
Pd-catalysed Carbonylations of Anilines in Ionic Liquids ...
Upload
khangminh22Category
view
3download
0
1
1
2
3
Ribozyme-catalysed RNA synthesis using triplet building blocks 4
5
6
7
Authors: James Attwater1, Aditya Raguram
1†, Alexey S. Morgunov1, Edoardo Gianni
1 & 8
Philipp Holliger1*
9
10
Affiliations: 11
1 MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge Biomedical Campus, 12
Cambridge, CB2 0QH, UK. 13
† Current address: Harvard University, Department of Chemistry and Chemical Biology, 12 14
Oxford Street, Cambridge, MA 02138, USA. 15
*Correspondence to: Philipp Holliger ([email protected]). 16

2
Abstract 17
RNA-catalyzed RNA replication is widely believed to have supported a primordial 18
biology. However, RNA catalysis is dependent upon RNA folding, and this yields structures that 19
can block replication of such RNAs. To address this apparent paradox we have re-examined the 20
building blocks used for RNA replication. We report RNA-catalysed RNA synthesis on structured 21
templates when using trinucleotide triphosphates (triplets) as substrates, catalysed by a general 22
and accurate triplet polymerase ribozyme that emerged from in vitro evolution as a mutualistic 23
RNA heterodimer. The triplets cooperatively invaded and unraveled even highly stable RNA 24
secondary structures, and support non-canonical primer-free and bidirectional modes of RNA 25
synthesis and replication. Triplet substrates thus resolve a central incongruity of RNA 26
replication, and here allow the ribozyme to synthesise its own catalytic subunit ‘+’ and ‘–’ strands 27
in segments and assemble them into a new active ribozyme. 28

3
Introduction 29
The premise that some RNA sequences can catalyse and template their own replication - 30
synthesizing both their own ‘+’ and ‘–’ strands - underpins current thinking about early genetic 31
systems (Crick, 1968; Orgel, 1968; Szostak et al., 2001). Any ancient ribozyme with such RNA 32
replicase capability seems to be lost, but efforts to recreate RNA self-replication in the laboratory 33
are ongoing (Martin et al., 2015) as a critical test of the “RNA world” hypothesis (Gilbert, 1986). 34
Early on, derivatives of naturally occurring self-splicing introns (Doudna et al., 1991; Green & 35
Szostak, 1992; Hayden & Lehman, 2006) as well as later in vitro evolved ligase ribozymes 36
(Lincoln & Joyce, 2009; Sczepanski & Joyce, 2014) were shown to be able to assemble one of 37
their own strands from cognate constituent RNA segments. A critical drawback of such systems is 38
their use of specific preformed building blocks of at least 8 nucleotides (nt) average length, 39
limiting their potential for open ended evolution, and precluding their replication from pools of 40
random-sequence oligonucleotide substrates (Green & Szostak, 1992; Doudna et al., 1993). 41
In a contrasting approach, RNA polymerase ribozymes (RPRs) have been developed that 42
can use general monomer building blocks (ribonucleoside 5’ triphosphates (NTPs)) in RNA-43
templated RNA synthesis (Johnston et al., 2001; Zaher & Unrau, 2007; Wochner et al., 2011; 44
Attwater et al., 2013b; Horning & Joyce, 2016), akin to the activity of modern proteinaceous 45
polymerases. However, even the most highly-evolved RPRs (Horning & Joyce, 2016) are 46
substantially impeded by template secondary structures. Such structures are ubiquituous in larger, 47
functional RNAs (including the RPRs themselves) and generally indispensable for function. The 48
strong inhibitory role of this central feature of RNA leads to an antagonism between the degree to 49
which an RNA sequence is able to fold into a defined three-dimensional structure to encode 50
function (such as catalysis) and the ease with which it can be replicated (Boza et al., 2014). This 51
ostensible “structure vs. replication” paradox would have placed stringent probability constraints 52

4
on the emergence of an RNA replicase and generally impeded the ability of RNA to function as 53
an early genetic polymer. 54
We wondered whether this paradox might be evaded through a re-consideration of 55
plausible building blocks for early RNA replication. Models of non-enzymatic polymerisation of 56
all four activated ribonucleotides – the presumed source of the first RNA sequences – yield pools 57
of di-, tri- and tetranucleotide- etc. length oligonucleotides (in decreasing abundance) dominating 58
the population alongside longer products (Monnard et al., 2003). Here we have examined 59
whether substrates of such lengths can support RNA-catalyzed RNA replication, by developing a 60
ribozyme capable of iterative templated ligation of 5’-triphosphorylated RNA trinucleotides 61
(henceforth called triplets). This heterodimeric triplet polymerase ribozyme demonstrated a 62
striking capacity to copy a wide range of RNA sequences, including highly structured, previously 63
intractable RNA templates, as well as its own catalytic domain and encoding template in 64
segments. Its characterization revealed emergent properties of triplet-based RNA synthesis, 65
including cooperative invasion and unraveling of stable RNA structures by triplet substrates, bi-66
directional (both 5’-γ’ and γ’-5’) and primer-free (triplet-initiated) RNA synthesis, and fidelity 67
augmented by systemic properties of the random triplet pools. 68

5
Results 69
In vitro evolution of triplet polymerase activity 70
We set out to explore the potential of short RNA oligonucleotides as substrates for RNA-71
catalyzed RNA replication. To do this we required a ribozyme capable of general, iterative RNA-72
templated oligonucleotide ligation. Previously-described RNA polymerase ribozymes such as the 73
‘Z’ RPR (Wochner et al., 2011) can use NTPs to iteratively extend a primer hybridized to an 74
RNA template, but do not accommodate oligonucleotides bound downstream of the primer or 75
accept them as substrates. However, we detected a weak templated ligation activity in a truncated 76
version of the Z RPR comprising its catalytic core domain (Zcore) (Figure 1a), which supported 77
incorporation of oligonucleotide substrates as short as 3 nt (Figure 1 - figure supplement 1), when 78
incubated in the eutectic phase of water ice (Attwater et al., 2010; Mutschler et al., 2015). 79
To be able to properly examine such RNA trinucleotide triphosphates (triplets) as 80
replication substrates, we first sought to convert Zcore into an effective triplet polymerase 81
ribozyme using in vitro evolution. We devised a selection strategy that required iterative 82
templated triplet ligation by ribozymes to achieve their in cis covalent linkage to a tagged primer 83
(Figure 1 - figure supplement 2). This enables their recovery, amplification and mutagenesis 84
before further rounds of selection to enrich the selection pool in improved triplet polymerase 85
ribozyme variants. 86
We initiated selections from a library of 1.5 × 1015
Zcore variants with a new random γ’ 87
N30 region under eutectic phase conditions that increase RNA half-life and enhance ribozyme 88
activity (Attwater et al., 2010). After 7 rounds of in-ice evolution (Attwater et al., 2013b) one-89
quarter of the selection pool comprised an improved ribozyme (type 0). Its core domain (0core, 90
Figure 1a) could catalyse the iterative polymerization of multiple triplets allowing us to begin to 91
investigate the properties of triplet-based RNA replication. 92

6
Significantly, we found that 0core could catalyze triplet polymerisation on a series of 93
structured templates, which had proven intractable to the parental Z RPR (Figure 1b). Here, 94
primer extension exhibited a steep sigmoidal dependence upon triplet concentrations (Figure 1c), 95
suggestive of a cooperative invasion and unraveling of template secondary structures by the 96
triplet substrates themselves. Although still inefficient, the fact that the nascent activity of the 97
0core ribozyme could already copy templates that had confounded an established RPR 98
encouraged us to continue to seek improved triplet polymerase ribozymes to leverage this 99
substrate behaviour. 100
101
Emergence of cooperativity and characterisation of a ribozyme heterodimer 102
We continued selections for a further 14 rounds. At this point, the type 0 ribozyme had 103
gone extinct, replaced by six new types of RNA each characterised by a unique γ’ domain (Figure 104
2a, Figure 2 - figure supplement 1). Type 1 RNAs were most abundant, comprising ~50% of pool 105
sequences but mysteriously were catalytically inactive with diverse mutations in their core 106
domains. In contrast, the type 2-6 RNAs all displayed triplet polymerase activity, but fell short of 107
the polyclonal activity of the selection pool (Figure 2 - figure supplement 2). To attempt to 108
explain this discrepancy, we explored potential interactions among the different pool lineages, 109
and found that addition of an equimolar amount of type 1 RNA substantially enhanced triplet 110
polymerase activity of all the other ribozyme types 2-6 (Figure 2b). 111
Dissecting type 1 RNA function, we found that 5’ truncation of the region that previously 112
contacted the primer/template duplex (Shechner et al., 2009) did not affect its cofactor activity 113
(Figure 3a, Figure 3 - figure supplement 1). As judged by gel mobility shift (Figure 3b) and 114
activity enhancement (Figure 3 - figure supplement 1), type 1 RNA appears to form a 1:1 115
heterodimeric complex directly with active triplet polymerase ribozymes. Our attention was 116
drawn to their selection construct-derived 5’ hairpin elements, which differed between active 117

7
triplet polymerases (‘cap+’, Figure 3a, Figure 2 - figure supplement 1) and the most common type 118
1 variants in the selection pool where this hairpin had acquired a mutation (yielding ‘cap–’, 119
Figure 3a). ‘cap–’ was dispensible for type 1’s cofactor activity, but when replacing ‘cap+’ in 120
active triplet polymerases it abolished both their activity enhancement by type 1 (Figure 3 - figure 121
supplement 1) and complex formation (Figure 3b). This points to the ‘cap+’ hairpin as the critical 122
site of interaction with type 1; ‘cap–’ in type 1 presumably served to deter its homodimerisation. 123
Indeed, transplanting the ‘cap+’ element could make the parental ribozymes (Zcore & Z 124
RPR) receptive to activity enhancement by type 1 RNA (Figure 2b, Figure 3 - figure supplement 125
2). The catalytically inert type 1 RNA thus represents a general, mutualistic RNA species. This 126
molecular symbiont appears to have emerged spontaneously during in vitro evolution by forming 127
a heterodimeric holoenzyme with triplet polymerase ribozymes, enhancing their activity and their 128
joint recovery prospects. 129
In complex with type 5 (the fastest enriching triplet polymerase ribozyme in the final 130
selection pool), type 1 boosts polymerization of triplets (or longer oligonucleotides) to enable 131
synthesis of long RNAs (Figure 3c). Here it became apparent that type 1 also obviates the need 132
for ribozyme-template tethering. Due to their poor affinity for primer/template duplex (Lawrence 133
& Bartel, 2003), RPRs generally depend upon such tethering to template (Attwater et al., 2010; 134
Wochner et al., 2011; Horning & Joyce, 2016), which enhances local ribozyme concentration 135
and promotes formation of the RPR-primer/template holoenzyme (Attwater et al., 2010; Attwater 136
et al., 2013a). In contrast, the triplet polymerase heterodimer appears to have a capacity for true 137
intermolecular, sequence-general interaction with primer-template duplexes, which enables 138
holoenzyme formation and copying of RNA templates without requiring specific ribozyme-139
template hybridization sites. 140
141
Secondary structure invasion by triplet substrates 142

8
We performed an additional five rounds of in vitro evolution to further evolve the type 5 143
triplet polymerase ribozyme (now in the presence of truncated type 1 RNA), diversifying the 144
previously-fixed γ’ domain reverse transcription primer binding sequence. This reselection 145
yielded a shorter final heterodimeric triplet polymerase holoenzyme, hereafter termed ‘t5+1’ 146
(Figure 4). This robust triplet polymerase activity now proved suitable for exploring the scope 147
and potential of triplet-based RNA replication. 148
As a first examination of t5+1
activity, we revisited triplet-based RNA synthesis on 149
structured templates. To provide a stringent test of template structure inhibition, we now 150
examined hairpin-containing templates (4S, 6S, 8S) with increasing RNA hairpin stability and 151
estimated TMs of up to 9γ˚C (8S). The latter had previously strongly arrested even the most 152
advanced mononucleotide RPRs at higher temperatures (Horning & Joyce, 2016). However, 153
using triplets as substrates t5+1
robustly copied all of these (Figure 5a), even when templates were 154
pre-folded allowing RNA secondary structures to form prior to triplet addition (Figure 5 - figure 155
supplement 1). The triplet concentration-dependent cooperative structure invasion and unraveling 156
(previously observed with the simpler 0core domain and partly wobble-paired RNA template 157
structures (Figure 1b, c)) was recapitulated with t5+1
and the highly stable 8S hairpin template 158
(Figure 5 - figure supplement 2). In contrast, dinucleotide triphosphate substrates yielded 159
extension only up to the structured region (Figure 5 - figure supplement 2). 160
We began to explore whether triplet-based RNA synthesis by t5+1
might exhibit the 161
generality required not just for synthesis of arbitrary structured sequences, but for replication of 162
functional sequences (requiring synthesis of both ‘+’ and ‘’ strands). Encouragingly, t5+1
could 163
synthesise both a functional fluorescent ‘+’ strand of the 52 nt Broccoli RNA aptamer (Filonov et 164
al., 2014) and its encoding ‘’ strand template from their 13 (+) & 12 () different constitutive 165
triplets (Figure 5b). 166
167

9
Ribozyme sequence self-synthesis and assembly 168
We next turned to the critical test of generality: could triplet substrates allow self-169
synthesis? As t5+1
currently lacks the efficiency to synthesize RNAs its own length, we divided 170
the catalytic t5 ribozyme into five segments , , , and . This segmentation strategy (akin to 171
that used by some RNA viruses e.g. influenza) could reduce tertiary structures (Doudna et al., 172
1991; Mutschler et al., 2015) and ease product separation during RNA replication (Szostak, 173
2012). Starting from ~8 nt RNA primers, t5+1
achieved synthesis of the +, +
, and + segments 174
from their constitutive triplets as well as all of the ‘’ strand segments -, -
, -, -
and -, but 175
required some triplets pre-linked (as e.g. hexanucleotides) for synthesis of full-length + and +
176
segments (Figure 6a). 177
Operating across 70 distinct ligation junctions in these reactions including AU-rich 178
sequences, t5+1
using triplet substrates demonstrates the sequence generality for self-synthesis. 179
Notably, the average extent of ligation per junction during synthesis of t5 ‘+’ and ‘’ strands 180
(78%) was similar to that observed when t5+1
used an unstructured model template (74%, Figure 181
3c) upon which the parental Z and other RPRs excel (Attwater et al., 2013b; Horning & Joyce, 182
2016). 183
At this point we tested whether the broad oligonucleotide ligation capacity of t5+1
(Figure 184
3c) might allow assembly of synthesised ‘+’ strand segments. Indeed, t5+1
could assemble these 185
into + and +
fragments, guided only by partially overlapping ‘’ strands (Figure 6b, Figure 6 186
- figure supplement 1). Through non-covalent association (Vaish et al., 2003; Mutschler et al., 187
2015), the ribozyme-synthesised + and +
fragments spontaneously reconstituted a new 188
catalytically active triplet polymerase ribozyme (with in vitro transcribed type 1 RNA). We found 189
that this synthesis product could regenerate fresh - segment using t5
+1 ribozyme-synthesised +
190
(left over from ribozyme assembly) as a template (Figure 6c), recapitulating elements of a self-191

10
replication cycle. However, while the t5+1
ribozyme displays a nascent capacity for templated 192
synthesis of its own catalytic domain ‘+’ strands (and ‘’ strands), efficiency of both segment 193
synthesis and assembly will need to be increased significantly to realise a full self-replication 194
cycle (which would also require synthesis and replication of the type 1 subunit). 195
196
Primer-free RNA synthesis 197
Templated ‘+’ strand self-synthesis is a central element of ribozyme self-replication. 198
However, a limitation of our above strategy in the context of triplet-based self-replication is the 199
continued requirement for some pre-synthesized longer oligonucleotides to act as primers and 200
occasional substrates (together providing here the equivalent of ~1/4 of triplet junctions pre-201
ligated). Specific oligonucleotide substrates were used in places to compete out inhibitory mutual 202
hybridisation between ‘’ strand template and corresponding ‘+’ strand unstructured elements in 203
the t5 ribozyme (Figure 6 - figure supplement 2). In vitro selections that stabilise the ribozyme 204
tertiary structure (Figure 6 - figure supplement 3) may contribute to attenuating this requirement. 205
Additionally, more concentrated triplet substrates can successfully compete with ribozyme 206
unstructured elements for hybridization to ‘’ strand templates (Figure 6 - figure supplement 2). 207
The majority of specific oligonucleotides, however, were provided as primers to initiate 208
syntheses, as required by all RPRs akin to the activity of replicative polymerases in biology. As a 209
consequence of this, the priming sequence would effectively be excluded from evolution. 210
Furthermore, RNA oligonucleotides able to act as specific primers are unlikely to be prevalent in 211
prebiotic substrate pools, and their depletion during successive replication cycles could lead to 212
sequence loss at genome ends. This ‘primer problem’ has previously been noted in the context of 213
nonenzymatic replication (Szostak, 2012) as one of the fundamental obstacles to RNA self-214
replication. 215

11
Unexpectedly, triplet substrates provide a route to bypass the ‘primer problem’. We 216
observed that t5+1
can extend primers bidirectionally, in both the canonical 5’-γ’ as well as the 217
reverse γ’-5’ directions (Figure 7a). This not only allows completion of RNA synthesis from 218
either template end but initiation from anywhere along a template potentially allowing non-219
classical hierarchical or distributive RNA replication schemes as previously proposed (Szostak, 220
2011, 2012). Given this flexibility, we wondered if t5+1
even had a requirement for a primer 221
oligonucleotide. Indeed, the t5+1
ribozyme could achieve ‘primer free’ RNA synthesis (whereby 222
synthesis is presumably initiated by ligation of adjacent triplets anywhere on the template), as 223
exemplified here for the + segment (Figure 7b), as well as ‘primer free’ RNA replication as 224
shown for the ‘+’ and ‘’ strands of the segment, which can be replicated using triplets alone 225
(Figure 7c). 226
Thus the capacity of triplet substrates to pre-organise themselves on a template not only 227
enables replication of structured templates but allows complete copying of some RNA sequences 228
exclusively from triplet building blocks, suggesting an alternative to the canonical end-primed 229
replication strategies inspired by PCR. Such a ribozyme operating in a more distributive 230
polymerisation mode might be able to replicate RNA sequences directly from the putative pools 231
of short random RNA oligonucleotides furnished by prebiotic chemistry. 232
233
Fidelity of triplet-based RNA synthesis 234
Next we investigated the consequences of using defined modern analogues of such 235
prebiotic pools as a source of substrates for the t5+1
triplet polymerase ribozyme. Random 236
sequence triplet pools (‘pppNNN’, comprising equimolar amounts of all 64 triplets) could be used 237
as substrates by t5+1
in segment syntheses in place of defined triplet sets (Figure 6 – figure 238

12
supplement 4). Furthermore, extension activity remained robust upon pool supplementation with 239
noncanonical dinucleotide and mononucleotide substrates (Figure 6 – figure supplement 5). 240
However, a replicase must incorporate the correct template-complementary substrate from 241
random sequence pools, or genetic information may become irretrievably corrupted during 242
replication (Eigen, 1971). Sequence fidelity is therefore a critical parameter of RNA replication. 243
The fidelity challenge is exacerbated in triplet-based RNA replication by the need to discriminate 244
between 64 distinct substrates; indeed, a previous investigation into the incorporation of 245
individual trinucleotides indicated that misincorporations could outstrip cognate incorporation for 246
some triplets (Doudna et al., 1993). 247
In order to assess the fidelity of triplet polymerase ribozymes of widely differing activity, 248
we identified the triplets incorporated from random ppp
NNN triplet pools using 12 different 249
compositionally representative NNN triplet sequences as templates in a consistent sequence 250
context (5’-GGG-NNN-GGG-3’), whose collation allowed an estimation of ribozyme 251
misincorporation tendencies. On average, the starting Zcore ribozyme exhibited ~91% fidelity per 252
position (Figure 8a), lower than that described for RPRs (92% (Horning & Joyce, 2016) – 97% 253
(Attwater et al., 2013b)). Furthermore, its accuracy exhibited a pronounced downward gradient 254
from the 1st (5’) to the 3
rd (γ’) triplet position, highlighting escalating risks to fidelity of synthesis 255
founded on longer building blocks. 256
To investigate if ribozymes could exhibit higher triplet incorporation fidelity, we had 257
included a persistent adaptive pressure for fidelity during in vitro evolution, spiking in an excess 258
of mispairing γ’-deoxy ‘terminator’ triplets from round 9 onwards, precluding recovery of 259
ribozymes that incorporated these mispairs (Figure 2 - source data 1, Figure 4 - source data 1, 260
Figure 2 - figure supplement 2). This yielded reshaped and improved fidelity profiles in the 261
‘surviving’ type 2-6 ribozymes (Figure 8a). Notably, the final t5+1
ribozyme achieves an average 262
positional fidelity of 97.4% using ppp
NNN in this sequence context, higher than the best RPR 263

13
fidelity with NTPs under comparable eutectic conditions (Attwater et al., 2013b). Deep 264
sequencing of internal triplet positions of a defined sequence (+ segment) synthesised by t5
+1 265
using ppp
NNN indicated similar fidelity could be achieved during longer product synthesis (Table 266
1). 267
268
Molecular basis of triplet polymerase ribozyme fidelity 269
Having established that accurate triplet-based copying is possible (in at least some 270
sequence contexts), we sought to understand how the triplet polymerase ribozyme achieves it. 271
Investigating the fidelity contributions of different t5+1
ribozyme components, we found that the 272
type 1 RNA cofactor did not contribute; rather, fidelity gains appeared to be mediated by the 273
newly-evolved t5 ‘’ γ’-domain, as its deletion (yielding the truncated ‘’ ribozyme) reverted 274
its fidelity profile towards that of Zcore (Figure 8 - figure supplement 1). Presence of the 275
domain did not uniformly increase fidelity, but selectively reduced the most acute errors at the 2nd
276
and 3rd
triplet positions (with over 10-fold reductions for some errors, Figure 8b, Figure 8 - figure 277
supplement 2). Overall error rates at the 2nd
and 3rd
triplet positions were reduced by 4-fold and 9-278
fold compared to Zcore (Figure 8a), though slightly increased (1.3-fold) at the 1st triplet position 279
due to a localised asymmetric tolerance of G:U wobble pairing (Figure 8b). The domain fidelity 280
function is contingent upon the presence of a downstream triplet, operating only with basal 281
fidelity for final triplet incorporation (Figure 8 - figure supplement 3). 282
Dissecting the molecular determinants of the fidelity phenotype, we found that using 283
triplet substrates modified at the 3rd
position with a 2-thiouracil in place of a uracil (disrupting 284
minor groove hydrogen bonding) rendered the fidelity domain unable to discriminate 285
mismatches (Figure 8c, Figure 8 - figure supplement 3). Previously, a similar replacement of a 286
uracil 2-keto group with a 2-thio modification had been shown to impair Z RPR activity when 287

14
present upstream in the primer/template region (Attwater et al., 2013a), where Z is thought to rely 288
upon sequence-general minor groove contacts through an ‘A-minor’ motif (Shechner et al., 289
2009). Modification at the 3rd
triplet position reverts ’s divergent effects on fidelity at the 290
adjacent 2nd
and the distal 1st triplet positions (Figure 8 - figure supplement 3); disruption of this 291
minor groove contact site thus abolishes overall fidelity domain operation. sensitivity to minor 292
groove composition may be critical to its recognition of cognate Watson-Crick base pairs, 293
reminiscent of Tetrahymena group I intron folding (Battle & Doudna, 2002) and the decoding 294
centre of the ribosome (which also tolerates wobble pairing at the analogous (5’) triplet position) 295
(Ogle et al., 2001). 296
297
Systems-level properties of triplet pools 298
An important contribution to triplet fidelity also appears to arise from unexpected 299
behaviours of the triplet substrates themselves. We observed that in some direct pair-mispair 300
triplet contests, inclusion of their complementary triplets caused a striking (~3-fold) drop in 301
misincorporation errors (Figure 9). A potential explanation may arise from differential formation 302
of triplet:anti-triplet dimers in the reaction: for example, more extensive ppp
GCC:ppp
GGC (than 303
pppACC:
pppGGU) dimer formation would selectively reduce the effective concentration of free 304
pppGCC vs.
pppACC upon inclusion of their complementary
pppGGC and
pppGGU. 305
These pairwise reductions were recapitulated in the presence of random ppp
NNN substrate 306
pools (Figure 9). Indeed, counterintuitively, raising ppp
NNN concentrations from 0.5 to 5 με each 307
almost halved the overall error rate (Figure 9 - figure supplement 1). Although diverse effects 308
upon individual misincorporations were observed, this fidelity enhancement was driven by 309
pronounced reductions in errors where the mismatched triplet has a high GC content compared to 310
the cognate triplet, including common G-U wobble mispairs (Figure 9 - figure supplement 1). 311
Dimer formation among ppp
NNN substrate pools would be expected to selectively buffer the free 312

15
concentrations of the more strongly-pairing GC-rich triplets, which could promote both fidelity 313
and sequence generality through normalization of triplet availability against template (and 314
complementary triplet) binding strength. 315
Indeed, more efficient, higher fidelity segment synthesis was observed when partially 316
mimicking this outcome using an ppp
NNN pool formulated with a reduced G content (Figure 6 – 317
figure supplement 4, Table 1). In a prebiotic scenario, substrate pool composition would have 318
been determined by the abundance and nontemplated polymerization tendencies of the different 319
nucleotides; large biases in these could skew triplet compositions or deplete a triplet (resulting in 320
mismatch incorporation). However, the potential for replication to proceed in different triplet 321
registers may provide a degree of resilience towards such biases. 322

16
Discussion 323
Here we describe the discovery and characterization of a ribozyme (t5+1
) with a robust 324
ability to polymerize RNA trinucleotide triphosphate (triplet) substrates. Unusually, this triplet 325
polymerase ribozyme comprises a heterodimer of a catalytic triplet polymerase subunit (t5) and a 326
non-catalytic RNA cofactor (type 1), which enhances triplet polymerase activity and abrogates 327
the need for template tethering. Such a quaternary structure - involving a heterodimer of a full-328
length and a truncated subunit - is reminiscent of the processivity factors of proteinaceous 329
polymerases such as the heterodimeric p66/p51 HIV reverse transcriptase holoenzyme (Huang et 330
al., 1992). There are multiple examples of dimerization in RNA evolution - such as the VS 331
ribozyme (Suslov et al., 2015), retroviral RNA genome dimerization (Paillart et al., 2004), in 332
vitro evolved heterodimeric RNA liposome binders (Vlassov et al., 2001), and recently the 333
homodimeric CORN fluorescent RNA aptamer (Warner et al., 2017). However, the spontaneous 334
emergence of a general, mutualistic RNA cofactor has not previously been observed for 335
ribozymes and may suggest an underappreciated dimension to the evolutionary dynamics of 336
ribozyme pools under stringent adaptive pressures. Indeed, the extinction of previously dominant 337
species in the selection that were unable to benefit from type 1 enhancement (e.g. type 0, see 338
Figure 3 - figure supplement 2) and succession with cooperative RNA species (Vaidya et al., 339
2012) illustrates the potential for such symbioses to shape RNA molecular ecologies. 340
The t5+1
ribozyme’s principal current shortcoming is its low catalytic efficiency. In the 341
optimal context for mononucleotide polymerase ribozymes, this triplet polymerase heterodimer 342
yields ~4-fold more unligated junctions than the RPR tC19Z (Attwater et al., 2013b), which itself 343
is 240-fold slower than the currently most advanced RPR 24-3 (Horning & Joyce, 2016). Yet 344
despite this modest catalytic power, t5+1
displays much enhanced generality in RNA synthesis and 345
now achieves both copying of previously intractable structured RNA templates, and templated 346

17
synthesis and assembly of an active ‘+’ strand copy of its catalytic domain, suggesting key 347
contributions of the triplet substrates themselves. 348
Indeed, one of the main findings of our work are the compelling advantages triplet 349
substrates appear to offer for sequence general RNA replication. For instance, when binding 350
templates, triplets incur a lower entropic cost per position compared to canonical 351
mononucleotides (thus aiding copying of sequences rich in weakly pairing A and U bases), with 352
particularly helpful stability contributions from intra-triplet base stacking (Eigen, 1971). 353
Furthermore, energetically favourable inter-triplet stacking interactions appear to instigate 354
cooperative binding and unfolding of even highly stable RNA template structures (Figures 1b, 5a) 355
upon reaching the required substrate concentration threshold. In our work, this process is aided by 356
the cold temperature and solute concentration effect of eutectic ice phase formation (Attwater et 357
al., 2010; Mutschler et al., 2015). Counterintuitively, a general solution to the copying of 358
structured RNAs arises not from conditions that disfavour base-pairing (which would also hinder 359
substrate binding), but rather from conditions that promote it. 360
Together these favourable molecular traits serve to pre-organize the template towards a 361
double-stranded RNA duplex with triplet junctions poised for ligation. A triplet/template duplex 362
presents a more ordered, regular target for sequence-general ribozyme docking (by e.g. the 363
domain) than a single stranded template (variably prone to secondary structure formation or 364
sequence-specific interactions with the ribozyme (Wochner et al., 2011)). Such general duplex 365
interactions also underlie other notable features observed in our triplet-based RNA synthesis such 366
as in trans template binding (Figure 3c) as well as the capacity for bidirectional (5’-γ’ / γ’-5’) and 367
primer-free RNA synthesis (Figure 7). 368
Contrary to expectations RNA-catalyzed triplet polymerisation can proceed with a fidelity 369
matching or exceeding even the best mononucleotide RNA polymerase ribozymes (Attwater et 370
al., 2013b; Horning & Joyce, 2016). t5+1
ribozyme fidelity is due to both a readout of cognate 371

18
minor groove interactions by the ribozyme domain (Figure 8) and an unanticipated fidelity 372
boost arising from systems-level properties of triplet pools, that appear to normalize the 373
availability of free triplet (and potentially longer oligonucleotide) substrates against their base-374
pairing strength (Figure 9). Though further work will be required to characterize triplet pool 375
properties, they likely involve formation of cognate or near-cognate triplet:anti-triplet interaction 376
networks, as formation of tRNA dimers via cognate anticodon:anticodon interactions has been 377
observed in a similar concentration range (Eisinger & Gross, 1975). 378
While phylogenetically unrelated, mechanistic analogies between the triplet polymerase 379
ribozyme and the ribosome are apparent. Both are RNA heterodimers that operate in a triplet 380
register along a single-stranded RNA template, whilst enforcing a minor-groove mediated pattern 381
of triplet / anticodon readout (including tolerance of 5’ wobble pairing), suggestive of convergent 382
adaptive solutions to the challenges of replication and decoding. It has long been speculated that 383
the decoding centre of the small ribosomal subunit might have had its origins in an ancestral RNA 384
replicase, but the implied triplet-based character of such a replicase was conspicuously discordant 385
with modern mononucleotide-based replication (Weiss & Cherry, 1993; Poole et al., 1998; 386
Noller, 2012). The utility of triplets as substrates for RNA synthesis and self-synthesis described 387
herein suggests that these early ideas deserve to be reconsidered. In the context of initial 388
uncorrelated evolution of the small and large ribosomal subunits (Petrov et al., 2015), an early 389
reliance upon triplets in RNA replication could have inadvertently supplied a decoding center for 390
translation. 391
In conclusion, the unexpected emergent properties of triplets – including cooperative 392
binding and unfolding of structured RNA templates, enhanced incorporation of AU-rich 393
substrates, and error attenuation (resulting from triplet pool interaction networks) – argue that 394
short RNA oligonucleotides represent predisposed substrates for RNA-catalyzed RNA 395
replication. Some of these benefits might also extend to anticodon behaviour in early translation, 396

19
and to the non-enzymatic replication of RNA (Szostak, 2012), where downstream trinucleotides 397
have recently been shown to enhance incorporation of preceding activated mononucleotides both 398
through stacking and positioning effects (Vogel et al., 2005; Zhang et al., 2018) and the 399
formation of a highly reactive intermediate (Prywes et al., 2016; O'Flaherty et al., 2018). Taken 400
together, the interaction of triplet substrate pools with RNA templates promotes uncoupling of an 401
RNA’s sequence (i.e. information content, and associated folding tendencies) from its 402
replicability, thereby enhancing RNA’s capacity to serve as an informational polymer. 403

20
Materials and Methods 404
Templated RNA-catalysed RNA synthesis 405
Standard ribozyme activity assays (modified where specified) comprise 5 pmol of each 406
ribozyme annealed in 2.5 μl water (80˚C 2 min, 17˚C 10 min), with 2 μl of 1 M MgCl2 and 0.5 μl 407
of 1 M tris•HCl pH 8.3 (at β5˚C, pH raised to 9.2 at 7˚C) then added on ice, and left for >5 min 408
to ensure folding. This was added to 5 pmol each of primer and template and 50 pmol of each 409
triplet pre-annealed in 5 μl water, then frozen on dry ice (10 min) and incubated at 7˚C in a R4 410
series TC120 refrigerated cooling bath (Grant) to allow eutectic phase formation and reaction. 411
Final pre-freezing concentrations of components are displayed throughout (in this 412
example, yielding 0.5 με ribozyme/primer/template, 5 με each triplet, 200 mM MgCl2, 50 mM 413
tris•HCl pH 8.3). Supercooled reactions (Figure 1c) remained liquid by omitting the dry-ice 414
freezing step, maintaining these concentrations. Ice crystal formation upon eutectic phase 415
equilibration, however, concentrates all solutes ~ 4-5 fold (Attwater et al., 2010) to their final 416
operational levels and cooling elevates tris-buffered pH to ~9.2. 417
Some substrate mixes (e.g. ppp
NNN) led to a higher final reaction volume, but eutectic 418
phase equilibration restored standard operational concentrations, also applicable to the four-fold-419
diluted extensions with the fragmented ribozyme (Figure 6c). These used 2 pmol each 420
ribozyme/fragment annealed in 3.25 μl 62 mM MgCl2, 15 mM tris•HCl pH 8.3 (γ7˚C 5 min, 421
ramped to 4˚C at 0.1˚C/s, 4˚C 10 min), with pre-annealed primer/template/substrate 422
(0.5/0.5/5 pmol) added in 0.75 μl water. These reactions, and preparative syntheses (Figure 6 - 423
figure supplement 1, Figure 7c), were supercooled at 7˚C followed by ice crystal addition for 424
quick freezing and optimal activity. 425
Figure 1c extensions were set up by adding buffer, then RNAs (preannealed together, 426
0.1 με final concentrations) to triplets. RNAs for + syntheses were chilled on ice instead of 427
annealing, with ribozyme/MgCl2/tris•HCl pH 8.3 mixed with the other RNAs at 7˚C. 428

21
Oligonucleotide substrates were added equimolar to template binding sites in the 429
primer/template/substrate anneal. NTPs, on the other hand, were added with the MgCl2/tris•HCl 430
pH 8.3 to the ribozyme polymerase. 431
432
Extension product separation 433
At the end of standard incubations, reactions were thawed and 2 μl aliquots added to stop 434
buffer (1 μl 0.44 M EDTA (pH 7.4), with urea to a 6 M final concentration and a 10-20 fold molar 435
excess over template of complementary competing oligonucleotide (see Supplementary file 3) to 436
prevent long product/template reannealing). Samples were denatured (94˚C 5 min) and RNAs 437
separated by 8 M urea 1 TBE denaturing PAGE. 438
To avoid using potentially confounding competing oligonucleotide when purifying 439
extension products, reactions with a biotinylated primer or template (stopped as above) could be 440
purified by bead capture using MyOne C1 (Invitrogen) streptavidin-coated paramagnetic 441
microbeads (using 5 μg pre-washed beads per pmol biotinylated RNA) in 0.5-0.8 bead buffer 442
(BB: 200 mM NaCl, 10 mM tris•HCl pH 7.4 (at β5˚C), 1 mM EDTA, 0.1% Tween-20). After 443
washing twice in BB to remove unbound components, beads were incubated (1 min) in 25 mM 444
NaOH, 1 mM EDTA, 0.05% Tween-20 to denature the duplexes (Horning & Joyce, 2016). To 445
recover biotinylated extension products (e.g. Figure 5b left panels, Figure 6 - figure supplement 1 446
+/+
, Figure 7c left panel) the supernatant was discarded, and beads were washed first in BB 447
with 200 mM tris•HCl pH 7.4, then in BB, then heated (94˚C 4 min) in 95% formamide, 10 mM 448
EDTA to release primers for urea-PAGE. To recover extension products bound to biotinylated 449
templates (e.g. Figure 6 - figure supplement 1 +, +
, +, Figure 7c right panel) the supernatant 450
was removed, neutralized with 500 mM tris•HCl pH 7.4, spin-concentrated using Ultracel 3K 451
filters (Amicon), recovered and denatured in 6M urea/10 mM EDTA before urea-PAGE. + 452
synthesised in Figure 6 - figure supplement 1 was not spin-concentrated, leading to a lower 453

22
recovery yield; + synthesis was denatured directly from the ligation reaction in 60% formamide 454
with excess EDTA. 455
For gel mobility shift assays (Figure 3b), ribozymes were mixed at 0.5 με, pre-annealed 456
and buffer added on ice as for extension reactions, then mixed with 5 loading buffer (50% 457
glycerol, 250 mM tris•HCl pH 8.3, 125 mM MgCl2) for separation by native PAGE (0.5 TB, 8% 458
59:1 acrylamide:bisacrylamide, 25 mM MgCl2, run in a Hoefer SE600 Chroma (upper chamber: 459
0.5TB 50 mM NaOAc, lower chamber: 0.5TB 25 mM Mg(OAc)2) kept at 4˚C in a circulator 460
bath for 6-8 h at 10 W), then SYBR Gold stained. 461
462
Extension product detection, quantification and purification 463
Fluorescent primer extension products were detected using the appropriate laser 464
wavelength on a Typhoon Trio scanner (GE); gel densitometry allowed quantification of RNA 465
synthesis efficiency. Gel contrasts in figures were linearly adjusted to optimize display of bands 466
of differing intensities. 467
The gel in Figure 5b (middle panel) was washed thrice (5 min) in water, incubated with 468
10 με DFHBI-1T ligand in buffer for 20 min to fold full-length broccoli aptamer (as in (Filonov 469
et al., 2015)) and scanned. The ligand was then eluted in three 1 TBE washes (leaving negligible 470
background fluoresence), and stained in 1 TBE with SYBR Gold (1:10000), washed again, and 471
re-scanned to detect all RNA products (left panel); scans were aligned via an adjacent Cy5-472
labelled primer extension lane (not shown). 473
Full-length product yields in the Figure 6 - figure supplement 1 plus-strand syntheses were 474
calculated by running samples of bead-eluted products (or raw reaction for +) alongside known 475
amounts of the positive controls indicated, followed by SYBR-Gold staining. To purify, bead-476
eluted products were run similarly, and excised using UV shadowing. Products were then eluted 477
from the gel fragments in 10 mM tris•HCl pH 7.4, and Spin-X column filtrate (Costar) 478

23
precipitated in 75% ethanol with 1 μl 1% glycogen carrier (omitted for +). Recovered full-length 479
product yields were calculated similarly to reaction yields for +/+
/+, or using A260s for +
, 480
+, +
. 481
482
Fragment sequencing 483
Purified ribozyme- and TGK-synthesized +/+
fragments were sequenced by first 484
ligating a γ’ adaptor (10 U/μl T4 RNA Ligase 2 truncated KQ in 1 RNA ligase buffer (NEB) 485
with 15% PEG-8000 and 2 με AdeHDVLig at 10˚C overnight). These reactions were bound to 486
MyOne C1 microbeads, washed with BB to remove unligated adaptor, and reverse transcribed 487
(50˚C 30 min) with 1 με HDVrec primer using Superscript III (Invitrogen). Beads were washed 488
again then PCR amplified (5 cycles with a 40˚C annealing step, then 20 cycles with a 50˚C 489
annealing step) using GoTaq HotStart master mix (Promega) and 0.8 με each of primers 490
P3HDV, and P5Xα8 or P5X 7, applying tags for high-throughput sequencing (Illumina MiSeq or 491
HiSeq) after PCR product agarose gel purification. + syntheses’ cDNAs were amplified with 492
P3HDV and P5X6. 493
494
Fidelity assay 495
To estimate RNA synthesis fidelity, ribozymes extended primers using ppp
NNN on 496
templates encoding CCC-XXX-CCC, where XXX were 12 different triplet sequences evenly 497
exploring base composition and distribution (see Supplementary file 3; for XXX = ACC, template 498
encodes CCC-ACC-UCC to avoid a terminal run of Gs). 499
Each primer/template pair (0.45/0.525 pmol per reaction) was annealed in 4 mM MgCl2, 500
1 mM tris•HCl pH 7.4 (80˚C 2 min, ramped to 4˚C at 0.1˚C/s, then kept on ice). The 12 pairs 501
were combined in 0.27M MgCl2/67 mM tris•HCl pH 8.3 on ice to discourage primer-template 502
assortment (of which sequencing later revealed negligible levels). 36 pmol of each triplet 503

24
(equivalent to 5 με final concentration after considering eutectic phase equilibration effects upon 504
this more dilute reaction) in ppp
NNN were added to a reaction vessel in 10.8 μl water, to which 505
5.4 μl of the primer/template/buffer mix was added followed by 7.2 pmol of ribozyme pre-506
annealed (80˚C 2 min 17˚C 10 min, ice >5 min) in 1.8 μl water (f.c. equivalent 1 με, in excess 507
over the 0.875 με template to which some ribozymes could tether to enhance extension). 508
Reactions were frozen and incubated (7 d at ˚C) as described above. 509
Reactions were stopped with 3.6 μl 0.44 M EDTA and 10.5 pmol of each template 510
competing oligonucleotide (migrating above product, with marker mutations to ensure exclusion), 511
denatured with 6 M urea, and urea-PAGE separated. After alignment with a fluorescence scan of 512
the gel, a region of the sample lane corresponding to primers extended by +4 to +14 nt was 513
excised (encompassing 2-4 triplet additions), and extension products were eluted, precipitated in 514
77% ethanol with 1 μl 1% glycogen carrier, washed in 85% ethanol and resuspended in water. 515
These extension products were γ’ adaptor ligated as for fragment sequencing. Products 516
were reverse transcribed (0.2 adaptor ligation reaction, 1 με HDVrec primer in Superscript III 517
reaction, 50˚C 30 min) and then PCR-amplified (1/30th
reverse transcription mix, 0.8 με each or 518
primers P3HDV and P5GGGX) for sequencing as above (yielding 2105-410
6 sequences per 519
ribozyme assay). 520
After processing and γ’ adaptor trimming, sequences corresponding to primer extended by 521
CCC + 1-3 additional triplets were collated for analysis. Variations in upstream primer sequences 522
(see Supplementary file 3) allowed the partner template to be identified for each sequenced 523
product; the triplet incorporated after the first CCC was counted. Separately, 10 μl extensions by 524
t5+1
of each primer/template alone with its encoded triplet and ppp
CCC (and ppp
UCC for the ACC 525
pair) were combined for purification and sequencing as above, to allow isolation of the ribozyme-526
mediated errors resulting from inclusion of the other 62 (61 for ACC) triplets in the reaction 527
(versus errors from sequencing, recombination etc.). The counts of cognate triplet (C) & each 528

25
error triplet (E) in the positive control (p) reduced error counts in the experimental samples (x) to 529
yield ribozyme-mediated error counts (Er) thusly: Er = Ex - Ep*(Cx/Cp) (not reducing Ex below 0, 530
and reallocating all reductions to Cr; ppp
CCC counts (and ppp
UCC for the ACC template) remained 531
uncorrected). 532
For each template, counts were then collated at the 1st/2
nd/3
rd positions to yield base-533
specific mutation rates for each position (Figure 8 - figure supplement 2, Figure 8 - source data 534
1). Across the 12 triplets, A, C, G, and U were encoded at each position three times; linear 535
averages were calculated to map the position’s error profile (Figure 8b) and geometric means of 536
the four nucleobases yielded the position’s overall fidelity (Figure 8a, Figure 8 - figure 537
supplement 1). 538
539
Triphosphorylated triplet synthesis 540
Triplets (and some other short oligonucleotides) were prepared from NTPs by T7 RNA 541
polymerase run-off transcription of a 5’ single-stranded DNA overhang downstream of a DNA 542
duplex T7 promoter sequence. In most cases the 5’ overhang encoded (was the reverse 543
complement of) the desired oligonucleotide. These oligonucleotides were short enough to 544
synthesise during the abortive initiation stage of transcription, attenuating sequence constraints on 545
the first bases of the transcript. However, T7 RNA polymerase exhibited tendencies to skip the 546
first (or even second) base (most severe for U>C>A>G before 2nd
position purines: encoding 547
CGU yielded some ppp
GU, encoding UAC yielded just ppp
AC) or use oligonucleotides generated 548
during transcription to re-initiate (e.g. encoding GAG yielded ppp
GAGAG, encoding AAA yielded 549
pppA6-9, encoding UCC yielded
pppCCC, encoding CGC yielded some
pppGCGC; this tendency was 550
most severe when the oligonucleotide could be accommodated opposite the final template bases 551
of the promoter). 552

26
These tendencies could be subverted by encoding additional first bases (usually without 553
providing the corresponding NTP). This initiated the oligonucleotide at the second position where 554
skipping tendencies were lower (e.g. encoding CUAG without CTP yielded ppp
UAG, encoding 555
UUAC yielded some ppp
UAC), and reduced recruitment as initiators of products with bases not 556
complementary to the introduced first position template base (e.g. encoding CGAG without CTP 557
yielded ppp
GAG, encoding CAA without CTP yielded ppp
AA and ppp
AAA, encoding AUCC 558
without ATP yielded ppp
UCC, encoding UCGC without UTP yielded ppp
CGC). 559
Each 30 μl transcription reaction contained 72 nmol of each desired product base as an 560
NTP (Roche) (e.g. for ppp
UCC, 72 nmol UTP, 144 nmol CTP) in 1 MegaShortScript kit buffer 561
with 1.5 μl MegaShortScript T7 enzyme (ThermoFisher). Also present were 15 pmol of each 562
DNA oligonucleotide forming the transcription duplex target (see Supplementary file 2). The 563
reactions were incubated overnight at γ7˚C, stopped with 3 μl 0.44 M EDTA and 17 μl 10 M 564
urea, and separated by electrophoresis (35 W, 4.5 h) on a 35180.15 cm 30% 19:1 565
acrylamide:bis-acrylamide 3 M urea tris-borate gel. Products were identified through their relative 566
migrations (reflecting overall composition, fastest to slowest: C>UA>G) by UV shadowing. 567
Triplet bands were excised and eluted overnight in 10 mM tris•HCl pH 7.4, and filtrate (Spin-X) 568
precipitated with 0.3 M sodium acetate pH 5.5 in 85% ethanol. Pellets were washed in 85% 569
ethanol, resuspended in water, and UV absorbances measured with a Nanodrop ND-1000 570
spectrophotometer. Oligocalc (Kibbe, 2007) was used to calculate sequence-specific 571
concentrations and yields. ppp
NNN was generated by combination of equal amounts of each of the 572
64 triplet stocks in a lo-bind microcentrifuge tube (Eppendorf). 573
γ’-deoxy triphosphorylated ‘terminator’ triplets were transcribed as above but using a γ’ 574
deoxynucleoside 5’ triphosphate (Trilink biotechnologies) for the last position, migrating faster 575
during PAGE than the equivalent all-RNA triplet. Triplets with 2-thiouridine residues were 576
transcribed as for their corresponding U, replacing UTP with U2S
TP (Jena Bioscience); 577

27
incorporation and migration were similar between the two, and their concentrations were 578
calculated from A260nm by comparison to the A260nm of mixtures of the component ribonucleotides 579
with UTP vs. U2S
TP. Triplets with β’-fluoro, β’-deoxy positions could also be transcribed, with 580
lower efficiency, by substituting the corresponding triphosphate (Trilink Biotechnologies). The 581
biotinylated ppp
GAU^^B triplet used in segment synthesis (Figure 7c) was transcribed as for 582
pppGAU, replacing UTP with biotin-16-aminoallyluridine-5’-triphosphate (Trilink 583
Biotechnologies), quantified via by comparison to the A290nm of mixtures of the component 584
ribonucleotides. 585
Longer triphosphorylated oligonucleotides used in ribozyme self-synthesis were generated 586
similarly, but using ~200 ng of fully double stranded DNA as a template. Candidate product 587
bands were purified and the desired oligonucleotide identified by ribozyme-catalysed in-frame 588
incorporation and, for some, fragment sequencing. 589
590
RNA oligonucleotide/ribozyme preparation 591
Transcriptions were performed on ~ 15 ng/μl dsDNA using MegaShortScript enzyme and 592
buffer (ThermoFisher) with 7.8 mM of each NTP, or, to yield a 5’ monophosphate on the product 593
to avoid aberrant ligation (‘GεP transcription’), 10 mM GMP and 2 mM of each NTP. 594
dsDNA templates for some of these (in Supplementary file 3) were generated (‘fill-in’) 595
using three cycles of mutual extension (GoTaq HotStart, Promega) between the associated DNA 596
oligonucleotide and 5T7 (or, where indicated, HDVrt for defined γ’ terminus formation (Schurer 597
et al., 2002)) followed by column purification (QiaQuick, Qiagen). 598
Some 5’ biotinylated RNAs were synthesized using the TGK polymerase (Cozens et al., 599
2012) (56 μg/ml, in 1 Thermopol buffer (NEB) supplemented with 3 mM MgCl2) to extend 5’ 600
biotinylated RNA primers (0.75 με) on DNA templates (1 με) using 2.5 mM of each NTP 601

28
(94˚C 30 s, 45˚C 2 min, 65˚C 30 min, 45˚C 2 min, 65˚C 30 min, then all repeated). Biotinylated 602
products were bead-purified as above. 603
γ’ biotinylation of RNAs was achieved in two stages: γ’ azidylation (at 2 με with 25 U/μl 604
yeast poly-A polymerase (ThermoFisher) and 0.5 mM β’-azido-β’-deoxycytidine triphosphate 605
(Trilink Biotechnologies) for 1 h at γ7˚C) with subsequent acidic phenol/chloroform extraction 606
and 75% ethanol precipitation, then copper-catalysed biotin-(PEG)4-alkyne (ThermoFisher) 607
cycloaddition (Winz et al., 2012) with subsequent 75% ethanol precipitation followed by 608
resuspension and buffer exchange in Ultracel 3K filters (Amicon) to remove residual biotin-609
alkyne. 610
611
Selection library synthesis 612
Round 1 libraries were synthesised by mutual extension of 4 nmol of oligonucleotides 613
1baN30 and 1GMPfo or 1GTPfo at 1 με each in 1 isothermal amplification buffer (NEB) with 614
250 με each dNTP, annealed (80˚C 3 min, 65 ˚C 5 min) before addition of 0.4 U/μl Bst 2.0 615
(NEB) and 30 min 65˚C incubation. 616
After purification, 375 μg of each DNA (~1.51015
molecules) were transcribed in 5 ml 617
transcription reactions (36 mM tris•HCl pH 7.9 (at 25˚C), 1.8 mM spermidine, 9 mM DTT, 618
10.8 mM MgCl2, 2 mM each NTP, 1% 10 MegaShortScript buffer, 2% 1:9 619
MegaShortScript:NEB T7 RNA polymerase, γ7˚C overnight). These were treated with DNase, 620
acid phenol/chloroform extracted and 73% ethanol precipitated prior to urea-PAGE purification, 621
elution, filtering (Spin-X) and re-precipitation, yielding the 1GTP Zcore selection construct 622
(Supplementary file 1). 10 mM GMP was present in transcriptions of the 1GMP construct, and for 623
future transcriptions of the GMP construct selection branch and rounds 8-18; round 19-21 and 624
reselection libraries were transcribed without GMP. Most subsequent selection rounds were 625

29
transcribed in 1/10th
scale transcriptions with 15 μg of DNA (~61013
molecules) derived from 626
amplification of recovered PCR products (see later). 627
For round 8, 700 pmol DNA was formed with Tri3CUUQ, amplifying round 7 merged 628
output (50 pmol), round 7 merged output recombined by StEP (Zhao & Zha, 2006) (200 pmol), 629
and 0core ribozyme with the starting γ’ 30N library domain added (450 pmol, but extinct at the 630
end of selection). DNA encoding type 5s amplified with AACAt5s was used to generate 631
reselection libraries by PCR amplification using primers TriGAA7GAAM and 632
T5ba13N/T5ba20N/T5ba28N; 5 pmol of the three dsDNA products were transcribed to generate 633
reselection constructs. 634
635
In vitro evolution cycle 636
An outline of the selection strategy is shown in Figure 1 - figure supplement 2, with 637
detailed lists of selection oligonucleotides and extension parameters in Figure 1 - source data 1, 638
Figure 2 - source data 1, Figure 4 - source data 1 and Supplementary file 3. Selection construct 639
was first annealed with equimolar dual-5’ biotinylated primer in water (80˚C 2-4 min, 17˚C 10 640
min), then chilled extension buffer and triplets were added before freezing and 7˚C incubation. 641
At the end of incubation the reaction was thawed on ice. To link the primer γ’ hydroxyl to 642
the 5’ monophosphate of GMP constructs, selection constructs were buffer-exchanged directly 643
after thawing using a PD-10 column (GE) in a cold room, into 3 ml ligation mix (optimised to 644
prevent ligation over gaps) (2 mM MgCl2, 50 mM tris•HCl pH 7.4, 0.1 mM ATP, 1 mM DTT, 645
2 με HO
GCG (Rounds 1-7) or 2 με HO
CUG (Rounds 8-18) with 30 U/ml T4 RNA Ligase 2 646
(NEB)). After incubation at 4˚C for 1 h, these were stopped with 2.2 mM EDTA and acid 647
phenol/chloroform treated. 648
Constructs were then precipitated with glycogen carrier and 0.3 M sodium acetate in 649
isopropanol (55%) before resuspension and denaturation (94˚C 4 min, in 6M urea 10 mM EDTA 650

30
with a 3 excess of competing oligonucleotide against the primer). For the reselection rounds, 651
constructs were then treated with polynucleotide kinase (NEB) before denaturation to resolve the 652
HDV-derived β’, γ’-cyclic phosphates and allow later adaptor ligation. 653
Constructs were urea-PAGE separated alongside FITC-labelled RNAs equivalent to 654
successfully ligated constructs. The gel region in the construct lane alongside these was excised to 655
exclude the bulk unreacted construct (judged by UV shadowing). Biotinylated (primer-linked) 656
constructs were eluted overnight into BB with 100 μg MyOne C1 beads. After 30 μm filtering 657
(Partec Celltrics) of the supernatant to remove gel fragments, the beads were washed in BB then 658
denaturing buffer (8 M urea, 50 mM tris•HCl pH 7.4, 1 mM EDTA, 0.1% Tween-20, 10 με 659
competing oligonucleotide, 60˚C 2 min) to confirm covalent linkage of construct to primer, 660
before further BB washing and transfer (to a fresh microcentrifuge tube to minimize downstream 661
contamination). At this stage in the reselection, γ’ adaptors were then ligated to bead-bound 662
constructs as above for 2 h (with buffer/enzyme added after bead resuspension in other reaction 663
components including 0.04% Tween-20), and beads BB washed and transferred again. 664
Bead-bound constructs were now reverse-transcribed using 1 με RTri (or HDVRec for 665
the reselection) by resuspension in a Superscript III reaction with added 0.02% Tween-20 (50˚C 666
30 min). Beads were BB washed and the RNA-bound cDNA γ’ end blocked by incubation with 667
terminal deoxynucleotidyl transferase (ThermoFisher) and 0.2 mM dideoxy-ATP (TriLink) with 668
0.02% added Tween-20 (γ7˚C 30 min), and beads were BB washed and transferred again. 669
cDNAs were eluted (10 μl 0.1 M NaOH 0.1% Tween-20 20 min), neutralized and plus 670
strands regenerated with 0.2 με rescue oligonucleotide in an IsoAmp II universal tHDA kit 671
(NEB) reaction (65˚C 60 min) to read through the structured product region. Whilst at this 672
temperature, reactions were stopped with 5 mM EDTA and 1 vol BB with 50-100 μg beads to 673
bind the nascent biotinylated plus strands at room temperature. These beads were then BB 674

31
washed, NaOH washed again to discard cDNAs (and recover only correctly-primed plus strands), 675
and washed and transferred again. 676
Each 50 μg beads were then subjected to plus strand recovery PCR in a 100 μl GoTaq 677
HotStart reaction with 0.5 με each RTri (or HDVrec for reselection) and RecInt (rounds 1-5) / 678
RecIntQ (rounds 6-9) / RecIntL (rounds 10-14, 19-21 & reselection) / RecIntQL (rounds 15-18). 679
The product was agarose size-purified, A260nm quantified and added to construct synthesis PCR in 680
3 molar amount of the anticipated recovered RNA (judged by test extensions) that yielded it. 681
This final PCR for construct transcription in the subsequent selection round used 1 με of 682
the indicated construct synthesis primer, plus 1 με RTri (or HDVrt for the reselection), in GoTaq 683
HotStart reactions or (where indicated in source data) the GeneMorph II kit for mutagenesis 684
(Agilent). 685

32
References and Notes 686
Attwater, J., Tagami, S., Kimoto, M., Butler, K., Kool, E. T., Wengel, J., Herdewijn, P., Hirao, I., 687
& Holliger, P. (2013a). Chemical fidelity of an RNA polymerase ribozyme. Chemical 688
Science. doi: 10.1039/C3SC50574J 689
Attwater, J., Wochner, A., & Holliger, P. (2013b). In-ice evolution of RNA polymerase ribozyme 690
activity. Nature Chemistry, 5 (12), 1011-1018. doi: 10.1038/nchem.1781 691
Attwater, J., Wochner, A., Pinheiro, V. B., Coulson, A., & Holliger, P. (2010). Ice as a 692
protocellular medium for RNA replication. Nat Commun, 1 (6), 76. doi: 693
10.1038/ncomms1076 694
Battle, D. J., & Doudna, J. A. (2002). Specificity of RNA-RNA helix recognition. Proc Natl Acad 695
Sci U S A, 99 (18), 11676-11681. doi: 10.1073/pnas.182221799 696
Boza, G., Szilagyi, A., Kun, A., Santos, M., & Szathmary, E. (2014). Evolution of the division of 697
labor between genes and enzymes in the RNA world. PLoS Comput Biol, 10 (12), 698
e1003936. doi: 10.1371/journal.pcbi.1003936 699
Cozens, C., Pinheiro, V. B., Vaisman, A., Woodgate, R., & Holliger, P. (2012). A short adaptive 700
path from DNA to RNA polymerases. Proc Natl Acad Sci U S A, 109 (21), 8067-8072. 701
doi: 10.1073/pnas.1120964109 702
Crick, F. H. (1968). The origin of the genetic code. J Mol Biol, 38 (3), 367-379. 703
Doudna, J. A., Couture, S., & Szostak, J. W. (1991). A multisubunit ribozyme that is a catalyst of 704
and template for complementary strand RNA synthesis. Science, 251 (5001), 1605-1608. 705
Doudna, J. A., Usman, N., & Szostak, J. W. (1993). Ribozyme-catalyzed primer extension by 706
trinucleotides: a model for the RNA-catalyzed replication of RNA. Biochemistry, 32 (8), 707
2111-2115. 708
Eigen, M. (1971). Selforganization of matter and the evolution of biological macromolecules. 709
Naturwissenschaften, 58 (10), 465-523. 710
Eisinger, J., & Gross, N. (1975). Conformers, Dimers, and Anticodon Complexes of Transfer-711
Rna-Glu2 (Escherichia-Coli). Biochemistry, 14 (18), 4031-4041. doi: Doi 712
10.1021/Bi00689a016 713
Filonov, G. S., Kam, C. W., Song, W., & Jaffrey, S. R. (2015). In-gel imaging of RNA processing 714
using broccoli reveals optimal aptamer expression strategies. Chem Biol, 22 (5), 649-660. 715
doi: 10.1016/j.chembiol.2015.04.018 716
Filonov, G. S., Moon, J. D., Svensen, N., & Jaffrey, S. R. (2014). Broccoli: rapid selection of an 717
RNA mimic of green fluorescent protein by fluorescence-based selection and directed 718
evolution. J Am Chem Soc, 136 (46), 16299-16308. doi: 10.1021/ja508478x 719
Gilbert, Walter. (1986). Origin of life: The RNA world. Nature, 319 (6055), 618-618. 720
Green, R., & Szostak, J. W. (1992). Selection of a ribozyme that functions as a superior template 721
in a self-copying reaction. Science, 258 (5090), 1910-1915. 722
Hayden, E. J., & Lehman, N. (2006). Self-assembly of a group I intron from inactive 723
oligonucleotide fragments. Chem Biol, 13 (8), 909-918. doi: 724
10.1016/j.chembiol.2006.06.014 725

33
Horning, D. P., & Joyce, G. F. (2016). Amplification of RNA by an RNA polymerase ribozyme. 726
Proc Natl Acad Sci U S A, 113 (35), 9786-9791. doi: 10.1073/pnas.1610103113 727
Huang, S. C., Smith, J. R., & Moen, L. K. (1992). Contribution of the p51 subunit of HIV-1 728
reverse transcriptase to enzyme processivity. Biochem Biophys Res Commun, 184 (2), 729
986-992. 730
Johnston, W. K., Unrau, P. J., Lawrence, M. S., Glasner, M. E., & Bartel, D. P. (2001). RNA-731
catalyzed RNA polymerization: accurate and general RNA-templated primer extension. 732
Science, 292 (5520), 1319-1325. 733
Kibbe, W. A. (2007). OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids 734
Res, 35 (Web Server issue), W43-46. 735
Lawrence, M. S., & Bartel, D. P. (2003). Processivity of ribozyme-catalyzed RNA 736
polymerization. Biochemistry, 42 (29), 8748-8755. 737
Lincoln, T. A., & Joyce, G. F. (2009). Self-sustained replication of an RNA enzyme. Science, 323 738
(5918), 1229-1232. 739
Markham, N. R., & Zuker, M. (2005). DINAMelt web server for nucleic acid melting prediction. 740
Nucleic Acids Res, 33 (Web Server issue), W577-581. doi: 10.1093/nar/gki591 741
Martin, L. L., Unrau, P. J., & Muller, U. F. (2015). RNA synthesis by in vitro selected ribozymes 742
for recreating an RNA world. Life (Basel), 5 (1), 247-268. doi: 10.3390/life5010247 743
Monnard, P. A., Kanavarioti, A., & Deamer, D. W. (2003). Eutectic phase polymerization of 744
activated ribonucleotide mixtures yields quasi-equimolar incorporation of purine and 745
pyrimidine nucleobases. J Am Chem Soc, 125 (45), 13734-13740. 746
Mutschler, H., Wochner, A., & Holliger, P. (2015). Freeze-thaw cycles as drivers of complex 747
ribozyme assembly. Nature Chemistry, 7 (6), 502-508. doi: 10.1038/nchem.2251 748
Noller, H. F. (2012). Evolution of protein synthesis from an RNA world. Cold Spring Harb 749
Perspect Biol, 4 (4), a003681. doi: 10.1101/cshperspect.a003681 750
O'Flaherty, D. K., Kamat, N. P., Mirza, F. N., Li, L., Prywes, N., & Szostak, J. W. (2018). 751
Copying of Mixed-Sequence RNA Templates inside Model Protocells. J Am Chem Soc. 752
doi: 10.1021/jacs.8b00639 753
Ogle, J. M., Brodersen, D. E., Clemons, W. M., Jr., Tarry, M. J., Carter, A. P., & Ramakrishnan, 754
V. (2001). Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science, 755
292 (5518), 897-902. doi: 10.1126/science.1060612 756
Orgel, L. E. (1968). Evolution of the genetic apparatus. J Mol Biol, 38 (3), 381-393. 757
Paillart, J. C., Shehu-Xhilaga, M., Marquet, R., & Mak, J. (2004). Dimerization of retroviral RNA 758
genomes: an inseparable pair. Nat Rev Microbiol, 2 (6), 461-472. doi: 10.1038/nrmicro903 759
Petrov, A. S., Gulen, B., Norris, A. M., Kovacs, N. A., Bernier, C. R., Lanier, K. A., Fox, G. E., 760
Harvey, S. C., Wartell, R. M., Hud, N. V., & Williams, L. D. (2015). History of the 761
ribosome and the origin of translation. Proc Natl Acad Sci U S A, 112 (50), 15396-15401. 762
doi: 10.1073/pnas.1509761112 763
Poole, A. M., Jeffares, D. C., & Penny, D. (1998). The path from the RNA world. J Mol Evol, 46 764
(1), 1-17. 765
Prywes, N., Blain, J. C., Del Frate, F., & Szostak, J. W. (2016). Nonenzymatic copying of RNA 766
templates containing all four letters is catalyzed by activated oligonucleotides. Elife, 5. 767
doi: 10.7554/eLife.17756 768

34
Rohatgi, R., Bartel, D. P., & Szostak, J. W. (1996). Kinetic and mechanistic analysis of 769
nonenzymatic, template-directed oligoribonucleotide ligation. J Am Chem Soc, 118 (14), 770
3332-3339. 771
Schurer, H., Lang, K., Schuster, J., & Morl, M. (2002). A universal method to produce in vitro 772
transcripts with homogeneous 3' ends. Nucleic Acids Res, 30 (12), e56. 773
Sczepanski, J. T., & Joyce, G. F. (2014). A cross-chiral RNA polymerase ribozyme. Nature, 515 774
(7527), 440-442. doi: 10.1038/nature13900 775
Shechner, D. M., Grant, R. A., Bagby, S. C., Koldobskaya, Y., Piccirilli, J. A., & Bartel, D. P. 776
(2009). Crystal structure of the catalytic core of an RNA-polymerase ribozyme. Science, 777
326 (5957), 1271-1275. 778
Suslov, N. B., DasGupta, S., Huang, H., Fuller, J. R., Lilley, D. M., Rice, P. A., & Piccirilli, J. A. 779
(2015). Crystal structure of the Varkud satellite ribozyme. Nat Chem Biol, 11 (11), 840-780
846. doi: 10.1038/nchembio.1929 781
Szostak, J. W. (2011). An optimal degree of physical and chemical heterogeneity for the origin of 782
life? Philos Trans R Soc Lond B Biol Sci, 366 (1580), 2894-2901. doi: 783
10.1098/rstb.2011.0140 784
Szostak, J. W. (2012). The Eightfold Path to non-enzymatic RNA replication. J. Syst. Chem., 3. 785
Szostak, J. W., Bartel, D. P., & Luisi, P. L. (2001). Synthesizing life. Nature, 409 (6818), 387-786
390. 787
Tagami, S., Attwater, J., & Holliger, P. (2017). Simple peptides derived from the ribosomal core 788
potentiate RNA polymerase ribozyme function. Nature Chemistry, 9 (4), 325-332. doi: 789
10.1038/nchem.2739 790
Vaidya, N., Manapat, M. L., Chen, I. A., Xulvi-Brunet, R., Hayden, E. J., & Lehman, N. (2012). 791
Spontaneous network formation among cooperative RNA replicators. Nature, 491 (7422), 792
72-77. doi: 10.1038/nature11549 793
Vaish, N. K., Jadhav, V. R., Kossen, K., Pasko, C., Andrews, L. E., McSwiggen, J. A., Polisky, 794
B., & Seiwert, S. D. (2003). Zeptomole detection of a viral nucleic acid using a target-795
activated ribozyme. Rna, 9 (9), 1058-1072. 796
Vlassov, A., Khvorova, A., & Yarus, M. (2001). Binding and disruption of phospholipid bilayers 797
by supramolecular RNA complexes. Proc Natl Acad Sci U S A, 98 (14), 7706-7711. doi: 798
10.1073/pnas.141041098 799
Vogel, S. R., Deck, C., & Richert, C. (2005). Accelerating chemical replication steps of RNA 800
involving activated ribonucleotides and downstream-binding elements. Chem Commun 801
(Camb) (39), 4922-4924. 802
Warner, K. D., Sjekloca, L., Song, W., Filonov, G. S., Jaffrey, S. R., & Ferre-D'Amare, A. R. 803
(2017). A homodimer interface without base pairs in an RNA mimic of red fluorescent 804
protein. Nat Chem Biol, 13 (11), 1195-1201. doi: 10.1038/nchembio.2475 805
Weiss, Robert, & Cherry, Joshua. (1993). 3 Speculations on the Origin of Ribosomal 806
Translocation. 807
Winz, M. L., Samanta, A., Benzinger, D., & Jaschke, A. (2012). Site-specific terminal and 808
internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-809
free strain-promoted click chemistry. Nucleic Acids Res, 40 (10), e78. doi: 810
10.1093/nar/gks062 811

35
Wochner, A., Attwater, J., Coulson, A., & Holliger, P. (2011). Ribozyme-catalyzed transcription 812
of an active ribozyme. Science, 332 (6026), 209-212. 813
Zaher, H. S., & Unrau, P. J. (2007). Selection of an improved RNA polymerase ribozyme with 814
superior extension and fidelity. Rna, 13 (7), 1017-1026. 815
Zhang, W., Tam, C. P., Zhou, L., Oh, S. S., Wang, J., & Szostak, J. W. (2018). Structural 816
Rationale for the Enhanced Catalysis of Nonenzymatic RNA Primer Extension by a 817
Downstream Oligonucleotide. J Am Chem Soc. doi: 10.1021/jacs.7b11750 818
Zhao, H., & Zha, W. (2006). In vitro 'sexual' evolution through the PCR-based staggered 819
extension process (StEP). Nat Protoc, 1 (4), 1865-1871. 820
Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic 821
Acids Res, 31 (13), 3406-3415. 822
823
824
Acknowledgments: We thank S. Thomsen, M. McKie and M. Sharrock for important 825
discussions, and B.T. Porebski and S.L. James for assistance with sequencing analysis. This work 826
was supported by a Homerton College junior research fellowship (J.A.), a Harvard-Cambridge 827
summer fellowship (A.R.) and by the Medical Research Council (J.A., E.G., A.M., P.H.; 828
programme no. MC_U105178804). 829
830
Conditions for selections are included as source data 1 for Figures 1, 2 and 4. Numerical data for 831
Figures 1c and 3b are included in Figure 1 - source data 2 and Figure 3 - source data 1 832
respectively. Numerical data and calculations for Figure 8 and its supplements 1 and 2 are 833
supplied as Figure 8 - source data 1. Numerical data and calculations for Figure 8 - figure 834
supplement 3 are supplied as Figure 8 - source data 2. A more extensive selection of substrate 835
concentration-dependent error rates is supplied in Figure 9 - source data 1. Sequences of 836
ribozymes, triplet synthesis templates, and oligonucleotides used in this study are supplied in 837
Supplementary files 1, 2 and 3 respectively. 838

36
Figure legends 839
Figure 1. Monomer polymerisation and triplet polymerisation. 840
(A) Scheme outlining initial derivation of a triplet polymerase activity from a mononucleotide 841
polymerase ribozyme via directed evolution. Z RPR truncation effects are shown in Figure 1 - 842
figure supplement 1, the selection cycle is outlined in Figure 1 - figure supplement 2, and the 843
selection conditions of rounds 1-7 are listed in Figure 1 - source data 1. Below, modes of action 844
and secondary structures of the mononucleotide polymerase ribozyme (Z RPR) and a triplet 845
polymerase ribozyme (0core), both depicted surrounding primer (tan) / template (grey) duplexes 846
with a mononucleoside triphosphate (NTP) or trinucleotide triphosphate (triplet) substrate present 847
(red). Here the templates are hybridised to the ribozyme upstream of the primer binding site, 848
flexibly tethered to enhance local concentration and activity (via L repeats of an AACA sequence, 849
e.g. L = 5 in templates SR1-4 below). Z RPR residues comprising its catalytic core (Zcore) are 850
black; mutations in 0core arising from directed evolution of Zcore are in teal. 851
(B) Primer extension by the Z RPR using monomers (1 mM NTPs) or by 0core using triplets 852
(5 με ppp
AUA & ppp
CGC) on a series of 6-nucleotide repeat templates (SR1-4, examples below) 853
with escalating secondary structure potential that quenches Z RPR activity beyond the shortest 854
template SR1 (7˚C ice 17 d, 0.5 με/RNA). Extension by the triplet polymerase ribozyme 0core 855
can overcome these structure tendencies up to the longest template SR4. 856
(C) Triplet concentration dependence of extension using templates SR1 (grey circles) and SR3 857
(black triangles) by 0core (ppp
AUA & ppp
CGC, 0.1 με of primer A10, template and ribozyme, 858
7˚C supercooled 15 d, ± s.d., n = 3); shown below is a model of cooperative triplet-mediated 859
unfolding of template SR3 structure to explain the sigmoidal triplet concentration dependence 860
(red curve) of extension upon it. Numerical values are supplied in Figure 1 - source data 2. 861
Figure 1 - source data 1. Selection conditions of rounds 1-7. 862
Figure 1 - source data 2. Triplet concentration-dependent extension values. 863
864
Figure 2. Emergence of cooperativity during in vitro evolution. 865
(A) Composition of the round 21 selection pool as a % of total pool sequences; selection 866
conditions of rounds 8-21 are listed in Figure 2 - source data 1. Secondary structures of ribozyme 867

37
type 1-6 archetypes are shown in Figure 2 - figure supplement 1, and comparison of their 868
activities to that of the polyclonal selection pool is shown in Figure 2 - figure supplement 2. 869
(B) Primer extension with triplets by these emergent triplet polymerase ribozyme types 2-6 (‘Rz’, 870
alongside the starting Zcore ribozyme with ‘cap+’ sequence from selection, see Figure 3a), alone 871
or with added truncated type 1 (+1, see Figure 3a) which boosted their triplet polymerase 872
activities (0.5 με Rzs/A10 primer/SR3 template, 5 με ppp
AUA & ppp
CGC, 7˚C ice 16 h). 873
Figure 2 - source data 1. Selection conditions of rounds 8-21. 874
875
Figure 3. Heterodimer formation and behaviour. 876
(A) Secondary structures of the most common type 1 and type 5 clones from the selection, with 877
in vitro-selected γ’ domain and core mutations coloured orange (type 1) or blue (type 5). 878
5’ truncation of type 1 (faded, including its putative primer/template interacting region), yielding 879
the minimal type 1 variant ‘1’, maintained its enhancement activity (see Figure 3 - figure 880
supplement 1). The effects of transplanting the indicated 5’ hairpin ‘cap+’ element from type 5 to 881
other ribozymes are shown in Figure 3 - figure supplement 2. The inset shows type 5 ‘cap+’ 882
hairpin element alteration to ‘cap–’ (yielding type 5cap-
). 883
(B) Gel mobility shift characteristic of complex formation resulting from mixing of type 5 884
ribozyme with type 1 RNA (equimolar, or with the indicated equivalents). Type 5cap-
loses this 885
shift and its susceptibility to type 1 activity enhancement (Figure 3 - figure supplement 1). Below, 886
shifted band intensities with increasing type 1 addition are plotted (quantified relative to the 887
indicated type 5+1
lane intensities, n = 4 ± s.d.), signifying 1:1 heterodimer formation; numerical 888
values are supplied in Figure 3 - source data 1. 889
(C) Type 1 enhancement allows type 5 variants to synthesise long RNAs using triphosphorylated 890
oligonucleotide (11 nt) or short triplet (3 nt) substrates (Sub, 3.6 με or 5 με each, substrate 891
sequences in grey beside lanes; 0.4 με primer A11/template I-8, 2 με each Rz, 7˚C ice for 892
16 d). This activity is independent of template tethering (Wochner et al., 2011), as comparable 893
synthesis is achieved by versions of type 5 whose 5’ regions allow or avoid hybridisation to the 894
template (type 5cis
or type 5trans
respectively, schematic above, sequences in Supplementary file 895
1). The average extent of ligation at the end of the reaction amongst all junctions in a lane is 896
shown beneath each lane. 897
Figure 3 - source data 1. Relative intensities of shifted bands when varying type 1 equivalents. 898

38
899
Figure 4. A trans-acting heterodimeric triplet polymerase. 900
Top, scheme outlining derivation of the final t5+1
triplet polymerase archetype from the type 1 901
and type 5 RNAs (shown in Figure 3a) by reselection using the conditions in Figure 4 - source 902
data 1. Below, the secondary structure of this ribozyme heterodimer, 135 nt (1) & 153 nt (t5) 903
long, is depicted operating in trans on a non-tethered primer/template duplex. Type 5 γ’ domain 904
bases that re-emerged after randomisation during reselection are coloured black in the t5 γ’ 905
domain. Ribozyme development is summarized in Figure 4 - figure supplement 1; all ribozyme 906
sequences are listed in Supplementary file 1. 907
Figure 4 - source data 1. Selection conditions of rounds 1-5 of the reselection. 908
909
Figure 5. Triplet-mediated structured and functional template copying. 910
(A) Extension on three structured hairpin templates (4S, 6S, 8S) with increasing stability. Top, the 911
mfold-predicted (Markham & Zuker, 2005) structure and TM of the most stable 8S template; 912
below, primer extensions on these templates by t5+1
triplet polymerase (with 5 με each encoded 913
triplet) or type 1-enhanced Z polymerase ribozyme (Z RPRcap+ +1
, with 1 mM each NTP) (2 με 914
ribozyme, 0.5 με 4S, 6S or 8S template and primer A9, 7˚C ice 25 d). The self-complementary 915
region in each template is indicated between each pair of lanes (shaded by triplet), with the 916
encoded triplet substrate sequences at the left (in grey, with 5’ template overhangs in brackets). 917
Syntheses using different substrate compositions and concentrations are shown in Figure 5 - 918
figure supplement 1 and Figure 5 - figure supplement 2. While all hairpin templates are robustly 919
copied by t5+1
, synthesis by the Z RPR is completely arrested by the 6S and 8S hairpins. 920
(B) Synthesis of the broccoli aptamer. The native secondary structure is shown above (Tan: bases 921
from ‘+’ strand synthesis primer. Green: bases from triplets. Outlined green: primer binding site 922
for the ‘’ strand synthesis). Below left, t5+1
-catalysed synthesis of fluorescent broccoli aptamer, 923
run alongside standard (Broc+, synthesized by in vitro transcription), and stained for RNA with 924
SYBR Gold (magenta) or folded with DFHBI-1T ligand (green fluorescence) (2 με t5+1
, 925
1 με BBrc10/TBrc, 5 με each triplet (in grey), -7˚C ice 22 d). Below right, ‘’ strand synthesis 926
on Broc+ standard (0.5 με without ligand in ribozyme extension buffer, 0.5 με FBrcb6 primer, 927
2 με t5+1
, 5 με each triplet (in grey), 7˚C ice 38 d). t5+1
is able to synthesize both full-length 928
functional (fluorescent) Broccoli ‘+’ and encoding ‘–’ strands. 929

39
930
Figure 6. Ribozyme self-synthesis and assembly of its own catalytic domain. 931
(A) t5+1
-catalysed syntheses of the five catalytic domain ‘+’ and ‘−’ segments via triplet extension 932
of primers (grey) in 7˚C ice. Triplets are coloured by segment and shown alongside the lanes; 933
longer oligonucleotide substrates (faded) were provided for α+ and
+ syntheses to combat 934
ribozyme-template pairing as shown in Figure 6 - figure supplement 2 and Figure 6 - figure 935
supplement 3. The triplets were supplied at 5 με (α+ to
+), 10 με ( −
to −), or 20 με (α−
) each, 936
with 0.5 με primer/template (P/T; 1 με for F 6/T ) and oligonucleotides equimolar to template 937
sites. Use of substrates of more heterogenous compositions and lengths are shown in Figure 6 - 938
figure supplement 4 and Figure 6 - figure supplement 5 respectively. Densitometry gave yields of 939
full-length products (boxed, by % of total primer), and geometric mean of the final extents of 940
ligation (78%) across all 70 junctions in this self-synthesis context. These segment sequences 941
derive from t5b, a t5 variant with a neutral signature mutation (Supplementary file 1). 942
(B) Secondary structure representation of a t5 catalytic domain (+/+
, t5b sequence), formed 943
via non-covalent assembly of t5+1
-synthesised ‘+’ strand fragments in Figure 6 - figure 944
supplement 1, coloured by segment and synthesis substrate as in (A). 945
(C) Activity of ribozyme-synthesised +/+
(B), compared to protein-synthesised +/+
946
and full-length t5b equivalents. These were assayed for synthesis of a -
strand segment on a 947
ribozyme-synthesised + template, with added in vitro transcribed type 1 (2 με each Rz, 5 με 948
triplets, 0.5 με P/T, 7˚C 0.25× ice 10 d). The ribozyme-synthesized and assembled +/+
949
ribozyme is as active as in vitro transcribed equivalents, and can efficiently utilize ribozyme-950
synthesized RNA (+) as a template. 951
952
Figure 7. Triplet-initiated template sequence copying. 953
(A) Extension by t5+1
of fluorescein-labelled primers bound to either the γ’ (A10) or 5’ (pppba) 954
ends of a template (T8GAA, 10 με ppp
GAA, 0.5 με/RNA, 7˚C ice 69 h), demonstrating 955
extension in either 5’-γ’ (A10) or γ’-5’ (pppba) directions. 956
(B) Synthesis of + on T template via t5
+1-catalysed polymerisation of the substrates indicated 957
on the right (2 με t5+1
, 5 με each triplet, 0.5 με template (lane 2: 0.5 με hexanucleotide), 7˚C 958
ice 9 d). Extension products in lanes 2-5 were eluted from template, PAGE-separated and SYBR-959

40
Gold stained alongside in vitro transcribed full-length segment control (lane 1). Lane 3 shows 960
full-length synthesis of +
segment from triplets alone. 961
(C) Triplet based replication of the segment. Top left, synthesis of + from the indicated 962
substrates (1 μM t5+1
, 5 με each triplet, 2 με THP template (lane 2: 2 με Bio7 primer), 7˚C 963
ice 7 d). Biotinylated extension products in lanes 2-4 were isolated from template, PAGE-964
separated and SYBR-Gold stained alongside in vitro transcribed staining marker (M+m1, lane 1). 965
This indicated 10% yield (per template) of full-length synthesis of + segment from triplets alone 966
(the final band in lane 4), which was purified for use as a template in - segment synthesis 967
(bottom right, 1 με t5+1
, 5 με each triplet, 0.05 με template, with 0.05% Tween-20, 7˚C ice 968
27 d). Extension products in lanes 5 & 6 were eluted from template, PAGE-separated and SYBR-969
Gold stained alongside in vitro transcribed full-length segment control (M -m1, lane 7). This 970
indicated 6% yield (per template) of full-length synthesis of - segment from triplets alone (the 971
final band in lane 6). 972
973
Figure 8. Fidelity of ribozyme-catalysed triplet polymerisation. 974
(A) To estimate its fidelity, each ribozyme was provided with an equimolar mix of all 64 triplet 975
substrates (ppp
NNN at 5 με each) for primer extension using templates containing twelve 976
representative trinucleotide sequences (NNN). Deep sequencing of extension products 977
identified the triplets added opposite each template trinucleotide, yielding position-specific error 978
tendencies; the overall fidelity was calculated as a geometric mean of positional errors at each 979
triplet position (n and s.d. of this value shown for ribozymes assayed multiple times, see Figure 8 980
- source data 1 for analysis of collated errors). The triplet polymerases exhibit diverse fidelity 981
profiles; fidelity profiles of other type 5 variants are shown in Figure 8 - figure supplement 1. 982
(B) Collation of error rates by base type and position for type 5 with (t5+1
) and without (α +1) 983
the fidelity domain. Positional and overall fidelities are calculated as geometric means (see 984
Figure 8 - source data 1); individual positional fidelities are plotted in Figure 8 - figure 985
supplement 2. 986
(C) Schematic summary of effects of triplet minor groove modification upon the fidelity 987
phenotype. In the depicted trinucleotide RNA duplex segment, spheres represent minor groove 988
groups potentially available for hydrogen bonding in a sequence-general manner. For three of 989
these groups (highlighted in black), we assayed whether their modification in substrates (2F = β’ 990

41
fluoro, 2S = 2-thio) affected the fidelity domain’s mismatch discrimination capabilities (detailed 991
in Figure 8 - figure supplement 3, with data and calculations in Figure 8 - source data 2). These 992
groups are labelled with the fraction of fidelity phenotype retained () when discriminating 993
between the indicated modified substrates. Colour reflects the impact of that group’s modification 994
upon the fidelity phenotype, with red denoting a strong disruptive effect, and yellow weak or 995
negligible effects. 996
Figure 8 - source data 1. Analysis of collated errors by ribozymes in the fidelity assay. 997
Figure 8 - source data 2. Calculation of residual fidelity phenotypes in Figure 8c. 998
999
Figure 9. Substrate pool interactions improve triplet fidelity. 1000
Applying the fidelity assay (Figure 8a, using t5+1
) to single templates with only an encoded triplet 1001
and a mispairing one as substrates (at 5 με each), we observed that relative mispair incorporation 1002
was proportionally reduced (by 61% (left) and 73% (right)) upon introduction of complementary 1003
triplets. Using all 64 triplets (ppp
NNN) has an analogous effect upon these pair/mispair 1004
comparisons with fidelity progressively improved upon increasing overall ppp
NNN concentrations, 1005
with example effects on other triplets and overall fidelity presented in Figure 9 - figure 1006
supplement 1, and comprehensive error rates and ratios in Figure 9 - source data 1. 1007
Figure 9 - source data 1. Collated error rates and ratios at different substrate concentrations. 1008

42
Figure supplement legends 1009
Figure 1 - figure supplement 1. Templated ligase activity from a mononucleotide 1010
polymerase. 1011
Extension of primer A11 on template HTI by the Z RPR (Wochner et al., 2011) and a γ’ 1012
truncation of it (Zcore, Figure 1a) at 7˚C in ice for 8 d (0.5 με of each RNA), adding as 1013
substrates either a long oligonucleotide (‘11 nt’, pppGCGAAGCGUGU at 0.5 με), a triplet 1014
(‘γ nt’, pppGCG at 5 με), or NTPs (‘1 nt’, at 1 mM each). Gel densitometry was used to calculate 1015
the percentage of primer extended (shown below each lane). Z RPR has only a very limited 1016
template ligation activity (slightly exceeding background nonenzymatic ligation (Rohatgi et al., 1017
1996)). In contrast, γ’ truncation (which abolished mononucleotide polymerase activity) 1018
accommodated and enhanced templated ligation. 1019
1020
Figure 1 - figure supplement 2. Selection scheme for in vitro evolution of triplet polymerase 1021
activity. 1022
Illustration of the in vitro evolution strategy to enrich iterative templated ligase activity from 1023
libraries of selection constructs. Centre, an example selection construct is shown comprising 1024
Zcore with a γ’ 30 nt random domain followed by a fixed hairpin-forming reverse transcription 1025
site (1). This construct is flexibly tethered at its 5’ in cis (Tagami et al., 2017; Attwater et al., 1026
2010) (via L repeats (3-8) of a flexible AACA linker) to a template region (grey) bound to 5’-dual 1027
biotin modified primer (tan). This construct requires successive polymerisation of two ppp
UCG 1028
triplet substrates (red) in ˚C ice followed by self-ligation to its triphosphorylated 5’ terminus to 1029
attach itself to the biotinylated primer (2). Active triplet polymerases are then identified and size-1030
purified by denaturing PAGE to deplete unreacted ribozymes and primer (3), then captured on 1031
streptavidin beads and subjected to a denaturing wash to ensure covalent linkage to primer. 1032
Remaining dual-purified bead-linked ribozymes were then reverse transcribed (4). cDNAs were 1033
eluted with NaOH and neutralised, before acting as templates for ‘+’ strand DNA synthesis from 1034
5’ biotinylated ‘rescue’ oligonucleotides that only prime correctly on the complete synthesised 1035
sequence (5), ensuring that constructs that deviated from the specified templated triplet 1036
polymerization fail to prime on cDNA preventing their ‘+’ strand synthesis. ‘+’ strands were then 1037
captured on fresh streptavidin beads, stripped of cDNAs, reamplified and diversified to generate 1038
selection construct for a new round of selection (6). See Materials and Methods for protocol 1039

43
details and Figure 1 - source data 1, Figure 2 - source data 1, Figure 4 - source data 1 and 1040
Supplementary file 3 for oligonucleotides used in selections. 1041
1042
Figure 2 - figure supplement 1. Secondary structures of type 1-6 ribozymes. 1043
The archetypes shown are active truncations of the most common/enriched variants of each type. 1044
Types 2-6 are depicted hybridised to a flexibly tethered primer (tan) / template (grey) / substrate 1045
(red) duplex analogous to the selection construct (Figure 1 - figure supplement 2), supporting 1046
extension on the SR1-4 templates (Figure 2b). Coloured bases deviate from Zcore or derive from 1047
the random sequence region. Type 1, the most common type in the output, displayed a high 1048
degree of sporadic mutation in the catalytic core domain, consistent with a loss of catalytic 1049
function. Shown here are the four most common core mutations, which were each present in 1050
>45% of type 1 sequences. 1051
1052
Figure 2 - figure supplement 2. Clonal versus polyclonal activity. 1053
Type 1 & 2 RNAs, and the indicated polyclonal ribozyme pools late in the selection, were 1054
transcribed as selection constructs generated with Tri8AUAM (then annealed to primer A10). 1055
Their abilities to incorporate eight ppp
AUA triplets then self-ligate in this selection construct 1056
context were compared (left, 0.1 με each RNA, 2 με ppp
AUA, in 7˚C ice for 5 d; right, 0.2 με 1057
each RNA, 2 με ppp
AUA and ppp
AUGγ’d
, in 7˚C ice for 9 d). 1058
Left: Despite being the most active type (Figure 2b), type 2 activity fell far short of the round 19 1059
pool polyclonal activity. 1060
Right: The incorporation of wobble pairing γ’-deoxy ‘terminator’ triplet ppp
AUGγ’d
(see results 1061
section on fidelity, yielding a faster-migrating band than incorporation of the cognate ppp
AUA 1062
triplet) was decreased sharply after round 19, as calculated by densitometry and averaging 1063
amongst the ligation junctions in each lane, indicative of enrichment of higher fidelity triplet 1064
polymerases in the selection pool. 1065
1066
Figure 3 - figure supplement 1. Parameters of type 1 activity enhancement. 1067
(A) PAGE of primer extension reactions comprising combinations of type 5 and type 1 variants 1068
(0.2 με primer A10, template SR3, and type 5 variant, here annealed together and combined with 1069

44
0.2 με of separately-annealed type 1 variant after buffer addition; 7˚C in ice for 63 h, 2 με 1070
each ppp
AUA, ppp
AUGγ’d
, ppp
CGC). Full-length type 1 enhanced the activity of type 5 as did 5’ 1071
truncated type 1 (‘1’, Figure 3a) without its duplex-interacting A-minor motif (Shechner et al., 1072
2009) and its ‘cap’ sequence (originally present on some type 1 sequences in the selection). 1073
Replacing the type 5 ribozyme’s selected 5’ ‘cap+’ sequence with ‘cap-’ (type 5cap-
) abolished its 1074
enhancement by type 1 RNA. 1075
(B) Stoichiometry of type 1 enhancement of type 3 activity (0.5 με primer A10, template SR3, 1076
2 με each ppp
AUA, ppp
GUAγ’d
, ppp
CGC in -7˚C ice for 20 h); quantification of average extension 1077
per primer in each lane is consistent with the 1:1 stoichiometry implied by type 1’s ~50% 1078
abundance in the round 21 selection pool. 1079
1080
Figure 3 - figure supplement 2. Type 1 enhancement of parental ribozymes. 1081
Left, transplanting the conserved 5’ ‘cap+’ sequence from types 2-6 allows the Zcore but not the 1082
0core ribozyme triplet polymerase activity to be enhanced by type 1; shown are PAGE of 1083
extensions of primer A10 on template CCCMisAUG by the indicated ribozyme cores (5 με of 1084
pppAUG and
pppCCC, 0.5 με of each RNA with type 1 annealed and added separately, 7˚C in ice 1085
for 22 h). Right, RNA polymerase activity of Z RPR using NTPs is enhanced by type 1 addition, 1086
but only when modified with the 5’ ‘cap+’ sequence (Z RPRcap+
), extending primer A10 on 1087
tethered template HTI (0.5 με of each RNA, 4 mM of each NTP, at 4˚C for 68 h). 1088
1089
Figure 4 - figure supplement 1. Summary of ribozyme development in this work. 1090
Left, map of triplet polymerase selection and engineering. Ribozyme sequences are listed in 1091
Supplementary file 1. Right, secondary structures of the central ribozymes in this work. 1092
1093
Figure 5 - figure supplement 1. Substrates for structured template copying. 1094
Primer extension reactions are shown using sets of defined triplet substrates (above the lanes), or 1095
random pools of all 64 triplets (ppp
NNN), each at the indicated concentrations on three templates 1096
with progressively longer, more stable secondary structure elements (right, TMs estimated using 1097
(Zuker, 2003)) (0.5 με primer A9, 0.5 με 4S, 6S, 8S templates, 2 με t5+1
, 7˚C ice for 25 d). 1098
Structure invasion and primer extension across the structured template region (beginning at the 1099

45
dotted red line on the image) occurred in all cases (with increased efficiency at higher substrate 1100
concentrations for the 8S template (5µM)), including extensions where template secondary 1101
structures were allowed to form in the absence of triplets (*, triplets added last after 30 min 1102
supercooled at -7˚C). 1103
1104
Figure 5 - figure supplement 2. Substrate concentration dependence of structured template 1105
copying. 1106
Extension on the 8S structured template with a defined set of complementary triplets (left) or 1107
dinucleotides (right), at the indicated substrate concentrations (0.5 με primer A9, template 8S, 1108
2 με t5+1
, 7˚C ice for 25 d). Left, quantification of the extent of extension into the structured 1109
template region below each lane (measured by the fraction of third triplet addition to primers 1110
already extended by two) recapitulates a sigmoidal relationship with triplet concentration. Right, 1111
dinucleotides (like mononucleotides) exhibit negligible extension into the template hairpin 1112
structure. 1113
1114
Figure 6 - figure supplement 1. Ribozyme catalytic domain self-synthesis and assembly. 1115
Top, conditions and yields for self-synthesis and assembly of the catalytic t5 domain (as α + and 1116
+ fragments, Figure 6b) by t5
+1. 1117
To obtain maximal amounts of fully synthesized and assembled t5b ribozyme for activity testing 1118
(Figure 6c), we implemented t5 ‘+’ strand synthesis using sequence-specified triplet substrates for 1119
segment syntheses (as shown). The synthesis scale corresponds to the limiting component present. 1120
*: Unlike other segment ligation steps, the ligation was carried out using freeze-thaw cycling 1121
(Mutschler et al., 2015) (2.5 h at γ0˚C, 21 h at 7˚C, then 0.5 h at γ7˚C), which modestly 1122
improved yield. 1123
Below, scheme of t5+1
-catalysed t5 synthesis and assembly reactions. 1124
PAGE separations of syntheses (xt) alongside purified reference segments and fragments (in bold) 1125
were stained with SYBR Gold to quantify boxed full-length products. These were excised for use 1126
in subsequent assembly steps (illustrated by dashed black arrows). Synthetic schemes of colour-1127
coded segments are shown beside corresponding lanes, denoting ‘+’ strand synthesis (bold primer 1128
and dashes) on ‘–’ strand template. Fully synthesized segments are shown as bold lines. Assembly 1129
reactions involve use of fully synthesized segments as the final substrate in a primer extension 1130

46
reaction (α + , + ) or direct templated ligation of synthesized segments ( + ). The α + and 1131
+ fragments associate spontaneously to form an active triplet polymerase ribozyme (Figure 1132
6b), tested for activity in Figure 6c. The synthesised segment sequences were derived from the t5b 1133
variant of t5, which differs by one neutral signature mutation from t5 in the α segment, and by 1134
another neutral signature mutation from t5a in the segment (mutations highlighted in 1135
Supplementary file 1); these neutral mutations, not present in the t5 and t5a used to synthesise the 1136
segments (above), allowed verification of the synthetic origin of the product fragments by 1137
sequencing, which revealed the correct signature mutations in each fragment ruling out 1138
contamination of the synthesized ‘+’ strand products by the synthesizing (‘+’ strand) triplet 1139
polymerase ribozyme. 1140
1141
Figure 6 - figure supplement 2. Substrate competition attenuates inhibitory +/ -
pairing 1142
during self-synthesis. 1143
(A) Longer oligonucleotide substrates are required to maximise synthesis of full-length + 1144
segment (0.5 µM F 9 primer/T template/oligonucleotide substrate and 5 µM each triplet (left, 1145
omitted where equivalent oligonucleotide substrate was present), 13 d in 7˚C ice). Replacing 1146
two triplets with a preformed hexanucleotide substrate (ppp
UGAAUG) boosts full-length +
1147
product synthesis by t5+1
(left panel), but not by the type 6s+1
ribozyme - with a different 1148
accessory domain (see Figure 2 - figure supplement 1, sequence in Supplementary file 1) - which 1149
synthesizes + independent of this hexanucleotide (right panel, allowing some full-length
+ 1150
synthesis with only triplets). This likely reflects unfavourable competition between the t5 1151
ribozyme’s own + domain and triplet substrates for complementary pairing to the
- RNA used as 1152
template. Consistent with this hypothesis, increasing t5+1
concentrations with this template is 1153
counterproductive (middle panel). For both ribozymes, using a preformed nonanucleotide 1154
substrate ppp
UUUUUCAUG at the end of the template boosted full-length product over use of a 1155
pppUUCAUG hexanucleotide +
pppUUU triplet, or just the three constituent triplets (S=9 vs. 6/3 1156
vs. 3/3/3); this is not due to the individual triplet sequences as t5+1
can efficiently incorporate 1157
pppUUU and
pppUUC (see Figure 5b, Figure 6a). 1158
(B) Doubling triplet concentrations to 10 µM can attenuate t5+1’s requirement for the 1159
pppUGAAUG hexanucleotide substrate during
+ synthesis. This suggests that at higher triplet 1160
concentrations they successfully compete with this part of t5 for - template hybridisation (0.5 µM 1161
F 9 primer/T template/longer oligonucleotide substrates, 13 d in 7˚C ice). With a fourfold 1162

47
excess of primer/template duplex and substrates (right lane), the amount of full-length segment 1163
generated exceeds the ribozyme added, evidence of multiple turnover of full-length + product. 1164
1165
Figure 6 - figure supplement 3. Ribozyme stabilisation attenuates inhibitory +/ -
pairing 1166
during self-synthesis. 1167
Left, + synthesis using 5 με of each complementary triplet substrate (hex) or 1 με of 1168
pppUGACAU (+hex) replacing the final two triplets (0.25 με primer F 7/template HT , 0.5 με 1169
each ribozyme, 6 d in 7˚C ice). Type 5 with the initially isolated domain (type 5s) needs 1170
hexamer substrate for + synthesis, but with the reselected domain (t5
a) does not. See 1171
Supplementary file 1 for ribozyme sequences. 1172
Right, model of substrate/ribozyme competition for template binding. The region at the end of the 1173
segment exhibits irregular pairing in the initially isolated domain (above), and appears 1174
vulnerable to - template pairing during synthesis with triplet substrates. Reselection 1175
strengthened base-pairing in the corresponding domain stem (below), replacing a G-U wobble 1176
with a cognate G-C pair and a U.U mispair with cognate U-A pair, stabilizing the - domain 1177
junction and potentially reducing competition with triplets during segment synthesis. 1178
1179
Figure 6 - figure supplement 4. Ribozyme segment synthesis with random substrate pools. 1180
For maximum self-synthesis yield, we had used specific triplet substrate sets. Here we compare 1181
synthesis by 0.5 µM t5+1
of the five t5 ‘+’ segments (30 d in 7˚C ice) using specific triplets vs. 1182
random triplet pools or reduced G-content random triplet pools. Reactions included 5 µM of each 1183
triplet in specific triplet sets (‘tri’, as in Figure 6a ‘+’ syntheses), random ppp
NNN, or a low-G 1184
pppNNN. The low-G
pppNNN had the same overall triplet concentration as
pppNNN, but individual 1185
triplets were five-fold less common for each constituent G (~10/2/0.4/0.08 µM for 0/1/2/3 Gs per 1186
triplet; some primer/template (P/T) concentrations were reduced here to ensure excess substrate 1187
over template). Synthesis is compared with and without some longer oligonucleotide substrates 1188
(indicated below, equimolar to template sites, replacing the corresponding triplets in ‘tri’ substrate 1189
mixes). Importantly, for all ‘+’ strand segments, full-length products are generated using ppp
NNN 1190
with yields approaching those obtained when using specific triplets (calculated by densitometry, 1191
above the gel); intriguingly the low-G ppp
NNN pools often gave superior yields, possibly helped 1192
by the higher concentrations of weaker-binding AU-rich triplets therein. 1193

48
1194
Figure 6 - figure supplement 5. Ribozyme segment synthesis with mixed length substrate 1195
pools. 1196
+ (left) or
+ (right) syntheses were performed (using 0.5 µM each of t5
+1 and the 1197
primer/templates above, 15 d in 7˚C ice) with their constitutive triplet sets alone (*, see Figure 1198
6a) or the random-sequence substrate mixes indicated. Introduction of dinucleotide and 1199
mononucleotide substrates (ppp
NN, ppp
N i.e. NTPs) decreases full-length product band intensity 1200
but not ligations performed (here quantified whilst assuming each extension product was formed 1201
from the fewest substrates possible), with increasing numbers of extension products deviating 1202
from the starting triplet register. Dinucleotides and mononucleotides appear poor substrates in the 1203
absence of triplets. 1204
1205
Figure 8 - figure supplement 1. Fidelity of type 5 variants. 1206
Positional error rates and fidelities (determined as in Figure 8a) for type 5 ribozyme variants; the 1207
overall fidelity was calculated as a geometric mean of positional errors at each triplet position (n 1208
and s.d. of this value shown for ribozymes assayed multiple times, see Figure 8 - source data 1). 1209
Fidelity of the initial type 5 isolate (tethered to the assay templates) is modestly improved in the 1210
absence of type 1. However, with type 1, fidelity is improved when operating fully in trans 1211
(type5s +1
). Reselection and stabilisation of the domain yields a further fidelity improvement 1212
(t5+1
) but truncation (α +1) reverts the pattern of error tendencies along the triplet towards that 1213
of the starting core. See Supplementary file 1 for ribozyme sequences. 1214
1215
Figure 8 - figure supplement 2. Fidelity domain influence upon template- and position-1216
specific error rates. 1217
At each position in each of the twelve triplet templates (top) tested in the fidelity assay 1218
(Figure 8a), three different errors are possible after analysis of sequence composition of added 1219
triplets (see Figure 8 - source data 1). Errors are represented by discs, coloured by mutation (e.g. 1220
A to G : green; A to C : blue; G to C : blue). Disc size is proportional to error frequency without 1221
the fidelity domain (its occurrence using +1). The effect of the fidelity domain upon these 1222
error tendencies is plotted on the y-axis (by comparison of error rates between t5+1
and +1). 1223
Many of the most significant errors (using +1) exhibit the greatest proportional reductions 1224

49
with the fidelity domain, including substantial (>10-fold) reductions in several misincorporations 1225
at the third position. Improvements in fidelity are variable but focused on the 2nd
and 3rd
triplet 1226
positions, while the 1st triplet position shows an increase specifically in A to G mutations, 1227
indicative of a tolerance for G-U wobble pairing. 1228
1229
Figure 8 - figure supplement 3. Determination of residual fidelity phenotype when using 1230
minor groove-modified substrates. 1231
Top: framework for estimation of residual fidelity phenotype (). Shown are model logistic 1232
curves for incorporation of a triplet e.g. GAU (at a fixed concentration, opposite γ’-AUC-5’) 1233
versus increasing concentrations of a mismatching triplet e.g. GGU. A ribozyme ‘+’ with a 1234
fidelity function (e.g. t5a, blue curve) will incorporate equal amounts of the matched and 1235
mismatched triplets at a different concentration of the mismatched triplet compared to a ribozyme 1236
‘−’ without a fidelity function (e.g. α , red curve). x+ and x− represent the lns of these 1237
concentrations for + and −; their separation (x+ - x−) is a proxy for the strength of the ‘+’ fidelity 1238
phenotype. We measured the relative incorporation (W) of triplet pairs by the two triplet 1239
polymerases at test concentrations (t) of mismatched triplet, chosen to maximise the difference in 1240
the ribozymes’ resulting fractional incorporations (marked by a blue dot vs. a red dot on 1241
respective curves). If a triplet modification (*, right) interferes with mismatch discrimination by 1242
‘+’, it would shift the ‘+’ relative incorporation curve (blue) towards that of ‘−’ (red), reducing 1243
the difference in fractional incorporation. Assuming curve steepness (k) remains constant, the 1244
residual phenotype () for that modification is described by the new separation (x+* - x−*) as a 1245
proportion of the original separation (x+ - x−); numerical values and calculations from the 1246
measurements described below are supplied in Figure 8 - source data 2. 1247
Middle: measurements of ratios of incorporation vs. misincorporation (W) for unmodified and 1248
modified triplet pairs (at the indicated test concentrations) by the t5a +1
triplet polymerase 1249
compared to the fidelity domain-truncated α +1 (at 0.5 με each). The expected triplet 1250
additions are in grey along the left of each gel; the average W from n independent experiments, 1251
calculated via densitometry of products containing slower-migrating G misincorporations, is 1252
shown below each lane along with the average . Primers (P), templates (T) and additional triplets 1253
were at 0.5/0.5/5 με, incubated for three days in 7˚C ice. Left: presence of a 2-thiouracil (2SU) 1254
at the 3rd
position of the triplet abolishes the fidelity domain’s ability to discriminate against a 2nd
1255
position wobble pair (P: A10, T: CCCMisGAU, +5 με ppp
CCC). Centre-left: the same 1256

50
modification (2SU) also abolished the fidelity domain’s preference for G misincorporation at the 1257
1st position (P: F 7, T: T AGU, +5 με
HOCUG). Centre-right: in contrast, a 2SU at the 1
st 1258
position of the triplet exerted no clear influence upon 3rd
position wobble discrimination (P: A10, 1259
T: CCCMisUGA, +5 με ppp
CCC). Right: replacement of a β’ hydroxyl group with a β’ fluoro 1260
(β’F) at the 1st triplet position likewise had no effect upon 3
rd position mismatch discrimination 1261
(P: F 7, T: T AGU, +5 με HO
CUG). 1262
Below: Measurement of fidelity phenotype in the presence or absence of a downstream triplet. 1263
Residual phenotypes indicate attenuation or abolishment of 1st position (left panel, P F 7, T 1264
T AGU), 2nd position (middle panel, P F 7, T T GAU), and 3
rd position (right panel, P F 7, T 1265
T AGU) fidelity effects from downstream triplet absence. No effects are seen (upon 3rd
position 1266
discrimination) from the presence or absence of a 5’ triphosphate on the downstream triplet (ppp). 1267
In the absence of a triplet bound downstream on the template, the fidelity phenotype is severely 1268
compromised, suggesting this adjacent triplet:template duplex plays a critical role in positioning 1269
the fidelity domain relative to the incoming triplet. 1270
1271
Figure 9 - figure supplement 1. Influence of random triplet pool concentrations upon triplet 1272
misincorporation tendencies. 1273
Left: 0.2 µM t5+1
was tested in the fidelity assay at a series of ppp
NNN concentrations. The 1274
observed overall error rate (black circles) in these reactions halved as ppp
NNN concentrations 1275
were increased to 7 µM each (after which extension activity began to reduce and error rates 1276
increase). Diverse patterns were observed in the concentration dependence of individual triplet 1277
positional errors, of which a selection are displayed here (×) coloured according to whether they 1278
raise (red-yellow) or lower (blue) the GC content of the expected triplet, and coloured more 1279
intensely for higher GC content triplets. 1280
Right: For a broader set of errors with 0.5 µM t5+1
(those with >0.3% incidence at 5 µM ppp
NNN, 1281
x-axes) the changes in their incidences from raising concentrations of each ppp
NNN from 2 µM to 1282
10 µM substrate are plotted on the y-axes. The frequencies of several mutations that increase 1283
triplet GC content in GC-rich triplets are reduced at 10 µM ppp
NNN concentrations (top panel). 1284
Conversely, the frequencies of some mutations that reduce GC content are increased at 10 µM 1285
pppNNN concentrations (bottom panel); these suggest relative depletion of available GC rich 1286
triplets as overall triplet concentrations rise. Such pool behaviour can explain the reduction of 1287

51
overall error rates in ~5 µM ppp
NNN triplet pools, as misincorporations that increase GC content 1288
(e.g. G opposite U) are responsible for more errors than those that decrease it (bottom left). 1289
Values and ratios for a wider range of errors are supplied in Figure 9 - source data 1. 1290

52
G C C A U C A A A G C U U G A G A G C A U C U U Internal
triplets’ average:
10 με each tri: 93.3 99.4 99.5 97.8 99.8 99.4 99.6 97.9 99.1 99.3 99.5 99.1 99.8 98.9 99.5 98.2 96.0 96.3 98.79
10 με each PPPNNN: 92.8 97.0 98.8 99.2 99.5 99.3 99.1 97.8 98.6 99.4 99.1 98.7 94.3 81.4 96.6 97.4 59.8 42.3 96.65
10 με average, low-G PPPNNN: 97.3 98.2 99.5 97.3 99.7 99.4 99.6 97.8 99.0 99.3 98.9 98.8 97.6 97.3 98.8 97.4 73.2 43.2 98.56
Table 1. Sequencing of ribozyme-synthesised + segment. 1291
Shown are the individual base fidelities (%) along the + sequences (top) synthesised by t5
+1, 1292
using the six specific triplets (tri), or random (ppp
NNN) or compositionally-biased random (low-G 1293
pppNNN, see Figure 6 - figure supplement 4) substrate pools, from F6 primer (the first six 1294
positions at the left) with template T (1 με each RNA, 13 d 7˚C ice). For their sequencing, 1295
extension products were eluted from templates, and full-length products were gel-purified, ligated 1296
to adaptor, reverse-transcribed and PCR amplified. For compositional analysis, a small percentage 1297
of unrelated amplified products were excluded (those with >9 mutations vs. the expected + 1298
sequence; similar levels were excluded if a >6 mutation threshold was applied, 0.2%/0.2% & 1299
3.7%/4.2% & 3.7%/3.8% for tri & ppp
NNN & low-G ppp
NNN). These sequences mostly appeared 1300
to derive from off-target priming and extension of F6 on the ribozyme in the presence of 1301
pppNNN. 1302
The sequencing of products generated from specific triplets provides an estimate of background 1303
error arising from amplification and sequencing. The final triplet constitutes an error hot-spot - 1304
likely to mutate to a more mutationally stable triplet during self-replication - exacerbated in 1305
pppNNN samples by the inability of the fidelity domain to operate in the absence of a downstream 1306
triplet (Figure 8 - figure supplement 3). The geometric average of internal triplet position 1307
fidelities is used to gauge overall t5+1
fidelity during RNA synthesis. While overall fidelity drops 1308
from defined to random triplets (98.8% to 96.7%), much of this loss in fidelity can be recovered 1309
by adjusting the triplet composition to a low-G random pool, where reductions in G-U wobble 1310
pairing more than compensate for increases in rarer misincorporations opposite template C. 1311
1312
Supplementary file 1. Ribozyme sequences. 1313
All sequences are written in a 5’-to-γ’ direction, generated by GMP transcription of the 1314
corresponding PCR-generated dsDNA of the sequence downstream of 5T7 sequence duplex. All 1315
transcripts were PAGE-purified. 1316

53
HDV ribozyme sequences (blue) were transcribed in series with reselected type 5 ribozymes and 1317
cleave themselves off during transcription (Schurer et al., 2002) to yield precise γ’ ends with β’, 1318
γ’-cyclic phosphates; this presence of group did not affect type 5 activity. 1319
Sequences corresponding to the 5’ ‘cap+’ or ‘cap’ regions from the selection, presenting a target 1320
for type 1 interaction, are coloured light green, with alternative arbitrary inert 5’ hairpin-forming 1321
sequences in dark green. Single stranded sequences capable of hybridization with sites at the γ’ 1322
(or 5’, type 5cis
) ends of certain templates to endow flexible ribozyme-template duplex tethering 1323
(Wochner et al., 2011; Attwater et al., 2010) are coloured yellow. Accessory domains γ’ of the 1324
catalytic core are in bold. 1325
1326
Supplementary file 2. Transcription of triplets. 1327
Each target was transcribed from oligonucleotides 5T7 (Supplementary file 3) and 1328
5’-(var)-TATAGTGAGTCGTATTAATTTCGCGGGCGAGATCGATC-γ’, where the (var) 1329
overhang encodes (is the DNA reverse complement of) the sequence indicated below. Guide 1330
yields: >15 nmol = ***, 10-15 nmol = **, 5-10 nmol = *, <5 nmol = ~. †: These transcriptions 1331
yield one main product suitable for use as markers to identify comigrating triplets with a similar 1332
G/AU/C content. 1333
1334
Supplementary file 3. Oligonucleotide sequences. 1335
All sequences are written in a 5’-to-γ’ direction. DNA sequences are coloured grey. RNA 1336
sequences are coloured black, with the exception of hammerhead ribozyme (brown) and HDV 1337
ribozyme (blue) sequences transcribed in series that cleave themselves off during transcription 1338
(Schurer et al., 2002) to yield precise 5’ and γ’ ends respectively. All RNAs were denaturing 1339
PAGE-purified, and DNAs were not, unless otherwise noted (‘(non)-GP’). Competing 1340
oligonucleotides, listed by templates, were purified using RNeasy columns (Qiagen). Primer and 1341
oligonucleotide binding sites on templates are underlined. 1342

AAAC
CUU
GGAG
AC
UG
CC
GA
CG
G
GC
UU
CAAC G
AG
AA
UCU
CAUCU
UCG
GUAG
A
C
G
AAA
AA
U
CAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
A
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G
P5’
0core
C
G
C UU
G AA
AAA
G
CG
UCA
GCGA
C
U
AA3’5’
AAAC
CUU
GGAG
A
CG
UCA
CU
GC
C
GA
CG
GA
GC
UUA
AC G
AG
AA
UU
CU
CAUCU
UCG
GUAG
C
A
C
G
AAA
AA
U
CCA
GGU
ACA
UGU
GG
CC
C
G
C U U
G A A
AAA
G
G
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
AACA( )L
3’
3’
5’
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
UAG
U
A
AC
UG
A
C
G
C G C
G C G
AA GC
CA
AA
AAA
GC
CC
CA
AU
CA
CC
GG
CGC
GCG
UUC
G
U
UAG
AUC
GGG
CCC
AAC
AUAC
UC
C
AG
G
AU
U
GC
P5’
Z RPR / Zcore
Z RPR Zcore Round 7library
+ 3’ N30
domaintype 0 0core
25%
selectiontruncation
A
B
Templates: SR1 SR1 SR2 SR3 SR4
Z RPR
noNTPs
Primer
Substrates: 1 mMNTPs
0core triplet polymerase
notriplets
SR1 SR1 SR2 SR3 SR4
5 μM pppAUA,5 μM pppCGC
[each triplet] (μM)
Superc
oole
d e
xte
nsio
n(t
riple
ts a
dded p
er
prim
er)
Template SR3
5’ P
CUA GU
GCG
G
U
AU
AU
U
G
GC
Template SR1
5’ PCUA GU G CUA GU G CUGAU CUG G AU G 5’ P
CGCAUA
PP
PP
PP
CGCAUA
PP
PP
PP
CGCAUA
PP
PP
PP+pppAUA
0.25
0.20
0.15
0.10
0.05
0
0 10 20 30 40
+pppCGC
C
NPPP
exte
nsio
n
Template SR1 ( )
Template SR3 ( )
5’
Nprimer
template
RPR
PPP N
PPPN
PPP
N
PPP
5’
NNN
tripletpolymerase
PPPNNN
PPPNNN
PPP
truncation

type 2 type 3 type 4 type 5 type 61 type 1
Round 7 library
Round 21 library
selection
53% 11% 0.5% 30% 6% 0.1%
truncation
Pri
Zcorecap+Rz+1 +1 +1 +1 +1
:
AUA
CGC
AUA
CGC
AUA
CGC
type 2 type 3 type 4 type 5 type 61+1
trip
lets
added
A
B

Type 1 / 1 (truncation)
AAG
U
GGAG
AC
C
G
CG
G
CAC G
AG
AA
C
CAUCU
UCG
GUAG
C
A
C
G
U
3’
A
CG
UGG
ACC
GC
CC
UA
CU
A
CGC
GCG
GAU
A
U
C
G
C
U
G
AAUGCAU
U
C
ACG G
CCACCAC
AUGAUCUGAAGAGCCUU
GG GUUUUUU
G
UCA
AG
UCA
CC
CU
C
AAA
AA
CCAAUAGG
P5’
U
G
C U G
G A CU
A
UU
UAC
A
AAAC
CUU
GGAG
C
A
A
UCA
CU
GC
C
GA
CG
G
GC
UU
CAAC G
AG
AG
UU
CU
CAUCU
UCG
GUAG
C
A
C
G
AAA
AA
U
CCAACAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
A3’CA G
U
U
UU
UG
UAC
U
GG AAAGUAA
UUG
GCC
G
UU
AU
GC
CG
GU
A AAA
C
G
CU
U
G
C
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G
P5’
U
G
C U G
G A CU
A
UU
UCC
Type 5
B
+1type 5
+1type 5cap-1
cap-
Rz: -
11 3 3 11 11
Pri
+88nt
C
0.1 72 74 66 71
type 5
cis
+1
type 5
trans +
1
type 5
cis
+1
type 5
trans +
1
GCG
AAG
CGU
GUG
CGA
AGC
GUG
UGC
GAA
GCG
UGU
GC
GA
AG
CG
UG
Ure
peate
d
repeate
d
substrate length (nt)
average ligationper junction (%)
cap+
GUAC
Type 5cap-
type 5 + 1
0.25 0.5 0.75 0.9
equivalents of 1:
1.1 1.25 1.5 2
5’
NNN
PPP
type 5
cis
+1
type 5
trans +
1
P
0 0.5 1 1.5 20
0.5
1.0
Equivalents of 1 added to type 5
Shifte
d b
and inte
nsity
- r
ela
tive to
Shift1
type 5
1

AAAC
CUU
GGAG
C
A
A
UCA
CU
GC
C
GA
CG
G
GC
UU
CAAC G
AG
AG
UU
CU
CAUCU
UCG
GUAG
C
A
C
G
AAA
AA
U
CAA
A
G
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
3’
5’
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
t5UUUC
C
A
GA
UC
U
CU
AG
G
3’
CA G
U
U
CA
UG
UAC
U
GAGGUAA
UUG
GCC
G
UU
AU
GC
CG
GU
A AAA
C
UU
1
CAA G
CUU
GGAG
AC
UG
CC
GAC
GG
CGAG
AA
CCU
CAUCU
UCG
GUAG
A
C
G
U
A
C G
UGG
ACC
G CA
CGC
GCG
GAU
A
G
CG
UCA
AG
CU
CAA
C
U5’
C
3’
C
CU
CA
AUGCAU
U
C
ACG G
CCACCAC
AUGAUCUGAAGAGCCUU
GG GUUUUUU
5’
P
P
5’
type 5
1
type 1
Round 5library
t5
t5+1
engineering
in reselection+ mix of 3’ N
13/N
20/N
28
- 5’ template hybridisation site- 3’ 20 nt reverse transcription sitetruncation

GGU
GUG
GAG
CGG
ACUCCU
UCU
(C)
-
-
-
-
- (
( (
( -
- -
- ) ) )
)- -
- -
-
GGU
GUG
GAG
UGC
GGA
CAC
UCC
UUC
-
-
-
- -
(
( (
(
( (
- -
- -
))) )
)
) - - - - -(UC)
GGU
GUG
GAG
UGU
GCG
GAC
ACA
CUC
-
-
-
-
- (
(
( (
(
( (
( -
- -
- ) )
)
) ) ) ) )
- - -
- -
CUU
CUC
Pri Pri Pri
4S 6S 8S
B
Rz: t5+1 t5+1 t5+1
CUGC C A A C C5’
GACGGUUGG C C A C A3’
UA G
UGC
AC
U
CCG
GC
GA
GCU
C
5’ AA AG G
GGUPPP
P
TM ~ 93˚C
Template 8S
A
Broc+
Synth w/t5+1
SYBRGold
DFHBI-1Tligand
CGG
GUC
CAG
AUA
UUC
GUA
UCU
GUC
AGUGUG
UAGGAG
GGCUCC+1
CG
UAUA
UA
G
UC
U
GU
CA
CUG
G
GGG CUC
GU
A
CC
GG
C
GG A GG CU
GU GU
GA A
UG GA
5’
3’
DFHBI-1Tligand
t5+1
-tri t5+1
+tri
Pri
CCA
CAC
UCU
ACU
CGA
CAG
AUA
CGAAUAUCUGGACCCGACCGUCUC+1
Bro
c+
as tem
pla
te:
Broc+
Synth w/t5+1
‘+’ strandsynthesis
‘-’ strandsynthesis
Z RPRcap+ +1 Z RPRcap+ +1 Z RPRcap+ +1

GG
UU
UC
GC
CG
CA
GC
GG
AG
GG
C
GA
GUUUUUCAUG
UGAAUG
CG
UC
UU
GU
GC
GC
CU
UC
AU
GA
GU
GA
GC
UA
AA
GACAAA
AA
AGACAAA
UC
UC
GA
UC
U
UG
A C
AU
CC
AC
AA
UC
UC
GU
CA
AC
GC
GGAUGCA
GCAAAA
GCCAUC
GGAUCUUCU
GCGAUAG
days:
[t5+1](µM)
0 0 0 0 02 2 2 2 2 20.5 0.5 0.5 0.5
7 1 7 7 1 7 7 1 7 7 1 7 7 1 7
P/T: Fα9/TαHP Fβ6/Tβ Fγ7/TγHP Fδ7/Tδ Fε6/Tε
yield:(%)
27 19 17 10 15
Pri
α+ β+ γ+ δ+ ε+
:
A
GC
AU
CU
GC
CG
CU
UC
C
UC
AC
UC
GG
CU
UU
UC
A
UC
AU
UU
CG
CU
GG
CG
UG
AA
UG
AG
GU
GCCACCGAAG
CAUGAAAAA
AAGAUGCUC
AGAUUUGUCU
AUGUCAU
0 0 0 0 02 2 2 2 2 22 2 2 2
25 3 25 25 3 25 25 3 25 25 3 25 25 3 25
Cyα10ba/αβ Fβ9ba/β+ Cyγ10ba/Fγ Fδ7ba/δ+ Fε9ba/εHP
6.1 29 36 51 10
UU
GG
AU
GA
UC
GA
GA
AC
CC
GC
C
GA
UA
GC
UA
UC
GC
AC
GC
AU
UU
UA
AG
UG
CC
AC
GC
G
α− β− γ− δ− ε−
days:
[t5+1](µM)
P/T:
Pri
:
yield:(%)
B
AAAC
CUU
GGAG
C
A
A
UCA
CUGCC
GA
CG
G
GC
UU
CAAC G
AG
AG
UU
CU
CAUCU
UG
G
UAG
C
A
C
G
AAA
AA
U
CAG
A
3’
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
UU
UC
C
A
GA
UC
U
CUAGG
CA GU
U
CA
U
G
UAC
U
GAGGUA
A
UUG
GCC
G
UU
AUGC
CG
GU
AAAA
C
UU
bio5’
bio5’3’
α+
β+
γ+
δ+
ε+
t5b
Pri
GGU
UGA
GAA
CGU
UGG
CGC
UAU
CGC
t5 variant:
t5+
1-s
ynth
esis
ed δ
+
C
δ−
αβ+,γδε+ none
protein t5+1synthesised by:
αβ+,γδε+
αβ+,γδε+

Lane: 1 2 3 4 5
Lane 1:
Lane 2:
Lane 3:
Lane 4:
Lane 5: No Rz,
pppGCCAUCAAAGCUUGAGAGCAUCUU alone (β+ sequence)
pppCUUpppCAUpppGAGpppUGApppGCUpppAAApppGCCAUC
pppCUUpppCAUpppGAGpppUGApppGCUpppAAApppGCC pppAUC
pppCUUpppCAUpppGAGpppUGApppGCUpppAAApppAUC
pppCUUpppCAUpppGAGpppUGApppGCUpppAAApppAUC
A10 PPPba
PP
P
3’
5’ 5’
Pri
Pri5’
5’
3’
5’
3’ e
xte
nsio
n
3’
5’ e
xte
nsio
n
Pri
Pri
A
B
PPPba
A10
C
Lane: 1 2 3 4
5 6 7
Lane 1:
Lane 2:
Lane 3:
Lane 4:
pppGAUGCAGAGGCGGCAGCCUUCGGUGGC>p alone (γ+ sequence)
pppGGUpppUUCpppGCCpppGCApppGCGpppGAGBio-GGAUGCA pppGGC
pppGGUpppUUCpppGCCpppGCApppGCGpppGAG pppGGCpppGCApGAU
Bio
pppGGUpppGCCpppGCApppGCGpppGAG pppGGCpppGCApGAU
Bio
Lane 5: pppUGCpppCUCpppCGCpppUGCpppGGCpppGAApppACCpppGCC
Bio
p
Lane 6: pppUGCpppCUCpppCGCpppUGCpppGGCpppGAA pppAUCpppACCpppGCC
Bio
p
Lane 7:pppGCCACCGAAGGCUGCCGCCUCUGCAUC alone (γ− sequence) Lane:
β+ s
ynth
esis
γ+ s
ynth
esis
γ− synth
esis
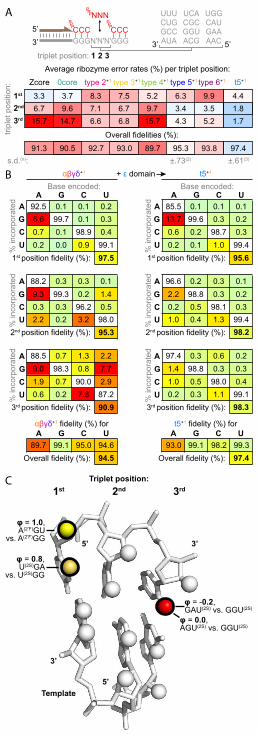
B
A
3.3 3.7 8.3 7.5 5.2 9.96.3
6.7 9.6 7.1 6.7 9.7 3.53.4
15.7 14.7 6.6 6.8 15.7 5.24.3
91.3 90.5 92.7 93.0 89.7 93.895.3
Zcore 0core t5+1
Average ribozyme error rates (%) per triplet position:
1st
2nd
3rd
4.4
1.8
1.7
97.4
s.d.(n): ±.73(2) ±.61(3)
Overall fidelities (%):
85.5 0.1 0.1 0.1
13.7 99.6 0.3 0.2
0.6 0.2 98.7 0.3
0.2 0.1 1.0 99.4
95.6
96.6 0.2 0.3 0.1
2.2 98.8 0.3 0.2
0.2 0.5 98.1 0.3
1.0 0.4 1.3 99.4
98.2
97.4 0.3 0.6 0.2
1.4 98.8 0.3 0.3
1.0 0.5 98.0 0.4
0.2 0.3 1.1 99.1
98.3
93.0 99.1 98.2 99.3
97.4
Base encoded:A G C U
t5+1
1st position fidelity (%):
3rd position fidelity (%):
A
G
C
U
% incorp
ora
ted
A
G
C
U
A
G
C
U
A G C U
92.5 0.1 0.1 0.2
6.6 99.7 0.1 0.3
0.7 0.1 98.9 0.4
0.2 0.0 0.9 99.1
97.5
88.5 0.7 1.3 2.2
9.0 98.3 0.8 7.7
1.9 0.7 90.0 2.9
0.6 0.2 7.8 87.2
90.9
A G C UBase encoded:
αβγδ+1 + ε domain
88.2 0.3 0.3 0.1
9.3 99.3 0.2 1.4
0.3 0.3 96.2 0.5
2.2 0.2 3.2 98.0
95.32nd position fidelity (%):
% incorp
ora
ted
% incorp
ora
ted
89.7 99.1 95.0 94.6
94.5Overall fidelity (%):
αβγδ+1 fidelity (%) for
A G C U
t5+1 fidelity (%) for
type 2+1 type 3+1
type 4+1 type 5+1 type 6+1
1st position fidelity (%):
3rd position fidelity (%):
A
G
C
U
A
G
C
U
A
G
C
U
2nd position fidelity (%):
trip
let positio
n:
Overall fidelity (%):
% incorp
ora
ted
% incorp
ora
ted
% incorp
ora
ted
GGGN'N'N'GGG
CCCPPP
CCCPPP
NNNCUGGCCAUA
UUUCGCGGUACG
UCACAUGAAAAC
UGG
3’ 5’3’
5’
C Triplet position:
1st 2nd 3rd
Template
5’ 3’
3’
5’
φ = 0.8,U(2S)GA
vs. U(2S)GG
φ = 1.0, A(2’F)GU
vs. A(2’F)GG
φ = -0.2,GAU(2S) vs. GGU(2S)
φ = 0.0,AGU(2S) vs. GGU(2S)
1 2 3triplet position:
PPP

2 μM pppNNN27%
pppACC, pppGCC31%
pppACCEncoded:
Mispair:
pppGAC
Incorporationof mispair vs.
encoded triplet
pppGCC pppGGC
pppACC, pppGCC,pppGGU, pppGGC
12%
5 μM pppNNN14%
10 μM pppNNN8%
2 μM pppNNN1.6%
pppGAC, pppGGC2.7%
pppGAC, pppGGC,pppGUC, pppGCC
0.71%
5 μM pppNNN0.74%
10 μM pppNNN0.69%
mispairpair + mispair
(%)
( )

Pri
Substrate (nt): 1 3 11 1 3 11 1 3
Ribozyme: none Z RPR Zcore
Extension (%): .01 .07 .07 35 .58 .22 .62 2.2
11
2.9
Gprimer
template
RPR
PPP G
PPPC
PPP
CGCUUCGCACCUAAGUAAC5’
U
PPP
A
PPP
GCGAAGCGUGU
PPP
CGCUUCGCACCUAAGUAAC5’
GCG
PPP
CGCUUCGCACCUAAGUAAC5’
1 nt substrates (NTPs)
3 nt substrate (triplet)
11 nt substrate (oligomer)
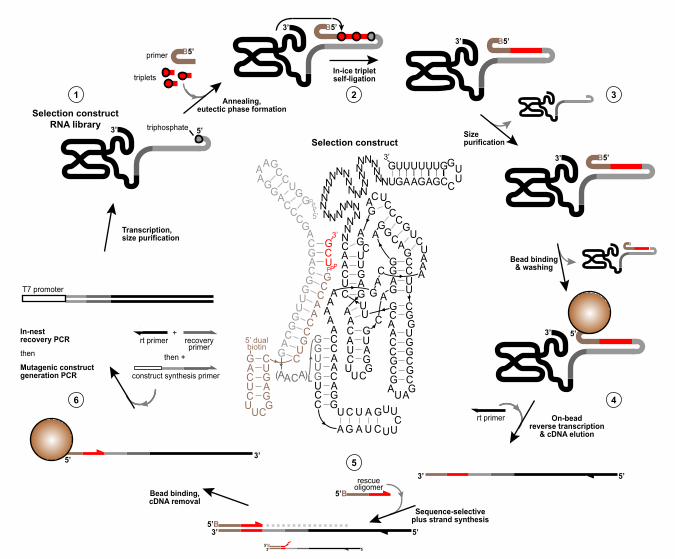
AAAC
CUU
GGAG
AC
UG
CC
GA
CG
G
GC
UU
CAAC G
AG
AA
UU
CU
CAUCU
UCG
GUAG
A
C
G
AAA
AA
U
CCAACAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
A
3’
GGUUGUCC
AACA( )L
U
AACC
CC
C
GA
GGUUGGC
G
C
PPP
G
Selection construct
CG
UCA
C
U
A
C U A
G A U
G
CU
UU
UC
UGAAGAGCCUU
GGGUUUUUU
NNN
NNNN
NNNNNNNNNN
NNNN
NNN N
NNN
NN
AGC
UCG
5’
AG
C
GAA
A
GG
UC
C
CC
AG
G
P
PP
3’
CU
GA
GAG
CUC
GCU
U
5’ dualbiotin
In-ice tripletself-ligation
Bead binding& washing
Annealing,eutectic phase formation
Sizepurification
On-beadreverse transcription
& cDNA elution
Sequence-selectiveplus strand synthesis
Bead binding,cDNA removal
In-nestrecovery PCR
Transcription,size purification
B5’
B5’
B5’
B
3’ 5’
3’ 5’
rt primer
rescueoligomer
5’B
5’B
3’ 5’5’B
3’5’
5’
T7 promoter
+recoveryprimer
B5’primer
triplets
Selection construct
RNA library triphosphate
construct synthesis primer
rt primer
then +
5’3’
3’
3’
3’
3’
Mutagenic constructgeneration PCR
then
1 2 3
4
5
6

AAAC
CUU
GGAG
C
A
A
UCA
CUGCC
GACGG
GC
UU
CAAC G
AG
AG
UU
CU
CAUCUUCG
GUAG
C
A
C
G
AAA
AA
U
CCAACAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
A3’CA GU
U
UU
UG
UAC
U
GG AAAGU
AA
UUG
GCCG
UU
AUGC
CG
GU
AAAA
C
G
CUU
G
C
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G
P5’
U
G
CU G
GA CU
A
UU
UCC
Type 5
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G AAAC
CUU
GGAG
A
CG
UCA
CUGUC
GACAG
AGC
UU
CAAC G
AG
AA
UU
UU
CAUCUUCG
GUAG
C
A
C
G
AAA
AA
U
CA
UAGG
3’
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
C
G
CU G
GA CU
A
UU
UCC
AC
GAAUU
CG U
GUC
A
A AUC
GU
AUGGC
C
CU
U
C
A
GG
A
G
GA
GAA
UUU
C
G
A
P5’
Type 6
AAAC
CUU
GGAG
A
CG
UCA
CUGUC
GACAG
AGC
UU
CAAC G
AG
AG
UU
CU
CAUCUUCG
GUAG
C
A
C
G
AAA
AA
C
CCAGCAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
U
G
CC G
GG CU
A
UU
UCC
CC
G
GCG
C
AUU
UCCCC
ACU
3’
P5’
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G
Type 4
Type 2
AAAC
CUU
GGAG
A
CG
UCA
CUGUC
GACAG
AGC
UU
CAAC G
AG
AG
UU
CU
CAUCUUCG
GUAG
C
A
C
G
AAA
AA
U
CCAAUAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
UG
ACA
GGC
GGC
G
CA
U
AUUAG
UA
UAC
A3’
UA
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G
P5’
U
G
CU G
GA CU
A
UU
UCC
Type 3
AAAC
CUU
GGAG
A
CG
UCA
CUGAA
GACU
GAGC
UU
CAAC G
AG
AA
UU
CU
CAUCUUCG
GUAG
C
A
C
G
AAA
AA
C
CCA
GGU
AUA
UGU
GG
CC
AACA( )L
3’
A
CG
UGG
ACC
GCA
CGC
GCG
GCU
A
G
U
3’
AGU
C
G
AC A
UGC
GG
C
A
U
U
G
AU
UUAUAU
G
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
P5’
U
G
CU G
GA CU
A
UU
UCC
Type 1
AAG
U
GGAG
AC
C
G
CGG
CAC G
AG
AA
C
CAUCUUCG
GUAG
C
A
C
G
U
3’
A
CG
UGG
ACC
GC
CC
UA
CU
A
CGC
GCG
GAU
A
U
C
G
C
U
G
AAU
GCAU
U
C
ACG G
CCACCACAUGAUCUGAAGAGC
CUUGG GUUUUUU
G
UCA
AG
UCA
CC
CU
C
AAA
AA
CCAAUAGG
P5’
U
G
CU G
GA CU
A
UU
UAC

Pri
Polyclonal output : R1959
R2089
R2190
R21 no AUG3’dpppAUA incorp. (%):
Full length
self-ligation%: .07 .35 .30 .71
AUG3’dAUA
Pri
R19polyclonal
type 21Rz:

+1type 5
+type 1
type 5cap-
+type 1
B
.05 .5.250[type 1] (μM):
Pri
.044
.115
.136
.005
triplets addedper primer
A
Pri
[type 3] (μM): .25 .25 .25 .25
:

Zcore
cap+
+ty
pe 1
0core
Zcore
cap+
Zcore
0core
cap+
0core
cap+
+ty
pe 1
Pri
Z R
PR
cap+
+1
Z R
PR
+
1
Z R
PR
Z R
PR
cap+
Pri
Rz: Rz:

type 2 type 3 type 4 type 5 type 6
1
type 1
Round 21 library
Round 5library
t5
t5+1
selection(see Figure 2 -source data 1)
engineering
in reselection(see Figure 4 -source data 1)
53% 11% 0.5% 30% 6% 0.1%
5’ truncation(see Figure 3a)
Z RPR Zcore
Round 7 library
+ 3’ N30
domain(see Figure 1 - figure supplement 2)
type 0
0core
25%
selection(see Figure 1 -source data 1)
3’ truncation(see Figure 1a)
truncation(see Figure 1a)
+ mix of 3’ N13
/N20
/N28
- 5’ template hybridisation site- 3’ 20 nt reverse transcription site
(see Figure 2 -figure supplement 1)
association(see Figure 4)
AAAC
CUU
GGAG
AC
UG
CC
GA
CG
G
GC
UU
CAAC G
AG
AA
UU
CU
CAUCU
UCG
GUAG
A
C
G
AAA
AA
U
CCAACAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
A
3’
GGUUGUCC
AACA( )L
U
AACC
CC
C
GA
GGUUGGC
G
C
PPP
G
Zcore starting library
CG
UCA
C
U
A
C U A
G A U
G
CU
UU
UC
UGAAGAGCCUU
GGGUUUUUU
NNN
NNNN
NNNNNNNNNN
NNNN
NNN N
NNN
NN
AGC
UCG
5’
AG
C
GAA
A
GG
UC
C
CC
AG
G
P
PP
3’
CU
GA
GAG
CUC
GCU
U
5’ dualbiotin
AAAC
CUU
GGAG
AC
UG
CC
GA
CG
G
GC
UU
CAAC G
AG
AA
UCU
CAUCU
UCG
GUAG
A
C
G
AAA
AA
U
CAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
A
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G
P5’
0core
C
G
C UU
G AA
AAA
G
CG
UCA
GCGA
C
U
AA3’
AAAC
CUU
GGAG
C
A
A
UCA
CU
GC
C
GA
CG
G
GC
UU
CAAC G
AG
AG
UU
CU
CAUCU
UCG
GUAG
C
A
C
G
AAA
AA
U
CCAACAGG
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
A3’CA G
U
U
UU
UG
UAC
U
GG AAAGUAA
UUG
GCC
G
UU
AU
GC
CG
GU
A AAA
C
G
CU
U
G
C
GGUUGUCC
AACA( )L
3’
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
5’
5’
G
P5’
U
G
C U G
G A CU
A
UU
UCC
Type 5
AAAC
CUU
GGAG
C
A
A
UCA
CU
GC
C
GA
CG
G
GC
UU
CAAC G
AG
AG
UU
CU
CAUCU
UCG
GUAG
C
A
C
G
AAA
AA
U
CAA
A
G
U
AACC
CC
C
GA
GGUUGGC
G
C
NNN
NNN
PPP
3’
5’
A
CG
UGG
ACC
GCA
CGC
GCG
GAU
A
G
t5UUUC
C
A
GA
UC
U
CU
AG
G
3’
CA G
U
U
CA
UG
UAC
U
GAGGUAA
UUG
GCC
G
UU
AU
GC
CG
GU
A AAA
C
UU
1
CAA G
CUU
GGAG
AC
UG
CC
GAC
GG
CGAG
AA
CCU
CAUCU
UCG
GUAG
A
C
G
U
A
C G
UGG
ACC
G CA
CGC
GCG
GAU
A
G
CG
UCA
AG
CU
CAA
C
U5’
C
3’
C
CU
CA
AUGCAU
U
C
ACG G
CCACCAC
AUGAUCUGAAGAGCCUU
GG GUUUUUU
5’
P
P
5’
Type 1
AAG
U
GGAG
AC
C
G
CG
G
CAC G
AG
AA
C
CAUCU
UCG
GUAG
C
A
C
G
U
3’
A
CG
UGG
ACC
GC
CC
UA
CU
A
CGC
GCG
GAU
A
U
C
G
C
U
G
AAUGCAU
U
C
ACG G
CCACCAC
AUGAUCUGAAGAGCCUU
GG GUUUUUU
G
UCA
AG
UCA
CC
CU
C
AAA
AA
CCAAUAGG
P5’
U
G
C U G
G A CU
A
UU
UAC

Pri
(μM): 5* 25 25 5* 25 25 5* 25 25
pppGGU pppGUGpppGAG pppUGUpppGCG pppGACpppACA pppCUC
pppCUU pppNNN
pppGGU pppGUGpppGAG pppUGCpppGGA pppCACpppUCC pppUUC pppNNN
pppGGU pppGUGpppGAG pppCGGpppACU pppCCU
pppUCU pppNNNSubstr
ate
s 4S 6S 8S
Structured template:
CUGC C A A C C5’
GACGGUUGG C C A C A3’
UA G
UGC
AC
U
CCG
GC
GA
GCU
C
5’ AA AG G
GGUPPP
P
TM ~ 93˚C
CUGC C A A C C5’
GACGGUUGG C C A C A3’
UA G
CU
CCG
GC
GA
GCU
C
5’ AA AG G
GGUPPP
P
TM ~ 89˚C
CUGC C A A C C5’
GACGGUUGG C C A C A3’
U
CCG
GC
GA
GCU
C
5’ AA AG G
GGUPPP
P
TM ~ 82˚C
Template 8S
Template 4S
Template 6S

0[each triplet]
(μM)0.5 21 3 105:
0 0.03 0.220.07 0.43 0.70.6extension ≥ 3extension ≥ 2 :
Pri
0 2 53 10 4020: 80[each dinucleotide]
(μM)
pppGG pppUG pppGA pppGUpppGC pppAC pppUC pppCU
pppGGU pppGUG pppGAGpppUGU pppGCG pppGACpppACA pppCUC pppCUU
Pri
Substrates:

Synthesis
Scale (pmol),
Volume (μl)
2000,1000
170,340
3000,6000
4000,2000
400,800
115,460
Ribozyme
2 μM t5+1
2 μM t5+1
0.5 μM t5+1
2 μM t5+1
0.5 μM t5a+1
0.5 μM t5a+1
Primer
2 μMpppGCCAUC
1 μMBioα9
0.5 μMpppGCAAAA
2 μMpppGCGAUAG
0.5 μMδ+
0.5 μMBioγ7
Incubation
time (d)
14
24
22
20
6*
11
Recovered
product (pmol)
183 β+
19 αβ+
521 ε+
432 δ+
115 δε+
5.2 γδε+
Triplets
10 μM pppAAA, pppGCU, pppUGA,pppGAG, pppCAU, pppCUU
10 μM pppCGA, pppUCU, pppAAA
5 μM pppCGC, pppGUG,pppCUU, pppCGU, pppGAG
5 μM pppCGC, pppCAA,pppCGU, pppUCU, pppCCA
none
5 μM pppGAG, pppGCG, pppGCA,pppGCC, pppUUC, pppGGU, pppGGC
βα+β
εδ
δεγ+δε
Template
2 μMTβ
1 μMTαL
0.5 μMTε
2 μMTδ
0.5 μMδεsplint
0.5 μMTγL
Oligomers
none
2 μM pppGACAAA0.5 μM β+
0.5 μM pppUGAAUG0.5 μM pppUUUUUCAUG
2 μMpppUGACAU
0.5 μMε+
0.25 μMδε+
Full-length
product (%)
31
14
21
15
36
8
αβ+ xt
β+ xt
31%
β+ 14%
αβ+ γδε+
xt ε+
21%
15%
xt δ+
8%
xt γδε+ xt δε+
36%
δ+
ε+
δε+
β α+β γ+δε δε δ ε

0.5 µM t5+1 2 µM t5+1 2 µM type 6s+1Ribozyme:
3’ terminalsubstrates
333
63 9
333
63 9
333
63 9
333
63 9
333
63 9
333
63 9
0.5 µMpppUGAAUG
0.5 µMpppUGAAUG
0.5 µMpppUGAAUG
0.5 µM t5+1
0.5 µMpppUGAAUG
9:
2 µM P, T, 9,pppUGAAUG
BA
[tri] (µM): 5 10 5 10 5
full-lengthproduct (%)
: 29 34 13 30 33
pppGUG
pppCUU
pppCGU
pppUGA
pppAUG
pppGAG
pppUUU
pppUUC
pppAUG
3’CA G
U
U
UU
UG
UAC
U
GCC
G
UU
AU
GC
CG
GU
A AAA
C
GG
5’core
5’ AUGUCAUGGUUGAGAACGUUGGCGCUAUCGC
UGAPPP
CAUPPP
UGACAUPPPC
CCAPPP
CCAPPP
δ template:
-hex: +hex:
Pri
Fulllength
CGC
CAA
CGU
UCU
CAA
UGACAU
CCA
CGC
CAA
CGU
UCU
CAA
UGACAU
CCA
-hex +hex
t5a +
1
type 5
s +
1
3’CA G
U
U
UG
UAC
U
GCC
G
UU
AU
GC
CG
GU
A AAA
C
5’core
5’ AUGUCAUGGUUGAGAACGUUGGCGCUAUCGC
UGAPPP
CAUPPP
UGACAUPPPC
CCAPPP
CCAPPP
δ template:
U
CA
-hex: +hex:
t5a +
1
type 5
s +
1
t5a +1
type 5s +1
Rz:

Full-length (%): 17 5.4 16 10 3.4 15 4.4 1.6 4.3 25 19 18 28 14 29 16 7.2 17 18 10 14 7.5 2.9 .76
tri
pppN
NN
pppN
NN
low
-G
tri
pppN
NN
pppN
NN
low
-G
tri
pppN
NN
pppN
NN
low
-G
tri
pppN
NN
pppN
NN
low
-G
tri
pppN
NN
pppN
NN
low
-G
tri
pppN
NN
pppN
NN
low
-G
tri
pppN
NN
pppN
NN
low
-G
tri
pppN
NN
pppN
NN
low
-G
Substrate:
P/T: Fα9/BTαHP Fβ6/Tβ Fγ7/BTγHP Fδ7/Tδ Fε6/Tε
Oligomers: pppGACAAA pppCAUCUU pppUGACAUpppUGAAUG
pppUUUUUCAUG
[P/T] (μM): .5 .5 .5 .35 .5 .5 .5.35 1 1 .35 .5 .5 .5 .5 .5 .5 .5 .5 .5 .5 .5.35 .35

Fγ7/TγHPFδ7/Tδ
[each pppNNN] (μM): *5 5 5 5 5 0 0
[each pppNN] (μM): 0 0 5 20 40 40 0
[each pppN] (μM): 0 0 5 80 320 320 0
*5 5 5 5 5 0 0
0 0 5 20 40 40 0
0 0 5 80 320 320 0
Average ligationsper primer 1.54 0.98 0.95 0.91 0.91 0.03 0 2.16 2.81 2.83 2.86 2.94 1.02 0:
Extension producttriplet register (%) 100 100 86 72 58 16 100 100 96 84 70 2:
Pri

6.3 5.5 5.9 4.4
3.4 2.1 2.7 1.8
4.3 3.7 3.1 1.7
95.3 96.2 96.1 97.4
type 5+1 t5+1
±.61(3)
type 5 type 5s +1
±1.1(3)±.73(2)s.d.(n):
αβγδ+1
2.5
4.7
9.1
94.5
±.31(4)
Overall fidelities (%):
Rz:
1st
2nd
3rd
trip
let base:
Average error rates (%) per triplet position:

10
1
0.1
0.01
1st position errors 2nd position errors 3rd position errors
A^^
Adr
A``
G^`
G`d
Gr^
Cdd
C`^
Crr
U^r
Ud`
Urd ^ A^
^ Gr
^ C`
d A`
d Cd
d U^
` Gd
` C^
` Ur
rAr
r G`
r Ud
^^A
^dU
^`C
d^C
d`G
drA
`dG
``A
`rU
r^U
rdC
rrGEncoded triplet:
t5+
1
αβγδ
+1ra
tio o
f m
isin
corp
ora
tion r
ate
s o
f=
A=
G=
C=
U
ε in
cre
ases
err
or
ε decre
ases
err
or

fractio
nal G
AU
incorp
ora
tion
ln [GGU]
xt
x− x+
0
1
fractional G
AU*
incorp
ora
tion
ln [GGU*]
xt
x−*x+*0
1
Modified triplets (*)Unmodified triplets
at = t, W = W* =GGU incorporation
GAU incorporation
GGU* incorporation
GAU* incorporation
[GGU]and
[GGU*]
ln = k(x+- x−)
W−W
+
( ) ln = k(x+*
- x−*) φ = = (x+*
- x−*)
(x+- x−)
If φ is , the modification * the fidelity phenotype (for GAU vs. GGU)
~0
<1
~1
>1
abolishesattenuatesdoesn’t affectenhances
ln(W−/W+)
ln(W−/W+)
* *( )**
W−W
+
+GGU
CCC
+GAU
CCC
GAU
t5a +1
Pri
αβγδ+1
GA
U2S
GG
U2S
Match (1 μM):
Mis- match (4 μM):
GA
U
GA
U2S
GA
U
GG
U2S
GG
U
GG
U
W, n=4: .66 2.4 1.7 1.9
+GGU+AGU
AGU
t5a +1
Pri
αβγδ+1
AG
U2S
GG
U2S
Match (1 μM):
Mis- match (4 μM):
AG
U
AG
U2S
AG
U
GG
U2S
GG
U
GG
U
W, n=3: 1.3 .79 .71 .77
φ = -0.2, ± 0.23 (s.d.) φ = 0.0, ± 0.18 (s.d.)
+UGG
CCC
+UGA
CCC
UGA
t5a +1
Pri
αβγδ+1
U2SG
AU
2SG
G
Match (1 μM):
Mis- match (4 μM):
UG
A
U2SG
A
UG
A
U2SG
G
UG
G
UG
G
W, n=4: .36 .27 .73 .45
φ = 0.8, ± 0.52 (s.d.)
+AGG+AGU
AGU
t5a +1
Pri
αβγδ+1
A2’FG
UA
2’FG
GMatch (1 μM):
Mis- match (9 μM):
AG
U
A2’FG
U
AG
U
A2’FG
G
AG
G
AG
G
W, n=3: 1.5 1.9 7.1 9.1
φ = 1.0, ± 0.19 (s.d.)
CUG CUG
t5a +1 αβγδ+1 t5a +1 αβγδ+1 t5a +1 αβγδ+1
Pri
CUG
AGU
+GGU+AGU
CUG
GAU
+GGU+GAU
CUG
AGU
+AGG+AGU
Pri Pri
Downstream triplet (5 μM):
HOC
UG
-
HOC
UG
-
HOC
UG
-
HOC
UG
-
HOC
UG
-
HOC
UG
-
pppC
UG
pppC
UG
W, n=3: 1.7 .84 1.1 .69 .65 4.7 3.0 5.3 2.3 2.4 9.4 11 9.5 14
φ = 0.5, ± 0.18 (s.d.) φ = 0.0, ± 0.54 (s.d.)
W, n=3:W, n=3:
φ = 0.3, ± 0.04 (s.d.) φppp = 0.9, ± 0.09 (s.d.)
1 μ
M G
AU
4 μ
M G
GU
1 μ
M A
GU
9 μ
M A
GG
1 μ
M A
GU
4 μ
M G
GU
vs.5’
AAC GU C
UC G
pppAGU pppAGG
OH
5’ AAC GU C
pppAGU pppAGG

0.1
1
10
0.3 30
2 to 3 1 to 2 0 to 1
Mutation incidence with 5 μM pppNNN (%)
Mutation effect upontriplet GC content:
Muta
tion incid
en
ce w
ith 1
0 μ
M p
ppN
NN
Muta
tion incid
ence w
ith 2
μM
pppN
NN
3
0.1
1
10
0.3 30
Mutation incidence with 5 μM pppNNN (%)
3
Mu
tation incid
en
ce
with 1
0 μ
M p
ppN
NN
Muta
tion incid
ence w
ith 2
μM
pppN
NN
Average error contribution (%)of classes of substitutionsby effect on GC content:
+GC =GC -GC
1.42 0.14 0.44
Err
or
occurr
ence (
%)
10
1
40
0.4
4
1 100.5 52
pppNNN concentration(μM of each triplet)
3 to 2 2 to 1 1 to 0Mutation effect upontriplet GC content:
: CC
G
A : GU
G
A : A A
G
A
: G G
U
C: A C
U
C : Overall error rate
0.5 μMpppNNN 2.78 0.28 0.59
5 μMpppNNN
Copyright © 2022 FDOKUMEN




















