
Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the...
11
Pms2 is a genetic enhancer of trinucleotide CAG . CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion Ma ´rio Gomes-Pereira, M. Teresa Fortune, Laura Ingram, John P. McAbney and Darren G. Monckton * Institute of Biomedical and Life Sciences, University of Glasgow, Anderson College Complex, 56 Dumbarton Road, Glasgow G11 6NU, UK Received May 10, 2004; Revised and Accepted June 4, 2004 The expansion of CAG . CTG repeat sequences is the cause of several inherited human disorders. Longer alleles are associated with an earlier age of onset and more severe symptoms, and are highly unstable in the germline and soma with a marked tendency towards repeat length gains. Germinal expansions underlie anticipation; whereas age-dependent, tissue-specific, expansion-biased somatic instability probably contrib- utes toward the progressive nature and tissue-specificity of the symptoms. The mechanism(s) of repeat instability is not known, but recent data have implicated mismatch-repair (MMR) gene mutS homologues in driving expansion. To gain further insight into the expansion mechanism, we have determined the levels of somatic mosaicism of a transgenic expanded CAG . CTG repeat in mice deficient for the Pms2 MMR gene. Pms2 is a MutL homologue that plays a critical role in the downstream processing of DNA mis- matches. The rate of somatic expansion was reduced by 50% in Pms2-null mice. A higher frequency of rare, but very large, deletions was also detected in these animals. No significant differences were observed between Pms2 1/1 and Pms2 1/2 mice, indicating that a single functional Pms2 allele is sufficient to generate normal levels of somatic mosaicism. These findings reveal that as well as MMR enzymes that directly bind mismatched DNA, proteins that are subsequently recruited to the complex also play a central role in the accumulation of repeat length changes. These data suggest that somatic expansion results not by replication slippage, single stranded annealing or simple MutS-mediated stabilization of secondary structures, but by inappropriate DNA MMR. INTRODUCTION The expansion of unstable simple repeat sequences within the human genome has been identified as the genetic basis of a number of complex disorders including myotonic dystrophy (DM), Huntington disease (HD), Friedreich ataxia, fragile-X syndrome and several of the spinocerebellar ataxias (1). Most of these diseases are associated with trinucleotide CAG . CTG repeat expansions. Such trinucleotide repeat sequences are usually polymorphic within the general popu- lation and are relatively stable when transmitted from parent to child. However, once into the expanded disease-associated range, the repeats become dramatically unstable in both the germline and soma (1). Mutations are strongly biased toward further expansion with longer alleles associated with a more severe phenotype and an earlier age of onset. Expansion- biased germline instability therefore provides the molecular basis for the phenomenon of anticipation—the decreasing age of onset and increased severity of clinical symptoms observed in successive generations of an affected family. Somatic instability of trinucleotide repeats involves a dynamic process in which the repeat size increases slowly with age, at diverse rates in different tissues, in a process that appears to be mediated by multiple small length gains (2–4). Interestingly, for some disorders at least, there is a cor- relation between the tissue specificity of somatic expansion and the affected tissue. Most notably, very large somatic expansions are observed in the skeletal muscle of DM type 1 patients (5–7) and in the affected brain regions of HD patients (8). Age-dependent, expansion-biased, tissue-specific Human Molecular Genetics, Vol. 13, No. 16 # Oxford University Press 2004; all rights reserved *To whom correspondence should be addressed. Tel: þ44 1413306213; Fax: þ44 1413306871; Email: [email protected] Human Molecular Genetics, 2004, Vol. 13, No. 16 1815–1825 doi:10.1093/hmg/ddh186 Advance Access published on June 15, 2004 by guest on May 21, 2014 http://hmg.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the...

Pms2 is a genetic enhancer of trinucleotideCAG.CTG repeat somatic mosaicism: implicationsfor the mechanism of triplet repeat expansion
Mario Gomes-Pereira, M. Teresa Fortune, Laura Ingram, John P. McAbney
and Darren G. Monckton*
Institute of Biomedical and Life Sciences, University of Glasgow, Anderson College Complex,
56 Dumbarton Road, Glasgow G11 6NU, UK
Received May 10, 2004; Revised and Accepted June 4, 2004
The expansion of CAG.CTG repeat sequences is the cause of several inherited human disorders. Longeralleles are associated with an earlier age of onset and more severe symptoms, and are highly unstable inthe germline and soma with a marked tendency towards repeat length gains. Germinal expansions underlieanticipation; whereas age-dependent, tissue-specific, expansion-biased somatic instability probably contrib-utes toward the progressive nature and tissue-specificity of the symptoms. The mechanism(s) of repeatinstability is not known, but recent data have implicated mismatch-repair (MMR) gene mutS homologuesin driving expansion. To gain further insight into the expansion mechanism, we have determined thelevels of somatic mosaicism of a transgenic expanded CAG.CTG repeat in mice deficient for the Pms2MMR gene. Pms2 is a MutL homologue that plays a critical role in the downstream processing of DNA mis-matches. The rate of somatic expansion was reduced by �50% in Pms2-null mice. A higher frequency of rare,but very large, deletions was also detected in these animals. No significant differences were observedbetween Pms2 1/1 and Pms2 1/2 mice, indicating that a single functional Pms2 allele is sufficient to generatenormal levels of somatic mosaicism. These findings reveal that as well as MMR enzymes that directly bindmismatched DNA, proteins that are subsequently recruited to the complex also play a central role in theaccumulation of repeat length changes. These data suggest that somatic expansion results not by replicationslippage, single stranded annealing or simple MutS-mediated stabilization of secondary structures, but byinappropriate DNA MMR.
INTRODUCTION
The expansion of unstable simple repeat sequences within thehuman genome has been identified as the genetic basis of anumber of complex disorders including myotonic dystrophy(DM), Huntington disease (HD), Friedreich ataxia, fragile-Xsyndrome and several of the spinocerebellar ataxias (1).Most of these diseases are associated with trinucleotideCAG.CTG repeat expansions. Such trinucleotide repeatsequences are usually polymorphic within the general popu-lation and are relatively stable when transmitted from parentto child. However, once into the expanded disease-associatedrange, the repeats become dramatically unstable in both thegermline and soma (1). Mutations are strongly biased towardfurther expansion with longer alleles associated with a more
severe phenotype and an earlier age of onset. Expansion-biased germline instability therefore provides the molecularbasis for the phenomenon of anticipation—the decreasingage of onset and increased severity of clinical symptomsobserved in successive generations of an affected family.Somatic instability of trinucleotide repeats involves adynamic process in which the repeat size increases slowlywith age, at diverse rates in different tissues, in a processthat appears to be mediated by multiple small length gains(2–4). Interestingly, for some disorders at least, there is a cor-relation between the tissue specificity of somatic expansionand the affected tissue. Most notably, very large somaticexpansions are observed in the skeletal muscle of DM type1 patients (5–7) and in the affected brain regions of HDpatients (8). Age-dependent, expansion-biased, tissue-specific
Human Molecular Genetics, Vol. 13, No. 16 # Oxford University Press 2004; all rights reserved
*To whom correspondence should be addressed. Tel: þ44 1413306213; Fax: þ44 1413306871; Email: [email protected]
Human Molecular Genetics, 2004, Vol. 13, No. 16 1815–1825doi:10.1093/hmg/ddh186Advance Access published on June 15, 2004
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

somatic instability may therefore account for the progressivenature of the symptoms and the variable tissue-specific patho-genesis associated with the different disorders.
Understanding the molecular mechanisms that drive trinu-cleotide repeat expansion in the soma is therefore of greatinterest as it might be intimately associated with disease devel-opment and progression. The most favoured mechanism oftriplet repeat mutation, the DNA polymerase slippage model,predicts that repeat size variability arises during DNA replica-tion in a cell division-dependent manner (9). DNA polymerasestalling and slippage might be facilitated by the propensity oftrinucleotide repeat sequences to fold into alternative non-B-DNA structures such as hairpins (10) and slipped-strandedDNA (S-DNA) (11). Slipped-stranded intermediate DNAspecies (SI-DNA) formed during DNA replication slippage(12) should be repaired by the DNA mismatch-repair(MMR) pathway, thus preventing expansions and deletionsduring replication. However, at large expanded repeat tractsthey may occur so frequently that they overwhelm the capacityof the system. Non-repaired SI-intermediates may then giverise to repeat length changes following some other repairevent or a second round of replication. As a result, mutationrates would be expected to increase with MMR genemutations and higher cell division rates. Support for such amodel is provided by the observation of elevated geneticinstability at short simple DNA repeat sequences in hereditarynon-polyposis colorectal cancer (HNPCC) tumours that aredefective for MMR (13).
MMR has been reported to have at least two effects on thestability of long expanded CAG.CTG repeat arrays cloned intoEscherichia coli. Although mutations in MMR genes reducethe frequency of large contractions in very long repeattracts, smaller length change mutations are elevated as inHNPCC (14–17). In Saccharomyces cerevisiae, more frequentsmall expansions and deletions are observed in MMR deficientstrains, but the frequency of large contractions is not affected(18). Although the analysis of trinucleotide repeat dynamics inmicrobial models has thus provided support for the replicationslippage model, relatively little is understood about the mol-ecular pathways that control triplet repeat instability incomplex mammalian cells.
Mouse models have been used to investigate the dynamicnature of triplet repeat sequences, providing an accuratemodel of the age-dependent, expansion-biased, tissue-specificsomatic instability observed in man (19–24). Monitoring ofsomatic mosaicism in tissues collected from these mice hasrevealed the lack of an obvious correlation between levels oftrinucleotide instability and the rates of cell turnover, with highlevels of somatic mosaicism being observed in post-mitotictissues such as brain and muscle (19,20,22,25). Moreover,the levels of trinucleotide repeat instability in homogeneousmouse cell lines do not correlate with cell division ratesin vitro (26). Considered together, these observations haveraised serious reservations about the relevance of thereplication slippage model in mammalian cells, and suggestthat the expansion mechanism is not entirely dependent onDNA replication.
In bacteria, post-replication MMR is mediated by threemajor proteins MutS, MutL and MutH (27). A MutS dimerrecognizes the physical mismatch and recruits a MutL
dimer, which in turn recruits MutH. MutH utilizes the methyl-ation status of newly synthesized DNA to distinguish the tem-plate and daughter strands and to nick the daughter strand. Thenick is then used to initiate excision of the incorrectly syn-thesized DNA allowing error-free resynthesis. In eukaryotesMMR is more complex. The current mammalian model impli-cates at least six MutS and MutL homologues (13) and amechanism of strand discrimination that probably involves adirect physical link to the replication fork via PCNA (28).The major MutS homologue is MSH2, which can form aheterodimer with either MSH6, to form MutSa, or withMSH3, to form MutSb (13). MutSa is critical for the recog-nition of single base mismatches and MutSb for the recog-nition of small insertion/deletion loops, such as those foundin SI-DNA. Recruitment of a second heterodimer consistingof MLH1 and either PMS2 (MutLa) or MLH3 (MutLb) isthought to be essential for subsequent excision and resynth-esis, resulting in DNA repair. In contrast to the predictionsresulting from the replication slippage model, murine MutShomologues Msh2 and Msh3 are required for the developmentof expansion-biased trinucleotide somatic instability in trans-genic mouse models carrying expanded CAG.CTG repeattracts (24,29,30). Interestingly, Msh6 activity suppresses theaccumulation of repeat length variation, possibly by compet-ing more effectively than Msh3 for Msh2 and thus limitingthe amount of MutSb complex (24). These observationssuggest a key role for MutS homologues in triplet repeatexpansion in mammalian cells.
How MutS homologues Msh2 and Msh3 mediate expan-sions at CAG.CTG repeat loci has not yet been determined.It has been suggested that this occurs via a process that doesnot involve MMR, but instead relies on the stabilization andprotection from repair of CAG.CTG hairpin structures(29,31). Consistent with this model, MSH2 has been shownto bind to both S-DNA and SI-DNA in vitro (12). However,in these experiments the absence of either MSH6 or MSH3,the two MSH2 partners in vivo, make the biological relevanceof this observation unclear. MutS homologues are alsoinvolved in additional DNA repair pathways, and it has beensuggested that expansion could be mediated via mechanismssuch as single stranded annealing (SSA) repair of double-strand breaks (30), which are independent of MutL homol-ogues (32). Currently unknown, but critical to gainingfurther insight into the mechanism, is the question ofwhether there is a requirement for MutL homologues in thetriplet repeat expansion pathway. One of the three MutL hom-ologues in mammals is PMS2. Mutations in PMS2 have beenassociated with dramatic microsatellite instability both innormal and tumour tissues of Turcot syndrome patients(33,34) and in tumour tissue of HNPCC patients (35). Inorder to gain a better understanding of the function of thePms2 protein in the MMR pathway of higher eukaryotes,Pms2 knock-out mice have been generated by gene targetingprocedures (36). Pms2-null homozygotes develop spontaneouslymphomas and sarcomas, whereas mice heterozygous for thePms2 disruption develop normally, with no predisposition totumour formation (36,37). As expected, spontaneous microsa-tellite mutation rates of di- and mono-nucleotide sequences areraised up to 100-fold in many tissues of Pms2 nullizygousmice, confirming a key role for Pms2 in the maintenance of
1816 Human Molecular Genetics, 2004, Vol. 13, No. 16
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
genomic stability in multiple, if not all, cell lineages (36,38).In an attempt to clarify the role of MutL homologues as modi-fiers of trinucleotide repeat dynamics in mammalian cells, wehave crossed Pms2-deficient mice (36) with mice carrying anexpanded CTG.CAG repeat transgene (39), and have usedsensitive small pool polymerase chain reaction (SP-PCR)detection methods (4) to investigate somatic mosaicism.
RESULTS
Pms2 deficiency reduces the rate of somatic expansionof a large CAG.CTG repeat tract
In order to assess the involvement of Pms2 in the metabolismof expanded trinucleotide repeats we crossed the previouslydescribed Pms2-null allele (36) into mice carrying theDmt-D transgene, which shows high levels of tissue-specific,age-dependent, expansion-biased somatic mosaicism (19).Mice carrying a single copy of the Dmt-D transgene and allthree genotypes at the Pms2 locus (þ/þ, þ/2 and 2/2)were allowed to age so that somatic expansions mightaccumulate. Complete Pms2 deficiency is associated with asevere cancer predisposition phenotype (37) and in ourcolony Pms2 2/2 mice rarely survived beyond 5 monthsof age and never beyond 8 months of age before developingtumours. Tissues from Pms2 2/2 and age-matched littermatecontrols (Pms2 þ/þ and Pms2 þ/2) were collected and analysedfor somatic mosaicism using sensitive SP-PCR techniques (4).In order that we might detect both increases and/or decreasesin the rate of expansion, four different tissues were selected tocover the range of trinucleotide repeat instability that presentsin Dmt-D transgenic mice (19). Lung and heart were chosen torepresent those tissues that show low levels of repeat instabil-ity and in which an increase in the rate of expansion might bedistinguished readily. Kidney was selected as the tissue wherethe transgene exhibits the most dramatic repeat lengthchanges, and hence the most appropriate target for detectinga decrease in the rate of expansion. Brain was also included,because it not only displays relatively high levels of repeatinstability, but is also one of the most commonly affectedtissues in the CAG.CTG expansion disorders. The degree ofsomatic mosaicism of the expanded CAG.CTG repeat Dmt-Dtransgene normally detectable in mice aged �5 months isrelatively low. Nonetheless, our analyses revealed consistentlylower rates of somatic expansion of the expanded CAG.CTGrepeat Dmt-D transgene in the kidney (Fig. 1A) and brain ina Pms2 2/2 genetic background. Because of the very lowlevels of mosaicism present in the lung and heart of younganimals it was not possible to determine if loss of Pms2 simi-larly suppressed expansion in these tissues (data not shown).However, comparison of repeat length distributions in one8-month-old Pms2 2/2 animal and its Pms2 þ/2 littermatedid reveal similar suppression of expansions in lung andkidney (Fig. 1B), in addition to heart and brain (Fig. 2A andB, see later). Although the degree of somatic mosaicism inthe Pms2 2/2 mice was lower than in their littermate controls,it was nonetheless readily detectable and biased toward expan-sion. In order to quantify the degree of modification of expan-sion dynamics we used single molecule PCR to determineprecise repeat length distributions in the lung and kidney
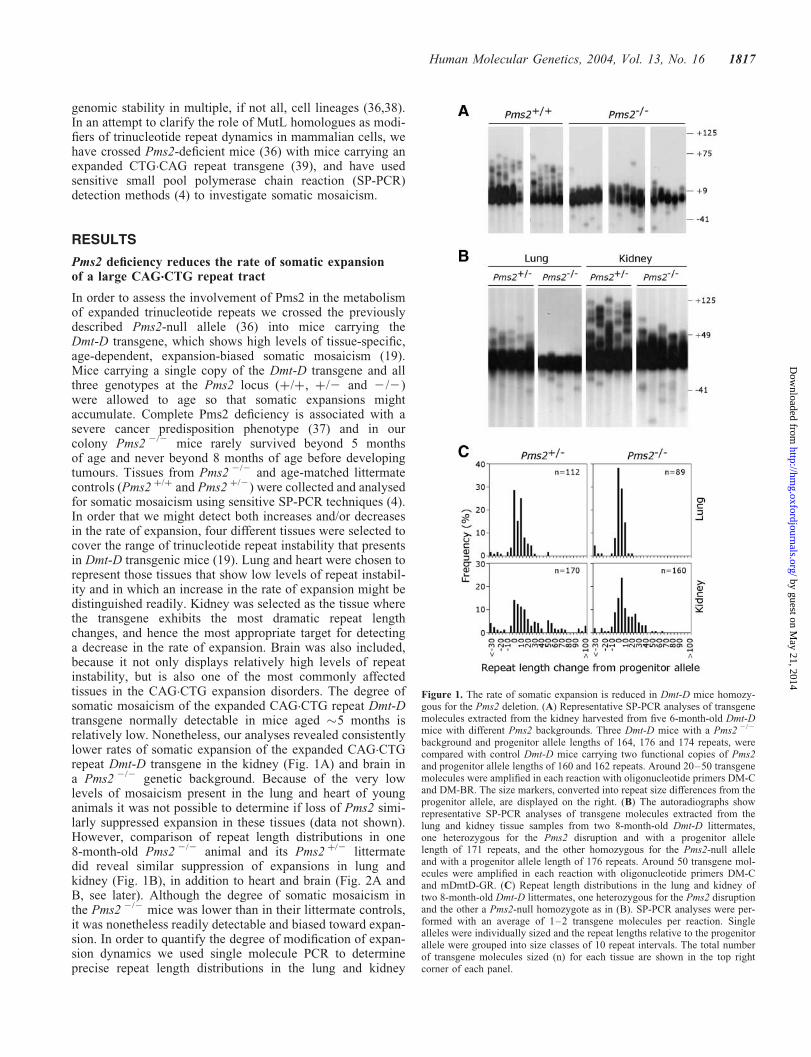
Figure 1. The rate of somatic expansion is reduced in Dmt-D mice homozy-gous for the Pms2 deletion. (A) Representative SP-PCR analyses of transgenemolecules extracted from the kidney harvested from five 6-month-old Dmt-Dmice with different Pms2 backgrounds. Three Dmt-D mice with a Pms2 2/2
background and progenitor allele lengths of 164, 176 and 174 repeats, werecompared with control Dmt-D mice carrying two functional copies of Pms2and progenitor allele lengths of 160 and 162 repeats. Around 20–50 transgenemolecules were amplified in each reaction with oligonucleotide primers DM-Cand DM-BR. The size markers, converted into repeat size differences from theprogenitor allele, are displayed on the right. (B) The autoradiographs showrepresentative SP-PCR analyses of transgene molecules extracted from thelung and kidney tissue samples from two 8-month-old Dmt-D littermates,one heterozygous for the Pms2 disruption and with a progenitor allelelength of 171 repeats, and the other homozygous for the Pms2-null alleleand with a progenitor allele length of 176 repeats. Around 50 transgene mol-ecules were amplified in each reaction with oligonucleotide primers DM-Cand mDmtD-GR. (C) Repeat length distributions in the lung and kidney oftwo 8-month-old Dmt-D littermates, one heterozygous for the Pms2 disruptionand the other a Pms2-null homozygote as in (B). SP-PCR analyses were per-formed with an average of 1–2 transgene molecules per reaction. Singlealleles were individually sized and the repeat lengths relative to the progenitorallele were grouped into size classes of 10 repeat intervals. The total numberof transgene molecules sized (n) for each tissue are shown in the top rightcorner of each panel.
Human Molecular Genetics, 2004, Vol. 13, No. 16 1817
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
from an 8-month-old Pms2 2/2 mouse and a Pms2 þ/2 litter-mate control (Fig. 1C). These data revealed that repeatlength distributions in the two animals were highly statisticallysignificantly different from each other for both lung(P ¼ 0.0007, two-tailed Mann–Whitney U-test) and kidney(P ¼ 0.0038, two-tailed Mann–Whitney U-test). The medianexpansion rates in Pms2 þ/2 and Pms2 2/2 mice wereþ0.029 and þ0.010 repeats per day in lung, and þ0.053and þ0.028 repeats per day in kidney, respectively. Thus,Pms2 deficiency results in an �50% reduction in the rate ofsomatic expansion.
Pms2 deficiency increases the frequency of rare largesomatic deletions of a CAG.CTG repeat tract
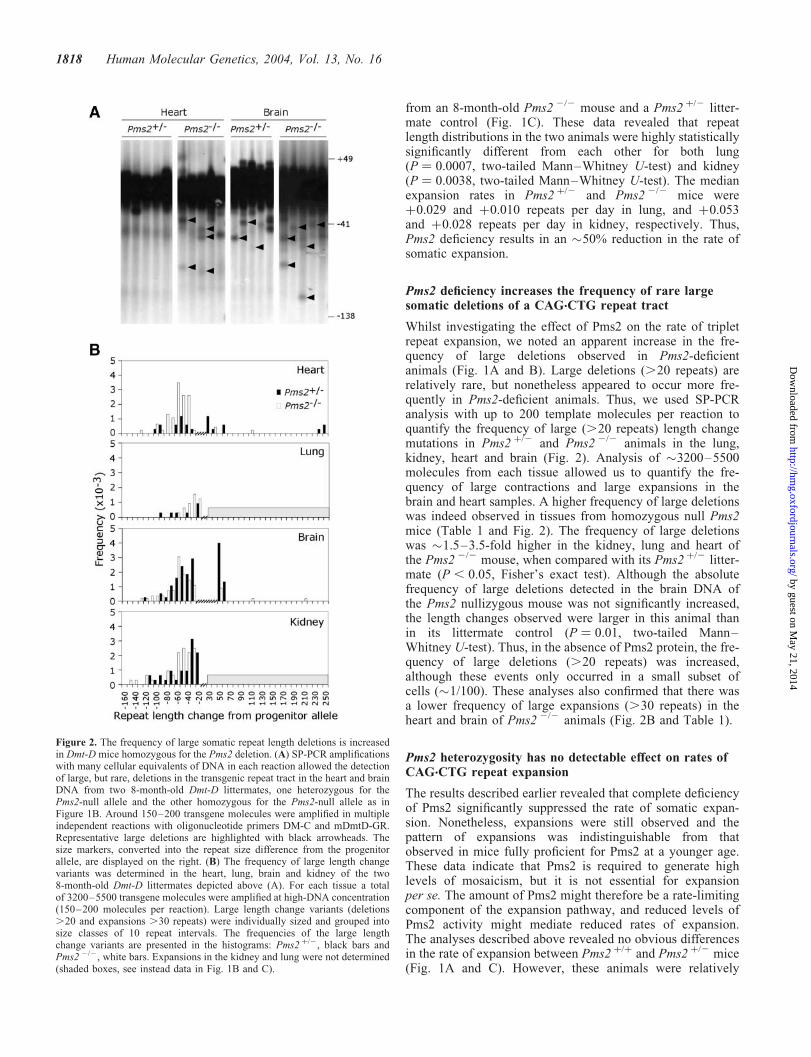
Whilst investigating the effect of Pms2 on the rate of tripletrepeat expansion, we noted an apparent increase in the fre-quency of large deletions observed in Pms2-deficientanimals (Fig. 1A and B). Large deletions (.20 repeats) arerelatively rare, but nonetheless appeared to occur more fre-quently in Pms2-deficient animals. Thus, we used SP-PCRanalysis with up to 200 template molecules per reaction toquantify the frequency of large (.20 repeats) length changemutations in Pms2 þ/2 and Pms2 2/2 animals in the lung,kidney, heart and brain (Fig. 2). Analysis of �3200–5500molecules from each tissue allowed us to quantify the fre-quency of large contractions and large expansions in thebrain and heart samples. A higher frequency of large deletionswas indeed observed in tissues from homozygous null Pms2mice (Table 1 and Fig. 2). The frequency of large deletionswas �1.5–3.5-fold higher in the kidney, lung and heart ofthe Pms2 2/2 mouse, when compared with its Pms2 þ/2 litter-mate (P , 0.05, Fisher’s exact test). Although the absolutefrequency of large deletions detected in the brain DNA ofthe Pms2 nullizygous mouse was not significantly increased,the length changes observed were larger in this animal thanin its littermate control (P ¼ 0.01, two-tailed Mann–Whitney U-test). Thus, in the absence of Pms2 protein, the fre-quency of large deletions (.20 repeats) was increased,although these events only occurred in a small subset ofcells (�1/100). These analyses also confirmed that there wasa lower frequency of large expansions (.30 repeats) in theheart and brain of Pms2 2/2 animals (Fig. 2B and Table 1).
Pms2 heterozygosity has no detectable effect on rates ofCAG.CTG repeat expansion
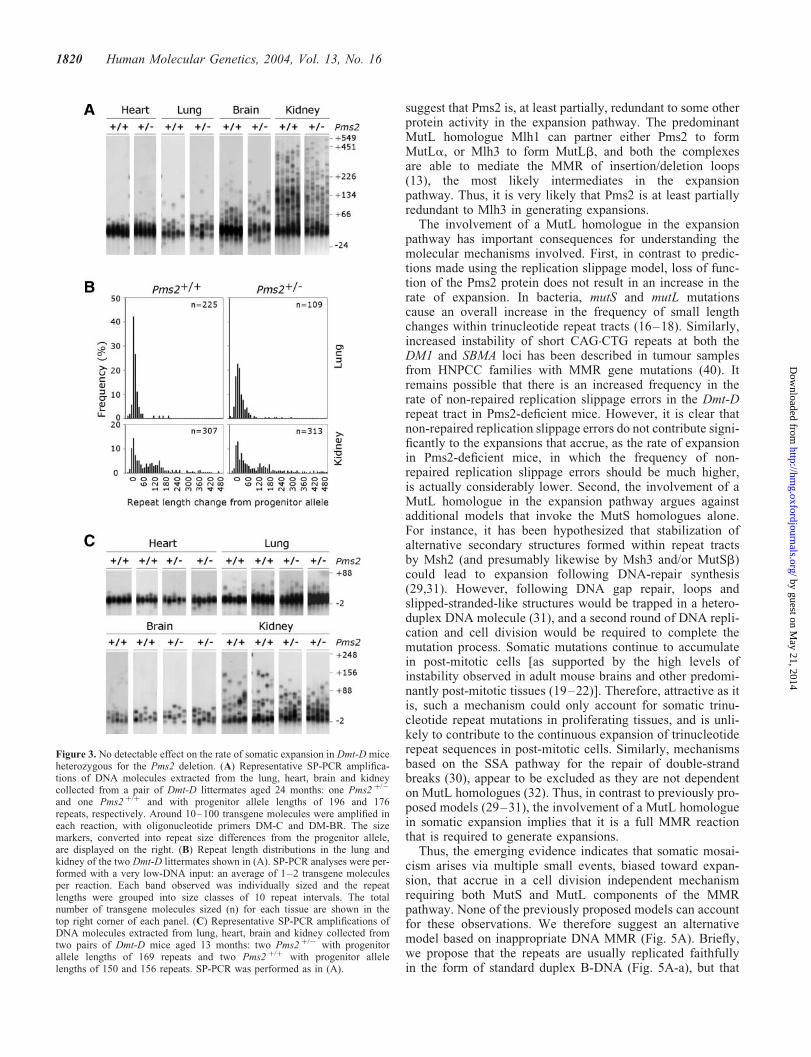
The results described earlier revealed that complete deficiencyof Pms2 significantly suppressed the rate of somatic expan-sion. Nonetheless, expansions were still observed and thepattern of expansions was indistinguishable from thatobserved in mice fully proficient for Pms2 at a younger age.These data indicate that Pms2 is required to generate highlevels of mosaicism, but it is not essential for expansionper se. The amount of Pms2 might therefore be a rate-limitingcomponent of the expansion pathway, and reduced levels ofPms2 activity might mediate reduced rates of expansion.The analyses described above revealed no obvious differencesin the rate of expansion between Pms2 þ/þ and Pms2 þ/2 mice(Fig. 1A and C). However, these animals were relatively
Figure 2. The frequency of large somatic repeat length deletions is increasedin Dmt-D mice homozygous for the Pms2 deletion. (A) SP-PCR amplificationswith many cellular equivalents of DNA in each reaction allowed the detectionof large, but rare, deletions in the transgenic repeat tract in the heart and brainDNA from two 8-month-old Dmt-D littermates, one heterozygous for thePms2-null allele and the other homozygous for the Pms2-null allele as inFigure 1B. Around 150–200 transgene molecules were amplified in multipleindependent reactions with oligonucleotide primers DM-C and mDmtD-GR.Representative large deletions are highlighted with black arrowheads. Thesize markers, converted into the repeat size difference from the progenitorallele, are displayed on the right. (B) The frequency of large length changevariants was determined in the heart, lung, brain and kidney of the two8-month-old Dmt-D littermates depicted above (A). For each tissue a totalof 3200–5500 transgene molecules were amplified at high-DNA concentration(150–200 molecules per reaction). Large length change variants (deletions.20 and expansions .30 repeats) were individually sized and grouped intosize classes of 10 repeat intervals. The frequencies of the large lengthchange variants are presented in the histograms: Pms2 þ/2, black bars andPms2 2/2, white bars. Expansions in the kidney and lung were not determined(shaded boxes, see instead data in Fig. 1B and C).
1818 Human Molecular Genetics, 2004, Vol. 13, No. 16
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

young when tested and had not accumulated high levels ofsomatic mosaicism. Therefore, by analysis of repeat lengthvariation in older animals we sought to determine if Pms2 het-erozygotes manifest a haploinsufficiency phenotype. Repeatlength variability of the Dmt-D transgene was determined bySP-PCR in DNA samples collected from two 24-month-oldmice, one with a Pms2 þ/2 background and the other with aPms2 þ/þ background. Although high levels of mosaicismwere detected in all the tissues analysed (heart, lung, brainand kidney), no obvious qualitative differences in the expan-sion profiles were observed (Fig. 3A). To search for a moresubtle effect, we performed a detailed quantitative analysisof the degree of repeat length variation in the lung andkidney using single molecule SP-PCR (Fig. 3B). The medianrate of expansion in the lung cells was essentially indistin-guishable (þ0.024 and þ0.021 repeats per day in Pms2 þ/þ
and Pms2 þ/2 mice, respectively, P ¼ 0.54, two-tailedMann–Whitney U-test). Surprisingly though, these datarevealed that the median rate of expansion was actuallyslightly higher in the kidney cells of the Pms2 þ/2 mouse(þ0.095 repeats per day) relative to the Pms2 þ/þ littermatecontrol (0.077 repeats per day). However, these repeatlength distributions were also not statistically significantlydifferent from each other (P ¼ 0.85 two-tailed Mann–Whitney U-test). The slightly higher rate of expansionobserved in the kidney of the Pms2 þ/2 mouse was probablymediated by the longer Dmt-D allele it inherited (196repeats versus 176 repeats in the Pms2 þ/þ animal). Thus,Pms2 heterozygosity appeared to have no effect on the rateof somatic expansion. In order to confirm this result, we exam-ined the repeat length distributions in an additional two pairsof Pms2 þ/þ and Pms2 þ/2 littermate controls from whichtissues were harvested at 13 months of age (Fig. 3C). Onceagain, no detectable differences were observed betweenPms2 þ/þ and Pms2 þ/2 animals.
Given the substantial effects observed with Pms2deficiency, we were surprised that we could not detect evena relatively subtle effect in the Pms2 heterozygotes. A possibleexplanation could be that although the Pms2 heterozygotes are50% deficient at the gene level, feedback mechanisms mayoperate to increase transcription levels and/or mRNA/proteinstability resulting in equivalent steady-state levels of Pms2
protein in both Pms2 þ/þ and Pms2 þ/2 animals. To test thisidea we determined Pms2 protein levels in Pms2 þ/þ,Pms2 þ/2 and Pms2 2/2 cells. Western blot analysis ofwhole cell protein lysates, collected from cultured mouseembryonic fibroblasts, was performed (Fig. 4A) and Pms2protein levels were quantified relative to b-tubulin. Asexpected, the analysis revealed that whereas nullizygousmice lack Pms2, mice heterozygous for the Pms2 deletionshowed a �50% reduction in the protein levels, relative tomice carrying two functional Pms2 alleles (P ¼ 0.001, t-test)(Fig. 4B). Thus, a 50% reduction in the steady-state proteinlevels in Pms2 þ/2 mice does not lead to a detectable differ-ence in the expansion rate relative to Pms2 wild-type mice.Indeed, these results once again highlight the remarkablydeterministic nature of the expansion pathway in vivo.
DISCUSSION
Age-dependent, tissue-specific, expansion-biased somaticmosaicism is an integral part of the unusual genetic phenom-ena that characterize human disorders associated with theexpansion of simple sequence repeats. In addition to posinga fascinating scientific question, somatic mosaicism alsovery likely contributes toward the tissue-specificity and pro-gressive nature of the symptoms. As such, a thorough under-standing of the molecular mechanisms that generateinstability is of interest, not only in terms of the insights itmay provide into basic biological processes, but alsobecause it may have direct clinical relevance. Recently, theuse of transgenic mouse models to understand the processesinvolved have started to yield important new insights. Mostnotably, several studies have revealed a critical role forthe MMR MutS homologues Msh2 and Msh3, both of whichare absolutely required to generate expansions (24,29–31).We have shown here for the first time that a MutL homologue,Pms2, is similarly required to generate normal levels of expan-sion. In contrast to the essential role of Msh2 and Msh3 in theexpansion pathway, Pms2 does not appear to be absolutelyrequired. Expansions still accrue in Pms2-deficient mice andthe overall pattern of expansion is not changed. Rather theabsolute rate of expansion is reduced by �50%. These data
Table 1. Frequency and magnitude of large length change mutations of the Dmt-D transgene in different Pms2 genetic backgrounds
Tissue Pms2 Large deletions (.20) Large expansions (.30)
genotype No.(mutants/cellsanalysed)
Frequency(�1023)(per cell)
Median lengthchange (repeatsper cell)
No.(mutants/cellsanalysed)
Frequency(�1023)(per cell)
Median lengthchange (repeatsper cell)
Heart þ/2 12/3408 3.5 20.18 10/3408 2.9 þ0.152/2 57/4602 12.4 (P � 0.01)� 20.72 3/4602 0.7 (P ¼ 0.02)� þ0.03
Brain þ/2 41/3760 10.9 20.52 20/3760 5.3 þ0.292/2 58/5545 10.5 20.61 7/5545 1.3 (P � 0.01)� þ0.06
Lung þ/2 5/3200 1.6 20.10 ND ND ND2/2 14/3200 4.4 (P ¼ 0.03)� 20.14 ND ND ND
Kidney þ/2 31/3200 9.7 20.35 ND ND ND2/2 47/3200 14.7 (P ¼ 0.04)� 20.69 ND ND ND
�Statistically significant differences between the frequency of length change variants in Pms2 þ/ 2 versus Pms2 2/ 2 mice are indicated.
Human Molecular Genetics, 2004, Vol. 13, No. 16 1819
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
suggest that Pms2 is, at least partially, redundant to some otherprotein activity in the expansion pathway. The predominantMutL homologue Mlh1 can partner either Pms2 to formMutLa, or Mlh3 to form MutLb, and both the complexesare able to mediate the MMR of insertion/deletion loops(13), the most likely intermediates in the expansionpathway. Thus, it is very likely that Pms2 is at least partiallyredundant to Mlh3 in generating expansions.
The involvement of a MutL homologue in the expansionpathway has important consequences for understanding themolecular mechanisms involved. First, in contrast to predic-tions made using the replication slippage model, loss of func-tion of the Pms2 protein does not result in an increase in therate of expansion. In bacteria, mutS and mutL mutationscause an overall increase in the frequency of small lengthchanges within trinucleotide repeat tracts (16–18). Similarly,increased instability of short CAG.CTG repeats at both theDM1 and SBMA loci has been described in tumour samplesfrom HNPCC families with MMR gene mutations (40). Itremains possible that there is an increased frequency in therate of non-repaired replication slippage errors in the Dmt-Drepeat tract in Pms2-deficient mice. However, it is clear thatnon-repaired replication slippage errors do not contribute signi-ficantly to the expansions that accrue, as the rate of expansionin Pms2-deficient mice, in which the frequency of non-repaired replication slippage errors should be much higher,is actually considerably lower. Second, the involvement of aMutL homologue in the expansion pathway argues againstadditional models that invoke the MutS homologues alone.For instance, it has been hypothesized that stabilization ofalternative secondary structures formed within repeat tractsby Msh2 (and presumably likewise by Msh3 and/or MutSb)could lead to expansion following DNA-repair synthesis(29,31). However, following DNA gap repair, loops andslipped-stranded-like structures would be trapped in a hetero-duplex DNA molecule (31), and a second round of DNA repli-cation and cell division would be required to complete themutation process. Somatic mutations continue to accumulatein post-mitotic cells [as supported by the high levels ofinstability observed in adult mouse brains and other predomi-nantly post-mitotic tissues (19–22)]. Therefore, attractive as itis, such a mechanism could only account for somatic trinu-cleotide repeat mutations in proliferating tissues, and is unli-kely to contribute to the continuous expansion of trinucleotiderepeat sequences in post-mitotic cells. Similarly, mechanismsbased on the SSA pathway for the repair of double-strandbreaks (30), appear to be excluded as they are not dependenton MutL homologues (32). Thus, in contrast to previously pro-posed models (29–31), the involvement of a MutL homologuein somatic expansion implies that it is a full MMR reactionthat is required to generate expansions.
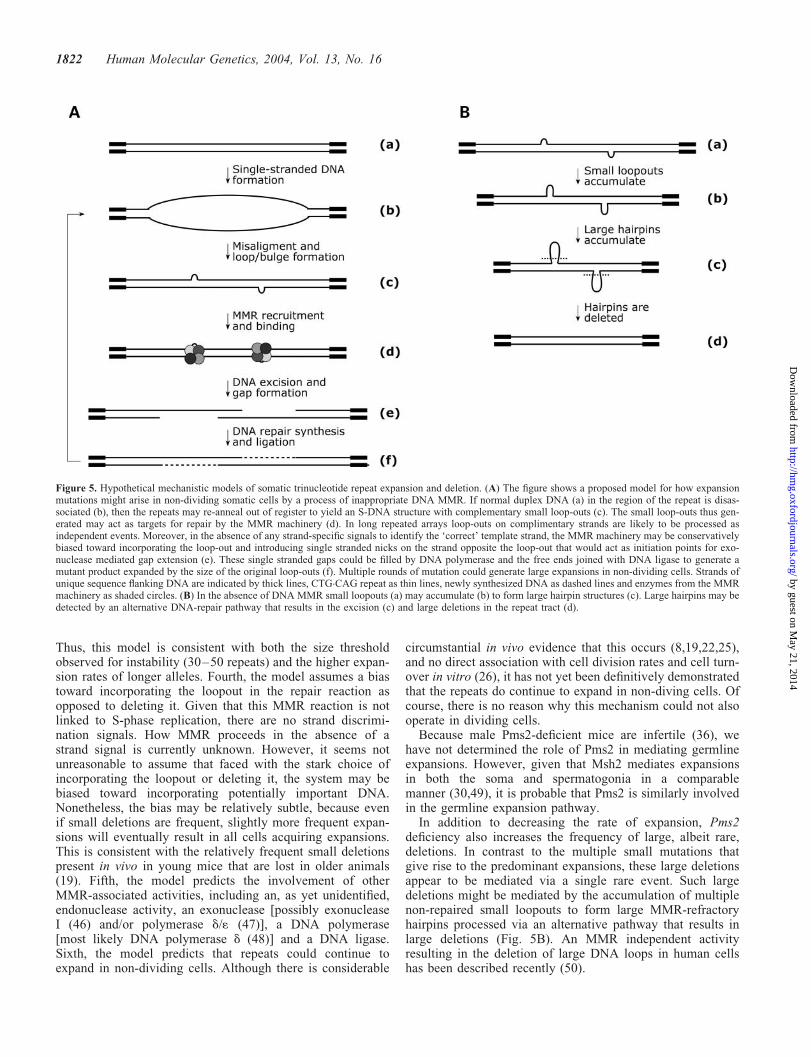
Thus, the emerging evidence indicates that somatic mosai-cism arises via multiple small events, biased toward expan-sion, that accrue in a cell division independent mechanismrequiring both MutS and MutL components of the MMRpathway. None of the previously proposed models can accountfor these observations. We therefore suggest an alternativemodel based on inappropriate DNA MMR (Fig. 5A). Briefly,we propose that the repeats are usually replicated faithfullyin the form of standard duplex B-DNA (Fig. 5A-a), but that
Figure 3. No detectable effect on the rate of somatic expansion in Dmt-D miceheterozygous for the Pms2 deletion. (A) Representative SP-PCR amplifica-tions of DNA molecules extracted from the lung, heart, brain and kidneycollected from a pair of Dmt-D littermates aged 24 months: one Pms2 þ/2
and one Pms2 þ/þ and with progenitor allele lengths of 196 and 176repeats, respectively. Around 10–100 transgene molecules were amplified ineach reaction, with oligonucleotide primers DM-C and DM-BR. The sizemarkers, converted into repeat size differences from the progenitor allele,are displayed on the right. (B) Repeat length distributions in the lung andkidney of the two Dmt-D littermates shown in (A). SP-PCR analyses were per-formed with a very low-DNA input: an average of 1–2 transgene moleculesper reaction. Each band observed was individually sized and the repeatlengths were grouped into size classes of 10 repeat intervals. The totalnumber of transgene molecules sized (n) for each tissue are shown in thetop right corner of each panel. (C) Representative SP-PCR amplifications ofDNA molecules extracted from lung, heart, brain and kidney collected fromtwo pairs of Dmt-D mice aged 13 months: two Pms2 þ/2 with progenitorallele lengths of 169 repeats and two Pms2 þ/þ with progenitor allelelengths of 150 and 156 repeats. SP-PCR was performed as in (A).
1820 Human Molecular Genetics, 2004, Vol. 13, No. 16
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

at some point during either the G0 or G1 stage of the cell cyclethe duplex is melted (Fig. 5A-b). The repeat sequences thenre-anneal out of register forming S-DNA structures wherethe predominant species are small mismatched loopouts of1–3 repeat units (Fig. 5A-c). Subsequently, these small loop-outs are recognized by the MutSb complex (Msh2 and Msh3)and recruit either the MutLa or MutLb complex (Fig. 5A-d) aspart of an MMR reaction in which the loopouts are incorpor-ated (Fig. 5A-e) and give rise to expansions (Fig. 5A-f). Theexpansion process could be reinitiated by the formation ofnew S-DNA structures allowing multiple mutations to accumu-late in non-dividing cells. In this way cells could accumulatevery large expansions as a result of frequent small gains.
This proposed mechanism makes several assumptions andtestable predictions. First, the repeats adopt S-DNA structures.Although such structures have not yet been reported in vivo,these conformations form readily in vitro and are very stable(11). Their formation in vivo might be mediated by transcrip-tion or DNA breathing. The absence of a correlation betweenexpression levels and somatic instability of a DM transgeneargue against a direct role for transcription in mediatingrepeat length changes (25). Spontaneous DNA breathing of alinear DNA molecule at physiological temperatures wouldbe unlikely to result in the production of a large enoughmelting domain to facilitate S-DNA formation. However,negative supercoiling can dramatically lower the meltingthreshold and facilitate large-scale opening of the duplex(41). Negative superhelical torsion can be generated by chro-matin remodelling (42) and this presents a possible mechan-ism whereby S-DNA formation might be mediated in non-dividing cells. Second, the loopouts are recognized and
repaired by the MutSb heterodimer (Msh2 and Msh3) com-plexed with MutLa or MutLb. This predicts that Mlh1 willbe essential for expansion and that Mlh3 will be partiallyredundant to Pms2 in mediating expansions. The small loop-outs of 1–3 repeat units (3–9 nt) hypothesized are commonlyobserved in S-DNA formed in vitro (43) and are within therange that can be processed by the MutSb complex (44). Inthe absence of a functional MMR pathway S-DNA structuresmay remain quiescent within the cell or even accumulate, con-sistent with the stabilization reported here and observed inMsh2- and Msh3-null animals (24,29). In theory, larger loop-outs capable of forming intrastrand hairpins could also form.However, such large loopouts would be predicted to be resist-ant to processing by the MMR machinery and would result inlarger length changes than are usually observed. Third, indi-vidual loopouts on opposite strands are repaired as separateevents. If the excision domain (Fig. 5A-e) for one loopoutincorporated the loopout on the opposite strand, then the netlength change would be zero. The size of the excisiondomain in mammalian cells is not known. However, in vitroexperiments using human nuclear extracts and nick-directedreconstitution of a MMR have revealed that the exonucleasetypically removes an additional 60-230 bp DNA beyond themismatch (45), thus defining the minimum size of the excisiondomain. Normal length alleles, although capable of formingS-DNA structures (11), would therefore be unlikely to repairopposing loops as separate events and would thus not beliable to expand via this mechanism. Moreover, longerexpanded alleles are even more likely to form S-DNA struc-tures with more distantly separated opposing loops andwould be expected to show increased levels of instability.
Figure 4. Pms2 protein levels are reduced in Pms2 þ/2 cells. (A) Western blot analysis of whole cell protein lysates extracted from cultured mouse embryonicfibroblasts. Two independent fibroblast cultures, derived from two independent Dmt-D embryos, were established for each genetic background: Pms2 þ/þ,Pms2 þ/2 and Pms2 2/2. Four protein samples collected from each culture were separated on a 12% SDS–PAGE gel and electroblotted. The blots wereprobed with anti-Pms2 and anti-b-tubulin antibodies. Pms2 protein is absent in nullizygotes, whereas heterozygotes for the deletion exhibit lower levels ofPms2 than Dmt-D mice carrying two functional Pms2 alleles. (B) The graphs show the quantitative analysis of Pms2 protein expression levels relative tob-tubulin, in Pms2 þ/þ and Pms2 þ/2 cells. Two replicate cultures derived from independent Dmt-D embryos were studied for each genetic background andfour replicate protein samples of the same protein lysate were electrophoresed in the same gel (A). The reduction in Pms2 protein levels in heterozygousmice for the Pms2 deletion is highly significant (P ¼ 0.001, two-tailed t-test).
Human Molecular Genetics, 2004, Vol. 13, No. 16 1821
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Thus, this model is consistent with both the size thresholdobserved for instability (30–50 repeats) and the higher expan-sion rates of longer alleles. Fourth, the model assumes a biastoward incorporating the loopout in the repair reaction asopposed to deleting it. Given that this MMR reaction is notlinked to S-phase replication, there are no strand discrimi-nation signals. How MMR proceeds in the absence of astrand signal is currently unknown. However, it seems notunreasonable to assume that faced with the stark choice ofincorporating the loopout or deleting it, the system may bebiased toward incorporating potentially important DNA.Nonetheless, the bias may be relatively subtle, because evenif small deletions are frequent, slightly more frequent expan-sions will eventually result in all cells acquiring expansions.This is consistent with the relatively frequent small deletionspresent in vivo in young mice that are lost in older animals(19). Fifth, the model predicts the involvement of otherMMR-associated activities, including an, as yet unidentified,endonuclease activity, an exonuclease [possibly exonucleaseI (46) and/or polymerase d/1 (47)], a DNA polymerase[most likely DNA polymerase d (48)] and a DNA ligase.Sixth, the model predicts that repeats could continue toexpand in non-dividing cells. Although there is considerable
circumstantial in vivo evidence that this occurs (8,19,22,25),and no direct association with cell division rates and cell turn-over in vitro (26), it has not yet been definitively demonstratedthat the repeats do continue to expand in non-diving cells. Ofcourse, there is no reason why this mechanism could not alsooperate in dividing cells.
Because male Pms2-deficient mice are infertile (36), wehave not determined the role of Pms2 in mediating germlineexpansions. However, given that Msh2 mediates expansionsin both the soma and spermatogonia in a comparablemanner (30,49), it is probable that Pms2 is similarly involvedin the germline expansion pathway.
In addition to decreasing the rate of expansion, Pms2deficiency also increases the frequency of large, albeit rare,deletions. In contrast to the multiple small mutations thatgive rise to the predominant expansions, these large deletionsappear to be mediated via a single rare event. Such largedeletions might be mediated by the accumulation of multiplenon-repaired small loopouts to form large MMR-refractoryhairpins processed via an alternative pathway that results inlarge deletions (Fig. 5B). An MMR independent activityresulting in the deletion of large DNA loops in human cellshas been described recently (50).
Figure 5. Hypothetical mechanistic models of somatic trinucleotide repeat expansion and deletion. (A) The figure shows a proposed model for how expansionmutations might arise in non-dividing somatic cells by a process of inappropriate DNA MMR. If normal duplex DNA (a) in the region of the repeat is disas-sociated (b), then the repeats may re-anneal out of register to yield an S-DNA structure with complementary small loop-outs (c). The small loop-outs thus gen-erated may act as targets for repair by the MMR machinery (d). In long repeated arrays loop-outs on complimentary strands are likely to be processed asindependent events. Moreover, in the absence of any strand-specific signals to identify the ‘correct’ template strand, the MMR machinery may be conservativelybiased toward incorporating the loop-out and introducing single stranded nicks on the strand opposite the loop-out that would act as initiation points for exo-nuclease mediated gap extension (e). These single stranded gaps could be filled by DNA polymerase and the free ends joined with DNA ligase to generate amutant product expanded by the size of the original loop-outs (f). Multiple rounds of mutation could generate large expansions in non-dividing cells. Strands ofunique sequence flanking DNA are indicated by thick lines, CTG.CAG repeat as thin lines, newly synthesized DNA as dashed lines and enzymes from the MMRmachinery as shaded circles. (B) In the absence of DNA MMR small loopouts (a) may accumulate (b) to form large hairpin structures (c). Large hairpins may bedetected by an alternative DNA-repair pathway that results in the excision (c) and large deletions in the repeat tract (d).
1822 Human Molecular Genetics, 2004, Vol. 13, No. 16
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

Given the significant role we have revealed for Pms2 in theexpansion pathway and the observations that Pms2 levelsappear to be rate limiting, we were slightly surprised thatwere unable to detect a haploinsufficiency phenotype forrepeat expansion in Pms2 þ/2 mice. This result was particu-larly surprising as we were able to demonstrate that levelsof Pms2 protein were indeed reduced in Pms2 heterozygousanimals. A possible explanation for this result is that theamount of Mlh1 in normal cells is rate limiting with regardto the formation of MutLb complexes. With 50% lowerlevels of Pms2 in the cell, more Mlh1 would be available toform active MutLb complexes, thus compensating for thedecreased amount of MutLa in loopout repair. Such aneffect would be analogous to the situation proposed regardingthe relative amounts of MutSa and MutSb in normal cells andthose deficient for Msh6 (24,44,51). However, it would also benecessary to postulate that the absolute amount of Mlh3 is alsorate limiting and the increased MutLb activity is unable tofully compensate for Pms2 deficiency, accounting for theobserved decrease in expansion rate in Pms2-null cells.
In summary, we have established that the Pms2 gene is amajor component of the expansion pathway. Pms2 is thus anexcellent candidate modifier gene for both the expansionpathway and consequently of disease severity in patients,and may also be a target for therapeutic intervention in therepeat expansion disorders.
MATERIALS AND METHODS
Mouse breeding and genotyping
The Pms2-null allele generated by Baker et al. (36) wasobtained on a mixed 129Sv/C57Bl6 background. It was back-crossed onto an FVB/N background for five generations andsubsequently crossed with Dmt-D transgenic mice (39) alsomaintained on a FVB/N background. Genotyping for Pms2and Dmt-D was performed as previously described (36,39).All mice analysed were hemizygous for the Dmt-D transgeneand the progenitor allele length inherited by each mouse wasdetermined by the analyses of tail DNA collected at weaning.
SP-PCR analysis
DNA purification, SP-PCR amplifications and PCR productelectrophoretic analyses were performed as described pre-viously (26). SP-PCR was performed using oligonucleotideprimers DM-C and DM-BR (4), or DM-C and mDmtD-GR(50AAAGGCAGGCATGGTTAGATTGAC30).
Establishment of cultured mouse embryonic fibroblasts
Primary mouse embryonic fibroblast cultures were isolatedfrom 14-day-old Dmt-D positive mouse embryos. Briefly,the head and the liver were removed from each embryo andthe remainder digested with 3 ml of pre-warmed trypsin–EDTA solution for 10 min in a humidified 5% CO2 incubatorat 378C. To disassociate the cells the embryo bodies weretransferred into a 5 ml syringe and passed through an18-gauge needle into a 25 cm2 tissue culture flask. The diges-tion was continued for 20–30 min and then stopped by the
addition of 20 ml of pre-warmed culture medium The contentsof the flask were transferred into a 50 ml conical tube andlarge pieces of tissue allowed to settle for 2 min. The disso-ciated cell-containing supernatant was transferred to a cleantube and the cells collected by centrifugation at 200g for5 min. Dissociated cells were resuspended in 5 ml of freshmedium and plated on a 25 cm2 tissue culture flask and trans-ferred to an incubator at 378C with 5% CO2. The remaininglarge pieces of tissue were further digested by adding 5 mlof fresh trypsin–EDTA and continuing the incubation for anadditional 20–30 min. Dissociated cells from the secondarydigest were collected as described and plated together withpreviously dissociated cells. Adherent mouse embryonic fibro-blasts thus derived were maintained in culture using standardcell culture techniques and DMEM medium supplementedwith 10% (v/v) fetal calf serum, non-essential amino acids,0.1 mM b-mercaptoethanol, 100 U ml21 penicillin and100 mgml21 streptomycin.
Protein sample preparation and western blotting
Protein was extracted from cultured mouse embryonic fibro-blasts using EBC lysis buffer [50 mM Tris–HCl, 120 mM
NaCl, 0.5% (v/v) NP-40] containing protease inhibitors (pro-tease inhibitor cocktail for mammalian tissues, Sigma, cat.no. P8340). Aliquots of 100 mg of whole cell protein lysatewere resolved by electrophoresis through a NuPAGE 4–12%Bis–Tris Gel (Invitrogen, cat. no. NP0321) and electroblottedonto Millipore Immobilon-P membranes (Millipore, cat. no.IPVH00010) at 30 V for 2 h in an XCellII Blot Module(Novex, cat. no. EI9051) in NuPAGE Transfer Buffer (Invitro-gen, cat. no. NP0006-1). The membranes were blocked for twohours at room temperature in 5% (w/v) dried milk in TBST(20 mM Tris–HCl pH 7.6, 137 mM NaCl, 0.06% (v/v)Tween-20), then incubated overnight at 48C in primary anti-body. The membranes were washed four times for 15 mineach in TBST, incubated for 1 h in secondary antibody atroom temperature, and washed three times for 15 min eachin TBST at room temperature. Antibody binding was visual-ized using SuperSignal West Pico Chemiluminescent Sub-strate (Pierce, cat. no. 34080). Pms2 was detected using1 mg ml21 mouse anti-Pms2 monoclonal antibody (BD Phar-Mingen, cat. no. 556415) and 8 ng ml21 goat anti-mouseIgG HRP-conjugated antibody (Jackson Laboratories, cat.no. 115-035-003) diluted 1:50 000. b-Tubulin was detectedusing a 0.5 mg ml21 rabbit anti-b-tubulin polyclonal antibody(Santa Cruz Biotechnology, cat. no. sc9104) and 16 ng ml21
goat anti-rabbit IgG HRP-conjugated antibody (Santa CruzBiotechnology, cat. no. sc-2004). Densitometric analysis ofprotein levels was performed using Kodak Digital Science1D software using exposures in the linear range of signalintensity.
ACKNOWLEDGEMENTS
We would like to thank Michael Liskay and Sean Baker forproviding the Pms2-deficient mice. We are also very gratefulto Peggy F. Shelbourne and the Dynamic Mutation Group atthe University of Glasgow for helpful discussion during the
Human Molecular Genetics, 2004, Vol. 13, No. 16 1823
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

course of this work, and to the Biological Services for excel-lent animal care and assistance with colony maintenance. Wewould also like to acknowledge the Lister Institute (UK), Mus-cular Dystrophy Association (USA), the Medical ResearchCouncil (UK) and the Association Francaise Contre Les Myo-pathies (France) for financial support. M.G.P. was supportedby awards from the Fundacao para a Ciencia e Tecnologiaand Fundacao Calouste Gulbenkian (Portugal).
REFERENCES
1. Richards, R.I. (2001) Dynamic mutations: a decade of unstableexpanded repeats in human genetic disease. Hum. Mol. Genet., 10,2187–2194.
2. Wong, L.-J.C., Ashizawa, T., Monckton, D.G., Caskey, C.T. andRichards, C.S. (1995) Somatic heterogeneity of the CTG repeat inmyotonic dystrophy is age and size dependent. Am. J. Hum. Genet., 56,114–122.
3. Martorell, L., Monckton, D.G., Gamez, J., Johnson, K.J., Gich, I.,Lopez de Munain, A. and Baiget, M. (1998) Progression of somatic CTGrepeat length heterogeneity in the blood cells of myotonic dystrophypatients. Hum. Mol. Genet., 7, 307–312.
4. Monckton, D.G., Wong, L.-J.C., Ashizawa, T. and Caskey, C.T. (1995)Somatic mosaicism, germline expansions, germline reversions andintergenerational reductions in myotonic dystrophy males: small poolPCR analyses. Hum. Mol. Genet., 4, 1–8.
5. Anvret, M., Ahlberg, G., Grandell, U., Hedberg, B., Johnson, K. andEdstrom, L. (1993) Larger expansions of the CTG repeat in musclecompared to lymphocytes from patients with myotonic dystrophy. Hum.Mol. Genet., 2, 1397–1400.
6. Ashizawa, T., Dubel, J.R. and Harati, Y. (1993) Somatic instability ofCTG repeat in myotonic dystrophy. Neurology, 43, 2674–2678.
7. Thornton, C.A., Johnson, K.J. and Moxley, R.T. (1994) Myotonicdystrophy patients have larger CTG expansions in skeletal muscle than inleukocytes. Ann. Neurol., 35, 104–107.
8. Kennedy, L., Evans, E., Chen, C.M., Craven, L., Detloff, P.J., Ennis, M.and Shelbourne, P.F. (2003) Dramatic tissue-specific mutation lengthincreases are an early molecular event in Huntington disease pathogenesis.Hum. Mol. Genet., 12, 3359–3367.
9. Richards, R.I. and Sutherland, G.R. (1994) Simple DNA is not replicatedsimply. Nat. Genet., 6, 114–116.
10. Gacy, A.M., Goellner, G., Juranic, N., Macura, S. and McMurray, C.T.(1995) Trinucleotide repeats that expand in human disease form hairpinstructures in vitro. Cell, 81, 533–540.
11. Pearson, C.E. and Sinden, R.R. (1996) Alternative structures in duplexDNA formed within the trinucleotide repeats of the myotonic dystrophyand fragile X loci. Biochemistry, 35, 5041–5053.
12. Pearson, C.E., Ewel, A., Acharya, S., Fishel, R.A. and Sinden, R.R. (1997)Human MSH2 binds to trinucleotide repeat DNA structures associatedwith neurodegenerative diseases. Hum. Mol. Genet., 6, 1117–1123.
13. Peltomaki, P. (2001) Deficient DNA mismatch repair: a common etiologicfactor for colon cancer. Hum. Mol. Genet., 10, 735–740.
14. Jaworski, A., Rosche, W.A., Gellibolian, R., Kang, S.M., Shimizu, M.,Bowater, R.P., Sinden, R.R. and Wells, R.D. (1995) Mismatch repair inEscherichia coli enhances instability of (CTG)(n ) triplet repeats fromhuman hereditary diseases. Proc. Natl Acad. Sci. USA, 92, 11019–11023.
15. Schumacher, S., Fuchs, R.P. and Bichara, M. (1998) Expansion of CTGrepeats from human disease genes is dependent upon replicationmechanisms in Escherichia coli: the effect of long patch mismatch repairrevisited. J. Mol. Biol., 279, 1101–1110.
16. Schmidt, K.H., Abbott, C.M. and Leach, D.R. (2000) Two opposingeffects of mismatch repair on CTG repeat instability in Escherichia coli.Mol. Microbiol., 35, 463–471.
17. Parniewski, P., Jaworski, A., Wells, R.D. and Bowater, R.P. (2000)Length of CTG.CAG repeats determines the influence of mismatch repairon genetic instability. J. Mol. Biol., 299, 865–874.
18. Schweitzer, J.K. and Livingston, D.M. (1997) Destabilization of CAGtrinucleotide repeat tracts by mismatch repair mutations in yeast. Hum.Mol. Genet., 6, 349–355.
19. Fortune, M.T., Vassilopoulos, C., Coolbaugh, M.I., Siciliano, M.J. andMonckton, D.G. (2000) Dramatic, expansion-biased, age-dependent,tissue-specific somatic mosaicism in a transgenic mouse model of tripletrepeat instability. Hum. Mol. Genet., 9, 439–445.
20. Seznec, H., Lia-Baldini, A.S., Duros, C., Fouquet, C., Lacroix, C.,Hofmann-Radvanyi, H., Junien, C. and Gourdon, G. (2000) Transgenicmice carrying large human genomic sequences with expanded CTG repeatmimic closely the DM CTG repeat intergenerational and somaticinstability. Hum. Mol. Genet., 9, 1185–1194.
21. Mangiarini, L., Sathasivam, K., Mahal, A., Mott, R., Seller, M. andBates, G.P. (1997) Instability of highly expanded CAG repeats in micetransgenic for the Huntington’s disease mutation. Nat. Genet., 15,197–200.
22. Kennedy, L. and Shelbourne, P.F. (2000) Dramatic mutation instability inHD mouse striatum: does polyglutamine load contribute to cell-specificvulnerability in Huntington’s disease? Hum. Mol. Genet., 9, 2539–2544.
23. Lorenzetti, D., Watase, K., Xu, B., Matzuk, M.M., Orr, H.T. andZoghbi, H.Y. (2000) Repeat instability and motor incoordination inmice with a targeted expanded CAG repeat in the Sca1 locus.Hum. Mol. Genet., 9, 779–785.
24. van Den Broek, W.J., Nelen, M.R., Wansink, D.G., Coerwinkel, M.M.,te Riele, H., Groenen, P.J. and Wieringa, B. (2002) Somatic expansionbehaviour of the (CTG)(n ) repeat in myotonic dystrophy knock-in mice isdifferentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum.Mol. Genet., 11, 191–198.
25. Lia, A.S., Seznec, H., Hofmann Radvanyi, H., Radvanyi, F., Duros, C.,Saquet, C., Blanche, M., Junien, C. and Gourdon, G. (1998) Somaticinstability of the CTG repeat in mice transgenic for the myotonicdystrophy region is age dependent but not correlated to the relativeintertissue transcription levels and proliferative capacities. Hum. Mol.
Genet., 7, 1285–1291.
26. Gomes-Pereira, M., Fortune, M.T. and Monckton, D.G. (2001) Mousetissue culture models of unstable triplet repeats: in vitro selection forlarger alleles, mutational expansion bias and tissue specificity, but noassociation with cell division rates. Hum. Mol. Genet., 10, 845–854.
27. Modrich, P. (1991) Mechanisms and biological effects of mismatch repair.Annu. Rev. Genet., 25, 229–253.
28. Umar, A., Buermeyer, A.B., Simon, J.A., Thomas, D.C., Clark, A.B.,Liskay, R.M. and Kunkel, T.A. (1996) Requirement for PCNA in DNAmismatch repair at a step preceding DNA resynthesis. Cell, 87, 65–73.
29. Manley, K., Shirley, T.L., Flaherty, L. and Messer, A. (1999) Msh2
deficiency prevents in vivo somatic instability of the CAG repeat inHuntington disease transgenic mice. Nat. Genet., 23, 471–473.
30. Savouret, C., Brisson, E., Essers, J., Kanaar, R., Pastink, A., te Riele, H.,Junien, C. and Gourdon, G. (2003) CTG repeat instability and sizevariation timing in DNA repair-deficient mice. EMBO J., 22, 2264–2273.
31. Kovtun, I.V. and McMurray, C.T. (2001) Trinucleotide expansion inhaploid germ cells by gap repair. Nat. Genet., 27, 407–411.
32. Sugawara, N., Paques, F., Colaiacovo, M. and Haber, J.E. (1997) Role ofSaccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc. Natl Acad. Sci. USA, 94,9214–3219.
33. De Rosa, M., Fasano, C., Panariello, L., Scarano, M.I., Belli, G.,Iannelli, A., Ciciliano, F. and Izzo, P. (2000) Evidence for a recessiveinheritance of Turcot’s syndrome caused by compound heterozygousmutations within the PMS2 gene. Oncogene, 19, 1719–1723.
34. Miyaki, M., Nishio, J., Konishi, M., Kikuchi-Yanoshita, R., Tanaka, K.,Muraoka, M., Nagato, M., Chong, J.M., Koike, M., Terada, T., et al.(1997) Drastic genetic instability of tumors and normal tissues in Turcotsyndrome. Oncogene, 15, 2877–2881.
35. Nicolaides, N.C., Papadopoulos, N., Liu, B., Wei, Y.F., Carter, K.C.,Ruben, S.M., Rosen, C.A., Haseltine, W.A., Fleischmann, R.D.,Fraser, C.M. et al. (1994) Mutations of two PMS homologues inhereditary nonpolyposis colon cancer. Nature, 371, 75–80.
36. Baker, S., Bronner, C.E., Zhang, L., Plug, A.W., Robatzek, M.,Warren, G., Elliott, E.A., Yu, J., Ashley, T., Arnheim, N. et al. (1995)Male mice defective in the DNA mismatch repair gene PMS2 exhibitabnormal chromosome synapsis in meiosis. Cell, 82, 309–319.
37. Prolla, T.A., Baker, S.M., Harris, A.C., Tsao, J.L., Yao, X., Bronner, C.E.,Zheng, B., Gordon, M., Reneker, J., Arnheim, N. et al. (1998) Tumoursusceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1and Pms2 DNA mismatch repair. Nat. Genet., 18, 276–279.
1824 Human Molecular Genetics, 2004, Vol. 13, No. 16
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

38. Narayanan, L., Fritzell, J.A., Baker, S.M., Liskay, R.M. and Glazer, P.M.
(1997) Elevated levels of mutation in multiple tissues of mice deficient in
the DNA mismatch repair gene Pms2. Proc. Natl Acad. Sci. USA, 94,3122–3127.
39. Monckton, D.G., Coolbaugh, M.I., Ashizawa, K., Siciliano, M.J. and
Caskey, C.T. (1997) Hypermutable myotonic dystrophy CTG repeats intransgenic mice. Nat. Genet., 15, 193–196.
40. Wooster, R., Cletonjansen, A.M., Collins, N., Mangion, J., Cornelis, R.S.,
Cooper, C.S., Gusterson, B.A., Ponder, B.A.J., Vondeimling, A.,Wiestler, O.D. et al. (1994) Instability of short tandem repeats
(microsatellites) in human cancers. Nat. Genet., 6, 152–156.
41. Lilley, D.M. (1988) DNA opens up—supercoiling and heavy breathing.Trends Genet., 4, 111–114.
42. Havas, K., Flaus, A., Phelan, M., Kingston, R., Wade, P.A., Lilley, D.M.
and Owen-Hughes, T. (2000) Generation of superhelical torsion by ATP-dependent chromatin remodeling activities. Cell, 103, 1133–1142.
43. Pearson, C.E., Wang, Y.H., Griffith, J.D. and Sinden, R.R. (1998)
Structural analysis of slipped-strand DNA (S-DNA) formed in(CTG)n.(CAG)n repeats from the myotonic dystrophy locus. Nucl. Acids
Res., 26, 816–823.
44. Genschel, J., Littman, S.J., Drummond, J.T. and Modrich, P. (1998)Isolation of MutSbeta from human cells and comparison of the mismatch
repair specificities of MutSbeta and MutSalpha. J. Biol. Chem., 273,19895–19901.
45. Genschel, J. and Modrich, P. (2003) Mechanism of 50-directed excision inhuman mismatch repair. Mol. Cell, 12, 1077–1086.
46. Genschel, J., Bazemore, L.R. andModrich, P. (2002) Human exonuclease Iis required for 50 and 30 mismatch repair. J. Biol. Chem., 277,13302–13311.
47. Wang, H. and Hays, J.B. (2002) Mismatch repair in human nuclearextracts. Time courses and ATP requirements for kineticallydistinguishable steps leading to tightly controlled 50 to 30 and aphidicolin-sensitive 30 to 50 mispair-provoked excision. J. Biol. Chem., 277,26143–26148.
48. Longley, M.J., Pierce, A.J. and Modrich, P. (1997) DNA polymerase deltais required for human mismatch repair in vitro. J. Biol. Chem., 272,10917–10921.
49. Savouret, C., Garcia-Cordier, C., Megret, J., te Riele, H., Junien, C. andGourdon, G. (2004) MSH2-dependent germinal CTG repeat expansionsare produced continuously in spermatogonia from DM1 transgenic mice.Mol. Cell. Biol., 24, 629–637.
50. McCulloch, S.D., Gu, L. and Li, G.M. (2003) Nick-dependent and-independent processing of large DNA loops in human cells. J. Biol.Chem., 278, 50803–50809.
51. de Wind, N., Dekker, M., Claij, N., Jansen, L., van Klink, Y., Radman, M.,Riggins, G., van der Valk, M., van’t Wout, K. and te Riele, H. (1999)HNPCC-like cancer predisposition in mice through simultaneous loss ofMsh3 and Msh6 mismatch-repair protein functions. Nat. Genet., 23,359–362.
Human Molecular Genetics, 2004, Vol. 13, No. 16 1825
by guest on May 21, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from




















