
Recent Developments on Rotaxane-Based Shuttles: An Update to 2010
34
Current Organic Chemistry, 2012, 16, 127-160 127 1875-5348/12 $58.00+.00 © 2012 Bentham Science Publishers Recent Developments on Rotaxane-Based Shuttles: An Update to 2010 Antonio Rescifina, *,a Ugo Chiacchio, a Antonino Corsaro, a and Giovanni Romeo b a Dipartimento di Scienze Chimiche, Università di Catania, Viale Andrea Doria 6, 95125 Catania, Italy b Dipartimento Chimico-Farmaceutico, Università di Messina, Viale SS. Annunziata 6, 95168 Messina, Italy Abstract: This update reviews the major developments on rotaxane-based shuttles during the period of August 2008 to October 2010 and is organized similarly to the previous review [1]. The progress in the field of chemical controlled shuttles can be summarized as a significant increase of the shuttling rates, second-generation pH-switchable Pd(II)-complexed rotaxanes, and new synthetic approaches, especially those based on Diels-Alder cy- cloadditions. The new synthetic routes allow ready access to rotaxanes that feature the template site fully incorporated into the interlocked product. This template site is unusual for active-template reactions and produces shuttles that exhibit entropy-driven translational isomerism with remarkably improved positional discrimination as was shown by the incorporation of a hydrogen-bonding TEG-station into an imine-bridged rotaxane. With respect to photochemically powered molecular switches, the most important result originated from the synthesis of a degenerate, donor-acceptor, light-gated, STOP-GO molecular shuttle. For electrochemically controllable bistable rotaxanes, the first example of a molecular shuttle with three different stations, which dramatically increase the shuttling process rate, has been reported. Finally, the last study in this review involves redox-driven switching and electrochromic responses of LC films containing a bistable [2]rotaxane. The presence of a polymer electrolyte provides new prospects for the further development of rotaxane-based molecular ma- terials by exploiting the dynamic, anisotropic, and coherent properties of liquid crystals. Keywords: Mechanically interlocked molecules; Click chemistry; Molecular devices; Nanoscale machines. INTRODUCTION Due to the expeditious propagation of new and more sophisti- cated interlocked machines, it is unsurprising that an update of the previously published review [1], which focused on [2]rotaxane-based shuttles, is desired. Since the first account reviewed the literature up to July 2008, this update covers that pub- lished between August 2008 and October 2010. Moreover, because there is a plethora of literature related to this field, this update fo- cuses on the most relevant results. To facilitate the reader, the organization of this update follows that of the original review and includes sections regarding chemically, photochemically, and elec- trochemically induced molecular switching. Mechanically interlocked molecules attract considerable atten- tion as the basis for molecular devices and nanoscale machines long after the first reported syntheses of these chemical curiosities in the mid-1960s [2]; [n]rotaxanes (rota meaning wheel, axis meaning axle) are one common form. The number “n” enclosed in square Address correspondence to this author at the Dipartimento di Scienze Chimiche, Università di Catania, Viale Andrea Doria 6, 95125 Catania, Italy; Tel: +390957385017; Fax: +3906233208980; E-mail: [email protected] brackets describes the number of molecular parts involved in mechanical bonding; for example, a [2]rotaxane is formed when a thread, dumbbell, or axle component is mechanically interlocked with a ring component. If the axle component is blocked at both ends by bulky functional groups to prevent dissociation of the ring component at ambient temperature (Fig. 1), the system is called a rotaxane. However, if there are no stoppers on the axle or the stop- pers are not of sufficient size to preclude dissociation at ambient temperature, the molecular assembly is referred to as a [2]pseudorotaxane. A [3]rotaxane is typically formed when two rings are threaded onto an axle component although two axles with one ring also satisfies the definition. There are three common methods of preparing rotaxanes: cap- ping, clipping, and slipping; however, a recent route via a template leads to improved yields over those achieved using statistical syn- theses. The value of rotaxanes as molecular abacuses or transport agents and their capacity to shuttle (i.e., molecular motion) and switch (i.e., molecular logic) has caused research to flourish over the past decade: much effort has been dedicated to improving the synthetic methodologies to enable enhanced functions [3]. Rotaxanes have emerged as a prototype for a class of molecular Fig. (1). Schematic representation of a [2]rotaxane. Stoppers Stations Ring Thread
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Recent Developments on Rotaxane-Based Shuttles: An Update to 2010

Current Organic Chemistry, 2012, 16, 127-160 127
1875-5348/12 $58.00+.00 © 2012 Bentham Science Publishers
Recent Developments on Rotaxane-Based Shuttles: An Update to 2010
Antonio Rescifina,*,a
Ugo Chiacchio,a Antonino Corsaro,
a and Giovanni Romeo
b
aDipartimento di Scienze Chimiche, Università di Catania, Viale Andrea Doria 6, 95125 Catania, Italy
bDipartimento Chimico-Farmaceutico, Università di Messina, Viale SS. Annunziata 6, 95168 Messina, Italy
Abstract: This update reviews the major developments on rotaxane-based shuttles during the period of August 2008 to October 2010 and
is organized similarly to the previous review [1].
The progress in the field of chemical controlled shuttles can be summarized as a significant increase of the shuttling rates,
second-generation pH-switchable Pd(II)-complexed rotaxanes, and new synthetic approaches, especially those based on Diels-Alder cy-
cloadditions. The new synthetic routes allow ready access to rotaxanes that feature the template site fully incorporated into the
interlocked product. This template site is unusual for active-template reactions and produces shuttles that exhibit entropy-driven
translational isomerism with remarkably improved positional discrimination as was shown by the incorporation of a hydrogen-bonding
TEG-station into an imine-bridged rotaxane.
With respect to photochemically powered molecular switches, the most important result originated from the synthesis of a degenerate,
donor-acceptor, light-gated, STOP-GO molecular shuttle. For electrochemically controllable bistable rotaxanes, the first example of a
molecular shuttle with three different stations, which dramatically increase the shuttling process rate, has been reported.
Finally, the last study in this review involves redox-driven switching and electrochromic responses of LC films containing a bistable
[2]rotaxane. The presence of a polymer electrolyte provides new prospects for the further development of rotaxane-based molecular ma-
terials by exploiting the dynamic, anisotropic, and coherent properties of liquid crystals.
Keywords: Mechanically interlocked molecules; Click chemistry; Molecular devices; Nanoscale machines.
INTRODUCTION
Due to the expeditious propagation of new and more sophisti-
cated interlocked machines, it is unsurprising that an update of the
previously published review [1], which focused on
[2]rotaxane-based shuttles, is desired. Since the first account
reviewed the literature up to July 2008, this update covers that pub-
lished between August 2008 and October 2010. Moreover, because
there is a plethora of literature related to this field, this update fo-
cuses on the most relevant results. To facilitate the reader, the
organization of this update follows that of the original review and
includes sections regarding chemically, photochemically, and elec-
trochemically induced molecular switching.
Mechanically interlocked molecules attract considerable atten-
tion as the basis for molecular devices and nanoscale machines long
after the first reported syntheses of these chemical curiosities in the
mid-1960s [2]; [n]rotaxanes (rota meaning wheel, axis meaning
axle) are one common form. The number “n” enclosed in square
Address correspondence to this author at the Dipartimento di Scienze Chimiche,
Università di Catania, Viale Andrea Doria 6, 95125 Catania, Italy;
Tel: +390957385017; Fax: +3906233208980; E-mail: [email protected]
brackets describes the number of molecular parts involved in
mechanical bonding; for example, a [2]rotaxane is formed when a
thread, dumbbell, or axle component is mechanically interlocked
with a ring component. If the axle component is blocked at both
ends by bulky functional groups to prevent dissociation of the ring
component at ambient temperature (Fig. 1), the system is called a
rotaxane. However, if there are no stoppers on the axle or the stop-
pers are not of sufficient size to preclude dissociation at ambient
temperature, the molecular assembly is referred to as a
[2]pseudorotaxane. A [3]rotaxane is typically formed when two
rings are threaded onto an axle component although two axles with
one ring also satisfies the definition.
There are three common methods of preparing rotaxanes: cap-
ping, clipping, and slipping; however, a recent route via a template
leads to improved yields over those achieved using statistical syn-
theses. The value of rotaxanes as molecular abacuses or transport
agents and their capacity to shuttle (i.e., molecular motion) and
switch (i.e., molecular logic) has caused research to flourish over
the past decade: much effort has been dedicated to improving the
synthetic methodologies to enable enhanced functions [3].
Rotaxanes have emerged as a prototype for a class of molecular
Fig. (1). Schematic representation of a [2]rotaxane.
Stoppers
Stations
Ring
Thread

128 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
machines as a result of their ability to undergo translational iso-
merization between two or more structures (Fig. 2), which origi-
nates from the translational molecular motion of the macrocyclic
ring along the acyclic component. The equilibration and dynamic
behavior back and forth along the acyclic component similar to the
movement of a bus or train between stations is known as shuttling
and can occur up to 40,000 times per second. Molecular recognition
sites are built into both components to produce thermodynamic
sinks, which form points along the linear component where two or
more components predominantly reside. It is vital for both synthesis
and functionality that the components of a rotaxane system are elec-
tronically complementary. This is the reason for the highly colored
nature of most rotaxanes.
Metal complexing sites, which often feature different denticities
(i.e., bidentate and tridentate), are popular options as “stations” on
axle components with the molecular shuttling driven by electro-
chemical stimulation. Sauvage and co-workers have shown that
stations with the same denticity may also be incorporated into the
axle structure and result in [2]rotaxanes that function via electro-
chemical means [4]. In this example, the axle binding sites include
a highly shielding phenanthroline ligand and a non-sterically hin-
dered bipyridine chelate; a bisquinoline unit in the macrocyclic ring
affords the complementarity via copper complexation (Fig. 3). Al-
though its preparation involves a multi-step synthesis, the electro-
chemically induced shuttling between the two stations is a fast,
clean process and is promising for the development of multi-state
machines in the future.
The successful achievement of this relatively complicated in-
terlocked system arises from the design of the macrocycle. The
positioning of a ligand in the macrocyclic structure provides two
functions: (1) it acts as a ligation site for a transition metal that will
act as a catalyst for the bond formation of the axle components, and
(2) it provides a template for the successive covalent bond forma-
tion of interlocked axle components. This high yielding methodol-
ogy may be applicable to other higher-order interlocked molecular
assemblies, which is important for the development of increasingly
complex molecular machinery. Interest has also been expressed for
potential biological applications of mechanically interlocked rotax-
anes due to the encapsulation or protection of the axle component
provided by the macrocyclic ring. As an example, Leigh and
co-workers reported a rotaxane comprising a pentapeptide axle and
a macrocycle, which protects the axle against peptidase-catalyzed
hydrolysis for several days [5]. The use of oligopeptide axle com-
ponents was reported by Moretto et al. who enabled characteristic
shuttling between stations upon the change of the solvents (Fig. 4)
[6].
Fig. (2). Rotaxanes are able to shuttle between two different states indicating switching (ON/OFF) or binary processes (0/1). Shuttling is a form of transla-
tional isomerism. The challenge is the control of this motion.
Fig. (3). Copper-complexing [2]rotaxane possessing two different bidentate stations. The diphenylbiisoquinoline ligand (DPBIIQ) site in the macrocyclic ring
structure is complementary to either the diphenylphenanthroline (DPP) or bipyridine (BIPY) sites along the threading molecule and shuttling is possible upon
electrochemically-driven complexation events with copper.
Shuttling
State
OFF or 0
ON or 1
N N
O O
O O
N N
Cu
DPP BIPY
DPBIIQ
N N
O O
O O
N N
Cu
Station 1DPP-Cu-DPBIIQ
Station 2BIPY-Cu-DPBIIQ

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 129
CHEMICALLY INDUCED SWITCHING OF BISTABLE
ROTAXANES
Research devoted to elucidating the effect of structural varia-
tions on the positional bias and kinetics of shuttling, which is im-
portant for the development of rotaxane systems with levels of mo-
tion control beyond that of simple switches, has led to a second
generation of palladium(II)-complexed molecular shuttles. These
shuttles feature structural changes to the size and shape of the mac-
rocycle [7] resulting in significantly increased shuttling rates and
improved co-conformational bias compared to the original system.
A notable feature of the first-generation system, e.g., rotaxane 1
(Scheme 1), was its metastability, i.e., a change of the chemical
state of the thread does not immediately cause a change in the posi-
tion of the macrocycle. Although this allows the system to be held
in an out-of-equilibrium state, which is difficult to achieve using
molecular shuttles that rely on weak non-covalent interactions,
elevated temperatures and extended times are required (up to 16 h
at 383 K to reach equilibrium) to overcome the energy barrier to
shuttling. Furthermore, the positional bias of the Pd-macrocycle
between the 4-dimethylaminopyridine (DMAP) and pyridine (Py)
Fig. (4). Schematic representation of a [2]rotaxane molecular machine possessing an oligopeptide axle that results in a helical structure.
Scheme 1. Operation conditions and positional bias of (a) first generation 1 and (b) second generation 2 molecular shuttles.
CHCl3, CH3CN,
NNO
R OR
NMe2
PdN N
O O
O O
tBu
But tBu
1
R =
NNO
R OR
NMe2
PdN N
O O2
O O
a)
b)
+
1:8383 K,16 h
DMF-d7
+
6:1383 K,1.5 h
DMF-d7
TsOH – TsOH TsOH – TsOH
+
1:32323 K, 4 hDMF-d7
+
12:1323 K,2 hDMF-d7
TsOH – TsOH TsOH – TsOH
N
N
N HN
NH
N
+ +

130 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
stations was modest in both the neutral (ca. 6:1) and protonated (ca.
1:8) states of the rotaxane thread (Scheme 1a). In the
second-generation molecular shuttle, e.g., rotaxane 2 (Scheme 1b),
the benzylic amide macrocycle is replaced by a bisanilide ring,
which is larger and has a different shape. The alteration of the steric
environment around the palladium(II) centre in rotaxane 2 leads to
greatly enhanced rates of macrocycle shuttling and improved
positional bias in both chemical states of the thread. In addition to
the minimum energy co-conformers of the two switched systems,
two metastable out-of-equilibrium positional isomers were suffi-
ciently stable to be isolated and characterized using 1H NMR spec-
troscopy.
Rotaxane 2 was prepared via a threading-and-stoppering pro-
cedure. Initially, a palladium-macrocycle-acetonitrile complex was
treated with an unstoppered DMAP end-thread precursor followed
by the reaction of the resulting pseudorotaxane with excess
phenol-based stoppers under Mitsunobu conditions to afford the
[2]rotaxane.
The associative nature of palladium(II) substitution reactions
and the faster shuttling rate of 2 than that of 1 suggest that the
bisanilide macrocycle provides a less sterically demanding envi-
ronment around the metal ion and thus easier access for incoming
coordinating solvent molecules or TsO– counter-anions.
Active-template synthesis involves the construction of me-
chanically interlocked structures with the metal ion acting as both a
template to entwine or thread the components and a catalyst to
capture the interlocked final product by covalent bond formation
[8]. During this process, the metal often changes coordination
strength and the preferred geometry of its ligands several times.
Despite the complexity of this mechanism, several different
metal-catalyzed reactions have already proven suitable for the ac-
tive-metal template synthesis of both rotaxanes and catenanes in-
cluding copper(I)-catalyzed terminal alkyne-azide cycloaddition
(i.e., the CuAAC “click” reaction), palladium- and copper-catalyzed
alkyne homocouplings and heterocouplings, and
palladium-catalyzed oxidative Heck couplings and Michael addi-
tions.
However, unlike traditional “passive” metal-template methods,
permanent intrinsic recognition motifs are not required on each of
the components that will be interlocked during active-template syn-
theses, i.e., the assembly can be traceless. This means that one of
the most widely exploited features of rotaxane template assembly,
i.e., that the intercomponent recognition motif remains in the rotax-
ane to provide preferred binding sites for the ring on the thread as is
used in molecular shuttles, is not inherently present using ac-
tive-template syntheses.
To solve this issue, Leigh’s group reported the active-template
synthesis of rotaxanes using Lewis acid–catalyzed Diels-Alder
cycloaddition [9]. The reaction proceeds in the presence of either
Zn(II) or Cu(II) salts with weakly coordinating triflate anions and
generates[2]rotaxanes in up to 91% yields. The active-template
reaction “selects” 2-substituted cyclopentadienes over 1-substituted
cyclopentadienes, which results in the generation of one rotaxane
isomer with 90%–99% selectivity as opposed to the four isomers
produced in the case of non-interlocked threads. Unusually for an
active-template reaction, the template site is fully incorporated into
the interlocked product; therefore, metal-chelated molecular shut-
tles can readily be prepared. This is exemplified by the synthesis of
a molecular shuttle in which the position of the macrocycle can be
switched by varying the coordination requirements of the metal ion
(e.g., trigonal-bipyramidal Zn(II) or square planar Pd(II)).
Rotaxane formation from dienophile 4, diene 5, and complexes
of macrocycle 3 with various Lewis acids, which were formed in
situ in CH2Cl2, was investigated [9]. The addition of acryloyl imi-
dazolidone 5 at room temperature and diene 5 at –78 °C to the
CH2Cl2 solution of the macrocycle-Lewis acid complexes was fol-
lowed by stirring at –78 °C for 20 h and at room temperature for a
further 4 h. Initial experiments with ZnCl2 were unsuccessful,
which is likely because the two chloride ligands are too strongly
coordinated to the metal. However, the use of Zn(OTf)2 afforded
[2]rotaxane 6 in 8% yield. Changing the Lewis acid from zinc tri-
flate to copper triflate increased the yield of 6 to 47%, whereas
increasing the amounts of 4 and 5 and extending the reaction time
(48 h at –78 °C followed by 48 h at room temperature) improved
the yield of 6 to 42% with Zn(OTf)2 and 83% with Cu(OTf)2. Using
the same optimized reaction conditions with only 30 mol% copper
triflate reduced the yield of 6 to 37%, which indicates that 6 se-
questers the metal ion thereby preventing turnover of the active
template to any significant extent during the reaction.
The proposed mechanism for rotaxane formation is shown in
Scheme 2. Replacement of the two labile ligands with oxygen at-
oms of the acryloyl imidazolidone unit affords threaded complex II;
in this complex, the activated double bond must react with
stoppered cyclopentadiene 5 on the macrocycle face opposite the
dienophile stopper group. Demetalation with ammonia [for Cu(II)]
or Na4EDTA [for Zn(II)] generates the metal-free rotaxane, 6.
The utility of the metal binding site in the rotaxane-forming Di-
els-Alder cycloaddition remains essentially unchanged in the ro-
taxane product, as was demonstrated during the synthesis of more
complex rotaxanes. Molecular shuttle 10:10 was prepared from
Py-containing dienophile building block 9 and diene 5 (Scheme 3).
The 42% yield of [2]rotaxane 10:10 was remarkably good, and a
high (1,4)/(1,3) ratio (95:5) was maintained despite the presence of
the Py unit, which can compete with the macrocycle for complexa-
tion with the Lewis acid. In this reaction, the building blocks offer
an unhindered site for the Lewis acid–catalyzed reaction to form the
thread. Additionally, the Py unit and -carbonyl imidazolidone
functionalities, which persist in [2]rotaxane 10:10 , could be used as
binding sites with different preferred coordination geometries to
control the position of the metal-coordinated macrocycle on the
thread (Scheme 4).
As with the simpler rotaxane, 6:6 , the room-temperature 1H
NMR spectrum of 10:10 (298 K, 9:1 CD2Cl2/CD3CN) shows an
upfield shift of the aliphatic and Py signals with respect to those of
the non-interlocked thread. This is particularly evident for the Hf–h,
H , and Hk–m protons, which indicates that the macrocycle moves
freely but spends most of its time on the less-hindered portions of
the thread. Upon the addition of Zn(OTf)2 to the NMR tube con-
taining 10:10 , the 1H NMR spectrum increased in complexity sug-
gesting that multiple coordinated species were present and likely
interchanging. However, the saturation of the rotaxane binding sites
by adding excess Zn(OTf)2 led to a greatly simplified spectrum.
This spectrum shows significant upfield shifts of the -carbonyl
imidazolidone moiety signals (i.e., H , Hv, Hu, and Hz) and down-
field shifts of the Py moiety (i.e., Hj–n) with respect to the metal-
free rotaxane. This indicates that the macrocycle is positioned over
the -carbonyl imidazolidone unit as part of a trigonal bipyramidal–
coordinated Zn(II) complex and the thread Py group is coordinated

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 131
Scheme 2. Diels-Alder active-metal-template synthesis of [2]rotaxane 6 from macrocycle 3, dienophile 4, and diene 5.
MO
N
O
O O3
O
N
O
O O
L L
MO
N
O
O O
O O
NNRO
O O
NN
RO
MO
N
O
O O
O O
NN
RO
OR
R = (t-BuC6H4)3C(C6H4)
M = Cu(II), Zn(II)
L = Cl, OTf
I
4
II
ML2
O O
NNRO
OR
OR
H
O
N
O
O O
O O
NN
RO
ORH
2 TfO
L = TfO– 2 L
2 TfO
5(2) only
O O
NNRO
+
OR
OR
OR
[1,5]-shift
5(2)
5'(1)
5(2):5'(1)55:45
7(1,4):7'(1,3)95:5
8(1,2): major8'(1,5): minor
1
2
1
1
H
H
3
4
2
5
7
8
7:861:39
NH3[Cu(II)]or
Na4EDTA[Zn(II)]
[Cu(NH3)6](OTf)2or
Na2[Zn(EDTA)] + 2 NaOTf
1 3
4
M = Zn: 42%M = Cu: 83%
6(1,4):6'(1,3)9:1
non-interlockedthread production

132 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
N
RO
NN
O
O
N
RO
N
O
O
H
OR
N
RO
NN
O
O
H
OR
N
RO
NN
O
O
H
OR
OR
3, 5, Cu(OTf)2
–78 °C, 48 h
then r.t. 48 h
42%
10(1,4):10'(1,3)
95:5
+ non-interlocked threads 11 and 12
42%
11(1,4):11'(1,3)
86:14
12(1,2): major
12'(1,5): minor
11:12
68:32
R = (t-BuC6H4)C(C6H4)
O
N
O
OO
N
9
Scheme 3. Assembly of molecular shuttle 10:10 from the Diels-Alder active-metal-template reaction of macrocycle 3, dienophile 9, and diene 5.
to a second Zn(II) ion (structure [10:10 Zn2]L2(OTf)4 in Scheme
3a). Demetalation (Na4EDTA, RT, 1 h) of the zinc complex liber-
ated metal-free rotaxane 10:10 .
The addition of PdCl2 to 10:10 results in the formation of a
different rotaxane complex, i.e., [10:10 Pd]Cl2 (Scheme 3c). How-
ever, in the 1H NMR spectrum of [10:10 Pd]Cl2, the signals of the
protons adjacent to the thread Py unit (Hj and Hn) are shielded by
the aromatic rings of the macrocycle and are consistent with the
chemical shifts of a previously reported palladium-complexed ro-
taxane [10] while the signals of the -carbonyl imidazolidone moi-
ety are similarly shifted to those of the non-interlocked thread.
Demetalation of [10:10 Pd]Cl2 with KCN (MeOH/CH2Cl2 1:1, RT,
1 h) again affords the metal-free [2]rotaxane, 10:10 .
This mechanism can be exploited to construct molecular shut-
tles in which the position of the macrocycle can be controlled by
the coordination requirements of the metal ions coordinated to the
rotaxane. Such metal ion–switchable systems historically require
lengthy syntheses (10–20 steps); therefore, the active-template Di-
els-Alder reaction is a valuable addition to the methods for the as-
sembly of interlocked molecular-level architectures.
Novel examples of molecular shuttles (13a,b) that exhibit en-
tropy-driven translational isomerism with remarkable positional
discrimination feature an imine-bridged attached to triethylene gly-
col ether (TEG) stations. These compounds can be completely
transformed to hydrolyzed [2]rotaxanes under hydrolytic conditions
due to hydrogen-bond formation between the macrocycle and the

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 133
TEG station [11]. Analysis of the thermodynamic parameters
revealed that imine-bond hydrolysis and the formation of hydrogen
bonds between the macrocycle and the station are thermodynami-
cally matched processes because they are both enthalpically favored
and accompanied by a loss of entropy. The combination of
imine-bonding and hydrogen-bonding stations in a rotaxane system
is key to the observed entropy-driven positional switching of the
macrocycle.
N
RO
NN
O
O
H
OR
R = (t-BuC6H4)C(C6H4)
O
N
O
OO
N
RO
N N
OO
H OR
Zn
O
N
O
OO
PdCl Cl
2 TfO
ZnL L
TfO OTf
N
RO
N
O
O
H
OR
O
N
O
OO
N
Zn(OTf)2 EDTA
KCN PdCl2
[10:10'Zn2]L2(OTf)4
L = H2O, solvent or pyridine
from another [2]rotaxane
A
B
C
D
E
F
G
HI
J
K
a–e
f
g
h
i
j
k
lm
n op
q
r
s
t u
v
yw
x
z–
10(1,4):10'(1,3)
95:5
[10:10'Pd]Cl2
a)
b)
c)
Scheme 4. Determination of the macrocycle position by the metal coordination geometry in molecular shuttle 10:10 . (a) Macrocycle position upon coordina-
tion to Zn(II): [10:10 Zn2]L2(OTf)4. (b) Metal-free rotaxane 10:10 . (c) Macrocycle position upon coordination to Pd(II): [10:10 Pd]Cl2.

134 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
Imine bonds can be equilibrated with an amine-aldehyde pair
under hydrolytic conditions; their cleavage under dynamic acidic
hydrolytic conditions is assisted by the formation of hydrogen
bonds between the resulting primary ammoniums (–NH3
+) and suit-
able, newly attached hydrogen-bonding acceptors [12].
From the perspective of thermodynamic contributions, imine
bonding and hydrogen bonding must work synergistically to enable
entropy-driven positional switching of the macrocycle in a rotaxane
structure (Scheme 5). Hydrogen bond formation is enthalpically
favored and is accompanied by a loss of entropy, whereas imine
bond formation is entropically favored in a rotaxane structure. Gi-
useppone and Lehn reported that the equilibrium ratio of the imine
form in dynamic combinatorial libraries (DCLs) increased with
increasing temperature under low acidic conditions [13]. Thus,
imine bond cleavage and hydrogen bond formation are thermody-
namically matched processes (i.e., enthalpically favored and en-
tropically disfavored processes). Therefore, the reverse processes of
hydrogen bond cleavage and imine bonding must also be matched
processes (i.e., enthalpically disfavored and entropically favored
processes). Accordingly, it is expected that the incorporation of
both imine bridging and hydrogen bonding stations on a rotaxane
axle would result in a novel rotaxane-based molecular shuttle that
exhibits entropy-driven translational isomerism [14].
Shuttles 13a,b, which feature TEG units as hydrogen-bonding
stations, were prepared by introducing end groups with TEG units,
17b, into imine-bridged pseudorotaxane 16, which was prepared
from a hydrindacene axle and a macrocyclic diamine using a
threading method directed by imine bond formation (Scheme 6).
Neutral dithioacetalized [2]rotaxanes 16b were obtained by treating
the solution of 13b with ethanedithiol under acidic hydrolytic con-
ditions.
Subsequently, the dynamic equilibrium between 13a,b and
14a,b·2H2+
with TEG stations under acidic hydrolytic conditions
was investigated to determine whether the preferred position of a
macrocycle could be switched from the imine-bridging hydrin-
dacene station to the TEG station on the rotaxane axle (Scheme 7).
Upon addition of TFA to a solution of 13a, which features a
TEG and xylylene (XYL) station, in wet CDCl3, the 1H NMR spec-
trum revealed the disappearance of the signals derived from
bis-imine 13a and the appearance of a new set of signals including
CHO signals (Ha,a ) at 9.4 ppm even at 298 K that were assigned
to a single species. The signals for the protons around the TEG
station (Hi–n, Ho, and Hh) of the newly generated species appeared
upfield of those of bis-imine 13a, whereas those for the protons
around the XYL station (Hh ,o and Hq,t) remained unchanged. Thus,
the newly generated species was assigned as a [2]rotaxane,
14a·2H2+
, in which the macrocycle is located at the TEG station on
the axle due to hydrogen-bond formation. The exclusive generation
of [2]rotaxane 14b·2H2+
from 13b was observed under acidic hy-
drolytic conditions at 295 K.
The rate (k) and energy barrier ( G‡) for the shuttling of the
macrocycle of 14b·2H2+
were determined to be 574 s–1
and 12.5 ±
0.2 kcal mol–1
at 273 K (Tc), respectively, using the coalescence
method via variable-temperature (VT) NMR. When the acidic solu-
tions containing 4a,b·2H2+
were subjected to dehydrating condi-
tions, bis-imines 13a,b were quantitatively regenerated.
In addition to the successful hydrolytically controlled switching
of bis-imines 13 and [2]rotaxanes 14·2H2+
, it was found that they
could also be switched by simply changing the temperature under
acidic hydrolytic conditions. In particular, 13a,b/14a,b·2H2+
can be
completely switched from one TEG station to another as a function
of temperature, thereby demonstrating entropy-driven positional
switching of the macrocycle in a molecular shuttle.
The release of water molecules accompanied by the in-
tramolecular imine-bond formation from 14a,b·2H2+
to 13a,b seem-
ingly contributes to the entropy gain during heating. Thus, the equi-
Scheme 5. Thermodynamic interplay of imine- and hydrogen-bonding in a rotaxane-based molecular shuttle exhibiting entropy-driven translational isomerism.
O O
N
O O O O
H < 0 S < 0
H > 0 S > 0
NH3
CHO
NH3
CHO
H < 0 S < 0
H > 0 S > 0
thermodinamically matched
imine-bonding cleavage hydrogen-bonding
OOOO
N
N
OOO
CHO
CHO NH3
H3NO
low T
high T
imine-bridged rotaxane 13a hydrogen-bonded [2]rotaxane 14a•2H2+
translocation of the macrocycle by temperature
entropy-driven switching

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 135
OTBSTBSO
N
N
CH2OOCH2
CH2OOCH2
(CH2)4
(CH2)4
A X
17a: X = Br, A =
17b: X = I, A =
CH2H2C
O
O
N
N
CH2OOCH2
CH2OOCH2
(CH2)4
(CH2)4
A2 A1
R
R R
CHO
NH3
OHC
NH3
OO
OO
(CH2)6
(CH2)6
A2 A1R R
NH2
NH2
OO
OO
(CH2)6
(CH2)6
A2 A1R R
SS
S S
TBAF
Cs2CO3
THF/DMF
16
13a,b; a = 25%, b = 76%
TFA, wet CDCl3
+ H2O– H2O
[mono-imine 15]
+ H2O – H2O
(CH2SH)2
for 14b
16b; 66%
14a,b•2H2+
Ha
O O
O O O O
t
s r
q
i j k l m n
O O O O
i j k l m n
O O O O
n' m' l' k' j' i'
A2 A1
a:
b:
2
2
2
R =
17a,b
o' h' g h o p
AB
C
D
E
Ha
Hb Hc d e
f
Scheme 6. Synthesis of imine-bridged rotaxanes 13a,b, hydrogen-bonded [2]rotaxanes 14a,b•2H2+
, and dithioacetalized [2]rotaxanes 16a,b.

136 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
Scheme 7. Transformation between imine-bridged rotaxanes 13a,b and [2]rotaxanes 14a,b under acidic hydrolysis/dehydration conditions.
librium ratio of 13a,b/14a,b, which determines the preferred posi-
tion of the macrocycle between the imine-bridging and hydro-
gen-bonding stations, could be completely reversed as a function of
temperature (the ratios of 13a,b/14a,b·H2+
= <5/95 at 273 K and
>95/5 at 373 K).
The changes in both enthalpy and entropy between 13a and
14a·2H2+
( H = –16.7 ± 0.7 kcal mol–1
and S = –52.2 ± 2.1 cal
mol–1
K–1
) were larger than those for the analogs incorporating the
non-hydrogen-bonding XYL station [10]. This difference originates
from the incorporation of a TEG station instead of a XYL station,
i.e., the additional enthalpic stabilization and loss of entropy due to
hydrogen bonding of the macrocycle with the TEG station in
14a·2H2+
. The conformational restriction of the TEG moiety that
accompanies hydrogen bonding might contribute to the negative
value of the S term. Consequently, the incorporation of a TEG
station with hydrogen-bonding abilities into imine-bridged rotax-
anes not only biases the 13a/14a·2H2+
equilibrium towards the hy-
drolyzed [2]rotaxane under acidic hydrolytic conditions but also
enables entropy-driven positional switching of the macrocycle by
supplying additional enthalpic and entropic differences.
Chemically driven shuttles, excluding those controlled by
acid-base proton transfer (i.e., pH-controlled reactions), remain
scarce. Catalytic reactions are considered to be prime candidates for
integrating this type of control into synthetic molecular machines
that work via an energy ratchet mechanism.
Berna et al. synthesized azo-functionalized hydrogen bond–
assembled [2]rotaxanes 20 and 21 that comprise a dumbbell com-
ponent, which contains an azodicarboxamide and a succinic amide
ester binding site, threaded through a tetrabenzylic amide macrocy-
cle (Scheme 8) [15]. These binding sites can be reversibly and effi-
ciently interconverted to their hydrazo forms through hydrogena-
tion-dehydrogenation of the nitrogen-nitrogen bond. This type of
chemically switchable control element has been implemented in
stimuli-responsive molecular shuttles that function through a re-
versible azo/hydrazo interconversion and produce large net
positional changes with good discrimination between the binding
sites of the macrocycle in both states of the shuttle. These molecu-
lar shuttles operate by two different mechanisms: (1) a discrete
mode through two reversible and independent chemical events and,
importantly, (2) a continuous regime through a catalyzed ester
bond–formation reaction in which the shuttle acts as an organo-
catalyst. In the latter mechanism, the incorporation of both states of
the shuttle into the simple chemical reaction network promotes
dynamic translocation of the macrocycle between two nitrogen- and
carbon-based stations of the thread, which allows an energetically
uphill esterification process to occur.
Molecular shuttles 20 and 21 were prepared as outlined in
Scheme 8. Threads 18 and 19 were synthesized by standard proce-
dures from diphenyl hydrazodicarboxylate, which was sequentially
aminated with commercially available 2,2-diphenylethylamine or
dibenzylamine and 2,2-diphenylethyl 3-(12-aminododecylcar-
bamoyl)propanoate, which already contains a succinic amide ester
station. The obtained hydrazo compounds, [2H]-18 and [2H]-19,
were dehydrogenated with NBS/Py to form azo threads 18 and 19
in 32 and 51% overall yields, respectively. Compounds 18 and 19
were subjected to rotaxane-forming reagents and conditions (i.e.,
p-xylylenediamine, isophthaloyl dichloride, triethylamine, chloro-
form; 4 h, high dilution) to produce molecular shuttles 20 and 21 in
34 and 45% yields, respectively.
Molecular shuttle 20 was quickly, cleanly, and quantitatively
converted into its hydrazo derivative [2H]-20 by reduction with
hydrazine: hydrogenation was complete in less than five minutes
and a rapid loss of the initial orange color of the azo compounds
was observed concomitant with the translocation of the macrocycle
to the succinic amide ester station.
OOOO
N
N
OOO
CHO
CHO NH3
H3NO
low T
high T
A2
A2
H
+ H2O
H
– H2O
13a,b
14a,b•2H2+
[15a,b•H+]
H
+ H2O
H
– H2O
13a,b:14a,b>95:5 at 373 K
<5:95 at 273 K
fixed
high temperature
low temperature slow shuttling
a:
b:OO
OO
O O
A2

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 137
A comparable series of shifts occur in the 1H NMR spectra of
molecular shuttle 21/[2H]-21. The translocation of the macrocycle
is fully reversed upon oxidation with NBS/Py to regenerate the
azodicarboxamide site and trigger ring movement to this station via
biased Brownian motion.
The successful implementation of energy ratchet mechanisms in
molecular-level structures has become essential to the development
of functional molecular machines that are more complex than sim-
ple switches. Thus, a chemically driven machine that works cycli-
cally to generate synthetically useful covalent bonds was designed
as a novel strategy to incorporate a new type of control over the
switching periodicity of the potential energy surface of a bistable
[2]rotaxane. In particular, the Mitsunobu protocol [16] takes ad-
vantage of the transformation of the azodicarboxamide function
PhOHN
NH
OPh
O
O
R1N
HN
NH
NH
HN
OPh
R2
O
O
Ph
O
O
( )7
R1N N
N NH
HN
O
Ph
R2
O
O
Ph
O
O
( )7
R1N N
N NH
HN
O
Ph
R2
O
O
Ph
O
O
( )7
R1N
HN
NH
NH
HN
O
Ph
R2
O
O
Ph
O
O
( )7
[2H]-18: R1 = Ph2CHCH2; R2 = H
[2H]-19: R1 = R2 = PhCH2
18: R1 = Ph2CHCH2; R2 = H
19: R1 = R2 = PhCH2
20: R1 = Ph2CHCH2; R2 = H
21: R1 = R2 = PhCH2
[2H]-20: R1 = Ph2CHCH2; R2 = H
[2H]-21: R1 = R2 = PhCH2
NH
NH
HN
HN
O
O
O
O
NH
NH
HN
HN
O
O
O
O
a,b
c
d
e c
Scheme 8. Synthesis of bistable molecular shuttles based on azodicarboxamide binding sites 7 and 8 and their corresponding 1,2-hydrazodicarboxamides
[2H]-20 and [2H]-21. Reagents and conditions: (a) Ph2CHCH2NH2 or (PhCH2)2NH, Et3N, CHCl3; (b) Ph2CHCH2O2C(CH2)2CONH(CH2)12NH2
(2,2-diphenylethyl-3-(12-aminododecylcarbamoyl)propanoate), Et3N, CHCl3, [2H]-18, 35%; [2H]-19, 54%; (c) NBS, Py, CH2Cl2, 18, 91%; 19, 95%; 20, 86%;
21, 93%; (d) isophthaloyl dichloride, p-xylylenediamine, Et3N, CHCl3, 20, 34%; 21, 45%; (e) N2H4 ·H2O, CHCl3, [2H]-20, 88%; [2H]-21, 92%.

138 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
integrated in the thread through the catalytic formation of ester
bonds [15]. During this process, the macrocycle continuously
translocates between the nitrogen- and carbon-based stations as-
sisted by triphenylphosphine, which acts as a chemical activator of
the azo group, and iodosobenzene diacetate, which is a stoichiomet-
ric oxidant of the resulting hydrazo unit (Scheme 9). Fortunately,
molecular shuttle 21 is a successful organocatalyst for the ester
bond formation and gives the product in 65% yield.
Although the hydrogen bonds between the azodicarboxamide
station and the macrocycle persist even in relatively polar solvents
such as THF or acetonitrile [17], these solvents may compete with
the hydrogen-bond donors on the thread. In such cases, the
Brownian macrocycle escapes from the azo station and allows
phosphane attack to form the corresponding Morrison-Brunn-
Huisgen betaine [18] like III (Scheme 9). This betaine was detected
by high-resolution mass spectrometry. The addition of triphenyl-
phosphane to the azodicarboxamide station alters its affinity for the
macrocycle and notably increases the steric bulk between the amide
groups. Accordingly, this intermediate follows the well known
mechanistic pathway of the Mitsunobu reaction to give the ester
and hydrazo [2]rotaxane [2H]-21, which, after in situ reoxidation,
recycles co-conformer 21 using excess bis(acetoxy)iodobenzene;
therefore, the catalytic wheel of ester bond formation is ready to
begin again. From a thermodynamic perspective, it should be noted
that, although the esterification reaction is endergonic, the oxidation
of phosphane promoted by the reduction of the azodicarboxamide
binding site makes the overall process exergonic. In these reaction
conditions, the molecular shuttle operates in a cyclic manner as
long as the fueling chemicals are accessible.
These molecular shuttles operate by two different mechanisms:
(1) a discrete mode through two reversible and independent chemi-
cal events that trigger reduction and oxidation reactions and (2) a
continuous mode via a catalyzed ester bond formation reaction in
which the shuttle acts as an organocatalyst.
Two novel multilevel switchable [2]rotaxanes containing
ammonium and 1,2,3-triazole (TZ) stations have been constructed
using a Cu(I)-catalyzed azide-alkyne cycloaddition reaction [19]
(Schemes 10, 11). In the protonated form, the macrocycle of
[2]rotaxane, which contains a C6-chain bridge between the two
hydrogen bonding stations, exhibits high selectivity for the ammo-
nium cation (Scheme 12). Interestingly, the macrocycle is able to
interact with two recognition stations when the bridge between
them is short. Upon deprotonation of both [2]rotaxanes, the macro-
cycle moves towards the TZ recognition site due to the hydrogen
bond interaction between the TZ nitrogen atoms and the amide
groups in the macrocycle. Upon addition of chloride anions, the
conformation of [2]rotaxane changes because of the cooperative
recognition of the chloride anion by favorable hydrogen-bond do-
nors from both the macrocycle isophthalamide and thread TZ CH
protons (Scheme 13).
TZ, which is generated by a click reaction, is still rare as a mo-
lecular station [20]. In this system, the click reaction is not only a
key step of stopping the reaction but also provides a potential alter-
Bn
N N
N NH
HN
O
Ph
Bn
O
O
Ph
O
O
( )9 Bn
N N
NH
N
HN
O
Ph
Bn
O
O
Ph
O
O
( )9
NH
NH
HN
HN
O
O
O
O
NH
NH
HN
HN
O
O
O
O
PPh3
Bn
N N
NH
NH
HN
O
Ph
Bn
O
O
Ph
O
O
( )9
NH
NH
HN
HN
O
O
O
O
PPh3
Bn
NHN
NH
NH
HN
O
Ph
Bn
O
O
Ph
O
O
( )9
NH
NH
HN
HN
O
O
O
O
PPh3
21
[2H]-21
III
ArCO2H
ROH
ArCO2
PhI(OAc)2
PhI + 2 AcOH
ArCO2R + O=PPh3
Scheme 9. Proposed cyclic chemical functioning of the shuttle 21 by means of an ester bond formation reaction between an acid, ArCO2H (Ar = 4-NO2C6H4),
and an alcohol, ROH (R = C6H5CH2) mediated by a classical Mitsunobu protocol with triphenylphosphane followed by an in situ oxidation of the hydrazo
generated shuttle [2H]-21.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 139
RO
O
Br
OHC
O
OHC
O
ROHBr Br
RO N3
RO NH2
t-Bu
t-Bu
t-Bu
R =
RO NH2
O
O
O
OO
O
O
O
NH
HNO
O
OH
OH
Br
b) NaN3
a)
Pd/C
MeOH/EtOAc
a)b)
a) toluene/reflux b) MeOH/NaBH4
c) CF3CO2H, acetone, then NH4PF6, water
25
26
27
28
29
24
H-22
H-23
N
O
O
O
N
N
N
OR
RO NH2
O
O
O
N
N
N
OR
PF6
PF6
PF6
b c
d
e
f gh
i
jk
l
m
n o
p
HH
OO
O
O
O
NH
HNO
O
25
CH2Cl2
CuBF4(CH3CN)4
A B
C
D E
F
G
H
I
J
K
Scheme 10. Synthesis of H-22 and H-23.

140 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
RO
OO
O
O
O
NH
HNO
O
H-22
22
N
O
O
O
N
N
N
OR
RO NH
O
O
O
N
N
OR
PF6
HH
O
O
O
O
O
N
N
O
O
H
N
H
H
CF3CO2H i-Pr2NEt
Scheme 11. Movement process of multistable rotaxane H-22 under acid/base stimuli.
native binding site, which is directly introduced by a coupling pro-
cedure.
The large dipole (5D) of TZ, which is oriented with the positive
end toward the H atom, contributes to its unexpectedly strong hy-
drogen-bonding capabilities.
Shuttle H-22 was synthesized according to Scheme 10. In
CH2Cl2, macrocycle 24 was assembled with monostoppered
ammonium-containing compound 29 to form a pseudorotaxane.
Covalent capture of the threaded intermediate using a click reaction
at room temperature in the presence of [Cu(CH3CN)4]BF4 catalyst
afforded thread H-23 and [2]rotaxane H-22 in 50% and 35% yields,
respectively, after chromatographic column purification.
The position of macrocycle 24 in the rotaxane was determined
by comparing the 1H NMR spectra of the uncomplexed dumb-
bell-shaped thread and the rotaxane since the XYL parts of the
macrocycle shield the encapsulated regions of the thread. The 1H
NMR spectra of thread H-23, rotaxane H-22, and macrocycle 24 in
acetonitrile confirm the interlocked structure. As expected, the pro-
tons adjacent to the ammonium unit are upfield shifted (He: 0.24
ppm; Hd: 0.14 ppm) relative to those in free H-23 due to the aro-
matic shielding effect of the macrocycle, whereas no shifts of the
signals corresponding to the protons near the TZ ring is evident.
This confirms that the macrocycle, 24, in rotaxane H-22 is pre-
dominantly localized on the alkyl ammonium region of the thread
under acidic conditions.
Upon addition of i-Pr2NEt to H-22, the ammonium group is
neutralized, which breaks the hydrogen bonds between the poly-
ether moiety and the ammonium group (Scheme 12). Upon addition
of CF3COOH to deprotonated rotaxane 22, the methylene protons,
Hl and Hn, appear at 4.98 and 4.45 ppm, respectively, while the
signal for the TZ ring resonates at 7.75 ppm; this suggests that 24
shuttles completely to the NH2
+ recognition site following reproto-
nation.
To verify the hydrogen bonding between macrocycle 24 and the
TZ ring, rotaxane H-30, which contains a short bridge between the
two recognition sites, was synthesized using a similar reaction
process (Scheme 11).
Interestingly, in contrast to rotaxane H-22, aromatic shielding
of the proton adjacent to the TZ ring (Hh: 0.25 ppm) in H-30 is
observed. Moreover, the ring amide protons, HD, experience a
downfield shift of 0.25 ppm in H-30 in comparison to 24, which is
ascribed to the formation of hydrogen bonds between the amide
group and the hydrogen-bond acceptor, i.e., the TZ nitrogen atoms.
These features indicate that the isophthalamide group interacts
with the TZ ring to some extent due to the hydrogen bonds between
them. The signals corresponding to the phenylene protons (Hf: 0.3
ppm and Hg: 0.4 ppm) and the macrocycle benzyl groups (HF:
0.21 ppm and HG: 0.31 ppm) experience a significant upfield shift
due to the aromatic shielding between them. Therefore, macrocycle
24 also forms a sandwich structure with the phenylene spacer in
thread H-31 via weak – stacking interactions. Thus, the 1H NMR
spectra support the fact that the isophthalamide group interacts with
TZ while the polyether moiety interacts with the ammonium cation.
Essentially, the macrocycle spans two potential hydrogen-bonding

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 141
recognition stations and forms a short bridge between them
(Scheme 13).
Deprotonation of H-30 with i-Pr2NEt resulted in significant
changes to the 1H NMR spectrum in acetonitrile: He was shifted
upfield by 0.46 ppm due to the deprotonation of the neighboring
ammonium center and shuttling of the macrocycle.
Upon addition of CF3COOH to fully deprotonated H-30, the
signals for Hj and the TZ hydrogen, Hi, shift downfield by 0.19 and
0.16 ppm relative to those for the same protons in 30, which indi-
cates that the isophthalamide group of 24 interacts with TZ and the
polyether moiety switches between the two recognition sites via a
de-/re-protonation process. Upon addition of chloride anions in the
form of tetrabutylammonium (TBA) salts to a solution of
[2]rotaxane 30, dramatic changes in the 1H NMR spectrum were
observed. The isophthalamide protons, HC and HD, shift downfield
by 0.68 and 1.54 ppm, respectively, which is due to the formation
of hydrogen bonds between them. The signal for the TZ proton
undergoes a downfield shift of 0.2 ppm due to hydrogen bonding to
the chloride anion. These phenomena are attributed to the coopera-
RO
t-Bu
t-Bu
t-Bu
R =
O
O O
O
O
N
N
O
O
a) toluene/reflux
b) MeOH/NaBH4
c) CF3CO2H, acetone,
then NH4PF6, water
24
H-30
H-31
N
O
NN
N
OR
RO NH2
O
N
N
N
OR
PF6
PF6
b c
d
e
f gh
i
j k
l
H H
OO
O
O
O
NH
HNO
O
25
CH2Cl2
CuBF4(CH3CN)4
A B
C
D E
F
G
H
I
J
K
26
OHC O
O
NH2RO
PF6
H
H
Scheme 12. Synthesis of H-30 and H-31.

142 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
tive recognition of the chloride anion by favorable hydrogen-bond
donors from both the macrocycle isophthalamide protons (HC and
HD) and the thread TZ proton. In addition, proton Hh is shifted
downfield by 0.14 ppm relative to the corresponding signal in 30;
however, the signal is still less than 5.12 ppm. This suggests that
the shielding effect of the macrocycle is reduced due to the confor-
mational change upon cooperative recognition of the chloride anion
(Scheme 13).
To demonstrate that chloride anions are the most effective ani-
ons for altering the conformation of 30, the addition of TBA salts of
bromide and iodide anions was also investigated by 1H NMR spec-
tra.
Based on the Job-plot determination of 1:1 binding stoichiome-
try, the association constants for all anions were obtained by fitting
the changes in the chemical shift of proton C to the binding iso-
therm. Accordingly, the strength of anion association increased in
the order of I–<Br
–<Cl
–, which is consistent with the increasing
hydrogen bond–acceptor ability of the anions (I–<Br
–<Cl
–). It also
reflects that the size and shape of the interlocked binding cavity
generated by 30 is ideally matched to the chloride anion. These data
suggest that the macrocycle isophthalamide protons (HC and HD)
and thread TZ proton surround the chloride anion through coopera-
tive hydrogen-bonding interactions, which leads to conformational
alteration of the macrocycle in 30.
Harada et al. reported unprecedented control of the shuttling of
rotors by reeling an axis molecule into the cavity of a host mole-
cule. Thus, a [2]rotaxane was designed with an -cyclodextrin
( -CD) moiety that functions as both a stopper and a rotor [21].
This [2]rotaxane forms a pseudo[2]rotaxane in D2O due to tumbling
of an altropyranose unit of the altro- -CD stopper. The conforma-
tional change of the [2]rotaxane resembles the action of a reel,
which rotates to reel the decamethylene chain into the cavity of the
altro- -CD stopper.
[2]Rotaxane 32 was prepared as outlined in Scheme 14. The
starting material is an axis molecule with an altro- -CD group (al-
tro- -CD stopper) and a stilbene unit linked via a decamethylene
group. This axis molecule undergoes condensation with adaman-
tane carboxylic acid in the presence of the -CD rotor in aqueous
solution to afford the rotaxane products.
The location and direction of the rotor on the axis were deter-
mined using 2D NOESY and 2D ROESY NMR spectroscopy. The
resonance peaks of 32 were assigned using COSY, TOCSY, and
ROESY measurements. When the 1D NMR spectra of [2]rotaxane
and a dumbbell molecule in DMSO-d6 were compared, only the
signals for the protons of the decamethylene group of the
[2]rotaxane were shifted; the protons of the stilbene and adamantyl
groups of the [2]rotaxane did not shift in this solvent. A dumbbell
molecule without a rotor molecule does not show ROE correlation
RO
O
O O
O
O
N
N
O
O
H-30
30
N
O
NN
N
OR
RO NH
ON
NNOR
PF6
H H
H
H
O
O O
O
O
N
N
O
O
HH
RO NH
ON
NNOR
O
OO
O
O
NN
O
O
HH
H
H
Cl
CF3CO2H
i-Pr2NEt
Cl
30-Cl
Scheme 13. Movement process of multistable rotaxane H-30 under different
stimuli.
Scheme 14. Preparation of [2]rotaxanes 32 and 33. DMT-MM = (4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 143
peaks between the inner protons of the altro- -CD stopper and the
protons of the axis group. The 2D ROESY NMR spectrum of 32 in
DMSO-d6 shows no correlation between the protons of the stilbene
group and the inner protons (C3-H, C5-H, and C6-H) of the -CD
rotor. However, the protons of the decamethylene group strongly
correlate to the protons of the -CD rotor. These results indicate
that the decamethylene group was incorporated within the -CD
rotor when [2]rotaxane 32 was dissolved in DMSO-d6.
The 2D NOESY NMR spectrum of 32 in D2O shows a clear
correlation between the C3-H inner proton of the -CD rotor and
protons A and B of the stilbene group adjacent to the decamethylene
group. However, only a weak correlation between the C3-H proton
Scheme 15. Formation of pseudo[2]rotaxane 34 from [2]rotaxane 32 via tumbling an altropyranose unit.
OAc
OAc
AcO
OAcN
N
O
R
N3
t-Bu
t-BuNH2
PF6
PF6+
OAc
OAc
AcO
OAcN
N
O
Rt-Bu
t-Bu
PF6
PF6
N
N
N
O
O
O
O
O
O
O
O
NH2
DB24C8
[Cu(CH3CN)4]PF6
2,6-lutidine
dry CH2Cl2, 24 h, r.t.
35a: R = H
35b: R = Me (50:50 cis/trans)
37a: R = H, 75%
37b: R = Me, 75% (50:50 cis/trans)
36
Scheme 16. Synthesis of rotaxanes 37.

144 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
and protons C–F of the stilbene group is evident. The C5-H and
C6-H protons correlate to protons C–E and proton E, respectively.
A correlation between proton F and the protons on the adamantyl
group was also observed. These results suggest that the -CD rotor
is located on the stilbene group and the narrow rim of the -CD
rotor is directed toward the adamantyl stopper. It should be noted
that the inner protons of the altro- -CD stopper show strong corre-
lation to the protons of the decamethylene group. This provides
evidence that [2]rotaxane 32 forms pseudo[2]rotaxane 34 in D2O
through tumbling of the altro- -CD stopper to include the de-
camethylene axis.
The conformational change of 32 via tumbling of an altro-
pyranose unit is shown in Scheme 15. The free activation energy
( G‡
288 K) for the conformational change from 32 to 34 was deter-
mined by observing the time-resolved UV spectral changes for the
process in D2O at 288 K. Assuming a two-site exchange process,
the G‡
288 K value was calculated to be 89.4 kJ·mol–1
using single
exponential fitting. The calculated G‡ value for the tumbling of
the altropyranose residue is higher than the reported value for
calix-[4]arene tumbling ( G‡ = 65.7 kJ·mol
–1) [22]. The glu-
copyranose units of permethylated CDs easily tumble because hy-
drogen bonds between neighboring permethylated glucopyranose
units cannot be formed by the deprotonation of hydroxyl groups. In
contrast, normal glucopyranose and altropyranose units are unable
to tumble easily due to the formation of hydrogen bonds between
neighboring units. Regardless, the G‡ value for this tumbling
process has not yet been determined.
A new class of rotaxanes that incorporate dibenzo[24]crown-8
(DB24C8) as the macrocycle and feature three different molecular
stations, i.e., anilinium, N-methyltriazolium, and either a mono- or a
disubstituted pyridinium amide, in the thread have been reported
[23].
These rotaxanes (37) [23] were synthesized from mono- or di-
substituted mannosyl pyridinium amide azide 35 and ammonium
compound 36 by copper(I)-catalyzed Huisgen alkyne-azide 1,3-
dipolar cycloaddition, which is also known as CuAAC click chem-
istry (Scheme 16). The reaction between DB24C8, azido compound
35, alkyne 36, Cu(CH3CN)4PF6, and 2,6-lutidine was performed in
dichloromethane at ambient temperature over 24 h. In the case of
rotaxane 37b, a 1:1 mixture of two stereoisomers was observed due
to cis/trans isomerism of the disubstituted amide.
A low pH, the macrocycle resides exclusively around the
anilinium moiety. Deprotonation of 37 was executed by adding
excess diisopropylethylamine (DIEA) at room temperature (Scheme
17) and results in a large-amplitude displacement of DB24C8 from
Me
O
OAc
OAc
AcO
OAcN
N
O
Rt-Bu
t-Bu
PF6
PF6
N
N
N
O
O
O
O
O
O
O
O
NH2
37a: R = H
37b: R = Me
O
OAc
OAc
AcO
OAcN
N
O
Ht-Bu
t-Bu
PF6
N
N
N
O
O
O
O
O
O
O
O
NH
38a
37a: R = H, 75%
37b: R = Me, 75% (50:50 cis/trans)
O
OAc
OAc
AcO
OAcN
N
O
Rt-Bu
t-Bu
PF6
PF6
N
N
N
O
O
O
O
O
O
O
O
NH2
Me
PF6
N
NO
Me t-Bu
t-Bu
N
N
N
NH
OAcO
OAc
OAc
AcO
O
OO
O
OO
O
O
O
O
O O
O
O
OPF6
O
OAc
OAc
AcO
OAcN
N
O
Ht-Bu
t-Bu
PF6
N
N
N
NH
Me
PF6
O
OAc
OAc
AcO
OAcN
N
O
Met-Bu
t-Bu
PF6
N
N
N
NH
PF6
O
O O
O
O
OO
O O
1) HCl
2) NH4PF6, CH2Cl2/H2ODIEA 1) HCl
2) NH4PF6, CH2Cl2/H2O
DIEA
1) HCl
2) NH4PF6, CH2Cl2/H2ODIEA
1) HCl
2) NH4PF6, CH2Cl2/H2O
DIEA
38a (50:50 cis/trans)
40b (42:58 cis/trans)40a
1) MeI, 3 d, r.t.
2) NH4PF6, CH2Cl2/H2O, 30 min
4C1
1C4
1
234
5
6
7
8
910
11
12
13
14
15
16
17
18 19
20
21
22
23
24
25
2627 28
29
3033
Scheme 17. Synthesis of 39a,b, and molecular machinery by deprotonation/protonation of rotaxanes 37a,b, and 39a,b.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 145
one end of the molecule to the other. The macrocycle is located
around the mono- or di-substituted pyridinium amide station in 38a
and 38b, respectively (Scheme 17).
The location of the macrocycle around the pyridinium moiety
differs slightly depending on the substitution of the amide. Indeed,
in the mono-substituted series 38a, DB24C8 interacts with H8 and
H11 via hydrogen bonding. However, in the di-substituted series
38b, DB24C8 does not interact with H8; instead, it interacts with
the H7 hydrogen atoms, which are closer to the cationic charge. In
this latter case, flipping of the chair-like mannopyranosyl ring from
the 1C4 to the
4C1 conformation was observed as a result of turning
off the reverse anomeric effect [24].
Subsequently, the introduction of a triazolium moiety between
the two previously studied molecular stations was envisaged. Re-
gioselective N-alkylation of the 1,4-disubstituted TZ moiety with
iodomethane followed by cation exchange provided three-station
triazolium rotaxanes 39a and 39b in 93% and 91% yields, respec-
tively (Scheme 17). Each [2]rotaxane molecular machine, 39a and
39b, contains three different sites for interaction with DB24C8:
anilinium, triazolium, and mono- or disubstituted pyridinium am-
ide.
Upon deprotonation of both rotaxanes 39a and 39b (Scheme
17), the macrocycle is displaced from its initial anilinium position;
however, its behavior differs depending on the degree of substitu-
tion of the pyridinium amide station.
In the di-substituted pyridinium amide series, the molecular
machine behaves as a pH-sensitive bistable [2]rotaxane and the
macrocycle shuttles between the anilinium and triazolium stations.
The macrocycle initially resides around the anilinium station in
39b. After deprotonation, the position of the macrocycle was de-
duced by comparing the 1H NMR spectra of rotaxanes 39b and 40b.
The significant upfield shifts for H25 ( = –1.06 ppm), H28 ( =
–0.88 ppm), and H30 ( = –0.69 ppm) in rotaxane 40b result from
the deprotonation of the anilinium moiety and shuttling of the mac-
rocycle. Concurrently, the signal for H18 is shifted downfield in 40b
( = +0.73 ppm). These two observations unambiguously
demonstrate the ability of rotaxane 40b to behave as a pH-sensitive
bistable molecular machine. After deprotonation, the macrocycle
resides on the triazolium station due to its better binding affinity for
DB24C8 than for the disubstituted pyridinium amide. This study
discussed the higher affinity of DB24C8 for triazolium than for the
disubstituted pyridinium amide station.
Similarly, in the mono-substituted pyridinium amide series, the
macrocycle initially resides on the anilinium station in protonated
rotaxane 39a; however, the macrocycle shuttles very differently
after deprotonation. The shuttling in rotaxane 40a at room tem-
perature results in a degenerate molecular machine: the macrocycle
continuously oscillates between the triazolium and monosubstituted
pyridinium amide stations. This result suggests a similar affinity of
the two stations for DB24C8. This is in contrast to rotaxane 40b,
which features a disubstituted pyridinium amide, wherein the mac-
rocycle shuttles around the sole triazolium station.
Kinetic and thermodynamic studies demonstrated that the two
translational isomers have very similar free enthalpy values at room
temperature. However, lowering the temperature increases G and
thus forces DB24C8 to reside more frequently around the energeti-
cally favorable pyridinium amide station. Around 223 K, oscillation
between the two stations ceases and DB24C8 is located primarily
around the monosubstituted pyridinium amide station, as in a bista-
ble molecular machine.
Additionally, hydrogen-bonding interactions between DB24C8
and the monopyridinium amide station in the deprotonated rotaxane
impair the rotation of the threaded pyridinium amide bond, i.e.,
DB24C8 acts as a molecular brake (Fig. 5).
Finally, the implementation of molecular rotaxanes for the gen-
eration of controllable nanostructures was reported [25]. This is
important to better understand the solid-state applications of mo-
lecular shuttles.
[2]Rotaxane 47 was prepared by Li et al. [25] according to
Scheme 18. The tetracyanobutadiene (TCBD) unit was chosen as
the stopper and aggregation core because its derivatives easily form
regular nanostructures due to intermolecular dipole–dipole interac-
tions [26]. Thread 46 was prepared in a high yield using a
[2+2]cycloaddition reaction that occurs at room temperature and in
apolar solvents without the help of a catalyst, which was developed
Diederich [27]. The treatment of 46 with p-xylylene diamine and
highly diluted isophthaloyl dichloride provided 47 in 5% yield. The
low yield of this process is ascribed to the weaker interactions be-
tween the peptide units and macrocycle precursors in the clipping
process: the less electron-donating aniline N-atom significantly
reduces the hydrogen-bond accepting ability of its adjacent car-
bonyl group.
The position of the macrocycle was determined by comparing
the chemical shifts of the protons in the rotaxane with those in the
corresponding thread. The 1H NMR spectra of thread 46 and rotax-
ane 47 confirm the interlocked structure and show that the macro-
cycle is primarily localized on the peptide region of the thread of 47
(Scheme 18).
When CD3OD is added to a CDCl3 solution of 47, minor
changes occurred in the 1H NMR spectrum. The methylene reso-
nances of the Hd and Hf protons of the peptide station have the same
chemical shifts as those in neat CDCl3, which indicates that the
macrocycle still resides at the peptide station in CDCl3/CD3OD
(1/1, v/v). This suggests that the intramolecular hydrogen bonds
Fig. (5). Schematic representation of the role of DB24C8 as a molecular brake for rotation of the C9–C10 bond in a) uncomplexed 39a, b) 38b, and c) 38a and
40a.
+ Py + Py+ Py
C7 C8
C9C10
O10
N11
Fast motion Fast motion Slow motiona) b) c)
Molecular brake

146 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
between the macrocycle and the peptide are stronger than the in-
termolecular hydrogen bonds between the peptide and CD3OD.
This is attributed to the excellent fit between the thread and macro-
cycle in terms of both the steric interactions and complementary
positioning of the hydrogen-bonding amide groups on the two
components.
The macrocycle can be dissociated from the peptide by
dissolving the rotaxane in a highly polar solvent: the solvent
solvates the hydrogen-bonding sites of the macrocycle and peptide
and breaks the intramolecular hydrogen bonds. In the DMSO-d6
solvent, the methylene resonances of Hf and Ho experience a sig-
nificant upfield shift of 0.52 and 0.51 ppm, respectively, and the
signals assigned to the alkyl chain exhibit a clear upfield shift,
which could be attributed to the shielding effect of the macrocycle.
The signal for Hd in 47 remains at the same chemical shift as in 46.
All these features indicate that the macrocycle departs from the
peptide station and resides on the alkyl chain in DMSO (Scheme
19).
The aggregation of 46 and 47 in different solvents was
determined to demonstrate the effect of the shuttling of the macro-
cycle on the aggregation behavior. One drop of solutions of 46 and
47 in different solvents was evaporated on silicon slides to assess
RNH2
Ph
Ph
Ph
Cl
O
Cl
I OH
NMe2
NH2
H2N
Cl Cl
OO
CNNC
NC CN
1)
2)
PPh3
THF/H2ONaN3
R
NH
O
N3 R
NH
O
NH2
R
NH
O
HN
O
O I( )
10
R
NH
O
HN
O
Br( )
10
Br(CH2)10CO2H
EDC, DMAP
RNH
O
HN
O
NMe2O
NH
O
HN
O
NMe2
O
NC
CN
CN
NC
Pd(PPh3)4
CuI
R
NH
O
HN
O
NMe2
O
NC
CN
CN
NC
41 42
4344
45
46
47
R =
NH
NH
HN
HN
O
O
O
O
R
a b
cd
ef
g
h
i
j
k
l
m
n
o p q
r st
u
v
w
x
y
z
Scheme 18. The synthesis route of 46 and 47.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 147
the nanostructures in the solid state. The morphologies of the ag-
gregates of 46 and 47 on the substrate were examined by scanning
electron microscopy (SEM) (Fig. 6). Figs. 6a and 6b show the typi-
cal morphologies of the 46 and 47 nanostructures formed in a
CHCl3/n-C6H14 (1:1, v/v) solvent, which was used to facilitate
molecular aggregation. The addition of n-C6H14 to a CHCl3 solution
does not affect the location of the macrocycle because it has a lower
polarity. Large-scale spherical particles with uniform diameters of
about 300 nm and interlaced fibers were observed for 46 and 47,
respectively. Figs. 6c and 6d present the morphologies of the 46
and 47 nanostructures formed in a CHCl3/MeOH (1;1, v/v) solvent.
Spherical nanoparticles with diameters of about 500 nm were ob-
served for 46 and 47.
Interestingly, most of the particles feature open holes on their
surfaces, which indicates that they may have hollow interiors. The
wall thicknesses of the vesicles of 46 and 47 were estimated to be
about 100 nm. This is sufficient to contain approximately 12 bilay-
ers with molecular dimensions of 4 nm, as estimated by 3D mo-
lecular structural modeling of 46 and 47. These results indicate that
the membrane structures of the hollow spherical vesicles of both 46
and 47 in methanol are most likely shell-like complex multilayer
structures. In DMSO, nanobundles and worm-like nanoparticles
were observed for 46 and 47, respectively, as shown in Figs. 6e and
6f.
UV-Vis absorption spectra provide information on the mecha-
nism of the formation of the nanostructures. Dilute CHCl3 solutions
of 46 and 47 display two intramolecular charge-transfer (ICT)
bands centered at 384 and 470 nm. When 46 and 47 aggregate into
stable and well-suspended nanostructures in CHCl3/hexane (1:1,
v/v), the two ICT bands of 46, which are assigned to the TCBD
unit, undergo a bathochromic shift to 389 and 480 nm while those
of 47 undergo a bathochromic shift to 389 and 490 nm. These
red-shifted absorption spectra indicate that J-type aggregation
occurs in both compounds; the larger bathochromic shift of 47 in-
dicates that its nanoaggregates feature more extended or ordered
stacking. In CHCl3/MeOH (1:1, v/v), the first ICT band of both 46
and 47 exhibits a blue-shift of 5 nm compared with those in CHCl3
while the second ICT bands of both 46 and 47 remain at the same
position as those in CHCl3. These results suggest that 46 and 47
experience similar H-type aggregation under these conditions. To
study the aggregation behavior of 46 and 47 in DMSO, a mixed
solvent of DMSO/H2O (1:1, v/v) was adopted to facilitate
aggregation. The absorption spectrum of 46 was red-shifted by 8
nm compared to that of 47 and high energy tails were evident only
in the spectrum of 46, which suggests that the nanoaggregates of 46
have more extended or ordered stacking under these conditions.
Additionally, the spectra of 46 and 47 in DMSO/H2O (1:2, v/v) are
similar to those in DMSO/H2O (1:1, v/v), which indicates that 46
and 47 are fully aggregated in DMSO/H2O (1:1, v/v).
To elucidate the distinct morphologies of 46 and 47 in different
solvents, a mechanism that correlates the molecular structures with
the morphologies of the nanostructures is proposed. In 47, the mac-
rocycle resides in the peptide station, which obstructs hydrogen
bonding between the peptide and TCBD. Additionally, molecules
RNH
O
HN
O
NMe2
O
NC
CN
CN
NC
NH
NH
HN
HN
O
O
O
O
R
NH
O
HN
O
NMe2
O
NC
CN
CN
NC
NH
NH
HN
HN
O
O
O
O
MeOH or
CH2Cl2DMSO
Scheme 19. The shuttling movement process of 47 in different solvents.

148 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
of 47 exhibit linear structures in solution, which facilitates the ag-
gregation of the TCBD units. The extended intermolecular di-
pole–dipole interactions between TCBD units induce 1D molecular
stacking. As a result, the TCBD units in rotaxane 47 aggregate in an
orderly fashion to form 1D nanofibers. For the formation of hollow
vesicle structures, the coexistence of hydrophilic and hydrophobic
units in a single low weight molecule or blocks with different solu-
bilities in copolymers or codendrimers are considered essential
[28]. For 46 and 47, the hydrophobic tetraphenyl unit and long alkyl
chain at one end of the molecule favor the non-polar CHCl3 solvent
while the other end favors the polar MeOH solvent due to the pres-
ence of a TCBD moiety, which is polar as a result of the in-
tramolecular charge transfer [27]. Accordingly, in the combined
solvent system, 46 and 47 are amphiphilic molecules that can as-
semble into layered structures that close to form vesicles. In
DMSO, all intramolecular hydrogen bonds are broken because of
the high polarity of the solvent; therefore, the only driving force for
aggregation is the dipole–dipole interactions of the TCBD units.
However, in 47, the macrocycle moves along the long alkyl chain
and hinders these interactions. Upon evaporation of the solvent, the
van der Waals driving forces between the independent molecules
induce the formation of disordered worm-like nanoparticles.
PHOTOCHEMICALLY POWERED MOLECULAR
SWITCHES
Degenerate [2]rotaxanes, which feature two identical binding
sites, generally exhibit equilibrium dynamics and have free energies
of activation ( G‡) for the shuttling process as low as 10 kcal mol
–1
[29]. This G‡ value can be significantly increased by inserting
“speed bumps” in the form of steric [30] and/or electrostatic [31]
barriers into the linkage between the two identical binding sites.
Stoddart et al. demonstrated that light can be used in conjunc-
tion with thermal energy to raise and lower the free energy barrier,
thus imparting STOP and GO instructions to the molecular shuttle
[32].
This shuttle was based on a 4,4 -azobiphenyloxy (ABP) unit
that acts as a binding site for a tetracationic cyclobis(paraquat-p-
phenylene) (CBPQT4+
) ring. Independently, four methyl groups and
four fluorine atoms were introduced at the 3,5,3 ,5 -positions of the
ABP unit to curtail binding by the CBPQT4+
ring via steric and
electronic interactions, respectively. The first approach forms a gate
(i.e., ABP-Me4) that remains closed, whereas the second approach
affords a gate (i.e., ABP-F4) that closes and opens upon irradiation
with UV light and visible light, respectively. The ABP-F4 gate can
also open in response to thermal energy in its surroundings.
Fig. (6). SEM images of 46 nanoscale aggregates in CHCl3/n-C6H14 (1:1, v/v) (a), in CHCl3/MeOH(1:1, v/v) (c), and in DMSO (e). The SEM images of 47
nanometer-scale aggregates in CHCl3/n-C6H14 (1:1, v/v) (b), in CHCl3/MeOH (1:1, v/v) (d), and in DMSO (f). The insets in (c) and (d) are the corresponding
TEM images.
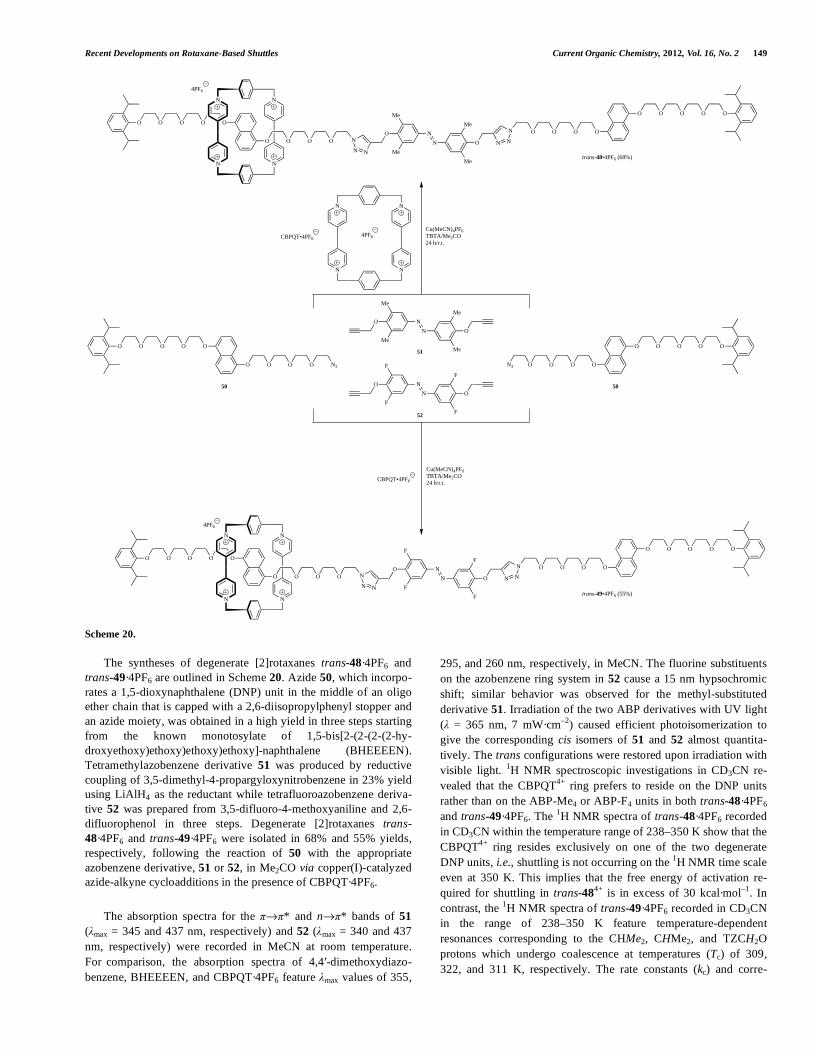
Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 149
Scheme 20.
The syntheses of degenerate [2]rotaxanes trans-48·4PF6 and
trans-49·4PF6 are outlined in Scheme 20. Azide 50, which incorpo-
rates a 1,5-dioxynaphthalene (DNP) unit in the middle of an oligo
ether chain that is capped with a 2,6-diisopropylphenyl stopper and
an azide moiety, was obtained in a high yield in three steps starting
from the known monotosylate of 1,5-bis[2-(2-(2-(2-hy-
droxyethoxy)ethoxy)ethoxy)ethoxy]-naphthalene (BHEEEEN).
Tetramethylazobenzene derivative 51 was produced by reductive
coupling of 3,5-dimethyl-4-propargyloxynitrobenzene in 23% yield
using LiAlH4 as the reductant while tetrafluoroazobenzene deriva-
tive 52 was prepared from 3,5-difluoro-4-methoxyaniline and 2,6-
difluorophenol in three steps. Degenerate [2]rotaxanes trans-
48·4PF6 and trans-49·4PF6 were isolated in 68% and 55% yields,
respectively, following the reaction of 50 with the appropriate
azobenzene derivative, 51 or 52, in Me2CO via copper(I)-catalyzed
azide-alkyne cycloadditions in the presence of CBPQT·4PF6.
The absorption spectra for the * and n * bands of 51
( max = 345 and 437 nm, respectively) and 52 ( max = 340 and 437
nm, respectively) were recorded in MeCN at room temperature.
For comparison, the absorption spectra of 4,4 -dimethoxydiazo-
benzene, BHEEEEN, and CBPQT·4PF6 feature max values of 355,
295, and 260 nm, respectively, in MeCN. The fluorine substituents
on the azobenzene ring system in 52 cause a 15 nm hypsochromic
shift; similar behavior was observed for the methyl-substituted
derivative 51. Irradiation of the two ABP derivatives with UV light
( = 365 nm, 7 mW·cm–2
) caused efficient photoisomerization to
give the corresponding cis isomers of 51 and 52 almost quantita-
tively. The trans configurations were restored upon irradiation with
visible light. 1H NMR spectroscopic investigations in CD3CN re-
vealed that the CBPQT4+
ring prefers to reside on the DNP units
rather than on the ABP-Me4 or ABP-F4 units in both trans-48·4PF6
and trans-49·4PF6. The 1H NMR spectra of trans-48·4PF6 recorded
in CD3CN within the temperature range of 238–350 K show that the
CBPQT4+
ring resides exclusively on one of the two degenerate
DNP units, i.e., shuttling is not occurring on the 1H NMR time scale
even at 350 K. This implies that the free energy of activation re-
quired for shuttling in trans-484+
is in excess of 30 kcal·mol–1
. In
contrast, the 1H NMR spectra of trans-49·4PF6 recorded in CD3CN
in the range of 238–350 K feature temperature-dependent
resonances corresponding to the CHMe2, CHMe2, and TZCH2O
protons which undergo coalescence at temperatures (Tc) of 309,
322, and 311 K, respectively. The rate constants (kc) and corre-
N
N
O
N
N N
OOOO
OOOOO
O
N
NN
O O O O
O O O O O
N
N
N
N
N
N
N
N
N
N
O
O
N
N
O
O
N
N
O
N
N N
OOOO
OOOOO
O
N
NN
O O O O
O O O O O
N
N
N
N
Me
Me
Me
Me
Me
Me
Me
Me
F
F
F
F
F
F
F
F
O O O O
O O O O O
N3OOOO
OOOOO
N3
Cu(MeCN)4PF6
TBTA/Me2CO
24 h/r.t.
51
52
50 50
Cu(MeCN)4PF6
TBTA/Me2CO
24 h/r.t.
CBPQT•4PF6
CBPQT•4PF6
trans-48•4PF6 (68%)
trans-49•4PF6 (55%)
4PF6
4PF6
4PF6

150 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
sponding G‡
c values calculated at these temperatures are listed in
Table 1. At 15.6 kcal·mol–1
, the free energy of activation ( G‡
c) for
the shuttling process is similar in magnitude to the G‡
c values
observed for other degenerate molecular shuttles [33] where the
links between the two identical recognition units are, for example,
polyether chains or terphenyl linkers. The 1H NMR spectrum of
trans-49·4PF6 recorded at 309 K showed that the Tc of the HCHMe2
probe protons dramatically changed upon irradiation with UV light
for 14 min. New well-resolved signals appeared for cis-494+
indi-
cating that the cis configuration of the ABP-F4 gate stops the shut-
tling of the CBPQT4+
ring.
In the 1H NMR of cis-49
4+, the two singlets at 5.16 and 5.22
ppm and two doublets at 1.13 and 1.17 ppm, which correspond to
the TrCH2O and CHMe2 protons, respectively, indicate that the
CBPQT4+
ring resides on only one of the two DNP units along the
dumbbell component. Irradiation of cis-494+
with a halogen lamp
for 20 min effected an 80% reversion to trans-494+
; its characteris-
tically broad resonances are commensurate with slow shuttling of
the CBPQT4+
ring at 309 K on the 1H NMR time-scale. By
alternatively irradiating gate 494+
with UV and visible light to
switch between its trans and cis isomers, the shuttling of the
CBPQT4+
ring undergoes a repeating STOP-GO sequence. Com-
plete isomerization of cis-494+
back to its trans isomer can also be
achieved by heating cis-49·4PF6 at 350 K for 60 min. An alternating
process of UV light and heat can also be applied.
The rate of the thermal gate-opening process at 350 K was de-
termined using 1H NMR spectroscopy. The first order rate constant,
kc t, was determined to be 0.124 s–1
at 350 K, which results in a
G‡ value of ca. 22 kcal·mol
–1. This free energy barrier is consis-
tent with cis-to-trans thermal relaxation of electron-deficient
azobenzene derivatives [34] including that of the corresponding
dumbbell, which was found to be ca. 20 kcal·mol–1
, as determined
from the kc t value of 0.098 s–1
at 350 K.
Tian et al. [35] designed a novel thermo- and photo-driven
[2]rotaxane, (Z)-60, comprising two fluorescent groups, i.e., por-
phyrin and fluorene, which are used as stoppers at the two ends of
the dumbbell, and two stations, i.e., fumaramide and succinimide,
which are inserted on the thread; the double bond of the fumara-
mide station undergoes photo-isomerization. Finally, two symmet-
rical reactive points such as alkyne groups are situated on the ring
of the rotaxane and are ideal for further substitution. With the active
alkyne groups appended on the ring, different compounds can be
introduced by, e.g., “click” reactions and Sonogashira coupling, to
change the optical properties of the porphyrin and fluorene moieties
on the thread. Additionally, the two symmetrical alkyne groups can
undergo polymerization.
The synthesis of thread 59 in the Z form is shown in Scheme
21. 5-(4-Aminophenyl)-10,15,20-triphenyl-porphyrin (53) reacts
with succinic anhydride to obtain succinic derivative 54. Derivative
54 is coupled with (8-amine-oxtal)-carbamic acid tert-butyl ester
(55) using EDCI/DMAP. After deprotection, the tert-butyl ester is
transformed to a free amine, which finally couples with fluorene
derivative 58.
Rotaxane (Z)-60 was assembled by slow addition of isophtha-
loyl chloride and p-xylylenediamine in the presence of Et3N
(Scheme 21). Since the double bond of the fumarimide is in the Z
form, this structure could not form hydrogen bonds with the ring
[17]. Therefore, the ring of rotaxane (Z)-60 resides on the suc-
cinimide group, which is close to the porphyrin part and distant
from the fluorene moiety. The E form of the fumarimide portion of
the thread is more suitable to form hydrogen bonds with the ring
[17] so the ring shuttles from the porphyrin to the fluorene.
Comparison of the 1H NMR spectra of rotaxanes (E)-60 and
(Z)-60 with their respective threads, (E)-59 and (Z)-59, reveals the
location of the ring on the thread. The 1H NMR spectrum of rotax-
ane (Z)-60 in CDCl3 features an upfield shift of the protons of the
succinimide moiety from 3.30 (Hi) and 2.70 (Hj) ppm to 1.90
and 1.88 ppm, respectively, compared with the same protons in
(Z)-59. This effect was observed due to shielding of the benzyl
aromatic rings on the macrocycle over the thread. A shift of the
amide protons near the succinimide moiety to the aromatic region,
i.e., from 6.10 (He) and 5.95 (Hf) ppm to 7.00 and 6.58 ppm,
respectively, was observed in the spectrum of rotaxane (Z)-60 in
CDCl3; this is caused by hydrogen bonding with the ring. Mean-
while, the signals corresponding to Ha, Hb, Hg, and Hh, which are
associated with the fumaramide potion, did not shift. These results
prove that the macrocycle of rotaxane (Z)-60 resides on the suc-
cinimide moiety. In contrast, the differences in the 1H NMR spectra
of (E)-59 and (E)-60 are opposite to those described above for
(Z)-59 and (Z)-60. This confirms that the macrocycle of rotaxane
(E)-60 resides on the fumaramide moiety. Additionally, there are no
differences among the absorption and fluorescent spectra of rotax-
anes (Z)-60 and (E)-60 and their threads, (Z)-59 and (E)-59, in
CH2Cl2.
Using thread (Z)-59 and exchanging the shuttling macrocycle
with a benzylic-amide macro-ring incorporating two Py groups, the
same authors exploited the properties of the porphyrin moieties to
coordinate with a variety of metal ions. By synthesizing an inter-
locked molecule that is able to interact with the Py groups of an-
other rotaxane monomer via intermolecular metal coordination, a
self-organized rotaxane nanostructure was achieved [36].
This molecular shuttle, (Z)-61, was prepared in 25% yield from
thread (Z)-59 (Scheme 22) and then converted into (E)-61 by heat-
ing. This compound can reconvert to (Z)-61 by irradiation at 254
nm. The addition of Zn2+
to a solution of rotaxane (Z)-61/(E)-61
leads to the replacement of the central hydrogen atoms in the por-
phyrin moiety with the metal center. Thus, the zinc porphyrin moi-
ety of one rotaxane monomer axially connects to a pyridyl unit of
another monomer. Since there are symmetrical pyridyl moieties on
either side of the macrocycle, the self-assembled supramolecular
structure extends to form a large network.
Table 1. Kinetic and Thermodynamic Parameters Obtained from Temperature-Dependent 1H NMR Spectra of Degenerate [2]Rotaxane 49·4PF6
Recorded in CD3CN.
Proton (ppm) (Hz) kc (s–1
) Tc (K) G‡ (kcal·mol
–1)
c
HCHMe2 3.35 86 191 322a 15.6
HCHMe2 1.18 31 69 309b 15.5
HTrCH2O 5.36 25 56 311b 15.7
a Calibrated using neat ethylene glycol. b Calibrated using neat MeOH.
c Value ±0.1.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 151
R NH2
N
NH N
HN
Ph
Ph
Ph
NH
O
OH
O
R NH
O
HN
O
( )6
R
OO O
i
HN BOC
H2N ( )6 NH
BOC
ii
R NH
O
HN
O
( )6
NH2
R
NH
O
HN
O
( )
HN
6
O
O NH
OH
O
O
NH
( )6
( )6
( )6
( )6
iii
R =
R
NH
O
HN
O
( )
HN
6
O
O
NH
( )6
( )6
iv
v
vi
R
NH
O
O
( )
HN
6
O
O NH
( )6
( )6
R
NH
O
HN
O
( )
O
O
NH
( )6
( )6
v
vi
O
Cl
O
Cl
O
vii
HN
HN
NH
NH
O
O
O
O
O
O
NH
NH
HN
HN
O
O
O
O
O
O
HN
HN
53 5455
56
57
58
(Z)-59 (E)-59
(Z)-60 (E)-60
a
bc de
f g
h
i
j
6
Scheme 21. Synthesis of bistable rotoxanes (Z)-60 and (E)-60. Reagents and conditions: (i) CHCl3/Et3N, room temperature, 95%; (ii) DMAP, EDCI, CHCl3, 0
°C, 65%; (iii) trifluoroacetic acid, CH2Cl2, room temperature, 99%; (iv) DMAP, EDCI, CHCl3, 0 °C, 35%; (v) 1,1,2,2-tetrachloride ethylene, 400 K, 50%; (vi)
CH2Cl2, 254 nm, 30%; (vii) p-xylylenediamine, Et3N/CHCl3, 0 °C, 8%; DMAP = 4-dimethylaminopyridine, EDCI =
1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, Boc = tert-butoxycarbonyl.

152 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
N
NH N
HN
Ph
Ph
Ph
i
ii
R2
NH
O
HN
O
( )
HN
6
O
O NH
( )6
( )6
iii
R1 =
R2
NH
O
HN
O
( )
HN
6
O
O
NH
( )6
( )6
R1 or R2
NH
O
O
( )
HN
6
O
O NH
( )6
( )6
R1 or R2
NH
O
HN
O
( )6
O
O
NH
( )6
( )6
HN
HN
NH
NH
O
O
N
N
O
O
NH
NH
HN
HN
O
O
N
N
O
O
HN
HN
(Z)-59-Zn (E)-59-Zn
(Z)-61; R1
(Z)-61-Zn; R2
i
ii
i
ii
(Z)-59 (E)-59
N
N N
N
Ph
Ph
PhR2 = Zn;
(E)-61; R1
(E)-61-Zn; R2
Scheme 22. Synthesis of bistable rotaxane (Z)-61/(E)-61. (i) 400 K, 51/49 for (E)-59/(Z)-59 and 43/57 for (E)-61/(Z)-61 in 1,1,2,2-tetra-chloride ethylene; (ii)
254 nm, 25/75 for (E)-59/(Z)-59 and 18/82 for (E)-61/(Z)-61 in CH2Cl2; (iii) p-xylylene diamine, 3,5-pyridinedicarbonyl dichloride, Et3N/CHCl3, 0 °C, 25%.
Rotaxane (E)-61 was obtained in 43% yield by thermal iso-
merization of rotaxane (Z)-61 at 400 K in 1,1,2,2-tetrachloride eth-
ylene. Photoisomerization of rotaxane (E)-61, which was achieved
via irradiation at 254 nm, furnished 18% yield of (Z)-61.
After replacing the hydrogen atoms of the porphyrin moiety
with the zinc ion to form (E)-61-Zn and (Z)-61-Zn, the 1H NMR
signals broaden and the spectra were quite different from those of
the corresponding monomers. The protons of the benzylic amide
macrocycle, especially the HA protons on the Py groups, underwent
a significant upfield shift due to the shielding effects of the zinc
porphyrin, which attaches to the macrocycle ring. These changes
confirmed that the zinc porphyrin stopper of one rotaxane monomer
was axially connected to the pyridyl unit of another.
Furthermore, the optical properties of (E)-61-Zn and (Z)-61-Zn
were investigated with respect to those of their reference threads
(E)-59-Zn and (Z)-59-Zn. It is interesting that the fluorescence of
the metalized (E)-61/(Z)-61 assemblies was red-shifted as compared
to that of (E)-59-Zn/(Z)-59-Zn due to the formation of self-
assemblies of the rotaxane monomers. It is also notable that
assembled rotaxane (Z)-61 displays much stronger fluorescence
than (Z)-59-Zn. This suggests that the fluorescence of (Z)-59-Zn is
weaker due to the intermolecular – stacking of the porphyrin
zinc(II) moieties. Accordingly, the fluorescence intensifies when
the porphyrin zinc(II) segments are primarily axially coordinated to
the pyridyl residues via complexation and the – stacking of the
porphyrins is eliminated.
The interior morphologies of the metalized rotaxane (E)-61 and
(Z)-61 assemblies were studied using SEM. The leaner rotaxane
monomer, i.e., (E)-61, self-aggregates to some extent to form the
rod structures observed in Fig. 7A. On the other hand, annular
structures were formed by the self-aggregation of curly monomer
(Z)-61 (Fig. 7C). This reveals that both conformations of the two
isomers of the rotaxane form self-aggregated structures, which may
be the result of intermolecular H–H bonding and – stacking of
the chromophores at the two ends of the thread subunit. The
coordination-driven self-organization process of the rotaxane
monomers occurs after mixing zinc(II) ion with the rotaxane at a
1:1 molar ratio, which corresponds to the formation of axle-

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 153
macrocycle-type nanostructures. Fig. 7B shows the SEM image of
the self-organization of rotaxane (E)-61 after metal coordination: a
regular formed network is clearly evident.
The average diameter of a single annular cavity is ~120 nm
while the width of the edge of the hole is estimated to be ~20 nm,
as observed using SEM. However, the organization of metalized
rotaxanes (Z)-61 and (E)-61 differs.
Fig. 7D shows SEM images of rotaxane (Z)-61 after
metalization, which demonstrate an irregular assembly rather than
networks. The self-organized morphology is an enlarged result,
which also reflects the significant influence of the conformational
structures of the rotaxane monomers on the integrative morphology
of the nanostructure as a result of the axle-macrocycle-type coordi-
nation. Linear (E)-61 is prone to form small networks while the
structurally curly (Z)-61 forms block structures.
ELECTROCHEMICALLY CONTROLLABLE BISTABLE
ROTAXANES
The first molecular shuttle with three distinct stations was re-
ported by Sauvage et al. [37] (Scheme 23). In their paper, they
demonstrated that the introduction of a 2,2 -bipyridine (BIPY) sta-
tion between the two terminal chelating groups results in much
faster gliding motion of the metal-complexed ring. In this three-
station system, the distance between the terminal stations is ap-
proximately 23 Å. The chelating units on the axis are 2,9-diphenyl-
1,10-phenanthroline (DPP), BIPY, and 2,2 ,6 ,2 -terpyridine
(TERPY), which is a tridentate ligand. The ring includes an 8,8 -
diphenyl-3,3 -biisoquinoline (DPBIIQ) bidentate ligand. This en-
docyclic, non-sterically bulky chelate is a key component that fa-
vors fast translational or rotational motions within shuttle-like ro-
taxanes or pirouetting systems, respectively. The principle of
electrochemically driven motion relies on the relative stabilities of
the copper(I) and copper(II) complexes formed with the various
ligands.
The thermodynamic stability of copper(I) complexes increases
in the following order: [Cu(TERPY)(DPBIIQ)]+<[Cu(BIPY)
(DPBIIQ)]+<[Cu(DPP)(DPBIIQ)]
+. The stability sequence is re-
versed for copper(II) complexes. The relative stabilities of the com-
plexes within these two sequences reflect the electrochemical prop-
erties of the complexes and, in particular, their redox potentials.
Upon oxidizing or reducing a given thermodynamically stable
state, the complex switches from one form to another to attain the
most stable form of the compound, i.e., [Cu(DPP)(DPBIIQ)]+ (four-
coordinate) for the reduced state or [Cu(TERPY)(DPBIIQ)]2+
(five-
coordinate) for the oxidized state. The general principle of the shut-
tling rotaxane is represented in Fig. (8).
This system was further studied using cyclic voltammetry (CV).
By varying the potential scan rate, the rate constant for the rear-
rangement of four-coordinate 62(4)
2+ to five-coordinate 62(5)
2+ was
estimated to be 0.4 s–1
. Accordingly, an upper value of 50 s–1
was
estimated for the conversion of five-coordinate 62(5)
+ to four- coor-
dinate 62(4)
+ (Scheme 23). The rates of the translational motion over
a distance of 23 Å compare favorably with those of other related
shuttles with shorter distances. This illustrates the importance of the
intermediate energy states provided by appropriate ligands.
Fig. (7). Scanning electron micrograph of the rotaxane (E)-61 before (A) and after (B) metalization by adding zinc acetate; (Z)-61 before (C) and after (D)
metalization by adding zinc acetate.

154 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
O
N N
O O
OO
62(4)+
O
OO
O
(p-But-C6H4)3C N N
N
N N
OO
O
O
(p-But-C6H4)3C
Cu
N
N
62(5)2+
O
OO
O
(p-But-C6H4)3C N N
N
N N
OO
O
O
(p-But-C6H4)3C
N
N
N
N
OO
O
Cu2
+ e – e
Scheme 23. Structures of the four-coordinate Cu(I) [62(4)
+] and five-coordinate Cu(II) [62(5)
2+] molecular shuttles in their thermodynamically stable forms. The
subscripts 4 and 5 indicate the coordination number of the copper center, excluding possible solvent molecules or counterions.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 155
Fig. (8). Principle of the electrochemically driven molecular shuttle based on copper(I) and copper(II) coordination.
Redox-responsive bistable [2]rotaxanes incorporating CD rings
have been investigated [38] as they might be able to interface with
redox-active enzymes or even act as switching components in im-
plantable medical devices. Accordingly, bistable [2]rotaxane 65
[38], which features an -CD ring that moves in response to the
redox properties of a tetrathiafulvalene (TTF) unit located in the
dumbbell component, was obtained from aqueous solution (Scheme
24). The synthesis employs a recently developed thread-
ing-followed-by-stoppering approach [39] that relies on the effi-
ciency of copper(I)-catalyzed azide-alkyne cycloaddition.
1H NMR and UV-vis spectroscopy in conjunction with CV and
induced circular dichroism (ICD) experiments were employed to
show that the TZ ring, which is installed during the synthesis of 65,
serves as a second station for the -CD ring when the TTF unit,
which is the preferred location for the -CD ring, is oxidized either
to a radical cation or to a dication. According to the ICD spectrum
of 65, which reveals a positive Cotton effect peak around the ab-
sorption band of the TTF unit, the latter is included in the cavity of
the -CD ring [40].
The oxidization of 65 by the addition of Fe(ClO4)3 solution was
monitored by UV-vis spectroscopy. The peak for the TTF unit at
457 nm shifts to 432 nm after the addition of the oxidant (0.2
equiv), which indicates that the -CD ring moves away from the
TTF unit immediately after oxidation. A peak at 595 nm, which
indicates the presence of the TTF·+
radical cation in the [2]rotaxane
[41], emerges after the further addition of Fe(ClO4)3 (0.2–1.0
equiv).
CV experiments on both 65 and 68 demonstrate that 68 exhibits
two one-electron reversible oxidation processes at +0.17 and +0.54
V for the first and second oxidization peaks of the TTF unit, respec-
tively. In contrast, in the spectrum of 65, the first oxidization peak
for shifts dramatically to +0.32 V, whereas the second oxidization
peak is at +0.55 V, which is very similar to that observed for the
dumbbell. These results support those obtained using UV-vis spec-
troscopy, i.e., the -CD ring moves away from the TTF unit of the
[2]rotaxane upon generation of the TTF·+
radical cation.
In the reducing cycle, the first reduction peaks of the TTF2+
di-
cation at +0.46 and +0.48 V for 65 and 68, respectively, are at very
similar voltages. The difference between the second reduction
peaks for 65 and 68 of 0.14 V suggests that the -CD ring is already
sufficiently close to the TTF unit in the [2]rotaxane to influence the
reduction of the TTF·+
radical cation back its neutral form. The -
CD ring might also facilitate the desolvation of the TTF·+
radical
cation in aqueous solution [42]. This dynamic property affects the
design, synthesis, and fabrication of nanoscale systems and devices.
These findings have implications for the production of mechanized
nanoparticles for drug delivery systems that exploit the unique re-
dox states that are often present within diseased cells but not in
healthy cells.
To further elucidate the details of the mechanism in this redox
switchable molecular shuttle, a theoretical free-energy profile ap-
proach from all-atom molecular dynamics simulations was con-
ducted [43]. Employing an umbrella sampling technique, free en-
ergy profiles were calculated for three oxidation states (i.e., 0, +1,
+2) of the TTF unit both in vacuo and in water. While the free en-
ergy profiles in vacuo do not explain the experimental observations,
the free energy profiles in water are in good agreement with the
observed binding preferences, which reveals the importance of the
water environment in theoretical calculations.
In the neutral state, the -CD@TTF isomer is calculated to be
energetically favored by 2.5 kJ/mol over the -CD@TZ isomer; this
result is in fair accordance with the experimental value of 5.5 kJ/
mol. In the oxidized states, the -CD@TZ isomer becomes the pre-
ferred co-conformation. Additionally, the free energy profile re-
veals that the shuttling of the -CD ring from the TTF unit to the
TZ unit is barrierless for the oxidized +2 state, which suggests that
the response time of this rotaxane is only slightly dependent on the
oxidization of TTF. The interactions between water and the rotax-
ane components induces the shuttling of the -CD ring from the
– e
+ e
Cu Cu2
translation
translation
translation
translation
= == TERPY= BIPY= DPP = DPBIIQ

156 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
TTF unit to the TZ unit in the oxidized states despite the increased
attractive interactions between the -CD ring and the dumbbell.
To realize the full potential of these functional molecules at the
nanoscale level [44], Stoddart directly observed electrochemically
driven single-molecule station changes within bistable rotaxane
molecules anchored laterally on gold surfaces [45].
Rotaxane molecule 694+
, which has disulfide groups attached to
the stoppers on the dumbbell termini (Fig. 9), was designed to bind
to Au{111} at each end. The sample was prepared with the desired
coverage of 5–6 molecules/1000 Å2 for STM imaging with the
thread section of the dumbbell assembled parallel to the surface
[46]. The distance between the two stations, i.e., TTF and 1,5-
dioxynaphthalene (DNP), in the fully stretched dumbbell is 36 Å. In
situ STM images of the bistable rotaxane molecules attached to
Au{111} showed ~3 Å high and 10–12 Å wide protrusions. No
protrusions were observed when a bare gold surface and a control
molecule, which comprised the dumbbell compound without the
CBPQT4+
ring, were treated and imaged under the same conditions
as the bistable rotaxane. The dumbbell component was apparently
not sufficiently conductive to be resolved by STM. However, the
CBPQT4+
rings, which have a higher electrical conductivity and
greater height relative to the dumbbell, appear as protrusions in the
STM images. The protrusions are therefore assigned to be the
CBPQT4+
rings, which is consistent with prior STM studies [47].
To study the station changes in 694+
, the same surface region
was repeatedly imaged to track the positions of the protrusions us-
ing image processing routines that were developed previously [48].
The molecules were imaged at two different electrode potentials,
i.e., –0.12 and –0.53 V, to view the TTF station in the reduced (neu-
tral) state and oxidized (cationic) state, respectively. Significant
displacement was observed when the potential was stepped from –
0.12 to –0.53 V (vs. Ag/AgCl). The correlations between the poten-
tial change and the motion of the CBPQT4+
ring suggest that the
positive charge of the cationic TTF station repels the CBPQT4+
ring. Subsequently, the potential was stepped down from –0.53 to –
0.12 V and the protrusions were again displaced, which suggests
that the CBPQT4+
ring returns to its thermodynamically favored
position encircling the neutral TTF station [46].
Using electrochemical scanning tunneling microscopy
(ECSTM), electrochemically controlled station changes of individ-
ual bistable rotaxane molecules were observed in situ. The motions
of the CBPQT4+
rings correlated with the redox states of the TTF
O O O
HO2C
HO2C
S
S
S
S
O
CO2H
CO2H
N
N N
OO
68 (80%)
65 (23%)
O O O
RO2C
RO2C
S
S
S
S
N3OO O
CO2R
CO2R
O O O
HO2C
HO2C
S
S
S
S
O
CO2H
CO2H
N
N N
OO
63 (R = H)
66 (R = Me)
64 (R = H)
67 (R = Me)
+
-CD HO
OH
OOH
6
1) CuSO4•5H2O/ascorbic acid/DMF
2) MeONa/MeOH
1) CuSO4•5H2O/H2O
sodium ascorbate
Scheme 24. Syntheses of the [2]rotaxane 65 and the corresponding dumbbell compound 68.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 157
station and displayed partial reversibility. The trajectories of the
rings suggest after adsorption of the bistable rotaxane molecule on a
surface, the motion of the CBPQT4+
ring relative to the dumbbell is
affected by its local environment and the flexibility of the molecule:
bistable rotaxane molecules with rigid dumbbells enable consistent
and fully reversible motions as well as direct visualization of the
rings and the shafts of the molecules.
Once again, Stoddart introduced an innovative idea to induce
mechanical movement in molecular assemblies by redox stimuli
using the dynamic properties of liquid crystals (LC). Thus, he de-
veloped a new class of LC materials based on a bistable [2]rotaxane
in which dendritic bistable LC [2]rotaxane 70 (Fig. 10) with TTF
and DNP recognition units encircled by a single CBPQT4+
ring
responded to redox signals. This system was switched electro-
chemically in dilute solution, i.e., in a molecularly dispersed state
[49]. Recently, Stoddard described the redox-driven switching and
electrochromic response of LC films comprising bistable
[2]rotaxane 70 and a polymer electrolyte [50].
[2]Rotaxane 70 forms a smectic A phase in the ranging of
10–150 °C, as previously reported [49]. Therefore, LC films of 70
with polydomain morphologies were readily prepared by casting or
spin-coating onto indium-tin-oxide (ITO) glass electrodes at ambi-
ent temperature. The thickness of the films was estimated to be 2–3
mm by SEM. The shuttling behavior of 70 in the LC film was stud-
ied using CV. The results suggest that the -accepting CBPQT4+
ring initially encircles the -donating TTF unit. After oxidation of
the TTF recognition unit, which is represented by the right central
dark grey cylinder in Fig. (11), the CBPQT4+
ring moves away due
to Coulombic repulsion and resides around the DNP unit, which is
represented by the left central light grey cylinder in Fig. (11), even
in a condensed and coherent environment. In this case, each
CBPQT4+
ring moves an estimated 1.4 nm between the two recog-
Fig. (9). Structure and motion of a bistable rotaxane molecule, adsorbed on Au{111} investigated using electrochemical scanning tunneling microscopy.
Fig. (10). Molecular structure of the smectic A liquid-crystalline bistable [2]rotaxane 70 having forklike mesogenic stoppers whose macrocycle is mobile
along the dumbbell under the influence of redox stimuli.
O O OOO
OOOO
O
S
S
– e
+ e
694
~36Å~20Å
DNP TTF
S
S
S
SOOO
OO
NN
N
O
R
O
O O
N N
N
O
R
O
N
N
N
N
4PF6
70
R =
O
O
O O O C5H
11
F F
O O O C5H
11
F F
O O O C5H
11
F F
O

158 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
nition units along the linear portion of the dumbbell component in
the layered smectic A phase.
Electrochromic cells based on the LC films of 70 were fabri-
cated by combining the [2]rotaxane with a polymer electrolyte pre-
pared from a solution of polymethylmethacrylate (PMMA; 7 wt%,
Mw = 120,000 and 350,000), propylene carbonate (PC; 40 wt%),
and LiPF6 (3 wt%) in CH3CN (50 wt%). The resultant transparent
viscous solution was spread over an ITO counter electrode, which
was then stored for one day at room temperature to enable forma-
tion of the self-supporting film. For the fabrication of a
two-electrode electrochromic cell, the ITO counter electrode with
the polymer electrolyte was set on the LC film–coated side of the
ITO working electrode.
Spectroelectrochemical studies of the cells support redox-driven
shuttling behavior in the LC state. The UV-vis-NIR spectrum of the
cell recorded at 0 V vs. the counter electrode (CE) displays a broad
absorption band centered around 850 nm due to charge-transfer
(CT) interactions between the TTF unit and the CBPQT4+
ring (Fig.
11, upper part). The application of positive potentials up to +1.6 V
vs. CE leads to a decrease in the intensity of the CT absorption band
at 850 nm. Concurrently, new absorption peaks appear at 400 and
770 nm and are assigned to the -dimer of TTF·+
radical cations
[51] (Fig. 11, lower part). The small absorption evident at 530 nm is
characteristic of the CT complex of the DNP unit with the
CBPQT4+
ring. These observations confirm the mechanical move-
ment of the macrocycle within [2]rotaxane 70 as a result of oxida-
tion of the TTF unit. Since the formation of the monomeric TTF·+
radical cation ( max = 448 and 600 nm) has been observed by
one-electron oxidation in dilute solution of the [2]rotaxane 70, the
dimerization of the oxidized TTF units is enhanced by their
face-to-face organization in the layered LC nanostructure.
It is noteworthy that the LC undergoes a visible color change
from greenish brown to reddish purple within 10 s of the applica-
tion of the oxidizing bias. When negative potentials are applied, 70
is reduced to its original form and the LC film in the cell recovers
its initial greenish brown color, which indicates that the redox-
driven mechanical movement is reversible in the LC film. In the
reduction process, the electrochromic switching requires a longer
period of time (ca. 40–50 s) for relaxation from the metastable state
back to the initial ground state as a result of the attractive interac-
tions of the CBPQT4+
ring with the DNP unit.
This approach offers new prospects for the further development
of rotaxane-based molecular materials by exploiting the dynamic,
anisotropic, and coherent properties of liquid crystals.
CONCLUSIONS
In this updated review, major recent developments on
[2]rotaxane-based shuttles based on chemically, photochemically,
and electrochemically induced molecular switching were described.
Among the chemically controlled shuttles, a significant increase of
the shuttling rates was achieved using second-generation pH-
switchable Pd(II)-complexed rotaxanes while new synthetic ap-
proaches, especially those based on the Diels-Alder cycloaddition,
allow ready access to rotaxanes with template sites incorporated
into the interlocked product, which is unusual for active-template
reactions. Moreover, the incorporation of a hydrogen-bonding TEG
station into an imine-bridged rotaxane resulted in shuttles that ex-
hibit entropy-driven translational isomerism with remarkable im-
proved positional discrimination. Finally, a molecular reel based on
the shuttling of a rotor by tumbling of an altro- -CD macrocycle
upon the change of solvent has been realized.
With respect to photochemically powered molecular switches,
the most important result was the synthesis of a degenerate donor-
acceptor light-gated STOP-GO molecular shuttle. This approach
could permit the isolation and identification of metastable and
ground-state co-conformations of bistable donor-acceptor
[2]rotaxanes in the near future.
For electrochemically controllable bistable rotaxanes, the first
example of a molecular shuttle with three different stations was
reported. In this case, the presence of an intermediate BIPY group
between two terminal chelating groups, i.e., DPP and TERPY, dra-
matically influences the shuttling process rate. Moreover, the use of
Fig. (11). Schematic representation of the redox-driven mechanical movement induced in the LC film of 70 on the electrode surface.

Recent Developments on Rotaxane-Based Shuttles Current Organic Chemistry, 2012, Vol. 16, No. 2 159
use of ECSTM allowed the in situ observation of electrochemically
controlled station changes of individual bistable shuttles. For a
bistable rotaxane molecule adsorbed on a surface, the motion of the
CBPQT4+
macrocyclic ring relative to the dumbbell is affected by
its local environment and by the flexibility of the molecule.
The last reported study regards the redox-driven switching and
electrochromic response of LC films comprising a bistable
[2]rotaxane and a polymer electrolyte. This approach offers new
prospects for the further development of rotaxane-based molecular
materials by exploiting the dynamic, anisotropic, and coherent
properties of liquid crystals.
Although the community has witnessed tremendous progress
since the first proof-of-principle studies, there is still a long way to
go to achieve mature devices employing functional supramolecular
systems that are applicable in daily life. Therefore, more effort
needs to be exerted in this research field.
REFERENCES
[1] Rescifina, A.; Zagni, C.; Iannazzo, D.; Merino, P. Recent developments on
rotaxane-based shuttles. Curr. Org. Chem., 2009, 13, 448-481.
[2] (a) Vetter, W.; Schill, G. Directed synthesis of catena compounds. IX. mass
spectrum of a catena compound. Tetrahedron, 1967, 23, 3079-3093; (b)
Schill, G. Catenanes, Rotaxanes and Knots; Academic Press: New York,
1971.
[3] (a) Balzani, V.; Credi, A.; Raymo, F.M.; Stoddart, J.F. Machines at the
molecular level. Angew. Chem. Int. Ed., 2000, 39, 3348-3391; (b) Silvi, S.;
Venturi, M.; Credi, A. Artificial molecular shuttles: from concepts to devices.
J. Mater. Chem., 2009, 19, 2279-2294; (c) Balzani, V.; Credi, A.; Venturi, M.
Light powered molecular machines. Chem. Soc. Rev., 2009, 38, 1542-1550;
(d) Fang, L.; Olson, M.A.; Benitez, D.; Tkatchouk, E.; Goddard, W.A., III;
Stoddart, J.F. Mechanically bonded macromolecules. Chem. Soc. Rev., 2010,
39, 17-29; (e) Ma, X.; Tian, H. Bright Functional rotaxanes. Chem. Soc. Rev.,
2010, 39, 70-80; (f) Konstas, K.; Langford, S.J.; Latter, M.J. Advances to-
wards synthetic machines at the molecular and nanoscale level. Int. J. Mol.
Sci., 2010, 11, 2453-2472.
[4] Collin, J.-P.; Durola, F.; Lux, J. Sauvage, J.-P. A Copper-based shuttling
[2]rotaxane with two bidentate chelates in the axis: steric control of the mo-
tion. New J. Chem., 2010, 34, 34-43.
[5] Fernandes, A.; Viterisi, A.; Coutrot, F.; Potok, S.; Leigh, D.A.; Aucagne, V.;
Papot, S. Rotaxane-based propeptides: protection and enzymatic release of a
bioactive pentapeptide. Angew. Chem. Int. Ed., 2009, 48, 6443-6447.
[6] Moretto, A.; Menegazzo, I.; Crisma, M.; Shotton, E.J.; Nowell, H.; Mammi,
S.; Toniolo, C. A rigid helical peptide axle for a [2]rotaxane molecular ma-
chine. Angew. Chem. Int. Ed., 2009, 48, 8986-8989.
[7] Leigh, D.A.; Lusby, P.J.; McBurney, R.T.; Symes, M.D. Improved dynamics
and positional bias with a second generation palladium(II)-complexed mo-
lecular shuttle. Chem. Commun., 2010, 46, 2382-2384.
[8] Crowley, J.D.; Goldup, S.M.; Lee, A.L.; Leigh, D.A.; McBurney, R.T.
Active metal template synthesis of rotaxanes, catenanes and molecular shut-
tles. Chem. Soc. Rev., 2009, 38, 1530-1541.
[9] Crowley, J.D.; Hänni, K.D.; Leigh, D.A.; Slawin, A.M.Z. Diels-Alder ac-
tive-template synthesis of rotaxanes and metal-ion-switchable molecular
shuttles. J. Am. Chem. Soc., 2010, 132, 5309-5314.
[10] Crowley, J.D.; Leigh, D.A.; Lusby, P.J.; McBurney, R.T.; Perret-Aebi, L.-E.;
Petzold, C.; Slawin, A.M.Z.; Symes, M.D.A. Switchable palla-
dium-complexed molecular shuttle and its metastable positional isomers. J.
Am. Chem. Soc., 2007, 129, 15085-15090.
[11] Humehara, T.; Kawai, H.; Fujiwara, K.; Suzuki, T. Entropy- and hydro-
lytic-driven positional switching of macrocycle between imine- and hydro-
gen-bonding stations in rotaxane-based molecular shuttles. J. Am. Chem. Soc.,
2008, 130, 13981-13988.
[12] Chiu, S.-H.; Stoddart, J.F. Reversing a rotaxane recognition motif: thread-
ing oligoethylene glycol derivatives through a dicationic cyclophane. J. Am.
Chem. Soc., 2002, 124, 4174-4175.
[13] Giuseppone, N.; Lehn, J.-M. Protonic and temperature modulation of con-
stituent expression by component selection in a dynamic combinatorial li-
brary of imines. Chem. Eur. J., 2006, 12, 1715-1722.
[14] Bottari, G.; Dehez, F.; Leigh, D.A.; Nash, P.J.; Pérez, E.M.; Wong, J.K.Y.;
Zerbetto, F. Entropy-driven translational isomerism: a tristable molecular
shuttle. Angew. Chem. Int. Ed., 2003, 42, 5886-5889.
[15] Berná, J.; Alajarín, M.; Orenes, R.-A. Azodicarboxamides as template bind-
ing motifs for the building of hydrogen-bonded molecular shuttles. J. Am.
Chem. Soc., 2010, 132, 10741-10747.
[16] Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsun-
obu and related reactions: advances and applications. Chem. Rev., 2009, 109,
2551-2651.
[17] Altieri, A.; Bottari, G.; Dehez, F.; Leigh, D.A.; Wong, J.K.Y.; Zerbetto, F. A
pseudorotaxane on gold: formation of self-assembled monolayers, reversible
dethreading and rethreading of the ring, and ion-gating behavior. Angew.
Chem. Int. Ed., 2003, 42, 2296-2300.
[18] (a) Morrison, D.C Reactions of alkyl phosphites with diethyl azodicarboxy-
late. J. Org. Chem., 1958, 23, 1072-1074. (b) Brunn, E.; Huisgen, R. Struc-
ture and reactivity of the betaine derived from triphenylphosphine and di-
methyl azodicarboxylate. Angew. Chem. Int. Ed., 1969, 8, 513-515.
[19] Zheng, H.; Zhou, W.; Lv, J.; Yin, X.; Li, Y.; Liu, H.; Li, Y. A dual-response
[2]rotaxane based on a 1,2,3-triazole ring as a novel recognition station.
Chem. Eur. J., 2009, 15, 13253-13262.
[20] Barrell, M.J.; Leigh, D.A.; Lusby, P.J.; Slawin, A.M.Z. An ion-pair template
for rotaxane formation and its exploitation in an orthogonal interaction an-
ion-switchable molecular shuttle. Angew. Chem. Int. Ed., 2008, 47,
8036-8039.
[21] Yamauchi, K.; Miyawaki, A.; Takashima, Y.; Yamaguchi, H.; Harada, A. A
molecular reel: shuttling of a rotor by tumbling of a macrocycle. J. Org.
Chem., 2010, 75, 1040-1046.
[22] Gutsche, C.D.; Bauer, L.J. Calixarenes. 13. The conformational properties of
calix[4]arenes, calix[6]arenes, calix[8]arenes, and oxacalixarenes. J. Am.
Chem. Soc., 1985, 107, 6052-6059.
[23] Busseron, E.; Romuald, C.; Coutrot, F. Bistable or oscillating state depend-
ing on station and temperature in three-station glycorotaxane molecular ma-
chines. Chem. Eur. J., 2010, 16, 10062-10073.
[24] Perrin, C.L. Reverse anomeric effect: fact or fiction? Tetrahedron, 1995, 51,
11901-11935.
[25] Zhou, W.; Xu, J.; Zheng, H.; Yin, X.; Zuo, Z.; Liu, H.; Li, Y. Distinct nanos-
tructures from molecular shuttle: effects of shuttling movement on nanos-
tructural morphologies. Adv. Funct. Mater., 2009, 19, 141-149.
[26] Xu, J.; Liu, X.; Lv, J.; Zhu, M.; Huang, C.; Zhou, W.; Yin, X.; Liu, H.; Li,
Y.; Ye, J. Morphology transition and aggregation-induced emission of an in-
tramolecular charge-transfer compound. Langmuir, 2008, 24, 4231-4237.
[27] Michinobu, T.; Boudon, C.; Gisselbrecht, J.-P.; Seiler, P.; Frank, B.;
Moonen, N.N.P.; Gross, M.; Diederich, F. Donor-substituted
1,1,4,4-tetracyanobutadienes (TCBDs): new chromophores with efficient in-
tramolecular charge-transfer interactions by atom-economic synthesis. Chem.
Eur. J., 2006, 12, 1889-1905.
[28] Yang, M.; Wang, W.; Yuan, F.; Zhang, X.; Li, J.; Liang, F.; He, B.; Minch,
B.; Wegner, G. Soft vesicles formed by diblock codendrimers of poly(benzyl
ether) and poly(methallyl dichloride). J. Am. Chem. Soc., 2005, 127,
15107-15111.
[29] Kawaguchi, Y.; Harada, A. A cyclodextrin-based molecular shuttle contain-
ing energetically favored and disfavored portions in its dumbbell component.
Org. Lett., 2000, 2, 1353-1356.
[30] B lohradsk , M.; Elizarov, A.M.; Stoddart, J.F. Speed-controlled molecular
shuttles. Collect. Czech. Chem. Commun., 2002, 67, 1719-1728.
[31] Brough, B.; Northrop, B.H.; Schmidt, J.J.; Tseng, H.-R.; Houk, K.N.; Stod-
dart, J.F.; Ho, C.-M. Evaluation of synthetic linear motor-molecule actuation
energetics. Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 8583-8588.
[32] Coskun, A.; Friedman, D.C.; Li, H.; Patel, K.; Khatib, H.A.; Stoddart, J.F. A
light-gated STOP–GO molecular shuttle. J. Am. Chem. Soc., 2009, 131,
2493-2495.
[33] Kang, S.S.; Vignon, S.A.; Tseng, H.-R.; Stoddart, J.F. Molecular shuttles
based on tetrathiafulvalene units and 1,5-dioxynaphthalene ring systems.
Chem. Eur. J., 2004, 10, 2555-2564.
[34] Lunak, S., Jr.; Nepras, M.; Hrdina, R.; Mustroph, H. Excited states of azo
compounds. II. Vibrational structure of the electronic absorption spectra of
4,4 -di-substituted azobenzene derivatives. Chem. Phys., 1994, 184, 255-260.
[35] Ji, F.-Y.; Zhu, L.-L.; Ma, X.; Wang, Q.-C.; Tian, H. A new thermo- and
photo-driven [2]rotaxane. Tetrahedron Lett., 2009, 50, 597-600.
[36] Ji, F.-Y.; Zhu, L.-L.; Zhang, D.; Chen, Z.-F.; Tian, H. Coordination-driven
self-organization of switchable [2]rotaxane. Tetrahedron, 2009, 65,
9081-9085.
[37] Collin, J.-P.; Durola, F.; Lux, J.; Sauvage, J.-P. A rapidly shuttling cop-
per-complexed [2] rotaxane with three different chelating groups in its axis.
Angew. Chem. Int. Ed., 2009, 48, 8532-8535.
[38] Zhao, Y.-L.; Dichtel, W. R.; Trabolsi, A.; Saha, S.; Aprahamian, I.; Stoddart,
J.F. A redox-switchable -cyclodextrin-based [2]rotaxane. J. Am. Chem.
Soc., 2008, 130, 11294-11296.
[39] Aucagne, V.; Hänni, K.D.; Leigh, D.A.; Lusby, P.J.; Walker, D.B. Catalytic

160 Current Organic Chemistry, 2012, Vol. 16, No. 2 Rescifina et al.
“click” rotaxanes: a substoichiometric metal-template pathway to mechani-
cally interlocked architectures. J. Am. Chem. Soc., 2006, 128, 2186-2187.
[40] Zhang, X.; Nau, W.M. Chromophore alignment in a chiral host provides a
sensitive test for the orientation�–�intensity rule of induced circular dichro-
ism. Angew. Chem. Int. Ed., 2000, 39, 544-547.
[41] Yoshizawa, M.; Kumazawa, K.; Fujita, M. Room-temperature and solu-
tion-state observation of the mixed-valence cation radical dimer of
tetrathiafulvalene, [(TTF)2]+•
, within a self-assembled cage. J. Am. Chem.
Soc., 2005, 127, 13456-13457.
[42] Bergamini, J.-F.; Hapiot, P.; Lorcy, D. Direct versus indirect electron trans-
fers in host–guest-inclusion complexes: Example of the oxidation of
TTF– -CD complexes. J. Electroanal. Chem., 2006, 593, 87-98.
[43] Zhang, Q.; Tu, Y.; Tian, H.; Zhao, Y.-L.; Stoddart, J.F.; Ågren, H. Working
mechanism for a redox switchable molecular machine based on cyclodextrin:
a free energy profile approach. J. Phys. Chem., 2010, 114, 6561-6566.
[44] Green, J.E.; Choi, J.W.; Boukai, A.; Bunimovich, Y.; Johnston-Halperin, E.;
DeIonno, E.; Luo, Y.; Sheriff, B.A.; Xu, K.; Shin, Y.S.; Tseng, H.R.; Stod-
dart, J.F.; Heath, J.R. A 160-kilobit molecular electronic memory patterned
at 1011
bits per square centimetre. Nature, 2007, 445, 414-417.
[45] Ye, T.; Kumar, A.S.; Saha, S.; Takami, T.; Huang, T.J.; Stoddart, J.F.; Weiss,
P.S. Changing stations in single bistable rotaxane molecules under electro-
chemical control. ACS Nano, 2010, 4, 3697-3701.
[46] Liu, Y.; Flood, A.H.; Bonvallett, P.A.; Vignon, S.A.; Northrop, B.H.; Tseng,
H.R.; Jeppesen, J.O.; Huang, T.J.; Brough, B.; Baller, M.; Magonov, S.; So-
lares, S.D.; Goddard, W.A.; Ho, C.-M.; Stoddart, J.F. Linear artificial mo-
lecular muscles. J. Am. Chem. Soc., 2005, 127, 9745-9759.
[47] Shigekawa, H.; Miyake, K.; Sumaoka, J.; Harada, A.; Komiyama, M. The
molecular abacus: STM manipulation of cyclodextrin necklace. J. Am. Chem.
Soc., 2000, 122, 5411-5412.
[48] Weiss, P.S. Functional molecules and assemblies in controlled environments:
formation and measurements. Acc. Chem. Res., 2008, 41, 1772-1781.
[49] Aprahamian, I.; Yasuda, T.; Ikeda, T.; Saha, S.; Dichtel, W.R.; Isoda, K.;
Kato, T.; Stoddart, J.F. A liquid-crystalline bistable [2]rotaxane. Angew.
Chem. Int. Ed., 2007, 46, 4675-4679.
[50] Yasuda, T.; Tanabe, K.; Tsuji, T.; Coti, K.K.; Aprahamian, I.; Stoddart, J.F.;
Kato, T. A redox-switchable [2]rotaxane in a liquid-crystalline state. Chem.
Commun., 2010, 46, 1224-1226.
[51] Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, oligoacenenes,
and their buckminsterfullerene derivatives: the brick and mortar of organic
electronics. Chem. Rev., 2004, 104, 4891-4946.
Received: December 02, 2010 Revised: June 21, 2011 Accepted: August 08, 2011




















