
Role of tryptophan-208 residue in cytochrome c oxidation by ascorbate peroxidase from Leishmania...
9
Role of tryptophan-208 residue in cytochrome c oxidation by ascorbate peroxidase from Leishmania major -kinetic studies on Trp208Phe mutant and wild type enzyme Rajesh K. Yadav, Subhankar Dolai, Swati Pal, Subrata Adak ⁎ Division of Structural Biology and Bioinformatics, Indian Institute of Chemical Biology, 4 Raja S.C. Mullick Road, Kolkata-700 032, India article info abstract Article history: Received 15 January 2008 Received in revised form 8 February 2008 Accepted 11 February 2008 Available online 23 February 2008 Ascorbate peroxidase from L. Major (LmAPX) is a functional hybrid between cytochrome c peroxidase (CCP) and ascorbate peroxidase (APX). We utilized point mutagenesis to investigate if a conserved proximal tryptophan residue (Trp208) among Class I peroxidase helps in controlling catalysis. The mutant W208F enzyme had no effect on both apparent dissociation constant of the enzyme–cytochrome c complex and K m value for cytochrome c indicating that cytochrome c binding affinity to the enzyme did not alter after mutation. Surprisingly, the mutant was 1000 times less active than the wild type in cytochrome c oxidation without affecting the second order rate constant of compound I formation. Our diode array stopped-flow spectral studies showed that the substrate unbound wild type enzyme reacts with H 2 O 2 to form compound I (compound II type spectrum), which was quite different from that of compound I in W208F mutant as well as horseradish peroxidase (HRP). The spectrum of the compound I in wild type LmAPX showed a red shift from 409 nm to 420 nm with equal intensity, which was broadly similar to those of known Trp radical. In case of compound I for W208F mutant, the peak in the Soret region was decreased in heme intensity at 409 nm and was not shifted to 420 nm suggesting this type of spectrum was similar to that of the known porphyrin π- cation radical. In case of an enzyme–H 2 O 2 –ascorbate system, the kinetic for formation and decay of compound I and II of a mutant enzyme was almost identical to that of a wild type enzyme. Thus, the results of cytochrome c binding, compound I formation rate and activity assay suggested that Trp208 in LmAPX was essential for electron transfer from cytochrome c to heme ferryl but was not indispensable for ascorbate or guaiacol oxidation. © 2008 Elsevier B.V. All rights reserved. Keywords: Leishmania Peroxidase Cytochrome c Electron transfer Rapid kinetics 1. Introduction Throughout the life cycle of the protozoan parasite Leishmania it is exposed to a variety of reactive oxygen species such as superoxide anion radical, hydroxyl radical, and hydrogen peroxide [1–5], and their cytotoxic effects are removed by a specific enzyme system [6,7]. In most eukaryotes two classes of heme proteins designated as hydroperoxidases, that is, catalase and peroxidase, play a central role in protecting cells from oxidative damage. Many aspects of the oxidative defense system of Leishmania, particularly those that are involved in hydroperoxide metabolism, are distinct from pathways used by other organisms, for example, the parasite has been reported to lack catalase. A novel type of hydroperoxidase the so-called ascorbate peroxidase (LmAPX) has been recently proposed to exhibit peroxidatic activities in Leishmania [8]. This enzyme is distinct from mammalian peroxidases but has similarity with the enzyme present in the plant kingdom. The LmAPX mediated detoxification of H 2 O 2 has been shown to be carried out by the wide range of reducing electron donors including iodide, guaiacol (aromatic donor), ascorbate and cytochrome c [8,9]. However, SCN- a substrate for mammalian peroxidases, is acting as an inhibitor in this enzyme. This enzyme can also catalyze the oxidation of H 2 O 2 to molecular oxygen in the presence of low concentration of ascorbate at physiological pH [8,9]. Putatively, APX catalyzes the electron transfer from an oxidizable substrate to a H 2 O 2 molecule following the classical peroxidase mechanism [10–13]. First, in the presence of H 2 O 2 , a two-oxidizing equivalent higher intermediate, compound I, is produced. One- oxidizing equivalent is stored in a ferryl (Fe iv ) state with S = 1 and the second, as a porphyrin π-cation radical [14,15]. In contrast to APX, the second oxidizing equivalent of cytochrome c peroxidase (CCP) compound I is localized on a spatially removed paramagnetic species with S=1/2, namely, a protein-based tryptophan radical [16]. This intermediate is then sequentially reduced back by substrate molecules. A group of worker prepares a CCP-like HRP mutant, F221W (Phe221 to Trp), and shows from their EPR studies that the reaction of HRP mutant with H 2 O 2 produces typical signals of a Trp radical as found for CCP [17]. The observation of ferryl porphyrin π-cation radical in APX is somewhat unexpected, since APX contains a tryptophan at position 179, analogous to that known [14] to be the site of protein oxidation in Biochimica et Biophysica Acta 1784 (2008) 863–871 ⁎ Corresponding author. Tel.: +91 33 2473 6793; fax: +91 33 2473 5197. E-mail address: [email protected] (S. Adak). Abbreviations: APX, ascorbate peroxidase; LmAPX, Leishmania major ascorbate peroxidase; HRP, horseradish peroxidase; CCP, cytochrome c peroxidase 1570-9639/$ – see front matter © 2008 Elsevier B.V. All rights reserved. doi:10.1016/j.bbapap.2008.02.006 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbapap
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Role of tryptophan-208 residue in cytochrome c oxidation by ascorbate peroxidase from Leishmania...

Biochimica et Biophysica Acta 1784 (2008) 863–871
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbapap
Role of tryptophan-208 residue in cytochrome c oxidation by ascorbate peroxidasefrom Leishmania major-kinetic studies on Trp208Phe mutant and wild type enzyme
Rajesh K. Yadav, Subhankar Dolai, Swati Pal, Subrata Adak ⁎
Division of Structural Biology and Bioinformatics, Indian Institute of Chemical Biology, 4 Raja S.C. Mullick Road, Kolkata-700 032, India
a r t i c l e i n f o
⁎ Corresponding author. Tel.: +91 33 2473 6793; fax:E-mail address: [email protected] (S. Adak).Abbreviations: APX, ascorbate peroxidase; LmAPX
peroxidase; HRP, horseradish peroxidase; CCP, cytochro
1570-9639/$ – see front matter © 2008 Elsevier B.V. Aldoi:10.1016/j.bbapap.2008.02.006
a b s t r a c t
Article history:Received 15 January 2008Received in revised form 8 February 2008Accepted 11 February 2008Available online 23 February 2008
Ascorbate peroxidase from L. Major (LmAPX) is a functional hybrid between cytochrome c peroxidase (CCP)and ascorbate peroxidase (APX). We utilized point mutagenesis to investigate if a conserved proximaltryptophan residue (Trp208) among Class I peroxidase helps in controlling catalysis. The mutant W208Fenzyme had no effect on both apparent dissociation constant of the enzyme–cytochrome c complex and Km
value for cytochrome c indicating that cytochrome c binding affinity to the enzyme did not alter aftermutation. Surprisingly, the mutant was 1000 times less active than the wild type in cytochrome c oxidationwithout affecting the second order rate constant of compound I formation. Our diode array stopped-flowspectral studies showed that the substrate unbound wild type enzyme reacts with H2O2 to form compound I(compound II type spectrum), which was quite different from that of compound I in W208F mutant as well ashorseradish peroxidase (HRP). The spectrum of the compound I in wild type LmAPX showed a red shift from409 nm to 420 nm with equal intensity, which was broadly similar to those of known Trp radical. In case ofcompound I for W208F mutant, the peak in the Soret region was decreased in heme intensity at 409 nm andwas not shifted to 420 nm suggesting this type of spectrum was similar to that of the known porphyrin π-cation radical. In case of an enzyme–H2O2–ascorbate system, the kinetic for formation and decay of compoundI and II of a mutant enzymewas almost identical to that of awild type enzyme. Thus, the results of cytochromec binding, compound I formation rate and activity assay suggested that Trp208 in LmAPX was essential forelectron transfer from cytochrome c to heme ferryl but was not indispensable for ascorbate or guaiacoloxidation.
© 2008 Elsevier B.V. All rights reserved.
Keywords:LeishmaniaPeroxidaseCytochrome cElectron transferRapid kinetics
1. Introduction
Throughout the life cycle of the protozoan parasite Leishmania it isexposed to a variety of reactive oxygen species such as superoxideanion radical, hydroxyl radical, and hydrogen peroxide [1–5], and theircytotoxic effects are removed by a specific enzyme system [6,7]. Inmost eukaryotes two classes of heme proteins designated ashydroperoxidases, that is, catalase and peroxidase, play a centralrole in protecting cells from oxidative damage. Many aspects of theoxidative defense system of Leishmania, particularly those that areinvolved in hydroperoxide metabolism, are distinct from pathwaysused by other organisms, for example, the parasite has been reportedto lack catalase. A novel type of hydroperoxidase the so-calledascorbate peroxidase (LmAPX) has been recently proposed to exhibitperoxidatic activities in Leishmania [8]. This enzyme is distinct frommammalian peroxidases but has similarity with the enzyme presentin the plant kingdom. The LmAPXmediated detoxification of H2O2 has
+91 33 2473 5197.
, Leishmania major ascorbateme c peroxidase
l rights reserved.
been shown to be carried out by the wide range of reducing electrondonors including iodide, guaiacol (aromatic donor), ascorbate andcytochrome c [8,9]. However, SCN- a substrate for mammalianperoxidases, is acting as an inhibitor in this enzyme. This enzymecan also catalyze the oxidation of H2O2 to molecular oxygen in thepresence of low concentration of ascorbate at physiological pH [8,9].
Putatively, APX catalyzes the electron transfer from an oxidizablesubstrate to a H2O2 molecule following the classical peroxidasemechanism [10–13]. First, in the presence of H2O2, a two-oxidizingequivalent higher intermediate, compound I, is produced. One-oxidizing equivalent is stored in a ferryl (Feiv) state with S=1 and thesecond, as a porphyrin π-cation radical [14,15]. In contrast to APX, thesecond oxidizing equivalent of cytochrome c peroxidase (CCP)compound I is localized on a spatially removed paramagnetic specieswith S=1/2, namely, a protein-based tryptophan radical [16]. Thisintermediate is then sequentially reduced back by substratemolecules.A group of worker prepares a CCP-like HRPmutant, F221W (Phe221 toTrp), and shows from their EPR studies that the reaction of HRPmutantwith H2O2 produces typical signals of a Trp radical as found for CCP[17]. The observation of ferryl porphyrin π-cation radical in APX issomewhat unexpected, since APX contains a tryptophan at position179, analogous to that known [14] to be the site of protein oxidation in

864 R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
CCP. In sharp contrast to these reports, recently Hiner et al [18] hasshown from their UV/visible and EPR spectroscopies together withHPLC studies that the APX can indeed form a protein-based radicalanalogous to that found in the CCP. Thus, it is quite clear that the natureof the compound I intermediate has been an important point fordebate in APX work, which remains to be solved unambiguously.
Based on the nucleotide-derived protein sequence studies, theLmAPX is related to the Class I group of heme peroxidase [8].Comparison of the LmAPXwith the plant APX or CCP sequence revealsthat the LmAPX has several features that are clearly unique withrespect to other APX or CCP e.g. (a) the specific activity of ascorbateand cytochrome c oxidation by LmAPX is lower compared to the plantAPX and CCP respectively, and most interestingly (b) the crucialascorbate or cytochrome c binding residue of Class I peroxidase isabsent in this enzyme suggesting a different binding mechanism[8,19]. Several spectroscopic studies combined with site-directedmutagenesis [11,14,20] have revealed that the porphyrin π-cationradical containing compound I of ascorbate peroxidase catalyzes theoxidation of small organic and inorganic compounds, whereas CCP hasa radical center on the tryptophan residue and prefers cytochrome c asthe redox partner [15,21–25]. The formation of the Trp radical wouldfacilitate one electron transfer from cytochrome c to the porphyrin[26,27]. Thus, the Trp radical in compound I in CCP is a key factor forcytochrome c oxidation.
The X-ray structure, structure-based engineering and calorimetricstudies in CCP–cytochrome c complex has revealed that interproteinelectron transfer between CCP and cytochrome c is originally impliedto occur through a tight–binding complex with 1:1 stoichiometry [27–29]. However, there has been a discrepancy, other research workershave shown that the CCP can bind cytochrome c at two distinctdomains, one of which undoubtedly involves the surface areavisualized in the crystal structure, and that a 2:1 complex can formover a range of ionic strengths [30–32]. On the other hand, theelectron transfer from low-affinity binding site to CCP compound I stillis not understood in detail. To date, the role of proximal tryptophanresidue in high-affinity binding site deficient CCP has not beenidentified. LmAPX, which catalyses the oxidation of cytochrome c byH2O2, may be used as a model enzyme for high-affinity cytochrome cbinding site deficient CCP.
In the present work, mutagenesis is used to simplify themechanism of electron transfer reaction, by creating a LmAPX mutantthat lacks the site of stable protein radical formation. The mutation ofproximal Trp-208 to Phe (Trp191 to Phe in CCP) eliminates the site ofthe stable protein radical in compound I, but does not alter thecompound I formation rate with π-cation oxyferryl heme site. Ourpresent experiments have indicated that the rate of electron transferfrom ferrous cytochrome c to the oxyferryl center is decreasedsignificantly by this mutation although the rate of electron transferfrom ascorbate to oxyferryl group does not change significantly. Adetailed characterization of the electron transfer reaction of theLmAPX and W208F mutant is reported here.
2. Materials and methods
2.1. Materials
All reagents and materials were obtained from Sigma or sources previouslyreported [8,9,33,34].
2.2. Molecular biology
Wild type LmAPX (34 amino acid deleted from N-terminal sequence of LmAPXgene) and mutants containing a His6 tag attached to their N terminus were overexpressed in Escherichia coli strain BL21 (DE3) using a pTrcA vector and were purified asdescribed [33]. Restriction digestions, cloning, and bacterial growth were performedusing standard procedures [8]. Site directed mutagenesis was done using the QuickChange polymerase chain reaction in vitro mutagenesis kit from Stratagene.Incorporated mutations were confirmed by DNA sequencing at the Indian Institute of
Chemical Biology core facility. DNAs containing the desired mutations weretransformed into E. coli for protein expression. Oligonucleotides used to constructsite-directed mutants in the LmAPX were synthesized by Integrated DNA Technologies.Oligonucleotides were as follows: W208F-sense: 5′-CGGCTACCATGGGCCGTTCACACAC-GACAAGAACGG-3′; W208F-antisense: 5′-CCGTTCTTGTCGTGTGTGAACGGCCCATGG-TAGCCG-3′; W208Y-sense: 5′-CGGCTACCATGGGCCGTACACACACGACAAGAACGG-3′;and W208Y-antisense: 5′-CCGTTCTTGTCGTGTGTGTACGGCCCATGGTAGCCG-3′.
2.3. Expression and purification of wild type and mutants LmAPX
Wild type and mutant LmAPX were expressed in E. coli and purified as describedpreviously [8]. UV–visible spectra were recorded in a Shimadzu-2550 spectro-photometer using 50 mM phosphate buffer pH-7.4. The Soret peak at 409 nm wasused to quantitate the heme protein content using an extinction coefficient of101 mM−1 cm−1 [8].
2.4. Steady-state activity assays
The steady-state activity assays of wild type, W208F and W208Y were carried outusing ascorbate, guaiacol, and ferrocytochrome c as substrate using previouslypublished protocols [9]. All assays were performed at room temperature on aShimadzu-2550 UV–vis spectrophotometer using 230 nM LmAPX and 0.3 mM H2O2
in 50 mM potassium phosphate buffer (pH 7.4). Ascorbate peroxidase assays wereperformed using 50 to 500 μM sodium ascorbate. The oxidation of ascorbate wasmonitored at 290 nm using an ε290 of 2.8 mM−1cm−1 for oxidized ascorbate. Guaiacoloxidation was measured using 0.75 to 25 mM guaiacol at 470 nm using an ε470 of26.6 mM−1cm−1 for oxidized guaiacol. Ferrocytochrome c oxidation was measuredusing 5 μM to 100 μM ferrocytochrome c at 550 nm using an extinction coefficient ε550of 21 mM−1cm−1 for oxidized ferrocytochrome c.
2.5. Stopped-flow experiments
Transient spectra were recorded in a stopped-flow instrument from Hi-Tech Ltd.(KinetAsyst) equipped with a rapid-scanning diode array device (Hi-Tech MG-6560)designed to collect 300 complete spectrawithin 450 ms in each mixing. Rapid scanningexperiments involved mixing solutions containing native enzyme in 50 mM phosphatebuffer, pH 7.5 with varying concentration of H2O2 solutions. Formation of thecompound I was followed at 408 nm for the LmAPX and 402 nm for HRP. The kineticand spectral data of compound I in LmAPX was recorded at 10 °C due to rapiddisappearance of porphyrin radical at room temperature. Signal-to-noise ratios wereimproved by averaging 10 individual traces. The diode array data were then fit todifferent reactionmodels by a Specfit program fromHi-tech Ltd. to obtain the calculatednumber of species, their individual spectra, the concentration of each species versustime, and rate constants for each transition.
2.6. Sedimentation equilibrium
Sedimentation equilibrium experiments of the enzyme–cytochrome c complex wererecorded in a Beckman Coulter Proteome Lab™ XL-I (Protein Characterization System)analytical ultracentrifuge equipped with absorbance optics using different loadingconcentration in six-channel centerpieces (path length 1.2 cm and 3 mm column length).Enzymes and cytochrome c were extensively dialyzed against 50 mM potassiumphosphate buffer pH 7.5 at 4 °C. Sedimentation equilibrium was attained at rotor speedof 10,000 rpm after 22 h run at 22 °C, where two scans taken at 5 h intervalswere perfectlyoverlaid. Sedimentation equilibrium profiles were obtained by scanning at 430 nm. Partialspecific volume (0.7335 ml/g for enzyme and 0.744 mL/g for horse cytochrome c) fromtheir amino acid sequence, buffer density (1.00 g/ml), and buffer viscosity (0.01 mPa s)were calculated by using SEDNTERP software version 1.09 (freeware from www.jphilo.mailway.com). SEDFIT and SEDPHAT (freeware from www.analyticalultracentrifugation.com) were used for transforming and analyzing the sedimentation equilibrium data tocalculate apparent equilibrium dissociation constant. All analyses involved fitting a modelto absorbance versus cell radius data using nonlinear regression. The parameters of globalfitting were concentration, molecular weight, mass conservation, molar extinctioncoefficient of both proteins, cellmeniscus and bottom, and baseline. Extinction coefficientsof ferrocytochrome c and the LmAPX at 430 nm were 45.8 and 40.7 mM−1·cm−1
respectively. In each case, three different loading concentrations were analyzedsimultaneously.
3. Results
3.1. Primary structure analysis
Examination of the LmAPX primary sequence is indicated that thehigh level of sequence identity is present in this enzyme with respectto the CCP (~35%) and the Pea APX (~36%) (Fig. 1A). However it lacksthe yeast CCP residues (Asp34/Glu35, Tyr39 and Glu290) required forcytochrome c binding and also the crucial pea APX residue (Arg 172)necessary for ascorbate binding [27,35]. Although the proximal cation

Fig. 1. Comparative analysis of LmAPX, APX, and CCP sequences and structural elements. Panel A, align LmAPX (top), pea APX (middle), and yeast CCP (bottom) sequences withmapped residue function and primary structure as determined from the substrate bound pea APX and yeast CCP structures [19,27]. The amino acid residues of the proximal and distalsites of heme implicated in the redox activity of Class I superfamily peroxidases are denoted by bold face letters. The boxed region of LmAPX and CCP represent C-terminal insertion.The residues involved in electrostatic interactions of the dimer formation, Trp radical stabilization, ascorbate binding, cytochrome c and K+-binding site in peroxidase are representedby d, r, a, c and p respectively. Panel B represents a cartoon structure of the heme surrounding conserved amino acid residues in the LmAPX. Panel C shows the heme coordinationstructure of resting state, Compound I and Compound II with their Soret absorption wavelengths.
865R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
binding loop of the LmAPX is more similar with pea APX, yet thepresence of a sequence insertion near c-terminal and proximal Trpradical stabilizing Met residues of the CCP are more similar with theLmAPX. The cartoon structure of the heme peripheral Arg64, Trp67,His68, His192, Trp208 and Asp253 residues of the LmAPX is identicalto distal and proximal residues of the CCP or Pea APX (Fig. 1B). Severalspectroscopic and EPR studies combined with site-directed mutagen-esis in the yeast CCP have revealed that proximal Trp191 (correspond-ing to Trp208 of LmAPX) plays an obligatory role for protein radicalformation in the compound I of CCP. The schematic presentation of thenative and transient intermediates of the LmAPXwith their Soret bandhas been shown in Fig. 1C. The reaction of H2O2 with the resting ferricform of peroxidase (Soret band at 408 nm) leads to the formation ofcompound I, which contains either a stable oxyferryl porphyrin cationradical (Soret band at 409 nmwith lower intensity) [14,15] or a stableoxyferryl tryptophan cation radical (Soret band at 420 nm)[21,23,24,36]. Extensive spectroscopic studies have demonstratedthat the Soret band of compound II is similar to an oxyferryltryptophan cation radical of compound I species [37].
The optical spectrum of the W208F mutant closely resembled thatof wild type LmAPX at pH 7.5 in the absence of ascorbate but W208Ymutant shows low spin heme. The addition of H2O2 to W208F mutantcaused a spectral red shift from high spin to low spin, indicating thatthe mutant can react with H2O2 while W208Y mutant does not reactwith H2O2 (Fig. 2). The reduction of oxidized W208F by ascorbate
indicates that its heme iron ligation is the same as in the wild typeLmAPX. Cytochrome c binding affinities are determined by sedimen-tation equilibriummethod in the presence of 50mMphosphate bufferpH-7.5 (Fig. 3). When the final protein distribution at equilibrium isanalyzed as a single homogeneous species, satisfactory fits areobtained for sedimentation equilibrium distribution, as derived fromthe global fit of SEDPHAT programme. The global fit shows that the Kd
value of cytochrome c for wild type andW208F LmAPX are 7.7 μM and10 μM, respectively. Thus, cytochrome c binding is not significantlyperturbed by the mutation of Trp208 to Phe.
Table 1 compares the catalytic turnover numbers of wild typeLmAPX and the Trp208Phe mutant with regard to ascorbate, guaiacoland cytochrome c, but the LmAPX enzyme exhibits an oxidation rateof ascorbate or cytochrome c that is much slower than the plant APXor the yeast CCP, respectively [25,38]. In the steady state, substitutionof Trp208 with Phe is altered to a 1000 times slower rate ofcytochrome c oxidation, but does not significantly alter ascorbate orguaiacol oxidation, suggesting that the mutation only affects themacro-molecule oxidation. To understand how the mutationdecreases the rate of cytochrome c oxidation by the LmAPX, wehave first examined the Km value of the mutant in cytochrome coxidation. As shown in Table 1, the Km value of cytochrome c inmutant protein is similar to wild type LmAPX. This suggests that thecytochrome c binding to a catalytically active enzyme is notsignificantly altered by the mutation of Trp208 to Phe.
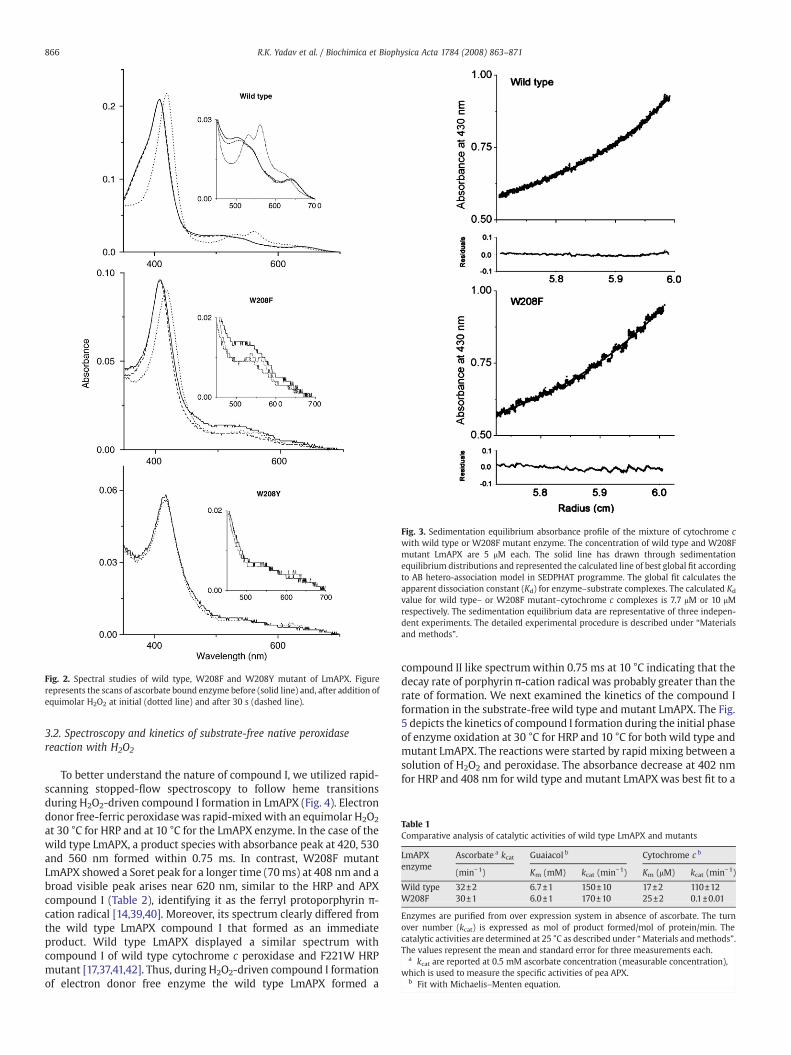
Fig. 3. Sedimentation equilibrium absorbance profile of the mixture of cytochrome cwith wild type or W208F mutant enzyme. The concentration of wild type and W208Fmutant LmAPX are 5 μM each. The solid line has drawn through sedimentationequilibrium distributions and represented the calculated line of best global fit accordingto AB hetero-association model in SEDPHAT programme. The global fit calculates theapparent dissociation constant (Kd) for enzyme–substrate complexes. The calculated Kd
value for wild type– or W208F mutant–cytochrome c complexes is 7.7 μM or 10 μMrespectively. The sedimentation equilibrium data are representative of three indepen-dent experiments. The detailed experimental procedure is described under “Materialsand methods”.
Table 1Comparative analysis of catalytic activities of wild type LmAPX and mutants
LmAPXenzyme
Ascorbate a kcat Guaiacol b Cytochrome c b
(min−1) Km (mM) kcat (min−1) Km (μM) kcat (min−1)
Wild type 32±2 6.7±1 150±10 17±2 110±12W208F 30±1 6.0±1 170±10 25±2 0.1±0.01
Enzymes are purified from over expression system in absence of ascorbate. The turnover number (kcat) is expressed as mol of product formed/mol of protein/min. Thecatalytic activities are determined at 25 °C as described under “Materials andmethods”.The values represent the mean and standard error for three measurements each.
a kcat are reported at 0.5 mM ascorbate concentration (measurable concentration),which is used to measure the specific activities of pea APX.
b Fit with Michaelis–Menten equation.
Fig. 2. Spectral studies of wild type, W208F and W208Y mutant of LmAPX. Figurerepresents the scans of ascorbate bound enzyme before (solid line) and, after addition ofequimolar H2O2 at initial (dotted line) and after 30 s (dashed line).
866 R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
3.2. Spectroscopy and kinetics of substrate-free native peroxidasereaction with H2O2
To better understand the nature of compound I, we utilized rapid-scanning stopped-flow spectroscopy to follow heme transitionsduring H2O2-driven compound I formation in LmAPX (Fig. 4). Electrondonor free-ferric peroxidasewas rapid-mixedwith an equimolar H2O2
at 30 °C for HRP and at 10 °C for the LmAPX enzyme. In the case of thewild type LmAPX, a product species with absorbance peak at 420, 530and 560 nm formed within 0.75 ms. In contrast, W208F mutantLmAPX showed a Soret peak for a longer time (70ms) at 408 nm and abroad visible peak arises near 620 nm, similar to the HRP and APXcompound I (Table 2), identifying it as the ferryl protoporphyrin π-cation radical [14,39,40]. Moreover, its spectrum clearly differed fromthe wild type LmAPX compound I that formed as an immediateproduct. Wild type LmAPX displayed a similar spectrum withcompound I of wild type cytochrome c peroxidase and F221W HRPmutant [17,37,41,42]. Thus, during H2O2-driven compound I formationof electron donor free enzyme the wild type LmAPX formed a
compound II like spectrumwithin 0.75 ms at 10 °C indicating that thedecay rate of porphyrin π-cation radical was probably greater than therate of formation. We next examined the kinetics of the compound Iformation in the substrate-free wild type and mutant LmAPX. The Fig.5 depicts the kinetics of compound I formation during the initial phaseof enzyme oxidation at 30 °C for HRP and 10 °C for both wild type andmutant LmAPX. The reactions were started by rapid mixing between asolution of H2O2 and peroxidase. The absorbance decrease at 402 nmfor HRP and 408 nm for wild type and mutant LmAPX was best fit to a
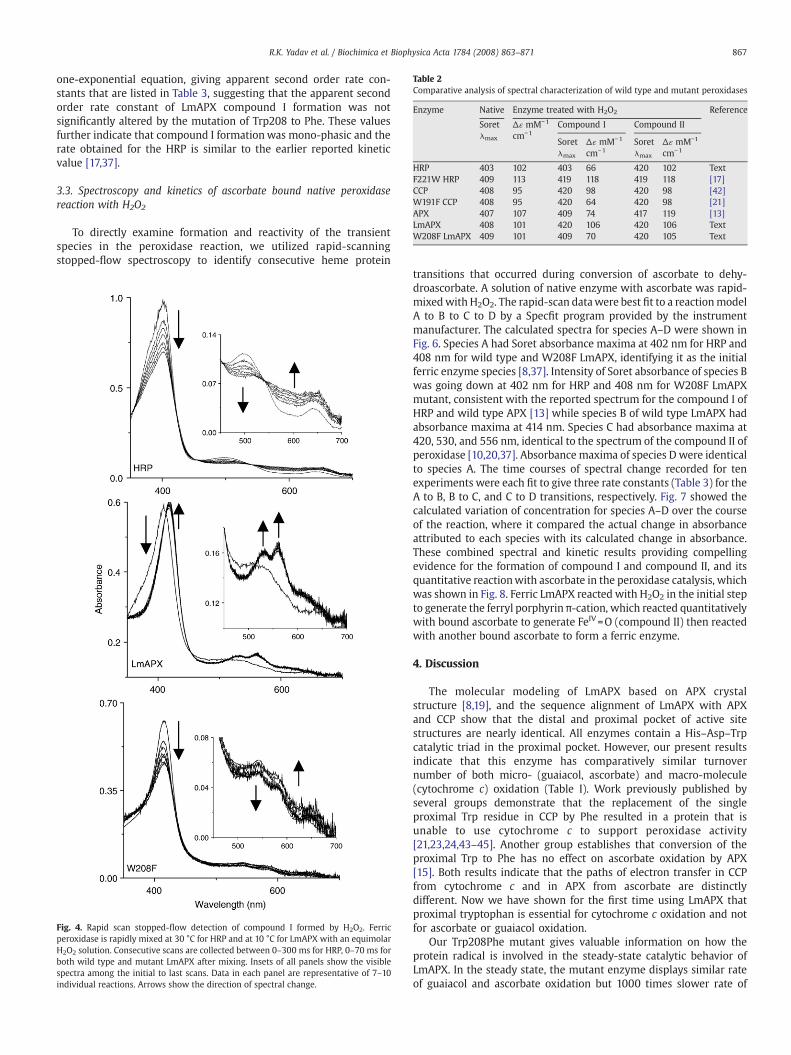
Table 2Comparative analysis of spectral characterization of wild type and mutant peroxidases
Enzyme Native Enzyme treated with H2O2 Reference
Soretλmax
Δɛ mM−1
cm−1Compound I Compound II
Soretλmax
Δɛ mM−1
cm−1Soretλmax
Δɛ mM−1
cm−1
HRP 403 102 403 66 420 102 TextF221W HRP 409 113 419 118 419 118 [17]CCP 408 95 420 98 420 98 [42]W191F CCP 408 95 420 64 420 98 [21]APX 407 107 409 74 417 119 [13]LmAPX 408 101 420 106 420 106 TextW208F LmAPX 409 101 409 70 420 105 Text
867R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
one-exponential equation, giving apparent second order rate con-stants that are listed in Table 3, suggesting that the apparent secondorder rate constant of LmAPX compound I formation was notsignificantly altered by the mutation of Trp208 to Phe. These valuesfurther indicate that compound I formationwas mono-phasic and therate obtained for the HRP is similar to the earlier reported kineticvalue [17,37].
3.3. Spectroscopy and kinetics of ascorbate bound native peroxidasereaction with H2O2
To directly examine formation and reactivity of the transientspecies in the peroxidase reaction, we utilized rapid-scanningstopped-flow spectroscopy to identify consecutive heme protein
Fig. 4. Rapid scan stopped-flow detection of compound I formed by H2O2. Ferricperoxidase is rapidly mixed at 30 °C for HRP and at 10 °C for LmAPX with an equimolarH2O2 solution. Consecutive scans are collected between 0–300 ms for HRP, 0–70 ms forboth wild type and mutant LmAPX after mixing. Insets of all panels show the visiblespectra among the initial to last scans. Data in each panel are representative of 7–10individual reactions. Arrows show the direction of spectral change.
transitions that occurred during conversion of ascorbate to dehy-droascorbate. A solution of native enzyme with ascorbate was rapid-mixedwith H2O2. The rapid-scan datawere best fit to a reactionmodelA to B to C to D by a Specfit program provided by the instrumentmanufacturer. The calculated spectra for species A–D were shown inFig. 6. Species A had Soret absorbance maxima at 402 nm for HRP and408 nm for wild type and W208F LmAPX, identifying it as the initialferric enzyme species [8,37]. Intensity of Soret absorbance of species Bwas going down at 402 nm for HRP and 408 nm for W208F LmAPXmutant, consistent with the reported spectrum for the compound I ofHRP and wild type APX [13] while species B of wild type LmAPX hadabsorbance maxima at 414 nm. Species C had absorbance maxima at420, 530, and 556 nm, identical to the spectrum of the compound II ofperoxidase [10,20,37]. Absorbance maxima of species D were identicalto species A. The time courses of spectral change recorded for tenexperiments were each fit to give three rate constants (Table 3) for theA to B, B to C, and C to D transitions, respectively. Fig. 7 showed thecalculated variation of concentration for species A–D over the courseof the reaction, where it compared the actual change in absorbanceattributed to each species with its calculated change in absorbance.These combined spectral and kinetic results providing compellingevidence for the formation of compound I and compound II, and itsquantitative reactionwith ascorbate in the peroxidase catalysis, whichwas shown in Fig. 8. Ferric LmAPX reacted with H2O2 in the initial stepto generate the ferryl porphyrin π-cation, which reacted quantitativelywith bound ascorbate to generate FeIV=O (compound II) then reactedwith another bound ascorbate to form a ferric enzyme.
4. Discussion
The molecular modeling of LmAPX based on APX crystalstructure [8,19], and the sequence alignment of LmAPX with APXand CCP show that the distal and proximal pocket of active sitestructures are nearly identical. All enzymes contain a His–Asp–Trpcatalytic triad in the proximal pocket. However, our present resultsindicate that this enzyme has comparatively similar turnovernumber of both micro- (guaiacol, ascorbate) and macro-molecule(cytochrome c) oxidation (Table I). Work previously published byseveral groups demonstrate that the replacement of the singleproximal Trp residue in CCP by Phe resulted in a protein that isunable to use cytochrome c to support peroxidase activity[21,23,24,43–45]. Another group establishes that conversion of theproximal Trp to Phe has no effect on ascorbate oxidation by APX[15]. Both results indicate that the paths of electron transfer in CCPfrom cytochrome c and in APX from ascorbate are distinctlydifferent. Now we have shown for the first time using LmAPX thatproximal tryptophan is essential for cytochrome c oxidation and notfor ascorbate or guaiacol oxidation.
Our Trp208Phe mutant gives valuable information on how theprotein radical is involved in the steady-state catalytic behavior ofLmAPX. In the steady state, the mutant enzyme displays similar rateof guaiacol and ascorbate oxidation but 1000 times slower rate of

Fig. 5. Kinetics of compound I formation in substrate-free peroxidase. Reactions are initiated by rapidmixing of an enzyme in 50mMphosphate buffer, pH 7.5 with an equal volume ofbuffer solution containing an equimolar concentration of H2O2. Figure shows the kinetics of absorbance change at 402 nm for HRP at 30 °C and 408 nm for wild type and W208FLmAPX at 10 °C. Smooth lines drawn through the kinetic traces are the calculated lines of best fit according to one-exponential equation. Data in each panel are representative of anaverage of 7–10 individual reactions. Insets show the plot of apparent mono-phasic rate constants (kobs) versus H2O2 concentration for the calculation of second order rate constant.
868 R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
cytochrome c oxidation compared to wild type enzyme. Remarkablythis is identical behavior with the analogous Trp191Phe CCP mutant,which displays a decrease in cytochrome c oxidation [21,23]. Thefollowing possibilities have been proposed to explain the role of thisresidue in cytochrome c oxidation. (i) It can control the secondarystructure of LmAPX. The spectroscopic data analysis indicates that thesecondary structure is not perturbed by the replacement of Trp toPhe. (ii) It can be involved in compound I/II formation required forelectron donor oxidation. The amino acid sequence of severalperoxidases at the heme distal side shows that both distal histidineand arginine residues are highly conserved. The core of LmAPX, APXand CCP, their active site and substrate access channel, are alsoconserved. The proposed model structure based on moleculardynamics calculation and energy minimization indicates a resem-blance of His-68 and Arg-64 of LmAPX to the His-52 and Arg-48 ofCCP. These residues are therefore implicated in compound Iformation in LmAPX. Our diode array spectrophotometric studiesindicate that the replacement of a single Trp residue by site-directedmutagenesis does not affect compound I formation and hence can beexcluded. (iii) The residue can be involved in the binding of thecytochrome c. The crystal structure of the CCP–cytochrome ccomplexes, the site specific crosslinked CCP–cytochrome c complexesand the site-directed mutagenesis studies have demonstrated thatthe two proteins are bound by favorable electrostatic interactionsbetween complementary sets of acidic residues on CCP and highlyconserved lysine residues on cytochrome c [45–48]. Their X-raycrystal structure of the CCP–cytochrome c complexes reveals that thenegatively charged residues Asp34 (or Glu35), and Glu290 interactwith positively charged lysine residues of cytochrome c [27,29,27,48].Our studies indicate that the both apparent dissociation constant and
Table 3Observed rate constants for transient species formation and decay
Enzyme k1a (M−1s−1)for
substrate-freeenzyme
Ascorbate bound enzyme b (s−1)
A→B (Fe3+→Fe5+) B→C (Fe5+→Fe4+) C→D (Fe4+→Fe3+)
HRP c 0.9±0.2×107 150±4 25±2 1.2±0.01LmAPX d 6.7±0.2×107 250±7 20±2 0.022±0.001W208F d 3.4±0.2×107 160±3 18±2 0.02±0.001
Reactions are initiated by rapidly mixing the enzyme with equimolar H2O2 and hemeoxidation as described under “Materials and methods”. The rates describe decay ofabsorbance at 402 nm for HRP or 408 nm for LmAPX versus time and are the averageobtained from two enzyme preparations. The data best fit to one-exponential curvein all cases to generate rate constant (kobs).
a Second order rate constants are calculated from linear curve of kobs versus H2O2
concentration.b The apparent rate constants are calculated from a Specfit program provided by
the instrument manufacturer, where the rapid-scan data are best fit to a reactionmodel A to B to C to D.
c Rate constant at 30 °C.d Rate constant at 10 °C.
Km value of the Trp208Phe mutant for cytochrome c is similar to wildtype LmAPX enzyme. Thus, the indole side chain of W208 is notessential for binding of cytochrome c.
Therefore another possibility is that the indole side chain of W208in LmAPX is needed to control the oxidation of cytochrome c byregulating the electron transfer mechanism from donor to heme-ferryl group (compound I/II) and, perhaps, similar to electron transfermechanisms of CCP. One, as already noted, is that the substrate for CCPis a protein rather than a small molecule. The other is that CCP isoxidized by H2O2 to a ferryl (FeIV=O) species and a protein radical [49],whereas most peroxidases, as typified by HRP, are oxidized to theferryl species plus a porphyrin radical cation [20]. These differencescomplicate comparisons of the electron transfer mechanisms of CCPwith those of other peroxidases. The proximal Trp is known to be thesite of free radical formation in CCP compound I. However, APX formsa porphyrin radical and not a Trp-centered radical, even though theHis–Asp–Trp triad structure is the same in both peroxidases [14].Remarkably, we found from our mutation studies that the indole sidechain of W208 governs electron transfer from cytochrome c to bothcompound I and II of the LmAPX. This role is unexpected and stands tobroaden our understanding of peroxidase.
The data presented for the first time in this work provide thecomplete kinetic and spectroscopic studies of wild type and W208mutant parasitic enzymes and differentiate the electron transferpathway between macro- (cytochrome c) and micro-molecule(ascorbate and guaiacol) oxidation. In light of these new data, it isappropriate to correlate the structural information of CCP with APXand to consider the broader mechanistic question of how LmAPX maymost effectively deal with oxidation of different types of substratesbound at different locations. For ascorbate and aromatic substratesoxidation, previous workers have assumed from their ascorbatebound APX and salicylhydroxamic acid (SHA) bound APX crystalstructures that electron transfer from ascorbate or aromatic donor tocompound I/II occurs directly through the 6-propionate or the π-orbitals of the porphyrin, respectively [19,50]. Therefore a porphyrinπ-cation intermediate is sufficient for the reduction of compound I inLmAPX, completely bypassing the need for involvement of W208 insmall molecule (both ascorbate and aromatic donor) oxidation andthose results are very consistent with the previous APX Trp179mutant[15]. On the other electron transfer pathway in LmAPX may involveproximal W208 for cytochrome c oxidation and these results areconsistent with CCP Trp191 mutant [23].
The most novel finding in this paper is that the characteristic peakof compound I in ascorbate bound LmAPX and substrate-free LmAPXare different. In the absence of a substrate, compound I of LmAPXshows a 420 nmpeakwith equal intensity as shown by the native stateof the enzyme but ascorbate bound LmAPX shows a peak at 414 nmwith decreasing Soret intensity. The formation of compound I ofW208F mutant, lacking proximal tryptophan residue, drasticallydecreases the intensity at the Soret region for a longer time in both
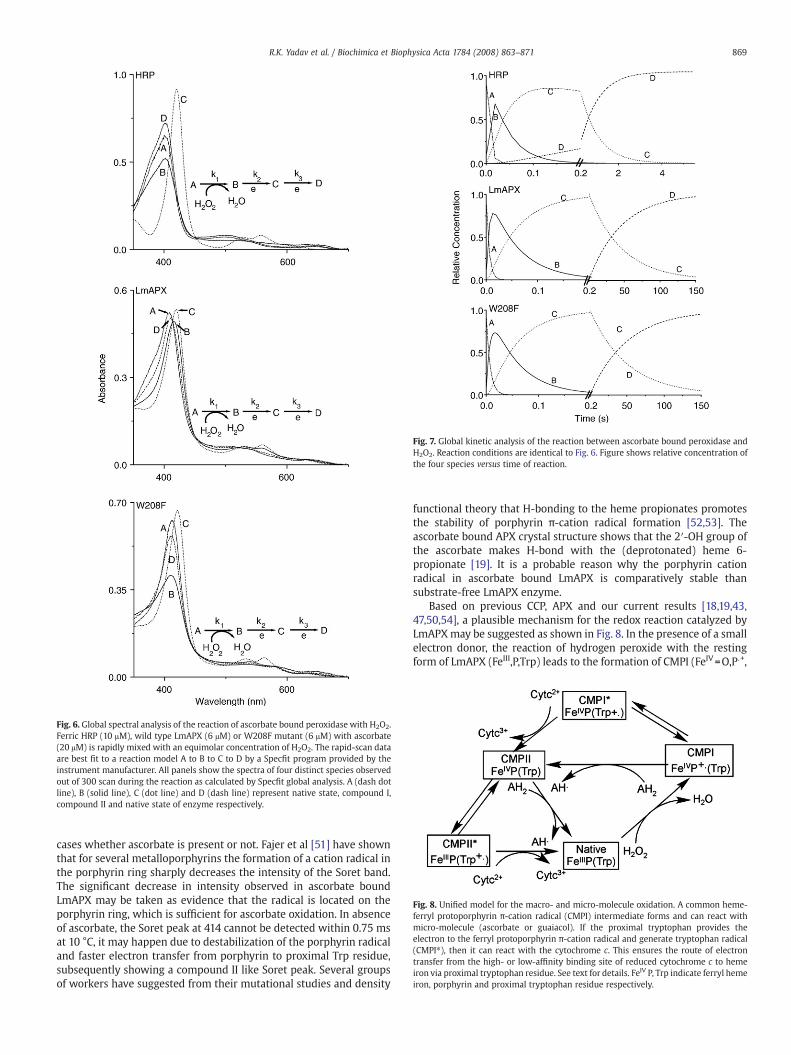
Fig. 6. Global spectral analysis of the reaction of ascorbate bound peroxidase with H2O2.Ferric HRP (10 μM), wild type LmAPX (6 μM) or W208F mutant (6 μM) with ascorbate(20 μM) is rapidly mixed with an equimolar concentration of H2O2. The rapid-scan dataare best fit to a reaction model A to B to C to D by a Specfit program provided by theinstrument manufacturer. All panels show the spectra of four distinct species observedout of 300 scan during the reaction as calculated by Specfit global analysis. A (dash dotline), B (solid line), C (dot line) and D (dash line) represent native state, compound I,compound II and native state of enzyme respectively.
Fig. 7. Global kinetic analysis of the reaction between ascorbate bound peroxidase andH2O2. Reaction conditions are identical to Fig. 6. Figure shows relative concentration ofthe four species versus time of reaction.
Fig. 8. Unified model for the macro- and micro-molecule oxidation. A common heme-ferryl protoporphyrin π-cation radical (CMPI) intermediate forms and can react withmicro-molecule (ascorbate or guaiacol). If the proximal tryptophan provides theelectron to the ferryl protoporphyrin π-cation radical and generate tryptophan radical(CMPI⁎), then it can react with the cytochrome c. This ensures the route of electrontransfer from the high- or low-affinity binding site of reduced cytochrome c to hemeiron via proximal tryptophan residue. See text for details. FeIV P, Trp indicate ferryl hemeiron, porphyrin and proximal tryptophan residue respectively.
869R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
cases whether ascorbate is present or not. Fajer et al [51] have shownthat for several metalloporphyrins the formation of a cation radical inthe porphyrin ring sharply decreases the intensity of the Soret band.The significant decrease in intensity observed in ascorbate boundLmAPX may be taken as evidence that the radical is located on theporphyrin ring, which is sufficient for ascorbate oxidation. In absenceof ascorbate, the Soret peak at 414 cannot be detected within 0.75 msat 10 °C, it may happen due to destabilization of the porphyrin radicaland faster electron transfer from porphyrin to proximal Trp residue,subsequently showing a compound II like Soret peak. Several groupsof workers have suggested from their mutational studies and density
functional theory that H-bonding to the heme propionates promotesthe stability of porphyrin π-cation radical formation [52,53]. Theascorbate bound APX crystal structure shows that the 2′-OH group ofthe ascorbate makes H-bond with the (deprotonated) heme 6-propionate [19]. It is a probable reason why the porphyrin cationradical in ascorbate bound LmAPX is comparatively stable thansubstrate-free LmAPX enzyme.
Based on previous CCP, APX and our current results [18,19,43,47,50,54], a plausible mechanism for the redox reaction catalyzed byLmAPXmay be suggested as shown in Fig. 8. In the presence of a smallelectron donor, the reaction of hydrogen peroxide with the restingform of LmAPX (FeIII,P,Trp) leads to the formation of CMPI (FeIV=O,PU+,

870 R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
Trp), which contains an oxyferryl heme FeIV=O and a porphyrin π-cation radical. A small molecule (ascorbate or guaiacol) initiallyreduces the π-cation radical of CMPI (FeIV=O,PU+,Trp) to producecompound II (FeIV=O,P,Trp). A second molecule of the electron donorthen reduces the oxyferryl heme in CMPII (FeIV=O, P,Trp) to form theresting enzyme (FeIII,P,Trp). However, in the presence of cytochrome c,the situation appears to alter. Indeed, one of the curious features ofLmAPX is that, in contrast to APX, its compound I is most probablycomparatively unstable and, leads to the generation of another speciesCompound I⁎ (FeIV=O, P,TrpU+), which contains an oxyferryl hemeFeIV=O and a TrpU+ radical cation located on the indole ring of Trp208.Ferrocytochrome c initially reduces the Trp208 radical cation in CMPI(FeIV=O,P,TrpU+) to produce compound II (FeIV=O,P,Trp). By internalelectron transfer in CMPII (FeIV=O,P,Trp) leads to regeneration of theTrp radical cation in CMPII⁎ (FeIII,P,TrpU+), which is then reduced bycytochrome c to form the native enzyme [43].
Although the pea APX forms a porphyrin radical, it is alsoimportant to address the question why LmAPX oxidizes cytochromec (like CCP) through proximal Trp and how the protein environmenthelps to destabilize a porphyrin radical in LmAPX. Several groups ofworkers from their mutational studies and density functional theoryhave provided an interesting explanation for stabilization or destabi-lization of a porphyrin radical in APX [52,53,55]. Those explanationsare (i) salt bridge or H-bonding to the heme propionates promotes thestability of porphyrin π-cation radical formation [52], and (ii) threeMet residues unique to the proximal pocket of CCP contain electron-rich sulfur atoms which helps in stabilizing the positive charge on theTrp191 radical [55]. LmAPX does not have an Arg residue (correspond-ing to R172 of APX), which can make H-bonding with at least onepropionate group. The proximal Met residues, M248 and M249 inLmAPX (corresponding to M230, M231 in CCP) may play an importantrole in the electrostatic stabilization of the Trp radical in LmAPX. Thus,those reasons can further support the possibility of an important roleof Trp residue in cytochrome c oxidation by LmAPX.
Acknowledgements
The work was supported by Grant SR/SO/BB-54/2005 from theDepartment of Science and Technology, Government of India.Additional financial support was obtained from CSIR network projects(MLP15 and NWP 0038), Government of India. R. K. Y and S. D. wereeach supported by individual fellowships from the Council of Scientificand Industrial Research, Government of India.
References
[1] J.Y. Channon, M.B. Roberts, J.M. Blackwell, A study of the differential respiratoryburst activity elicited by promastigotes and amastigotes of Leishmania donovani inmurine resident peritoneal macrophages, Immunology 53 (1984) 345–355.
[2] K.P. Chang, Human cutaneous Lieshmania in a mouse macrophage line:propagation and isolation of intracellular parasites, Science 209 (1980)1240–1242.
[3] K.R. Gantt, T.L. Goldman, M.L. McCormick, M.A. Miller, S.M. Jeronimo, E.T.Nascimento, B.E. Britigan, M.E. Wilson, Oxidative responses of human and murinemacrophages during phagocytosis of Leishmania chagasi, J. Immunol. 167 (2001)893–901.
[4] M.A. Miller, S.E. McGowan, K.R. Gantt, M. Champion, S.L. Novick, K.A. Andersen, C.J.Bacchi, N. Yarlett, B.E. Britigan, M.E. Wilson, Inducible resistance to oxidant stressin the protozoan Leishmania chagasi, J. Biol. Chem. 275 (2000) 33883–33889.
[5] H.W. Murray, C.F. Nathan, Macrophage microbicidal mechanisms in vivo: reactivenitrogen versus oxygen intermediates in the killing of intracellular visceralLeishmania donovani, J. Exp. Med. 189 (1999) 741–746.
[6] I. Fridovich, B. Freeman, Antioxidant defenses in the lung, Annu. Rev. Physiol. 48(1986) 693–702.
[7] I. Fridovich, Biological effects of the superoxide radical, Arch. Biochem. Biophys.247 (1986) 1–11.
[8] S. Adak, A.K. Datta, Leishmania major encodes an unusual peroxidase that is a closehomologue of plant ascorbate peroxidase: a novel role of the transmembranedomain, Biochem. J. 390 (2005) 465–474.
[9] S. Dolai, R.K. Yadav, A.K. Datta, S. Adak, Effect of thiocyanate on the peroxidase andpseudocatalase activities of Leishmania major ascorbate peroxidase, Biochim.Biophys. Acta 1770 (2007) 247–256.
[10] H.B. Dunford, J.S. Stillman, On the function and mechanism of action of peroxidases,Coord. Chem. Rev. 19 (1976) 187–251.
[11] D.A. Dalton, Ascorbate peroxidase, CRC Press, Boca Raton, 1990.[12] D.K. Jones, D.A. Dalton, F.I. Rosell, E.L. Raven, Class I heme peroxidases:
characterization of soybean ascorbate peroxidase, Arch. Biochem. Biophys. 360(1998) 173–178.
[13] L.A. Marquez, M. Quitoriano, B.A. Zilinskas, H.B. Dunford, Kinetic and spectralproperties of pea cytosolic ascorbate peroxidase, FEBS Lett. 389 (1996)153–156.
[14] W.R. Patterson, T.L. Poulos, D.B. Goodin, Identification of a porphyrin pi cationradical in ascorbate peroxidase compound I, Biochemistry 34 (1995) 4342–4345.
[15] H. Pappa, W.R. Patterson, T.L. Poulos, The homologous tryptophan critical forcytochrome c peroxidase function is not essential for ascorbate peroxidaseactivity, J. Biol. Inorg. Chem. 1 (1996) 61–66.
[16] J.E. Huyett, P.E. Doan, R. Gurbiel, A.L.P. Houseman, M. Sivaraja, D.B. Goodin, B.M.Hoffman, Compound ES of cytochrome c peroxidase contains Trp pi-cationradical: characterization by CW and pulsed Q-band ENDOR spectroscopy, J. Am.Chem. Soc. 117 (1995) 9033–9041.
[17] A. Morimoto, M. Tanaka, S. Takahashi, K. Ishimori, H. Hori, I. Morishima, Detectionof a tryptophan radical as an intermediate species in the reaction of horseradishperoxidase mutant (Phe-221 –N Trp) and hydrogen peroxide, J. Biol. Chem. 273(1998) 14753–14760.
[18] A.N. Hiner, J.I. Martinez, M.B. Arnao, M. Acosta, D.D. Turner, E. Lloyd Raven, J.N.Rodriguez-Lopez, Detection of a tryptophan radical in the reaction of ascorbateperoxidase with hydrogen peroxide, Eur. J. Biochem. 268 (2001) 3091–3098.
[19] K.H. Sharp, M. Mewies, P.C. Moody, E.L. Raven, Crystal structure of the ascorbateperoxidase–ascorbate complex, Nat. Struct. Biol. 10 (2003) 303–307.
[20] H.B. Dunford, Horseradish Peroxidase: Structure and Kinetic Properties, CRC press,Boca Raton, Florida, 1991.
[21] J.E. Erman, L.B. Vitello, J.M. Mauro, J. Kraut, Detection of an oxyferrylporphyrin pi-cation-radical intermediate in the reaction between hydrogenperoxide and a mutant yeast cytochrome c peroxidase. Evidence fortryptophan-191 involvement in the radical site of compound I, Biochemistry28 (1989) 7992–7995.
[22] D.B. Goodin, A.G. Mauk, M. Smith, The peroxide complex of yeast cytochrome cperoxidase contains two distinct radical species, neither of which resides atmethionine 172 or tryptophan 51, J. Biol. Chem. 262 (1987) 7719–7724.
[23] J.M. Mauro, L.A. Fishel, J.T. Hazzard, T.E. Meyer, G. Tollin, M.A. Cusanovich, J. Kraut,Tryptophan-191–phenylalanine, a proximal-side mutation in yeast cytochrome cperoxidase that strongly affects the kinetics of ferrocytochrome c oxidation,Biochemistry 27 (1988) 6243–6256.
[24] M. Sivaraja, D.B. Goodin, M. Smith, B.M. Hoffman, Identification by ENDOR ofTrp191 as the free-radical site in cytochrome c peroxidase compound ES, Science245 (1989) 738–740.
[25] G.D. DePillis, B.P. Sishta, A.G. Mauk, P.R. Ortiz de Montellano, Small substrates andcytochrome c are oxidized at different sites of cytochrome c peroxidase, J. Biol.Chem. 266 (1991) 19334–19341.
[26] B.C. Finzel, T.L. Poulos, J. Kraut, Crystal structure of yeast cytochrome c peroxidaserefined at 1.7-A resolution, J. Biol. Chem. 259 (1984) 13027–13036.
[27] H. Pelletier, J. Kraut, Crystal structure of a complex between electron transferpartners, cytochrome c peroxidase and cytochrome c, Science 258 (1992)1748–1755.
[28] G.C. Kresheck, L.B. Vitello, J.E. Erman, Calorimetric studies on the interaction ofhorse ferricytochrome c and yeast cytochrome c peroxidase, Biochemistry 34(1995) 8398–8405.
[29] M.A. Miller, L. Geren, G.W. Han, A. Saunders, J. Beasley, G.J. Pielak, B. Durham, F.Millett, J. Kraut, Identifying the physiological electron transfer site of cytochromec peroxidase by structure-based engineering, Biochemistry 35 (1996) 667–673.
[30] E.D. Stemp, B.M. Hoffman, Cytochrome c peroxidase binds two molecules ofcytochrome c: evidence for a low-affinity, electron-transfer-active site oncytochrome c peroxidase, Biochemistry 32 (1993) 10848–10865.
[31] J.S. Zhou, B.M. Hoffman, Stern–volmer in reverse: 2:1 stoichiometry of thecytochrome c–cytochrome c peroxidase electron-transfer complex, Science 265(1994) 1693–1696.
[32] M.R. Mauk, J.C. Ferrer, A.G. Mauk, Proton linkage in formation of the cytochromec–cytochrome c peroxidase complex: electrostatic properties of the high- andlow-affinity cytochrome binding sites on the peroxidase, Biochemistry 33 (1994)12609–12614.
[33] S. Adak, K.S. Aulak, D.J. Stuehr, Direct evidence for nitric oxide production by anitric-oxide synthase-like protein from Bacillus subtilis, J. Biol. Chem. 277 (2002)16167–16171.
[34] S. Adak, C. Crooks, Q. Wang, B.R. Crane, J.A. Tainer, E.D. Getzoff, D.J. Stuehr,Tryptophan 409 controls the activity of neuronal nitric-oxide synthase byregulating nitric oxide feedback inhibition, J. Biol. Chem. 274 (1999) 26907–26911.
[35] E.H. Bursey, T.L. Poulos, Two substrate binding sites in ascorbate peroxidase: therole of arginine 172, Biochemistry 39 (2000) 7374–7379.
[36] M.M. Fitzgerald, M.J. Churchill, D.E. McRee, D.B. Goodin, Small molecule binding toan artificially created cavity at the active site of cytochrome c peroxidase,Biochemistry 33 (1994) 3807–3818.
[37] H.B. Dunford, Heme peroxidases, John Wiley and Sons, Inc., New York, 1999.[38] L. Lad, M. Mewies, E.L. Raven, Substrate binding and catalytic mechanism in
ascorbate peroxidase: evidence for two ascorbate binding sites, Biochemistry. 41(2002) 13774–13781.
[39] D. Dolphin, A. Forman, D.C. Borg, J. Fajer, R.H. Felton, Compounds I of catalase andhorse radish peroxidase: pi-cation radicals, Proc. Natl. Acad. Sci. U. S. A. 68 (1971)614–618.

871R.K. Yadav et al. / Biochimica et Biophysica Acta 1784 (2008) 863–871
[40] J.E. Penner-Hahn, K.S. Eble, T.J. McMury, M. Renner, A.L. Balch, J.T. Groves, J.H.Dawson, K.O. Hodgson, J. Am. Chem. Soc. 108 (1986) 7819–7825.
[41] A.F. Coulson, J.E. Erman, T. Yonetani, Studies on cytochrome c peroxidase. XVII.Stoichiometry andmechanism of the reaction of compound ES with donors, J. Biol.Chem. 246 (1971) 917–924.
[42] T. Yonetani, H. Anni, Yeast cytochrome c peroxidase. Coordination and spin statesof heme prosthetic group, J. Biol. Chem. 262 (1987) 9547–9554.
[43] M.A. Miller, L. Vitello, J.E. Erman, Regulation of interprotein electron transfer byTrp191 of cytochrome c peroxidase, Biochemistry 34 (1995) 12048–12058.
[44] R.Q. Liu, S. Hahm, M. Miller, B. Durham, F. Millett, Photooxidation of Trp-191 incytochrome c peroxidase by ruthenium–cytochrome c derivatives, Biochemistry34 (1995) 973–983.
[45] M.A. Miller, R.Q. Liu, S. Hahm, L. Geren, S. Hibdon, J. Kraut, B. Durham, F. Millett,Interaction domain for the reaction of cytochrome c with the radical and theoxyferryl heme in cytochrome c peroxidase compound I, Biochemistry 33 (1994)8686–8693.
[46] H. Mei, K. Wang, S. McKee, X. Wang, J.L. Waldner, G.J. Pielak, B. Durham, F. Millett,Control of formation and dissociation of the high-affinity complex betweencytochrome c and cytochrome c peroxidase by ionic strength and the low-affinitybinding site, Biochemistry 35 (1996) 15800–15806.
[47] K. Wang, H. Mei, L. Geren, M.A. Miller, A. Saunders, X. Wang, J.L. Waldner, G.J.Pielak, B. Durham, F. Millett, Design of a ruthenium–cytochrome c derivative to
measure electron transfer to the radical cation and oxyferryl heme in cytochromec peroxidase, Biochemistry 35 (1996) 15107–15119.
[48] M. Guo, B. Bhaskar, H. Li, T.P. Barrows, T.L. Poulos, Crystal structure andcharacterization of a cytochrome c peroxidase–cytochrome c site-specific cross-link, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 5940–5945.
[49] H.R. Bosshard, H. Anni, T. Yonetani, Peroxidases in Chemistry and Biology,Academic Press, New York, 1990.
[50] K.H. Sharp, P.C. Moody, K.A. Brown, E.L. Raven, Crystal structure of the ascorbateperoxidase–salicylhydroxamic acid complex, Biochemistry 43 (2004) 8644–8651.
[51] J. Fajer, D.C. Borg, A. Forman, D. Dolphin, R.H. Felton, pi-Cation radicals anddications of metalloporphyrins, J. Am. Chem. Soc. 92 (1970) 3451–3459.
[52] T.P. Barrows, T.L. Poulos, Role of electrostatics and salt bridges in stabilizingthe compound I radical in ascorbate peroxidase, Biochemistry 44 (2005)14062–14068.
[53] V. Guallar, M.H. Baik, S.J. Lippard, R.A. Friesner, Peripheral heme substituentscontrol the hydrogen-atom abstraction chemistry in cytochromes P450, Proc. Natl.Acad. Sci. U. S. A. 100 (2003) 6998–7002.
[54] H. Mei, L. Geren, M.A. Miller, B. Durham, F. Millett, Role of the low-affinity bindingsite in electron transfer from cytochrome c to cytochrome c peroxidase,Biochemistry 41 (2002) 3968–3976.
[55] T.P. Barrows, B. Bhaskar, T.L. Poulos, Electrostatic control of the tryptophan radicalin cytochrome c peroxidase, Biochemistry 43 (2004) 8826–8834.




















