
Severe hyperthyroidism induces mitochondria-mediated apoptosis in rat liver
11
Severe Hyperthyroidism Induces Mitochondria- Mediated Apoptosis in Rat Liver Geeta Upadhyay, 1 Rajesh Singh, 1 Ashok Kumar, 1 Sanjeev Kumar, 2 Amit Kapoor, 2 and Madan M. Godbole 1 Thyrotoxicosis may be associated with a variety of abnormalities of liver function. The pathogenesis of hepatic dysfunction in thyrotoxicosis is unknown, but has been attributed to mitochondrial dysfunction. We studied the effect of altered thyroid function on the apopto- tic index in rat liver. Extensive DNA fragmentation and significantly increased caspase-3 activity (P < .001) and caspase-9 activation (P < .005) were observed in hyperthyroid rat liver; cell death by apoptosis was confirmed. In hyperthyroid rat liver, 60% of mitochondria exhibited disruption of their outer membranes and a decrease in the number of cristae. These findings, along with significant translocation of cytochrome c and second mitochondria- derived activator of caspases to cytosol (P < .005), suggest activation of a mitochondrial- mediated pathway. However, no change in the expression levels of Bcl-2, Bax, and Bcl-x L were found in hyperthyroidism. For in vitro experiments, rat liver mitochondria were isolated and purified in sucrose density gradients and were treated with triiodothyronine (T3; 2– 8 M). T3 treatment resulted in an abrupt increase in mitochondrial permeability transition. Using a cell-free apoptosis system, the apoptogenic nature of proteins released from mitochondria was confirmed by observing changes in nuclear morphologic features and DNA fragmentation. Proteins released by 6 M T3 contained significantly increased amounts of cytochrome c (P < .01) and induced apoptotic changes in 67% of nuclei. In conclusion, using in vivo and in vitro approaches, we provide evidence that excess T3 causes liver dysfunction by inducing apoptosis, as a result of activation of a mitochondria-depen- dent pathway. Thus, the results of this study provide an explanation for liver dysfunction associated with hyperthyroidism. (HEPATOLOGY 2004;39:1120 –1130.) T hyroid hormone (TH) affects all tissues and mod- ulates the rate of metabolic activity. Liver damage in hyperthyroid patients has been extensively re- ported since Habershon’s original report in 1874. 1–5 Liver function becomes compromised in 45% to 90% of thy- rotoxic patients; in most cases, the changes in the liver are characterized by some degree of fatty infiltration, and by cytoplasmic vacuolization, nuclear irregularity, and hy- perchromatism in hepatocytes. 6,7 The pathogenesis of he- patic dysfunction in severe hyperthyroidism is unknown. Possible thyroid–liver interactions include liver damage secondary to the systemic effects of excess thyroid hor- mone, direct toxic effects of thyroid hormone on the liver, an association of intrinsic liver disease and intrinsic thy- roid disease involving autoimmune mechanisms, changes in thyroid hormone metabolism secondary to intrinsic liver disease, and subclinical physiologic effects of thyroid hormone on functions of the liver. 5 The hepatic injury associated with hyperthyroidism varies from mild liver dysfunction associated with nonspecific histologic changes to severe central hepatic ischemia. Ultrastructural and functional changes in mitochon- dria, such as enlargement, a mass increase, and formation of megamitochondria, have been reported in the liver of hyperthyroid patients and in a rat model of hyperthyroid- ism. 7–13 Decrease in mitochondrial transmembrane po- tential and proton motive force and altered cellular oxidation-reduction occur during mitochondrial-medi- Abbreviations: TH, thyroid hormone; T3, triiodothyronine; AST, serum glu- tamic oxaloacetic transaminase; ALT, serum glutamic pyuric transaminase; SMAC, second mitochondria-derived activator of caspases; IAPs, inhibitor of apoptosis pro- teins; PT, permeability transition. From the Departments of 1 Endocrinology and 2 Microbiology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India. Received May 18, 2003; accepted January 18, 2004. Supported by an SGPGIMS intramural grant (no. 776/99), an SGPGIMS fellowship (to G.U.), and a CSIR fellowship (to R.S.). Address reprint requests to: Madan M. Godbole, Department of Endocrinology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Raebareli Road, Lucknow, India - 226 014. E-mail: [email protected]; fax: 91-522- 2668017/2668973. Copyright © 2004 by the American Association for the Study of Liver Diseases. Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hep.20085 1120
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Severe hyperthyroidism induces mitochondria-mediated apoptosis in rat liver

Severe Hyperthyroidism Induces Mitochondria-Mediated Apoptosis in Rat Liver
Geeta Upadhyay,1 Rajesh Singh,1 Ashok Kumar,1 Sanjeev Kumar,2 Amit Kapoor,2 and Madan M. Godbole1
Thyrotoxicosis may be associated with a variety of abnormalities of liver function. Thepathogenesis of hepatic dysfunction in thyrotoxicosis is unknown, but has been attributed tomitochondrial dysfunction. We studied the effect of altered thyroid function on the apopto-tic index in rat liver. Extensive DNA fragmentation and significantly increased caspase-3activity (P < .001) and caspase-9 activation (P < .005) were observed in hyperthyroid ratliver; cell death by apoptosis was confirmed. In hyperthyroid rat liver, 60% of mitochondriaexhibited disruption of their outer membranes and a decrease in the number of cristae. Thesefindings, along with significant translocation of cytochrome c and second mitochondria-derived activator of caspases to cytosol (P < .005), suggest activation of a mitochondrial-mediated pathway. However, no change in the expression levels of Bcl-2, Bax, and Bcl-xL
were found in hyperthyroidism. For in vitro experiments, rat liver mitochondria wereisolated and purified in sucrose density gradients and were treated with triiodothyronine(T3; 2–8 �M). T3 treatment resulted in an abrupt increase in mitochondrial permeabilitytransition. Using a cell-free apoptosis system, the apoptogenic nature of proteins releasedfrom mitochondria was confirmed by observing changes in nuclear morphologic featuresand DNA fragmentation. Proteins released by 6 �M T3 contained significantly increasedamounts of cytochrome c (P < .01) and induced apoptotic changes in 67% of nuclei. Inconclusion, using in vivo and in vitro approaches, we provide evidence that excess T3 causesliver dysfunction by inducing apoptosis, as a result of activation of a mitochondria-depen-dent pathway. Thus, the results of this study provide an explanation for liver dysfunctionassociated with hyperthyroidism. (HEPATOLOGY 2004;39:1120–1130.)
Thyroid hormone (TH) affects all tissues and mod-ulates the rate of metabolic activity. Liver damagein hyperthyroid patients has been extensively re-
ported since Habershon’s original report in 1874.1–5 Liverfunction becomes compromised in 45% to 90% of thy-rotoxic patients; in most cases, the changes in the liver arecharacterized by some degree of fatty infiltration, and by
cytoplasmic vacuolization, nuclear irregularity, and hy-perchromatism in hepatocytes.6,7 The pathogenesis of he-patic dysfunction in severe hyperthyroidism is unknown.Possible thyroid–liver interactions include liver damagesecondary to the systemic effects of excess thyroid hor-mone, direct toxic effects of thyroid hormone on the liver,an association of intrinsic liver disease and intrinsic thy-roid disease involving autoimmune mechanisms, changesin thyroid hormone metabolism secondary to intrinsicliver disease, and subclinical physiologic effects of thyroidhormone on functions of the liver.5 The hepatic injuryassociated with hyperthyroidism varies from mild liverdysfunction associated with nonspecific histologicchanges to severe central hepatic ischemia.
Ultrastructural and functional changes in mitochon-dria, such as enlargement, a mass increase, and formationof megamitochondria, have been reported in the liver ofhyperthyroid patients and in a rat model of hyperthyroid-ism.7–13 Decrease in mitochondrial transmembrane po-tential and proton motive force and altered cellularoxidation-reduction occur during mitochondrial-medi-
Abbreviations: TH, thyroid hormone; T3, triiodothyronine; AST, serum glu-tamic oxaloacetic transaminase; ALT, serum glutamic pyuric transaminase; SMAC,second mitochondria-derived activator of caspases; IAPs, inhibitor of apoptosis pro-teins; PT, permeability transition.
From the Departments of 1Endocrinology and 2Microbiology, Sanjay GandhiPostgraduate Institute of Medical Sciences, Lucknow, India.
Received May 18, 2003; accepted January 18, 2004.Supported by an SGPGIMS intramural grant (no. 776/99), an SGPGIMS
fellowship (to G.U.), and a CSIR fellowship (to R.S.).Address reprint requests to: Madan M. Godbole, Department of Endocrinology,
Sanjay Gandhi Postgraduate Institute of Medical Sciences, Raebareli Road,Lucknow, India - 226 014. E-mail: [email protected]; fax: �91-522-2668017/2668973.
Copyright © 2004 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.20085
1120

ated apoptosis.14,15 The changes reported in the liver mi-tochondria from thyrotoxic patients7–13 are similar tothose found in the mitochondria of apoptotic cells.
Mitochondria contain several apoptogenic proteins inthe intermembrane space.14,16 Cytochrome c is a key me-diator of apoptosis; its efflux into cytosol initiates theformation of apoptosome and activates the initiator,caspase-9, which leads to activation of a proteolytic cas-cade that results in cellular disassembly by the effector,caspase-3.15–17 Inhibitors of apoptosis proteins (IAPs)form heterodimers with caspases and inhibit their activa-tion.18 Translocation of secondary mitochondria-derivedactivator of caspases (SMAC) to cytosol is one of theessential steps for downstream activation of caspase. Itdoes so by binding to IAPs and releasing the caspases foractivation.18,19 Many proteins of the Bcl-2 family resideon the outer membrane of mitochondria, where they areanchored by a hydrophobic sequence of amino acids lo-cated within the COOH termini of proteins oriented to-ward the cytosol. An alteration in the ratio ofproapoptotic to antiapoptotic proteins of the Bcl-2 familymay modulate the release of apoptogenic proteins.15
Because of their biochemical functions, mitochondriaare a natural target for the calorigenic effects of TH.20 Theinteraction between mitochondria and TH in relation toother physiological functions is not understood. TH-in-duced apoptosis has been demonstrated in cultured Tlymphocytes and tadpole intestine in primary culture.21,22
However, the mechanism of action of TH on mitochon-dria during this process is unknown. Adult liver, being atriiodothyronine (T3)-responsive tissue, is probably suit-able for studying the role of T3 in initiating a mitochon-drial pathway of apoptosis in hyperthyroidism.
Here, we report that severe hyperthyroidism inducesextensive apoptosis in rat liver; hypothyroidism inhibitsapoptosis. Hyperthyroidism induces change in mito-chondrial morphologic features and the release of cyto-chrome c from mitochondria, without altering the levelsof Bcl-2, Bcl-xL, and Bax proteins. Using an in vitro apo-ptotic system, we also found that T3 acts directly on mi-tochondria and induces release of apoptogenic proteinsfrom them.
Materials and MethodsIn Vivo Studies
Animals. Four-month-old male Sprague Dawley ratswere used in all experiments. The animals were housed ina temperature- and light-controlled room (21-24°C, 12-hour:12-hour light:dark cycle); they were allowed a chowdiet and water ad libitum. Rats were divided into threegroups (n � 15 for each group). The first group (group 1)was given the chow diet and ordinary drinking water
alone (control); the second group (group 2) in additionwas given methimazole (Sigma, St. Louis, MO) at a con-centration of 0.025 % (wt/vol) in drinking water for 8weeks (hypothyroid)23; and the third group (group 3) inaddition received not methimazole, but T3 (Sigma) 100�g/100 g body weight subcutaneously for 10 days (hyper-thyroid).24 The intake of water and food, and the bodyweight of each animal, were recorded daily. Blood sam-ples were collected at the time that animals were sacrificedfor estimation of TH, serum aspartate aminotransferase(AST) and serum alanine aminotransferase (ALT). Allanimal procedures were undertaken in accordance withthe institutional guidelines for animal care and research.
Serum Total T3 (TT3), Total T4, AST, and ALT.Serum TT3 and total T4 were measured using standardradioimmunoassay kits (DPC kit, Los Angles, CA); ASTand ALT were measured using standard kits according tothe manufacturer’s instructions (Merck Gmbh, Darm-stadt, Germany).
DNA Fragmentation Analysis. Liver (0.5 �g) wasminced in 10 volumes of buffer A (20 mM Tris, pH 8.0,10 mM ethylenediaminetetraacetic acid [EDTA], and0.5% Triton-X-100) and incubated on ice for 2 hours.After centrifugation at 14,000g at 4°C for 20 minutes, thesupernatant was extracted with phenol/chloroform. DNAwas precipitated by ethanol and washed with 70% etha-nol. The pellet resuspended in buffer B (10 mM Tris, pH8.0, 1 mM EDTA), treated with RNase-A (100 �g/mL),and extracted with phenol/chloroform. DNA was precip-itated as described above and was analyzed in a 2% aga-rose gel.25
Preparation of Cytosolic and Mitochondrial Frac-tion. Liver tissue was minced in buffer C (0.32 M su-crose, 1 mM K-EDTA, 10 mM Tris HCl, pH 7.4,supplemented with a protease inhibitor cocktail [RocheApplied Sciences, Mannheim, Germany]), homogenizedand centrifuged at 1300g for 10 minutes at 4°C. Thesupernatant was collected, and the pellet was resuspendedin buffer C and centrifuged at 1300g for 10 minutes. Thesupernatant was pooled26 and centrifuged at 17000g for15 minutes to collect the mitochondrial fraction. Assaysfor marker enzymes, such as lactate dehydrogenase andcytochrome oxidase, confirmed the supernatant to be a cy-tosolic fraction and the pellet a mitochondrial fraction. Themitochondrial pellet was resuspended in buffer C and thecytosolic fraction was aliquoted and stored at �80°C forsubsequent analysis.26 The protein concentration was deter-mined using a standard method.27
Western Blotting. Cytosolic or mitochondrial frac-tion proteins (50 �g), or both, were subjected to 15%SDS-PAGE (sodium dodecyl sulfate-Polyacrylamide gelelectrophoresis) and transferred onto a nitrocellulose
HEPATOLOGY, Vol. 39, No. 4, 2004 UPADHYAY ET AL. 1121

membrane. Equal loading and transfer of proteins tomembrane were confirmed by Ponceau S staining. Mem-branes were incubated with primary antibodies followedby incubation with horseradish peroxidase-conjugatedsecondary antibody. The signals were detected using anenhanced chemiluminescence system (Amersham Bio-sciences, Buckinghamshire, UK). Antibodies to caspase-9and SMAC (gifts from Dr. X. Wang) and cytochrome c(7H8.2C12; gift from Dr. R. Jemmerson) were used. An-tibodies against Bcl-xL, Bax, and Bcl-2 and secondary an-tibodies were purchased from Santa Cruz Biotech (SantaCruz, CA). The relative amount of each protein was de-termined quantitatively. Blots from all the three replicateswere analyzed using an Alpha Imager microdensitometer(AlphaImager Corp., San Leandro, CA).
Colorimetric Assay for Caspase-3 Activation.Caspase activity was quantitated using the ApoAlertCaspase-3 colorimetric assay kit (Clontech, Palo Alto,CA) according to the manufacturer’s instructions.
Electron Microscopy. Liver was fixed in phosphatebuffered saline (PBS)-buffered 2.5% glutaraldehyde(Sigma) for 2 hours at 4°C. After dehydration in a series ofdiluted alcohols, blocks were placed in propylene oxidesolutions and were embedded in epoxy resin. Five blocksfrom each animal were prepared, and semithin sectionswere stained with toluidine blue and were examined bylight microscopy for orientation. Ultrathin sections of atleast three blocks per animal were stained with uranylacetate and lead citrate28 and were examined using aJEOL-1010 electron microscope (JEOL, Tokyo, Japan).
In Vitro Studies
Isolation of Mitochondria, Nuclei, and Cytosol.Mitochondria from rat liver were isolated as describedpreviously.12 Mitochondria were purified in 1.0 M/1.5 Msucrose density gradients and resuspended at a concentra-tion of 10 mg protein/mL in buffer D (400 mM Manni-tol, 50 mM Tris HCl, pH 7.4, bovine serum albumin(BSA) 5 mg/mL, KH2PO4).29 Liver nuclei were isolatedusing a previously described standard method.30 Purifiednuclei were resuspended in buffer E (10 mM PIPES (pi-perazine-N,N�-bis [2-ethanesulfonic acid]), pH 7.4, 80mM KCl, 20 mM NaCl, 5 mM Na-EGTA (ethyleneglycol-bis [�-aminoethyl ether]-N,N,N�,N�-tetraaceticacid), 250 mM sucrose, and 1 mM DTT (dithiothreitol))at a concentration of 8.5 � 107 nuclei/mL; the suspensionwas stored at �80°C in multiple aliquots. Cytosol fromHeLa-S3 cells was prepared as described previously.31
Mitochondrial Swelling. Mitochondria were washedin buffer F (0.175 M KCl, 0.025 M Tris HCl, pH 7.4)and were resuspended in the same buffer at a concentra-tion of 10 mg protein/mL.32 The mitochondria were
treated with different concentrations of T3; absorbancewas recorded at 520 nm after different time intervals in atemperature-controlled cell (Hitachi Koki Co., Tokyo,Japan).
Electron Microscopy of Isolated Mitochondria. Themitochondrial pellet was fixed in PBS-buffered 1% gluat-araldehyde/1% paraformaldehyde. The pellet was washedin PBS, fixed by osmium tetroxide (OsO4), and embed-ded in epoxy resin.32 Ultrathin sections were examinedusing a JEOL-1010 electron microscope (Tokyo, Japan).
Nuclear Morphologic Features and DNA Fragmen-tation. Aliquots of 50 �l of HeLa cytosol (250 �g), 6 �l ofliver nuclei, 1 mM MgCl2, and 1 mM dATP (deoxyade-nosine 5�-triphosphate) were incubated in the presence orabsence of T3-treated mitochondrial supernatant at 37°Cfor 2 hours. After propidium iodide staining, nuclear mor-phologic features were assessed using a fluorescence micro-scope (Olympus, Tokyo, Japan). After incubation, analiquot of 500 �l buffer G (100 mM Tris HCl, pH 8.5, 5mM EDTA, 0.2 M NaCl, 0.2% SDS, and 0.2 mg/mL pro-teinase K) was added to each reaction mixture; after incuba-tion of the mixtures at 37°C overnight, sodium chloride wasadded to achieve a final concentration of 1.5 M. Nucleardebris was centrifuged at 15,000g for 15 minutes. DNA wasprecipitated by ethanol, washed with 70% ethanol, and re-suspended in buffer H (10 mM Tris HCl, pH 7.5, 1 mMNa-EDTA, and 200 �g/mL DNase-free-RNase-A). DNAwas analyzed in a 2% agarose gel.31
In Vitro Assay for Cytochrome c Release. Mitochon-dria (50 �g protein in 20 mL), resuspended in protein-re-lease buffer I (220 mM mannitol, 68 mM sucrose, 20 mMHEPES KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1mM Na-EDTA, 1 mM Na-EGTA, 1 mM DTT, and 1 mMPMSF [phenylmethyl sulfonyl fluoride]) were treated withT3 at 30°C for 1 hour. The mixture was centrifuged at14,000g for 10 min at 4°C to generate mitochondrial pellets.The presence of cytochrome c was detected in supernatantsand pellets by Western blotting using an Enhanced Chemi-luminescene (ECL) kit and spectroscopic scanning (HitachiKoki Co).31
Statistical Analysis. Results are expressed as mean �SEM. Experimental groups were compared using a one-way ANOVA and Student’s t test. P values of 0.05 or lesswere considered to be significant.
ResultsIn Vivo Studies
Induction of Hypothyroidism and Hyperthyroid-ism. Serum levels of T3 were significantly lower (P �.05) in the hypothyroid group and significantly higher
1122 UPADHYAY ET AL. HEPATOLOGY, April 2004

(P � .001) in the hyperthyroid group than in the controlgroup (Fig. 1A). The weights of the hypothyroid andcontrol rats remained similar throughout the experimen-tal period, whereas the weights of hyperthyroid rats de-creased (P � .05; Fig. 1B).
Markers of Liver Toxicity. Serum ALT and AST areknown markers of liver injury. We found several-fold in-creases in the levels of ALT and AST in the hyperthyroidgroup (P � .001; Fig. 1C,D). In the hypothyroid group,ALT levels decreased (P � .001), whereas AST levels re-mained unchanged (Fig. 1D).
Hyperthyroidism and DNA Fragmentation. To ex-amine whether hyperthyroidism affects liver function byinducing apoptosis, we assessed DNA fragmentation, atypical marker of apoptosis. Dose-dependent extensiveDNA fragmentation occurred in liver tissue from rats
treated with T3; it was detected after even low doses of T3(Fig. 2, lanes 3–6). Maximum DNA fragmentation wasobserved in rats treated with 100 �g T3/100 g bodyweight; this dose was used in further studies. DNA frag-mentation was not detected in the hypothyroid and con-trol groups (Fig. 2).
Hyperthyroidism and Caspase-3 and Caspase-9 Ac-tivity. Caspase-3 is the main effector caspase that is in-volved in apoptosis. To confirm that apoptosis is inducedby hyperthyroidism, we applied a colorimetric assay forcaspase-3 to the cytosolic fraction prepared from rat liverof all three groups. Caspase-3 activity was significantlyhigher in the hyperthyroid group (P � .001) than in thecontrol group (Fig. 3A). Interestingly, we found lesscaspase-3 activity in the hypothyroid group than in thecontrol group (P � .01; Fig. 3A).
Fig. 1. Change in serum biochemistry and body weight in hypothyroid and hyperthyroid rats. (A) Serum TT3: Serum TT3 was determined in bloodsamples collected from control, hypothyroid, and hyperthyroid rats (n � 15 in each group) using a standard RIA method. *P � .05 control versushypothyroid; � P � .001 control versus hyperthyroid. (B) Weight of the rats in control, hypothyroid, and hyperthyroid groups after treatment. *P �.05 control versus hyperthyroid. (C,D) Serum ALT and AST levels: Blood was collected from rats at the end of the experimental period (n � 5 in eachgroup). *P � .05 control versus hypothyroid; � P � .001 control versus hyperthyroid. � P � .001 control versus hyperthyroid; data are expressedas means � SEM.
HEPATOLOGY, Vol. 39, No. 4, 2004 UPADHYAY ET AL. 1123

In hyperthyroidism, both the alteration in mitochon-drial morphologic features reported previously7–13 andthe increased caspase-3 activity found in this studystrongly suggest that initiator caspase-9 is activated via themitochondrial pathway. Accordingly, we undertookWestern blot analysis of caspase-9 activation using a poly-clonal antibody that recognizes the procaspase andcleaved subunits. A protein band of 35 kd, correspondingto procaspase-9, with no detectable cleavage products wasobserved in both euthyroid and hypothyroid groups. Inthe hyperthyroid group, the intensity of the procaspase-9band was not significantly greater than that in the control
group (Fig. 3B). However, high levels of cleaved subunitsof 20 kd and 12 kd were found in the hyperthyroid group(P � .01).
Hyperthyroidism and Translocation of ApoptoticMolecules From Mitochondria to Cytosol. Activationof caspase-9 in hyperthyroidism strongly suggests an in-volvement of mitochondria. Accordingly, we studied thetranslocation of apoptogenic molecules from mitochon-dria to cytosol.14,18,19 Cytochrome c was localized largelyin the mitochondrial fraction in all of the experimentalgroups (Fig. 4A). However, a significant amount of cyto-chrome c was translocated to cytosol in the hyperthyroidgroup (P � .001). At an early stage of apoptosis, SMAC isreleased from mitochondria into the cytosol, where it
Fig. 3. (A) Hyperthyroidism and caspase-3 activation. Cytosolic fractions were prepared from control, hypothyroid, and hyperthyroid rat liver.Caspase-3 activity was determined using the ApopAlert caspase-3 assay kit (Clontech). There was a substantial increase in caspase-3 activity inhyperthyroid rat liver; low levels were detected in hypothyroid group. The experiments were undertaken on three subgroups; each subgroup consistedof five rats in each group. Results are expressed as mean � SEM. *P � .05 control versus hypothyroid; P � .001 control versus hyperthyroid. (B)Hyperthyroidism and caspase-9 activation. Cytosolic fractions (50 g) from liver of control (C), hypothyroid (M), and hyperthyroid (H) rats weresubjected to 15% SDS-PAGE followed by Western blotting with a caspase-9 antibody that recognizes procaspase-9 and its cleaved subunits. Cleavedsubunits of caspase-9 were significantly increased in hyperthyroid rats. Equal loading and transfer of proteins on membrane was confirmed byPonceau S staining (lower panel). The experiment was repeated three times.
Fig. 2. Hyperthyroidism and DNA fragmentation in rat liver. Rats weregiven normal saline (control), 12.5, 25, 50 or 100 �g T3/100 g bodyweight subcutaneously for 10 days (hyperthyroid), or methimazole indrinking water (hypothyroid). DNA was extracted from liver tissue asdescribed in the Materials and Methods and analyzed in a 2% agarosegel (lane M, 1 kbp DNA ladder; lane1, control; lane2, hypothyroid; lanes3–6, 12.5, 25, 50, 100 �g T3/100 g body weight, respectively). Theexperiment was repeated four times.
Fig. 4. Altered thyroid function and translocation of cytochrome c andSMAC from mitochondria into cytosol. Cytosolic and mitochondrial frac-tions were prepared from rat liver (control, C; hypothyroid: M; andhyperthyroid: H). The fractions were subjected to 15% SDS-PAGE andprobed with anti-cytochrome c (A) and anti-SMAC (B). Release ofcytochrome c from mitochondria into cytosol was detected in hyperthy-roid rats; expression and translocation of SMAC occurred in both hypo-thyroid and hyperthyroid rats. Equal loading was confirmed by PonceauS staining (C). The experiment was repeated three times.
1124 UPADHYAY ET AL. HEPATOLOGY, April 2004
binds with IAPs and inhibits their antiapoptoticactivity.14,18,19 To investigate the role of SMAC, we alsostudied its translocation. SMAC was localized in the cy-tosolic fraction in both the hypothyroid and hyperthyroidgroups. In the control group SMAC was detected in nei-ther mitochondrial pellets nor cytosol (Fig. 4B).
Hyperthyroidism, Mitochondrial Integrity, andLevels of Proteins of the Bcl-2 Family. The Bcl-2 fam-ily of proteins are located on outer membrane of mito-chondria; they maintain mitochondrial integrity14,17 andcontrol the release of apoptogenic proteins that initiatethe caspase pathway. To study the role of proteins of the
Bcl-2 family, we assessed expression of Bcl-2, Bcl-xL, andBax by Western blotting. The levels of antiapoptotic pro-teins, such as Bcl-2 and Bcl-xL, and the proapoptotic pro-tein, Bax, were not significantly changed either in eitherthe hypothyroid or hyperthyroid group (Fig. 5).
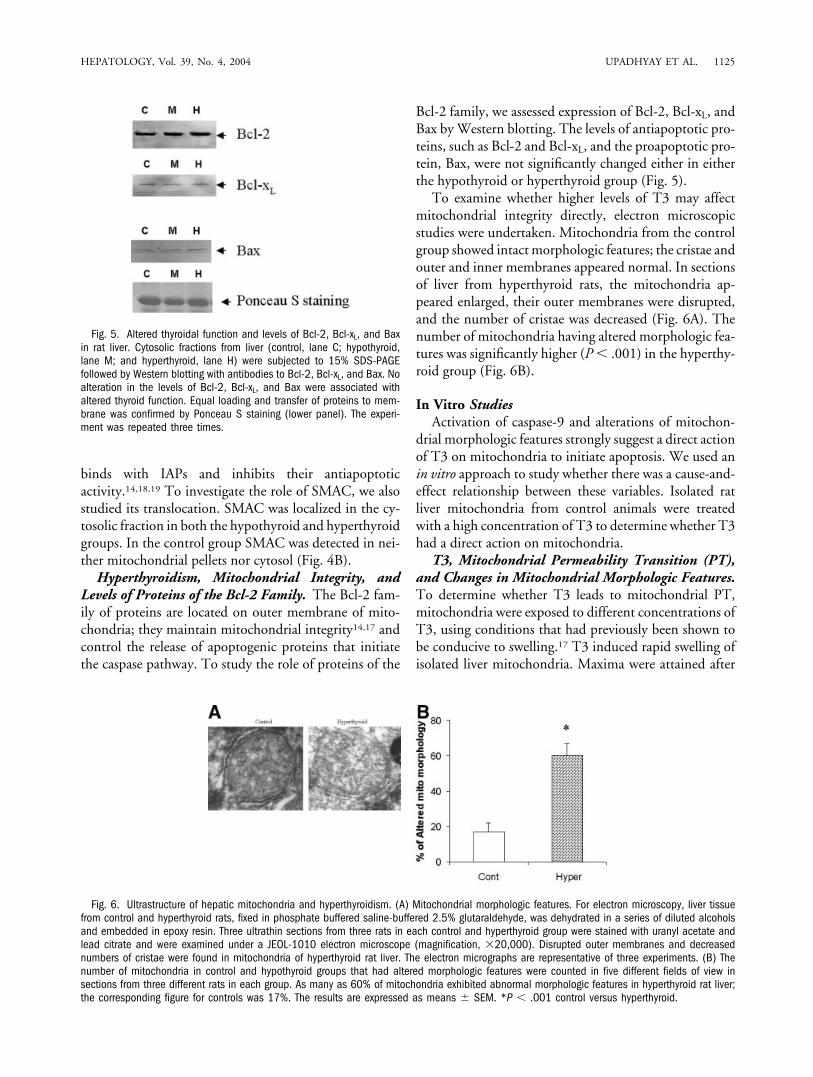
To examine whether higher levels of T3 may affectmitochondrial integrity directly, electron microscopicstudies were undertaken. Mitochondria from the controlgroup showed intact morphologic features; the cristae andouter and inner membranes appeared normal. In sectionsof liver from hyperthyroid rats, the mitochondria ap-peared enlarged, their outer membranes were disrupted,and the number of cristae was decreased (Fig. 6A). Thenumber of mitochondria having altered morphologic fea-tures was significantly higher (P � .001) in the hyperthy-roid group (Fig. 6B).
In Vitro StudiesActivation of caspase-9 and alterations of mitochon-
drial morphologic features strongly suggest a direct actionof T3 on mitochondria to initiate apoptosis. We used anin vitro approach to study whether there was a cause-and-effect relationship between these variables. Isolated ratliver mitochondria from control animals were treatedwith a high concentration of T3 to determine whether T3had a direct action on mitochondria.
T3, Mitochondrial Permeability Transition (PT),and Changes in Mitochondrial Morphologic Features.To determine whether T3 leads to mitochondrial PT,mitochondria were exposed to different concentrations ofT3, using conditions that had previously been shown tobe conducive to swelling.17 T3 induced rapid swelling ofisolated liver mitochondria. Maxima were attained after
Fig. 5. Altered thyroidal function and levels of Bcl-2, Bcl-xL, and Baxin rat liver. Cytosolic fractions from liver (control, lane C; hypothyroid,lane M; and hyperthyroid, lane H) were subjected to 15% SDS-PAGEfollowed by Western blotting with antibodies to Bcl-2, Bcl-xL, and Bax. Noalteration in the levels of Bcl-2, Bcl-xL, and Bax were associated withaltered thyroid function. Equal loading and transfer of proteins to mem-brane was confirmed by Ponceau S staining (lower panel). The experi-ment was repeated three times.
Fig. 6. Ultrastructure of hepatic mitochondria and hyperthyroidism. (A) Mitochondrial morphologic features. For electron microscopy, liver tissuefrom control and hyperthyroid rats, fixed in phosphate buffered saline-buffered 2.5% glutaraldehyde, was dehydrated in a series of diluted alcoholsand embedded in epoxy resin. Three ultrathin sections from three rats in each control and hyperthyroid group were stained with uranyl acetate andlead citrate and were examined under a JEOL-1010 electron microscope (magnification, �20,000). Disrupted outer membranes and decreasednumbers of cristae were found in mitochondria of hyperthyroid rat liver. The electron micrographs are representative of three experiments. (B) Thenumber of mitochondria in control and hypothyroid groups that had altered morphologic features were counted in five different fields of view insections from three different rats in each group. As many as 60% of mitochondria exhibited abnormal morphologic features in hyperthyroid rat liver;the corresponding figure for controls was 17%. The results are expressed as means � SEM. *P � .001 control versus hyperthyroid.
HEPATOLOGY, Vol. 39, No. 4, 2004 UPADHYAY ET AL. 1125

incubation for 12 minutes; swelling was sustained there-after (Fig. 7A).
To examine whether a change in PT results in struc-tural alterations, electron microscopic assessment wasundertaken. Mitochondria treated with T3 (6 �M) ex-hibited maximum change in PT. Untreated mitochondriaexhibited regular cristae and integrated morphologic fea-tures. Mitochondria treated with 6 �M T3 were swollen;the number of cristae was reduced and outer membraneswere enlarged and ruptured. The experiment was under-taken in duplicate, and approximately 80% of mitochon-dria exhibited abnormal morphologic features (Fig. 7B).
Nuclear Morphologic Features and DNA Fragmen-tation. The induction in PT suggests that proteins arereleased from mitochondria. To examine the apoptogenicnature of such proteins, liver nuclei were incubated withproteins released from mitochondria that had beentreated with 6 �M T3, in the presence of HeLa cell cy-tosol and dATP. The nuclei underwent dramatic mor-phologic changes and irregular condensations ofchromatin were observed (Fig. 8A). As soon as we hadconfirmed the release of apoptogenic proteins, the assaywas repeated after treatments at other concentrations ofT3. The percentage of nuclei undergoing apoptosis alsowas maximal after exposure to 6 �M T3; 67% of suchnuclei exhibited apoptotic morphologic features (Fig.8B). We found that morphologic changes in nuclei andfragmentation of chromatin DNA occurred simulta-neously (Fig. 8C). DNA fragmentation also increased asthe T3 concentration increased, up to a maximum at 6�M; it decreased at even higher concentrations of T3.
Presence of Cytochrome c in Released Factors. Cy-tochrome c is a protein that is known to be released inresponse to apoptotic stimuli.14 We applied an in vitrocytochrome c release assay. The intensity of the cyto-chrome c band increased in the supernatant and the in-tensity of the cytochrome c band in the mitochondrialpellet decreased as concentrations of T3 increased. Theintensity of the cytochrome c band in supernatants afterexposure to T3 concentrations in the range of 2 to 8 �Mwas significantly greater than that in basal supernatants inthe absence of exposure to T3 (P � .01; Fig. 9A). Spec-troscanning also confirmed the presence of cytochrome cin supernatants (Fig. 9B).
DiscussionThere is increasing evidence that apoptosis plays a ma-
jor role in various pathologic conditions. Earlier, mor-phologic changes in the liver of patients withthyrotoxicosis were attributed to necrotic cell death.3,4
However, at the time, the process of apoptosis had notbeen investigated. Here, we provide experimental evi-dence that high levels of TH activate the mitochondrialpathway of apoptosis in liver tissue.
To investigate the pathogenic effect of TH on liver, weinduced hypothyroidism and hyperthyroidism in rats andstudied the mechanism of apoptosis in the liver. Thechanges in serum levels of TT3 indicated that severe hy-perthyroidism and hypothyroidism had been induced(Fig. 1). In hyperthyroid rats, we found increased DNA
Fig. 7. (A) T3 and permeability transition in isolated mitochondria. Mitochondria isolated from rat liver were treated with different concentrationsof T3 (0, 2, 4, 6, and 8 �M) at 20°C. Mitochondrial swelling was observed by measuring decrease in optical density at 520 nm with time. Opticaldensity normalized to 100 and was plotted against time. A significant increase in mitochondrial permeability transition (PT) occurred at T3concentrations 4, 6, and 8 �M; a maximal increase in PT occurred at a T3 concentration of 6 �M. The data shown are representative of four separateexperiments. *P � .01; P � .001. (B) Ultrastructural changes in T3-treated mitochondria. Mitochondria isolated from rat liver were treated with T3(6 �M); morphologic features were assessed by electron microscopy (magnification, �18,000). T3-treated mitochondria exhibited less cristae andmore swelling and rupturing of outer membranes than normal mitochondria. These data are representative of four separate experiments.
1126 UPADHYAY ET AL. HEPATOLOGY, April 2004

fragmentation, which suggested increased apoptosis (Fig.2). Caspases, which are cysteine proteases, cleave differentintracellular targets, which results in cell shrinkage, chro-matin condensation, and DNA fragmentation.16 To con-firm that hyperthyroidism induce apoptosis, we alsostudied caspase-3 activation. We observed significant in-creases in caspase-3 activity in hyperthyroid rats (Fig. 3A),suggesting that apoptosis is caspase dependent.
Caspase-3 is activated by either death receptor or amitochondria-mediated pathway.18 However, widely rec-ognized calorigenic effects of TH in mammals32 and otherpreviously reported effects of TH on liver mitochon-dria4–10 prompted us to study mitochondrial metabolismin hyperthyroidism. The activation of initiator caspase-9in hyperthyroid rats strongly suggests an involvement of amitochondrial pathway (Fig. 3B). During apoptosis, mi-
Fig. 8. (A) Triiodothyronine-induced release of proteins and the initiation of apoptosis in vitro. Mitochondria isolated from rat liver were treatedwith different concentrations of T3 (0, 2, 4, 6, and 8 �M) at 30°C for 1 hour. After treatment, reaction mixtures were centrifuged and supernatantscontaining proteins that were released from mitochondria were collected. Aliquots of HeLa-cytosol (250 �g in 50 L) and 6 �L of liver nuclei wereincubated at 37°C for 2 hours either in the absence (a) or presence (c–f) of supernatant or cytochrome c (b). Nuclei were stained with propidiumiodide (PI) and were examined under a fluorescence microscope. Apoptotic morphologic changes in nuclei were assessed. (a) Normal morphologicfeatures; (b) positive control; (c–f) various stages of apoptosis in nuclei in the presence of supernatant after treatment with 6 �M T3. The data arerepresentative of four separate experiments. (B) The number of apoptotic nuclei in the presence of supernatant was counted in five different fieldsof view. A significant increase in the number of apoptotic nuclei was found in the presence of supernatants correlated with the concentrations of T3used before and was found in the presence of supernatant collected after treatment with T3; the magnitude of the increase in apoptotic nucleicorrelated with the T3 concentration was used. The results are expressed as means � SEM. The experiment was repeated three times. *P � .001.(C) T3-induced release of proteins from mitochondria and DNA fragmentation. DNA was extracted from rat liver nuclei of untreated (nuclei � cytosol,lane 1); negative control (nuclei � cytosol � supernatant from untreated mitochondria, lane 2); positive control (nuclei � cytosol � cytochromec, lane 3), and treated (nuclei � cytosol � supernatant collected from mitochondria treated with 2, 4, 6, and 8 �M T3, lanes 4–7, respectively).The DNA was extracted by a salt precipitation method and was analyzed in a 2% agarose gel. Appreciable DNA fragmentation occurred in DNAextracted from T3-treated nuclei (lanes 4–7).
HEPATOLOGY, Vol. 39, No. 4, 2004 UPADHYAY ET AL. 1127

tochondrial proteins located in the intermembrane spaceare released into the cytosol, where they activate thecaspase-9 pathway, as found in this study. Efflux of cyto-chrome c into cytosol is a primary event that leads to theformation of apoptosomes and activation of the caspasecascade. The release of cytochrome c was observed only inhyperthyroid rats (Fig. 4). In hypothyroid rats, the ab-sence of release of cytochrome c and DNA fragmentation,and a significant decrease in ALT levels, indicate thathypothyroidism inhibits apoptosis. SMAC in cytosol(Fig. 4) inactivate the IAPs that inhibit apoptosis, therebyrendering hepatic cells liable to undergo apoptosis.18,19
We observed the release of SMAC in both hypothyroidand hyperthyroid rats. Translocation of cytochrome c andSMAC, together with caspase-3 and caspase-9 activation,supports our hypothesis that an excess of TH causes mas-sive apoptosis via a mitochondria-mediated pathway. Inhypothyroid rats, however, release of SMAC from mito-chondria may not be sufficient to induce apoptosis, as norelease of cytochrome c was apparent. Evidence presentedhere and our earlier reports33,34 clearly suggest that ade-quate levels of TH are required for hepatocytes and othercell types to mediate their differentiated functions and toprevent them from undergoing changes that make apo-ptosis liable to occur.
To understand further the mechanism of hyperthy-roidism-induced release of cytochrome c and SMAC, westudied factors controlling the integrity of mitochondria.The Bcl-2 family of proteins are known to regulate mito-chondrial-mediated apoptosis.11,13 However, we foundno change in the expression of either antiapoptotic Bcl-2and Bcl-xL proteins or the proapoptotic Bax proteins inhyperthyroidism, suggesting that T3 may act directly onmitochondria to induce apoptotic effects (Fig. 5). The
alteration in mitochondrial morphologic features ob-served in hyperthyroid rat liver in this study, is consistentwith earlier observations (Fig. 6)7,11–13 and further sup-ports our hypothesis of a direct action of T3 on mitochon-dria.
Our in vivo observations suggest that T3 may act di-rectly on mitochondria, altering its structure and induc-ing release of apoptogenic proteins. However, themechanism of action of T3 on liver cells in vivo is com-plicated; T3 acts on both the nucleus and the mitochon-dria. Accordingly, an in vitro system became our methodof choice to study direct actions of T3 on liver mitochon-dria. In many aspects of cell biology and metabolism, invitro reconstitution of complicated pathways has facili-tated considerably the assessment of cause-and-effect re-lationships between various cellular components. Ahigher concentration of substrate is required for a directeffect to occur in a cell-free system.31 Therefore, we usedhigher concentrations of T3, which previously have beenshown to induce effects in isolated mitochondria.12,32
Our finding that T3 induced an increase in mitochondrialPT (Fig. 7A) is in agreement with the findings of oth-ers.11–13 The increase in mitochondrial PT is accompa-nied by electron-microscopic changes in mitochondrialmorphologic features (Fig. 7B). The T3-induced mito-chondrial swelling reported here is analogous to the in-crease in mitochondrial size observed histologically inliver tissue from hyperthyroid rats in this study and liverbiopsies from hyperthyroid patients reported previously.7
The change in PT results in the release of apoptogenicproteins from mitochondria.14 We investigated the re-lease and nature of those proteins in vitro. T3 inducesrelease of cytochrome c (Fig. 9A,B) and other proteins,having molecular weights in the range 20 kd to 50 kd
Fig. 9. (A) T3 and cytochrome c release from rat liver mitochondria. Mitochondrial supernatants and pellets were prepared as described. Releaseof cytochrome c was assessed by Western blotting. Significant release of cytochrome c was detected in supernatants after mitochondria had beentreated with 2, 4, 6, and 8 �M T3 (lanes 2–5). Lane 1 shows basal release of cytochrome c; lane C shows a positive control in which purifiedcytochrome c was loaded. Equal loading and transfer of proteins on membrane were confirmed by Ponceau S staining (lower panel). The experimentwas repeated three times. (B) Absorption spectrum of cytochrome c. Mitochondria isolated from rat liver were treated with T3 (6 �M). Aftercentrifugation, the supernatant was analyzed for the presence of cytochrome c by spectroscopic scanning. The supernatant had peaks of absorptionat 415, 520, and 549 nm, a pattern characteristic of the reduced form of cytochrome c.
1128 UPADHYAY ET AL. HEPATOLOGY, April 2004

(data not shown), that orchestrate the process of apoptosis(Fig. 8B,C). The formation of condensed chromatin bod-ies observed in vitro in liver nuclei exposed to proteinsreleased by T3-treated mitochondria (Fig. 8A,B) seems tobe analogous to nuclear irregularity and ballooning ofnuclei reported previously in liver histologic results ofthyrotoxic patients.1–8 The in vitro observations supportour hypothesis of a direct action of T3 on mitochondria,and also corroborate our in vivo findings of T3-induceddisruption of mitochondrial integrity and release of apop-togenic proteins by mitochondria. In addition, the resultsof this study support an earlier hypothesis that an intrinsicpathway of apoptosis prevails in hepatocytes.35
This study has provided insights into the pathogenesisof liver dysfunction associated with severe hyperthyroid-ism. No firm evidence has been provided on the nature ofthe hepatic injury in existing literature on hyperthyroid-ism.1–8 Data from this study suggest that hyperthyroid-ism induces apoptosis in the liver. Our results provideevidence that excess TH may be directly toxic to the liver.However, the study does not rule out the possibility ofliver damage occurring secondary to the systemic effectsof excess TH. Our in vitro and in vivo data together indi-cate that T3 acts directly on mitochondria; it induces PTand release of proteins that lead to activation downstreamof the caspase cascade, which, in turn, leads to apoptosis.However, the mechanisms of the change in PT and therelease of apoptotic proteins from T3-treated mitochon-dria currently are not understood. Adenine nucleotidetranslocator, a unit of the PT pore complex, was reportedto bind T3 with high affinity.36,37 TH receptors also havebeen identified in the matrix of rat liver mitochondria.38
Whether binding of T3 to mitochondrial T3 receptorsand adenine nucleotide translocator regulates the PT andrelease of apoptogenic proteins has not yet been investi-gated. Further studies should enable us to broaden ourunderstanding of action of T3, not only on the liver, butalso on other organs.
Acknowledgment: The authors thank Dr. X. Wang(University of Southwestern Texas Medical Centre, Dal-las, TX) for the kind gift of antibodies against SMAC andcaspase-9, Dr. R. Jemmerson (University of Minnesota)for anti-cytochrome c, and Dr. Shashi Wadhwa (All IndiaInstitute of Medical Sciences, New Delhi, India) for pro-viding the electron microscope facility. We also thank Dr.Cliona Stapleton, NIEHS, NIH, and Dr. S.K. Yaccha,SGPGIMS, Lucknow, for their critical evaluation of themanuscript.
References1. Habershon SO. Exophthalmic goiter, heart disease; jaundice: death. Lan-
cet 1874;1:510–512.
2. Gavin LA, Cavalieri RR. Interrelationship between thyroid gland and theliver. In: Zakim D, Boyer TD, eds. Hepatology: A Textbook of LiverDisease. Philadelphia: Saunders, 1982:508–515.
3. Inoue T, Tanigawa K, Furuya H, et al. A case of thyroid crisis complicatedwith acute hepatic failure. Nippon Naika Gakki Zasshi 1988;77:564–567.
4. Bhattacharyya A, Wiles PG. Thyrotoxic crisis presenting as acute abdo-men. J Roy Soc Med 1997;90:681–682.
5. Choudhary AM, Roberts I. Thyroid storm presenting with liver failure.J Clin Gastroentrol 1999;29:318–321.
6. Dooner HP, Parada J, Aliaga C, Hoyl C. The liver in thyrotoxicosis. ArchIntern Med 1967;120:25–32.
7. Klion FM, Segal R, Schaffner F. The effect of altered thyroid function onthe ultrastructure of the human liver. Am J Med 1971;50:317–324.
8. Kalderon B, Hermesh O, Bar-Tana J. Mitochondrial permeability transi-tion is induced in vivo by thyroid hormone treatment. Endocrinology1995;136:3552–3556.
9. Kalderon B, Hertz R, Bar-Tana J. Effect of thyroid hormone on redox andphosphate potentials in rat liver. Endocrinology 1992;131:400–407.
10. Seitz HJ, Muller MJ, Soboll S. Rapid thyroid-hormone effect on mito-chondrial and cytosolic ATP/ADP ratios in intact rat liver. Biochem J1985;227:149–153.
11. Sestoft L. Metabolic aspects of the calorigenic effect of thyroid hormone inmammals. Clin Endocrinol 1980;13:489–506.
12. Harper ME, Brand MD. The quantitative contributions of mitochondrialproton leak and ATP turnover reaction to the changed respiration rates ofhepatocytes from rats of different thyroid status. J Biol Chem 1993;268:14850–14860.
13. Gregory RB, Berry MN. On thyroid hormone induced increase in respi-ratory capacity of isolated rat hepatocytes. Biochim Biophys Acta 1991;1098:61–67.
14. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–1311.
15. Adrain C, Martin SJ. The mitochondrial apoptosome: a killer unleashed bythe cytochrome seas. Trends Biochem Sci 2001;26:390–397.
16. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science 1998;281:1312–16.
17. Hengartner MO. The biochemistry of apoptosis. Nature 2000;407:770–776.
18. Verhagen A, Ekert PG, Pakusch M, Silke J, Connoly LM, Reid GE, MoritzRL, et al. Identification of DIABLO, a mammalian protein that promotesapoptosis by binding and antagonizing IAP proteins. Cell 2000;102:43–53.
19. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein thatpromotes cytochrome c-dependent caspase activation by eliminating IAPinhibition. Cell 2000;102:33–42.
20. Malkevitch NV, Dedukhova VI, Simonian RA, Skulachev VP, StarkovAA. Thyroxine induces cyclosporine A-insensitive Ca�2 dependent revers-ible permeability transition pore in rat liver mitochondria. FEBS Lett1997;412:173–178.
21. Mihara S, Suzuki N, Wakisaka S, Suzuki S, Sekita N, Yamamoto S, SaitoN, et al. Effects of thyroid hormone on apoptotic cell death of humanlymphocytes. J Clin Endocrinol Metabol 1999;84:1378–1385.
22. Su Y, Shi Y, Stolow MA, Shi YB. Thyroid hormone induces apoptosis inprimary cell cultures of tadpole intestine: cell type specificity and effects ofextracellular matrix J Cell Biol 1997;139:1533–1543.
23. Calvo R, Obregon MJ, Ruiz de Ona C, Escobar del Rey E, Morreal deEscobar G. Congenital hypothyroidism as studied in rats. Critical role ofmaternal thyroxine but not of 3,5,3� triiodothyronine in the protection ofthe fetal brain. J Clin Invest 1990;86:889–899.
24. Francavilla A, Carr BI, Azzrone A, Polimeno L, Wang Z, Van Thiel DH,Subbotin V, et al. Hepatocyte proliferation and gene expression induced bytriiodothyronine in vivo and in vitro. HEPATOLOGY 1994;20:1237–1241.
25. Linnik MD, Miller JA, Cavallo JS, Mason PJ, Thompson FY, Montgom-ery LR, Schroeder KK. Apoptotic DNA fragmentation in the rat cerebralcortex induced by permanent middle cerebral artery occlusion. Brain ResMol Brain Res 1995;32:116–124.
HEPATOLOGY, Vol. 39, No. 4, 2004 UPADHYAY ET AL. 1129

26. Vega-Nunez E, Alaverz AM, Menendez-Hurtado A, Santos A, Perez-Castillo A. neuronal mitochondrial morphology and transmembrane po-tential are severely altered by hypothyroidism during rat braindevelopment. Endocrinology 1997;138:3771–3778.
27. Lowry OH, Rosebrough NH, Farr AL, Randall RJ. Protein measurementswith the Folin phenol reagent. J Biol Chem 1951;193:265–275.
28. Feldmann G, Haouzi D, Moreau A, Durand-Schneider AM, Bringuier A,Berson A, Mansouri A, et al. Opening of the mitochondrial permeabilitytransition pore causes matrix expansion and outer membrane rupture inFas-mediated hepatic apoptosis in mice. HEPATOLOGY 2000:31:674–683.
29. Jurgensmeier J, Xie Z, Deveraux Q, Ellerby L, Bredsen D, Reed JC. Baxdirectly induces release of cytochrome c from isolated mitochondria. ProcNatl Acad Sci U S A 1998;95:4997–5002.
30. Blobel G, Potter VR. Nuclei from rat liver: Isolation method that com-bines purity with high yield. Science 1966;154:1662–1665.
31. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptoticprogram in cell-free extracts: requirement for dATP and cytochrome c.Cell 1996;86:147–157.
32. Scott A, Hunter FE Jr. Support of thyroxine-induced swelling of livermitochondria by generation of high energy intermediates at any one ofthree sites in electron transport. J Biol Chem 1966;242:1060–1065.
33. Singh R, Upadhyay G, Kumar S, Kapoor A, Kumar A, Tiwari M, GodboleMM. Hypothyroidism alters the expression of Bcl-2 family genes to induceenhanced apoptosis in the developing cerebellum. J Endocrinol 2003;176:39–46.
34. Singh R, Upadhyay G, Godbole MM. Hypothyroidism alters mitochon-drial morphology and induces release of apoptogenic proteins during de-veloping rat cerebellum. J Endocrinol 2003;176:321–326.
35. Li S, Zhou Y, He X, Kim TH, Kuharsky DK, Rabinowich H, Chen J, et al.Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrialrelease of SMAC in Fas-mediated hepatocyte apoptosis. J Biol Chem 2002;277:26912–26920.
36. Sterling K. Thyroid hormone action: identification of the mitochondrialthyroid hormone receptor as adenine nucleotide translocase. Thyroid1991;1:167–171.
37. Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, VieiraHL, Prevost MC, et al. Bax and adenine nucleotide translocator coop-erate in the mitochondrial control of apoptosis. Science 1998:281:2027–2031.
38. Wrutnaik CC, Cassar-Malek I, Marchal S, Rascle A, Heusser S, KellerJM, Flechon J, et al. A 43 kDa protein related to c-Erb A-1 is located inthe mitochondrial matrix of rat liver. J Biol Chem 1995;270:16347–16354.
1130 UPADHYAY ET AL. HEPATOLOGY, April 2004




















