
Sign Learning Kink-based (SiLK) Quantum Monte Carlo for ...
10
Louisiana State University Louisiana State University LSU Digital Commons LSU Digital Commons Faculty Publications Department of Physics & Astronomy 1-7-2016 Sign Learning Kink-based (SiLK) Quantum Monte Carlo for Sign Learning Kink-based (SiLK) Quantum Monte Carlo for molecular systems molecular systems Xiaoyao Ma Louisiana State University Randall W. Hall Dominican University of California Frank Löffler Louisiana State University Karol Kowalski Environmental Molecular Sciences Laboratory Kiran Bhaskaran-Nair Louisiana State University See next page for additional authors Follow this and additional works at: https://digitalcommons.lsu.edu/physics_astronomy_pubs Recommended Citation Recommended Citation Ma, X., Hall, R., Löffler, F., Kowalski, K., Bhaskaran-Nair, K., Jarrell, M., & Moreno, J. (2016). Sign Learning Kink-based (SiLK) Quantum Monte Carlo for molecular systems. Journal of Chemical Physics, 144 (1) https://doi.org/10.1063/1.4939145 This Article is brought to you for free and open access by the Department of Physics & Astronomy at LSU Digital Commons. It has been accepted for inclusion in Faculty Publications by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected].
-
Upload
khangminh22 -
Category
Documents
-
view
4 -
download
0
Transcript of Sign Learning Kink-based (SiLK) Quantum Monte Carlo for ...

Louisiana State University Louisiana State University
LSU Digital Commons LSU Digital Commons
Faculty Publications Department of Physics & Astronomy
1-7-2016
Sign Learning Kink-based (SiLK) Quantum Monte Carlo for Sign Learning Kink-based (SiLK) Quantum Monte Carlo for
molecular systems molecular systems
Xiaoyao Ma Louisiana State University
Randall W. Hall Dominican University of California
Frank Löffler Louisiana State University
Karol Kowalski Environmental Molecular Sciences Laboratory
Kiran Bhaskaran-Nair Louisiana State University
See next page for additional authors
Follow this and additional works at: https://digitalcommons.lsu.edu/physics_astronomy_pubs
Recommended Citation Recommended Citation Ma, X., Hall, R., Löffler, F., Kowalski, K., Bhaskaran-Nair, K., Jarrell, M., & Moreno, J. (2016). Sign Learning Kink-based (SiLK) Quantum Monte Carlo for molecular systems. Journal of Chemical Physics, 144 (1) https://doi.org/10.1063/1.4939145
This Article is brought to you for free and open access by the Department of Physics & Astronomy at LSU Digital Commons. It has been accepted for inclusion in Faculty Publications by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected].

Authors Authors Xiaoyao Ma, Randall W. Hall, Frank Löffler, Karol Kowalski, Kiran Bhaskaran-Nair, Mark Jarrell, and Juana Moreno
This article is available at LSU Digital Commons: https://digitalcommons.lsu.edu/physics_astronomy_pubs/3575

THE JOURNAL OF CHEMICAL PHYSICS 144, 014101 (2016)
Sign Learning Kink-based (SiLK) Quantum Monte Carlofor molecular systems
Xiaoyao Ma,1 Randall W. Hall,2,3 Frank Löffler,4 Karol Kowalski,5 Kiran Bhaskaran-Nair,1,4
Mark Jarrell,1,4 and Juana Moreno1,41Department of Physics and Astronomy, Louisiana State University, Baton Rouge, Louisiana 70803, USA2Department of Natural Sciences and Mathematics, Dominican University of California, San Rafael,California 94901, USA3Department of Chemistry, Louisiana State University, Baton Rouge, Louisiana 70803, USA4Center for Computation and Technology, Louisiana State University, Baton Rouge, Louisiana 70803, USA5William R. Wiley Environmental Molecular Sciences Laboratory, Battelle, Pacific Northwest NationalLaboratory, Richland, Washington 99352, USA
(Received 31 July 2015; accepted 16 December 2015; published online 5 January 2016)
The Sign Learning Kink (SiLK) based Quantum Monte Carlo (QMC) method is used to calculatethe ab initio ground state energies for multiple geometries of the H2O, N2, and F2 molecules. Themethod is based on Feynman’s path integral formulation of quantum mechanics and has two stages.The first stage is called the learning stage and reduces the well-known QMC minus sign problemby optimizing the linear combinations of Slater determinants which are used in the second stage, aconventional QMC simulation. The method is tested using different vector spaces and compared to theresults of other quantum chemical methods and to exact diagonalization. Our findings demonstratethat the SiLK method is accurate and reduces or eliminates the minus sign problem. C 2016 AIPPublishing LLC. [http://dx.doi.org/10.1063/1.4939145]
I. INTRODUCTION
The development of accurate and computationallytractable ab initio methods for studying correlated electronicsystems ranging from single molecules to bulk materials1 isan area of wide interest. Feynman’s path integral formulationof quantum mechanics2 has long attracted attention due toits ability to include exact correlation and finite temperatureeffects, as well as providing a method that can simultaneouslytreat electronic and geometric degrees of freedom. Thepath integral formulation is one of a number of methodscommonly referred to as Quantum Monte Carlo (QMC)-basedalgorithms.3
In general, the use of QMC-based algorithms are hinderedby the so-called minus sign problem in which the fluctuatingsign of the fermionic density matrix leads to statisticalerrors that scale exponentially with inverse temperatureand system size. The minus sign problem4,5 remains agreat challenge in condensed matter physics and quantumchemistry.
In quantum chemistry, there are a number of methodsused to include electron correlation. Commonly used methodsinclude density functional theory (DFT),6 configurationinteraction (CI),7 many body perturbation theory (MBPT),8–10
and coupled cluster (CC).11–18 The CC method has beenregarded as the “gold” standard.11 These approaches, whilevery useful, have well-known deficiencies such as theapproximate inclusion of correlation (DFT), size inconsistency(truncated CI, such as with single and double excitations orwith single, double, and triple excitations), or non-variationalenergies (CC). Therefore, it is important to investigatealternative approaches.
There are three major numerical methods used tostudy strongly correlated many body systems. These areexact diagonalization, density matrix renormalization group(DMRG),19 and QMC. Exact diagonalization is only feasiblefor small systems since it scales exponentially with thesystem size. DMRG has become useful for certain classesof molecules with an order of 50 strongly correlatedelectrons.20–22
The Monte Carlo method was first introduced anddeveloped by Fermi, Teller, and Metropolis.23–25 QMC, unlikeexact diagonalization and DMRG, is a scalable method thatcan be applied to multi-dimensional lattice systems. However,QMC does have the minus sign problem in fermionic andfrustrated quantum systems.
A variety of methods have been proposed to alleviate theminus sign problem in QMC. These include auxiliary fieldMonte Carlo,26 shifted contour auxiliary field Monte Carlo,27
and fixed node diffusion Monte Carlo.3,28 More recently, aresummation path integral approach,29,30 which is similar tothe SiLK method, phaseless auxiliary-field QMC,31 a finitetemperature version of diffusion Monte Carlo,32,33 and fullconfiguration interaction QMC34,35 have been developed.
The Sign-Learning Kink (SiLK) QMC algorithmoriginally developed by Hall36,37 can be used to overcomethe minus sign problem. SiLK has previously been used tostudy the 3 × 3 Hubbard model and atoms using a small basisset.36,37 An approximate version of the method has been usedto study small molecules.38 This method uses a novel learningprocess to overcome the minus sign problem.
The goal of this work is to investigate the ability ofthe SiLK method to reduce the sign problem and accuratelycalculate potential energy surfaces in model systems with
0021-9606/2016/144(1)/014101/8/$30.00 144, 014101-1 © 2016 AIP Publishing LLC

014101-2 Ma et al. J. Chem. Phys. 144, 014101 (2016)
relatively small basis sets. Investigation of the scalability ofthe method is left for future work. Therefore, SiLK QMCcalculations are performed on H2O, N2, and F2 at a numberof different geometries. The results of the calculations arecompared to the results to exact diagonalization and a varietyof quantum chemistry methods and demonstrate that the SiLKmethod is accurate and that it reduces the minus sign problemfor all geometries.
II. SILK FORMALISM AND ALGORITHM
A. SiLK formalism
Assume there are a finite set of states composed of Slaterdeterminants {αi} formed from orthogonal, one electron spinorbitals. With Hamiltonian, H , and β = 1/kBT , the canonicalpartition function Q can be written as
Q = Tr�e−βH
=
j
α j |e−βH |α j
�. (1)
Using
e−βH = (e−βH/P)P, (2)
and the identity
1 =ji
|α ji⟩⟨α ji |, (3)
the partition function becomes
Q =
j1, j2, ..., jP
α j1
�����exp
(− β
PH
) ����� α j2
×
α j2
�����exp
(− β
PH
) ����� α j3
· · ·
×α jP
�����exp
(− β
PH
) ����� α j1
. (4)
P is introduced as a discretization variable that allowsfor the evaluation of the matrix elements by expanding theexponential, vide infra. For a given set of {α ji}, some ofthe matrix elements in Eq. (4) may be diagonal. Thus, termsappearing in the summand may be classified by the numberof off-diagonal matrix elements. In the SiLK formalism,off-diagonal matrix elements are referred to as kinks. Byanalytically summing over the diagonal matrix elements inEq. (4), we obtain a kink-based version of the partitionfunction. Defining
x j ≡α j
�����exp
(− β
PH
) ����� α j
≈
α j
�����1 − β
PH�����α j
+ O
(1P2
)≈ exp
(− β
P⟨α j |H |α j⟩
)+ O
(1P2
), (5)
ti j ≡αi
�����exp
(− β
PH
) ����� α j
≈
αi
�����1 − β
PH�����α j
+ O
(1P2
)≈ exp
(− β
P⟨αi |H |α j⟩
)− 1 + O
(1P2
), when i , j, (6)
the result of this analytical summation is36
Q = limP→∞
Q (P) , (7)
Q (P) =j
xPj +
Pn=2
Pn*.,
ni=1
ji
+/-*,
nk=1
t jk, jk+1+-
× S��
x j
,n,m,
�s j�, (8)
where n is the number of kinks, m is the number of distinctα j’s in a given set of states with n kinks, s j is the number oftimes the state α j appears in a given set of states with n kinks,and
S��
x j
,n,m,
�s j�=
mj=1
1�s j − 1
�!
ds j−1
dxs j−1j
xs j−1j
×ml=1
xP−n+m−1l
i,l(xl − xi) , (9)
where S may be evaluated recursively. Due to the derivativesin Eq. (9), it is possible for S to be negative. In addition, theoff-diagonal matrix elements t jk, jk+1 can also be negative.
Fig. 1 depicts the types of kink configurations thatappear in this sum over states. The top figure without kinkscorresponds to the case where only diagonal matrix elementsoccur such that j1 = j2, . . .. The second case contains two
FIG. 1. Examples of different kink configurations that occur in Eq. (4)when P= 5. Horizontal lines correspond to diagonal matrix elements andslanted lines correspond to off-diagonal matrix elements that are referred toas kinks. The lines at the beginning and end of a kink configuration wraparound due to the Trace operation required by the partition function. The zerokink configuration contains matrix elements for a single state, the two kinkconfiguration contains matrix elements for just two states, etc. Note that thenumber of kinks and the number of states are not necessarily equal to eachother as it is seen in the four kink configuration.

014101-3 Ma et al. J. Chem. Phys. 144, 014101 (2016)
“kinks” where two identical off-diagonal matrix elementsare introduced. This so-called kink expansion was used incondensed matter physics by Anderson39 and later in thechemical physics literature by Wolynes.40
The first term is non-negative. The n = 2 is also non-negative since the off-diagonal matrix elements appear as|t j1, j2|2 and with s1 and s2 = 1, S > 0. Therefore the signproblem is due to terms with n ≥ 3. The SiLK methoduses a learning algorithm to construct new states as linearcombinations of the initial {αi} states that minimize themagnitude of the contributions from terms with n ≥ 3 andthereby reduces or eliminates the sign problem.
Eq. (8) has the form of a grand canonical partition functionand thus Monte Carlo methods may be used to evaluate thepartition function and its properties. Writing this equation as
Q(P) =P
n=0
{αi}
ρ(n,{αi}), (10)
a Monte Carlo simulation will involve sampling differentstates and inserting and removing kinks. The average energyof the system can be evaluated using ⟨E⟩ = − d
dβln Q,
⟨E⟩ = −P
n=0
{αi}ddβ
ρ(n,{αi})Pn=0
{αi}ρ(n,{αi})
= −P
n=0
{αi}|ρ(n,{αi})||ρ(n,{αi})|
ddβ
ρ(n,{αi})Pn=0
{αi}
|ρ(n,{αi})||ρ(n,{αi})| ρ(n,{αi})
= −
1|ρ |
ddβ
ρ|ρ |
⟨sign(ρ)⟩|ρ | , (11)
where kink configurations are sampled from |ρ|.
B. SiLK algorithm
Simulations are performed in two stages. The first stageis a “learning” period and is used to construct an improveddescription of the states of the system. We choose the lowestenergy Hartree-Fock state as the initial state. As the grandcanonical simulation proceeds, additional states are insertedand removed and a list is maintained of states that haveappeared. At fixed intervals (30 iterations in our calculations)or when the number of kinks present at the end of a MonteCarlo pass exceeds a specified number (9 in our case), theHamiltonian is diagonalized in the sub-space of the states thathave appeared since the last diagonalization (or the start ofthe simulation). The state is then set to the lowest energy state(a zero-kink configuration) and the simulation is continued.The learning period ends when there are only zero and twokinks configurations present for an extended number of MonteCarlo passes. At this point, the expectation is that the partitionfunction will be dominated by kink configurations with asmall number of kinks (dominated by configurations with 0 or2 kinks) as the current set of states will better approximate theground state of the system than the initial ones. As currentlyimplemented, the learning stage can be thought of as usingthe simulation to construct CI states. In the present work, thelearning period ranged from 8000 to 119 000 passes.
The second stage in the simulation is the data acquisitionduring which the states are not modified and the grandcanonical simulation proceeds in the standard way. If thenumber of kinks increases dramatically during this stage(perhaps due to the simulation exploring a previouslyunexplored region of phase space), a diagonalization isperformed and the second stage is restarted. In the calculations,where additional diagonalizations were performed, thediagonalization made an insignificant change in the groundand excited state energies. Between 1000 and 2000 MonteCarlo passes were used in the second stage.
The Monte Carlo algorithm consists of two types ofmoves: change of state and insertion/removal of states. Theformer is performed in the standard way using the Metropolisalgorithm. The latter uses the Metropolis algorithm as follows.A potential new kink configuration c′ is sampled basedon the current kink configuration c using the normalizedconditional probability T(c′|c) and accepted with probabilityA(c′|c) = min
1, ρ(c
′)T (c |c′)ρ(c)T (c′|c)
. If there are n states in the current
kink configuration, there are n + 1 places to insert a new stateinto the kink configuration (as state 1, state 2, . . . , state n + 1).There are n ways to remove a state. We set
T(c′|c) = Tremove(c′|c) + Tadd(c′|c), (12)
with the probability of removing the state at location k in thelist of states
Tremove(c′ = {n − 1, k}|c)=
|ρ(1,2, . . . , k − 1, k + 1, . . . ,n − 1)|D(c′|c) , (13)
and with the probability of adding α j at location k,
Tadd(c′ = {n + 1, k,α j}|c)=
|ρ(1,2, . . . , k − 1,α j, k, k + 1, . . . ,n + 1)|D(c′|c) , (14)
with D(c′|c) the normalization for the probability,
D(c′|c) =k
|ρ(1,2, . . . , k − 1, k + 1, . . . ,n − 1)|
+k
α j
|ρ(1,2, . . . , k − 1,α j, k, k + 1, . . . n + 1)|.
(15)
The acceptance probability is then
A(c′|c) = min1,
D(c′|c)D(c|c′)
. (16)
III. RESULTS AND DISCUSSION
We use the SiLK algorithm to calculate the energiesof H2O, N2, and F2 at selected bond lengths and bondangles for H2O. The Cartesian Gaussian DZ basis set41–44
is used in all calculations. Computer memory constraintsimposed by the current SiLK implementation dictates thenumber of determinants that can be used in the calculations.The reason is that at present the CI coefficients for theground and excited states must be stored. Future work willfocus on alleviating the memory issues. We therefore use
014101-4 Ma et al. J. Chem. Phys. 144, 014101 (2016)
either the full vector space of determinants generated by allpossible excitations of the Hartree-Fock determinant (FullConfiguration Interaction, FCI) or the restricted vector spacesgenerated by the HF determinant and either all possible singleand double excitations (SD) or all possible single, doubleand triple excitations (SDT) of the HF determinant. In allcases, a comparison of the SiLK method to the exact resultwithin the vector space is made to assess, as mentionedin the Introduction, the ability of SiLK to provide accurateresults and alleviate the sign problem. At each geometry,a Hartree-Fock computation using the NWChem ab initiopackage45 is used to generate the initial molecular orbitalsfrom which the determinants are created. Determinantscorresponding to excited states are generated by excitations ofall molecular orbitals except the core orbitals (the frozen coreapproximation). Symmetry is used to restrict the determinantsto those with the same symmetry as the ground Hartree-Fockstate. For the calculations presented here, we use C2v spatialsymmetry for H2O and D2h spatial symmetry for N2 andF2, respectively. We use T = 1 K (β = 3 × 105 hartree−1).Exact energies are obtained by numerical diagonalization forthe SD and SDT vector spaces. A series of calculationswith increasing values for P were performed until aconvergence in the energy was obtained. The values of Pchosen for the reporting of data ranged from 2 × 107 to2 × 1010. The FCI calculations for H2O is performed usingMolpro.46,47
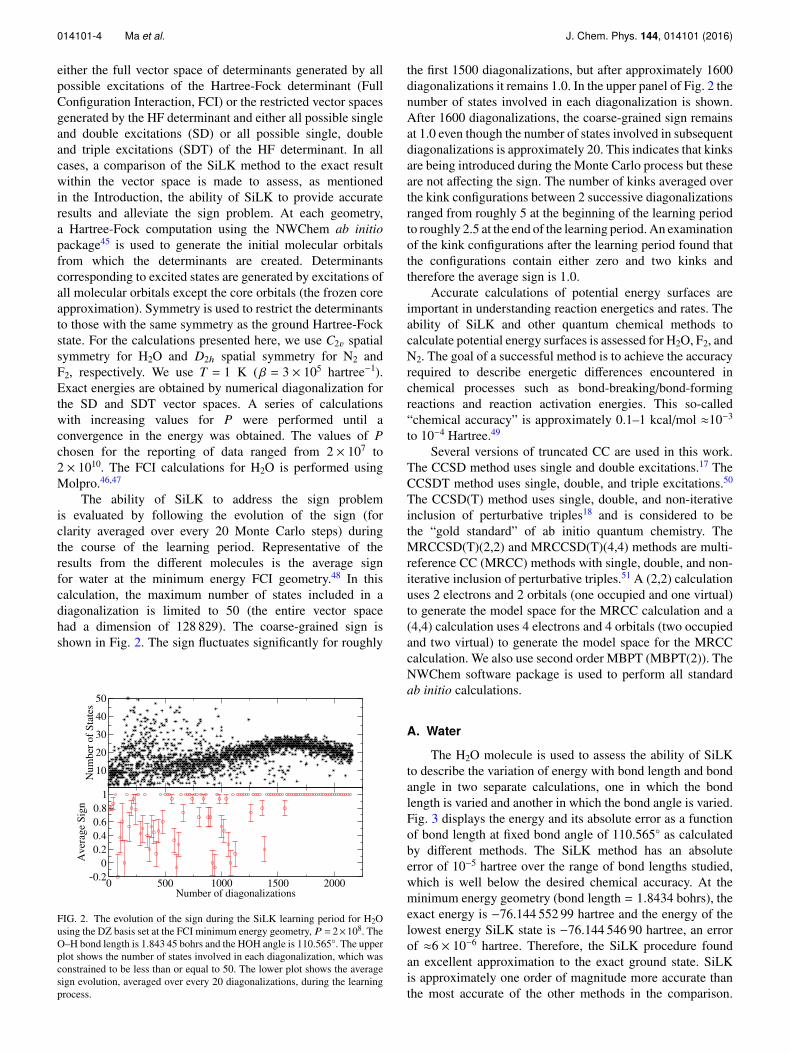
The ability of SiLK to address the sign problemis evaluated by following the evolution of the sign (forclarity averaged over every 20 Monte Carlo steps) duringthe course of the learning period. Representative of theresults from the different molecules is the average signfor water at the minimum energy FCI geometry.48 In thiscalculation, the maximum number of states included in adiagonalization is limited to 50 (the entire vector spacehad a dimension of 128 829). The coarse-grained sign isshown in Fig. 2. The sign fluctuates significantly for roughly
FIG. 2. The evolution of the sign during the SiLK learning period for H2Ousing the DZ basis set at the FCI minimum energy geometry, P = 2×108. TheO–H bond length is 1.843 45 bohrs and the HOH angle is 110.565◦. The upperplot shows the number of states involved in each diagonalization, which wasconstrained to be less than or equal to 50. The lower plot shows the averagesign evolution, averaged over every 20 diagonalizations, during the learningprocess.
the first 1500 diagonalizations, but after approximately 1600diagonalizations it remains 1.0. In the upper panel of Fig. 2 thenumber of states involved in each diagonalization is shown.After 1600 diagonalizations, the coarse-grained sign remainsat 1.0 even though the number of states involved in subsequentdiagonalizations is approximately 20. This indicates that kinksare being introduced during the Monte Carlo process but theseare not affecting the sign. The number of kinks averaged overthe kink configurations between 2 successive diagonalizationsranged from roughly 5 at the beginning of the learning periodto roughly 2.5 at the end of the learning period. An examinationof the kink configurations after the learning period found thatthe configurations contain either zero and two kinks andtherefore the average sign is 1.0.
Accurate calculations of potential energy surfaces areimportant in understanding reaction energetics and rates. Theability of SiLK and other quantum chemical methods tocalculate potential energy surfaces is assessed for H2O, F2, andN2. The goal of a successful method is to achieve the accuracyrequired to describe energetic differences encountered inchemical processes such as bond-breaking/bond-formingreactions and reaction activation energies. This so-called“chemical accuracy” is approximately 0.1–1 kcal/mol ≈10−3
to 10−4 Hartree.49
Several versions of truncated CC are used in this work.The CCSD method uses single and double excitations.17 TheCCSDT method uses single, double, and triple excitations.50
The CCSD(T) method uses single, double, and non-iterativeinclusion of perturbative triples18 and is considered to bethe “gold standard” of ab initio quantum chemistry. TheMRCCSD(T)(2,2) and MRCCSD(T)(4,4) methods are multi-reference CC (MRCC) methods with single, double, and non-iterative inclusion of perturbative triples.51 A (2,2) calculationuses 2 electrons and 2 orbitals (one occupied and one virtual)to generate the model space for the MRCC calculation and a(4,4) calculation uses 4 electrons and 4 orbitals (two occupiedand two virtual) to generate the model space for the MRCCcalculation. We also use second order MBPT (MBPT(2)). TheNWChem software package is used to perform all standardab initio calculations.
A. Water
The H2O molecule is used to assess the ability of SiLKto describe the variation of energy with bond length and bondangle in two separate calculations, one in which the bondlength is varied and another in which the bond angle is varied.Fig. 3 displays the energy and its absolute error as a functionof bond length at fixed bond angle of 110.565◦ as calculatedby different methods. The SiLK method has an absoluteerror of 10−5 hartree over the range of bond lengths studied,which is well below the desired chemical accuracy. At theminimum energy geometry (bond length = 1.8434 bohrs), theexact energy is −76.144 552 99 hartree and the energy of thelowest energy SiLK state is −76.144 546 90 hartree, an errorof ≈6 × 10−6 hartree. Therefore, the SiLK procedure foundan excellent approximation to the exact ground state. SiLKis approximately one order of magnitude more accurate thanthe most accurate of the other methods in the comparison.

014101-5 Ma et al. J. Chem. Phys. 144, 014101 (2016)
FIG. 3. Potential energy curve of H2O molecule as a function of OH bondlength. A comparison of results obtained using MBPT(2), CCSD, CCSD(T),CCSDT, MRCCSD(T)(2,2), MRCCSD(T)(4,4), and SiLK formalisms. Thebottom plot displays the absolute error in the calculated energy for thedifferent methods. P = 2×108 is used for bond lengths in the range [1.34-3.64] bohrs and P = 2×109 is used for bond lengths in the range [3.74-4.34] bohrs.
Notably, SiLK is accurate at the longer bond lengths (roughlytwo orders of magnitude more accurate than any other method)which is crucial to a description of bond dissociation andbond breaking processes. None of the other methods (exceptMRCCSD(T)(4,4)) achieves chemical accuracy over the entirerange of bond lengths studied.
Then we use these methods to calculate the energy asa function of bond angle for the H2O molecule with bondlength 1.843 45 bohrs. Fig. 4 displays the energies andabsolute errors for bond angles ranging from 95◦ to 125◦.The SiLK method is approximately two orders of magnitudemore accurate than the most accurate of the other methods.All methods except MBPT(2) and CCSD achieve chemicalaccuracy.
It is also instructive to consider calculations restricted tojust single and double excitations as sometimes computationsbased on such restricted vector spaces can yield useful results
FIG. 4. Potential energy curves of H2O FCI vector space for the DZ basis.43
A comparison of results obtained by SiLK with results from MBPT(2),CCSD, CCSD(T), CCSDT, MRCCSD(T)(2,2), and MRCCSD(T)(4,4). Bot-tom plot displays the absolute error of energy. P = 2×108 is used for allangles in SiLK QMC.
FIG. 5. Potential energy curves for H2O molecule using the DZ basis and theSD and SDT vector spaces. The exact results obtained from exact diagonal-ization and the SiLK results are shown. P = 2×1010 is used for all the bondlengths in the SD and the SDT vector spaces.
using significantly fewer computational resources. Therefore,the SiLK algorithm is used to calculate the energies andabsolute errors of the H2O molecule as a function of bondlength and bond angle. Figs. 5 and 6 show their comparisonwith exact results. The SiLK method is able to reproducethe exact results to 10−5 hartree, well within chemicalaccuracy.
B. Nitrogen
N2 has a triple bond, which provides a challenging testfor ab initio methods due to its large electronic correlation.52
Due to memory limitations, the SiLK calculations wererestricted to the Hartree-Fock determinant plus either theSD and SDT vector spaces. Fig. 7 shows that the SiLK QMCresults converge to the exact result over a wide range ofbond lengths. At the minimum energy geometry (bond length= 2.168 bohrs), the exact energy is−109.085 809 5 hartree andthe energy of the lowest energy SiLK state is −109.085 809 4hartree, an error of ≈1 × 10−7 hartree. Therefore, the SiLK
FIG. 6. Potential energy curves for the H2O molecule within the SD andSDT vector spaces and the DZ basis. Exact results obtained from exactdiagonalization and SiLK results are shown. P = 2×1010 is used for all theangles in the SD vector space. Within the SDT space, P = 2×1010 is usedfor angles in the range [95.565, 120.565] and P = 2×107 is used for the125.565◦ calculation in SiLK QMC.

014101-6 Ma et al. J. Chem. Phys. 144, 014101 (2016)
FIG. 7. Potential energy curves for N2 molecule as a function of bond lengthusing the SD and SDT vector spaces. The exact results obtained from exactdiagonalization and SiLK results are shown. P = 2×1010 is used for all bondlengths.
procedure found an excellent approximation to the exactground state. The results demonstrate that the SiLK methodis suitable for multi-reference systems such as N2 where morethan a single determinant is strongly coupled in the groundelectronic state.
C. Fluoride
Electron correlations are difficult to include in thesimulation of F2 as many determinants contribute smallbut important contributions to the total energy.53 Thisphenomenon is often referred to as dynamic correlation.54
Therefore, the SiLK method is applied to the F2 molecule.As with Nitrogen, due to memory limitations, the SiLKcalculations is limited to the SD and SDT vector spaces.Fig. 8 shows that the SiLK QMC results converge to the exactresults and demonstrate that SiLK is capable of accuratelyincluding dynamic correlation. At the minimum energygeometry (bond length = 2.868 16 bohrs), the exact energyis −198.949 416 9 hartree and the energy of the lowest energySiLK state is −198.949 416 9 hartree, an error of <1 × 10−7
FIG. 8. Potential energy curves for F2 molecule as a function of bond lengthand using the Hartree-Fock determinant plus either the SD and SDT vectorspaces. The exact results obtained from exact diagonalization and the SiLKresults are shown. P = 2×1010 is used.
FIG. 9. The length of the learning period as a function of the number ofSlater determinants. The results for the SD, SDT, and FCI vector spaces forall molecules and geometries are shown.
hartree. Therefore, the SiLK procedure found an excellentapproximation to the exact ground state.
D. Scaling analysis
It is beyond the scope of this work to make a thoroughanalysis of the scaling of the SiLK method as the memoryrequirements of the current implementation of the SiLKmethod prohibit the use of a wide range of basis set size.However, it is important to assess the scaling of the SiLKalgorithm with the size of the basis set and vector space.No truncation methods, such as a truncation in the spaceof the single-particle density matrix,55 will improve theefficiency of the algorithm for certain systems. Therefore,a scaling analysis is presented for the current work usingthe relatively limited size of the vector spaces. If theSiLK algorithm increases too quickly with the size of thevector space, the computational requirements for the SiLKmethod will make its use in the present form intractable. Thedependence of the length of the learning period on the sizeof the vector space (number of determinants) is presentedin Fig. 9. As expected, there is an increase in the size ofthe learning period. However, a wider range of vector spacesizes is necessary to fully understand and quantify the scalingbehavior.
IV. CONCLUSIONS
The minus sign problem in Quantum Monte Carlosimulations of frustrated or correlated electronic systems isa challenging problem. It has even been suggested that ageneral solution of this problem is NP-complete.4 Therefore,one should not expect an effective solution for all the MonteCarlo simulations which have the minus sign problem. Inthis paper, we demonstrate that SiLK QMC can reduce theminus sign problem by using a learning stage that includes adiagonalization procedure. In this paper, we demonstrate thatthe energies obtained by the SiLK QMC match the results fromexact diagonalization and surpass the accuracy obtained usingother quantum chemistry methods, particularly for geometries

014101-7 Ma et al. J. Chem. Phys. 144, 014101 (2016)
relatively far from equilibrium. In addition, SiLK can beapplied to systems that require a multiple reference stateapproach. An intriguing possibility for future work is to usethe SiLK learning procedure in combination with other QMCalgorithms to reduce the minus sign problem.
As the learning stage progresses, the states become morecomplicated linear combinations of determinants so that moreevaluations of matrix elements are required, thereby increasingthe computational expense. However, at the same time, thenumber of non-zero matrix elements between these statesdecreases. So, further optimization is possible by storing often-needed matrix elements in memory. For example, storing theoff-diagonal matrix elements between the ground and excitedstates yields a large speed up, since these are the only matrixelements required once the learning stage reaches the pointwhere mostly zero and two kink configurations appear in thesimulation. It is also possible to halt the learning stage at anearlier point, when the ground SiLK state is not as accuratean approximation to the exact ground state, but when thesign problem is alleviated but not eliminated and relies onthe Monte Carlo sampling to provide the exact energy. Thiswould reduce the computational effort required to evaluatethe matrix elements since fewer diagonalizations will haveoccurred. An investigation of the efficacy of a shorter learningperiod is left for future work.
The SiLK method requires the knowledge of the off-diagonal matrix elements of the Hamiltonian. As the sizeof the system increases, the number of off-diagonal matrixelements increases factorially and it is not possible to storethe matrix elements or the CI coefficients for the ground andexcited states that would allow for on-the-fly evaluation of thematrix elements. As such, without a procedure to accuratelytruncate the number of determinants used to describe theground and excited state wavefunctions, the use of the allpossible determinants in a SiLK calculation will be limitedto relatively small systems. However, SiLK can certainly beused when determinants are restricted to, for example, singleand double excitations. Such truncated sets of determinantsare often sufficient for the study of chemical systems. In caseswhere restrictions to single and double excitations are notsufficient, more sophisticated methods of truncation, such asthe one developed by Maurits,55 will be needed.
The SiLK QMC is a versatile method to calculate theground state energy of molecular systems. Since the pathintegral formulation uses the canonical partition function it ispossible to use the SiLK method to simulate the motion of theatoms at a finite temperature. Future work will investigate theuse of the SiLK method in finite temperature simulations.
ACKNOWLEDGMENTS
This work is funded by the NSF EPSCoR LA-SiGMA project under Award No. EPS-1003897 and theUS Department of Energy under Contract No. DE-AC06.76RLO-1830. R.W.H. acknowledges support fromDominican University of California’s Lillian L. Y. WangYin, Ph.D. Endowed Professorship of Chemistry. This workused the high performance computing resources providedby Louisiana State University (http://www.hpc.lsu.edu) and
the computational resources of the Environmental MolecularSciences Laboratory at Pacific Northwest National Laboratory(PNNL), which is sponsored by the Department of EnergyOffice of Biological and Environmental Research.
1A. Dreuw and M. Head-Gordon, Chem. Rev. 105, 4009 (2005).2R. P. Feynman, A. R. Hibbs, and D. F. Styer, Quantum Mechanics and PathIntegrals: Emended Edition (Dover Publications, 2005).
3W. Foulkes, L. Mitas, R. Needs, and G. Rajagopal, Rev. Mod. Phys. 73, 33(2001).
4M. Troyer and U.-J. Wiese, Phys. Rev. Lett. 94, 170201 (2005).5E. Loh, Jr., J. Gubernatis, R. Scalettar, S. White, D. Scalapino, and R. Sugar,Phys. Rev. B 41, 9301 (1990).
6R. O. Jones and O. Gunnarsson, Rev. Mod. Phys. 61, 689 (1989).7R. Harrison and N. Handy, Chem. Phys. Lett. 95, 386 (1983).8C. Møller and M. S. Plesset, Phys. Rev. 46, 618 (1934).9N. Handy, P. Knowles, and K. Somasundram, Theor. Chim. Acta 68, 87(1985).
10P. Pendergast and E. F. Hayes, J. Comput. Phys. 26, 236 (1978).11R. J. Bartlett and M. Musiał, Rev. Mod. Phys. 79, 291 (2007).12F. Coester, Nucl. Phys. 7, 421 (1958).13F. Coester and H. Kümmel, Nucl. Phys. 17, 477 (1960).14J. Cížek, J. Chem. Phys. 45, 4256 (1966).15J. Cížek and J. Paldus, Int. J. Quantum Chem. 5, 359 (1971).16J. Paldus, J. Cížek, and I. Shavitt, Phys. Rev. A 5, 50 (1972).17G. D. Purvis III and R. J. Bartlett, J. Chem. Phys. 76, 1910 (1982).18K. Raghavachari, G. W. Trucks, J. A. Pople, and M. Head-Gordon, Chem.
Phys. Lett. 157, 479 (1989).19S. R. White, Phys. Rev. B 48, 10345 (1993).20G. K.-L. Chan and S. Sharma, Annu. Rev. Phys. Chem. 62, 465 (2011).21K. H. Marti and M. Reiher, Z. Phys. Chem. 224, 583 (2010).22S. Wouters and D. Van Neck, Eur. Phys. J. D 68, 1 (2014).23N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E.
Teller, J. Chem. Phys. 21, 1087 (1953).24N. Metropolis and S. Ulam, J. Am. Stat. Assoc. 44, 335 (1949).25E. Fermi and R. Richtmyer, “Note on census-taking in Monte-Carlo calcu-
lations,” Technical Report No. AECD-3164; LADC-946, Los Alamos Sci-entific Laboratory, 1948.
26N. Rom, E. Fattal, A. K. Gupta, E. A. Carter, and D. Neuhauser, J. Chem.Phys. 109, 8241 (1998).
27R. Baer, M. Head-Gordon, and D. Neuhauser, J. Chem. Phys. 109, 6219(1998).
28D. M. Ceperley and B. Alder, Phys. Rev. Lett. 45, 566 (1980).29A. J. Thom and A. Alavi, J. Chem. Phys. 123, 204106 (2005).30A. J. Thom, G. H. Booth, and A. Alavi, Phys. Chem. Chem. Phys. 10, 652
(2008).31M. Suewattana, W. Purwanto, S. Zhang, H. Krakauer, and E. J. Walter, Phys.
Rev. B 75, 245123 (2007).32E. W. Brown, B. K. Clark, J. L. DuBois, and D. M. Ceperley, Phys. Rev. Lett.
110, 146405 (2013).33E. W. Brown, J. L. DuBois, M. Holzmann, and D. M. Ceperley, Phys. Rev. B
88, 081102 (2013).34G. H. Booth, A. J. Thom, and A. Alavi, J. Chem. Phys. 131, 054106 (2009).35G. H. Booth, A. Grüneis, G. Kresse, and A. Alavi, Nature 493, 365 (2013).36R. W. Hall, J. Chem. Phys. 116, 1 (2002).37R. W. Hall, Chem. Phys. Lett. 362, 549 (2002).38R. W. Hall, J. Chem. Phys. 122, 164112 (2005).39P. W. Anderson, G. Yuval, and D. Hamann, Phys. Rev. B 1, 4464 (1970).40R. Chiles, G. Jongeward, M. Bolton, and P. Wolynes, J. Chem. Phys. 81,
2039 (1984).41D. Feller, J. Comput. Chem. 17, 1571 (1996).42K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J.
Chase, J. Li, and T. L. Windus, J. Chem. Inf. Model. 47, 1045 (2007).43T. H. Dunning, Jr., J. Chem. Phys. 53, 2823 (1970).44T. H. Dunning, P. J. Hay, and H. Schaefer, Modern Theoretical Chemistry
(Plenum Press, New York, 1977), Vol. 3, p. 1.45M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. Van
Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus et al., Comput. Phys.Commun. 181, 1477 (2010).
46H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, and M. Schütz, WileyInterdiscip. Rev.: Comput. Mol. Sci. 2, 242 (2012).
47H.-J. Werner and P. J. Knowles, with contributions from J. Almlöf, R. D.Amos, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert,

014101-8 Ma et al. J. Chem. Phys. 144, 014101 (2016)
S. T. Elbert, C. Hampel, R. Lindh, A. W. Lloyd, W. Meyer, A. Nicklass,K. Peterson, R. Pitzer, A. J. Stone, P. R. Taylor, M. E. Mura, P. Pulay, M.Schütz, H. Stoll, and T. Thorsteinsson, , version 2015.1, a package ofab initio programs, 2015, see http://www.molpro.net.
48P. Saxe, H. F. Shaefer, and N. C. Handy, Chem. Phys. Lett. 79, 202 (1981).49P. A. Bash, L. L. Ho, A. D. MacKerell, D. Levine, and P. Hallstrom, Proc.
Natl. Acad. Sci. U. S. A. 93, 3698 (1996).50J. Noga and R. J. Bartlett, J. Chem. Phys. 86, 7041 (1987).
51K. Bhaskaran-Nair, J. Brabec, E. Aprà, H. J. van Dam, J. Pittner, and K.Kowalski, J. Chem. Phys. 137, 094112 (2012).
52K. Kowalski and P. Piecuch, J. Chem. Phys. 113, 5644 (2000).53K. Kowalski and P. Piecuch, Chem. Phys. Lett. 344, 165 (2001).54D. K. Mok, R. Neumann, and N. C. Handy, J. Phys. Chem. 100, 6225
(1996).55Y. Lu, M. Höppner, O. Gunnarsson, and M. W. Haverkort, Phys. Rev. B 90,
085102 (2014).




















