
Regulation of cardiovascular remodeling by the counter-regulatory axis of the renin-angiotensin...
16
Author Proof 1 10.2217/FCA.12.75 © 2013 Future Medicine Ltd ISSN 1479-6678 Future Cardiol. (2013) 9(1), 1–xxx Future Cardiology part of The renin–angiotensin system The renin–angiotensin system (RAS) is integrally involved in the development of various cardiovascular diseases (CVDs) such as hypertension and end-organ damage, diabetes, atherosclerosis and cardiac remodeling, including hypertrophy and fibrosis, major risk factors for heart failure. The classical RAS pathway involves the release of renin from the kidneys into the circulation in response to low volume states and high sodium content within the distal tubules. In the blood, renin catalyzes the liver-derived angiotensinogen to form the decapeptide angiotensin I (Ang I), which then is hydrolyzed by angiotensin-converting enzyme (ACE) to the active octapeptide Ang II. Physiologically, Ang II has important roles in blood pressure homeostasis via its effects on the kidney to conserve sodium, stimulation of aldosterone release, actions on smooth muscle to mediate vasoconstriction and sympathetic activation in the CNS, all actions that maintain blood pressure. Ang II also has important roles in individual tissues that promote cell signaling leading to growth and cell differentiation. Chronic disruption of the RAS, however, leads to overactivity of Ang II, and an array of pathophysiological effects, for example, raised blood pressure, adverse cardiac remodeling, vasoconstriction, oxidative stress and abnormalities in renal sodium handling. The effects of Ang II are mediated by the angiotensin type 1 and type 2 receptors (AT 1 R and AT 2 R, respectively) (FIGURE 1 & TABLE 1) . Both are transmembrane G protein-coupled receptors (GPCRs) that tend to have opposing actions. AT 1 R is mainly responsible for the classical pathophysiological actions of Ang II, whereas Ang II stimulation of the AT 2 R is reported to antagonize the pathological effects of AT 1 R. Cardiovascular remodeling & the role of Ang II Cardiac remodeling occurs within the injured heart resulting from pathological hemodynamic overload from CVDs such as congenital heart disease, chronic hypertension or myocardial infarction (MI). The adaptive function of the heart is initiated in response to injury as it compensates for the increased hemodynamic load, resulting in an increase in left ventricular mass. Cardiac remodeling begins fairly rapidly and often occurs within the first few hours following an infarct and is regarded as an intermediary process to several adverse cardiovascular Regulation of cardiovascular remodeling by the counter- regulatory axis of the renin– angiotensin system Carolyn Clarke 1 , Monica Flores-Muñoz 1 , Clare A McKinney 1 , Graeme Milligan 2 & Stuart A Nicklin* 1 1 Institute of Cardiovascular & Medical Sciences, College of Medical, Veterinary & Life Sciences, University of Glasgow, BHF Glasgow Cardiovascular Research Centre, 126 University Place, University of Glasgow, G12 8TA, UK 2 Institute of Molecular Cell & Systems Biology, College of Medical, Veterinary & Life Sciences, University of Glasgow, UK *Author for correspondence: Tel.: +44 141 330 2521 n Fax: +44 141 330 6997 n [email protected] The counter-regulatory axis of the renin–angiotensin system (RAS) is a novel therapeutic target in cardiovascular disease. Pathophysiological effects mediated via angiotensin II (Ang II) are well established in regulation of blood pressure, cardiac and vascular remodeling, and renal sodium handling, which lead to disorders such as hypertension and associated end-organ damage, atherosclerosis and heart failure. The counter-regulatory axis of the RAS is centered on the angiotensin-converting enzyme 2/angiotensin-1–7 (Ang-[1–7])/Mas receptor axis and has been shown to inhibit many detrimental phenotypes in cardiovascular disease. More recently, an alternative peptide, Ang-(1–9), has been reported as a potential new member of this axis. This review will discuss the cardiovascular regulatory roles of Ang-(1–7) and Ang-(1–9) in the counter- regulatory axis of the RAS and the potential for new therapeutic approaches in cardiovascular disease. Keywords n angiotensin converting enzyme-2 n angiotensin-(1–7) n angiotensin-(1–9) n cardiac remodeling n Mas n renin–angiotensin system n vascular dysfunction Review
Transcript of Regulation of cardiovascular remodeling by the counter-regulatory axis of the renin-angiotensin...

Author Pro
of
110.2217/FCA.12.75 © 2013 Future Medicine Ltd ISSN 1479-6678Future Cardiol. (2013) 9(1), 1–xxx
Futu
re C
ard
iolo
gy
part of
The renin–angiotensin systemThe renin–angiotensin system (R AS) is integrally involved in the development of various cardiovascular diseases (CVDs) such as hypertension and end-organ damage, diabetes, atherosclerosis and cardiac remodeling, including hypertrophy and fibrosis, major risk factors for heart failure. The classical RAS pathway involves the release of renin from the kidneys into the circulation in response to low volume states and high sodium content within the distal tubules. In the blood, renin catalyzes the liver-derived angiotensinogen to form the decapeptide angiotensin I (Ang I), which then is hydrolyzed by angiotensin-converting enzyme (ACE) to the active octapeptide Ang II. Physiologically, Ang II has important roles in blood pressure homeostasis via its effects on the kidney to conserve sodium, stimulation of aldosterone release, actions on smooth muscle to mediate vasoconstriction and sympathetic activation in the CNS, all actions that maintain blood pressure. Ang II also has important roles in individual tissues that promote cell signaling leading to growth and cell differentiation. Chronic disruption of the RAS, however, leads to overactivity of Ang II, and an array of pathophysiological effects, for
example, raised blood pressure, adverse cardiac remodeling, vasoconstriction, oxidative stress and abnormalities in renal sodium handling. The effects of Ang II are mediated by the angiotensin type 1 and type 2 receptors (AT
1R
and AT2R, respectively) (Figure 1 & Table 1).
Both are transmembrane G protein-coupled receptors (GPCRs) that tend to have opposing actions. AT
1R is mainly responsible for the
classical pathophysiological actions of Ang II, whereas Ang II stimulation of the AT
2R is
reported to antagonize the pathological effects of AT
1R.
Cardiovascular remodeling & the role of Ang II
Cardiac remodeling occurs within the injured heart resulting from pathological hemodynamic overload from CVDs such as congenital heart disease, chronic hypertension or myocardial infarction (MI). The adaptive function of the heart is initiated in response to injury as it compensates for the increased hemodynamic load, resulting in an increase in left ventricular mass. Cardiac remodeling begins fairly rapidly and often occurs within the first few hours following an infarct and is regarded as an intermediary process to several adverse cardiovascular
Regulation of cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system
Carolyn Clarke1, Monica Flores-Muñoz1, Clare A McKinney1, Graeme Milligan2 & Stuart A Nicklin*1
1Institute of Cardiovascular & Medical Sciences, College of Medical, Veterinary & Life Sciences, University of Glasgow, BHF Glasgow Cardiovascular Research Centre, 126 University Place, University of Glasgow, G12 8TA, UK 2Institute of Molecular Cell & Systems Biology, College of Medical, Veterinary & Life Sciences, University of Glasgow, UK *Author for correspondence: Tel.: +44 141 330 2521 n Fax: +44 141 330 6997 n [email protected]
The counter-regulatory axis of the renin–angiotensin system (RAS) is a novel therapeutic target in cardiovascular disease. Pathophysiological effects mediated via angiotensin II (Ang II) are well established in regulation of blood pressure, cardiac and vascular remodeling, and renal sodium handling, which lead to disorders such as hypertension and associated end-organ damage, atherosclerosis and heart failure. The counter-regulatory axis of the RAS is centered on the angiotensin-converting enzyme 2/angiotensin-1–7 (Ang-[1–7])/Mas receptor axis and has been shown to inhibit many detrimental phenotypes in cardiovascular disease. More recently, an alternative peptide, Ang-(1–9), has been reported as a potential new member of this axis. This review will discuss the cardiovascular regulatory roles of Ang- (1–7) and Ang- (1–9) in the counter-regulatory axis of the RAS and the potential for new therapeutic approaches in cardiovascular disease.
Keywords
n angiotensin converting enzyme-2 n angiotensin-(1–7) n angiotensin-(1–9) n cardiac remodeling n Mas n renin–angiotensin system n vascular dysfunction
Revie
w

Author Pro
of
Future Cardiol. (2013) 9(1)2 future science group
outcomes. This maladaptive response leads to reduced left ventricular performance, cardiac impairment and an increased risk of cardiac events, which eventually culminates in heart failure. At the cellular level, although most of these changes occur in cardiomyocytes, they can also occur in cardiac fibroblasts, in addition to endothelial and smooth muscle cells of the cardiac vasculature. For example, Ang II, via the AT
1R, activates phospholipase C and the
ERK1/2 pathway through Gaq/11
to induce cardiomyocyte hypertrophy [1]. Ang II also
regulates extracellular matrix remodeling in cardiac fibroblasts by increasing the synthesis of collagen and fibronectin, promoting the accumulation of proteoglycans and increasing expression of TGF-b [2] (Figure 2).
Downstream signaling at the AT2R is less
well understood. Although it is thought to act through the Gai
-subunit, evidence is not entirely clear as it has been suggested that it also encodes a 5-amino acid motif that impairs G-protein coupling [3,4]. Blockade of AT
2R in Ang II-infused rats increased left
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin
Table 1. Actions of the renin–angiotensin system receptors and their functional ligands.
AT1R AT2R Mas
Actions Ligand Actions Ligand(s) Actions Ligand
Vasoconstriction Ang II Vasodilation Ang II Vasodilation Ang-(1–7)
Hypertrophy Anti-hypertrophic Ang-(1–9) Anti-hypertrophic
Fibrosis Anti-fibrotic Anti-fibrotic
Proliferation Anti-proliferative Anti-proliferative
Inflammation Anti-inflammatory Anti-inflammatory
ECM remodeling Apoptotic Anti-thrombotic
NADPH oxidase activation
AT1R AT2R AT2RMas
Angiotensinogen (1–255)
Renin
Ang I (1–10)
Ang II (1–8) Ang-(1–7)
Ang-(1–9)
ACE
ACE
ACE2
ACE2
NEP NEPTOP
POP
Cp A, Cx A
Figure 1. The renin–angiotensin system. The renin–angiotensin system is a network of interconverted peptides, mainly generated by the actions of ACE and ACE2. Individual angiotensin peptides mediates cardiovascular effects via a defined set of receptors. ACE: Angiotensin-converting enzyme; Ang: Angiotensin; AT
1R: Ang II type 1 receptor; AT
2R: Ang II
type 2 receptor; Cp A: Cathepsin A; Cx A: Carboxypeptidase A; NEP: Neutral endopeptidase 11; POP: Prolyl endopeptidase; TOP: Thimet oligopeptidase.

Author Pro
of
www.futuremedicine.com 3future science group
ventricular growth, increased protein synthesis and augmented protein kinase C translocation to the membrane [5]. Furthermore, lentivirus-mediated overexpression of the AT
2R in stroke-
prone spontaneously hypertensive rats (SHRSP) diminished left ventricular wall thickness and collagen without affecting blood pressure [6]. Aortic banding in transgenic mice overexpressing AT
2R reduced left ventricular myocyte diameter
as well as decreasing collagen levels [7]. Although typical hypertrophy markers were not regulated, the expression of phospholamban, as well as the ratio between sarcoplasmic reticulum Ca2+ ATPase (SERCA2a) and inducible nitric oxide (NO) synthase (iNOS), were higher, indicating a role for AT
2R in hypertrophy regulation [7]
(Figure 3). Activation of AT2R has also been
associated with increased NO signaling and has been shown to induce endothelial NOS (eNOS) activity in diabetic mice via interaction with MAPK signaling pathways [8], indicating a role for AT
2R in improved endothelial and vascular
function in disease states.Dysregulation of the RAS is also one of
the key contributing factors to remodeling of the vasculature, with the majority of the pathological processes involved (e.g., vascular smooth muscle cell [VSMC] growth and migration, oxidative stress and remodeling of the extracellular matrix) being mediated by Ang II signaling at AT
1R. Through AT
1R, Ang
II activates the Gaq/11 subunit, transactivates the
EGF receptor (EGFR), phosphorylates Rho-kinase and induces hypertrophy in VSMC [9]. Ang II signaling at AT
2R counteracts AT
1R
signaling by activating serine/threonine and tyrosine phosphatases to inactivate MAPK signaling in proliferation and migration and coupling to the bradykinin type 2 receptor (B
2R)
to induce NO production and vasodilation (reviewed in [3,4]). Within the vasculature, biologically active Ang II is also responsible for proliferation of endothelial cells, production and accumulation of collagen and stimulation of hormones such as endothelin in the vessel wall. Ang II through these actions may also contribute to increased vascular stiffness.
With the multitude of effects of Ang II in individual tissues and systems in the body it is not surprising that the RAS is important in the etiology of hypertension and CVD, and its importance in clinical medicine is highlighted by the efficacy of pharmacological agents that inhibit the synthesis (ACE inhibitors) or activity (AT
1R antagonists) of Ang II. ACE inhibitors
and AT1R antagonists are well tolerated and
effective anti-hypertensive medications, which lower blood pressure and reduce morbidity and mortality associated with CVD [10]. In addition, unlike other antihypertensive treatments, RAS-blocking therapeutics have direct tissue-protective properties that aid in the prevention of end-organ damage, highlighting their power in medicine [11]. With the identification of new facets to the RAS, it has become clear that other therapeutic targets exist that may further the clinicians armory in reducing CVD morbidity and mortality.
The counter-regulatory axis of the RASHistorically, the RAS was considered a linear process with Ang II as the major active peptide. However, the conventional view of the RAS has undergone significant changes based on two fundamental discoveries. The first was the expression of the RAS in specific tissues,
Cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system Review
AngII
AT1R αβγ
(p) ERK1/2(p) JNK(p) p38
Fibrosis
TGF-β1 NFκB
ANPBNP
Hypertrophy
Figure 2. Signaling of Ang II at the AT1R in cardiomyocytes and fibroblasts. Ang II through coupling to AT
1R promotes
phosphorylation of ERK1/2, JNK and p38 molecules as well as promoting TGF-b production to induce fibrosis. Ang II also increases levels of NFkB and expression of hypertrophy markers ANP and BNP. ANP: Atrial natriuretic peptide; AT
1R: Ang II
type 1 receptor; BNP: Brain natriuretic peptide.
Author Pro
of
Future Cardiol. (2013) 9(1)4 future science group
highlighting the existence of a local RAS in which Ang II could be generated within individual tissues, without recruitment from the systemic circulation. Second, new novel components of the RAS were identified within specific tissues, most specifically the enzyme ACE2, a homologue of ACE but which differs greatly in its substrate specificity [12,13]. ACE2 catalyzes the conversion of Ang II to angiotensin-1–7 (Ang-[1–7]), with 400-fold greater affinity for Ang II than Ang I [12]. However, Ang-(1–7) can also form less efficiently through ACE2 hydrolysis of Ang I to angiotensin-1–9 [Ang-(1–9)], which is subsequently hydrolyzed to Ang-(1–7) via ACE [14], or alternatively via direct conversion by the action of prolyl endopeptidase, neutral endopeptidase or thimet oligopeptidase, a process that is independent of ACE [15,16]. ACE2 is key to the generation of Ang-(1–7), which antagonizes detrimental Ang II signaling via the receptor Mas, and together the ACE2/Ang-(1–7)/Mas axis defines the counter-regulatory axis of the RAS. Together with recent identification of other potential functional members of this axis, the number of new therapeutic targets has expanded (box 1 & Table 1).
Cardiac ACE2/Ang-(1–7)/Mas axisACE2 knockout results in a severe cardiac contractility defect and increased Ang II levels
in the kidneys, heart and plasma, providing evidence that ACE2 functions as a key cardiac function regulator and modulates the RAS via altering Ang I and Ang II levels [17]. In MI the loss of ACE2 promotes fibroblast proliferation by activating ERK1/2 and JNK1/2, as well as inf lammation by increasing neutrophil infiltration to the infarct zone, leading to an increase in inflammatory components such as cytokines including IL-6, and a deficiency in matrix metalloproteinase (MMP)2, which leads to disruption of the extracellular matrix [18]. It was also found that in these mice the lack of ACE2 induced the upregulation of hypoxic genes and levels of Ang II were increased. Furthermore, infusion of Ang-(1–7) or administration of the AT
1R antagonist irbesartan is cardioprotective
in heart failure in ACE2-knockout mice [19]. These studies suggest a protective role for ACE2 in heart failure. However, other studies have also shown a negative role. Masson et al. demonstrated cardiac fibrosis and deficits in ejection fraction and fractional shortening in SHRSP overexpressing ACE2 mediated by adeno-associated virus serotype 6 delivery [20]. In transgenic mice, cardiac-specific overexpression of ACE2 produced severe and progressive malfunction of rhythm and conduction, which caused ventricular tachycardia and sudden death
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin
AngII
AT2R αβγ
PKC
Fibrosis Hypertrophy
??? ???
SERCA2a, PLB
Ang-(1–9)
AT2R αβγ
Collagen I Rho kinases
Fibrosis Hypertrophy
???
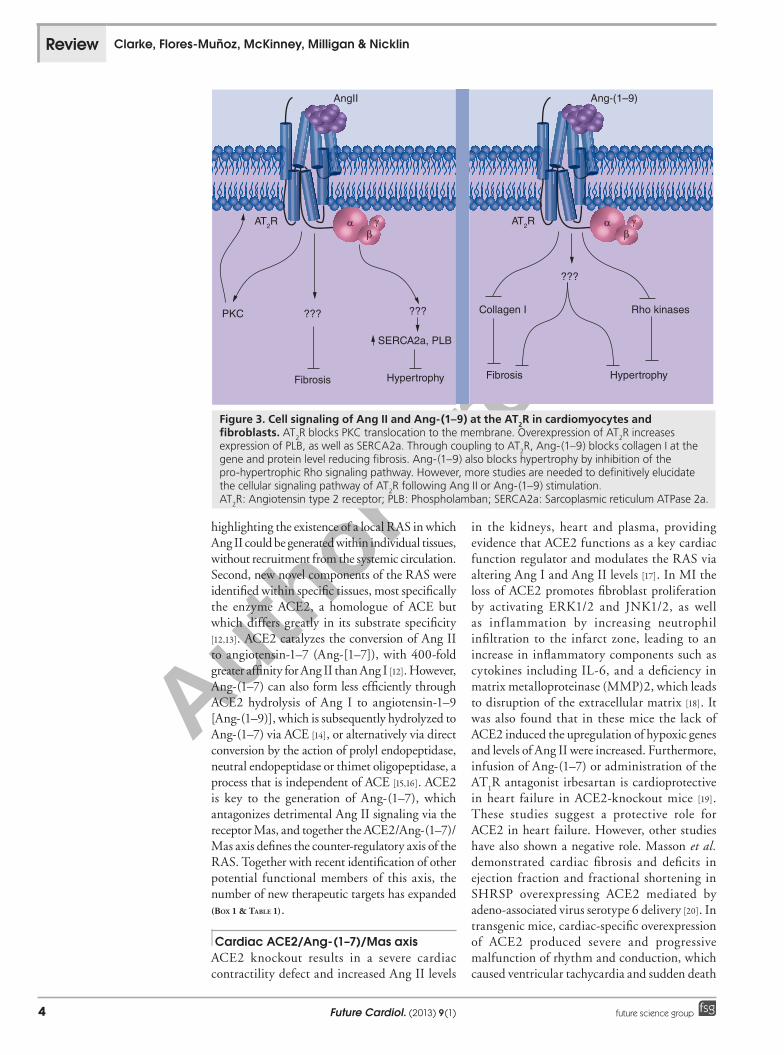
Figure 3. Cell signaling of Ang II and Ang-(1–9) at the AT2R in cardiomyocytes and fibroblasts. AT
2R blocks PKC translocation to the membrane. Overexpression of AT
2R increases
expression of PLB, as well as SERCA2a. Through coupling to AT2R, Ang-(1–9) blocks collagen I at the
gene and protein level reducing fibrosis. Ang-(1–9) also blocks hypertrophy by inhibition of the pro-hypertrophic Rho signaling pathway. However, more studies are needed to definitively elucidate the cellular signaling pathway of AT
2R following Ang II or Ang-(1–9) stimulation.
AT2R: Angiotensin type 2 receptor; PLB: Phospholamban; SERCA2a: Sarcoplasmic reticulum ATPase 2a.

Author Pro
of
www.futuremedicine.com 5future science group
[21]. These disturbances in the ACE2 transgenic mouse hearts were attributed to ACE2-mediated gap junction remodeling with associated downregulation of gap junction proteins connexin-40 and connexin-43 [21]. It is likely that these conflicts could also be due to issues of timing of expression and cellular localization of ACE2 overexpression. Therefore, further research is required to address these conflicting data regarding ACE2 in cardiovascular physiology and pathophysiology.
This ACE2/Ang-(1–7)/Mas axis was shown to have a critical role in the vasculature and heart, exerting mainly protective effects. Ang-(1–7), a seven amino acid peptide that mediates most of its actions through engagement of the GPCR encoded by the Mas protooncogene, is regarded as the main effector peptide of the counter-regulatory RAS [22]. Ang-(1–7) is present in the myocardium of the intact human heart and is derived from multiple angiotensin-processing pathways (Figure 1) [14–16,23–25]. Work originating from Ferrario and colleagues established the initial and basic counter-regulatory mechanisms of the Ang-(1–7)/ACE2/Mas axis in opposition to the pathological actions of Ang II and key findings of this work are expertly reviewed in [26,27].
One of the most studied actions of Ang-(1–7) is in cardiac remodeling. Many publications have demonstrated that Ang-(1–7) has some effect in at least one of the components of cardiac remodeling, including hypertrophy and/or fibrosis, and with use of the pharmacological Mas antagonist A-779, signaling via this receptor has been shown to be integral (Figure 4) [28]. Many of these early studies were observational studies that reported on downstream phenotypes following systemic Ang-(1–7) infusion. The development of transgenic models expressing Ang-(1–7) in individual tissues has expanded our knowledge. For example, Ang-(1–7) transgenic rats administered with either isoproterenol or deoxycorticosterone acetate (DOCA) to induce hypertension were protected against fibrosis and cardiac hypertrophy [29,30]. In addition, local expression of an Ang-(1–7) fusion protein driven by the a-myosin heavy chain promoter in the heart was found to improve myocardial contractility and attenuate cardiac remodeling in either isoproterenol or DOCA-induced models of hypertension in transgenic Ang-(1–7) mice and rats [31,32]. Recent data has also suggested that Ang-(1–7) may mediate some of its beneficial cardiac effects via stimulating endothelial progenitor cells [33].
Overactivation of the RAS is largely involved in the progression of diabetes-associated CVD (reviewed recently [34,35]), however, there has been recent interest in the role of the counter-regulatory RAS in various in vivo models of diabetes-induced cardiac remodeling and dysfunction. For example, in a mouse model of diabetes (Akita mice) deficient of ACE2, it was shown that the loss of ACE2 results in cardiac myopathy [36]. This was as a result of the development of systolic dysfunction, and increased activation of NADPH oxidase and MMPs, promoting increased oxidative stress and degradation of the extracellular matrix. Interestingly, these changes were associated with increased plasma and tissue levels of Ang II, and inhibited by irbesartan, suggesting the effects are Ang II/AT
1R dependent.
Furthermore, this demonstrates a protective role for ACE2 in this setting via suppressed production and activity of Ang II [36]. Additionally, it was recently shown that oral administration of the ACE2 activator 1-[[2-(dimethylamino)ethyl]amino]-4-(hydroxymethyl)-7-[[(4-methylphenyl)sulfonyl]oxy]-9H-xanthen-9-one (XNT) to rats that had streptozotocin-induced diabetes resulted in reduced hyperglycemia, cardiac dysfunction, hypertrophy and fibrosis in comparison to control animals [37]. In the same diabetic rat model it was found that administration of Ang-(1–7) or the Mas receptor agonist AVE099 reduces diabetes-induced cardiac dysfunction [38]. These findings were recently extended to provide further insight into the mechanisms involved in the protective effects observed and it was found that selective inhibition of ACE2 by the specific inhibitor DX600
Cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system Review
Box 1. Therapeutic advances in the counter-regulatory renin–angiotensin system.
ACE2 activators�n XNT, a synthetic small molecule, which despite positive cardiovascular effects
in vivo, is unlikely to reach clinical trial due to solubility issues. �n APN01, a clinical derivative of human recombinant ACE2, is currently under clinical
investigation by Apeiron Biologics.Ang-(1–7) formulations�n HPbCD/Ang-(1–7) and cAng-(1–7) are synthetically modified stable formulations
of Ang-(1–7). HPbCD/Ang-(1–7) can be effectively delivered orally, however, the pulmonary route may be more promising for cAng-(1–7 ). Both are being explored in the clinic.
Mas agonists�n AVE 0991 and CGEN-856S are synthetic nonpeptide agonists of Mas that elicit
similar cardioprotective effects as Ang-(1–7) in vivo. The clinical potential of CGEN-856S as a cardiovascular therapeutic is currently being explored by BioLineRx Ltd.
AT2R agonist�n Compound 21 is an oral agonist of AT
2R that has beneficial cardiovascular effects
in several animal models. Compound 21 is a potential therapeutic option in myocardial infarction and heart failure to abrogate adverse cardiac remodeling.
Author Pro
of
Future Cardiol. (2013) 9(1)6 future science group
resulted in increased cardiac dysfunction, which was further exacerbated by dual administration with the Mas receptor antagonist A-779 [39]. Taken together this data suggests that ACE2 has a protective effect in the heart owing to increased Ang-(1–7) production and signaling at the Mas receptor as well as reduced Ang II production and activity at AT
1R.
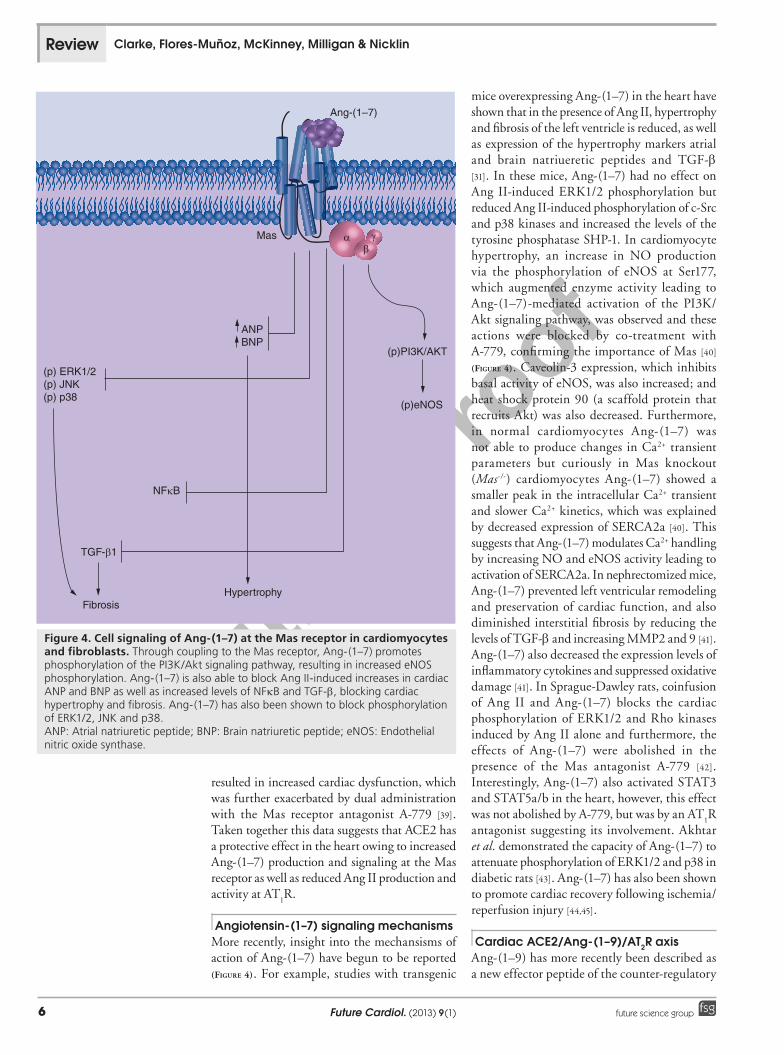
Angiotensin-(1–7) signaling mechanismsMore recently, insight into the mechansisms of action of Ang-(1–7) have begun to be reported (Figure 4). For example, studies with transgenic
mice overexpressing Ang-(1–7) in the heart have shown that in the presence of Ang II, hypertrophy and fibrosis of the left ventricle is reduced, as well as expression of the hypertrophy markers atrial and brain natriueretic peptides and TGF-b [31]. In these mice, Ang-(1–7) had no effect on Ang II-induced ERK1/2 phosphorylation but reduced Ang II-induced phosphorylation of c-Src and p38 kinases and increased the levels of the tyrosine phosphatase SHP-1. In cardiomyocyte hypertrophy, an increase in NO production via the phosphorylation of eNOS at Ser177, which augmented enzyme activity leading to Ang-(1–7)-mediated activation of the PI3K/Akt signaling pathway, was observed and these actions were blocked by co-treatment with A-779, confirming the importance of Mas [40] (Figure 4). Caveolin-3 expression, which inhibits basal activity of eNOS, was also increased; and heat shock protein 90 (a scaffold protein that recruits Akt) was also decreased. Furthermore, in normal cardiomyocytes Ang-(1–7) was not able to produce changes in Ca2+ transient parameters but curiously in Mas knockout (Mas-/-) cardiomyocytes Ang-(1–7) showed a smaller peak in the intracellular Ca2+ transient and slower Ca2+ kinetics, which was explained by decreased expression of SERCA2a [40]. This suggests that Ang-(1–7) modulates Ca2+ handling by increasing NO and eNOS activity leading to activation of SERCA2a. In nephrectomized mice, Ang-(1–7) prevented left ventricular remodeling and preservation of cardiac function, and also diminished interstitial fibrosis by reducing the levels of TGF-b and increasing MMP2 and 9 [41]. Ang-(1–7) also decreased the expression levels of inflammatory cytokines and suppressed oxidative damage [41]. In Sprague-Dawley rats, coinfusion of Ang II and Ang-(1–7) blocks the cardiac phosphorylation of ERK1/2 and Rho kinases induced by Ang II alone and furthermore, the effects of Ang-(1–7) were abolished in the presence of the Mas antagonist A-779 [42]. Interestingly, Ang-(1–7) also activated STAT3 and STAT5a/b in the heart, however, this effect was not abolished by A-779, but was by an AT
1R
antagonist suggesting its involvement. Akhtar et al. demonstrated the capacity of Ang-(1–7) to attenuate phosphorylation of ERK1/2 and p38 in diabetic rats [43]. Ang-(1–7) has also been shown to promote cardiac recovery following ischemia/reperfusion injury [44,45].
Cardiac ACE2/Ang-(1–9)/AT2R axisAng-(1–9) has more recently been described as a new effector peptide of the counter-regulatory
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin
Ang-(1–7)
αβγ
ANPBNP
HypertrophyFibrosis
NFκB
TGF-β1
(p) ERK1/2(p) JNK(p) p38
(p)PI3K/AKT
(p)eNOS
Mas
Figure 4. Cell signaling of Ang-(1–7) at the Mas receptor in cardiomyocytes and fibroblasts. Through coupling to the Mas receptor, Ang-(1–7) promotes phosphorylation of the PI3K/Akt signaling pathway, resulting in increased eNOS phosphorylation. Ang-(1–7) is also able to block Ang II-induced increases in cardiac ANP and BNP as well as increased levels of NFkB and TGF-b, blocking cardiac hypertrophy and fibrosis. Ang-(1–7) has also been shown to block phosphorylation of ERK1/2, JNK and p38. ANP: Atrial natriuretic peptide; BNP: Brain natriuretic peptide; eNOS: Endothelial nitric oxide synthase.

Author Pro
of
www.futuremedicine.com 7future science group
RAS, counteracting the actions of Ang II [46]. ACE2 generates Ang-(1–9) via the cleavage of the terminal amino acid leucine from Ang I [13] (Figure 1). Alternatively, formation of Ang-(1–9) in human heart extracts/homegenates is also possible through the activity of carboxypeptidase A or cathepsin A, which convert Ang I to Ang-(1–9) [47,48]. Ang-(1–9) is also formed in the heart of ACE- and ACE2-null mice through the cleavage of Ang I by carboxypeptidase A [49]. Ang-(1–9) can consequently generate Ang-(1–7) through the cleavage of two N-terminal amino acids, phenylalanine and histidine, by ACE (Figure 1). These findings demonstrated that the generation of Ang-(1–9) from Ang I is the central pathway of Ang-(1–9) formation in cardiomyocytes, in addition to suggesting that Ang-(1–9) is an active peptide that may mediate its effects through inhibition of Ang II formation [47,49]. Furthermore, circulating levels of Ang-(1–9) and ACE2 are elevated in a rat model of early MI [50]. These results raised the possibility of an additional axis featuring Ang-(1–9) and ACE2 counteracting that of ACE catalyzing the formation of Ang II in the classical RAS. In humans, circulating Ang-(1–9) levels are low; however, in patients with heart failure the myocardium forms as much as 170 nM of Ang I, of which 85% is converted to equivalent levels of Ang-(1–9) and Ang II [47]. Ang-(1–9) has also been shown to potentiate bradykinin B
2 receptor
activity, by enhancing the release of arachidonic acid in human atria and ventricles induced by a B
2 agonist [51,52]. It also enhances intracellular
Ca2+ concentration and NO synthesis triggered by exposure of endothelial cells to bradykinin [51]. Importantly, it has also been shown that Ang-(1–9) is an active peptide and that it is more potent in potentiating the effects of bradykinin than Ang-(1–7) [48,51]. These studies were the first to suggest Ang-(1–9) as an active peptide.
Ocaranza et al. provided the first evidence of an anti-hypertrophic role for Ang-(1–9) in cultured neonatal cardiac myocytes and a rat MI model [46]. They also reported an increase in Ang-(1–9) plasma levels when rats that had undergone MI were infused with AT
1R
antagonists or ACE inhibitors, suggesting that Ang-(1–9) may be an endogenous component of the counter-regulatory RAS. Furthermore, it was established that the action of Ang-(1–9) does not occur through Mas and is thus independent to that of Ang-(1–7). Later in vitro studies with primary adult rabbit cardiomyocytes and rat neonatal H9c2 cardiomyocytes demonstrated that Ang-(1–9) mediates its anti-hypertrophic
effects via engagement of the AT2R, highlighting
this peptide as an important additional member of the counter-regulatory RAS with receptor-specific effects [53] (Figure 3). The AT
2R antagonist
PD123,319 blocked the anti-hypertrophic effects of Ang-(1–9), while the Mas antagonist A779 did not, and the opposite effect was observed for Ang-(1–7). These findings indicated that Ang-(1–9) signals independently of Ang-(1–7) and its anti-hypertrophic effects are produced via direct AT
2R activity. Furthermore, it has also been
proposed that inhibition of the pro-hypertrophic Rho signaling pathway may be implicated in the downstream effects of Ang-(1–9) as inhibition of Rho kinase in DOCA-sodium hypertensive rats significantly increased Ang-(1–9) plasma levels [54] (Figure 3). This was the first study to elucidate potential downstream signaling partners of Ang-(1–9), which will be an important area for future research.
The counter-regulatory effects of Ang-(1–9) infusion were also recently studied in SHRSP. Infusion of Ang-(1–9) using osmotic mini-pumps had a negligible effect on blood pressure, cardiac function and left ventricular mass, however, cardiac fibrosis was reduced by almost 50%, which was attributed to a decrease in perivascular and interstitial deposits of collagen fibers, specifically collagen I and not collagen III [55]. These changes were blocked by antagonizing the AT
2R with PD123,319 with
similar effects observed in primary neonatal rat cardiac fibroblasts, demonstrating that Ang-(1–9) was also able to mediate an antiproliferative effect on fibroblasts via AT
2R [55] (Figure 3).
These data provide further evidence to support an anti-fibrotic role for the action of AT
2R,
which is independent of changes in systemic blood pressure and cardiac hypertrophy [56,57]. Further work on the function of Ang-(1–9) at AT
2R is required since there is still a possibility
for conversion to Ang-(1–7), which has been shown to function via both Mas and AT
2R
in the setting of AT1R blockade [58,59]. Hence
clear delineation of the functions of these two peptides will further highlight their roles in the RAS.
Vascular ACE2/Ang-(1–7)/Mas axisIn addition to its protective effects in the heart, the ACE2/Ang-(1–7)/Mas axis of the RAS has been shown to play an important role in the vasculature by inhibiting the pathological actions of Ang II. ACE2 is expressed in the vasculature of humans and rodents, in both endothelial and smooth muscle cells and its activity has been
Cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system Review

Author Pro
of
Future Cardiol. (2013) 9(1)8 future science group
associated with improved vascular function and reduced vascular remodeling [13,60–62]. The link between ACE2 activity and reduced atherosclerosis has been of great interest over the last few years. In 2008 it was reported that equivalent ACE2 expression was present in both non-diseased and atherosclerotic human carotid arteries, although enzyme activity was altered in atherosclerosis [62]. ACE2 activity was decreased in advanced but stable atherosclerotic lesions in comparison to early-stage atherosclerosis or unstable lesions containing thrombosis, demonstrating differential activity of ACE2 at different stages of the disease. While it is currently unclear how this differential activity is regulated, it may be that ACE2 is increased in the early stages of atherosclerosis and in unstable lesions in an attempt to control the disease pathology and lessen injury. Additionally, overexpression of ACE2 using adenoviral vectors has been shown to reduce atherosclerotic lesion size in ApoE-/- mice [63], stabilize existing atherosclerotic lesions and inhibit the progression of lesion development at early stages of atherosclerosis, but not at advanced stages of the disease [64], suggesting a key role for this enzyme in atherosclerosis. The role of ACE2 in atherosclerosis was further investigated recently in ApoE-/- mice that were also deficient in ACE2. In this study it was found that ACE2 deficiency increased atherosclerotic lesion size in ApoE-/- mice associated with increased inflammation, expression of MMP 2 and 9 and growth of VSMC [65]. In summary, ACE2 plays an important, protective role in the vasculature and is a novel therapeutic target for vascular disease.
Various in vitro studies have shown that Ang-(1–7) interaction with the Mas receptor inhibits proliferation and migration of VSMCs, acts as a vasodilator and inhibits thrombosis via a number of different mechanisms including release of prostaglandins such as prostaglandin I2 and E2, which results in increased cAMP levels and inhibition of cyclooxygenases and various MAPK pathways, including inhibition of ERK1/2 [66,67] (Figure 5). These anti-proliferative and anti-migratory effects have been found to contribute to reduced vascular remodeling in a number of in vivo models of vascular disease. For example, Ang-(1–7) acting via the Mas receptor has been shown to reduce neointimal formation in a rat balloon injury model [68], and following stent implantation in rats [69]. It has also been associated with reduced vascular remodeling following angioplasty in rabbits [70] and reduced atherosclerotic lesion size
in ApoE-/- mice [71]. Additionally, Ang-(1–7) signaling at the Mas receptor improves vascular dysfunction associated with diabetes-induced transactivation of EGFR, an important tyrosine kinase receptor involved in VSMC growth and migration, and vascular remodeling [43]. In streptozotocin-induced diabetic rats it was found that administration of Ang-(1–7) inhibited glucose- and Ang II-induced phosphorylation of the EGFR to similar levels as an EGFR antagonist (AG1478), associated with a reduction in Src-dependent EGFR transactivation. In this study the protective effects of Ang-(1–7) were inhibited in the presence of the Mas receptor antagonist A-779 [43]. Furthermore, the nonpeptide agonist of the Mas receptor, AVE0991, has recently been shown to inhibit VSMC proliferation [72]. These studies clearly show a protective role for Ang-(1–7) acting via the Mas receptor in vascular remodeling and highlight this axis as a novel therapeutic target that could be exploited clinically to improve vascular diseases underpinned by remodeling.
Moreover, Ang-(1–7), acting via the Mas receptor, also acts as a vasodilator and improves vascular endothelial function [73,74]. This has been shown to be due to increased production of NO via reciprocal phosphorylation/dephosphorylation at serine 1177/threonine495 of the eNOS enzyme, increasing its activity and leading to sustained Akt signaling (Figure 5) [75]. It is also worth highlighting that Ang-(1–7) has also been reported to modulate vascular function by acting at AT
2R, when co-incubated in the
presence of an AT1R antagonist [58,59], therefore
indicating that Ang-(1–7) function requires more research.
Vascular ACE2/Ang-(1–9)/AT2R axisTo date, the majority of the effects of ACE2 in the vasculature have been attributed to the generation of Ang-(1–7). However, following the recent identification of Ang-(1–9) as a functional peptide of the counter-regulatory RAS [53] and the previously reported beneficial effects of this peptide in the vasculature [48] it would not be unreasonable to suggest that some of the vasculoprotective effects of ACE2 discussed in the previous section could also be due to the production and activity of Ang-(1–9), although further work is required to provide conclusive evidence. Ocaranza et al. have suggested that production of Ang-(1–9) as opposed to Ang-(1–7) by ACE2 is the main peptide involved in counter-regulating the effects of Ang II and have recently shown
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin

Author Pro
of
www.futuremedicine.com 9future science group
that stimulation of the ACE2/Ang-(1–9) axis by inhibition of the RhoA/Rho-associated, coiled-coil containing protein kinase (ROCK) signaling pathway (which is integrally involved in vascular remodeling), resulting in increased activity and expression of ACE2 in the aorta and increased Ang-(1–9) plasma levels, reduced blood pressure and vascular remodeling [50,54]. Additionally, inhibition of the RhoA/ROCK signaling pathway resulted in reduced ACE activity and Ang II levels, and increased expression of eNOS, identifying that the ACE2/Ang-(1–9) axis may be a novel protective interaction in the vasculature.
Recently it was reported that Ang-(1–9) infusion in SHRSP improved aortic vasorelaxation and NO bioavailability, via AT
2R
[55]. While the mechanisms involved are currently unknown it was previously demonstrated that Ang-(1–9) may increase NO bioavailability by stimulating bradykinin release in cardiac endothelial cells (Figure 5) [48]. Additionally, Ang-(1–9) infusion resulted in an increase in
NADPH oxidase 4 expression in a PD123,319-sensitive manner [55]. In a recent study it was demonstrated that transgenic overexpression of NADPH oxidase 4 in the mouse endothelium promoted vasodilation via the release of hydrogen peroxide independent of NO activity, suggesting this may be one potential mechanism [76]. Taken together, these recent findings have identified another potential mechanism involved in the Ang-(1–9) /AT
2R interaction in the vasculature
that requires further investigation.
Recent therapeutic approaches in the counter-regulatory RAS axis
Angiotensin-converting enzyme 2Two approaches to augmenting endogenous ACE2 activity have been pursued. One potential, novel anti-hypertensive agent is 1-[(2-dimethylamino) ethylamino]-4-(hydroxymethyl)-7-[(4-methylphenyl) sulfonyl oxy]-9H-xanthene-9-one (XNT), a small molecule activator of ACE2, which has been shown to reverse hypertension-induced cardiac
AT2R
MKP-1 PP2ASHP-1
BK2R
PKA
eNOSP
NO
VASODILATION MAPK
↓ PROLIFERATION↓MIGRATION
AT2R
MKP-1 PP2ASHP-1
BK2R
PKA
eNOSP
eNOSP
NO
VASODILATION MAPK
↓ PROLIFERATION↓MIGRATION
Mas
PI3K
AktP
NO
eNOSP
PLA2
VASODILATION
AA
PGI2
cAMP
MAPK
↓ PROLIFERATION↓MIGRATION
PGE2
Mas
PI3K
AktP
AktP
NO
eNOSP
eNOSP
PLA2
VASODILATION
AA
PGI2
cAMP
MAPK
↓ PROLIFERATION↓MIGRATION
PGE2
BK2R AT2RMas
PKA
eNOS
eNOS
NO
NO
Vasodilation
Vasodilation
MKP-1 SHP-1 PP2A
PGI2 PGE2
cAMP
MAPK
PLA2
MAPK
ProliferationMigration
ProliferationMigration
PI3K
AktAA
P
P
P
Figure 5. Signaling via the AT2R and Mas receptor in the vasculature. Activation of vascular AT2R results in NO production by
stimulating phosphorylation of eNOS via crosstalk with BK2R, promoting vasodilation. Activation of protein phosphatases including
MKP-1, SHP-1 and PP2A has been shown to inhibit vascular smooth muscle cell proliferation and migration via inhibition of MAPK signaling pathways [3, 4]. Mas receptor signaling promotes vasodilation via activation of the PI3K/Akt signaling pathway resulting in increased phosphorylation of eNOS and production of NO. Additionally, stimulation of PLA
2 activity via Mas receptor activation increases
the release of AA, which in turn produces the prostanoids PGI2 and PGE
2. PGI
2 and PGE
2 increase levels of cAMP which promotes
vasodilation and inhibition of MAPK activity, and therefore reduce vascular smooth muscle cell proliferation and migration. Mas receptor signaling also directly inhibits MAPK activity to produce its anti-proliferative and anti-migratory effects [66, 67, 75]. AA: Arachidonic acid; AT
2R: Angiotensin type 2 receptor; BK2R: bradykinin type 2 receptor; eNOS: Endothelial nitric oxide synthase; NO:
Nitric oxide.
Cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system Review

Author Pro
of
Future Cardiol. (2013) 9(1)10 future science group
fibrosis in spontaneously hypertensive rats [77]. Furthermore, chronic treatment with XNT effectively prevented pulmonary remodeling, right ventricular hypertrophy and fibrosis in a monacrotaline-induced pulmonary hypertension rat model [78]. In addition, ACE2 activation via XNT also reduced platelet attachment and had an anti-thrombotic effect in the vessels of the spontaneously hypertensive rat [79]. More recently, chronic oral administration of XNT improved cardiac function, whilst attenuating cardiac hypertrophy and fibrosis in a model of diabetes-induced cardiac dysfunction [37]. Unfortunately, it is unlikely that XNT will reach clinical trials due to a lack of efficient solubility. However, human recombinant ACE2 shows promise in a preclinical model of pressure overload heart failure [80], and a clinical derivative of ACE2, APN01, co-developed by Apeiron (Vienna, Austria) and GlaxoSmithKline, is currently being investigated in clinical trials for treatment of heart failure and acute respiratory distress syndrome (www.apeiron-biologics.com). Therefore, augmentation of ACE2 activity remains an appealing therapeutic target. It does remain to be seen whether application of recombinant ACE2 can provide sustained benefit, as clearly immune responses to the administered protein may only facilitate application in acute treatment settings. However, gene therapy applications such as those already described experimentally may offer future potential for treatment of chronic conditions [63,64].
Angiotensin-(1–7)A stable oral formulation of Ang-(1–7), hydroxypropyl b-cyclodextrin (HPbCD)/Ang-(1–7), has been developed [81], which is critical for therapeutic application as the natural form of Ang-(1–7) is rapidly inactivated via ACE activity. This agent produced an anti-thrombotic effect in Mas-deficient mice [82] and also cardioprotective effects in rat models of MI and isoproterenol-induced heart failure [83]. Moreover, the inclusion of Ang-(1–7) in cyclodextrin (HPbCD) prevented the degradation of Ang-(1–7) within the GI tract, which is a common problem associated with orally administered peptides [83]. This peptide is highly hydrosoluble, which may lead to washing out effects; however this may be advantageous as data suggests that HPbCD is secreted in the feces whilst the biologically active Ang-(1–7) enters the circulation [83]. Furthermore, the long-term administration of this oral Ang-(1–7) formulation is feasible as the beneficial
cardioprotective effects of this peptide continued to reduce the maladaptive cardiac remodeling response induced by MI [84].
A further oral formulation is cyclic Ang-(1–7) (cAng-[1–7]), an analogue of Ang-(1–7), which contains an enzymatically introduced thioether-ring bridged to Ang-(1–7) [85]. cAng-(1–7) is stable in plasma and tissue homogenates from different organs as it is fully resistant against ACE degradation. Cyclized Ang-(1–7) induced relaxation of precontracted rat aorta rings, which was abolished by the Ang-(1–7) receptor antagonist, D-Pro(7)-Ang-(1–7), demonstrating that this analogue of Ang-(1–7) also reacts with Mas [85]. Recently, chronic intravenous infusion of cAng-(1–7) in a rat model of MI reduced left ventricular mass and diastolic pressure whilst also improving aortic endothelial function [86]. Similarly, with oral administration of cAng-(1–7), intravenous infusion appears to have superior pharmacologic properties to that of natural Ang-(1–7). In contrast to intravenous and subcutaneous administration, with oral delivery, bioavailability of cAng-(1–7) was poor, however, pulmonary delivery looked promising [87]. Further improvement of this formulation is required for safe and effective oral administration in the clinical setting.
It is worth highlighting that, while not a cardiovascular application, Ang-(1–7) has been tested as a first-in-class antiangiogenic drug with activity for treating cancer that is linked to reduction of circulating plasma placental growth factor (PIGF) levels [88]. A Phase II study of single-agent Ang-(1–7) in patients with sarcoma is ongoing. Seminal studies showed that Ang-(1–7) inhibits tumor angiogenesis and proliferation in xenograft tumors via a reduction in angiogenic factors, including VEGF and PIGF, to attenuate vascularization [89,90]. Proliferation of cancer-associated fibroblasts and tumour fibrosis were also reduced by Ang-(1–7) treatment, partially due to reduced TGF-b and an increase in the MAPK phosphatase, DUSP1 [91]. Furthermore, in the clinic, Ang-(1–7) also has therapeutic benefits after myelosuppressive cancer treatment. Following chemotherapy and radiotherapy, Ang-(1–7) infusion enhanced hematopoietic recovery with levels of multi-lineage hematopoietic progenitors in bone marrow restored to baseline and increased leukocyte and platelet numbers [92,93]. TXA127, an injectable agent containing Ang-(1–7) as the active ingredient, reduced thrombocytopenia in a previous clinical study [94] and is currently in Phase II clinical trials for
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin

Author Pro
of
www.futuremedicine.com 11future science group
cancer therapy following hematopoietic stem cell transplantation in humans.
Mas agonistsThe first synthetic nonpeptide agonist developed to mimic the effects of Ang-(1–7) was AVE 0991, an agonist of Mas [95]. It is an insoluble compound and can be delivered orally [96]. AVE 0991 is protective against cardiac dysfunction and remodeling in an isoproterenol-induced model of heart failure and also in diabetic rats [96,97]. Endothelial function in normotensive rats was also improved with AVE 0991 administration by increasing the hypotensive effect of bradykinin [98]. In a rat model of MI, improved cardiac function and attenuated ventricular remodeling was attributed to AVE 0991-mediated inhibition of inflammatory cytokines TGF-b and TNF-a [99]. In addition to improving cardiac parameters, AVE 0991 has also recently been shown to have beneficial effects on atherogenesis in ApoE-/- mice [100], whilst also reducing dyslipidaemia in a rat model of diabetes [101].
Other Mas-specific agonists include CGEN-856, its derivative CGEN-856S and CGEN-857, which were identified by a computational discovery platform for screening naturally occurring GPCR ligands [102]. These compounds are unrelated in sequence to angiotensin or its associated peptides, and CGEN-856S in particular was identified as having similar beneficial cardiac effects to that of Ang-(1–7) and appears to bind the Mas receptor at the same binding domain as Ang-(1–7) [103]. CGEN-856S induced vasodilation in rat and mouse thoracic aortic rings, promoted an antiarrhythmogenic effect in isolated rat hearts, and had an antihypertensive effect in spontaneously hypertensive rats [103]. The possibility of CGEN-856S as a cardiovascular therapeutic is currently being commercially explored by BioLineRx Ltd.
AT2R agonistsMost of the prescribed medication to inhibit the RAS is dominated by inhibitors of AT
1R,
however, work has also focused on AT2R,
culminating in the development of the AT2R
agonist, Compound 21 (C21). C21 is an orally active drug and selective agonist for AT
2R,
which was first studied in a rat model of MI [104]. A total of 7 days after MI, rats treated with C21 showed diminished scar size and significant improvements in systolic and diastolic function. The histological ana lysis of the infarct tissue showed that C21 effects on MI may be due to anti-apoptotic and anti-inflammatory effects
[104]. In contrast to these findings, Jehle et al. showed no effect of C21 on a mouse model of occlusion–reperfusion [105]. In this study, the left anterior descending coronary artery of mice treated with C21 via osmotic minipumps was occluded for 1 h followed by reperfusion. A total of 28 days after MI, C21-treated mice showed a detrimental effect on cardiac function, with increased left ventricular end diastolic and end systolic volume index and a decrease in left ventricular end-systolic and end-diastolic pressure.
C21 has also been shown to have a role in vascular remodeling. Infusion of C21 into SHRSP for 6 weeks reduced vascular stiffness as well as reducing collagen content in aortic media and myocardium [106]. In this model, C21 also reduced the expression of myosin heavy chain-b and a-skeletal muscle actin, suggesting a role for C21 in cardiac hypertrophy. In accordance with this, Paulis et al. also showed a protective effect of C21 in N(w)-nitro-l-arginine-methyl ester (L-NAME)-induced hypertension in rats [107]. C21 was able to blunt the increase in wall thickness and stiffness of the aorta as well as reduce the pulse wave velocity induced by L-NAME. However, in both cases C21 did not reduce blood pressure, highlighting the tissue specificity of the RAS.
ConclusionThe dysregulation of the RAS is important in the development of various CVDs, with the role of Ang II, AT
1R and ACE well recognized within
the cardiovascular system. The ACE2/Ang-(1–7)/Mas axis is established as a central counter-regulatory pathway of the classical RAS, and recently the ACE2/Ang-(1–9 )/AT
2R axis has
emerged as another potential counter-regulatory pathway. In general, the counter-regulatory pathways of the RAS protect the cardiovascular system from the pathophysiological effects of the classical RAS. However, further understanding of the cell signaling of these peptides through their respective receptors is required for the development of effective therapeutic targets. Ang-(1–7) signaling via Mas is well established and recent evidence indicates that Ang-(1–9) signals through the AT
2R, but little is known
about the downstream molecular mechanism of action for either AT
2R or Ang-(1–9). In
addition, further translational knowledge of the different components of the counter-regulatory RAS is critical. Minimal patient information is available regarding how levels change in the clinical cardiovascular setting, which would be
Cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system Review

Author Pro
of
Future Cardiol. (2013) 9(1)12 future science group
essential for the future development of this new avenue of cardiovascular research.
Future perspectiveOver the past decade, the counter-regulatory RAS has featured the ACE2/Ang-(1–7)/Mas axis as the central pathway that counteracts the pathophysiological effects of the RAS. The past few years have seen the emergence of the ACE2/Ang-(1–9)/AT
2R counter-regulatory axis with
cardioprotective effects, opposing the classical RAS. However, there still remains a great deal to investigate to further increase our understanding in this important area of cardiovascular research. Although important data has been generated via peptide infusion to rodent models and observational data generated for changing levels of RAS components in patients on drug therapy,
there remains a lack of definitive evidence on how changing endogenous RAS peptide and enzyme levels influence CVD phenotypes. This will be important for future research. Currently, specific Ang-(1–7) formulations are in clinical trials for pre-eclampsia and MI, and the outcome of these trials is eagerly anticipated in order to determine the precise cardiovascular benefits of Ang-(1–7) for the patient. More trials, however, are required to establish whether Ang-(1–7) is an effective clinical therapeutic in CVDs such as atherosclerosis and coronary artery disease. Ang-(1–9) remains to be tested using these approaches.
In addition to functional interactions between the RAS peptides and receptors, there is mounting evidence to suggest that the receptors can interact directly, and that these functional interactions are important for receptor function/
Executive summary
The counter-regulatory axis of the renin–angiotensin system�n The pathophysiological effects of the renin–angiotensin system (RAS) are opposed by protective effects of the counter-regulatory axis.
Cardiac ACE2/Ang-(1–7)/Mas axis�n The cardiac ACE2/Ang-(1–7)/Mas axis is the central counter-regulatory pathway, and its cardioprotective effects are mediated through
the action of Ang-(1–7) via its receptor, Mas.�n Ang-(1–7) signaling via Mas attenuates cardiac remodeling, reduces levels of inflammatory cytokines and TGF-β, and phosphorylation
of ERK1/2 and p38.
Cardiac ACE2/Ang-(1–9)/AT2R axis�n The cardiac ACE2/Ang-(1–9)/Mas axis has recently been proposed as an additional counter-regulatory axis through the actions of
Ang-(1–9), via its receptor AT2R.
�n Ang-(1–9) has an anti-hypertrophic role in neonatal cardiomyocytes and in a rat myocardial infarction model, and inhibits cardiac fibrosis in a rat model (SHRSP) of essential hypertension.
Vascular ACE2/Ang-(1–7)/Mas axis�n ACE2 activity improves vascular function and reduces vascular remodeling and atherosclerosis development.�n Ang-(1–7) exerts anti-proliferative and anti-migratory effects on vascular smooth muscle cells.�n Increased nitric oxide production, stimulated by Ang-(1–7), improves vascular endothelial function.
Vascular ACE2/Ang-(1–9)/AT2R axis�n Inhibition of the RhoA/ROCK signaling pathway stimulates increased aortic ACE2 activity and circulating Ang-(1–9) levels, reducing
blood pressure and vascular remodeling.�n Ang-(1–9) infusion in SHRSP improves aortic vasorelaxation and nitric oxide bioavailability, via AT
2R.
Recent therapeutic approaches in the counter-regulatory RAS axisAngiotensin converting enzyme 2 �n The ACE2 activator, XNT, improves cardiac function, and prevents cardiac fibrosis and hypertrophy in animal models. Human
recombinant ACE2 is currently in trials for heart failure.Angiotensin-(1–7)�n HPβCD and cAng-(1–7) are modified formulations of Ang-(1–7) with cardioprotective properties that can be administered effectively
via oral or pulmonary delivery, respectively.Mas agonists�n The Mas agonists, AVE0991 and CGEN-856S, have protective effects within the heart and vasculature in preclinical studies.
AT2R agonists�n An oral AT
2R agonist, C21, reduces infarct size in myocardial infarction and reduces vascular stiffness in hypertensive rats.
Conclusion�n The counter-regulatory pathway of the RAS, featuring the ACE2/Ang-(1–7)/Mas axis and the recently identified ACE2/Ang-(1–9)/AT
2R
axis, protects the cardiovascular system from the (patho)physiological effects of the RAS and is a therapeutic target in cardiovascular disease.
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin

Author Pro
of
www.futuremedicine.com 13future science group
Financial & competing interests disclosureWork in the authors’ laboratory is supported by the British Heart Foundation (PG/11/43/28901) and Medical Research Council (G0901161), and a Medical Research Council Doctoral Training Grant PhD Studentship. The authors have no other relevant affili-ations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
signaling and to our understanding of the RAS. For example, AT
1R forms heterodimers
with receptors from the kinin–kallikrein [108], adrenergic [109] and dopaminergic [110] systems, and recently the cannabanoid family [111], resulting in enhanced Ang II potency at the AT
1R. Conversely, direct receptor interactions
between AT1R and AT
2R, Mas or apelin receptors
have been reported to antagonize AT1R activity
[112–115]. This area remains relatively unexplored but may have the potential to drive discovery of new pharmacological interventions in the RAS. Furthermore, the recent identification of new RAS peptides, including Ang-(1–12) [116] and the identification of Ang III as an AT
2R agonist
in the kidney [117], suggests that it is conceivable that other new RAS peptides will continue to be discovered over the next decade.
The RAS has come a long way in the 100 or so years since actions attributable to Ang II were
first recognized and drugs that antagonize Ang II generation or function are current mainstays in the treatment of varied cardiovascular diseases. Many new and exciting discoveries in the RAS have arisen in the past decade so it will be exciting to observe how this field develops over the next 10 years.
ReferencesPapers of special note have been highlighted as:n of interestnn of considerable interest
1. Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 15(1), 75–83 (2009).
2. Huang F, Thompson JC, Wilson PG, Aung HH, Rutledge JC, Tannock LR. Angiotensin II increases vascular proteoglycan content preceding and contributing to atherosclerosis development. J. Lipid Res. 49(3), 521–530 (2008).
3. Mukoyama M, Nakajima M, Horiuchi M, Sasamura H, Pratt RE, Dzau VJ. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J. Biol. Chem. 268(33), 24539–24542 (1993).
4. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292(1), C82–C97 (2007).
5. Bartunek J, Weinberg EO, Tajima M, Rohrbach S, Lorell BH. Angiotensin II type 2 receptor blockade amplifies the early signals of cardiac growth response to angiotensin II in hypertrophied hearts. Circulation 99(1), 22–25 (1999).
6. Metcalfe BL, Huentelman MJ, Parilak LD et al. Prevention of cardiac hypertrophy by angiotensin II type-2 receptor gene transfer. Hypertension 43(6), 1233–1238 (2004).
7. Yan X, Schuldt AJ, Price RL et al. Pressure overload-induced hypertrophy in transgenic mice selectively overexpressing AT2 receptors
in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 294(3), H1274–H1281 (2008).
8. Taguchi K, Matsumoto T, Kobayashi T. Angiotensin II type 2 receptor- dependent increase in nitric oxide synthase activity in the endothelium of db/db mice is mediated via a MEK pathway. Pharmacol. Res. 66, 41–50 (2012).
9. Ohtsu H, Higuchi S, Shirai H et al. Central role of Gq in the hypertrophic signal transduction of angiotensin II in vascular smooth muscle cells. Endocrinology 149(7), 3569–3575 (2008).
10. McMurray JJ. Val-HeFT: do angiotensin-receptor blockers benefit heart failure patients already receiving ACE inhibitor therapy? Nat. Clin. Pract. Cardiovasc. Med. 2(3), 128–129 (2005).
11. Steckelings UM, Paulis L, Unger T, Bader M. Emerging drugs which target the renin-angiotensin-aldosterone system. Expert Opin. Emerg. Dr. 16(4), 619–630 (2011).
12. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme – cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 275(43), 33238–33243 (2000).
13. Donoghue M, Hsieh F, Baronas E et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 87(5), E1–E9 (2000).
14. Vickers C, Hales P, Kaushik V et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 277(17), 14838–14843 (2002).
15. Keidar S, Kaplan M, Gamliel-Lazarovich A. ACE2 of the heart: From angiotensin I to angiotensin (1–7). Cardiovasc. Res. 73(3), 463–469 (2007).
16. Trask AJ, Averill DB, Ganten D, Chappell MC, Ferrario CM. Primary role of angiotensin-converting enzyme-2 in cardiac production of angiotensin-(1–7) in transgenic Ren-2 hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 292(6), H3019–H3024 (2007).
17. Crackower MA, Sarao R, Oudit GY et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417(6891), 822–828 (2002).
18. Kassiri Z, Zhong J, Guo D et al. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ. Heart Fail. 2(5), 446–455 (2009).
19. Patel VB, Bodiga S, Fan D et al. Cardioprotective effects mediated by angiotensin II type 1 receptor blockade and enhancing angiotensin 1–7 in experimental heart failure in angiotensin-converting enzyme 2-null mice. Hypertension 59(6), 1195–1203 (2012).
20. Masson R, Nicklin SA, Craig MA et al. Onset of experimental severe cardiac fibrosis is mediated by overexpression of angiotensin-converting enzyme 2. Hypertension 53(4), 694–700 (2009).
21. Donoghue M, Wakimoto H, Maguire CT et al. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J. Mol. Cell Cardiol. 35(9), 1043–1053 (2003).
22. Santos RA, Simoes e Silva AC, Maric C et al. Angiotensin-(1–7) is an endogenous ligand
Cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system Review

Author Pro
of
Future Cardiol. (2013) 9(1)14 future science group
for the G protein-coupled receptor Mas. Proc. Natl Acad. Sci. USA 100(14), 8258–8263 (2003).
23. Zisman LS, Meixell GE, Bristow MR, Canver CC. Angiotensin-(1–7) formation in the intact human heart: in vivo dependence on angiotensin II as substrate. Circulation 108(14), 1679–1681 (2003).
24. Averill DB, Ishiyama Y, Chappell MC, Ferrario CM. Cardiac angiotensin-(1–7) in ischemic cardiomyopathy. Circulation 108(17), 2141–2146 (2003).
25. Welches WR, Brosnihan KB, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase Life Sci. 52(18), 1461–1480 (1993).
26. Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1–7): an evolving story in cardiovascular regulation. Hypertension 47(3), 515–521 (2006).
27. Ferrario CM. ACE2: more of Ang-(1–7) or less Ang II? Curr. Opin. Nephrol. Hypertens. 20(1), 1–6 (2011).
28. Santos RA, Campagnole-Santos MJ, Baracho NC et al. Characterization of a new angiotensin antagonist selective for angiotensin-(1–7): evidence that the actions of angiotensin-(1–7) are mediated by specific angiotensin receptors. Brain Res. Bull. 35(4), 293–298 (1994).
29. Nadu AP, Ferreira AJ, Reudelhuber TL, Bader M, Santos RA. Reduced isoproterenol-induced renin-angiotensin changes and extracellular matrix deposition in hearts of TGR(A1–7)3292 rats. J. Am. Soc. Hypertens. 2(5), 341–348 (2008).
30. Santiago NM, Guimaraes PS, Sirvente RA et al. Lifetime overproduction of circulating Angiotensin-(1–7) attenuates deoxycorticosterone acetate-salt hypertension-induced cardiac dysfunction and remodeling. Hypertension 55(4), 889–896 (2010).
31. Mercure C, Yogi A, Callera GE et al. Angiotensin(1–7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ. Res. 103(11), 1319–1326 (2008).
nn� The first paper to show a direct effect of Ang-(1–7) on the heart by transgenic overexpression and highlighted downstream signaling pathways.
32. Ferreira AJ, Santos RA, Bradford CN et al. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension 55(2), 207–213 (2010).
33. Wang Y, Qian C, Roks AJ et al. Circulating rather than cardiac angiotensin-(1–7)
stimulates cardioprotection after myocardial infarction. Circ. Heart Fail. 3(2), 286–293 (2010).
n� First demonstration that Ang-(1–7) may modulate endothelial progenitor cell function and homing to the infarcted heart.
34. Ribeiro-Oliveira A Jr, Nogueira AI, Pereira RM, Boas WW, Dos Santos RA, Simoes e Silva AC. The renin–angiotensin system and diabetes: an update. Vasc. Health Risk Manag. 4(4), 787–803 (2008).
35. Hsueh WA, Wyne K. Renin–angiotensin–aldosterone system in diabetes and hypertension. J. Clin. Hypertens. (Greenwich) 13(4), 224–237 (2011).
36. Patel VB, Bodiga S, Basu R et al. Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: a critical role of the angiotensin II/AT1 receptor axis. Circ. Res. 110(10), 1322–1335 (2012).
37. Murca TM, Moraes PL, Capuruco CA et al. Oral administration of an angiotensin-converting enzyme 2 activator ameliorates diabetes-induced cardiac dysfunction. Regul. Pept. 177(1–3), 107–115 (2012).
38. Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1–7) prevents diabetes-induced cardiovascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 292(1), H666–H672 (2007).
39. Yousif MH, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF. Characterization of Angiotensin-(1–7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol. Res. 66(3), 269–275 (2012).
40. Dias-Peixoto MF, Santos RA, Gomes ER et al. Molecular mechanisms involved in the angiotensin-(1–7)/Mas signaling pathway in cardiomyocytes. Hypertension 52(3), 542–548 (2008).
nn� Detailed analysis of the cell signaling mechanisms for Ang-(1–7).
41. Li Y, Wu J, He Q et al. Angiotensin (1–7) prevent heart dysfunction and left ventricular remodeling caused by renal dysfunction in 5/6 nephrectomy mice. Hypertens. Res. 32(5), 369–374 (2009).
42. Giani JF, Gironacci MM, Munoz MC, Pena C, Turyn D, Dominici FP. Angiotensin-(1 7) stimulates the phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: role of the AT1 and Mas receptors. Am. J. Physiol. Heart Circ. Physiol. 293(2), H1154–H1163 (2007).
43. Akhtar S, Yousif MH, Dhaunsi GS, Chandrasekhar B, Al-Farsi O, Benter IF. Angiotensin-(1–7) inhibits epidermal growth factor receptor transactivation via a Mas
receptor-dependent pathway. Br. J. Pharmacol. 165(5), 1390–1400 (2012).
44. Zhang Y, Lu J, Shi J et al. Central administration of angiotensin-(1–7) stimulates nitric oxide release and upregulates the endothelial nitric oxide synthase expression following focal cerebral ischemia/reperfusion in rats. Neuropeptides 42(5–6), 593–600 (2008).
45. Al-Maghrebi M, Benter IF, Diz DI. Endogenous angiotensin-(1–7) reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats. Pharmacol. Res. 59(4), 263–268 (2009).
46. Ocaranza MP, Lavandero S, Jalil JE et al. Angiotensin-(1–9) regulates cardiac hypertrophy in vivo and in vitro. J. Hypertens. 28(5), 1054–1064 (2010).
47. Kokkonen JO, Saarinen J, Kovanen PT. Regulation of local angiotensin II formation in the human heart in the presence of interstitial fluid. Inhibition of chymase by protease inhibitors of interstitial fluid and of angiotensin-converting enzyme by Ang-(1–9) formed by heart carboxypeptidase A-like activity. Circulation 95(6), 1455–1463 (1997).
48. Jackman HL, Massad MG, Sekosan M et al. Angiotensin 1–9 and 1–7 release in human heart: role of cathepsin A. Hypertension 39(5), 976–981 (2002).
49. Garabelli PJ, Modrall JG, Penninger JM, Ferrario CM, Chappell MC. Distinct roles for angiotensin-converting enzyme 2 and carboxypeptidase A in the processing of angiotensins within the murine heart. Exp. Physiol. 93(5), 613–621 (2008).
50. Ocaranza MP, Godoy I, Jalil JE et al. Enalapril attenuates downregulation of Angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 48(4), 572–578 (2006).
51. Erdos EG, Jackman HL, Brovkovych V, Tan F, Deddish PA. Products of angiotensin I hydrolysis by human cardiac enzymes potentiate bradykinin. J. Mol. Cell Cardiol. 34(12), 1569–1576 (2002).
52. Marcic B, Deddish PA, Jackman HL, Erdos EG. Enhancement of bradykinin and resensitization of its B2 receptor. Hypertension 33(3), 835–843 (1999).
53. Flores-Munoz M, Smith NJ, Haggerty C, Milligan G, Nicklin SA. Angiotensin1–9 antagonises pro-hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J. Physiol. 589(Pt 4), 939–951 (2011).
n� First demonstration that Ang-(1–9) is a functional agonist at AT
2R.
54. Ocaranza MP, Rivera P, Novoa U et al. Rho
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin

Author Pro
of
www.futuremedicine.com 15future science group
kinase inhibition activates the homologous angiotensin-converting enzyme-angiotensin-(1–9) axis in experimental hypertension. J. Hypertens. 29(4), 706–715 (2011).
55. Flores-Munoz M, Work LM, Douglas K et al. Angiotensin-(1–9) attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rat via the angiotensin type 2 receptor. Hypertension 59(2), 300–307 (2012).
56. Yan X, Price RL, Nakayama M et al. Ventricular-specific expression of angiotensin II type 2 receptors causes dilated cardiomyopathy and heart failure in transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 285(5), H2179–H2187 (2003).
57. D’amore A, Black MJ, Thomas WG. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension 46(6), 1347–1354 (2005).
58. Bosnyak S, Widdop RE, Denton KM, Jones ES. Differential mechanisms of ang (1–7)-mediated vasodepressor effect in adult and aged candesartan-treated rats. Int. J. Hypertens. 2012, 192567 (2012).
59. Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1–7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 45(5), 960–966 (2005).
60. Hamming I, Timens W, Bulthuis MI, Lely AT, Navis GJ, Van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203(2), 631–637 (2004).
61. Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1–7) expression in the aorta of spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 289(3), H1013–H1019 (2005).
62. Sluimer JC, Gasc JM, Hamming I et al. Angiotensin-converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 215(3), 273–279 (2008).
63. Lovren F, Pan Y, Quan A et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 295(4), H1377–H1384 (2008).
64. Dong B, Zhang C, Feng JB et al. Overexpression of ACE2 enhances plaque stability in a rabbit model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28(7), 1270–1276 (2008).
65. Thomas MC, Pickering RJ, Tsorotes D et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 107(7), 888–897 (2010).
nn� Demonstrated the importance of ACE2 activity in the development of atherosclerosis.
66. Jaiswal N, Tallant EA, Jaiswal RK, Diz DI, Ferrario CM. Differential regulation of prostaglandin synthesis by angiotensin peptides in porcine aortic smooth muscle cells: subtypes of angiotensin receptors involved. J. Pharmacol. Exp. Ther. 265(2), 664–673 (1993).
67. Zhang F, Hu Y, Xu Q, Ye S. Different effects of angiotensin II and angiotensin-(1–7) on vascular smooth muscle cell proliferation and migration. PLoS One 5(8), e12323 (2010).
68. Strawn WB, Ferrario CM, Tallant EA. Angiotensin-(1–7) reduces smooth muscle growth after vascular injury. Hypertension 33(1 Pt 2), 207–211 (1999).
69. Langeveld B, Van Gilst WH, Tio RA, Zijlstra F, Roks AJ. Angiotensin-(1–7) attenuates neointimal formation after stent implantation in the rat. Hypertension 45(1), 138–141 (2005).
70. Zeng W, Chen W, Leng X, He JG, Ma H. Chronic angiotensin-(1–7) administration improves vascular remodeling after angioplasty through the regulation of the TGF-b/Smad signaling pathway in rabbits. Biochem. Biophys. Res. Commun. 389(1), 138–144 (2009).
71. Tesanovic S, Vinh A, Gaspari TA, Casley D, Widdop RE. Vasoprotective and atheroprotective effects of angiotensin (1–7) in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 30(8), 1606–1613 (2010).
72. Sheng-Long C, Yan-Xin W, Yi-Yi H et al. AVE0991, a nonpeptide compound, attenuates angiotensin II-induced vascular smooth muscle cell proliferation via induction of heme oxygenase-1 and downregulation of p-38 MAPK phosphorylation. Int. J. Hypertens. 2012, 958298 (2012).
73. Brosnihan KB, Li P, Ferrario CM. Angiotensin-(1–7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension 27(3 Pt 2), 523–528 (1996).
74. Faria-Silva R, Duarte FV, Santos RA. Short-term angiotensin(1–7) receptor MAS stimulation improves endothelial function in normotensive rats. Hypertension 46(4), 948–952 (2005).
75. Sampaio WO, Souza Dos Santos RA, Faria-Silva R, Da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1–7) through
receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 49(1), 185–192 (2007).
76. Ray R, Murdoch CE, Wang M et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 31(6), 1368–1376 (2011).
77. Ferreira AJ, Shenoy V, Qi Y et al. Angiotensin-converting enzyme 2 activation protects against hypertension-induced cardiac fibrosis involving extracellular signal-regulated kinases. Exp. Physiol. 96(3), 287–294 (2011).
78. Ferreira AJ, Shenoy V, Yamazato Y et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am. J. Resp. Crit. Care 179(11), 1048–1054 (2009).
79. Fraga-Silva RA, Sorg BS, Wankhede M et al. ACE2 activation promotes antithrombotic activity. Mol. Med. 16(5–6), 210–215 (2010).
80. Zhong J, Basu R, Guo D et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 122(7), 717–728, 718 p following 728 (2010).
nn� Demonstrated that recombinant ACE2 infusion into a disease model could abrogate adverse cardiac remodeling.
81. Lula I, Denadai AL, Resende JM et al. Study of angiotensin-(1–7) vasoactive peptide and its b-cyclodextrin inclusion complexes: complete sequence-specific NMR assignments and structural studies. Peptides 28(11), 2199–2210 (2007).
82. Fraga-Silva RA, Costa-Fraga FP, De Sousa FB et al. An orally active formulation of angiotensin-(1–7) produces an antithrombotic effect. Clinics (Sao Paulo) 66(5), 837–841 (2011).
83. Marques FD, Ferreira AJ, Sinisterra RD et al. An oral formulation of angiotensin-(1–7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension 57(3), 477–483 (2011).
84. Marques FD, Melo MB, Souza LE et al. Beneficial effects of long-term administration of an oral formulation of angiotensin-(1–7) in infarcted rats. Int. J. Hypertens. 2012, 795452 (2012).
n� Showed for the first time that oral administration of a drug formulation of Ang-(1–7) provided sustained therapeutic benefit in myocardial infarction.
85. Kluskens LD, Nelemans SA, Rink R et al. Angiotensin-(1–7) with thioether bridge: an angiotensin-converting enzyme-resistant, potent angiotensin-(1–7) analog. J.
Cardiovascular remodeling by the counter-regulatory axis of the renin–angiotensin system Review

Author Pro
of
Future Cardiol. (2013) 9(1)16 future science group
Pharmacol. Exp. Ther. 328(3), 849–854 (2009).
86. Durik M, Van Veghel R, Kuipers A et al. The effect of the thioether-bridged, stabilized Angiotensin-(1–7) analogue cyclic ang-(1–7) on cardiac remodeling and endothelial function in rats with myocardial infarction. Int. J. Hypertens. 2012, 536426 (2012).
87. De Vries L, Reitzema-Klein CE, Meter-Arkema A et al. Oral and pulmonary delivery of thioether-bridged angiotensin-(1–7). Peptides 31(5), 893–898 (2010).
88. Petty WJ, Miller AA, Mccoy TP, Gallagher PE, Tallant EA, Torti FM. Phase I and pharmacokinetic study of angiotensin-(1–7), an endogenous antiangiogenic hormone. Clin. Cancer Res. 15(23), 7398–7404 (2009).
89. Krishnan B, Torti FM, Gallagher PE, Tallant EA. Angiotensin-(1–7) reduces proliferation and angiogenesis of human prostate cancer xenografts with a decrease in angiogenic factors and an increase in sFlt-1. Prostate doi:10.1002/pros.22540 (2012) (Epub ahead of print).
90. Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA. Angiotensin-(1–7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol. Cancer Ther. 8(6), 1676–1683 (2009).
91. Cook KL, Metheny-Barlow LJ, Tallant EA, Gallagher PE. Angiotensin-(1–7) reduces fibrosis in orthotopic breast tumors. Cancer Res. 70(21), 8319–8328 (2010).
92. Rodgers K, Xiong SQ, Dizerega GS. Effect of angiotensin II and angiotensin(1–7) on hematopoietic recovery after intravenous chemotherapy. Cancer Chemother. Pharmacol. 51(2), 97–106 (2003).
93. Rodgers KE, Xiong S, Dizerega GS. Accelerated recovery from irradiation injury by angiotensin peptides. Cancer Chemother. Pharmacol. 49(5), 403–411 (2002).
94. Rodgers KE, Oliver J, Dizerega GS. Phase I/II dose escalation study of angiotensin 1–7 [A(1–7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother. Pharmacol. 57(5), 559–568 (2006).
95. Santos RA, Ferreira AJ. Pharmacological effects of AVE 0991, a nonpeptide angiotensin-(1–7) receptor agonist. Cardiovasc. Drug Rev. 24(3–4), 239–246 (2006).
96. Ebermann L, Spillmann F, Sidiropoulos M et al. The angiotensin-(1–7) receptor agonist AVE0991 is cardioprotective in diabetic rats. Eur. J. Pharmacol. 590(1–3), 276–280 (2008).
97. Ferreira AJ, Oliveira TL, Castro ML et al.
Isoproterenol-induced impairment of heart function and remodeling are attenuated by the nonpeptide angiotensin-(1–7) analogue AVE 0991. Life Sci. 81(11), 916–923 (2007).
98. Carvalho MB, Duarte FV, Faria-Silva R et al. Evidence for Mas-mediated bradykinin potentiation by the angiotensin-(1–7) nonpeptide mimic AVE 0991 in normotensive rats. Hypertension 50(4), 762–767 (2007).
99. Zeng WT, Chen WY, Leng XY et al. Impairment of cardiac function and remodeling induced by myocardial infarction in rats are attenuated by the nonpeptide angiotensin-(1–7) analog AVE 0991. Cardiovasc. Ther. 30(3), 152–161 (2012).
100. Jawien J, Toton-Zuranska J, Gajda M et al. Angiotensin-(1–7) receptor Mas agonist ameliorates progress of atherosclerosis in apoE-knockout mice. J. Physiol. Pharmacol. 63(1), 77–85 (2012).
101. Singh K, Sharma K, Singh M, Sharma P. Possible mechanism of the cardio-renal protective effects of AVE-0991, a non-peptide Mas-receptor agonist, in diabetic rats. J. Renin Angiotensin Aldosterone Syst. 13(3), 334–340 (2012).
102. Shemesh R, Toporik A, Levine Z et al. Discovery and validation of novel peptide agonists for G-protein-coupled receptors. J. Biol. Chem. 283(50), 34643–34649 (2008).
103. Savergnini SQ, Beiman M, Lautner RQ et al. Vascular relaxation, antihypertensive effect, and cardioprotection of a novel peptide agonist of the MAS receptor. Hypertension 56(1), 112–120 (2010).
104. Kaschina E, Grzesiak A, Li J et al. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118(24), 2523–2532 (2008).
nn� The first report of an orally administered drug that antagonizes AT
2R and leads to
therapeutic effects.
105. Jehle AB, Xu Y, Dimaria JM et al. A nonpeptide angiotensin II type 2 receptor agonist does not attenuate postmyocardial infarction left ventricular remodeling in mice. J. Cardiovasc. Pharmacol. 59(4), 363–368 (2012).
106. Rehman A, Leibowitz A, Yamamoto N, Rautureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 59(2), 291–299 (2012).
107. Paulis L, Becker ST, Lucht K et al. Direct angiotensin II type 2 receptor stimulation in
Nomega-nitro-l-arginine-methyl ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension 59(2), 485–492 (2012).
108. Abdalla S, Abdel-Baset A, Lother H, el Massiery A, Quitterer U. Mesangial AT1/B2 receptor heterodimers contribute to angiotensin II hyperresponsiveness in experimental hypertension. J. Mol. Neurosci. 26(2–3), 185–192 (2005).
109. Barki-Harrington L. Oligomerisation of angiotensin receptors: novel aspects in disease and drug therapy. J. Renin Angiotensin Aldosterone Syst. 5(2), 49–52 (2004).
110. Zeng C, Yang Z, Wang Z et al. Interaction of angiotensin II type 1 and D5 dopamine receptors in renal proximal tubule cells. Hypertension 45(4), 804–810 (2005).
111. Rozenfeld R, Gupta A, Gagnidze K et al. AT1R-CB(1)R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J. 30(12), 2350–2363 (2011).
112. Kostenis E, Milligan G, Christopoulos A et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 111(14), 1806–1813 (2005).
113. Canals M, Jenkins L, Kellett E, Milligan G. Up-regulation of the angiotensin II type 1 receptor by the MAS proto-oncogene is due to constitutive activation of G(q)/G(11) by MAS. J. Biol. Chem. 281(24), 16757–16767 (2006).
114. Abdalla S, Lother H, Abdel-Tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 276(43), 39721–39726 (2001).
115. Siddiquee K, Hampton J, Mcanally D, May LT, Smith LH. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br. J. Pharmacol. (2012).
116. Trask AJ, Jessup JA, Chappell MC, Ferrario CM. Angiotensin-(1–12) is an alternate substrate for angiotensin peptide production in the heart. Am. J. Physiol. Heart Circ. Physiol. 294(5), H2242–H2247 (2008).
117. Kemp BA, Bell JF, Rottkamp DM et al. Intrarenal Angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension 60(2), 387–395 (2012).
Review Clarke, Flores-Muñoz, McKinney, Milligan & Nicklin




















