
Resistance to thromboembolism in PI3Kg-deficient mice
20
The FASEB Journal express article 10.1096/fj.00-0810fje. Published online July 9, 2001. Resistance to thromboembolism in PI3Kγ-deficient mice Emilio Hirsch*, Ornella Bosco † , Philippe Tropel*, Muriel Laffargue ‡ , Ronan Calvez ‡ , Fiorella Altruda*, Matthias P. Wymann ‡§ , Giuseppe Montrucchio †§ *Dipartimento di Genetica, Biologia e Biochimica, Università di Torino, 10126 Torino, Italy; † Dipartimento di Fisiopatologia Clinica, Università di Torino, 10126 Torino, Italy; and ‡ Institute of Biochemistry, University of Fribourg, CH-1700 Fribourg, Switzerland § Authors contributed equally to this work. Corresponding author: Emilio Hirsch, Dipartimento di Genetica, Biologia e Biochimica, Università di Torino, Via Santena 5 bis, 10126 Torino, Italy. E-mail: [email protected] ABSTRACT Platelet aggregation and subsequent thrombosis are the major cause of ischemic diseases such as heart attack and stroke. ADP, acting via G protein-coupled receptors (GPCRs), is an important signal in thrombus formation and involves activation of phosphoinositide 3-kinases (PI3K). When platelets from mice lacking the G protein-activated PI3Kγ isoform were stimulated with ADP, aggregation was impaired. Collagen or thrombin, however, evoked a normal response. ADP stimulation of PI3Kγ-deficient platelets resulted in decreased PKB/Akt phosphorylation and α IIb β 3 fibrinogen receptor activation. These effects did not influence bleeding time but protected PI3Kγ-null mice from death caused by ADP-induced platelet-dependent thromboembolic vascular occlusion. This result demonstrates an unsuspected, well-defined role for PI3Kγ downstream of ADP and suggests that pharmacological targeting of PI3Kγ has a potential use as antithrombotic therapy. Key words: platelet aggregation • thrombosis • G protein-coupled receptors latelet activation after contact with a subendothelial matrix induces adhesion to the exposed surface, aggregation, and granule release, culminating in the formation of a hemostatic plug. This process is tightly controlled by soluble agonists such as thrombin, thromboxane A 2 , and ADP released from damaged endothelial and red blood cells (1). These events ensure normal hemostasis, but aberrant platelet activation also contributes to the initiation of numerous ischemic diseases such as disseminated thromboembolic vascular occlusion, unstable angina, myocardial infarction, and stroke. Platelet activation involves cytoskeletal rearrangements and granule secretion and converts the fibrinogen receptor—the integrin α IIb β 3 —from a low to a high affinity binding state. In these processes, important roles are played by phosphoinositide 3-kinases (PI3Ks). In fact, P
Transcript of Resistance to thromboembolism in PI3Kg-deficient mice

The FASEB Journal express article 10.1096/fj.00-0810fje. Published online July 9, 2001.
Resistance to thromboembolism in PI3Kγ-deficient mice
Emilio Hirsch*, Ornella Bosco†, Philippe Tropel*, Muriel Laffargue‡, Ronan Calvez‡, Fiorella Altruda*, Matthias P. Wymann‡§, Giuseppe Montrucchio†§ *Dipartimento di Genetica, Biologia e Biochimica, Università di Torino, 10126 Torino, Italy; †Dipartimento di Fisiopatologia Clinica, Università di Torino, 10126 Torino, Italy; and ‡Institute of Biochemistry, University of Fribourg, CH-1700 Fribourg, Switzerland § Authors contributed equally to this work. Corresponding author: Emilio Hirsch, Dipartimento di Genetica, Biologia e Biochimica, Università di Torino, Via Santena 5 bis, 10126 Torino, Italy. E-mail: [email protected] ABSTRACT Platelet aggregation and subsequent thrombosis are the major cause of ischemic diseases such as heart attack and stroke. ADP, acting via G protein-coupled receptors (GPCRs), is an important signal in thrombus formation and involves activation of phosphoinositide 3-kinases (PI3K). When platelets from mice lacking the G protein-activated PI3Kγ isoform were stimulated with ADP, aggregation was impaired. Collagen or thrombin, however, evoked a normal response. ADP stimulation of PI3Kγ-deficient platelets resulted in decreased PKB/Akt phosphorylation and αIIbβ3 fibrinogen receptor activation. These effects did not influence bleeding time but protected PI3Kγ-null mice from death caused by ADP-induced platelet-dependent thromboembolic vascular occlusion. This result demonstrates an unsuspected, well-defined role for PI3Kγ downstream of ADP and suggests that pharmacological targeting of PI3Kγ has a potential use as antithrombotic therapy. Key words: platelet aggregation • thrombosis • G protein-coupled receptors
latelet activation after contact with a subendothelial matrix induces adhesion to the exposed surface, aggregation, and granule release, culminating in the formation of a hemostatic plug. This process is tightly controlled by soluble agonists such as thrombin,
thromboxane A2, and ADP released from damaged endothelial and red blood cells (1). These events ensure normal hemostasis, but aberrant platelet activation also contributes to the initiation of numerous ischemic diseases such as disseminated thromboembolic vascular occlusion, unstable angina, myocardial infarction, and stroke. Platelet activation involves cytoskeletal rearrangements and granule secretion and converts the fibrinogen receptor—the integrin αIIbβ3—from a low to a high affinity binding state. In these processes, important roles are played by phosphoinositide 3-kinases (PI3Ks). In fact,
P

wortmannin, a PI3K inhibitor (2), impairs thrombin and phorbol miristate acetate (PMA)-stimulated aggregation (3, 4). The products of PI3Ks, PtdIns(3,4,5)P3, and PtdIns(3,4)P2 serve as docking sites for pleckstrin homology (PH) domain-containing proteins (e.g., PKB/Akt, the Tec family protein kinases, and guanine nucleotide exchange factors [5, 6]). It has been suggested that cytoskeletal rearrangements, membrane fusion events, and activation of αIIbβ3 integrins are downstream of these signaling molecules in platelets (7, 8). Platelets express three of the four cell surface receptor-activated “class I” PI3K isoforms, namely PI3Kα, PI3Kβ, and PI3Kγ (7), but not PI3Kδ (as shown in our work). Class IA PI3Ks α, β, and δ are translocated via the two SH2 domains of their p85 adapter protein to phosphorylated Tyr-X-X-Met motives. PI3Kγ, however, is activated by Gβγ subunits of heterotrimeric G proteins in vitro, and thus it has been suggested that it relays signals from G protein-coupled receptors (GPCRs) (9–11). Many soluble platelet stimuli, including ADP, thromboxane A2, and thrombin, exert their physiological actions through GPCRs (12). ADP, which is locally secreted by platelets or released by damaged cells, plays an important role in reinforcing the effects of other agonists (13). Stimulation of mouse platelets with ADP alone triggers a reversible aggregation profile in vitro, but is not sufficient to induce release of dense α-granules (14). ADP causes a rapid rise in cytosolic calcium and inhibition of adenylate-cyclase (15). These processes are governed by at least two distinct ADP receptors that are members of the P2 class of nucleotide and nucleoside receptors. These molecules (P2Y1 and P2Y12) are GPCRs coupled to mobilization of calcium and inhibition of adenylyl cyclase, respectively (12, 16, 17). P2Y1 has been cloned and inactivated in mice by gene targeting. The lack of P2Y1 receptor impairs platelet aggregation in response to ADP but does not affect ADP-dependent inhibition of cAMP increase (18, 19). P2Y12, the other G protein-coupled ADP receptor (also known as P2Tac), inhibits cAMP synthesis and is probably linked to a member of the Gi family (12, 17). Coupling of class P2 ADP receptors to produce PtdIns(3,4,5)P3 and PtdIns(3,4)P2 is controversial. Although thrombin and thromboxane A2-dependent 3-phosphatidyl inositol (PPI) generation has been clearly established (7, 20), some reports indicate that, in human platelets, ADP alone induces negligible (21) or small amounts (8) of PtdIns(3,4)P2. It has been demonstrated, however, that ADP-dependent PI3K activity can be readily detected in stimulated adherent platelets (22) and is essential for spreading (23) and sustaining thrombin-induced aggregation (24, 25). In this latter process, ADP-dependent PI3K activation appears to occur late in the process and was correlated to cytoskeleton reorganization (4, 24). Thus far, the importance of PI3K signaling for thrombocyte function has not been demonstrated in vivo, and there is no consensus on which PI3K isoforms are activated by GPCR ligands. Using PI3Kγ-deficient mice (26), we show here for the first time that PI3Kγ mediates platelet responses and thus protects animals from platelet-dependent vascular thromboembolism. MATERIALS AND METHODS Animals

PI3Kγ-deficient mice were generated as previously described (26). Mice used for experiments were 8-to 12-week-old wild-type, PI3Kγ-/- of either sex; and on 129sv background mice of different genotypes were age-matched or litter mates. Animal experiments were carried out in accordance with institutional guidelines as well as with Italian and Swiss government regulations. Histology For in situ detection of LacZ activity, spleens from PI3Kγ-/- mice were dissected, fixed in 4% paraformaldehyde, and snap-frozen in liquid nitrogen. Cryostat sections (12 µm thick) were incubated overnight at 37°C in X-Gal solution: 1 mg/ml X-Gal (5-bromo-4-chloro-3-indoyl-β-D-galactopyranoside, Sigma Chemical Co., St. Louis, MO), 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, 0.001% Deoxycholate (Sigma), 0.01% NP-40 (Sigma). Preparation of platelets for Western blot and aggregation analysis Male and female mice (20–30 g) were anesthetized by intraperitoneal injection of 20 µl/g of Avertin (1.2% tribromoethanol/amylalcohol in 0.9% saline solution). Whole blood was collected from the inferior vena cava into heparinized syringes at a final concentration of 5 U/ml of heparin. For each experiment, platelets from blood were pooled from five or six mice of each genotype. Platelet-rich plasma (PRP) was prepared by centrifuging whole blood at 90 g for 6 min. After the upper layer was collected, platelet-poor plasma (PPP) was obtained by centrifugation of the remaining blood at 1500 g for 6 min. PRP was adjusted to 2 × 105 platelets/µl using PPP as diluent. To obtain washed platelets (WPs), 700 µl of blood was drawn into a syringe containing 200 µl acid citrate dextrose (ACD) (85 mM trisodium citrate, 75 mM citric acid, 110 mM D-glucose), 500 µl saline, and 50 ng/ml prostaglandine E1 (PGE1). The mixture from 3–4 mice for each genotype was pooled into 15-ml tubes and centrifuged at 90 g for 10 min to obtain the PRP. For maximal platelet recovery, the red blood cell pellet was diluted with 1–2 ml of citrate glucose saline (CGS) (0.038% trisodium citrate, 0.6% D-glucose, 0.72% NaCl, pH 7.0) containing PGE1 at 25 ng/ml and centrifuged as above. Supernatant was collected, pooled with PRP, made up to a volume of 15 ml with CGS, and centrifuged at 1500 g for 6 min. The pelleted platelets were resuspended in Ca2+ – Mg2+ free Tyrode’s buffer, counted, diluted to 2 × 105 platelets/µl in the same buffer, and incubated on ice for 30 min before use. CaCl2 (2 mM) and MgCl2 (1 mM) were added before aggregation assays.
Western blot analysis Washed platelets were resuspended in 150 mM NaCl, 5 mM KCl, 5 mM KH2PO4, 10 mM HEPES pH 7.4 supplemented with 1 mM MgCl2 and 1 mM CaCl2 before stimulation at a concentration of 5 × 105 cells/µl. Cells were subsequently stimulated with the indicated agonists, centrifuged, and subjected to standard denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (7.5%) followed by electrotransfer to PVDF membranes. PI3K

isoforms were detected with monoclonal antibodies to PI3Kα and PI3Kγ (kindly obtained from A. Klippel, Atugen, Germany, and R. Wetzker, University of Jena, Germany, respectively) and rabbit polyclonal antibodies to PI3Kβ and δ (donated by B. Vanhaesebroek, Ludwig Institute, London, U.K.). All the above antibodies were previously shown to detect their targets in granulocytes (26). PKB/Akt was detected with a polyclonal antibody kindly provided by B. Hemmings Friedrich Miescher Institut, Basel, Switzerland, and PKB/Akt phosphorylated on Ser473 was revealed with antibodies from New England Biolabs (Beverly, MA), as suggested by the manufacturer. Antibodies against αIIb fibrinogen receptor and focal adhesion kinase were kindly provided by R. Berthier, CEA, Grenoble, France, and G. Tarone, University of Torino, Italy, respectively. Measurement of platelet aggregation Platelet aggregation was measured in a lumi-aggregometer (Chronolog, Haverton, PA). Platelet aggregation in whole blood was assessed by recording the increase in electrical impedance in 1 ml sample of blood diluted onefold with Tyrode’s buffer containing 2 U/ml of heparin and stirred at 1000 rpm at 37°C. Aggregation response in whole blood was induced with ADP (0.2–10 µM) and PMA (100 nM). Aggregation was quantified as maximum amplitude (ohms). For measuring platelet aggregation in PRP or WP, 300 µl of platelet suspension was added to the aggregometer chamber and aggregation was optically monitored after stimulation with ADP (1–10 µM), THR (0.05–1 U/ml), collagen (1–10 µg/ml), and PMA (100 nM). Fibrinogen (200 µg/ml final) was added prior to ADP stimulation of WP. To determine the aggregation response, light transmission was continually compared to a reference consisting of PPP or Tyrode’s buffer alone. To compare the different responses, maximal aggregation was quantified by Weiss’s formula (OD0 – ODm) × 100/ OD0, where OD0 is the initial light transmission value and ODm is the minimum light transmittance. Measurement of ATP, cAMP, and Ca2+ release ATP release was measured by standard methods, using the firefly luciferase (7.3 µg/ml final) and luciferin (800 U/ml final). Luminescence created by platelet-secreted ATP was monitored using the lumi-aggregometer, and measures were compared with the data obtained by adding an ATP standard. For cAMP measurements, washed platelets (8 × 104/µl) were stimulated at 30-s intervals with either Tyrode’s buffer without Ca2+ or Mg2+, or agonists in a final volume of 250 µl. The reaction was stopped by centrifugation (10,000 g, 10 s) and substitution of the supernatant with 50 µl ethanol. Intracellular cAMP was determined using a commercial kit (TRK 432, Amersham, Piscataway, NJ). Intracellular calcium fluxes were measured as described (19). Briefly, washed platelets were loaded with fura-2/AM and monitored by a spectrofluorometer using excitation wavelengths of 340 and 38 nm. Fluorescence emission was detected at 510 nm. Assessment of platelet fibrinogen binding in vitro

Whole blood (500 µl) was collected as described above in a final concentration of 25 U/ml heparin. Diluted PRP was obtained by centrifugation (85 g, 5 min) of blood samples after adding a half-volume of modified Tyrode’s buffer. An aliquot of diluted PRP (5 µl) was mixed with modified Tyrode’s buffer containing 1.25 mM MgCl2 and with Oregon green-labeled fibrinogen (150 µg/ml final; Molecular Probes, Eugene, OR) to reach a volume of 90 µl. After incubation for 3 min at room temperature, platelets were stimulated with 10 µl ADP (1 µM final; Sigma) or collagen (1 µg/ml final; Helena Labs, Beaumont, TX), for 1 and 10 min, respectively. Platelets were then fixed in 1% paraformaldehyde in PBS for 30 min. After two washes, labeled platelets were analyzed by flow cytometry (Becton-Dickinson, Franklin Lakes, NJ), using a gated exclusion of aggregates. Assessment of bleeding time Male and female mice (20–30 g) were anesthetized with Avertin. A 2–3-mm fragment of the tail tip was amputated with a sharp scalpel blade, and the tail was immediately immersed in 0.9% isotonic saline solution at 37°C. Bleeding time was recorded as time for the blood flow to cease for at least 1 min. Where necessary, bleeding was manually stopped at a 10-min time point. In vivo thrombosis models Acute pulmonary thromboembolism was induced according to Nordöy and Chandler (27). The jugular vein of anesthetized PI3Kγ-null and wild-type mice was surgically exposed and connected to a plastic catheter. ADP at 300 mg/kg of body weight was injected within 60 s. The mortality of mice in each group was determined 30 min after ADP infusion. Radiolabeled platelets Washed platelets were prepared as described above. Approximately 35 µCi of 111In-oxine (Amersham) was added dropwise to 10 ml of the platelet suspension, which was then incubated at room temperature for 10 min. Subsequently, the platelet suspension was centrifuged at 1400 g for 10 min and the platelet pellet was resuspended in sterile saline. The functionality of 111In-labeled platelets was assessed using the aggregometer and 1-mM ADP as a stimulus. PI3Kγ-null and wild-type 111In-labeled platelet suspension (2.107 thrombocytes in 200 µl) was injected in the inferior vena cava of mice of the corresponding genotype. After 1 min, a 300-mg/kg concentration of ADP was injected within 60 s through the jugular vein. Mice were killed after 1 min, and samples of lungs, blood, and muscle were obtained. Tissue samples were then weighed and placed in a gamma spectrometer (Hewlett-Packard model 5400) to determine the amount of incorporated 111In radioactivity. RESULTS PI3Kγ-null platelets show impaired aggregation after stimulation with the GPCR agonist ADP

PI3Kγ is known to be expressed in all leukocyte populations and in human platelets (7). PI3Kγ-null mice (26) were used to establish the expression pattern of PI3Kγ in the murine megakaryocytic lineage. In these mice, a LacZ reporter gene was inserted in the PI3Kγ first coding exon, thus allowing the detection of PI3Kγ transcription driven by the endogenous gene-controlling regions. As shown in Figure 1A, reporter gene activity was detected in splenic megakaryocytes and appeared stronger in these cells than in other splenocytes. According to this expression pattern, PI3Kγ protein was detectable in mature murine thrombocytes (Fig. 1B). In PI3Kγ-null mice, platelets lacked immunoreactive PI3Kγ; but expression of other class IA PI3Ks, such as p110α and β, was not affected (Fig. 1B). In addition, the PI3Kδ isoform, which is absent in wild-type murine platelets, was not up-regulated in PI3Kγ-null thrombocytes (Fig. 1B). Mice lacking PI3Kγ did not show altered numbers of circulating platelets (26), which indicates that PI3Kγ does not play a major role in thrombopoiesis. Nevertheless, accumulation of PI3Kγ might be necessary in mature thrombocytes to respond to aggregating stimuli. The role of PI3Kγ in relaying signals from natural GPCR agonists such as thrombin and ADP is still poorly understood. Given the multiple functions of thrombin, which cleaves and activates multiple receptors, we first analyzed the role of PI3Kγ in ADP-induced aggregation. Analysis of the aggregation response in whole blood revealed that ADP at low doses (1 and 0.5 µM) stimulated a reversible aggregation in both genotypes. However, in PI3Kγ-null mice, maximal aggregation was reduced: After stimulation with 0.5 µM ADP, maximal aggregation of PI3Kγ-null platelets in whole blood was significantly lower (54%) than that of wild-type whole blood (Fig. 2A and B). On the contrary, PMA, a stimulus that is known to bypass the need of PI3Kγ (8), did not induce significantly different responses in wild-type and PI3Kγ-null whole blood (Fig. 2B). To confirm that the defect in ADP-induced aggregation was due to an abnormal platelet response, we evaluated aggregation in PRP and in WPs. Determination of ADP-dependent aggregation in PI3Kγ-null PRP indicated a 30–40% reduction in the response at concentrations ranging from 0.6 to 10 µM (Fig. 2C). In addition, the platelet shape change did not appear to be disturbed, but the kinetics of the ADP response were altered: PI3Kγ-null platelets (in PRP) aggregated and disaggregated more slowly than their wild-type counterparts. Analysis of multiple pools of animals indicated that in PI3Kγ-null PRP, maximal aggregation determined by an intermediate dose of ADP (3 µM) was significantly lower (40%; P<0.05) than that in wild-type PRP (Fig. 2D). This defect could be detected independently of the presence of plasma: Washed PI3Kγ-null platelets showed a 34% reduction (P<0.05) even at relatively high ADP doses (10 µM). Similar to what we observed in whole-blood aggregation tests, PMA (100 nM) elicited equal responses in the two genotypes (Fig. 2D). We further examined the specificity of the PI3Kγ-null platelet defect by testing the ability of mutant platelets to respond to other physiological agonists that act dependently or independently of GPCRs. Collagen is known to bind different receptors of differing natures (the glycoprotein VI and the integrin α2β1) and to possibly activate tyrosine kinase-dependent PI3Ks (28). Therefore, stimulation with collagen at 1–5 µg/ml resulted in equal maximal aggregation in both genotypes (Fig. 2E). Similarly, stimulation with the GPCR agonist thrombin elicited comparable responses in PI3Kγ-null and wild-type washed platelets, even at subthreshold doses (0.04 U/ml;

Fig. 2F). Analysis of secreted ATP after stimulation with both collagen (2 µg/ml; wild-type: 1.83±0.84 nmol, PI3Kγ-null: 1.20±0.12 nmol; mean±SD; n=5) and thrombin (0.1 U/ml; wild-type: 3.46±0.64 nmol, PI3Kγ-null: 4.05±0.93 nmol; mean±SD; n=5) did not reveal significant differences between the two genotypes. Taken together, these results clearly demonstrate that PI3Kγ is dispensable for signal transduction triggered by collagen and thrombin and that the enzyme plays a specific role in ADP signaling. To demonstrate that this pathway is truly independent of other signals known to influence platelet aggregation, we studied intracellular Ca2+ fluxes and cAMP production after wild-type and mutant platelets were stimulated with ADP. As shown in Figure 3A, ADP stimulation causes an identical increase in intracellular Ca2+ concentration in platelets of both genotypes. Similarly, ADP was equally able to inhibit PGE1-stimulated accumulation of cAMP in wild-type and mutant platelets (Fig. 3B). These observations prove that the lack of PI3Kγ does not interfere with P2Y1-induced Ca2+ fluxes and P2Y12-dependent adenylyl cyclase inhibition.
PKB phosphorylation and fibrinogen binding is decreased in ADP-stimulated PI3Kγ-null platelets To understand the biochemical mechanism leading to impaired aggregation in ADP-stimulated PI3Kγ-null platelets, we checked whether activation of possible downstream targets of PI3K was defective. The serine/threonine protein kinase PKB/Akt is present in platelets and is known to be activated in response to 3-PPI production (29). It was interesting that ADP stimulation raised the level of phosphorylated PKB in wild-type platelets as soon as 2 min after exposure to the agonist. In contrast, PI3Kγ-null platelets showed a significant reduction of activated PKB (70%; P<0.05) under the same conditions (Fig. 4A and B). In agreement with aggregation studies, stimulation with thrombin resulted in PKB activation in platelets of both genotypes (Fig. 4A and B). These findings further confirm the crucial involvement of PI3Kγ in ADP but not in thrombin-triggered signal transduction and suggest that the ADP-induced aggregation defect in PI3Kγ-null platelets might depend on impaired PKB activation. Next, we studied the nature of the ADP receptors involved in PI3Kγ-dependent PKB phosphorylation using wild-type platelets and specific inhibitors that block either the P2Y1 or the P2Y12 receptor. Adding the ATP analogue AR-C69931MX, which specifically blocks the Gi-coupled P2Y12 receptor (19), completely inhibited ADP but not thrombin-induced PKB phosphorylation (Fig. 4C). Conversely, the presence of the AMP analogue A3P5P (17), which blocks only the Gq-coupled P2Y1 receptor, did not significantly alter the ADP-induced PKB activation (Fig. 4D). These results demonstrate that the Gi-coupled P2Y12 receptor is specifically involved in triggering the PI3Kγ-dependent signaling pathway in platelets. After platelet activation by agonists, the aggregation process is mediated by a conformational switch of the αIIbβ3 integrin to its high-affinity binding state for fibrinogen (30). The precise molecular mechanism involved in this process (termed inside-out signaling) is still poorly understood, but it was found that wortmannin can block the αIIbβ3 conformational switch (8). We therefore tested αIIbβ3-mediated increase of fibrinogen binding in wild-type and PI3Kγ-null platelets. After stimulation with 1 µM ADP, wild-type platelets showed a 10-fold increase in

fluorescent-labeled fibrinogen binding (Fig. 5A and B). In contrast, in platelets lacking PI3Kγ, the response to ADP was reduced by ~30% (P<0.05) at two distinct doses (Fig. 5B). This effect was specifically dependent on ADP signaling, because platelets of both genotypes could bind equally well to labeled fibrinogen after collagen stimulation (Fig. 5C). In agreement with a specific signaling defect, possible differences in expression of the αIIb fibrinogen receptor subunit between wild-type and mutant platelets were ruled out by Western blot analysis, which displayed identical αIIb total expression levels in the two genotypes (Fig. 5D). Reduced thrombosis in PI3Kγ-null mice with normal bleeding time To test whether the lack of PI3Kγ could impair primary hemostasis in vivo, we determined the bleeding times of wild-type and PI3Kγ-deficient mice after amputation of the tail tip. The time required for the hemorrhage to cease was equal in wild-type and mutant mice (319±37 vs. 300±37 s; mean±SE, n=37 each, P =0.8, two-tailed Student's t test). During thrombotic plug formation, intravascular platelet aggregation plays an essential role. To determine the in vivo impact of the defective ADP-induced aggregation response in PI3Kγ-null platelets, wild-type and mutant mice were challenged with an ADP-dependent acute thrombotic process (27). After wild-type mice were exposed to ADP (300 mg/kg), 11 out of 24 animals died from respiratory arrest caused by occlusion of the pulmonary microcirculation (Fig. 6A). In contrast, when the same doses of agonist were used, 20 out of 24 PI3Kγ-null mice survived after 30 min. To confirm that the decrease in mortality was due to a platelet aggregation defect, we measured in the lungs of mice of both genotypes the accumulation of radioactively labeled platelets after ADP-induced thromboembolism. The jugular vein of wild-type and mutant animals was catheterized, and the abdominal vena cava was surgically exposed. Before perfusion of ADP, 111In-labeled platelets derived from animals of the corresponding genotype were injected in the inferior vena cava. One minute after ADP exposure, lungs were excised and platelet accumulation was quantified as counts per minute per milligram of wet weight. ADP administration led to a twofold increase in the amount of 111In accumulation. Mice lacking PI3Kγ, however, accumulated ~33% fewer platelets when compared with wild-type animals (Fig. 6B; P=0.0001), thereby suggesting that the protection from ADP-induced thromboembolism is caused by defective ADP-stimulated platelet aggregation. DISCUSSION Platelet activation and aggregation are crucial steps for both the control of hemostasis and the development of thrombotic diseases. Understanding the signal transduction pathways that lead to thrombocyte activation can provide fundamental knowledge to treat pathologies such as stroke and myocardial infarction. To gain insight into the role of signaling enzymes of the PI3K family, we analyzed platelet function in mice lacking a particular PI3K isoform, PI3Kγ, which is highly expressed in platelets and is known to be activated by GPCRs (7). PI3Kγ-null platelets showed impaired response to a selective GPCR agonist like ADP, and PI3Kγ-null mice appeared to be protected from thromboembolism but displayed normal bleeding time.

There are several indications that PI3Kγ is specifically activated by GPCR agonists (9, 10, 26). PI3Kγ-null platelets showed decreased aggregation after stimulation with ADP but not with another GPCR agonist like thrombin. Thrombin, however, has complex multiple signaling functions, and it is conceivable that, for example, its protease activity relays other intracellular signals that might overcome the lack of PI3Kγ. Consistent with this view, thrombin triggers the activation of Src-like tyrosine kinases, which subsequently induce PtdIns(3,4,5)P3 production via phosphotyrosine-dependent p85 associated PI3Kα or β (31). The presence of normal ADP-induced intracellular calcium fluxes and adenylyl cyclase inhibition indicated that PI3Kγ is involved in a novel signaling pathway that is necessary for full platelet aggregation after ADP stimulation. Downstream of ADP receptors, PI3Kγ was necessary for phosphorylation and activation of the Akt/PKB, a serine/threonine kinase involved in several cellular processes, including adhesion and migration (32). Analysis of PKB activation in the presence of specific inhibitors of the two ADP receptors showed that P2Y12 plays a crucial role in triggering PKB phosphorylation. All of these findings revealed that PI3Kγ is activated by a Gi- but not by a Gq-coupled receptor. Such evidence is in perfect agreement with previous reports indicating PI3Kγ downstream of other Gi-coupled heptahelical transmembrane receptors such as chemokine receptors (33). To explain the cellular mechanisms by which PI3Kγ determines an ADP-dependent full aggregation of platelets, we found that PI3Kγ plays a significant role in the ADP-mediated activation of the αIIbβ3 integrin fibrinogen receptor, a key factor in the aggregation process. Recent evidence indicates that the cytoplasmic tail of the β3 subunit of the fibrinogen receptor is a possible downstream target of PKB (34). PKB specifically acts on a threonine, which, according to in vitro experiments, may be important for outside-in signaling by the fibrinogen receptor. The in vivo role of this mechanism is not yet defined. Nonetheless, other reports indicated that serine/threonine phosphorylation of the β3 subunit promotes inside-out signaling and consequent exposure of fibrinogen/von Willebrand factor binding sites (35). It is thus possible to speculate that, in platelets lacking PI3Kγ, the effect on αIIbβ3 might be related to the impaired activation of PKB. Although the dose–response assessment of ADP-induced fibrinogen binding and aggregation showed that the two processes are similarly inhibited at distinct ADP concentrations (Fig. 2B and 5B), the difference in ADP-stimulated fibrinogen binding is probably not sufficient to completely explain the observed aggregation defect. Besides reduced αIIbβ3 integrin function, the lack of PI3Kγ might cause other, yet undetected, perturbations that prevent full-scale aggregation responses. Although the lack of PI3Kγ showed interesting effects in preventing thromboembolism, the mutation had limited consequences for in vivo hemostasis. The finding that bleeding time after cutting the tail tip is normal in PI3Kγ-null mice is in agreement with the observation that PI3Kγ-null mice have no tendency to bleed spontaneously. In the tail-bleeding assay, time for hemorrhage to cease is thought to depend on the coagulation cascade and thrombin generation (14). The fact that the thrombin response in PI3Kγ-null platelets is normal might therefore explain the unaltered bleeding time. It is noteworthy that the lack of a P2Y1 ADP receptor leads to a slight increase of the bleeding time (18, 19); our results rule out a possible involvement of

PI3Kγ in the onset of such a defect and are consistent with the separation between anti-thrombotic and anti-hemostatic effects observed with antagonists of the P2T (P2Y12) receptor (19). Thromboembolism is a major cause of pathological processes such as atherosclerosis, occlusion of vascular grafts, or acute restenosis after angioplasty. In this process, thrombin plays a major role; nevertheless, the importance of ADP is demonstrated by the current use of ADP receptor inhibitors as anti-aggregant agents. Currently, inhibitors of the P2Y12 receptor, such as the thienopyridines ticlopidine and clopidogrel, are used as antithrombotic therapy (36). Treating patients with these drugs, particularly ticlopidine, can, however, induce critical adverse effects. Life-threatening neutropenia and less critical gastrointestinal symptoms are seen in 40–50% of ticlopidine-treated patients (36). Clopidogrel (34), however, can induce bleeding and thrombotic thrombocytopenic purpura (37). Although the ADP-induced thromboembolism model is probably not representative of any human thrombotic process, the data obtained with PI3Kγ-null mice indicate that inhibitors of this enzyme might accomplish the tasks of ADP receptor antagonists. Furthermore, the finding that PI3Kγ-null mice appear healthy in standard conditions but are protected against acute thromboembolism indicates PI3Kγ as a target for antithrombotic drugs that will eventually lack major adverse effects. In conclusion, although platelets express several PI3K isoforms, our results defined a relevant role of PI3Kγ for the full response to only selective GPCR agonists such as ADP. The lack of PI3Kγ did not interfere with thrombin-induced platelet aggregation or bleeding time. We previously suggested that PI3Kγ could be an interesting target for new anti-inflammatory drugs in humans (26). The fact that PI3Kγ-null mice do not show alteration of hemostasis suggests that future use of specific PI3Kγ inhibitors as therapeutic agents will not increase the risk of hemorrhage. In addition, our findings indicate that treatment with such compounds could help to prevent thrombosis. ACKNOWLEDGMENTS We wish to thank O. Azzolino for her assistance and G. Camussi and F. Di Cunto for critically reading the manuscript. We are grateful to AstraZeneca R&D Charnwood, Loughborough, U.K., for the supply of AR-C69931MX. This work was supported by Progetto Finalizzato Biotecnologie, CNR (project biotechnology) to FA, the Swiss National Science Foundation (grant 3100-50506.97) and a research grant from Novartis, Horsham. PT and ML are recipients of an “Association pour la Recherche sur le Cancer” fellowship. REFERENCES 1. Kunapuli, S. P., and Daniel, J. L. (1998) P2 receptor subtypes in the cardiovascular
system. Biochem. J. 336, 513–523 2. Arcaro, A., and Wymann, M. P. (1993) Wortmannin is a potent phosphatidylinositol 3-
kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 296, 297–301

3. Yatomi, Y., Hazeki, O., Kume, S., and Ui, M. (1992) Suppression by wortmannin of
platelet responses to stimuli due to inhibition of pleckstrin phosphorylation. Biochem. J. 285, 745–751
4. Kovacsovics, T. J., Bachelot, C., Toker, A., Vlahos, C. J., Duckworth, B., Cantley, L. C.,
and Hartwig, J. H. (1995) Phosphoinositide 3-kinase inhibition spares actin assembly in activating platelets but reverses platelet aggregation. J. Biol. Chem. 270, 11358–11366
5. Wymann, M. P., and Pirola, L. (1998) Structure and function of phosphoinositide 3-
kinases. Biochim. Biophys. Acta 1436, 127–150 6. Leevers, S. J., Vanhaesebroeck, B., and Waterfield, M. D. (1999) Signalling through
phosphoinositide 3-kinases: the lipids take centre stage. Curr. Opin. Cell Biol. 11, 219–225 7. Rittenhouse, S. E. (1996) Phosphoinositide 3-kinase activation and platelet function.
Blood 88, 4401–4414 8. Zhang, J., Shattil, S. J., Cunningham, M. C., and Rittenhouse, S. E. (1996)
Phosphoinositide 3-kinase gamma and p85/phosphoinositide 3-kinase in platelets. Relative activation by thrombin receptor or beta-phorbol myristate acetate and roles in promoting the ligand-binding function of alphaIIbbeta3 integrin. J Biol. Chem. 271, 6265–6272
9. Stoyanov, B., Volinia, S., Hanck, T., Rubio, I., Loubtchenkov, M., Malek, D., Stoyanova,
S., Vanhaesebroeck, B., Dhand, R., Nurnberg, B., Gierschik, P., Seedorf, K., Hsuan, J. J., Waterfield, M. D., and Wetzker, R. (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269, 690–693
10. Stephens, L. R., Eguinoa, A., Erdjument-Bromage, H., Lui, M., Cooke, F., Coadwell, J.,
Smrcka, A. S., Thelen, M., Cadwallader, K., Tempst, P., and Hawkins, P. T. (1997) The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89, 105–114
11. Zhang, J., Benovic, J. L., Sugai, M., Wetzker, R., Gout, I., and Rittenhouse, S. E. (1995)
Sequestration of a G-protein beta gamma subunit or ADP-ribosylation of Rho can inhibit thrombin-induced activation of platelet phosphoinositide 3-kinases. J. Biol. Chem. 270, 6589–6594
12. Brass, L. F. (1999) More pieces of the platelet activation puzzle slide into place. J. Clin.
Invest. 104, 1663–1665 13. Cattaneo, M., Canciani, M. T., Lecchi, A., Kinlough-Rathbone, R. L., Packham, M. A.,
Mannucci, P. M., and Mustard, J. F. (1990) Released adenosine diphosphate stabilizes thrombin-induced human platelet aggregates. Blood 75, 1081–1086

14. Tsakiris, D. A., Scudder, L., Hodivala-Dilke, K., Hynes, R. O., and Coller, B. S. (1999) Hemostasis in the mouse (Mus musculus): a review. Thromb. Haemost. 81, 177–188
15. Daniel, J. L., Dangelmaier, C., Jin, J., Ashby, B., Smith, J. B., and Kunapuli, S. P. (1998)
Molecular basis for ADP-induced platelet activation. I. Evidence for three distinct ADP receptors on human platelets. J. Biol. Chem. 273, 2024–2029
16. Hollopeter, G., Jantzen, H. M., Vincent, D., Li, G., England, L., Ramakrishnan, V.,
Yang, R. B., Nurden, P., Nurden, A., Julius, D., and Conley, P. B. (2001) Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 409, 202–207
17. Cattaneo, M., and Gachet, C. (1999) ADP receptors and clinical bleeding disorders.
Arterioscler. Thromb. Vasc. Biol. 19, 2281–2285 18. Fabre, J. E., Nguyen, M., Latour, A., Keifer, J. A., Audoly, L. P., Coffman, T. M., and
Koller, B. H. (1999) Decreased platelet aggregation, increased bleeding time and resistance to thromboembolism in P2Y1-deficient mice. Nat. Med. 5, 1199–1202
19. Ingall, A. H., Dixon, J., Bailey, A., Coombs, M. E., Cox, D., McInally, J. I., Hunt, S. F.,
Kindon, N. D., Teobald, B. J., Willis, P. A., Humphries, R. G., Leff, P., Clegg, J. A., Smith, J. A., Tomlinson, W. (1999) Antagonists of the platelet P2T-receptor: A novel approach to antithrombotic therapy. J. Med. Chem. 42, 213–220
20. Kucera, G. L., and Rittenhouse, S. E. (1990) Human platelets form 3-phosphorylated
phosphoinositides in response to alpha-thrombin, U46619, or GTP gamma S. J. Biol. Chem. 265, 5345–5348
21. Gachet, C., Payrastre, B., Guinebault, C., Trumel, C., Ohlmann, P., Mauco, G.,
Cazenave, J. P., Plantavid, M., and Chap, H. (1997) Reversible translocation of phosphoinositide 3-kinase to the cytoskeleton of ADP-aggregated human platelets occurs independently of Rho A and without synthesis of phosphatidylinositol (3,4)-bisphosphate. J. Biol. Chem. 272, 4850–4854
22. Gironcel, D., Racaud-Sultan, C., Payrastre, B., Haricot, M., Borchert, G., Kieffer, N.,
Breton, M., and Chap, H. (1996) alphaIIb beta 3-integrin mediated adhesion of human platelets to a fibrinogen matrix triggers phospholipase C activation and phosphatidylinositol 3',4'-biphosphate accumulation. FEBS Lett., 389, 253–256
23. Heraud, J. M., Racaud-Sultan, C., Gironcel, D., Albiges-Rizo, C., Giacomini, T., Roques,
S., Martel, V., Breton-Douillon, M., Perret, B., and Chap, H. (1998) Lipid products of phosphoinositide 3-kinase and phosphatidylinositol 4',5'-bisphosphate are both required for ADP-dependent platelet spreading. J. Biol. Chem. 273, 17817–17823
24. Trumel, C., Payrastre, B., Plantavid, M., Hechler, B., Viala, C., Presek, P., Martinson, E.
A., Cazenave, J. P., Chap, H., and Gachet, C. (1999) A key role of adenosine diphosphate in

the irreversible platelet aggregation induced by the PAR1-activating peptide through the late activation of phosphoinositide 3-kinase. Blood 94, 4156–4165
25. Selheim, F., Idsoe, R., Fukami, M. H., Holmsen, H., and Vassbotn, F. S. (1999)
Formation of PI 3-kinase products in platelets by thrombin, but not collagen, is dependent on synergistic autocrine stimulation, particularly through secreted ADP. Biochem. Biophys. Res. Commun. 263, 780–785
26. Hirsch, E., Katanaev, V. L., Garlanda, C., Azzolino, O., Pirola, L., Silengo, L., Sozzani,
S., Mantovani, A., Altruda, F., and Wymann, M. P. (2000) Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287, 1049–1053
27. Nordöy, A., and Chandler, A. B. (1964) Platelet thrombosis induced by adenosine
diphosphate in rat. Scand. J. Haemat. 1, 16–25 28. Watson, S. P. (1999) Collagen receptor signaling in platelets and megakaryocytes.
Thromb. Haemost. 82, 365–376 29. Banfic, H., Downes, C. P., and Rittenhouse, S. E. (1998) Biphasic activation of
PKBalpha/Akt in platelets. Evidence for stimulation both by phosphatidylinositol 3,4-bisphosphate, produced via a novel pathway, and by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 11630–11637
30. Sims, P. J., Ginsberg, M. H., Plow, E. F., and Shattil, S. J. (1991) Effect of platelet
activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J. Biol. Chem. 266, 7345–7352
31. Gutkind, J. S., Lacal, P. M., and Robbins, K. C. (1990) Thrombin-dependent association
of phosphatidylinositol-3 kinase with p60c-src and p59fyn in human platelets. Mol. Cell Biol. 10, 3806–3809
32. Chan, T. O., Rittenhouse, S. E., and Tsichlis, P. N. (1999) AKT/PKB and other D3
phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68, 965–1014
33. Wymann, M. P., Sozzani, S., Altruda, F., Mantovani, A., and Hirsch, E. (2000) Lipids on
the move: phosphoinositide 3-kinases in leukocyte function. Immunol. Today 21, 260–264 34. Kirk, R. I., Sanderson, M. R., and Lerea, K. M. (2000) Threonine phosphorylation of the
beta 3 integrin cytoplasmic tail, at a site recognized by PDK1 and Akt/PKB in vitro, regulates Shc binding. J. Biol. Chem. 275, 30901–30906
35. van Willigen, G., Hers, I., Gorter, G., and Akkerman, J. W. (1996) Exposure of ligand-
binding sites on platelet integrin alpha IIB/beta 3 by phosphorylation of the beta 3 subunit. Biochem. J. 314, 769–379

36. Quinn, M. J., and Fitzgerald, D. J. (1999) Ticlopidine and clopidogrel. Circulation 100, 1667–1672
37. Bennett, C. L., Connors, J. M., Carwile, J. M., Moake, J. L., Bell, W. R., Tarantolo, S. R.,
McCarthy, L. J., Sarode, R., Hatfield, A. J., Feldman, M. D., Davidson, C. J., and Tsai, H. M. (2000) Thrombotic thrombocytopenic purpura associated with clopidogrel. N. Engl. J. Med. 342, 1773–1737
Received January 2, 2001; revised May 7, 2001.

Fig. 1
Figure 1. PI3Kγ is expressed by murine megakaryocytes and platelets. A) Insertion of the LacZ reporter gene cassette in the PI3Kγ locus results in β-galactosidase-positive megakaryocytes in spleen sections. Arrowheads indicate spots of LacZ staining in the cytoplasm of a PI3Kγ-deficient megakaryocyte. Scale bar represents 40 µm. B) Western blot analysis of PI3Kα, β, δ, and γ in platelets derived from wild-type (+/+) and PI3Kγ-null (-/-) mice. An equal amount of loaded proteins is shown by the detection of gelsolin.
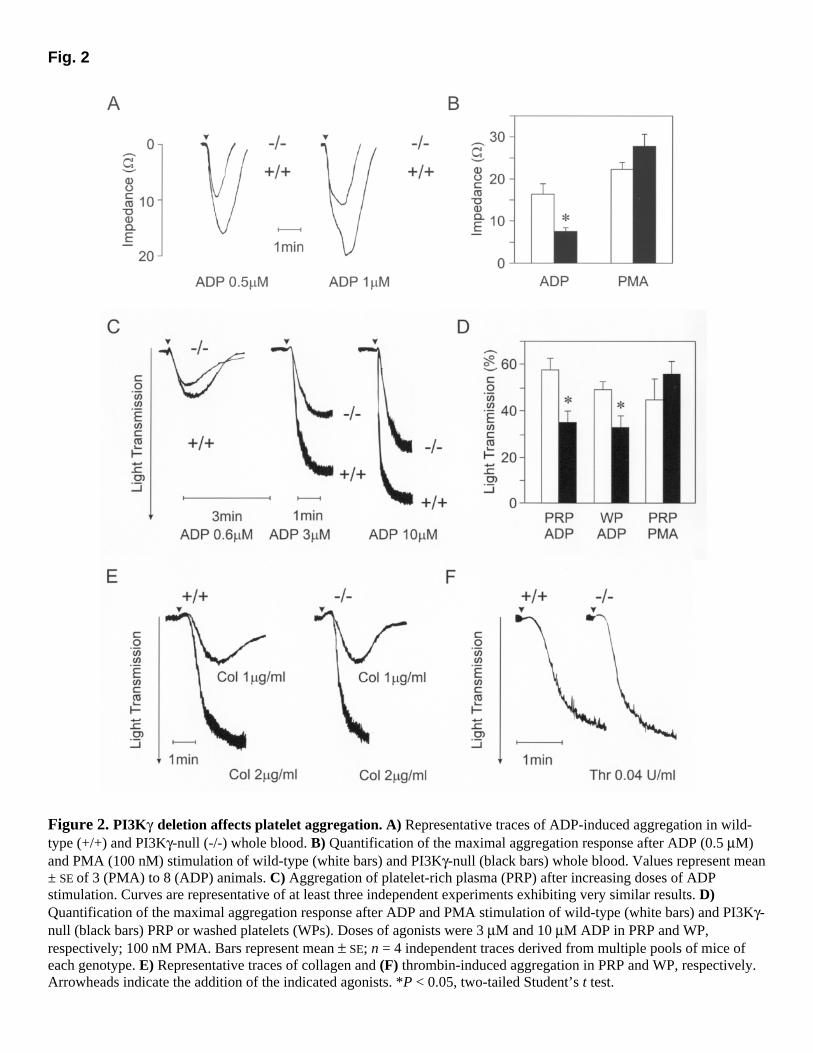
Fig. 2
Figure 2. PI3Kγ deletion affects platelet aggregation. A) Representative traces of ADP-induced aggregation in wild-type (+/+) and PI3Kγ-null (-/-) whole blood. B) Quantification of the maximal aggregation response after ADP (0.5 µM) and PMA (100 nM) stimulation of wild-type (white bars) and PI3Kγ-null (black bars) whole blood. Values represent mean ± SE of 3 (PMA) to 8 (ADP) animals. C) Aggregation of platelet-rich plasma (PRP) after increasing doses of ADP stimulation. Curves are representative of at least three independent experiments exhibiting very similar results. D) Quantification of the maximal aggregation response after ADP and PMA stimulation of wild-type (white bars) and PI3Kγ-null (black bars) PRP or washed platelets (WPs). Doses of agonists were 3 µM and 10 µM ADP in PRP and WP, respectively; 100 nM PMA. Bars represent mean ± SE; n = 4 independent traces derived from multiple pools of mice of each genotype. E) Representative traces of collagen and (F) thrombin-induced aggregation in PRP and WP, respectively. Arrowheads indicate the addition of the indicated agonists. *P < 0.05, two-tailed Student’s t test.

Fig. 3
Figure 3. The lack of PI3Kγ does not alter ADP-dependent intracellular calcium fluxes and adenylyl cyclase inhibition. A) Intracellular calcium mobilization in response to ADP in both wild-type and PI3Kγ-null platelets. B) Inhibition of adenylyl cyclase by ADP. Cyclic AMP levels were measured in platelets stimulated with vehicle (Vh), 1 µM PGE1, and PGE1 plus 5 µM ADP.

Fig. 4
Figure 4. PI3Kγ is needed for P2Y12-dependent activation of PKB/Akt. A) Western blot analysis of phosphorylated PKB/Akt (P-PKB) and total PKB in wild-type (+/+) and PI3Kγ-null (-/-) platelets after 2 min of stimulation with ADP (3 µM) or thrombin (Thr, 1 U/ml). B) Values represent the amount of PKB phosphorylated on Ser 473 (P-PKB), as quantified by densitometry (3 independent experiments; values represent mean ± SE; *P<0.05, two-tailed Student’s t test). C) Western blot analysis of phosphorylated PKB/Akt (P-PKB) and total PKB in wild-type platelets after inhibition of P2Y12 by pre-incubation with AR-C69931 (kindly provided by AstraZeneca, Loughborough, U.K.) for 20 min followed by a 2-min stimulation with either ADP or thrombin at the given concentrations. In these conditions, ADP-dependent PKB phosphorylation is lost. D) The same as panel C but with a 20-min preincubation with the P2Y1-specific inhibitor A3P5P (adenosine 3’,5’-diphosphate; Sigma).
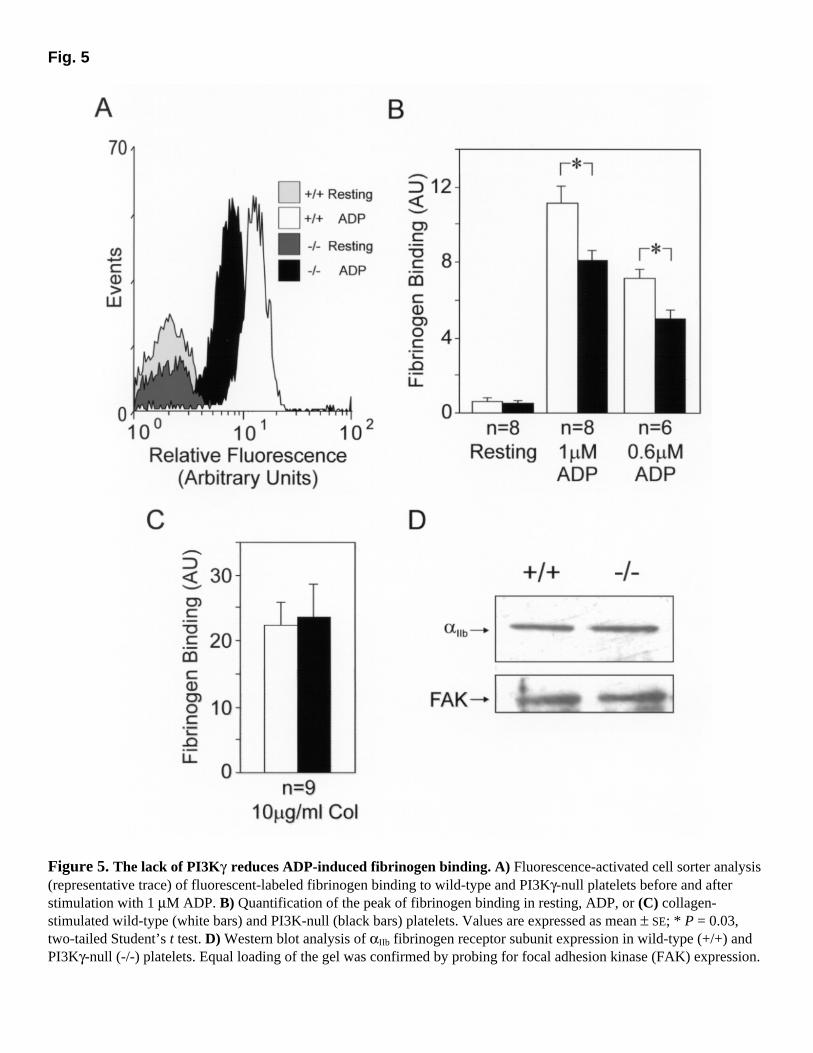
Fig. 5
Figure 5. The lack of PI3Kγ reduces ADP-induced fibrinogen binding. A) Fluorescence-activated cell sorter analysis (representative trace) of fluorescent-labeled fibrinogen binding to wild-type and PI3Kγ-null platelets before and after stimulation with 1 µM ADP. B) Quantification of the peak of fibrinogen binding in resting, ADP, or (C) collagen-stimulated wild-type (white bars) and PI3K-null (black bars) platelets. Values are expressed as mean ± SE; * P = 0.03, two-tailed Student’s t test. D) Western blot analysis of αIIb fibrinogen receptor subunit expression in wild-type (+/+) and PI3Kγ-null (-/-) platelets. Equal loading of the gel was confirmed by probing for focal adhesion kinase (FAK) expression.
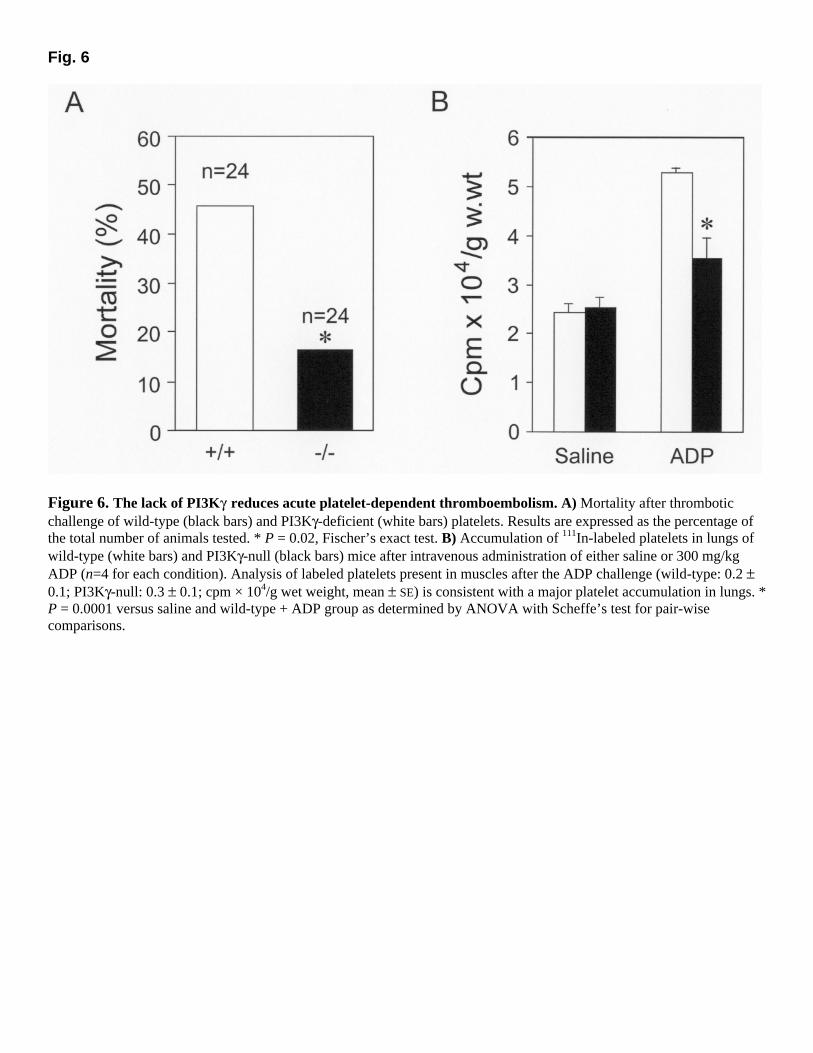
Fig. 6
Figure 6. The lack of PI3Kγ reduces acute platelet-dependent thromboembolism. A) Mortality after thrombotic challenge of wild-type (black bars) and PI3Kγ-deficient (white bars) platelets. Results are expressed as the percentage of the total number of animals tested. * P = 0.02, Fischer’s exact test. B) Accumulation of 111In-labeled platelets in lungs of wild-type (white bars) and PI3Kγ-null (black bars) mice after intravenous administration of either saline or 300 mg/kg ADP (n=4 for each condition). Analysis of labeled platelets present in muscles after the ADP challenge (wild-type: 0.2 ± 0.1; PI3Kγ-null: 0.3 ± 0.1; cpm × 104/g wet weight, mean ± SE) is consistent with a major platelet accumulation in lungs. * P = 0.0001 versus saline and wild-type + ADP group as determined by ANOVA with Scheffe’s test for pair-wise comparisons.




















