hesitancy and dysprosody.
Electroencephalogram was unremarkable. He could no longer work and required
assistance for most instrumental activities of daily living. The patient deteriorated
during the following two years and he developed widespread spontaneous and action
myoclonus, progressive ataxia, more severe pyramidal signs and eventual akinetic
Page 10 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
11
mutism. He was clinically diagnosed as possible CJD although he would be diagnosed
as probable CJD following the new proposed criteria (Zerr et al., 2009). He died at the
age of 50 years of a respiratory tract infection. Post-mortem examination was performed
15 hours after death, only the brain was removed.
Genotyping of PRNP
PRNP genotyping showed a heterozygous D178N mutation and M129V polymorphism
by sequencing DNA extracted from both PBC and brain cells. The unaffected parents of
the proband were not carriers of the mutation. The genotypes were 129MV-178DD in
his father and 129VV-178DD in his mother. Paternity was confirmed by microsatellite
typing (LR=2.9 E9).
To further characterize this chromosome harboring the mutation, we analysed four
microsatellite loci and five single nucleotide polymorphisms surrounding the PRNP
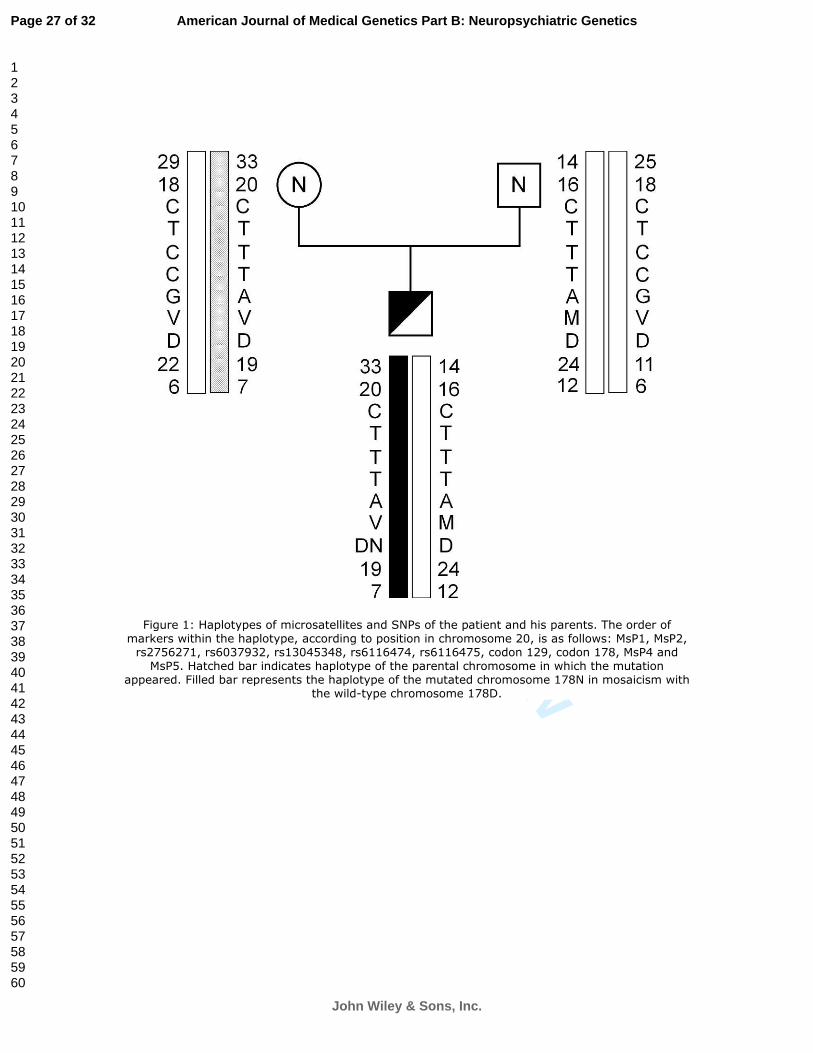
gene (Figure 1). Disease-associated haplotype proved to be 33-20-C-T-T-T-A-129V-
178N-19-7.
Presence of three different alleles
The D178N mutation was determined to be in ligation phase with 129V by double
enzyme digestion of DNA extracted from PBC. Bands corresponding to the mutant
129V-178N and wild-type 129M-178D alleles were detected. In addition, two soft
bands appeared. One of them (300bp) corresponded to the 129M-178N allele but the
presence of this allele could not be confirmed by other techniques, suggesting it may
have been the consequence of partial digestions. The other band (152bp) corresponded
to the wild type 129V-178D allele (Figure 2). Cloning of PCR products of DNA
Page 11 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
12
extracted from PBC containing both 129 and 178 positions confirmed the presence of
three different alleles by sequencing 72 colonies: 129M-178D (40/72), 129V-178N
(30/72) and 129V-178D (2/72). Furthermore, enzymatic digestion with BsaA1 allowed
the selection of the 129V allele population. To confirm the presence of 129V-178D
allele, the fraction corresponding to 129V population was selected and sequenced. As a
result, the electropherogram of DNA extracted from PBC and brain tissue showed a
weak peak of G nucleotide corresponding to 178D overlapping the main A peak
corresponding to mutant 178N allele (Figure 3). The PRNP sequence of the parents
clearly showed only the wild type G peak.
Allele dosage
AFLP analyses failed to estimate the percentage of the two alleles, probably because of
the relaxed sequence recognition shown by Tth111I restriction endonuclease.
Quantitative PCR SNP detection technology (see Material and methods) showed a good
correlation coefficient between expected and observed values (R2=0.991). Then the
proportion of mutated allele in the 129V fraction, which is equal to the percentage of
cells carrying mutation assuming normal chromosomal composition of cells, was
obtained. The analysis showed that 96.7% (±5.5%) of PBC and 97.5% (± 3.3%) of brain
cells were mutated. Controls carrying only mutated or wild type alleles resulted in
99.999% (± 6.7 E-06%) and 2.4 E-10% (± 1.8 E-10%) of mutated alleles, respectively.
The sample from the father showed results within the range of the wild type of controls
(3.7 E-10% of mutated alleles).
Neuropathological study
Page 12 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
13
The brain weighed 1.390 g. The macroscopical examination did not show major
pathologic features. The microscopical examination disclosed variable neuron loss in
the cerebral cortex, accompanied by spongiform change characterized by small,
confluent microvacuoles (Figure 4 A-E). Lesions were more marked in the entorhinal
cortex followed by the frontal, temporal, parietal, occipital and insular cortices, and then
the subiculum. Discrete and focal spongiform degeneration was found in the CA1
region of the hippocampus, whereas the dentate gyrus was spared. The cerebellum
showed moderate loss of Purkinje cells and moderate spongiform change in the
molecular layer (Figure 4 F). Massive spongiform change occurred in the caudate and
putamen with confluent vacuoles and severe neuron loss (Figure 4 G, H). This was in
contrast with the apparent preservation of the globus pallidus (Figure 4 I). The thalamus
showed mild spongiosis.
Astrocytic gliosis was found in the cerebral cortex (Figure 5 A) but astrocytes were
practically absent in the caudate and putamen (Figure 5 B). αB-crystallin
immunohistochemistry disclosed large numbers of immunoreactive neurons in the deep
layers of the frontal and temporal cortex, whereas only weakly immunoreactive cellular
debris was present in the putamen (Fig. 5 C-E). Moderate to severe microgliosis was
similar to that seen in other CJD cases (data not shown).
PrPSc
immunohistochemistry revealed accumulation of PrP resistant to proteinase K in
the cerebral cortex, basal ganglia and cerebellum. PrPSc
deposition was characterized by
globular confluent deposits forming structures reminiscent of morulae. Isolated deposits
were manifested as globules, mainly in the inner layers of the cortex. Similar deposits
were seen in the different regions of the cerebral cortex, including neocortex and
subiculum, and in the cerebellum (Figure 5 F-I). These structures differed from more
common PrPres
deposits in the different forms of sCJD (Ironside et al., 2008). Florid and
Page 13 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
14
kuru-like plaques were absent. A fine, punctate PrP pattern was additionally observed in
sections not treated with proteinase K (data not shown).
β-amyloid plaques were absent. Neurofibrillary tangles and other hyper-phosphorylated
tau inclusions were not observed in any region. α-synuclein inclusions were absent.
TDP-43 immunohistochemistry disclosed no abnormalities in the cellular localization of
the antibody.
PrP typing
Immunoblotting showed positive signal for PrPSc
in all regions analyzed (Figure 6).
Occipital and temporal cortices were the regions with the most intense PrPSc
signal. The
molecular weight of the non-glycosylated band was 21 kDa and it was under-
represented compared with the glycosylated bands (Figure 6 and Supplementary Figure
3). Differences in the prion protein band intensity correlated with PrPSc
deposition as
revealed by immunohistochemistry; higher expression levels were found in the cerebral
cortex, whereas the striatum and cerebellum showed lower PrPSc
expression. Samples of
the proband were processed in parallel with samples from typical CJD PrP type I and
type II, FFI (PRNP D178N-129M) and Gerstmann-Sträussler-Scheinker disease (PRNP
Y218N). As seen in Figure 6, the pattern of the present PRNP D178N 129V mutation
differs from the pattern of other sporadic and inherited prion diseases.
Page 14 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
15
DISCUSSION
Here we report a case of apparently sporadic CJD carrying a de novo D178N mutation
in PRNP. The parents, whose paternity was genetically confirmed, did not carry the
mutation. Some authors have suggested that the global distribution of the D178N
mutation is determined by recurrent mutational events because of the hot-spot nature of
the sequence (Dagvadorj et al., 2002). The characterization in this case of
microsatellites-SNPs haplotypes formed by markers located close to or within PRNP
(expanding 107,3kbp around codon 178) may allow future comparisons with other
gCJD patients with D178N-129V, to clarify a founder effect.
Clinically, this patient did not differ from other cases of sCJD. The neuropathological
study revealed marked involvement of the caudate and putamen, and widespread
distribution of typical lesions in the cerebral cortex, as previously reported in gCJD
D178N-129V (Ironside et al., 2008; Parchi et al., 1996; Parchi et al., 2003). The
characteristics of the PrPSc
deposition are different from what is seen in the distinct
types of sCJD (Ironside et al., 2008). It is here characterized by the presence of small
globules, often confluently forming globular masses reminiscent of morulae that at a
low magnification may resemble patches. Prion typing also differs from typical patterns
encountered in sCJD. As previously reported in gCJD D178N-129V (Haik et al., 2004;
Parchi et al., 1999; Parchi et al., 2003; Parchi et al., 2009), the PrPres
pattern resembles
type I because of the size of the lower band of about 21 kDa, but this lower band is
under-represented in comparison with the glycosylated forms. This pattern is also
similar to that seen in gCJD E200K-129M because of the under-representation of the
lower band, but it is clearly distinct from the FFI profile (D178N-129M) and gCJD
E200K-129V in which the low band has 19 kDa in which the low and under-represented
band has 19 kDa (Gambetti et al., 2003; Parchi et al., 2003). Thus, the E200K and
Page 15 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
16
D178N mutations have opposite PrP types depending on the presence of valine or
methionine in codon 129.
In the present case, the D178N mutation in PRNP is mainly coupled with valine at
codon 129 (129V-178N), but there is a fraction of 129V allele population associated
with wild-type 178D (129V-178D). Therefore, this patient carried three different alleles:
the wild types 129M-178D and 129V-178D, and the mutant 129V-178N. The existence
of these three alleles was confirmed by robust techniques such as the cloning and
sequencing of PCR products. Furthermore, control samples were included in the
different analysis methods and the results were coherent. The mosaic nature implies that
this de novo mutation is a post-zygotic mutation. To our knowledge this patient
represents the first case of a defined somatic mosaicism of a mutation in PRNP.
Recently, it has been suggested that mosaicism is common in all genes throughout the
genome and that a few genetically altered cells in the body may influence diseases such
as cancer or neurodegeneration (Frank ,2009). Somatic mosaicism with development of
specific disease-associated mutations has been suggested as a potential cause of several
hereditary disorders. Thus, somatic mosaicism has been reported in 10-20% of sporadic
cases of retinoblastoma (Sippel et al., 1998), as well as in Duchenne muscular
dystrophy (Passos-Bueno et al., 1992; van Essen et al., 1992) and other diseases
(Ketterling et al., 1999; Leuer et al., 2001; Verhoef et al., 1999).
In our case, approximately 97% of cells of mesoderm-derived PBC and ectoderm-
derived brain cells harbored the D178N mutation. Unfortunately, no endoderm was
available for genetic study. The high, and similar, percentage of mutated cells among
cells developing from different embryonic layers suggests that the mutation occurred at
an early stage of embryogenesis. This is an important issue, as the offspring of a patient
suffering from gCJD has a 50% probability of inheriting the mutation. Yet the
Page 16 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
17
probability of transmission of a de novo PRNP mutation differs depending on pre- or
post-zygotic origin of the mutation. In the former case, 50% of the offspring will carry
the mutation, whereas in the latter the percentage depends on the distribution and
percentage of mutated cells. Therefore, the mutation analysis of PRNP in DNA
extracted from different germ layers becomes necessary. This notion could be
particularly useful for the study of possible mutations in apparently sporadic cases.
Regarding neurodegenerative diseases, including sCJD, it is important to obtain DNA
from ectodermal tissues such as the oral cavity mucosa (oral epithelial cells), hair roots
and cerebrospinal fluid, as they are embryologically related with the neural tissue.
Detection of somatic mosaicism with a low percentage of mutated cells may require
more precise techniques such as PCR product cloning and sequencing.
In conclusion, we report the first case of an apparently sporadic CJD with a somatic
mosaicism of a mutation in PRNP. In light of this case, the sequencing of PRNP is
compulsory even when the family history is negative, in order to give proper genetic
counseling in CJD cases.
Page 17 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
18
ACKNOWLEDGEMENTS
We thank Dr. Amets Saenz for her helpful advice and Concha Vidales for her technical
contribution. The technical work of Mrs. Margarita Carmona and Rosa Blanco is greatly
appreciated. We thank T. Yohannan for editorial assistance. The authors declare no
conflict of interests. This work was supported by Ilundain Fundazioa, Diputación Foral
de Gipuzkoa [expediente 76/08], the Basque Government (SAIOTEK program) and
Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas
(CIBERNED). This work was also supported by Neuroprion (EU 2004 Food-CT-2004-
506579) and Brain Net II (EU 2004-CT-2004-503039).
Page 18 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
19
REFERENCES
Bai RK, Wong LJ. 2004. Detection and quantification of heteroplasmic mutant
mitochondrial DNA by real-time amplification refractory mutation system quantitative
PCR analysis: a single-step approach. Clin Chem 50:996-1001.
Budka H, Head MW, Ironside JW. 2003. Sporadic Creutzfeldt-Jakob disease. In:
Dickson D, editor. Neurodegeneration: The molecular pathology of dementia and
movement disorders. Basel: ISN Neuropath Press; p. 287-97.
Dagvadorj A, Petersen RB, Lee HS, Cervenakova L, Shatunov A, Budka H, Brown P,
Gambetti P, Goldfarb LG. 2002. Spontaneous mutations in the prion protein gene
causing transmissible spongiform encephalopathy. Ann Neurol 52:355-359.
Dearmond SJ, Prusiner SB. 1995. Etiology and pathogenesis of prion diseases. Am J
Pathol 146:785-811.
Egeland T, Mostad PF, Mevag B, Stenersen M. 2000. Beyond traditional paternity and
identification cases. Selecting the most probable pedigree. Forensic Sci Int 110:47-59.
Frank SA. 2009. Somatic evolutionary genomics: Mutations during development cause
highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl
Acad Sci U S A.
Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. 2003. Sporadic and familial CJD:
classification and characterisation. Br Med Bull 66:213-239.
Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P,
Julien J, Vital C, Pendelbury WW, . 1992. Fatal familial insomnia and familial
Page 19 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
20
Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism.
Science 258:806-808.
Haik S, Peoc'h K, Brandel JP, Privat N, Laplanche JL, Faucheux BA, Hauw JJ. 2004.
Striking PrPsc heterogeneity in inherited prion diseases with the D178N mutation. Ann
Neurol 56:909-910.
Hill AF, Joiner S, Beck JA, Campbell TA, Dickinson A, Poulter M, Wadsworth JD,
Collinge J. 2006. Distinct glycoform ratios of protease resistant prion protein associated
with PRNP point mutations. Brain 129:676-685.
Ironside JW, Ghetti B, Head MW, Piccardo P, Will RG. 2008. Prion diseases. In: Love
S, Louis DN, Ellison DW, editors. Greenfield's Neuropathology. London: Hodder
Harnold; p. 1197-274.
Ironside JW, Head MW. 2008. Biology and neuropathology of prion diseases. Handb
Clin Neurol 89:779-797.
Ironside JW, Head MW, Will RG. 2003. Variant Creutzfeldt-Jakob disease. In: Dickson
D, editor. Neurodegeneration: The molecular pathology of dementia and movement
disorders. Basel: ISN Neuropath Press; p. 310-6.
Ketterling RP, Vielhaber E, Li X, Drost J, Schaid DJ, Kasper CK, Phillips JA, III,
Koerper MA, Kim H, Sexauer C, Gruppo R, Ambriz R, Paredes R, Sommer SS. 1999.
Germline origins in the human F9 gene: frequent G:C-->A:T mosaicism and increased
mutations with advanced maternal age. Hum Genet 105:629-640.
Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, Van DC, Collins SJ,
Boyd A, Giulivi A, Coulthart M, asnerie-Laupretre N, Brandel JP, Zerr I, Kretzschmar
Page 20 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
21
HA, de Pedro-Cuesta J, Calero-Lara M, Glatzel M, Aguzzi A, Bishop M, Knight R,
Belay G, Will R, Mitrova E. 2005. Genetic prion disease: the EUROCJD experience.
Hum Genet 118:166-174.
Leuer M, Oldenburg J, Lavergne JM, Ludwig M, Fregin A, Eigel A, Ljung R, Goodeve
A, Peake I, Olek K. 2001. Somatic mosaicism in hemophilia A: a fairly common event.
Am J Hum Genet 69:75-87.
Parchi P, Capellari S, Chen SG, Gambetti P. 2003. Familial Creutzfeldt-Jakob disease.
In: Dickson D, editor. Neurodegeneration: The molecular pathology of dementia and
movement disorders. Basel: ISN Neuropath Press; p. 298-306.
Parchi P, Capellari S, Chin S, Schwarz HB, Schecter NP, Butts JD, Hudkins P, Burns
DK, Powers JM, Gambetti P. 1999. A subtype of sporadic prion disease mimicking fatal
familial insomnia. Neurology 52:1757-1763.
Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson
DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. 1996. Molecular basis of
phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 39:767-778.
Parchi P, Gambetti P. 1995. Human prion diseases. Curr Opin Neurol 8:286-293.
Parchi P, Notari S, Weber P, Schimmel H, Budka H, Ferrer I, Haik S, Hauw JJ, Head
MW, Ironside JW, Limido L, Rodriguez A, Strobel T, Tagliavini F, Kretzschmar HA.
2009. Inter-laboratory assessment of PrPSc typing in creutzfeldt-jakob disease: a
Western blot study within the NeuroPrion Consortium. Brain Pathol 19:384-391.
Passos-Bueno MR, Bakker E, Kneppers AL, Takata RI, Rapaport D, den Dunnen JT,
Zatz M, van Ommen GJ. 1992. Different mosaicism frequencies for proximal and distal
Page 21 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
22
Duchenne muscular dystrophy (DMD) mutations indicate difference in etiology and
recurrence risk. Am J Hum Genet 51:1150-1155.
Prusiner SB. 1991. Molecular biology of prion diseases. Science 252:1515-1522.
Ricketts MN, Pergami P. 2003. Iatrogenic prion disorders. In: Dickson D, editor.
Neurodegeneration: The molecular pathology of dementia and movement disorders.
Basel: ISN Neuropath Press; p. 307-9.
Rodriguez-Martinez AB, Barreau C, Coupry I, Yague J, Sanchez-Valle R, Galdos-
Alcelay L, Ibanez A, Digon A, Fernandez-Manchola I, Goizet C, Castro A, Cuevas N,
varez-Alvarez M, de Pancorbo MM, Arveiler B, Zarranz JJ. 2005. Ancestral origins of
the prion protein gene D178N mutation in the Basque Country. Hum Genet 117:61-69.
Rodriguez-Martinez AB, fonso-Sanchez MA, Pena JA, Sanchez-Valle R, Zerr I,
Capellari S, Calero M, Zarranz JJ, de Pancorbo MM. 2008. Molecular evidence of
founder effects of fatal familial insomnia through SNP haplotypes around the D178N
mutation. Neurogenetics 9:109-118.
Sippel KC, Fraioli RE, Smith GD, Schalkoff ME, Sutherland J, Gallie BL, Dryja TP.
1998. Frequency of somatic and germ-line mosaicism in retinoblastoma: implications
for genetic counseling. Am J Hum Genet 62:610-619.
van Essen AJ, Abbs S, Baiget M, Bakker E, Boileau C, van BC, Bushby K, Clarke A,
Claustres M, Covone AE, . 1992. Parental origin and germline mosaicism of deletions
and duplications of the dystrophin gene: a European study. Hum Genet 88:249-257.
Verhoef S, Bakker L, Tempelaars AM, Hesseling-Janssen AL, Mazurczak T, Jozwiak S,
Fois A, Bartalini G, Zonnenberg BA, van Essen AJ, Lindhout D, Halley DJ, van den
Page 22 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
23
Ouweland AM. 1999. High rate of mosaicism in tuberous sclerosis complex. Am J Hum
Genet 64:1632-1637.
Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, Breithaupt
M, Varges D, Meissner B, Ladogana A, Schuur M, Haik S, Collins SJ, Jansen GH,
Stokin GB, Pimentel J, Hewer E, Collie D, Smith P, Roberts H, Brandel JP, van Duijn
C, Pocchiari M, Begue C, Cras P, Will RG, Sanchez-Juan P. 2009. Updated clinical
diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 132(Pt10):2659-2668.
Zheng L, Longfei J, Jing Y, Xinqing Z, Haiqing S, Haiyan L, Fen W, Xiumin D,
Jianping J. 2008. PRNP mutations in a series of apparently sporadic neurodegenerative
dementias in China. Am J Med Genet B Neuropsychiatr Genet 147B:938-944.
Page 23 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
24
Figure legends
Figure 1: Haplotypes of microsatellites and SNPs of the patient and his parents. The
order of markers within the haplotype, according to position in chromosome 20, is as
follows: MsP1, MsP2, rs2756271, rs6037932, rs13045348, rs6116474, rs6116475,
codon 129, codon 178, MsP4 and MsP5. Hatched bar indicates haplotype of the parental
chromosome in which the mutation appeared. Filled bar represents the haplotype of the
mutated chromosome 178N in mosaicism with the wild-type chromosome 178D.
Figure 2: Double-enzyme digestion to determine ligation phase between 129MV and
178DN. A: The scheme shows the expected size of the fragments after double digestion
with enzymes HpYCH4 IV and Tth111 I depending on the amino acid encoded by
codon 129 to which the mutation D178N is associated; B: 3% agarose gel stained with
ethidium bromide of double digestion of amplified fragment of PRNP gene. 239bp and
61bp bands correspond to 129M-178D allele, and 213bp and 87bp bands to 129V-178N
allele. But there are two additional soft bands: a 300bp band that could result from
partial digestions and a 152bp band that corresponds to 129V-178D allele.
Figure 3: Electropherogram of codon 178 (in the box) and the flanking region of the
PRNP gene obtained by the sequencing the PCR fraction corresponding to 129V alleles.
This fraction is selected by digestion of PRNP PCR by BsaA1 restriction enzyme. A)
DNA extracted from PBC; B) DNA extracted from brain tissue.
Page 24 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
25
Figure 4: Neuron loss and spongiform change in the frontal (A), occipital (B), temporal
(C) cortex, subiculum (D), entorhinal cortex (E), cerebellum (F), caudate (G) and
putamen (H). Severe spongiform change is found in the caudate and putamen compared
with the other regions. Tissue shrinkage resulting from cell loss is apparent in the
entorhinal cortex. This is in striking contrast with the apparently normal morphology of
the globus pallidus (I). Paraffin sections, stained with hematoxylin and eosin, x400.
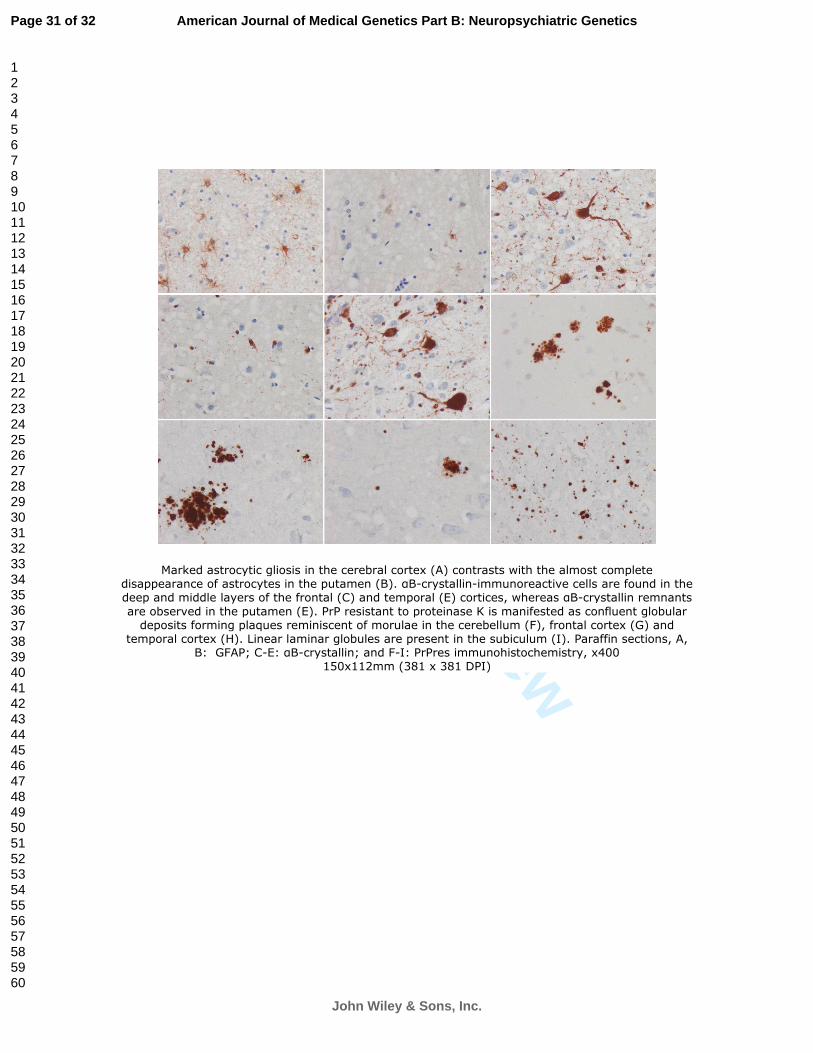
Figure 5: Marked astrocytic gliosis in the cerebral cortex (A) contrasts with the almost
complete disappearance of astrocytes in the putamen (B). αB-crystallin-immunoreactive
cells are found in the deep and middle layers of the frontal (C) and temporal (E)
cortices, whereas αB-crystallin remnants are observed in the putamen (E). PrP resistant
to proteinase K is manifested as confluent globular deposits forming plaques
reminiscent of morulae in the cerebellum (F), frontal cortex (G) and temporal cortex
(H). Linear laminar globules are present in the subiculum (I). Paraffin sections, A, B:
GFAP; C-E: αB-crystallin; and F-I: PrPres
immunohistochemistry, x400.
Figure 6: PrP blotting of sCJD type I, frontal cortex (I), sCJD type II, frontal cortex
(II), a case of Gerstmann-Sträussler-Scheinker (GSS) bearing the PRNP Y218N
mutation and one case of Fatal Familial Insomnia (FFI) bearing the 129 Met/Met PRNP
D178N mutation run in parallel with the case carrying the de novo PRNP D178N
mutation: 1: occipital cortex; 2: striatum; 3: cerebellum; 6: parietal cortex; 8: frontal
cortex.
Note that the lower band in the de novo mutation corresponds to PrP type I (about 21
kDa) and differs from that seen in FFI (type II; about 19 kDa). The PrP pattern in the de
novo mutation is reminiscent of type I of sporadic CJD.
Page 25 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
Table 1: Primer sequences (5′–3′), amplicon lengths (bp) and PCR conditions used for
PRNP amplification and sequencing (PRNP Fw and Rev), determination of the linkage
phase of codons 178-129 (PORF L and H) and PRNP product cloning and sequencing
(PGEM-T Fw and Rev).
Primer names Primer sequence (5’-3’) amplicon
length (bp)
PCR conditions
5’PRNP Fw TGTAAAACGACGGCCAGTGTCCTAAGTGCTTCAGA
5’PRNP Rev CAGGAAACAGCTATGACCTAGTACACTTGGTTGGGGTA
683
3’PRNP Fw TGTAAAACGACGGCCAGTGAACAAGCCGAGTAAGCCAA
3’ PRNP Rev CAGGAAACAGCTATGACCGGGGCTTGACCAGCATCTCA
692
96ºC, 3 min; 5 cycles (94ºC,
40’’; 55ºC, 40’’; 72ºC,60’’); 30
cycles (94ºC, 40’’:60ºC, 40’’,
72ºC, 60’’); 72ºC, 4 min
PRNP Fw GCCAAAAACCAACATGAAGC
PRNP Rev CATGCTCGATCCTCTCTGG
385 98ºC, 30s; 35 cycles (98ºC, 10s;
touch-down 63ºC-58ºC, 30s;
72ºC, 30s); 72ºC, 7 min
PORF-L CCTCTTCATTTTGCAGAGCA
PORF-H GATGGTGAAAACAGGAAGACC
300 95°C, 5 min; 35 cycles (94°C, 30
s; 63°C, 30 s; 72°C, 30 s); and
72°C, 5 min
pGEM-T Fw GGGCGAATTGGGCCCGACGT
pGEM-T Rev CCAAGCTATTTAGGTGACAC
535 95°C, 5 min; 35 cycles (95°C, 30
s; 60°C, 30 s; 72°C, 45 s); and
72°C, 5 min
Page 26 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
For Peer Review
Figure 1: Haplotypes of microsatellites and SNPs of the patient and his parents. The order of markers within the haplotype, according to position in chromosome 20, is as follows: MsP1, MsP2, rs2756271, rs6037932, rs13045348, rs6116474, rs6116475, codon 129, codon 178, MsP4 and MsP5. Hatched bar indicates haplotype of the parental chromosome in which the mutation
appeared. Filled bar represents the haplotype of the mutated chromosome 178N in mosaicism with the wild-type chromosome 178D.
Page 27 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
Figure 2: Double-enzyme digestion to determine ligation phase between 129MV and 178DN. A: The scheme shows the expected size of the fragments after double digestion with enzymes HpYCH4 IV and Tth111 I depending on the amino acid encoded by codon 129 to which the mutation D178N is associated; B: 3% agarose gel stained with ethidium bromide of double digestion of amplified
fragment of PRNP gene. 239bp and 61bp bands correspond to 129M-178D allele, and 213bp and 87bp bands to 129V-178N allele. But there are two additional soft bands: a 300bp band that could
result from partial digestions and a 152bp band that corresponds to 129V-178D allele. 254x190mm (96 x 96 DPI)
Page 28 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
Figure 3: Electropherogram of codon 178 (in the box) and the flanking region of the PRNP gene obtained by the sequencing the PCR fraction corresponding to 129V alleles. This fraction is selected
by digestion of PRNP PCR by BsaA1 restriction enzyme. A) DNA extracted from PBC; B) DNA extracted from brain tissue. 254x190mm (96 x 96 DPI)
Page 29 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
Neuron loss and spongiform change in the frontal (A), occipital (B), temporal (C) cortex, subiculum (D), entorhinal cortex (E), cerebellum (F), caudate (G) and putamen (H). Severe spongiform change
is found in the caudate and putamen compared with the other regions. Tissue shrinkage resulting from cell loss is apparent in the entorhinal cortex. This is in striking contrast with the apparently
normal morphology of the globus pallidus (I). Paraffin sections, stained with hematoxylin and eosin, x400
150x112mm (381 x 381 DPI)
Page 30 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
For Peer Review
Marked astrocytic gliosis in the cerebral cortex (A) contrasts with the almost complete disappearance of astrocytes in the putamen (B). αB-crystallin-immunoreactive cells are found in the deep and middle layers of the frontal (C) and temporal (E) cortices, whereas αB-crystallin remnants
are observed in the putamen (E). PrP resistant to proteinase K is manifested as confluent globular deposits forming plaques reminiscent of morulae in the cerebellum (F), frontal cortex (G) and
temporal cortex (H). Linear laminar globules are present in the subiculum (I). Paraffin sections, A, B: GFAP; C-E: αB-crystallin; and F-I: PrPres immunohistochemistry, x400
150x112mm (381 x 381 DPI)
Page 31 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
PrP blotting of sCJD type I, frontal cortex (I), sCJD type II, frontal cortex (II), a case of Gerstmann-Sträussler-Scheinker (GSS) bearing the PRNP Y218N mutation and one case of Fatal Familial
Insomnia (FFI) bearing the 129 Met/Met PRNP D178N mutation run in parallel with the case carrying the de novo PRNP D178N mutation: 1: occipital cortex; 2: striatum; 3: cerebellum; 6: parietal
cortex; 8: frontal cortex. Note that the lower band in the de novo mutation corresponds to PrP type I (about 21 kDa) and differs from that seen in FFI (type II; about 19 kDa). The PrP pattern in the de novo mutation is
reminiscent of type I of sporadic CJD, yet the the unglycosylated band is under-represented (compare lane 1 with lane 8 both corresponding to the frontal cortex), and this is particularly
marked in the striatum (lane 2). 254x190mm (96 x 96 DPI)
Page 32 of 32
John Wiley & Sons, Inc.
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960




















