
Regional changes in the intrarenal insulin-like growth factor-I axis in diabetes
8
Kidney Internationa4 Vol. 51(1997), pp. 811—818 Regional changes in the intrarenal insulin-like growth factor-I axis in diabetes FERNANDO C. FERVENZA, TANNY TSAO, ANDREW R. HOFFMAN, and RALPH RABKIN Research Service, Veterans Affairs Medical Center, Palo Alto, and Department of Medicine, Stanford University, Stanford, California, USA Regional changes in the intrarenal insulin-like growth factor-I axis in diabetes. Since insulin-like growth factor-I (IGF-I) has been shown to promote renal growth and as kidney IGF-I content increases during the early days after the onset of diabetes, it is likely that this growth factor contributes to initial diabetic renal hypertrophy. However, it is unclear whether IGF-I contributes to the continued renal growth that occurs in diabetes. Since IGF-I action is mediated through its receptor and as its bioavailability is regulated by IGF binding proteins (IGFBP), we postu- lated that changes in IGF-I receptor binding or IGFBP production may favor a role for IGF-I in diabetic renal growth when kidney IGF-I levels have returned to normal. To test this thesis, we studied kidneys of rats after seven days of streptozotocin diabetes. In diabetic cortex and medulla, growth hormone receptor mRNA levels and IGF-I and IGF-I receptor mRNA and protein product levels were unchanged. In cortex IGFBP-1 mRNA levels were increased while IGFBP-2 and -4 mRNA levels decreased. In medulla the only change was a fall in IGFBP-1 mRNA levels. Using Western ligand blot we observed an increase in a 32 kDa plasma membrane-associated TGFBP. Insulin therapy reversed all changes except the elevated cortical IGFBP-l mRNA levels, indicating the pres- ence of regional heterogeneity in the IGFBP response to diabetes in the kidney. However, the lack of change in IGF-I, IGF-I receptor and growth hormone receptor gene expression and protein products after one week of diabetes argues against a role for IGF-I in sustaining diabetic renal growth beyond the initial growth phase. Diabetes mellitus is now the leading cause of end-stage renal failure in the USA. A feature of early diabetes is renal enlarge- ment and hyperfunction [1]. In many instances, this appears to be the forerunner of structural damage leading to loss of renal function. In rats, diabetic nephromegaly is almost invariably followed by pathologic changes. This lead to the concept that identification of factors that drive the kidney to enlarge in diabetes will contribute significantly to our understanding of the pathogcnesis of diabetic nephropathy. Insulin-like growth factor-I (IGF-I) is among the many factors that could potentially play a role in diabetic kidney enlargement and hyperfunction [2—5]. This peptide growth factor promotes renal growth [4, 6] and increases renal blood flow and glomerular filtration [7]. It is present in the circulation largely derived from the liver, hut is also produced within kidney and other tissues mainly under the influence of growth hormone (GH). In early experimental diabetes in the rat, Received for publication April 8, 1996 and in revised form August 15, 1996 Accepted for publication September 3, 1996 © 1997 by the International Society of Nephrology kidney IGF-I content increases transiently [8, 9] though the mechanism for this increase is unknown. When this increase is blocked by treatment with a somatostatin analogue, then renal hypertrophy does not occur [10]. For these reasons it has been suggested that IGF-I may play an important role in the develop- ment of diabetic renal hypertrophy and, perhaps, diabetic kidney disease. Other components of the IGF-I axis may also play a role in mediating diabetic renal hypertrophy. These include the growth hormone (GH) and IGF-I receptors and the insulin-like growth factor binding proteins (IGFBP). These binding proteins have a high affinity for IGF-I and, by sequestering the growth factor, IGFBPs affect hormone delivery to cells and thereby modulate its bioactivity [11, 12]. Six distinct IGFBP have been identified and all are expressed in the kidney, although IGFBP-6 is present in very low abundance [Ii, 13]. These proteins are produced in tissues throughout the body and several, especially IGFBP-3, are present in high concentration in the circulation. In diabetes the production of IGFBPs in a variety of tissues including kidney is altered, with some IGFBPs increasing, some decreasing and some remaining unchanged [3, 12]. Thus, it is possible that local alterations in the intrarenal content of IGF-I and its modulating IGFBPs and perhaps the IGF-I receptor could result in enhanced local IGF-I action, which could then contribute to the development of renal hypertrophy and ultimately to renal pathology. As a first step in pursuing this hypothesis, we studied rats with early streptozotocin (STZ) induced diabetes and have characterized the regional changes in expression of genes that encode proteins comprising the intrarenal IGF-I axis [4]. Included in this study are the GH and IGF-I receptors, IGF-l and IGFBP-i through -5. Methods Male Sprague-Dawley rats weighing —200 g and allowed free access to food and water were studied. Rats were divided into three groups: diabetics, insulin treated diabetics and non-diabetic controls. Diabetes was induced by i.v. injection of 55 mg/kg of streptozotocin (Sigma Chemicals, St. Louis, MO, USA). Control rats received vehicle alone. Two days later blood glucose levels were checked and only rats with glucose levels above 250 mg/dl were included in the study. One group of diabetic animals was treated with insulin beginning two days after the injection of STZ. Long-acting, heat-treated ultralente insulin (Novo, Denmark) was given at an initial dose of 10 IU s.c. followed by 2 to 6 IU daily according to the blood glucose level measured by a glucometer 81!
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Regional changes in the intrarenal insulin-like growth factor-I axis in diabetes

Kidney Internationa4 Vol. 51(1997), pp. 811—818
Regional changes in the intrarenal insulin-like growth factor-Iaxis in diabetes
FERNANDO C. FERVENZA, TANNY TSAO, ANDREW R. HOFFMAN, and RALPH RABKIN
Research Service, Veterans Affairs Medical Center, Palo Alto, and Department of Medicine, Stanford University, Stanford, California, USA
Regional changes in the intrarenal insulin-like growth factor-I axis indiabetes. Since insulin-like growth factor-I (IGF-I) has been shown topromote renal growth and as kidney IGF-I content increases during theearly days after the onset of diabetes, it is likely that this growth factorcontributes to initial diabetic renal hypertrophy. However, it is unclearwhether IGF-I contributes to the continued renal growth that occurs indiabetes. Since IGF-I action is mediated through its receptor and as itsbioavailability is regulated by IGF binding proteins (IGFBP), we postu-lated that changes in IGF-I receptor binding or IGFBP production mayfavor a role for IGF-I in diabetic renal growth when kidney IGF-I levelshave returned to normal. To test this thesis, we studied kidneys of ratsafter seven days of streptozotocin diabetes. In diabetic cortex and medulla,growth hormone receptor mRNA levels and IGF-I and IGF-I receptormRNA and protein product levels were unchanged. In cortex IGFBP-1mRNA levels were increased while IGFBP-2 and -4 mRNA levelsdecreased. In medulla the only change was a fall in IGFBP-1 mRNAlevels. Using Western ligand blot we observed an increase in a 32 kDaplasma membrane-associated TGFBP. Insulin therapy reversed all changesexcept the elevated cortical IGFBP-l mRNA levels, indicating the pres-ence of regional heterogeneity in the IGFBP response to diabetes in thekidney. However, the lack of change in IGF-I, IGF-I receptor and growthhormone receptor gene expression and protein products after one week ofdiabetes argues against a role for IGF-I in sustaining diabetic renal growthbeyond the initial growth phase.
Diabetes mellitus is now the leading cause of end-stage renalfailure in the USA. A feature of early diabetes is renal enlarge-ment and hyperfunction [1]. In many instances, this appears to bethe forerunner of structural damage leading to loss of renalfunction. In rats, diabetic nephromegaly is almost invariablyfollowed by pathologic changes. This lead to the concept thatidentification of factors that drive the kidney to enlarge indiabetes will contribute significantly to our understanding of thepathogcnesis of diabetic nephropathy. Insulin-like growth factor-I(IGF-I) is among the many factors that could potentially play arole in diabetic kidney enlargement and hyperfunction [2—5]. Thispeptide growth factor promotes renal growth [4, 6] and increasesrenal blood flow and glomerular filtration [7]. It is present in thecirculation largely derived from the liver, hut is also producedwithin kidney and other tissues mainly under the influence ofgrowth hormone (GH). In early experimental diabetes in the rat,
Received for publication April 8, 1996and in revised form August 15, 1996Accepted for publication September 3, 1996
© 1997 by the International Society of Nephrology
kidney IGF-I content increases transiently [8, 9] though themechanism for this increase is unknown. When this increase isblocked by treatment with a somatostatin analogue, then renalhypertrophy does not occur [10]. For these reasons it has beensuggested that IGF-I may play an important role in the develop-ment of diabetic renal hypertrophy and, perhaps, diabetic kidneydisease.
Other components of the IGF-I axis may also play a role inmediating diabetic renal hypertrophy. These include the growthhormone (GH) and IGF-I receptors and the insulin-like growthfactor binding proteins (IGFBP). These binding proteins have ahigh affinity for IGF-I and, by sequestering the growth factor,IGFBPs affect hormone delivery to cells and thereby modulate itsbioactivity [11, 12]. Six distinct IGFBP have been identified and allare expressed in the kidney, although IGFBP-6 is present in verylow abundance [Ii, 13]. These proteins are produced in tissuesthroughout the body and several, especially IGFBP-3, are presentin high concentration in the circulation.
In diabetes the production of IGFBPs in a variety of tissuesincluding kidney is altered, with some IGFBPs increasing, somedecreasing and some remaining unchanged [3, 12]. Thus, it ispossible that local alterations in the intrarenal content of IGF-Iand its modulating IGFBPs and perhaps the IGF-I receptor couldresult in enhanced local IGF-I action, which could then contributeto the development of renal hypertrophy and ultimately to renalpathology. As a first step in pursuing this hypothesis, we studiedrats with early streptozotocin (STZ) induced diabetes and havecharacterized the regional changes in expression of genes thatencode proteins comprising the intrarenal IGF-I axis [4]. Includedin this study are the GH and IGF-I receptors, IGF-l and IGFBP-ithrough -5.
Methods
Male Sprague-Dawley rats weighing —200 g and allowed freeaccess to food and water were studied. Rats were divided intothree groups: diabetics, insulin treated diabetics and non-diabeticcontrols. Diabetes was induced by i.v. injection of 55 mg/kg ofstreptozotocin (Sigma Chemicals, St. Louis, MO, USA). Controlrats received vehicle alone. Two days later blood glucose levelswere checked and only rats with glucose levels above 250 mg/dlwere included in the study. One group of diabetic animals wastreated with insulin beginning two days after the injection of STZ.Long-acting, heat-treated ultralente insulin (Novo, Denmark) wasgiven at an initial dose of 10 IU s.c. followed by 2 to 6 IU dailyaccording to the blood glucose level measured by a glucometer
81!

812 Fervenza et al: IGF-I and diabetes
(Companion 2; Medisense, Waltham, MA, USA). Serum glucoseon day 2 and day 7 were measured with a Glucose Analyzer 2(Beckman, Fullerton, CA, USA). Seven days after STZ or vehicleinjections rats were anacsthetized, the right kidneys were re-moved, weighed, separated into cortex and medulla and frozen inliquid nitrogen and stored at —80°C until processing. Left kidneyswere perfused in Situ with phosphate buffered saline (37°C) untilfree of blood, and collected and stored as above.
Preparation of crude kidney membranes
Crude plasma membranes were isolated separately from cortexand medulla as previously described [14]. Briefly, kidneys werehomogenized in 8% buffered sucrose followed by differentialcentrifugation and collection of a 47,000 g pellet. The pellet wasresuspended in calcium-free Krebs-Ringer Hepes buffer (KRH,pH 7.4) and stored at —80°C. Membrane protein content wasmeasured with the Bio-Rad method (Bio-Rad Laboratories, Rich-mond, CA, USA).
Kidney membrane IGF-I receptor binding assay
IGF-I binding was measured exactly as previously describedusing recombinant human IGF-l (a gift from Genentech Inc.,South San Francisco, CA, USA), radiolabcled with '251-Na [141.In brief, 100 j.tg of crude plasma membrane protein were incu-bated with 10 ' M '251-IGF-l in calcium-free KRH containing0.5% BSA, 2 mM bacitracin and 5 mrvi N-ethylmaleimide at 4°Covernight. Membrane-bound and free 1251-IGF-l were separatedby centrifugation, the pellet was washed and the radioactivitycounted. Non-specific 1251-IGF-I binding was determined fromincubation with 10_6 M of unlabeled IGF-I.
Western ligand blot
Cortical and medullary plasma membranes prepared fromsaline perfused kidneys were used. Membrane protein, 100 jtg,was electrophoresed in 12% polyacrylamide gel. Separated pro-teins were electroblotted onto nitrocellulose filters (Biorad Semi-Dry Transfer Cell) and then incubated with a solution containingone million cpm of 1251-IGF-l and 1251-IGF-II overnight at 4°Cand visualized by autoradiography [15]. Measurement was bydensitometty, though it should be recognized that this is not trulyquantitative because of variations in the transfer of proteins to thenitrocellulose.
Radioimmunoassay of serum and kidney IGF-I
As before [14], IGF-l was measured by radioimmunoassay inacid-ethanol extracts prepared from serum and saline perfusedwhole kidneys with a commercial assay kit (Nichols Institute, SanJuan Capistrano, CA, USA). Recovery of IGF-1 added to homog-enate of kidneys from control, untreated diabetics and treateddiabetic rats averaged 99 2.8%, 98 4.8% and 88 1.5%,respectively. Recovery from serum averaged 91 2.6%, 884.1% and 95 2.6%, respectively.
RNA extraction
Total RNA was isolated separately from the cortex and medullaby an acid guanidinium thiocyanate-phenol chloroform single stepmethod [161.
Preparation of radiolabeled probes
For the solution hybridization/RNase protection assay, radiola-beled antisense IGF-I, IGF-1 and GH receptor and 18S ribo-probes were prepared with 32P-CTP [14]. The IGF-l riboprobe, a376 bp Sau3A-EcoRl fragment, was designed to detect both theEa and Eb IGF-I mRNAs [17]. The IGF-I receptor riboprobecontained 280 bases complementary to the IGF-I receptormRNA. The GH receptor riboprobe contained 137 bases (posi-tion 811 to 948) complementary to the rat GH receptor mRNA(Dr. L.S. Mathews, University of Michigan) [181. The 18S ribo-probe contains 80 bases complementary to a highly conservedregion of the human RNA [19]. For Northern blot analysis thecDNA probes were labeled with 50 jsCi 32P-dCTP by a randomprimer method (DNA labeling kit 70200; United States Biochem-ical Corporation, Cleveland, OH, USA). Purification of radiola-beled cDNA probes were achieved through a G-50 Sephadexcolumn.
The cDNA probes for the five IGFBPs studied were preparedfrom rat cDNAs (Dr. S. Shimasaki, Whittier Institute, La Jolla,CA, USA). Probes were prepared by synthesizing the correspond-ing cDNA by PCR using the cloned cDNA as a template [11]. TheIGFBP-1 probe consisted of 407 bp corresponding to the codingregion spanning nucleotide position 486 to 892 in the rat IGFBP-1cDNA sequence. The IGFBP-2 probe was comprised of 342 bpspanning nucleotide position 670 to 1011. The IGFBP-3 probecomprised 636 bp spanning nucleotide positions 196 to 831. TheIGFBP-4 probe consisted of 354 bp spanning nucleotide position487 to 840. The IGFBP-5 probe comprised 259 bp spanningnucleotide position 856 to 1114.
Northern blot analysis
Total RNA was size-separated by formaldehyde agarose gelelectrophoresis [141. Briefly, RNA was denatured with 0.55 Mformaldehyde. Samples containing 20 jsg RNA were loaded andelectrophoresed on a denaturing agarose gel containing 0.55 Mformaldehyde and stained with ethidium bromide. RNA wastransferred to nitrocellulose filters, fixed to the filters by UVcross-link (UV 1800 Stratalinker; Stratgene, La Jolla, CA, USA)and photographed under tJV light. Filters were then prehybrid-ized in a solution containing 50% formamide, 1.5 X SSPE, 2.5 ><Denharts solution, 50 pg salmon sperm DNA, 0.2% SDS at 42°Cfor four hours. The mRNA levels of IGFBP-1, -2, -3, -4, and -5were detected by hybridizing 32P-radiolabeled eDNA probes tothe mRNA at 42°C overnight. Washed filters were then exposedto Kodak XAR films (Kodak, Rochester, NY, USA) for two toseven days. The abundance of each IGFBP mRNA was measuredby scanning densitometry (Ultrascan XL; Pharmacia LKB Bio-technology, Inc., Alameda, CA, USA) and adjusted for the levelsof the 18S rRNA readings obtained from photographic negativesof the nitrocellulose filters [14].
Solution hybridizationiRNAse protection assay
These assays were performed as previously described [14J. inbrief, labeled antisense IGF-i, IGF-l and GH receptor and 18SRNAs were synthesized using 32P-Iabeled CTP. Total RNA, 20jig, was hybridized with 32P-CTP labeled antisense IGF-I or IGF-Ireceptor RNA probes overnight at 42°C in 75% formamide/400mM NaCI. After hybridization, the mixture was added to RNAsedigestion buffer containing RNAse A (25 pg/mI) and RNAse T1
Fervenza Ct al: IGF-I and diabetes 813
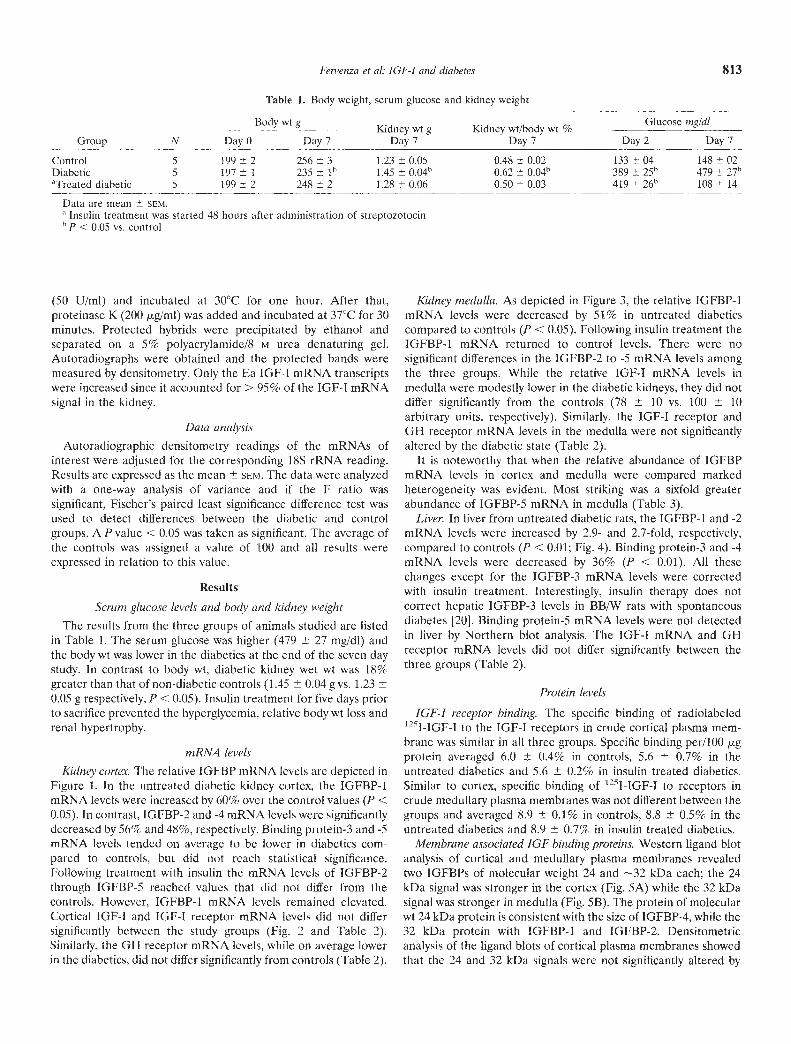
Table 1. Body weight, serum glucose and kidney weight
Group NBody
Day 0
wt g—-—--Day 7
.Kidney wt g
Day 7
.Kidney wt/body wt %
Day 7
Glucose mg/dI
Day 2 Day 7
Control 5 199 2 256 3 1.23 0.05 0.48 0.02 133 04 148 02Diabetic 5 197 1 235 l' 1.45 0.04k' 0.62 0.04t 389 25 479 27b°Treated diabetic 5 199 2 248 2 1.28 0.06 0.50 0.03 419 26 108 14
Data are mean SEM."Insulin treatment was started 48 hours after administration of streptozotocinh P < 0.05 vs. control
(50 U/ml) and incubated at 30°C for one hour. After that,proteinase K (200 tgIml) was added and incubated at 37°C for 30minutes. Protected hybrids were precipitated by ethanol andseparated on a 5% polyacrylamide/8 M urea denaturing gel.Autoradiographs were obtained and the protected bands weremeasured by densitometry. Only the Ea IGF-I mRNA transcriptswere increased since it accounted for > 95% of the IGF-I mRNAsignal in the kidney.
Data analysisAutoradiographic densitometry readings of the mRNAs of
interest were adjusted for the corresponding 18S rRNA reading.Results are expressed as the mean SEM. The data were analyzedwith a one-way analysis of variance and if the F ratio wassignificant, Fischer's paired least significance difference test wasused to detect differences between the diabetic and controlgroups. A P value < 0.05 was taken as significant. The average ofthe controls was assigned a value of 100 and all results wereexpressed in relation to this value.
Results
Serum glucose levels and body and kidney weightThe results from the three groups of animals studied are listed
in Table 1. The serum glucose was higher (479 27 mg/dl) andthe body wt was lower in the diabetics at the end of the seven daystudy. In contrast to body wt, diabetic kidney wet wt was 18%greater than that of non-diabetic controls (1.45 0.04 g vs. 1.230.05 g respectively, P < 0.05). Insulin treatment for five days priorto sacrifice prevented the hyperglycemia, relative body wt loss andrenal hypertrophy.
mRNA levels
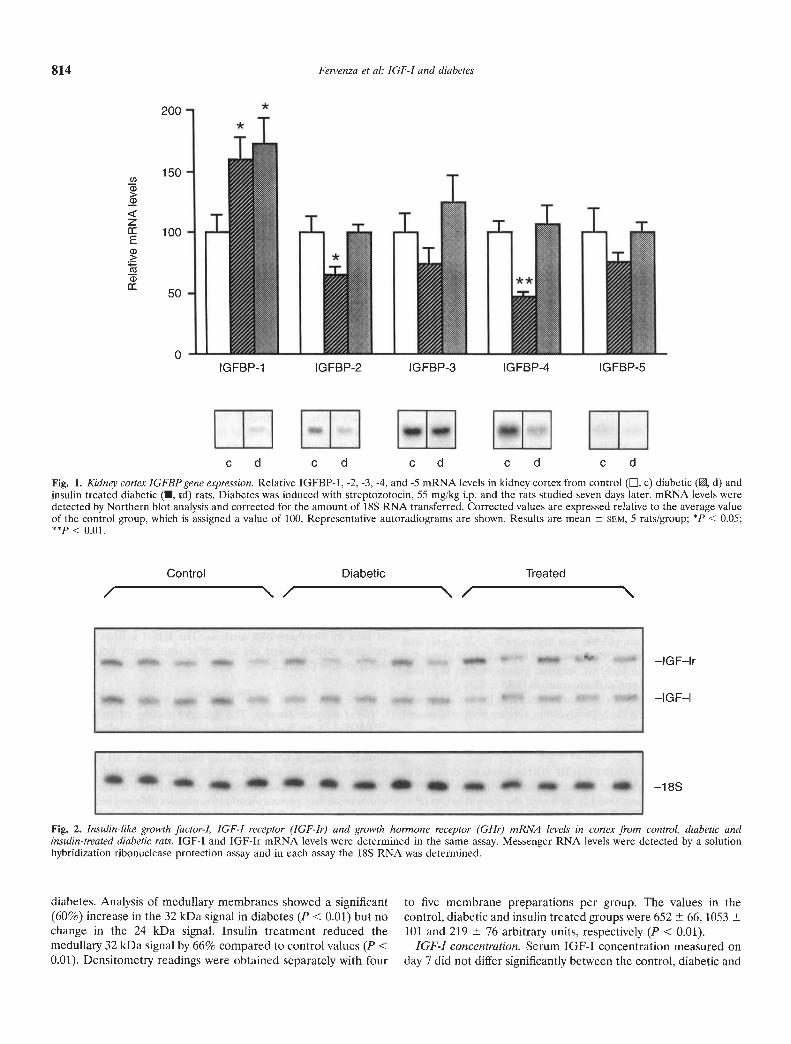
Kidney cortex. The relative IGFBP mRNA levels are depicted inFigure 1. In the untreated diabetic kidney cortex, the IGFBP-1mRNA levels were increased by 60% over the control values (P <0.05). In contrast, IGFBP-2 and -4 mRNA levels were significantlydecreased by 56% and 48%, respectively. Binding protein-3 and -5mRNA levels tended on average to be lower in diabetics com-pared to controls, but did not reach statistical significance.Following treatment with insulin the mRNA levels of IGFBP-2through IGFUP-5 reached values that did not differ from thecontrols. However, IGFBP-1 mRNA levels remained elevated.Cortical IGF-l and IGF-I receptor mRNA levels did not differsignificantly between the study groups (Fig. 2 and Table 2).Similarly, the GI-l receptor mRNA levels, while on average lowerin the diabetics, did not differ significantly from controls (Table 2).
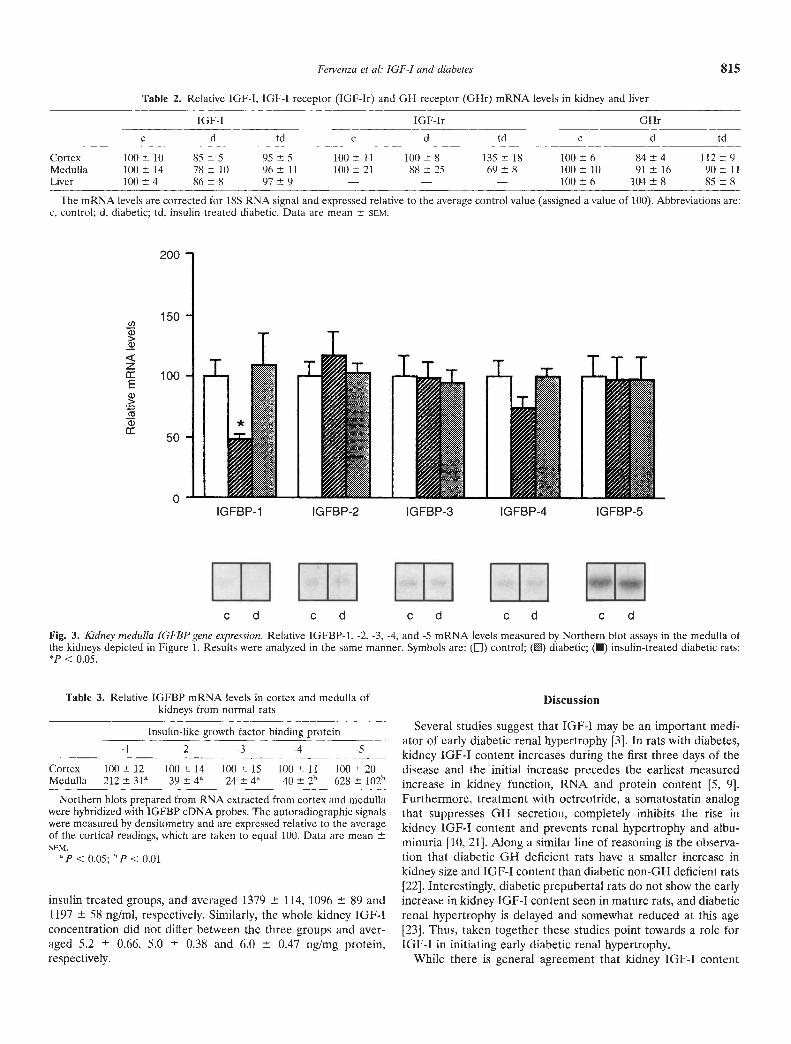
Kidney medulla. As depicted in Figure 3, the relative IGFBP-1mRNA levels were decreased by 51% in untreated diabeticscompared to controls (P < 0.05). Following insulin treatment theIGFBP-1 mRNA returned to control levels. There were nosignificant differences in the IGFBP-2 to -5 mRNA levels amongthe three groups. While the relative IGF-I mRNA levels inmedulla were modestly lower in the diabetic kidneys, they did notdiffer significantly from the controls (78 10 vs. 100 10
arbitrary units, respectively). Similarly, the IGF-I receptor andGH receptor mRNA levels in the medulla were not significantlyaltered by the diabetic state (Table 2).
It is noteworthy that when the relative abundance of IGFBPmRNA levels in cortex and medulla were compared markedheterogeneity was evident. Most striking was a sixfold greaterabundance of IGFBP-5 mRNA in medulla (Table 3).
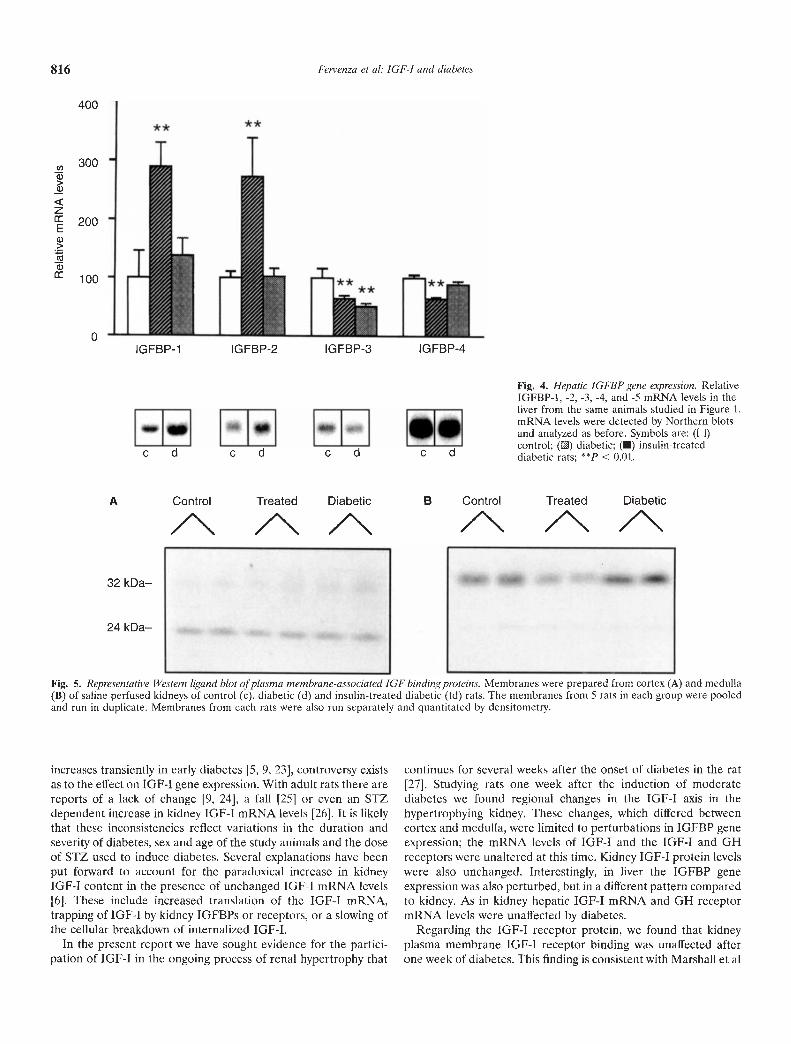
Liver. In liver from untreated diabetic rats, the IGFBP-1 and -2mRNA levels were increased by 2.9- and 2.7-fold, respectively,compared to controls (P < 0.01; Fig. 4). Binding protein-3 and -4mRNA levels were decreased by 36% (P < 0.01). All thesechanges except for the IGFBP-3 mRNA levels were correctedwith insulin treatment. Interestingly, insulin therapy does notcorrect hepatic IGFBP-3 levels in BB/W rats with spontaneousdiabetes [20]. Binding protein-S mRNA levels were not detectedin liver by Northern blot analysis. The IGF-I mRNA and GHreceptor mRNA levels did not differ significantly between thethree groups (Table 2).
Protein levels
IGF-I receptor binding. The specific binding of radiolabeled12511GF1 to the IGF-I receptors in crude cortical plasma mem-brane was similar in all three groups. Specific binding per/100 jtgprotein averaged 6.0 0.4% in controls, 5.6 0.7% in theuntreated diabetics and 5.6 0.2% in insulin treated diabetics.Similar to cortex, specific binding of 125l-IGF-1 to receptors incrude medullary plasma membranes was not different between thegroups and averaged 8.9 0.1% in controls, 8.8 0.5% in theuntreated diabetics and 8.9 0.7% in insulin treated diabetics.
Membrane associated JGF binding proteins. Western ligand blotanalysis of cortical and medullary plasma membranes revealedtwo IGFBPs of molecular weight 24 and —32 kDa each; the 24kDa signal was stronger in the cortex (Fig.A) while the 32 kDasignal was stronger in medulla (Fig. SB). The protein of molecularwt 24 kDa protein is consistent with the size of IGFBP-4, while the32 kDa protein with IGFBP-i and IGFBP-2. Densitometricanalysis of the ligand blots of cortical plasma membranes showedthat the 24 and 32 kDa signals were not significantly altered by
814 Fervenza et a!: IGF-I and diabetes
200
150()ci)>ci)
z100
50
0IGFBP-1 IGFBP-2 IGFBP-3 IGFBP-4 IGFBP-5
c d c d c d c d c d
Fig. 1. Kidney cortex IGFBP gene expression. Relative IGFBP-1, -2, -3, -4, and -5 mRNA levels in kidney cortex from control (, c) diabetic (, d) andinsulin treated diabetic (U, td) rats. Diabetes was induced with streptozotocin, 55 mg/kg i.p. and the rats studied seven days later. mRNA levels weredetected by Northern blot analysis and corrected for the amount of 18S RNA transferred. Corrected values are expressed relative to the average valueof the control group, which is assigned a value of 100. Representative autoradiograms are shown. Results are mean 5EM, 5 rats/group; * < 0.05;
< 0.01.
Control Diabetic Treated
—IGF—lr
—IGF—l
—1 8S
Fig. 2. Insulin-like growth factor-I, IGF-I receptor (IGF-Ir) and growth hormone receptor (GI-Ir) mRNA levels in cortex from control, diabetic andinsulin-treated diabetic rats. IGF-I and IGF-Ir mRNA levels were determined in the same assay. Messenger RNA levels were detected by a solutionhybridization ribonuclease protection assay and in each assay the 18S RNA was determined.
diabetes. Analysis of medullary membranes showed a significant to five membrane preparations per group. The values in the(60%) increase in the 32 kDa signal in diabetes (P < 0.01) but no control, diabetic and insulin treated groups were 652 66, 1053change in the 24 kDa signal. Insulin treatment reduced the 101 and 219 76 arbitrary units, respectively (P < 0.01).medullary 32 kDa signal by 66% compared to control values (P < IGF-I concentration. Serum IGF-I concentration measured on0.01). Densitometry readings were obtained separately with four day 7 did not differ significantly between the control, diabetic and
I I H HH __ I I
a
55 a a a a a a S a a a a a
111111 I I I I I I __
Fervenza et al: JGF-I and diabetes 815
Table 2. Relative IGF-I, IGF-I receptor (IGF-Ir) and GH receptor (GHr) mRNA levels in kidney and liver
IGF-I IGF-Ir
c
GHr
d tdc d td c d td
Cortex 100± 10 85±5 95±5 100± Il 100±8 135±18 100±6 84±4 112±9Medulla 100± 14 78±10 96±11 100± 21 88±25 69±8 100±10 91±16 90±11Liver 100± 4 86±8 97±9 — — — 100±6 104±8 85±8
The mRNA levels are corrected for 185 RNA signal and expressed relative to the average control value (assigned a value of 100). Abbreviations are:c, control; d, diabetic; td, insulin treatcd diabetic. Data are mean SCM.
IGFBP-1 IGFBP-2 IGFBP-3 IGFBP-4 IGFBP-5
c d c d c d c d c d
Fig. 3. Kidney medulla IGFBP gene expression. Relative IGFBP-1, -2, -3, -4, and -5 mRNA levels measured by Northern blot assays in the medulla ofthe kidneys depicted in Figure 1. Results were analyzed in the same manner. Symbols are: (El) control; () diabetic; ( insulin-treated diabetic rats;'P C 0.05.
U)C)>C)
z100
CC
C)I
Table 3. Relative IGFBP mRNA levels in cortex and medulla ofkidneys from normal rats
Insulin-like growth factor binding protein-l -2 -3 -4 -5
Cnrtcx 100 12 100 14 lOt) 15 tOO 11 100 20Medulla 212 3l 39 4" 24 4" 40 2 628 102h
Northern blots prepared from RNA cxtracted from cortex and medullawere hybridized with IOFBP eDNA probes. Thc autoradiographic signalswere measured by densitometry and are expressed relative to the avcragcof the cortical readings, which are taken to equal 100. Data are meanSCM.
"p < 0.05; 5P C 0.01
insulin treated groups, and averaged 1379 114, 1096 89 and1197 58 ng/ml, respectively. Similarly, the whole kidney IGF-Iconcentration did not differ between the three groups and aver-aged 5.2 0.66, 5.0 0.38 and 6.0 0.47 ng!mg protein,respectively.
Discussion
Several studies suggest that IGF-I may be an important medi-ator of early diabetic renal hypertrophy [31. In rats with diabetes,kidney IOF-I content increases during the first three days of thedisease and the initial increase precedes the earliest measuredincrease in kidney function, RNA and protein content [5, 9].Furthermore, treatment with octreotride, a somatostatin analogthat suppresses GM secretion, completely inhibits the rise inkidney IGF-I content and prevents renal hypertrophy and albu-minuria [10, 21]. Along a similar line of reasoning is the observa-tion that diabetic OH deficient rats have a smaller increase inkidney size and [OF-I content than diabetic non-OH deficient rats[22]. Interestingly, diabetic prepubertal rats do not show the earlyincrease in kidney IOF-I content seen in mature rats, and diabeticrenal hypertrophy is delayed and somewhat reduced at this age[23]. Thus, taken together these studies point towards a role forIGF-I in initiating early diabetic renal hypertrophy.
While there is general agreement that kidney IGF-I content
200
150
50
0
TTT
** **
a)ci)
cCzIEci)>ccici)I
Fig. 4. Hepatic IGFBP gene expression. RelativeIGFBP-1, -2, -3, -4, and -5 mRNA levels in theliver from the same animals studied in Figure 1.mRNA levels were detected by Northern blotsand analyzed as before. Symbols are: (U)control; () diabetic; (•) insulin-treateddiabetic rats; cc 0.01.
A Control Treated Diabetic B Control Treated Diabetic/\ /\ /\ /\ /N /\
Fig. 5. Representative Western ligand blot of plasma membrane-associated IGF binding proteins. Membranes were prepared from cortex (A) and medulla(B) of saline perfused kidneys of control (c), diabetic (d) and insulin-treated diabetic (td) rats. The membranes from 5 rats in each group were pooledand run in duplicate. Membranes from each rats were also run separately and quantitated by densitometry.
increases transiently in early diabetes [5, 9, 23], controversy existsas to the effect on JOF-I gene expression. With adult rats there arereports of a lack of change [9, 24], a fall [25] or even an STZdependent increase in kidney IGF-T mRNA levels [26]. It is likelythat these inconsistencies reflect variations in the duration andseverity of diabetes, sex and age of the study animals and the doseof STZ used to induce diabetes. Several explanations have beenput forward to account for the paradoxical increase in kidneylOP-I content in the presence of unchanged IGF-I mRNA levels[6]. These include increased translation of the IOF-I mRNA,trapping of lOP-I by kidney IGFBPs or receptors, or a slowing ofthe cellular breakdown of internalized IGF-I.
In the present report we have sought evidence for the partici-pation of IOF-I in the ongoing process of renal hypertrophy that
continues for several weeks after the onset of diabetes in the rat[27]. Studying rats one week after the induction of moderatediabetes we found regional changes in the TOP-I axis in thehypertrophying kidney. These changes, which differed betweencortex and medulla, were limited to perturbations in IGFBP geneexpression; the mRNA levels of lOP-I and the lOP-I and OHreceptors were unaltered at this time. Kidney IGF-I protein levelswere also unchanged. Interestingly, in liver the IOFBP geneexpression was also perturbed, but in a different pattern comparedto kidney. As in kidney hepatic IGF-I mRNA and OH receptormRNA levels were unaffected by diabetes.
Regarding the IOF-T receptor protein, we found that kidneyplasma membrane IOF-I receptor binding was unaffected afterone week of diabetes. This finding is consistent with Marshall et al
816 Pervenza et al: IGF-i and diabetes
400
300
200
100
0IGFBP-1 IGFBP-2 IGFBP-3 IGFBP-4
c d c d c d c d
32 kDa—
24 kDa—
— S

Fervenza et al: IGF-I and diabetes 817
[5] who also studied kidney IGF-I receptor binding after a week ofdiabetes. In contrast, Werner and associates [28] studying 14-day-old diabetic rats noted a 2.5-fold increase in kidney IGF-Ireceptor mRNA levels and a similar increase in receptor binding.These differences likely reflect the changing response of thekidney to diabetes over time.
In kidney cortex, where the IGFBP-1 gene is normally ex-pressed in glomeruli and distal convoluted tubules [13], we founda significant increase in IGFBP-1 mRNA levels in the diabetickidneys which exceeded the non-diabetic values by 60%. Incontrast, cortical IGFBP-2 and IGFBP-4 mRNA levels fell by56% and 48%, respectively. In the cortex the genes for IGFBP-2and IGFBP-4 are normally expressed in glomeruli and proximaltubules, respectively [13]. In renal medulla the only significantchange noted in the diabetic kidney was a 51% fall in the IGFBP-1mRNA level. This gene is normally detected in the thick ascend-ing limb of Henle. In liver there was a near threefold increase inIGFBP-1 and IGFBP-2 mRNA levels and a 35% decrease inIGFBP-4 mRNA levels. In liver we noted an increase in theIGFBP-1 and IGFBP-2 mRNA levels and this is consistent withan earlier report [29]. Insulin treatment corrected the hypergly-cemia and reversed all the changes in the kidney and liver, exceptthat IGFBP-1 mRNA levels in kidney cortex remained elevated.Since insulin treatment normalized JGFBP-1 levels in renalmedulla and in liver, it is likely that the lack of cortical IGFBP-1response reflects a direct local effect of STZ rather than asecondary effect of diabetes. Of note, when the relative abundanceof the individual IGFBPs in cortex and medulla were compared,heterogeneity of abundance was apparent. Most striking was theIGFBP-5 mRNA level that was sixfold greater in medulla. Thesefindings are consistent with our in situ hybridization study [13].
The changes in kidney IGFBP expression noted above arelargely consistent with a report by Landau et a! [27] who studiedthe diabetic kidney by means of in situ hybridization. In ratsstudied seven days after the induction of STZ diabetes, theseauthors found a twofold increase in the IGFBP-1 mRNA level inthe cortex while the mRNA levels in the inner stripe of the outermedulla were reduced markedly. These changes persisted over thesix months of study. There was also a significant reduction incortical IGFBP-4 mRNA levels. Finally, there were modestchanges in IGFBP-5 mRNA levels that were decreased in cortexand increased in outer medulla.
Because of the altered IGFBP gene expression in the kidney thepotential impact on IGF-I action requires consideration. Ingeneral, circulating and tissue IGFBPs act as carriers for IGF-Iwith the protein bound IGF-I forming a reservoir from which thefree IGF-l is slowly released. This process reduces the immediatehioavailability of IGF-l and thus the IGFBPs are largely inhibi-tory. However, under selected conditions some IGFBPs canactually enhance the mitogenic activity of IGF-I action on certaincell types such as fibroblasts and osteoblasts [30, 311. There is littleinformation on the function of IGFBPs function in kidney, thoughthe marked anatomical of IGFBP gene expression in the kidneysuggests diversity of function [131. In studies with cultured rabbitproximal tubule cells we noted that endogenous IGFBPs andexogenous recombinant human IGFBP-3 inhibited IGF-l inducedmitogenesis [32]. Similarly, recombinant human IGFBP-3 andIGFBP-1 inhibited mitogenesis in cultured proximal-like oppo-sum kidney cells [33]. In contrast, there is a preliminary reportthat indicates that IGFBP-2 enhances IGF-I stimulated DNA
synthesis in MDCK cells, a cell line derived from the distalnephron [34]. Accordingly, until the complex interactions betweenIGF-I, the binding proteins and the different cell types of thekidney are unravelled the true significance of the changes inIGFBP gene expression induced by diabetes will remain obscure.
We did not find any significant changes in cortical plasmamembrane-associated IGFBPs after one week of moderate dia-betes. However, there was a significant, 60% increase in the 32kDa IGFBP associated with medullary plasma membrane. Thisprotein is consistent with the kidney plasma membrane associatedimmunoreactive IGFBP-5 identified by Hise et a! [35]. Since ourassay cannot detect IGFBPs in the interstitial or intracellularcompartments, it is conceivable that changes may be present inthese compartments that are below the sensitivity of the assay.While there are reports of increased IGFBPs in the diabetickidney, it is unclear whether these proteins are produced in thekidney or derived from the systemic circulation [36—38]. Forexample, Flyvbjerg et al [38] reported a transient early increase inthe 30 kDa and 38 to 47 kDa IGFBP species in the kidney thatreturned to basal levels after four days of diabetes. Since theserum levels levels were also elevated at that time, these IGFBPsmay have originated from the circulation.
In this study we have noted that there are regional changes inthe expression of IGFBP genes and of a 32 kDa medullary plasmamembrane-associated IGFBP in the kidney after one week ofdiabetes. However, it is difficult to conceive how these localchanges can account for the diffuse renal hypertrophy that isoccurring at this time. Thus, taken together with the lack ofchange in IGF-I and IGF-I receptor gene and protein expression,we conclude that it is unlikely that IGF-I contributes to the globalrenal hypertrophy that is present at this stage of diabetes.Whether IGF-I participates at a later stage when the structuralchanges of diabetic kidney disease develops remains to be estab-lished. In this regard Muchaneta-Kubara, Cope and El Nahas [39]described the biphasic pattern of IGF-I expression in the remnantkidney following renal ablation, which suggested that IGF-I maybe involved in the early trophic response and again later in theprocess of renal scarring.
Acknowledgments
This study was supported by the Juvenile Diabetes Foundation Inter-national and the Department of Veterans Affairs.
Reprint requests to Ralph Rabkin, M.D., Department of Veterans AffairsMedical Center (154L), 3801 Miranda Avenue, Palo Alto, California 94304,USA.E-mail: [email protected]
References
1. MORGENSEN CE, ANDERSEN Mi: Increased kidney size and glomeru-lar filtration rate in early juvenile diabetes. Diabetes 22:706—712, 1973
2. STRIKER GE, PETEN EP, CARONE MA, Pvscv CM, SCHMIDT K, YANGC-W, ELLIOT SJ, STRIKER JL: The kidney disease of diabetes mellitus:A cell and molecular biology approach. Diabetes/Metabolism Rev9:37—56, 1993
3. RABKIN R, FERVENZA FC: Renal hypertrophy and kidney disease indiabetes. Diabetes/Metabolism Rev 1996 (in press)
4. HAMMERMAN MR, MILLER SB: The growth hormone insulin-likegrowth factor axis in the kidney revisited. Am J Physiol 265:F1—F14,1993

818 Fervenza et al: IGF-I and diabetes
5. MARSHALL SM, FLYVBJERG A, FRYSTYK J, KORSGAARD, ORSKOV LH:Renal insulin-like growth factor I and growth hormone receptorbinding in experimental diabetes and after unilateral nephrectomy.Diabetologia 34:632—639, 1991
6. FLYVBJERG A: The role of insulin-like growth factor I in initial renalhypertrophy in experimental diabetes, in Growth Hormone and Insu-lin-like Growth Factor I, edited by FLYVBJERG A, New York, JohnWhiley & Sons Ltd, 1993, pp 271—306
7. HIRsCHBERG R, KOPPLE JD: Evidence that insulin-like growth factorI increases renal plasma flow and glomerular filtration rate in fastedrats. J Clin Invest 83:326—330, 1989
8. SAYED-AHMED N, MUCHANETA-KUBARA EC, BESBAS N, SH0RTLANDJ, COPE GH, EL NAHAS AM: Insulin-like growth factor-I and experi-mental diabetic kidney disease. Exp Nephrol 1:364—371, 1993
9. FLYVBJERG A, BORNFELDT KE, MARSHALL SM, ARNOVIST HJ, 0RS-KOv H: Kidney IGF-I mRNA in initial renal hypertrophy in experi-mental diabetes in rats. Diaberologia 33:334—338, 1990
10. FLYVBJERG A, MARSHALL SM, FRYSTYK J, HANSEN KW, HARRIS AG,H: Octreotide administration in diabetic rats: Effects on renal
hypertrophy and urinary albumin excretion. Kidney mt 41:805—812,1992
11. SHIMASAKI S, LING N: Identification and molecular characterization ofinsulin-like growth factor binding proteins (IGFBP-1, -2, -3, -4, -5 and-6). Progr Growth Factor Res 3:243—266, 1991
12. JONES JI, CLEMMONS DR: Insulin-like growth factors and their bindingproteins: Biological actions. Endocrine Rev 16:3—34, 1995
13. RABKIN R, BRODY M, Lu LH, CHAN C, SHAI-IEEN AM, GILLETIT N:Expression of the genes encoding the rat renal insulin-like growthfactor-I system. JAm Soc Nephrol 6:1511—1518, 1995
14. TsAo T, WANG J, FERVENZA FC, Vu TH, JIN IH, HOFFMAN AR,RABKIN R: Renal growth hormone-insulin-like growth factor-I systemin acute renal failure. Kidney mt 47:1658—1668, 1995
15. HOSSENSLOPP P, SEURIN D, SEGOVIA-QUINSON B, HARDOUIN 5, BIN-oux M: Analysis of serum insulin-like growth factor binding proteinsusing Western ligand blotting; use of the method for titration of thebinding proteins and competitive binding studies. Anal Biochem154:138—143, 1986
16. CHOMCZYNSKI P, SACCHI N: Single-step method of RNA isolation byacid guanidinium thiocyanate-phenol-chloroform extraction. AnalBiochem 162:156—159, 1987
17. LOWE WL JR, LASKY SR, LEROITH D, ROBERTS CT JR: Distributionand regulation of rat insulin-like growth factor I messenger ribonu-cleic acids encoding alternative carboxyterminal E-peptides: Evidenceof differential processing and regulation in liver. Mol Endocrinol2:528—535, 1988
18. MATHEWS LS, ENBERG B, NORSTEDT G: Regulation of rat growthhormone receptor gene expression.JBiol Chem 264:9905—9910, 1989
19. GONZALEZ IL, SCHMICKEL RD: The human 18S ribosomal RNA gene:Evolution and stability. Am Hum Genet 38:419—427, 1986
20. Luo J, MURPHY LI: Differential expression of the insulin-like growthfactor binding proteins in spontaneously diabetic rats. J Mol Endocri-nol 8:155—163, 1992
21. FLYVBJERG A, FRYSTYK J, THORLACIUS-USSING 0, 0RSKOV H: Soma-tostatin analogue administration prevents increase in kidney somato-medin C and initial renal growth in diabetic and uniphrcctomized rats.Diaberologia 32:26 1—265, 1989
22. FLYVBJERG A, FRYSTYK J, ØSTERBY R, 0RSKOV H: Kidney IGF-I andrenal hypertrophy in GH-deficient diabetic dwarf rats. Am J Physiol262:E956—E962, 1992
23. BACH LA, STEVENSON JL, ALLEN TJ, JERUMS G, HERINGTON AC:Kidney insulin-like growth factor-I mRNA levels are increased inpostpuberal diabetic rats. J Endocrinol 129:5—10, 1991
24. PHILLIP M, WERNER H, PALESE T, KOWARSKI AA, STANNARD B, BACHLA, LEROITH D, ROBERTS CT JR: Differential accumulation ofinsulin-like growth factor-I in kidneys of pre- and postpubertalstreptozotocin-diabetic rats. J Mol Endocrinol 12:215—224, 1994
25. FAGIN JA, ROBERTS CT JR, LEROITH D, BROWN AL: Coordinatedecrease of tissue insulin-like growth factor I posttranscriptionalalternative mRNA transcripts in diabetes mellitus. Diabetes 38:428—434, 1989
26. CATANESE VM, SCIAV0LIN0 PJ, LANGO MN: Discordant, organ-specific regulation of insulin-like growth factor-I messenger ribonu-cleic acid in insulin-deficient diabetes in rats. Endocrinology 132:496—503, 1993
27. LANDAU D, CHIN E, BONDY C, DOMENE H, ROBERTS CT JR, GRON-BARK H, FLY VBJERG A, LEROITH D: Expression of insulin-like growthfactor binding proteins in the rat kidney: Effects of long-term diabetes.Endocrinology 136:1835—1842, 1995
28. WERNER H, SHEN-ORR Z, STANNARD B, BURGUERA B, ROBERTS CTJR, LEROITH D: Experimental diabetes increases insulin-like growthfactor I and II receptor concentration and gene expression in kidney.Diabetes 39:1490—1497, 1990
29. UNTERMAN TG, OEHLER DT, BECKER RE: Identification of a type Iinsulin-like growth factor binding protein (IGF-BP) in serum fromrats with diabetes mellitus. Biochem Biophys Res Commun 163:882—887, 1989
30. CONOVER CA: Potentiation of insulin-like growth factor (IGF) actionby IGF-binding protein-3: Studies of underlying mechanism. Endocri-nology 130:3191—3199, 1992
31. ANDRESS DL, BIRNBAUM RS: Human osteoblast-derived insulin-likegrowth factor (IGF) binding protein-5 stimulates osteoblast mitogen-esis and potentiates IGF action. J Biol Chem 267:22467—22472, 1992
32. YAP J, TsAo T, STEWART T, M0IILER M, COOK J, RABKIN R:Insulin-like growth factor binding protein (BP) secretion by rabbitproximal tubular cells (RPTC). (abstract) J Am Soc Nephrol 3:483,1992
33. FAWCETr J, RABKIN R: The processing of insulin-like growth factor-Iby a cultured kidney cell line is altered by IGF-binding protein-3.Endocrinology 136:1340—1347, 1995
34. KOBAYASHI 5, Ai T, HISHIDA A, CLEMMONS DR: Insulin-likegrowth factor (IGF)-binding protein enhances the mitogenic responseto IGF-I in MDCK cells. (abstract) JAm Soc Nephrol 3:471, 1992
35. HISE MK, MANTZOURIS NM, LAHN JS, SHEIKH MS, SI-lAo Z-M,FONTANA JA: IGF binding protein 5 and IGF-I receptor regulation inhypophysectomized rat kidneys. Am J Physiol 266:F147—F154, 1994
36. GELATO MC, ALEXANDER D, MARSH K: Differential tissue regulationof insulin-like growth factor binding proteins in experimental diabetesmellitus in the rat. Diabetes 41:1511—1519, 1992
37. BACH LA, Cox AJ, MENDELSOHN FAO, HERINGTON AC, WERTHERGA, JERUMS G: Focal induction of IGF binding proteins in proximaltubules of diabetic rat kidney. Diabetes 41:499—507, 1992
38. FLYVBJERG A, KESSLER U, DoRiA N, FUNK B, ØRSKOV H, KIESS W:Transient increase in renal insulin-like growth factor binding proteinsduring initial kidney hypertrophy in experimental diabetes in rats.Diabetologia 35:589—593, 1992
39. MUCHANETA-KUBARA EC, CoPE GH, EL NAHAS AM: Biphasic ex-pression of IOF-I following subtotal renal ablation. Exp Ncphrol3:165—172, 1995




















