
Rare mutations in SLC12A1 and SLC12A3 protect against hypertension by reducing the activity of renal...
9
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited. Rare mutations in SLC12A1 and SLC12A3 protect against hypertension by reducing the activity of renal salt cotransporters Rocı ´o Acun ˜a a , Lilia Martı ´nez-de-la-Maza a , Jose ´ Ponce-Coria a , Norma Va ´ zquez a , Pene ´ lope Ortal-Vite a , Diana Pacheco-Alvarez b , Norma A. Bobadilla a and Gerardo Gamba a Objectives Screening for variants in SLC12A1 and SLC12A3 genes, encoding the renal Na R :Cl S (NCC) and Na þ :K þ :2Cl S (NKCC2) cotransporters, respectively, in 3125 members of the Framingham Heart Study (FHS) revealed that carrying a rare mutation in one of these genes was associated with a significant reduction in blood pressure, in the risk of arterial hypertension, and of death due to cardiovascular disease. Because near 60% of the rare mutations identified have not been related to Bartter’s or Gitelman’s disease, the consequence of such mutations on cotransporter activity is unknown. Methods We used the heterologous expression system of Xenopus laevis oocytes, microinjected with wild-type or mutant NCC or NKCC2 cRNAs, to examine the effect of these inferred NCC and NKCC2 mutations on the cotransporters’ functional properties. Cotransporter activity was defined as the diuretic-sensitive radioactive tracer uptake and response to known modulators was assessed. Results Basal NCC activity was significantly reduced in all NCC mutants and, excluding NCC-S186F, response to WNK3, WNK4, or intracellular chloride depletion was conserved. Similarly, basal activity was reduced in six out of nine NKCC2 mutants and response to WNK3 was maintained. No effect on protein expression was seen, except for NCC-S186F, which was significantly reduced. Conclusions The rare NCC or NKCC2 mutations found in the FHS significantly reduced the basal activity of the cotransporters. This observation supports that even a small, but chronic reduction of NCC or NKCC2 function results in a lower blood pressure and decreased risk of hypertension in otherwise healthy individuals in the general population. J Hypertens 29:000– 000 Q 2011 Wolters Kluwer Health | Lippincott Williams & Wilkins. Journal of Hypertension 2011, 29:000–000 Keywords: blood pressure, distal convoluted tubule, genetic variation, NCC, NKCC2, thiazide diuretics, thick ascending limb Abbreviations: DCT, distal convoluted tubule; FHS, Framingham Heart Study; GWAS, genome-wide association studies; NCC, the renal Na þ :Cl S cotransporter; NKCC2, the renal specific Na þ :K þ :2Cl S cotransporter; ROMK, rat outer medulla potassium channel; SLC12A1, solute carrier cotransporter family 12 member A1; SLC12A3, solute carrier cotransporter family 12 member A3; TAL, thick ascending limb of Henle’s loop; WNK3, with no lysine kinase 3; WNK4, with no lysine kinase 4 a Molecular Physiology Unit, Instituto Nacional de Cardiologı ´a Ignacio Cha ´ vez, Instituto Nacional de Ciencias Me ´ dicas y Nutricio ´ n Salvador Zubira ´ n, and Instituto de Investigaciones Biome ´ dicas, Universidad Nacional Auto ´ noma de Me ´ xico Tlalpan and b Escuela de Medicina, Universidad Panamericana, Mexico City, Mexico Correspondence to Gerardo Gamba, MD, PhD, Molecular Physiology Unit, Vasco de Quiroga No. 15, Tlalpan 14000, Mexico City, Mexico Tel: +5255 5513 3868; fax: +5255 5655 0382; e-mail: [email protected] or [email protected] Received 22 July 2010 Revised 20 September 2010 Accepted 21 October 2010 Introduction Sodium reabsorption by renal transport mechanisms has a critical role in long-term blood pressure control, which is illustrated by rare monogenic syndromes altering salt handling in the kidney and considerably affecting blood pressure regulation [1,2]. The bumetanide-sensitive Na þ :K þ :2Cl cotransporter (NKCC2) in the apical mem- brane of the thick ascending limb of Henle’s loop (TAL) and the thiazide-sensitive Na þ :Cl cotransporter (NCC) in the distal convoluted tubule (DCT) are two major salt reabsorption pathways in the nephron, accounting for the reabsorption of 35% of the salt filtered at the glomerulus. The activity of both cotransporters has an effect on final urinary salt excretion, consequently influen- cing long-term blood pressure levels. Loss-of-function mutations in the NKCC2 gene, SLC12A1, or in the NCC gene, SLC12A3, cause type I Bartter’s syndrome and Gitelman’s syndrome, respectively, with both diseases featuring arterial hypotension along with electrolyte abnormalities. The renal salt wasting and hypotension accompanying these two rare autosomic recessive diseases represent a lower extreme in the spectrum of salt reabsorption and blood pressure, and the study of these blatant phenotypes has provided critical insight into the role of salt transport mechanisms in blood pressure maintenance. The more subtle phenotype associated with the heterozygous state of either of these Mendelian diseases has been less examined, although carriers of Gitelman’s syndrome Original article 1 0263-6352 ß 2011 Wolters Kluwer Health | Lippincott Williams & Wilkins DOI:10.1097/HJH.0b013e328341d0fd
Transcript of Rare mutations in SLC12A1 and SLC12A3 protect against hypertension by reducing the activity of renal...

C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
Original article 1
Rare mutations in SLC12A1 an
d SLC12A3 protect againsthypertension by reducing the activity of renalsalt cotransportersRocıo Acunaa, Lilia Martınez-de-la-Mazaa, Jose Ponce-Coriaa, Norma Vazqueza,Penelope Ortal-Vitea, Diana Pacheco-Alvarezb, Norma A. Bobadillaa andGerardo GambaaObjectives Screening for variants in SLC12A1 and
SLC12A3 genes, encoding the renal NaR:ClS (NCC) and
Naþ:Kþ:2ClS (NKCC2) cotransporters, respectively, in 3125
members of the Framingham Heart Study (FHS) revealed
that carrying a rare mutation in one of these genes was
associated with a significant reduction in blood pressure, in
the risk of arterial hypertension, and of death due to
cardiovascular disease. Because near 60% of the rare
mutations identified have not been related to Bartter’s or
Gitelman’s disease, the consequence of such mutations on
cotransporter activity is unknown.
Methods We used the heterologous expression system of
Xenopus laevis oocytes, microinjected with wild-type or
mutant NCC or NKCC2 cRNAs, to examine the effect of
these inferred NCC and NKCC2 mutations on the
cotransporters’ functional properties. Cotransporter activity
was defined as the diuretic-sensitive radioactive tracer
uptake and response to known modulators was assessed.
Results Basal NCC activity was significantly reduced in all
NCC mutants and, excluding NCC-S186F, response to
WNK3, WNK4, or intracellular chloride depletion was
conserved. Similarly, basal activity was reduced in six out of
nine NKCC2 mutants and response to WNK3 was
maintained. No effect on protein expression was seen,
except for NCC-S186F, which was significantly reduced.
Conclusions The rare NCC or NKCC2 mutations
found in the FHS significantly reduced the basal
opyright © Lippincott Williams & Wilkins. Unauth
0263-6352 � 2011 Wolters Kluwer Health | Lippincott Williams & Wilkins
activity of the cotransporters. This observation supports
that even a small, but chronic reduction of NCC or
NKCC2 function results in a lower blood pressure and
decreased risk of hypertension in otherwise healthy
individuals in the general population. J Hypertens 29:000–
000 Q 2011 Wolters Kluwer Health | Lippincott Williams &
Wilkins.
Journal of Hypertension 2011, 29:000–000
Keywords: blood pressure, distal convoluted tubule, genetic variation, NCC,NKCC2, thiazide diuretics, thick ascending limb
Abbreviations: DCT, distal convoluted tubule; FHS, Framingham HeartStudy; GWAS, genome-wide association studies; NCC, the renal Naþ:ClS
cotransporter; NKCC2, the renal specific Naþ:Kþ:2ClS cotransporter; ROMK,rat outer medulla potassium channel; SLC12A1, solute carrier cotransporterfamily 12 member A1; SLC12A3, solute carrier cotransporter family 12member A3; TAL, thick ascending limb of Henle’s loop; WNK3, with no lysinekinase 3; WNK4, with no lysine kinase 4
aMolecular Physiology Unit, Instituto Nacional de Cardiologıa Ignacio Chavez,Instituto Nacional de Ciencias Medicas y Nutricion Salvador Zubiran, and Institutode Investigaciones Biomedicas, Universidad Nacional Autonoma de MexicoTlalpan and bEscuela de Medicina, Universidad Panamericana, Mexico City,Mexico
Correspondence to Gerardo Gamba, MD, PhD, Molecular Physiology Unit, Vascode Quiroga No. 15, Tlalpan 14000, Mexico City, MexicoTel: +5255 5513 3868; fax: +5255 5655 0382;e-mail: [email protected] or [email protected]
Received 22 July 2010 Revised 20 September 2010Accepted 21 October 2010
IntroductionSodium reabsorption by renal transport mechanisms has a
critical role in long-term blood pressure control, which is
illustrated by rare monogenic syndromes altering salt
handling in the kidney and considerably affecting blood
pressure regulation [1,2]. The bumetanide-sensitive
Naþ:Kþ:2Cl� cotransporter (NKCC2) in the apical mem-
brane of the thick ascending limb of Henle’s loop (TAL)
and the thiazide-sensitive Naþ:Cl� cotransporter (NCC)
in the distal convoluted tubule (DCT) are two major
salt reabsorption pathways in the nephron, accounting
for the reabsorption of 35% of the salt filtered at the
glomerulus. The activity of both cotransporters has an
effect on final urinary salt excretion, consequently influen-
cing long-term blood pressure levels. Loss-of-function
mutations in the NKCC2 gene, SLC12A1, or in the
NCC gene, SLC12A3, cause type I Bartter’s syndrome
and Gitelman’s syndrome, respectively, with both diseases
featuring arterial hypotension along with electrolyte
abnormalities.
The renal salt wasting and hypotension accompanying
these two rare autosomic recessive diseases represent a
lower extreme in the spectrum of salt reabsorption and
blood pressure, and the study of these blatant phenotypes
has provided critical insight into the role of salt transport
mechanisms in blood pressure maintenance. The more
subtle phenotype associated with the heterozygous state
of either of these Mendelian diseases has been less
examined, although carriers of Gitelman’s syndrome
orized reproduction of this article is prohibited.
DOI:10.1097/HJH.0b013e328341d0fd

C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
2 Journal of Hypertension 2011, Vol 00 No 00
mutations have been reported to display lower blood
pressure than controls [3,4]. Recently, Ji et al. [5] studied
the effect of heterozygosity for rare independent
mutations in salt-handling proteins on blood pressure.
This group developed criteria to identify NCC, NKCC2
and ROMK (a potassium channel involved in renal
sodium reabsorption) rare variants under purifying selec-
tion, by searching for low-frequency mutations in amino
acids under complete phylogenetic conservation, which
were then corroborated by protein prediction programs.
By applying these criteria to coding DNA sequence
variants in 3125 participants of the Framingham Heart
Study (FHS), 49 heterozygous carriers (representing
1.5% of the studied population) of 30 different NCC,
NKCC2, or ROMK mutations were detected. As a group,
carriers were found to have lower blood pressure than the
general population and their noncarrier siblings, as well as
a decreased risk of developing hypertension and
mortality from cardiovascular disease.
Out of 30 variants described, 18 (five in NCC and nine in
NKCC2) had not been previously reported as disease-
causing mutations and, hence, are inferred to reduce
protein activity by affecting the cotransporters’ primary
structure. Carriers for these variants had a similar effect on
blood pressure and hypertension prevalence as carriers
harboring biochemically proven loss-of-function muta-
tions in the same study [5], suggesting, indeed, decreased
activity in cotransporters with inferred mutations. None-
theless, because the effect of these variants on the cotran-
sporters’ functional properties was not analyzed, experi-
mental testing is necessary to demonstrate that these
inferred mutations impact cotransporter activity [6]. If
so, this may be evidence that, in the general population,
slightly decreased renal salt reabsorption influences blood
pressure regulation. Additionally, the functional and bio-
chemical corroboration of the criteria used by Ji et al. [5] to
detect rare independent mutations in the general popu-
lation would support the use of this methodology in
genomic studies of polygenic diseases [7].
MethodsSite-directed mutagenesisMutations describedby Ji et al. were reproduced in ratNCC
and rat NKCC2 by site-directed mutagenesis (Quik-
Change; Stratagene) on corresponding amino acids, using
previously described rNCC/pSPORT1 and rNKCC2/
pSPORT1 as a template [8]. All primers were custom made
(Sigma) and all mutations were corroborated by automatic
DNA sequencing. To avoid undesired mutations, a fully
sequenced restriction fragment harboring the wanted
mutation was subcloned into wild-type NCC or NKCC2.
In-vitro rNCC and rNKCC2 cRNA translationTo prepare cRNA, all rNCC and rNKCC2 clones were
digested at the 30-end using Not I and Xba I (Invitrogen),
respectively. cRNA was transcribed in vitro using a T7
opyright © Lippincott Williams & Wilkins. Unautho
RNA polymerase mMESSAGE kit (Ambion). Transcrip-
tion product integrity was confirmed on agarose gels, and
concentration was determined by absorbance reading at
260 nm (DU 640; Beckman, Fullerton, California, USA)
and by densitometric analysis of the corresponding band
in ethidum bromide-stained agarose gels. cRNA was
stored frozen in aliquots at �808C until use.
Xenopus oocytes expression systemIn the present study, we used the heterologous expression
system in Xenopus laevis oocytes to assess the effect of rare
mutations in NCC or NKCC2 on the activity and some
functional properties of the cotransporters. This expres-
sion system has been shown to be an excellent tool for a
robust and reproducible expression of NCC and NKCC2
in our hands [8–18] and in other laboratories [19–24]. In
contrast, both NCC and NKCC2 functional expression in
transfected mammalian cells has been unsuccessful in
many laboratories and the few reports obtained [25,26],
exhibit a low level of activity over basal uptake and thus,
are not appropriate for detection of subtle differences
between wild-type and mutant cotransporter activity.
Xenopus laevis frog oocytes were surgically harvested from
anesthetized adult females under 0.17% tricaine and
incubated for 1 h under vigorous shaking in calcium-free
frog Ringer-ND96 (mmol/l: 96 NaCl, 2 KCl, 1 MgCl, and
5 HEPES/Tris, pH 7.4), supplemented with collagenase
B (2 mg/ml). The oocytes were manually defolliculated
and incubated overnight in ND96 at 188C containing
5 mg/100 ml of gentamicin. The following day, mature
oocytes were injected with 50 nl of water with or without
cRNA from wild-type or mutant rNCC at a concentration
of 0.5 mg/ml (i.e. 25 ng cRNA/oocyte) or rNKCC2 at
0.25 mg/ml (i.e. 12.5 ng cRNA/ooctye). After injection,
oocytes were incubated for 3 days at 188C or, when
specified, for 2 days at 298C in ND96 with gentamicin.
FunctionalanalysisoftheNaþ:Cl�cotransporterwasdeter-
mined by assessing tracer 22Naþ uptake (New England
Nuclear) in groups of at least 10 oocytes. 22Naþ uptake was
measured using our usual protocol: a 30-min incubation
period in an isotonic Kþ and Cl�-free medium (mmol/l: 96
Naþ gluconate, 6.0 Ca2þ gluconate, 1.0 Mg2þ gluconate,
5 HEPES/Tris, pH 7.4) with 1 mmol/l ouabain, 100 mmol/l
bumetanide, and 100 mmol/l amiloride, followed by a 60-
min uptake in a Kþ-free isotonic medium. The isotonic
medium contained (in mmol/l) 40 NaCl, 56 N-methyl-D-
glucamine(NMDG)-Cl,1.8CaCl2,1MgCl2,5HEPES,pH
7.4,and1 mmol/louabainaswellas100 mmol/lbumetanide,
100 mmol/l amiloride,and2.5 mCi 22Naþ. Todetermine the
thiazide-sensitive portionof theobserved uptake, a parallel
group of oocytes for each experimental group was exposed
to the same uptake protocol in the presence of 100 mmol/l
metolazone in both incubation periods.
Functional analysis of the Naþ:Kþ:2Cl� cotransporter
was measured by assessing 86Rbþ uptake in groups of
rized reproduction of this article is prohibited.

C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
Characterization of rare NCC and NKCC2 mutants Acuna et al. 3
at least 10 oocytes according to our usual protocol for
NKCC2: a 30-min incubation period in the same isotonic
Kþ and Cl� -free medium described previously, followed
by a 60-min uptake in an isotonic medium containing
Naþ, Kþ and Cl� (mmol/l: 90 NaCl, 10 KCl, 1.8 CaCl2, 1
MgCl, and 5 HEPES/Tris, pH 7.4 and 1 mmol/l ouabain).
As for NCC, to determine the bumetanide-sensitive
portion of the observed uptake, for every experimental
group a corresponding group of oocytes was exposed to
the uptake protocol in the presence of 100 mmol/l bume-
tanide in both periods of incubation. Because X. laevisoocytes express an endogenous NKCC1-type Naþ-Kþ-
2Cl– cotransporter, every experiment included control
water-injected oocytes with and without bumetanide.
All uptakes were performed at 328C. At the end of the
uptake period, oocytes were washed five times in ice-cold
uptake solution without the isotope to remove extracellu-
lar fluid tracer. After the oocytes were dissolved in 10%
SDS, tracer activity was determined for each oocyte by
b-scintillation counting.
Western blottingWestern blotting was used to compare wild-type and
mutant rNCC and rNKCC2 total protein in cRNA-
injected oocytes. In brief, groups of 15 oocytes injected
with water or cRNA were homogenized in 5 ml/oocyte of
homogenization buffer, centrifuged twice at 100g for
10 min at 48C, and the supernatant was recollected. Oocyte
protein (three oocytes/lane) was heated in sample buffer
containing 6% SDS, 15% glycerol, 0.3% bromophenol
blue, 150 mmol/l Tris, pH 7.6, and 2% b-mercaptoethanol
and resolved by SDS-PAGE (7.5%). The proteins were
transferred to a polyvinylene difluoride membrane (Amer-
sham Pharmacia Biotech) at 10 V for 60 min in a buffer
containing 25 mmol/l Tris, 190 mmol/l glycine, 0.1% SDS,
and 20% (vol/vol) methanol. Prestained Rainbow markers
(Amersham) were used as molecular mass standards.
SNAP i.d. protein detection system (Millipore) was used
for blocking, antibody addition and membrane washing.
Bands were detected by using the Immun-Star Chemilu-
minescent Protein Detection System (Bio-Rad).
Statistical analysisStatistical significance is defined as two-tailed P< 0.05,
and the results are presented as means �SE of normal-
ized data, taking wild-type NCC or NKCC2 basal activity
as 100%. The significance of the differences between
groups was tested by one-way ANOVA with Dunn’s
correction for multiple comparisons.
The study was approved by an institutional review com-
mittee.
ResultsThe activity of NCC mutants is reducedJi et al. [5] described 15 independent NCC mutations
associated with lower hypertension prevalence in the
opyright © Lippincott Williams & Wilkins. Unauth
FHS cohort, only five of which had not been previously
studied as biochemical loss-of-function or as Gitelman’s
disease-causing mutations. The putative localization of
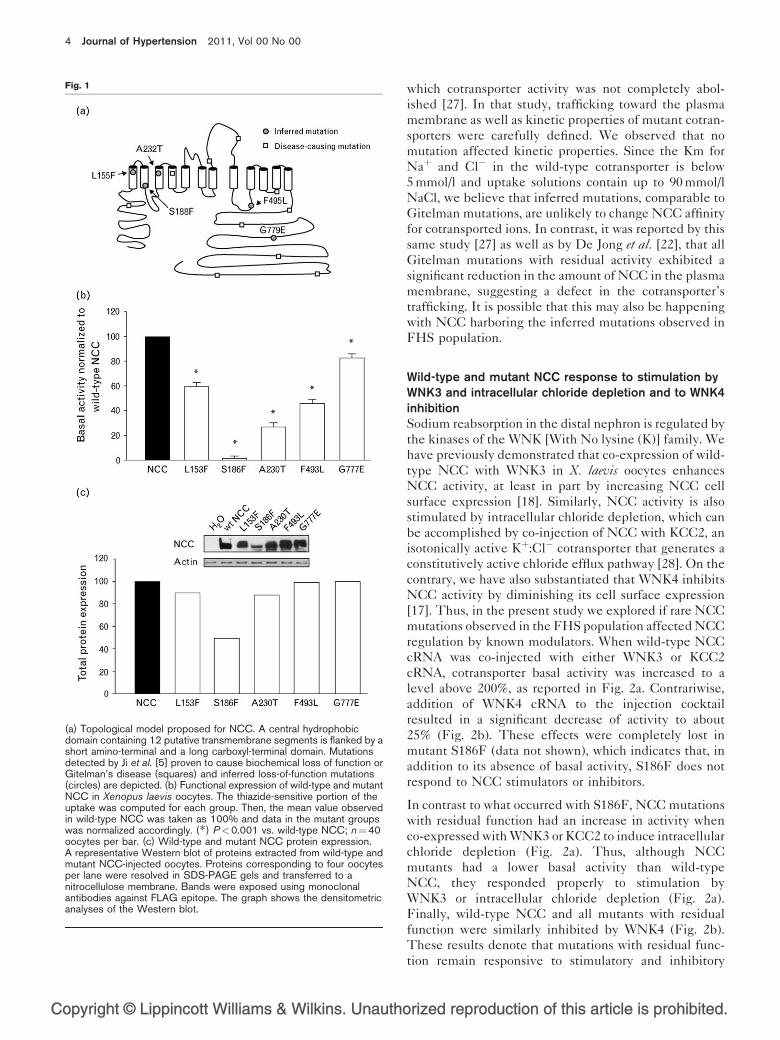
these mutations is depicted in the topological model of
NCC in Fig. 1a. Because of the high degree of identity at
the protein level between human and rat NCC (near to
90%), with all mutations involving conserved residues,
along with our previous experience studying mutations
causing Gitelman’s syndrome in rat NCC [27], we gener-
ated and characterized these independent mutations in
rat NCC.
A representative experiment of functional expression in
X. laevis oocytes injected with wild-type or mutant NCC
cRNA is presented as the uptake of 22Naþ in the absence
or presence of metolazone in Supplementary Figure S1,
http://links.lww.com/HJH/A58. Several experiments
were performed and NCC function is reported as meto-
lazone-sensitive 22Naþ uptake, taking wild-type NCC
uptake as 100% (Fig. 1b). Only NCC mutation S186F
exhibited no significant activity, whereas the four remain-
ing mutants (L153F, A230T, F493L and G777E) had a
lower, albeit significant function, although it varied
among the different studied mutants. As shown in
Fig. 1b, the basal activity of NCC mutations is: G777E
(78%) > L153F (59%) > F493L (46%) > A230T (26%)
> S186F (0%). To further support our results, we repro-
duced mutation G779E, located in a less conserved
region of the NCC C-terminal domain, both in rat and
in human NCC with a similar outcome (Supplemental
Figure S2, http://links.lww.com/HJH/A59).
Wild-type and mutant NCC protein expression is similarThe monomeric form of wild-type and mutant NCC was
detected at the expected weight of 110 kDa in the
immunoblot. A representative western blot with total
protein expression measured by densitometry and nor-
malized to wild-type NCC protein is depicted in Fig. 1c.
Total protein expression does not vary substantially
between wild-type NCC and L153F, A230T, F493L,
and G777E mutants, suggesting that decreased protein
expression is not the source of these mutants’ lower
function. In contrast, mutation S186F resulted in a sig-
nificantly lower protein expression when compared to
wild-type NCC. Additionally, wild-type NCC and
mutants L153F, A230T, F493L and G777E show the
characteristic diffuse band visible above the better-
defined protein monomer band at 110 kDa in the immu-
noblot, corresponding to the glycosylated form of the
cotransporter. In this regard, we have previously ascer-
tained that N-glycosylation of NCC at residues N404 and
N424 is necessary for proper folding and protein traffick-
ing to the plasma membrane [16,27]. This glycosylation
band is absent in S186F NCC mutant, implying that the
lack of glycosylation in this variant would prevent its
transport to plasma membrane. In this regard, we have
previously studied NCC disease-causing mutations in
orized reproduction of this article is prohibited.
Copyright © Lippincott Williams & Wilkins. Unautho
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
4 Journal of Hypertension 2011, Vol 00 No 00
Fig. 1
(a) Topological model proposed for NCC. A central hydrophobicdomain containing 12 putative transmembrane segments is flanked by ashort amino-terminal and a long carboxyl-terminal domain. Mutationsdetected by Ji et al. [5] proven to cause biochemical loss of function orGitelman’s disease (squares) and inferred loss-of-function mutations(circles) are depicted. (b) Functional expression of wild-type and mutantNCC in Xenopus laevis oocytes. The thiazide-sensitive portion of theuptake was computed for each group. Then, the mean value observedin wild-type NCC was taken as 100% and data in the mutant groupswas normalized accordingly. (�) P<0.001 vs. wild-type NCC; n¼40oocytes per bar. (c) Wild-type and mutant NCC protein expression.A representative Western blot of proteins extracted from wild-type andmutant NCC-injected oocytes. Proteins corresponding to four oocytesper lane were resolved in SDS-PAGE gels and transferred to anitrocellulose membrane. Bands were exposed using monoclonalantibodies against FLAG epitope. The graph shows the densitometricanalyses of the Western blot.
which cotransporter activity was not completely abol-
ished [27]. In that study, trafficking toward the plasma
membrane as well as kinetic properties of mutant cotran-
sporters were carefully defined. We observed that no
mutation affected kinetic properties. Since the Km for
Naþ and Cl� in the wild-type cotransporter is below
5 mmol/l and uptake solutions contain up to 90 mmol/l
NaCl, we believe that inferred mutations, comparable to
Gitelman mutations, are unlikely to change NCC affinity
for cotransported ions. In contrast, it was reported by this
same study [27] as well as by De Jong et al. [22], that all
Gitelman mutations with residual activity exhibited a
significant reduction in the amount of NCC in the plasma
membrane, suggesting a defect in the cotransporter’s
trafficking. It is possible that this may also be happening
with NCC harboring the inferred mutations observed in
FHS population.
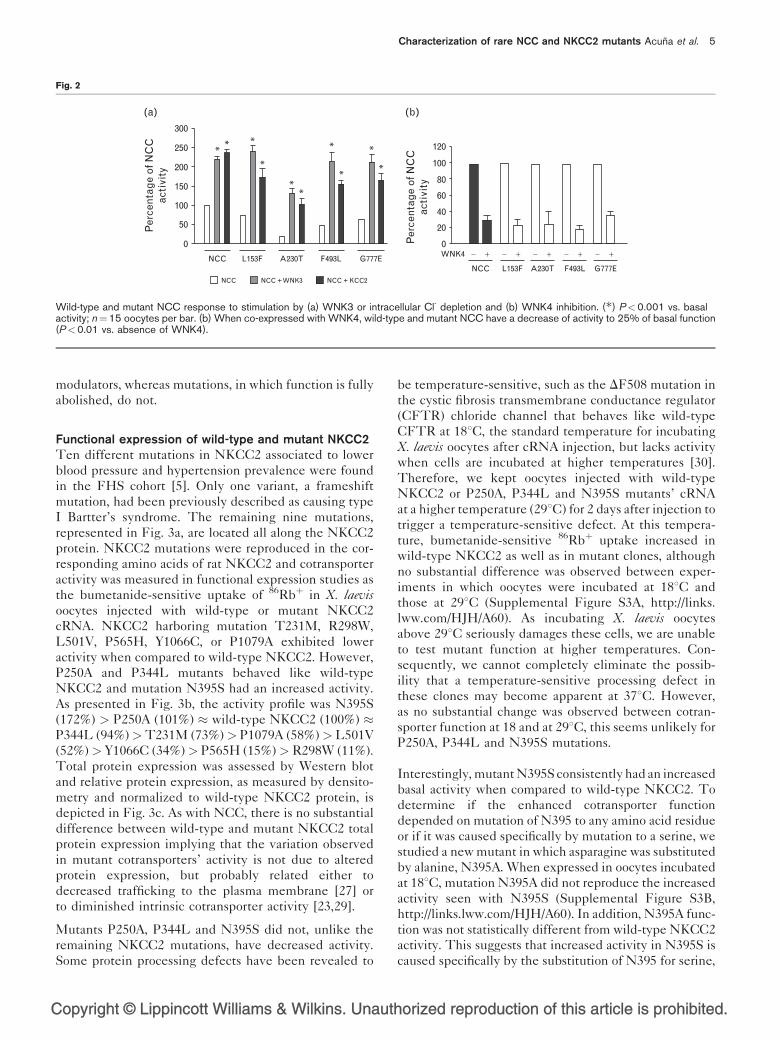
Wild-type and mutant NCC response to stimulation byWNK3 and intracellular chloride depletion and to WNK4inhibitionSodium reabsorption in the distal nephron is regulated by
the kinases of the WNK [With No lysine (K)] family. We
have previously demonstrated that co-expression of wild-
type NCC with WNK3 in X. laevis oocytes enhances
NCC activity, at least in part by increasing NCC cell
surface expression [18]. Similarly, NCC activity is also
stimulated by intracellular chloride depletion, which can
be accomplished by co-injection of NCC with KCC2, an
isotonically active Kþ:Cl� cotransporter that generates a
constitutively active chloride efflux pathway [28]. On the
contrary, we have also substantiated that WNK4 inhibits
NCC activity by diminishing its cell surface expression
[17]. Thus, in the present study we explored if rare NCC
mutations observed in the FHS population affected NCC
regulation by known modulators. When wild-type NCC
cRNA was co-injected with either WNK3 or KCC2
cRNA, cotransporter basal activity was increased to a
level above 200%, as reported in Fig. 2a. Contrariwise,
addition of WNK4 cRNA to the injection cocktail
resulted in a significant decrease of activity to about
25% (Fig. 2b). These effects were completely lost in
mutant S186F (data not shown), which indicates that, in
addition to its absence of basal activity, S186F does not
respond to NCC stimulators or inhibitors.
In contrast to what occurred with S186F, NCC mutations
with residual function had an increase in activity when
co-expressed with WNK3 or KCC2 to induce intracellular
chloride depletion (Fig. 2a). Thus, although NCC
mutants had a lower basal activity than wild-type
NCC, they responded properly to stimulation by
WNK3 or intracellular chloride depletion (Fig. 2a).
Finally, wild-type NCC and all mutants with residual
function were similarly inhibited by WNK4 (Fig. 2b).
These results denote that mutations with residual func-
tion remain responsive to stimulatory and inhibitory
rized reproduction of this article is prohibited.
C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
Characterization of rare NCC and NKCC2 mutants Acuna et al. 5
Fig. 2
300
(a) (b)
Per
cent
age
of N
CC
acti
vity
Per
cent
age
of N
CC
acti
vity
120
100
80
60
40
20
0
250
200
150
100
50
0
NCC
NCC NCC + WNK3 NCC + KCC2
L153F A230T F493L G777ENCC L153F A230T F493L G777E
WNK4 − + − + − + − + − +
* * *
*
**
*
* *
*
Wild-type and mutant NCC response to stimulation by (a) WNK3 or intracellular Cl- depletion and (b) WNK4 inhibition. (�) P<0.001 vs. basalactivity; n¼15 oocytes per bar. (b) When co-expressed with WNK4, wild-type and mutant NCC have a decrease of activity to 25% of basal function(P<0.01 vs. absence of WNK4).
modulators, whereas mutations, in which function is fully
abolished, do not.
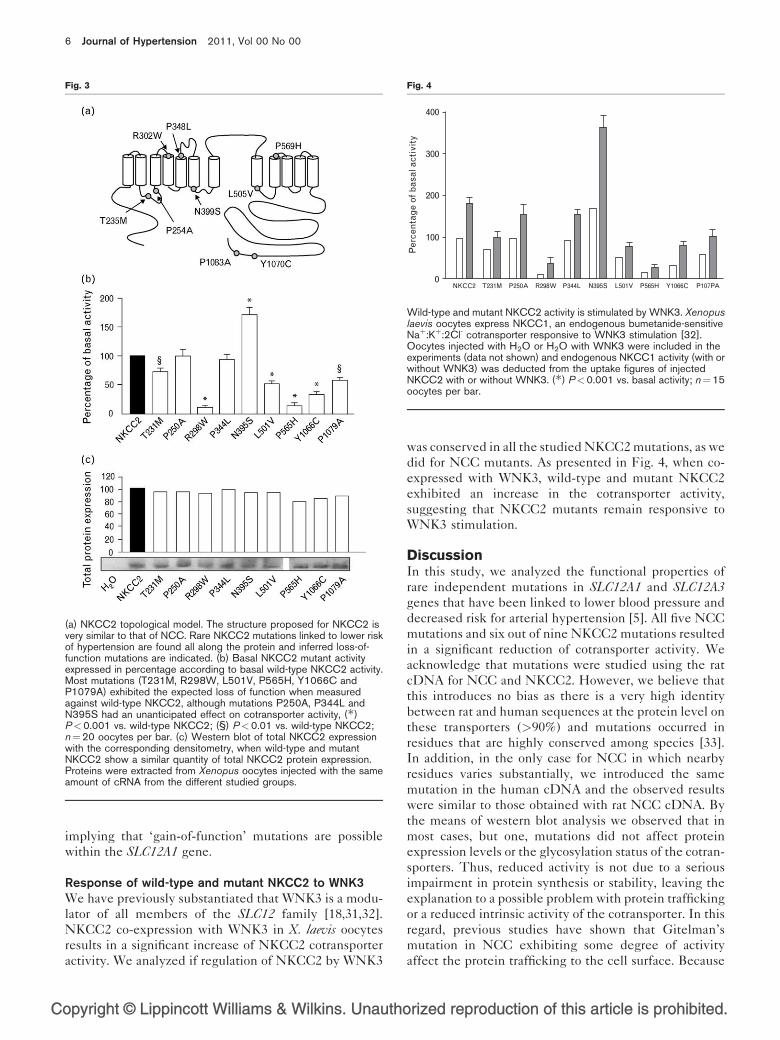
Functional expression of wild-type and mutant NKCC2Ten different mutations in NKCC2 associated to lower
blood pressure and hypertension prevalence were found
in the FHS cohort [5]. Only one variant, a frameshift
mutation, had been previously described as causing type
I Bartter’s syndrome. The remaining nine mutations,
represented in Fig. 3a, are located all along the NKCC2
protein. NKCC2 mutations were reproduced in the cor-
responding amino acids of rat NKCC2 and cotransporter
activity was measured in functional expression studies as
the bumetanide-sensitive uptake of 86Rbþ in X. laevisoocytes injected with wild-type or mutant NKCC2
cRNA. NKCC2 harboring mutation T231M, R298W,
L501V, P565H, Y1066C, or P1079A exhibited lower
activity when compared to wild-type NKCC2. However,
P250A and P344L mutants behaved like wild-type
NKCC2 and mutation N395S had an increased activity.
As presented in Fig. 3b, the activity profile was N395S
(172%) > P250A (101%) � wild-type NKCC2 (100%) �P344L (94%)>T231M (73%)> P1079A (58%)>L501V
(52%)> Y1066C (34%)> P565H (15%)>R298W (11%).
Total protein expression was assessed by Western blot
and relative protein expression, as measured by densito-
metry and normalized to wild-type NKCC2 protein, is
depicted in Fig. 3c. As with NCC, there is no substantial
difference between wild-type and mutant NKCC2 total
protein expression implying that the variation observed
in mutant cotransporters’ activity is not due to altered
protein expression, but probably related either to
decreased trafficking to the plasma membrane [27] or
to diminished intrinsic cotransporter activity [23,29].
Mutants P250A, P344L and N395S did not, unlike the
remaining NKCC2 mutations, have decreased activity.
Some protein processing defects have been revealed to
opyright © Lippincott Williams & Wilkins. Unauth
be temperature-sensitive, such as the DF508 mutation in
the cystic fibrosis transmembrane conductance regulator
(CFTR) chloride channel that behaves like wild-type
CFTR at 188C, the standard temperature for incubating
X. laevis oocytes after cRNA injection, but lacks activity
when cells are incubated at higher temperatures [30].
Therefore, we kept oocytes injected with wild-type
NKCC2 or P250A, P344L and N395S mutants’ cRNA
at a higher temperature (298C) for 2 days after injection to
trigger a temperature-sensitive defect. At this tempera-
ture, bumetanide-sensitive 86Rbþ uptake increased in
wild-type NKCC2 as well as in mutant clones, although
no substantial difference was observed between exper-
iments in which oocytes were incubated at 188C and
those at 298C (Supplemental Figure S3A, http://links.
lww.com/HJH/A60). As incubating X. laevis oocytes
above 298C seriously damages these cells, we are unable
to test mutant function at higher temperatures. Con-
sequently, we cannot completely eliminate the possib-
ility that a temperature-sensitive processing defect in
these clones may become apparent at 378C. However,
as no substantial change was observed between cotran-
sporter function at 18 and at 298C, this seems unlikely for
P250A, P344L and N395S mutations.
Interestingly, mutant N395S consistently had an increased
basal activity when compared to wild-type NKCC2. To
determine if the enhanced cotransporter function
depended on mutation of N395 to any amino acid residue
or if it was caused specifically by mutation to a serine, we
studied a new mutant in which asparagine was substituted
by alanine, N395A. When expressed in oocytes incubated
at 188C, mutation N395A did not reproduce the increased
activity seen with N395S (Supplemental Figure S3B,
http://links.lww.com/HJH/A60). In addition, N395A func-
tion was not statistically different from wild-type NKCC2
activity. This suggests that increased activity in N395S is
caused specifically by the substitution of N395 for serine,
orized reproduction of this article is prohibited.
C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
6 Journal of Hypertension 2011, Vol 00 No 00
Fig. 3
(a) NKCC2 topological model. The structure proposed for NKCC2 isvery similar to that of NCC. Rare NKCC2 mutations linked to lower riskof hypertension are found all along the protein and inferred loss-of-function mutations are indicated. (b) Basal NKCC2 mutant activityexpressed in percentage according to basal wild-type NKCC2 activity.Most mutations (T231M, R298W, L501V, P565H, Y1066C andP1079A) exhibited the expected loss of function when measuredagainst wild-type NKCC2, although mutations P250A, P344L andN395S had an unanticipated effect on cotransporter activity, (�)P<0.001 vs. wild-type NKCC2; (§) P<0.01 vs. wild-type NKCC2;n¼20 oocytes per bar. (c) Western blot of total NKCC2 expressionwith the corresponding densitometry, when wild-type and mutantNKCC2 show a similar quantity of total NKCC2 protein expression.Proteins were extracted from Xenopus oocytes injected with the sameamount of cRNA from the different studied groups.
Fig. 4
400
300
200
100
0NKCC2 T231M P250A R298W P344L N395S L501V P565H Y1066C P107PA
Per
cent
age
of b
asal
act
ivit
y
Wild-type and mutant NKCC2 activity is stimulated by WNK3. Xenopuslaevis oocytes express NKCC1, an endogenous bumetanide-sensitiveNaþ:Kþ:2Cl- cotransporter responsive to WNK3 stimulation [32].Oocytes injected with H2O or H2O with WNK3 were included in theexperiments (data not shown) and endogenous NKCC1 activity (with orwithout WNK3) was deducted from the uptake figures of injectedNKCC2 with or without WNK3. (�) P<0.001 vs. basal activity; n¼15oocytes per bar.
implying that ‘gain-of-function’ mutations are possible
within the SLC12A1 gene.
Response of wild-type and mutant NKCC2 to WNK3We have previously substantiated that WNK3 is a modu-
lator of all members of the SLC12 family [18,31,32].
NKCC2 co-expression with WNK3 in X. laevis oocytes
results in a significant increase of NKCC2 cotransporter
activity. We analyzed if regulation of NKCC2 by WNK3
opyright © Lippincott Williams & Wilkins. Unautho
was conserved in all the studied NKCC2 mutations, as we
did for NCC mutants. As presented in Fig. 4, when co-
expressed with WNK3, wild-type and mutant NKCC2
exhibited an increase in the cotransporter activity,
suggesting that NKCC2 mutants remain responsive to
WNK3 stimulation.
DiscussionIn this study, we analyzed the functional properties of
rare independent mutations in SLC12A1 and SLC12A3genes that have been linked to lower blood pressure and
decreased risk for arterial hypertension [5]. All five NCC
mutations and six out of nine NKCC2 mutations resulted
in a significant reduction of cotransporter activity. We
acknowledge that mutations were studied using the rat
cDNA for NCC and NKCC2. However, we believe that
this introduces no bias as there is a very high identity
between rat and human sequences at the protein level on
these transporters (>90%) and mutations occurred in
residues that are highly conserved among species [33].
In addition, in the only case for NCC in which nearby
residues varies substantially, we introduced the same
mutation in the human cDNA and the observed results
were similar to those obtained with rat NCC cDNA. By
the means of western blot analysis we observed that in
most cases, but one, mutations did not affect protein
expression levels or the glycosylation status of the cotran-
sporters. Thus, reduced activity is not due to a serious
impairment in protein synthesis or stability, leaving the
explanation to a possible problem with protein trafficking
or a reduced intrinsic activity of the cotransporter. In this
regard, previous studies have shown that Gitelman’s
mutation in NCC exhibiting some degree of activity
affect the protein trafficking to the cell surface. Because
rized reproduction of this article is prohibited.

C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
Characterization of rare NCC and NKCC2 mutants Acuna et al. 7
the reduction of activity in mutants analyzed in this study
is not as dramatic as in Gitelman’s or Bartter’s mutations,
the analysis of cell surface expression of the cotransporter
would require fine techniques that are beyond the scope
of this study.
Our results support the proposal that even a small, but
chronic reduction of NCC or NKCC2 function entails a
higher urinary salt excretion, resulting in a lower blood
pressure and decreased risk of hypertension in otherwise
healthy individuals in the general population. Thus, our
study complements the observations in the FHS popu-
lation by providing a mechanism to explain the reduced
blood pressure and/or protection against hypertension in
otherwise healthy individuals harboring the inferred
mutations. In addition, there has been a debate for years
about the mechanisms by which therapy with loop or
thiazide diuretics decreases blood pressure [34]. Our
observations suggest that the low blood pressure
observed in FHS participants is due to a reduced activity
of the transporters that are the target for these diuretics,
supporting the proposal that it is the chronic natriuretic
effect of these drugs what causes the decrease in blood
pressure in treated hypertensive patients.
Three NKCC2 mutations did not exhibit loss of function
when compared to wild-type NKCC2. The finding that
NKCC2-N395S is a mutation with increased, instead of
lower, activity may be due to the strategy used by Ji et al.[5] since their methodology sought for mutations in which
amino acid substitutions would affect protein function,
without distinguishing its direction (i.e. between loss and
gain-of-function mutations). Although most mutations
are likely to be disruptive, some may enhance protein
function, which could be the case for NKCC2-N395S.
Supporting this conclusion is our observation that sub-
stituting N395 for another residue did not increase
NKCC2 activity. Blood pressure in carriers of mutation
NKCC2-N395S is slightly lower than expected, which we
believe could be due to chance or to other factors not
taken into account. When patients were analyzed as a
group, the impact of all mutations on blood pressure
levels and protection against hypertension was striking.
However, as only two out of 37 patients in which
mutations in SLC12A1 or SLC12A3 were detected, har-
bored this mutation, the effect of these individuals on the
data of the entire carrier group may be minimal.
The effect of residue substitution in NKCC2-P250A and
NKCC2-P344L may not be visible in X. laevis oocytes
due either to temperature-dependent processing defects
[30], to the lack of the physiological context of the TAL
(for instance, absence of apical-basal polarity in oocytes)
or it may be a false-positive. On one hand, unfortunately,
all attempts until now to express functional NCC or
NKCC2 in polarized epithelia have been unsuccessful.
Consequently, it is still possible that these two mutants
exhibit decreased function in the TAL cells. On the other
opyright © Lippincott Williams & Wilkins. Unauth
hand, the criteria used in the FHS analysis [5] anticipate a
false-positive rate of 10%, which may vindicate NKCC2-
P250A and NKCC2-P344L as tolerated mutations
with no effect on cotransporter function. Remarkably,
five out of five inferred mutations in NCC and six out
nine inferred mutations in NKCC2 indeed exhibited
decreased functional activity. Therefore, even if
NKCC2-P250A and NKCC2-P344L mutants were fully
functional in the TAL, we believe that our results corro-
borate the methodology and criteria used by Ji et al. [5],
which was effective in detecting NCC and NKCC2
mutations with a biochemical effect that correlated with
a physical and epidemiological observation, with a poten-
tial false-positive rate of 10%.
Interestingly, there is a noteworthy difference in levels of
activity between cotransporters harboring mutations pre-
viously described as disease-causing (for NCC as well as
for NKCC2) and cotransporters carrying most of the
inferred mutations analyzed in the present study. Bart-
ter’s or Gitelman’s disease-causing mutations expressed
in X. laevis oocytes exhibit activity below 50% of wild-
type cotransporter function, most frequently between 0
and 25% [19,22,23,27]. In contrast, some of the mutations
characterized in this study display levels of activity well
over 50%. Conceivably, these NCC and NKCC2
mutations with high residual activity may not have been
described in association with Gitelman’s or Bartter’s
syndrome, as they might induce a subclinical phenotype
(such as slightly lower blood pressure or a decreased risk
of hypertension), unlikely to trigger genetic work-up, or
mild forms of disease [35]. Thus, in contrast to the study
of a genetic disease, in which the point of departure is a
symptomatic patient who prompts the discovery of a
specific mutation, the methodology proposed by Ji
et al. [5] progresses from genotype to phenotype, allowing
detection of mutations that might otherwise go unnoticed.
Despite the difference in biochemical activity between
cotransporters harboring proven and inferred mutations,
interestingly there is no disparity in blood pressure
between both groups in the FHS [5]. This is analogous
to what occurs with Gitelman’s syndrome, a highly
heterogeneous disease in which the same NCC mutation
can lead to a wide range of severity in age of onset and
clinical manifestations, including blood pressure levels.
Thus, although rare NCC or NKCC2 mutations have a
clear impact on blood pressure in the general population,
these results hint to the complexity of blood pressure
maintenance. Additional elements, such as epigenetics,
other modulatory genes or environmental factors, may
modify the severity of the phenotype produced by NCC
or NKCC2 dysfunction.
In combination with previously published studies, our
findings support the ‘rare variant, common disease’
model for arterial hypertension; NCC and NKCC2 rare
variants, not associated to disease but exhibiting loss of
orized reproduction of this article is prohibited.

C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
8 Journal of Hypertension 2011, Vol 00 No 00
function, are linked to a lower blood pressure and
decreased prevalence of arterial hypertension in the
general population. Although certain genetic variants
may be rare, it is expected that each individual may be
carrying an average number of ten or more rare variants in
his or her whole genome [36,37]. For instance, if 1.5% of
3125 studied individuals carried a rare variant in a hyper-
tension-associated gene, when only three genes were
taken into account, it is possible that a significant pro-
portion of the general population may carry a rare variant
in one or more of the multiple genes involved in blood
pressure maintenance.
The ‘rare variant, common disease’ model does not
exclude the possibility that common allelic variants also
play a significant role in the genetics of arterial hyperten-
sion. Indeed, genome-wide association studies (GWAS)
have identified common variants in the atrial and brain
natriuretic peptides with modest effects on blood pres-
sure levels and hypertension prevalence [38]. In another
example, a recent GWAS identified the gene STK39encoding SPAK kinase as a hypertension-associated gene
[39]. Interestingly, SPAK is expressed in the TAL and
DCT [40] and has a significant role in modulating the
activity of NKCC2 and NCC. Therefore, both theories
not only coexist, but may also overlap, as the effect of rare
variants could be modulated by common variants.
In conclusion, corroboration of the method and criteria
used by Ji et al.[5] to identify rare genetic variants altering
protein function and correlating with a clinical obser-
vation in the present study, supports its application for
detecting both alleles causing disease [41], and alleles
with a milder effect on disease predisposition. With
whole-exome sequencing [42], this approach may be used
to identify rare variants in multiple genes, including
genes causing Mendelian diseases with altered blood
pressure levels (such as other types of Bartter’s syndrome
or Gordon’s disease), as well as in genes or locus detected
by GWAS to be linked to changes in blood pressure (such
as ANP and BNP genes NPPA and NPPB or SPAK gene
STK39). Furthermore, this methodology may prove use-
ful in fields beyond blood pressure maintenance and
arterial hypertension, by detecting rare variants associ-
ated with disease predisposition or subclinical traits.
AcknowledgementsThe work was supported in part by the Leducq Founda-
tion Transatlantic Network on Hypertension, the
National Institutes of Health Grant DK-64635, and El
Consejo Nacional de Ciencia y Tecnologıa (CONACYT-
Mexico) Grant 59992 (to G.G.).
There are no conflicts of interest.
References1 Guyton AC. Blood pressure control: special role of the kidneys and body
fluids. Science 1991; 252:1813–1816.
2 Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of humanhypertension. Cell 2001; 104:45–556.
opyright © Lippincott Williams & Wilkins. Unautho
3 Cruz DN, Simon DB, Nelson-Williams C, Farhi A, Finberg K, Burleson L,et al. Mutations in the Na-Cl cotransporter reduce blood pressure inhumans. Hypertension 2001; 37:1458–1464.
4 Fava C, Montagnana M, Rosberg L, Burri P, Almgren P, Jonsson A, et al.Subjects heterozygous for genetic loss of function of the thiazide-sensitivecotransporter have reduced blood pressure. Hum Mol Genet 2008;17:413–418.
5 Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, Simon DB, et al. Rareindependent mutations in renal salt handling genes contribute to bloodpressure variation. Nat Genet 2008; 40:592–599.
6 Devuyst O. Salt wasting and blood pressure. Nat Genet 2008; 40:495–496.
7 Wagner CA. How much is blood pressure in the general populationdetermined by rare mutations in renal salt-transporting proteins? J Nephrol2008; 21:632–634.
8 Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger MA,Hebert SC. Molecular cloning, primary structure and characterization oftwo members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 1994;269:17713–17722.
9 Meade P, Hoover RS, Plata C, Vazquez N, Bobadilla NA, Gamba G, HebertSC. cAMP-dependent activation of the renal-specific Naþ-Kþ-2Cl-cotransporter is mediated by regulation of cotransporter trafficking. Am JPhysiol Renal Physiol 2003; 284:F1145–F1154.
10 Plata C, Meade P, Hall AE, Welch RC, Vazquez N, Hebert SC, Gamba G.Alternatively spliced isoform of the apical Na-K-Cl cotransporter geneencodes a furosemide sensitive Na-Cl cotransporter. Am J Physiol RenalPhysiol 2001; 280:F574–F582.
11 Plata C, Meade P, Vazquez N, Hebert SC, Gamba G. Functional propertiesof the apical Naþ-Kþ-2Cl- cotransporter isoforms. J Biol Chem 2002;277:11004–11012.
12 Ponce-Coria J, San Cristobal P, Kahle KT, Vazquez N, Pacheco-Alvarez D,De Los Heros P, et al. Regulation of NKCC2 by a chloride-sensingmechanism involving the WNK3 and SPAK kinases. Proc Natl Acad SciU S A 2008; 105:8458–8463.
13 Gamba G, Saltzberg SN, Lombardi M, Miyanoshita A, Lytton J, Hediger MA,et al. Primary structure and functional expression of a cDNA encoding thethiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc NatlAcad Sci USA 1993; 90:2749–2753.
14 Monroy A, Plata C, Hebert SC, Gamba G. Characterization of the thiazide-sensitive Na(þ)-Cl(-) cotransporter: a new model for ions and diureticsinteraction. Am J Physiol Renal Physiol 2000; 279:F161–F169.
15 Vazquez N, Monroy A, Dorantes E, Munoz-Clares RA, Gamba G. Functionaldifferences between flounder and rat thiazide-sensitive Na- Clcotransporter. Am J Physiol Renal Physiol 2002; 282:F599–F607.
16 Hoover RS, Poch E, Monroy A, Vazquez N, Nishio T, Gamba G, Hebert SC.N-glycosylation at two sites critically alters thiazide binding and activity ofthe rat thiazide-sensitive Na(þ):Cl(-) cotransporter. J Am Soc Nephrol2003; 14:271–282.
17 Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, et al.Molecular pathogenesis of inherited hypertension with hyperkalemia: theNa-Cl cotransporter is inhibited by wild-type but not mutant WNK4. ProcNatl Acad Sci U S A 2003; 100:680–684.
18 Rinehart J, Kahle KT, De Los HP, Vazquez N, Meade P, Wilson FH, et al.WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl-cotransporters required for normal blood pressure homeostasis. Proc NatlAcad Sci U S A 2005; 102:16777–16782.
19 Kunchaparty S, Palcso M, Berkman J, Velazquez H, Desir GV, Bernstein P,et al. Defective processing and expression of thiazide-sensitive Na-Clcotransporter as a cause of Gitelman’s syndrome. Am J Physiol 1999;277:F643–F649.
20 Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulatethiazide-sensitive Na-Cl cotransport. J Clin Invest 2003; 111:1039–1045.
21 De Jong JC, Willems PH, Mooren FJ, van den Heuvel LP, Knoers NV,Bindels RJ. The structural unit of the thiazide-sensitive NaCl cotransporteris a homodimer. J Biol Chem 2003; 278:24302–24307.
22 De Jong JC, Van Der Vliet WA, van den Heuvel LP, Willems PH, Knoers NV,Bindels RJ. Functional expression of mutations in the human NaClcotransporter: evidence for impaired routing mechanisms in Gitelman’ssyndrome. J Am Soc Nephrol 2002; 13:1442–1448.
23 Starremans PG, Kersten FF, Knoers NV, van den Heuvel LP, Bindels RJ.Mutations in the human Na-K-2Cl cotransporter (NKCC2) identified inBartter syndrome type I consistently result in nonfunctional transporters.J Am Soc Nephrol 2003; 14:1419–1426.
24 Gimenez I, Forbush B. Regulatory phosphorylation sites in the N-terminusof the renal Na-K-Cl cotransporter (NKCC2). Am J Physiol Renal Physiol2005; 289:F1341–F1345.
rized reproduction of this article is prohibited.

C
CE: Namrta; HJH/202096; Total nos of Pages: 9;
HJH 202096
Characterization of rare NCC and NKCC2 mutants Acuna et al. 9
25 De Jong JC, Willems PH, van den Heuvel LP, Knoers NV, Bindels RJ.Functional expression of the human thiazide-sensitive NaCl cotransporter inMadin-Darby canine kidney cells. J Am Soc Nephrol 2003; 14:2428–2435.
26 Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, CampbellDG, et al. Activation of the thiazide-sensitive Naþ-Cl- cotransporter by theWNK-regulated kinases SPAK and OSR1. J Cell Sci 2008; 121:675–684.
27 Sabath E, Meade P, Berkman J, De Los Heros P, Moreno E, Bobadilla NA,et al. Pathophysiology of functional mutations of the thiazide-sensitiveNa-Cl cotransporter in Gitelman disease. Am J Physiol Renal Physiol2004; 287:F195–F203.
28 Pacheco-Alvarez D, San Cristobal P, Meade P, Moreno E, Vazquez N,Munoz E, et al. The Na-Cl cotransporter is activated and phosphorylated atthe amino terminal domain upon intracellular chloride depletion. J BiolChem 2006; 281:28755–28763.
29 Moreno E, Tovar-Palacio C, De Los Heros P, Guzman B, Bobadilla NA,Vazquez N, et al. A single nucleotide polymorphism alters the activity of therenal Naþ:Cl- cotransporter and reveals a role for transmembrane segment4 in chloride and thiazide affinity. J Biol Chem 2004; 279:16553–16560.
30 Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ.Processing of mutant cystic fibrosis transmembrane conductanceregulator is temperature-sensitive. Nature 1992; 358:761–764.
31 De Los Heros P, Kahle KT, Rinehart J, Bobadilla NA, Vazquez N, SanCristobal P, et al. WNK3 bypasses the tonicity requirement for K-Clcotransporter activation via a phosphatase-dependent pathway. Proc NatlAcad Sci U S A 2006; 103:1976–1981.
32 Kahle KT, Rinehart J, De Los Heros P, Louvi A, Meade P, Vazquez N, et al.WNK3 modulates transport of Cl- in and out of cells: implications forcontrol of cell volume and neuronal excitability. Proc Natl Acad Sci U S A2005; 102:16783–16788.
opyright © Lippincott Williams & Wilkins. Unauth
33 Gamba G. Molecular physiology and pathophysiology of the electroneutralcation-chloride cotransporters. Physiol Rev 2005; 85:423–493.
34 Hughes AD. How do thiazide and thiazide-like diuretics lowerblood pressure? J Renin Angiotensin Aldosterone Syst 2004;5:155–160.
35 Pressler CA, Heinzinger J, Jeck N, Waldegger P, Pechmann U, Reinalter S,et al. Late-onset manifestation of antenatal Bartter syndrome as a result ofresidual function of the mutated renal Naþ-Kþ-2Cl- co-transporter. J AmSoc Nephrol 2006; 17:2136–2142.
36 Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, et al.The complete genome of an individual by massively parallel DNAsequencing. Nature 2008; 452:872–876.
37 Bodmer W, Bonilla C. Common and rare variants in multifactorialsusceptibility to common diseases. Nat Genet 2008; 40:695–701.
38 Arora P, Newton-Cheh C. Blood pressure and human geneticvariation in the general population. Curr Opin Cardiol 2010; 25:229–237.
39 Wang Y, O’Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, et al.Whole-genome association study identifies STK39 as a hypertensionsusceptibility gene. Proc Natl Acad Sci U S A 2009; 106:226–231.
40 Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanovic S, et al.Role of the WNK-activated SPAK kinase in regulating blood pressure.EMBO Mol Med 2010; 2:63–75.
41 Lifton RP. Individual genomes on the horizon. N Engl J Med 2010;362:1235–1236.
42 Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Geneticdiagnosis by whole exome capture and massively parallel DNA sequencing.Proc Natl Acad Sci U S A 2009; 106:19096–19101.
orized reproduction of this article is prohibited.




















