
CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5 ...
10
ORIGINAL ARTICLE CoMFA and docking study of 2,N 6 -disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines as novel PfDHFR enzyme inhibitors for antimalarial activity Pritam N. Dube a, * , Santosh Mokale a , Prasanna Datar b a Department of Pharmaceutical Chemistry, Y. B. Chavan College of Pharmacy, Aurangabad 431001, M.S., India b Sinhgad Institute of Pharmacy, Narhe, Pune 411041, M.S., India Received 27 October 2013; accepted 22 February 2014 KEYWORDS PfDHFR; 1,3,5-Triazine; CoMFA; Docking Abstract A three-dimensional quantitative structure–activity relationship (3D-QSAR) study was performed on 1,3,5-triazine derivatives which were based on Ala16Val + Ser108Thr mutant DHFR inhibitors of Plasmodium falciparum clone (FCR-3). Comparative Molecular Field Analysis (CoMFA) was carried out for designing novel PfDHFR enzyme inhibitors. It is shown that the steric and electrostatic properties by CoMFA contours can be related to the PfDHFR inhibitory activity. Glide-XP of Schro¨dinger was used for docking of PfDHFR inhibitors into the putative binding sites of the PfDHFR. ª 2014 Production and hosting by Elsevier B.V. on behalf of Faculty of Pharmacy, Cairo University. 1. Introduction Malaria is one of the most widespread diseases in the world. According to WHO estimates 40% of the world’s populations presently live under malarial threat. Around 300 and 500 mil- lion cases of malaria occur annually, leading to 1–3 million deaths. 1 Its control is globally a high priority task. Although effective antimalarial agents have been known for a long time, the alarming spread of drug resistant strains of Plasmodium falciparum, which is the most lethal parasite species, undergoes the urgency and continuous need for the discovery of new ther- apeutics. A major initiative in this direction is to find enzyme targets that are critical to the disease process or essential for the survival of the parasite. Identification and design of novel chemical entities specifically affecting these targets could lead to better drugs for the treatment of malaria. 2 The widespread occurrence of malaria could be attributed to the development of resistance of the parasite to the available antimalarial drugs such as chloroquine, cycloguanil and pyrimethamine. These drugs have been used clinically in the treatment of malaria, especially to P. falciparum malaria for longer period of time. But due to the emergence of drug-resistant parasite in many countries, there is need of new and effective drugs for treat- ment of malaria. 3–8 The dihydrofolate reductase (DHFR) domain of a bi-functional enzyme which is known as dihydrofolate reductase-thymidylate synthase (DHFR-TS) is one of the * Corresponding author. Tel.: +91 9503981237. E-mail address: [email protected] (P.N. Dube). Peer review under responsibility of Faculty of Pharmacy, Cairo University. Production and hosting by Elsevier Bulletin of Faculty of Pharmacy, Cairo University (2014) xxx, xxx–xxx Cairo University Bulletin of Faculty of Pharmacy, Cairo University www.elsevier.com/locate/bfopcu www.sciencedirect.com 1110-0931 ª 2014 Production and hosting by Elsevier B.V. on behalf of Faculty of Pharmacy, Cairo University. http://dx.doi.org/10.1016/j.bfopcu.2014.02.003 Please cite this article in press as: Dube PN et al. CoMFA and docking study of 2,N 6 -disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines as novel PfDHFR enzyme inhibitors for antimalarial activity, Bulletin Facult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/ j.bfopcu.2014.02.003
-
Upload
khangminh22 -
Category
Documents
-
view
6 -
download
0
Transcript of CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5 ...

Bulletin of Faculty of Pharmacy, Cairo University (2014) xxx, xxx–xxx
Cairo University
Bulletin of Faculty of Pharmacy, Cairo University
www.elsevier.com/locate/bfopcuwww.sciencedirect.com
ORIGINAL ARTICLE
CoMFA and docking study of 2,N6-disubstituted
1,2-dihydro-1,3,5-triazine-4,6-diamines as novel PfDHFR
enzyme inhibitors for antimalarial activity
Pritam N. Dubea,*, Santosh Mokale
a, Prasanna Datar
b
a Department of Pharmaceutical Chemistry, Y. B. Chavan College of Pharmacy, Aurangabad 431001, M.S., Indiab Sinhgad Institute of Pharmacy, Narhe, Pune 411041, M.S., India
Received 27 October 2013; accepted 22 February 2014
* Corresponding author. Tel.E-mail address: pritamdube@
Peer review under responsibi
University.
Production an
1110-0931 ª 2014 Production
http://dx.doi.org/10.1016/j.bfop
Please cite this article in pressnovel PfDHFR enzyme inhj.bfopcu.2014.02.003
: +91 95gmail.co
lity of F
d hostin
and hosti
cu.2014.
as: Dubibitors f
KEYWORDS
PfDHFR;
1,3,5-Triazine;
CoMFA;
Docking
Abstract A three-dimensional quantitative structure–activity relationship (3D-QSAR) study was
performed on 1,3,5-triazine derivatives which were based on Ala16Val + Ser108Thr mutant DHFR
inhibitors of Plasmodium falciparum clone (FCR-3). Comparative Molecular Field Analysis
(CoMFA) was carried out for designing novel PfDHFR enzyme inhibitors. It is shown that the
steric and electrostatic properties by CoMFA contours can be related to the PfDHFR inhibitory
activity. Glide-XP of Schrodinger was used for docking of PfDHFR inhibitors into the putative
binding sites of the PfDHFR.ª 2014 Production and hosting by Elsevier B.V. on behalf of Faculty of Pharmacy, Cairo University.
1. Introduction
Malaria is one of the most widespread diseases in the world.According to WHO estimates 40% of the world’s populations
presently live under malarial threat. Around 300 and 500 mil-lion cases of malaria occur annually, leading to 1–3 milliondeaths.1 Its control is globally a high priority task. Although
effective antimalarial agents have been known for a long time,the alarming spread of drug resistant strains of Plasmodium
03981237.m (P.N. Dube).
aculty of Pharmacy, Cairo
g by Elsevier
ng by Elsevier B.V. on behalf of F
02.003
e PN et al. CoMFA and dockingor antimalarial activity, Bulletin
falciparum, which is the most lethal parasite species, undergoesthe urgency and continuous need for the discovery of new ther-apeutics. A major initiative in this direction is to find enzyme
targets that are critical to the disease process or essential forthe survival of the parasite. Identification and design of novelchemical entities specifically affecting these targets could leadto better drugs for the treatment of malaria.2 The widespread
occurrence of malaria could be attributed to the developmentof resistance of the parasite to the available antimalarial drugssuch as chloroquine, cycloguanil and pyrimethamine. These
drugs have been used clinically in the treatment of malaria,especially to P. falciparum malaria for longer period of time.But due to the emergence of drug-resistant parasite in many
countries, there is need of new and effective drugs for treat-ment of malaria.3–8
The dihydrofolate reductase (DHFR) domain of a
bi-functional enzyme which is known as dihydrofolatereductase-thymidylate synthase (DHFR-TS) is one of the
aculty of Pharmacy, Cairo University.
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/

2 P.N. Dube et al.
well-established drug targets in P. falciparum. It catalyzesNADPH-dependent reduction of 7,8-dihydrofolate (DHF) to5,6,7,8-teterahydrofolate (THF) that plays a crucial role in
many biochemical processes such as folate metabolism andDNA synthesis. Thus, inhibition of DHFR causes interruptionof DNA formation which ultimately results in death of the
parasitic cells. This observation has been exploited to designantimalarial agents that inhibit the normal function ofPfDHFR enzyme.9–12 It is a specific target for type-2 anti-fo-
lates such as pyrimethamine and cycloguanil.13 These antima-larial drugs selectively inhibit PfDHFR enzyme withoutaffecting the corresponding enzyme in humans. However,due to point mutations at amino acid residues 16, 51, 59, 108
and 164 in the active site of PfDHFR enzyme, the therapeuticvalues of cycloguanil and pyrimethamine have dramaticallydeclined in many parts of the world.14,15 Such mutations de-
crease the optimal binding interactions of the drugs and aminoacid residues in the active site of PfDHFR enzyme which resultin reduction of efficacy.16
Computer-aided drug design approaches such as pharma-cophore mapping, docking, homology modeling and quantita-tive structure–activity relationships (QSAR) models17–20 are
some of the approaches which have been employed inunderstanding parasitic drug resistance, and also in designingantimalarial drug candidates for inhibition of drug-resistantP. falciparum parasite.
Two of the most widely used 3D-QSAR approaches arebased on Comparative Molecular Field Analysis (CoMFA)21
and Comparative Molecular Similarity Index Analysis (CoM-
SIA).22 In CoMFA model, the steric and electrostatic fields arecomputed based on 3D-structures of ligands in the training set.Partial Least Square (PLS) is then used to derive an equation
that correlates the biological activity with the different probesat various lattice points. Different color-coded contour mapssurrounding the ligands give insights about favorable and
unfavorable ligand–receptor interactions, and also used asguides for designing novel leads. Molecular docking studywas carried out to know the possible interaction betweenDHFR and title compounds.
2. Results and discussion
2.1. Molecular alignment and model generation
The compounds selected for the 3D-QSAR analysis and their
estimated biological activities are listed in Table 1. The 28compounds were used in the 3D-QSAR analysis. Among thedata set used, only 20 molecules were included in the training
set and the remaining 08 molecules were used as test set. Thechoice of compounds as training and test set was performedrandomly. The statistic results of CoMFA models were con-
structed by the minimum energy conformer-based alignment.
2.2. Statistical analyses
The CoMFA model with the highest external predictive ability
(r2pred = 0.740) was selected as the best model. The training setcompounds used to generate this model, and the test set com-pounds are given in Table 1. The cross-validation coefficient,
r2cv, and the non-cross validate coefficient, r2ncv, of this modelwere found to be 0.656 and 0.996, respectively, for optimum
Please cite this article in press as: Dube PN et al. CoMFA and dockingnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletinj.bfopcu.2014.02.003
number of components (ONC) 6. The standard error ofestimation (SEE) of this model was 0.033. The summary ofstatistics of the best CoMFA model is given in Table 2. The
relative contributions of steric and electrostatic fields 76.7%and 23.3%, respectively, indicate that steric interactions arethe major contributors for the variation of biological activities
of triazine derivatives used in the study.The schematic representation showing the relative positions
of Val16, Thr108 and Asp54 in the active site of Ala16Val +
Ser108Thr mutant P. falciparum parasite is given in Fig. 1whereas the conformation of pyrimethamine bound in the ac-tive site of X-ray crystallographic structure of double mutant(S108N+ C59R) PfDHFR (Fig. 2). As shown in Fig. 2, the
para-chloro phenyl ring of pyrimethamine was displaced froma plane which was perpendicular to the plane of the 2,4-diami-nopyrimidine ring. Similar displacement also occurs in the
p-chloro phenyl ring of cycloguanil which results in loss ofits binding interactions and affinity in the active site of Ala16-Val + Ser108Thr mutant enzyme as compared to that of the
wild type.As indicated in Table 2, the predictive correlation coefficient
(r2pred = 0.740) was reasonable to suggest that our model is a
satisfactory model with good predictive ability. A high r2 value(0.999) of bootstrapping which was carried out at 100 runs alsosupports the statistical validity of the model.23,24 The summaryof statistics of the CoMFA studies is given in Table 2.
The comparison between the experimental and predictedactivities of the training set and test set compounds (Table 3)indicated that the CoMFA models show good predictive abil-
ities as demonstrated by the small residual values of both setsof compounds. Fig. 3 shows the graph of predicted vs. exper-imental activities of the training and test set compounds. The
linearity of the graphs also indicates that both the CoMFA(Fig. 3(A)) models are predictive enough to be used as guidesin designing new molecules.
2.3. Contour map analyses
Having obtained a valid CoMFA model, it is possible to iden-tify regions of space around the molecules where changes with
altering substitution would lead to an increase or a decrease inbiological activity. This has been achieved by using a CoMFAcontour map for the model, as shown in Fig. 4(a) (steric con-
tributions) and in Fig. 4(b) (electrostatic contributions).
2.3.1. Visual inspection of 3D contour maps generated by
CoMFA
The CoMFA steric and electrostatic maps of triazine deriva-tives used in the study are depicted in Fig. 4 along with themost active compound (16.10) displayed in the background.
In these contour maps, the green contours show the regionswhere sterically bulkier groups are associated with increasein biological activity. On the other hand, yellow contours indi-
cate the regions where such bulkier groups lower biologicalactivities of ligands.25 For the selected best CoMFA model,green contours were observed near 30-substituent of 5-arylring and at about three C–C bonds away from C6 atom of
the 1,3,5-triazine ring (Fig. 4(a)). The green regions show thatincreasing steric bulk at these positions results in increased bio-activities of triazine-derivatives. This is consistent with the fact
that those compounds with higher pIC50 values bear bulky
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/
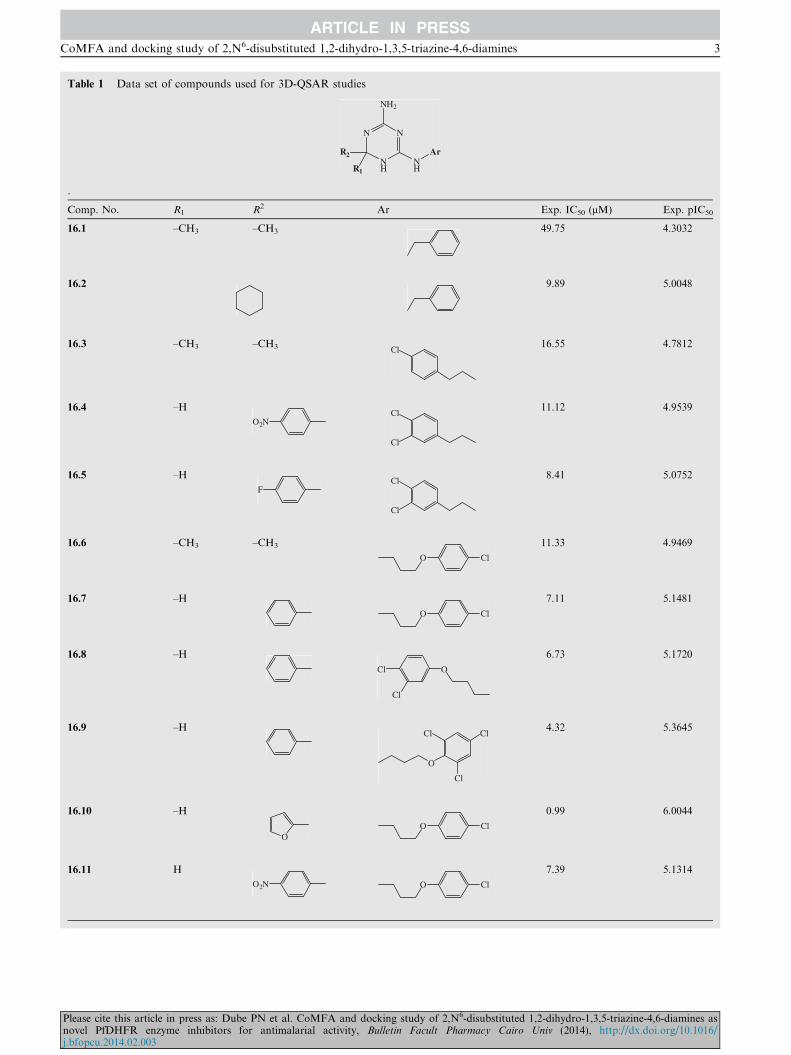
Table 1 Data set of compounds used for 3D-QSAR studies
NN
NH
NH
R2
R1
NH2
Ar
.
Comp. No. R1 R2 Ar Exp. IC50 (lM) Exp. pIC50
16.1 –CH3 –CH3 49.75 4.3032
16.2 9.89 5.0048
16.3 –CH3 –CH3 Cl16.55 4.7812
16.4 –H
O2NCl
Cl
11.12 4.9539
16.5 –H
FCl
Cl
8.41 5.0752
16.6 –CH3 –CH3
O Cl
11.33 4.9469
16.7 –H
O Cl
7.11 5.1481
16.8 –H
OCl
Cl
6.73 5.1720
16.9 –H
O
Cl
Cl
Cl4.32 5.3645
16.10 –H
OO Cl
0.99 6.0044
16.11 H
O2N O Cl
7.39 5.1314
CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines 3
Please cite this article in press as: Dube PN et al. CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletin Facult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/j.bfopcu.2014.02.003

Table 1 (continued).
Comp. No. R1 R2 Ar Exp. IC50 (lM) Exp. pIC50
16.12 H
ClO Cl
6.66 5.1765
16.13 –H
O Cl
4.59 5.3382
16.14
O Cl
7.94 5.1002
16.15 –H
H3CO O Cl
6.66 5.1765
16.16 –H
N O Cl
44.75 4.3492
16.17 –H
F O Cl
6.21 5.2069
16.18 –H
F3C O Cl
5.54 5.2565
16.19 –H
S Cl
1.30 5.8861
16.20 –CH3 –CH3
S Cl
7.84 5.1057
16.21 –H
O
Cl4.53 5.3439
16.22 –H
O
Cl
Cl
4.52 5.3449
16.23 –H
O
F10.85 4.9646
16.24 –H
O NO2
3.87 5.4123
4 P.N. Dube et al.
Please cite this article in press as: Dube PN et al. CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletin Facult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/j.bfopcu.2014.02.003

Figure 1 The schematic representation of the relative orienta-
tions of Val16, Thr108 and Asp54 in the active site of
A16V + S108T mutant PfDHFR enzyme.
Figure 2 The conformation of pyrimethamine bound in the
active site of X-ray crystallographic structure of double mutant
(S108N+ C59R) PfDHFR enzyme (1J3J.pdb).
Table 1 (continued).
Comp. No. R1 R2 Ar Exp. IC50 (lM) Exp. pIC50
16.25 –H
O OCH3
12.31 4.9097
16.26 –CH3 –CH3
O Cl
7.20 5.1427
16.27 –H
O Cl
2.71 5.5670
16.28 –H
O
Cl1.36 5.8665
Table 2 Summary of CoMFA model statistics.
NOC r2cv r2bs r2ncv F-test SEE r2pred PRESS Field Contribution
S E
6 0.656 0.999 0.996 562.55 0.033 0.740 1.312 76.7 23.3
S: steric; E: electrostatic; r2pred: predictive correlation coefficient; ONC: optimum number of components; r2cv: cross-validated correlation
coefficient; r2ncv: conventional correlation coefficient; SEE: standard error of estimation; F-value: F-test value; PRESS: predicted residual sum of
squares of test set molecules.
CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines 5
substituents either at C6- or the meta-position of the 5-arylring. Some compounds also have bulky substituents at both
positions.Compound 16.10 bears cyclic ring i.e. furan substituent at
their C6-positions, and show highest anti-plasmodial activities
(pIC50 > 5.70) compared to cycloguanil as demonstrated bytheir high pIC50 values of 6.004. Though it has benzene substi-tuent at their C6-positions, 16.19 and 16.28 have high activity.
This observation could be attributed to the fact that the 19-Clsubstituent favorably interacts with the enzyme’s active site toenhance inhibitory activity and helps to reduce the unfavorableeffect of 27-Cl substituent.
Please cite this article in press as: Dube PN et al. CoMFA and dockingnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletinj.bfopcu.2014.02.003
On the other hand, compounds which belong to no steri-cally bulky substituents fall in the above mentioned green
regions. These compounds generally exhibit low activities(pIC50 < 4.8). Even though they have dimethyl and dimethyl-amine substituents at C6-position of the triazine ring,compounds 16.1, 16.3 and 16.16 show low activities. This
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/
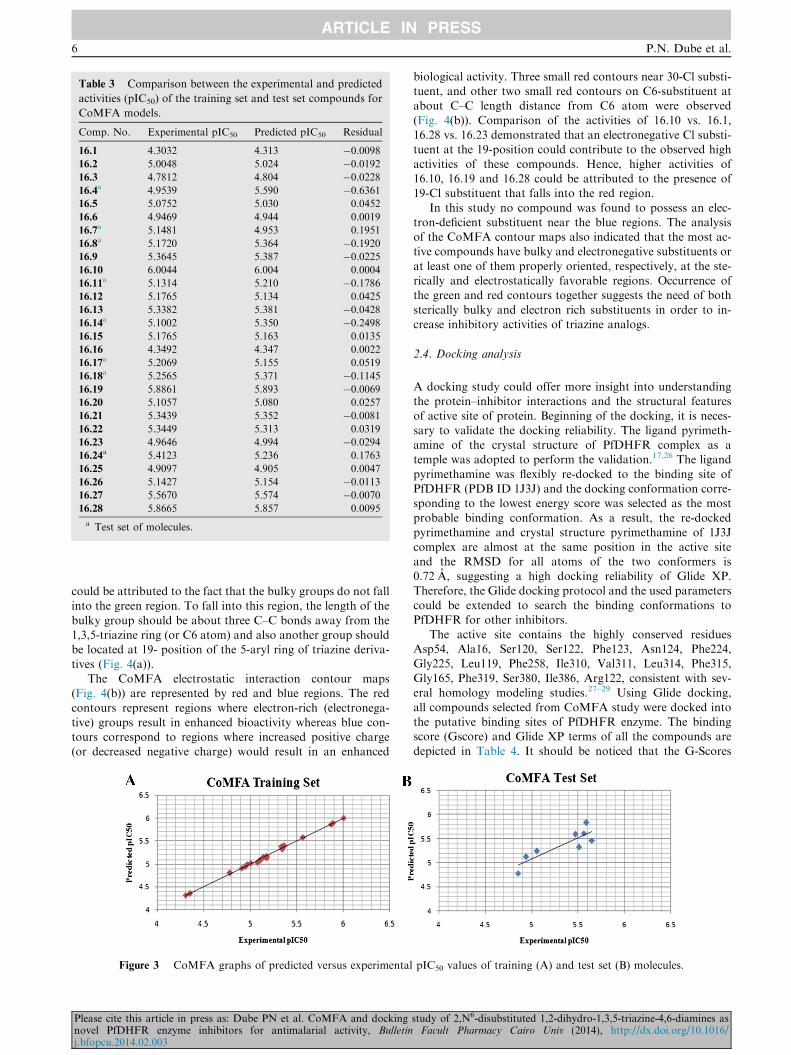
Table 3 Comparison between the experimental and predicted
activities (pIC50) of the training set and test set compounds for
CoMFA models.
Comp. No. Experimental pIC50 Predicted pIC50 Residual
16.1 4.3032 4.313 �0.009816.2 5.0048 5.024 �0.019216.3 4.7812 4.804 �0.022816.4
a 4.9539 5.590 �0.636116.5 5.0752 5.030 0.0452
16.6 4.9469 4.944 0.0019
16.7a 5.1481 4.953 0.1951
16.8a 5.1720 5.364 �0.1920
16.9 5.3645 5.387 �0.022516.10 6.0044 6.004 0.0004
16.11a 5.1314 5.210 �0.1786
16.12 5.1765 5.134 0.0425
16.13 5.3382 5.381 �0.042816.14
a 5.1002 5.350 �0.249816.15 5.1765 5.163 0.0135
16.16 4.3492 4.347 0.0022
16.17a 5.2069 5.155 0.0519
16.18a 5.2565 5.371 �0.1145
16.19 5.8861 5.893 �0.006916.20 5.1057 5.080 0.0257
16.21 5.3439 5.352 �0.008116.22 5.3449 5.313 0.0319
16.23 4.9646 4.994 �0.029416.24a 5.4123 5.236 0.1763
16.25 4.9097 4.905 0.0047
16.26 5.1427 5.154 �0.011316.27 5.5670 5.574 �0.007016.28 5.8665 5.857 0.0095
a Test set of molecules.
6 P.N. Dube et al.
could be attributed to the fact that the bulky groups do not fallinto the green region. To fall into this region, the length of the
bulky group should be about three C–C bonds away from the1,3,5-triazine ring (or C6 atom) and also another group shouldbe located at 19- position of the 5-aryl ring of triazine deriva-
tives (Fig. 4(a)).The CoMFA electrostatic interaction contour maps
(Fig. 4(b)) are represented by red and blue regions. The red
contours represent regions where electron-rich (electronega-tive) groups result in enhanced bioactivity whereas blue con-tours correspond to regions where increased positive charge(or decreased negative charge) would result in an enhanced
Figure 3 CoMFA graphs of predicted versus experimental
Please cite this article in press as: Dube PN et al. CoMFA and dockingnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletinj.bfopcu.2014.02.003
biological activity. Three small red contours near 30-Cl substi-tuent, and other two small red contours on C6-substituent atabout C–C length distance from C6 atom were observed
(Fig. 4(b)). Comparison of the activities of 16.10 vs. 16.1,16.28 vs. 16.23 demonstrated that an electronegative Cl substi-tuent at the 19-position could contribute to the observed high
activities of these compounds. Hence, higher activities of16.10, 16.19 and 16.28 could be attributed to the presence of19-Cl substituent that falls into the red region.
In this study no compound was found to possess an elec-tron-deficient substituent near the blue regions. The analysisof the CoMFA contour maps also indicated that the most ac-tive compounds have bulky and electronegative substituents or
at least one of them properly oriented, respectively, at the ste-rically and electrostatically favorable regions. Occurrence ofthe green and red contours together suggests the need of both
sterically bulky and electron rich substituents in order to in-crease inhibitory activities of triazine analogs.
2.4. Docking analysis
A docking study could offer more insight into understandingthe protein–inhibitor interactions and the structural features
of active site of protein. Beginning of the docking, it is neces-sary to validate the docking reliability. The ligand pyrimeth-amine of the crystal structure of PfDHFR complex as atemple was adopted to perform the validation.17,26 The ligand
pyrimethamine was flexibly re-docked to the binding site ofPfDHFR (PDB ID 1J3J) and the docking conformation corre-sponding to the lowest energy score was selected as the most
probable binding conformation. As a result, the re-dockedpyrimethamine and crystal structure pyrimethamine of 1J3Jcomplex are almost at the same position in the active site
and the RMSD for all atoms of the two conformers is0.72 A, suggesting a high docking reliability of Glide XP.Therefore, the Glide docking protocol and the used parameters
could be extended to search the binding conformations toPfDHFR for other inhibitors.
The active site contains the highly conserved residuesAsp54, Ala16, Ser120, Ser122, Phe123, Asn124, Phe224,
Gly225, Leu119, Phe258, Ile310, Val311, Leu314, Phe315,Gly165, Phe319, Ser380, Ile386, Arg122, consistent with sev-eral homology modeling studies.27–29 Using Glide docking,
all compounds selected from CoMFA study were docked intothe putative binding sites of PfDHFR enzyme. The bindingscore (Gscore) and Glide XP terms of all the compounds are
depicted in Table 4. It should be noticed that the G-Scores
pIC50 values of training (A) and test set (B) molecules.
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/
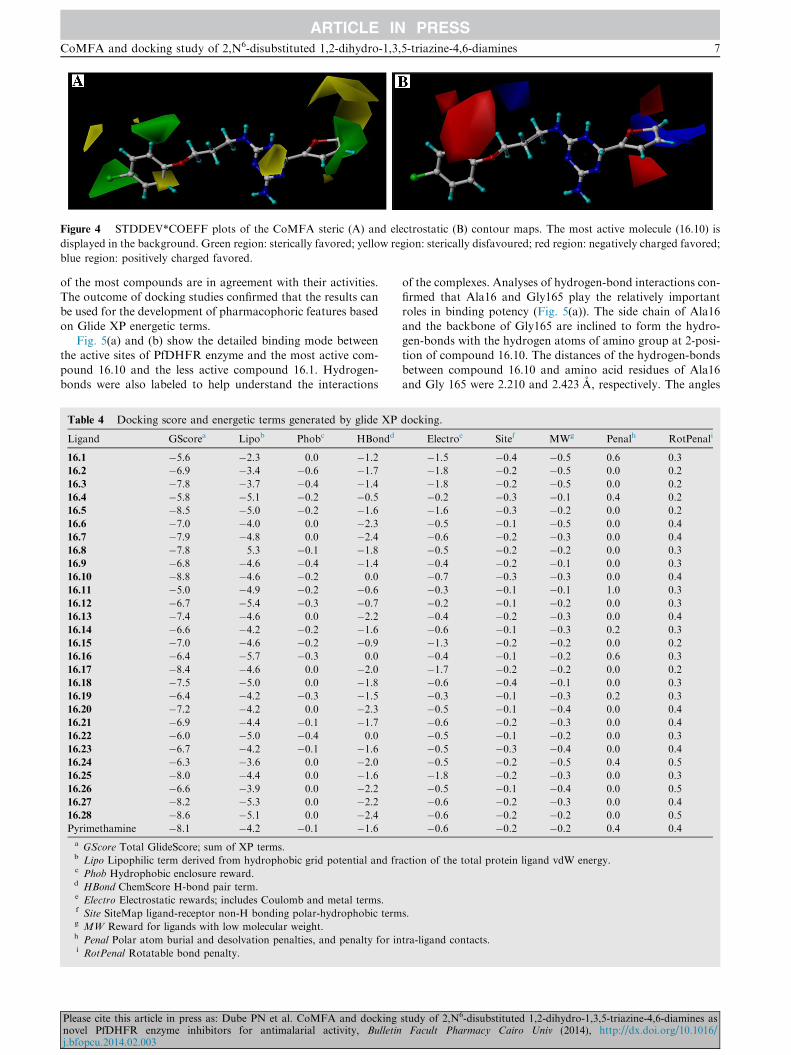
Figure 4 STDDEV*COEFF plots of the CoMFA steric (A) and electrostatic (B) contour maps. The most active molecule (16.10) is
displayed in the background. Green region: sterically favored; yellow region: sterically disfavoured; red region: negatively charged favored;
blue region: positively charged favored.
CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines 7
of the most compounds are in agreement with their activities.The outcome of docking studies confirmed that the results canbe used for the development of pharmacophoric features based
on Glide XP energetic terms.Fig. 5(a) and (b) show the detailed binding mode between
the active sites of PfDHFR enzyme and the most active com-
pound 16.10 and the less active compound 16.1. Hydrogen-bonds were also labeled to help understand the interactions
Table 4 Docking score and energetic terms generated by glide XP
Ligand GScorea Lipob Phobc HBondd
16.1 �5.6 �2.3 0.0 �1.216.2 �6.9 �3.4 �0.6 �1.716.3 �7.8 �3.7 �0.4 �1.416.4 �5.8 �5.1 �0.2 �0.516.5 �8.5 �5.0 �0.2 �1.616.6 �7.0 �4.0 0.0 �2.316.7 �7.9 �4.8 0.0 �2.416.8 �7.8 5.3 �0.1 �1.816.9 �6.8 �4.6 �0.4 �1.416.10 �8.8 �4.6 �0.2 0.0
16.11 �5.0 �4.9 �0.2 �0.616.12 �6.7 �5.4 �0.3 �0.716.13 �7.4 �4.6 0.0 �2.216.14 �6.6 �4.2 �0.2 �1.616.15 �7.0 �4.6 �0.2 �0.916.16 �6.4 �5.7 �0.3 0.0
16.17 �8.4 �4.6 0.0 �2.016.18 �7.5 �5.0 0.0 �1.816.19 �6.4 �4.2 �0.3 �1.516.20 �7.2 �4.2 0.0 �2.316.21 �6.9 �4.4 �0.1 �1.716.22 �6.0 �5.0 �0.4 0.0
16.23 �6.7 �4.2 �0.1 �1.616.24 �6.3 �3.6 0.0 �2.016.25 �8.0 �4.4 0.0 �1.616.26 �6.6 �3.9 0.0 �2.216.27 �8.2 �5.3 0.0 �2.216.28 �8.6 �5.1 0.0 �2.4Pyrimethamine �8.1 �4.2 �0.1 �1.6a GScore Total GlideScore; sum of XP terms.b Lipo Lipophilic term derived from hydrophobic grid potential and frac Phob Hydrophobic enclosure reward.d HBond ChemScore H-bond pair term.e Electro Electrostatic rewards; includes Coulomb and metal terms.f Site SiteMap ligand-receptor non-H bonding polar-hydrophobic termg MW Reward for ligands with low molecular weight.h Penal Polar atom burial and desolvation penalties, and penalty for ini RotPenal Rotatable bond penalty.
Please cite this article in press as: Dube PN et al. CoMFA and dockingnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletinj.bfopcu.2014.02.003
of the complexes. Analyses of hydrogen-bond interactions con-firmed that Ala16 and Gly165 play the relatively importantroles in binding potency (Fig. 5(a)). The side chain of Ala16
and the backbone of Gly165 are inclined to form the hydro-gen-bonds with the hydrogen atoms of amino group at 2-posi-tion of compound 16.10. The distances of the hydrogen-bonds
between compound 16.10 and amino acid residues of Ala16and Gly 165 were 2.210 and 2.423 A, respectively. The angles
docking.
Electroe Sitef MWg Penalh RotPenali
�1.5 �0.4 �0.5 0.6 0.3
�1.8 �0.2 �0.5 0.0 0.2
�1.8 �0.2 �0.5 0.0 0.2
�0.2 �0.3 �0.1 0.4 0.2
�1.6 �0.3 �0.2 0.0 0.2
�0.5 �0.1 �0.5 0.0 0.4
�0.6 �0.2 �0.3 0.0 0.4
�0.5 �0.2 �0.2 0.0 0.3
�0.4 �0.2 �0.1 0.0 0.3
�0.7 �0.3 �0.3 0.0 0.4
�0.3 �0.1 �0.1 1.0 0.3
�0.2 �0.1 �0.2 0.0 0.3
�0.4 �0.2 �0.3 0.0 0.4
�0.6 �0.1 �0.3 0.2 0.3
�1.3 �0.2 �0.2 0.0 0.2
�0.4 �0.1 �0.2 0.6 0.3
�1.7 �0.2 �0.2 0.0 0.2
�0.6 �0.4 �0.1 0.0 0.3
�0.3 �0.1 �0.3 0.2 0.3
�0.5 �0.1 �0.4 0.0 0.4
�0.6 �0.2 �0.3 0.0 0.4
�0.5 �0.1 �0.2 0.0 0.3
�0.5 �0.3 �0.4 0.0 0.4
�0.5 �0.2 �0.5 0.4 0.5
�1.8 �0.2 �0.3 0.0 0.3
�0.5 �0.1 �0.4 0.0 0.5
�0.6 �0.2 �0.3 0.0 0.4
�0.6 �0.2 �0.2 0.0 0.5
�0.6 �0.2 �0.2 0.4 0.4
ction of the total protein ligand vdW energy.
s.
tra-ligand contacts.
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/

Figure 5 Model of compound 16.10 (A) and 16.1 (B) docked into the binding site of PfDHFR. Important residues are shown as sticks.
8 P.N. Dube et al.
of the two hydrogen-bonds were 168.60� and 150.24�, respec-tively. Similarly, the side chain of Asp54 formed a hydrogen-
bond with the hydrogen atom of amino group of compound16.1, the distance and angle of the hydrogen-bond were2.835 A and 166.20�, respectively. However, we have not ob-
served the hydrogen-bond between compound 16.1 and thebackbone of Ala16 and Gly165 (Fig. 5(b)).
3. Materials and methods
3.1. Data set
A data set of 28 triazine derivatives was used for the 3D-QSAR study.30 The compounds were evaluated for their
in vitro anti-plasmodial activities of cycloguanil-resistant P.falciparum clone harboring Ala16Val + Ser108Thr mutantPfDHFR enzyme. The reported growth inhibition constant
(IC50) values were converted into corresponding pIC50 usingthe formula-
pIC50 ¼ � log IC50
pIC50 values were used for generation of the CoMFA model.The reported IC50 values are given in the original literatures.30
The selection of test and trainee sets was performed randomly.The training set contains 20 and test set contains 08 compounds.
Please cite this article in press as: Dube PN et al. CoMFA and dockingnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletinj.bfopcu.2014.02.003
3.2. Molecular modeling
All molecular modeling studies were carried out on SYBYL-X1.3 (SYBYL-X 1.3, Molecular Modeling Software, Tripose
Inc., 1699).31 Since no bound ligand or crystal structure ofAla16Val + Ser108Thr mutant PfDHFR enzyme was avail-able, the lowest energy conformer of the most active compound
(16.10) was used as a template for proposition of the bioactiveconformation. This is a common approach in ligand based 3D-QSAR studies in the absence of 3D-structural information ofthe receptor or enzyme.32–34 A preliminary minimization was
performed on 16.10 to remove close atom or bad van der Waalscontacts by using 1000 cycle minimization with standard Triposforce field. A 0.005 kcal/mol energy gradient convergence crite-
rion using Powell’s method was also used as a final step of en-ergy minimization. To obtain the lowest energy conformer,16.10 was then subjected to simulated annealing to heat the
molecule at 700 K for 1000 fs followed by annealing the mole-cule to 200 K for 1000 fs.25,35 The resulting conformer with thelowest potential energy was saved and subsequently used as atemplate. All the remaining molecules were constructed based
on this template, and subjected to 1000 cycle minimization withthe standard Tripos force field and 0.005 kcal/mol A energygradient convergence criterion using Powell’s method. All the
molecules were assigned with Gasteiger–Huckle charges.36
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/

CoMFA and docking study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines 9
3.3. CoMFA model generation
A Comparative Molecular Field Analysis (CoMFA) was car-ried out to evaluate steric and electrostatic fields using theQSAR module, and was scaled using the CoMFA standard
in SYBYL-X 1.3. All the analyses were performed by placingaligned training set molecules in a 3D grid with a spacing of2.0 A. The van der Waals potential and Coulombic termswhich represent steric and electrostatic fields, respectively,
were calculated using the standard Tripos force fields. A sp3
hybridized carbon was used as a probe atom to generate thesteric (Lennard–Jones potential) field and a charge of +1.0
to generate the electrostatic (Coulomb potential) field. The ste-ric and electrostatic contributions were set at a default cutoffenergy value of 30 kcal/mol. Several CoMFA models were
developed by permutation of molecules between training andtest sets.
3.4. Partial Least Square (PLS) analysis
PLS method was used to correlate CoMFA fields with biolog-ical activities of compounds. In this study the cross-validation(r2cv) was carried out using leave-one-out (LOO) method which
removes one compound from the data set, and its propertiespredicted from the model developed using the rest of com-pounds. In order to increase the speed of the analyses and to
improve noise-to-signal ratio, all the cross-validations wereperformed using a column filter value (d) of 2.0 kcal/mol byomitting those lattice points whose energy variation is below
this threshold value. After obtaining an optimum number ofcomponents in the cross-validation analysis step, a final non-cross validated analysis was performed using the optimal num-ber of components obtained from LOO cross-validation step.
To further assess the robustness and the statistical validity ofthe obtained models, bootstrapping analysis for 100 runs wasperformed. Finally, the model quality was evaluated in terms
of the LOO cross-validated correlation coefficient q2, the noncross-validation correlation coefficient r2, the predictive corre-lation coefficient r2pred, the standard error of estimate S value,
and the F-statistic for analysis for F value.
3.5. Predictive ability of CoMFA models
To assess the predictive abilities of the CoMFA model derivedby the training set, biological activities of an external test setcomposed of 08 compounds were predicted. The predictiveability of the model is expressed by the predictive correlation
coefficient r2pred, calculated by the equation as shown below-
r2pred ¼SD� PRESS
SD
where SD is the sum of the squared deviations between the bio-logical activities of the test set and mean activities of the train-ing set compounds; PRESS is the sum of the squared
deviations between the actual and predicted activities of thetest compounds.21
3.6. Molecular docking studies
Molecular docking studies were performed inMaestro 9.3 usingGlide v5.8 (Schrodinger, LLC, New York, NY, 2010). All
Please cite this article in press as: Dube PN et al. CoMFA and dockingnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletinj.bfopcu.2014.02.003
compounds were built usingMaestro build panel and optimizedto lower energy conformers using Ligprep v2.5 which usesOPLS_2005 force field. Epik v2.3 was used to generate ionized
state of all compounds at target pH 7.0 ± 2.0. The coordinatesfor PfDHFR enzyme (PDB ID 1J3J) were taken from RCSBProtein Data Bank and prepared for docking using ‘protein
preparation wizard’ in Maestro v9.3. Water molecules in thestructures were removed and termini were capped by addingACE and NMA residue. The bond orders and formal charges
were added for hetero groups and hydrogens were added to allatoms in the structure. Side chains that are not close to the bind-ing cavity and do not participate in salt bridges were neutralized.After preparation, the structure was refined to optimize the
hydrogen bond network using OPLS_2005 force field. Thishelps in reorientation of side chain hydroxyl group. Theminimi-zationwas terminated when the energy converged or the RMSD
reached a maximum cutoff of 0.30 A. Grids were then definedaround refined structure by centering on ligand using defaultbox size. The extra precision (XP) docking mode for all com-
pounds was performed on generated grid of protein structure.37
The final evaluation of ligand–protein binding was donewith Glide score (docking score).GScore = a · vdW+ b ·Coul + Lipo+Hbond+Metal + BuryP+RotB+ Sitewhere,vdW: – Van der Waal energy; Coul: – Coulomb energy; Lipo: –Lipophilic contact term; HBond: – Hydrogen-bonding term;Metal: – Metal-binding term; BuryP: – Penalty for buried polar
groups; RotB: – Penalty for freezing rotatable bonds; Site: –Polar interactions at the active site; and the Coefficients ofvdW and Coul are: a = 0.065, b = 0.130.
4. Conclusions
The main objective of this study was to develop statistically
robust CoMFA models with good correlation and predictivepowers using anti-plasmodial activity data of triazinederivatives. The CoMFA model was found to give better
result as demonstrated by its statistical indices (r2cv = 0.656,r2ncv = 0.996, r2bs = 0.999 and r2pred = 0.740). The constructed3D-QSAR models and structure–activity relationship (SAR)
analyses of the compounds used in the study suggested thatan electronegative and bulky substituent at 30-position andone bulky substituent at C6-position of triazine analogsare required to design novel inhibitors of Ala16 Val +
Ser108Thr mutant PfDHFR enzyme. The docking studiesprovide useful information for understanding the hydrogen-bonding interactions between the inhibitors and the active
site residues of the Ala16 Val + Ser108Thr mutant PfDHFRenzyme. The CoMFA model and the docking studies mayafford the guidance for designing new potential DHFR
inhibitors.
Conflict of interest
The author(s) confirm that this article content has no conflictsof interest.
Acknowledgment
All authors thank to Dr. K.G. Bothara, Sinhgad Institute of
Pharmacy, Pune, India for their valuable suggestions on revis-ing and improving the work.
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/

10 P.N. Dube et al.
References
1. Nwaka S, Hudson A. Innovative lead discovery strategies for
tropical diseases. Nat Rev Drug Discov 2006;5:941–55.
2. Philips RS. Current status of malaria and potential control. Clin
Microbiol Rev 2001;14:208–26.
3. Baird JK. Effectiveness of antimalarial drugs. New Engl J Med
2005;352:1565–77.
4. Winstaly P, Ward S, Snow R, Breckenridge A. Therapy of
falciparum malaria in Sub-Saharan Africa: from molecule to
policy. Clin Microbiol Rev 2004;17:612–37.
5. Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S.
Antimalarial drug discovery; efficacy model for compound
screening. Nat Rev Drug Discov 2004;3:509–20.
6. Ridley RG. Medical need, scientific opportunity and the drive for
antimalarial drugs. Nature 2002;415:686–93.
7. Talisuna OA, Bloland P, D’Alessandro U. History, dynamics and
public health importance malaria parasite. Clin Microbiol Rev
2004;17:235–54.
8. Sharma YD. Genetic alteration in drug resistance markers of P.
falciparum. Ind J Med Res 2005;121:13–22.
9. Braun PD, Barglow KT, Lin YM, Akompong T, Briesewitz R,
Ray GT, Haldar K, Wandless TJ. A bifunctional molecule that
displays context-dependent cellular activity. J Am Chem Soc
2003;125:7575–80.
10. Anderson AC. Targeting DHFR in parasitic protozoa. Drug
Discov Today 2005;10:121–8.
11. Da Cuhan EF, Ramlho TC, Maia ER, de Alenncastro RB. The
search for new DHFR inhibitors: a review of patents. January
2001–February 2005. Expert Opin Ther Patents 2005;15:1–20.
12. Kompis IM, Islam K, Then RL. DNA and RNA synthesis:
antifolates. Chem Rev 2006;105:593–620.
13. Warhurst DC. Resistance to antifolates in P. falciparum, the
causative agent of tropical malaria. Sci Prog 2002;85:89–111.
14. Peterson DS, Milhous WK, Wellems TE. Molecular basis of
differential resistance to cycloguanil and pyrimethamine in P.
falciparum malaria. Proc Natl Acad Sci USA 1990;87:3018–22.
15. Sirawaraporn W, Sathitkul T, Sirawaraporn R, Yuthavong Y,
Santi DV. Antifolate-resistant mutants of P. falciparum dihydro-
folate reductase. Proc Natl Acad Sci USA 1997;94:1124–9.
16. Rastelli G, Sirawaraporn W, Sompornpisut P, Vilaivan T,
Kamchonwongpaisan S, Quarrell R, Lowe G, Thebtaranonth Y,
Yuthavong Y. Interaction of pyrimethamine, cycloguanil,
WR99210 and their analogues with PfDHFR: structural basis of
antifolate. Bioorg Med Chem 2000;8:1117–28.
17. Yuthavong Y, Vilainvan T, Chareonsethakul N, Kam-
chonwongpaisan S, Sirawaraporn W, Quarrell R, Lowe G.
Development of a lead inhibitor for the A16V + S108T mutant
of DHFR from cycloguanil-resistant strain (T9/94) of P. falcipa-
rum. J Med Chem 2000;43:2738–44.
18. Rastelli G, Pacchioni S, Sirawaraporn W, Sirawaraporn R,
Parenti MD, Ferrari AM. Docking and database screening reveal
new classes of P. falciparum dihydrofolate reductase inhibitors. J
Med Chem 2003;46:2834–45.
19. Portela C, Afonso CM, Pinto MM, Ramos MJ. Computational
studies of new potential antimalarial compounds––stereoelectronic
complementarity with the receptor. J Comput-Aided Mol Des
2003;17:583–95.
20. Parenti MD, Pacchioni S, Ferrari AM, Rastelli G. Three-dimen-
sional quantitative structure–activity relationship analysis of a set
of P. falciparum dihydrofolate reductase inhibitors using a phar-
macophore generation approach. J Med Chem 2004;47:4258–67.
Please cite this article in press as: Dube PN et al. CoMFA and dockingnovel PfDHFR enzyme inhibitors for antimalarial activity, Bulletinj.bfopcu.2014.02.003
21. Cramer RD, Patterson DE, Bunce JD. Effect of shape on binding
of steroids to carrier proteins. J Am Chem Soc 1988;110:
5959–67.
22. Klebe G, Abraham U, Mietzner T. Molecular similarity indices in
a comparative analysis (COMSIA) of drug molecules to correlate
and predict their biological activity. J Med Chem 1994;37:
4130–46.
23. Salo OMH, Savinainen JR, Parkkari T, Nevalainen T, Kakkonen
J, Gynther J, Laitinen JT, Jarvinen T, Poso A. 3D-QSAR studies
on cannabinoid CB1 receptor agonists: G-protein activation as
biological data. J Med Chem 2006;49:554–66.
24. Barakat MT, Dean PM. Molecular structure matching by simu-
lated annealing. I. A comparison between different cooling
schedules. J Comput-Aided Mol Des 1990;4:295–316.
25. Cramer RD, Bunce JD, Patterson DE, Frank IE. Cross-valida-
tion, bootstrapping, and partial least squares compared with
multiple regressions in conventional QSAR studies. Quant Struct-
Act Relat 1988;7:18–25.
26. Delfino RT, Santos-Filho OA, Figueroa-Villar JD. Molecular
modeling of wildtype and antifolate resistant mutant P. falciparum
DHFR. Biophys Chem 2002;98:287–300.
27. Santos-Filho OA, Mishra RK, Hopfinger AJ. Free energy force
field (FEFF) 3D-QSAR analysis of a set of P. falciparum
dihydrofolate reductase inhibitors. J Comput-Aided Mol Des
2001;15:787–810.
28. Yuvaniyama AJ, Chitnumsub P, Kamchonwongpaisan S, Vanic-
hatanankul J, Sirawaraporn W, Taylor P, Walkinshaw MD,
Yuthavong Y. Insights into antifolate resistance from malarial
DHFR-TS structures. Nat Struct Biol 2003;10:357–65.
29. Franc TCC, de Medeiros ALR, dos Santos ECP, Santos-Filho
JDA, Figueroa-Villar JDA. A Complete homology model of the
P. falciparum bifunctional enzyme dihydrofolate reductase-thymi-
dylate synthase. A model to design new antimalarials. J Braz
Chem Soc 2004;15:450–4.
30. Gravestock D, Rousseau AL, Lourens ACU, Moleele SS, van Zyl
PA, Steenkamp PA. Expeditious synthesis and biological evalu-
ation of novel 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-
diamines as potential antimalarials. Eur J Med Chem
2011;46:2022–30.
31. SYBYL-X 1.3. Molecular Modeling Software, Tripose Inc., South
Hanley Road, St. Louis, MO 631444, USA, 1699.
32. Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-
QSAR studies of indole derivatives as phosphodiesterase IV
inhibitors. Eur J Med Chem 2003;38:975–82.
33. Aboye TL, Sobhia ME, Bharatam PV. 3D-QSAR studies of
pyruvate dehydrogenase kinase inhibitors based on a divide and
conquer strategy. Bioorg Med Chem 2004;12:2709–15.
34. Avila CM, Romeiro NC, da Silva GM, Sant’Anna CM, Barreiro
CA, Fraga CA. Development of new CoMFA and CoMSIA 3D-
QSAR models for anti-inflammatory phthalimide-containing
TNFalpha modulators. Bioorg Med Chem 2006;14:6874–85.
35. Sippl W. Development of biologically active compounds by
combining 3D-QSAR and structure-based design methods. J
Comput-Aided Mol Des 2002;16:825–30.
36. Murthy VS, Kulkarni VM. 3D-QSAR CoMFA and CoMSIA on
protein tyrosine phosphatase 1B inhibitors. Bioorg Med Chem
2002;10:2267–82.
37. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood
TA, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision
glide: docking and scoring incorporating a model of hydrophobic
enclosure for protein-ligand complexes. J Med Chem
2006;49:6177–96.
study of 2,N6-disubstituted 1,2-dihydro-1,3,5-triazine-4,6-diamines asFacult Pharmacy Cairo Univ (2014), http://dx.doi.org/10.1016/















![Structural requirements of pyrido[2,3-d]pyrimidin-7-one as CDK4/D inhibitors: 2D autocorrelation, CoMFA and CoMSIA analyses](https://static.fdokumen.com/doc/165x107/6333b4b7ce61be0ae50ec7c1/structural-requirements-of-pyrido23-dpyrimidin-7-one-as-cdk4d-inhibitors-2d.jpg)




