
Soil remediation by Fenton-like process: Phenol removal and soil organic matter modification
8
Please cite this article in press as: A. Romero, et al., Soil remediation by Fenton-like process: Phenol removal and soil organic matter modification, Chem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022 ARTICLE IN PRESS G Model CEJ-7852; No. of Pages 8 Chemical Engineering Journal xxx (2011) xxx–xxx Contents lists available at ScienceDirect Chemical Engineering Journal journal homepage: www.elsevier.com/locate/cej Soil remediation by Fenton-like process: Phenol removal and soil organic matter modification Arturo Romero a,∗ , Aurora Santos a , Tomás Cordero b , José Rodríguez-Mirasol b , Juana María Rosas a , Fernando Vicente a a Dpto Ingenieria Quimica, Facultad de Ciencias Químicas, Universidad Complutense Madrid, Ciudad Universitaria S/N, 28040 Madrid, Spain b Dpto Ingenieria Quimica, Facultad de Ciencias, Universidad de Málaga, Campus de Teatinos S/N, 29013 Málaga, Spain article info Article history: Received 23 December 2010 Received in revised form 2 March 2011 Accepted 7 March 2011 Keywords: Fenton’s like oxidation Hydrogen peroxide Soil organic matter Soil remediation Sorption abstract In situ chemical oxidation (ISCO) seems to be a powerful technology for soil remediation. In this work the application of Fenton’s like oxidation treatment (H 2 O 2 + natural ferrous species present in soil) has been evaluated to two different soils (S1 and S2) contaminated with phenol (q PhOH S1o = 360 mg g −1 , q PhOH S2o = 160 mg g −1 ). They were categorized as calcareous loamy sand soils with different soil organic mat- ter content (SOM S1 = 15.1%, SOM S2 = 10%). A refractory fraction of PhOH remained in the solid phase after treatment with H 2 O 2 (C H 2 O 2 o = 1 and 2%), probably due to an entrapment phenomenon of the contami- nant and because of the competition of SOM for the oxidant. Consequently, lower remediation efficiencies are obtained as SOM increases. It has also been paid attention to the effect of the oxidant on the SOM. It was found that H 2 O 2 does not produce any important modification of both SOM content and distribution. This was in agreement with the oxygen obtained after hydrogen peroxide had reacted. Only a slight degradation of the humic and fulvic acids and a partial removal of the quinone and carbonyl groups were noticed, while carbonate groups and most of the SOM remain the same. However, a decrease in the sorption capacity of PhOH in the treated soils is observed. Therefore, it can be assumed that the oxidation of SOM could modify the external surface of SOM. Moreover, the oxidation of SOM by H 2 O 2 could create more hydrophilic sites and thus decreases the sorption ability. © 2011 Published by Elsevier B.V. 1. Introduction The remediation of contaminated soils is a significant prob- lem because they are complex media and many contaminants are sorbed. Therefore, both adsorption studies and remediation techniques have been studied in recent years [1,2]. The increas- ing contamination of soil by organic compounds has promoted the development and application of several remediation processes [2,3]. In situ chemical oxidation (ISCO) shows current alterna- tive for contaminated soils remediation by organic compounds. At present, the most common field applications have been based on Fenton’s reagent whereby H 2 O 2 is applied with an iron catalyst creating a hydroxyl free radical (OH • ). This OH • is capable of oxi- dizing complex organic compounds [4,5]. The oxidation reactions are commonly quick and appear to be concluded within few hours [6]. ∗ Corresponding author. Tel.: +34 913944171; fax: +34 913944171. E-mail addresses: [email protected] (A. Romero), [email protected] (A. Santos), [email protected] (T. Cordero), [email protected] (J. Rodríguez-Mirasol), [email protected] (J.M. Rosas), [email protected] (F. Vicente). In fact, despite the optimal pH for Fenton’s process is very low, the organic pollutants mineralization is reported to occur also at neutral pH in presence of iron [7–10]. The approach of the Fen- ton process allows the use of pH-neutral conditions and avoids acidification of the soil that could produce a strongly negative envi- ronmental impact. In many sites, there is an abundance of naturally occurring heterogeneous forms of Fe which serves as the predom- inant source of catalyst for the Fenton-like process [11,12]. The obtained results demonstrated that one of the main draw- backs of an in situ Fenton treatment relies in the instability of H 2 O 2 , when it gets in touch with naturally occurring chemical species present in surface soil (organic and inorganic soil components) [13]. Soil organic matter (SOM) has been found to consume and compete for the oxidant during Fenton oxidation reaction [14]. Therefore, a high oxidant demand in the remediation process can be due to non-productive catalyzed decomposition of H 2 O 2 to O 2 and H 2 O. Decomposition of H 2 O 2 in soil in batch has already been described in literature [13], but less discussion has been paid to the changes produced by the Fenton’s reagent onto the soil. The oxidation treatment may also have an effect on the soil quality [15]. Moreover, change of soil properties with oxidant application could also affect the remediation process while fate and transport 1385-8947/$ – see front matter © 2011 Published by Elsevier B.V. doi:10.1016/j.cej.2011.03.022
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Soil remediation by Fenton-like process: Phenol removal and soil organic matter modification

G
C
Sm
AJa
b
a
ARRA
KFHSSS
1
latit[tpFcda[
(j
1d
ARTICLE IN PRESSModel
EJ-7852; No. of Pages 8
Chemical Engineering Journal xxx (2011) xxx–xxx
Contents lists available at ScienceDirect
Chemical Engineering Journal
journa l homepage: www.e lsev ier .com/ locate /ce j
oil remediation by Fenton-like process: Phenol removal and soil organicatter modification
rturo Romeroa,∗, Aurora Santosa, Tomás Corderob, José Rodríguez-Mirasolb,uana María Rosasa, Fernando Vicentea
Dpto Ingenieria Quimica, Facultad de Ciencias Químicas, Universidad Complutense Madrid, Ciudad Universitaria S/N, 28040 Madrid, SpainDpto Ingenieria Quimica, Facultad de Ciencias, Universidad de Málaga, Campus de Teatinos S/N, 29013 Málaga, Spain
r t i c l e i n f o
rticle history:eceived 23 December 2010eceived in revised form 2 March 2011ccepted 7 March 2011
eywords:enton’s like oxidationydrogen peroxideoil organic matteroil remediation
a b s t r a c t
In situ chemical oxidation (ISCO) seems to be a powerful technology for soil remediation. In this workthe application of Fenton’s like oxidation treatment (H2O2 + natural ferrous species present in soil) hasbeen evaluated to two different soils (S1 and S2) contaminated with phenol (qPhOH S1o = 360 mg g−1, qPhOH
S2o = 160 mg g−1). They were categorized as calcareous loamy sand soils with different soil organic mat-ter content (SOMS1 = 15.1%, SOMS2 = 10%). A refractory fraction of PhOH remained in the solid phase aftertreatment with H2O2 (CH2O2o = 1 and 2%), probably due to an entrapment phenomenon of the contami-nant and because of the competition of SOM for the oxidant. Consequently, lower remediation efficienciesare obtained as SOM increases.
It has also been paid attention to the effect of the oxidant on the SOM. It was found that H2O2 does
orption not produce any important modification of both SOM content and distribution. This was in agreementwith the oxygen obtained after hydrogen peroxide had reacted. Only a slight degradation of the humicand fulvic acids and a partial removal of the quinone and carbonyl groups were noticed, while carbonategroups and most of the SOM remain the same. However, a decrease in the sorption capacity of PhOH inthe treated soils is observed. Therefore, it can be assumed that the oxidation of SOM could modify theexternal surface of SOM. Moreover, the oxidation of SOM by H2O2 could create more hydrophilic sites
rptio
and thus decreases the so. Introduction
The remediation of contaminated soils is a significant prob-em because they are complex media and many contaminantsre sorbed. Therefore, both adsorption studies and remediationechniques have been studied in recent years [1,2]. The increas-ng contamination of soil by organic compounds has promotedhe development and application of several remediation processes2,3]. In situ chemical oxidation (ISCO) shows current alterna-ive for contaminated soils remediation by organic compounds. Atresent, the most common field applications have been based onenton’s reagent whereby H2O2 is applied with an iron catalyst
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
reating a hydroxyl free radical (OH•). This OH• is capable of oxi-izing complex organic compounds [4,5]. The oxidation reactionsre commonly quick and appear to be concluded within few hours6].
∗ Corresponding author. Tel.: +34 913944171; fax: +34 913944171.E-mail addresses: [email protected] (A. Romero), [email protected]
A. Santos), [email protected] (T. Cordero), [email protected] (J. Rodríguez-Mirasol),[email protected] (J.M. Rosas), [email protected] (F. Vicente).
385-8947/$ – see front matter © 2011 Published by Elsevier B.V.oi:10.1016/j.cej.2011.03.022
n ability.© 2011 Published by Elsevier B.V.
In fact, despite the optimal pH for Fenton’s process is very low,the organic pollutants mineralization is reported to occur also atneutral pH in presence of iron [7–10]. The approach of the Fen-ton process allows the use of pH-neutral conditions and avoidsacidification of the soil that could produce a strongly negative envi-ronmental impact. In many sites, there is an abundance of naturallyoccurring heterogeneous forms of Fe which serves as the predom-inant source of catalyst for the Fenton-like process [11,12].
The obtained results demonstrated that one of the main draw-backs of an in situ Fenton treatment relies in the instability of H2O2,when it gets in touch with naturally occurring chemical speciespresent in surface soil (organic and inorganic soil components) [13].Soil organic matter (SOM) has been found to consume and competefor the oxidant during Fenton oxidation reaction [14]. Therefore,a high oxidant demand in the remediation process can be due tonon-productive catalyzed decomposition of H2O2 to O2 and H2O.
Decomposition of H2O2 in soil in batch has already been
on-like process: Phenol removal and soil organic matter modification,
described in literature [13], but less discussion has been paid tothe changes produced by the Fenton’s reagent onto the soil. Theoxidation treatment may also have an effect on the soil quality[15]. Moreover, change of soil properties with oxidant applicationcould also affect the remediation process while fate and transport

IN PRESSG
C
2 ineering Journal xxx (2011) xxx–xxx
osoc
FueeS[
oTabctia
2
2
AHpoaps
2
asrTte
aecSfe
2
amb
bITaonr
Table 1Operating conditions of PhOH degradation by Fenton-like reaction. VL/W = 2 mL g−1,T = 20 ◦C.
RUN Soil (qPhOH)o (mg kg−1) (CH2O2 )o mg L−1 molesH2O2 /molesPhOH
1 S1 360 10,000 152
ARTICLEModel
EJ-7852; No. of Pages 8
A. Romero et al. / Chemical Eng
f organic contaminants can be modified. There are very limitedtudies regarding the change of SOM and the following effectsn sorption and desorption characteristics to hydrophobic organicontaminants after Fenton oxidation [6,16].
The aim of this work is to evaluate the effectiveness of theenton-like treatment in two soils with higher SOM content at nat-ral neutral pH, contaminated with PhOH. The PhOH presence innvironmental matrices is of great concern to their high toxicitynvironmental persistence. For this reason, it has been listed by thepanish Protection Agency as priority environmental contaminants17].
The influence of the oxidant agent on the SOM and the impactf this interaction on sorption of pollutants have been studied.hat is the reason why PhOH has been used as contaminant modelnd sorption of this pollutant before and after treatment of soilsy H2O2 has been analyzed. The SOM characteristics, content andomposition, were evaluated before and after the treatment withhe oxidant reagent. Implications for the environmental impact ofn situ chemical oxidation and availability of organic contaminantsre discussed.
. Materials and methods
.1. Reagents
PhOH was selected as a target pollutant and was purchased fromldrich Company. The hydrogen peroxide 30% (w/w) from Riedel deaën was used in the oxidation experiments. Titanium (IV) oxysul-hate solution from Riedel de Haën was used in the determinationf hydrogen peroxide. Methanol from Aldrich Company was useds solvent extractant. All the suspensions and solutions were pre-ared with Milli-Q water (>18 M� cm) purified with a deionizingystem.
.2. Soil samples
The soils selected for this study (S1 and S2) were categorizeds loamy sand at neutral pH. The main difference between the twooil samples was the SOM content (15.1 and 10% for S1 and S2,espectively). The soils were classified as calcaric Fluvisols (FLca).his is a type of soil that develops the basins of the main rivers thatraverse limestone material, which is the dominant material in theastern half of the Iberian Peninsula [18].
The pH was measured in 1:2.5 soil/water suspensions [19], withvalue of 7.4 and 7.8 for soil S1 and S2, respectively. The CaCO3
quivalent (10.7% for S1 and 7.3% for S2) was determined by cal-imeter Bernard method. Total Fe (28,750 and 34,390 mg kg−1
soil for1 and S2 expressed as Fe oxide) and Mn (460 and 540 mg kg−1
soilor S1 and S2 expressed as Mn oxide) were determined by acidxtraction/atomic absorption spectroscopy (EPA 3050B Method).
.3. Soil characterization
The SOM content in the soils was determined by incineration ofknown weight of sample placed in a ceramic crucible in an electricuffle for 3 h at 550 ◦C, obtaining the corresponding SOM content
y mass difference (NEN 5754 Method).Soil contains two different forms of carbon: total organic car-
on (TOC) and inorganic carbon (IC). The total carbon (TC) andC are measured by a Shimadzu TOC-V CSH solid analyzer. For
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
C measurements, the sample is introduced in a ceramic cruciblend heated at 900 ◦C in a high frequency furnace to react with thexygen stream. The determination of IC concentration in soils isecessary to prepare the samples for measurement after completeemoval of IC with acid. Add to the crucible 4 mL of concentrated
2 S1 360 20,000 3043 S2 160 10,000 3414 S2 160 20,000 682
H3PO4 to eliminate all carbonates and take measurement at 200 ◦C.The TOC concentration is determined by difference:
TOC = TC − IC (1)
Labile and recalcitrant pools of carbon in SOM were evaluatedfrom acid hydrolysis to quantify changes in C fractions after H2O2treatment. In this study, we used the two-step acid hydrolysis pro-cedure with H2SO4 to determine labile and recalcitrant C pools[20].
The porous structure of the samples was characterized by N2adsorption–desorption at −196 ◦C and CO2 adsorption at 0 ◦C, per-formed in a SA 3100 surface area analyzer (Coulter). Samples werepreviously outgassed for at least 8 h at room temperature to avoidsoil degradation. From the N2 isotherm, the apparent surface area(SgN2 ) was determined applying the BET equation. The pore vol-ume (Vp) and mean pore radio (r̄p) were obtained also from the N2isotherm data. The narrow micropore surface area (ACO2
DR ) was cal-culated by the Dubinin–Radushkevich (DR) method, applied to theCO2 adsorption isotherm.
Temperature programmed desorption (TPD) and temperatureprogrammed oxidation (TPO) profiles were obtained in a customquartz fixed bed reactor placed inside an electrical furnace, usingsample mass of approximately 150 mg. The samples were heatedfrom room temperature up to 900 ◦C at a heating rate of 10 ◦C/minin helium or air flow (200 cm3 STP/min), respectively. The amountsof CO and CO2 desorbed from the samples were monitored withnondispersive infrared (NDIR) gas analyzers (Siemens ULTRAMAT22).
Non-isothermal thermogravimetric analyses were carried outin a gravimetric thermobalance system, CI electronics. The ther-mobalance automatically measures the weight of the sample andthe temperature as a function of time. Experiments were carried outin inert atmosphere (N2), and in air atmosphere, for a total flow rateof 150 cm3 STP/min, using sample mass of approximately 10 mg.The sample temperature was increased from room temperature upto 900 ◦C at a heating rate of 10 ◦C/min.
2.4. Fenton-like oxidation experiments
Some experiments were performed to evaluate the effective-ness of Fenton’s like oxidation to remediate soils contaminatedwith phenol (PhOH). The Fe present in soil is available for reactionwith H2O2.
Contaminated soils were artificially prepared by using uncon-taminated control soils (S1 and S2). The soils were spiked with asolution of PhOH dissolved in methanol. Then, the mixture wascompletely stirred and blended to ensure homogeneity. Finally, themixture was laid until the methanol was completely evaporated.During the ventilation, the mixture was stirred each day. The totalresidual mass of the contaminants in the soils were determinedby ultrasonic solvent extraction by using methanol as the solvent.
on-like process: Phenol removal and soil organic matter modification,
The target concentrations of PhOH in the spiked soils are shown inTable 1.
The PhOH oxidation experiments in both soils (S1 and S2) wereperformed in batch reactor systems, at room temperature (20 ◦C)and at natural soil pH. H2O2 solution was used as oxidant. The soil-

ING
C
ineeri
oa
tusAT
rtpscdmc
2
ipcraTdAHb
tct
2
(ftppsiaic
2
mm[
limfihdn
ARTICLEModel
EJ-7852; No. of Pages 8
A. Romero et al. / Chemical Eng
xidant slurries were stirred (50 ua) during the reaction period onshaking water bath UNITRONIC, supplied by SELECTA.
Soil samples (1 g) were put in contact with 2 mL aqueous solu-ion in 15 mL glass vials. The Fenton’s like reaction was startedpon addition of H2O2. Then 66 �L or 134 �L of the stock 30% H2O2olution was added, giving a final H2O2 concentration of 1% or 2%.ll the tests performed and the dosages used are summarized inable 1.
Vials were examined at different times. Each reaction vial rep-esents one time point. The caps were not sealed during reactionime to permit evacuation of the generated gas in order to avoidossible explosions due to gas accumulation in the vials [21]. Theoil and aqueous phase were separated before the extraction. Afterentrifugation, the supernatant and soil extracts were analyzed toetermine PhOH by means of a gas chromatograph (GC). Deter-ination of the remaining hydrogen peroxide in supernatant was
arried out as described below.
.5. Decomposition of H2O2 by soil
For the sake of comparison, H2O2 degradation was also stud-ed. The experiments were carried out for each soil, without anyollutant, through batch experiments, and at the same previousonditions. The uncontaminated soil weight to liquid phase volumeatio (W/VL) was 0.5 g mL−1. The experiments were performed withn initial H2O2 concentration of 2%. The soil pH was not adjusted.he samples were collected at different reaction times and imme-iately centrifuged for 5 min in a CENTROLIT SELECTA centrifuge.fter centrifugation, the supernatant was analyzed to determine2O2 concentration. The rate of O2 gas generation was determinedy a gasometer.
The soils were analyzed before and after the treatment withhe oxidant ((CH2O2 )o = 2%) to study the chemical changes asso-iated with the decomposition of the SOM by the characterizationechniques above mentioned.
.6. Sorption equilibrium experiments
The adsorption study was carried out in batch reactors, stirred50 ua) at 20 ◦C. A volume of 35 mL of contaminant solution at dif-erent initial concentrations was mixed with 10 g of soil. After 48 hhe equilibrium was reached. Analysis of organic contaminant waserformed by gas chromatography. The amount of organic com-ound adsorbed on soil (qe in mg g−1) was obtained by ultrasonicolvent extraction using methanol as solvent. These data were val-dated by the amount calculated by mass balance derived from thenalysis of the liquid phase. After three extraction steps, pollutantn the methanolic phase obtained after extraction of samples waslose to 95% of the initial pollutant in the soil.
.7. Analytical methods
Hydrogen peroxide concentration in the supernatant waseasured by using a UV-1603 spectrophotometer, supplied by Shi-adzu, after colour development with titanium sulphate technique
22].Residual PhOH in the supernatant was analyzed by an Agi-
ent Technologies 6890N gas chromatograph, fitted with a flameonization detector and equipped with a HP5 high resolution chro-
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
atography column (30 m length × 0.32 mm i.d.). The initial andnal oven temperatures were 110 ◦C and 135 ◦C, respectively. Theeating rate of the program was 5 ◦C min−1. The injector and theetector temperatures were 300 ◦C, and 250 ◦C, respectively. Theitrogen carrier gas flow rate was 25 mL min−1.
PRESSng Journal xxx (2011) xxx–xxx 3
3. Results and discussion
3.1. Remediation of contaminated soils by the Fenton-likeoxidation
Operating conditions of PhOH degradation by Fenton-like reac-tion are collected in Table 1. The initial PhOH concentrations inboth soils are higher than the standard regulatory stablished bythe RD9/2005. Phenol adsorbed amounts increase as the SOM con-tent does, which is due to the contaminants adsorption onto soilsis directly related to the SOM [23–25].
The theoretical H2O2 demand for the total mineralization ofthe sorbed PhOH has been calculated as 911 mg L−1 for S1 and405 mg L−1 for S2, according to the stoichiometric equation (2).
C6H5OH + 14H2O2 → 6CO2 + 17H2O (2)
Therefore, as can be seen in Table 1, H2O2 concentrations, used inthis work, are much higher than the theoretical H2O2 demand, forthe mineralization of the pollutant adsorbed, due to non-specificlosses caused by its decomposition in the presence of reactive mate-rials in soils [26].
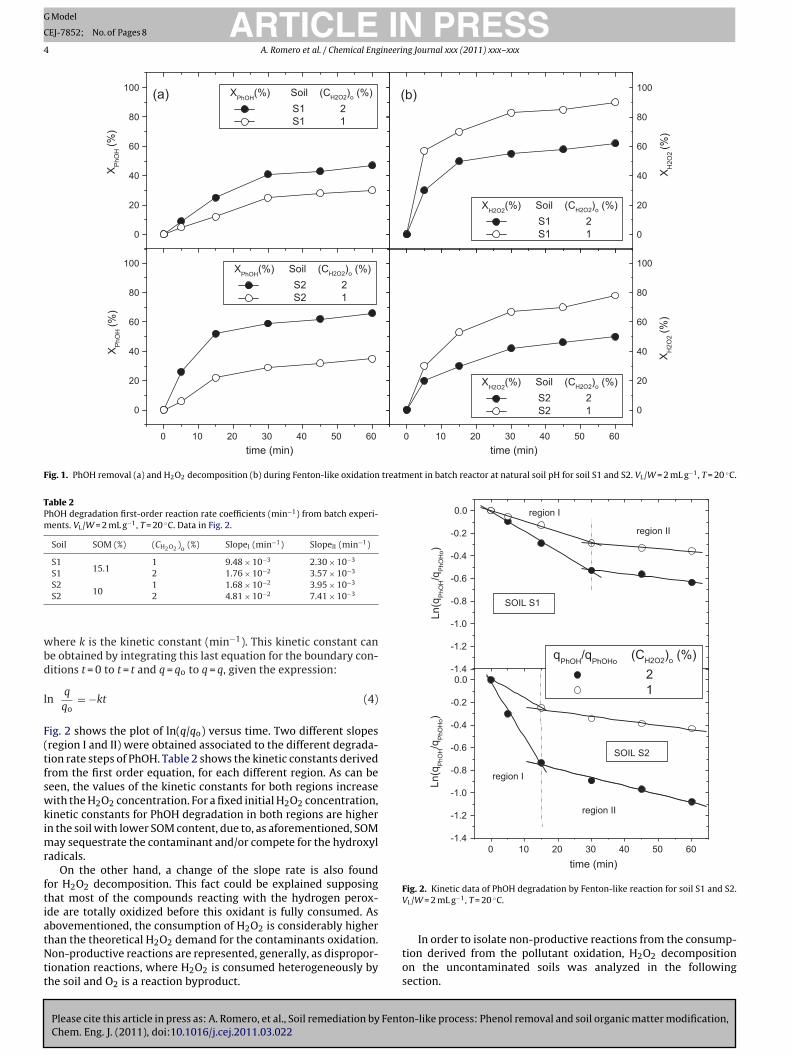
PhOH removal and H2O2 conversion during Fenton oxidationtreatment are shown in Fig. 1. PhOH removal increases with higherconcentrations of H2O2 for both soils. For a fixed H2O2 concentra-tion, PhOH degradation is higher in the soil with lower SOM content.According to the literature, contaminants become sequestrated inthe soil matrix, being sorption strength one of the most impor-tant factors to determine the susceptibility of the contaminantsto remediation processes. Even in soils with low organic content,contaminants may remain entrapment in pores and voids [27].
In this sense, Watts et al. studied the Fenton-like oxidation ofhexachlorobenzene in a silica sand and a natural soil and theyreported that the aggressive Fenton-like reaction conditions mayalter the hexachlorobenzene sorption characteristics and increaseits rate of desorption, due to perhydroxyl radicals promoted thedesorption of hydrophic organic compounds from solid surfaces,resulting in an enhanced coupled desorption–oxidation mecha-nism [28].
On the other hand, Ndjou’ou and Cassidy observed the pro-duction and temporary accumulation of surfactants accompanyingthe treatment of fuel oil-contaminated soil with MFC in labora-tory slurry reactors. The authors hypothesized that the surfactantsproduced were the products of partial oxidation of hydrocarbonsand/or native organics (e.g., carboxylic acids, aldehydes, ketones)having surfactant-like properties. These surfactants can improvedesorption of hydrophobic contaminants in soils, thus enhancingthe ISCO treatment [29].
Both theories support the idea that Fenton-like oxidation mod-ified the sorption properties of the soil, improving the remediationprocess. However, despite this process can favor desorption ofhydrophobic contaminants, some authors have reported that sus-ceptibility of PAH to chemical oxidation was a function of TOC[30]. Moreover, SOM may compete with the contaminant for thehydroxyl radicals (OH•), obtaining lower remediation efficienciesas SOM increases.
The removal rate of PhOH is characterized by a very fast ini-tial rate (t < 30 min for S1 and t < 15 min for S2), in which most ofthe contaminant oxidation takes place, followed by a much slowerdegradation rate, in agreement with the results reported by Kang[11]. The first step may be associated to the fast oxidation of themost accessible or lower entrapped contaminant.
on-like process: Phenol removal and soil organic matter modification,
The following first-order kinetic expression was used to describethe degradation kinetic of PhOH:
−dqPhOH
dt= rPhOH = k · qPhOH (3)
ARTICLE IN PRESSG Model
CEJ-7852; No. of Pages 8
4 A. Romero et al. / Chemical Engineering Journal xxx (2011) xxx–xxx
6050403020100
0
20
40
60
80
100
XH2O2
(%) Soil (CH2O2
)o (%)
S2 2
S2 1
XH
2O
2 (%
)
0
20
40
60
80
100(b)
XH2O2
(%) Soil (CH2O2
)o (%)
S1 2
S1 1
XH
2O
2 (%
)
0
20
40
60
80
100X
PhOH(%) Soil (C
H2O2)
o (%)
S1 2
S1 1
XP
hO
H (
%)
(a)
6050403020100
0
20
40
60
80
100X
PhOH(%) Soil (C
H2O2)
o (%)
S2 2
S2 1
XP
hO
H (
%)
time (min) time (min)
Fig. 1. PhOH removal (a) and H2O2 decomposition (b) during Fenton-like oxidation treatment in batch reactor at natural soil pH for soil S1 and S2. VL/W = 2 mL g−1, T = 20 ◦C.
Table 2PhOH degradation first-order reaction rate coefficients (min−1) from batch experi-ments. VL/W = 2 mL g−1, T = 20 ◦C. Data in Fig. 2.
Soil SOM (%) (CH2O2 )o (%) SlopeI (min−1) SlopeII (min−1)
S115.1
1 9.48 × 10−3 2.30 × 10−3
−2 −3
wbd
l
F(tfswkimr
ftiatNtt
-1.4
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
SOIL S1
region II
L
n(q
PhO
H/q
PhO
Ho)
region I
0 10 20 30 40 50 60
-1.4
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
region II
qPhOH
/qPhOHo
(CH2O2
)o (%)
2
1
Ln
(qP
hO
H/q
PhO
Ho)
time (min)
region I
SOIL S2
S1 2 1.76 × 10 3.57 × 10S2
101 1.68 × 10−2 3.95 × 10−3
S2 2 4.81 × 10−2 7.41 × 10−3
here k is the kinetic constant (min−1). This kinetic constant cane obtained by integrating this last equation for the boundary con-itions t = 0 to t = t and q = qo to q = q, given the expression:
nq
qo= −kt (4)
ig. 2 shows the plot of ln(q/qo) versus time. Two different slopesregion I and II) were obtained associated to the different degrada-ion rate steps of PhOH. Table 2 shows the kinetic constants derivedrom the first order equation, for each different region. As can beeen, the values of the kinetic constants for both regions increaseith the H2O2 concentration. For a fixed initial H2O2 concentration,
inetic constants for PhOH degradation in both regions are highern the soil with lower SOM content, due to, as aforementioned, SOM
ay sequestrate the contaminant and/or compete for the hydroxyladicals.
On the other hand, a change of the slope rate is also foundor H2O2 decomposition. This fact could be explained supposinghat most of the compounds reacting with the hydrogen perox-de are totally oxidized before this oxidant is fully consumed. Asbovementioned, the consumption of H2O2 is considerably higher
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
han the theoretical H2O2 demand for the contaminants oxidation.on-productive reactions are represented, generally, as dispropor-
ionation reactions, where H2O2 is consumed heterogeneously byhe soil and O2 is a reaction byproduct.
Fig. 2. Kinetic data of PhOH degradation by Fenton-like reaction for soil S1 and S2.VL/W = 2 mL g−1, T = 20 ◦C.
on-like process: Phenol removal and soil organic matter modification,
In order to isolate non-productive reactions from the consump-tion derived from the pollutant oxidation, H2O2 decompositionon the uncontaminated soils was analyzed in the followingsection.
ARTICLE IN PRESSG Model
CEJ-7852; No. of Pages 8
A. Romero et al. / Chemical Engineering Journal xxx (2011) xxx–xxx 5
Table 3Content of TC, C fractions pools, TOC, SOM and structural parameters of S1 and S2 before (BO) and after (AO) oxidation. (CH2O2 )o = 2%.
Soil TC (%) Labile and recalcitrant pools TOCTotal (%) SOM (%) SgN2 (m2/g) SCO2DR (m2/g) Vp (cm3/g) r̄mp (Å) wheeler
LP I (%) LP II (%) R (%)
S1BO 7.75 ± 0.01 2.05 0.70 3.53 6.28 ± 0.01 15.1S1AO 7.66 ± 0.01 1.68 0.83 3.62 6.13 ± 0.01 16.4S2BO 5.89 ± 0.01 1.52 0.67 2.17 4.36 ± 0.01 10S2AO 5.35 ± 0.01 0.98 0.72 2.19 3.89 ± 0.01 11.3
0.0
0.2
0.4
0.6
0.8
1.0
6050403020100
0.0
0.2
0.4
0.6
0.8
1.0
O2 production (mol/mol)
S1
S2
2·(
nO
2/n
H2
O2
o)
(mo
l/m
ol)
XH
2O
2
XH2O
2
Fp
3
uvOco
idcta
pitSmhr
H
p[
Aa0vtha
time (min)
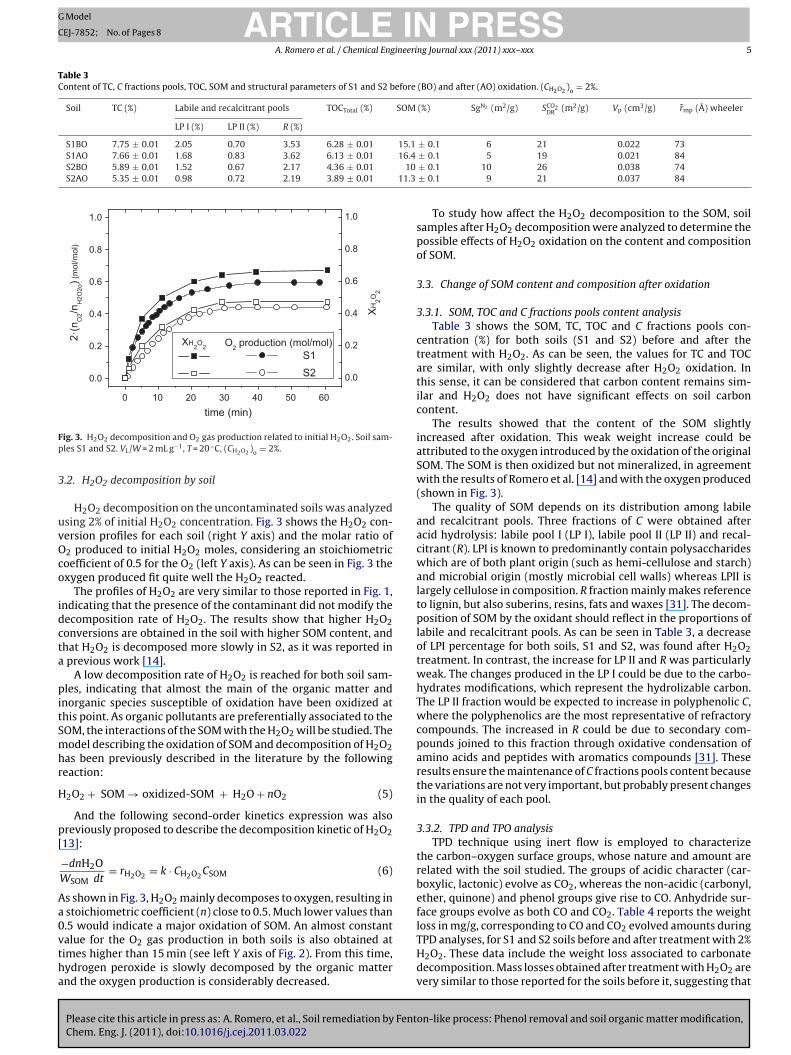
ig. 3. H2O2 decomposition and O2 gas production related to initial H2O2. Soil sam-les S1 and S2. VL/W = 2 mL g−1, T = 20 ◦C, (CH2O2 )o = 2%.
.2. H2O2 decomposition by soil
H2O2 decomposition on the uncontaminated soils was analyzedsing 2% of initial H2O2 concentration. Fig. 3 shows the H2O2 con-ersion profiles for each soil (right Y axis) and the molar ratio of2 produced to initial H2O2 moles, considering an stoichiometricoefficient of 0.5 for the O2 (left Y axis). As can be seen in Fig. 3 thexygen produced fit quite well the H2O2 reacted.
The profiles of H2O2 are very similar to those reported in Fig. 1,ndicating that the presence of the contaminant did not modify theecomposition rate of H2O2. The results show that higher H2O2onversions are obtained in the soil with higher SOM content, andhat H2O2 is decomposed more slowly in S2, as it was reported inprevious work [14].
A low decomposition rate of H2O2 is reached for both soil sam-les, indicating that almost the main of the organic matter and
norganic species susceptible of oxidation have been oxidized athis point. As organic pollutants are preferentially associated to theOM, the interactions of the SOM with the H2O2 will be studied. Theodel describing the oxidation of SOM and decomposition of H2O2
as been previously described in the literature by the followingeaction:
2O2 + SOM → oxidized-SOM + H2O + nO2 (5)
And the following second-order kinetics expression was alsoreviously proposed to describe the decomposition kinetic of H2O213]:
−dnH2OWSOM dt
= rH2O2 = k · CH2O2 CSOM (6)
s shown in Fig. 3, H2O2 mainly decomposes to oxygen, resulting instoichiometric coefficient (n) close to 0.5. Much lower values than
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
.5 would indicate a major oxidation of SOM. An almost constantalue for the O2 gas production in both soils is also obtained atimes higher than 15 min (see left Y axis of Fig. 2). From this time,ydrogen peroxide is slowly decomposed by the organic matternd the oxygen production is considerably decreased.
± 0.1 6 21 0.022 73± 0.1 5 19 0.021 84± 0.1 10 26 0.038 74± 0.1 9 21 0.037 84
To study how affect the H2O2 decomposition to the SOM, soilsamples after H2O2 decomposition were analyzed to determine thepossible effects of H2O2 oxidation on the content and compositionof SOM.
3.3. Change of SOM content and composition after oxidation
3.3.1. SOM, TOC and C fractions pools content analysisTable 3 shows the SOM, TC, TOC and C fractions pools con-
centration (%) for both soils (S1 and S2) before and after thetreatment with H2O2. As can be seen, the values for TC and TOCare similar, with only slightly decrease after H2O2 oxidation. Inthis sense, it can be considered that carbon content remains sim-ilar and H2O2 does not have significant effects on soil carboncontent.
The results showed that the content of the SOM slightlyincreased after oxidation. This weak weight increase could beattributed to the oxygen introduced by the oxidation of the originalSOM. The SOM is then oxidized but not mineralized, in agreementwith the results of Romero et al. [14] and with the oxygen produced(shown in Fig. 3).
The quality of SOM depends on its distribution among labileand recalcitrant pools. Three fractions of C were obtained afteracid hydrolysis: labile pool I (LP I), labile pool II (LP II) and recal-citrant (R). LPI is known to predominantly contain polysaccharideswhich are of both plant origin (such as hemi-cellulose and starch)and microbial origin (mostly microbial cell walls) whereas LPII islargely cellulose in composition. R fraction mainly makes referenceto lignin, but also suberins, resins, fats and waxes [31]. The decom-position of SOM by the oxidant should reflect in the proportions oflabile and recalcitrant pools. As can be seen in Table 3, a decreaseof LPI percentage for both soils, S1 and S2, was found after H2O2treatment. In contrast, the increase for LP II and R was particularlyweak. The changes produced in the LP I could be due to the carbo-hydrates modifications, which represent the hydrolizable carbon.The LP II fraction would be expected to increase in polyphenolic C,where the polyphenolics are the most representative of refractorycompounds. The increased in R could be due to secondary com-pounds joined to this fraction through oxidative condensation ofamino acids and peptides with aromatics compounds [31]. Theseresults ensure the maintenance of C fractions pools content becausethe variations are not very important, but probably present changesin the quality of each pool.
3.3.2. TPD and TPO analysisTPD technique using inert flow is employed to characterize
the carbon–oxygen surface groups, whose nature and amount arerelated with the soil studied. The groups of acidic character (car-boxylic, lactonic) evolve as CO2, whereas the non-acidic (carbonyl,ether, quinone) and phenol groups give rise to CO. Anhydride sur-face groups evolve as both CO and CO2. Table 4 reports the weight
on-like process: Phenol removal and soil organic matter modification,
loss in mg/g, corresponding to CO and CO2 evolved amounts duringTPD analyses, for S1 and S2 soils before and after treatment with 2%H2O2. These data include the weight loss associated to carbonatedecomposition. Mass losses obtained after treatment with H2O2 arevery similar to those reported for the soils before it, suggesting that

ARTICLE IN PRESSG Model
CEJ-7852; No. of Pages 8
6 A. Romero et al. / Chemical Engineering Journal xxx (2011) xxx–xxx
Table 4Mass loss in mg g−1 during TPD, TPO and TGA analyses in both inert and air flow, forthe soils S1 and S2, before (BO) and after (AO) oxidation. (CH2O2 )o = 2%.
TGA (mg g−1) TPD (mg g−1) TPO (mg g−1)
Inert flow Air flow Inert flow Air flow
S1BO 182 218 211 375S1AO 203 246 227 425S2BO 168 146 148 236S2AO 175 149 149 224
Ft
tc
oima
ipb(f8t4Tcmw
fTsos
ig. 4. CO2 evolution as a function of temperature for soil S1, before and afterreatment with 2% of H2O2, (a) during TPD analysis and (b) during TPO analysis.
he treatment does not significantly modify the soil organic matterontent.
CO evolution (not shown), mainly associated to decompositionf carbonyl and quinone groups at high temperatures (700–900 ◦C),s lower than the one observed for CO2. Hydrogen peroxide treat-
ent on both soils produces the partial removal of these carbonylnd quinone groups.
Fig. 4a shows CO2 evolution as a function of temperature innert flow for S1 before and after treatment with 2% of H2O2. Bothrofiles are very similar, characterized by decomposition of car-oxyl and lactone groups between 300 and 500 ◦C and anhydride500–700 ◦C), present in the fatty acid, peptides, carbohydrates andulvic and humic acids of the SOM, and carbonates between 700 and00 ◦C. The peak associated to carbonates decomposition remainshe same after H2O2 treatment and corresponds approximately to5 and 33 mg/g of CO2 evolved for soils S1 and S2, respectively.hese results suggest that both the amount and distribution ofarbon–oxygen groups of soil organic matter are not significantlyodified after treatment with hydrogen peroxide, in agreementith the results of Bissey et al. [32].
CO2 evolution as a function of temperature during TPO analysisor soil S1 before and after H O treatment, is shown in Fig. 4b.
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
2 2able 3 also reports mass loss during the TPO analyses for bothoils before and after H2O2 treatment. In the presence of oxygen, therganic matter is almost completely burnt, yielding carbon dioxide,ome CO and water. This allows the organic carbon content to be
Fig. 5. Mass loss rate as a function of temperature for soil S1 before and after treat-ment with 2% of H2O2, (a) in an inert flow and (b) in an air flow.
determined from the amount of carbon dioxide between 250 and650 ◦C. Similar results are obtained by the TGA experiments in airatmosphere (Fig. 4b) [33].
The CO2 mass evolved from the TPO experiment shown inTable 4 for S2 before and after H2O2 treatment are very simi-lar. However, S1 presents an appreciable CO2 evolution after thetreatment, probably due to the incorporation of oxygen in theorganic matter during the treatment with H2O2, as it was pre-viously reported [14]. This trend is not observed in S2 probablydue to the lower organic matter content of this soil. From thecomparison of the CO2 profiles presented in Fig. 4a and b can bededuced that the CO2 peak at low temperatures can be associatedto oxidative decomposition of simple organic matter as fatty acids,peptides, carbohydrates (cellulose and hemicellulose). CO2 evolu-tion between 350 and 600 ◦C can be related to fulvic and humicacids oxidation. While the peak at high temperatures is related toboth carbonates decomposition and refractory organic matter com-bustion [34]. It is important to point out that CO2 evolution up to600 ◦C is considerably higher for S1 than the one observed for S2(not shown) and S1 presents higher organic matter content thanS2. These results indicate that S1 contains higher contributions ofboth simple organic matter and fulvic and humic acid than S2. Verysimilar profiles were obtained for both soils before and after H2O2treatment, indicating that the mild conditions used in this treat-ment does not produce any significant degradation of the organicmatter.
3.3.3. TG analysis
on-like process: Phenol removal and soil organic matter modification,
Fig. 5a shows the mass loss rate as a function of temperature forsoil S1 before and after treatment with 2% H2O2, in an inert flowof nitrogen. The profiles obtained by TG analysis are very similar tothose obtained by TPD analysis.
IN PRESSG
C
ineering Journal xxx (2011) xxx–xxx 7
ottatiaa3dg
sbfiystsuI3s[ttibttc
3
ffcmhvttotbso
3
hrscsfnatc(o
d
0.00
0.02
0.04
0.06
0.08
0.10
qe (
mg
·g-1)
Treat. S1: qe=1.57·10
-3·C
0.80
e
original S1
treated S1
Orig. S1: qe=2.13·10
-3·C
0.81
e
a
100806040200
0.00
0.02
0.04
0.06
0.08
0.10
qe (
mg
·g-1)
Ce (mg·L
-1)
Treat. S2: qe=1.17·10
-3·C
0.58
e
original S2
treated S2
Orig. S2: qe=1.66·10
-3·C
0.64
e
b
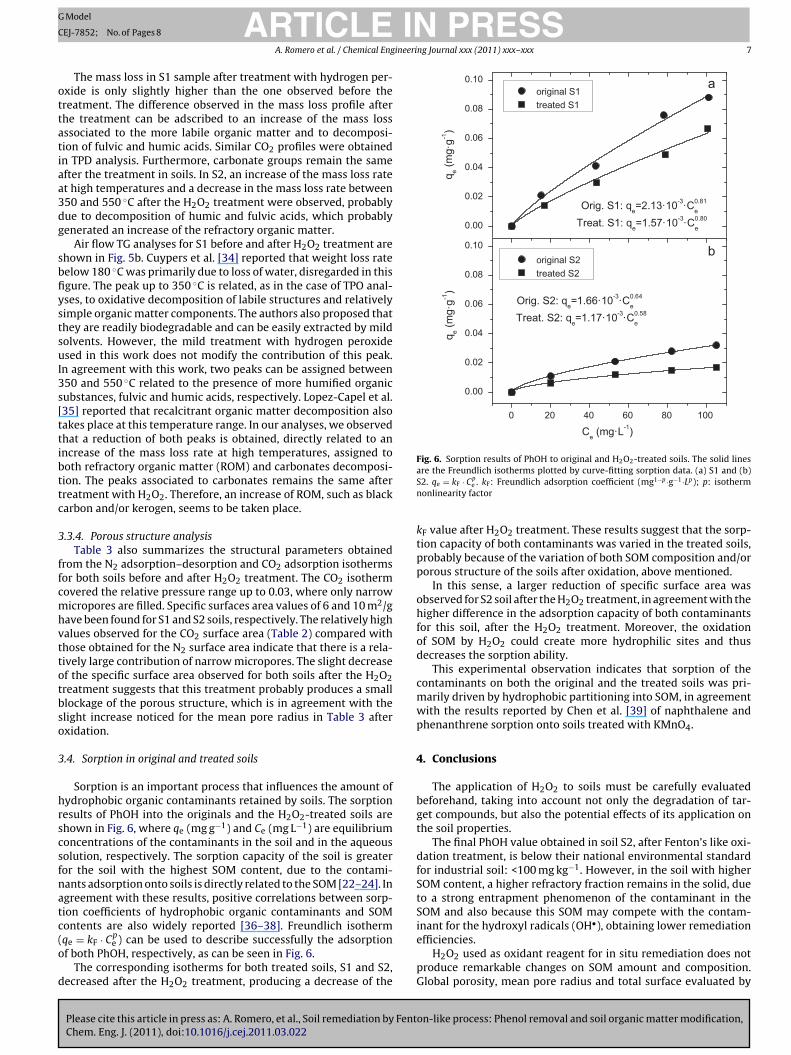
Fig. 6. Sorption results of PhOH to original and H O -treated soils. The solid lines
ARTICLEModel
EJ-7852; No. of Pages 8
A. Romero et al. / Chemical Eng
The mass loss in S1 sample after treatment with hydrogen per-xide is only slightly higher than the one observed before thereatment. The difference observed in the mass loss profile afterhe treatment can be adscribed to an increase of the mass lossssociated to the more labile organic matter and to decomposi-ion of fulvic and humic acids. Similar CO2 profiles were obtainedn TPD analysis. Furthermore, carbonate groups remain the samefter the treatment in soils. In S2, an increase of the mass loss ratet high temperatures and a decrease in the mass loss rate between50 and 550 ◦C after the H2O2 treatment were observed, probablyue to decomposition of humic and fulvic acids, which probablyenerated an increase of the refractory organic matter.
Air flow TG analyses for S1 before and after H2O2 treatment arehown in Fig. 5b. Cuypers et al. [34] reported that weight loss rateelow 180 ◦C was primarily due to loss of water, disregarded in thisgure. The peak up to 350 ◦C is related, as in the case of TPO anal-ses, to oxidative decomposition of labile structures and relativelyimple organic matter components. The authors also proposed thathey are readily biodegradable and can be easily extracted by mildolvents. However, the mild treatment with hydrogen peroxidesed in this work does not modify the contribution of this peak.
n agreement with this work, two peaks can be assigned between50 and 550 ◦C related to the presence of more humified organicubstances, fulvic and humic acids, respectively. Lopez-Capel et al.35] reported that recalcitrant organic matter decomposition alsoakes place at this temperature range. In our analyses, we observedhat a reduction of both peaks is obtained, directly related to anncrease of the mass loss rate at high temperatures, assigned tooth refractory organic matter (ROM) and carbonates decomposi-ion. The peaks associated to carbonates remains the same afterreatment with H2O2. Therefore, an increase of ROM, such as blackarbon and/or kerogen, seems to be taken place.
.3.4. Porous structure analysisTable 3 also summarizes the structural parameters obtained
rom the N2 adsorption–desorption and CO2 adsorption isothermsor both soils before and after H2O2 treatment. The CO2 isothermovered the relative pressure range up to 0.03, where only narrowicropores are filled. Specific surfaces area values of 6 and 10 m2/g
ave been found for S1 and S2 soils, respectively. The relatively highalues observed for the CO2 surface area (Table 2) compared withhose obtained for the N2 surface area indicate that there is a rela-ively large contribution of narrow micropores. The slight decreasef the specific surface area observed for both soils after the H2O2reatment suggests that this treatment probably produces a smalllockage of the porous structure, which is in agreement with thelight increase noticed for the mean pore radius in Table 3 afterxidation.
.4. Sorption in original and treated soils
Sorption is an important process that influences the amount ofydrophobic organic contaminants retained by soils. The sorptionesults of PhOH into the originals and the H2O2-treated soils arehown in Fig. 6, where qe (mg g−1) and Ce (mg L−1) are equilibriumoncentrations of the contaminants in the soil and in the aqueousolution, respectively. The sorption capacity of the soil is greateror the soil with the highest SOM content, due to the contami-ants adsorption onto soils is directly related to the SOM [22–24]. Ingreement with these results, positive correlations between sorp-ion coefficients of hydrophobic organic contaminants and SOM
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
ontents are also widely reported [36–38]. Freundlich isothermqe = kF · Cp
e ) can be used to describe successfully the adsorptionf both PhOH, respectively, as can be seen in Fig. 6.
The corresponding isotherms for both treated soils, S1 and S2,ecreased after the H2O2 treatment, producing a decrease of the
2 2
are the Freundlich isotherms plotted by curve-fitting sorption data. (a) S1 and (b)S2. qe = kF · Cp
e . kF: Freundlich adsorption coefficient (mg1−p·g−1·Lp); p: isothermnonlinearity factor
kF value after H2O2 treatment. These results suggest that the sorp-tion capacity of both contaminants was varied in the treated soils,probably because of the variation of both SOM composition and/orporous structure of the soils after oxidation, above mentioned.
In this sense, a larger reduction of specific surface area wasobserved for S2 soil after the H2O2 treatment, in agreement with thehigher difference in the adsorption capacity of both contaminantsfor this soil, after the H2O2 treatment. Moreover, the oxidationof SOM by H2O2 could create more hydrophilic sites and thusdecreases the sorption ability.
This experimental observation indicates that sorption of thecontaminants on both the original and the treated soils was pri-marily driven by hydrophobic partitioning into SOM, in agreementwith the results reported by Chen et al. [39] of naphthalene andphenanthrene sorption onto soils treated with KMnO4.
4. Conclusions
The application of H2O2 to soils must be carefully evaluatedbeforehand, taking into account not only the degradation of tar-get compounds, but also the potential effects of its application onthe soil properties.
The final PhOH value obtained in soil S2, after Fenton’s like oxi-dation treatment, is below their national environmental standardfor industrial soil: <100 mg kg−1. However, in the soil with higherSOM content, a higher refractory fraction remains in the solid, dueto a strong entrapment phenomenon of the contaminant in theSOM and also because this SOM may compete with the contam-inant for the hydroxyl radicals (OH•), obtaining lower remediation
on-like process: Phenol removal and soil organic matter modification,
efficiencies.H2O2 used as oxidant reagent for in situ remediation does not
produce remarkable changes on SOM amount and composition.Global porosity, mean pore radius and total surface evaluated by

ING
C
8 ineeri
nH
cci
A
fpS
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
ARTICLEModel
EJ-7852; No. of Pages 8
A. Romero et al. / Chemical Eng
itrogen adsorption show only slight changes after treatment with2O2.
However, changes on the oxidation state of the SOM surfaceould alter the hydrophobicity of the SOM surface. These changesould also explain the decrease noticed for the adsorption capabil-ty of the soil after treatment with H2O2.
cknowledgements
The authors acknowledge financial support for this researchrom the Comunidad Autónoma de Madrid provided throughoutroject CARESOIL (S2009AMB-1648) and from Spanish Ministry ofcience and innovation, project CTM2006-317.
eferences
[1] J.P. Gao, J. Maguhn, P. Spitzauer, A. Kettrup, Sorption of pesticides in thesediment of the Teufelesweither pond (southern Germany). I. Equilibriumassessments, effect of organic carbon content and pH, Water Res. 32 (1998)1662–1672.
[2] F.I. Khan, T. Husain, R. Hejazi, An overview and analysis of site remediationtechnologies, J. Environ. Manage. 7 (2004) 122–195.
[3] M.T. Alcántara, J. Gómez, M. Pazos, M.A. Sanromán, Combined treatmentof PAHs contaminated soils using the sequence extraction with surfactant-electrochemical degradation, Chemosphere 70 (2008) 1438–1444.
[4] W. Huang, W.J. Weber Jr., A distributed reactivity model for sorption by soils andsediments. 10. Relationships between desorption, hysteresis, and the chemicalcharacteristics of organic domains, Environ. Sci. Technol. 31 (1997) 2562–2569.
[5] N.K. Daud, M.A. Ahmad, B.H. Hameed, Decolorization of Acid Red 1 dye solutionby Fenton-like process using Fe–montmorillonite K10 catalyst, Chem. Eng. J.165 (2010) 111–116.
[6] H.-W. Sun, Q.-S. Yan, Influence of Fenton oxidation on soil organic matter andits sorption and desorption of pyrene, J. Hazard. Mater. 144 (2007) 164–170.
[7] R.J. Watts, P.C. Stanton, J. Howsawkeng, A.L. Teel, Mineralization of a sorbedpolycyclic aromatic hydrocarbon in two soils using catalyzed hydrogen perox-ide, Water Res. 36 (2002) 4283–4292.
[8] V. Flotron, C. Delteil, Y. Padellec, V. Camel, Removal of sorbed polycyclic aro-matic hydrocarbons from soil, sludge and sediment samples using the Fenton’sreagent process, Chemosphere 59 (2005) 1427–1437.
[9] M. Lu, Z.Z. Zhang, W. Qiao, Y.M. Guan, M. Xiao, C. Peng, Removal of residual con-taminants in petroleum-contaminated soil by Fenton-like oxidation, J. Hazard.Mater. 179 (2010) 604–611.
10] A. Rastogi, S.R. Al-Abed, D.D. Dionysiou, Effect of inorganic, synthetic and nat-urally occurring chelating agents on Fe(II) mediated advanced oxidation ofchlorophenols, Water Res. 43 (2009) 684–694.
11] N. Kang, I. Hua, Enhanced chemical oxidation of aromatic hydrocarbons in soilsystems, Chemosphere 61 (2005) 909–922.
12] S.-H. Kong, R.J. Watts, J.-H. Choi, Treatment of petroleum-contaminated soilsusing iron mineral catalyzed hydrogen peroxide, Chemosphere 37 (1998)1473–1482.
13] R. Baciocchi, M.R. Boni, L. D’Aprile, Hydrogen peroxide lifetime as an indicatorof the efficiency of 3-chlorophenol Fenton’s and Fenton-like oxidation in soils,J. Hazard. Mater. B96 (2003) 305–329.
14] A. Romero, A. Santos, F. Vicente, S. Rodriguez, A. Lopez Lafuente, In situ oxi-dation remediation technologies: kinetic of hydrogen peroxide decomposition
Please cite this article in press as: A. Romero, et al., Soil remediation by FentChem. Eng. J. (2011), doi:10.1016/j.cej.2011.03.022
on soil organic matter, J. Hazard. Mater. 170 (2009) 627–632.15] C. Sirguey, P.T. Souza e Silva, C. Schwartz, M.O. Siminnot, Impact of chemical
oxidation on soil quality, Chemosphere 72 (2008) 282–289.16] C.K.-J. Yeh, Y.-A. Kao, C.-P. Cheng, Oxidation of chlorophenols in soil at nat-
ural pH by catalyzed hydrogen peroxide: the effect of soil organic matter,Chemosphere 46 (2002) 67–73.
[
[
PRESSng Journal xxx (2011) xxx–xxx
17] RD 9/2005, relationship of potentially soil polluting activities and the criteriaand standards for the declaration of contaminated soils, BOE no. 15, 2005, pp.1833–1843.
18] A.L. Lafuente, C. González, J.R. Quintana, A. Vázquez, A. Romero, Mobility ofheavy metals in poorly developed carbonate soils in the Mediterranean region,Geoderma 145 (2008) 238–244.
19] L.P. Van Reeuwijk, Procedure for Soil Analysis, fourth ed., International SoilReference and Information Center, Wageningen, Netherlands, 1993.
20] P. Rovira, V.R. Vallejo, Labile and recalcitrant pools of carbon and nitrogenin organic matter decomposing at different depths in soil: an acid hydrolysisapproach, Geoderma 107 (2002) 109–141.
21] R. Mecozzi, L. Di Palma, C. Merli, Experimental in situ chemical peroxidation ofatrazine in contaminated soil, Chemosphere 62 (2006) 1481–1489.
22] W.C. Schumb, C.N. Stratterfield, R.L. Wentworth, Hydrogen Peroxide, AmericanChemical Society, Rienholt Publishing, New York, 1995.
23] C.T. Chiou, S.E. McGroddy, D.E. Kile, Partition characteristics of polycyclic aro-matic hydrocarbons on soils and sediments, Environ. Sci. Technol. 32 (1998)264–269.
24] W.J. Weber Jr., P.M. McGinley, L.E. Katz, A distributed reactivity model for sorp-tion by soils and sediments. 1. Conceptual basis and equilibrium assessments,Environ. Sci. Technol. 26 (1992) 1955–1962.
25] A.S. Gunasekara, B.S. Xing, Sorption and desorption of naphthalene by soilorganic matter: importance of aromatic and aliphatic components, J. Environ.Qual. 32 (2003) 240–246.
26] J.X. Ravikumar, M.D. Gurol, Chemical oxidation of chlorinated organics byhydrogen peroxide in the presence of sand, Environ. Sci. Technol. 28 (1994)394–400.
27] S. Jonsson, Y. Persson, S. Frenkki, B. Van Bavel, S. Lundstedt, P. Haglund, M.Tysklind, Degradation of polycyclic aromatic hydrocarbons (PAHs) in contam-inated soils by Fenton’s reagent: a multivariate evaluation of the importanceof soil characteristics and PAH properties, J. Hazard. Mater. 149 (2007) 86–96.
28] R.J. Watts, S. Kong, M. Dippre, W.T. Barnes, Oxidation of sorbed hexachloroben-zene in soils using catalyzed hydrogen peroxide, J. Hazard. Mater. 39 (1994)33–47.
29] A.C. Ndjou’ou, D.P. Cassidy, Surfactant production accompanying the modifiedFenton oxidation of hydrocarbons in soil, Chemosphere 65 (2006) 1610–1615.
30] B.W. Bogan, V. Trbovic, Effect of sequestration on PAH degradability with Fen-ton’s reagent: roles of total organic carbon, humin, and soil porosity, J. Hazard.Mater. B100 (2003) 285–300.
31] P. Rovira, V.R. Vallejo, Labile, recalcitrant, and inert organic matter in Mediter-ranean forest soils, Soil Biol. Biochem. 39 (2007) 202–215.
32] L.L. Bissey, J.L. Smith, R.J. Watts, Soil organic matter-hydrogen peroxidedynamics in the treatment of contaminated soils and groundwater using cat-alyzed H2O2 propagations (modified Fenton’s reagent), Water Res. 40 (2007)2477–2484.
33] F. Gaál, I. Szöllósy, M. Arnold, F. Paulik, Determination of the organic mat-ter, metal carbonate and mobile water in soils, J. Therm. Pyrol. 42 (1994)1007–1016.
34] C. Cuypers, T. Grotenhuis, K.G.J. Nierop, E. Maneiro Franco, A. de Jager, W.Rulkens, Amorphous and condensed organic matter domains: the effect ofpersulfate oxidation on the composition of soil/sediment organic matter,Chemosphere 48 (2002) 919–931.
35] E. Lopez-Capel, G.D. Abbott, K.M. Thomas, D.A.C. Manning, Coupling of thermalanalysis with quadrupole mass spectrometry and isotope ratio mass spectrom-etry for simultaneous determination of evolved gases and their carbon isotopiccomposition, J. Anal. Appl. Pyrol. 75 (2006) 82–89.
36] A. Farenhorst, Importance of soil organic matter fractions in soil-landscape andregional assessments of pesticide sorption and leaching in soil, Soil Sci. Soc. Am.J. 70 (2006) 1005–1012.
37] B.T. Mader, K.U. Goss, S.J. Eisenreich, Sorption of nonionic, hydrophobic organic
on-like process: Phenol removal and soil organic matter modification,
chemicals to mineral surfaces, Environ. Sci. Technol. 31 (1997) 1079–1086.38] S. Mitra, P.C. Bhowmik, B. Xing, Sorption of isoxaflutole by five different
soils varying in physical and chemical properties, Pestic. Sci. 55 (1999)935–942.
39] W. Chen, L. Hou, X. Luo, L. Zhu, Effects of chemical oxidation on sorption and des-orption of PAHs in typical Chinese soils, Environ. Pollut. 157 (2009) 1894–1903.




















