
Demonstration of the Recurrence of Marfan-like Skeletal and Cardiovascular Manifestations Due to...
18
911 Letters to the Editor Table 1 Prevalence of AP in Asian Music Students, Stratified by Type of Music Program Type of Music Program (No. of Students Surveyed) No. (%) of Students with AP Conservatory (73) 36 (49.3) University music program (152) 39 (25.7) Liberal arts college (12) 1 (8.3) All programs combined (237) 76 (32.1) Am. J. Hum. Genet. 65:911–913, 1999 Absolute Pitch: Prevalence, Ethnic Variation, and Estimation of the Genetic Component To the Editor: Absolute pitch (AP), also known as “perfect pitch,” is a distinct cognitive ability possessed by a minority of musicians. The essential feature of this trait is the ca- pacity to recognize and name the pitch of a musical note or ambient sound without the use of a reference pitch and with a minimum of deliberation. Elegant studies by Miyazaki (1988) have provided a method of measuring this ability, and Baharloo et al. (1998) recently reported on the characterization of a population of AP possessors, using a modification of this approach. These studies have emphasized that, although there is some variation in levels of accuracy in AP possessors, musicians with this ability are nevertheless distinct from those who do not possess AP. As such, AP ability is one of the few cognitive phenotypes that exhibit a clear qualitative difference be- tween those who possess it and those who do not. Although informal prevalence estimates for AP (<1: 1,500 among amateur music students) have been sug- gested (Profita and Bidder 1988), the study by Baharloo et al. (1998) represents the only published data con- cerning the prevalence of the AP phenotype. In a survey of 612 highly accomplished musicians, Baharloo et al. (1998) observed a prevalence rate of 15%. We have now completed a survey of 2,707 music students at music conservatories as well as at university and college music programs in the United States. We surveyed student pop- ulations ranging in size from 20 to 390 students (mean [5 SD] ), using a two-page questionnaire ask- 104 5 78 ing about the presence of AP in the students and in their family members. We assumed AP ability to be present if students reported both the ability to perceive tones in an absolute manner and the ability to sing a note when given the letter name, but without a reference pitch. In our experience (see below), this correlates reasonably well with AP ability on objective testing, as has been observed by others (Takeuchi and Hulse 1991; Baharloo et al. 1998). We observed large variations in AP prevalence among different student populations (range 0%–35%). There was a significant association ( ) between the type P ! .001 of institution or music program and the prevalence of AP in the students: conservatory (24.6% with AP), uni- versity-based school of music (7.3% with AP), or liberal arts/state university music program (4.7% with AP). We also noted a strong correlation between the prevalence of AP and the percentage of students in these schools who reported their ethnic background as “Asian or Pa- cific Islander” ( , , Spearman rank cor- r= .81 P ! .0001 relation coefficient). This raised the possibility that AP is more prevalent in Asian students in general. The prevalence data in table 1 (Asian students) and table 2 (non-Asian students) indicate that AP is signifi- cantly more prevalent in Asian students compared with all other ethnic groups (non-Asian) combined (32.1% vs. 7.0%, ). Furthermore, the higher rate of AP P ! .001 in Asian students is observed in all types of educational institutions. Even among non-Asians, however, the rate of AP was significantly higher in students at major music conservatories (table 2). A multivariate logistic regres- sion indicated that Asian ethnicity and attendance at a conservatory were independently associated with AP in the student populations. Asian ethnic background had a relative risk (RR) of 5.0 (95% CI 3.6–7.0), whereas attendance at a music conservatory (vs. other music pro- grams) had (95% CI 2.6–4.8). There were in- RR = 3.5 sufficient numbers of Hispanic or African American stu- dents to perform a meaningful subgroup analysis within the non-Asian group. Most of the individuals in the non- Asian group were white, and there were no obvious trends among the other broad ethnic groupings. As has been reported by others, we also observed a significant association between AP and the age at which an individual first began playing music. For the AP group as a whole, the mean age of starting musical activities
-
Upload
univ-paris7 -
Category
Documents
-
view
1 -
download
0
Transcript of Demonstration of the Recurrence of Marfan-like Skeletal and Cardiovascular Manifestations Due to...

911
Letters to the Editor
Table 1
Prevalence of AP in Asian Music Students, Stratified byType of Music Program
Type of Music Program(No. of Students Surveyed)
No. (%) of Studentswith AP
Conservatory (73) 36 (49.3)University music program (152) 39 (25.7)Liberal arts college (12) 1 (8.3)
All programs combined (237) 76 (32.1)
Am. J. Hum. Genet. 65:911–913, 1999
Absolute Pitch: Prevalence, Ethnic Variation, andEstimation of the Genetic Component
To the Editor:Absolute pitch (AP), also known as “perfect pitch,” isa distinct cognitive ability possessed by a minority ofmusicians. The essential feature of this trait is the ca-pacity to recognize and name the pitch of a musical noteor ambient sound without the use of a reference pitchand with a minimum of deliberation. Elegant studies byMiyazaki (1988) have provided a method of measuringthis ability, and Baharloo et al. (1998) recently reportedon the characterization of a population of AP possessors,using a modification of this approach. These studies haveemphasized that, although there is some variation inlevels of accuracy in AP possessors, musicians with thisability are nevertheless distinct from those who do notpossess AP. As such, AP ability is one of the few cognitivephenotypes that exhibit a clear qualitative difference be-tween those who possess it and those who do not.
Although informal prevalence estimates for AP (<1:1,500 among amateur music students) have been sug-gested (Profita and Bidder 1988), the study by Baharlooet al. (1998) represents the only published data con-cerning the prevalence of the AP phenotype. In a surveyof 612 highly accomplished musicians, Baharloo et al.(1998) observed a prevalence rate of 15%. We have nowcompleted a survey of 2,707 music students at musicconservatories as well as at university and college musicprograms in the United States. We surveyed student pop-ulations ranging in size from 20 to 390 students (mean[5 SD] ), using a two-page questionnaire ask-104 5 78ing about the presence of AP in the students and in theirfamily members. We assumed AP ability to be presentif students reported both the ability to perceive tones inan absolute manner and the ability to sing a note whengiven the letter name, but without a reference pitch. Inour experience (see below), this correlates reasonablywell with AP ability on objective testing, as has beenobserved by others (Takeuchi and Hulse 1991; Baharlooet al. 1998).
We observed large variations in AP prevalence amongdifferent student populations (range 0%–35%). There
was a significant association ( ) between the typeP ! .001of institution or music program and the prevalence ofAP in the students: conservatory (24.6% with AP), uni-versity-based school of music (7.3% with AP), or liberalarts/state university music program (4.7% with AP). Wealso noted a strong correlation between the prevalenceof AP and the percentage of students in these schoolswho reported their ethnic background as “Asian or Pa-cific Islander” ( , , Spearman rank cor-r = .81 P ! .0001relation coefficient). This raised the possibility that APis more prevalent in Asian students in general.
The prevalence data in table 1 (Asian students) andtable 2 (non-Asian students) indicate that AP is signifi-cantly more prevalent in Asian students compared withall other ethnic groups (non-Asian) combined (32.1%vs. 7.0%, ). Furthermore, the higher rate of APP ! .001in Asian students is observed in all types of educationalinstitutions. Even among non-Asians, however, the rateof AP was significantly higher in students at major musicconservatories (table 2). A multivariate logistic regres-sion indicated that Asian ethnicity and attendance at aconservatory were independently associated with AP inthe student populations. Asian ethnic background hada relative risk (RR) of 5.0 (95% CI 3.6–7.0), whereasattendance at a music conservatory (vs. other music pro-grams) had (95% CI 2.6–4.8). There were in-RR = 3.5sufficient numbers of Hispanic or African American stu-dents to perform a meaningful subgroup analysis withinthe non-Asian group. Most of the individuals in the non-Asian group were white, and there were no obvioustrends among the other broad ethnic groupings.
As has been reported by others, we also observed asignificant association between AP and the age at whichan individual first began playing music. For the AP groupas a whole, the mean age of starting musical activities

912 Letters to the Editor
Table 2
Prevalence of AP in Non-Asian Music Students, Stratifiedby Type of Music Program
Type of Music Program(No. of Students Surveyed)
No. (%) of Studentswith AP
Conservatory (276) 50 (18.1)University music program (1,844) 107 (5.8)Liberal arts college (350) 16 (4.5)
All programs combined (2,470) 173 (7.0)
was years, whereas, for the non-AP group, the5.4 5 2.8mean age was years ( ). This same7.9 5 3.2 P ! .0001trend was observed for Asian students as well as non-Asian students.
The issue of familial aggregation of AP is importantfor assessing the genetic contribution to this phenotype.In a small study (Gregersen and Kumar 1996), we es-timated ls (recurrence risk to sibs divided by populationprevalence) at ∼20, whereas the data of Baharloo et al.(1998) suggested a ls of ∼7 (Gregersen 1998). Becauseour current survey populations have such highly variablerates of AP, we estimated ls using the recurrence risk insibs of unaffecteds as the denominator in the RR cal-culation. For this survey population, the recurrence ratefor AP in siblings was reported as 14.1% for probandswith AP and 1.7% in the siblings of subjects who didnot have AP, leading to an estimate of . By thisl = 8.3s
method, the ls estimate for Asians was 11.1. Of course,this approach to estimating ls will tend to underestimateits value, since the background prevalence of AP in thegeneral population is undoubtedly much lower than itis in the sibs of unaffected musicians. Notably, the prev-alence of AP in the parents of AP probands is also higherthan in the parents of music students without AP (6.5%vs. 1.6%), similar to our previous report (Gregersen andKumar 1996).
These data indicate that estimates of the prevalenceof AP are highly dependent on the selection of the pop-ulation under study. Although AP may occur in non-musicians, the method of ascertainment of the AP phe-notype restricts prevalence surveys to musically educatedpopulations. This fact makes it especially difficult to sep-arate the environmental from the genetic factors thatpredispose to AP, since exposure to music is both re-quired for ascertainment as well as implicated in thedevelopment of the phenotype. In addition, the presenceof AP almost certainly increases the probability that mu-sical education will be pursued, and it may well provokeeducational activities at an earlier age, thus confoundingthe interpretation of the association between early-child-hood musical activities and AP. Our data also suggestthat more-professionally oriented music schools are es-pecially likely to attract or admit individuals with AP,
independent of the ethnic background of students inthese schools.
There are several possible reasons for the markedlyincreased prevalence of AP in students of Asian back-ground. The presence of AP in a child may provokemore-serious parental efforts at music education in cer-tain cultural groups and may lead to preferentialselection of this population into higher levels of musiceducation. Alternatively, certain childhood educationalsystems (for example, the Yamaha method in Japan) mayfoster the development of AP. We do not currently haveinformation on our study population concerning child-hood exposure of the Asian students to these methods.Finally, the possibility that certain Asian populationsmay have a higher prevalence of AP susceptibility genesshould be considered.
Because these data are derived from a survey, the re-sults must be treated as preliminary. In our experience,self report for AP is a very good indicator of AP ability;180% of 173 subjects who reported AP have passed arigorous test of their pitch-naming ability (E. Kowalskyand P. K. Gregersen, unpublished data). However, thereliability of reporting on AP ability in sibs or parentsis uncertain and needs to be validated. It would also bevaluable to obtain data on early-childhood music ex-posure and education from sibs of AP probands, to bet-ter control for the influence of environment on the de-velopment of AP. Familial aggregation of AP appears tobe common, yet the measurement of background prev-alence is not possible in the general population. Thus,more-extensive contact with the family members of alarge number of AP subjects will be required, to providefurther epidemiological evidence for genetic predispo-sition to AP, independent of environment. On the otherhand, many subjects report the spontaneous appearanceof AP in very early childhood. It is likely that inheritanceplays a significant role in AP, perhaps in the setting ofenvironmental exposure to music during a “critical” pe-riod (Goodman and Schatz 1993).
PETER K. GREGERSEN,1 ELENA KOWALSKY,1
NINA KOHN,2 AND ELIZABETH WEST MARVIN3
Division of 1Biology and Human Genetics and2Biostatistics, North Shore University Hospital,Manhasset, NY; and 3Department of Music Theory,Eastman School of Music, Rochester, NY
References
Baharloo S, Johnston PA, Service SK, Gitschier J, Freimer NB(1998) Absolute pitch: an approach for identification of ge-netic and nongenetic components. Am J Hum Genet 62:224–231
Goodman CS, Schatz CJ (1993) Developmental mechanismsthat generate precise patterns of neuronal connectivity. Cell72/Neuron 10 Suppl:77–98

Letters to the Editor 913
Gregersen PK (1998) Instant recognition: the genetics of pitchperception. Am J Hum Genet 62:221–223
Kumar S, Gregersen PK (1996) The genetics of perfect pitch.Am J Hum Genet Suppl 59:A179
Miyazaki K (1988) Musical pitch identification by absolutepitch possessors. Percept Psychophysiol 44:501–512
Profita J, Bidder GT (1988) Perfect pitch. Am J Med Genet29:763–771
Takeuchi AH, Hulse SH (1991) Absolute-pitch judgements ofblack and white-key pitches. Music Percept 9:27–46
Address for correspondence and reprints: Dr. Peter K. Gregersen, Division ofBiology and Human Genetics, North Shore University Hospital, 350 CommunityDrive, Manhasset, NY 11030. E-mail: [email protected]
q 1999 by The American Society of Human Genetics. All rights reserved.0002-9297/1999/6503-0035$02.00
Am. J. Hum. Genet. 65:913–917, 1999
Extremely Skewed X-Chromosome Inactivation IsIncreased in Women with Recurrent SpontaneousAbortion
To the Editor:Recurrent spontaneous abortion (RSA), defined as threeor more consecutive losses at <20 wk gestation (Stirrat1990), affects 1%–2% of couples trying to have a family(Stray-Pedersen and Lorentzen-Styr 1979; Roman1984). Although spontaneous abortion occurs quite fre-quently in humans, affecting ∼15% of all clinically rec-ognized pregnancies (Warburton and Fraser 1964; Ed-monds et al. 1982; Wilcox et al. 1988), the observedrate of RSA is much higher than the expected rate of0.3% due to chance alone. This suggests the presenceof factors that may predispose particular couples to mul-tiple pregnancy losses. Nearly 60% of RSA cases can bepotentially explained by identifiable autoimmune, en-docrine, anatomical, or infectious factors or by struc-tural chromosome rearrangements in one partner (Ste-phenson 1996). However, 140% of RSA is stillunexplained. We suggest that a significant proportion ofthe unexplained cases of RSA may be caused by a geneticmutation or chromosomal abnormality that would notbe discovered by routine investigation.
X-chromosome inactivation (XCI) is the processwhereby one of the two X chromosomes present in eachcell of female mammals is inactivated during early em-bryogenesis, to achieve dosage compensation with males(Lyon 1961). Generally, in a given cell type in humans,the maternal X chromosome is inactivated approxi-mately equally as often as the paternal X chromosome(Belmont 1996). However, extremely skewed XCI, de-fined in this letter as 190% inactivation of one allele, is
observed in ∼2% of newborns and ∼4.5% of 28–32-year-old women (Busque et al. 1996). This extremelyskewed XCI pattern may be due to a number of possiblecauses: (1) chance; (2) a mutation in the XIST gene thatis found on the X chromosome and is thought to becritical in the inactivation process (Plenge et al. 1997);(3) selection against cells with a growth disadvantagebecause of a deletion or mutation on one of the X chro-mosomes (Pegoraro et al. 1997) or to an X-autosometranslocation (Gaal and Laszlo 1977); and (4) a reduc-tion in the fetal precursor-cell pool size, as has beensuggested to occur in twinning (Bamforth et al. 1996;Goodship et al. 1996). Trisomy mosaicism has also re-cently been shown to be associated with extremelyskewed XCI (Lau et al. 1997). Extremely skewed XCI(190% inactivation of one allele) was found in the ma-jority (11 of 18) of prenatally detected mosaic caseswhen the trisomic cell line was of meiotic origin andabsent from most fetal tissues (Lau et al. 1997; W.P.Robinson and M.S. Penahererra, unpublished data).Skewing is hypothesized to result from a reduction inthe number of embryonic precursor cells, because of se-lection against the trisomic cells shortly after XCI.
At least three causes of skewed XCI are expected tobe associated with an increased risk of spontaneousabortion: (1) some deletions or mutations on the X chro-mosome may be lethal to male fetuses carrying the ab-normal X chromosome (Pegoraro et al. 1997); (2)X-autosome translocations can lead to RSA, becausesome gametes may be deleted and/or duplicated for por-tions of each chromosome that are involved in the re-arrangement (Byrne and Ward 1994); and (3) trisomymosaicism may also be associated with RSA if the germ-line is affected, since recurrent aneuploidy may result(Kohn and Shohat 1987; Gersdorf et al. 1990; Satge etal. 1996). Although it is impossible to determine howoften the germline is mosaic in individuals with a normalphenotype and blood karyotype, one case was reportedin which trisomy 16 was found in placenta and oocytesbut in no other fetal tissue (Stavropoulos et al. 1998).To evaluate the degree to which mosaicism or other ge-netic factors associated with extremely skewed XCI maycontribute to RSA, we screened women with RSA inorder to determine their XCI status and compared themwith controls of similar age.
Patients were ascertained through the Recurrent Preg-nancy Loss Clinic at British Columbia’s Women’s Hos-pital and Health Centre. Between September of 1997and December of 1998, all new patients with a historyof RSA who were seen by the one of the authors (M.D.S.)were offered participation in this study. RSA was definedas three or more consecutive pregnancy losses prior to20 wk gestation, with each pregnancy documented bya positive result on serum or urinary hCG, ultrasound,or pathology. Ethics approval was obtained from the

914 Letters to the Editor
Table 1
Proportion of Females with RSA Who Showed ExtremelySkewed XCI Compared with Control Females
GROUP
NO. (%) OBSERVED WITH
!90% Skewing 190% Skewing
Females with RSA 62 (82) 14 (18)Control females 105 (95) 6 (5)
NOTE.—For comparison by x2, P ! .001.
University of British Columbia Clinical Research EthicsBoard, and the consent form was thoroughly reviewedwith each patient. A single tube of peripheral blood wascollected from consenting women, and DNA was ex-tracted by use of a standard protocol. Every effort wasmade to collect these blood samples when samples forother standard blood work were being collected, ac-cording to the RSA protocol of Stephenson (1996). Con-trols were mothers who had donated blood previously,each having had at least one full-term pregnancy.
The mean number of consecutive spontaneous abor-tions per patient was 4.1, with a range of 3–11. Patients’ages were 19–45 years, with a mean of 33.6 years. Forthe control group, the age at which the blood was drawnwas available for 86 of the 111 individuals informativeat the androgen-receptor (AR) locus; the controls’ ageswere 20–49 years, with a mean age of 35.0 years. Resultsof karyotype analysis using standard Giemsa banding at550-band resolution, in patients with RSA, their repro-ductive partners, and prior spontaneous abortions weretaken from patients’ charts; a total of 49 aborted preg-nancies from patients with RSA who were informativefor XCI status underwent karyotype analysis.
The degree of skewed XCI was estimated by an assaybased on a methylation-sensitive HpaII restriction sitelocated near the human AR gene. This site is known tobe methylated on the inactive X chromosome and to beunmethylated on the active X chromosome (Allen et al.1992). When HpaII was used to digest the genomic DNAprior to PCR, the AR allele was amplified only from theinactive X chromosome, since the sequence to be am-plified on the active X chromosome was cleaved by therestriction enzyme. A trinucleotide CAG-repeat poly-morphism located within the amplified region was usedto distinguish between the two X chromosomes. Foreach patient, two PCR reactions were performed—onewith genomic DNA digested with HpaII and one withoutHpaII. The second reaction served as an internal controlto establish a baseline level of amplification of each al-lele, specific to the individual. This measure correctedfor any preferential amplification of one allele over theother, in a given patient. Genomic DNA from males wasused as a digestion control, since their X chromosomeis always active (unmethylated) and therefore shouldhave been completely digested by HpaII and should haveyielded no amplification product. Products were sepa-rated by PAGE and were visualized by silver staining.Quantification of the resulting bands was performed asdetailed elsewhere (Lau et al. 1997). The analysis wasrepeated for any patient in whom skewing was 170%,in order to verify the result. Thus, the degree of skewingreported in these cases was an average of two or threeindependent tests; the mean difference between two es-timates of skewing for the same patient was four per-centage points.
XCI status was informative in 76 of the 98 patientswith RSA and in 111 of the 137 female controls. Thisfrequency may be less than that in other reports becausesome heterozygous cases were considered to be unin-formative if the two bands were too close to resolveadequately for densitometric analysis. Extremely skewedXCI, defined as 190% amplification of one allele, wasfound in 14 (18%) of the 76 informative females withRSA and in just 6 (5%) of the 111 controls ( ;P ! .001x2 test) (see table 1). The mean rate of skewing in thecontrol group was similar to the 4.5% rate of skewingobserved by Busque et al. (1996) in women 28–32 yearsof age. This rate was not significantly altered if controlsfor whom no age data were available were excluded.
These results show that factors associated with ex-tremely skewed XCI account for a significant proportion(i.e., as much as 18%) of couples with RSA. Similarresults have been reported, by Lanasa et al. (1999), forwomen who have experienced two or more spontaneousabortions. What is the explanation for extreme skewingin these patients with RSA? A pattern of extremelyskewed XCI is commonly seen in females who carrybalanced translocations involving one X chromosomeand an autosome. The normal X chromosome is usuallypreferentially inactivated, presumably to maintain a bal-anced chromosomal complement in each cell (Gaal andLaszlo 1977). Theoretically, X-chromosome rearrange-ments could also lead to recurrent pregnancy loss. How-ever, in a study that reported karyotype results in 1,142couples with recurrent abortion there was not a singlerearrangement involving the X chromosome (Portnoi etal. 1988). Thus, this is too rare a finding to account forthe significant number of patients with RSA whom weobserved to have extremely skewed XCI. Furthermore,in our study, all 14 of the patients with RSA who hadextreme skewing had normal results on karyotype anal-ysis—that is, 46,XX (see table 2).
Extremely skewed XCI may also result from a mu-tation on one of the two X chromosomes because ofselective advantage of those cells that have the normalX chromosome active. In such cases, any male conceptusinheriting the abnormal X chromosome would mostlikely be aborted, because of the presence of only thedefective copy of the locus in question. One such familyhas been reported in which 100% of the females exhib-

Letters to the Editor 915
Table 2
Karyotype Data on Female Patients withRSA and on Their Male ReproductivePartners
Gender and Karyotype No.
Females:46,XXa 8646,XX,t(2;12)(q13;q24.31) 146,XX,t(8;12)(q22;q22) 146,XX,inv(2)(p11.2q13) 146,XX/45,Xb 1NA 8
Total 98Males:
46,XY 81NA 17
Total 98
a All 14 patients with RSA who had ex-tremely skewed XCI also had a 46,XXkaryotype.
b A 45,X karyotype was found in 4/100lymphocytes, and further examinationshowed a 46,XX karyotype in 30/30 skin fi-broblast cells.
Table 3
Karyotypes of Spontaneous Abortions among Female Patients withRSA Who Have Extremely Skewed XCI, versus Those Who Do NotHave Such Skewing
TYPE OF
ABORTUS
KARYOTYPED
NO. OF ABORTUSES FROM PATIENTS WHO HAVE
!90% Skewing 190% Skewing
46,XY 14 146,XX 11 1Aneuploid 15a 7a
Total 40 9
a (by Fisher’s exact test comparing proportion of abnormalP = .03karyotypes in the two groups).
iting extremely skewed XCI showed the presence of anX chromosome with an inherited deletion (Pegoraro etal. 1997). The women in this family had a spontaneous-abortion rate more than twice that in their female rel-atives who did not have the deletion or skewing. Theyalso had a greater proportion of live-born females thanof live-born males. There is currently no efficient methodthat can screen for mutations on the X chromosome thataffect viability. However, it would be expected that, ifthis were the cause of RSA in our group of patients withextremely skewed XCI, then a high rate of 46,XY kar-yotypes should be seen among their spontaneous abor-tions. Of the karyotyped spontaneous-abortion speci-mens that were euploid, 1 of the 2 abortuses frompatients with skewing was male, compared with 14males among the 25 normal abortuses from patients withRSA and without skewing (table 3).
It should be noted that, although the frequency ofpregnancy loss would be higher in carriers of an X-chromosome mutation or deletion than in the generalpopulation, in theory only 25% of the conceptuses (i.e.,one-half of the males) would be at risk of being abortedbecause of the mutation; this is because all of the femalesand half of the males should be protected from the mu-tation, by the presence of the normal X chromosome. Ifit is assumed that the population rate of abortion dueto independent causes is 15%, then the joint probabilityof pregnancy loss would be ∼36%. Thus, only a smallnumber (i.e., ∼5%) of women with such an X-chro-mosome mutation might be expected to have three ormore consecutive spontaneous abortions and no livebirths. Thus, an X-chromosome mutation might be rel-
atively more common among women with either only afew losses or several losses combined with some livebirths than it is among women who experience a largernumber of losses and no successful full-term pregnancies.It is therefore interesting to note that the mean numberof spontaneous abortions (4.1) in the 14 patients whohad skewed XCI was equal to that of the entire groupwith RSA.
Finally, a factor that may be associated with both ex-tremely skewed XCI and RSA is aneuploidy mosaicism.Although aneuploidy is the leading cause of randomspontaneous abortion, accounting for ∼50% of kary-otyped losses (Boue et al. 1975; Hassold and Jacobs1984), it is difficult to evaluate whether recurrence ofthe same aneuploidy occurs more often than would beexpected by chance, since karyotype information oneach loss is lacking in most RSA cases. Nonetheless,many cases of gonadal mosaicism for a trisomy havebeen reported, many of which are ascertained by themultiple recurrence of Down syndrome (Kohn and Sho-hat 1987; Nielsen et al. 1988; Gersdorf et al. 1990; Sachset al. 1990; Pangalos et al. 1992; Tseng et al. 1994; Satgeet al. 1996), and mosaicism is found in the lymphocytesof one of the two parents in 4% of families with Downsyndrome (Uchida and Freeman 1985). However, evenif germline mosaicism were a cause of RSA, we wouldnot necessarily expect the same trisomy to recur, sinceit has been shown that the presence of an unpaired chro-mosome in mouse oocytes can cause both disruption ofmeiosis and missegregation of other chromosomes (Huntet al. 1995). There is also evidence from rare cases offertile 45,X women that the same effect occurs in hu-mans (Warburton 1989). In our study, seven of ninespontaneous-abortion specimens from the group ofwomen with extremely skewed XCI were found to beaneuploid, whereas, among the women with nonskewedXCI, just 15 (27%) of 40 spontaneous-abortion speci-mens were aneuploid ( ; see table 3). AlthoughP = .03both skewed XCI and aneuploidy increase with maternalage (Hassold and Jacobs 1984; Busque et al. 1996), themean age of the patients with extremely skewed XCI

916 Letters to the Editor
was lower (32.4 years) than that of the group withoutskewing (33.9 years). Thus, age cannot explain thehigher rate of aneuploidy in the spontaneous-abortionspecimens from patients with extremely skewed XCI.
It is estimated that 1%–2% of first-trimester preg-nancies assessed by chorionic-villus sampling are mosaic(Vejerslev and Mikkelsen 1989). Although the abnormalcell line is often assumed to be confined to the placenta,this is difficult to prove. Even when mosaicism is foundin amniotic fluid, the aneuploidy is frequently absentfrom fetal/newborn blood (Hsu et al. 1997), and trisomymosaicism for most chromosomes is unlikely to be de-tected by routine blood karyotyping. Although analysisof skin fibroblasts may detect a greater proportion ofmosaic cases, it is still possible for trisomic cells to befound in oocytes even when no other fetal tissues areaffected (Stavropoulos et al. 1998). Alternatively, we canlook at indirect indicators, such as skewed XCI, to pro-vide clues as to whether an individual may be the prod-uct of a pregnancy associated with mosaicism.
Pregnancy loss is a devastating issue for many couples,and identification of an etiology is very important forcounseling couples with RSA as to their treatment op-tions. Clearly, genetic factors associated with extremelyskewed XCI are important in at least some patients withRSA and, most likely, involve either an X-linked mu-tation or germline mosaicism. Further review of the med-ical histories and pedigrees may provide clues as towhich etiology is involved in a particular case. A largerepidemiological study documenting the outcome of allpregnancies and including karyotype data from thespontaneous abortions is also necessary to clarify themechanism involved and, subsequently, to improvecounseling of patients with RSA, in regard to futurepregnancy outcomes.
Acknowledgments
The authors would like to thank the patients with RSA andthe patients’ partners, for their participation in this study. Theauthors would also like to thank Maria Serena Penaherreraand Fabiana Bernasconi-Quadroni, for expert technical assis-tance, and Jennifer Oakes, for assistance with patients’ chartsand consent procedures. This study was supported by BritishColumbia Health Research Foundation grant 98(96-2).
KARAN K. SANGHA,1,3 MARY D. STEPHENSON,2,4
CAROLYN J. BROWN,1 AND WENDY P. ROBINSON1,3
Departments of 1Medical Genetics and 2Obstetrics andGynecology, University of British Columbia; 3BCResearch Institute for Children’s & Women’s Health;and 4Children’s & Women’s Health Centre of BritishColumbia, Vancouver
References
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, BelmontJW (1992) Methylation of HpaII and HhaI sites near thepolymorphic CAG repeat in the human androgen-receptorgene correlates with X chromosome inactivation. Am J HumGenet 51:1229–1239
Bamforth F, Machin G, Innes M (1996) X-chromosome in-activation is mostly random in placental tissues of femalemonozygotic twins and triplets. Am J Med Genet 61:209–215
Belmont JW (1996) Genetic control of X inactivation and pro-cesses leading to X-inactivation skewing. Am J Hum Genet58:1101–1108
Boue J, Boue A, Lazar P (1975) Retrospective and prospectiveepidemiological studies of 1500 karyotyped spontaneoushuman abortions. Teratology 12:11–26
Busque L, Mio R, Mattioli J, Brais E, Brais N, Lalonde Y,Maragh M, et al (1996) Nonrandom X-inactivation patternsin normal females: lyonization ratios vary with age. Blood88:59–65
Byrne JLB, Ward K (1994) Genetic factors in recurrent abor-tion. Clin Obstet Gynecol 37:693–704
Edmonds DK, Lindsay KS, Miller JF, Williamson E, Wood PJ(1982) Early embryonic mortality in women. Fertil Steril 38:447–453
Gaal M, Laszlo J (1977) X inactivation pattern in an unbal-anced X-autosome translocation with gonadal dysgenesis.Hum Hered 27:396–402
Gersdorf E, Utermann B, Utermann G (1990) Trisomy 18 mo-saicism in an adult woman with normal intelligence andhistory of miscarriage. Hum Genet 84:298–299
Goodship J, Carter J, Burn J (1996) X-inactivation patternsin monozygotic and dizygotic female twins. Am J Med Genet61:205–208
Hassold TJ, Jacobs PA (1984) Trisomy in man. Annu RevGenet 18:69–97
Hsu LY, Yu MT, Neu RL, Van Dyke DL, Benn PA, BradshawCL, Shaffer LG, et al (1997) Rare trisomy mosaicism di-agnosed in amniocytes, involving an autosome other thanchromosomes 13, 18, 20, and 21: karyotype/phenotype cor-relations. Prenat Diagn 17:201–242
Hunt P, LeMaire R, Embury P, Sheean L, Mroz K (1995) Anal-ysis of chromosome behavior in intact mammalian oocytes:monitoring the segregation of a univalent chromosome dur-ing female meiosis. Hum Mol Genet 4:2007–2012
Kohn G, Shohat M (1987) Trisomy 18 mosaicism in an adultwith normal intelligence. Am J Med Genet 26:929–931
Lanasa MC, Hogge WA, Hoffman EP (1999) The X chro-mosome and recurrent spontaneous abortion: the signifi-cance of transmanifesting carriers. Am J Hum Genet 64:934–938
Lau AW, Brown CJ, Penaherrera M, Langlois S, Kalousek DK,Robinson WP (1997) Skewed X-chromosome inactivationis common in fetuses or newborns associated with confinedplacental mosaicism. Am J Hum Genet 61:1353–1361
Lyon MF (1961) Gene action in the X-chromosome of themouse (Mus musculus L.). Nature 190:372–373
Nielsen KG, Poulsen H, Mikkelsen M, Steuber E (1988) Mul-

Letters to the Editor 917
tiple recurrence of trisomy 21 Down syndrome. Hum Genet78:103–105
Pangalos CG, Talbot CC Jr, Lewis JG, Adelsberger PA, Peter-son MB, Serre J-L, Rethore M-O, et al (1992) DNA poly-morphism analysis in families with recurrence of free tri-somy 21. Am J Hum Genet 51:1015–1027
Pegoraro E, Whitaker J, Mowery-Rushton P, Surti U, LanasaM, Hoffman EP (1997) Familial skewed X inactivation: amolecular trait associated with high spontaneous-abortionrate maps to Xq28. Am J Hum Genet 61:160–170
Plenge RM, Hendrich BD, Schwartz C, Arena JF, Nau-mova A, Sapienza C, Winter RM, et al (1997) A pro-moter mutation in the XIST gene in two unrelated fam-ilies with skewed X-chromosome inactivation. NatGenet 17:353–356
Portnoi MF, Joye N, van den Akker J, Morlier G, TaillemiteJL (1988) Karyotypes of 1142 couples with recurrent abor-tion. Obstet Gynecol 72:31–34
Roman E (1984) Fetal loss rates and their relation to pregnancyorder. J Epidemiol Community Health 38:29–35
Sachs ES, Jahoda MG, Los FJ, Pijpers L, Wladimiroff JW(1990) Trisomy 21 mosaicism in gonads with unexpectedlyhigh recurrence risks. Am J Med Genet Suppl 7:186–188
Satge D, Geneix A, Goburdhun J, Lasne-Desmet P, RosenthalC, Arnaud R, Malet P (1996) A history of miscarriages andmild prognathism as possible mode of presentation of mo-saic trisomy 18 in women. Clin Genet 50:470–473
Stavropoulos DJ, Bick D, Kalousek DK (1998) Molecular cy-togenetic detection of confined gonadal mosaicism in a con-ceptus with trisomy 16 placental mosaicism. Am J HumGenet 63:1912–1914
Stephenson MD (1996) Frequency of factors associated withhabitual abortion in 197 couples. Fertil Steril 66:24–29
Stirrat GM (1990) Recurrent miscarriage. I. Definition andepidemiology. Lancet 336:673–675
Stray-Pedersen B, Lorentzen-Styr A (1979) The prevalence oftoxoplasma antibodies among 11 736 pregnant women inNorway. Scand J Infect Dis 11:159–165
Tseng LH, Chuang SM, Lee TY, Ko TM (1994) RecurrentDown’s syndrome due to maternal ovarian trisomy 21 mo-saicism. Arch Gynecol Obstet 255:213–216
Uchida IA, Freeman VC (1985) Trisomy 21 Down syndrome:parental mosaicism. Hum Genet 70:246–248
Vejerslev LO, Mikkelsen M (1989) The European collabora-tive study on mosaicism in chorionic villus sampling: datafrom 1986 to 1987. Prenat Diagn 9:575–588
Warburton (1989) The effect of maternal age on the frequencyof trisomy: change in meiosis or in utero selection? In: Has-sold TJ, Epston CJ (eds) Molecular and cytogenetic studieson non-disjunction. Alan R Liss, New York, pp 165–181
Warburton D, Fraser FC (1964) Spontaneous abortion risksin man: data from reproduction histories collected in a med-ical genetics unit. Am J Hum Genet 16:1–25
Wilcox AJ, Weinberg CR, O’Connor JF, Baird DD, SchlattererJP, Canfield RE, Armstrong EG, et al (1988) Incidence ofearly loss of pregnancy. N Engl J Med 319:189–194
Address for correspondence and reprints: Dr. Wendy P. Robinson, BC ResearchInstitute for Children’s and Women’s Health, 3086 – 950 West 28th Avenue,Vancouver, B.C., Canada V5Z 4H4. E-mail: [email protected]
q 1999 by The American Society of Human Genetics. All rights reserved.0002-9297/1999/6503-0036$02.00
Am. J. Hum. Genet. 65:917–921, 1999
Demonstration of the Recurrence of Marfan-likeSkeletal and Cardiovascular Manifestations Due toGermline Mosaicism for an FBN1 Mutation
To the Editor:Marfan syndrome (MFS [MIM 154700]) is a dominantlyinherited disease of connective tissue. Cardinal manifes-tations involve the eye (lens dislocation and myopia),skeleton (dolichostenomelia, arachnodactyly, anteriorchest deformity, spinal curvature, and joint laxity) andcardiovascular system (aortic root dilation and dissec-tion, mitral valve prolapse, and mitral and aortic valveregurgitation). Striae distensae and inguinal hernia arefrequent findings in the integument, and pneumothoraxand dural ectasia occur in some patients (Pyeritz andMcKusick 1979; First International Symposium on theMarfan Syndrome 1989). If untreated, the syndromeshortens life expectancy mainly because of cardiovas-cular complications. The disorder is characterized byconsiderable variation in the distribution and severity oforgan system involvement between families, leading tothe definition of diagnostic criteria listed first in the Ber-lin nosology (Beighton et al. 1988) and subsequentlyrevised in the Ghent nosology (de Paepe et al. 1996). In1986, Sakai and colleagues identified a 350-kD glyco-protein called “fibrillin,” which represents the majorstructural component of connective tissue microfibrils.By using an anti-fibrillin antibody, Godfrey, Hollister,and their colleagues demonstrated a reduction of mi-crofibrils in immunofluorescence studies of cultured der-mal fibroblasts in patients with MFS (Godfrey et al.1990; Hollister et al. 1990). Subsequent studies of fi-brillin synthesis, secretion, and incorporation into theextracellular matrix showed abnormalities in most butnot all MFS fibroblast strains (Milewicz et al 1992; Col-lod et al. 1994). Finally, mutations in the FBN1 gene,encoding fibrillin, have been demonstrated to result inMFS or associated phenotypes (Dietz et al. 1991; Hay-ward et al. 1994; Kainulainen et al. 1994; Lonnqvist etal. 1994; Sood et al. 1996). The FBN1 gene is ∼200 kbin size, with a coding sequence fragmented into 65 exons(Corson et al. 1993; Pereira et al. 1993; Biery et al. 1999)located on chromosome 15q21.1 (Magenis et al. 1991).

918 Letters to the Editor
It encodes a large glycoprotein composed of repeatedmodules, 47 of which are homologous to human epi-dermal growth factor (EGF) (“EGF-like modules”) andare interspersed by seven modules displaying high ho-mology to transforming growth factor b1–binding pro-tein (TGFb1-bp), (“8-cysteine modules”) (Corson et al.1993; Pereira et al. 1993). To date, 1160 FBN1 muta-tions in patients with MFS or associated phenotypeshave been reported or submitted in the Marfan Database(Collod et al. 1996; Collod-Beroud et al. 1997, 1998).However, hardly any predictions of the resulting phe-notype can yet be made on the basis of the nature of aspecific mutation.
The prevalence of MFS has been estimated at 1/5,000,and >25% of patients represent sporadic cases. Thishigh mutation rate should be associated with cases ofgermline mosaicism, as has been reported in other con-nective-tissue disorders or other genetic disorders witha high mutation rate. Therefore, it was surprising that,until recently (Montgomery et al. 1998; Rantamaki etal. 1999), no instance of somatic or germline mosaicismhad been reported in MFS. Furthermore, since FBN1mutations are also associated with phenotypes overlap-ping MFS, mosaicism could also be identified in thesesubtypes. In this report, we demonstrate somatic mo-saicism of a FBN1 genomic mutation in the father oftwo siblings who presented with the typical skeletal andcardiovascular features observed in the Marfan syn-drome.
The proband, MS48MA307, was identified at theCentre Hospitalier in Amiens (by M.M. and Y.M.), andhis family was investigated at the Marfan Clinic at Am-broise Pare Hospital, Boulogne (by G.J.). The diagnosticcriteria used were those reported by Beighton et al.(1988). The parents of MS48MA307 and his brother,MS48MA308, were unaffected. Patient MS48MA307,a 16-year-old boy, presented dilation of the ascendingaorta (46 mm at the sinuses of Valsalva, 8 SD above themean when standardized to age and body surface area),mitral valve prolapse with regurgitation, highly archedpalate, arachnodactyly (positive wrist and thumb signs),tall stature (199 cm, 14 SD, 76 kg), and scoliosis. His9-year-old brother (MS48MA308) displayed dilation ofthe ascending aorta (32 mm at the sinuses of Valsalva,6 SD above the mean when standardized to age and bodysurface area), arachnodactyly (positive wrist and thumbsigns), dolichostenomelia (arm span–to-height ratio1.05), tall stature (144 cm, 13 SD, 31 kg), highly archedpalate, and joint hypermobility. No other typical anom-aly of MFS (including ectopia lentis) was found in eithersubject. Both parents were examined thoroughly, andthe diagnosis of MFS was excluded for both. Blood sam-ples were collected from the four family members andfrom 150 unrelated French subjects. DNA was extractedfrom white blood cells (Henry et al. 1984). Informed
consent was obtained for all individuals. Sense and an-tisense primers designed from flanking intron sequenceswere used for PCR amplification of exons 1–65 of FBN1and were described, along with PCR amplification con-ditions, in Nijbroek et al. (1995). SSCP analysis of theFBN1 gene from white blood cells revealed an abnormalpattern for the 419-bp fragment of exon 24 for patientMS48MA307. This abnormal pattern was also identifiedin his brother, MS48MA308, but was absent in themother. However, the father presented a very slight ab-normal pattern (fig. 1a). Paternity and maternity hadbeen tested previously and indirectly by analysis ofhighly polymorphic markers on chromosomes 3, 5, and15 (data not shown). The abnormal fragment from theSSCP gel was cut out of the gel, and DNA was elutedin water and reamplified by PCR. The PCR product waspurified with the Promega Wizard Prep kit and was di-rectly sequenced on both strands by means of a cycle-sequencing kit (Pharmacia). Sequencing revealed that thetwo boys carried the identical heterozygous 2954 GrAtransition that results in a GlyrGlu change at codon985 (G985E) (fig. 1d). There is a compelling body ofevidence to suggest that G985E is indeed a disease-pro-ducing mutation. First, this alteration was not observedduring screening of 306 chromosomes. Second, the mu-tation substitutes an uncharged for a negatively chargedamino acid of much higher molecular weight. Finally,the mutational event occurs in the 8-cysteine module 3at a position conserved in the bovine, murine, and por-cine sequences.
Since MFS is characterized by a high mutation rate,the recurrence of the disease in the sibs could have beendue to two unrelated de novo mutations. However, sincemutations in the FBN1 gene are essentially private, thepresence of an identical mutation in the brothers sug-gested that the most likely hypothesis was that the mu-tation had been inherited from one of the parents. Sincethe mutation creates a new TaqI restriction site resultingin two fragments of 202 and 217 bp, it could easily belooked for in the family (fig. 1b). After transfer on Hy-bond N1 membrane (Amersham) and hybridizationwith the sense primer, the 217-bp fragment resultingfrom digestion was found in the father’s white bloodcell DNA (MS48MA305), at a very low level, but wasnot found in that from the mother (MS48MA306) or inthree controls (fig. 1c). The finding of the alteration inthe father’s white blood cells and the recurrence of thedisease in his children implied somatic and germline mo-saicism in the father. Careful reassessment of clinicalexamination of the father (performed systematically be-fore the identification of the mosaicism) revealed no skel-etal or ocular sign but minor findings: discreet dilationof the ascending aorta (43 mm, 12 SD when standard-ized to age and body surface area [193 cm, 75 kg, atage 41 years]) and minimal aortic regurgitation. This

Letters to the Editor 919
Figure 1 a, DNA single-strand analysis by nondenaturing PAGE (SSCP) of a 419-bp PCR product including exon 24. Aberrant migrationof PCR product is found in subjects MS47MA307 and MS47MA308 compared with their parents and the normal control. b, The GrA transitioncreates a TaqI site within the 419-bp PCR product of exon 24, resulting in two fragments of 202 and 217 bp. TaqI digestion confirmed theG985E mutation in the two brothers MS47MA307 and MS47MA308. c, The digestion products were migrated and then were hybridized withthe sense primer. Only the PCR product resistant to digestion can be found for normal controls 1, 2, and 3 and for the mother (MS47MA306).The heterozygous three-banded restriction enzyme pattern after TaqI digestion is present for MS47MA307 and the father MS47MA305, afteroverexposure. d, The normal (for MS47MA306) and abnormal (for MS47MA307) fragments from the SSCP gel (in a, above) were cut, elutedin water, and reamplified by PCR. Sequencing for MS47MA307 compared with normal sequence (MS47MA306) revealed a GrA transition atnucleotide position 2954, resulting in a GlyrGlu change at codon 985 (G985E).
mutation probably arose at an early mitotic stage inembryonic development, as reflected by distribution insomatic and germ cell tissues.
The MFS-like phenotype associated with the G985Emutation in exon 24 is not associated with ocular anom-alies. Of interest, the Marfan Database (Collod-Beroudet al. 1998), when sorted for mutations in MFS patientswho have no ocular anomaly, indicates that half (9/19)of these mutations are located in exons 23–29. This con-trasts with mutations associated with the complete clas-sic MFS, which are widely distributed throughout thegene. Furthermore, study of the distribution of muta-tions identified in 8-cysteine modules after their align-ment by their consensus sequence indicates that theG985E mutation affects a residue close to three consec-
utive cysteines. This region harbors three other muta-tions (5137ins4 [Dietz et al. 1993], C1721Y [Collod-Beroud et al. 1998], and V984I [Collod-Beroud et al.1998]) identified in probands that do not have ectopialentis. The 8-cysteine modules are found only in fibrillinsand latent TGFb1-bp, and their function in fibrillins isstill unclear. The absence of ectopia lentis and, therefore,the probable absence of major zonular alteration in sub-jects carrying mutations in this region of the 8-cysteinemodules would tend to indicate the absence of a specificfunction of this module in the zonule.
Our observation shows that somatic and germline mo-saicism are associated with MFS-like features and shouldbe looked for in parents of sporadic cases presentingwith these MFS-like features. In effect, if mild or isolated

920 Letters to the Editor
features of the disease are found in one of the parents,genetic counseling should take into account the possiblepresence of the disease in another child. Somatic mo-saicism could also explain the mild and incomplete fea-tures often seen in patients referred to MFS clinics fordiagnosis. Again, caution is warranted in the follow-upof these patients and in evaluation of the risk of trans-mission.
Acknowledgments
This work was supported by grants from Fondation deFrance, Universite Rene Descartes Paris V, Ministere del’Education Nationale, de l’Enseignement Superieur, de la Re-cherche et de l’Insertion Professionnelle (ACC-SV2), Facultede Medecine Necker, Association Francaise contre les Myopa-thies (A.F.M.) and Projet Hospitalier de Recherche Clinique(PHRC AOM 96070). G.C.B. is supported by a grant fromFondation pour la Recherche Medicale.
GWENAELLE COLLOD-BEROUD,1
MARILYN LACKMY-PORT-LYS,1 GUILLAUME JONDEAU,2
MICHELE MATHIEU,4 YVES MAINGOURD,5
MONIQUE COULON,1 MICHEL GUILLOTEL,1
CLAUDINE JUNIEN,1, 3 AND CATHERINE BOILEAU1, 3
1INSERM U383, Hopital Necker-Enfants Malades,Universite Rene Descartes, Paris; 2Service deCardiologie et 3Laboratoire Central de Biochimie,d’Hormonologie et de Genetique Moleculaire, HopitalAmbroise Pare, Boulogne; and 4Centre de GenetiqueClinique and 5Unite de Cardiologie Pediatrique, CHUd’Amiens, Hopital Nord, Amiens, France
Electronic-Database Information
Accession numbers and URLs for data in this article are asfollows:
Marfan Database, http://www.umd.necker.fr/ (for FBN1 muta-tions)
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for MFS [MIM 154700)
References
Beighton P, De Paepe A, Danks D, Finidori G, Gedde-Dahl T,Goodman R, Hall JG, et al (1988) International nosologyof heritable disorders of connective tissue. Am J Med Genet29:581–594
Biery NJ, Eldadah ZA, Moore CS, Stetten G, Spencer F, DietzHC (1999) Revised genomic organization of FBN1 and sig-nificance for regulated gene expression. Genomics 56:70–77
Collod G, Babron MC, Jondeau G, Coulon M, WeissenbachJ, Dubourg O, Bourdarias JP, et al (1994) A second locusfor Marfan syndrome maps to chromosome 3p24.2-p25.Nat Genet 8:264–268
Collod G, Beroud C, Soussi T, Junien C, Boileau C (1996).
Software and database for the analysis of mutations in thehuman FBN1 gene. Nucleic Acids Res 24:137–140
Collod-Beroud G, Beroud C, Ades L, Black C, Boxer M, BrockDJ, Godfrey M, et al (1997). Marfan Database (second edi-tion): software and database for the analysis of mutationsin the human FBN1 gene. Nucleic Acids Res 25:147–150
Collod-Beroud G, Beroud C, Ades L, Black C, Boxer M, BrockDJ, Holman KJ et al (1998) Marfan database (third edition):new mutations and new routines. Nucleic Acids Res 26:229–233
Corson GM, Chalberg SC, Dietz HC, Charbonneau NL, SakaiLS (1993) Fibrillin binds calcium and is coded by cDNAsthat reveal a multidomain structure and alternatively splicedexons at the 5′ end. Genomics 17:476–484
De Paepe A, Devereux RB, Dietz HC, Hennekam RC, PyeritzRE (1996) Revised diagnostic criteria for the Marfan syn-drome. Am J Med Genet 62:417–426
Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Cor-son GM, Puffenberger EG, et al (1991) Marfan syndromecaused by a recurrent de novo missense mutation in thefibrillin gene. Nature 352:337–339
Dietz H, McIntosh I, Sakai L, Corson G, Chalberg S, PyeritzR, Francomano C (1993) Four novel FBN1 mutations: sig-nificance for mutant transcript level and EGF-like domaincalcium binding in the pathogenesis of Marfan syndrome.Genomics 17:468–475
First International Symposium on the Marfan syndrome.(1989) Baltimore, July 8–10, 1988. Am J Med Genet 32:239–251
Godfrey M, Menashe V, Weleber RG, Koler D, Bigley RH,Lovrien E, Zonana J, et al (1990) Cosegregation of elastin-associated microfibrillar abnormalities with the Marfan phe-notype in families. Am J Hum Genet 46:652–660
Hayward C, Porteous ME, Brock DJ (1994) A novel mutationin the fibrillin gene (FBN1) in familial arachnodactyly. MolCell Probes 8:325–327
Henry I, Uzan G, Nicolas H, Kaplan JC, Marguerie C, KahnA, Junien C (1984) The genes coding for alpha-, beta-, andgamma-chains of fibrinogen map to 4q2. Am J Hum Genet36: 760–768
Hollister DW, Godfrey M, Sakai L (1990) Immunohistologicabnormalities of the microfibrillar-fiber system in the Mar-fan syndrome. N Engl J Med 323:152–159
Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltonen L(1994) Mutations in the fibrillin gene responsible for dom-inant ectopia lentis and neonatal Marfan syndrome. NatGenet 6:64–69
Lonnqvist L, Child A, Kainulainen K, Davidson R, PuhakkaL, Peltonen L (1994) A novel mutation of the fibrillin genecausing ectopia lentis. Genomics 19:573–576
Magenis RE, Maslen CL, Smith L (1991) Localisation of thefibrillin (FBN) gene to chromosome 15, band q21.1. Gen-omics 11:346–351
Milewicz D, Pyeritz RE, Crawford ES, Byers P (1992) Marfansyndrome: defective synthesis, secretion, and extracellularmatrix, formation of fibrillin by cultured dermal fibroblasts.J Clin Invest 89:79–86
Montgomery RA, Geraghty MT, Bull E, Gelb BD, Johnson M,McIntosh I, Francomano CA, et al (1998) Multiple molec-

Letters to the Editor 921
ular mechanisms underlying subdiagnostic variants of Mar-fan syndrome. Am J Hum Genet 63:1703–1711
Nijbroek G, Sood S, McIntosh I, Francomano CA, Bull E,Pereira L, Ramirez F, et al (1995) Fifteen novel FBN1 mu-tations causing Marfan syndrome detected by heteroduplexanalysis of genomic amplicons. Am J Hum Genet 57:8–21
Pereira L, D’Alesio M, Ramirez F, Lynch JR, Sykes B, PangilianT, Bonadio J, et al (1993) Genomic organization of the se-quence coding for fibrillin, the defective gene product inMarfan Syndrome. Hum Mol Genet 2:961–968
Pyeritz RE, McKusick VA (1979) Marfan syndrome: diagnosisand management. N Engl J Med 300:772–777
Rantamaki T, Kaitila I, Syvanen AC, Lukka M, Peltonen L(1999) Recurrence of Marfan syndrome as a result of pa-rental germ-line mosaicism for an FBN1 mutation. Am JHum Genet 64:993–1001
Sakai L, Keene DR, Engvall E (1986) Fibrillin, a new 350-kDglycoprotein, is a component of extracellular microfibrils. JCell Biol 103:2499–2509
Sood S, Eldadah ZA, Krauss WL, McIntosh I, Dietz HC(1996). Mutation in fibrillin-1 and the Marfanoid-cranio-synostosis (Shprintzen-Goldberg) syndrome. Nat Genet 12:209–211
Address for correspondence and reprints: Dr. C. Boileau, INSERM U383,Hopital Necker-Enfants Malades, Clinique Maurice Lamy, 149-161, rue de Svres,75743 Paris Cedex 15, France. E-mail: [email protected]
q 1999 by The American Society of Human Genetics. All rights reserved.0002-9297/1999/6503-0037$02.00
Am. J. Hum. Genet. 65:921–924, 1999
The Jewish Ashkenazi Founder Mutations in theBRCA1/BRCA2 Genes Are Not Found at an IncreasedFrequency in Ashkenazi Patients with Prostate Cancer
To the Editor:BRCA1 and BRCA2, the predisposing genes for breastcancer (BC) and ovarian cancer (OC), have been sug-gested to increase the risk of prostate cancer (PrC) inmale carriers (Ford et al. 1994; Thorlacius et al. 1996;Struewing et al. 1997); however, no direct evidence existsto confirm this hypothesis. A population with a highcarrier frequency of BRCA1 and BRCA2 germinal mu-tations allows a direct approach to studying the roleBRCA1 and BRCA2 play in the development of PrC; ifgerminal mutations in BRCA1 and BRCA2 increase therisk of PrC in carriers, it is to be expected that the carrierfrequency in PrC patients will be higher than in the gen-eral population, as was demonstrated in female patientsdiagnosed with BC and OC (Ford et al. 1995; Claus etal. 1996, Abeliovich et al. 1997).
In the Ashkenazi Jewish population, three foundermutations, 185delAG and 5382insC in the BRCA1 gene
and 6174delT in the BRCA2 gene, exist at a high fre-quency (2.5%) (Struewing et al. 1995; Oddoux et al.1996; Roa et al. 1996; Fodor et al. 1998). To assess thecontribution of the BRCA1/BRCA2 germinal mutationsto PrC morbidity, we analyzed the Ashkenazi foundermutations in two groups (with the same age distribution)of Ashkenazi men, a group of unselected PrC patients,and a control group of men with no history of cancer.The study was designed around the fact that, in familiesknown to segregate BRCA1/BRCA2 mutations, menwith PrC were noted sporadically. It was thus assumedthat, if BRCA1 and BRCA2 play a role in the devel-opment of PrC, they do so as risk modifiers rather thanas major dominant genes, and therefore will not be con-fined to familial cases.
Patients diagnosed with adenocarcinoma of the pros-tate ( ) were recruited from the oncology outpa-n = 87tient clinic at Sharett Institute, Hadassah Hebrew Uni-versity Hospital, with no preselection. The patientssigned an informed-consent form approved by the hos-pital’s ethics committee. Each patient was interviewedregarding his family history. Clinical and pathologicalrecords were the sources of the clinical data.
The control group included 87 healthy men with nohistory of cancer. These men were approached in Jeru-salem-area homes for the elderly and were asked to par-ticipate in the study; if they agreed, they signed an in-formed-consent form. Their blood samples were keptanonymous, labeled only with the patients’ ages andorigins (table 1). The median age was 71 years at thetime of diagnosis for the patients with PrC and 72 yearsat the time of blood sampling for the control group (table2). The mutations were analyzed as described elsewhere(Abeliovich et al. 1997).
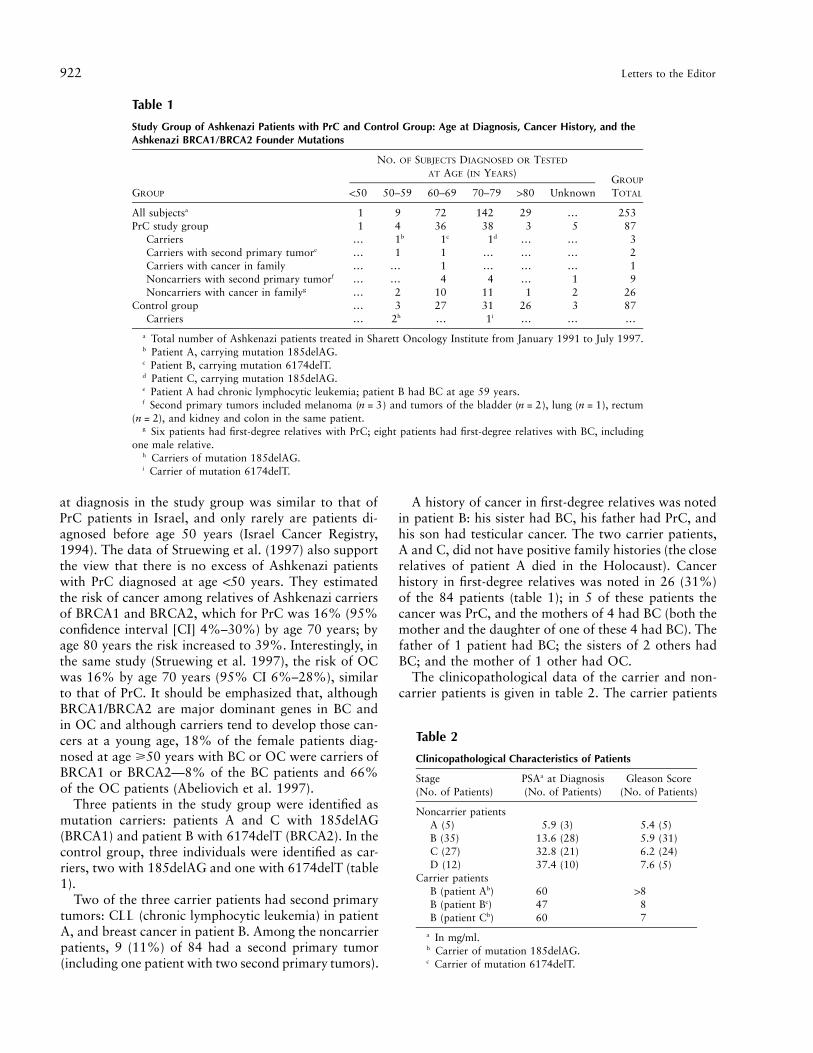
The risk of developing PrC is age-dependent and isdetermined by differential exposure to environmentalfactors. In addition, positive family history is a majorrisk factor for developing PrC at an early age (Steinberget al. 1990; Spitz et al. 1991; Whittemore et al. 1995).It is assumed that ∼10% of all cases of PrC and half ofthe cases diagnosed at an early age (!60 years) are dom-inantly inherited. Linkage analyses in families with mul-tiple cases of PrC pointed to a PrC-susceptibility gene(or group of genes) on chromosome 1 (Smith et al. 1996;Gronberg et al. 1997a; Berthon et al. 1998; Schaid etal. 1998), and, recently, an X-linked gene was suggested(Xu et al. 1998). It can be argued that BRCA1 andBRCA2 markedly reduce the age at onset of PrC andthat therefore the effect of BRCA1/BRCA2 will beshown only in patients diagnosed with PrC at age !60years, whereas in our study only five patients were as-certained in this age group. However, since 2.5% ofAshkenazi males are BRCA1/BRCA2 carriers, it wouldbe expected that an excess of Ashkenazi men will de-velop PrC at age !60 years. The stratification of the ages
922 Letters to the Editor
Table 1
Study Group of Ashkenazi Patients with PrC and Control Group: Age at Diagnosis, Cancer History, and theAshkenazi BRCA1/BRCA2 Founder Mutations
GROUP
NO. OF SUBJECTS DIAGNOSED OR TESTED
AT AGE (IN YEARS)GROUP
TOTAL!50 50–59 60–69 70–79 180 Unknown
All subjectsa 1 9 72 142 29 ) 253PrC study group 1 4 36 38 3 5 87
Carriers ) 1b 1c 1d ) ) 3Carriers with second primary tumore ) 1 1 ) ) ) 2Carriers with cancer in family ) ) 1 ) ) ) 1Noncarriers with second primary tumorf ) ) 4 4 ) 1 9Noncarriers with cancer in familyg ) 2 10 11 1 2 26
Control group ) 3 27 31 26 3 87Carriers ) 2h ) 1i ) ) )
a Total number of Ashkenazi patients treated in Sharett Oncology Institute from January 1991 to July 1997.b Patient A, carrying mutation 185delAG.c Patient B, carrying mutation 6174delT.d Patient C, carrying mutation 185delAG.e Patient A had chronic lymphocytic leukemia; patient B had BC at age 59 years.f Second primary tumors included melanoma ( ) and tumors of the bladder ( ), lung ( ), rectumn = 3 n = 2 n = 1
( ), and kidney and colon in the same patient.n = 2g Six patients had first-degree relatives with PrC; eight patients had first-degree relatives with BC, including
one male relative.h Carriers of mutation 185delAG.i Carrier of mutation 6174delT.
Table 2
Clinicopathological Characteristics of Patients
Stage(No. of Patients)
PSAa at Diagnosis(No. of Patients)
Gleason Score(No. of Patients)
Noncarrier patientsA (5) 5.9 (3) 5.4 (5)B (35) 13.6 (28) 5.9 (31)C (27) 32.8 (21) 6.2 (24)D (12) 37.4 (10) 7.6 (5)
Carrier patientsB (patient Ab) 60 18B (patient Bc) 47 8B (patient Cb) 60 7
a In mg/ml.b Carrier of mutation 185delAG.c Carrier of mutation 6174delT.
at diagnosis in the study group was similar to that ofPrC patients in Israel, and only rarely are patients di-agnosed before age 50 years (Israel Cancer Registry,1994). The data of Struewing et al. (1997) also supportthe view that there is no excess of Ashkenazi patientswith PrC diagnosed at age !50 years. They estimatedthe risk of cancer among relatives of Ashkenazi carriersof BRCA1 and BRCA2, which for PrC was 16% (95%confidence interval [CI] 4%–30%) by age 70 years; byage 80 years the risk increased to 39%. Interestingly, inthe same study (Struewing et al. 1997), the risk of OCwas 16% by age 70 years (95% CI 6%–28%), similarto that of PrC. It should be emphasized that, althoughBRCA1/BRCA2 are major dominant genes in BC andin OC and although carriers tend to develop those can-cers at a young age, 18% of the female patients diag-nosed at age >50 years with BC or OC were carriers ofBRCA1 or BRCA2—8% of the BC patients and 66%of the OC patients (Abeliovich et al. 1997).
Three patients in the study group were identified asmutation carriers: patients A and C with 185delAG(BRCA1) and patient B with 6174delT (BRCA2). In thecontrol group, three individuals were identified as car-riers, two with 185delAG and one with 6174delT (table1).
Two of the three carrier patients had second primarytumors: CLL (chronic lymphocytic leukemia) in patientA, and breast cancer in patient B. Among the noncarrierpatients, 9 (11%) of 84 had a second primary tumor(including one patient with two second primary tumors).
A history of cancer in first-degree relatives was notedin patient B: his sister had BC, his father had PrC, andhis son had testicular cancer. The two carrier patients,A and C, did not have positive family histories (the closerelatives of patient A died in the Holocaust). Cancerhistory in first-degree relatives was noted in 26 (31%)of the 84 patients (table 1); in 5 of these patients thecancer was PrC, and the mothers of 4 had BC (both themother and the daughter of one of these 4 had BC). Thefather of 1 patient had BC; the sisters of 2 others hadBC; and the mother of 1 other had OC.
The clinicopathological data of the carrier and non-carrier patients is given in table 2. The carrier patients

Letters to the Editor 923
were diagnosed at ages 57, 62, and 73 years (average64 years). The average level of prostate serum antigen(PSA) in the carrier patients was 55.8 mg/ml, higher thanthe average (23.6) in noncarrier patients at all stages;the difference in the PSA level was highly significant( ). The three carrier patients were diagnosed at2x 1 30stage B with Gleason scores of 7, 8, and 18, higher thanthe average (5.9) for the noncarrier patients at stage Band similar to the average at stage D. The clinicopath-ological records of the patients indicated that the tumorsin the three carriers were highly proliferative. This maysuggest that mutations in BRCA1 and BRCA2 may havesome role in the progression of the disease. A similarobservation was made of PrC in patients who belong toHPC1-linked families (Gronberg et al. 1997b) and inBRCA1-associated breast cancers (Eisinger et al. 1996;Marcus et al. 1996; Blackwood and Weber 1998; Rob-son et al. 1998). However, this conclusion is based onthree patients and should be confirmed in a larger num-ber of patients.
The frequency of carriers in the study group of PrCpatients and in the group of healthy men was 3.4%(95% CI 1.48%–5.4%), which is within the range ofthe population frequency (2.5%) (Fodor et al. 1998). Inorder to detect a minor difference between the twogroups, a much larger sample was needed. Instead, wechose a different approach in which we calculated theexpected percentage of carriers of BRCA1/BRCA2 foun-der mutations among the PrC patients, on the basis ofthe existent risk figures: 16% by age 70 years and 39%by age 80 years (Struewing et al. 1997). Assuming thatwe follow Ashkenazi men from age 50 years throughage 80 years, we further assumed that the rate of carriersis 2.5% and that among the carriers the average risk ofdeveloping PrC prior to age 80 years is ∼20%. We wouldthen expect that every year 33 of 100,000 new Ashke-nazi patients with PrC would be carriers of any of theBRCA1/BRCA2 founder mutations. Israeli data showthat the number of new cases among Ashkenazi men atthis age (50–80 years), is ∼260 in 100,000 (Israel CancerRegistry, 1994); hence the carriers would be ∼13% ofthe patients (33/260). We had 87 patients, and thereforeexpected 11 carriers in our study group, but observed3. The difference between the expected and observedresult is highly significant ( in the exact-bi-P ! .0005nomial test). The size of the sample enables a power of>80% for detecting a difference in carriers of 2.5% inthe control group and at least 12.5% in the patientsgroup. It is interesting to note that the strong associationfound among Israeli females between ethnic origin andbreast cancer is not evident for prostate cancer. The age-standardized rate of breast cancer among Jewish womenborn in Europe or America (i.e., having an Ashkenaziorigin) is 1.57 times that of Jewish women born in NorthAfrica (non-Ashkenazi origin), whereas the respective
rate for men having prostate cancer is 0.9 (Israel CancerRegistry, 1994). The age-adjusted rate of PrC (per100,000) in Israeli Jewish men by place of birth is 32.2for those born in Europe and North America (AshkenaziJews), 32.5 for men born in Africa and Asia, and 43.5for men born in Israel (Israel Cancer Registry, 1994).We therefore suggest that the contribution of BRCA1/BRCA2 germinal mutations to PrC morbidity is negli-gible. Our conclusion is in agreement with other studiesin which PrC patients were tested directly (Langston etal. 1996; Johannesdottir et al. 1996; Wilkens et al. 1999)and with some of the epidemiological studies (Isaacs etal. 1995; McCahy et al. 1996). However, our conclusioncontradicts other epidemiological studies (Arason et al.1993; Ford et al. 1994; Thorlacius et al. 1996; Struewinget al. 1997), in which the data were based on infor-mation received about first-degree relatives of carriers,while the PrC patients themselves were not analyzed. Itwould be interesting to explore the possibility of othersources of variation, such as environmental factors thataffect BRCA1/BRCA2 carriers to a greater extent thannoncarriers and to which men in Israel are not exposed.
AYALA HUBERT,1 TAMAR PERETZ,1 ORLY MANOR,2
LUNA KADURI,1 NAOMI WIENBERG,3 ISRAELA LERER,3
MICHAL SAGI,3 AND DVORAH ABELIOVICH3
1Sharett Institute of Oncology, 2School of PublicHealth and Community Medicine, and 3Departmentof Human Genetics, Hadassah Hebrew UniversityHospital, Hadassah Hebrew University MedicalSchool, Jerusalem
References
Abeliovich D, Kaduri L, Lerer I, Weinberg N, Amir G, SagiM, Zlotogora J, et al (1997) The founder mutations185delAG and 5382insC in BRCA1 and 6174delT inBRCA2 appear in 60% of ovarian cancer and 30% of early-onset breast cancer patients among Ashkenazi women. AmJ Hum Genet 60:505–514
Arason A, Barkadottier RB, Egilsson V (1993) Linkage anal-ysis of chromosome 17q markers and breast-ovarian cancerin Icelandic families, and possible relationship to prostaticcancer. Am J Hum Genet 52:711–717
Blackwood AM, Weber BL (1998) BRCA1 and BRCA2: frommolecular genetics to clinical medicine. J Clin Oncol 16:1969–1977
Berthon P, Valeri A, Cohen-Akenine A, Drelon E, Paiss T, WohrG, Latil A, et al (1998) Predisposing gene for early-onsetprostate cancer, localized on chromosome 1q42.2- 43. AmJ Hum Genet 62:1416–1424
Claus EB, Schildkraut JM, Thompson WD, Risch NJ (1996)The genetic attributable risk of breast and ovarian cancer.Cancer 77:2318–2324
Eisinger F, Stoppa-Lyonnet D, Longy M, Kerangueven F, No-guchi T, Bailly C, Vincent-Salomon A, et al (1996) Germline

924 Letters to the Editor
mutation at BRCA1 affects the histoprognostic grade in he-reditary breast cancer. Cancer Res 56:471–474
Fodor FH, Weston A, Bleiweiss IJ, McCurdy LD, Walsh MM,Tartter PI, Brower ST, et al (1998) Frequency and carrierrisk associated with common BRCA1 and BRCA2 mutationsin Ashkenazi Jewish breast cancer patients. Am J Hum Genet63:45–51
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE, BreastCancer Linkage Consortium (1994) Risks of cancer inBRCA1-mutation carriers. Lancet 343:692–695
Ford D, Easton DF, Peto J (1995) Estimates of the gene fre-quency of BRCA1 and its contribution to breast and ovariancancer incidence. Am J Hum Genet 57:1457–1462
Gronberg H, Damber L, Damber J-E, Iselius L (1997a) Seg-regation analysis of prostate cancer in Sweden: support todominant inheritance. Am J Epidemiol 146:552–557
Gronberg H, Isaacs SD, Smith JR, Carpten JD, Bova SG, FreijeD, Xu J, et al (1997b) Characteristics of prostate cancer infamilies potentially linked to the hereditary prostate cancer1 (HPC1) locus. JAMA 278:1251–1255
Isaacs SD, Kiemeney LALM, Baffoe-Bonnie A, Beaty TH,Walsh PC (1995) Risk of cancer in relatives of prostate can-cer probands. J Natl Cancer Inst 87:991–996
Israel Cancer Registry (1994) Prostate cancer. In: Cancer inIsrael. Ministry of Health, State of Israel, pp 19–21
Johannesdottir G, Gudmundsson J, Bergthorsson JT, ArasonA, Agnarsson BA, Eiriksdottir G, Johannsson OT, et al(1996) High prevalence of the 999del5 mutation in Icelandicbreast and ovarian cancer. Cancer Res 56:3663–3665
Langston AA, Stanford JL, Wicklund KG, Thompson JD, Bla-zej RG, Ostrander EA (1996) Germ-line BRCA1 mutationsin selected men with prostate cancer. Am J Hum Genet 58:881–885
Marcus JN, Watson P, Page DL, Narod SA, Lenoir GM, ToninP, Linder-Stephenson L, et al (1996) Hereditary breast can-cer: patholobiology, prognosis, and BRCA1 and BRCA2gene linkage. Cancer 77:697–709
McCahy PJ, Harris CA, Neal DE (1996) Breast and prostatecancer in the relatives of men with prostate cancer. Br J Urol78:552–556
Oddoux C, Struewing JP, Clayton CM, Neuhausen S, BrodyLC, Kaback M, Haas B, et al (1996) The carrier frequencyof the BRCA2 6174delT mutation in Ashkenazi Jewish in-dividuals is approximately 1%. Nat Genet 14:188–190
Roa BB, Boyd AA, Volcik K, Richards CS (1996) AshkenaziJewish population frequencies for common mutations inBRCA1 and BRCA2. Nat Genet 14:185–187
Robson M, Gilewski T, Haas B, Levin D, Borgen P, Rajan P,Hirschaut Y, et al (1998) BRCA-associated breast cancer inyoung women. J Clin Oncol 16:1642–1649
Schaid DJ, McDonnell SK, Blute ML, Thibodeau SN (1998)Evidence for autosomal dominant Inheritance of prostatecancer. Am J Hum Genet 62:1425–1438
Smith JR, Freije D, Carpten JD, Gronberg H, Xu J, Isaacs SD,Brownstein MJ, et al (1996) Major susceptibility locus forprostate cancer on chromosome 1 suggested by genome-wide search. Science 274:1371–1374
Spitz MR, Currier RD, Fueger JJ, Babaian RJ, Newell GR(1991) Familial patterns of prostate cancer: a case-controlanalysis. J Urol 146:1305–1307
Steinberg GD, Carter BS, Beaty TH, Childs B, Walsh PC (1990)Family history and the risk of prostate cancer. Prostate 17:337–347
Struewing JP, Abeliovich D, Peretz T, Avishai N, Kaback MM,Collins FS, Brody LC (1995) The carrier frequency of theBRCA1 185delAG mutation is approximately 1 percent inAshkenazi Jewish individuals. Nat Genet 11:198–200
Struewing JP, Hartge P, Wacholder S (1997) The risk of cancerassociated with specific mutations of BRCA1 and BRCA2among Ashkenazi Jews. N Engl J Med 336:1401–1408
Thorlacius S, Olafsadottir G, Tryggvadottir L, Neuhausen S,Jonasson JG, Tavtigian SV, Tulinius H, et al (1996) A singleBRCA2 mutation in male and female breast cancer familiesfrom Iceland with varied cancer phenotype. Nat Genet 13:117–119
Wilkens EP, Freije D, Xu J, Nusskern DR, Suzuki H, IsaacsSD, Wiley K, et al (1999) No evidence for a role of BRCA1or BRCA2 mutations in Ashkenazi Jewish families with he-reditary prostate cancer. Prostate 39:280–284
Whittemore AS, Wu AH, Kolonel LN, John EM, GallagherRP, Howe GR, West DW, et al (1995) Family history andprostate cancer risk in black, white, and Asian men in UnitedStates and Canada. Am J Epidemiol 141:732–740
Xu J, Meyers D, Freije D, Isaacs S, Wiley K , Nusskern D,Ewing C, et al (1998) Evidence for a prostate cancer sus-ceptibility locus on the X chromosome. Nat Genet 20:175–179
Address for correspondence and reprints: Dr. Dvorah Abeliovich, Departmentof Human Genetics, Hadassah Hospital, P.O. Box 12 000, Ein Kerem, Jerusalem91120, Israel. E-mail: [email protected]
q 1999 by The American Society of Human Genetics. All rights reserved.0002-9297/1999/6503-0038$02.00
Am. J. Hum. Genet. 65:924–926, 1999
An HFE Intronic Variant Promotes Misdiagnosis ofHereditary Hemochromatosis
To the Editor:Hereditary hemochromatosis (HH; MIM 235200), anautosomal recessive disorder of iron metabolism, canresult in numerous clinical complications and is esti-mated to affect ∼1/300 individuals of northern Europeanorigin (Merryweather-Clarke et al. 1997). Two muta-tions—C282Y and H63D—that contribute to HH havebeen identified (Feder et al. 1996), and screening for theC282Y mutation, in particular, is routinely done to iden-tify carriers and affected individuals. Biochemical mark-ers indicate a relatively clear distinction between thesetwo groups, with minimal clinical consequences for het-erozygotes (Bulaj et al. 1996). We initiated screening forthe C282Y mutation, using the primer sequences pro-vided by Feder et al. (1996) and subsequent restrictiondigestion of PCR products (Jazwinska et al. 1996). Re-

Letters to the Editor 925
Figure 1 PCR amplification and SnaBI digestion of DNA (Ja-zwinska et al. 1996) from individuals referred for HH testing. Lane1, C282Y carrier. Lane 2, Normal homozygote. Lane 3, C282Y ho-mozygote. Lane 4, Individual with an anomalous pattern with traceamounts of undigested PCR product. All 25-ml PCR reactions wereperformed in parallel, with use of 150 ng DNA template and 1.25 UPLATINUMyTaq (GIBCO-BRL), in 20 mM Tris-HCl (pH 8.4), 50mM KCl, and 1.5 mM MgCl2. PCR conditions were as follows: 947Cfor 2 min; then 30 cycles at 947C for 30 s, 657C for 30 s, and 727Cfor 30 s; and a final extension at 727C for 10 min, by means of aGeneAmp PCR System 9600 (Perkin-Elmer). PCR products were di-gested with SnaBI at 377C for 3 h and were resolved by use of 1.5%agarose gel.
Figure 2 DNA sequence of HFE exon 4, with flanking introns.The relative locations of the C282Y mutation (GrA) in exon 4 andthe 5569 GrA (892148 GrA) polymorphism in intron 4 are shown.Sequences and locations are highlighted for Feder et al. (1996) forwardand reverse primers and for the two alternative flanking primers (HCS:F and HCS:R) that were used to amplify this region. Conditions forPCR with HCS:F and HCS:R followed those outlined for the Feder etal. primer set (fig. 1), with an annealing temperature of 557C. The486-bp HCS PCR product was cleaved into 320- and 166-bp fragmentsby SnaBI in the presence of the C282Y mutation. The intronic poly-morphism was confirmed, by sequencing and by MseI digestion ofHCS PCR products, in all samples that showed an anomalous SnaBIdigestion pattern by use of Feder et al. (1996) PCR products.
cently, we have identified anomalous results in some in-dividuals while screening for the presence of the C282Ymutation. We initially identified eight individuals, sevenof whom were unrelated, who appeared to be C282Yhomozygotes with trace amounts of undigested DNA(fig. 1). It was assumed that these individuals were ho-mozygotes with some form of sample contamination.Clinical histories of these individuals did not includeprevious blood transfusion or tissue transplantation. In-creased amounts of restriction enzyme and incubationtime, as well as resampling of these individuals, did notresolve the anomalous results. An increase in the strin-gency of the PCR conditions, achieved either by increas-ing the annealing temperature or by decreasing theamount of genomic template, reduced the amount ofamplified normal product to generate a C282Y homo-zygote pattern (results not shown). Biochemical data onserum iron levels, serum ferritin levels, and transferrinsaturation were available for two of these individuals—a35-year-old man and a 71-year-old woman—and valueswere below the affected range. In one particular family,two sibs showed this pattern, yet, when their parentswere tested, only the mother was found to be a C282Ycarrier. In this instance, a combination of two indepen-dent incidents of nonpaternity and sample contamina-tion would be required to explain the results.
We reanalyzed these cases, using two different ap-proaches: (1) the Baty et al. (1998) amplification re-
fractory mutation system (ARMS), which includes analternative reverse primer with the Feder et al. (1996)forward primer, and (2) a modification of the previousprotocol (Jazwinska et al. 1996) to incorporate two al-ternative primers that flank the Feder et al. primer sites(fig. 2). With both techniques, we found that all eightsamples gave a clear carrier pattern for C282Y (resultsnot shown). In all cases, sequencing across the Feder etal. primer sites revealed a GrA substitution at nucleotideposition 5569 (GenBank accession number Z92910) onthe non-C282Y allele. This sequence variant is locatedin intron 4, 5 bases from the 3′ terminus of the reverse-primer site identified by Feder et al., and is not predictedto disrupt normal hemochromatosis gene (HFE) splic-ing (fig. 2). In all eight samples, this mutation was seenon a non-C282Y, non-H63D chromosome. PCR usingthis reverse primer can result in dramatically reducedamplification of the polymorphic allele, such that aC282Y carrier can appear to be a C282Y homozygote.The relative intensity of the non-C282Y PCR productis inversely proportional to the stringency of the PCRconditions. Sequencing of both alleles in the eight in-dividuals in the present study allowed clear assignmentof carrier, rather than affected, status.
The 5569 GrA substitution introduces an MseI re-striction site. We used this enzyme, in conjunction withthe Feder et al. (1996) forward primer and the HCS:

926 Letters to the Editor
R primer, to screen 48 individuals (45 of whom wereunrelated) whom we had previously identified asC282Y homozygotes. In this group, we identified oneadditional unrelated individual with the C282Y mu-tation and the 5569 GrA polymorphism. Closer ex-amination of the assay on the basis of which the pre-vious diagnosis was made in this individual revealedan extremely faint normal band that had been inter-preted to result from partial digestion. In total, there-fore, the polymorphism has been found in 8 of 202unrelated individuals who were referred for HH testing.An estimate of the allele frequency can be made on thebasis of the C282Y carrier frequency. We found these8 polymorphism carriers among a total of 43 unrelatedC282Y (non-H63D) carriers. Our estimated popula-tion frequency of this allele is, therefore, 8/43 (=.186).Consequently, in our population, this polymorphismhad the potential to result in ∼19% of C282Y heter-ozygotes being misidentified as homozygotes.
We identified the 8 polymorphism carriers, in addi-tion to 44 unrelated C282Y homozygotes, from ourtotal sample of 202 unrelated individuals referred fortesting. If the assumption of homozygosity, along withaccess to parental genotypes, had been made in all in-dividuals with the polymorphism, as well as in thosewith the homozygosity, this would have led to an es-timate of ∼8/52 (=.154) for nonparentage, of whichhalf of these cases, or 8%, would have been assumedto result from nonpaternity. The frequency of this poly-morphism is high enough to warrant concern that theinterpretation of homozygosity in these cases will resultin an overestimate of the C282Y-allele frequency, a mis-diagnosis of this condition, and an incorrect assump-tion of nonpaternity in some families. In our hands,the polymorphism promoted misinterpretation of a re-striction-digestion–based assay, but any form of anal-ysis (including allele-specific oligonucleotide hybridi-zation, ARMS, or direct sequencing) that incorporatesthe Feder et al. (1996) reverse primer is equally proneto misdiagnosis. It is recommended that all laboratoriesusing the Feder et al. reverse primer to test for theC282Y mutation confirm C282Y-homozygote resultsby using a flanking primer set and MseI digestion.
Acknowledgments
The authors thank Drs. Diane Cox, Nancy Carson, SherrylTaylor, and Marsha Speevak for their helpful comments andadvice.
MARTIN J. SOMERVILLE, KATHLEEN A. SPRYSAK,MARK HICKS, BASIL G. ELYAS, AND
LEANNE VICEN-WYHONY
Department of Medical GeneticsUniversity of AlbertaEdmonton
Electronic-Database Information
Accession numbers and URLs for data in this article are asfollows:
GenBank, http://www.ncbi.nlm.nih.gov/Web/Genbank (forZ92910)
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for HH [MIM 235200])
References
Baty D, Terron Kwiatkowski A, Mechan D, Harris A, PippardMJ, Goudie D (1998) Development of a multiplex ARMStest for mutations in the HFE gene associated with hereditaryhaemochromatosis. J Clin Pathol 51:73–74
Bulaj ZJ, Griffen LM, Jorde LB, Edwards CQ, Kushner JP(1996) Clinical and biochemical abnormalities in peopleheterozygous for hemochromatosis. N Engl J Med 335:1799–1805
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA,Basava A, Dormishian F, et al (1996) A novel MHC classI-like gene is mutated in patients with hereditary haemo-chromatosis. Nat Genet 13:399–408
Jazwinska EC, Cullen LM, Busfield F, Pyper WR, Webb SI,Powell LW, Morris CP, et al (1996) Haemochromatosis andHLA-H. Nat Genet 14:249–251
Merryweather-Clarke AT, Pointon JJ, Shearman JD, RobsonKJ (1997) Global prevalence of putative haemochromatosismutations. J Med Genet 34:275–278
Address for correspondence and reprints: Dr. Martin J. Somerville, 8-26 Med-ical Sciences Building, Department of Medical Genetics, University of Alberta,Edmonton, Alberta, Canada T6G 2H7. E-mail: [email protected]
q 1999 by The American Society of Human Genetics. All rights reserved.0002-9297/1999/6503-0039$02.00
Am. J. Hum. Genet. 65:926–928, 1999
No Mutations in the Coding Region of the PRKCGGene in Three Families with Retinitis PigmentosaLinked to the RP11 Locus on Chromosome 19q
To the Editor:Retinitis pigmentosa (RP) and allied degenerations of theretina are genetically heterogeneous, with well over 50loci implicated so far through gene identifications orlinkage-based chromosomal assignments. Among thesegenes, the dominantly inherited RP11 locus (MIM600138) on chromosome 19q is noteworthy becausesome carriers develop RP that is symptomatic at age !20years, whereas others are asymptomatic and show nofunduscopic or electroretinographic signs of disease evenat age 170 years (Berson et al. 1969; Berson and Si-monoff 1979; Evans et al. 1995; Nakazawa et al. 1996;McGee et al. 1997). On the basis of its chromosomal

Letters to the Editor 927
assignment, the PRKCG gene is a candidate for RP11.This gene encodes a form of protein kinase C that isexpressed in the retina. Last year, Al-Maghtheh et al.(1998) described two families with RP11-linked domi-nant RP in which a missense change (Arg659Ser) inPRKCG cosegregated with disease. Only one of thesetwo families (RP1907) clearly exhibited asymptomatic,obligate carriers who transmitted the disease to off-spring. The authors failed to discover a mutation inPRKCG in three other families with reduced penetranceshowing linkage to this region. Nevertheless, the authorsspeculated that PRKCG could be the RP11 gene.
In response to that report, we have undertaken ananalysis of the PRKCG gene in three additional familieswith dominant RP with reduced penetrance. All threefamilies have unaffected, obligate carriers, and we pre-viously reported linkage data pointing to RP11 as thecause of RP in these families (McGee et al. 1997). Anaffected individual from each family was chosen for thecurrent study (the patients were individuals III-2 fromfamily 1295, IV-8 from family 2474, and IV-34 fromfamily 1562) (McGee et al. 1997), as well as DNA froman unrelated control individual without RP and withouta family history of retinal degeneration. We amplifiedeach of the 18 exons of the PRKCG gene individually,using PCR from leukocyte DNA obtained from theseindividuals. Primer pairs were the same as those reportedelsewhere (Al-Maghtheh et al. 1998) except for exons5, 10, and 11, for which we used primers (sense/anti-sense) as follows: exon 5, 5′ portion, TGAGGTGCT-ACCCGCAGCTT / CAGTTACGTGGATCTCATCT;exon 5, 3′ portion, AGGCTGCGAGATGAACGTGC /AGGCGAGGGGGCGGGGCCTC; exon 10, GGCT-GTGTAAGGTCTAAGTG / CACAGGAGCCCAGTCT-CTTC; exon 11, CTGGGTTCCCAACATGGACT / CT-TGCCTCTCCCTAAACTCA. The amplified fragmentswere sequenced directly by means of standard methods.
None of the patients had a defect in codon 659. Fur-thermore, none of the patients had an abnormality inthe coding region or the flanking-intron splice-acceptoror -donor sites, except for one patient who heterozy-gously carried a silent change in codon 24 (Ala24Ala,GCTrGCC). In all eight gene copies carried by the threepatients and the control individual, the sequence of co-dons Phe19 and Ser148 in exons 1 and 5, respectively,was different from that published elsewhere (TTT in-stead of TTC, and TCC instead of TCT, respectively),suggesting that the previously reported sequence(GenBank accession number M13977) is an allelic var-iant, a sequencing artifact, or an error. The previouslyreported (Al-Maghtheh et al. 1998) silent polymorphismat codon Asn189 was encountered, with two of the threepatients being heterozygotes (carrying the sequencesAAT and AAC for that codon) and the third patientbeing a homozygote for the sequence AAT. We previ-
ously documented a recombination event in the vicinityof RP11 in one branch of family 1562 (McGee et al.1997); however, the intragenic polymorphisms were notinformative in this branch of that family and did notallow us to determine on which side of the crossover thePRKCG locus lies.
Our analysis provides no evidence that PRKCG is theRP11 gene. Although our data cannot exclude the path-ogenicity of the Arg659Ser missense change, the possi-bility that it is a rare, nonpathogenic variant remainsplausible, since none of the three families analyzed hereand only two of the five families analyzed elsewhere (Al-Maghtheh et al. 1998) carry an anomaly that wouldchange the sequence of the encoded protein. Anotherformal possibility is that there are two RP genes in thisregion, but it would be necessary that both RP loci ex-hibit reduced penetrance. In either case, it appears thatan RP gene in this region remains to be identified.
Acknowledgments
Supported by grants from the National Eye Institute(EY08683, EY11655, and EY00169), the Foundation FightingBlindness, and the Massachusetts Lions Eye Research Fund,Inc., and by private donations to the Taylor Smith Laboratoryand the Ocular Molecular Genetics Institute. T.P.D. is a Re-search to Prevent Blindness Senior Scientific Investigator.
THADDEUS P. DRYJA,1,2 JENNIFER MCEVOY,1
TERRI L. MCGEE,1 AND ELIOT L. BERSON2
1Ocular Molecular Genetics Institute and 2Berman-Gund Laboratory for the Study of RetinalDegenerations, Harvard Medical School,Massachusetts Eye and Ear Infirmary, Boston
Electronic-Database Information
Accession numbers and URLs for data in this article are asfollows:
GenBank, http://www.ncbi.nlm.nih.gov/Entrez/nucleotide.html (for sequence data reported previously [accession num-ber M13977])
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for the RP11 locus [MIM600138])
References
Al-Maghtheh M, Vithana EN, Inglehearn CF, Moore T, BirdAC, Bhattacharya SS (1998) Segregation of a PRKCG mu-tation in two RP11 families. Am J Hum Genet 62:1248–1252
Berson EL, Gouras P, Gunkel RD, Myrianthopoulos NC(1969) Dominant retinitis pigmentosa with reduced pene-trance. Arch Ophthalmol 81:226–234
Berson EL, Simonoff EA (1979) Dominant retinitis pigmentosawith reduced penetrance: further studies of the electroretin-ogram. Arch Ophthalmol 97:1286–1291

928 Letters to the Editor
Evans K, Al-Maghtheh M, Fitzke FW, Moore AT, Jay M, In-glehearn CF, Arden GB, et al (1995) Bimodal expressivityin dominant retinitis pigmentosa genetically linked to chro-mosome 19q. Br J Ophthalmol 79:841–846
McGee TL, Devoto M, Ott J, Berson EL, Dryja TP (1997)Evidence that the penetrance of mutations at the RP11 locuscausing dominant retinitis pigmentosa is influenced by agene linked to the homologous RP11 allele. Am J Hum Ge-net 61:1059–1066
Nakazawa M, Xu S, Gal A, Wada Y, Tamai M (1996) Variableexpressivity in a Japanese family with autosomal dominantretinitis pigmentosa closely linked to chromosome 19q. ArchOphthalmol 114:318–322
Address for correspondence and reprints: Dr. Thaddeus P. Dryja, Massachu-setts Eye and Ear Infirmary, 243 Charles Street, Boston, MA 02114. E-mail:[email protected]
q 1999 by The American Society of Human Genetics. All rights reserved.0002-9297/1999/6503-0040$02.00




















