
Reaction of rhenium and carbon at high pressures and temperatures
10
This article is protected by German copyright law. You may copy and distribute this article for your personal use only. Other use is only allowed with written permission by the copyright holder. Reaction of rhenium and carbon at high pressures and temperatures Erick A. Juarez-Arellano * ,I , Bjo ¨rn Winkler I , Alexandra Friedrich I , Dan J. Wilson I , Monika Koch-Mu ¨ller II , Karsten Knorr III, 1 , Sven C. Vogel IV , James J. Wall IV , Helmut Reiche IV , Wilson Crichton V , Mayahuel Ortega-Aviles VI and Miguel Avalos-Borja VII I Institut fu ¨r Geowissenschaften, Universita ¨t Frankfurt, Altenho ¨ferallee 1, 60438 Frankfurt a. M., Germany II GeoForschungsZentrum, Telegrafenberg, Sektion 4.1, 14473 Potsdam, Germany III Institut fu ¨r Geowissenschaften/Mineralogie/Kristallographie, Christian-Albrechts-Universita ¨t zu Kiel, Olshausenstraße 40, 24098 Kiel, Germany IV Los Alamos National Laboratory, Lujan Center, Mail Stop, H805, NM 87545 Los Alamos, USA V ESRF, BP, 220, 38043 Grenoble CEDEX, France VI IMP, LMEUAR, Eje Central La ´zaro Ca ´rdenas 152, 07730 Me ´xico City, Me ´xico VII CCMC, UNAM, 2681 Ensenada, B.C., Me ´xico Received May 30, 2008; accepted July 25, 2008 Carbides / High pressures and temperatures / Synchrotron radiation / Analytical electron microscopy / Density functional theory (DFT) / X-ray diffraction / Powder diffraction structure analysis Abstract. A combined X-ray diffraction, EELS and DFT study of the reaction of rhenium with carbon at high-(P, T ) conditions up to P max ¼ 67 GPa and T max ¼ 3800 K is pre- sented. A hexagonal rhenium carbide, ReC x , was identified as the stable phase at high-(P, T ) conditions. A composi- tion of ReC 0:5 is proposed. No evidence for a cubic ReC polymorph with rocksalt structure, as suggested in the lit- erature, or for any other phase was found at the P-T condi- tions explored. A preliminary P-T rhenium-carbon phase diagram has been derived and properties such as bulk moduli and elastic stiffness coefficients were obtained. 1. Introduction Among the metallic elements, rhenium has one of the highest known bulk moduli (B 0 ¼ 360–372 GPa [1, 2]) and one of the highest shear moduli (179 GPa [1]), and the second highest melting temperature (3458 K [3]). Rhe- nium crystallizes in the space group P 6 3 m mc with unit cell parameters a ¼ 2.7609 A and c ¼ 4.476 A [4]. It does not undergo a temperature-induced phase transition up to melting and at ambient temperature it remains stable to pressures of at least 216 GPa [2]. These properties make rhenium one of the strongest and most stable poly- crystalline materials, suitable for applications such as, for example, gasket material in diamond anvil cells (DACs). Due to its chemical, thermal and structural stability, rhe- nium has been proposed as a pressure calibrant for heated DACs [5]. However, as no previous in-situ studies have been reported, little is known about the reactivity of rhe- nium at high (P , T ) conditions and hence the present study aims to fill this gap. This knowledge is relevant for in-situ laser heated diamond anvil cell synthesis experi- ments using rhenium as a gasket material. It is well established that rhenium does not form stoi- chiometric carbides at ambient pressure. The phase dia- gram of the system Re––C shows limited solubility of car- bon into rhenium [6, 7]. The maximum solubility is reached at 11.7 at% C at the eutectic temperature (2773 K) and it falls sharply with temperature (4.2 at% C at 2073 K). No intermediate phase exists, although Kharkova and Velikanova [7] mentioned the existence of some addi- tional reflections in the vicinity of the solidus that could be related to the formation of an unstable intermediate phase. The lattice parameters of rhenium as a function of carbon saturation at ambient conditions has been reported by Kharkova and Velikanova [7]. They found that the lat- tice parameters of Re increase in the 2.58–8.6 at% C re- gion from a ¼ 2.760 A to a ¼ 2.803 A, and from c ¼ 4.447 A to c ¼ 4.461 A. However, all these measure- ments were performed on quenched samples using X-ray diffraction. This implies that rhenium carbide phases which might only be stable at high temperatures would not have been detected. Also, X-ray diffractograms would be completely dominated by the contributions from the rhenium and any ordering of carbon on sublattices would not be detectable. In contrast, at high pressures the formation of rhenium carbides has been reported by Popova et al. [8–10]. They synthesized a hexagonal ReC, which they suggested had a g 0 -MoC type structure, with lattice parameters a ¼ 2.8403 A, c ¼ 9.8543 A, at synthesis conditions above 6 GPa and 1073 K [8]. However, no further structural de- 492 Z. Kristallogr. 223 (2008) 492–501 / DOI 10.1524/zkri.2008.0054 # by Oldenbourg Wissenschaftsverlag, Mu ¨nchen * Correspondence author (e-mail: [email protected]) 1 Present address: Bruker AXS GmbH, Karlsruhe, Germany
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Reaction of rhenium and carbon at high pressures and temperatures

Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
Reaction of rhenium and carbon at high pressuresand temperatures
Erick A. Juarez-Arellano*, I, Bjorn WinklerI, Alexandra FriedrichI, Dan J. WilsonI, Monika Koch-MullerII, Karsten KnorrIII, 1,Sven C. VogelIV, James J. WallIV, Helmut ReicheIV, Wilson CrichtonV, Mayahuel Ortega-AvilesVI
and Miguel Avalos-BorjaVII
I Institut fur Geowissenschaften, Universitat Frankfurt, Altenhoferallee 1, 60438 Frankfurt a. M., GermanyII GeoForschungsZentrum, Telegrafenberg, Sektion 4.1, 14473 Potsdam, GermanyIII Institut fur Geowissenschaften/Mineralogie/Kristallographie, Christian-Albrechts-Universitat zu Kiel, Olshausenstraße 40, 24098 Kiel,
GermanyIV Los Alamos National Laboratory, Lujan Center, Mail Stop, H805, NM 87545 Los Alamos, USAV ESRF, BP, 220, 38043 Grenoble CEDEX, FranceVI IMP, LMEUAR, Eje Central Lazaro Cardenas 152, 07730 Mexico City, MexicoVII CCMC, UNAM, 2681 Ensenada, B.C., Mexico
Received May 30, 2008; accepted July 25, 2008
Carbides / High pressures and temperatures /Synchrotron radiation / Analytical electron microscopy /Density functional theory (DFT) / X-ray diffraction /Powder diffraction structure analysis
Abstract. A combined X-ray diffraction, EELS and DFTstudy of the reaction of rhenium with carbon at high-(P, T )conditions up to Pmax ¼ 67 GPa and Tmax ¼ 3800 K is pre-sented. A hexagonal rhenium carbide, ReCx, was identifiedas the stable phase at high-(P, T ) conditions. A composi-tion of ReC0:5 is proposed. No evidence for a cubic ReCpolymorph with rocksalt structure, as suggested in the lit-erature, or for any other phase was found at the P-T condi-tions explored. A preliminary P-T rhenium-carbon phasediagram has been derived and properties such as bulkmoduli and elastic stiffness coefficients were obtained.
1. Introduction
Among the metallic elements, rhenium has one of thehighest known bulk moduli (B0 ¼ 360–372 GPa [1, 2])and one of the highest shear moduli (179 GPa [1]), andthe second highest melting temperature (3458 K [3]). Rhe-
nium crystallizes in the space group P63
mmc with unit
cell parameters a ¼ 2.7609 �A and c ¼ 4.476 �A [4]. It
does not undergo a temperature-induced phase transitionup to melting and at ambient temperature it remains stableto pressures of at least 216 GPa [2]. These propertiesmake rhenium one of the strongest and most stable poly-crystalline materials, suitable for applications such as, forexample, gasket material in diamond anvil cells (DACs).
Due to its chemical, thermal and structural stability, rhe-nium has been proposed as a pressure calibrant for heatedDACs [5]. However, as no previous in-situ studies havebeen reported, little is known about the reactivity of rhe-nium at high (P, T ) conditions and hence the presentstudy aims to fill this gap. This knowledge is relevant forin-situ laser heated diamond anvil cell synthesis experi-ments using rhenium as a gasket material.
It is well established that rhenium does not form stoi-chiometric carbides at ambient pressure. The phase dia-gram of the system Re––C shows limited solubility of car-bon into rhenium [6, 7]. The maximum solubility isreached at 11.7 at% C at the eutectic temperature (2773 K)and it falls sharply with temperature (4.2 at% C at2073 K). No intermediate phase exists, although Kharkovaand Velikanova [7] mentioned the existence of some addi-tional reflections in the vicinity of the solidus that couldbe related to the formation of an unstable intermediatephase. The lattice parameters of rhenium as a function ofcarbon saturation at ambient conditions has been reportedby Kharkova and Velikanova [7]. They found that the lat-tice parameters of Re increase in the 2.58–8.6 at% C re-gion from a ¼ 2.760 �A to a ¼ 2.803 �A, and fromc ¼ 4.447 �A to c ¼ 4.461 �A. However, all these measure-ments were performed on quenched samples using X-raydiffraction. This implies that rhenium carbide phaseswhich might only be stable at high temperatures wouldnot have been detected. Also, X-ray diffractograms wouldbe completely dominated by the contributions from therhenium and any ordering of carbon on sublattices wouldnot be detectable.
In contrast, at high pressures the formation of rheniumcarbides has been reported by Popova et al. [8–10]. Theysynthesized a hexagonal ReC, which they suggested hada g0-MoC type structure, with lattice parametersa ¼ 2.8403 �A, c ¼ 9.8543 �A, at synthesis conditions above6 GPa and 1073 K [8]. However, no further structural de-
492 Z. Kristallogr. 223 (2008) 492–501 / DOI 10.1524/zkri.2008.0054
# by Oldenbourg Wissenschaftsverlag, Munchen
* Correspondence author(e-mail: [email protected])
1 Present address: Bruker AXS GmbH, Karlsruhe, Germany

Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
tails were given. The cell parameters of the hexagonal poly-morph were confirmed by Kharkova and Velikanova [7].Again, the atomic coordinates were not reported. The synth-esis of a cubic polymorph with NaCl-type structure and alattice parameter of 4.005 �A was reported for pressuresabove 17 GPa and 1300 K [9, 10]. All diffraction experi-ments, again, were performed on quenched samples.
To further investigate the reaction of rhenium with car-bon, we undertook high temperature solid state synthesis,laser heated diamond anvil cell experiments and multi-an-vil press synthesis experiments. We analysed samples withX-ray diffraction, electron energy loss spectroscopy(EELS) and made observations by scanning electron mi-croscopy (SEM) and energy dispersive X-ray spectroscopy(EDS). The experimental studies are complemented withdensity functional theory (DFT) based model calculations.
2. Experimental
2.1 Rhenium and carbon at high temperature,multi-anvil press based synthesis and X-raydiffraction
2.1.1 Rhenium and carbon at high temperature
A mechanical mixture of approximately equal parts by vol-ume of rhenium and graphite was compressed into a pel-let and was loaded in a high temperature furnace at LosAlamos National Laboratory. The mixture was heated upto 2450 K for about one hour and cooled to room tem-perature in about 20 minutes. This sample served as a re-ference for carbon-saturated Re and allowed us to checkearlier X-ray diffraction results.
2.1.2 Multi-anvil press based synthesis
The synthesis was carried out in a multi-anvil apparatus atthe GeoForschungsZentrum Potsdam using a 14=8 assembly(octahedron length/truncation length) at 12 GPa and1673 K for one hour. The assembly was calibrated atroom temperature against the phase transitions in Bi metal[11, 12]. Calibrations at higher temperature were based onthe following phase transitions: CaGeO3: garnet-perovskite[13]; SiO2: coesite – stishovite [14]; Mg2SiO4: a� btransition [15]. A mechanical mixture of rhenium and gra-phite was loaded into a MgO sleeve which was enclosedin a MgO-based octahedral pressure medium within aLaCrO3 heater. The temperature was controlled using aW5%Re–W26%Re thermocouple. No pressure correctionwas applied to the emf.
2.1.3 X-ray diffraction
The recovered samples were ground and placed in a cap-illary (0.5 mm inner diameter). Powder X-ray diffractionpatterns were recorded at room temperature with a Panaly-tical XPert/MPD diffractometer. CuKa radiation from a Cuanode operating at 45 kV and 40 mA was used. The phaseidentification was performed with the program XPertHighScore Plus. Subsequent Le Bail fittings and Rietveldrefinements, using the program FULLPROF [16], wereperformed using a pseudo-Voigt profile function (Tables 1
and 2). The zero point, scale factor, unit cell parameters,half-width and pseudo-Voigt parameters of the peaks, posi-tional and isotropic displacement parameters were refined.A linear interpolation of 20 fixed points was used for thebackground and an absorption correction was applied.
2.2 Laser heated diamond anvil cell experiments
The reaction between rhenium and graphite was inves-tigated by in situ high-(P, T ) diffraction experiments at theID27 beamline of the European Synchrotron Radiation Fa-cility (ESRF). Monochromatic radiation (l ¼ 0.3738 �A)and a MarCCD detector were used. The sample-to-detec-tor distance of 249.669 mm was determined from a Si re-ference sample. Diffraction data were collected at up toabout 2q ¼ 24� giving a maximum sin q/l of 0.556 �A
�1.
A rhenium (Re) foil (thickness �20 mm) and powderedgraphite were used as starting materials. The starting mate-rials were compressed using a diamond-anvil cell (DAC)
Reaction of rhenium and carbon at high pressures and temperatures 493
Table 1. Lattice parameters of the structures discussed here.
Phase S.G. a/�A c/�A V/�A3
ReaP
63
mmc 2.803 4.461 30.3 [7]
Re P63
mmc 2.7812(1) 4.4612(1) 29.884(1) b c
RedP
63
mmc 2.7486 9.7537 63.82 DFT
ReC P63
mmc 2.840(1) 9.85(1) 68.85 [8]
ReCx P63
mmc 2.8425(1) 9.8599(2) 68.990(2) c
(I)- Re4C2 P63
mmc 2.85 10.07 70.84 DFT
MgO Fm�33m 4.2135(1) –– 74.807(2) c
MgO Fm�33m 4.2114 –– 74.69 [29]
D-HexeP
63
mmc 2.5239(3) 4.1213(8) 22.735(6) c
D-HexeP
63
mmc 2.52 4.12 22.66 [32]
a: Re saturated with carbon (8.6 at% C).b: High-T carbon saturated.c: From Rietveld refinement, this study.d: Re in AABB stacking sequence (g0-MoC type structure).e: Diamond-Hexagonal.
Table 2. Reliability factors and atomic positions of the recoveredsample from the multi-anvil press synthesis.
Phase ReCx Rea (I)-Re4C2 MgO D-Hexb
Exp.c DFT DFT Exp.c Exp.c
Rbragg 5.35 –– –– 5.77 27.2
Atom Red Re Re/C Mg/O C
x=a 1=31=3
1=3=1=3 0=1=2
1=3
y=b 2=32=3
2=3=2=3 0=1=2
2=3
z=c 0.1084(1) 0.108 0.111=1=4 0=1=2 0.12(4)
B=�A2
0.58(2) –– –– 0.6/0.6 0.6
Occup. 1.0 1.0 1.0 1.0 1.0
a: Re in the hypothetical g0-MoC type structure, AABB stackingsequence b: Diamong-Hexagonal c: Global factors: Rp ¼ 3.71,Rwp ¼ 4.85, Rexp ¼ 3.25, c2 ¼ 2.20 d: No carbon position was refined.

Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
with rhenium gaskets (pre-indented to a thickness of about40 mm) which had a 110 mm hole. Compressed argon wasloaded as the pressure-transmitting medium using a pres-sure vessel. Pressure was determined using the equation ofstate of argon (Ar-EOS) measured at ambient temperatures[17]. Dewaele et al. [18] have determined that the un-certainties in pressure at high temperature (Ar meltingpoint) using the ambient temperature Ar-EOS is DP ¼ �4GPa. The starting materials were laser heated from oneside at different pressures by a Nd : YAG laser, providing amaximum power of 80 W. The temperature was measuredat the center of the hot spot by analyzing the pyrometricsignals. The uncertainties in measured temperatures (100–300 K) were estimated by comparing the results of threeanalysis methods of the thermal emission spectrum re-corded between 550 and 750 nm [19]. The diffractionimages were processed, corrected for distortion by theCCD and integrated using FIT2D [20]. Intense and welldefined single-crystal diffraction spots from individual lar-ger grains of the sample, from the crystallized pressuremedium, and from the diamonds were masked manuallyand excluded from the integration. The background of theintegrated powder diffraction patterns was extracted usingthe program DATLAB [21]. Le Bail fits were performedusing the program FULLPROF [16] in order to obtain unitcell parameters. A linear interpolation between approxi-mately 30 manually selected points for the backgroundand a pseudoVoigt profile function were used. Diffractiondata were collected at different pressures without heating;at pressures of 4, 18–20 and 56–67 GPa prior to heating,during heating between 1800 and 3800 K; and on tem-perature quenched samples. For each temperature run, anew sample position was selected.
2.3 Electron microscopy characterization
SEM observations were made using a JEOL 5300 scanningelectron microscope on the multi-anvil press recovered sam-ple. The ground, uncoated sample was mounted on alumi-num stubs with double-sided sticking carbon tape. The EDSanalysis was performed with a Kevex Superdry EDS systemmounted on the same JEOL 5300 microscope. Data acquisi-tion was done through a 4 PI multichannel processor inter-face. Great care was taken to avoid counts from the carbonsticking tape by acquiring EDS spectra at the center of largegrains. Backscattered electron images were acquired with aGW detector and an additional 4 PI interface.
For the TEM and EELS work, the sample was groundin an agate mortar with ethanol. A drop of that suspensionwas placed on a lacey carbon coated copper grid and driedunder a lamp for a few minutes. The microscope was aTecnai G2 F30 operated at 300 kV, with a spherical-aber-ration coefficient (Cs) of 1.2 mm. EELS was performedwith a Gatan Image Filter system mounted on the sameinstrument.
2.4 Density functional theory
Density functional theory (DFT) calculations have beenused extensively to model structure-property relations oftransition metal carbides (e.g. [22, 23]). For our calcu-
lations we have employed the CASTEP code [24]. Thecode is an implementation of Kohn-Sham DFT based on aplane wave basis set in conjunction with pseudopotentials.The plane-wave basis set is unbiassed (as it is not atom-centered) and does not suffer from the problem of basis-set superposition error unlike atom-centred basis sets. Italso makes converged results straightforward to obtain inpractice, as the convergence is controlled by a single ad-justable parameter, the plane wave cutoff, which we set to750 eV. All pseudopotentials were ultrasoft, and were gen-erated using the PBE exchange-correlation functional [25]to allow for a fully consistent treatment of the core andvalence electrons. The rhenium atom was described withcore (in parentheses) and valence regions of ([Kr]4d104f 14) 5s25p66s2, while the carbon was simply ([He])2s22p2. The Brillouin-Zone integrals were performed usingMonkhorst-Pack grids [26] with spacings between gridpoints of less than 0.02 �A
�1. Simultaneous geometry opti-
misation of unit cell and internal co-ordinates was per-formed so that forces were converged to 0.005 eV/�A andthe stress residual to 0.005 GPa.
In order to assess the reliability of the Re pseu-dopotential, the structure of hexagonal Re (space group
P63
mmc) was computed. The experimental lattice para-
meters are aexp ¼ 2.7609(4) �A and cexp ¼ 4.476(4) �A [4].The theoretical values are in excellent agreement withthese values, as atheo ¼ 2.770 �A and ctheo ¼ 4.487 �A. AllRe atoms are located on special positions, and hence thegeometry optimization provides no further insight into thereliability of the pseudopotential. Therefore, we computedthe elastic stiffness coefficients, as the elastic behaviour of acrystal is intimately linked to the interatomic interactions[27]. The results are compared to experimental data in Table3. Clearly, the elastic stiffness coefficients are reproducedwithin the usual range [28] and we are therefore confidentin the predictive power of the Re pseudopotential. Thecarbon potential was tested by comparing calculated toobserved properties for diamond. Here, equally good agree-ment was observed (aexp ¼ 3.568 �A, atheo ¼ 3.567 �A).
3. Results and discussion
The mixture of rhenium and graphite loaded in the hightemperature furnace did not form a quenchable new phaseup to the maximum temperature reached (2450 K). The
494 E. A. Juarez-Arellano, B. Winkler, A. Friedrich et al.
Table 3. Comparison of experimentally determined and computed val-ues for the elastic stiffness coefficients, cij, and bulk moduli, B. Allvalues are given in GPa.
Reexp Retheo (I)-Re4C2
at 4 K – [42] this study this study
c11 634.4 612(2) 668(2)
c33 701.6 666(1) 800(5)
c44 169.1 157(1) 245(1)
c12 266.0 268(2) 200(2)
c13 202.0 216(1) 225(2)
B 367.8 365(1) 378(1)

Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
rhenium lattice parameters obtained from the heated mix-ture agree with the values of rhenium saturated with car-bon (8.6 at% C) from Kharkova and Velikanova [7] (Table 1).
The laser heated diamond anvil cell experiments werethe first high-(P, T ) set of experiments performed duringthe current study. However, due to the presence of toomany phases with similar unit cell parameters and crystalstructures (like hcp-rhenium, fcc-argon, hcp-argon, possi-ble fcc-ReC, hcp-ReC and the possible formation of com-pounds only stable at high-(P, T ) conditions) the analysisbecame too complicated to proceed. In order to obtain asample with fewer phases, a multianvil press synthesis wasundertaken. The analysis of the products allowed us toindex the patterns recorded in the DAC at high-(P, T ) con-ditions. Hence, we first describe the results of the multi-anvil press synthesis, followed by the DAC results.
3.1 Structural characterization of the recoveredsample from the multianvil synthesis
Three phases were identified in the recovered multi-anvilpress sample: diamond, ReC and MgO, which was an im-purity from the surrounding capsule material. Neither rhe-nium nor graphite was detected in the X-ray diffractionpattern (Fig. 1). The refined cell parameters and the spacegroup of MgO agree well with the values reported in theliterature [29] (Table 1). MgO can hence be used as aninternal reference. The analysis of the diamond reflectionswas not as straightforward as for MgO. From the diffrac-togram, we observed only two reflections not overlappingwith other phases, the 2.06 �A and the 1.26 �A reflectionsthat we initially thought were related to the (111) and(220) cubic diamond planes, respectively. The Rietveld re-finement gave a cell parameter of a ¼ 3.5688(2) �A whichseems to be in reasonable agreement with values reportedearlier (e.g. a ¼ 3.5671 �A [30]).
However, a detailed analysis of the residuals showedthat although the description of the 2.06 �A reflection wasunproblematic (Fig. 2a), the 1.26 �A reflection was not
well described. Hence, we searched for an alternativestructural model. According to the pressure-temperaturecarbon phase diagram [31], at 12 GPa and 1673 K (thesynthesis conditions in this work) hexagonal diamond isthe stable phase. A fit based on the hexagonal diamondmodel resulted in a significantly improved description ofthe X-ray diffraction pattern (Figs. 2a–b). The cell para-meters obtained agree well with those given by Bundy andKasper [32] (Table 1).
The reflections due to the main phase, ReCx, were in-dexed using the TREOR program [33]. The best solutionhad a figure-of-merit of M20 ¼ 65 and F20 ¼ 27 [34]. Aftera Le Bail fit using the program Fullprof [16] the cell param-eters obtained were a ¼ 2.8425(1) �A and c ¼ 9.8584(1) �A.These lattice parameters agree well with those given byPopova [8] (Table 1).
According to Popova [8], the hexagonal phase has a g0-
MoC type structure with space group P63
mmc. Neither Po-
pova [8] nor subsequent studies citing this article discussthe atomic positions in ReC or in g0-MoC type structures.In the literature, there currently is a discussion about
Reaction of rhenium and carbon at high pressures and temperatures 495
Fig. 1. Observed (+), calculated (–) and difference (at the bottom) X-ray powder diffraction pattern from the sample synthesized in a multi-anvil press at 12 GPa and 1673 K. These data were taken at ambientconditions. Vertical marks correspond, from top to bottom, to theposition of the allowed Bragg reflections of ReCx, MgO and hexago-nal diamond, respectively.
a�
b�Fig. 2. Non-overlapping diamond reflections at 2.06 �A (2q � 43.9�)(a) and at 1.26 �A (28 75.2�) (b). Observed (þ) and calculated (�) X-ray diffraction pattern. Vertical marks correspond to the position ofthe allowed Bragg reflections.

Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
whether to consider the metastable g0-MoC phase as a partof the molybdenum-carbon phase diagram [35, 36]. It hasbeen shown that oxygen is required in order to stabilizethis metastable phase and, therefore, this phase should beconsidered as an oxycarbide and not as a true binary car-bide [37, 38]. The distinguishing feature of the g0-MoCphase is an AABB stacking sequence of the metal atomsalong [001] (Fig. 3), that differs from the ABAB sequencein hexagonal Mo2C or pure rhenium (Fig. 3); and fromthe AAAA sequence in the hexagonal g-MoC phase. Theatomic position of molybdenum in g0-MoC has been dis-cussed only by Kuo and Hagg [39], while no data werefound relating to the atomic positions of the carbon atoms.
In order to derive a starting model for the Rietveld re-finement, we performed DFT calculations. Using the vo-lume obtained in the Le Bail fitting and assuming that thedensity of the quenched high-P phase would be not todissimilar to the density of Re at ambient pressure wetested the small number of arrangements for 4 Re atomsin a unit cell with a � 2.85 �A and c � 9.86 �A and the
space group symmetry P63
mmc (International Tables No.
194). The most stable structure had Re atoms on 4 f posi-tion, with z ¼ 0.108. This position is similar to that pro-posed for the Mo atoms in g0-MoC, where Kuo and Hagg[39] suggested a value close to 1=8. We then performed aRietveld refinement, obtaining a value of z ¼ 0.1084(1), inexcellent agreement with the DFT prediction. Therefore, therhenium AABB structural motif proposed by Popova [8] isfully confirmed here. The very satisfactory fit is shown inFig. 1. The final refined lattice parameters, reliability factorsand atomic positions are given in Tables 1 and 2.
The X-ray diffraction data could not be used to locatethe carbon atoms, as the scattering contribution of carbonis very small compared to the Re contribution (Re has 75electrons, carbon has 6 electrons). It could also have beenpossible that instead of rhenium carbide we had a highpressure pure Re-polymorph and that all carbon reacted tohexagonal diamond. However, the presence of a high pres-sure pure Re-polymorph seems unlikely for two reasons.First, while the calculations for pure rhenium at ambientpressure are in excellent agreement with the experimentaldata, the c-lattice of the computed high-P compound is
significantly smaller than the experimental values. In anearlier study, we have shown that the incorporation of car-bon into Ti leads to a noticeable expansion of the c-latticeparameter [23]. The second indication comes from theelastic stiffness calculations. While the proposed ReAABB
structure was fully optimized and hence resulted in aforce-free structure, the attempts to calculate elastic stiff-ness coefficients show that this structure does not corre-spond to a local minimum of the total energy. We employthe stress-strain method for the calculation of elastic stiff-ness coefficients, and the distortions applied to obtain c44
invariably led to results which could not be analyzed interms of a linear relation between the applied strain andthe computed stress. The structure only became stablewith respect to small distortions after the introduction ofcarbon. Furthermore, the c-lattice parameter expanded sig-nificantly thereby bringing the theoretical lattice para-meters into closer agreement with the experimental obser-vations. This already seems to be a clear indication of theincorporation of carbon into the ReAABB lattice.
Once we had established the arrangement of the rhe-nium atoms, we investigated the most probable atomic po-sition of carbon. The modeling technique we used is basedon periodic boundary conditions and hence structures withpartial occupancies and/or substoichiometric compositioncannot be easily studied. However, in a series of simula-tions, we introduced increasing amounts of carbon into theRe-lattice and performed full geometry optimisations. Foreach relaxed structure, we also computed the reaction en-thalpy for its formation from Re and diamond. A fewstructures are shown in Fig. 3. Two different configura-tions were tested for a Re4C2 stoichiometry, one locatingthe carbon on the position 2b (0, 0, 1=4) and the other onthe position 2a (0, 0, 0), labeled as (I) and (II), respec-tively.
The incorporation of carbon into the Re-lattice leads toa significant extension of the c-lattice (Fig. 4). This beha-vior was expected, as we have observed a similar resultduring the incorporation of carbon into titanium [23].From Fig. 4, we can see that the expansion of the c-latticeparameter not only depends on the amount of carbon butalso on the atomic configuration. Comparing the energiesof formation of the simulated structures, it can be seen
496 E. A. Juarez-Arellano, B. Winkler, A. Friedrich et al.
Fig. 3. Full geometry optimizations ofincreasing amounts of carbon into theAABB Re-lattice. Rhenium is shownin black spheres and carbon in greyspheres. Only Re––C bonds are plottedfor clarity of visualization.

Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
that the relative stability is dependent upon the carboncontent and that the most stable configuration is (I)-Re4C2
(ReC0:5) (Fig. 5). This configuration is about 0.23 eV(about 22 kJ mol�1 per Re atom) more stable than Re4C3
(ReC0:75), and about 0.45 eV (about 43 kJ mol�1 per Reatom) more stable than ReAABB. Lu et al. [38] found ex-perimentally that the composition of g0-MoC was near toMoC0.75. This implies that findings for ReCx may not bedirectly transferable to the g0-MoC system. By a directcomparison of the ReCx lattice parameters with those ob-tained by DFT and by considering their energies of forma-tion, we believe that ReCx � 0:5 (Re4C2) is probably thecomposition of the ReCx phase we have synthesized in themulti-anvil press and that the carbon atoms are located atthe position 2b (0, 0, 1=4). Very likely, x is slightly smallerthan 0.5, but this would be difficult to establish by a com-parison to DFT-data. Instead, to confirm this, neutron dif-fraction studies are required, and these are currentlyplanned.
To further clarify the nature of the recovered samplefrom the multi-anvil press synthesis and to further confirmthe incorporation of carbon, electron microscopy character-ization was performed.
3.2 Electron microscopy characterization of therecovered sample from the multi-anvilpress synthesis
When we recovered the sample from the multi-anvil presssynthesis, we observed that the sample was made up oftwo compounds; one soft and transparent (MgO sleeve)and the other one dense, hard, and black (the reacted sam-ple). The transparent compound was removed mechani-cally. For the electron microscopy characterization, a smallpiece of the black compound was used. A general view,representative of the sample, is shown in Fig. 6. The sam-ple consists of an agglomeration of two kinds of homoge-neous particles with diameters of less than 5 mm.
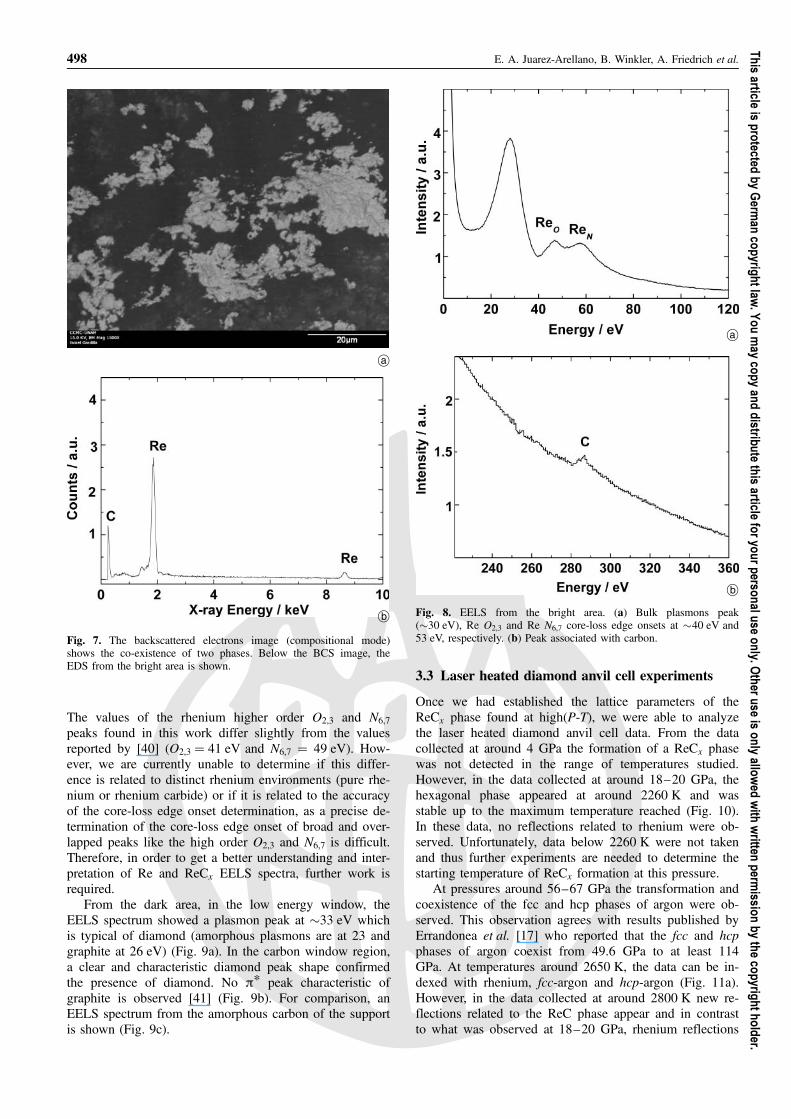
An image of the same area using backscattered elec-trons (BCS) in compositional mode shows that the sam-ple is made up of two compounds (Fig. 7). In this mode,brighter areas correspond to higher atomic number thandarker areas. EDS from the bright area indicate a rhe-nium rich compound while the dark regions indicatespure carbon. Even though magnesium does not appear inthe EDS of Fig. 7, it is also present in some areas of thesample, mainly in the dark area. The sensitivity of EDSanalysis is higher for heavy atoms than for light atoms,whose concentrations are generally underestimated. Fivespots standardless analysis, performed in five differentbright areas, gave an average carbon content of 15 at%C. Thus, semi-quantitatively, this gives a lower limit ofthe carbon content in ReCx.
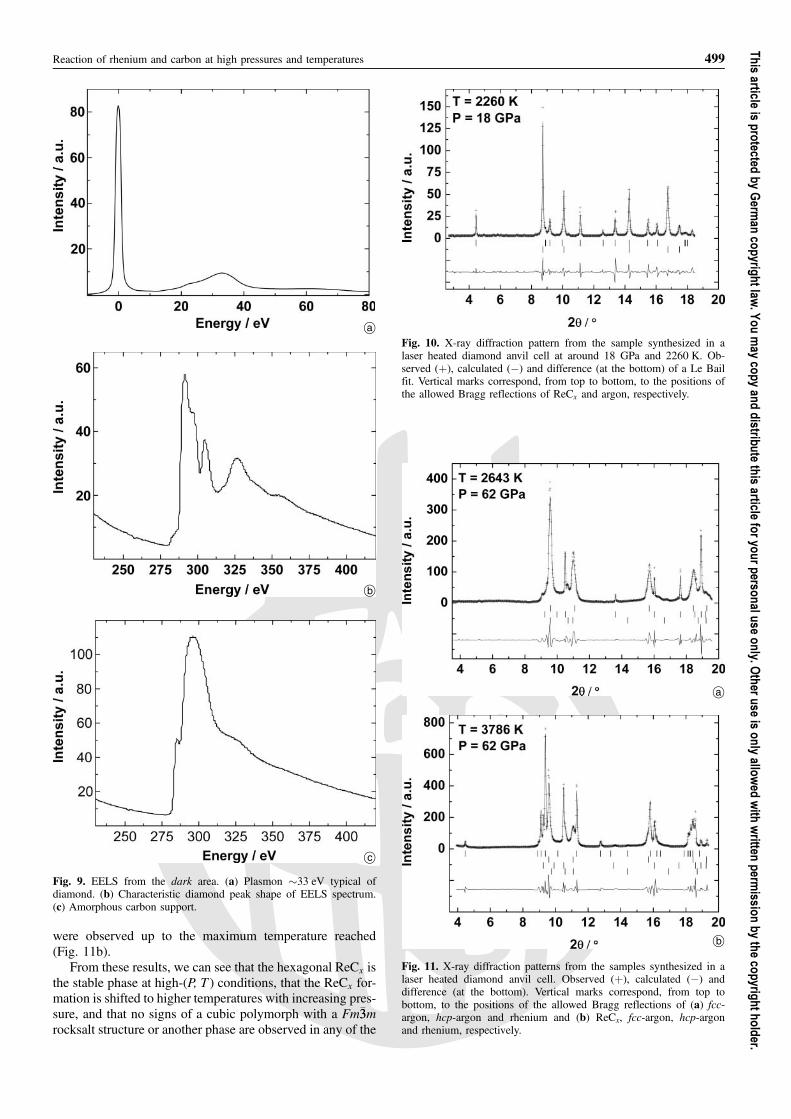
From the bright area, the EELS spectrum with no sup-port interference and in the low energy window shows thezero-loss peak and the bulk plasmon peak (�30 eV). Therhenium higher order O2,3 and N6,7 core-loss edge onsetswere located at 40 eV and 53 eV, respectively (Fig. 8a). Inthe carbon window region, a tiny peak is observed (Fig. 8b).
Reaction of rhenium and carbon at high pressures and temperatures 497
Fig. 4. The effect of increasing amounts of carbon into the Re-latticeparameters, obtained by DFT calculations. The black-triangle andblack-circle represent the experimental lattice parameters of ReCx andRe [4], respectively. All lattice parameters were normalized so as tocorrespond to a cell with 4 Re atoms. The dashed line is a guide tothe eye.
Fig. 5. Energies of formation, for the reaction 4 Re þ xCDiamond ¼Re4Cx, as a function of carbon content obtained by DFT calculations.
Fig. 6. SEM image (secondary electrons) of the recovered samplefrom the multi-anvil press synthesis. The sample is formed of homo-geneous particles with diameters of less than 5 mm.
Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
The values of the rhenium higher order O2,3 and N6,7
peaks found in this work differ slightly from the valuesreported by [40] (O2,3 ¼ 41 eV and N6,7 ¼ 49 eV). How-ever, we are currently unable to determine if this differ-ence is related to distinct rhenium environments (pure rhe-nium or rhenium carbide) or if it is related to the accuracyof the core-loss edge onset determination, as a precise de-termination of the core-loss edge onset of broad and over-lapped peaks like the high order O2,3 and N6,7 is difficult.Therefore, in order to get a better understanding and inter-pretation of Re and ReCx EELS spectra, further work isrequired.
From the dark area, in the low energy window, theEELS spectrum showed a plasmon peak at �33 eV whichis typical of diamond (amorphous plasmons are at 23 andgraphite at 26 eV) (Fig. 9a). In the carbon window region,a clear and characteristic diamond peak shape confirmedthe presence of diamond. No p* peak characteristic ofgraphite is observed [41] (Fig. 9b). For comparison, anEELS spectrum from the amorphous carbon of the supportis shown (Fig. 9c).
3.3 Laser heated diamond anvil cell experiments
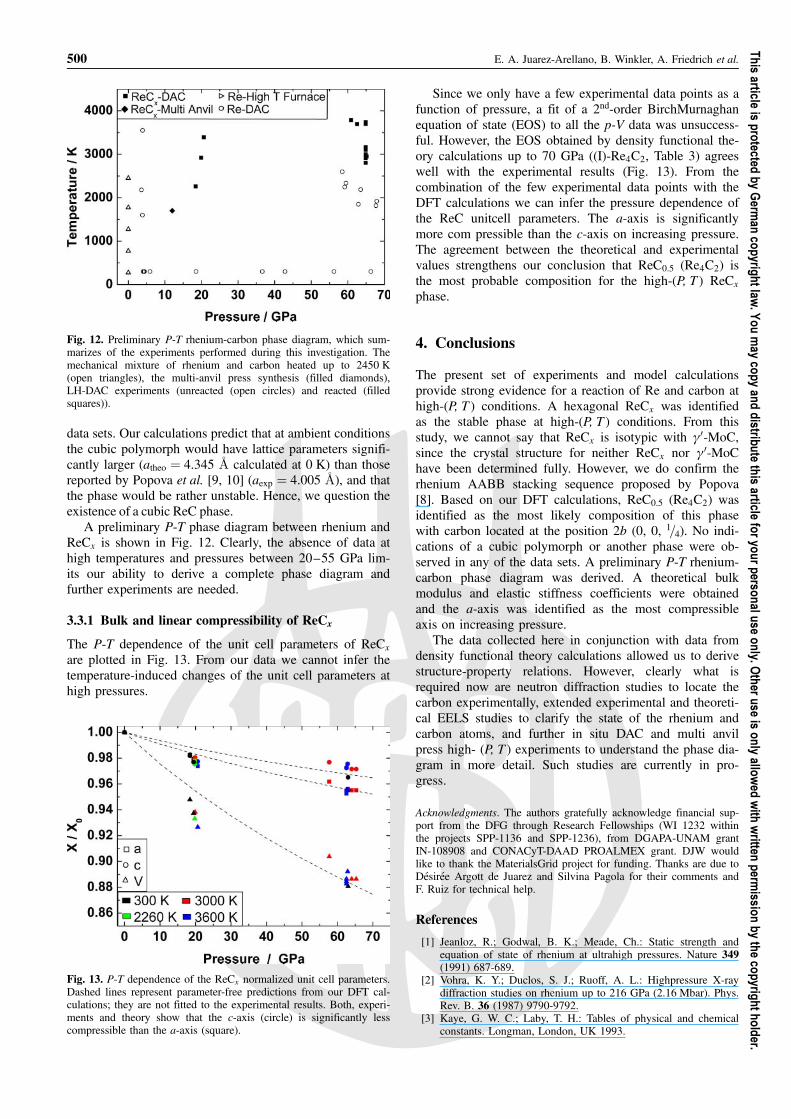
Once we had established the lattice parameters of theReCx phase found at high(P-T), we were able to analyzethe laser heated diamond anvil cell data. From the datacollected at around 4 GPa the formation of a ReCx phasewas not detected in the range of temperatures studied.However, in the data collected at around 18–20 GPa, thehexagonal phase appeared at around 2260 K and wasstable up to the maximum temperature reached (Fig. 10).In these data, no reflections related to rhenium were ob-served. Unfortunately, data below 2260 K were not takenand thus further experiments are needed to determine thestarting temperature of ReCx formation at this pressure.
At pressures around 56–67 GPa the transformation andcoexistence of the fcc and hcp phases of argon were ob-served. This observation agrees with results published byErrandonea et al. [17] who reported that the fcc and hcpphases of argon coexist from 49.6 GPa to at least 114GPa. At temperatures around 2650 K, the data can be in-dexed with rhenium, fcc-argon and hcp-argon (Fig. 11a).However, in the data collected at around 2800 K new re-flections related to the ReC phase appear and in contrastto what was observed at 18–20 GPa, rhenium reflections
498 E. A. Juarez-Arellano, B. Winkler, A. Friedrich et al.
a�
b�
Fig. 7. The backscattered electrons image (compositional mode)shows the co-existence of two phases. Below the BCS image, theEDS from the bright area is shown.
a�
b�
Fig. 8. EELS from the bright area. (a) Bulk plasmons peak(�30 eV), Re O2,3 and Re N6,7 core-loss edge onsets at �40 eV and53 eV, respectively. (b) Peak associated with carbon.
Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
were observed up to the maximum temperature reached(Fig. 11b).
From these results, we can see that the hexagonal ReCx isthe stable phase at high-(P, T ) conditions, that the ReCx for-mation is shifted to higher temperatures with increasing pres-sure, and that no signs of a cubic polymorph with a Fm�33mrocksalt structure or another phase are observed in any of the
Reaction of rhenium and carbon at high pressures and temperatures 499
a�
b�
c�
Fig. 9. EELS from the dark area. (a) Plasmon �33 eV typical ofdiamond. (b) Characteristic diamond peak shape of EELS spectrum.(c) Amorphous carbon support.
Fig. 10. X-ray diffraction pattern from the sample synthesized in alaser heated diamond anvil cell at around 18 GPa and 2260 K. Ob-served (þ), calculated (�) and difference (at the bottom) of a Le Bailfit. Vertical marks correspond, from top to bottom, to the positions ofthe allowed Bragg reflections of ReCx and argon, respectively.
a�
b�
Fig. 11. X-ray diffraction patterns from the samples synthesized in alaser heated diamond anvil cell. Observed (þ), calculated (�) anddifference (at the bottom). Vertical marks correspond, from top tobottom, to the positions of the allowed Bragg reflections of (a) fcc-argon, hcp-argon and rhenium and (b) ReCx, fcc-argon, hcp-argonand rhenium, respectively.
Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
data sets. Our calculations predict that at ambient conditionsthe cubic polymorph would have lattice parameters signifi-cantly larger (atheo ¼ 4.345 �A calculated at 0 K) than thosereported by Popova et al. [9, 10] (aexp ¼ 4.005 �A), and thatthe phase would be rather unstable. Hence, we question theexistence of a cubic ReC phase.
A preliminary P-T phase diagram between rhenium andReCx is shown in Fig. 12. Clearly, the absence of data athigh temperatures and pressures between 20–55 GPa lim-its our ability to derive a complete phase diagram andfurther experiments are needed.
3.3.1 Bulk and linear compressibility of ReCx
The P-T dependence of the unit cell parameters of ReCx
are plotted in Fig. 13. From our data we cannot infer thetemperature-induced changes of the unit cell parameters athigh pressures.
Since we only have a few experimental data points as afunction of pressure, a fit of a 2nd-order BirchMurnaghanequation of state (EOS) to all the p-V data was unsuccess-ful. However, the EOS obtained by density functional the-ory calculations up to 70 GPa ((I)-Re4C2, Table 3) agreeswell with the experimental results (Fig. 13). From thecombination of the few experimental data points with theDFT calculations we can infer the pressure dependence ofthe ReC unitcell parameters. The a-axis is significantlymore com pressible than the c-axis on increasing pressure.The agreement between the theoretical and experimentalvalues strengthens our conclusion that ReC0.5 (Re4C2) isthe most probable composition for the high-(P, T ) ReCx
phase.
4. Conclusions
The present set of experiments and model calculationsprovide strong evidence for a reaction of Re and carbon athigh-(P, T ) conditions. A hexagonal ReCx was identifiedas the stable phase at high-(P, T ) conditions. From thisstudy, we cannot say that ReCx is isotypic with g0-MoC,since the crystal structure for neither ReCx nor g0-MoChave been determined fully. However, we do confirm therhenium AABB stacking sequence proposed by Popova[8]. Based on our DFT calculations, ReC0.5 (Re4C2) wasidentified as the most likely composition of this phasewith carbon located at the position 2b (0, 0, 1=4). No indi-cations of a cubic polymorph or another phase were ob-served in any of the data sets. A preliminary P-T rhenium-carbon phase diagram was derived. A theoretical bulkmodulus and elastic stiffness coefficients were obtainedand the a-axis was identified as the most compressibleaxis on increasing pressure.
The data collected here in conjunction with data fromdensity functional theory calculations allowed us to derivestructure-property relations. However, clearly what isrequired now are neutron diffraction studies to locate thecarbon experimentally, extended experimental and theoreti-cal EELS studies to clarify the state of the rhenium andcarbon atoms, and further in situ DAC and multi anvilpress high- (P, T ) experiments to understand the phase dia-gram in more detail. Such studies are currently in pro-gress.
Acknowledgments. The authors gratefully acknowledge financial sup-port from the DFG through Research Fellowships (WI 1232 withinthe projects SPP-1136 and SPP-1236), from DGAPA-UNAM grantIN-108908 and CONACyT-DAAD PROALMEX grant. DJW wouldlike to thank the MaterialsGrid project for funding. Thanks are due toDesiree Argott de Juarez and Silvina Pagola for their comments andF. Ruiz for technical help.
References
[1] Jeanloz, R.; Godwal, B. K.; Meade, Ch.: Static strength andequation of state of rhenium at ultrahigh pressures. Nature 349(1991) 687-689.
[2] Vohra, K. Y.; Duclos, S. J.; Ruoff, A. L.: Highpressure X-raydiffraction studies on rhenium up to 216 GPa (2.16 Mbar). Phys.Rev. B. 36 (1987) 9790-9792.
[3] Kaye, G. W. C.; Laby, T. H.: Tables of physical and chemicalconstants. Longman, London, UK 1993.
500 E. A. Juarez-Arellano, B. Winkler, A. Friedrich et al.
Fig. 12. Preliminary P-T rhenium-carbon phase diagram, which sum-marizes of the experiments performed during this investigation. Themechanical mixture of rhenium and carbon heated up to 2450 K(open triangles), the multi-anvil press synthesis (filled diamonds),LH-DAC experiments (unreacted (open circles) and reacted (filledsquares)).
Fig. 13. P-T dependence of the ReCx normalized unit cell parameters.Dashed lines represent parameter-free predictions from our DFT cal-culations; they are not fitted to the experimental results. Both, experi-ments and theory show that the c-axis (circle) is significantly lesscompressible than the a-axis (square).

Th
is a
rticle
is p
rote
cte
d b
y G
erm
an
co
pyrig
ht la
w. Y
ou
may c
op
y a
nd
dis
tribu
te th
is a
rticle
for y
ou
r pers
on
al u
se o
nly
. Oth
er u
se is
on
ly a
llow
ed
with
writte
n p
erm
issio
n b
y th
e c
op
yrig
ht h
old
er.
[4] Wasilewski, R. J.: Physical and mechanical properties of rhe-nium. In Transactions of the Metallurgical Society of Aime 221(1961) 1081-1082.
[5] Zha, C. S.; Bassett, W. A.; Shim, S. H.: Rhenium, an in situpressure calibrant for internally heated diamond. Review ofScientific Instruments 75 (2004) 2409-2418.
[6] Hughes, J. E.: A survey of the Rhenium-Carbon system. J. less-common metals 1 (1959) 377-381.
[7] Kharkova, A. M.; Velikanova, T. Y.: Structure of alloys of thesystem rhenium-carbon in the region rich with rhenium.Poroshkovaya Metallurgiya 12-300 (1987) 52-56.
[8] Popova, S. V.; Boiko, L. G.: A new rhenium carbide formed byhigh-pressure treatment. High Temperature high pressure 3(1971) 237-238.
[9] Popova, S. V.; Fomicheva, L. N.; Khvostantsev, L. G.: Synthesisand superconducting properties of cubic rhenium monocarbide.Pis ’ma v Zhurnal Eksperimentalnoi, Teoreticheskoi Fiziki. 16(1972) 609-610.
[10] Popova, S. V.: The crystal structure of new superconducting ma-terials obtained by high pressure treatment. Acta Crystallogr. A31 (1975) 99- 99.
[11] Lloyd, E. C.: Editorial introduction and summary to: accuratecharacterization of the high pressure environment. U.S. NationalBureau Standards Special Publication 326 (1971) 1-3.
[12] Piermarini, G. J.; Block, S.: Ultra high pressure diamond-anvilcell and several semiconductor phase transition pressures in rela-tion to fixed point pressure scale. Reviews of Scientific Instru-ments 46 (1975) 973-980.
[13] Susaki, J. I.; Akaogi, M.; Akimoto, S.; Shimoura, O.: Garnet-perowskite transformation in CaGeO3: in situ X-ray measure-ments using synchrotron radiation. Geophysical Research Letters12 (1985) 729-732.
[14] Akaogi, M.; Yusa, H.; Shiraishi, K.; Suzuki, T.: Thermodynamicproperties of alpha-quartz, coesite, and stishovite and equili-brium phase relations at high pressures and high temperatures. J.Geophys. Res 100 (1995) 337-347.
[15] Morishima, H.; Kato, T.; Suto, M.; Ohtani, E.; Urakawa, S.;Utsumi, W.; Shimomura, O.; Kikegawa, T.: The phase boundarybetween a- and b-Mg2SiO4 determined by in situ X-ray obser-vation. Science 265 (1994)1202-1203.
[16] Rodriguez-Carvajal, J.: Recent advances in magnetic structuredetermination by neutron powder diffraction. Physica B 192(1993) 55-69.
[17] Errandonea, D.; Boehler, R.; Japel, S.; Mezouar, M.; Benedetti,L. R.: Structural transformation of compressed solid Ar: An X-ray diffraction study to 114 GPa. Phys. Rev. B 73 (2006)092106 (1-4).
[18] Dewaele, A.; Mezouar, M.; Guignot, N.; Loubeyre, P.: Meltingof lead under high pressure studies using second-scale timeresolved X-ray diffraction. Phys. Rev. B 76 (2007) 144106 (1-5).
[19] Benedetti, L. R.; Loubeyre, P.: Temperature gradients, wave-length-dependent emissivity, and accuracy of high and very-hightemperatures measured in the laser-heated diamond cell. HighPressure Research 4 (2004) 423-445.
[20] Hammersley, A. P.; Svensson, S. O.; Hanfland, M.; Fitch, A. N.;Hauserman, D.: Two-dimensional detector software: from realdetector to idealised image or two-theta scan. High Pressure Re-search 14 (1996) 235-248.
[21] Syassen, K.: DATLAB. Version 1.37d. MPI/FKF Stuttgard, Ger-many 2005.
[22] Lopez de la Torre, L.; Winkler, B.; Schreuer, J.; Knorr, K.; Ava-los-Borja, M.: Elastic properties of tantalum carbide (TaC). So-lid State Communications 134 (2005) 245-250.
[23] Winkler, B.; Wilson, D. J.; Vogel, S. C.; Brown, D. W.; Sis-neros, T. A.; Milman, V.: In situ observation of the formation ofTiC from the elements by neutron diffraction. J. Alloys Com-pounds 441 (2007) 374-380.
[24] Clark, S. J.; Segall, M. D.; Pickard, C. J.; Hasnip, P. J.; Probert,M. J.; Refson, K.; Payne, M. C.: First principles methods usingCASTEP. Z. Kristallogr. 220 (2005) 567-570.
[25] Perdew, J. P.; Burke, K.; Ernzerhof, M.: Generalized gradientapproximation made simple. Phys. Rev. Lett. 77 (1996) 3865-3868.
[26] Monkhorst, H.; Pack, J. D.: Special points for Brillouin-zoneintegrations. Phys. Rev. B 13 (1976) 5188-5192.
[27] Schreuer, J.: Exploration of structure-property relationships.Chimia 55 (2001) 562-569.
[28] Winkler, B.; Hytha, M.; Warren, M. C.; Milman, V.; Gale, J. D.;Schreuer, J.: Calculation of the elastic constants of the Al2SiO5polymorphs andalusite, sillimanite and kyanite. Z. Kristallogr.216 (2001) 67-70.
[29] Schmahl, N. G.; Barthel, J.; Eikerling, G. F.: RontgenographisheUntersuchungen an den Systemen MgO-CuO und NiO-CuO. Z.Anorg. Allg. Chemie 332 (1964) 230–237.
[30] Yamanaka, T.; Morimoto, S.: Isotope effect on anharmonic ther-mal vibration and K-refinemant of 12C and 13C diamond. ActaCrystallogr. B 52 (1996) 232-238.
[31] Bundy, F. P.: Pressure-Temperature phase diagram of elementalcarbon. Physica A 156 (1989) 169-178.
[32] Bundy, F. P.; Kasper, J. S.: Hexagonal carbon – a new form ofcarbon. J. Chemical Physics 46 (1967) 3437–3446.
[33] Werner, N. R.; Eriksson, L.; Westdahl. M.: TREOR, a semi-ex-haustive trial-and-error powder indexing program for all symme-tries. J. Applied Crystallogr. 18 (1985) 367–370.
[34] DeWolff, P. M.: A simplified criterion for the reliability of apowder pattern indexing. J. Applied Crystallogr. 1 (1968) 108–113.
[35] Rudy, E.; Windisch, S.; Stosick, A. J.; Hoffmann, J. R.: The con-stitution of binary molybdenum-carbon alloys. Transactions of theMetallurgical Society of Aime 239 (1967) 1247-1267.
[36] Velikanova, T. Ya.; Kulli, V. Z.; Khaenko, B. V.: Solid-statetransformations and phase-equilibria in the molybdenum carbonsystem. Soviet Powder Metallurgy and Metal Ceramics 27(1988) 891–896.
[37] Nowotny, H.; Parthe, E.; Kieffer, R.; Benesovsky, F.: Das Dreis-toffsystem: MolybdanSilizium-Kohlenstoff. Monatshefte furChemie 85 (1954) 255-272.
[38] Lu, J.; Hugosson, H.; Eriksson, O.; Nordstrom, L.; Jansson, U.:Chemical vapor deposition of molybdenum carbides: aspects ofphase stability. Thin Solid Films 370 (2000) 203-212.
[39] Kuo, K.; Hagg, G.: A new molybdenum carbide. Nature 170(1952) 245–246.
[40] Carpenter, R. W.; Chizmeshya, V. G.: Structure of Low-LossEELS in Rhenium. Microscroscopy and Microanalysis 11 (Sup-plement 2) (2005) 734-735.
[41] Duarte-Moller, A.; Espinosa-Magana, F.; Martinez-Sanchez, R.;Avalos-Borja, M.; Hirata, G. A.; Cota-Araiza, L.: Study of dif-ferent forms of carbon by analytical electron microscopy. J.Electron Spectroscopy and Related Phenomena 104 (1999) 61–66.
[42] Wern, H.: Single Crystal Elastic Constants and Calculated BulkProperties. Logos Verlag, Berlin 2004.
Reaction of rhenium and carbon at high pressures and temperatures 501




















