
Theoretical investigation of the relative stabilities of singlet and triplet disulfides
9
Theoretical investigation of the relative stability of Na+He n (n = 2–24) clusters: Many- body versus delocalization effects Noureddine Issaoui, Kawther Abdessalem, Houcine Ghalla, Saud Jamil Yaghmour, Florent Calvo, and Brahim Oujia Citation: The Journal of Chemical Physics 141, 174316 (2014); doi: 10.1063/1.4900873 View online: http://dx.doi.org/10.1063/1.4900873 View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/141/17?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Many-body effects on the structures and stability of Ba2+Xe n (n = 1–39, 54) clusters J. Chem. Phys. 141, 154308 (2014); 10.1063/1.4896607 Diffusion Monte Carlo: A powerful tool for studying quantum many-body systems Am. J. Phys. 82, 980 (2014); 10.1119/1.4890824 An intramolecular vibrationally excited intermolecular potential for He–OCS: Globally tested by simulation of vibrational shifts for OCS in He N N = 1 − 100 Clusters J. Chem. Phys. 137, 234310 (2012); 10.1063/1.4772186 Theoretical investigation of nonadiabatic and internal temperature effects on the collision-induced multifragmentation dynamics of Na 5 + cluster ions J. Chem. Phys. 123, 074331 (2005); 10.1063/1.1991857 Theoretical study of the finite-temperature spectroscopy in van der Waals clusters. III. Solvated chromophore as an effective diatomics J. Chem. Phys. 118, 8763 (2003); 10.1063/1.1566952 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 41.229.253.14 On: Fri, 07 Nov 2014 15:10:10
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Theoretical investigation of the relative stabilities of singlet and triplet disulfides

Theoretical investigation of the relative stability of Na+He n (n = 2–24) clusters: Many-body versus delocalization effectsNoureddine Issaoui, Kawther Abdessalem, Houcine Ghalla, Saud Jamil Yaghmour, Florent Calvo, and BrahimOujia Citation: The Journal of Chemical Physics 141, 174316 (2014); doi: 10.1063/1.4900873 View online: http://dx.doi.org/10.1063/1.4900873 View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/141/17?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Many-body effects on the structures and stability of Ba2+Xe n (n = 1–39, 54) clusters J. Chem. Phys. 141, 154308 (2014); 10.1063/1.4896607 Diffusion Monte Carlo: A powerful tool for studying quantum many-body systems Am. J. Phys. 82, 980 (2014); 10.1119/1.4890824 An intramolecular vibrationally excited intermolecular potential for He–OCS: Globally tested by simulation ofvibrational shifts for OCS in He N N = 1 − 100 Clusters J. Chem. Phys. 137, 234310 (2012); 10.1063/1.4772186 Theoretical investigation of nonadiabatic and internal temperature effects on the collision-inducedmultifragmentation dynamics of Na 5 + cluster ions J. Chem. Phys. 123, 074331 (2005); 10.1063/1.1991857 Theoretical study of the finite-temperature spectroscopy in van der Waals clusters. III. Solvated chromophore asan effective diatomics J. Chem. Phys. 118, 8763 (2003); 10.1063/1.1566952
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10

THE JOURNAL OF CHEMICAL PHYSICS 141, 174316 (2014)
Theoretical investigation of the relative stability of Na+Hen (n = 2–24)clusters: Many-body versus delocalization effects
Noureddine Issaoui,1,a) Kawther Abdessalem,1 Houcine Ghalla,1 Saud Jamil Yaghmour,2
Florent Calvo,3 and Brahim Oujia1,2
1Faculty of Sciences, Quantum Physics Laboratory, University of Monastir, Monastir 5079, Tunisia2Faculty of Science, King Abdul-Aziz University, Jeddah, Kingdom of Saudi Arabia3University of Grenoble Alpes, LIPHY, F-38000 Grenoble, France and CNRS, LIPHY,F-38000 Grenoble, France
(Received 28 May 2014; accepted 20 October 2014; published online 7 November 2014)
The solvation of the Na+ ion in helium clusters has been studied theoretically using optimizationmethods. A many-body empirical potential was developed to account for Na+–He and polarizationinteractions, and the most stable structures of Na+Hen clusters were determined using the basin-hopping method. Vibrational delocalization was accounted for using zero-point energy corrections atthe harmonic or anharmonic levels, the latter being evaluated from quantum Monte Carlo simulationsfor spinless particles. From the static perspective, many-body effects are found to play a minor role,and the structures obtained reflect homogeneous covering up to n = 10, followed by polyicosahedralpacking above this size, the cluster obtained at n = 12 appearing particularly stable. The cationicimpurity binds the closest helium atoms sufficiently to negate vibrational delocalization at smallsizes. However, this snowball effect is obliterated earlier than shell completion, the nuclear wave-functions of 4HenNa+ with n = 5–7, and n > 10 already exhibiting multiple inherent structures. Thedecrease in the snowball size due to many-body effects is consistent with recent mass spectrometrymeasurements. © 2014 AIP Publishing LLC. [http://dx.doi.org/10.1063/1.4900873]
I. INTRODUCTION
The determination of the electronic structure is of a greatimportance in the calculation of the resonance lines pressureof alkali-rare gas systems. Indeed, the resonance lines pres-sure are extremely important in the spectra of brown dwarfsand they can be considered as a source of opacity in extra so-lar giant planet atmospheres.1–3 The accuracy of the calcula-tions of these profiles can lead to develop effective diagnosticsof the temperature and the composition of the atmosphere.4, 5
The potential energy curves are also essential to understandthe structure and the stability of alkali-rare gas clusters, whichhave been used as model compounds in a number of stud-ies aimed to get insight into the molecular mechanisms ofsolvation.6 The interactions among rare-gas atoms and be-tween those atoms and the alkali impurity are responsiblefor the most stable cluster structures detected experimentallythrough higher abundances in mass spectra, and the competi-tion between those interactions dictates the generic behavior.If the impurity is a cation, it tends to become embedded in therare-gas cluster essentially homogeneously,7 whereas in neu-tral form the absence of polarization forces can result in theimpurity floating on the inert cluster.8
One particular case of rare-gas solvent is helium, whichpresents particularly weak interactions with other neutralatoms, including itself, and has thus been suggested as anideal cryostat for exploring spectroscopy and reactivity underextremely cold conditions.9 Due to vibrational delocalization,
a)E-mail: [email protected]
pure helium clusters are naturally found as liquid dropletspossibly presenting superfluidity.10 Doping such clusters withimpurities, however, can relocalize some atoms if the inter-actions are strong enough, at least in the vicinity of the im-purity, an effect known as “snowballing.”11 In the case ofions, this effect is caused by electrostriction and the increasein the binding strength due to polarization forces. The in-terplay between interactions and vibrational delocalizationhas been explored in the case of ionic alkaline earth and al-kali impurities by several groups in the past12–15 and espe-cially Gianturco and co-workers16–20 who notably focused onlithium16 and potassium.17 Using quantum chemistry meth-ods and empirical pairwise potentials, these authors predictedthe first solvation shell of the Li+ ion to be completed atonly 10 atoms,16 at variance with the icosahedral shell com-pletion occurring at 12 atoms for potassium17 and generallynotorious in heavier van der Waals clusters. These authorsalso emphasized the importance of nuclear quantum effectson the binding energy of such clusters18, 19 and confirmedthat shell completion is associated with snowballing at largersizes.
In the case of sodium, earlier calculations by Reatto andco-workers in the liquid and cluster phases13 have suggested asnowball size of 10, but later efforts from the same group14, 15
revised this number to 12, in agreement with more recentquantum Monte Carlo simulations19 by Marinetti et al. Thisnumber corresponds to a complete icosahedral shell, oftenmet as a magic number in van der Waals clusters, and cal-culations have even suggested that icosahedral order couldextend beyond the first solvation shell.15 Despite consen-
0021-9606/2014/141(17)/174316/8/$30.00 © 2014 AIP Publishing LLC141, 174316-1
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10

174316-2 Issaoui et al. J. Chem. Phys. 141, 174316 (2014)
sus on the snowball size of 12 atoms among theoreticians,recent mass spectrometry measurements carried out in theScheier group21 have found a significant drop in the abun-dances of 4HenNa+ clusters at n > 9. At present, the reasonsfor this intriguing discrepancy remain unclear. The variationsin the snowball size across the series of alkali cations indicatethe key role of the interactions. Because the practical solu-tion to the vibrational Schrödinger equation requires inten-sive evaluations of the potential energy, pair potentials haveusually been considered for this task.12–19 In the case of thehelium solvent, the role of more accurate interactions beyondtwo-body terms has mostly been addressed at the ab initiolevel16–18, 22 or with explicit three-body contributions betweeninduced dipoles.19 Those interactions were found to be ratherinsignificant on the stable structures,19, 20 which is not sur-prising in light of the weak polarizability of the helium atom,whereas heavier rare gases could react differently especiallyin presence of dications.23–25 However, such effects on the in-teractions have not been included in the determination of thevibrational wavefunction so far. Many-body forces could im-pact nuclear delocalization because helium-helium binding isso weak that it could compete with these higher-order elec-trostatic effects, especially near the first solvation shell wherethe induced dipoles are strongest.
The purpose of the present article is to revisit theinterplay between interaction forces and nuclear quantum ef-fects in Na+Hen clusters across the regime where snowballingtakes place, namely, for n = 1–24. We have constructed a po-larizable potential based on pairwise diatomic energy curvesand a self-consistent many-body contribution between in-duced dipoles, and used it with the basin-hopping optimiza-tion method in order to locate the most stable structures at0 K. Those minima turn out to be anisotropic at very smallsizes, then homogeneous around 6–10 atoms, and finallyicosahedrally packed for large enough clusters. The influenceof nuclear quantum effects was investigated specifically byconsidering harmonic zero-point energies (ZPE) and, morerigorously, by performing quantum Monte Carlo simulationsof the ground state wavefunction. Those simulations indicatethat delocalization occurs at sizes earlier than previously pre-dicted, the nuclear wavefunction showing multiple nearly de-generate isomers for n = 5 and 6 and, more importantly, atsizes 11 and onward. Repeating the simulations but with thepair potential confirms the spreading of the wavefunction overmultiple isomers at the small sizes mentioned, but reevaluatesthe onset of appearance of a liquid layer covering a solid-like structure above icosahedral shell filling, in agreementwith earlier studies also employing pair potentials.13–15, 19
Our finding of a slightly lower onset of snowballingthan previously reported agrees with mass spectrometryexperiments.21
The article is organized as follows. In Sec. II, we brieflydescribe the potential energy developed to model Na+Henclusters, and the computational methods employed to locatetheir putative global minima and to evaluate the ground stateenergies. Section III presents our results and discusses therelative stabilities and the extent of vibrational delocaliza-tion based on specific energy indicators. Some concludingremarks finally end the article in Sec. IV.
II. THEORETICAL METHODS
Our computational investigation of the structures and en-ergies of Na+Hen clusters is based on an explicit potentialenergy surface that allows systematic sampling of the energylandscapes for unbiased global optimization through basin-hopping as well as diffusion Monte Carlo (DMC).
A. Many-body potential
To model Na+Hen clusters, we follow here a stan-dard approach26 by decomposing the potential energy of the(n+1)-atom system into its dominant contributions of short-range repulsion, long-range dispersion, as well as electrostaticinteractions and polarization. The total potential energy isthus written as
V =n∑
i=1
VNa+−He(r0i) +n∑
i=1
∑i<j
VHe−He(rij ) + Vpol, (1)
where rij denotes the distance between particles i and j, thesodium ion having index 0. In this equation, the first termrefers to the pair interaction between the sodium cation andneutral helium atoms, which is explicitly modeled by the fol-lowing expression:
V (R) =[A + A′
r12
]exp(−br) − C6/r6
−C8/r8 − C10/r10 − C12/r12 (2)
and thus contains 7 adjustable parameters A, A′, b, C6, C8,C10, C12. The repulsive term A′/r12 was added for practicalpurposes only in order to avoid the attractive catastrophe atshort distances. For the He–He potential, we used a well es-tablished potential given by Tang and Toennies.27 The polar-ization potential Vpol is expressed as
Vpol = −αHe
2
n∑i=1
�Ei ·→E0
i , (3)
where→Ei and
→E0
i , respectively, denote the total and bare elec-tric fields on helium atom i, αHe = 9.34 Å3 being the atomicpolarizability of helium. The total electric fields contain thecontributions from induced dipoles, and this leads to the tradi-tional self-consistent solution to the many-body electrostatic
problem, in which→Ei and the induced dipole moment
→μi at
site i satisfy the equations
→Ei =
→E0
i +∑
j
Tij
→μj with
→μj = αHe
→Ej , (4)
Tij being the dipole tensor whose component (k,l = x, y, z)reads
T klij = 1
r3ij
[3rk
ij rlij
r2ij
− δkl
], (5)
δkl denoting the Kronecker symbol. The equations are solvedself-consistently to yield the set of dipole moments and elec-tric fields on each helium atom.
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10

174316-3 Issaoui et al. J. Chem. Phys. 141, 174316 (2014)
FIG. 1. Upper panel: Electronic ground state potential energy of Na+He ver-sus internuclear distance, as obtained from the present model and comparedwith reference data from Ref. 28. Lower panel: Potential energy of Na+He2as a function of the He–Na+–He angle, as obtained from the many-body andpairwise potentials. The shaded area depicts the probability distribution of theangle predicted from the diffusion Monte Carlo trajectories with the many-body potential.
While being of course more rigorous, the many-body po-tential is also computationally more demanding than a sim-ple pair potential, and we have considered the truncation ofthe expression of Eq. (1) into two-body contributions only, anapproximation that is equivalent to replacing Vpol by
V(pair)
pol = −αHe
2
n∑i=1
1
r4i0
. (6)
This approximation is exact only for the diatomicNa+–He, but tends to overestimate the binding energy inlarger clusters due to the neglect of the contribution of in-duced dipoles on the electric fields.
The parameters A, A′, b, C6, C8, C10, C12 were fittedto reproduce the experimental potential energy curve of Sol-dan and co-workers,28 and they read A = 2.1, A′ = 108,b = 3.93747, C6 = 19.637, C8 = −173.21, C10 = −5134.08,C12 = 33508, all in atomic units. The quality of the ad-justment can be gauged by looking at Fig. 1 in which wehave represented the resulting energy curve together with thereference data.28 Table I reports the spectroscopic constantsobtained with this potential and compares them with otheravailable data.29, 30 The agreement is usually very good, espe-cially for the equilibrium distance where the residual discrep-ancy is lower than 0.02 Å. The potential also quantitatively
TABLE I. Spectroscopic constants of Na+He obtained with the present pairpotential and compared with available data: equilibrium distance Re, poten-tial well depth De, vibrational constant ωe, anharmonic constant ωexe, androtational constant Be.
Re (Å) De (cm−1) ωe (cm−1) ωexe (cm−1) Be (cm−1) Ref.
2.322 330 128.803 13.34 . . . 282.317 318 129 . . . . . . 292.343 325 . . . . . . . . . 302.322 327 128 13.14 0.9163 Present work
agrees with the one calculated by Marinetti and co-workers19
at the coupled-cluster level with single, double, and perturba-tive triple excitations.
B. Geometry optimization
The lowest-energy structures of the Na+Hen clusterswere located using the basin-hopping global optimizationmethod,31 which has a long and successful historical recordin clusters science32, 33 and has been used for doped heliumclusters in the past.16–19 This stochastic algorithm performsa random walk on the transformed energy landscape consist-ing of the local minima obtained after each large amplitudecollective random displacement. Five series of 105 minimiza-tions were performed for each cluster size.
C. Vibrational delocalization
Because helium compounds are so weakly bound andlikely to exhibit strong nuclear quantum effects, it is neces-sary to correct the binding energy for the contribution of ZPE.We have considered the harmonic approximation to the ZPEby extracting the vibrational frequencies of all global minimafrom the diagonalization of the dynamical matrix. This ap-proximation, which will be refered to as quantum harmonic,works well for mildly delocalized systems such as argon oreven neon,34 however it is questionable for helium and wehave considered instead a more accurate anharmonic evalua-tion based on DMC.35
We have simulated the vibrational wavefunction of4HenNa+ clusters at T = 0 by performing simple (un-guided) DMC without exchange symmetry, hence ignoringthe bosonic character of the solvent atoms. Two thousandinitial walkers were propagated, the diffusion equation be-ing integrated over 20 K−1 subsequently to an equilibrationtime of 10 K−1, and three time steps of 0.5 × 10−4, 10−4, and1.5 × 10−4 K−1 were used for the integration. For the presentfirst-order integrator, the final energies obtained from 10 in-dependent samples were linearly extrapolated to a zero timestep. Expectation values and density profiles were obtained byaveraging over the snapshots via descendant weighting.36
III. RESULTS AND DISCUSSION
We first discuss the general structural features of the clus-ters from the purely static perspective of global minima. Thelowest-energy structures of Na+Hen clusters obtained withthe basin-hopping method and our polarizable potential aredepicted in Fig. 2 as a function of size. Solvation of thesodium cation by helium atoms proceeds first on the side untiln = 6, above which it becomes more isotropic. Thisanisotropic pattern is naturally expected as the consequenceof He–He interactions being attractive, even after includingthe contribution between induced dipoles, helium atoms cov-ering progressively the impurity. Once size 7 is reached, amore homogeneous and isotropic solvation shell is found asthe result of longer He–He bonds that accommodate favor-ably with such a low surface density. Size 11 marks the on-set of the icosahedral motif, which is complete at size 12 and
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10

174316-4 Issaoui et al. J. Chem. Phys. 141, 174316 (2014)
FIG. 2. Lowest-energy structures of Na+Hen clusters predicted by the basin-hopping algorithm. The sodium ion is shown as a blue sphere, the heliumatoms of the first and higher solvation shells being represented as dark andlight gray spheres, respectively.
remains above this size as additional helium atoms tend to capthe icosahedron. However, in contrast with pure van der Waalsclusters37 that are dominated by polyicosahedral growth, theheterogeneity in the He–He and Na+–He interactions makesthe growth more isotropic around the impurity, a feature thatcan also be seen as a manifestation of geometric frustration.This is, for example, seen on the 19-particle cluster whosemost stable structure differs from the elongated and five-fold symmetric double icosahedron. Frustration can also benoted in the smaller clusters, which do not display symme-tries as high as pure van der Waals clusters. The 7-particlesystem, for instance, shows significant distortions and be-longs to the C3 point group instead of having the fivefoldsymmetry of the pure van der Waals compound37 or the oc-tahedral symmetry arising when the ion-rare gas interactiondominates.19 In larger clusters, the extra helium atoms cap thefirst shell over existing triangular facets, but they do so by sur-rounding the cationic impurity at the expense of He–He firstneighbor bonds that remain disfavored over Na+–He secondneighbor bonds. The growth pattern near n = 24 is notablytypical of polyicosahedral growth37, 38 arising from the pre-dominance of interactions to a central particle. Repeating thecalculations with the pair potential does not change theseoverall structures, but we do not exclude that larger clustersmay be slightly affected due to the very large number of min-ima they support.
From a more quantitative point of view, it is interest-ing to discuss the specific case of the triatomic Na+He2, for
which the polarizable potential predicts a very slight elonga-tion of the Na+–He bond distance (from 2.33 to 2.34 Å) oncemany-body effects are taken into account. This elongation re-flects the slightly less attractive potential due to the repulsionbetween induced dipoles on neighboring atoms, and is con-sistent with the ab initio results of Sapse and co-workers.22
Potential energy curves were calculated as a function of theHe–Na+–He angle by fixing the Na+–He distances to theircorresponding value in the minimum structure. They are rep-resented in Fig. 1 for the two potentials having or neglect-ing the self-consistent interaction between induced dipoles.The curves for both potentials are particularly flat, with welldepths lower than 10 wavenumbers and comparable to thebinding energy of a He2 bond. More importantly, their rel-ative difference is of the same order of magnitude near theminimum, suggesting that this minor correction could be ofconsequence on the relative stability of isomers.
With such shallow minima, the trimer is likely to un-dergo strong nuclear delocalization along its bending angle.The probability distribution of this angle was evaluated fromquantum Monte Carlo simulations with the polarizable poten-tial, it is superimposed in Fig. 1 to the potential energy curves.Clearly, this distribution spreads over more than 40◦ aroundthe minimum value of 80◦, indicating significant delocaliza-tion effects, even though the barrier at the linear configurationis not crossed.
The binding energy of the clusters is represented in Fig. 3as a function of their size, in the static, quantum harmonic andanharmonic approximations. The static results are insightfulbecause they exhibit clearly two regimes with different slopesat small (n < 10) and large (n > 12) sizes. The former regimeis associated with the predominance of Na+–He bonds thatare much stronger than the newly formed He–He bonds oncethe first solvation shell is filled. Just before the icosahedralshell is completed, the binding energy shows a clear acci-dent, the cluster at n = 11 being rather instable. Those en-ergies and their variations, which are in good agreement withthe work by Marinetti and co-workers,19 confirm the inter-play between size mismatch and shell completion known in
FIG. 3. Binding energy E(n) of Na+Hen clusters in the static limit (blackcircles) and after including harmonic (red squares) and anharmonic (blue tri-angles) zero-point corrections. The classical results obtained with the pairpotential are also indicated.
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10
174316-5 Issaoui et al. J. Chem. Phys. 141, 174316 (2014)
heterogeneous systems. The pairwise approximation for theinteraction potential does not significantly alter those con-clusions except for a general overestimation of the cohesionenergies. However, this difference approaches and exceedsthe helium-helium binding strength approximately once theicosahedron is complete. Although we have not found majorstructural transitions except at n = 6, we anticipate that vi-brational delocalization could have different signatures in thepair and many-body approximations.
As expected, the inclusion of zero-point energy correc-tions significantly alters the binding energies, especially inlarger clusters where the helium solvent plays an increas-ing role. However, the two regimes identified in the classi-cal limit remain clearly visible only with different slopes, andthe accident near shell completion at n = 12 remains as well.The harmonic approximation is found to be remarkably ac-curate for small clusters built up to the first solvation shell,as the result of relatively strong bonds and a higher massassociated with Na+–He vibrations. Once the shell is com-plete, additional helium atoms have to bind to other heliumatoms, the resulting bonds being much weaker and increas-ingly prone to delocalization. The general overestimation ofzero-point energies by the harmonic approximation becomesexcessive in this regime, to the extent that the binding energyexhibits a net decrease in modulus with addition of extra he-lium atoms. This unphysical result marks the limit of valid-ity of the harmonic approximation, which is not suitable todescribe strongly delocalized helium compounds. The resultsfrom quantum Monte Carlo simulations are at variance withthis decreasing trend, the ground state energies being now re-evaluated as always binding (i.e., negative), as they should.The increasing importance of the zero-point contribution inNa+Hen clusters can also be appreciated by considering theenergy difference �E(n) = E(n + 1) − E(n) associated withthe dissociation of a helium atom from the Na+Hen+1 clus-ter, a quantity that is usually sensitive to particularly stable orunstable sizes. The variations of these dissociation energiesare represented in Fig. 4 as a function of cluster size, again inthe classical, quantum harmonic and anharmonic approxima-tions.
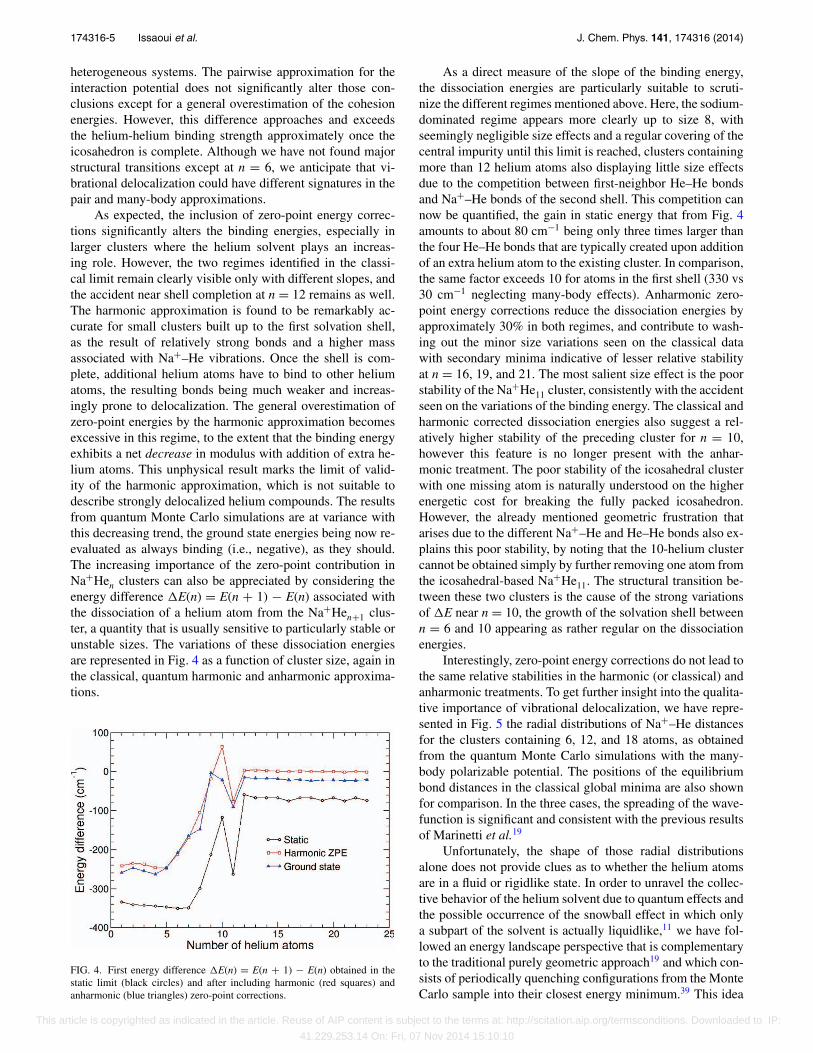
FIG. 4. First energy difference �E(n) = E(n + 1) − E(n) obtained in thestatic limit (black circles) and after including harmonic (red squares) andanharmonic (blue triangles) zero-point corrections.
As a direct measure of the slope of the binding energy,the dissociation energies are particularly suitable to scruti-nize the different regimes mentioned above. Here, the sodium-dominated regime appears more clearly up to size 8, withseemingly negligible size effects and a regular covering of thecentral impurity until this limit is reached, clusters containingmore than 12 helium atoms also displaying little size effectsdue to the competition between first-neighbor He–He bondsand Na+–He bonds of the second shell. This competition cannow be quantified, the gain in static energy that from Fig. 4amounts to about 80 cm−1 being only three times larger thanthe four He–He bonds that are typically created upon additionof an extra helium atom to the existing cluster. In comparison,the same factor exceeds 10 for atoms in the first shell (330 vs30 cm−1 neglecting many-body effects). Anharmonic zero-point energy corrections reduce the dissociation energies byapproximately 30% in both regimes, and contribute to wash-ing out the minor size variations seen on the classical datawith secondary minima indicative of lesser relative stabilityat n = 16, 19, and 21. The most salient size effect is the poorstability of the Na+He11 cluster, consistently with the accidentseen on the variations of the binding energy. The classical andharmonic corrected dissociation energies also suggest a rel-atively higher stability of the preceding cluster for n = 10,however this feature is no longer present with the anhar-monic treatment. The poor stability of the icosahedral clusterwith one missing atom is naturally understood on the higherenergetic cost for breaking the fully packed icosahedron.However, the already mentioned geometric frustration thatarises due to the different Na+–He and He–He bonds also ex-plains this poor stability, by noting that the 10-helium clustercannot be obtained simply by further removing one atom fromthe icosahedral-based Na+He11. The structural transition be-tween these two clusters is the cause of the strong variationsof �E near n = 10, the growth of the solvation shell betweenn = 6 and 10 appearing as rather regular on the dissociationenergies.
Interestingly, zero-point energy corrections do not lead tothe same relative stabilities in the harmonic (or classical) andanharmonic treatments. To get further insight into the qualita-tive importance of vibrational delocalization, we have repre-sented in Fig. 5 the radial distributions of Na+–He distancesfor the clusters containing 6, 12, and 18 atoms, as obtainedfrom the quantum Monte Carlo simulations with the many-body polarizable potential. The positions of the equilibriumbond distances in the classical global minima are also shownfor comparison. In the three cases, the spreading of the wave-function is significant and consistent with the previous resultsof Marinetti et al.19
Unfortunately, the shape of those radial distributionsalone does not provide clues as to whether the helium atomsare in a fluid or rigidlike state. In order to unravel the collec-tive behavior of the helium solvent due to quantum effects andthe possible occurrence of the snowball effect in which onlya subpart of the solvent is actually liquidlike,11 we have fol-lowed an energy landscape perspective that is complementaryto the traditional purely geometric approach19 and which con-sists of periodically quenching configurations from the MonteCarlo sample into their closest energy minimum.39 This idea
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10

174316-6 Issaoui et al. J. Chem. Phys. 141, 174316 (2014)
FIG. 5. Distribution of Na+–He distances in selected Na+Hen clusters,n = 6, 12, 18, in their global minima (vertical lines pointing up) or obtainedfrom diffusion Monte Carlo simulations (continuous curves). For n = 12, thediscrete distances are also shown for a metastable minimum (b) depicted inFig. 6 as vertical lines pointing down.
is similar to the concept of inherent structures in liquid statetheory,40 in which energy minimizations are performed in or-der to clean the instantaneous configurations from the thermalnoise, thereby revealing a diversity of local minima that con-trasts with the much more limited set of minima associatedwith the crystal state. In the present case, 10 000 configura-tions were locally minimized for each cluster size, leading toa set of energies {Eα} each associated with a specific mini-mum α, and keeping also track of the number of occurrences{gα} of each minimum. From those data an energetic delo-calization index δE was defined as the standard fluctuation√
〈E2〉 − 〈E〉2 of the set of minima energies weighted withtheir corresponding statistical factor gα . The variations of thisindex are shown in Fig. 6 as a function of cluster size.
The delocalization index does not capture the extent offloppiness to atomic motion, and in particular is unable toquantify the crossing of saddle states connecting permuta-tional isomers. Such a situation could take place in the small-est clusters that only support one energy minimum, for whichδE necessarily vanish. From Fig. 6, this indicator takes anonzero value at sizes n = 5–7, and inspection of the set ofcorresponding inherent structures reveals that the wavefunc-tions contain two or three minima only. In the three cases,these few isomers are present in proportions that are suf-ficiently mixed to have a signature on the energy fluctua-tions. In the 5-helium cluster, the fourfold symmetric globalminimum is only 1 cm−1 lower than another isomer ob-tained by capping the global minimum of Na+He4 within its
FIG. 6. Energy delocalization index δE(n) measuring the fluctuations of in-herent structure energies among the vibrational wavefunction obtained fromquantum Monte Carlo simulations with many-body and pairwise potentials.For the three sizes n = 6, 12, and 18, some of those metastable minima aredepicted.
mirror plane. In the 6-helium cluster, the C3 global minimumis nearly as frequent among the statistics of inherent structuresas a second, less symmetric (C2 point group) distorted octa-hedron which lies approximately 0.5 cm−1 higher in energyand can be seen as a capped Na+He5 (see inset of Fig. 6). Athird isomer lying 2 wavenumbers above the global minimumis found as well but contributes less than 1%. In Na+He7, asecond isomer with slightly different surface arrangement isfound 1.4 cm−1 above the global minimum. Such low energydifferences, added to that the pathways connecting these sim-ilar structures are likely to be very flat owing to only minordifferences in the relative orientations of the helium atomsaround the impurity, explain why the quantum Monte Carlosimulations sample these various isomers. In the much larger18-helium cluster, many different inherent structures popu-late the vibrational wavefunction, leading to a high value ofδE close to 20 cm−1, the vast majority of those structures cor-responding to icosahedra capped with 6 atoms. This clusteris clearly in a snowball state11 where nuclear delocalizationprimarily affects the outer parts of the helium cluster (whichbehave as a liquid) in equilibrium with a more localized(rigidlike) first shell.
More surprising is the observation that the energy delo-calization index is nonzero at 11 helium atoms, and especiallyat n = 12, that is also exactly at the icosahedral shell com-pletion that was expected to be more stable. A more detailedlook into the corresponding inherent structures shows a hand-ful of minima, all based on a doubly capped 10-helium shell,two of those minima being depicted in Fig. 6. This amount ofdelocalization is now quite significant with a similar patternas in the Na+He11 cluster whose wavefunction shows severalsingly capped 10-helium shell minima in addition to the in-complete icosahedron global minimum. This extent of delo-calization in the cluster at n = 12 explains the much lowerrelative stability in the energy difference of Fig. 4. This valueagrees with early conclusions from Galli and co-workers13
and is close to experimental values,21 but is at variancewith subsequent results from the same group14, 15 as well as
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10

174316-7 Issaoui et al. J. Chem. Phys. 141, 174316 (2014)
results of Marinetti and co-workers19 who included the ef-fects of bosonic exchange statistics in their quantum MonteCarlo simulations. These authors based their calculations onpair potentials, hence we have repeated ours in the same pair-wise approximation. The variations of the resulting energy de-localization index with increasing cluster size, superimposedin Fig. 6, show the same overall trend but significantly lowervalues, the onset of spreading over different energy isomerstaking place now at n = 14 with the appearance of multipledoubly capped icosahedra. Inspection of snapshots from thewavefunction of Na+He13 shows that the capping atom floatsfreely, having a liquidlike behavior although the same inher-ent structure is visited. Delocalization in the small Na+Henclusters with n = 5–7 is similar with the pair and many-body potentials, the few isomers remaining close to each otherwithin less than one or two wavenumbers. In larger clusters,the generally lower extent of delocalization is consistent withthe slightly stronger binding in the pair approximation, it con-firms the results of Marinetti et al.19 and additionally suggeststhat the discrepancies in the results with the many-body po-tential are not caused by the neglect of exchange statistics inour quantum Monte Carlo sampling but by many-body inter-actions whose effect is sufficient to alter the relative isomerenergies.
The present results suggest that nuclear quantum effectsin Na+Hen clusters depend sensitively on the global poten-tial energy surface, as the consequence of the many-bodyeffects becoming comparable to He–He bonding in largerclusters. These findings confirm the recent mass spectrome-try measurements,21 in which no special stability was foundfor Na+He12, while a lower snowball size of about 9 wasevaluated from those experiments. A few additional com-ments can also be made in comparison with helium coat-ing other alkali cationic impurities, for which computationalresults are available.13–19 Gianturco and co-workers showedhow the solvation pattern markedly depends on the impurity,shell completion being reached at n = 10, 12, and 15 forlithium, sodium, and potassium, respectively,16–19 and theygenerally found that snowballing takes place right after thiscompletion. However, these conclusions are again challengedby mass spectrometry21 in which the onset of snowballingwas found to be lower than predicted. Although we have notstudied ions other than sodium, our overall conclusions gen-erally confirm these earlier computational results, but shed anew light onto the possible combined importance of many-body forces between induced dipoles and nuclear quantumeffects, in a regime where most isomers are separated by afew wavenumbers only, and indicate that this small contribu-tion could be sufficient to shift snowballing to lower sizes.Geometric frustration arising due to the very different inter-actions between the atoms also contributes to delocalizing thewavefunction, because no optimal shell filling is reached andmany distorted isomers can interconvert more easily than in ahomogeneous system. Because bosonic exchange is expectedto further enhance delocalization, it would be interesting torepeat the present simulations with a more accurate quan-tum Monte Carlo framework accounting for indistinguish-able particles, and to extend the size range of interest fartherbeyond the first shell completion, to see how the snowball
effect in larger droplets affects this first shell and revisit theextent of icosahedral order suggested in a recent path-integralinvestigation.15 Because the binding energy does not simplydepend on the alkali impurity,19 it is also unclear how theseresults will convey to the Li+ or K+ ions, and revisiting theprevious conclusions reached in Refs. 15–19 by taking many-body effects into account seems necessary in the light of ex-perimental discrepancies.21 While the interplay between elec-trostatic forces and nuclear delocalization has been notablyinvestigated in the past,13–20 less efforts have been devotedto the next order effects of polarization with the exceptionof the lead dication24 for which additional contributions suchas spin-orbit coupling were reported. In this respect, it wouldbe interesting to consider lighter dicationic impurities such asMg2+ or Ba2+ and the impact of many-body forces on thesize and stability of the snowballs. In addition, the strongerbinding they would exert on helium atoms would be proneto easier detection of relative abundances in mass spectrome-try. Beyond atomic ions, ionic molecules or clusters could un-dergo much richer and size-dependent behavior, as suggestedby recent experiments and calculations on fullerenes.41
IV. SUMMARY AND CONCLUDING REMARKS
Among the rich variety of van der Waals compounds, he-lium clusters doped with ionic impurities stand as model sys-tems able to display a range of behaviors from well localized,classical-like to fully delocalized, fluid-like depending on thedetails of the interactions and on size itself. The present studywas theoretically devoted to the sodium cation solvated by upto 24 helium atoms, and notably aimed at revisiting the roleof electrostatic and polarization forces on the stable structuresand, more importantly, on the extent of vibrational delocaliza-tion and the possible occurrence of the so-called snowballingeffect at the onset of shell completion. Our modeling is basedon a newly developed potential that mainly comprises pairinteractions that were either taken from the literature (He–He) or fitted to reproduce spectroscopic data (Na+–He), butalso includes many-body polarization forces between induceddipoles, the contribution of the dipoles to the electric fieldsitself being accounted for by the usual self-consistent proce-dure. Although the many-body effects are small with respectto the direct polarization energy of the helium atoms by thecation, the corresponding interaction energies are comparablein magnitude to the weak helium-helium binding once the firstsolvation shell is formed, and this leads to a notable overes-timation of the total binding energy by the pairwise approxi-mation in larger clusters.
As far as topology is concerned, the most stable struc-tures found with the pair potential are essentially preservedupon inclusion of many-body forces, and exhibit three dis-tinct regimes where helium atoms cover the impurity first onthe side (n < 6), then more isotropically (n = 6–12) with theformation of a perfect icosahedral shell, and finally wherethey grow as additional layers in a still isotropic fashion. Theground state vibrational wavefunction explored with quan-tum Monte Carlo simulations reveals multiple inherent struc-tures at sizes 5–7 that lie within a couple of wavenum-bers from each other. This near-degeneracy is mainly caused
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10

174316-8 Issaoui et al. J. Chem. Phys. 141, 174316 (2014)
by the geometric frustration arising from the differentNa+–He and He–He bond lengths at equilibrium. As clus-ter size increases the uniform shell of helium atoms becomesmore crowded and accommodates better with the longer He–He bond, resulting in a more classical (localized) behavior.Only above 10 helium atoms do the solvation shell gets delo-calized again, multiple inherent structures with different shellarrangements and floating capping atoms being found. Thepairwise approximation seems to delay the onset of outer de-localization and snowballing above size 12, in agreement withearlier results.14, 15, 19 However, our results of a lower snowballonce many-body effects are included agree with recent massspectrometry experiments,21 indicating that the transition topartially delocalized structures is more sensitive to the detailsof the interactions than the global minima themselves.
The importance of nuclear delocalization could impactthe relative cluster stabilities, and our present findings of ashifted onset of snowballing contribute to elucidating the roleof many-body forces on the collective quantum behavior ofsuch doped helium clusters. Additional work could be de-voted to other alkali cations, but also to the neutral counter-part NaHen of the present systems, which in absence of polar-ization forces should exhibit more pronounced delocalizationeffects. Additional issues related to post-ionization processesare of interest as well,7 as the nuclear rearrangement follow-ing the loss of the valence sodium electron could lead to par-tial or even complete localization if appropriate numbers ofhelium atoms are thermally released. The dynamics of suchrearrangements, with the possible localization or delocaliza-tion of the nuclei upon evaporation, could be tackled usingtheoretical methods such as ring-polymer molecular dynam-ics. Anionic impurities would be of interest as well, as likelycandidates exhibiting strong polarization forces and for whichexperimental data are available.42 More generally, the inter-play between localization and cluster size could show furtherunexpected features in higher ionic charges, and we expecteven richer behaviors for dicationic impurities, starting withlarger snowballs. This is left for future efforts.
1R. Jayawardhana, Science 303, 322 (2004).2K. Lodders, Science 303, 323 (2004).3A. Burrows, W. B. Hubbard, J. I. Lunine, and J. Liebert, Rev. Mod. Phys.73, 719 (2001).
4N. F. Allard, F. Allard, P. Hauschildt, J. F. Kielkopf, and L. Machin, Astron.Astrophys. 411, L473 (2003).
5A. Burrows and M. Volobuyev, Astrophys. J. 583, 985 (2003).6W. H. Breckenridge, C. Jouvet, and B. Soep, in Advances in Metal Semicon-ductors and Clusters, edited by M. A. Duncan (JAI Press, Inc., Greenwich,CT, 1995), Vol. 3.
7F. Stienkemeier, J. Higgins, C. Callegari, S. I. Kanorsky, W. E. Ernst, andG. Scoles, Z. Phys. D 38, 253 (1996).
8D. Bellet and W. H. Breckenridge, Chem. Rev. 102, 1595 (2002).9J. P. Toennies and A. F. Vilesov, Angew. Chem. Int. 43, 2622 (2004); F.Stienkemeier and K. K. Lehmann, J. Phys. B 39, R127 (2006); S. Goyal,D. L. Schutt, and G. Scoles, Phys. Rev. Lett. 69, 933 (1992).
10M. Hartmann, R. E. Miller, J. P. Toennies, and A. Vilesov, Phys. Rev. Lett.75, 1566 (1995).
11K. R. Atkins, Phys. Rev. 116, 1339 (1959); N. Takahashi, T. Shigematsu,S. Shimizu, K. Horie, Y. Hirayama, H. Izumi, and T. Shimoda, Physica B284, 89 (2000).
12T. MacFarland, S. A. Vitiello, L. Reatto, G. V. Chester, and M. H. Kalos,Phys. Rev. B 50, 13577 (1994).
13D. E. Galli, M. Buzzacchi, and L. Reatto, J. Chem. Phys. 115, 10239(2001).
14M. Rossi, M. Verona, D. E. Galli, and L. Reatto, Phys. Rev. B 69, 212510(2004).
15D. E. Galli, D. M. Ceperley, and L. Reatto, J. Phys. Chem. A 115, 7300(2011).
16C. Di Paola, F. Sebastianelli, E. Bodo, I. Baccarelli, and F. A. Gianturco, J.Chem. Theory Comput. 1, 1045 (2005).
17E. Yurtsever, E. Yildirim, M. Yurtsever, E. Bodo, and F. A. Gianturco, Eur.Phys. J. D 43, 105 (2007).
18E. Bodo, E. Coccia, D. López-Durán, and F. A. Gianturco, Phys. Scr. 76,C104 (2007).
19F. Marinetti, E. Coccia, E. Bodo, F. A. Gianturco, E. Yurtsever, M.Yurtsever, and E. Yildirim, Theor. Chem. Acc. 118, 53 (2007).
20F. Sebastianelli, E. Bodo, I. Baccarelli, C. Di Paola, F. A. Gianturco, andM. Yurtsever, Comput. Mater. Sci. 35, 261 (2006).
21L. An der Lan, P. Bartl, C. Leidlmair, R. Jochum, S. Denifl, O. Echt, and P.Scheier, Chem. Eur. J. 18, 4411 (2012).
22A. M. Sapse, A. Dumitra, and D. C. Jain, J. Clust. Sci. 14, 21 (2003).23W. Gaied and M. B. Hadj Rhouma, Int. J. Quantum Chem. 111, 652 (2011).24P. Slavicek and Lewerenz, Phys. Chem. Chem. Phys. 12, 1152 (2010).25K. Abdessalem, H. Habli, H. Ghalla, S. J. Yaghmour, F. Calvo, and B.
Oujia, J. Chem. Phys. 141, 154308 (2014).26A. J. Stone, The Theory of Intermolecular Forces (Oxford University Press,
Oxford, 2002).27K. T. Tang and J. P. Toennies, J. Chem. Phys. 118, 4976 (2003).28P. Soldan, E. P. F. Lee, and T. G. Wright, Mol. Phys. 97, 139 (1999).29N. Geum and G.-H. Jeung, Chem. Phys. Lett. 333, 314 (2001).30E. A. Mason and H. W. Schamp, Ann. Phys. (N.Y) 4, 233 (1958).31D. J. Wales and J. P. K. Doye, J. Phys. Chem. A 101, 5111 (1997).32D. J. Wales, Energy Landscapes (Cambridge University Press, Cambridge,
2003).33F. Calvo, Comput. Mater. Sci. 45, 8 (2009).34F. Calvo, J. P. K. Doye, and D. J. Wales, J. Chem. Phys. 114, 7312 (2001).35R. N. Barnett and K. B. Whaley, Phys. Rev. A 47, 4082 (1993).36J. Casulleras and J. Boronat, Phys. Rev. B 52, 3654 (1995).37J. A. Northby, J. Chem. Phys. 87, 6166 (1987).38J. P. K. Doye, D. J. Wales, and R. S. Berry, J. Chem. Phys. 103, 4234
(1995).39F. Calvo, F. Y. Naumkin, and D. J. Wales, J. Chem. Phys. 135, 124308
(2011).40F. H. Stillinger and T. A. Weber, Phys. Rev. A 25, 978 (1982).41F. F. da Silva, P. Waldburger, S. Jaksch, A. Mauracher, S. Denifl, O. Echt,
T. D. Märk, and P. Scheier, Chem. Eur. J. 15, 7101 (2009).42C. Leidlmair, Y. Wang, P. Bartl, H. Schöbel, S. Denifl, M. Probst, M. Al-
camí, F. Martín, H. Zettergren, K. Hansen, O. Echt, and P. Scheier, Phys.Rev. Lett. 108, 076101 (2012); F. Calvo, Phys. Rev. B 85, 060502 (2012).
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
41.229.253.14 On: Fri, 07 Nov 2014 15:10:10









![Cholesterol Hydroperoxides Generate Singlet Molecular Oxygen [O 2 ( 1 Δ g )]: Near-IR Emission, 18 O-Labeled Hydroperoxides, and Mass Spectrometry](https://static.fdokumen.com/doc/165x107/63335dc0ce61be0ae50e9c78/cholesterol-hydroperoxides-generate-singlet-molecular-oxygen-o-2-1-d-g-near-ir.jpg)










