
Serial Regulation of Transcriptional Regulators in the Yeast Cell Cycle
Upload
khangminh22Category
view
0download
0
ROLE OF TGIF IN CELL CYCLE CONTROL AND
ESTABLISHMENT OF LATERALITY
by
L Y N N MAR
M.Sc, University of Toronto, 1999
A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
in
THE FACULTY OF GRADUATE STUDIES
(MEDICAL GENETICS)
THE UNIVERSITY OF BRITISH COLUMBIA
August 2006
© Lynn Mar, 2006

ABSTRACT
Holoprosencephaly (HPE) is the most common structural anomaly of the human
brain, resulting from incomplete cleavage of the developing forebrain during
embryogenesis. Haploinsufficient mutations in TG-Interacting Factor (TGIF) were
previously identified in a subset of HPE families and sporadic patients, and this gene is
located within a region of Chromosome 18 that is associated with non-random
chromosomal aberrations in HPE patients. TGIF is a transcription factor that contains a
three amino acid loop extension (TALE) homeodomain and functions both as a co-
repressor of the TGF-f3 pathway and as a competitor of the retinoic acid pathway. Mice
made deficient for 7gz/exhibited laterality defects and growth retardation, and
developed kinked tails. Analysis of Tgif'1' mouse embryonic fibroblasts (MEFs) in
vitro demonstrated that Tgif regulates proliferation and progression through the Gi cell
cycle phase. Wild-type human TGIF was able to rescue this proliferative defect in
MEFs. In contrast, a subset of human Tgif mutations detected in HPE patients was
unable to rescue the proliferative defect. However, an absence of Tgif did not alter the
normal inhibition of proliferation caused by treatment with TGF-fi or retinoic acid.
Developmental control of proliferation by Tgif may play a role in the pathogenesis of
HPE.
ii

TABLE OF CONTENTS
Page No.
Abstract ii
Table of Contents iii
List of Tables vi
List of Figures vii
List of Abbreviations viii
Acknowledgements ix
Dedication x
1. CHAPTER 1 INTRODUCTION 1 1.1. Developmental genetics and embryology 1
1.1.1. Holoprosencephaly 2 1.1.1.1. Multifactorial etiology 3 1.1.1.2. Candidate genes in human 4 1.1.1.3. Forebrain development 5
1.1.1.3.1. Anterior visceral endoderm induces anterior neural fate 7 1.1.1.3.2. Node derived anterior definitive endoderm reinforces and
further elaborates forebrain patterning 9 1.1.1.3.3. Node derived prechordal plate provides ventral midline
patterning centre 11 1.1.1.3.4. Regional pattering of the forebrain 12 1.1.1.3.5. Potential disruptions by mutations in HPE genes 15
1.1.2. Asymmetric body plan 16 1.1.2.1. Breaking of symmetry 19 1.1.2.2. Asymmetric gene expression in the node and lateral plate mesoderm... 21 1.1.2.3. Asymmetric gene expression maintained by the embryonic midline 22 1.1.2.4. Organ positioning and morphogenesis 23 1.1.2.5. Retinoic acid and symmetrical development of somites 23
1.2. TG-Interacting Factor 24 1.2.1. TGIF is a TALE HD 24 1.2.2. TGIF is a RA antagonist 26 1.2.3. TGIF is a TGF-0 co-repressor 28 1.2.4. TGIF mutations identified in HPE patients 31 1.2.5. Evolution conservation 32
1.3. Mammalian cell cycle 33 1.4. Hypothesis 35 1.5. Objectives 35
i n

2. CHAPTER 2 MATERIALS AND METHODS..; 36 2.1. Generation of 7gz/mutant mice 36
2.1.1. Isolation of murine Tgifgenomic clone 36 2.1.2. Construction of the Tgif targeting vector 36 2.1.3. Embryonic stem cell line targeting and identification of homologous
recombinant clones 37 2.1.4. Identification of Tgif"mutant alleles by Southern blot analysis 37 2.1.5. Identification of Tgif 'mutant alleles by PCR 39 2.1.6. Production of chimeras and Tgif mutant mice from targeted ES cells 39 2.1.7. Mouse breeding, genetic backgrounds, and congenic strains 39
2.2. Expression analysis of Tgif using in situ RNA hybridization 40 2.3. Analysis of Tgif mutant mice 41
2.3.1. Determination of the rate of viability and fertility 41 2.3.2. Morphological examination of adults 42 2.3.3. Morphological examination of embryos 42
2.4. Cell cycle analysis of Tgif mutant mouse embryonic fibroblasts 42 2.4.1. Generation of mouse embryonic fibroblasts 42 2.4.2. Expression of Tgif in mouse embryonic fibroblasts 43 2.4.3. Growth curve analysis 43 2.4.4. Cell cycle analysis by BrdU incorporation and flow cytometry 43 2.4.5. Proliferation assay by 3H-thymidine incorporation 44 2.4.6. Proliferation assay of synchronized mouse embryonic fibroblasts 44
2.5. Analysis of in vivo proliferation by somite staging 44 2.5.1. Analysis of in vivo proliferation by BrdU incorporation 45
2.6. Expression of human TGIF by retrovirus in mouse embryonic fibroblasts 45 2.6.1. Expression of wild-type human TGIF by retrovirus 45 2.6.2. Expression of human TGIF containing mutations identified in HPE patients.46 2.6.3. Expression of transduced TGIF protein 46
2.7. Analysis of Proliferation response TGF-0, retinoic acid and U0126 46
3. CHAPTER 3 GENERATION AND CHARACTERIZATION OF TGIF MICE 48
3.1. 7g7/expression during embryogenesis 48 3.2. Generation of Tgif mutant mice 51
3.2.1. Isolation of 7g7/genomic clone 51 3.2.2. Construction of Tgif targeting vector 51 3.2.3. Screen for clones in which the Tgif locus was targeted 53 3.2.4. Targeted Tgif alleles are null alleles and produce a fused lacZ transcript 54 3.2.5. Production of chimeras and Tgif'mutant mice from targeted ES cells 54
3.3. Analysis of Tgif mutant mice 56 3.3.1. Analysis of Tgif mutant adult mice on mixed 129/Sv/CDl genetic
background 56 3.3.2. Analysis of the Tgif mutant adult mice with a 129/Sv congenic genetic
background 57 3.3.3. Analysis of the Tgif mutant adult mice with a C57BL6 congenic genetic
background 61 3.3.4. Morphological examination of embryos 61 3.3.5. Abnormal laterality determination 62
3.4. Summary 64
iv

4. CHAPTER 4 ROLE OF TGIF IN CELL CYCLE CONTROL.... 65 4.1. Generation of Tgif mouse embryonic fibroblasts 65 4.2. Cell cycle defects in mouse embryonic fibroblasts 65
4.2.1. Growth curve analysis ; 65 4.2.2. Cytometric analysis of mouse embryonic fibroblasts cell cycle 69 4.2.3. Proliferation assay by 3[H]-thymidine incorporation 69 4.2.4. Proliferation of synchronized mouse embryonic fibroblasts 72 4.2.5. Rescue of the proliferation defect by re-expression of human TGIF
in mutant mouse embryonic fibroblasts 72 4.3. Proliferation defects in vivo 73
4.3.1. Growth retardation in embryonic day 8 embryos 73 4.3.2. BrdU incorporation in embryonic day 8 embryos 73
4.4. Assaying the proliferative function of human TGIF mutations in mouse embryonic fibroblasts 77
4.5. Proliferation response to signaling pathways 80 4.5.1. Proliferation in response to TGF-R 80 4.5.2. Proliferation in response to retinoic acid 81 4.5.3. Proliferation in response to Ras/MAPK inhibitor, U0126 82
4.6. Summary 85
5. CHAPTER 5 DISCUSSION 86 5.1. Mutant mice did not exhibit HPE, but did exhibit HPE related defects 86
5.1.1. Lack of HPE phenotype 87 5.1.1.1. Genetic background 88
5.1.2. HPE-related defects 89 5.1.3. Future directions 92
5.2. Mutant mice exhibited laterality defects 94 5.2.1. Molecular basis of laterality defects... : 9 6 5.2.2. Future directions 97
5.3. Role of TGIF in cell cycle control 98 5.3.1. Proliferation during neural development 100 5.3.2. Future directions 103
5.4. Final comments 105
References 107
V

L IST O F T A B L E S
Table 3.1 Phenotypes resulting from rgz/heterozygote matings on 129Sv/CDl genetic background at 3 weeks of age 58
Table 4.1 Phenotypes resulting from rgz/heterozygote matings on 129Sv/CDl genetic background at 3 weeks of age 76
vi

LIST OF FIGURES
Figure 1.1 Forebrain patterning in the mouse embryo 6
Figure 1.2 Molecular network controlling dorsal-ventral patterning of the
telencephalon 13
Figure 1.3 Genetic pathway for the determination of left-right asymmetry 18
Figure 1.4 Tgif functional domains 25
Figure 1.5 The retinoic acid signal transduction pathway 27
Figure 1.6 The TGF-P signal transduction pathway 29
Figure 1.7 The cell cycle 34
Figure 3.1 Tgif expression in the early embryo 49
Figure 3.2 Tgi/expression in the developing limbs 50
Figure 3.3 Gene targeting of the mouse Tgif locus 52
Figure 3.4 Targeted ES cells express Tgif-lacZ fusion transcript 55
Figure 3.5 Tgif mutant mice on the 129/Sv/CDl genetic background developed pleiotropic phenotypes 59
Figure 3.6 Tgif mutant mice on the 129/Sv/CDl genetic background developed
laterality defects 60
Figure 3.7 Tgif 'mutant embryos displayed abnormal left-right patterning 63
Figure 4.1 MEF cells normally express Tgif but mutant cells do not 66
Figure 4.2 Tgif1' MEFs exhibited reduced growth due to a delay in GI phase
progression 67
Figure 4.3 Cell cycle analysis of proliferation defect in Tgif1' MEFs 70
Figure 4.4 Tgif1' mutant embryos exhibited growth retardation 74
Figure 4.5 Expression of a subset of mutated human TGIF rescued the proliferative defect of MEFs 78
Figure 4.6 Analysis of MEF proliferation in response to TGF-P, 9-cis RA, all-trans-RA and U0126 82
vii

LIST OF ABBREVIATIONS
7 A A D - 7-amino-actinomycin D
A N R - anterior neural ridge
A V E - anterior visceral endoderm
Bmp - bone morphogenetic protein
BrdU - bromodeoxyuridine
C P M - counts per minute
CtBP - carboxyl terminal binding protein
En - embryonic day n; e.g. E8, embryonic day 8
Fgf - fibroblast growth factor
H D A C - histone deacetylase
H P E - Holoprosencephaly
INK - c-Jun N-terminal Kinase
L P M - lateral plate mesoderm
M A P K - mitogen-activated protein kinase
M E F - mouse embryonic fibroblasts
O C T - optimum cutting temperature
PCR - polymerase chain reaction
R A - retinoic acid
Shh - sonic hedgehog
T A L E H D - three amino acid loop extension homeodomain
TF - transcription factor
TGF -P - transforming growth factor-(3
TG1F - T G - Interacting Factor
V E - visceral endoderm
vm

A C K N O W L E D G E M E N T S
First and foremost, I thank Pamela Hoodless for giving me the opportunity to work on this exciting project and for always being supportive. Members o f my committee, Connie Eaves, M u r i e l Harris, and Rob K a y were invaluable and provided excellent advice and guidance.
Members o f the lab: Ani ta Charters, Rebecca Cu l lum, Sam Lee, Rachel Montpetit, Kris ten M c K n i g h t , Elizabeth Wederell , and M o n a W u were wonderful people to work with. Special thanks go to Robin Dickinson who always has time to talk about science and experiments, and w i l l always give his best advice. Juan H o u arrived at the lab just in time to give me sage advice on embryology. It is always fun and stimulating to converse with Pavle Vrl j icak.
The Terry Fox Laboratories is a wonderful learning environment for me. Maura Gasparetto, my favorite F A C S teacher, 1 can't thank you enough. Danny Chu i is a valuable resource o f E S and mouse information. M i k e Hughes and Frann Antignano taught me how to do tritium incorporation and counting, and fix the many leaks. Patty Rosten helped me with many molecular biology problems and is always fun to have a laugh with. Rewa Grewal performed the crucial blastocyst injections. Bob Argiropoulos and Ke i th Humphries helped with the retroviral transductions. F ina l ly superb F A C S support was provided by Gary de Jong, Gayle Thornbury, Lindsey Laycock, and C a m Smith.
The B C Cancer Research Centre is also a source o f wonderful people. Dan Doxsee was generous with his time and expertise with the cell counter. Brad Coe for statistics advice. The J A F and A R C cared for al l my mice.
I have been lucky to have had great role models in Sabine Cordes and Bernhard Weber.
Thank you, Timon, Cathie, Irma, Geraldine, Kat , Mel inda , and Jon for your friendship. I enjoyed the company o f fellow students Dale Robinson, Iris Cheung, and. S i lv i a Bakovic .
I also wish to acknowledge the Effie I. Lefeaux Scholarship in Menta l Retardation and the Albert B . and M a r y Steiner award for travel.
M y m o m Sui and my dad Eddy have always supported through many endeavours. I 'm grateful and hope to make you proud. Steven Quayle, my constant companion, was by my side through all the ups and downs.
Thank you a l l .
ix

D E D I C A T I O N
To my parents
x

1. CHAPTER 1 INTRODUCTION*
1.1. DEVELOPMENTAL GENETICS AND EMBRYOLOGY
Congenital malformations affect 4% of newborns, which equates to more than
150,000 affected newborns a year in the US (U.S. Department of Health and Human
Services). After accidents, birth defects are the leading cause of death in children. For
these patients, morbidity is also dramatically higher during infancy and childhood than
in the average population (Epstein 2003).
A major goal of research into congenital malformations is to provide a basis for
improved genetic counselling for the families in the short term, and ultimately to create
therapies for those directly affected. In addition, such research provides insights into
the mechanisms of development. Such discoveries can have significant impacts on the
understanding of birth defects and other diseases, thus aiding in the development of
drug targets, gene therapy, and cell, tissue and organ transplantation techniques.
The pathogenesis of congenital malformations has been investigated in many
model organisms, but most of all in the mammalian model, mice (Bier and McGinnis
2003). In mice, technology is available to re-create virtually any mutation in any gene
identified in humans (Glaser et al. 2005). Countless studies have demonstrated
similarities between mice and humans during embryonic development, as well as
between genes that regulate development. Such models demonstrate accurate disease
progression, and the accessibility of all developmental stages and tissues makes it
possible to investigate the functions of the genes that are altered in heritable human
diseases. Significantly, this may also allow testing and development of therapeutic
strategies. Nevertheless there are also important differences between mice and human
* A version of this thesis has been published. Mar, L and Hoodless, PA. Embryonic fibroblasts from mice lacking Tgif were defective in cell cycling. Mol Cell Biol. 26(11):4302-4310.
1

since most mouse models fail to reproduce all the features observed in the
corresponding human conditions(Watase and Zoghbi 2003).
The work described here focuses on two types of congenital defects,
holoprosencephaly (HPE), a structural malformation of the forebrain, and laterality
defects resulting from a failure to properly establish the left-right axis in the developing
embryo. Interestingly, disruptions to forebrain patterning sometimes arise concurrently
with laterality defects, thus giving insights into the mechanism of both processes. HPE
naturally occurs in lower vertebrates, including zebrafish, Xenopus, sheep and mice
(Capdevila et al. 2000; Muenke and Beachy 2000). This is consistent with observations
indicating early brain development is similar in vertebrates, although different species
acquire unique features later during development (Wilson and Houart 2004).
1.1.1. Holoprosencephaly
HPE is the most common birth defect affecting forebrain development (Cohen
2003). Its frequency is 1 in 10,000 to 20,000 amongst newborns, but a higher
frequency of 1 in 250 during fetal development indicates a substantial number of the
prenatal embryos are lost (Matsunaga and Shiota 1977). High mortality is associated
with this disease during infancy, but a significant number of HPE individuals continue
to live for many years (Redlinger-Grosse et al. 2002). The frequency of HPE is
actually thought to be higher since mild HPE is believed to be present in asymptomatic
individuals with reduced cognitive abilities.
The defining hallmark of HPE is the failure of the forebrain to divide into two
separate hemispheres and ventricles, resulting from the loss of midline structures
(Muenke and Beachy 2000). HPE encompasses a continuum of brain malformations.
At the most severe end of the spectrum, alobar HPE is characterized by a single
2

ventricle with no separation between the cerebral hemispheres. Semilobar HPE occurs
when the left and right frontal and parietal lobes are fused but the interhemispheric
fissure is present posteriorly. Lobar HPE is characterized by the separation of most of
the right and left cerebral hemispheres and lateral ventricles, but with a fusion of the
most rostral aspects of the telencephalon, especially ventrally. HPE is most commonly
accompanied by craniofacial anomalies including cyclopia, cleft lip and palate, and eye
defects in about 80% of individuals (Cohen 2003).
1.1.1.1. Multifactorial etiology
HPE can be caused by exposure to teratogens or other environmental agents
(Cohen 2003). For example, maternal diabetes was shown to increase the risk for HPE
by 200-fold (OMIM %236100). Low cholesterol is also being investigated as a
potential teratogen (Edison and Muenke 2003). Finally, alcohol and retinoic acid (RA)
were shown to increase the risk of HPE in humans and in animal models (Lammer et al.
1985; Sulik et al. 1995; Cohen and Shiota 2002).
Complex genetic anomalies are also observed in HPE individuals (Cohen 2003).
25-50% of individuals have numerical or structural chromosomal abnormalities that
were either inherited or occurred de novo. Mutations in single genes can also cause
HPE, or other complex syndromes that include HPE.
Nonsyndromic HPE cases that are inherited as a monogenic disease are
amenable for genetic investigations. More than 12 chromosomal regions have been
mapped in sporadic and familial HPE cases (Cohen 2003). Candidate genes were then
identified based on mutations found in patients. Many varieties of mutations were
found, including submicroscopic and small deletions, nonsense, missense and
frameshift mutations (Cohen 2003; Bendavid et al. 2005a). Currently, 25% of HPE
3

newborns and 22% of HPE fetuses with normal karyotypes have mutations in known
candidate genes.
1.1.1.2. Cand ida te genes in human
Heterozygous mutations in the four genes, SHH, SIX3, ZIC2 and TGIF, are most
frequently identified in patients (Wallis and Muenke 2000). For instance, SHH (Sonic
hedgehog) mutations were identified in 17% of familial cases and 3.7% of sporadic
cases (Roessler et al. 1996; Roessler et al. 1997). Mutations were also identified in
other members of the SHH pathway. GLI2, a downstream transcription factor, was
mutated in 1.8% of patients (Roessler et al. 2003). Four case reports also demonstrated
mutations in PTCH, a SHH receptor (Ming et al. 2002). Finally, 2-4% of patients with
Smith-Lemli-Opitz syndrome, an HPE related syndromic disease, have mutations in
DHCR7, a cholesterol reductase associated with cholesterol modification of SHH (Irons
2003).
Six3 mutations were identified in 3-4% of patients (Wallis et al. 1999).
Interestingly, mutations were identified at a moderately higher percent (5.3%) in fetuses
relative to patients, suggesting Six3 function is critical during the fetal period (Bendavid
et al. 2005b).
A number of mutations have also been identified in members of the Nodal
pathway. 2-6% of patients have haploinsufficient mutations in TGIF, a transcriptional
repressor shown to modulate Nodal signaling (Gripp et al. 2000). Additionally, 4-6%
of affected newborns and 8.5%) of affected fetuses have mutations in Zic2, a
transcription factor that potentially regulates Nodal signaling (Brown et al. 1998; Nagai
et al. 2000; Houston and Wylie 2005). Heterozygous mutations in TDGF1, a co-factor
critical for Nodal signaling, were identified in 0.5% of patients (de la Cruz et al. 2002).
4

Finally, heterozygous missense variants of FOXH1, a transcription factor that
transduces Nodal signals, were also identified in HPE (Ming and Muenke 2002).
Intriguingly, HPE demonstrates considerable variability and incomplete
penetrance for all known loci; the penetrance for TGIF mutations or deletions is only
10% (Nanni et al. 1999; Aguilella et al. 2003). A discussion of the potential
mechanism of these candidate genes during normal forebrain and HPE development
will be presented after normal forebrain development is reviewed.
1.1.1.3. Forebrain development
The vertebrate brain arises from the embryonic neural plate. The forebrain, or
prosencephalon, develops from the anterior neural plate and gives rise to the cerebral
cortex, basal ganglia, eye, thalamus and hypothalamus, all of which are neural
structures affected in HPE patients (Muenke and Beachy 2000).
By the time the neural plate arises at the anterior end of the mouse embryo on
Embryonic day 7 (E7), many patterning events critical for the neural plate have already
occurred. Thus, an understanding of earlier stages is required. At E6 the mouse
embryo consists of the epiblast, a radial cup-shaped layer of epithelial cells that will
give rise to the entire embryo; a surrounding layer of visceral endoderm (VE); and
extra-embryonic tissues located proximal to the epiblast (Figure 1.1). First, neural fate
is induced by the anterior visceral endoderm (AVE) located at the future anterior end of
the embryo. Simultaneously, gastrulation initiates at the diametrically opposite side to
the AVE, marking the future posterior end. Gastrulation, the process whereby
epithelial cells ingress and generate mesoderm, initiates and gives rise to the primitive
streak. The primitive streak begins at the rim of the epiblast cup and proceeds to the
distal tip. The antero-posterior polarity of the embryo is thus established. The
5

PROXIMAL
ANTERIOR
anterior visceral endoderm
POSTERIOR
primitive streak
DISTAL
Figure 1.1 Forebrain induction and patterning in the mouse embryos. Anterior visceral
endoderm (red cells; visceral endoderm, pale green) rotates to the future anterior region
and induces the overlying epiblast or embryonic ectoderm (blue) to become forebrain.
Anterior definitive endoderm and axial mesoderm derived from node (orange) and
primitive streak (purple) further refine and pattern the forebrain at later stages. Extra
embryonic tissues (white) are located proximal Iy. See text for details. Adapted from
Beddington and Robertson, 1998.
6

derivatives of the anterior primitive streak - the node, anterior definitive endoderm, and
axial mesoderm - reinforce and further refine the existing patterns within the neural
plate. At later stages, additional patterning centres, such as the anterior neural ridge,
are generated. Together with the existing patterning centers, they regulate the
morphogenesis of the forebrain. A variety of forebrain defects has been characterized
in gene disruption experiments, and functional analyses of these genes have given
insights to each successive developmental process. Excellent, in depth reviews of this
field are available (Beddington and Robertson 1999; Wilson and Houart 2004).
1.1.1.3.1. Anterior visceral endoderm induces anterior neural fate
One of the most celebrated experiments in developmental biology was
published in 1927 by Spemann and Mangold. This experiment revealed the presence of
cells that have the ability to pattern the entire vertebrate organism. By grafting a small
clump of cells, the organizer, from one amphibian blastula into another, they
demonstrated that these cells have the ability to co-opt and organize a complete
secondary axis (Gilbert 2003).
In mammals this grafting experiment was never possible; graft experiments with
the equivalent organizer were able to duplicate an entire trunk, but the resultant
secondary axis never included the head (Beddington 1994; Tam and Steiner 1999).
However, more recent grafting experiments using the organizer, plus the AVE, have
been able to duplicate an entire axis, including the head, in mouse embryos (Tam and
Steiner 1999). The converse removal of the A V E resulted in a loss of anterior neural
tissues (Thomas and Beddington 1996). Together, these experiments demonstrate that
the A V E is essential for inducing anterior neural ectoderm in mammals.
7

The neural plate normally forms by E7.5 and expresses neural markers such as
Six3 or Otx2, but mutants with A V E defects never express neural markers, indicating
that neurectoderm was never fully induced (Rhinn et al. 1998; Shawlot et al. 1999).
The A V E initially forms at the distal tip of the embryo but arrives at the prospective
head region through rotation (Figure 1.1). To promote head formation, posterior axis
development is suppressed (Piccolo et al. 1999; Robertson et al. 2003; Wilson and
Houart 2004); trunk and other posterior structures develop from intricate and complex
interactions between the Nodal, Wnt, BMP, and RA pathways. The A V E antagonizes
these pathways by inducing the expression of several antagonist genes: Cerrl acts as a
multifunctional antagonist against the Nodal, Wnt and BMP signaling pathways; lefty 1
is a Nodal antagonist; and noggin and chordin are both BMP antagonists (Piccolo et al.
1999; Bachiller et al. 2000; Perea-Gomez et al. 2002). Consistent with this model,
transplant experiments using posteriorizing factors such as RA, and mutants disrupted
for genes in posteriorizing pathways, developed similar neural plate induction defects
as A V E mutants (Ang et al. 1994; Liu et al. 1999).
One of the key signaling pathways regulating the expression of A V E genes is
Nodal, a secreted signaling factor belong to the Transforming Growth Factor - P (TGF^
/?) family of ligands. Nodal signaling directly initiates the expression of lefty 1 and
Cerrl in the visceral endoderm at the distal tip of the epiblast (Brennan et al. 2001).
Nodal also turns on the expression of Cripto, which directs the orthogonal movement of
distal visceral endoderm to the prospective anterior end (Varlet et al. 1997; Ding et al.
1998). Consistent with these observations, disruptions to the Nodal pathway resulted in
a failure to activate anterior neural fate (Ding et al. 1998; Brennan et al. 2002;
Yamamoto et al. 2004; Takaoka et al. 2006). Interestingly, in Cripto mutants the A V E
failed to rotate and remained at the distal tip, and the epiblast cells adjacent to the A V E
8

began to express anterior neural markers (Ding et al. 1998). This result further
demonstrated signals emanating from the A V E induced head formation.
1.1.1.3.2. Node-derived anterior definitive endoderm reinforces and further
elaborates forebrain patterning
Concurrent with the rotation of distal visceral endoderm to the anterior end,
proximal epiblast rotates to the prospective posterior end to initiate gastrulation and
primitive streak formation (Ding et al. 1998; Brennan et al. 2001). The node arises at
the anterior end of the primitive streak as it elongates from the rim of the epiblast cup
and reaches the distal tip. The node in the mouse embryo is the equivalent structure to
the organizer in the frog blastula, even though it lacks head induction activity, as
discussed above. However, the function of the node is necessary for head formation as
evidenced by forebrain mutants such as Wnt3 and Arkadia that developed a normal
A V E and initiated proper expression of neural plate markers, but lacked the node (Liu
et al. 1999; Episkopou et al. 2001). This finding is not surprising as the node gives rise
to structures that will have critical functions later during forebrain patterning. The
ventral cells of the node give rise to the prechordal plate, notochord and definitive
endoderm, while the dorsal cells of the node contribute to the floor plate along the
ventral midline of the embryo (Placzek and Briscoe 2005).
As gastrulation proceeds in the embryo, the A V E is displaced by anterior
definitive endoderm (ADE) (Thomas and Beddington 1996). The ADE migrates to the
head region and lines the ventral neural plate. Curiously, the ADE expresses many of
the same genes as the AVE, including Cerrl, Liml/Lhxl and Hex, prompting the idea
that the function of the ADE is to maintain the expression and function previously
established by the A V E (Wilson and Houart 2004). Mutants with disrupted ADE
9

generally acquired anterior neural tissue at E7, but by E7.5 - E8 the expression of these
neural markers was lost, and, consequently, anterior neural defects developed (Shawlot
et al. 1999; Martinez Barbera et al. 2000; Shawlot et al. 2000; Hallonet et al. 2002).
Nodal signaling is also critical in generating a wide variety of cells during
gastrulation in the anterior primitive streak, including the ADE lineage (Robertson et al.
2000; Tremblay et al. 2000; Vincent et al. 2003; Chu et al. 2004). Consistent with this
idea, disruptions to transducers of the Nodal signaling pathway, including ALK4,
ActRlIA, ActRIIB, Smad2, Smad3, Smad4, and FoxHl, resulted in anterior primitive
streak defects (Ding et al. 1998; Heyer et al. 1999; Song et al. 1999; Hoodless et al.
2001; Yamamoto et al. 2001; Norris et al. 2002; Vincent et al. 2003; Chu et al. 2004;
Dunn et al. 2004). In fact, Nodal plays multiple roles in the establishment of anterior-
posterior patterning in the epiblast of the mouse embryo. As a morphogen, various
levels of Nodal signaling convey different instructions. To illustrate, high Nodal
signaling is necessary for definitive endoderm formation while lower levels are
sufficient for A V E and primitive streak formation (Vincent et al. 2003; Dunn et al.
2004). Consequently, a small reduction of Nodal signaling in some mutants listed
above caused ADE defects, but a greater reduction in signaling led to A V E defects.
Conversely higher than normal levels of Nodal signaling can also cause developmental
abnormalities. Ectopic Nodal signaling in mutants that express reduced Nodal
antagonists, including Drapl, lefty], and lefty2, caused disruptions to A V E formation
(Iratni et al. 2002; Perea-Gomez et al. 2002). It is important to note that the forebrain is
the part of the embryo most sensitive to disturbances of Nodal signaling. The
biochemistry of the Nodal signaling pathway will be discussed in greater detail below.
10

1.1.1.3.3. Node derived prechordal plate provides ventral midline patterning
centre
The node also generates axial mesoderm that migrates along the length of the
embryo beneath the midline of the neural plate. Some A D E intercalates into the axial
mesoderm to form the axial mesendoderm, a rod-shaped structure that later replaces the
A D E . Axial mesendoderm underlying the prosencephalon from this stage on is referred
to as the prechordal plate, while that underlying the caudal axis is referred to as the
notochord.
The probable function of the axial mesendoderm, like the A V E and A D E before
it, is in the maintenance of neural identity through the continual opposition of posterior
signals from the epiblast. As well, the prechordal plate and notochord now produce
additional signals that further refine the pattern along the mediolateral axis that will
later translate into the dorsoventral axis as the neural plate folds into the neural tube
(Shimamura and Rubenstein 1997; Placzek and Briscoe 2005). The anterior neural tube
is now called the prosencephalon or forebrain, and will give rise to the telencephalon
and diencephalon. The prechordal plate has distinct gene expression and temporal
patterns from the notochord. For instance, gsc is expressed by the prechordal plate but
not by the notochord and, while chordin and noggin are both expressed in the
notochord early at the 3 somite stage, their expression in the prechordal plate appears
later at the 5 somite stage (Belo et al. 1997; Anderson et al. 2002).
The focus from this point will be on mutants that affect the prechordal plate and
the forebrain. Mutants lacking prechordal plate initiate early neural marker expression
normally, but the established patterns degenerate over time, indicating prechordal
mesendoderm maintains existing expression patterns (Shawlot et al. 1999; Camus et al.
11

2000). In addition, mutants develop expanded dorsal cell fate at the expense of ventral
midline and ventral cell fate (Filosa et al. 1997; Nishioka et al. 2005).
1 . 1 . 1 . 3 . 4 . Regional patterning of the forebrain
By early somite stages the rostro-caudal and dorsoventral axes within the
forebrain are subdivided into the rostral telencephalon and eye field, the caudal
diencephalons, which will contribute to the prethalamus and pretectum, and ventrally to
the hypothalamus. These divisions arise through expression of region-specific markers:
rostral expression of Fgf8, FoxGJ, Rx, Hesx and Six3, caudal expression of Otx2 in the
forebrain-midbrain region, dorsal-medial expression of BMP and Wnt family members
(Grove et al. 1998; Lee et al. 2000), and ventral expression ofShh and Foxa2IHnf3B in
the notochord and floorplate. Expression of these markers precedes the appearance of
morphological structures at E l 1, including the medial ganglionic eminence (MGE) at
the ventral-most location, the lateral ganglionic eminence (LGE) at the ventral-lateral
location, and the cortex at the dorsal-most location. The mechanism of forebrain
patterning is dynamic and complex and is still being investigated. Detailed reviews are
available (Hebert 2005; Lupo et al. 2006).
Three interdependent regional patterning centres organize and further refine
forebrain morphogenesis (Figure 1.2). The anterior neural ridge (ANR) is a
morphologically defined structure located at the junction of the anterior neural plate
and the non-neural ectoderm and represents the rostral patterning center. Fgf8
signalling from the ANR was discovered to induce and maintain forebrain fate
(Shimamura and Rubenstein 1997). Reducing Fgf8 levels caused a smaller
telencephalon to develop from reduced rate of neurogenesis, and was preceded by
12
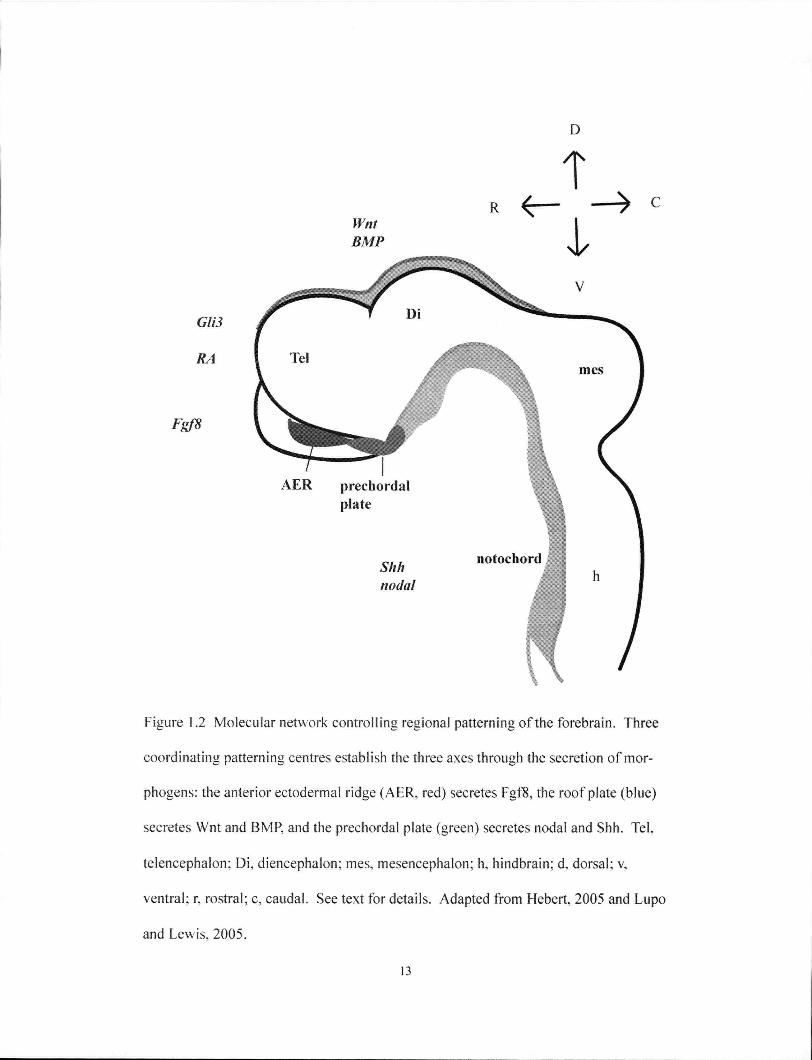
D
T
Figure 1.2 Molecular network controlling regional patterning of the forebrain. Three
coordinating patterning centres establish the three axes through the secretion of mor-
phogens: the anterior ectodermal ridge (AER, red) secretes Fgf8, the roof plate (blue)
secretes Wnt and BMP, and the prechordal plate (green) secretes nodal and Shh. Tel.
telencephalon; Di , diencephalon; mes, mesencephalon; h, hindbrain: d. dorsal; v.
ventral; r, rostral; c, caudal. See text for details. Adapted from Hebert, 2005 and Lupo
and Lewis, 2005.
13

reduced expression of Foxgl and Six3 (Anderson et al. 2002; Storm et al. 2003; Storm
et al. 2006).
The activities of the ventral patterning centre are located in the prechordal plate
and floorplate, which secrete Shh (Figure 1.2). Mutations in Shh in both mice and
humans can cause HPE (Roessler et al. 1996; Zhang et al. 2006). The Shh protein is
both necessary and sufficient for the development of ventral telencephalic structures,
such as the MGE and LGE, and the expression of associated neural markers (Gunhaga
et al. 2000; Watanabe et al. 2005). Interestingly, Shh is required during MGE
specification earlier on and during LGE specification at a slightly later stage (Kohtz et
al. 1998). A repressive transcription mediator of the dorsal Shh signal in the forebrain
is GU3. Mice deficient for GH3 exhibit a dorsal to ventral transformation, consistent
with its function as a repressor of Shh signalling. Surprisingly, though, dorsoventral
patterning is established normally in the absence of both Shh and GU3 (Rallu et al.
2002). This result showed that Shh and GU3 are crucial for dorsoventral patterning of
the telencephalon, but their functions are dispensable when both are eliminated. Shh's
function during telencephalon patterning is dependent on timing, but additional
mechanisms are clearly also involved.
Nodal signalling is also suspected to contribute to ventral patterning, since
nodal is expressed in the prechordal mesoderm at this stage (Figure 1.2). In mice,
Nodal's role is not yet well understood since many of the mutants are severely affected
by disruptions to earlier events. However, in zebrafish, Nodal mutants lack ventral
structures in the diencephalons (Mathieu et al. 2002). Currently, detailed understanding
of Nodal in this process is limited, but it is thought to function through multiple
mechanisms: through direct regulation and through co-operation with the Shh pathway,
as well as through pathways that are independent of Shh (Lupo et al. 2006).
14

Dorsal signals from the roof plate and dorsal-medial cells require members of
the B M P and Wnt families to mediate patterning of dorsal fates (Furuta et al. 1997;
Grove et al. 1998; Lee et al. 2000)(Figure 1.2). B M P has the ability to induce dorsal
midline fate as evidenced by its induction of the expression of Msx, a dorsal forebrain
marker (Furuta et al. 1997). In contrast, disruption to Bmprla resulted in the loss of the
dorsal-medial structure, the choroid plexus (Hebert et al. 2002). In addition, members
of the Wnt family are expressed dorsally and were shown to specify dorsal rather than
ventral telencephalic identity in chick explants (Braun et al. 2003; Gunhaga et al.
2003). Finally, ES cells induced to form telencephalon neurons can be induced to
adopt a dorsal fate by Wnt signalling (Watanabe et al. 2005).
It is worth noting again that the local patterning centers and their associated
pathways are interdependent, a conclusion that is drawn from multiple studies that
witnessed the breakdown of other patterning centres from initial disruptions to one
signalling centre (Figure 1.2) (Dou et al. 1999; Camus et al. 2000; Ohkubo et al. 2002;
Ribes et al. 2006; Storm et al. 2006). R A has been shown to coordinate these
patterning centres. Raldh2, a regulator of steady R A levels, is expressed in the
forebrain neuroepithelium and the overlying surface ectoderm, and its loss resulted in
defective morphogenesis of various forebrain derivatives (Ribes et al. 2006). It plays a
role in maintaining Fg/8 signals from the A N R and Shh signalling from the ventral
patterning centre. Thus, R A signalling is integral to the FGF-SHH signalling loop
circuit.
1.1.1.3.5. Potential disruptions caused by mutations in HPE genes
As discussed earlier, mutations in SHH, SIX3, ZIC2 and TGIF were identified in
H P E patients. The Shh pathway is critical as a ventral patterning morphogen;
15

disruptions to this pathway cause dorsoventral transformation in mice, consistent with
the involvement in HPE (Cohen 2003).
Six3 is an antagonist of the Wnt pathway (Braun et al. 2003; Lagutin et al.
2003). Antagonizing Wnt signaling is essential for establishing the anterior forebrain.
Consequently, a reduction in Six3 activity resulted in the loss of anterior forebrain
structures (Lagutin et al. 2003). Later, Wnt signals become localized to the dorsal
midline and play a role in patterning the dorsoventral axis (Grove et al. 1998). Six3
may also modulate Wnt signaling during this process. Disruptions to anterior forebrain
patterning and later dorsoventral patterning can both lead to HPE.
Zic2 mutations are found in HPE patients, and interestingly this group of
patients did not develop craniofacial abnormalities (Wallis et al. 1999). Mouse models
indicate dorsal patterning has been disrupted (Nagai et al. 2000).
Disruptions to both Zic2 and Tgif may affect Nodal signaling and cause
abnormal forebrain patterning (Hoodless et al. 2001; Houston and Wylie 2005; Lupo et
al. 2006). Knowledge of the precise functions played by Nodal during forebrain
patterning are not known, but evidence points to disturbances in Shh expression in the
prechordal plate (Rohr et al. 2001). Alternatively, Tgif may modulate the RA pathway
inappropriately. The RA pathway is now known to be an important coordinator of
forebrain patterning centres. Potential molecular mechanisms by which Tgif 'may affect
Nodal and RA signaling will be discussed below.
1.1.2. Asymmetric body plan
Superficially, mammals appear bilaterally symmetric, with a symmetric face,
head, torso and bilateral appendages. Upon reflection and examination, though,
mammals are obviously pseudo-bilateral. Many internal organs possess invariant left-
16

or right- handedness, known as situs. Specifically, the heart and spleen are
predominantly in the left side of the thoracic cavity, the stomach is in the left side of the
abdominal cavity, and so on. Furthermore, it is fascinating to discover language, sense
of humour, taste and smell, as well as many other aspects of our brain functions, are
located in asymmetric CNS organs (McManus 2002; Sun et al. 2005; Sun and Walsh
2006).
Congenital laterality disorders are found in humans at a frequency of 1 in 8500
(Capdevila et al. 2000; Purandare et al. 2002; Bisgrove et al. 2003). Situs inversus, a
complete reversal of the internal organs, has a natural frequency of 1 in 10,000 but is
present in 20% of Kartagener syndrome patients (Capdevila et al. 2000; Bisgrove et al.
2003). While people with this condition have minor sinus and pulmonary infections,
and males are sterile, they have a normal life expectancy. However, situs ambiguus,
resulting in the reversal of only some of the organs, is a source of significant health
problems. There is a wide spectrum of situs ambiguus disorders, including right
isomerism, or asplenia syndrome, where bilateral right-sidedness occurs; left
isomerism, or polysplenia, where bilateral left-sidedness occurs; as well as a host of
complex phenotypes that require careful examination and diagnosis (Bisgrove et al.
2003; Levin 2005).
Genetic analysis and experimental perturbations in human and animal models
revealed a complex cascade of molecular events that establish laterality early during
embryogenesis before the onset of organogenesis. The following review focuses on
left-right determination in mice, although the mechanism of left-right patterning is
fundamentally conserved in vertebrates, and any significant or relevant differences will
be noted (Figure 1.3). Briefly, a break in bilateral symmetry before or around the
formation of the node initiates this process. Subsequently, situs-specific gene
17

L E F T RIGHT midline
barrier
Node
leftness
Figure 1.3 Genetic pathway for the determination of left-right asymmetry. See text for
details. Adapted from Hamada, 2002.
18

expression patterns become established within or near the node during gastrulation.
Left-right patterning information is then transferred from the node to the lateral plate
mesoderm (LPM), where left-sided expression of Nodal becomes stabilized. The
establishment of asymmetric expression is unilaterally restricted by midline structures.
Finally, patterning information from the LPM is transferred to organ primoridia where
asymmetric morphogenesis takes place. Excellent and comprehensive reviews of these
events are available (Bisgrove et al. 2003; Hornstein and Tabin 2005; Levin 2005; Raya
and Belmonte 2006).
1.1.2.1. Breaking of symmetry
One of the most controversial and intellectually captivating questions in the
laterality field is what mechanism initiates bilateral asymmetry? There is now little
dispute that nodal cilia, located within the node, rotate in a clockwise fashion and
generate a right to left fluid flow across the node (see videos at
http://www.cell.eom/cgi/content/full/95/6/829/DCl) (Nonaka et al. 1998). The nodal
flow model was proposed after the identification of numerous genes involved in human
and mouse with left-right patterning defects were found to affect genes encoding
dyneins, or motor proteins, and other proteins involved in the ultrastructure and motion
of the nodal cilia. Nodal flow and cilia movements can be visualized by electron
microscopy and high resolution microscopy (Nonaka et al. 1998; Okada et al. 2005).
Sophisticated methods were used to monitor and experiment with the velocity and
direction of fluids within the node (Nonaka et al. 2002; Okada et al. 2005). Altogether,
these experiments showed a strong correlation between loss of cilia movement,
alteration in nodal flow, and resultant aberrant laterality development.
19

The functional effect of the uni-directional fluid flow is still under dispute
(Tabin and Vogan 2003). An attractive model postulates the leftward transport of
morphogen proteins by nodal flow (Nonaka et al. 1998). Recently, nodal vesicular
parcels were visualized as being released within the node, transported by the nodal flow
and subsequently fragmented, presumably leading to the release of the contents of the
disrupted parcels (Tanaka et al. 2005). These were shown to contain the putative
morphogens Shh and RA, and the release of these parcels required Fgf signalling
(Tanaka et al. 2005). Consistent with this model, Fg/8, Shh, Ihh, and RA signalling
have been shown to regulate asymmetric Nodal expression and early asymmetric
patterning (Meyers and Martin 1999; Tsukui et al. 1999; Zhang et al. 2001).
An alternative model also starts with a unidirectional fluid flow but is based on
a physical deformation of mechano-sensory cilia that provide directional cues, leading
to subsequent asymmetric downstream events (McGrath et al. 2003; Tabin and Vogan
2003). This two-cilia model was proposed upon discovery of immotile cilia that
surround motile cilia within the node (McGrath et al. 2003; Tabin and Vogan 2003). In
contrast to the nodal flow model, which cannot adequately explain why mutants that
lacked cilia have the same laterality phenotype as mutants that had immotile cilia, the
two cilia model can account for these different defects. The two cilia model predicts
mutants with immotile cilia, but functional mechanosensory cilia, such as iv, developed
situs inversus and situs solitus (normal situs) because small, stochastic signals
randomly defined left from right. Conversely, mutants, such as Kif3B, that completely
lacked cilia developed a situs ambiguus phenotype when there was neither directional
information nor subsequent signalling activity. Furthermore, mechano-sensory cilia
were shown to trigger Ca + + influx, and asymmetric Ca + + influxes were previously
associated with symmetry breaking events (McGrath et al. 2003; Raya et al. 2004).
20

Current controversies mostly surround the speculation that cilia-induced flow
may only be transducing an earlier asymmetric signal rather than initiating asymmetry
de novo. This idea arises from discoveries in zebrafish and chick that asymmetric
activities, namely H+/K+-ATPase channel activity, Ca + + flux and accumulation, and
Notch signalling, preceded node formation and the appearance of cilia in the node
(Hornstein and Tabin 2005). Perhaps these seemingly contradictory observations
indicate the initial steps are different in vertebrates, arising from different evolutionary
pressures on the different gastrulas. Although nodal flow is universal in all vertebrates
(Bisgrove et al. 2000), it may be the primary mechanism used for symmetry breaking in
mammals (Kawakami et al. 2005). Other notable differences exist, such as the
differences in expression pattern and function of Sonic hedgehog and Fgf8, between
mammals and.other vertebrates during laterality determination (Yoshioka et al. 1998;
Meyers and Martin 1999; Tsukui et al. 1999).
1.1.2.2. Asymmetric gene expression in the node and lateral plate mesoderm
Regardless of the precise nature of the initial symmetry breaking events,
expression of the TGF- P family member Nodal becomes enhanced on the left side of
the node during gastrulation in all vertebrates (Figure 1.3)(Brennan et al. 2002; Hamada
et al. 2002). The left-sided Nodal signal from the node is likely relayed to the left LPM
through diffusion, and is later amplified and stabilized. The expression of Nodal in the
LPM is critical for initiating downstream target genes, such as lefty 1, lefty2 and pitx2.
Accurate regulation of these expression patterns is achieved through complex positive
and negative feedback loops. Nodal expression is maintained by amplification through
a positive autoregulatory loop mechanism. The threshold, range and duration of Nodal
signalling is restricted by numerous factors, including SPC1, SPC4, Drapl, and lefty2,
21

through negative feedback loops (Meno et al. 1997; King and Brown 1999; Constam
and Robertson 2000a; Constam and Robertson 2000b). Regulatory elements
participating in these feedback loops have also been identified. For instance, the
expression of Nodal, lefty 1 and pitx2 in the L P M were co-ordinately regulated by an
asymmetric enhancer that contains binding sites for the forkhead transcription factor
FoxHl (Hamada et al. 2002). Ultimately, the precise regulation of Nodal levels and
expression pattern is essential for the downstream target genes that orchestrate
asymmetric morphogenesis.
1.1.2.3. Asymmetric gene expression maintained by the embryonic midline
The importance of the embryonic midline, which includes the notochord and the
neural floorplate, during laterality determination, was appreciated early on. Surgical
removal of notochords in frogs led to aberrant cardiac looping (Danos and Yost 1996);
right-handed looping of the heart tube is the earliest visible manifestation of
asymmetry. The notochord is a rod-like structure derived from mesodermal cells that
ingress through the node during gastrulation. The neural floorplate is located at the
base of the developing neural tube, and is induced by and directly adjacent to the
notochord. Many midline mutants in zebrafish and mice also develop situs
abnormalities. For example, Shh mutants did not maintain notochord and neural
floorplate and developed multiple laterality defects (Meyers and Martin 1999).
Experimental results indicate the midline prevents the contra-lateral spread of
asymmetric gene expression patterns as they become established. Thus, midline
mutants developed randomized Nodal expression and exhibited situs ambiguus.
Midline cells provide a barrier between the left and right sides through a number of
mechanisms. To prevent Nodal expression from crossing over to the right side of the
22

midline, lefty 1, a homologue of lefty2, and an antagonist of Nodal, is expressed in the
floorplate (Meno et al. 1998; Bisgrove et al. 2003; Levin 2005). The midline is also
rich in extracellular matrix which have the ability to insulate activities such as left-
sided Ca + + signals from propagating to the right side of the embryo (McGrath et al.
2003). Interestingly, this property of the midline is underlined by the common
observation that among two conjoined twins, only the twin on the right developed
laterality defects (Levin 2005). Apparently, asymmetric signals from one embryo
cannot cross over the midline, but are able to influence the neighboring embryo.
1.1.2.4. Organ positioning and morphogenesis
The last steps in laterality morphogenesis are the simplest conceptually, but the
precise molecular pathway is not yet well characterized. This step instructs organ
morphogenesis that obeys situs information. For example, during late neurulation
stages, the linear heart tube, formed from paired cardiac progenitors in the left and right
LPM, undergoes right-sided looping. Another fascinating example is embryonic
turning in mouse and chick embryos; events that bring the embryo from a dorsally
flexed position to a ventrally flexed or fetal position. The only gene known thus far to
play a role in these events ispitx2 (Ryan et al. 1998).
1.1.2.5. Retinoic acid and symmetrical development of somites
Somites are paired structures that give rise to a number of bilateral structures,
including the muscles and the skeleton. In the mouse embryo somites are physically
adjacent to tissues that develop in response to asymmetric patterning information. As
discussed, the Nodal, RA, Wnt, Notch, Shh, Fgf and BMP pathways play critical roles
in laterality determination. These pathways are used repeatedly during many other
23

developmental processes, from limb development to facial patterning. How do tissue
progenitors respond to the same signals when they are used both during laterality
determination and during somite development? Recently it was discovered that
symmetric development of somites is not a default state. While this is not yet well
understood, during symmetric somite development RA signalling was shown to buffer
and prevent laterality signals from ectopically activating somitogenesis asymmetrically
(Kawakami et al. 2005; Vermot and Pourquie 2005). Disruptions to the RA pathway
can cause laterality disturbances to both synchronized somite development and other
organs. Likewise, inhibition to the RA pathway, using antagonists in embryos at the
headfold stage, led to bilateral expression of pitx2 in the L P M (Vermot and Pourquie
2005).
In summary, forebrain patterning and laterality determination involve
overlapping developmental mechanisms and signalling pathways.
1.2. T G - INTERACTING FACTOR (TGIF)
The discovery of Tgi/mutations in HPE patients was particularly exciting since
Tgif'has been shown to modulate both the Nodal and retinoic acid pathways (Gripp et
al. 2000). As discussed, the Nodal and RA pathways have long been associated with
the development of HPE in patients and in animal models.
1.2.1. TGIF is a T A L E HD
TGIF is a 272 amino acid transcription factor that belongs to the three amino
acid loop extension (TALE) superclass of homeobox genes (Figure 1.4) (Burglin 1997).
Homeobox proteins are an evolutionarly conserved family of transcription factors that
contain a highly conserved stretch of 60 amino acids that fold into a helix-turn-helix
24

H P E Mutations
C/3
* *
u — a>
* * * *
272
C t B P Binding Domain
H o n i e o d o m a i n Smad2/3 & H D A C Interaction Domain
Repressive Domain & Sin3 Interaction Domain ( E G F / M A P K phosphorylation)
Figure 1.4 TGIF functional domains. HPE patient mutations are indicated by asterices
above the domains.
25

motif. Members of the TALE family, which includes PBC and ME1S class proteins,
form heteromeric complexes with each other to regulate transcription of
developmentally important genes, such as the Hox genes and Pax6 (Mann and Affolter
1998). As co-factors, they provide specificity and cooperativity for Hox proteins.
TALE proteins can also serve as co-factors to other transcription factors (Sagerstrom
2004). Finally, TALE proteins may also penetrate inactive chromatin and anchor Hox
or other transcription factor to regulate activity. Although TGIF has not been shown to
bind PBC proteins (Chang et al. 1997; Knoepfler et al. 1997), TGIF was shown to
mutually compete with Meis for DNA binding and inhibit D1A dopamine receptor
(Yang et al. 2000a). The homeodomain is located between residues 35-97 (Wotton et
al. 1999a).
1.2.2. TGIF is a RA antagonist
TGIF was first implicated as an antagonist in the RA pathway because it
competed for DNA binding with the RXRa retinoic acid receptor (Bertolino et al.
1995). The RA pathway is an important morphogen for multiple processes during
development (Chambon 1996; Mark et al. 2006). Signalling of this pathway is initiated
by binding of RA to nuclear receptors, the RARs (a, p\ and y), which then regulate the
transcription of target genes by heterodimerizing with retinoid X receptor (RXR)
(Figure 1.5) (Chambon 1996). TGIF directly competes for DNA binding with RXR
responsive elements found within the promoter regions of genes such as the cellular
retinol-binding protein II (Bertolino et al. 1995).
More recently, TGIF was also shown to function as a transcriptional co-
repressor independent of direct DNA binding. TGIF directly bound to the ligand
binding domain of the RXR family of receptors, and this binding led to the repression
2 6
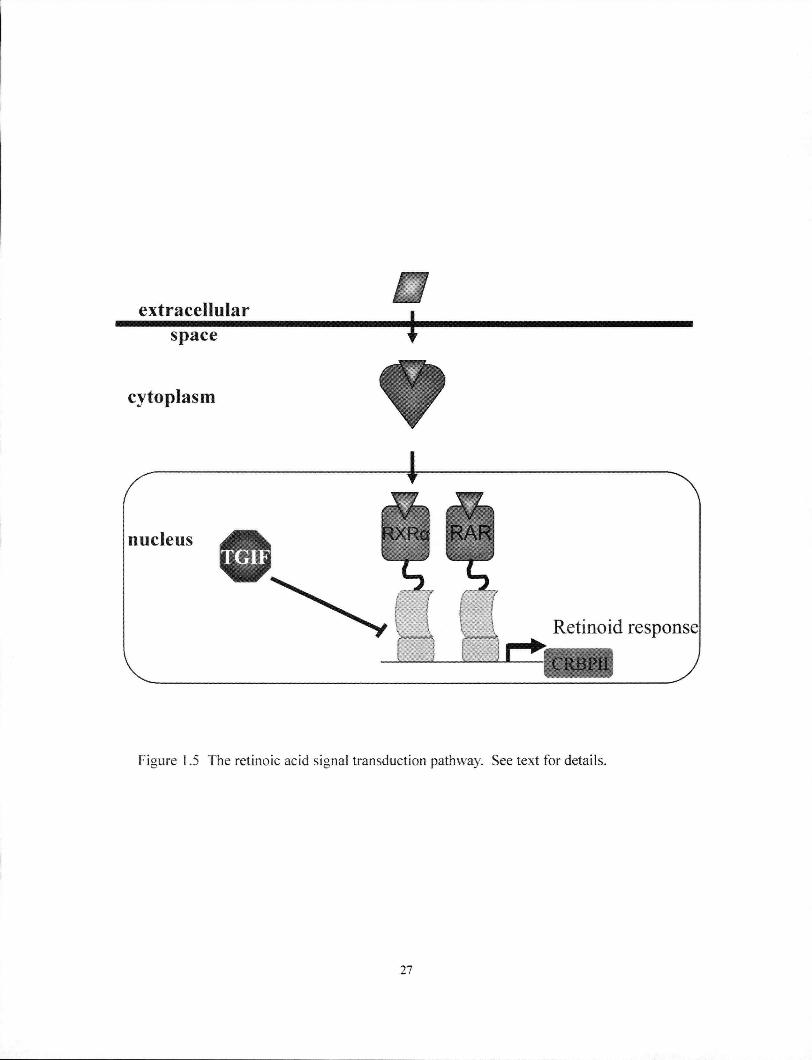
Figure 1.5 The retinoic acid signal transduction pathway. See text for details.
27

of downstream transcription via the recruitment of CtBP complexes (Melhuish and
Wotton 2000; Bartholin et al. 2006). Furthermore, interaction between TGIF and
RXRa could be competed by the addition of RA (Bartholin et al. 2006).
It is worth repeating here that the RA pathway has previously been implicated in
the development of HPE. Prenatal exposure to RA in humans and model organisms can
increase the incidence of this developmental defect (Schneider et al. 2001; Ming and
Muenke 2002). Consistent with this, RARa-RARy double mutants, and Raldh2
mutants, display telencephalic abnormalities (Lohnes et al. 1994; Ribes et al. 2006).
1.2.3. TGIF is a TGF-P co-repressor
TGIF was also shown to be a transcriptional co-repressor for TGF-P responsive
transcription through its interactions with Smad2 (Wotton et al. 1999a). The TGF-P
pathway plays important roles during cell cycle regulation, growth, differentiation and
other fundamental processes during embryogenesis and in the adult (Massague et al.
2005). Signalling events at the cell surface are initiated by the binding of ligands and
the formation of type I and type II receptor complex (Figure 1.6). The serine/threonine
kinase domain of the type II receptor subsequently phosphorylates the type I receptor.
For example, Nodal binds to Activin Receptor II (ActRII), which phosphorylates
Activin Receptor I (ALK4). Nodal then propagates its signal through the Type I
receptor by phosphorylation of the receptor (R)-regulated Smad proteins, Smad2 or
Smad3, in the cytosol. Upon activation, R-smads form complexes with the common
Smad, Smad4. Smad complexes then translocate into the nucleus and regulate
transcription by forming heteromeric complexes with DNA binding proteins such as
FoxHl. The level of transcriptional activity is regulated by the availability and
28

extracellular space
Figure 1.6 TGFP signal transduction pathway. See text for details. I, type I receptor; II, type II
receptor.
29

recruitment of transcriptional activators, such as p300/CBP, or transcription repressors,
such as TGIF, that become incorporated into the Smad protein complex.
TGIF represses TGF-P responsive transcription through multiple mechanisms:
by directly competing with the co-activator p300/CBP for Smad2 interaction, and by
recruiting histone deacetylase (HDAC) complex, the carboxyl terminal binding protein
(CtBP) complex, and the Sin3 complex (Wotton et al. 1999b; Melhuish and Wotton
2000; Wotton et al. 2001). Al l three of these complexes lead to modifications of the
chromatin in the neighbouring regions, effectively "closing" the DNA and limiting
access by transcription activating factors. Consistent with this, the chick orthologue of
TGIF, AKR, was also shown to be a transcriptional repressor (Ryan et al. 1995).
Two adjacent domains between residues 137-192 that interact with Smad2 and
Smad3, and that recruit histone deacetylase (HDAC) protein complexes (Wotton et al.
1999a). The N-terminal residues 24-28 are capable of recruiting CtBP protein
complexes (Melhuish and Wotton 2000). The extreme carboxyl terminus domain
between residue 192 and 272 that binds Sin3 protein complexes (Melhuish and Wotton
2000; Wotton et al. 2001).
It is relevant to note here that the TGF-p/Nodal pathway has also previously
been implicated in the development of HPE: (1) mutations in FOXH1, a transcription
factor in the TGF-P pathway, have been identified in HPE patients (Ming and Muenke
2002); and (2) mice deficient for genes in the TGF-P pathway, such as the compound
mutant Smad2+/~; Nodat1', develop HPE (Nomura and Li 1998).
TGF-P also activates the c-Jun N-terminal Kinase (JNK) pathway, which then
suppresses Smad2 signaling by recruiting Tgif, which competes with Smad2 for binding
to p300 (Pessah et al. 2001). A final interesting property of TGIF is its stability is
enhanced by the Ras/MAP kinase pathway (Lo et al. 2001). Increased stability of the
30

TGIF protein can be achieved via phosphorylation of Erk MAP kinase sites, T235 and
T239, near the C-terminus. Differential splicing results in two rgz/isoforms that
produce proteins differing by 20 amino acids at the N-terminus, but does not affect any
known functional domains.
1.2.4. T G I F mutations identified in H P E patients
More than 12 chromosomal regions have been mapped in sporadic and familial
HPE cases. One region, mapping to 18pl 1.3 in humans, contains TGIF. Heterozygous
deletions and mutations of TGIF have been associated with HPE (Overhauser et al.
1995; Gripp et al. 2000; Chen et al. 2002; Aguilella et al. 2003; Bendavid et al. 2005a;
Bendavid et al. 2005b; Chen et al. 2006). The incidence of TGIF microdeletions -
deletions that encompass the entire gene but are only detectable at the sub-karyotypic
level - occurs at the same frequency (1-3%) as point mutations in HPE patients.
However, patients at the mildest end of the HPE spectrum do not carry TGIF
microdeletions (Bendavid et al. 2005a; Bendavid et al. 2005b).
Interestingly, most point mutations affect known functional domains of TGIF
and may provide insights into the mechanism of HPE. For example, the nonsense
mutation, Y59X, produces a protein that terminates within the homeodomain (Aguilella
et al. 2003). Multiple missense mutations, such as S28C, P63R, R90C, T151A, and
T162F, probably disrupt the CtBP binding domain, homeodomain, Smad2/3 interacting
domain or the HDAC interacting domain, respectively (Gripp et al. 2000; Chen et al.
2002). The mutations Q107L and V126A (Aguilella et al. 2003; Chen et al. 2006) are
not located in known domains but may prevent protein interactions or proper folding of
the TGIF protein. A series of polymorphisms have also been reported in patients, but
their significance is not yet known (Wallis and Muenke 2000).
31

1.2.5. Evolutionary conservation
Tgif is conserved in eukaryotes from yeast to mammals (Burglin 1997). TALE
proteins are characterized by an extension of three amino acids between a-helices 1 and
2 within the homeodomain. Within the TGIF class, the homeodomain is distinguished
by the presence of an Ala residue at the 27 th position; in all other members of the TALE
superclass, a Pro is found at this position.
TOS8 is the yeast ortholog of Tgif While its precise function is not known,
TOS8 is directly downstream of the Swi-4-Swi6 cell cycle box binding factor (SBF)
and Mlul binding factor (MBF), which are transcription factors that regulate the start of
the yeast cell cycle, and therefore TOS8 presumably functions during G i / S progression
(Horak et al. 2002). The Drosophila genome contains 2 orthologues, achintya and
vismay, and both orthologues play redundant roles during meiosis and spermatogenesis
(Ayyar et al. 2003; Wang and Mann 2003). A chick orthologue named Avian Knotted-
Related was identified as a negative regulator of apoVLDLll transcription (Ryan et al.
1995). Orthologues in Xenopus and zebrqfish have also been identified, but are only
well conserved over the homeodomain.
There are 3 paralogues in mice and humans: Tgif2 (Imoto et al. 2000; Melhuish
et al. 2001), Texl (Lai et al. 2002), and Tgiflx/y (Bianco-Arias et al. 2002). Tgif2 lacks
the N-terminus CtBP binding domain, but the HDAC and Sin3 repressive domains were
shown to be functionally similar to Tgif (Melhuish et al. 2001). Tgiflx/y is located in a
shared region among the sex chromosomes and evolved by retrotransposition of Tgif2.
Consequently, Tgif2 and Tgiflx/y contain the same domains (Bianco-Arias et al. 2002).
The most divergent paralogue is Texl which functions during spermatogenesis (Lai et
al. 2002). The human Texl is not yet identified but is presumed to also be present in
the genome.
32

1.3. M A M M A L I A N C E L L C Y C L E
The importance of proliferation during development is evidenced by the
dramatic growth that occurs during embryogenesis. Despite this, our understanding of
the mechanisms by which patterning coordinates proliferation with other processes is
superficial.
The cell cycle generates daughter cells by cyclic progression through 4 phases:
G i , when cells are resting in the diploid stage; synthesis (S), when cells replicate DNA
to become tetraploid cells; G2, when cells generate sufficient materials for generating
two independent cells; and mitosis (M), when cells partition equivalent genetic and
other materials into the daughter cells (Figure 1.7)(Hunter 2000; Nurse 2000). Cells
can exist in an extended Gi phase referred to as Go when progression into S-phase is
blocked (Lodish et al. 2000).
Checkpoints ensure that each of these phases proceed in an orderly manner and
respond appropriately to both intrinsic signals, such as DNA damage or developmental
programs, as well as extrinsic signals, such as TGFp\ retinoic acid and Fgf. The
restriction (R) point is arguably the most important checkpoint and regulates the
commitment of a cell to undergo another round of cell division. Briefly, cyclic
expression of cyclin proteins leads to the formation of a cyclin-dependent kinase
(CDK)-cyclin complex, which has the ability to phosphorylate retinoblastoma (Rb) and
bypass the R point. The presence of the active CDK-cyclin complex is regulated at
multiple levels, including protein stability and a host of cyclin-kinase inhibitor proteins
such as p57K i p 2, p27K i p l and p21 C i p l (Sherr and Roberts 1995).
Many mammalian cell cycle proteins have been identified over the last 30 years.
The complex regulation of a multitude of these proteins by intrinsic and extrinsic cues
in single cells and cell lines is now better understood. However, the integration of
33
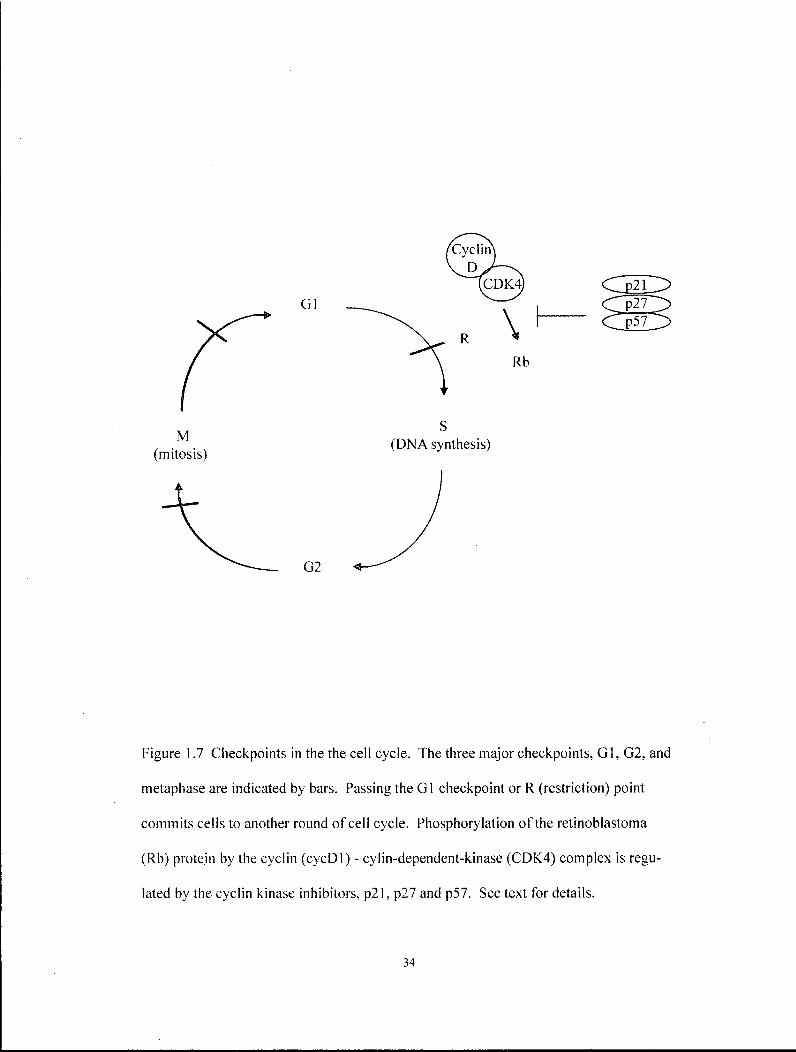
Figure 1.7 Checkpoints in the the cell cycle. The three major checkpoints, GI , G2, and
metaphase are indicated by bars. Passing the GI checkpoint or R (restriction) point
commits cells to another round of cell cycle. Phosphorylation of the retinoblastoma
(Rb) protein by the cyclin (cycDl) - cylin-dependent-kinase (CDK4) complex is regu
lated by the cyclin kinase inhibitors, p21, p27 and p57. See text for details.
34

intrinsic genetic programs and extrinsic signals is likely more complicated in
multicellular organisms. Yet the idea that the function of some cell cycle proteins is
vital has been challenged by the viability of mice that are depleted of essential cell
cycle genes (Kozar et al. 2004; Malumbres et al. 2004; Hinds 2006). Nevertheless,
evidence is emerging that in multicellular organisms related family members previously
believed to be functionally redundant may serve specialized functions during
development (Dyer and Cepko 2001).
1.4. HYPOTHESES
The hypotheses that were addressed are: (1) the loss of Tgif'will cause HPE or
related forebrain defects in mice; (2) the loss of Tgif will cause other developmental
defects that result from abnormal patterning of the primary axes; and (3) Tgif carries
out these functions by repressing target genes downstream of the TGF-P, RA and
Ras/MAPK pathways.
1.5. OBJECTIVES
To investigate these hypotheses, the following experiments were planned: (1) to
create mice with a disrupted Tgif, (2) to examine the mutant mice obtained for forebrain
and other developmental defects; (3) to determine potential defects at the cellular level
that might explain any developmental abnormalities seen; (4) to determine potential
defects at the molecular level; (5) to determine the effect of Tgif disruption on TGF-P,
RA and Ras/MAPK signalling.
35

2. CHAPTER 2 MATERIALS AND METHODS
2.1. GENERATION OF TGIF MUTANT MICE
2.1.1. Isolation of murine Tgf/genomic clone
The murine 129S6/SvEvTac genomic bacterial artificial chromosome (BAC)
library (generously donated by K. Humphries) was screened using Tgif'EST BF133915
as a hybridization probe. Multiple independent BAC clones containing Tgif md its
surrounding sequences were thus isolated.
Mouse genome sequences available through UCSC (http://genome.ucsc.edu/),
Ensembl (http://www.ensembl.6rg), and NCBI (http://www.ncbi.nlm.nih.gov) indicated
a 7.6 kilobase (kb) EcoRl fragment within BAC 49612 included exons 2 and 3 of Tgif,
which contain the majority of the coding sequences. Subsequent cloning generated the
genomic plasmid clone used to prepare the Tgif targeting construct (see below).
Restriction mapping was performed to confirm that the desired ^//"sequences were
present in the subcloned plasmid.
2.1.2. Construction of the Tgif targeting vector
The targeting vector was designed to replace exons 2 and 3, which encode
nearly all of the open reading frame sequences of Tgif, with the reporter gene lacZ
fused in frame to the remaining N-terminal 7 amino acids (Hasty 1993). The residual 7
amino acids are likely not functional. The construct also included a PGK-neo positive
selectable marker gene cassette that allowed enrichment of targeting events. The
targeting vector consisted of a 5' homology region, which was the 3.4 kb Xhol-SacIL
fragment, and a 3' homology region, which was the 1.5 kb EcoRl-Hindlll fragment.
Construction of the targeting vector (Figure 3.3) was as follows. First, a 7.0 kb
EcoRI-XhoI fragment was subcloned from the 7.6 kb EcoRl plasmid. Next, exons 2
36

and 3, which were contained within a 2.3 kb SacII-Hindlll fragment, were excised and
replaced with lacZ and PGK-neo. lacZ was constructed such that it inserted in frame
downstream of the remaining exon 2. The targeting vector was sequence verified.
2.1.3. Embryonic stem cell line targeting and identification of homologously
recombined clones
A brief description, including minor modifications, of the protocols used for ES
cell maintenance and targeting protocols are as follows (Wurst 1993). ES cell culture
media consisted of Dulbecco's Modified Essential Medium (DMEM) with high
glucose, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 uM
monothioglycerol, 2 mM L-glutamine, 15 % fetal calf serum, 50 ug/mL each of
penicillin and streptomycin, and leukaemia inhibitory factor (LIF) that was prepared by
L. Sly. The gene targeting vector was linearized with EcoRl and 20 ug was
electroporated using Bio-Rad Gene Pulsar (0.34 kV, 250 mF) into 1 x 107 CCE ES cells
(generously donated by E. Robertson). ES cells were then selected with 180 ug/mL of
active G418 for neomycin expression for five days. Resistant colonies were plucked
into 96-well plates and expanded in culture. Duplicate plates were prepared; one was
frozen at -80°C as stock, while the other was used to prepare genomic DNA for
identification of homologous recombinants.
2.1.4. Identification of 7g// mutant alleles by Southern blot analysis
Southern blot analysis was used as the primary screen for homologous
recombination events. First, genomic DNA from individual ES cell lines was prepared.
ES cells in 96-well plates were lysed in 10 mM Tris (pH 7.5), 10 mM EDTA, 10 mM
NaCl, 0.5 % sarcocyl or SDS, and 1 mg/mL proteinase K (Sigma) at 65°C for 12-16
37

hours. DNA in each well was precipitated with 100 uL of chilled 75 mM NaCl: 100 %
ethanol (ETOH) for 1 hour, washed three times with 70 % ETOH, and the DNA pellets
were allowed to dry. Second, restriction digested genomic DNA was transferred to
nylon membranes. Genomic DNA was digested with EcoRl, and these DNA fragments
were separated on a 0.7 % agarose gel at 20 V for 12 hours. DNA in the agarose gel
was transferred to Nylon Membrane (Bio-Rad Zeta-Probe Genomic Tested Blotting
Membrane) overnight in lOx SSC, and fixed to the membrane by vacuum-drying for 1
hour at 80°C, or by UV cross-linking. Third, radioactive probes were hybridized to
genomic DNA that was fixed to the membrane. The probe was labelled with [32P]
dCTP by random priming using Ready to Go™ DNA Labelling Beads (-dCTP)
(Pharmacia). Membranes were pre-hybridized in 9 mL of hybridization buffer: 1.5x
SSPE, 1 % SDS, and 1 % skim milk. Denatured probe was added and hybridized for
16 hours at 58°C. Excess probe was washed off at 65°C for two 30 minute intervals for
each wash solution, 0.3x SSC and 0.1 % SDS. Fourth, results of the hybridization
experiment were visualized by exposing membranes to a Phosphorlmager cassette for 1
night, scanned on a STORM 860 Phosphorlmager, and the resulting signal analyzed
using ImageQuant (Molecular Dynamics, Sunnyvale CA).
To demonstrate correct and unique homologous recombination events occurred
in the identified clones, homologous recombination on both the 5' and 3' homologous
arms was verified by Southern blot analysis. Correct targeting of the 5' arm was
demonstrated when the 5' external probe, a 0.3 kb Xhol fragment, detected the
endogenous 7.6 kb EcoRl wild-type fragment, and a modified 13 kb fragment. At the
3' arm, the external probe, a 0.5 kb Xbal fragment, detected the endogenous 7 kb Pstl
fragment, and the modified 5.5 kb Pstl fragment. Two CCE clones, B2 and C6, were
identified after screening 144 clones.
38

2.1.5. Identification of Tgif mutant alleles by P C R
An alternative PCR strategy was devised for ear-punch and visceral yolk sac
DNA to rapidly determine the genotype of mice and embryos. A 418 bp fragment
within exon 2 was only present on the wild-type allele and was amplified by primers
TGIF 105 (5'-ttccctgctggtgaaagcaa-3') and TGIF 207 (5'-tgttcatacagccagtctcg-3')- The
mutant allele consisted of the remains of exon 2 fused to the lacZ gene, and this
junction was amplified only by TGIF 105 and LacZ 201 (5'-ggcctcttcgatattacgcc-3') to
yield a 160 bp PCR product.
2.1.6. Production of chimeras and 7gi/mutant mice from targeted ES cells
Two independently-targeted ES cell lines, CCE derived B2 and C6, were
injected into the inner cell mass of embryonic day E3.5 C57BL/6 host blastocysts to
generate chimeric mice (Papaioannou 1993). Two independently targeted cell lines
were used to generate chimeras in order to eliminate artefacts introduced by gene
targeting. Germline transmission from two chimeric males was apparent by coat colour
and confirmed by genomic blot analysis. The phenotypes observed in mice derived
from both ES cell lines were similar and their combined results are presented. Multiple
chimeric mice were generated and used for germline transmission of the mutated Tgif
allele to their progenies.
2.1.7. Mouse breeding, genetic backgrounds, and congenic strains
All mice were bred and maintained at the Joint Animal Facility (JAF) at the
British Columbia Cancer Research Centre. Inbred strains 129/SV and C57BL/6, and
outbred strain CD1 were obtained from the JAF.
39

Routine testing for viral pathogens and mycoplasma was carried out on the
mouse colonies by JAF staff. Experiments complied with all relevant federal
guidelines and were approved by the institutional animal care and use committee (see
Appendix).
Chimeric males were mated to CD 1 females to generate mice with a mixed
129/Sv/CDl background. The colony with this mixed genetic background was
established by mating of mice of the appropriate genotype within the colony.
The same chimeras were also used to mate to 129/Sv female mice to generate
mice with the inbred 129/Sv genetic background. Since the mutant allele of the
heterozygote originated from ES cells, and the ES cells were derived from 129/Sv mice,
a colony with the inbred 129/Sv genetic background was established after one cross to
129/Sv females. This colony was subsequently maintained by sister-brother mating of
heterozygote mice.
The same chimeras were also used to mate to C57BL/6 female mice to generate
mice and embryos of a mixed 129/Sv/C57BL/6 background. A congenic C57BL/6
strain was generated after 10 generations of breeding heterozygote 129/Sv/C57BL/6
mice to C57BL/6 mice.
2.2. E X P R E S S I O N A N A L Y S I S O F T G I F U S I N G I N S I T U R N A
H Y B R I D I Z A T I O N
In situ RNA hybridization is a standard technique used to characterize RNA
expression pattern in wholemount embryos. The method used is briefly outlined below
but has been previously described in detail (Belo et al. 1997). First, embryos at the
appropriate stages were prepared for cRNA hybridization. Mouse embryos were fixed
in 4 % paraformaldehyde and methanol (MeOH). Embryos were then bleached in 6%
40

H 2 0 2 :PBT (PBS + 0.1 % Tween-20) for 1 hour at 4°C. Embryos were finally
permeated by 10 ug/mL proteinase K digestion. Second, cRNA probes incorporated
with digoxigenin were prepared. In situ RNA probes were transcribed using linearized
template, digoxigenin-labelled nucleotides, and the appropriate RNA polymerase, SP6,
T7 or T3, according to the manufacturer's instructions. RNA probes were purified
through ProbeQuant G-50 microcolumns (Amersham) to remove unincorporated
ribonucleotides. Hybridization took place at 60°C for over 16 hours. Third, excess
probe was washed off. Fourth, digoxigenin was localized using antibody conjugated to
alkaline phosphatase (Roche) overnight. Fifth, staining using B M Purple (Roche)
substrate was used to localize the RNA probes.
The expression of Tgif 'was characterized using antisense RNA probes
synthesized from EST BF133915 and subcloned fragments and hybridized to embryos
and tissues of interest between E6 - E l 2.
2.3. ANALYSIS OF TGIF MUTANT MICE
2.3.1. Determination of the rate of viability and fertility
Experimental genotype and sex ratios were compared to the expected
Mendelian ratios using the Chi-squared test. Statistical analyses utilizing the Chi-
squared tests were performed using the Analyze-It plug-in for Microsoft Excel.
Fertility of mutant mice was determined by mating mutant mice to heterozygote
or wild-type littermates. Transmission of the mutant allele indicated both mutant male
and female mice were fertile.
41

2.3.2. Morphological examination of adults
Mouse phenotyping was guided by Necropsy of the Mouse by V. Covelli
(http://www.eulep.org/Necropsy_of_the_Mouse/index_2004.php). Examination of
weight, eye, and tail were carried out in the JAF in a level II biohazard laminar flow
hood. A mouse was considered growth retarded if its weight was 20 % less than the
mean weight value of its litter. Fisher's exact test was used to determine the
significance of observed phenotypes (Table 3.1)(Zar 1984).
Adult mice were sacrificed by asphyxiation in CO2 gas. The sidedness and
morphology of internal organs including the heart, lungs, liver, spleen, stomach, gut
and brain were macroscopically examined and photographed using a Nikon Coolpix
950 digital camera.
2.3.3. Morphological examination of embryos
Embryos were collected from natural timed matings of Tgif' mice. Noon on
the day of the vaginal plug was designated as 0.5 days post coitum (dpc). Embryos
were dissected at E8.0 - E8.5. Morphology of the embryo was visually inspected, with
particular emphasis on the forebrain. The surrounding yolk sacs were used to prepare
DNA for genotyping.
2.4. C E L L C Y C L E ANALYSIS OF TGIF MUTANT MOUSE EMBRYONIC
FIBROBLASTS
2.4.1. Generation of mouse embryonic fibroblasts
Primary mouse embryonic fibroblasts (MEF) were isolated from Tgif^/+, Tgif'
and Tgif'1' E l 3.5 embryos obtained by heterozygous matings (Nagy et al. 2003).
Briefly, carcasses were first dissected free of limbs, internal organs, and brain, then
42

minced to a slurry, and finally the minced tissues were digested gently to a cell
suspension by trypsinization over a 30 minute interval. Cells were subsequently
maintained in DMEM supplemented with 10 % fetal bovine serum (FBS) and
antibiotics (Stem Cell Technologies). Experiments were carried out only with early
passage (<8) cells from Tgif^/+ and Tgif1' littermates.
2.4.2. Expression of Tgif in mouse embryonic fibroblasts
Total RNA was isolated from MEFs using TRIZOL® Reagent (Invitrogen)
according to the manufacturer's protocol. Reverse transcription was performed using
SuperScriptll (Invitrogen) with 1 ug of template RNA. Subsequent PCR reactions were
performed using 1 ug of the resulting cDNA as template. The wild-type transcript was
amplified with primers TGIF-RT (5'-atgaaaagcaagaagggtct-3') and TGIF 207 (5'-
tgttcatacagccagtctcg-3').
2.4.3. Growth curve analysis
Experiments were carried out by comparing cells at identical passage numbers
obtained from littermate embryos. In 12-well culture plates, 2 x 104 MEFs of each
genotype were plated, fed, and counted on a daily basis using a Coulter Counter. Two
experiments were performed using independently derived MEFs.
2.4.4. Cell cycle analysis using BrdU incorporation and flow cytometry
1 x 106 MEFs at log phase were labelled with bromodeoxyuridine (BrdU) and 7-
amino-actinomycin D (7AAD) following the manufacturer's instructions (BD
Biosciences Pharmingen). BrdU is a halogenated thymidine analogue that is only
incorporated into DNA synthesized during S-phase, while 7AAD is a DNA binding dye
43

used to distinguish cells in all phases of the cell cycle. BrdU labelling time was
optimized for 45 minutes, the length of time required for all cells in S phase to have
incorporated BrdU. FACS results were acquired on a Cytopeia Influx and analyzed
using FlowJo flow cytometry software. Statistical analysis of data used single factor
ANOVA using the Analyze-It plug-in for Microsoft Excel. This set of experiments was
repeated five times, and each replicate examined a different and independently derived
group of MEFs.
2.4.5. Proliferation assay by f3H]-thymidine incorporation
Cell proliferation assays were performed using 5000 cells/well in 96-well plates
in triplicate. 1 uCi/well [3H]-thymidine was added to the cells, incubated for 2 hours,
harvested, and [3H]-thymidine incorporation quantified using a p-counter (LKB Wallac,
Turku, Finland).
2.4.6. Proliferation assay of synchronized mouse embryonic fibroblasts
5000 MEFs per well were grown in DMEM lacking serum for 2 days to induce
quiescence (Herrera et al. 1996). Re-addition of 10 % FBS at defined timepoints
stimulated synchronous re-entry into Gi . DNA synthesis was then assessed using [3H]-
thymidine incorporation as described. Two experiments were each performed in
triplicate, using cells derived from independent MEF lines.
2.5. ANALYSIS OF IN VIVO PROLIFERATION BY SOMITE STAGING
Embryos were collected as described above. Embryos were dissected at E8.0 -
E8.5. Developmental progression was precisely scored by headfold morphology, early
44

or late, or by counting the number of somites, which was typically between 0-10
somites. The surrounding yolk sacs were used to prepare DNA for genotyping.
2.5.1. Analysis of in vivo proliferation by BrdU incorporation
Congenic C57BL/6J Tgif" females pregnant with E8.0 - E8.25 embryos
received an intraperitoneal injection of 400 ug BrdU per gram weight and were
euthanized 1 hour later, as described previously (Hebert et al. 2002). Embryos were
collected and staged as described above, fixed in 4 % paraformaldehyde (PFA)
overnight and frozen in optimum cutting temperature (OCT) medium. Frozen 10 um
sections were stained with anti-BrdU conjugated alexa 549 (Molecular Probes) and
Hoechst. The fraction of BrdU incorporated cells was determined by counting the
number of positive cells and dividing by the total number of Hoechst positive cells.
Only neuroectodermal cells were counted. Numbers were also compared in dorsal 1/3,
ventral 1/3 and mid 1/3 sections. At least three adjacent sections were counted, and
three sets of matched mutant and control embryos were analyzed.
2.6. EXPRESSION OF HUMAN TGIF BY RETROVIRUS IN MOUSE
EMBRYONIC FIBROBLASTS
2.6.1. Expression of wild-type human TGIF by retrovirus
Human TGIF cDNA was subcloned upstream of an IRES-yFP cassette in the
murine stem cell virus (MSCV) retroviral vector to generate MSCV-7/G/F-IRES-7FP
(Buske et al. 2001). Tgif was flag-tagged at the N-terminus to allow for western blot
analyses.
High titer helper-free recombinant MSCV-TGIF-IRES-YFP retroviruses were
generated and tested according to protocols developed by G. Nolan
45

(www.stanford.edu/group/nolan). Briefly, Phoenix Eco cell lines were transfected
using FuGENE (Roche). Supernatant from the Phoenix viral producer cells
supplemented with 6 ug of ammonium sulphate per millilitre was used for infection of
the MEFs.
YFP-expressing cells were enriched through a Cytopeia Influx sorter, and
proliferation was measured by [3H]-thymidine incorporation as described above.
2.6.2. Expression of human TGIF containing mutations identified in HPE
patients
Site-directed mutagenesis using a modified restriction site protocol generated
the 7 point mutations previously identified in patients (Tao 1994). Al l mutations were
sequence verified. Patient mutations were generated in MSCV-retroviral constructs,
expressed in Tgif1' MEFs, and proliferation was assessed. Five experiments were each
performed in sextuplicate using MEFs derived from independent TgiJ*/+ and Tgif''
embryos.
2.6.3. Expression of transduced TGIF protein
The level of transduced TGIF protein expression was assessed by standard
Western blot analyses of MEF cell extracts and probed with an anti-flag antibody.
TGIF generally migrated as a doublet of 30-32 kDa; the upper band likely represents
the phosphorylated form (Lo et al. 2001).
2.7. ANALYSIS OF PROLIFERATION RESPONSE TO TGF-P, RETINOIC
ACID AND U0126
46

5000 MEFs per well were grown in the presence of 0.6 pg/mL - 10 ng/mL
TGF-P for 24 hours the day after plating. DNA synthesis was measured by [3H]-
thymidine incorporation as described. Fresh 9-cis-RA and all-trans-RA were added to
5000 MEFs per well on a daily basis for 5 days. [3H]-thymidine incorporation was
performed on a daily basis. U0126, an inhibitor of MEK, ranging from 0.01 - 100 uM,
was added to 5000 MEFs per well and [3H]-thymidine incorporation was performed
after 24 hours of culture. Three experiments were each performed in triplicate using
MEFs derived from independent Tgif+ and Tgif1' embryos. Normalization was
conducted by dividing the incorporation value by the maximum incorporation level.
47

3. C H A P T E R 3 G E N E R A T I O N A N D C H A R A C T E R I Z A T I O N O F T G I F
N U L L M I C E
3.1. T G I F E X P R E S S I O N D U R I N G E M B R Y O G E N E S I S
Expression patterns of genes can lead to informed hypotheses for function.
Wholemount in situ RNA hybridization is a standard technique used to visualize RNA
expression pattern in relation to the anatomy of the mouse embryo. The ease of
performing parallel experiments on numerous embryos at various ages, and from
different genotypes allows meaningful comparisons within an experiment. Notably,
this technique detects dynamic spatial changes in expression during development or
changes to expression by genetic modifications.
To examine the expression pattern of Tgif, antisense RNA probe synthesized
from EST BF133915 was hybridized to embryos or tissues between the ages of E6 -
E12. Tgif expression at E6, prior to gastrulation, was ubiquitously low throughout the
epiblast (Figure 3.1 A). During head fold stages (E8.0-E8.25), Tgif expression was
detected in the anterior neuroectoderm at a low and ubiquitous level and at a slightly
higher level along the midline of the developing forebrain and the lateral mesoderm
exiting the primitive streak near the tail bud (Figure 3.1B,C,E). 7g//"continues to be
expressed throughout the forebrain until neural tube closure (Figure 3.1D,E,G). Later
during embryogenesis, Tgif expression was also in the hindbrain and branchial arches
from E8 - E9.5 (Figure 3.1F,G). Interestingly, the expression of Tgif 'was found in
proliferating cells of the developing limbs, including the ectodermal layer and the tips
of the digits (Figure 3.2). Similar expression patterns were detected using N and C
terminal portions of the gene. Thus, the expression pattern of Tgif is consistent with its
potential function in the developing forebrain.
4 8

A
Figure 3.1 rgz/expression in the early embryo. (A) Tgif'xs expressed as early as embry
onic day 6; a, anterior; p, posteior. (B, C) Respectively, dorsal and ventral views of the
developing forebrain at E8.25 demonstrating Tgif is expressed at a low and ubiquitous
level, but at a higher level in the midline cells (arrow). Anterior is at the top. (D,E)
Dorsal and ventral views of E8.5 embryos with 7g//expression persistent in the neural
plate as it folds into the neural tube; note the lack of expression in the heart (h). Lateral
views of E8.5 (F) and E9.5 (G) embryos expressing Tgif in the lateral mesoderm (lm)
that exited the primitive streak, developing branchial arches (ba) and hindbrain (hb).
49
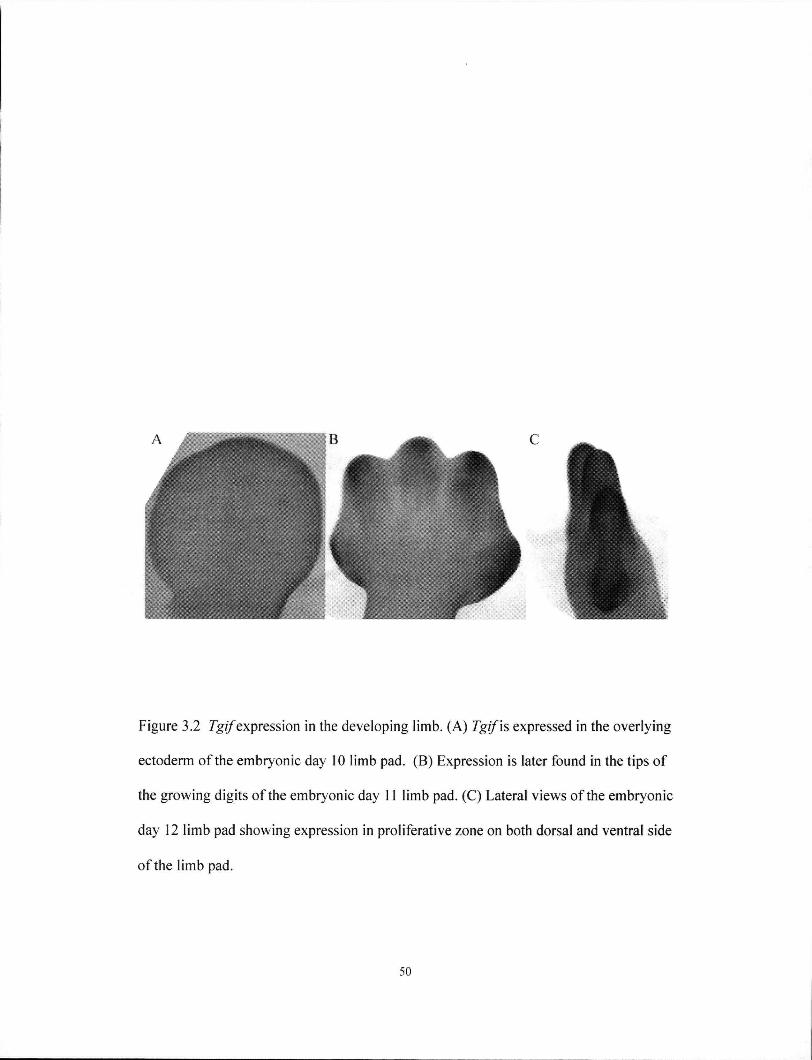
Figure 3.2 Tgz/expression in the developing limb. (A) Tgif'xs expressed in the overlying
ectoderm of the embryonic day 10 limb pad. (B) Expression is later found in the tips of
the growing digits of the embryonic day 11 limb pad. (C) Lateral views of the embryonic
day 12 limb pad showing expression in proliferative zone on both dorsal and ventral side
of the limb pad.
50

3.2. GENERATION OF TGIF MUTANT MICE
Genetic manipulation in mice is one of the most important experimental systems
to study the in vivo function of genes in mammals. Numerous kinds of mutations have
been created for thousands of genes to provide unprecedented insights, and the results
have made great contributions to our understanding of development and disease (Bier
and McGinnis 2003). Thus 1 undertook the generation of mice that lack Tgif.
3.2.1. Isolation of a Tgif genomic clone
Multiple BAC clones containing Tgif and the surrounding sequences were
identified by screening a 129S6/SvEvTac genomic BAC library. The gene structure
and sequence of Tgif 'was available through mouse genome sequence databases.
Analysis indicated a 7.6 kb EcoRl fragment from BAC 19612 was sufficient for
preparing the Tgif targeting construct because it included exons 2 and 3 of Tgif, which
contained the majority of the coding sequences (Figure 3.3A) and the necessary
flanking sequences for homologous recombination. This genomic fragment was
subcloned into a plasmid and verified by restriction mapping.
3.2.2. Construction of Tgif targeting vector
To ensure the function of Tgif 'was completely disrupted, nearly all of the coding
sequences were removed (Hasty 1993). The targeting vector was designed to replace
exons 2 and 3 of Tgif'with the reporter gene lacZ fused in frame to the remaining N -
terminal 7 amino acids (Figure 3.3A). The remaining 7 amino acids are likely not
functional. The construct also included a PGK-neo positive selectable marker gene
cassette that allowed for enrichment of targeting events. Sufficient sequence for
efficient homologous recombination was provided by 3.4 kb of 5' homology region,
51
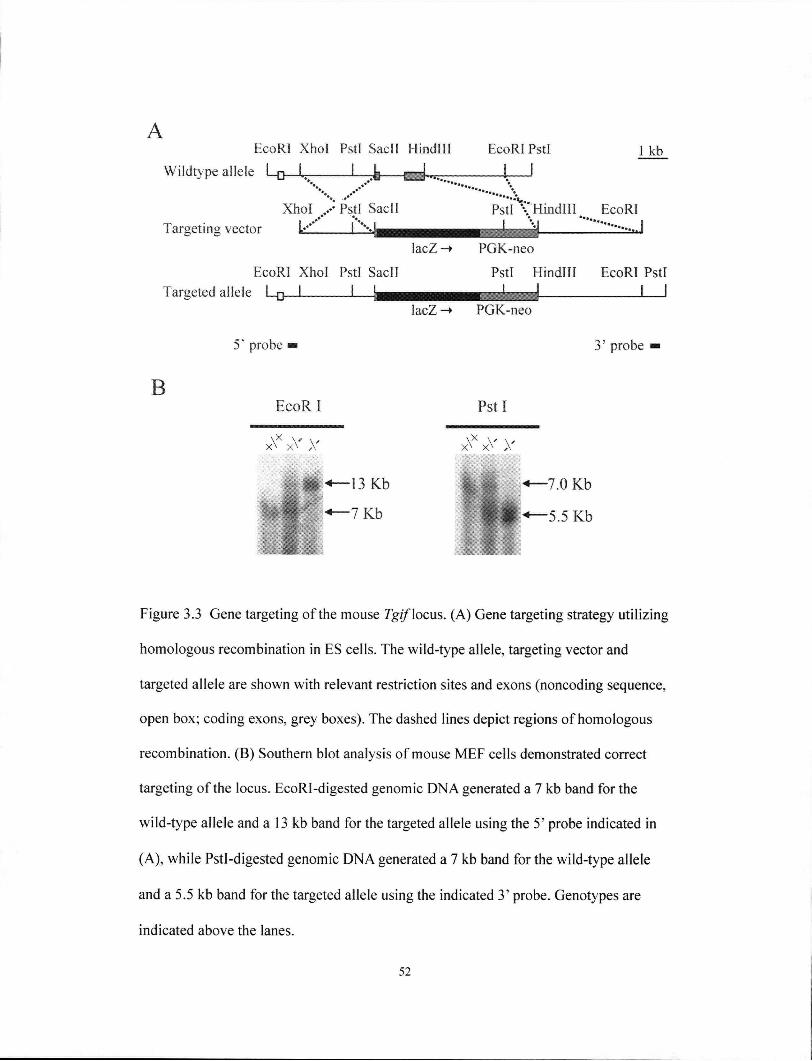
A EcoRl Xhol Pstl SacII Hindlll EcoRl Pstl 1 kb
Wildtype allele L___L. L 1 ,.«? \
Xhol .xPstl SacII Psti'X'Hindlll EcoRl Targeting vector iillt 1 VI
lacZ-» PGK-neo
EcoRl Xhol Pstl SacII Pstl Hindlll EcoRl Pstl
Targeted allele L _ _ J ^---h^mmmmmamamdm^ I I lacZ -» PGK-neo
B
5* probe — 3' probe
EcoR I Pst 1
x \ x x \ ' y x \ X x V y
-13 Kb «—7.0 Kb
'7 Kb |H <«—5.5 Kb
Figure 3.3 Gene targeting of the mouse Tgif \ocus. (A) Gene targeting strategy utilizing
homologous recombination in ES cells. The wild-type allele, targeting vector and
targeted allele are shown with relevant restriction sites and exons (noncoding sequence,
open box; coding exons, grey boxes). The dashed lines depict regions of homologous
recombination. (B) Southern blot analysis of mouse M E F cells demonstrated correct
targeting of the locus. EcoRI-digested genomic D N A generated a 7 kb band for the
wild-type allele and a 13 kb band for the targeted allele using the 5' probe indicated in
(A), while Pstl-digested genomic DNA generated a 7 kb band for the wild-type allele
and a 5.5 kb band for the targeted allele using the indicated 3' probe. Genotypes are
indicated above the lanes.
52

and 1.5 kb of 3' homology region (Figure 3.3A). The design of the targeting vector
also allowed for subsequent strategies to unambiguously discriminate recombinant
clones by Southern blot analysis.
3.2.3. Screening for clones in which the 7g/yiocus was targeted
Mouse ES cell lines are derived from pre-implantation embryos. When ES cells
are injected back into pre-implantation embryos to create a chimera they can remain
multipotent and contribute to all tissues (Papaioannou 1993). Significantly, ES cells
can be cultured in vitro extensively in an undifferentiated state and therefore allow gene
targeting experiments. This characteristic is the cornerstone of genetic manipulation
technology in mice. Equally important is the ability of ES cells to contribute to the
germline through a chimera, and thus perpetuate mutations produced by gene targeting
in ES cells.
The targeting vector was linearized and electroporated into ES cells to allow
homologous recombination to occur (Wurst 1993). However, foreign DNA is more
likely to integrate randomly into the host genome, and the clones in which the desired
genetic change (i.e. homologous recombination at the 7g-//locus) occurs need to be
identified. Therefore, colonies that survive the positive selection were then clonally
isolated and screened.
A number of clones survived G418 selection, and 144 clones were selected for
screening. Correctly targeted 7_7/clones were identified by Southern blot analysis
using Pstl digestion and a 0.3 kb 3' external probe that hybridized to the wild-type 7 kb
allele and the targeted 5.5 kb allele (Figure 3.3B). The 5' arm of homology was also
examined for correct targeting using a 0.3 kb Xhol external probe that detected the
wild-type 7 kb EcoRl fragment and the mutated 13 kb fragment. This screen identified
53

two homologously recombined clones. The frequency of 1/72 is within the normal
range of homologous recombination events.
A PCR strategy was designed for ear-punch and visceral yolk sac DNA to
routinely and rapidly determine the genotype of mice and embryos. The wild-type
allele was detected using primers that amplified a 418 bp fragment within exon 2. The
mutant allele was detected using primers that amplified a 160 bp PCR product that
included the junction between the remaining exon 2 and the fused lacZ gene.
3.2.4. Targeted Tgif allele is a null allele and produced a fused lacZ transcript
To confirm that 7g//'expression was disrupted, RNA was isolated from wild-
type and targeted ES cells and assayed using reverse-transcriptase (RT)-PCR (Figure
3.4). The Tgif-lacZ fusion transcript was detected only in heterozygous ES cells
demonstrating Tgif'was disrupted as expected.
3.2.5. Production of chimeras and Tgif mutant mice from targeted ES cells
Multiple chimeric mice were generated from the injection of two independently-
targeted ES cell lines, CCE-derived B2 and C6, into the inner cell mass of E3.5
C57BL6 host blastocysts. Two independently targeted cell lines were used to generate
chimeras in order to eliminate mutations that may have occurred accidentally in the ES
cells during culture, cloning or gene targeting. Germline transmission from two
chimeric males was confirmed by genomic blot analysis. The phenotypes observed in
mice derived from both ES cell lines were similar and the combined results are
presented below. Experiments were performed using mice derived from two chimeric
males to eliminate the possibility that any effects seen were due to mutations unrelated
to the targeted locus in the ES cells.
54
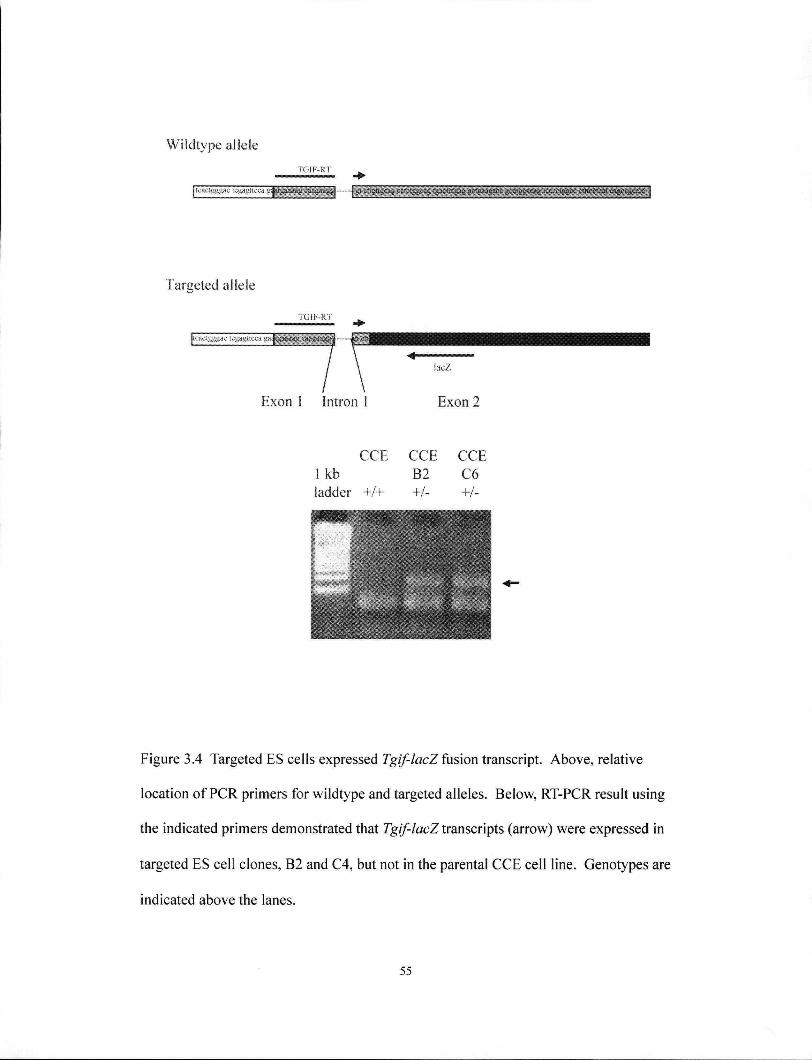
Wildtype allele
T G I F - R T
Targeted allele
TGIF-RT
jtcacigggac tcgagucca ga^||
i a c Z
Exon 1 Intron 1 Exon 2
C C E C C E C C E 1 kb B2 C6 ladder +/+ +/- +/-
Figure 3.4 Targeted ES cells expressed Tgif-lacZ fusion transcript. Above, relative
location of PCR primers for wildtype and targeted alleles. Below, RT-PCR result using
the indicated primers demonstrated that Tgif-lacZ transcripts (arrow) were expressed in
targeted ES cell clones, B2 and C4, but not in the parental C C E cell line. Genotypes are
indicated above the lanes.
55

3 . 3 . A N A L Y S I S O F T G I F M U T A N T M I C E
3 . 3 . 1 . Analysis of Tgi/mutant adult mice on mixed 129/Sv/CDl genetic
background
A mixed 129/Sv/CDl genetic background colony was first established. This
breeding scheme took advantage of the hybrid vigour of FI mice and is the fastest
means of producing mutant homozygotes for analysis.
The genotype and sex of mice that were generated from heterozygote matings
were examined. Although reduced viability in heterozygote mice was anticipated, as
has been observed in humans, a X 2 analysis of 80 F2 offspring at 3 weeks of age
demonstrated a normal Mendelian ratio [25 Tgif* (31%), 36 Tgif (45%), 19 Tgif
(24%); p=0.43], demonstrating Tgif was not essential for embryo viability. Fertility
was also examined since the loss of achintya and vismay, two Drosophila orthologs,
were important during Drosophila spermatogenesis (Mann and Affolter 1998; Ayyar et
al. 2003). In contrast, both mutant male and female mice were fertile.
Adult mice were examined using a guide prepared by V. Covelli on the website,
Necropsy of the Mouse. Attention was paid to the hallmark of human HPE, varying
degree of separation of the cerebral hemispheres, and other accompanying
characteristics including craniofacial anomalies, developmental delay, seizures, and
perinatal and neonatal lethality. The results of these examinations were as follows.
Live mice examined externally for the appearance of fur, face, spacing between eyes,
nasal opening, teeth and limbs, showed obvious differences between wild-type and
mutant mice. Neither activity nor gait was noticeably different. Examination of the
structure and size of the brain in mice did not identify any overt abnormalities. The two
hemispheres and nasal bulbs were always present and distinctly separate.
56

However additional analysis of 92 Tgif' mice revealed that 19% were growth
retarded (Table 3.1; Figure 3.5A). For this analysis, the weight of a mouse had to be
20% less than the mean weight value of its litter to be considered growth retarded. In
addition, some mutant mice exhibited kinked tails and eye defects (Figure 3.5 B-E;
Table 3.1). Some mutant mice exhibited microcephaly (Figure 3.5F), but the frequency
of this phenotype will have to be examined further in the future. None of these defects
were observed in control littermates (Table 3.1).
Significantly, 13% of Tgif1' mice developed laterality defects, with both situs
inversus and situs ambiguus being observed after examining 67 mutant mice (Figure
3.6 A-D; Table 3.1). Situs ambiguus most often involved the incorrect positioning of
the heart and occasionally involved abnormal lung lobe numbers (3 cases) or
polysplenia (1 case). Cardiac situs was not examined to determine the isomerism class,
but together with polysplenia, it is likely situs ambiguus cases were left isomerism.
Overall, 43% of 92 mutant mice examined displayed at least one of the defects
listed above (Table 3.1). However, each individual defect was observed in only a
limited number of homozygous mutant mice, indicating a low penetrance of each defect
identified.
3.3.2. Analysis of the Tgif mutant adult mice with a 129/Sv congenic genetic
background
After initial characterization of the effects of Tgif deletion, this genotype was
introduced into different inbred genetic backgrounds. The rationale was that the
penetrance of the mutation might be higher in an inbred genetic background (LeCouter
et al. 1998; Hide et al. 2002). A Tgif-\29Sv congenic strain was thus created from
matings between the chimeras and the 129/Sv strain.
57
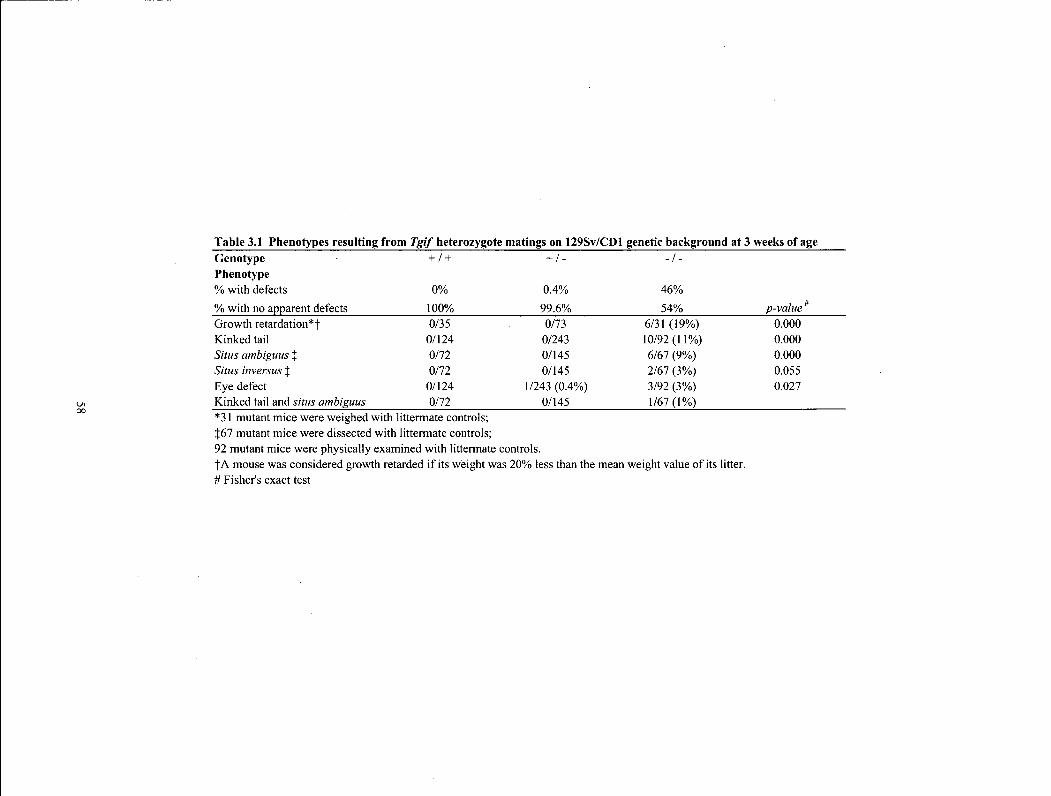
Table 3.1 Phenotypes resulting from Tgif heterozygote matings on 129Sv/CDl genetic background at 3 weeks of age Genotype + / + + / - - / -Phenotype % with defects 0% 0.4% 46% % with no apparent defects 100% 99.6% 54% p-value* Growth retardation*f 0/35 0/73 6/31 (19%) 0.000 Kinked tail 0/124 0/243 10/92(11%) 0.000 Situs ambiguus % 0/72 0/145 6/67 (9%) 0.000 Situs inversus % 0/72 0/145 2/67 (3%) 0.055 Eye defect 0/124 1/243 (0.4%) 3/92 (3%) 0.027 Kinked tail and situs ambiguus 0/72 0/145 1/67(1%) *31 mutant mice were weighed with littermate controls; {67 mutant mice were dissected with littermate controls; 92 mutant mice were physically examined with littermate controls. fA mouse was considered growth retarded if its weight was 20% less than the mean weight value of its litter. # Fisher's exact test
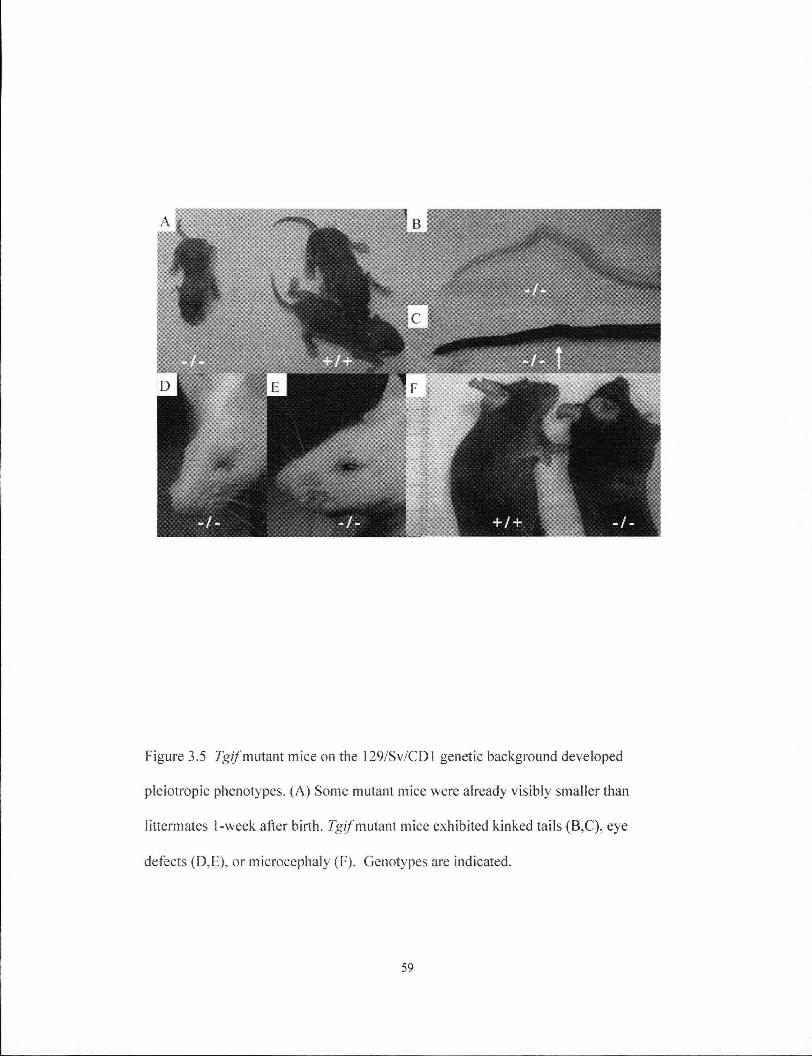
Figure 3.5 7g/7 mutant mice on the 129/Sv CI) 1 genetic background developed
pleiotropic phenotypes. (A) Some mutant mice were already visibly smaller than
littermates 1-week after birth. Tgif mutant mice exhibited kinked tails (B.C), eye
defects (D,E), or microcephaly (F). Genotypes are indicated.
59
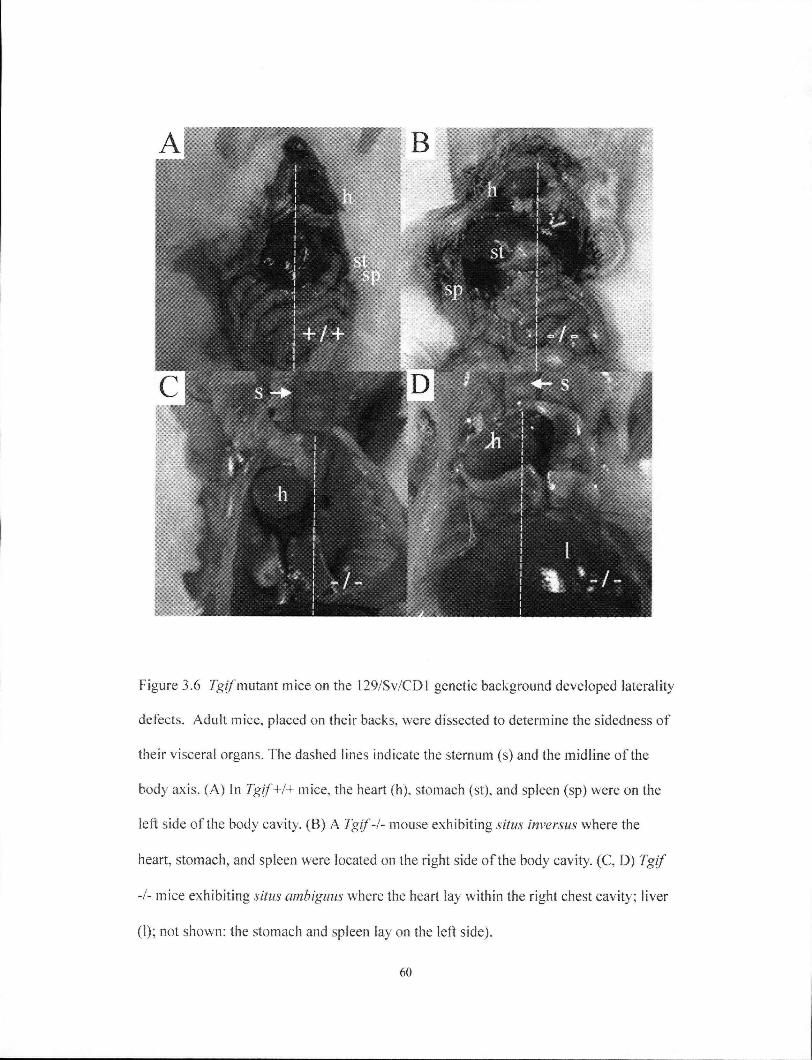
Figure 3.6 Tgif mutant mice on the 129/Sv/CDl genetic background developed laterality
defects. Adult mice, placed on their backs, were dissected to determine the sidedness of
their visceral organs. The dashed lines indicate the sternum (s) and the midline of the
body axis. (A) In Tgif+/+ mice, the heart (h). stomach (st), and spleen (sp) were on the
left side of the body cavity. (B) A Tgif-I- mouse exhibiting situs inversus where the
heart, stomach, and spleen were located on the right side of the body cavity. (C, D) Tgif
-I- mice exhibiting situs ambiguus where the heart lay within the right chest cavity; liver
(1); not shown: the stomach and spleen lay on the left side).
60

The genotype and sex of mice derived from heterozygous mating was
determined at 3 weeks of age. x2 analysis demonstrated a normal Mendelian ratio [21
Tgif+ (21 %), 56 Tgif' (57 %), 22 Tgif (22 %); p=0.43], showing mice inheriting
the homozygous Tgif mutation on the 129/Sv genetic background were also viable.
Mutant mice were also fertile on this background.
129 substrains are notoriously poor breeders and difficult to maintain.
Therefore, only limited analyses were performed after it became apparent that mutant
mice were viable and fertile with no overt defects.
3.3.3. Analysis of the Tgif mutant adult mice with a C57BL/6 congenic genetic
background
A congenic C57BL/6 Tgif null strain was also derived in an attempt to increase
the penetrance of the observed phenotypes (Zhang et al. 2006). This strain was
produced by 10 generations of backcrossing of Tgif'" mice onto C57BL/6J mice.
Contrary to expectation, Tgif mutations on the C57BL/6 genetic background did not
result in an HPE phenotype. A slight reduction in the expected number of mutants was
observed, although the number of mice examined was too small to demonstrate a
statistically significant difference [22 Tgif+ (22 %), 59 Tgif (60 %), 17 Tgif (17
%); p=0.10]. Al l mutant mice displayed normal tails and growth. Examination of 26
mutant mice also revealed normal situs of the internal organs. From this result it was
concluded that the phenotypes observed were dependent on the genetic background.
3.3.4. Morphological examination of embryos
Developing forebrains were examined for developmental progress.
Morphological landmarks including narrow forebrain, width and apposition of bilateral
61

structures such as the optic sulci, optic pits, and thickening of the ANR can be
examined as early as E8 (4 - 5 somites) (Juriloff et al. 1985; Hentges et al. 1999;
Lagutin et al. 2003). Over 20 mutant embryos between E8 - E10 were examined for
these landmarks, but significant differences were not detected. However, mutant
embryos were often developmentally delayed as early as E8 (see Chapter 4).
3.3.5. A b n o r m a l l a t e r a l i t y d e t e r m i n a t i o n
Kinked tails are an indication that midline patterning was disrupted during
development (Table 1). Additionally, the occurrence of situs inversus and situs
ambiguus suggested that early patterning events in the Tgif1' mice may be disrupted.
As described earlier, left-right determination and patterning take place early during
embryogenesis. After the breaking of symmetry, expression of the TGF-P family
member Nodal becomes enhanced on the left side of the node during gastrulation
(Hamada et al. 2002). The left-sided Nodal signal is relayed to the left LPM where
downstream target genes such as lefty2, pitx2, and nodal itself are activated. The left-
sided signals and pathways are barred from the right side by midline mesodermal cells.
These molecular determinants precede overt morphological changes.
To determine whether abnormal molecular patterning had led to the observed
laterality defects, I examined the expression pattern of Nodal, lefty2 and pitx2 in 20 Tgif
+ / + and 20 Tgif1' mice (Figure 3.7). Nodal expression was examined in embryos at the
early headfold, late headfold, and 0 - 5 somite stages. Iefty2 was examined in embryos
at the 3-8 somite stages, while pitx2 was examined in embryos at the 5-12 somite
stages. Nodal and lefty2 were normally expressed in all embryos tested for each
marker. However, 1 mutant embryo (out of 20) did exhibit bilateral pitx2 expression,
62

wildtype mutant
Figure 3.7 7g//mutant embryos displayed abnormal left-right patterning. Both wild-
type and mutants displayed normal nodal and lefty2 expression patterns, but 1/20
mutant embryos examined showed bilateral pitx2 expression. *, region of right sided
pitx2 expression. Dashed lines indicate the midline of the embryo; 1, left; r, right.
63

while all wild-type embryos displayed pitx2 expression only in the left L P M (Figure
3.7). The expression ofpitx2 is present but weaker on the right. The detection of
bilateral pitx2 expression in early embryos is consistent with the situs ambiguous
phenotype observed in adult mutant mice. The frequency of bilateral pitx2 (5%) was
also similar to the frequency of situs ambiguous in the adult (9%). Although the
frequencies of situs ambiguous and bilateral pitx2 may appear low, considering that the
natural incidence of laterality defects is 1 in 10,000, the incidence is 300 fold higher
than expected. Bilateral expression of pitx2 suggests the loss of Tgif may disrupt
laterality determination at a molecular level upstream of pitx2 expression.
Unfortunately, the low penetrance of this phenotype precluded further studies to
determine the exact mechanism by which this occurred.
3.4 S U M M A R Y
Heterozygous and homozygous deletions at the Tgif locus were generated in
mice derived from multiple genetic backgrounds. Overt evidence of HPE was not
observed on any of these backgrounds, and all mutant mice were both viable and fertile.
Low penetrance, pleiotropic phenotypes were observed only on the 129/Sv/CDl genetic
background, suggesting these defects were dependent on the genetic background.
Amongst the phenotypes observed, Tgif'' mice were more frequently growth retarded
and exhibited evidence of defects in laterality determination. Such altered laterality
patterning signals may cause situs inversus and situs ambiguous observed in the adult
mice. These results indicate Tgif 'may function early during left-right patterning of the
developing embryo.
64

4. CHAPTER 4 ROLE OF TGIF IN CELL CYCLE CONTROL
4.1. GENERATION OF TGIF MOUSE EMBRYONIC FIBROBLASTS
Immortalized cell lines have been used historically for cell cycle studies, but
these cells acquire spontaneous and undefined genetic alterations during the process of
immortalization (D'Abaco and Olson 2000). Mammalian cell cycle studies now
commonly use primary cell lines, mostly MEFs (Herrera et al. 1996; D'Abaco and
Olson 2000; Malumbres et al. 2004). The advantages to using MEFs include the power
of genetic analysis, the ability to generate genetically modified mice disrupted for one
or multiple genes, and the ease of M E F preparation. Characterization of MEFs derived
from mice with a disruption in Rb (Herrera et al. 1996), cell cycle proteins and
regulators (Malumbres et al. 2000; Sotillo et al. 2001; Kozar et al. 2004; Malumbres et
al. 2004), and many others have provided valuable insights into cell cycle regulation
(Ciemerych and Sicinski 2005).
Tgif is normally expressed in MEFs. Using reverse transcriptase (RT)-PCR,
Tgif transcripts were robustly detected in wild-type and heterozygous MEFs, while no
transcript was observed in mutant MEFs (Figure 4.1).
4.2. CELL CYCLE DEFECTS IN MOUSE EMBRYONIC FIBROBLASTS
4.2.1. Growth curve analysis
To address the role of Tgif in cell cycle regulation, MEFs were generated from
Tgif + and Tgif1' embryos for functional investigation of 7g//" activity. To compare
their proliferative capability, equal numbers of early passage primary MEFs were
plated and counted on a daily basis for 12 days. Mutant cells proliferated at a slower
rate and accumulated to a lower density compared with control cells (Figure 4.2A).
65

Wi id type allele
TGJF-RT
, * i ( , " - r , |tggcaaqaqa aqga gaqaq . " ,.aj*u^tLCACjdtt>: • y L : : ; : ^ : ; : ua^r,; . : / : , ;^ <•} i;cagagoa gaqaaaggais tqtccca j
Targeted allele
T01F-RT j 'caagqr.jac tcgagrtcca gj|tgaa-)-;g r gcacjqj 4?tc
Exon 1 intron Exon 2
RT _ + - + - +
160 bp
Figure 4.1 M E F cells normally express Tgif, but mutant cells do not. RT-PCR result
using the indicated primers above demonstrated that Tgif transcripts were expressed in
Tgif+/+ and Tgif +/-, but not Tgif-I- MEFs. RT indicates presence (+) or absence (-) of
reverse transcriptase in each sample.
66
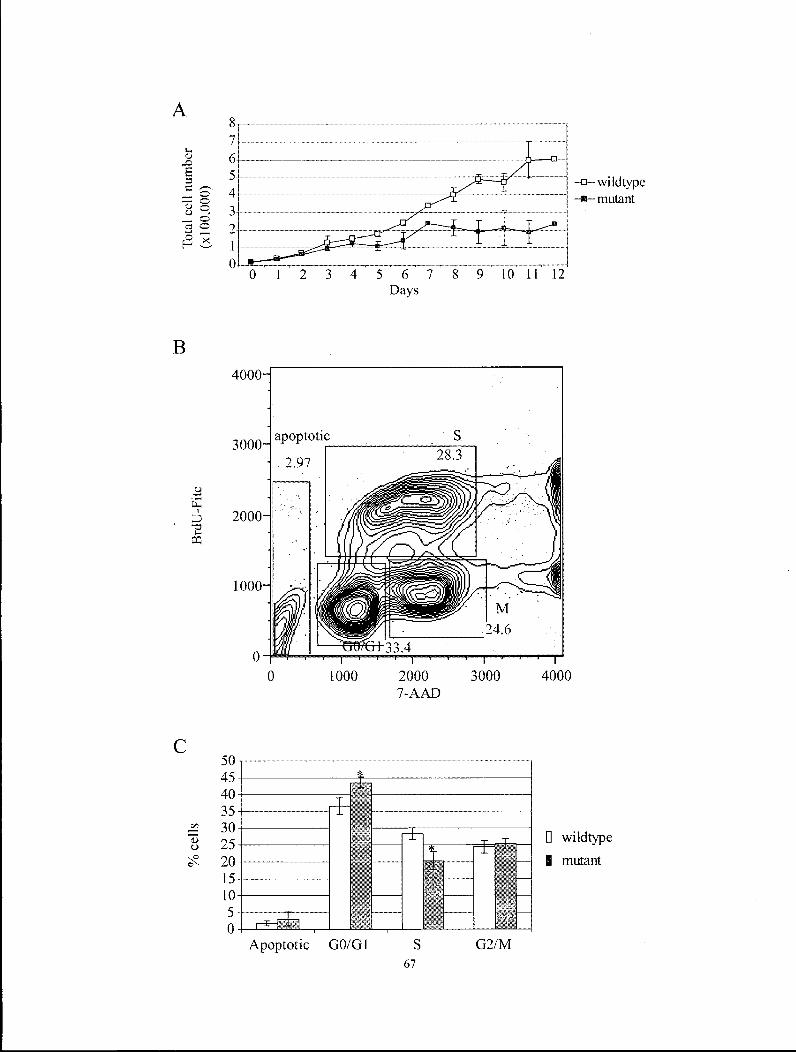
B
400(H
7-AAD
c 50-45-40-35-30-25-20-15-10-5-0-
Apoptotic G0/G1
J L D wildtype
I mutant
G2/M 67

Figure 4.2 Tgif' MEFs exhibited reduced growth due to a delay in GI phase
progression. (A) Equal numbers of MEFs were plated and counted daily for 12 days.
Tgif1' cells proliferated at a lower rate and accumulated to a lower density than Tgif+/+
cells. Data are pooled from two experiments using independently derived MEFs. (B)
Representative FACS profile of cells labelled with BrdU and 7AAD. Apoptotic cells
contained little DNA and stained low for 7AAD; cells in Go/Gi phase were negative for
BrdU and diploid for DNA content; cells in S phase were BrdU positive and
intermediate between diploid and tetraploid DNA content; cells in M phase were
negative for BrdU and tetraploid for DNA content. Cells were gated accordingly and
their relative percentages to the total number of cells are indicated. (C) FACS analysis
of cells labelled with BrdU and 7AAD described above indicated Tgif1' cells
accumulated in GO/GI phase and showed delayed entry into S phase. * />=0.003 and *
p=0.002 between Tgif* (n=8) and Tgif' (n=8) cells with regard to the percentage of
cells in GO/GI and S phase respectively using single factor ANOVA. A representative
result is shown from 5 experiments using independently derived MEFs.
68

4.2.2. Cytometric analysis of mouse embryonic fibroblast cell cycle
To investigate the cause of the proliferation defect, flow cytometry was used to
characterize cell cycle progression. MEFs were labelled with 7AAD, a fluorescent dye
that binds stoichiometrically to DNA and thus allows cell in the Go/Gi, S and G2/M
phases of the cell cycle to be distinguished (Tlsty et al. 1995; Herrera et al. 1996; Kozar
et al. 2004)(Figure 4.2B). However, the precise number of cells in S phase can be
difficult to distinguish from those in Gi and G2 phases of the cell cycle. Therefore,
BrdU, a thymidine analog, was incorporated into MEFs undergoing DNA synthesis to
selectively label cells in S phase. Cells in S phase could then be precisely distinguished
by their binding of fluorescently labelled antibodies specific for BrdU (Figure 4.2B).
A higher percentage of Tgif1' MEFs was observed in the G0/G1 phase of the cell
cycle (36 % Tgif* vs 44 % Tgif; p-value=0.003, n=8; Figure 4.2C); a similarly
reduced percentage of Tgif cells was detected in the S phase (28 % Tgif* vs 20 %
Tgif'; p-value=0.002, n=8). The percentages of cells undergoing apoptosis or in G2/M
phase were comparable between Tgif* and Tgif cells. These results indicate the
decreased proliferation of Tgif MEFs was due to prolonged Gi progression.
4.2.3. Proliferation assay by [3H]-thymidine incorporation
To determine whether Tgif MEFs were impaired in their ability to respond to
mitogens that regulate the restriction point, proliferation was assessed by measuring
[3H]-thymidine incorporation into MEF DNA following stimulation of log phase cells
with increasing levels of mitogen (0.1% - 10% FBS). The proliferation rate was
directly proportional to the concentration of FBS for both Tgif* and Tgif' cells,
suggesting Tgif cells were able to sense increasing levels of mitogen (Figure 4.3A).
69
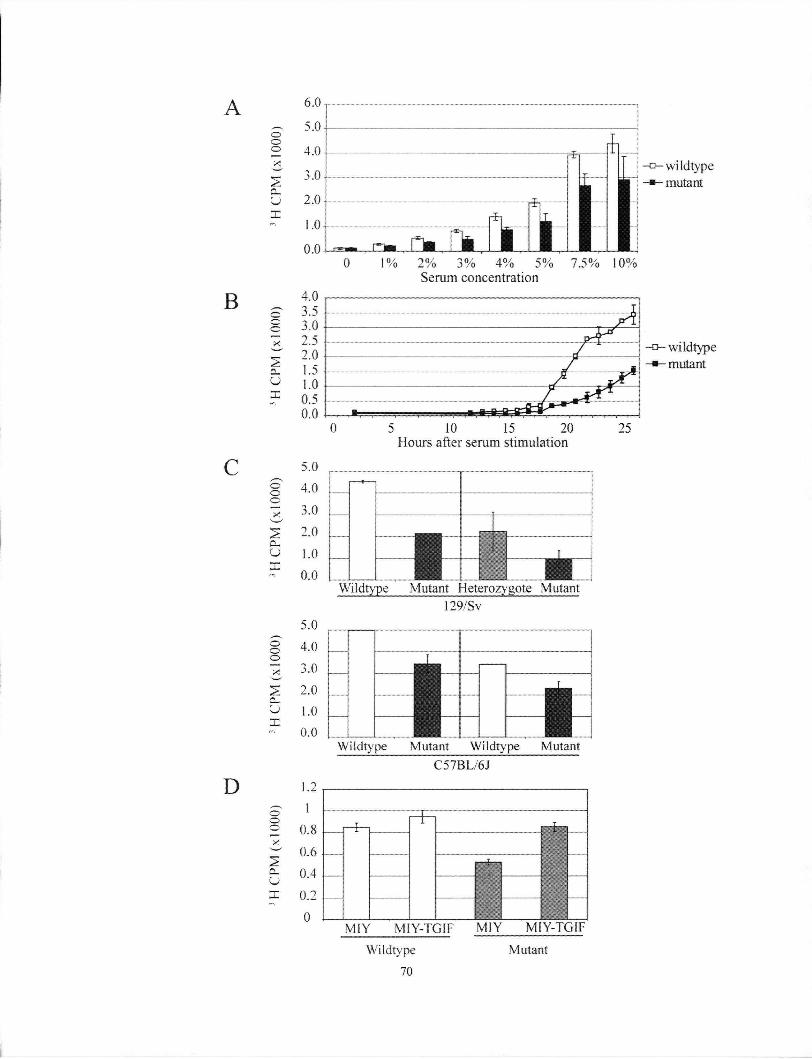
A
B
C
D
CD —
a c
u x
o
Cu U
o o
U
o o
—
6.0
5.0
4.0
3.0
2.0
1.0
0.0
Xi \\
5.0 4.0 3.0 2.0 1.0 0.0
5.0 4.0 3.0 2.0 1.0 0.0
•wild type •mutant
2% 3% 4% 5% 7.5% 10% Serum concentration
wildtype mutant
5 10 15 20 Hours after serum stimulation
I Wildtype Mutant Heterozygote Mutant
129/Sv
Wildtype Mutant Wildtype Mutant C57BL/6J
MIY MlY-TGIF MIY M I Y - T G 1 F
Wildtype 70
Mutant

Figure 4.3 Cell cycle analysis of proliferation defects in Tgif1' MEFs. (A) Tgif* and
Tgif1' cells were cultured in media supplemented with increasing concentrations of
serum. DNA synthesis was measured by [3H]-thymidine incorporation for 2 hours.
Proliferation rate was directly proportional to the concentration of serum. Mutants
consistently proliferated at a slower rate than wild-type controls. (B) Tgif* and Tgif1'
cells were growth arrested by serum starvation, then serum was added to stimulate
synchronous re-entry into the cell cycle. Fewer mutant MEFs re-entered the cell cycle
at all time points. Consequently, the total number of mutant MEFs entering into cell
cycle was lower. Representative results from two experiments are shown, each
performed in triplicate, using cells derived from independent MEFs. (C) Analysis of
proliferation in MEFs derived from congenic 129/Sv and C57BL/6J embryos. MEF
lines derived from two independent litters of each genotype are shown. One litter from
the congenic 129/Sv genetic background did not include any Tgif* embryos, and Tgif
*'~ MEFs were used for analysis instead. Mutant MEFs displayed lower proliferation
relative to wild-type and heterozygote controls (p<0.002). Experiments were performed
in triplicate using cells derived from independent embryos. (D) Tgif*1* and Tgif'1'
MEFs were infected with MSCV-retroviruses expressing human TGIF and YFP (MIY-
TGIF) or YFP alone (MIY). Infected cells were enriched by FACS and DNA synthesis
was measured by [ H]-thymidine incorporation as described above. Expression of
human TGIF restored the proliferation rate of mutant MEFs to normal levels.
Expression of human TGIF in wild-type MEFs induced a small but consistent increase
in proliferation rate. Data shown here were obtained with MEFs derived from embryos
on the 129/Sv/CDl genetic background. Error bars indicate standard deviation. CPM,
counts per minute.
71

4.2.4. Proliferation of synchronized mouse embryonic fibroblasts
To further characterize the Gi entry of the mutant MEFs, cells were arrested at
Go/Gi and allowed to enter S phase synchronously. Serum starvation is an effective
method for inducing Go/Gi arrest in a variety of normal and untransformed cell lines,
including MEFs (Krek and DeCaprio 1995; Herrera et al. 1996; Kozar et al. 2004;
Malumbres et al. 2004). Serum-starved cells are healthy for many days in this
quiescent state. The re-addition of high concentrations of serum then induces them to
leave the Go state and progress synchronously through the subsequent cell cycle stages.
Wild type and mutant cells were fed media supplemented with 10% FBS and DNA
synthesis was subsequently assessed using 3H-thymidine incorporation as described.
Approximately two-fold more quiescent Tgif+/+ MEFs entered S phase relative
to Tgif'' MEFs at the indicated times after serum stimulation (Figure 4.3B).
Consequently, the total number of mutant cells that re-entered the cell cycle over time
was lower. Taken together, these results indicate that Tgif'' MEFs do not progress as
rapidly through G| as Tgif* MEFs, consistent with the results of the FACS analysis.
A proliferation defect was also observed in MEFs derived from congenic
129/Sv and C57BL/6J embryos (Figure 4.3C), indicating the loss of Tgif caused a
proliferation defect in multiple genetic backgrounds.
4.2.5. Rescue of the proliferation defect by re-expression of human TGIF in
mutant mouse embryonic fibroblasts
To determine whether the proliferative defect observed in Tgif MEFs could be
rescued, a cDNA for human TGIF was expressed in mutant MEFs, which were then
tested for proliferative capability. Retroviral constructs were generated to express
human TGIF cDNAs upstream of an IRES-YFP sequence. The infection efficiency of
72

MEFs was 50-60%, and cells expressing TGIF were enriched by fluorescence activated
cell sorting (FACS) for YFP expression. Expression of human TGIF was able to rescue
the proliferation defect in mutant MEFs (Figure 4.3D). Over-expression of TGIF in
wild-type MEFs resulted in a small, but consistent, increase in proliferation.
4.3. P R O L I F E R A T I O N D E F E C T S IN V I V O
4.3.1. Growth retardation was observed in embryonic day 8 embryos
To assess whether proliferation defects were present in vivo, embryos were
dissected between E8.0 and E8.25, the stage during which dorsal-ventral patterning is
occurring in the forebrain. Embryos were precisely staged with respect to their
morphology and somite number. Upon examination of 15 wild-type and 17 mutant
embryos from 7 litters, mutant embryos were determined to be developmentally
delayed by 2 - 3 somites (Figure 4.4A, 0.001<p<0.002 using the Mann-Whitney test).
Consistent with this observation, some neonates and adult mutant mice were growth
retarded (Figure 3.5; Table 1). Together, these observations suggest reduced
proliferation in the absence of Tgif led to a growth retardation phenotype in vivo as
early as E8.
4.3.2. BrdU incorporation rate in embryonic day 8 embryos
Using FACS analysis, it was determined that 28% of wild-type MEFs were in S
phase compared to 20%> of mutant MEFs (Figure 4.2C). To determine whether
embryonic cells in vivo also showed reduced proliferation, in vivo BrdU incorporation
experiments were conducted to compare the proportion of cells in S phase between the
wild-type and mutant embryos at E8. The experiments were conducted on E8 embryos
because staging using somite number indicated this was an appropriate stage.
73
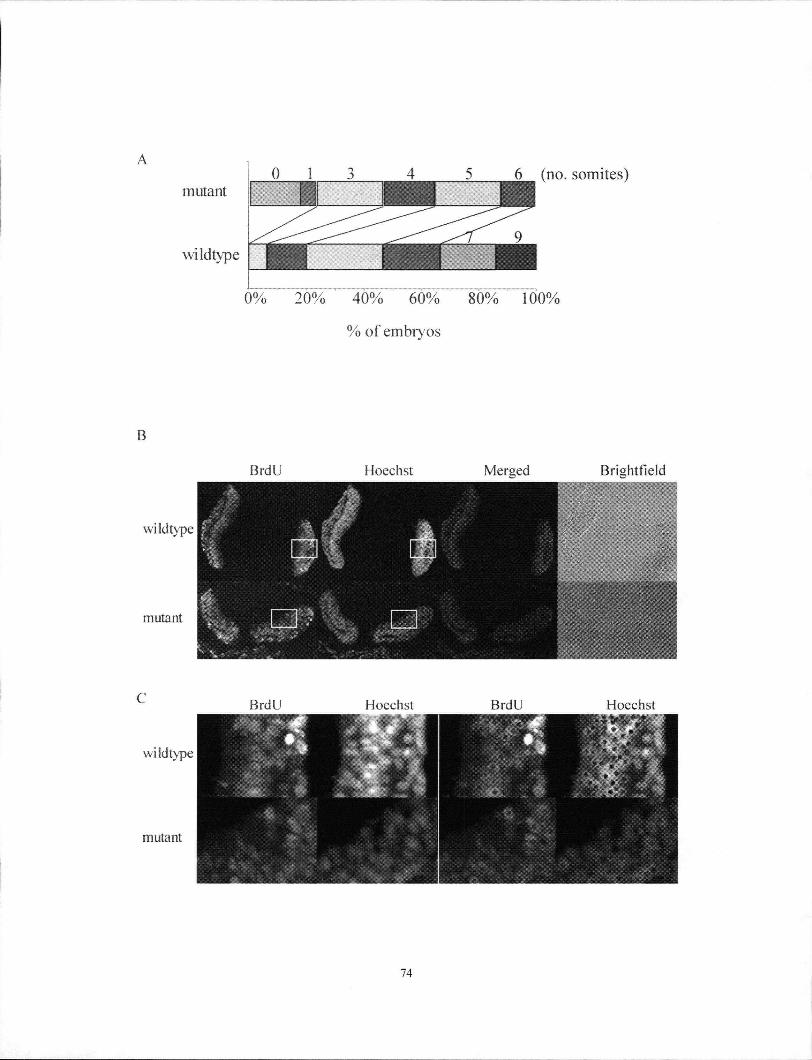
74

Figure 4.4 Tgif' mutant embryos exhibited growth retardation. (A) Examination of the
developmental stage of Tgif/+ and Tgif'' embryos (n=15 and 17, respectively, from 7
litters) at E8-E8.25 according to their somite numbers. Mutant embryos were
determined to be developmentally delayed by 2-3 somites (0.00l<p<0.002 using the
Mann-Whitney test). (B, C) In vivo BrdU incorporation rate was similar between wild-
type and mutant forebrains at E8.0-E8.25. Embryos labelled with BrdU were used to
determine the percentage of cells in S phase in vivo; all nuclei were counter stained
with Hoechst. The fraction of BrdU incorporated cells was determined by counting the
number of BrdU positive cells (red) and dividing by the total number of Hoechst
positive cells (green). At least three adjacent sections were counted, and three sets of
matched mutant and control embryos were analyzed. The boxed regions in (B) were
enlarged in (C) and spots were placed over positively stained cells to determine the
percentage of BrdU positive cells. The embryos shown here were on the C57BL/6J
genetic background.
75
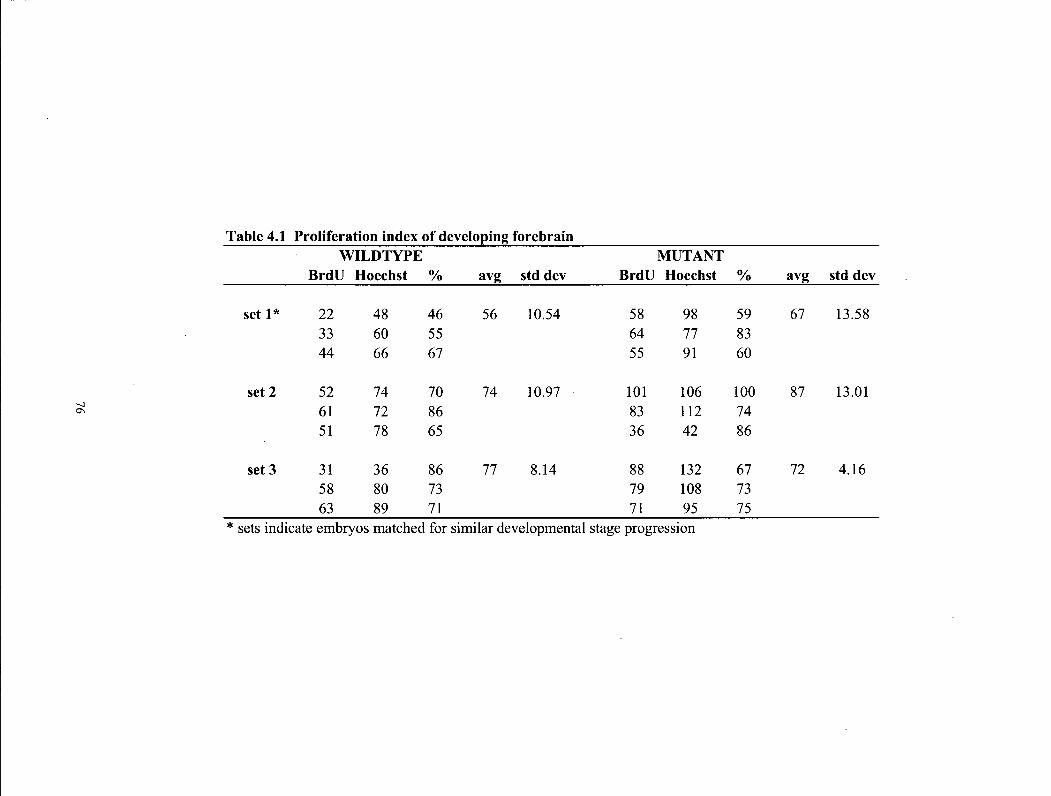
Table 4.1 Proliferation index of developing forebrain WILDTYPE MUTANT
BrdU Hoechst % avg std dev BrdU Hoechst % avg std dev
set 1* 22 48 46 56 10.54 58 98 59 67 13.58 33 60 55 64 77 83 44 66 67 55 91 60
set 2 52 74 70 74 10.97 101 106 100 87 13.01 61 72 86 83 112 74 51 78 65 36 42 86
set 3 31 36 86 77 8.14 88 132 67 72 4.16 58 80 73 79 108 73 63 89 71 71 95 75
* sets indicate embryos matched for similar developmental stage progression

Littermate embryos at the same somite stage were carefully matched, and 10 um frontal
sections spanning the developing forebrain, midbrain and hindbrain were compared.
The proportion of BrdU positive cells within the dorsal, mid and ventral regions of the
neural tube was also compared. The sensitivity of this technique was approximately 5-
10% since the proportion BrdU positive cells in adjacent sections generally deviated
within this range.
A significant difference was not detected in the proportion of cells that
incorporated BrdU between wild-type and mutant forebrains (Figure 4.4B, C, Table
4.1). The sensitivity of in vivo BrdU incorporation may be too low to detect the small
difference in proliferation predicted from the in vitro MEF assays. Alternatively, it is
also possible that in vivo proliferation was not significantly different between wild-type
and mutant embryos at the developmental stage examined, as cell cycle is normally
tightly regulated during in vivo development. Consistent with these observations, the
E8 embryo or telencephalon would be predicted to be significantly smaller if the
reduction in proliferation observed in MEFs also affected most embryonic tissues, or
the entire telencephalon.
4.4. ASSAYING THE PROLIFERATIVE FUNCTION OF H U M A N TGIF
MUTATIONS IN MOUSE EMBRYONIC FIBROBLASTS
To test whether mutations identified in HPE patients (Figure 4.5A) (Gripp et al.
2000; Chen et al. 2002; Aguilella et al. 2003) were able to rescue the proliferation
defect, TGIF point mutations were re-created in the retroviral constructs, expressed in
Tgif1' MEFs, and proliferation assessed. Not surprisingly, the Y59X mutation did not
rescue the proliferative defect, presumably because the protein pre-terminated within
the homeodomain (Figure 4.5B). Notably, the other point mutations within the
77

H P E Mutations
272
CtBP Homeodomain Smad2/3 & HDAC Repressive Domain Binding Interaction Domain & Sin3 Interaction Domain Domain ( E G F / M A P K
phosphorylation)
78

Figure 4.5 Expression of a subset of mutated human TGIF rescued the proliferative
defect of MEFs. (A) Diagram of HPE patient mutations, and their positions relative to
the functional domains of TGIF, which were generated in MSCV-retroviral constructs.
(B) Tgif1' MEFs were infected with MSCV-retroviruses expressing human TGIF and
YFP (MYY-TGIF), YFP alone (MIY) or human TGIF containing the patient mutations
from (A). Infected cells were enriched by FACS and DNA synthesis was measured by
[3H]thymidine incorporation as described. Expression of human TGIF restored the
proliferation rate of mutant MEFs to normal levels. Mutations affecting the
homeodomain were unable to rescue the proliferative defect. Representative results
from 5 experiments are shown; each experiment was performed in sextuplicate using
cells derived from independent Tgif* and Tgif1' embryos. Error bars represent
standard deviation of the mean. * indicates p<0.001 between wildtype and mutant TGIF
transduction; § indicates p<0.001 between MIY and mutant TGIF transduction using
Student's t-test. Cpm, counts per minute. (C) Tgif'~ MEFs were infected with MSCV-
retroviral constructs containing flag-tagged TGIF as described. Western blot analysis of
MEF cell extracts showed similar expression levels for all mutant proteins. TGIF
generally migrated as a doublet of 30-32 kDa; the upper band likely represents the
phosphorylated form.
79

homeodomain, P63R and R90C, were also unable to compensate for the loss of Tgif. In
contrast, other mutations, S28C, Q107L, T151A, and T162F, which are located within
the CtBP and SMAD2 and HDAC interaction domains, rescued the proliferative defect
in mutant MEFs. These results confirm a proliferation function for Tgif that requires
the homeodomain.
During construction of the retroviral expression construct, Tgif was flag-tagged
at the N-terminus to allow for western blot analyses. The level of TGIF protein
expression for all mutant proteins was assessed using MEF cell extracts and standard
Western blot using antibody to the flag epitope. The level of protein expression was
similar between wild-type and mutant TGIFs, with the exception of Y59X, which did
not produce a detectable protein at the expected size (Figure 4.5C). This suggests the
ability of each mutant protein to rescue the proliferative defect is likely due to the
modifications to their activity resulting from the point mutations.
4.5. PROLIFERATION RESPONSE TO SIGNALLING PATHWAYS
4.5.1. Proliferation in response to TGF-P
TGF-P is a cytokine known to exhibit potent anti-proliferative activity
(Massague et al. 2000). As discussed earlier, Tgif 'was previously shown to repress the
transcriptional response of the TGF-P pathways; the relative level of Tgif incorporated
into Smad protein complexes can regulates the level of downstream target gene
expression (Figure 1.6) (Bertolino et al. 1995; Wotton et al. 1999a). Given the role of
Tgif in regulating TGF-P signalling, I hypothesized that Tg/J-dependent regulation of
downstream genes is responsible for enabling TGF-P signalling to inhibit proliferation.
In that case, mutant MEFs lacking Tgif would be more sensitive to the anti-proliferative
effect of TGF-p. To investigate this hypothesis, the rate of [3H]-thymidine
80

incorporation into MEFs was assessed after treatment with TGF-p\ Tgif*1* MEFs
exhibited a typical inhibition of proliferation in response to TGF-P treatment: growth
inhibition was detected at TGF-P concentrations as low as 0.08 ng/mL, or 3 pM, and
reached a maximum 2-fold inhibition at 0.6 ng/mL, or 25 pM (Figure 4.6A) (Sirard et
al. 2000). Tgif1' MEFs proliferated more slowly compared with Tgif*1* MEFs in
response to increasing concentrations of TGF-p. Since Tgif1' cells normally
proliferated at a lower rate, the level of [3H]-thymidine uptake was normalized to the
maximum incorporation level. Through this transformation it became apparent that the
degree of growth inhibition between Tgif*1* and Tgif1' MEFs was similar at similar
concentrations of TGF-fi (Figure 4.6B). This result indicates Tgif does not regulate
proliferation by modulating TGF-P response genes in MEFs.
4.5.2. Proliferation in response to retinoic acid signaling
Similar to TGF-P, RA also functions to arrest proliferation (Goyette et al. 2000).
Additionally, TGIF was reported to compete for DNA binding with the RXRa retinoic
acid receptor (Bertolino et al. 1995). Therefore, I hypothesized the expression of Tgif
may play a role in regulating RA-mediated transcription of downstream cell cycle
genes. It would then be predicted that the rate of proliferation in MEFs lacking Tgif
would mimic exposure to increased RA. To investigate this possibility, growth arrest in
response to treatment with 9-cis RA and aW-trans-KA in MEFs was assessed. Similar
to keratinocytes, treatment of Tgif*1* MEFs with 10"12 M 9-cis-RA caused a reduction
in DNA synthesis, with a maximal effect seen at 10"6 M , which caused an 80%
reduction in proliferation (Goyette et al. 2000). A\l-trans-RA was also effective in
inhibiting proliferation with a maximal effect at 10"6 M , that resulted in a 70%>
reduction in proliferation. When normalized, these dose-response experiments again
81
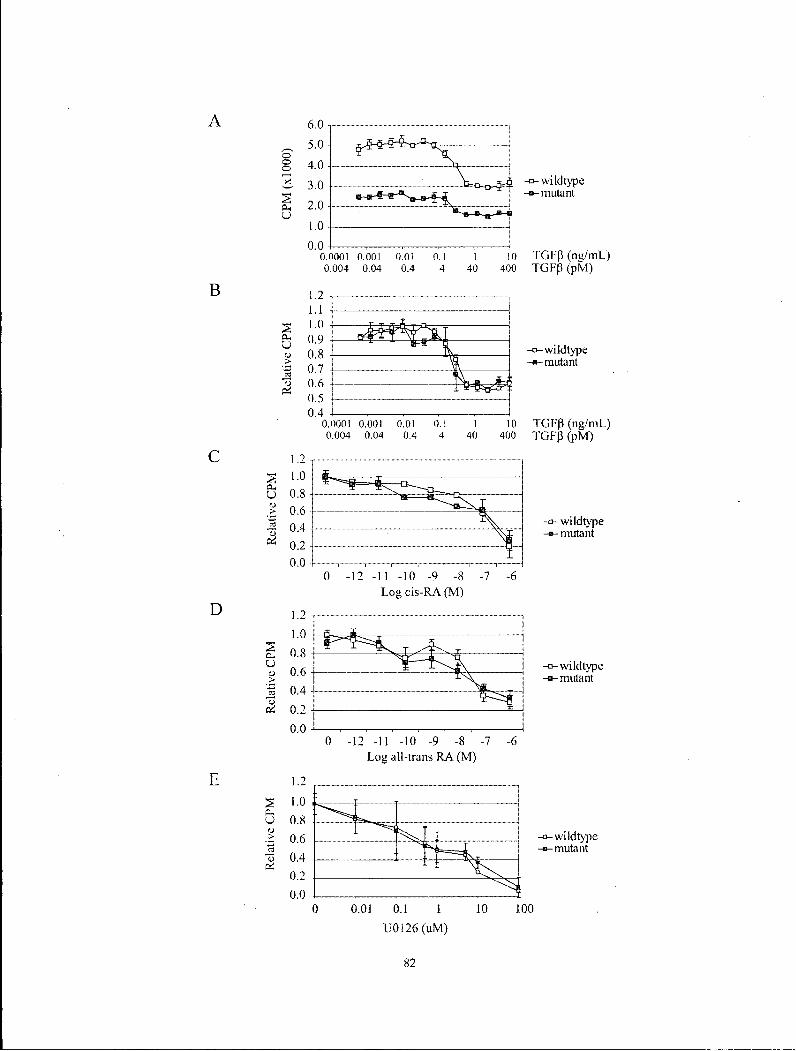
1.2 1.0
s ft.
0.8 -o 0.6 -0.6 ->• 03 0.4 -
0.2 -0.0 -
-12 -11 -10 -9 -8 -7 Log cis-RA (M)
-wildtype -mutant
TGFP (na/mL) TG1::P (pM)
-wildtype - mutant
TGFp (ng/mL) TGFP (pM)
-o- wildtype -B-mutant
-wildtype -mutant
-12 -11 -10 -9 -8 -7 Log all-trans RA (M)
u 1.0 0.8 0.6 0.4 0.2 0.0
K J i -wildtype - mutant
0.01 0.1 1 U0126 (uM)
10 100
82

Figure 4.6 Analysis of MEF proliferation in response to TGF(3, 9-cis RA, all-rrara-RA
and U0126. (A) Primary Tgif* and Tgif1' MEF cells were grown in the presence of 0.6
pg/mL-10 ng/mL TGF-P for 24 hours and DNA synthesis was measured by
[ H]thymidine incorporation as described. Mutant MEFs displayed lower proliferation
at all concentrations of TGF-P relative to wild-type MEFs. (B) Results from (A) were
normalized to the maximum value of each genotype since mutant MEFs consistently
showed reduced proliferation. The proliferation inhibitory response was similar
between wild-type and mutant MEFs at all concentrations of TGFp. (C-D) Fresh 9-cis-
RA (C) and al\-trans-RA (D) were added to MEFs on a daily basis for 5 days and
[3H]thymidine incorporation was performed on day 6. Results were normalized as in
(B). The proliferative inhibitory response to 9-cis-RA and all-/rara'-RA was similar
between wild-type and mutant MEFs. (E) 0.01-100 uM U0126 was added to MEFs and
[ H]thymidine incorporation was performed after 24 hours of culture. The proliferative
inhibitory response to U0126 was similar between wild-type and mutant MEFs. A l l
results are from representative experiments. For each, three experiments were
performed in triplicate using cells derived from independent Tgif* and Tgif
embryos. Error bars represent standard deviation of the mean. Cpm, counts per minute.
83

showed similar growth arrest responses between Tgif*1* and Tgif1' MEFs (Figure
4.6C,D). Therefore, and contrary to expectation, Tgif does not appear to regulate
proliferation in MEFs by modulating the transcriptional levels of cell cycle genes
downstream of the RA pathway.
4.5.3. Proliferation in response to R a s / M A P K inhibitor, U0126
The Ras/MAP Kinase pathway is activated downstream of many mitogenic
factors, such as the Fgf (fibroblast growth factor) family (Lodish et al. 2000). TGIF
protein can be stabilized by phosphorylation of two sites near its C-terminus via the
Ras/MAP kinase pathway (Lo et al. 2001). It is possible that the Ras/MAP Kinase
pathway may modulate Tgif s stability, and hence its activity in regulating
proliferation. To address this possibility, U0126, an inhibitor of the kinase activity of
M E K in the Ras/MAP Kinase pathway, was used to inhibit the phosphorylation and
stability of Tgif (Lo et al. 2001). The reduction of Tgif activity by U0126 would be
predicted to reduce proliferation in wild-type cells, and therefore this treatment would
mimic mutant MEFs lacking Tgif. U0126 inhibited cell proliferation at 0.01 uM and
almost completely inhibited cell cycling at 100 uM. However, the proliferative
response to U0126 treatment or Ras/MAP kinase inhibition in Tgif*1* and Tgif1' MEFs
did not differ significantly (Figure 4.6E). Therefore, TGIF protein phosphorylation and
stability, as regulated by the Ras/MAP kinase pathway, did not have a significant
function in regulating proliferation in MEFs. Taken together, the pathways known to
be modulated by Tgif did not show an increased sensitivity to ligand in Tgif'' cells, and
the mutant MEFs displayed growth inhibition responses that were unaltered relative to
Tgif*'* cells.
84

4.7 S U M M A R Y
MEFs derived from Tgif1' cells consistently proliferated slower and
accumulated to a lower density than did wild-type cells. This decreased proliferation
was shown to result from reduced Gi/S progression of the cell cycle. Additionally,
mutant embryos exhibited a developmental delay consistent with the observation that
19% of mutant adults were growth retarded. However, in vivo proliferation within the
developing neuroectoderm was normal in the mutants at E8.
Re-expression of wild-type TGIF in MEFs rescued this proliferative defect,
confirming that the loss of Tgif 'was critical in regulating progression through the cell
cycle. However, TGIF proteins containing mutations in the homeodomain were unable
to rescue this phenotype, indicating the homeodomain is necessary for Tgif to function
during cell cycle regulation. Finally, the slow proliferation of mutant MEFs in standard
culture conditions was not the result of enhanced sensitivity to TGF-P or R A , or
reduced responsiveness to M A P K signalling.
85

5. C H A P T E R 5 DISCUSSION
5.1. M U T A N T M I C E DID N O T E X H I B I T H P E , B U T DID E X H I B I T H P E
R E L A T E D D E F E C T S
Medical genetics has provided abundant and significant insights into normal
development and disease progression (Epstein et al. 2003). One recent and notable
study associated the SLITRK1 gene with Tourette's Syndrome. This study is providing
insights into and understanding of neuronal growth, guidance and branching, as well as
generating hypotheses for the etiology of this disease (Abelson et al. 2005). Similarly,
this study of Tgif, a gene mutated in some HPE patients, has provided important
insights and generated hypotheses for future studies of forebrain development and the
etiology of HPE.
Deletion, nonsense and missense mutations in TGIF have been identified in a
subset of HPE patients (Overhauser et al. 1995; Gripp et al. 2000; Chen et al. 2002;
Aguilella et al. 2003; Bendavid et al. 2005a; Bendavid et al. 2005b). Significantly,
HPE can develop in patients carrying heterozygous mutations in TGIF, suggesting the
processes being disrupted are exquisitely sensitive to the level of TGIF. Paradoxically,
most carriers do not develop HPE, indicating a low level of penetrance for this disorder,
and that the manifestation of the phenotype may be dependent on genetic modifiers
(Nanni et al. 1999; Aguilella et al. 2003).
Here, I showed that mice lacking Tgif were viable and fertile without overt
evidence of an HPE phenotype. However, some mutant mice did develop multiple
defects including laterality and eye defects, growth retardation and kinked tails,
suggesting that Tgif 'may be involved in many processes during development. As of
May 2006, the characterization of Tgif mutant mice has been reported independently by
three other groups, and all indicating a lack of a HPE phenotype in the mutant mice
86

(Shen and Walsh 2005; Bartholin et al. 2006; Jin et al. 2006). Furthermore, those
groups did not report microcephaly, eye defects, kinked tails, or laterality defects with
mutant mice on C57BL/6 and 129 genetic backgrounds.
5.1.1. Lack of HPE phenotype
Mice generated through loss-of-function experiments often do not develop the
expected phenotypes, and there are many explanations. One unexplained phenomenon
in mammalian genetics is that the corresponding mutation in mice often results in a
much stronger phenotype in humans (Bier and McGinnis 2003). A classic example is
that heterozygous mutations in SHH result in HPE in humans, but comparable
abnormalities are only observed in homozygous Shh mice (Chiang et al. 1996).
Additional examples include Ptch and FoxHl (Goodrich et al. 1997; Hoodless et al.
2001). Likewise, heterozygous mutations in TGIF are found in patients, but
homozygous mutant mice described here developed only microforms, or associated
defects, of HPE. The basis for these differences in mouse and human CNS
development are beginning to be explored (Li et al. 2005).
The loss of Tgif cm potentially be compensated by rgz/paralogues. In mice,
three rgz/paralogues have been identified: Tgif2, Tex, and Tgiflx/y (Melhuish et al.
2001; Bianco-Arias et al. 2002; Lai et al. 2002). Tgif has been shown to possess many
similar biochemical properties as Tgif, and its expression has also been detected in the
developing nervous system (Jin et al. 2005). Tex and Tgiflx/y are also predicted to
contain many of the same functional domains as Tgif. In order to address the potential
roles of these paralogues, loss-of-function mice for these genes could be generated and
bred with Tgif1" mice to evaluate the potential redundancy amongst these genes.
87

As discussed earlier, the penetrance for heterozygous TGIF mutations for HPE
is only 10% in humans; the penetrance of mutations in the other HPE genes is similarly
incomplete (Dubourg et al. 2004). In this study I examined 80 adult mice: 25 were Tgif
+ / + (31%), 36 were Tgif*1' (45%), and 19 were Tgif1' (24%). It is possible that some
Tgif mutants developed HPE at low penetrance and died undetected as dams are less
likely to nurture their abnormal neonates than are humans. Consistent with this
possibility, a significant number of neonates can be lost (greater than 50%, or 15
mutant mice in this example) and not be detected statistically at a significance level of
95%). In order to detect a decrease in viability perinatally, pups could be examined for
HPE before they are born.
Another possibility is that the dissection methods used in this study were
insufficiently sensitive to detect mild HPE cases since forebrain defects in many human
HPE patients can be subtle and only be detected by MRI (Takahashi et al. 2004). A
detailed histological study could address this possibility. Detailed histology was
conducted by another group, and while HPE was not detected, the mice examined in
that study were on the C57BL/6J genetic background (Shen and Walsh 2005).
5.1.1.1.Genetic background
The most likely explanation for a lack of HPE defect in mice is the influence of
genetic background. The degree to which the function of other genes and alleles
contribute to a given phenotype is complex (Nadeau 2003). For example, one variant
of TGF-fil in patients caused severe impairment and lung disease relative to patients
with different alleles that exhibited only mild impairment (Drumrn et al. 2005).
Likewise, Tgif function in humans is possibly influenced by genetic background as
evidenced by HPE mutations that exhibit variable expressivity and a low degree of
88

correlation between genotype and phenotype. Furthermore, sometimes carriers of the
same mutation in the same family develop a wide spectrum of defects, ranging from no
defect to severe HPE (Aguilella et al. 2003). However, twin studies that can better
separate the effects of genetic and environmental influences have not yet been
performed.
Multigenic mutation hypotheses have been proposed for HPE and other
multifactorial diseases. It is thought that forebrain development and other biological
processes have evolved to be regulated by redundant mechanisms and that disruptions
to more than one gene or pathways would disturb the developmental program
sufficiently to cause abnormalities. This hypothesis was proposed when multiple HPE
patients including two patients from different families that carry mutations in both Shh
and TGIF were identified (Ming and Muenke 2002).
Interestingly, TGIF is located only 5 Mb from TWISTED GASTRULATION
(TSG), a member of the BMP pathway; BMP plays a role in regulating dorsal-ventral
patterning of the forebrain (Petryk et al. 2005). Recently, many patients were identified
with sub-karyotypic microdeletions that encompass TGIF (Bendavid et al. 2005a;
Bendavid et al. 2005b). Patients with microdeletions or large rearrangements over the
HPE4 region that affected both TGIF and TSG function may develop more severe
symptoms compared to patients with smaller rearrangements that only affected TGIF.
Studying the correlation between the severity of HPE symptoms and the extent of
chromosomal rearrangements in patients may shed light on the effect of disrupting one
or both genes.
The influence of genetic background in mice has long been recognized, but the
prohibitive costs of mouse genetics research likely precluded further investigations
(Bonyadi et al. 1997; LeCouter et al. 1998; Nadeau 2003). Both amino acid
8 9

polymorphisms and regulatory variations are known to cause variations in the temporal
or spatial patterns, and absolute level of expression in different inbred mouse strains
(Cowles et al. 2002). The loss of Cdo, a co-receptor for Shh, caused HPE only on
selected genetic backgrounds exemplifies this phenomenon (Zhang et al. 2006).
Tgif1' mice generated from this study displayed a variety of low penetrant
defects. In contrast, three other groups did not report these findings. Tgif 'is a simple, 3
exon-containing gene encompassed within 7 kb of genomic DNA. The targeting
constructs from all four groups were similarly designed to remove the majority of
coding sequences with only minor differences. I discovered the loss of Tgif resulted in
pleiotropic defects only on the 129/Sv/CDl background, while mutants on the
C57BL/6J and 129/Sv backgrounds did not develop any of the defects. The other three
groups did not report the phenotypes I observed, but they only analyzed mutant mice on
the C57BL/6J and 129/Sv/C57BL/6J genetic backgrounds (Shen and Walsh 2005;
Bartholin et al. 2006; Jin et al. 2006). I also examined a large number of mice, over
450, of which 92 were homozygous mutant, and detected phenotypes that occurred with
low penetrance. For example, thorough analysis of the weight of 139 mice revealed
19% of mutant mice displayed a growth retardation phenotype relative to littermate
controls. Interestingly, this result is in agreement with the decreased, but not
statistically significant, weights reported by one study that did examine the weight of
mice (Shen and Walsh 2005). This result was likely masked when the weights of 54
mice were averaged.
The precise mechanism by which genetic background can influence
manifestation of diseases such as HPE can vary. However, haploinsufficient mutations
in patients suggest a reduction in the level of Tgif, perhaps below a threshold level, in
90

the developing forebrain is sufficient to cause HPE development in a sensitive genetic
background (Nadeau 2001).
5.1.2. HPE-related defects
7_-*ymutant mice developed laterality and eye defects and kinked tails, as well
as growth retardation. Laterality defects are often associated with HPE and likely arose
from disruptions to a common developmental process (Roessler and Muenke 2001;
Cohen 2003). The forebrain is in part patterned by the ventral midline and the
prechordal plate, which instructs neural plate cells of their midline and lateral positions.
The same midline cells coordinate and instruct the surrounding mesodermal cells of
their position relative to the left-right axis. Consistent with this idea, disruptions to the
signalling pathways that transduce these cues lead to both laterality defects and HPE
(Roessler and Muenke 2001). Additionally, kinked tails are also indicative of midline
defects (Purandare et al. 2002). Finally, eye defects and growth retardation are often
exhibited by HPE patients (Cohen 2003).
An additional line of evidence from studies conducted in chick spinal cord
suggests Tgif may play a role during forebrain patterning. Tgif expression was detected
in the dorsal medial spinal cord, and a series of overexpression experiments indicated
Tgif repressed transcription of dorsal expressing genes that regulate the dorsoventral
identity of neurons (Knepper et al. 2006). Consistent with this hypothesis, abnormal
patterning of the forebrain dorsoventral axis can cause HPE. One caveat associated
with overexpression studies is the possibility that ectopic expression can cause artifacts
by competing for binding factors and disrupting protein complexes. For instance, since
Tgif normally interacts with a multitude of proteins, such as p300, high levels of Tgif
may quench and disrupt such protein complexes. Consequently, it is important to
91

verify that the same genes shown to be regulated by 7g//in overexpression studies are
also misregulated in loss-of-function animals, such as the Tgif1' mice. Analysis of
several forebrain and ventral markers was conducted in this study, as well as by another
group, but differences in the expression pattern of these ventral markers were not
detected (Jin et al. 2006).
5.1.3. Future Directions
To further address the functions of Tgif during forebrain development I propose
three genetic strategies in an attempt to uncover an HPE phenotype in Tgif mutant mice.
Genetic strategies are being proposed because Tgif1' mice are viable and fertile and can
thus be used strategically. This is a realistic and promising objective, since patients
with rgj/mutations develop HPE, and HPE can develop in mice. Developing an
additional HPE mouse model would be a valuable reagent that could provide insights
into the mechanism of forebrain patterning.
My first proposal is to identify an inbred genetic background in which Tgif
mutant mice develop HPE. Since mutant mice on the 129/Sv/CDl background
developed HPE related defects, and CD1 outbred mice were originally derived from
Swiss mice, Swiss inbred mice may be a good candidate inbred strain (Chia et al.
2005). A variety of other inbred strains are also available from the Jackson Lab, such
as the widely available and studied Balb/cJ, FBV, CBA, and C3H inbred strains. For
example, on the C57BL/6J genetic background, the retinoblastoma pi 30 null mice were
viable and fertile, but a plethora of functions were revealed in the Balb/cJ genetic
background. If this strategy was successful, markers for progressive steps during
forebrain patterning would be used to determine the precise function of Tgif during
HPE development. In addition, modifier genes in the affected strain could also be
92

identified using QTL analysis. Identification of modifier genes may also be
informative since they may play a role in regulating or interacting with Tgif during
forebrain patterning (Neumann et al. 1994; Bonyadi et al. 1997). Finally, penetrance
does not have to be 100% to identify such genetic modifiers. The major challenge in
this experiment would be the expense of housing multiple lines of mice. However, the
advantage of this experiment is the simplicity of the procedure.
The second strategy is to generate compound mutants. This strategy is
traditionally used by geneticists and has proven successful for many genes. A few
notable examples include Smad2+,';Nodat/', Smad2+l~;Smad3+l~', Cerl"l'\leftyVl', and gsc
'';HNF3B+/' double mutant mice (Filosa et al. 1997; Nomura and L i 1998; Perea-Gomez
et al. 2002; Dunn et al. 2004). The reason for this success is that multiple genetic
lesions can disrupt functional redundancy amongst genes and pathways that would
otherwise compensate and mask the effects of single mutations. Consequently, digenic
and multigenic hypotheses were proposed for many genetic diseases, including HPE
(Ming and Muenke 2002). Candidate mutations to introduce with the Tgif mutation
include genes in the Nodal, RA, BMP, Shh, Fgf and Wnt pathways. For example, Shh
mutant mice are an excellent candidate since two patients carried mutations in both Shh
and Tgif (Ming and Muenke 2002). Alternatively, Zic2 mutant mice is another
candidate since Zic2 is an important HPE gene that regulates Nodal signalling. Since
many mutants in these pathways are available, this strategy is feasible. The advantages
and disadvantages are similar to the last strategy.
The third strategy is to identify mutants using an ENU-mutagenesis screen
(Cordes 2005). ENU-mutagenesis is a method used to create high rates of point
mutations that can saturate the mouse genome. While the second strategy requires the
assessment of a good hypothesis and the selection of a likely candidate, a mutagenesis
93

screen identifies candidates based on function. With a clearly defined phenotypic
screening strategy, E N U mutagenesis screens have been used to successfully identify
novel genes and alleles of known genes that have provided valuable insights into
developmental mechanisms (Anderson 2000). If Tgif1' mice are sensitized to
developing forebrain defects, the introduction of an additional mutation may be
sufficient to further disrupt and allow HPE to develop. Identification of such genes
may give insights into Tgif function and forebrain patterning. On this note, many genes
that cause HPE are not yet known. Only 20% of non-syndromic, chromosomally-
normal HPE patients have known mutations. A screen may identify candidate HPE
genes and genes previously not suspected to be involved in forebrain patterning.
5.2. MUTANT MICE EXHIBITED L A T E R A L I T Y DEFECTS
Events determining left-right patterning take place early during embryogenesis
before the onset of organogenesis. Soon after the breaking of symmetry, expression of
the TGF -P family member Nodal becomes enhanced on the left side of the node during
gastrulation (Hamada et al. 2002). The left-sided Nodal signal is relayed to the left
L P M , where downstream target genes such as lefty 1, lefty2 and pitx2 are activated. The
left-sided signals and pathways are barred from the right side by midline mesodermal
cells.
Laterality defects were discovered in 13%> of mutant mice, with 2 mice showing
complete situs inversus, indicating that Tg/Jplays a role in laterality determination.
Expression analysis by in situ hybridization revealed that while changes to Nodal and
lefty2 expression were not detected, 1 out of 20 mutant embryos did exhibit bilateral
pitx2 expression. Bilateral pitx2 expression suggests the loss of Tg//may disrupt
laterality at a molecular level upstream of pitx2. The low penetrance of this defect
94

impeded further studies, but this result does suggest TGIF is a novel candidate gene for
laterality disorders in humans.
Laterality mutants can be classified based on the nature of the disruption to the
laterality pathway and the type of situs defect that results. Situs inversus is caused by
disruptions to the early symmetry breaking event. The classic mutant that is disrupted
during this process is situs inversus mice. At the molecular level, the defect is likely a
failure in generating a nodal flow. This defect leads to the right-sided expression of
genes normally expressed on the left side. In contrast, situs ambiguus phenotypes are
caused by perturbations to relatively later events when laterality information is
transduced inappropriately, causing laterality defects to each organ independently, or
else loss-of-midline defects. The consequences of such defects include bilateral, or
absence of, expression of genes normally expressed only on the left side. One example
of such a defect is mutants in the Nodal signal transduction pathway.
The varieties of laterality defects, both situs inversus and situs ambiguus, in Tgif
~'~ mice is unusual and intriguing because this suggests Tgif may function during
multiple stages of laterality determination. To my knowledge, only one mutant, Zic3,
exhibits both situs inversus and situs ambiguus (Purandare et al. 2002). Zic3 regulates
Nodal signalling, consistent with the fact that Zic3 mutants initiate Nodal expression
normally but fail to maintain it. Therefore, in Zic3 mutants, NodaVs function is
disrupted not only during early symmetry breaking, but also later when the leftness
signal is propagated to the organ primordia. It is relevant to note here that mutations in
Zic2, a paralogue of Zic3, are found in HPE patients.
In addition, situs amgiguus in rgz/mutants may originate from abnormal
midline development, an idea that is supported by the kinked tails phenotype
(Purandare et al. 2002). An example of a midline mutant is Shh (Meyers and Martin
95

1999). Midline mutants initiate the breaking of symmetry at the node normally, and
transfer asymmetric expression from the node to the LPM correctly. However, defects
first become apparent when asymmetric expression and differentiation is not
maintained when the midline structures are disrupted. Such defects are typically
examined using midline markers such as shh, hrachyury, HNF3B and lefty 1. shh was
used to examine the midline in Tgif mutant embryos, but expression was not different
from control embryos.
5.2.1. Molecular basis of laterality defects
Loss of Tgif 'may cause laterality defects through inappropriate modulation of
the Nodal and RA signalling pathways. TGIF has been shown to repress the level of
TGF-P-activated transcription through interaction with SMAD2 and SMAD3. Since
Smad2 and Smad3 mediate Nodal signalling, the loss of Tgif is predicted to increase the
sensitivity of cells to the level of Nodal (Wotton et al. 1999a). Therefore, ectopic
Nodal response may lead to inappropriate symmetry breaking in the node in Tgif
mutants. Later, ectopic Nodal response may also become propagated to both the left
and right sides of the embryo. I examined expression of Nodal by in situ hybridization
but did not detect differences in its expression pattern, but it is possible that in situ
results were not sufficiently sensitive to detect changes in Nodal expression level. A
more sensitive method would be to use the Nodal-lacZ transgenic reporter that can
detect subtle differences in Nodal levels (Meyers and Martin 1999; Brennan et al.
2001). Alternatively, given the low penetrance of this phenotype it may be necessary to
examine a greater number of embryos in order to characterize the molecular defect.
Nevertheless, an absence of changes to Nodal expression level is not unexpected, since
Nodal expression is carefully regulated by multiple redundant mechanisms (Meno et al.
96

1997; King and Brown 1999; Constam and Robertson 2000b; Constam and Robertson
2000a).
A second potential molecular mechanism by which Tgif'may disrupt laterality
determination is its ability to modulate RA signalling. Recent reports demonstrated that
RA and Shh are involved in breaking of the left-right symmetry (Tanaka et al. 2005;
Vermot and Pourquie 2005). Since TGIF has been shown to compete, and thus inhibit,
RA signalling and repress RA responsive transcription, the loss of Tgif may disrupt this
step of the laterality determination pathway (Bertolino et al. 1995; Bertolino et al. 1996;
Bartholin et al. 2006). The nodal flow model predicts the left-ward transport of
morphogen proteins such as Shh and RA (Tanaka et al. 2005). It is conceivable that the
loss of 7gz/may sensitize cells to low levels of RA on the right side and result in
subsequent activation of Nodal expression on both sides.
'Situs amgiguus in Tgif1" mice may also originate from abnormal midline
development. This possibility is supported by the appearance of kinked tails in these
mice (Purandare et al. 2002), and bilateral pitx2 expression. Consistent with this, Shh
and RA signalling have been shown to regulate lefty I expression in the left side of
prospective floor plate of neural tube (Tsukui et al. 1999). The function of lefty I in the
midline is crucial for restricting Nodal to the left LPM (Meno et al. 1998). Tgif 'is
expressed in the forebrain midline and may modulate lefty 1 expression. Alternatively,
Tgif 'may play a direct role in midline integrity.
5.2.2. Future directions
The challenge to further dissecting the mechanism of the laterality defect is the
low penetrance of this phenotype. To address this issue, I would apply a similar
strategy to what was outlined above. Perhaps the 7g7/mutation will contribute to a
97

higher penetrance of situs defects in other genetic backgrounds, in combination with
other laterality mutants, or in combination with novel mutations that can be generated
by ENU mutagenesis. If successful, marker analysis can better define the
temporalspatial disruption caused by the loss of Tgifs function during both laterality
and forebrain development.
5.3. R O L E OF TGIF DURING C E L L C Y C L E CONTROL
This study provides the first evidence of a proliferative function for Tgif in
mammalian cells. MEF cell lines lacking Tg/Jproliferated at a slower rate and did not
reach the same density as normal controls. Further cell cycle analysis revealed that
mutant MEFs were defective in Gi/S progression, and that re-expression of human
TGIF rescued this proliferation defect. Consistent with observations made in this
study, the yeast protein Tos8, a TALE homeodomain transcription factor related to
TGIF, participates in G|/S transition events (Horak et al. 2002). Furthermore,
expression of human TGIF (CPR1) restored cell cycle progression by overcoming
mating pheromone-induced Gi arrest in yeast (Edwards et al. 1997). Also of relevance,
the Drosophila TGIF orthologues achintya and vismay were shown to activate
downstream target genes whose functions were necessary during spermatogenesis and
meiosis (Ayyar et al. 2003; Wang and Mann 2003). This suggests that a role for Tgif in
the regulation of the cell cycle may be conserved in eukaryotes from yeast to mammals.
Accordingly, the expression patterns of Tgif support its role in proliferation. In
the mouse embryo 7g//expression was detected in proliferating cell populations,
including those within the forebrain, branchial arches, lung, limb buds, and tongue
(Bertolino et al. 1996). Tgif expression is also widespread in the developing central
nervous system undergoing dramatic growth and expansion (Bertolino et al. 1996; Shen
98

and Walsh 2005; Jin et al. 2006). Furthermore, Tgif may regulate the generation of
neuronal precursors in the neural tube (Knepper et al. 2006). Altogether, the expression
analyses currently available indicate Tgi/potentially plays a role in the proliferation of
multiple neuronal populations, as well as other tissues. Consistent with this idea, some
Tgif'' mice were growth retarded and mutant embryos were often developmentally
delayed as early as E8 when somite numbers were used to assess developmental
progress.
To determine whether embryonic cells in vivo also showed a reduced proportion
of cells in S phase, in vivo BrdU incorporation experiments were conducted. However,
assays of in vivo proliferation did not demonstrate a significant difference in the
number of proliferating cells in the developing forebrains of wild-type and mutant
embryos at E8. While in vivo BrdU incorporation was useful in mutant mice that
displayed dramatic proliferation defects, such as Pox6 (Warren et al. 1999), it may not
be sufficiently sensitive to detect small proliferation changes. Furthermore, only when
global proliferation is perturbed will a dramatic reduction in proliferation be detected;
isolated effects in specific progenitors are unlikely to be detected. Alternatively, in
vivo proliferation may not be altered by the loss of Tgif at the stage examined. Cell
cycle is tightly regulated in vivo during development, and other studies have failed to
detect in vivo proliferation defects after initially observing such defects in vitro. For
example, in vivo proliferation defects could not be demonstrated in mice lacking the D-
type cyclin-dependent kinases Cdk4 and Cdk6, and in mice lacking the D-cyclins,
cyclin DI, D2 and D3 (Kozar et al. 2004; Malumbres et al. 2004).
TGIF possesses many well characterized properties that link its functions to
many signalling pathways. Tgif can suppress TGF-P responsive transcription (Wotton
et al. 1999a). Similarly, Tgif competed with RA during transcription activation of RXR
99

responsive elements, such as the cellular retinol-binding protein II promoter, through
direct competition for DNA binding elements (Bertolino et al. 1995). More recently,
Tgif was shown to function as a competitive antagonist independent of direct DNA
binding; Tgif directly bound to the ligand binding domain of the RXR family of
receptors and repressed downstream transcription through recruitment of CtBP
complexes (Bartholin et al. 2006). TGIF protein stability can be modulated by the
Ras/MAP kinase pathway (Lo et al. 2001). However, while it was expected that the
loss of Tgif in mutant MEFs may cause hypersensitivity to TGF-P or RA, an increased
growth arrest in these cells was not observed.
Further studies utilizing TGIF mutations identified in HPE patients addressed
the mechanism of TGIF in regulating proliferation. These experiments indicated
mutations located in the homeodomain were not able to rescue the proliferative defect.
These results suggest a novel mechanism by which Tgif may regulate cell cycle genes
or downstream targets through DNA binding or protein-protein interactions through the
homeodomain. Given that only transcriptional repressive activity has been
demonstrated for TGIF, its target genes may be inhibitors of Gi/S progression.
5.3.1. P R O L I F E R A T I O N D U R I N G N E U R A L D E V E L O P M E N T
Proliferation plays an important and decisive role during development of a
multicellular organism; morphogenesis coordinates differentiation, controlled
proliferation and other processes. For example, prior to establishment of the primary
axis, the mouse embryo is radially symmetric. Nodal signalling and the localization of
its antagonists establish the anteroposterior axis through the appropriate regional
proliferation of visceral endoderm cells (Yamamoto et al. 2004). Thus, the
100

proliferation of distal visceral endoderm determines the location of the future anterior
side.
Proliferation defects are implicated in forebrain patterning and HPE etiology
(Xuan et al. 1995; Lukaszewicz et al. 2005). MRI studies of HPE patients
demonstrated a correlation between reduced size of the developing telencephalon and
the severity of HPE (Takahashi et al. 2004). Furthermore, proliferation function is
associated with most HPE genes. Shh is mutated most frequently in HPE patients
(Roessler et al. 1996); reduction in Shh signalling leads to HPE in mice and men. Shh
signalling patterns the dorsoventral axis of the forebrain and spinal cord, through the
coordination of three interdependent mechanisms: (1) region-specific expression of
multiple transcription factors along the axis (Briscoe et al. 2001; Xu et al. 2005); (2)
proliferation of neuronal progenitors (Britto et al. 2002); (3) sorting and partitioning of
similar neuronal progenitors (Wijgerde et al. 2002).
Six3 is a second HPE gene (Wallis et al. 1999). Recent reports showed Six3
promotes proliferation of neuronal precursors by competing for binding to Geminin
(Del Bene et al. 2004). Geminin normally suppresses cell cycling by binding to and
inactivating Cdtl, an important component of the replication complex that mediates
proliferation. Accordingly, over-expression of Geminin, or the loss of Six3, led to
inactivation of Cdtl activity and reduced proliferation, and subsequently the
development of cyclopia, a severe form of HPE, the loss of forebrain, as well as
premature differentiation of neurons.
The third HPE gene is Zic2. While the molecular functions of Zic2 are poorly
understood, the activity of Zicl, a paralogue of Zic2, is associated with the expansion
and differentiation of neuronal progenitors (Aruga et al. 2002). Interesting, Zic3,
another paralogue of Zic2, is involved in laterality determination and has been shown to
101

modulate Nodal signalling. Nodal signalling has been shown to regulate
morphogenesis in part through regulating proliferation (Yamamoto et al. 2004). In fact,
intriguing parallels can be drawn between Zic2 and Tgif. Reduction in the activity of
both genes can sensitize cells to Nodal signalling. Additionally, both transcription
factors have been shown to directly bind to the promoter of D1A dopamine receptor
and regulate its expression (Yang et al. 2000a; Yang et al. 2000b).
Finally, other pathways associated with HPE, such as megalin and the BMP4
pathway were shown to directly regulate proliferation and apoptosis in the developing
neural tube (Ohkubo et al. 2002; Spoelgen et al. 2005). In addition, the mouse mutant
flat-top exhibited a telencephalon regionalization defect caused by a proliferation defect
specific to the forebrain (Hentges et al. 1999). It is interesting to note they also
exhibited kinked neural tube and laterality defects. Unfortunately the function offlat
top is not currently known.
Therefore, based on the proliferative defect observed in rgz/mutants, it is
tempting to speculate that a reduction in Tg-z/activity may reduce the rate of
proliferation and cause HPE in patients. Since Nodal has been shown to promote
proliferation, Tgif may be mediating this function through the Nodal pathway.
Consistent with this hypothesis, Smadl and Smad4 mutants exhibited reduced
proliferation (Sirard et al. 1998). Alternatively, ectopic RA is associated with HPE and
reduced proliferation (Schneider et al. 2001). Interestingly, exencephaly incidence was
higher in embryos born from pregnant mutant mice exposed to teratogenic levels of RA
(Bartholin et al. 2006). Exencephaly is not a forebrain related birth defect, but results
from neural tube closure defects instead, and can result from disruptions to cell cycle
genes (Juriloff and Harris 2000).
102

However, in vitro studies with MEFs indicate that the proliferation defect was
not mediated by the nodal, RA, or MAPK signalling pathways. Whether these
signalling pathways may be modulated by Tgif in specific neuronal populations remains
to be addressed. Regardless, Tgif 'may function downstream of multiple signalling
pathways that regulate and coordinate cell cycle with other morphogenetic events. An
important example is string, a cell cycle regulator in Drosophila that coordinates cell
cycle regulation and morphogenesis through integrating inputs from numerous
signalling pathways (Skaer 1998).
5.3.2. Future directions
To address whether Tgif regulates neurogenesis in the forebrain and whether
proliferation is an important mechanism in achieving this, I propose three approaches.
A chick overexpression study indicated Tgif 'may regulate the identity of dorsal neurons
of the spinal cord (Knepper et al. 2006), suggesting neurons in the Tgif1' mice may not
have properly differentiated. To test this hypothesis, expression of dorsal expressing
genes in Tgif1' forebrains can be assessed to determine whether dorsal progenitors are
properly specified. If this hypothesis is correct, mutant forebrains would exhibit
reduced expression of dorsal genes and possibly a concomitant dorsal shift of ventral
markers.
A second approach is to direct ES cells to differentiate into telencephalic
progenitors. ES cells would have to be generated from wild-type and mutant
blastocysts (Hoodless et al. 2001). ES cells can be cultured to generate motor neurons,
telencephalic neurons, haematopoietic lineages, and many other lineages of interest
(Kennedy et al. 1997; Wichterle et al. 2002; Watanabe et al. 2005). Mutant ES cells
could be used to differentiate into dorsal, ventral and other intermediate telencephalic
103

neuronal progenitor populations in vitro to address whether Tgif 'plays a role during
differentiation. In vitro culturing methods are sometimes more sensitive than in vivo
conditions where defects may be masked by compensatory mechanisms. If mutant ES
cells exhibited abnormal neurogenesis, a variety of questions could be posed. For
example, is the differentiation of a specific neuronal progenitor regulated by Tgif? Do
the TGF-R, Nodal or M A P K signalling pathways modulate the activity of Tgif! What
is the relationship (epistatic, synergistic, parallel, sequential) of other signalling
pathways, such as Shh, BMP, and Wnt, with respect to regulating neurogeneis? Is the
effect achieved through proliferation? Do patient mutations disrupt these functions and
can give insights into HPE etiology? ES cells can form embryoid bodies when cultured
in suspension (Kennedy et al. 1997). I have previously created ES cell lines
overexpressing 7g//and have compared the embryoid bodies generated from wild-type
ES cells and ES cells overexpressing Tgif Embryoid bodies overexpressing Tgif
consistently larger than control embryoid bodies based on their size measurements (data
not shown), support the proliferation function of Tgi/discussed here, and further
suggest Tgif activity may promote neural progenitor cells.
The in vivo proliferation rate of wild-type and mutant telencephalons was
examined in this study, but a difference was not detected. The developing
telencephalon likely comprises a large variety of neuronal progenitor cells that are not
yet well characterized. Unless global proliferation is perturbed by the loss of Tgif, a
reduction in proliferation in specific lineages is unlikely to be detected. In contrast,
retinal neurogenesis is better characterized than in any other region of the CNS (Dyer
and Cepko 2001). Therefore, my final proposal is to further characterize cell cycle
defects in mutant retina. Defects would be expected in the retina since it is a forebrain-
derived neural tissue. Though not all mutant mice developed eye defects, many mice
104

may have subtle defects not revealed by this study. Consistent with this hypothesis,
HPE patients often developed eye defects. Initial experiments should include an
expression study of Tgif'in the developing retina to identify retinal populations most
likely to be disturbed in the mutants. Retinal progenitors are well characterized, as is
the process of their differentiation into postmitotic neurons and glia (Dyer and Cepko
2000). The expectation is mutant retina would have reduced numbers of retinal
progenitors due to defects in Gj/S progression, and later-born neurons may even fail to
be born.
5.4. F INAL COMMENTS
At the time this thesis project started relatively little was known about the
genetic program involved in the patterning, growth and differentiation of the forebrain.
Few molecular markers expressed in regionally restricted patterns were available for
analysis, even though many of the major signalling pathways were known to be
involved. However, the same signalling pathways play earlier roles during
development and prevented detailed analysis of forebrain defects in mutants, and
interpretations of observations were further hampered by overlapping expression and
functional redundancy of related family members.
This thesis project set out to determine whether the loss of Tgif 'in mice would
lead to HPE development and allow the characterization of the underlying molecular
mechanisms. The experimental system used provided the answer that HPE does not
develop in mice lacking one or both alleles of Tgif, unlike in humans. However, a
number of related defects were observed lending credence to Tgifs function in
forebrain development. A significant contribution from this project is the discovery of
Tgifs function during cell cycle, and to provide the novel hypothesis that Tgifs
105

function during cell cycle regulation plays a role in the etiology of HPE. Consistent
with this hypothesis, multiple studies in this field indicate proliferation is integral to
forebrain development. Preliminary evidence shown here indicates reduced
proliferation is a possible mechanism for HPE development, but this mechanism will
require further investigations.
The last five years have witnessed the identification of many patterning genes
and subregionally expressed genes that have provided a better understanding of the
genetic pathways during the patterning process. Sophisticated conditional strategies
have become useful for manipulating spatio-temporal gene expression and allow better
dissection of the underlying developmental mechanisms. This knowledge has
culminated in the ability to induce ES cells to differentiate into neurons that have
therapeutic potential. The task of identifying and detailing the molecular mechanisms
that pattern and generate the entire repertoire of neurons of our brain is still a
formidable task but is now closer to reality. Studies such as this will lead to better
understanding of the molecular pathways, may also give insights into teratogenic
causes during pregnancy, and will eventually reduce the incidence of HPE and related
disorders.
106

REFERENCES
Abelson, J.F., Kwan, K.Y. , O'Roak, B.J., Baek, D.Y., Stillman, A.A., Morgan, T.M., Mathews, C.A., Pauls, D.L., Rasin, M.R., Gunel, M. , Davis, N.R., Ercan-Sencicek, A.G., Guez, D.H., Spertus, J.A., Leckman, J.F., Dure, L.S.t., Kurlan, R., Singer, H.S., Gilbert, D.L., Farhi, A., Louvi, A., Lifton, R.P., Sestan, N. , and State, M.W. 2005. Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science 310(5746): 317-320.
Aguilella, C , Dubourg, C , Attia-Sobol, J., Vigneron, J., Blayau, M . , Pasquier, L., Lazaro, L., Odent, S., and David, V. 2003. Molecular screening of the TGIF gene in holoprosencephaly: identification of two novel mutations. Human Genetics 112(2): 131-134.
Anderson, K.V. 2000. Finding the genes that direct mammalian development: ENU mutagenesis in the mouse. Trends Genet 16(3): 99-102.
Anderson, R.M., Lawrence, A.R., Stottmann, R.W., Bachiller, D., and Klingensmith, J. 2002. Chordin and noggin promote organizing centers of forebrain development in the mouse. Development 129(21): 4975-4987.
Ang, S.L., Conlon, R.A., Jin, O., and Rossant, J. 1994. Positive and negative signals from mesoderm regulate the expression of mouse Otx2 in ectoderm explants. Development 120(10): 2979-2989.
Aruga, J., Tohmonda, T., Homma, S., and Mikoshiba, K. 2002. Zicl promotes the expansion of dorsal neural progenitors in spinal cord by inhibiting neuronal differentiation. Dev Biol 244(2): 329-341.
Ayyar, S., Jiang, J., Collu, A., White-Cooper, FL, and White, R.A. 2003. Drosophila TGIF is essential for developmentally regulated transcription in spermatogenesis. Development 130(13): 2841-2852.
Bachiller, D., Klingensmith, J., Kemp, C , Belo, J.A., Anderson, R.M., May, S.R., McMahon, J.A., McMahon, A.P., Harland, R.M., Rossant, J., and De Robertis, E.M. 2000. The organizer factors Chordin and Noggin are required for mouse forebrain development. Nature 403(6770): 658-661.
Bartholin, L., Powers, S.E., Melhuish, T.A., Lasse, S., Weinstein, M . , and Wotton, D. 2006. TGIF inhibits retinoid signaling. Mol Cell Biol 26(3): 990-1001.
Beddington, R.S. 1994. Induction of a second neural axis by the mouse node. Development 120(3): 613-620.
Beddington, R.S. and Robertson, E.J. 1999. Axis development and early asymmetry in mammals. Cell 96(2): 195-209.
Belo, J.A., Bouwmeester, T., Leyns, L., Kertesz, N. , Gallo, M . , Follettie, M . , and De Robertis, E.M. 1997. Cerberus-like is a secreted factor with neutralizing activity
1 0 7

expressed in the anterior primitive endoderm of the mouse gastrula. Mech Dev 68(1-2): 45-57.
Bendavid, C , Dubourg, C , Gicquel, I., Pasquier, L., Saugier-Veber, P., Durou, M.R., Jaillard, S., Frebourg, T., Haddad, B.R., Henry, C , Odent, S., and David, V. 2005a. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum Genet: 1-8.
Bendavid, C , Haddad, B.H., Griffin, A., Huizing, M. , Dubourg, C., Gicquel, I., Cavalli, L.R., Pasquier, L., Long, B., Ouspenskaia, M . , Odent, S., Lacbawan, F., David, V., and Muenke, M . 2005b. Multicolor FISH and quantitative PCR and detect submicroscopic deletions in holoprosencephaly patients with a normal karyotype. J Med Genet.
Bertolino, E., Reimund, B., Wildt-Perinic, D., and Clerc, R.G. 1995. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem 270(52): 31178-31188.
Bertolino, E., Wildt, S., Richards, G., and Clerc, R.G. 1996. Expression of a novel murine homeobox gene in the developing cerebellar external granular layer during its proliferation. Dev Dyn 205(4): 410-420.
Bier, E. and McGinnis, W. 2003. Model Organisms in the Study of Development and Disease, in Inborn Errors of Development The Molecular Basis of Clinical Disorders of Morphogenesis (ed. C.J. Epstein, R.P. Erickson, and A. Wynshaw-Boris), pp. 25-45. Oxford University Press.
Bisgrove, B.W., Essner, J.J., and Yost, H.J. 2000. Multiple pathways in the midline regulate concordant brain, heart and gut left-right asymmetry. Development 127(16): 3567-3579.
Bisgrove, B.W., Morelli, S.H., and Yost, H.J. 2003. Genetics of human laterality disorders: insights from vertebrate model systems. Annu Rev Genomics Hum Genet 4: 1-32.
Bianco-Arias, P., Sargent, C.A., and Affara, N.A. 2002. The human-specific Yp l 1.2/Xq21.3 homology block encodes a potentially functional testis-specific TGIF-like retroposon. Mamm Genome 13(8): 463-468.
Bonyadi, M . , Rusholme, S.A., Cousins, F.M., Su, H .C , Biron, C.A., Farrall, M . , and Akhurst, R.J. 1997. Mapping of a major genetic modifier of embryonic lethality in TGF beta 1 knockout mice. Nat Genet 15(2): 207-211.
Braun, M.M. , Etheridge, A., Bernard, A., Robertson, CP. , and Roelink, H. 2003. Wnt signaling is required at distinct stages of development for the induction of the posterior forebrain. Development 130(23): 5579-5587.
Brennan, J., Lu, C C , Norris, D.P., Rodriguez, T.A., Beddington, R.S., and Robertson, E.J. 2001. Nodal signalling in the epiblast patterns the early mouse embryo. Nature 411(6840): 965-969.
108

Brennan, J., Norris, D.P., and Robertson, E.J. 2002. Nodal activity in the node governs left-right asymmetry. Genes Dev 16(18): 2339-2344.
Briscoe, J., Chen, Y., Jessell, T.M., and Struhl, G. 2001. A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol Cell 7(6): 1279-1291.
Britto, J., Tannahill, D., and Keynes, R. 2002. A critical role for sonic hedgehog signaling in the early expansion of the developing brain. Nat Neurosci 5(2): 103-110.
Brown, S.A., Warburton, D., Brown, L.Y., Yu, C.Y., Roeder, E.R., Stengel-Rutkowski, S., Hennekam, R.C., and Muenke, M. 1998. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet 20(2): 180-183.
Burglin, T.R. 1997. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res 25(21): 4173-4180.
Buske, C , Feuring-Buske, M . , Antonchuk, J., Rosten, P., Hogge, D.E., Eaves, C.J., and Humphries, R.K. 2001. Overexpression of HOXA10 perturbs human lymphomyelopoiesis in vitro and in vivo. Blood 97(8): 2286-2292.
Camus, A., Davidson, B.P., Billiards, S., Khoo, P., Rivera-Perez, J.A., Wakamiya, M . , Behringer, R.R., and Tam, P.P. 2000. The morphogenetic role of midline mesendoderm and ectoderm in the development of the forebrain and the midbrain of the mouse embryo. Development 127(9): 1799-1813.
Capdevila, J., Vogan, K.J., Tabin, C.J., and Izpisua Belmonte, J.C. 2000. Mechanisms of left-right determination in vertebrates. Cell 101(1): 9-21.
Chambon, P. 1996. A decade of molecular biology of retinoic acid receptors. Faseb J 10(9): 940-954.
Chang, CP. , Jacobs, Y., Nakamura, T., Jenkins, N.A., Copeland, N.G., and Geary, M.L. 1997. Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol Cell Biol 17(10): 5679-5687.
Chen, CP. , Chern, S.R., Du, S.H., and Wang, W. 2002. Molecular diagnosis of a novel heterozygous 268C~>T (R90C) mutation in TGIF gene in a fetus with holoprosencephaly and premaxillary agenesis. Prenat Diagn 22(1): 5-7.
Chen, M . , Kuo, S.J., Liu, C.S., Chen, W.L., Ko, T.M., Chen, T.H., Chang, S.P., Huang, C.H., Chang, Y.Y. , and Wang, B.T. 2006. A novel heterozygous missense mutation 377T > C (V126A) of TGIF gene in a family segregated with holoprosencephaly and moyamoya disease. Prenat Diagn 26(3): 226-230.
Chia, R., Achilli, F., Festing, M.F., and Fisher, E.M. 2005. The origins and uses of mouse outbred stocks. Nat Genet 37(11): 1181-1186.
109

Chiang, C , Litingtung, Y., Lee, E., Young, K.E., Corden, J.L., Westphal, FL, and Beachy, P.A. 1996. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383(6599): 407-413.
Chu, G.C., Dunn, N.R., Anderson, D C , Oxburgh, L., and Robertson, E.J. 2004. Differential requirements for Smad4 in TGFbeta-dependent patterning of the early mouse embryo. Development 131(15): 3501-3512.
Ciemerych, M.A. and Sicinski, P. 2005. Cell cycle in mouse development. Oncogene 24(17): 2877-2898.
Cohen, M . M . 2003. SHH and Holoprosencephaly. in Inborn Errors of Development The Molecular Basis of Clinical Disorders of Morphogenesis (ed. C.J. Epstein, R.P. Erickson, and A. Wynshaw-Boris). Oxford University Press.
Cohen, M.M. , Jr. and Shiota, K. 2002. Teratogenesis of holoprosencephaly. Am J Med Genet 109(1): 1-15.
Constam, D.B. and Robertson, E.J. 2000a. SPC4/PACE4 regulates a TGFbeta signaling network during axis formation. Genes Dev 14(9): 1146-1155.
-. 2000b. Tissue-specific requirements for the proprotein convertase furin/SPCl during embryonic turning and heart looping. Development 127(2): 245-254.
Cordes, S.P. 2005. N-ethyl-N-nitrosourea mutagenesis: boarding the mouse mutant express. Microbiol Mol Biol Rev 69(3): 426-439.
Cowles, C.R., Hirschhorn, J.N., Altshuler, D., and Lander, E.S. 2002. Detection of. regulatory variation in mouse genes. Nat Genet 32(3): 432-437.
D'Abaco, G.M. and Olson, M.F. 2000. Mouse embryo fibroblasts: a genetic model system for studying Rho- and Ras-dependent cell cycle progression. Methods Enzymol 325: 415-425.
Danos, M . C and Yost, H.J. 1996. Role of notochord in specification of cardiac left-right orientation in zebrafish and Xenopus. Dev Biol 177(1): 96-103.
de la Cruz, J.M., Bamford, R.N., Burdine, R.D., Roessler, E., Barkovich, A.J., Donnai, D., Schier, A.F., and Muenke, M . 2002. A loss-of-function mutation in the CFC domain of TDGF1 is associated with human forebrain defects. Hum Genet 110(5): 422-428.
Del Bene, F., Tessmar-Raible, K., and Wittbrodt, J. 2004. Direct interaction of geminin and Six3 in eye development. Nature 427(6976): 745-749.
Ding, J., Yang, L., Yan, Y.T., Chen, A., Desai, N. , Wynshaw-Boris, A., and Shen, M . M . 1998. Cripto is required for correct orientation of the anterior-posterior axis in the mouse embryo. Nature 395(6703): 702-707.
Dou, C.L., L i , S., and Lai, E. 1999. Dual role of brain factor-1 in regulating growth and patterning of the cerebral hemispheres. Cereb Cortex 9(6): 543-550.
110

Drumm, M.L., Konstan, M.W., Schluchter, M.D., Handler, A., Pace, R., Zou, F., Zariwala, M . , Fargo, D., Xu, A., Dunn, J.M., Darrah, R.J., Dorfman, R., Sandford, A.J., Corey, M. , Zielenski, J., Durie, P., Goddard, K., Yankaskas, J.R., Wright, F.A., and Knowles, M.R. 2005. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med 353(14): 1443-1453.
Dubourg, C , Lazaro, L., Pasquier, L., Bendavid, C , Blayau, M . , Le Duff, F., Durou, M.R., Odent, S., and David, V. 2004. Molecular screening of SHH, Z1C2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum Mutat 24(1): 43-51.
Dunn, N.R., Vincent, S.D., Oxburgh, L., Robertson, E.J., and Bikoff, E.K. 2004. Combinatorial activities of Smad2 and Smad3 regulate mesoderm formation and patterning in the mouse embryo. Development 131(8): 1717-1728.
Dyer, M.A. and Cepko, C L . 2000. p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development 127(16): 3593-3605.
-. 2001. Regulating proliferation during retinal development. Nat Rev Neurosci 2(5): 333-342.
Edison, R. and Muenke, M . 2003. The interplay of genetic and environmental factors in craniofacial morphogenesis: holoprosencephaly and the role of cholesterol. Congenit Anom (Kyoto) 43(1): 1-21.
Edwards, M . C , Liegeois, N. , Horecka, J., DePinho, R.A., Sprague, G.F., Jr., Tyers, M . , and Elledge, S.J. 1997. Human CPR (cell cycle progression restoration) genes impart a Far- phenotype on yeast cells. Genetics 147(3): 1063-1076.
Episkopou, V., Arkell, R., Timmons, P.M., Walsh, J.J., Andrew, R.L., and Swan, D. 2001. Induction of the mammalian node requires Arkadia function in the extraembryonic lineages. Nature 410(6830): 825-830.
Epstein, C.J. 2003. Human Malformations and Their Genetic Basis, in Inborn Errors of Development The Molecular Basis of Clinical Disorders of Morphogenesis (ed. C.J. Epstein, R.P. Erickson, and A. Wynshaw-Boris), pp. 3-10. Oxford University Press.
Epstein, C.J., Erickson, R.P., and Wynshaw-Boris, A. 2003. Inborn Errors of Development The Molecular Basis of Clinical Disorders of Morphogenesis.
Filosa, S., Rivera-Perez, J.A., Gomez, A.P., Gansmuller, A., Sasaki, H., Behringer, R.R., and Ang, S.L. 1997. Goosecoid and HNF-3beta genetically interact to regulate neural tube patterning during mouse embryogenesis. Development 124(14): 2843-2854.
Furuta, Y., Piston, D.W., and Hogan, B.L. 1997. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124(11): 2203-2212.
Gilbert, S.F. 2003. Developmental Biology. Sinauer Associates.
I l l

Glaser, S., Anastassiadis, K., and Stewart, A.F. 2005. Current issues in mouse genome engineering. Nat Genet 37(11): 1187-1193.
Goodrich, L.V., Milenkovic, L., Higgins, K . M . , and Scott, M.P. 1997. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277(5329): 1109-1113.
Goyette, P., Feng Chen, C , Wang, W., Seguin, F., and Lohnes, D. 2000. Characterization of retinoic acid receptor-deficient keratinocytes. J Biol Chem 275(22): 16497-16505.
Gripp, K.W., Wotton, D., Edwards, M . C , Roessler, E., Ades, L., Meinecke, P., Richieri-Costa, A., Zackai, E.H., Massague, J., Muenke, M. , and Elledge, S.J. 2000. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet 25(2): 205-208.
Grove, E.A., Tole, S., Limon, J., Yip, L., and Ragsdale, C W . 1998. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in GH3-deficient mice. Development 125(12): 2315-2325.
Gunhaga, L., Jessell, T.M., and Edlund, T. 2000. Sonic hedgehog signaling at gastrula stages specifies ventral telencephalic cells in the chick embryo. Development 127(15): 3283-3293.
Gunhaga, L., Marklund, M . , Sjodal, M . , Hsieh, J.C, Jessell, T.M., and Edlund, T. 2003. Specification of dorsal telencephalic character by sequential Wnt and FGF signaling. Nat Neurosci 6(7): 701-707.
Hallonet, M . , Kaestner, K.H. , Martin-Parras, L., Sasaki, H., Betz, U.A., and Ang, S.L. 2002. Maintenance of the specification of the anterior definitive endoderm and forebrain depends on the axial mesendoderm: a study using HNF3beta/Foxa2 conditional mutants. Dev Biol 243(1): 20-33.
Hamada, H., Meno, C , Watanabe, D., and Saijoh, Y. 2002. Establishment of vertebrate left-right asymmetry. Nat Rev Genet 3(2): 103-113.
Hasty, P., Bradley, A. 1993. Gene targeting vectors for mammalian cells, in A Practical Approach (ed. A. Joyner), pp. 33-61. Oxford University Press, Oxford.
Hebert, J.M. 2005. Unraveling the molecular pathways that regulate early telencephalon development. Curr Top Dev Biol 69: 17-37.
Hebert, J.M., Mishina, Y., and McConnell, S.K. 2002. BMP signaling is required locally to pattern the dorsal telencephalic midline. Neuron 35(6): 1029-1041.
Hentges, K., Thompson, K., and Peterson, A. 1999. The flat-top gene is required for the expansion and regionalization of the telencephalic primordium. Development 126(8): 1601-1609.
112

Herrera, R.E., Sah, V.P., Williams, B.O., Makela, T.P., Weinberg, R.A., and Jacks, T. 1996. Altered cell cycle kinetics, gene expression, and GI restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol 16(5): 2402-2407.
Heyer, J., Escalante-Alcalde, D., Lia, M . , Boettinger, E., Edelmann, W., Stewart, C.L., and Kucherlapati, R. 1999. Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc Natl Acad Sci USA 96(22): 12595-12600.
Hide, T., Hatakeyama, J., Kimura-Yoshida, C , Tian, E., Takeda, N. , Ushio, Y. , Shiroishi, T., Aizawa, S., and Matsuo, I. 2002. Genetic modifiers of otocephalic phenotypes in Otx2 heterozygous mutant mice. Development 129(18): 4347-4357.
Hinds, P.W. 2006. A confederacy of kinases: Cdk2 and Cdk4 conspire to control embryonic cell proliferation. Mol Cell 22(4): 432-433.
Hoodless, P.A., Pye, M . , Chazaud, C , Labbe, E., Attisano, L., Rossant, J., and Wrana, J.L. 2001. FoxHl (Fast) functions to specify the anterior primitive streak in the mouse. Genes Dev 15(10): 1257-1271.
Horak, C.E., Luscombe, N.M., Qian, J., Bertone, P., Piccirrillo, S., Gerstein, M . , and Snyder, M . 2002. Complex transcriptional circuitry at the Gl/S transition in Saccharomyces cerevisiae. Genes Dev 16(23): 3017-3033.
Hornstein, E. and Tabin, C.J. 2005. Developmental biology: asymmetrical threat averted. Nature 435(7039): 155-156.
Houston, D.W. and Wylie, C. 2005. Maternal Xenopus Zic2 negatively regulates Nodal-related gene expression during anteroposterior patterning. Development 132(21): 4845-4855.
Hunter, T. 2000. Signaling-2000 and beyond. Cell 100(1): 113-127.
Imoto, I., Pimkhaokham, A., Watanabe, T., Saito-Ohara, F., Soeda, E., and Inazawa, J. 2000. Amplification and overexpression of TGIF2, a novel homeobox gene of the TALE superclass, in ovarian cancer cell lines. Biochem Biophys Res Commun 276(1): 264-270.
Iratni, R., Yan, Y.T., Chen, C , Ding, J., Zhang, Y., Price, S.M., Reinberg, D., and Shen, M . M . 2002. Inhibition of excess nodal signaling during mouse gastrulation by the transcriptional corepressor DRAP1. Science 298(5600): 1996-1999.
Irons, M . 2003. DHCR7 and the Smith-Lemli-Opitz (RSH) Syndrome and Cyclopamine Teratogenesis. in Inborn Errors of Development The Molecular Basis of Clinical Disorders of Morphogenesis (ed. C.J. Epstein, R.P. Erickson, and A. Wynshaw-Boris), pp. 229-239. Oxford University Press.
Jin, J.Z., Gu, S., McKinney, P., and Ding, J. 2006. Expression and functional analysis of Tgif during mouse midline development. Dev Dyn 235(2): 547-553.
113

Jin, L., Zhou, Y., Kuang, C , Lin, L., and Chen, Y. 2005. Expression pattern of TG-interacting factor 2 during mouse development. Gene Expr Patterns 5(4): 457-462.
Juriloff, D.M. and Harris, M.J. 2000. Mouse models for neural tube closure defects. Hum Mol Genet 9(6): 993-1000.
Juriloff, D.M., Sulik, K.K., Roderick, T.H., and Hogan, B.K. 1985. Genetic and developmental studies of a new mouse mutation that produces otocephaly. J Craniofac Genet Dev Biol 5(2): 121-145.
Kawakami, Y., Raya, A., Raya, R.M., Rodriguez-Esteban, C , and Belmonte, J.C. 2005. Retinoic acid signalling links left-right asymmetric patterning and bilaterally symmetric somitogenesis in the zebrafish embryo. Nature 435(7039): 165-171.
Kennedy, M . , Firpo, M . , Choi, K., Wall, C , Robertson, S., Kabrun, N. , and Keller, G. ( 1997. A common precursor for primitive erythropoiesis and definitive haematopoiesis. Nature 386(6624): 488-493.
King, T. and Brown, N.A. 1999. Developmental biology. Antagonists on the left flank. Nature 401(6750): 222-223.
Knepper, J.L., James, A.C., and Ming, J.E. 2006. TGIF, a gene associated with human brain defects, regulates neuronal development. Dev Dyn.
Knoepfler, P.S., Calvo, K.R., Chen, H., Antonarakis, S.E., and Kamps, M.P. 1997. Meisl and pKnoxl bind DNA cooperatively with Pbxl utilizing an interaction surface disrupted in oncoprotein E2a-Pbxl. Proc Natl Acad Sci U SA 94(26): 14553-14558.
Kohtz, J.D., Baker, D.P., Corte, G., and Fishell, G. 1998. Regionalization within the mammalian telencephalon is mediated by changes in responsiveness to Sonic Hedgehog. Development 125(24): 5079-5089.
Kozar, K., Ciemerych, M.A., Rebel, V.I., Shigematsu, H., Zagozdzon, A., Sicinska, E., Geng, Y. , Yu, Q., Bhattacharya, S., Bronson, R.T., Akashi, K., and Sicinski, P. 2004. Mouse development and cell proliferation in the absence of D-cyclins. Cell 118(4): 477-491.
Krek, W. and DeCaprio, J.A. 1995. Cell synchronization. Methods Enzymol 254: 114-124.
Lagutin, O.V., Zhu, C C , Kobayashi, D., Topczewski, J., Shimamura, K., Puelles, L., Russell, H.R., McKinnon, P.J., Solnica-Krezel, L., and Oliver, G. 2003. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev 17(3): 368-379.
Lai, Y.L. , L i , H., Chiang, H.S., and Hsieh-Li, H.M. 2002. Expression of a novel TGIF subclass homeobox gene, Texl, in the spermatids of mouse testis during spermatogenesis. Mech Dev 113(2): 185-187.
114

Larnrner, E.J., Chen, D.T., Hoar, R.M., Agnish, N.D., Benke, P.J., Braun, J.T., Curry, C.J., Fernhoff, P.M., Grix, A.W., Jr., Lott, I.T., and et al. 1985. Retinoic acid embryopathy. N EnglJ Med 313(14): 837-841.
LeCouter, J.E., Kablar, B., Whyte, P.F., Ying, C , and Rudnicki, M.A. 1998. Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related pi30 gene. Development 125(23): 4669-4679.
Lee, S.M., Tole, S., Grove, E., and McMahon, A.P. 2000. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127(3): 457-467.
Levin, M . 2005. Left-right asymmetry in embryonic development: a comprehensive review. Mech Dev 122(1): 3-25.
L i , X.J., Du, Z.W., Zarnowska, E.D., Pankratz, M. , Hansen, L.O., Pearce, R.A., and Zhang, S.C. 2005. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol 23(2): 215-221.
Liu, P., Wakamiya, M. , Shea, M.J., Albrecht, U., Behringer, R.R., and Bradley, A. 1999. Requirement for Wnt3 in vertebrate axis formation. Nat Genet 22(4): 361-365.
Lo, R.S., Wotton, D., and Massague, J. 2001. Epidermal growth factor signaling via Ras controls the Smad transcriptional co-repressor TGIF. Embo J 20(1-2): 128-136.
Lodish, H., Berk, A., Zipursky, S.L., Matsudaira, P., Baltimore, D., and Darnell, J.E. 2000. Molecular Cell Biology. W. H. Freeman & Co, New York.
Lohnes, D., Mark, M . , Mendelsohn, C , Dolle, P., Dierich, A., Gorry, P., Gansmuller, A., and Chambon, P. 1994. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 120(10): 2723-2748.
Lukaszewicz, A., Savatier, P., Cortay, V., Giroud, P., Huissoiid, C , Berland, M . , Kennedy, H., and Dehay, C. 2005. GI phase regulation, area-specific cell cycle control, and cytoarchitectonics in the primate cortex. Neuron 47(3): 353-364.
Lupo, G., Harris, W.A., and Lewis, K.E. 2006. Mechanisms of ventral patterning in the vertebrate nervous system. Nat Rev Neurosci 7(2): 103-114.
Malumbres, M . , Ortega, S., and Barbacid, M . 2000. Genetic analysis of mammalian cyclin-dependent kinases and their inhibitors. Biol Chem 381(9-10): 827-838.
Malumbres, M . , Sotillo, R., Santamaria, D., Galan, J., Cerezo, A., Ortega, S., Dubus, P., and Barbacid, M . 2004. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118(4): 493-504.
Mann, R.S. and Affolter, M . 1998. Hox proteins meet more partners. Curr Opin Genet Dev 8(4): 423-429.
115

Mark, M . , Ghyselinck, N.B., and Chambon, P. 2006. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol 46: 451-480.
Martinez Barbera, J.P., Clements, M . , Thomas, P., Rodriguez, T., Meloy, D., Kioussis, D., and Beddington, R.S. 2000. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 127(11): 2433-2445.
Massague, J., Blain, S.W., and Lo, R.S. 2000. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103(2): 295-309.
Massague, J., Seoane, J., and Wotton, D. 2005. Smad transcription factors. Genes Dev 19(23): 2783-2810.
Mathieu, J., Barth, A., Rosa, F.M., Wilson, S.W., and Peyrieras, N . 2002. Distinct and cooperative roles for Nodal and Hedgehog signals during hypothalamic development. Development 129(13): 3055-3065.
Matsunaga, E. and Shiota, K. 1977. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology 16(3): 261-272.
McGrath, J., Somlo, S., Makova, S., Tian, X., and Brueckner, M . 2003. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 114(1): 61-73.
McManus, C. 2002. Right Hand, Left Hand The Origins of Asymmetry in Brains, Bodies, Atoms and Cultures. Weidenfeld & Nicolson, London.
Melhuish, T.A., Gallo, C M . , and Wotton, D. 2001. TGIF2 interacts with histone deacetylase 1 and represses transcription. J Biol Chem 276(34): 32109-32114.
Melhuish, T.A. and Wotton, D. 2000. The interaction of the carboxyl terminus-binding protein with the Smad corepressor TGIF is disrupted by a holoprosencephaly mutation in TGIF. JBiol Chem 275(50): 39762-39766.
Meno, C , Ito, Y., Saijoh, Y., Matsuda, Y., Tashiro, K., Kuhara, S., and Hamada, H. 1997. Two closely-related left-right asymmetrically expressed genes, lefty-1 and lefty -2: their distinct expression domains, chromosomal linkage and direct neuralizing activity in Xenopus embryos. Genes Cells 2(8): 513-524.
Meno, C , Shimono, A., Saijoh, Y., Yashiro, K., Mochida, K., Ohishi, S., Noji, S., Kondoh, H., and Hamada, H. 1998. lefty-1 is required for left-right determination as a regulator of lefty-2 and nodal. Ce//94(3): 287-297.
Meyers, E.N. and Martin, G.R. 1999. Differences in left-right axis pathways in mouse and chick: functions of FGF8 and SHH. Science 285(5426): 403-406.
Ming, J.E., Kaupas, M.E., Roessler, E., Brunner, H.G., Golabi, M . , Tekin, M . , Stratton, R.F., Sujansky, E., Bale, S.J., and Muenke, M . 2002. Mutations in PATCHED-1, the
116

receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet 110(4): 297-301.
Ming, J.E. and Muenke, M . 2002. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Hum Genet 71(5): 1017-1032.
Muenke, M . and Beachy, P.A. 2000. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev 10(3)': 262-269.
Nadeau, J.H. 2001. Modifier genes in mice and humans. Nat Rev Genet 2(3): 165-174.
-. 2003. Modifier genes and protective alleles in humans and mice. Curr Opin Genet Dev 13(3): 290-295.
Nagai, T., Aruga, J., Minowa, O., Sugimoto, T., Ohno, Y. , Noda, T., and Mikoshiba, K. 2000. Zic2 regulates the kinetics of neurulation. Proc Natl Acad Sci USA 97(4): 1618-1623.
Nagy, A., Gertsenstein, M . , Vintersten, K., and Behringer, R.R. 2003. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
Nanni, L., Ming, J.E., Bocian, M . , Steinhaus, K., Bianchi, D.W., Die-Smulders, C , Giannotti, A., Imaizumi, K., Jones, K.L. , Campo, M.D., Martin, R.A., Meinecke, P., Pierpont, M.E., Robin, N.H., Young, I.D., Roessler, E., and Muenke, M . 1999. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet 8(13): 2479-2488.
Neumann, P.E., Frankel, W.N., Letts, V.A., Coffin, J.M., Copp, A.J., and Bernfield, M . 1994. Multifactorial inheritance of neural tube defects: localization of the major gene and recognition of modifiers in ct mutant mice. Nat Genet 6(4): 357-362.
Nishioka, N. , Nagano, S., Nakayama, R., Kiyonari, H., Ijiri, T., Taniguchi, K., Shawlot, W., Hayashizaki, Y. , Westphal, H., Behringer, R.R., Matsuda, Y., Sakoda, S., Kondoh, H., and Sasaki, H. 2005. Ssdpl regulates head morphogenesis of mouse embryos by activating the Liml-Ldbl complex. Development 132(11): 2535-2546.
Nomura, M . and L i , E. 1998. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393(6687): 786-790.
Nonaka, S., Shiratori, H., Saijoh, Y., and Hamada, H. 2002. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 418(6893): 96-99.
Nonaka, S., Tanaka, Y., Okada, Y., Takeda, S., Harada, A., Kanai, Y. , Kido, M . , and Hirokawa, N. 1998. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95(6): 829-837.
117

Norris, D.P., Brennan, J., Bikoff, E.K., and Robertson, E.J. 2002. The Foxhl-dependent autoregulatory enhancer controls the level of Nodal signals in the mouse embryo. Development 129(14): 3455-3468.
Nurse, P. 2000. A long twentieth century of the cell cycle and beyond. Cell 100(1): 71-78.
Ohkubo, Y., Chiang, C , and Rubenstein, J.L. 2002. Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience 111(1): 1-17.
Okada, Y., Takeda, S., Tanaka, Y., Belmonte, J.C., and Hirokawa, N. 2005. Mechanism of nodal flow: a conserved symmetry breaking event in left-right axis determination. Cell 121(4): 633-644.
Overhauser, J., Mitchell, H.F., Zackai, E.H., Tick, D.B., Rojas, K., and Muenke, M . 1995. Physical mapping of the holoprosencephaly critical region in 18pl 1.3. Am J Hum Genet 57(5): 1080-1085.
Papaioannou, V., Johnson, R. 1993. Production of chimeras and genetically defined offspring from targeted ES cells, in A Practical Approach (ed. A. Joyner). Oxford University Press, Oxford.
Perea-Gomez, A., Vella, F.D., Shawlot, W., Oulad-Abdelghani, M . , Chazaud, C , Meno, C , Pfister, V., Chen, L., Robertson, E., Hamada, H., Behringer, R.R., and Ang, S.L. 2002. Nodal antagonists in the anterior visceral endoderm prevent the formation of multiple primitive streaks. Dev Cell 3(5): 745-756.
Pessah, M . , Prunier, C , Marais, J., Ferrand, N. , Mazars, A., Lallemand, F., Gauthier, J.M., and Atfi, A. 2001. c-Jun interacts with the corepressor TG-interacting factor (TGIF) to suppress Smad2 transcriptional activity. Proc Natl Acad Sci USA 98(11): 6198-6203.
Petryk, A., Shimmi, O., Jia, X. , Carlson, A.E., Tervonen, L., Jarcho, M.P., O'Connor M , B., and Gopalakrishnan, R. 2005. Twisted gastrulation and chordin inhibit differentiation and mineralization in MC3T3-E1 osteoblast-like cells. Bone 36(4): 617-626.
Piccolo, S., Agius, E., Leyns, L., Bhattacharyya, S., Grunz, H., Bouwmeester, T., and De Robertis, E.M. 1999. The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature 397(6721): 707-710.
Placzek, M . and Briscoe, J. 2005. The floor plate: multiple cells, multiple signals. Nat Rev Neurosci 6(3): 230-240.
Purandare, S.M., Ware, S.M., Kwan, K .M. , Gebbia, M . , Bassi, M.T., Deng, J.M., Vogel, H., Behringer, R.R., Belmont, J.W., and Casey, B. 2002. A complex syndrome of left-right axis, central nervous system and axial skeleton defects in Zic3 mutant mice. Development 129: 2293-2302.
118

Rallu, M. , Machold, R., Gaiano, N. , Corbin, J.G., McMahon, A.P., and Fishell, G. 2002. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development 129(21): 4963-4974.
Raya, A. and Belmonte, J.C. 2006. Left-right asymmetry in the vertebrate embryo: from early information to higher-level integration. Nat Rev Genet 7(4): 283-293.
Raya, A., Kawakami, Y., Rodriguez-Esteban, C , Ibanes, M. , Rasskin-Gutman, D., Rodriguez-Leon, J., Buscher, D., Feijo, J.A., and Izpisua Belmonte, J.C. 2004. Notch activity acts as a sensor for extracellular calcium during vertebrate left-right determination. Nature 427(6970): 121-128.
Redlinger-Grosse, K., Bernhardt, B.A., Berg, K., Muenke, M . , and Biesecker, B.B. 2002. The decision to continue: the experiences and needs of parents who receive a prenatal diagnosis of holoprosencephaly. Am J Med Genet 112(4): 369-378.
Rhinn, M . , Dierich, A., Shawlot, W., Behringer, R.R., Le Meur, M . , and Ang, S.L. 1998. Sequential roles for Otx2 in visceral endoderm and neuroectoderm for forebrain and midbrain induction and specification. Development 125(5): 845-856.
Ribes, V., Wang, Z., Dolle, P., and Niederreither, K. 2006. Retinaldehyde dehydrogenase 2 (RALDH2)-mediated retinoic acid synthesis regulates early mouse embryonic forebrain development by controlling FGF and sonic hedgehog signaling. Development 133(2): 351-361.
Robertson, E.J., Norris, D.P., Brennan, J., and Bikoff, E.K. 2003. Control of early anterior-posterior patterning in the mouse embryo by TGF-beta signalling. Philos Trans R Soc LondB Biol Sci 358(1436): 1351-1357; discussion 1357.
Robertson, S.M., Kennedy, M. , Shannon, J.M., and Keller, G. 2000. A transitional stage in the commitment of mesoderm to hematopoiesis requiring the transcription factor SCL/tal-1. Development 127(11): 2447-2459.
Roessler, E., Belloni, E., Gaudenz, K., Jay, P., Berta, P., Scherer, S.W., Tsui, L.C., and Muenke, M . 1996. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet 14(3): 357-360.
Roessler, E., Belloni, E., Gaudenz, K., Vargas, F., Scherer, S.W., Tsui, L.C., and Muenke, M . 1997. Mutations in the C-terminal domain of Sonic Hedgehog cause holoprosencephaly. Hum Mol Genet 6(11): 1847-1853.
Roessler, E., Du, Y.Z., Mullor, J.L., Casas, E., Allen, W.P., Gillessen-Kaesbach, G., Roeder, E.R., Ming, J.E., Ruiz i Altaba, A., and Muenke, M . 2003. Loss-of-function mutations in the human GL12 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci USA 100(23): 13424-13429.
Roessler, E. and Muenke, M . 2001. Midline and laterality defects: left and right meet in the middle. Bioessays 23(10): 888-900.
119

Rohr, K.B., Barth, K.A:, Varga, Z.M., ana Wilson, S.W. 2001. The nodal pathway acts upstream of hedgehog signaling to specify ventral telencephalic identity. Neuron 29(2): 341-351.
Ryan, A.K., Blumberg, B., Rodriguez-Esteban, C , Yonei-Tamura, S., Tamura, K., Tsukui, T., de la Pena, J., Sabbagh, W., Greenwald, J., Choe, S., Norris, D.P., Robertson, E.J., Evans, R.M., Rosenfeld, M.G., and Izpisua Belmonte, J.C. 1998. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature 394(6693): 545-551.
Ryan, A.K., Tejada, M.L., May, D.L., Dubaova, M. , and Deeley, R.G. 1995. Isolation and characterization of the chicken homeodomain protein AKR. Nucleic Acids Res 23(16): 3252-3259.
Sagerstrom, C.G. 2004. PbX marks the spot. Dev Cell 6(6): 737-738.
Schneider, R.A., Hu, D., Rubenstein, J.L., Maden, M . , and Helms, J.A. 2001. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development 128(14): 2755-2767.
Shawlot, W., Min Deng, J., Wakamiya, M . , and Behringer, R.R. 2000. The cerberus-related gene, Cerrl, is not essential for mouse head formation. Genesis 26(4): 253-258.
Shawlot, W., Wakamiya, M. , Kwan, K . M . , Kania, A., Jessell, T.M., and Behringer, R.R. 1999. Liml is required in both primitive streak-derived tissues and visceral endoderm for head formation in the mouse. Development 126(22): 4925-4932.
Shen, J. and Walsh, C A . 2005. Targeted disruption of Tgif, the mouse ortholog of a human holoprosencephaly gene, does not result in holoprosencephaly in mice. Mol Cell Biol 25(9): 3639-3647.
Sherr, C J . and Roberts, J.M. 1995. Inhibitors of mammalian GI cyclin-dependent kinases. Genes Dev 9(10): 1149-1163.
Shimamura, K. and Rubenstein, J.L. 1997. Inductive interactions direct early regionalization of the mouse forebrain. Development 124(14): 2709-2718.
Sirard, C , de la Pompa, J.L., Elia, A., Itie, A., Mirtsos, C , Cheung, A., Hahn, S., Wakeham, A., Schwartz, L., Kern, S.E., Rossant, J., and Mak, T.W. 1998. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev 12(1): 107-119.
Sirard, C , Kim, S., Mirtsos, C , Tadich, P., Hoodless, P.A., Itie, A., Maxson, R., Wrana, J.L., and Mak, T.W. 2000. Targeted disruption in murine cells reveals variable requirement for Smad4 in transforming growth factor beta-related signaling. J Biol Chem 275(3): 2063-2070.
Skaer, H. 1998. Who pulls the string to pattern cell division in Drosophila? Trends Genet 14(9): 337-339.
120

Song, J., Oh, S.P., Schrewe, H., Nomura, M . , Lei, H., Okano, M. , Gridley, T., and L i , E. 1999. The type II activin receptors are essential for egg cylinder growth, gastrulation, and rostral head development in mice. Dev Biol 213(1): 157-169.
Sotillo, R., Dubus, P., Martin, J., de la Cueva, E., Ortega, S., Malumbres, M . , and Barbacid, M . 2001. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. Embo J70(23): 6637-6647.
Spoelgen, R., Hammes, A., Anzenberger, U., Zechner, D., Andersen, O.M., Jerchow, B., and Willnow, T.E. 2005. LRP2/megalin is required for patterning of the ventral telencephalon. Development 132(2): 405-414.
Storm, E.E., Garel, S., Borello, U., Hebert; J.M., Martinez, S., McConnell, S.K., Martin, G.R., and Rubenstein, J.L. 2006. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133(9): 1831-1844.
Storm, E.E., Rubenstein, J.L., and Martin, G.R. 2003. Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci U S A 100(4): 1757-1762.
Sulik, K.K. , Dehart, D.B., Rogers, J.M., and Chernoff, N . 1995. Teratogenicity of low doses of all-trans retinoic acid in presomite mouse embryos. Teratology 51(6): 398-403.
Sun, T., Patoine, C , Abu-Khalil, A., Visvader, J., Sum, E., Cherry, T.J., Orkin, S.H., Geschwind, D.H., and Walsh, C A . 2005. Early asymmetry of gene transcription in embryonic human left and right cerebral cortex. Science 308(5729): 1794-1798.
Sun, T. and Walsh, C A . 2006. Molecular approaches to brain asymmetry and handedness. Nat Rev Neurosci 7(8): 655-662.
Tabin, C.J. and Vogan, K.J. 2003. A two-cilia model for vertebrate left-right axis specification. Genes Dev 17(1): 1-6.
Takahashi, T.S., Kinsman, S., Makris, N. , Grant, E., Haselgrove, C , Mclnerney, S., Kennedy, D.N., Takahashi, T.A., Fredrickson, K., Mori, S., and Caviness, V.S. 2004. Holoprosencephaly—topologic variations in a liveborn series: a general model based upon MRI analysis. JNeurocytol 33(1): 23-35.
Takaoka, K., Yamamoto, M . , Shiratori, H., Meno, C , Rossant, J., Saijoh, Y. , and Hamada, H. 2006. The mouse embryo autonomously acquires anterior-posterior polarity at implantation. Dev Cell 10(4): 451-459.
Tam, P.P. and Steiner, K.A. 1999. Anterior patterning by synergistic activity of the early gastrula organizer and the anterior germ layer tissues of the mouse embryo. Development 126(22): 5171-5179.
Tanaka, Y., Okada, Y., and Hirokawa, N . 2005. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 435(7039): 172-177.
121

Tao, B. 1994. Mutagenesis by PCR. in PCR Technology Current Innovations (ed. H. Griffin and A. Griffin), pp. 69-83. CRC Press, Boca Raton.
Thomas, P. and Beddington, R. 1996. Anterior primitive endoderm may be responsible for patterning the anterior neural plate in the mouse embryo. Curr Biol 6(11): 1487-1496.
Tlsty, T., Briot, A., and Poulose, B. 1995. Analysis of cell cycle checkpoint status in mammalian cells. Methods Enzymol 254: 125-133.
Tremblay, K.D., Hoodless, P.A., Bikoff, E.K., and Robertson, E.J. 2000. Formation of the definitive endoderm in mouse is a Smad2-dependent process. Development 127(14): 3079-3090.
Tsukui, T., Capdevila, J., Tamura, K., Ruiz-Lozano, P., Rodriguez-Esteban, C , Yonei-Tamura, S., Magallon, J., Chandraratna, R.A., Chien, K., Blumberg, B., Evans, R.M., and Belmonte, J.C. 1999. Multiple left-right asymmetry defects in Shh(-/-) mutant mice unveil a convergence of the shh and retinoic acid pathways in the control of Lefty-1. Proc Natl Acad Sci USA 96(20): 11376-11381.
Varlet, I., Collignon, J., Norris, D.P., and Robertson, E.J. 1997. Nodal signaling and axis formation in the mouse. Cold Spring Harb Symp Quant Biol 62: 105-113.
Vermot, J. and Pourquie, O. 2005. Retinoic acid coordinates somitogenesis and left-right patterning in vertebrate embryos. Nature 435(7039): 215-220.
Vincent, S.D., Dunn, N.R., Hayashi, S., Norris, D.P., and Robertson, E.J. 2003. Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev 17(13): 1646-1662.
Wallis, D. and Muenke, M . 2000. Mutations in holoprosencephaly. Hum Mutat 16(2): 99-108.
Wallis, D.E., Roessler, E., Hehr, U., Nanni, L., Wiltshire, T., Richieri-Costa, A., Gillessen-Kaesbach, G., Zackai, E.H., Rommens, J., and Muenke, M . 1999. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet 22(2): 196-198.
Wang, Z. and Mann, R.S. 2003. Requirement for two nearly identical TGIF-related homeobox genes in Drosophila spermatogenesis. Development 130(13): 2853-2865.
Warren, N. , Caric, D., Pratt, T., Clausen, J.A., Asavaritikrai, P., Mason, J.O., Hill, R.E., and Price, D.J. 1999. The transcription factor, Pax6, is required for cell proliferation and differentiation in the developing cerebral cortex. Cereb Cortex 9(6): 627-635.
Watanabe, K., Kamiya, D., Nishiyama, A., Katayama, T., Nozaki, S., Kawasaki, H., Watanabe, Y. , Mizuseki, K., and Sasai, Y. 2005. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci 8(3): 288-296.
Watase, K. and Zoghbi, H.Y. 2003. Modelling brain diseases in mice: the challenges of design and analysis. Nat Rev Genet 4(4): 296-307.
122

Wichterle, H., Lieberam, I., Porter, J.A., and Jessell, T.M. 2002. Directed differentiation of embryonic stem cells into motor neurons. Cell 110(3): 385-397.
Wijgerde, M . , McMahon, J.A., Rule, M . , and McMahon, A.P. 2002. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev 16(22): 2849-2864.
Wilson, S.W. and Houart, C. 2004. Early steps in the development of the forebrain. Dev Cell 6(2): 167-181.
Wotton, D., Knoepfler, P.S., Laherty, C D . , Eisenman, R.N., and Massague, J. 2001. The Smad transcriptional corepressor TGIF recruits mSin3. Cell Growth Differ 12(9): 457-463.
Wotton, D., Lo, R.S., Lee, S., and Massague, J. 1999a. A Smad transcriptional corepressor. Cell 97(1): 29-39.
Wotton, D., Lo, R.S., Swaby, L.A., and Massague, J. 1999b. Multiple modes of repression by the Smad transcriptional corepressor TGIF. J Biol Chem 274(52): 37105-37110.
Wurst, W., Joyner, AL . 1993. Production of targeted embryonic stem cell clones, in A Practical Approach (ed. A. Joyner), pp. 1-31. Oxford University Press, Oxford.
Xu, Q., Wonders, CP. , and Anderson, S.A. 2005. Sonic hedgehog maintains the identity of cortical interneuron progenitors in the ventral telencephalon. Development 132(22): 4987-4998.
Xuan, S., Baptista, C.A., Balas, G., Tao, W., Soares, V.C., and Lai, E. 1995. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 14(6): 1141-1152.
Yamamoto, M . , Meno, C , Sakai, Y., Shiratori, H., Mochida, K., Ikawa, Y., Saijoh, Y. , and Hamada, H. 2001. The transcription factor FoxHl (FAST) mediates Nodal signaling during anterior-posterior patterning and node formation in the mouse. Genes Dev 15(10): 1242-1256.
Yamamoto, M . , Saijoh, Y., Perea-Gomez, A., Shawlot, W., Behringer, R.R., Ang, S.L., Hamada, H., and Meno, C. 2004. Nodal antagonists regulate formation of the anteroposterior axis of the mouse embryo. Nature 428(6981): 387-392.
Yang, Y., Hwang, C.K., D'Souza, U.M., Lee, S.H., Junn, E., and Mouradian, M . M . 2000a. Three-amino acid extension loop homeodomain proteins Meis2 and TGIF differentially regulate transcription. J Biol Chem 275(27): 20734-20741.
Yang, Y., Hwang, C.K., Junn, E., Lee, G., and Mouradian, M.M. 2000b. ZIC2 and Sp3 repress Spi-induced activation of the human D1A dopamine receptor gene. J Biol Chem 275(49): 38863-38869.
Yoshioka, H., Meno, C , Koshiba, K., Sugihara, M. , Itoh, H., Ishimaru, Y. , Inoue, T., Ohuchi, H., Semina, E.V., Murray, J.C., Hamada, H., and Noji, S. 1998. Pitx2, a
123

bicoid-type homeobox gene, is involved in a lefty-signaling pathway in determination of left-right asymmetry. Cell 94(3): 299-305.
Zar, J.H. 1984. Biostatistical Analysis. Prentice-Hall, Inc., Englewood Cliffs, N.J.
Zhang, W., Kang, J.S., Cole, F., Y i , M.J., and Krauss, R.S. 2006. Cdo functions at multiple points in the sonic hedgehog pathway, and cdo-deficient mice accurately model human holoprosencephaly. Dev Cell 10(5): 657-665.
Zhang, X . M . , Ramalho-Santos, M . , and McMahon, A.P. 2001. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell 106(2): 781-792.
124
Copyright © 2022 FDOKUMEN




















