
Rapid detection of neutralizing antibodies against bovine viral diarrhoea virus using quantitative...
8
Journal of Virological Methods 198 (2014) 56–63 Contents lists available at ScienceDirect Journal of Virological Methods jou rn al hom epage: www.elsevier.com/locate/jviromet Rapid detection of neutralizing antibodies against bovine viral diarrhoea virus using quantitative high-content screening Michael Eschbaumer ∗ , Sampson Law, Cristina Solis, Adam Chernick, Frank van der Meer, Markus Czub Faculty of Veterinary Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4N1, Canada Article history: Received 19 September 2013 Received in revised form 26 November 2013 Accepted 17 December 2013 Available online 2 January 2014 Keywords: BVDV Neutralization assay IN Cell Analyzer High-throughput Dose–response curves Nonlinear regression a b s t r a c t Bovine viral diarrhoea virus (BVDV) is an important cause of morbidity, mortality and economic losses in cattle worldwide. Humoral immunity to BVDV plays a major role in the protection against infection and disease. In vitro serum neutralization tests can quantify humoral responses, but standard protocols are time-consuming and labour-intensive. The objective of this study was to develop a highly sensitive assay based on high-content cell-by-cell screening that is faster and less subjective than the conventional protocols. It can detect a neutralizing antibody response within the first week after infection of an animal, takes less than 24 h to complete and excludes operator bias by automated data acquisition and analysis. © 2014 Elsevier B.V. All rights reserved. 1. Introduction Bovine viral diarrhoea virus (BVDV) is an important cause of morbidity, mortality and economic losses in dairy and beef cattle worldwide (Maclachlan and Dubovi, 2010). BVDV is an umbrella term for two distinct virus species, BVDV1 and BVDV2, within the family Flaviviridae, genus Pestivirus (Ridpath, 2008). Both species are divided further into several subgroups based on nucleotide sequence similarities (Flores et al., 2002; Vilˇ cek et al., 2004). With reference to their behaviour in cell culture, BVDV strains are separated into cytopathic (cp) and non-cytopathic (ncp) bio- types (Ridpath, 2008). Independent of the biotype or strain, acute BVDV infection can lead to enteric, respiratory or reproductive Abbreviations: BVDV, bovine viral diarrhoea virus; CCD, charge-coupled device; cp, cytopathic; DAPI, 4 ,6-diamidino-2-phenylindole; ELISA, enzyme-linked immunosorbent assay; FDA, Food and Drug Administration; FITC, fluorescein iso- thiocyanate; HCS, high-content screening; ID, infectious dose; mAb, monoclonal antibody; MDBK, Madin-Darby bovine kidney; NA, neutralizing antibody-bodies; ncp, non-cytopathic; ND, neutralizing dose; OIE, World Organisation for Animal Health; PBS, phosphate-buffered saline; PI, persistently infected; RT-PC, Rreverse transcription polymerase chain reaction; SD, standard deviation; TCID, tissue culture infectious dose. ∗ Corresponding author. Present address: Foreign Animal Disease Research Unit, USDA/ARS Plum Island Animal Disease Center, P.O. Box 848, Greenport NY 11944, United States. Tel.: +1 631 323 3368. E-mail addresses: [email protected], [email protected] (M. Eschbaumer). disease and is accompanied by immune suppression and secondary infections. Ncp strains can also establish persistent infections with immune tolerance in unborn calves. In persistently infected (PI) animals, ncp BVDV can mutate into antigenically identical cp BVDV and cause fatal mucosal disease (Ridpath, 2008). Humoral immunity plays a major role in protection against BVDV. Presence of neutralizing antibodies in blood correlates with protection afforded by BVDV vaccination (Saliki and Dubovi, 2004). This protection can be conferred to immunologically naïve animals through passive transfer of maternal antibodies (Howard et al., 1989; Shope et al., 1976). The main target of neutralizing anti- bodies (NA) is the highly immunogenic BVDV glycoprotein E2, which shows considerable variation among strains (Deregt et al., 1998). Serum NA titres are a useful indicator of vaccine efficacy (Saliki and Dubovi, 2004). Neutralization assays provide a means of assessing this protective response in an in vitro test (Onions, 1983). The Manual of Diagnostic Tests and Vaccines of the World Organisation for Animal Health (OIE) sets the standard for a BVDV microneutralization test (OIE, 2012), and similar protocols have been in use for decades (Gillespie et al., 1961). The test is straight- forward: a known quantity of virus is incubated with a serial dilution of the diagnostic specimen and then brought in contact with susceptible indicator cells. If virus replication causes visible damage to the cells, the test can be read directly; otherwise, viral antigen in infected cells must be visualized by a specific immune stain (OIE, 2012). 0166-0934/$ – see front matter © 2014 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.jviromet.2013.12.017
Transcript of Rapid detection of neutralizing antibodies against bovine viral diarrhoea virus using quantitative...

Rd
MFF
ARR2AA
KBNIHDN
1
mwtfasWatB
ditanHti
UU
(
0h
Journal of Virological Methods 198 (2014) 56– 63
Contents lists available at ScienceDirect
Journal of Virological Methods
jou rn al hom epage: www.elsev ier .com/ locate / jv i romet
apid detection of neutralizing antibodies against bovine viraliarrhoea virus using quantitative high-content screening
ichael Eschbaumer ∗, Sampson Law, Cristina Solis, Adam Chernick,rank van der Meer, Markus Czub
aculty of Veterinary Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4N1, Canada
rticle history:eceived 19 September 2013eceived in revised form6 November 2013ccepted 17 December 2013vailable online 2 January 2014
a b s t r a c t
Bovine viral diarrhoea virus (BVDV) is an important cause of morbidity, mortality and economic lossesin cattle worldwide. Humoral immunity to BVDV plays a major role in the protection against infectionand disease. In vitro serum neutralization tests can quantify humoral responses, but standard protocolsare time-consuming and labour-intensive. The objective of this study was to develop a highly sensitiveassay based on high-content cell-by-cell screening that is faster and less subjective than the conventionalprotocols. It can detect a neutralizing antibody response within the first week after infection of an animal,takes less than 24 h to complete and excludes operator bias by automated data acquisition and analysis.
eywords:VDVeutralization assay
N Cell Analyzerigh-throughput
© 2014 Elsevier B.V. All rights reserved.
ose–response curvesonlinear regression
. Introduction
Bovine viral diarrhoea virus (BVDV) is an important cause oforbidity, mortality and economic losses in dairy and beef cattleorldwide (Maclachlan and Dubovi, 2010). BVDV is an umbrella
erm for two distinct virus species, BVDV1 and BVDV2, within theamily Flaviviridae, genus Pestivirus (Ridpath, 2008). Both speciesre divided further into several subgroups based on nucleotideequence similarities (Flores et al., 2002; Vilcek et al., 2004).
ith reference to their behaviour in cell culture, BVDV strains
re separated into cytopathic (cp) and non-cytopathic (ncp) bio-ypes (Ridpath, 2008). Independent of the biotype or strain, acuteVDV infection can lead to enteric, respiratory or reproductiveAbbreviations: BVDV, bovine viral diarrhoea virus; CCD, charge-coupledevice; cp, cytopathic; DAPI, 4′ ,6-diamidino-2-phenylindole; ELISA, enzyme-linked
mmunosorbent assay; FDA, Food and Drug Administration; FITC, fluorescein iso-hiocyanate; HCS, high-content screening; ID, infectious dose; mAb, monoclonalntibody; MDBK, Madin-Darby bovine kidney; NA, neutralizing antibody-bodies;cp, non-cytopathic; ND, neutralizing dose; OIE, World Organisation for Animalealth; PBS, phosphate-buffered saline; PI, persistently infected; RT-PC, Rreverse
ranscription polymerase chain reaction; SD, standard deviation; TCID, tissue culturenfectious dose.∗ Corresponding author. Present address: Foreign Animal Disease Research Unit,SDA/ARS Plum Island Animal Disease Center, P.O. Box 848, Greenport NY 11944,nited States. Tel.: +1 631 323 3368.
E-mail addresses: [email protected], [email protected]. Eschbaumer).
166-0934/$ – see front matter © 2014 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.jviromet.2013.12.017
disease and is accompanied by immune suppression and secondaryinfections. Ncp strains can also establish persistent infections withimmune tolerance in unborn calves. In persistently infected (PI)animals, ncp BVDV can mutate into antigenically identical cp BVDVand cause fatal mucosal disease (Ridpath, 2008).
Humoral immunity plays a major role in protection againstBVDV. Presence of neutralizing antibodies in blood correlates withprotection afforded by BVDV vaccination (Saliki and Dubovi, 2004).This protection can be conferred to immunologically naïve animalsthrough passive transfer of maternal antibodies (Howard et al.,1989; Shope et al., 1976). The main target of neutralizing anti-bodies (NA) is the highly immunogenic BVDV glycoprotein E2,which shows considerable variation among strains (Deregt et al.,1998).
Serum NA titres are a useful indicator of vaccine efficacy(Saliki and Dubovi, 2004). Neutralization assays provide a meansof assessing this protective response in an in vitro test (Onions,1983). The Manual of Diagnostic Tests and Vaccines of the WorldOrganisation for Animal Health (OIE) sets the standard for a BVDVmicroneutralization test (OIE, 2012), and similar protocols havebeen in use for decades (Gillespie et al., 1961). The test is straight-forward: a known quantity of virus is incubated with a serialdilution of the diagnostic specimen and then brought in contact
with susceptible indicator cells. If virus replication causes visibledamage to the cells, the test can be read directly; otherwise, viralantigen in infected cells must be visualized by a specific immunestain (OIE, 2012).
Virolo
mivpsoarlp
sttp
2
2
i(ieao(t4ohf
l3wc−nAA
Caab
2
blTuwaocM(peK
M. Eschbaumer et al. / Journal of
Commonly, reading a serum neutralization test requires manualicroscopic examination, which is time-consuming and labour-
ntensive (Huang et al., 2010). Manual test evaluation based onisual readout is subjective, requires considerable skill and putsractical limits on the number of events that can be counted perample (Abai et al., 2007; Johnson et al., 2008). Another drawbackf conventional protocols is the long turnaround time. It often takes
week from the arrival of a sample in the laboratory until the testesults are available. This is due largely to the long incubation fol-owing the addition of the indicator cells – for example, the OIErotocol recommends 4–5 days (OIE, 2012).
The objective of this study was to develop an improved BVDVerum neutralization test using a high-content screening (HCS) sys-em, where that incubation time is reduced drastically and manualest evaluation is replaced by computerized image analysis and datarocessing.
. Materials and methods
.1. Serum samples
Fifteen 6-month-old BVDV-free beef calves were inoculatedntranasally with 1.6 × 107 median tissue culture infectious dosesTCID50) of BVDV2 subgenotype 2a strain “1373”, which had beensolated from an outbreak in Ontario, Canada, in the 1990s (Carmant al., 1998; Stoffregen et al., 2000). The initial BVDV antigen-nd antibody-negative status of all calves as well as the successf the infection was confirmed by real-time quantitative RT-PCRVetMAXTM-Gold BVDV Detection Kit, catalogue no. 4413938, lifeechnologies, Carlsbad, CA, USA) and ELISA (BVDV Total Ab Test, 99-4000, IDEXX, Westbrook, ME, USA) (data not shown). The calvesriginated from a closed herd that has been free of BVDV and bovineerpesvirus for generations. Ten other animals (calves N1–N10)
rom the same herd were used as negative controls.For five infected calves (calves 1–5), blood samples were col-
ected 7 days before (day −7) as well as 1, 3, 5, 7, 9, 11, 14 and0 days after infection. The other ten infected calves (calves 6–15)ere bled on days −7, 7, 9 and 14 only. All samples from all infected
alves were tested in the HCS assay, but only samples taken on days7, 7, 9 and 14 were tested with the standard OIE protocol. The tenegative control animals were bled once and tested in both assays.ll animal procedures were approved by the University of Calgary’snimal Care and Use Committee (permit no. AC12-143).
For further validation, six field samples (from farms in Alberta,anada; permit no. SHC10R-16) that had been positive in the BVDVntibody ELISA (BVDV Total Ab Test, IDEXX; data not shown) werelso tested in both neutralization assays. All sera were inactivatedy incubation at 56 ◦C for 1 h and stored at −20 ◦C until testing.
.2. Virus stocks and titrations
For the in-vitro tests, BVDV2a 1373 was grown in Madin-Darbyovine kidney (MDBK) cells (CCL-22, American Type Culture Col-
ection, Manassas, VA, USA), then aliquoted and stored at −80 ◦C.he infectivity of the frozen virus stock was determined beforese in the neutralization tests. Different dose finding methodsere used for the standard OIE protocol and the HCS assay and
re described in detail later on. Briefly, serial ten-fold dilutionsf the virus stock were prepared in triplicate on 96-well tissueulture plates (353075, BD Biosciences, Mississauga, ON, Canada),DBK cells were added and the plates were incubated for 96 h
standard OIE protocol) or 16 h (HCS assay). For the standard OIErotocol, the median tissue culture infectious dose (TCID50/96h) wasstimated with the Spearman–Kaerber formula (Spearman, 1908;ärber, 1931), corresponding to the reciprocal of the virus dilution
gical Methods 198 (2014) 56– 63 57
that leads to infection in 50% of replicate wells after a 96-h incuba-tion. For the HCS assay, the 90% infectious dose (ID90/16h), i.e. thereciprocal of the virus dilution that leads to 90% infected cells perwell at 16 h, was estimated by nonlinear regression (see Section2.6).
2.3. Serum neutralization tests
All neutralization tests were performed in triplicate on 96-welltissue culture plates. The plate layout was identical for the standardOIE protocol and the HCS assay; they differed only in the amountsof virus and cells that were used.
Serial fourfold dilutions of the serum samples were made intissue culture media (Dulbecco’s modified Eagle medium sup-plemented with 5% horse serum, 100 U/mL penicillin, 100 �g/mLstreptomycin and 0.25 �g/mL amphotericin B; 11995, 16050-130, 15240-062, life technologies). From an initial dilution of1/2 (0.5 × 40), the sera were diluted in seven steps to 1/32768(0.5 × 4−7), in a final volume of 75 �L. An identical volume of BVDV21373 diluted from the original stock was added to each serum dilu-tion. Each neutralization plate also contained 12 wells with virusbut without serum (no-serum controls) and 12 wells with neithervirus nor serum (no-virus controls).
The virus dose added to the serum dilutions differed betweenthe standard OIE protocol and the HCS assay. As suggested in theOIE manual, the standard protocol used 100 TCID50/96h, estimatedfrom a virus titration that had been incubated for 96 h. The HCSassay, on the other hand, used a higher dose, 1 ID90/16h, estimatedfrom a 16-h titration, corresponding to the shorter incubation timein the assay itself. After serum and virus had been mixed, the plateswere left at 37 ◦C for 1 h before cells were added. In the standardOIE protocol, 1 × 104 cells per well were added and the plates wereincubated for another 96 h. The HCS assay used 4 × 104 cells perwell and a 16-h incubation.
Sixteen or 96 h after the cells had been added, the plateswere washed once with phosphate-buffered saline (PBS; 137 mMNaCl, 2.7 mM KCl, 6.5 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4) andfixed for 30 min at room temperature in 4% (w/v) paraformalde-hyde (158127, Sigma–Aldrich, Oakville, ON, Canada) in PBS. Afteranother wash with PBS, all wells were filled with 100 �L PBS andthe plates were stored at 4 ◦C until staining.
2.4. Immunostaining
Fixed cells were incubated for 1 h at 37 ◦C with a monoclonalantibody (mAb) against the viral E2 glycoprotein (348, VMRD, Pull-man, WA, USA) (Deregt et al., 1998), diluted 1/1000 in PBS with0.1% (w/v) saponin (47036, Sigma–Aldrich) and 0.1% (w/v) bovineserum albumin (A7906, Sigma–Aldrich). This was followed by incu-bation with a secondary antibody conjugated to a fluorescent dye(Alexa Fluor® 488 goat anti-mouse IgG; A11019, life technologies)under the same conditions. Cells were washed three times with PBSwith 0.1% (v/v) Polysorbate 20 (TWEEN® 20; P1379, Sigma–Aldrich)between steps. Finally, cell nuclei were stained with 600 nM 4′,6-diamidino-2-phenylindole (DAPI; D3571, life technologies) in PBSfor 5 min at room temperature in the dark. The DAPI overlay wasremoved, wells were filled with 100 �L PBS and plates were storedat 4 ◦C until image acquisition.
2.5. Image acquisition and analysis
The plates were read by an IN Cell Analyzer 2000 automated
microscope (GE Healthcare, Little Chalfont, United Kingdom).Using a high-performance CCD camera (1040 × 1392 pixels,6.4 �m × 6.4 �m pixel size) and a Nikon 4×/0.20 plan apochromatobjective, one centred field of view covering 12.5% of the total
58 M. Eschbaumer et al. / Journal of Virological Methods 198 (2014) 56– 63
Fig. 1. Establishing criteria to determine the BVDV infection status of cells after a 16-h incubation. MDBK cells in 96-well plates were stained for BVDV glycoprotein E2(mAb 348/Alexa Fluor® 488; green) and nuclei were counterstained with DAPI (blue). Each plate contained negative and positive control wells (see panels A/B and C/D forrandomly chosen fields of view and the corresponding histograms of cell intensities). Panel B shows how the positive threshold is set based on the cell intensities in thenegative control wells. The histogram in panel D shows the events (cells) counted in the field of view in panel C (negatives in red, positives in green). Panel E is an enlargedsection of that field of view and in panel F, positive and negative cells (marked with “1” and “0”, respectively; colours as in D) have been differentiated automatically by theI terprew
abIsaif
rot1iiiboi
uactw
N Cell Investigator software using the threshold intensity shown in panel B. (For ineb version of this article.)
rea was evaluated in each well. On plates that had been incu-ated for 16 h, each field of view contained roughly 4000 cells.
mages were taken with two different excitation/emission filterets (350/455 nm for DAPI and 490/525 nm [fluorescein isothiocy-nate, FITC] for Alexa Fluor® 488). See Fig. 1A and C for samplemages. With one field of view per well, acquiring the images for aull 96-well plate takes less than 10 min.
The first step of the analysis is image segmentation, which sepa-ates objects of interest from the background as well as from eachther. Acquired images were processed with the IN Cell Inves-igator software (IN Cell Analyzer 1000 Workstation [3.7, build461], GE Healthcare). Cell nuclei were identified in the DAPI
mages by “top-hat” segmentation (Meyer, 1979), a method thats suitable for objects of uniform shape and size. In the FITCmages, the corresponding cytoplasmic regions were delineatedy a “global threshold” method (Pratt, 1978), which works bestn high-contrast images with even background intensity and candentify objects independent of their shape.
Infected, BVDV-E2-positive (Alexa-Fluor®-488-stained) andninfected, E2-negative (unstained) cells were differentiated by the
verage intensity of their cytoplasmic fluorescence. The no-virusontrols were analyzed first. Based on their fluorescence intensi-ies, a threshold value that separates infected from uninfected cellsas set as shown in Fig. 1B.tation of the references to colour in this figure legend, the reader is referred to the
The simple manual threshold adjustment used here (Fig. 1B)is possible as long as the cytoplasmic intensities in negative cellsare distributed normally and their intensity is distinctly lower thanthat of positive cells (high signal/noise ratio). Alternatively, thethreshold can be defined by following the “empirical rule” (Pecket al., 2009, p. 180). For example, it can be set to the mean (forpopulations distributed normally) or the mode (for populations dis-tributed non-symmetrically) of the cytoplasmic intensities in theno-virus control plus 2 or 3 standard deviations, but that was notdone in this study.
Cells whose average cytoplasmic intensity lies above the thresh-old are considered positive (see Fig. 1D and F). At the cell seedingdensities used in the HCS assay, the automatic analysis of theimages from a full 96-well plate takes less than 10 min. The INCell Investigator software exports the analysis results (cell countsand percentages of positive vs. negative cells for each well) in amachine-readable format (Microsoft Excel) for further processing.Raw images are stored for future reference.
The images presented in Fig. 1 were optimized first by adjus-ting brightness and contrast globally, then converted from 16-bit
to 8-bit using ImageJ 1.46r (U.S. National Institutes of Health,Bethesda, MD, USA). Subsequently, individual grayscale imageswere imported into the blue (DAPI filter) and green (FITC filter)channels of a blank RGB image file using Adobe Photoshop 9.0
Virolo
(i
2
e9pitd
tpi(uc(eef
rEee
t“pitw
2t
tkp
ponUatss
erTss
3
3
EM
M. Eschbaumer et al. / Journal of
Adobe Systems Inc., San Jose, CA, USA) to recreate a two-colourmage (Paddock et al., 1997).
.6. Test evaluation
A percentage of infected vs. uninfected cells was recorded forach well on the plates. For plates that had been incubated for6 h, the Spearman–Kaerber formula was used to estimate 50% end-oints, as suggested by the OIE. Wells were scored as negative (0%
nfected cells) or positive (≥1% infected cells). The estimated neu-ralizing doses (ND50) correspond to the reciprocal of the serumilution where 50% of the replicate wells were negative.
After 16 h of incubation in the HCS assay, neutralizing activi-ies were calculated by comparing the percentage of infected cellser well for a given sample dilution to the mean percentage of
nfected cells in the no-serum control wells on the same platereferred to as “%reduction”). Dose–response curves were fittedsing a four-parameter log-logistic model. The asymptotes wereonstrained to the range of plausible values (0–100% reduction)Abai et al., 2007; Herman et al., 2008). Among the available mod-ls, the constrained four-parameter log-logistic model was selectedmpirically, because it provided the narrowest confidence intervalsor the dose estimates (Findlay and Dillard, 2007).
With the fitted curves, 50% neutralizing doses (ND50; i.e., theeciprocal of the serum dilution that caused a 50% reduction in2-positive cells compared to the no-serum control at 16 h) werestimated for each sample. Estimates smaller than 1 were consid-red implausible and rejected.
All model fittings and calculations were done in the free R sta-istical environment (version 3.0.1) (R Core Team, 2013), using thedrc” package (version 2.3-7) (Ritz and Streibig, 2005). See the sup-lemental material for the R script that was used to create the plots
n Fig. 3 (S1), as well as a sample data file (S2). Using this script, neu-ralization data from a full 96-well plate can be analyzed in secondsithout further user input.
.7. Estimation of sensitivity, specificity and agreement betweenests
Overall, 60 samples (taken on days −7, 7, 9 and 14) from fif-een calves that had been infected experimentally, 10 samples fromnown BVDV-negative calves as well as 6 ELISA-positive field sam-les were tested in both assays.
Based on the known infection status of the experimental sam-les, the sensitivity and specificity of each assay, as well as theirverall percentage agreement, positive percent agreement andegative percent agreement were calculated as suggested by the.S. Food and Drug Administration (FDA, 2007). All samples takenfter BVDV infection (on days 7, 9 and 14) were assumed to con-ain specific NA. Due to their a priori unknown NA status, the fieldamples were not used for calculating the absolute sensitivity andpecificity, but only for assessing the agreement between the tests.
For days 9 and 14, the agreement between the two assays wasxamined further by calculating the Pearson product-moment cor-elation coefficient (Soper et al., 1917) using R (R Core Team, 2013).he coefficients were calculated separately for each day to con-erve sample independence, and the correlation was consideredignificant for p-values <0.05.
. Results
.1. MDBK cells infected with BVDV2 express E2 glycoprotein
Cells were fixed and stained after 16 h. The viral glycoprotein2 was found predominantly in the cytoplasmic compartment ofDBK cells (Fig. 1E). Earlier time points (i.e., incubation times
gical Methods 198 (2014) 56– 63 59
shorter than 16 h) have been evaluated, but these did not lead tosatisfactory staining intensities (data not shown). Uninfected cells(no-virus controls) had a low mean fluorescence intensity (Fig. 1Aand B) that was typically 2-fold lower than in positive cells (Fig. 1Cand D).
Fig. 1B shows the histogram of the fluorescence intensity ofall 40,731 cells in the 12 negative control wells on an exemplaryplate. The threshold was set manually to an intensity level aboveall events observed in the negative controls (here: 555 [arbitraryunits]). When the remainder of the plate was analyzed, all cellswith cytoplasmic intensity above the threshold were consideredE2-positive by the software (Fig. 1D and F).
3.2. Virus titration
Two measures of infectivity were estimated for the virus stock:the dilution at which 90% of cells were infected per well after 16 h(1/6.3, corresponding to 6.3 ID90/16h in 75 �L of the undiluted stock),and the dilution at which 50% of replicate wells were infected after96 h (1/104.67, or 4.7 × 104 TCID50/96h per 75 �L).
Accordingly, the virus stock was diluted 1/6.3 for use in the HCSassay, and 1/470 for use in the standard OIE protocol. In absoluteterms, the amount of virus per well used in the standard OIE pro-tocol (100 TCID50/96h per well) was considerably lower than in theHCS assay (1 ID90/16h), with the same stock being diluted 75-foldmore.
3.3. The HCS assay collects continuous data with high resolution
After 96 h of incubation (as per the standard OIE protocol),the plates showed a quantal (“all-or-none”) response. Wells wereeither infected completely (virtually all cells E2-positive) or not atall (no positive cells) (Fig. 2A). Accordingly, the Spearman–Kaerberformula was used to estimate 50% endpoints of neutralization. After16 h of incubation, the plates displayed a graded response (Fig. 2B).A percentage of infected vs. uninfected cells was recorded for eachwell and 50% neutralization endpoints were estimated using fittedcurves (see also Fig. 3).
In the example shown in Fig. 2, the standard assay detected neu-tralization only in the day 14 sample (with a titre of 1/645), but notin samples taken on day 7 or 9. The HCS assay detected neutral-ization in all three samples, with titres of 1/4.9, 1/25.2 and 1/9923,respectively.
3.4. BVDV-neutralizing activity increases exponentially over time
The ten uninfected calves were negative in both assays.Beginning on day 3 after infection, the HCS assay detected dose-dependent neutralizing activity in infected animals (Fig. 3). Theobserved neutralization increased exponentially after day 7 (Fig. 4).
3.5. Comparison of neutralization results from the standard OIEprotocol and the HCS assay
For calves 1–5, samples taken on days −7, 1, 3, 5, 7, 9, 11, 14and 30 after infection were tested in the HCS assay (see Fig. 4 forthe ND50 estimates). For comparison, the standard assay results forthese calves (for samples taken on days −7, 7, 9 and 14) are alsoshown in the figure. The standard OIE protocol detected NA laterand with lower magnitude than the HCS assay.
The absolute specificity and sensitivity of both assays was cal-
culated based on the known infection status of the experimentalsamples. Both tests had a specificity of 100% (25 of 25 NA-negativesamples were identified correctly). The sensitivity varied depend-ing on the time after infection when the samples had been taken,
60 M. Eschbaumer et al. / Journal of Virological Methods 198 (2014) 56– 63
Fig. 2. Comparison of neutralization plates incubated for 16 or 96 h. Images are adapted from screenshots from the IN Cell Investigator software. (A) Neutralization plate,9 alf 2 (c fectes out an
w(
ant(
4
uahsonfslemai
were possible only by flow cytometry (Cosma et al., 2004), whichhas the additional benefit of allowing a specific neutralization read-out after a short incubation of less than 24 h (Sashihara et al., 2009;
Table 1Absolute sensitivity of the assays. Based on the known infection status of samplesfrom an animal experiment, the sensitivity of each assay was calculated for samplestaken on days 7, 9 and 14 after infection. See S3 in the supplemental material forindividual results.
Assay Day Correctly identified positive samples (n/n) Sensitivity (%)
Standard 7 0/15 0.0Standard 9 9/15 60.0
6-hour incubation (standard OIE protocol). The plate contained three samples of controls as described in Section 2.3. Wells are shaded based on the percentage of inhades of grey). (B) Neutralization plate, 16-h incubation (HCS assay). The plate lay
ith the HCS assay showing greater sensitivity at earlier time pointssee Table 1).
Compared to the OIE protocol, the HCS assay showed an over-ll agreement of 72.7%, a positive agreement of 100.0% and aegative agreement of 50.0%. The ND50 estimates obtained withhe two assays showed significant correlation on days 9 and 14r = 0.640/p = 0.010 and r = 0.714/p = 0.003, respectively).
. Discussion
Serological tests can be used to detect exposure of individ-als or herds to BVDV, confirm vaccination protocol compliancend assess vaccine efficacy (Saliki and Dubovi, 2004). For non-aemagglutinating viruses, the neutralization assay is the onlytandard serological test that can measure the biological activityf antibodies in a sample directly (Onions, 1983). It is, however,ot used widely owing to its long turnaround time (days vs. hours
or other tests, e.g. ELISAs) and the need for subjective micro-copic evaluation (Hause et al., 2010; Willems et al., 2011), whicheads to lower precision compared to other assay formats (Gonda
t al., 2012). Microscopic evaluation can be replaced by colouri-etric or luminescent cell viability assays that are suitable forutomated reading and data analysis (Willems et al., 2011), but thiss applicable only for cp viruses and requires long incubation times
days 7, 9 and 14; three identical replicates each) as well as no-serum and no-virusd vs. uninfected cells, from 0% (white) to 100% (black) (5% increments, 20 differentd colour scheme is identical to (A).
for the development of strong cytopathic effects. Independent ofcytopathic effect, virus replication in a neutralization test can bedetected by PCR (Varada et al., 2013), ELISA (Huang et al., 2010) orby using reporter-enzyme-expressing viruses (Manischewitz et al.,2003; Kolokoltsov et al., 2006; Kaku et al., 2012). Like the cell via-bility assays, however, these methods can assess only the bulkresponse on a per-well basis (Sullivan et al., 2012). Until recently,quantitative measurements on large numbers of individual cells
Standard 14 15/15 100.0HCS 7 12/15 80.0HCS 9 15/15 100.0HCS 14 15/15 100.0

M. Eschbaumer et al. / Journal of Virological Methods 198 (2014) 56– 63 61
F s presd ll (50%d
Ctniim
ig. 3. Neutralizing activities of serum samples measured in the HCS assay. The plotilutions are plotted against the reduction in the percentage of infected cells per weata with a constrained four-parameter log-logistic function.
hen et al., 2010). Flow cytometry has been used to assess theotal antibody response to BVDV infection (Beer et al., 2006), but
ot for BVDV serum neutralization tests. The protocol presentedn this study offers the speed and precision of flow cytometry onntact cell monolayers. Like flow cytometry, high-throughput auto-
ated microscopy is capable of compiling well measurements from
ented are for calf 1; calves 2–5 showed similar results. The reciprocals of the serum reduction is marked with a broken horizontal line). Curves have been fitted to the
cell-by-cell metrics performed on thousands of cells, whichincreases the depth of the analysis greatly (Bushway et al., 2008).
Small differences in neutralizing activity can be detected withhigh confidence when many events (i.e., infected vs. non-infectedcells) per well (over 4000 in our example) are evaluated simulta-neously, leading to a greatly increased analytical sensitivity and
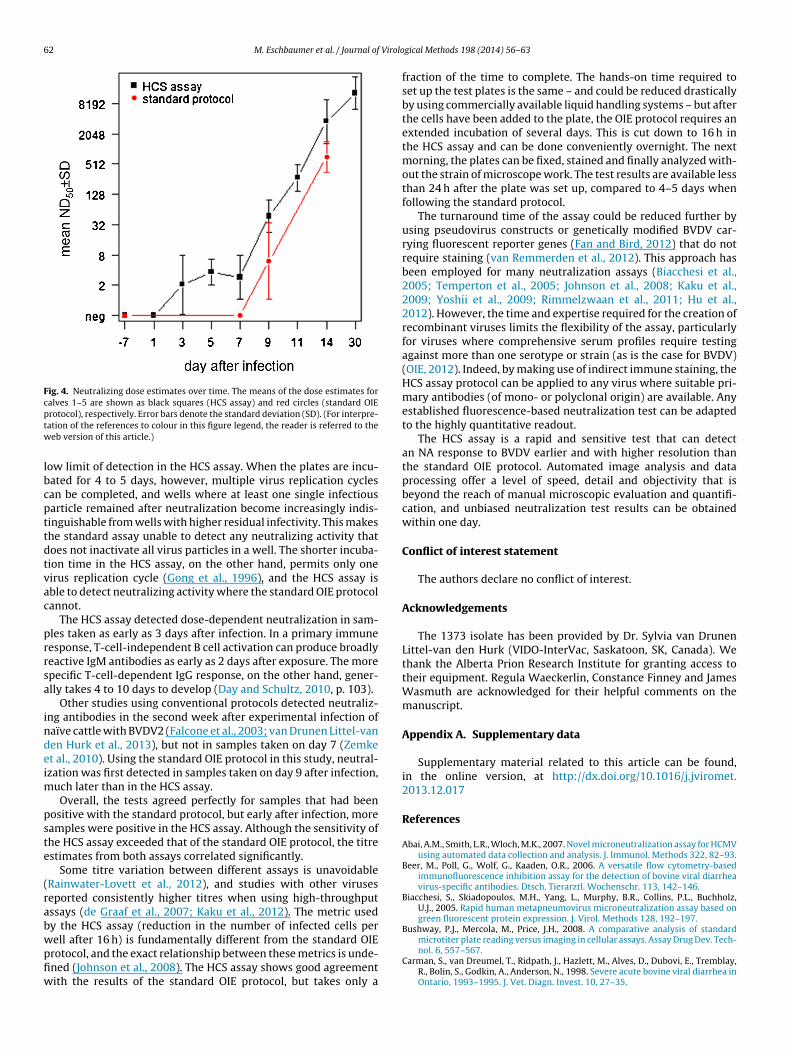
62 M. Eschbaumer et al. / Journal of Virolo
Fig. 4. Neutralizing dose estimates over time. The means of the dose estimates forcalves 1–5 are shown as black squares (HCS assay) and red circles (standard OIEptw
lbcpttdtvac
prrsa
indeim
pste
(rabwpfiw
rotocol), respectively. Error bars denote the standard deviation (SD). (For interpre-ation of the references to colour in this figure legend, the reader is referred to theeb version of this article.)
ow limit of detection in the HCS assay. When the plates are incu-ated for 4 to 5 days, however, multiple virus replication cyclesan be completed, and wells where at least one single infectiousarticle remained after neutralization become increasingly indis-inguishable from wells with higher residual infectivity. This makeshe standard assay unable to detect any neutralizing activity thatoes not inactivate all virus particles in a well. The shorter incuba-ion time in the HCS assay, on the other hand, permits only oneirus replication cycle (Gong et al., 1996), and the HCS assay isble to detect neutralizing activity where the standard OIE protocolannot.
The HCS assay detected dose-dependent neutralization in sam-les taken as early as 3 days after infection. In a primary immuneesponse, T-cell-independent B cell activation can produce broadlyeactive IgM antibodies as early as 2 days after exposure. The morepecific T-cell-dependent IgG response, on the other hand, gener-lly takes 4 to 10 days to develop (Day and Schultz, 2010, p. 103).
Other studies using conventional protocols detected neutraliz-ng antibodies in the second week after experimental infection ofaïve cattle with BVDV2 (Falcone et al., 2003; van Drunen Littel-vanen Hurk et al., 2013), but not in samples taken on day 7 (Zemket al., 2010). Using the standard OIE protocol in this study, neutral-zation was first detected in samples taken on day 9 after infection,
uch later than in the HCS assay.Overall, the tests agreed perfectly for samples that had been
ositive with the standard protocol, but early after infection, moreamples were positive in the HCS assay. Although the sensitivity ofhe HCS assay exceeded that of the standard OIE protocol, the titrestimates from both assays correlated significantly.
Some titre variation between different assays is unavoidableRainwater-Lovett et al., 2012), and studies with other viruseseported consistently higher titres when using high-throughputssays (de Graaf et al., 2007; Kaku et al., 2012). The metric usedy the HCS assay (reduction in the number of infected cells per
ell after 16 h) is fundamentally different from the standard OIErotocol, and the exact relationship between these metrics is unde-ned (Johnson et al., 2008). The HCS assay shows good agreementith the results of the standard OIE protocol, but takes only agical Methods 198 (2014) 56– 63
fraction of the time to complete. The hands-on time required toset up the test plates is the same – and could be reduced drasticallyby using commercially available liquid handling systems – but afterthe cells have been added to the plate, the OIE protocol requires anextended incubation of several days. This is cut down to 16 h inthe HCS assay and can be done conveniently overnight. The nextmorning, the plates can be fixed, stained and finally analyzed with-out the strain of microscope work. The test results are available lessthan 24 h after the plate was set up, compared to 4–5 days whenfollowing the standard protocol.
The turnaround time of the assay could be reduced further byusing pseudovirus constructs or genetically modified BVDV car-rying fluorescent reporter genes (Fan and Bird, 2012) that do notrequire staining (van Remmerden et al., 2012). This approach hasbeen employed for many neutralization assays (Biacchesi et al.,2005; Temperton et al., 2005; Johnson et al., 2008; Kaku et al.,2009; Yoshii et al., 2009; Rimmelzwaan et al., 2011; Hu et al.,2012). However, the time and expertise required for the creation ofrecombinant viruses limits the flexibility of the assay, particularlyfor viruses where comprehensive serum profiles require testingagainst more than one serotype or strain (as is the case for BVDV)(OIE, 2012). Indeed, by making use of indirect immune staining, theHCS assay protocol can be applied to any virus where suitable pri-mary antibodies (of mono- or polyclonal origin) are available. Anyestablished fluorescence-based neutralization test can be adaptedto the highly quantitative readout.
The HCS assay is a rapid and sensitive test that can detectan NA response to BVDV earlier and with higher resolution thanthe standard OIE protocol. Automated image analysis and dataprocessing offer a level of speed, detail and objectivity that isbeyond the reach of manual microscopic evaluation and quantifi-cation, and unbiased neutralization test results can be obtainedwithin one day.
Conflict of interest statement
The authors declare no conflict of interest.
Acknowledgements
The 1373 isolate has been provided by Dr. Sylvia van DrunenLittel-van den Hurk (VIDO-InterVac, Saskatoon, SK, Canada). Wethank the Alberta Prion Research Institute for granting access totheir equipment. Regula Waeckerlin, Constance Finney and JamesWasmuth are acknowledged for their helpful comments on themanuscript.
Appendix A. Supplementary data
Supplementary material related to this article can be found,in the online version, at http://dx.doi.org/10.1016/j.jviromet.2013.12.017
References
Abai, A.M., Smith, L.R., Wloch, M.K., 2007. Novel microneutralization assay for HCMVusing automated data collection and analysis. J. Immunol. Methods 322, 82–93.
Beer, M., Poll, G., Wolf, G., Kaaden, O.R., 2006. A versatile flow cytometry-basedimmunofluorescence inhibition assay for the detection of bovine viral diarrheavirus-specific antibodies. Dtsch. Tierarztl. Wochenschr. 113, 142–146.
Biacchesi, S., Skiadopoulos, M.H., Yang, L., Murphy, B.R., Collins, P.L., Buchholz,U.J., 2005. Rapid human metapneumovirus microneutralization assay based ongreen fluorescent protein expression. J. Virol. Methods 128, 192–197.
Bushway, P.J., Mercola, M., Price, J.H., 2008. A comparative analysis of standard
microtiter plate reading versus imaging in cellular assays. Assay Drug Dev. Tech-nol. 6, 557–567.Carman, S., van Dreumel, T., Ridpath, J., Hazlett, M., Alves, D., Dubovi, E., Tremblay,R., Bolin, S., Godkin, A., Anderson, N., 1998. Severe acute bovine viral diarrhea inOntario, 1993–1995. J. Vet. Diagn. Invest. 10, 27–35.

Virolo
C
C
D
d
D
F
F
F
F
F
G
G
G
H
H
H
H
H
J
K
K
K
K
M. Eschbaumer et al. / Journal of
hen, M., Chang, J.S., Nason, M., Rangel, D., Gall, J.G., Graham, B.S., Ledgerwood, J.E.,2010. A flow cytometry-based assay to assess RSV-specific neutralizing antibodyis reproducible, efficient and accurate. J. Immunol. Methods 362, 180–184.
osma, A., Bühler, S., Nagaraj, R., Staib, C., Hammarin, A.L., Wahren, B., Goebel, F.D.,Erfle, V., Sutter, G., 2004. Neutralization assay using a modified vaccinia virusAnkara vector expressing the green fluorescent protein is a high-throughputmethod to monitor the humoral immune response against vaccinia virus. Clin.Diagn. Lab. Immunol. 11, 406–410.
ay, M.J., Schultz, R.D., 2010. Veterinary Immunology: Principles and Practice, 1sted. CRC Press, Boca Raton, FL, USA.
e Graaf, M., Herfst, S., Schrauwen, E.J., van den Hoogen, B.G., Osterhaus, A.D., Fouch-ier, R.A., 2007. An improved plaque reduction virus neutralization assay forhuman metapneumovirus. J. Virol. Methods 143, 169–174.
eregt, D., Bolin, S.R., van den Hurk, J., Ridpath, J.F., Gilbert, S.A., 1998. Mapping of atype 1-specific and a type-common epitope on the E2 (gp53) protein of bovineviral diarrhea virus with neutralization escape mutants. Virus Res. 53, 81–90.
alcone, E., Cordioli, P., Tarantino, M., Muscillo, M., Sala, G., La Rosa, G., Archetti, I.L.,Marianelli, C., Lombardi, G., Tollis, M., 2003. Experimental infection of calveswith bovine viral diarrhoea virus type-2 (BVDV-2) isolated from a contaminatedvaccine. Vet. Res. Commun. 27, 577–589.
an, Z.C., Bird, R.C., 2012. Development of a reporter bovine viral diarrhea virus andinitial evaluation of its application for high throughput antiviral drug screening.J. Virol. Methods 180, 54–61.
indlay, J.W., Dillard, R.F., 2007. Appropriate calibration curve fitting in ligand bind-ing assays. AAPS J. 9, E260–E267.
lores, E.F., Ridpath, J.F., Weiblen, R., Vogel, F.S., Gil, L.H., 2002. Phylogenetic analysisof Brazilian bovine viral diarrhea virus type 2 (BVDV-2) isolates: evidence for asubgenotype within BVDV-2. Virus Res. 87, 51–60.
DA, 2007. Statistical Guidance on Reporting Results from Studies Evaluating Diag-nostic Tests. Office of Surveillance and Biometrics, Division of Biostatistics,Diagnostic Devices Branch, Center for Devices and Radiological Health, Foodand Drug Administration, U.S. Department of Health and Human Services.Issued on March 13, 2007. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071287.pdf (accessed18.11.13).
illespie, J.H., Coggins, L., Thompson, J., Baker, J.A., 1961. Comparison by neutraliza-tion tests of strains of virus isolated from virus diarrhea and mucosal disease.Cornell Vet. 51, 155–159.
onda, M.G., Fang, X., Perry, G.A., Maltecca, C., 2012. Measuring bovine viral diarrheavirus vaccine response: using a commercially available ELISA as a surrogate forserum neutralization assays. Vaccine 30, 6559–6563.
ong, Y., Trowbridge, R., Macnaughton, T.B., Westaway, E.G., Shannon, A.D., Gowans,E.J., 1996. Characterization of RNA synthesis during a one-step growth curve andof the replication mechanism of bovine viral diarrhoea virus. J. Gen. Virol. 77 (Pt11), 2729–2736.
ause, B.M., Oleson, T.A., Bey, R.F., Stine, D.L., Simonson, R.R., 2010. Antigeniccategorization of contemporary H3N2 Swine influenza virus isolates using ahigh-throughput serum neutralization assay. J. Vet. Diagn. Invest. 22, 352–359.
erman, R.A., Scherer, P.N., Shan, G., 2008. Evaluation of logistic and polynomialmodels for fitting sandwich-ELISA calibration curves. J. Immunol. Methods 339,245–258.
oward, C.J., Clarke, M.C., Brownlie, J., 1989. Protection against respiratory infectionwith bovine virus diarrhoea virus by passively acquired antibody. Vet. Microbiol.19, 195–203.
u, Q., Chen, W., Huang, K., Baron, M.D., Bu, Z., 2012. Rescue of recombinant pestedes petits ruminants virus: creation of a GFP-expressing virus and applicationin rapid virus neutralization test. Vet. Res. 43, 48.
uang, M.L., Chiang, P.S., Luo, S.T., Liou, G.Y., Lee, M.S., 2010. Development ofa high-throughput assay for measuring serum neutralizing antibody againstenterovirus 71. J. Virol. Methods 165, 42–45.
ohnson, M.C., Damon, I.K., Karem, K.L., 2008. A rapid, high-throughput vaccinia virusneutralization assay for testing smallpox vaccine efficacy based on detection ofgreen fluorescent protein. J. Virol. Methods 150, 14–20.
ärber, G., 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenver-suche. Arch. Exp. Path. Pharmak. 162, 480–487.
aku, Y., Noguchi, A., Marsh, G.A., Barr, J.A., Okutani, A., Hotta, K., Bazartseren, B.,Fukushi, S., Broder, C.C., Yamada, A., Inoue, S., Wang, L.F., 2012. Second genera-tion of pseudotype-based serum neutralization assay for Nipah virus antibodies:sensitive and high-throughput analysis utilizing secreted alkaline phosphatase.J. Virol. Methods 179, 226–232.
aku, Y., Noguchi, A., Marsh, G.A., McEachern, J.A., Okutani, A., Hotta, K., Bazartseren,B., Fukushi, S., Broder, C.C., Yamada, A., Inoue, S., Wang, L.F., 2009. A neutral-
ization test for specific detection of Nipah virus antibodies using pseudotypedvesicular stomatitis virus expressing green fluorescent protein. J. Virol. Methods160, 7–13.olokoltsov, A.A., Wang, E., Colpitts, T.M., Weaver, S.C., Davey, R.A., 2006. Pseudo-typed viruses permit rapid detection of neutralizing antibodies in human and
gical Methods 198 (2014) 56– 63 63
equine serum against Venezuelan equine encephalitis virus. Am. J. Trop. Med.Hyg. 75, 702–709.
Maclachlan, N.J., Dubovi, E.J., 2010. Fenner’s Veterinary Virology, 4th ed. AcademicPress, Saint Louis, MO, USA.
Manischewitz, J., King, L.R., Bleckwenn, N.A., Shiloach, J., Taffs, R., Merchlinsky, M.,Eller, N., Mikolajczyk, M.G., Clanton, D.J., Monath, T., Weltzin, R.A., Scott, D.E.,Golding, H., 2003. Development of a novel vaccinia-neutralization assay basedon reporter-gene expression. J. Infect. Dis. 188, 440–448.
Meyer, F., 1979. Iterative image transformations for an automatic screening of cer-vical smears. J. Histochem. Cytochem. 27, 128–135.
OIE, 2012. Bovine Viral Diarrhoea, Manual of Diagnostic Tests and Vaccines for Ter-restrial Animals., pp. 698–711.
Onions, D.E., 1983. The immune response to virus infections. Vet. Immunol.Immunopathol. 4, 237–277.
Paddock, S.W., Hazen, E.J., De Vries, P.J., 1997. Methods and applications of three-color confocal imaging. Biotechniques 22 (120–2), 124–126.
Peck, R., Olsen, C., Devore, J., 2009. Introduction to Statistics & Data Analysis, 3rd ed.Brooks/Cole, Belmont, CA, USA.
Pratt, W.K., 1978. Digital Image Processing. Wiley, New York, NY, USA.R Core Team, 2013. A Language and Environment for Sstatistical Computing. R Foun-
dation for Statistical Computing, Vienna, Austria.Rainwater-Lovett, K., Rodriguez-Barraquer, I., Cummings, D.A., Lessler, J., 2012. Vari-
ation in dengue virus plaque reduction neutralization testing: systematic reviewand pooled analysis. BMC Infect. Dis. 12, 233.
Ridpath, J.F., 2008. Bovine viral diarrhea virus. In: Mahy, B.W.J., van Regenmortel,M.H.V. (Eds.), Encyclopedia of Virology. , 3rd ed. Academic Press, Oxford, pp.374–380.
Rimmelzwaan, G.F., Verburgh, R.J., Nieuwkoop, N.J., Bestebroer, T.M., Fouchier, R.A.,Osterhaus, A.D., 2011. Use of GFP-expressing influenza viruses for the detectionof influenza virus A/H5N1 neutralizing antibodies. Vaccine 29, 3424–3430.
Ritz, C., Streibig, J.C., 2005. Bioassay analysis using R. J. Stat. Softw. 12 (5), 1–22.Saliki, J.T., Dubovi, E.J., 2004. Laboratory diagnosis of bovine viral diarrhea virus
infections. Vet. Clin. North Am. Food Anim. Pract. 20, 69–83.Sashihara, J., Burbelo, P.D., Savoldo, B., Pierson, T.C., Cohen, J.I., 2009. Human antibody
titers to Epstein-Barr Virus (EBV) gp350 correlate with neutralization of infectiv-ity better than antibody titers to EBV gp42 using a rapid flow cytometry-basedEBV neutralization assay. Virology 391, 249–256.
Shope, R.E., Muscoplat, C.C., Chen, A.W., Johnson, D.W., 1976. Mechanism of pro-tection from primary bovine viral diarrhea virus infection. I: The effects ofdexamethasone. Can. J. Comp. Med. 40, 355–359.
Soper, H.E., Young, A.W., Cave, B.M., Lee, A., Pearson, K., 1917. On the distribu-tion of the correlation coefficient in small samples Appendix II to the papersof “Student” and R. A. Fisher. A co-operative study. Biometrika 11, 328–413.
Spearman, C., 1908. The method of ‘right and wrong cases’ (’constant stimuli’) with-out gauss’s formulae. Br. J. Psychol. 2, 227–242.
Stoffregen, B., Bolin, S.R., Ridpath, J.F., Pohlenz, J., 2000. Morphologic lesions in type 2BVDV infections experimentally induced by strain BVDV2-1373 recovered froma field case. Vet. Microbiol. 77, 157–162.
Sullivan, K., Kloess, J., Qian, C., Bell, D., Hay, A., Lin, Y.P., Gu, Y., 2012. High throughputvirus plaque quantitation using a flatbed scanner. J. Virol. Methods 179, 81–89.
Temperton, N.J., Chan, P.K., Simmons, G., Zambon, M.C., Tedder, R.S., Takeuchi, Y.,Weiss, R.A., 2005. Longitudinally profiling neutralizing antibody response toSARS coronavirus with pseudotypes. Emerg. Infect. Dis. 11, 411–416.
van Drunen Littel-van den Hurk, S., Lawman, Z., Snider, M., Wilson, D., van den Hurk,J.V., Ellefsen, B., Hannaman, D., 2013. Two doses of bovine viral diarrhea virusDNA vaccine delivered by electroporation induce long-term protective immuneresponses. Clin. Vaccine Immunol. 20, 166–173.
van Remmerden, Y., Xu, F., van Eldik, M., Heldens, J.G., Huisman, W., Widjojoatmodjo,M.N., 2012. An improved respiratory syncytial virus neutralization assay basedon the detection of green fluorescent protein expression and automated plaquecounting. Virol. J. 9, 253.
Varada, J.C., Teferedegne, B., Crim, R.L., Mdluli, T., Audet, S., Peden, K., Beeler, J.,Murata, H., 2013. A neutralization assay for respiratory syncytial virus using aquantitative PCR-based endpoint assessment. Virol. J. 10, 195.
Vilcek, S., Durkovic, B., Kolesárová, M., Greiser-Wilke, I., Paton, D., 2004. Geneticdiversity of international bovine viral diarrhoea virus (BVDV) isolates: identifi-cation of a new BVDV-1 genetic group. Vet. Res. 35, 609–615.
Willems, T., Lefebvre, D.J., Neyts, J., De Clercq, K., 2011. Diagnostic performance andapplication of two commercial cell viability assays in foot-and-mouth diseaseresearch. J. Virol. Methods 173, 108–114.
Yoshii, K., Ikawa, A., Chiba, Y., Omori, Y., Maeda, J., Murata, R., Kariwa, H.,
Takashima, I., 2009. Establishment of a neutralization test involving reportergene-expressing virus-like particles of tick-borne encephalitis virus. J. Virol.Methods 161, 173–176.Zemke, J., König, P., Mischkale, K., Reimann, I., Beer, M., 2010. Novel BVDV-2 mutantsas new candidates for modified-live vaccines. Vet. Microbiol. 142, 69–80.




















