
The deactivation of singlet excited all-trans-1,6-diphenylhexa-1,3,5-triene by intermolecular charge...
18
ELSEVIER Chemical Physics 206 (1996) 193-210 Chemical Physics • e The deactivation of slngl t excited aU-trans- 1,6-diphenylhexa- 1,3,5-triene by intermolecular charge transfer processes. 1. Mechanisms of fluorescence quenching and of triplet and cation formation E Schael l, H.-G. Ltihmannsr6ben * lnstitut fiir Physikalische und Theoretische Chemie. TU Braunschweigo Hans-Sommer-Strasse 10, D-38106 Braunschweig, Germany Received 14 September 1995 Abstract The photophysical and fluorescence quenching properties of all-trans- 1,6-diphenylhexa- 1,3,5-triene (DPH) were inves- tigated in toluene and acetonitrile solution and the influences of the external heavy-atom effect (HAE) and of charge transfer (CT) processes on the fluorescence quenching mechanism were characterized. Particular emphasis was placed upon the elucidation of the electron transfer reactions in the quenching of DPH fluorescence by p-dicyanobenzene (p-DCB) in acetonitrile. It was shown that the combination of stationary and time-resolved fluorescence measurements, laser flash pho- tolysis and CIDNP experiments allows a comprehensive description of the primary and secondary electron transfer reactions in the fluorescence quenching process. In particular, the characterization of the kinetics of genuine charge recombination (CR) reactions of the DPH/p-DCB singlet and triplet ion pairs, of the spin evolution process and of the degenerate electron transfer between neutral and cationic DPH was achieved. 1. Introduction Photoinduced electron transfer is one of the sim- plest chemical reactions of electronically excited molecules and one of the central processes in photobi- ology. In many photobiological processes the family of polyene compounds plays an important role. Be- cause of their charge transfer properties and their ex- tended or-electron systems carotenoid polyenes have long been discussed as 'electron mediators' in pho- * Corresponding author. Present address: Institut fiir Lasertechnik, FH Ostfriesland, Constantiaplatz 4, D-26723 Emden, Germany. I Present address: Department of Chemistry, Technion - Israel Institute of Technology, Haifa 32000, Israel. tosynthesis [ 1]. Some experimental evidence for the capability of polyene molecules to transport electrons through membranes has been reported [ 2,3]. Another important process in photobiology involving polyenes is the visual process based upon the trans-cis photoi- somerization of retinal as key reaction. Furthermore, carotenoids are known to be important pigments in photosynthesis providing a protective function by quenching triplet chlorophyll and acting as accessory light harvesting molecules [4]. The lowest excited singlet state (SI) in//-carotene was shown to be a tAg state [5] that was considered to be responsible for the efficient singlet energy transfer observed between fl-carotene and chlorophyll in green ptants [6,7]. In the series of the a,o~-diphenylpolyenes, all-trans- 0301-0104/96/$15.00 (~) 1996 Elsevier Science B.V. All fights reserved Pil S0301-0104(96)00012-2
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of The deactivation of singlet excited all-trans-1,6-diphenylhexa-1,3,5-triene by intermolecular charge...

E L S E V I E R Chemical Physics 206 (1996) 193-210
Chemical Physics
• e The deactivation of slngl t excited aU-trans- 1,6-diphenylhexa- 1,3,5-triene by intermolecular charge
transfer processes. 1. Mechanisms of fluorescence quenching and of triplet and cation formation
E Schael l, H.-G. Ltihmannsr6ben * lnstitut fiir Physikalische und Theoretische Chemie. TU Braunschweigo Hans-Sommer-Strasse 10, D-38106 Braunschweig, Germany
Received 14 September 1995
Abstract
The photophysical and fluorescence quenching properties of all-trans- 1,6-diphenylhexa- 1,3,5-triene (DPH) were inves- tigated in toluene and acetonitrile solution and the influences of the external heavy-atom effect (HAE) and of charge transfer (CT) processes on the fluorescence quenching mechanism were characterized. Particular emphasis was placed upon the elucidation of the electron transfer reactions in the quenching of DPH fluorescence by p-dicyanobenzene (p-DCB) in acetonitrile. It was shown that the combination of stationary and time-resolved fluorescence measurements, laser flash pho- tolysis and CIDNP experiments allows a comprehensive description of the primary and secondary electron transfer reactions in the fluorescence quenching process. In particular, the characterization of the kinetics of genuine charge recombination (CR) reactions of the DPH/p-DCB singlet and triplet ion pairs, of the spin evolution process and of the degenerate electron transfer between neutral and cationic DPH was achieved.
1. Introduction
Photoinduced electron transfer is one of the sim- plest chemical reactions of electronically excited molecules and one of the central processes in photobi- ology. In many photobiological processes the family of polyene compounds plays an important role. Be- cause of their charge transfer properties and their ex- tended or-electron systems carotenoid polyenes have long been discussed as 'electron mediators' in pho-
* Corresponding author. Present address: Institut fiir Lasertechnik, FH Ostfriesland, Constantiaplatz 4, D-26723 Emden, Germany.
I Present address: Department of Chemistry, Technion - Israel Institute of Technology, Haifa 32000, Israel.
tosynthesis [ 1 ]. Some experimental evidence for the capability of polyene molecules to transport electrons through membranes has been reported [ 2,3]. Another important process in photobiology involving polyenes is the visual process based upon the trans-cis photoi- somerization of retinal as key reaction. Furthermore, carotenoids are known to be important pigments in photosynthesis providing a protective function by quenching triplet chlorophyll and acting as accessory light harvesting molecules [4]. The lowest excited singlet state (SI) in//-carotene was shown to be a tAg state [5] that was considered to be responsible for the efficient singlet energy transfer observed between fl-carotene and chlorophyll in green ptants [6,7].
In the series of the a,o~-diphenylpolyenes, all-trans-
0301-0104/96/$15.00 (~) 1996 Elsevier Science B.V. All fights reserved Pil S0301-0104(96)00012-2

194 E Schael. H.-G. LiJhmannsriiben/ Chemical Physics 206 (1996) 193-210
1,6-diphenylhexa- 1,3,5-triene (DPH) is the smallest chromophore in which the Si state is clearly assigned to be of lAg symmetry, and it is likely that this holds tbr all linear trans-polyene chromophores containing at least three conjugated double bonds [8]. DPH, which in contrast to most other polyene molecules is highly fluorescent, is widely used as fluorescent probe [ 9,10], and has recently attracted attention because of strong SI state optical nonlinearity [ 11 ]. The rodlike DPH molecule was also employed in investigations of mblecular assemblies, e.g. in nanotube aggregates of cyclodextrins [12] and in a study ;~,,f ,-;ir~.~c,~! c.~- ergy transfer through Langmuir-Blodgett-Kuhn mul- tilayers [ 13 ].
Several reviews summarize the results of numerous studies concerning the interesting photophysical prop- erties of DPH [ 8,14,15 ]" The unusual solvent shift ef- fects on the fluorescence and absorption bands, and the strong solvent and temperature dependence of the flu- orescence quantum yield (¢~°) and fluorescence life- time (r °) are well documented [ 16-22] 2. For ex- ample, increasing solvent polarity drastically changes q~o from high values in nonpolar solvents (e.g. up to 0.86 ih quinoline) to low values in polar solvents (e.g. ~'own to 0.17 in acetonitrile) [22], and the experi- inentaily obtained radiative lifetimes (r0 = r ° / ¢ ~°) are significantly longer (ca. by a factor of 5-20, de- pending on solvent [8] ) than those calculated from the Strickler-Berg formula (the results quoted are for room temperature).
These and other effects are now generally inter- preted to be due to the strong coupling between the close-lying Si ( lAg) and S2 (tBu) states of DPH. With a simple model Andrews and Hudson were able to explain e.g. the experimentally observed solvent de- pendence of the DPH fluorescence rate constant kr: (= I/r0) on the solvent polarizability [23]. The en- ergy gap between the two states was estimated to be ca. 1000 cm -I in hydrocarbon solvents [ 24 ]. In accor- dance with this interpretation, the low-lying Si state was observed directly in high resolution absorption spectra at low temperature [ 8] and in two-photon ex- citation spectra [25 ], and a weak thermally activated emission on the blue edge of the DPH fluorescence spectrum was assigned to result from an $2-S0 ra-
2 The superscript '0' in ¢,o, ¢o etc. is used here to denote molec- ular parameters of DPH in the absence of quencher molecules.
diative transition [ 26-28]. Similar to the situation in DPH, in a previous investigation we found two close- lying, strongly coupled excited singlet states to be re- sponsible for some unusual photophysical properties of periflanthene and 1,16-benzoperiflanthene [ 29].
Other important properties of DPH under investi- gation are the capability of formation of ground state s-cis conformers [ 30] and the trans-cis photoisomer- ization from the Si and $2 state [31,32].
Bimolecular reactions of singlet excited DPH (IDPH*) have only rarely been studied; in partic- ular, no systematic investigations of photoinduced electron and energy transfer processes seem to be available. Two works have dealt with the possible influence of the external heavy-atom effect (HAE) on DPH fluorescence quenching. However, differ- ing results were reported: Using several heavy-atom containing salts and solvents, including ethyl iodide, Song et al. observed no detectable quenching of DPH fluorescence [33]. As opposed to that, Chattopad- hyay et al. found a considerable quenching effect of ethyl iodide (with bimolecular rate constants of flu- orescence quenching of k~ -- 1.9 x 10 s M -I s -I in methanol and 1.7 x 108 M -I s -! in cyclohexane) accompanied by increasing yields of DPH triplet formation [34]. The external heavy-atom perturba- tion was also used by Ramamurthy et ai. to observe the formation of triplet DPH (3DPH*) and other triplet a,~-diphenylpolyenes in Tl+-exchanged zeo- lites [ 35]. In recent studies of Hirose and coworkers, reactions of I DPH* with N,N-diethylaniline and p- dicyanobenzene in various solvents were examined by resonance CARS spectroscopy [ 36-38 ]. Their exper- iments showed the formation of DPH anion (2DPH-) and cation (2DPH+) radicals. Basu et al. described the formation of exciplexes between DPH and N,N- diethylaniline in n-hexane and acetone [ 39,40]. The spectroscopy and reactivity of 2DPH+ was recently elucidated by Wang and McGimpsey [ 41 ].
In solvents containing no heavy-atoms the intersys- tem crossing (ISC) quantum yields of DPH are very low (e.g. 4 ° = 0.015 in degassed methanol at 295 K [ 34] ). However, the presence of heavy-atom contain- ing quenchers was found not only to strongly enhance DPH triplet formation but also to lead to the occur- rence of internal conversion (IC) as additional deac- tivation pathway of I DPH* [34]. These observations

E Schael, H.-G. LbTzmannsriiben/Chemical Physics 206 (1996) 193-210 ! 95
motiva,~ed us to elucidate the mechanism of DPH fluo- rescence quenching by intermolecular charge transfer (CT) processes because it is well known that back electron transfer reactions from the primarily produced ion pairs to the molecules in the locally excited (LE) triplet state and in the ground state can formally be regarded as quencher-induced ISC and IC [42]. The deactivation of I DPH* in acetonitrile and toluene so- lution by organic electron acceptors was investigated with stationary and time-resolved fluorescence spec- troscopy, laser flash photolysis and t H-CIDNP spec- troscopy. Special emphasis was placed upon the iden- tification of the primary products in the fluorescence quenching process and on the deactivation of the CT intermediates. In a subsequent paper we will present results of our investigations of the deactivation pro- cesses in a DPH exciplex system [43].
measurements were performed as described before [44] with a nitrogen-filled nanosecond flash lamp (Edinburgh Instruments) as excitation source. The excitation wavelength was 358 nm.
A Bruker WM 250 NMR spectrometer was used for the IH-CIDNP measurements and a 308 nm excimer laser (Lambda Physik) was used as excitation source. Experimental details can be found elsewhere [451.
The polarographic haifwave reduction potentials Erl / 2 ed were measured in acetonitrile with 0.I M tetrabutylammonium hexafluorophosphate (Fluka) as supporting electrolyte and against the fer- rocene/ferrocenium redox couple as internal standard. The potentials were converted to the SCE scale by adding +0.395 V [46].
3. Results and discussion
2. Experimental details
The substances used were commercially available (Aldrich, Fluka). DPH (Fluka, >99%) was used as received. The other substances were purified by recrys- tallisation, sublimation or distillation, if necessary. The solvents toluene and acetonitrile (Aldrich) were spec- troscopic grade and were used without purification. All samples were prepared in I x 1 cm cuvettes except |br laser flash spectroscopic measurements in acetoni- trile that were carried out in a flow system to prevent excessive formation of DPH isomers. DPH concentra- tions employed were in the range (1-10) x 10 -6 M. In cuvettes, the samples were deoxygenated by bubbling purified nitrogen through the solutions and sealed off. If not stated otherwise, measurements were performed at room temperature.
Fluorescence measurements were carried out with a quantum-corrected Perkin Elmer MPF-44 fluo- rescence spectrometer and absorption spectra were recorded with a Shimadzu UV-240 spectrometer. The transient absorption spectroscopy was carried out with a laser flash apparatus described elsewhere [42]. For the laser flash spectroscopic experiments an excimer-pumped dye laser was used. Excitation wavelength was 376 nm with typical excitation ener- gies of ca. 0.2 mJ/0.25 cm 2. The fluorescence decay functions were determined by the method of time- correlated single-photon counting (TCSPC). The
3.1. Primary and secondary electron transfer reactions in fluorescence quenching processes
Before consideriJlg photophysical properties and CT processes of DFH we will give a more general de- scription of primary and secondary electron transfer reactions. Scheme ! presents a simplified kinetic de- scription for the fluorescence quenching of an excited energy donor ~D* due to heavy-atom perturbation and primary, geminate electron transfer reactions in- duced by a ground state quencher molecule I A in polar solvents. Scheme ! encompasses the following processes: diffusion (rate constant kd), dissociation of the encounter complexes ( I D* . . . t A) (k_d), ISC induced by external perturbation from heavy-atom substituted quencher molecules (k~s c), forward and backward charge separation (CS) reactions (kcs, k-cs) , dissociation of the solvent-separated ion pairs 1,3(D±... A~=) into free ions (ksep), and the charge recombination (CR) reactions producing ground
G state (effective rate constant keR,e if) or triplet state
(k~R,eff) donor molecules. In comparison to the usual kinetic schemes of electron transfer reactions in solu- tion [47], Scheme 1 has been extended by the ISC process originating from the locally excited (LE) encounter complexes. This allows to distinguish the influence of the external HAE on LE complexes from the influence of the external HAE on CT intermedi- ates [481.

196 E Schael. H.-G. L6hmannsriiben/ Chemical Physics 206 (1996) i 93-210
ID* + t A kd
k-d
kcs ksep ( I D ' . . . ~ A ) ~ ~,a(D+...A:F) .._, 2D±+ 2Aa:
k - c s
a D ' + IA aD+ 1A
Scheme 1
I(D + . . Aa:) Z(D:~... A~:) I
2D:I: "1- 2A:F
( l - s ) k r J ~ k s e p ksep~~krec
I ( D + . . A:~) ~ Z(D±... A ~:)
ID + 1A 3 D * - I- 1A
Scheme 2
At this point a more detailed consideration of (i) the nature of CT intermediates, (ii) of triplet formation therefrom and (iii) of diffusion rate constants is use- ful: (i) While in polar solvents the structure of the CT intermediates is best described by a solvent separated ion pair (SSIP), in less polar solvents the structure is better approximated by a contact ion pair (CIP), i (D +A ~ ), or an exciplex [ I ( D +A ~ ) ~ I (DA) * ]. In less polar solvents separation into free ions is not observed. (ii) The SSIP has been designated with sin- glet and triplet multiplicity [ 1.3(D+... A ~:) ] since hyperfine-coupling (hfc)-induced spin evolution can lead from the initially formed singlet to the triplet ion pair 3. Scheme 2 illustrates the spin evolution process. Since the overall CR processes depicted in Scheme 1 are a combination of the spin evolution and the gen- uine CR reactions (Scheme 2), the experimentally ob- tained effective CR rate cons tan t s kcGR,eff and kTR,eff are determined by the kinetics of these processes. The spin evolution shown in Scheme 2 is responsible for chemi- cally induced nuclear polarization (CIDNP) phenom- ena in magnetic fields (see below). Formation of 3D* can thus occur, under the influence of heavy-atom per- turbation, with a spin-forbidden CR reaction from the singlet ion pair (not shown in Scheme 2), or, after hfc- induced spin 'evolution, with a spin-allowed CR reac- tion from the triplet ion pair (rate constant k~R). Due
3The terms 'spin', 'multiplicity' etc. refer to electron spin, whereas nuclear spin is denoted explicitly.
kcGrt
1D + IA
Scheme 3
¥
3D* + 1A
to the tight structure of CIP or exciplexes the hyper- fine interaction probably does not provide an effective mechanism for 3D* formation in less polar solvents. (iii) If transient effects are neglected (of. Section 3.3 ) approximate rate constants of diffusional motion can be obtained within the classical treatment advanced by v. Smoluchowski, Debye and Eigen for two un- charged species (Eqs. (1) and (2)) and two oppo- sitely charged ions (Eqs. (3) and (4)) [ 49-51 ]:
kd = 4~N°DDArDA , (I)
471" kd / k - d = -~" r3DA N ' , (2)
3 DDA rc ( 3 ) ksep = r a A [ e x p ( r c / r D A ) _ 1] '
47rNt DDArc
krec = 1 - e x p ( - - r c / r D A ) " (4)
Here, N' is Avogadro's number per mmol and krec is the rate constant for diffusion-controlled encountering (cf. Scheme 3). With a reaction distance of roA = 7
A, [47], a diffusion coefficient of DDA = DO + DA =
4.2 X 10 -5 cm 2 s -I (see below) and a Coulomb ra- dius of rc - e 2 / ( 4 z r ¢ o e k T ) - 15 A (parameters given for acetonitrile at room temperature) one obtains from

E Schael, H.-G. LiJhmannsriJben/Chemical Physics 206 (1996) 193-210 197
Eqs. ( I ) - ( 4 ) : kd = 2.2 x 10 t° M-Is -! , kd/k-d = 0.86 M -I , ksep = 7 x 109 s -I and krec = 5.4 x 10 I° M-I s- I . While alternative approaches, e.g. based on thermodynamics or random walk theory, indicate that Eq. (2) may underestimate k-d [ 54 ], we feel that the values obtained for kd and k-d are in general agree- ment with expectations based on currently available experimental results (cf. e.g. Refs. [ 52,53 ] ). Prob- lems arise with the diffusion rate constants of the charged species: Mataga et al. have measured directly the formation of free ions from the separation of ! 6 or- ganic donor/acceptor systems in acetonitrile. The re- ported ksep values are in the range of (0.5-2.5) x 109 s -I with an average of ca. ! x 109 s -i [55]. Similar results were reported by Peters et al. [56]. With re- alistic reaction distances the experimental values for ksep cannot be reproduced from Eq. (3), so that a consistent treatment of the diffusion processes seems not possible with Eqs. ( 1 ) - (4) . Instead of pursuing more elaborate theoretical treatments (cf. e.g. Refs. [57,58 ], and references therein), we have based the tbllowing evaluations of our experimental data on the average value of ksep = 1 x 109 s -I from the exper- imental work of Mataga et al. To our knowledge, a direct experimental determination of krec has not been performed for organic ions comparable to those con- sidered here. In analogy to the problem with ksep it cannot be excluded that the value for krec calculated with Eq. (4) is also too large. Lacking a more reli- able estimate, we have therefore regarded the value obtained from Eq. (4) as upper limit for the recombi- nation rate constant in acetonitrile: krec <~ 5.4 x 10 l° M-I s-l .
In addition to the processes encompassed in Scheme !, at least two subsequent reactions of the free ions have to be considered (secondary electron trans- fer reactions): (i) The homogeneous CR reactions, that provide a third, bimolecular pathway of 3D* for- mation and can be singled out by time-resolved triplet absorption measurements (Section 3.5). In Scheme 3, the homogeneous CR reactions are introduced as the ion recombination reactions (with rate constant krec and spin statistical factor s) followed by the spin evo- lution and genuine CR reactions (Scheme 2). From spin-statistical considerations one expects the forma- tion of triplet ion pairs with a probability of s - 0.75 and that of singlet ion pairs with ! - s = 0.25 in the iaomogeneous ion recombination. On the other hand,
the primary CS reaction in the fluorescence quench- ing process (Scheme I) leads initially to exclusive formation of singlet ion pairs. Thus a comparison of the kinetics and e.g. triplet formation yields of the primary and secondary electron transfer reactions should give information about the spin evolution and the genuine CR processes. (ii) Another secondary reaction of the free ions that has to be considered is the degenerate electron transfer reaction (DELT) between ion and parent molecules, e.g.
2D+ + ID k~,-~-d 2 (D+ . . .D) ~ I D + 2D+ k-d
Scheme 4
with kEx being the unimolecular rate constant for the electron exchange step, which is assumed to be ir- reversible for simplicity. The electron self-exchange reaction (Scheme 4), which produces no chemical change but shortens the lifetime of individual ions, was investigated by time-resolved CIDNP experiments (Section 3.6). The species with non-equilibrium pop- ulation in their nuclear spin states detected by CIDNP spectroscopy are denoted in bold face in Scheme 4.
In the following we use kinetic schemes analogous to Scheme 4 for the simplified kinetic description of (i) the influence of the HAE on the LE complex (Scheme 1, neglecting kcs and k-cs) , (ii) the ho- mogeneous CR reaction (Scheme 3, neglecting spin evolution and setting s = 1 ) and (iii) the DELT reaction (Scheme 4). Under steady-state conditions, the unimolecular rate constants of deactivation of the appropriate excited complexes (i.e. (i) deactivation rate constant k~s c of singlet LE complex; (ii) k~R, triplet SSIP; (iii) kEx, doublet encounter complex) can be then evaluated from experimentally accessible bimolecular rate constants (kobs) if the rate constants of diffusional motion (kd, k-d, krec, ksep) are known.
3.2. Photophysical properties of DPH
Photophysical properties of DPH in numerous sol- vents have been reported in the literature (cf. e.g. Refs. [ 17,22,34 ] ) and hence only a brief overview of some data relevant for the subsequent considerations will be given here. In Fig. 1 the absorption and fluorescence spectra of DPH in acetonitrile and toluene are shown. Clearly discernible are the different influences of the solvents on the absorption and the fluorescence bands

198 E Schael, H.-G. Liihmannsr6ben/Chemical Physics 206 (1996) 193-210
, f
t.o Acetonitrile 8
6
0.5 4 '? , , m s i 2"°
•
1.0 A, ~ 6 " '
05 4 @
2
16000 22000 28000 ' 34000 ~ l c m -I
Fig. I. Absorption and fluorescence spectra of DPH in acetonitrile (upper part) and toluene (lower part) at room temperature. The fluorescence spectra were scaled to unity at the absolute maxima and are given in relative quanta per unit time per unit spectral energy ( I~ ).
and the shoulder on the blue edge of the fluorescence spectra at ca. 26000 cm -I (ca. 385 nm). This is the emission that is attributed in the literature to the S2- So fluorescence [26-28]. It is important to note that since the absorption spectra in Fig. 1 correspond to the S0-$2 transitions the S~ state energies cannot sim- ply be determined from the points of intersection of the absorption and fluorescence spectra in Fig. 1. In the following we will therefore (for both acetonitrile and toluene) take the SI state energy obtained from two-photon excitation spectroscopy (in EPA at 77 K): E(SI) --3.11 eV (25050cm - I ) [25].
Some photophysical properties of DPH determined in this work together with literature values for the quantum yields of trans-cis isomerization are summa- rized in Table 1. The values of 1 "° in acetonitrile and @o in toluene are in accordance with earlier results [ 17,22], while our results for @o in acetonitrile and r ° in toluene slightly differ from previous publications in which @o = 0.15 [ 17] and 0.17 [ 22] in acetoni- trile, r ° = 11.2 ns [22] and 6.1 ns [61] in toluene were reported. The data in Table 1 can be taken to il- lustrate some peculiar aspects of the DPH photophys- ical behaviour which have already been described in more detail in the literature: In acetonitrile @o is about 3 times lower than in toluene because the polar sol- vent is thought to induce non-radiative transitions via intermediate twisted molecular structures [22]. This effect is also reflected by a ca. twofold increase of
tile isomerization quantum yields by changing the sol- vent from methylcyclohexane to acetonitrile [32], It is noted that the trans-cis photoisomerization of DPH occurs via a singlet mechanism, i.e. from I DPH* only and not from 3DPH* [ 31 ]. The sum of DPH fluores- cence, intersystem crossing and trans-cis isomeriza- tion quantum yields in acetonitrile is nmch less than
0 unity (@° + ¢,o r + ¢'trans-ei~ = 0.39 4- 0.03) so that it is obvious, that the deactivation of I DPH* is dominated by the non-radiative transition back to the ground state So.
The measured fluorescence decay functions were strictly monoexponential which indicates that the equi- libration of the SI and $2 state populations proceeds too fast to be observable on a nanosecond time scale [ 59,60]. The fluorescence rate constants kF, as calcu- lated from the experimental values of @o and ~.o, show the well-documented strong solvent dependence. This is caused by the different solvent shift effects on the S~ and $2 states of DPH, so that the energy separation and therefore the coupling between these states becomes a function of solvent polarizability [23,61 ]. Evidently, the significant solvent dependence of the DPH fluo- rescence behaviour makes it very important that in flu- orescence quenching experiments the quencher con- centrations are kept low enough to avoid changes in macroscopic sol vent properties.
3.3. Quenching of DPH fluorescence in acetonitrile and toluene
First, we will concentrate on the primary pho- toinduced electron transfer processes according to Scheme 1, and ISC from the LE complexes and the secondary reactions will be neglected. Under steady- state conditions the experimental rate constant of fluorescence quenching (kq F) is then given by
F kq = kd
1 + (k_,dks)ea(; , ' s /er + (k_d/kOs)e"C:~s/Rr ' (5)
G T with ks = ksep +kcR.eff+kca,eff and k°s = kcsea(;,*'s/st.
The free energy change of activation AG~cs can be evaluated from the standard free energy change for the charge separation reaction (AGcs) by the empirical free energy relationship of Rehm and Weller [ 47]"

E Schael. H.-G. Liihmannsriiben/Chemical Physics 206 (1996) 193-210
"ihble I
Photophysical properties of DPH in toluene and acetonitrile at room temperature
199
Toluene Acetonitrile
es (M - I c m - ' ) (Amax ( r im) ) a 711004-1600 (358) 825004-4500 (352) eT (M - I cm - I ) (Amax (nm)) a 102000 (425) 119000 (4!7) eg (M - I cm - ! ) (Amax (nm)) a - 143500 (590) Oo b 0.8 ! 4-0.11 0.264-0.02 tib~TC 0.044-0.01 0.0 ! 4-0.005 ¢ib(tlrans.--".c,s d 0.0594-0.005 e 0. ! 244.0.005 ~'~ (ns) f 7.44-0.1 4.14.0.1 r ~ ( # s ) g 90 4 0
kF ( s - I ) h (I.1 4-0.2) × 108 (64- I) x 107
" Extinction coefficient of singlet ground state (~s), triplet (eT) and cationic (~g) DPH at the wavelengths given in parentheses; estimated experimental uncertainties of ~T and ~g are ca. 4-10%. b Quantum yield of fluorescence. c Quantum yield of intersystem crossing. d Quantum yield for trans-cis photoisomerization from Ref. 1321. e In methylcyclohexane. f Fluorescence lifetime. g Triplet lifetime; lower limits because of possible interference of impurity quenching. h Rate constant of fluorescence as obtained from ¢ ,o /~ .
(6)
In our experiments photoexcited DPH was both the energy and electron donor (IDPH* = I D*), and all quencher molecules employed were the electron acceptors (A). If Coulombic terms are neglected,
I/2 + E~/2(A-/A) E(Si) is A G c s = Eox ( D / D ) - readily available from the DPH halfwave oxidation
I/2 + potential (Eox ( D / D ) = 1.02 V versus SCE [62]), the halfwave reduction potential of the quencher
molecules E~/f ( A - / A ) , and the DPH S! state energy
E(Si). AG~cs(0) =0.25/I = 0.25(Ai + Ao) is the free energy change of activation for AGcs= 0 ('intrinsic barrier') and is composed of the internal (Ai) and solvent (A~) reorganization energies.
The experimental rate constants of fluorescence F in case of stationary measurements were quenching kq
obtained from the modified Stern-Volmer equation derived by Weller [ 63 ]:
o o / o F = 1 + kF¢°[A] , (7)
e x p ( -- Vo v / OF l O° [ A ] )
V D -- ( 8 ) 4 ~ N t ( D D A ) 3/2 "
VD denotes a molar volume and DDA = DD + DA
the sum of the diffusion coefficients of donor (DD) and acceptor molecules (DA). We have used for all DPH/acceptor-pairs average diffusion coefficients of DDA =4 .2 x l0 -5 cm 2 s -I and DDA = 2.6X l0 -5 c m 2
s-! in acetonitrile and toluene, respectively (293 K) 4. The fluorescence decay functions of DPH in the pres- ence of quencher molecules in toluene were mono-
4 Diffusion coefficients were estimated from the empirical rela- tionship of Othmer and Thakar (OT) 164l for water, corrected by addition of 15% 147], and were then converted to acetonitrile and toluene with the viscosity relation. The applied values for the viscosities (7 ) are 7 /= !.002 cP for water 165], 7/= 0.359 cP for acetonitrile and 'r/= 0.586 cP for toluene (from data in Ref. [66l) . In this way, for DPH DD = !.5 x 10 -5 cm 2 s - I , and for the different acceptor molecules in Table I diffusion coefficients in the range of DA = ( ! .8-3 .5) x l0 -5 cm 2 s - ! were e.g. obtained for acetonitrile at 293 K. It was checked that the effect the 'indi- vidual' DA values on the k~ values obtained with Eqs. (7) and (8) is so minor (maximum variations ~< 5%) that the use of the OT-procedure for the individual acceptor molecules is not justified. In [67] for DPH in cyclohexane DD = ! .2 x l0 -5 cm 2 s - I at an unspecified temperature has been reported. If this is a room tem- perature value it would correspond to e.g. DD = 3.5 x 10 -5 cm 2 s - ! in acetonitrile. Recently reported experimental values of dif- fusion coefficients of aromatic compounds 1681 are also up to a factor of two larger than the values obtained with the OT proce- dure. The use of doubled DDA values leads to maximum increase
of k~ of ca. 10%.

200 E Schaei, H.-G. LiJhmannsriiben/ Chemical Physics 206 (! 996) 193-210
exponential (for two exceptions see below). It was therefore concluded that transient effects due to diffu- sional processes, which are expected to influence the fluorescence decay on sub-nanosecond 'amescale after excitation [ 69], were not resolved with our apparatus. rF denotes the fluorescence lifetime observed during the period after the transient diffusional effects.
The rate constants kq F from the time-resolved mea- surements are obtained from the linear Stern-Volmer equation:
__. . . F O r0 I + kqT"F[A ] , ( 9 ) 7" F
The bimolecular rate constants of the quenching of DPH fluorescence by the investigated acceptor molecules obtained from stationary and time-resolved measurements in acetonitrile and toluene are sum- marized in Table 2. Before the discussion of the re- sults the following experimentally-relevant points are noted: (i) It was carefully checked that addition of the quencher molecules did not significantly change the properties of the solvents. Measurements of the refractive indices (n~0) showed that even at maxi- mum quencher concentrations changes were usually below AnD = 0002, so that a variation of kF of DPH as a consequence of changing polarizabilities can be ruled out. (ii) Within the ranges of quencher concen- trations employed the Stern-Volmer plots showed no curvature. (iii) Upon irradiation in acetonitrile, DPH undergoes efficient trans-cis isomerization and the excitation energies had therefore to be kept very low. (iv) In absence and presence of quencher molecules, the DPH fluorescence decay was monoexponential in toluene, and the quenching rate constants obtained from time-resolved measurements were generally in accordance with the results from stationary measure- ments (cf. Table 2 and Fig. 2) revealing that no significant contributions from static quenching were detectable. However, it is notable that for the quencher molecules ethyl bromide (No. 10), ethyl benzoate (No. I 1 ) and 2-bromonaphthalene (No. 12) the kq ~ values obtained t?om stationary measurements seem to be systematically larger than those from time- resolved measurements. This may be due to otherwise undetected ground state complexation leading to static quenching of DPH fluorescence. (v) In the presence of the quencher molecules p-dicyanobenzene (No. 6) (in toluene) and l-cyanonaphthalene (No. 8) (in
" " I . . . . I . . . . I . . . . I . . . .
0 • o o - ~ . ~ o
% : \ ~-|
-3
I I I ~ I ,~
",, -I.5 -1 -0.5 0 0.5
AGes I eV
Fig. 2. Logarithmic plot of the rate constants of DPH fluores- cence quenching normalized to diffusion rate constant (k~q/kd) as a function of standard free energy change for charge separation (&Gcs) in toluene (o, A) and acetonitrile ( . ) . (e, o) from sta- tionary fluorescence measurements, (A) from fluorescence life- time measurements; the curve was calculated according to Eq. (5) with the parameters given by Rehm and Weller [471.
both solvents) spectral changes in the stationary fluorescence spectra were observed, and the DPH fluorescence decay functions in toluene were not mo- noexponential. We have shown that in these cases the formation of exciplexes takes place [43]. (vi) In the presence of several other quencher molecules, the fluorescence decay of DPH in acetonitrile was also not strictly monoexponential, even though no changes of the stationary fluorescence spectra were detectable. This effect, which possibly stems from product formation due to DPH trans-cis isomerization or direct excitation of the quencher molecules, was not investigated in detail, and fluorescence lifetime measurements in acetonitrile were not pursued.
In Fig. 2 the quenching rate constants k~ measured in acetonitrile and toluene versus the standard free en- ergy change AGes obtained for acetonitrile are shown together with the Rehm-Weller curve calculated from Eq. (5) with the original parameters given in Ref. [47]. To account for the different viscosities, the kq F values are normalized to diffusion rate constants kd -
2.2x 10 l° M-Is -! in acetonitrile and kd = 1.4x 10 I° M-Is -I in toluene, respectively. From the results in Fig. 2, the following observations can be made: (i) In both solvents, the kehm-Weller treatment gives an adequate description of the dependence of k F on AGes, i.e. the efficiency of DPH fluorescence quench- ing is correlated with the reduction potentials of the

Table 2 Bimolecular rate constan,~s of DPH fluorescence quenching in acetonitrile and toluene (k~), experimental rate constants of triplet formation (kq a') and triplet formation
T F efficienies (rrr = kq/k,i ) in toluene at room temperature
I~ Quencher molecule Acetonitrile Toluene
El/2 a c ~: k~ ( M - r/t -,~:ti (V) AGcs ( e V ) " kq F (M - I s - I ) k F (m - I s - ' ) k F ( M - ' s - ' ) d ' s - i ) e
I hexachloroacetone -0 .27 1701 - ! . 8 2 ( !.3 -1- 0.2) x 10 I° f 2 maleic anhydride -0 .84 - ! . 2 4 ( i . 9 4 - 0 . 2 ) x I0 m ( I .74 -0 .2 ) x I0 m (2 .34-0.2) × I0 I° < 5 × 107 < 0.01 3 hexabromobenzene -0 .86 -1 .22 (8.7 4- 0.9) x IO 9 (2.8 4- 0.3) x IO 9 0.32 4 fumaronitdle - I . 2 5 -0 .83 ( l . 6 : l :O . I ) x IO m ( I . 4 + O . l ) x lO l° ( ! . 44 -0 .1 ) x lO I° ( I . 5 + 0 . 2 ) x IO 9 O.II 5 fumarodiethylester - I . 4 5 -0 .63 (8.04-0.8) x IO 9 ( 1 . 4 + 0 . 2 ) x IO s 0.02 6 p-dicyanobenzene - l . 6 1 -0 .47 ( ! . 34 -0 .1 ) x IO i° ( 9 . 7 + l.O') x IO 9 g (7.34- !.2) x lO 9 h (5 .44-0.4) x iO '~ 0.56
7 i.,aaleic dtethylester - i . 8 7 171l -0.21 (2.1 4- 0.2) x IO 9 (2.3 4-0.2) x lO 9 (2.44- 0.2) x IO 9 8 I-cyanonaphthalene - I . 9 4 -0 .15 ( I . 24 -0 .1 ) x IO l° g (6.5-1-0.6) x IO a g (8 .04-2.0) x lO ~ h 9 p-dibromobenzcne -2 .10 1721 -0 .02 (6 .54-0.7) x IO 7 (3.34-0.5) × IO 7 0.51
10 ethyl bromide -2 .13 1721 0.05 (6 4- 2) x 107 (2.3 4-0.6) x 107 (7 q - l ) × 106 (7--1-2) x 106 I I I ethyl benzoate -2 .14 1731 0.06 (I .7 4- 0.3) x 107 (4.0 4- 2.0) × 106 12 2-bromonaphthalene -2 .16 0.08 (I .3- t -0.2) x 108 (5.55:: 1.0) x 107 (7.04- !.0) x 107 I 13 acrylonitrile -2 .17 0.09 < 2 x 107 < 2 x 107 ( i . 44 -0 .3 ) x 107 14 benzonitrile -2 .35 1741 0.27 ( ! . 84 -0 .4 ) x 107 ( I .44 -0 .3 ) x 107
a Halfwave reduction potential in acetonitrile (versus SCE). h Standard free energy change for CS as calculated from the Weller equation without work term. c Obtained with stationary measurements from integrated fluorescence spectra. d Obtained from fluorescence lifetime measurements. e Bimoleeular rate constant of DPH triplet formation. i This value may be slightly affected by static fluorescence quenching which was evident by the decrease of DPH absorbance with increasing quencher concentration.
g These values were obtained from the relative intensities of the short-wavelength fluorescence band at ca. 405 nm. h Obtained from the fluorescence decay function with the usual kinetic scheme for exciplex formation 1431.
I t~

202 E Schael, H.-G. Liihmannsriiben/ Chemical Physics 206 (1996) 193-210
quencher molecules employed. This provides solid experimental evidence that the fluorescence quench- ing is dominated by electron transfer processes. (ii) No significant influence of solvent polarity is dis- cernible in the DPH fluorescence quenching in ace- tonitrile and toluene. Attempts to estimate the solva- tion and Coulombic energies of solvent-separated ion pairs in polar and exciplexes in nonpolar solvents lead to energy differences of ca. 0.4 eV [75]. Experimen- tally, for the fluorescence quenching of phenanthrene and phenanthrene derivatives by amines in acetonitrile and cyclohexane even a difference in standard free en- ergy changes of 0.7 eV has been reported [ 76]. From our results in Fig. 2 it can be estimated that the ener- gies of the donor (DPH)/acceptor (A) CT interme- diates differ by less than ca. 0.2 eV in acetonitrile and toluene. In accordance to this observation, Ghoneim et al. reported very similar dependencies of kq F on AGes for the electron transfer quenching reactions of 9,10- dicyanoanthracene by various organic donors in hex- ane, i-fluoropentane and acetonitrile solutions [ 77]. We have made a similar observation for the quenching of the fluorescence of 1,7-diazaperylene by organic molecules [46]. These results might give an indica- tion that the energy differences between CT interme- diates in polar and nonpolar solvents have been over- estimated in earlier works.
3.4. Influence of the extenlal HAE on the fluoresce,we quenching of DPH
In order to evaluate the influence of the exter- nal HAE, the quenching of DPH fluorescence by xenon was also investigated. In benzene saturated with xenon, a quenching rate constant of kq F ,~, 7 x 107 M-i s-i was obtained 5 . In accordance with earlier observations on aromatic fluorophores [80], it was established that the heavy atom perturbation led exclusively to an increase of 3DPH* formation (k~" ~-, 6 x 107 M-i s- i ; cf. next section). The prob-
ability of the spin-forbidden ISC process I DPH* ---, 3DPH* scales approximately with the square of the effective spin-orbit-coupling parameter (~2) of the heavy atom perturber, if effects due to different orbital
5 Benzene was selected as solvent because the xenon saturation concentration of 0.126 M at 298 K was available from the literature 178,791.
overlaps between I DPH* and the HAE-perturbers are neglected: k~c cx s ¢2. It can be assumed that in the quenching of DPH fluorescence by xenon charge sep- aration processes will play no role. From the simpli- fied Scheme I and under steady-state conditions, the relationship between the experimental rate constant (kobs =) k F and kl'~c is then obtained (cf. Section 3.1):
kTc----" k d - - - ~ ~ kq ~ k ~ _ d j . ( 1 0 )
It is thus obvious that below the diffusion-controlled limit the rate constant of fluorescence quenching by the external HAE is also approximately proportional to ~2.
If we use ~ = 7094, 5060, 2460 cm-i for Xe, I and Br [52], the ratios of quenching rate constants will ca. be 8 : 4 : I. Hence, from the Xe quenching rate constant as a very rough estimate values of kq F 4 x l 0 7 M - I s - l andkq F ~ 9 x 1 0 6 M -! s -I (in ben- zene) are obtained for iodine and bromine substituted quencher molecules as contributions to the quench- ing of DPH fluorescence that are solely due to the influence of the external HAE on the LE complexes ('heavy-atom quenching'). Comparison with our re- sults in Table 2 indicates that such a 'pure heavy- atom quenching' of the DPH fluorescence was proba- bly observed only in the time-resolved measurements in toluene with ethyl bromide (no 10) as quencher molecule for which k~c ~, 8 x 106 s -Z is obtained. All the other kq F values indicate the predominance of CT processes in the quenching mechanism ( 'CT - quench- ing' ), as seems e.g. also to be the case in the DPH flu- orescence quenching by ethyl iodide investigated by Chattopadhyay et al. [ 34] (see above).
It is important to recognize this superposition of fluorescence quenching mechanisms and the interplay between heavy-atom substitution-induced changes in spin orbit coupling and redox properties. Heavy-atom substitution of a quencher molecule not only increases s ~, which leads to enhanced 'heavy-atom quenching',
~-.I/2 which leads to en- but usually also increases "-'red, hanced 'CT - quenching' in the case of reductive flu- orescence quenching. Separation of the two effects is thus difficult. This situation is even more compli- cated here, since the fluorescence properties of DPH depend on solvent polarizability, which is certainly
E Schael, H.-G. 1-~hmannsriiben/Chemical Physics 206 (1996) 193-210 203
1 5 . . . . ! . . . . I . . . . i . . . . i . . . . I . . . .
, .! 10
: j 350 400 450 - 500 550 600 . . . . 650
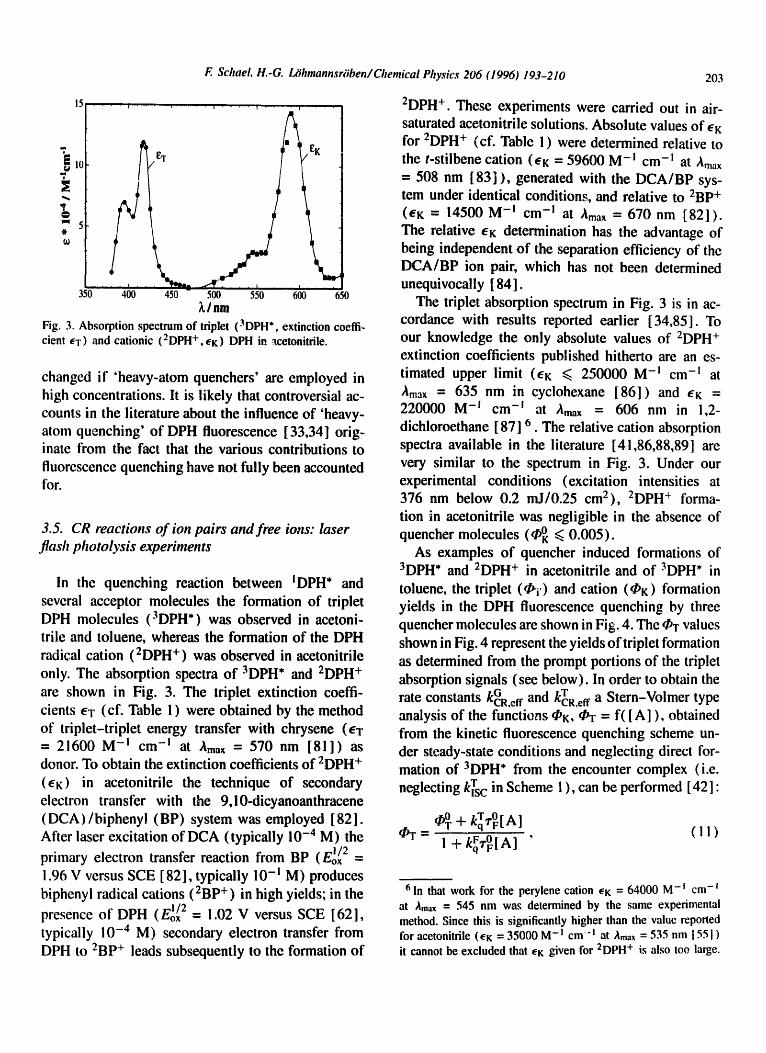
~,/nm Fig. 3. Absorption spectrum of triplet (3DPH*, extinction coeffi- cient eT) and cationic (2DPH+,eK) DPH in acetonitrile.
changed if 'heavy-atom quenchers' are employed in high concentrations. It is likely that controversial ac- counts in the literature about the influence of 'heavy- atom quenching' of DPH fluorescence [ 33,34] orig- inate from the fact that the various contributions to fluorescence quenching have not fully been accounted for.
3.5. CR reactions of ion pairs and free ions: laser flash photolysis experiments
In the quenching reaction between I DPH* and several acceptor molecules the formation of triplet DPH molecules (3DPH*) was observed in acetoni- trile and toluene, whereas the formation of the DPH radical cation (2DPH+) was observed in acetonitrile only. The absorption spectra of 3DPH* and 2DPH+ are shown in Fig. 3. The triplet extinction coeffi- cients eT (cf. Table 1 ) were obtained by the method of triplet-triplet energy transfer with chrysene (eT = 21600 M - i cm - i at Amax = 570 nm [81]) as donor. To obtain the extinction coefficients of 2DPH+ (6K) in acetonitrile the technique of secondary electron transfer with the 9,10-dicyanoanthracene (DCA)/biphenyl (BP) system was employed [ 821. After laser excitation of DCA (typically 10 -4 M) the
primary electron transfer reaction from BP (F_,o !/2 = !.96 V versus SCE [82], typically 10 - t M) produces biphenyi radical cations (2Bp+) in high yields; in the
~!/2 presence of DPH (,-,ox = 1.02 V versus SCE [62], typically 10 -4 M) secondary electron transfer from DPH to 2Bp+ leads subsequently to the formation of
2DPH+. These experiments were carried out in air- saturated acetonitrile solutions. Absolute values of eK for 2DPH+ (cf. Table I ) were determined relative to the t-stilbene cation (eK = 59600 M -I cm -I at Amax = 508 nm [83] ), generated with the DCA/BP sys- tem under identical conditions, and relative to 2Bp+ (eK = 14500 M -I cm -I at Amax = 670 nm [82]). The relative eK determination has the advantage of being independent of the separation efficiency of the DCA/BP ion pair, which has not been determined unequivocally [ 84].
The triplet absorption spectrum in Fig. 3 is in ac- cordance with results reported earlier [34,85]. To our knowledge the only absolute values of 2DPH+ extinction coefficients published hitherto are an es- timated upper limit (¢K <~ 250000 M-I cm-! at ~max = 635 nm in cyclohexane [86] ) and eK = 220000 M -I cm -I at Amax -- 606 nm in 1,2- dichloroethane [ 87 ] 6. The relative cation absorption spectra available in the literature [41,86,88,89] are very similar to the spectrum in Fig. 3. Under our experimental conditions (excitation intensities at 376 nm below 0.2 mJ/0.25 cm2), 2DPH+ forma- tion in acetonitrile was negligible in the absence of quencher molecules (O ° ~< 0.005).
As examples of quencher induced formations of 3DPH* and 2DPH+ in acetonitrile and of 3DPH* in toluene, the triplet ( ~ r ) and cation (¢'K) formation yields in the DPH fluorescence quenching by three quencher molecules are shown in Fig. 4. The qST values shown in Fig. 4 represent the yields of triplet formation as determined from the prompt portions of the triplet absorption signals (see below). In order to obtain the
G T rate constants keR,eff and kCR,e ff a Stern-Voimer type analysis of the functions ¢'K, OT = f( [ A ] ), obtained from the kinetic fluorescence quenching scheme un- der steady-state conditions and neglecting direct for- mation of 3DPH* from the encounter complex (i.e. neglecting k~s c in Scheme I ), can be performed [42]"
_- --I- kqT '°tA] , ( I I )
i +
6 in that work for the perylene cation eK = 64000 M-I cm- at 3max = 545 nm was determined by the same experimental method. Since this is significantly higher than the value reported for acetonitrile (eK = 35000 M -I cm ' i at ,~max = 535 nm 1551 ) it cannot be excluded that eK given for 2DPH+ is also too large.

204 E Schael. H.-G, Liihmam~sriiben/Chemical Physics 206 (1996) 193-210
0 . 6 , ! i i i
0.4 IIT= 0.56 (- 0.47 eY)
0 . . . . . . . 0 [ p - D ~
0.2
~T =0.11 (- 0.80 eV) /
0~ ~ _ ..- ~ ..- tiT < 0.01. (- 1.20 eV)
I ~ ~ ~ I ~ I
0 0.005 0. 1 0.015 0.02 [A]/M
Fig. 4. Dependence of triplet DPH formation effieiencies (~T) on the acceptor concentration in in toluene (p-dicyanobenzene ( s ) , fumaronitrile ( A ) , and maleie anhydride (1"1); the values of (AGcs) in acetonitrile are given in parantheses). Inset: Depen- dence of ~T and of the DPH cation formation efficiency (4~K) on the p-DCB concentration in acetonitrile. The curves were cal- culated with F_,qs. ( ! 1 ) and (12), respectively.
kq °tAJ ¢'K = ( 1 2 )
1 + F o kqrF[A]
T F K F = kCR,eff/ks, r/K • (with r/T kq/kq T = = k q / k q = ksep/ks ,
G ks = ksep q- kCR,e ff q- kTR,eff). 'r/T a n d r/K d e n o t e the
fractions of quenching events that result in triplet and cation |brmation.
Under laser irradiation DPH isomerization in ace- tonitrile is efficient; we have therefore concentrated the laser flash photolysis !nvestigations in that solvent on the pair DPH/p-dicyanobenzene (p-DCB) for which r/r = 0.06-t-0.01 and r/r = 0.354-0.05 in acetonitrile 7 and Or - 0.54 4- 0.07 in toluene was obtained. This means that in acetonitrile the CR reaction from the DPH/p-DCB ion pair back to the ground states with a standard free energy change of AG~R = -2 .63 eV proceeds with ca. 60% efficiency. With ksep = 1 x 109 s -I (cf. Section 3.1 ) the effective rate constants of charge recombination in acetonitrile were then avail-
G T able: kCR,eff = (1.7 4- 0.4) X 109 S -! and kcR,e ff = ( 1.7 4- 0.4) x 108 s - I . In connection with Scheme 2, two conclusions can be drawn from these results: (i) Initially only singlet ion pairs are formed and since the effective triplet formation is significantly less impor-
7Since the extinction coefficients of the radical anion 2(p. DCB) - are much smaller (~A ~ 6000 M - I cm - t at ~max = 520 nm in acetonitrile 1901 ) than ~K of 2DPH + and ~T ofaDPH *, 2 (p.DCB) - was not detected.
tant than the deactivation back to the singlet ground states (k~R,eff << kcCR,eff) it is obvious that the exper-
G imentally observed rate constant kcR,e ff corresponds approximately to the rate constant of the genuine CR
G reaction: kCR,eff ~ kGR . (ii) The hfc-induced spin evo- lution in larger aromatic molecules is usually consid- ered to take place on a timescale of ca. 10 ns [91 ], which is the timescale also observed here for the effec- tive triplet formation. The experimentally determined values of k~R,eff alone does therefore not allow to iden- tify either spin evolution orgenuine CR reaction as rate-determining process and thus no information on k~R is obtainable at this point. This is in accordance with preliminary experimental evidence in our previ- ous work indicating that the overall rate constant for the spin forbidden CR reaction was independent of the appropriate energy gap AG~R and in the range of (3.5 4- 2.5) x 108 s -I [42]. Such a behaviour is to be expected if the spin process is rate determining. It can thus tentatively be assumed that the value of k~R,eff obtained here for the DPH/p-DCB system also essentially reflects the kinetics of the spin evolution process.
While the transient absorption signal of 2DPH+ at 590 nm appeared immediately with the laser flash, the signal of 3DPH* at 417 nm in acetonitrile showed a prompt formation followed by a growing-in which reached a maximum on a/zs timescale. In toluene only prompt triplet formation was observed. Similar obser- vations were made before by Schomburg et al. [ 92] in the investigation of the systems pyrene/diethylaniline and pyrene/p-DCB in acetonitrile and it was shown that the delayed triplet formation resulted from the diffusion-controlled recombination reaction of the free ions with the experimental rate constant (kob.~ =) kr. From the initial second order decay of 2DPH+ this rate constant for the recombination reaction between 2DPH+ and 2(p-DCB) .... in acetonitrile was here de- termined to be kr = (~L3 4-1.1 ) x 1010 M - i s -I . This is in agreement with the value of kr = (4.2 4-0.3) x 10 I° M - I s -! for pyrene/p-DCB reported in Ref. [92]. As before, the experimental, bimolecular rate constant kr is used to calculate the corresponding unimolecular rate constant of the triplet ion pair deactivation:
ksep kr
k~R = kree - kr" ( ! 3)
To obtain Eq. (13), the spin evolution process and

E Schael. H.-G. LiihmannsriJben/Chemical Physics 206 (1996) 193-210 205
the formation of singlet ion pairs in Scheme 3 were neglected for simplicity. Due to the high experimental error in kr a more elaborated evaluation is not justified. In particular, as the upper experimental limit for kr exceeds the assumed value for krec only a lower limit for kTR is obtainable. With kr >t 3.2 x 10 I° M-! s - I, which is the lower limit of the experimental result, one obtains: k~: R t> I x 109 s -I .
In summary, the genuine CR reactions of the DPH/p-DCB singlet and triplet ion pairs proceed with rate constants of kcCR = ( 1.7 4- 0.4) x 109 s -I and k~: R ~> I x 109 s - i , while the kinetics of the spin evolution process is probably essentially described by the value o f kTR,eff -- ( 1.7 4- 0.4) x 108 s - l .
The value obtained for kcCR is in the same order of magnitude as e.g. rate constants of spin-allowed CR reactions of perylenes determined in the region of AGcCR = -2 .6 eV [42,931, which is in the Mar- cus inverted region of the electron transfer reaction. Previous theoretical analyses of L ltermolecuiar CR re- actions of organic compounds afforded values for the solvent reorganization energies in the range of ,~o = !. I-1.4 eV and for the matrix coupling element V in the order of 10-3 eV ( in acetonitrile at room tempera- ture, with fixed internal reorganization energies of,~i = 0.25 eV and mean vibrational frequencies of h~,,, = 1500 cm -I ) [42,82,93]. Our result for the deactiva- tion of the DPH/p-DCB ion pair leads us to suppose that the CR reactions of DPH can also approximately be described with these parameters.
Due to the lack of free ion formation in toluene individual rate constants for the deactivation of the DPH/acceptor contact ion pairs cannot be obtained directly from Eqs. ( I 1 ) and (12). In a forthcoming publication it will be shown that a comprehensive de- scription of the deactivation pathways in nonpolar sol- vents can be accomplished if additional spectroscopic information is available from the evaluation of the ex- ciplex decay kinetics [43]. As discussed above, the high yields of 3DPH* formation observed in the DPH fluorescence quenching in toluene (cf. Table 2) can- not solely be ascribed to 'heavy-atom quenching'. The r/T obtained from the experiments in toluene are listed in Table 2. A rough correlation between rtr and the fi'ee energy changes (obtained for acetonitrile) is ob- served: r/T decreases with increasing - A G c s , i.e. de-
G creasing -AGc~R (cf. Fig. 4). This indicates that kcR,eff
T increases relative to kcR.e ff with decreasing --AGGR . But additionally, the influence of the HAE on the de- activation processes of the CT intermediate has to be considered. For example, r/T of the quenching reaction with hexabromobenzene is 0.32 and r/T of the quench- ing reaction with maleic anhydride is < 0.0I, while AGcs for both reaction is comparable (AGcs= -0.86 and -0.84 eV, respectively). The enhancement of r/T in this case may be due to the HAE enhancement of
G k~R.e ff relative to kCR,e ft. That "r/T correlates roughly with AGcs but not with the spin-orbit-couplingparam- eter s ¢ provides again direct evidence that 'CT quench- ing' is predominant also in toluene.
For all acceptor molecules investigated, the depen- dence of the quantum yields of triplet or cation forma- tion on the quencher concentrations was in agreement with a Stern-Volmer type behaviour (cf. Fig. 4). This indicates bimolecular quenching mechanisms. No in- dication was found for other quenching mechanisms, as e.g. discussed by Hirose et al. [ 38 ] who suggested an electron transfer from a solvent molecule as initial step.
3.6. Degenerate electron transfer reactions: I H-CIDNP experiments
Time-resolved chemically induced nuclear polar- ization (CIDNP) provides a powerful tool in the in- vestigation of photoinduced electron transfer reactions [941. Especially, this method was employed for the determination of rate constants and activation barri- ers of electron self-exchange reactions (Scheme 4) [95,961.
The CIDNP effect is based on the formation of products with non-equilibrium populations in their nu- clear spin states (nuclear spin polarization) in chem- ical reactions carried out in magnetic fields. CIDNP is manifest in transient changes of nmr line intensities in emission or enhanced absorption. The qualitative explanation of this phenomenon was given by Cioss [ 97 ] and Kaptein and Osterhoff [ 98 ] : The spin evolu- tion occurring during the lifetime of the electron-spin- correlated radical pairs (cf. Scheme 2) proceeds with nuclear-spin-dependent probability. In this way, oppo- site nuclear spin polarizations are created in the singlet [ l ( D ~ . . . A T) ] and the triplet [3(D+ . .-A ~:) 1 ion pairs and are transferred to the reaction products orig- inating from these pairs. The observation of CIDNP

206 E Schael, H.-G. LiJhmannsriiben/ Chenffcal Physics 206 (1996) 193-210
in a reversible electron transfer reaction is thus evi- dence tbr the formation of solvent-separated ion pairs and tbr the spin evolution therein (cf. Section 3.1 ). In case of reversible electron transfer reactions, CIDNP is observable during the lifetime of the paramagnetic free ions [ 95 ] if the lifetimes of the singlet and triplet ion pairs are different [99].
In Fig. 5 a I H-NMR spectrum of DPH in CD3CN is shown (upper part) together with a I H-CIDNP spectrum obtained immediately after laser irradiation of a CD3CN solution with 1.4 x 10 -5 M DPH and 4.6 x 10 - 4 M p-DCB (lower part). In the CIDNP ex- periments a presaturation radio frequency pulse was employed before laser irradiation to remove all un- changing equilibrium magnetizations. No CIDNP was observable in the absence of p-DCB or from a DPH/p- DCB solution in benzene-d6. This indicates that a re- action between I DPH* and p-DCB was the origin of CIDNP and that only in acetonitrile the spin evolu- tion, which requires a considerable separation of the ions, was effective.
From the nmr spectra it is obvious that the resonance frequencies observed in the CIDNP spectra coincide with those of the DPH protons. The signs and relative intensities of the most intense CIDNP signals are in good agreement with spin densities and hfc constants obtained with AM! computations for the DPH cation. In addition, the relative CIDNP intensities are consis- tent with experimental values of the hfc constants for the DPH anion [ 100] 8. We conclude therefore, that the observed polarization pattern indicates the inter- mediacy of the DPH cation that is produced in high yields in the electron transfer reaction of DPH with p-DCB (see above).
Fig. 6 shows the time dependence of the logarith- mic CIDNP intensity. The linear plot indicates that a (pseudo) first order kinetics was observed. There are three mechanisms that can account for the CIDNP signal decay: (i) the electron self-exchange reaction (Scheme 4, pseudo first order kinetics with respect to 2DPH+ concentration), (ii) the homogeneous CR reaction (Scheme 3, second order kinetics) and (iii) spin-lattice relaxation of the ion protons. Since the latter process induces a loss of nuclear spin polariza-
x Since to our knowledge for the 2DPH+ protons hfc constants are not available in the literature they can only be very roughly compared to 2DPH- protons i 1011.
.o
N
_=
2
r I , , t I ~ , t I t ,
0 5 10 15 20 t/las
Fig. 6. Logarithmic plot of the relative CIDNP intensity in versus time t. IN was obtained from the integrals of all observed CIDNP signals.
tion in the free ions, an observed CIDNP signal would reach a non-zero level at long times (on the reaction time scale) if spin-lattice relaxation is effective [ 96]. Here, the observed polarization vanished after ca. 20 /zs so that the influence of spin-lattice relaxation can be assumed to be negligible. Because of the observed first order kinetics it is expected that the electron self- exchange reaction was responsible for the CIDNP sig- nal decay. From the slope of the signal decay in Fig. 6 the experimental bimolecular rate constant (kob,~ - ) kc = 9.8 x 109 M -! s -I at 301 K is obtained. Since it is difficult to rule out a small contribution of the ho- mogeneous CR reaction to the CIDNP signal decay because the absolute ion concentrations in the CIDNP experiments were not known, the experimental uncer- tainty in ke is estimated to be ca. 20%. The unimolec- ular rate constant for the electron exchange reaction was then calculated from:
k-d kc kEx = ~ (14)
kd -- kc '
The rate constant of the electron self-exchange reac- tion between DPH and 2DPH+ was obtained to be: kEx = (2.1 4- 0.8) x 10 l° s - l . This valve is in accor- dance with published experimental results for DELT reactions in organic systems [ 45,96,102,103 ]. How- ever, more data are required, e.g. from measurements of the temperature dependence, for a detailed discus- sion of kEx in the framework of electron transfer the- ories.

E Schael. H.-G. LiJhmannsriJben/Chemicai Physics 206 (1996) 193-210 207
H 2' H.~ H 1 HOrth= Hmeta
Hpara
Hortho I H meta H1,1'
-J- Hpara ~ H3,3 ,
I I I _J_ H2, 2' _I_
+
8 7.5 7 6.5 6 8 / ppm
Fig. 5. 250 MHz IH-NMR spectrum of DPH in CD3CN (upper part) and tH-CIDNP spectrum of 4.1 x 10 -5 M DPH and 46x 10 -3 M p-DCB in CD3CN obtained at 301 K immediately after laser irradiation (lower part). Also shown are the assignments of the signals.
4. C o n c l u s i o n s
In this work, the fluorescence quenching of sin- glet excited all-trans-l,6-diphenylhexa-l,3,5-triene (DPH) by electron accepting organic quencher molecules was investigated in toluene and acetoni- trile solutions. The influences of the external HAE and of CT processes on the fluorescence quenching mechanism were characterized. In both solvents, the Rehm-Weiler treatment gives an adequate description of the dependence of the bimolecular rate constant of fluorescence quenching (kq F) on the standard free energy change of charge separation (AGcs).
As primary products of the quenching reactions triplet (aDPH*) and cationic (2DPH+) DPH were
identified by means of time-resolved laser flash pho- tolysis in acetonitrile. In toluene only formation of 3DPH* was observed. The 3DPH* formation effien- ties in toluene depended mainly on AGcs and only to a minor extend on the spin-orbit coupling parameter (s ¢2) of the quencher. This provided indirect evidence that CT quenching was predominant also in toluene.
Particular emphasis was placed upon the eluci- dation of primary and secondary electron transfer reactions in the quenching of DPH fluorescence by p-dicyanobenzene (p-DCB) in acetonitrile. The ef- fective rate constants ( kcR,eff , G kTR,eff) of the charge re- combination (CR) reactions of the solvent-separated ion pair (SSIP) formed between DPH and p-DCB were determined. Together with the investigations

208 E Schael, H.-G. LiihmannsriJben/Chemical Physics 206 (1996) 193-210
of the secondary ion recombination reaction be- tween 2DPH + and anionic p-DCB ( 2 p - D C B - ) and
the CIDNP investigation of the degenerate electron transfer (DELT) reaction between DPH cation and parent molecules, the results allowed the character- ization of the following elementary reactions: The genuine CR reactions of the D P H / p - D C B singlet and triplet ion pairs proceed with rate constants of kCGR = ( 1.7 4- 0.4) X 109 S - I and k~R t> 1 x 109 s - l ,
the kinetics of the spin evolution process is prob-
ably essentially described by the value of kTR,eff =
( ! .7 4- 0.4) X 108 S - I (cf. Scheme 2), and the
rate constant of the DELT reaction was found to be
kEx = (2.1 4- 0.8) × 10 I° s - I . Our results for the D P H / p - D C B system indicate
thdt with the combination of stationary and time- resolved fluorescence measurements, laser flash pho- tolysis and CIDNP experiments a comprehensive description of the primary and secondary electron transfer reactions in the fluorescence quenching pro- cess can be achieved. Further studies are necessary for the investigation of other donor/acceptor systems and, in particular, for the elucidation of spin evolu-
tion processes, the characterization of the free energy dependences of the genuine CR reactions, and the in- fluences of solvent polarity, heavy-atom perturbation and solvent polarity thereon.
Acknowledgements
Thanks are due to M. Komfort and J. Kiister for
single-photon counting measurements, to Dr. G.:Eck- ert for his kind support with the CIDNP experi- ments and fruitful discussions, and to E Schrobs-
dorff for technical assistance. Financial support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged.
References
III J.R. Platt, Science 129 (1959) 372. 121 H.T. Tien, Bilayer lipid membranes (Marcel Dekker, New
York, 1974). 131 D.S. Berns, Photochem. Photobiol. 24 (1976) 117. 141 R.J. Cogdell and H.A. Frank, Biochim. Biophys. Acta 895
(1987) 63. 151 R.J. Thrash, H.L.-B. Fang and G.E. Leroi, J. Chem. Phys.
67 (1977) 5930.
I61
171
181
191
!101
l l l l
[121 i131
!141 !151
1161
1171
1181
1191 1201
I211
1221
1231
1241 1251
I261 I271
I281
1291
I301
1311 I321
1331
I341
1351
I361
R.J. Thrash, H.L.-B. Fang and G.E. Leroi, Photochem. Photobiol. 29 (1979) 1049. J.C. Cogdell, P.O. Andersson and T. Gillbro, J. Photochem. Photobiol. B i 5 (1992) 105. B.S. Hudson, B.E. Kohler and K. Schulten, in: Excited states, Vol.6, ed. E,C. Lim (Academic Press, New York, 1982). J.R. Lakowicz and EG. Prendergast, Science 200 (1978) 1399. R.H. Bisby, R.B. Cundail, L. Davenport, I.D. Johnson and E.W. Thomas, in: Fluorescent Probes, eds. G.S. Beddard and M.A. West (Academic Press, New York, 1981 ). D.C. Rodenberger, J.R. Heflin and A.E Garito, Nature 359 (1992) 309. Li Guang and L.B. McGown, Science 264 (1994) 249. S.P. Spooner and D.G. Whitten, J. Am. Chem. Soc. !16 (1994) 1240. M.T. Allen and D.G. Whitten, Chem. Rev. 89 (1989) 1691. J. Saltiel and Y.-P. Sun, in: Photochromism, molecules and systems, eds. H. DUn and H. Bouas-Laurent (Elsevier, Amsterdam, 1990). E.D. Cehelnik, R.B. Cundall, J.R. Lockwood and T.E Palmer, Chem. Phys. Letters 27 (1974) 586. E.D. Cehelnik, R.B. Cundall, J.R. Lockwood and T.E Palmer, J. Phys. Chem. 79 (1975) 1369. J.B. Birks and D.J.S. Birch, Chem. Phys. Letters 31 (1975) 608. J.B. Birks, Chem. Phys. Letters 54 (1978) 430. J.B. Birks, G.N.R. Tripathi and M.D. Lumb, Chem. Phys. 33 (1978) 185. L.A. Sklar, B.S. Hudson, M. Petersen and J. Diamond, Biochemistry 16 (1977) 813. S.L. Bondarev and S.M. Bachilo, J. Photochem. Photobiol. A 59 (1991) 273. J.R. Andrews and B.S. Hudson, J. Chem. Phys. 68 (1978) 4587. T. ltoh, Chem. Phys. Letters 159 (1989) 263. H.L.-B. Fang, R.J. Thrash and G.E. Lroi, Chem. Phys. Letters 57 (1978) 59. T. Itoh and B.E. Kohler, J. Phys. Chem. 91 (1987) 1760. P.C. Alford and T.E Pahner, Chem. Phys. Letters 86 (1982) 248. P.C. Aiford and T.E Palmer, J. Chem. Soc. Faraday "li'~ns. !! 79 (1983) 433. E Schael and H.-G. L6hmannsr6ben, J. Photochem. Photobiol. A 69 (1992) 27. J. Saltiel, D.E Sears, Jr.,Y.-E Sun and J.-O. Choi, J. Am. Chem. Soc. 114 (1992) 3607. H. G6mer, ,L Photochem. 19 (1982) 343. J. Saltiel, D.-H. Ko and S.A. Fleming, J. Am. Chem. Soc. ! 16 (1994) 4099. P.-S. Song, Q. Chae, M. Fujita and H.. Baba, J. Am. Chem. Soc. 98 (1976) 819. S.K. Chattopadhyay, P.K. Das and G.-L. Hug, J. Am. Chem. Soc. 104 (1982) 4507. V. Ramamurthy, J.V. Caspar, D.E Eaton, E.W. Kuo, and D.R. Corbin, J. Am. Chem. Soc. 114 (1992) 3882. M. Wanatabe, T. Kamisuki, N. Akamatsu and C. Hirose, Chem. Phys. Letters 170 (1990) 451.

E Schael, H.-G. 1.Shmannsrfiben/Chemical Physics 206 (1996) 193-210 209
371 T. Kamisuki and C. Hirose, J. Phys. Chem. 95 ( 1991 ) 5003. 381 T. Kamisuki, T. Dudev and C. Hirose, J. Phys. Chem. Soc.
95 ( 199 ! ) 5845. 391 A.G. Majumdar, B.B. Bhowm[k and S. Basu, Indian J. Chem.
30A ( ! 991 ) 964. 401 R. Dutta, S. Basu and M. Chrowdhury, Chem. Phys. Letters
182 ( 199 ! ) 429. 41 I Z. Wang and W.G. McGimpsey, J. Phys. Chem. 97 (1993)
3324, 5054. 421 E Lewitzka and H.-G. Lfhmannsdiben, Z. Physik. Chem.
NF 169 (1990) 203. 431 E Schaei, J. Kiister and H.-G. l.,6hmannsdiben, J. Inf. Rec.
Mats. 21 (1994) 525; manuscript in preparation. 441 H. Dreeskamp, T. Salthammer and A.G.E.l.~ufer, J.
Luminescence 44 (1989) 161. 451 M. Goez, Z. Physik. Chem. NF 169 (1990) 123. 461 T. Sander, H.-G. LOhmannsdiben and H. Langhals, J.
Photochem. Photobiol. A 86 (1995) 103. 1471 D. Rehm and A. Weller, Bet. Bunsenges. Physik. Chem. 73
(1969) 834. 1481 R.E. F/511, H.E.A. Kramer and U.E. Steiner, J. Phys. Chem.
94 (1990) 2476. 1491 M. v. Smoluehowski, Z. Physik. Chem. 92 (1917) 129. 1501 P. Debye, Trans. Electrochem. Soc. 82 (1942) 265. 1511 M. Eigen, Z. Physik. Chem. NF I (1954) 176. 1521 S.L. Murov, I. Carmichaei and G.L. Hug, Handbook of
photochemistry, 2rid Ed. (Marcel Dekker, New York, 1993 ). 1531 D. Mauzerall and S.G. Ballard, Ann. Rev. Phys. Chem. 33
(1982) 377. 1541 J. Saltiel and B.W. Atwater, Advan. Photochem. 14 (1988)
i. 1551 N. Mataga, T. Asahi, T. Kanda, T. Okada and T. Kakitani,
Chem. Phys. 127 (1988) 249. 1561 E. Driscoll, J.D. Simon and K.S. Peters, J. Am. Chem. Soc.
112 (1990) 7091. 1571 T. Niwa, K. Kikuchi, N. Matsusita, M. Nayashi, T. Katagiri,
Y. Takahashi and T. Miyashi, J. Phys. Chem. 97 (1993) ! 1960.
1581 Z. Schulten and K. Schulten, J. Chem. Phys. 66 (1977) 4616.
1591 T.C. Feider, K.-J. Choi and M.R. Topp, Chem. Phys. 64 (1982) 175.
1601 C. Rulli~re and A. Declrmy, Chem. Phys. Letters 135 (1987) 213.
1611 L.A. Brey, G.B. Schuster and H.G. Drickamer, J. Chem. Phys. 71 (1979) 2765.
162 ! A. Stanienda, Z. Physik. Chem. NF 33 (1962) 170. 1631 A. Weller, Z. Physik. Chem. NF 13 (1957) 335. 1641 D.E Othmer and M.S. Thakar, Ind. Eng. Chem. 45 ( ! 953)
589. 1651 R.C. West, ed., Handbook of chemistry and physics, 67th
Ed. (CRC Press, Boca Raton, 1986). 1661 J. Saltiei, P.T. Shannon, O.C. Zafiriou and A.K. Uriarte, J.
Am. Chem. 102 (1980) 6799. 1671 E. Francais, M. Bouchy J.C. Andre, N. Midoux and J.C.
Charpentier, Entropie 27 ( 1991 ) 39. 1681 Y. Fan, R. Quian, M. Cheng, M. Shi and J. Shi, Ber.
Bunsenges. Physik. Chem. 99 (1995) 1043.
1691 1701
1711
[721
1731
1741
I751 t761
[771
I781 1791
1801 I811
I821
I831
1841
1851
1861
1871
1881
1891
1901
1911 1921
1931
1941
1951
1961 [971 I981
W.R. Ware and J.S. Norms, J. Phys. Chem. 70 (1966) 3246. EM. Martens, J.W. Verhoeven, R.A. Gase, U.K. Pandit and Th.J. De Boer, Tetrahedron 34 (1978) 443. R.A. Caldwell, D. Creed, D.C. DeMarco, L.A. Melton, H. Ohta and P.H. Wine, J. Am. Chem. Soc. 102 (1980) 2369. C.K. Mann and K.K. Barnes, Electrochemical reactions in nonaqueous systems (Marcel Dekker, New York, 1970). K. lwai, E Takemura, M. Furue and S. Nozakura, Bull. Chem. Soc. J:~Fan 57 (1984) 763. D. Rehm and A. Weller, Z. Physik. Chem. NF 69 (1970) 183. A. Weiler, Z. Physik. Chem. NF 133 (1982) 93. J.-M. Chen, T.-I. Ho and C.-Y. Mou, J. Phys. Chem. 94 (1990) 2889. N. Ghoneim, C. Hammer, E. Haselbach, D. Pilloud and P. Suppan, J. Chem. Soc. Faraday Trans. 89 (1993) 4271. H.L. Clever, J. Phys. Chem. 62 (1958) 375. A.R. Horrocks, A. Kearvell, K. Tickle and E Wilkinson, Trans. Faraday Soc. 63 (1966) 3393. A. Kearvell and E Wilkinson, Mol. Cryst. 4 (1968) ,59. B. Amand and R. Bensasson, Chem. Phys. Letters 34 (1975) 44. I.R. Gould, D. Ege, J.E. Moser and S. Farid, J. Am. Chem. Soc. 112 (1990) 4290. ED. Lewis, E. Dykstra, I.R. Gould and S. Fad& J. Phys. Chem. 92 (1988) 7042. E. Vauthey, D. Pilloud, E. Haselbach, P. Suppan and P. Jacques, Chem. Phys. Letters 215 (1993) 264. R. Bensasson, E.J. Land, J. Lafferty, R.S. Sinclair and T.G. Truscott, Chem. Phys. Letters 41 (1976) 333. M. Almgren and J.K. Thomas, Photochem. Photobiol. 31 (1980) 329. Y. Yamamoto, T. Aoyama and K. Hayashi, J. Chem. Soc. Faraday Trans. I 84 (1988) 2209. V. Ramamurthy, J.V. Caspar and D.R. Corbin, J. Am. Chem. Soc. 1 ! 3 ( i 991 ) 594. T. Shida, Electronic spectra of radical ions (Elsevier, Amsterdam, 1988). H. Schomburg, H. Staerk and A. Weller, Chem. Phys. Letters 22 (1973) !. A. Weller, Z. Physik. Chem. NF 130 (1982) 129. H. Schomburg, H. Staerk and A. Weller, Chem. Phys. Letters 21 (1973) 433. T. Kircher and H.-G. LfhmannsriSben, GIT Fachz. Lab. 36 (1992) 914. G.L. Closs, RJ. Miller and O.D. Redwine, Accounts Chem. Res. 18 (1985) 196. G.L. Closs and E.V. Sitzmann, J. Am. Chem. Soc. 103 (1981) 3217. M. Goez, Chem. Phys. 147 (1990) 143. G.L. Closs, J. Am. Chem. Soc. 91 (1969) 4552. R. Kaptein and LJ. Oosterhoff, Chem. Phys. Letters 195 (1969) 214.
I991 J. Bargon, J. Am. Chem. Soc. 99 (1977) 8350. 11001 R. Schenk, W. Huber, E Schade and K. Miillen, Chem.
Bet. 121 (1988)2201. 11011 A. Carrington and A.D. McLachlan, Introduction to
magnetic resonance (Harper and Row, New York, 1967).

210 F. Schael, H.-G. LOhmannsrOben/ Chemical Physics 206 (1996) 193-210
11021 G. GramppandW. Jaenicke, Ber. Bunsenges.Physik.Chem. [ 103] E. Eberson, Advan. Phys. Org. Chem. 18 (1982) 79. 88 (1984) 335; J. Chem. $oc. Faraday Trans. I! 81 (1985) 1035: Ber. Bunsenges. Physik. Chem. 95 (1991) 904.
![SAR of a series of anti-HSV-1 acridone derivatives, and a rational acridone-based design of a new anti-HSV-1 3 H-benzo[ b]pyrazolo[3,4- h]-1,6-naphthyridine series](https://static.fdokumen.com/doc/165x107/631beba7665120b3330b99e5/sar-of-a-series-of-anti-hsv-1-acridone-derivatives-and-a-rational-acridone-based.jpg)







![The jet-cooled S 0→S 1 excitation spectrum of 1,6-epoxy-[10]annulene](https://static.fdokumen.com/doc/165x107/631e1beedc32ad07f3076a22/the-jet-cooled-s-0s-1-excitation-spectrum-of-16-epoxy-10annulene.jpg)
![Design, synthesis, and biological evaluation of substituted 2,3-dihydro-1 H-cyclopenta[ b]quinolin-9-ylamine related compounds as fructose-1,6-bisphosphatase inhibitors](https://static.fdokumen.com/doc/165x107/63332c96a290d455630a0469/design-synthesis-and-biological-evaluation-of-substituted-23-dihydro-1-h-cyclopenta.jpg)





![Tris(acetonitrile-κ N ){2,6-bis[(diphenylphosphanyl)amino]-4-ethoxy-1,3,5-triazine-κ 3 P , N 1 , P ′}iron(II) bis(tetrafluoridoborate) acetonitrile disolvate](https://static.fdokumen.com/doc/165x107/6323aaae03238a9ff60a8974/trisacetonitrile-k-n-26-bisdiphenylphosphanylamino-4-ethoxy-135-triazine-k.jpg)




